
Inhibition of 11β-hydroxysteroid dehydrogenase 1 by carbenoxolone affects glucose homeostasis and...
9
Inhibition of 11b-hydroxysteroid dehydrogenase 1 by carbenoxolone affects glucose homeostasis and obesity in db ⁄ db mice Nirav Dhanesha,* Amit Joharapurkar,* Gaurang Shah, † Samadhan Kshirsagar,* Vipin Dhote,* Ajay Sharma* and Mukul Jain* *Department of Pharmacology & Toxicology, Zydus Research Centre, Cadila Healthcare Limited, Moraiya, Ahmedabad and † KB Institute of Pharmaceutical Education and Research, Gandhinagar, Gujarat, India SUMMARY 1. One of the major causes of metabolic syndrome is elevated 11b-hydroxysteroid dehydrogenase 1 (11b-HSD1) in the liver and adipose tissue. High 11b-HSD1 expression contributes signi- ficantly to the diabetic phenotype in db ⁄ db mice. The purpose of the present study was to test the effect of the pharmacological inhibition of 11b-HSD1 inhibition by carbenoxolone in db ⁄ db mice, a genetic model of diabetes. 2. Inhibition of 11b-HSD1 by carbenoxolone was evaluated in liver homogenates obtained from untreated mice. At 0.4, 0.8, 1.6 and 3.2 lmol ⁄ L, carbenoxolone reduced the conversion of corti- sone to cortisol by 21%, 48%, 82% and 95%, respectively. 3. In another series of experiments in which female db ⁄ db mice were dosed orally with carbenoxolone (10, 25 and 50 mg ⁄ kg, twice daily) for 10 days, dose-dependent decreases were observed in 11b-HSD1 activity in the brain, adipose and liver. In the case of 10 mg ⁄ kg carbenoxolone, the effects were not significant. In addition, the bodyweight of female db ⁄ db mice was reduced by 10% and 13% following treatment with 10 and 50 mg ⁄ kg carbe- noxolone, respectively. Carbenoxolone treatment dose-depen- dently improved fat mass, energy expenditure, the serum lipid profile, serum leptin and insulin and glucose tolerance. Further- more, 50 mg ⁄ kg carbenoxolone reduced both phosphoenolpyr- uvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) activity in the liver by 75% and 52%, respectively. These decreases were associated with increased glucokinase protein expression and activity in the liver. 4. Carbenoxolone inhibition of 11b-HSD1 in the liver, adipose and brain significantly improves the symptoms of metabolic syn- drome in db ⁄ db mice. These improvements can be attributed to increased energy expenditure, decreased activity of the gluconeo- genic enzymes PEPCK and G6Pase in the liver and improved glucokinase function in the liver and pancreas. Key words: 11b-hydroxysteroid dehydrogenase 1, adipose, carbenoxolone, glucokinase, glucose homeostasis, liver. INTRODUCTION Glucocorticoids (mainly cortisol in humans and corticosterone in rodents) play a key role in regulating salt and water balance, blood pressure, immune function and metabolism. 1 Glucocorticoids enhance gluconeogenesis by activating key enzymes, such as phos- phoenolpyruvate carboxykinase (PEPCK) and glucose-6-phospha- tase (G6Pase). The in vivo effect of glucocorticoids appears to include both impaired insulin-dependent glucose uptake in the periphery and enhanced gluconeogenesis in the liver, in addition to decreased pancreatic insulin secretion. 2 Glucocorticoid excess, exem- plified by Cushing’s syndrome in humans, leads to insulin resistance and ⁄ or Type 2 diabetes, dyslipidaemia and a redistribution of fat to visceral depots, which is associated with increased cardiovascular risk. 3 These effects of excess cortisol depend, in part, on the opposing the actions of insulin (i.e. inducing a state of insulin resistance). 1 There are strong morphological and metabolic similarities between the rare Cushing’s syndrome due to endogenous or exogenous gluco- corticoid excess and metabolic syndrome. 4 Glucocorticoid receptor (GR) antagonists can ameliorate diabetes and insulin resistance, high- lighting the importance of excessive glucocorticoid action in the pathogenesis of Type 2 diabetes and metabolic syndrome. 5 However, whereas high circulating glucocorticoid levels cause Cushing’s syn- drome, there is little evidence of plasma cortisol excess in individuals with metabolic syndrome. 3,4 The glucocorticoid-activating enzyme 11b-hydroxysteroid dehydro- genase 1 (11b-HSD1) catalyses the conversion of inert 11-dehydro- corticosterone (cortisone in humans) into active corticosterone (cortisol) in vivo. 6 This enzyme is highly expressed in metabolically active tissues, such as the liver and adipose. 7 Transgenic mice overexpressing 11b-HSD1 in adipose tissue exhibit several features of metabolic syndrome, 8 whereas liver-specific overexpression of 11b-HSD1 in transgenic mice results in insulin resistance without obesity. 9 Thus, increased glucocorticoid action within tissues like the liver and adipose, mediated by 11b-HSD1, could explain the similarities between Cushing’s syndrome and metabolic syndrome in the absence of elevated circulating cortisol in humans. Conversely, mice with loss of 11b-HSD1 are protected from diet-induced visceral obesity and its metabolic consequences, probably via a decrease in the regeneration of glucocorticoids. 10,11 Correspondence: Amit Joharapurkar, Department of Pharmacology & Toxi- cology, Zydus Research Centre, Cadila Healthcare Limited, Sarkhej-Bavla NH no. 8A, Moraiya, Ahmedabad 382210, India. Email: amitjoharapur [email protected] Received 12 May 2011; revision 28 October 2011; accepted 3 November 2011. Ó 2011 The Authors Clinical and Experimental Pharmacology and Physiology Ó 2011 Blackwell Publishing Asia Pty Ltd Clinical and Experimental Pharmacology and Physiology (2012) 39, 69–77 doi: 10.1111/j.1440-1681.2011.05640.x
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Inhibition of 11β-hydroxysteroid dehydrogenase 1 by carbenoxolone affects glucose homeostasis and...

Inhibition of 11b-hydroxysteroid dehydrogenase 1 by carbenoxolone affectsglucose homeostasis and obesity in db ⁄dbmice
Nirav Dhanesha,* Amit Joharapurkar,* Gaurang Shah,† Samadhan Kshirsagar,* Vipin Dhote,*Ajay Sharma* and Mukul Jain*
*Department of Pharmacology & Toxicology, Zydus Research Centre, Cadila Healthcare Limited, Moraiya, Ahmedabad and†KB Institute of Pharmaceutical Education and Research, Gandhinagar, Gujarat, India
SUMMARY
1. One of the major causes of metabolic syndrome is elevated11b-hydroxysteroid dehydrogenase 1 (11b-HSD1) in the liverand adipose tissue. High 11b-HSD1 expression contributes signi-ficantly to the diabetic phenotype in db ⁄db mice. The purpose ofthe present study was to test the effect of the pharmacologicalinhibition of 11b-HSD1 inhibition by carbenoxolone in db ⁄dbmice, a genetic model of diabetes.2. Inhibition of 11b-HSD1 by carbenoxolone was evaluated in
liver homogenates obtained from untreated mice. At 0.4, 0.8, 1.6and 3.2 lmol ⁄L, carbenoxolone reduced the conversion of corti-sone to cortisol by 21%, 48%, 82% and 95%, respectively.3. In another series of experiments in which female db ⁄dbmice
were dosed orally with carbenoxolone (10, 25 and 50 mg ⁄kg,twice daily) for 10 days, dose-dependent decreases were observedin 11b-HSD1 activity in the brain, adipose and liver. In the caseof 10 mg ⁄kg carbenoxolone, the effects were not significant. Inaddition, the bodyweight of female db ⁄db mice was reduced by10% and 13% following treatment with 10 and 50 mg ⁄kg carbe-noxolone, respectively. Carbenoxolone treatment dose-depen-dently improved fat mass, energy expenditure, the serum lipidprofile, serum leptin and insulin and glucose tolerance. Further-more, 50 mg ⁄kg carbenoxolone reduced both phosphoenolpyr-uvate carboxykinase (PEPCK) and glucose-6-phosphatase(G6Pase) activity in the liver by 75% and 52%, respectively.These decreases were associated with increased glucokinaseprotein expression and activity in the liver.4. Carbenoxolone inhibition of 11b-HSD1 in the liver, adipose
and brain significantly improves the symptoms of metabolic syn-drome in db ⁄db mice. These improvements can be attributed toincreased energy expenditure, decreased activity of the gluconeo-genic enzymes PEPCK and G6Pase in the liver and improvedglucokinase function in the liver and pancreas.
Key words: 11b-hydroxysteroid dehydrogenase 1, adipose,carbenoxolone, glucokinase, glucose homeostasis, liver.
INTRODUCTION
Glucocorticoids (mainly cortisol in humans and corticosterone inrodents) play a key role in regulating salt and water balance, bloodpressure, immune function and metabolism.1 Glucocorticoidsenhance gluconeogenesis by activating key enzymes, such as phos-phoenolpyruvate carboxykinase (PEPCK) and glucose-6-phospha-tase (G6Pase). The in vivo effect of glucocorticoids appears toinclude both impaired insulin-dependent glucose uptake in theperiphery and enhanced gluconeogenesis in the liver, in addition todecreased pancreatic insulin secretion.2 Glucocorticoid excess, exem-plified by Cushing’s syndrome in humans, leads to insulin resistanceand ⁄or Type 2 diabetes, dyslipidaemia and a redistribution of fat tovisceral depots, which is associated with increased cardiovascularrisk.3 These effects of excess cortisol depend, in part, on the opposingthe actions of insulin (i.e. inducing a state of insulin resistance).1
There are strong morphological and metabolic similarities betweenthe rare Cushing’s syndrome due to endogenous or exogenous gluco-corticoid excess and metabolic syndrome.4 Glucocorticoid receptor(GR) antagonists can ameliorate diabetes and insulin resistance, high-lighting the importance of excessive glucocorticoid action in thepathogenesis of Type 2 diabetes and metabolic syndrome.5 However,whereas high circulating glucocorticoid levels cause Cushing’s syn-drome, there is little evidence of plasma cortisol excess in individualswith metabolic syndrome.3,4
The glucocorticoid-activating enzyme11b-hydroxysteroid dehydro-genase 1 (11b-HSD1) catalyses the conversion of inert 11-dehydro-corticosterone (cortisone in humans) into active corticosterone(cortisol) in vivo.6 This enzyme is highly expressed in metabolicallyactive tissues, such as the liver and adipose.7 Transgenic miceoverexpressing 11b-HSD1 in adipose tissue exhibit several features ofmetabolic syndrome,8 whereas liver-specific overexpression of11b-HSD1 in transgenic mice results in insulin resistance withoutobesity.9 Thus, increased glucocorticoid action within tissues likethe liver and adipose, mediated by 11b-HSD1, could explain thesimilarities between Cushing’s syndrome and metabolic syndrome inthe absence of elevated circulating cortisol in humans. Conversely,mice with loss of 11b-HSD1 are protected from diet-induced visceralobesity and its metabolic consequences, probably via a decrease in theregeneration of glucocorticoids.10,11
Correspondence: Amit Joharapurkar, Department of Pharmacology & Toxi-cology, Zydus Research Centre, Cadila Healthcare Limited, Sarkhej-BavlaNH no. 8A, Moraiya, Ahmedabad 382210, India. Email: [email protected]
Received 12 May 2011; revision 28 October 2011; accepted 3 November2011.
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology� 2011 Blackwell Publishing Asia Pty Ltd
Clinical and Experimental Pharmacology and Physiology (2012) 39, 69–77 doi: 10.1111/j.1440-1681.2011.05640.x

The db ⁄ db mouse is hyperleptinaemic and develops obesity andsevere Type 2 diabetes partly because of a functional defect in theleptin receptor.12 The phenotype of db ⁄db mice includes componentsof metabolic syndrome, including obesity, insulin resistance anddyslipidaemia characterized by hypertriglyceridaemia and hypercho-lesterolaemia. Recent studies have demonstrated that increasedexpression of GR and 11b-HSD1 enzyme in the liver contributes tothe phenotype of the db ⁄ db mouse model.13 Hence, the contributionof 11b-HSD1 inhibition to restoring normal physiology in metabolicsyndrome and diabetes requires further investigation.14
Carbenoxolone, a synthetic hemisuccinyl ester derivative of glyc-yrrhetinic acid, is a potent 11b-HSD1 inhibitor (IC50 110 nmol ⁄L).15
It has been shown to improve hepatic insulin sensitivity in normal16
and diabetic individuals.17 The purpose of the present study was toinvestigate the effects of chronic inhibition of 11b-HSD1 by carbe-noxolone on bodyweight, glucose homeostasis, lipid profile andactivity of key enzymes involved in regulating hepatic glucosemetabolism.
METHODS
Materials
Serum triglycerides, cholesterol and protein concentrations were measuredusing kits purchased from Pointe Scientific (Canton, MI, USA). Serum con-centrations of high-density lipoprotein (HDL) and non-esterified free fattyacids (FFA) were determined using commercially available kits (Randox Lab-oratories, Crumlin, UK). Serum concentrations of insulin and glucose weredetermined using kits purchased from Millipore (Billerica, MA, USA) andRanbaxy Fine Chemicals (New Delhi, India), respectively. Serum concentra-tions of leptin were measured using kits purchased from CrystalChem (Down-ers Grove, IL, USA). Serum potassium was estimated using an autoanalyser(Cobas c system; Roche, Indianapolis, IN, USA). Carbenoxolone disodium,NADPH, cortisol, cortisone, dithiothreitol (DTT), albumin, glucose-6-phos-phate dehydrogenase, malic dehydrogenase and amyloglucosidase werepurchased from Sigma-Aldrich (St Louis, MO, USA). All other reagentsused in the present study were of analytical grade.
Animals
All animal protocols (ZRC ⁄CPCSEA ⁄PH2324–2K8) were approved by theInstitutional Animal Ethics Committee (IAEC) at Zydus Research Centre.Female C57BL ⁄KsJ-obese (db ⁄ db) mice (10–12 weeks old), weighing 40–55g, were obtained from the Central Animal Research Facility of ZydusResearch Centre. Mice were housed in polypropylene cages in temperature(22–28�C)-controlled rooms, with lights on from 0630 to 1930 hours. Allmice had free access to water and standard mice chow (National Institute ofNutrition, Hyderabad, India).
Estimation of 11b-HSD1 inhibition by carbenoxolone in the liver
The activity of 11b-HSD1 was determined using the methods of Horigomeet al.18 with some modification. Livers were collected from untrreated db ⁄ dbmice after perfusion with phosphate-buffered saline (PBS; pH 7.4). Approxi-mately 300 g tissue was homogenized in PBS (containing 50 lg ⁄mL corti-sone and 100 lmol ⁄L NADPH) using a Polytron PT3100 tissue homogenizer(Kinematica, Bohemia, NY, USA). Liver homogenates were incubated for2 h at 37�C with different concentrations of carbenoxolone (0.4, 0.8, 1.6 and3.2 lmol ⁄L) and then centrifuged at 12 879 g for 10 min at 4�C. The resul-tant supernatant was filtered through a 45 lm syringe filter and the enzymereaction was terminated by the addition of 100 lL of 10% trichloroacetic acid.The reaction mixture was centrifuged at 17 530 g for 5 min at 4�C. Cortisoneand cortisol in the supernatant were assayed using HPLC. The HPLC system
consisted of an Agilant 1100 series and an automatic injector (Agilant Tech-nologies, Waldronn, Germany) equipped with a variable wavelength ultravio-let detector. Samples were analysed at 30�C using a J’sphere ODS-H80column (150 · 4.6 mm i.d.; particle size 4 lm; YMC Separation Technolo-gies, Milford, IN, USA). The mobile phase consisted of water : aceticacid : triethyl amine : acetonitrile (750 : 1 : 0.3 : 250) and was delivered inisocratic mode at a flow rate of 1 mL ⁄min. The detector was set to 254 nmand the percentage area of each peak (cortisol and cortisone) was determined.The resolution between two adjacent peaks was > 1.5 and measured by CHEM-
STATION software (Agilent Technologies, Santa Clara, CA, USA). The reten-tion times of cortisol (12.7 min) and cortisone (13.87 min) were confirmed byrunning a mixture of a standard solution of cortisol and cortisone with eachassay. The activity of 11b-HSD1 was estimated indirectly by calculating thearea under the cortisol peak as a percentage of the total area under the corti-sone and cortisol peaks.
Repeated dose treatment with carbenoxolone
Mice were treated with either vehicle (deionized water) or 10, 25 or50 mg ⁄ kg carbenoxolone (formulated in deionized water) twice daily (at0930 and 1800 hours) by oral gavage. The dosing volume was 10 mL ⁄ kg.Bodyweight and feed intake were recorded each day in the eveningthroughout the treatment period of 10 days. At the end of the treatmentperiod, energy expenditure was measured using an indirect open-circuitcalorimeter (Oxylet; Panlab, Cornella, Spain). Mice were acclimated tothe metabolic chambers for 24 h and data were collected for followingfull dark cycle.
Estimation of serum lipids
At the end of treatment period, mice were fasted and serum triglyceride,cholesterol, HDL and potassium levels were determined.
Oral glucose tolerance test and insulin tolerance test
Mice treated with repeated doses of carbenoxolone were subjected to an oralglucose tolerance test (OGTT) after an overnight fast. Mice were given a glu-cose load (1 g ⁄ kg) by oral gavage. Blood samples (200 lL) were collected at0, 30, 60 and 120 min. Serum insulin and glucose concentrations were deter-mined using commercially available kits (see above). The degree of insulinresistance and pancreatic b-cell function were estimated by the homeostasismodel assessment of insulin resistance (HOMA-IR) and HOMA-b, respec-tively, using the following equations:
HOMA-IR ¼ FSI� FSG=22:4
HOMA� b ¼ FSI� 20=ðFSG� 3:5Þ
where FSI is fasting serum insulin (in lU ⁄mL) and FSG is fasting serumglucose (in mmol ⁄L).
The insulin tolerance test (ITT) was performed in mice after an overnightfast. Briefly, mice were injected with human regular insulin (1 IU ⁄ kg, i.p.) at0 min and blood samples were collected at 0, 30, 60, 120 and 240 min.Serum glucose concentrations were determined using commercially availablekits.
Effects of 10 day carbenoxolone treatment on 11b-HSD1 activityin the liver, adipose tissue and brain
Two hours after mice had received a final dose of carbenoxolone in the morn-ing, they were killed and their liver, mesenteric fat and brain were collectedfor the estimation of enzyme activity, as described above. In this series ofexperiments, tissue homogenates were incubated without ex vivo carbenoxo-lone treatment.
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
70 N Dhanesha et al.

Estimation of glucokinase, PEPCK and G6Pase activity
Glucokinase (GK) activity was determined in hepatic and pancreatic homo-genates using a spectrophotometric assay, as described by Davidson andArion19 with slight modification, wherein the formation of glucose 6-phos-phate at 37�C was coupled to its oxidation by glucose-6-phosphate dehydro-genase and NAD+. The reaction mixture (final volume 1 mL) contained50 mmol ⁄L sodium HEPES, pH 7.4, 100 mmol ⁄L KCl, 7.5 mmol ⁄L MgCl2,5 mmol ⁄L ATP, 2.5 mmol ⁄L dithioerythritol, 10 mg ⁄mL albumin,1 mmol ⁄L NAD+, 5.5 U glucose-6-phosphate dehydrogenase (Leuconostocmesenteroides), 10 lL hepatic cytosol and 10 mmol ⁄L glucose.
Phosphoenolpyruvate carboxykinase activity was monitored in the direc-tion of oxaloacetate synthesis using the spectrophotometric assay described byBentle and Lardy20 with a slight modification. The reaction mixture (final vol-ume 1 mL) contained 50 mmol ⁄L sodium HEPES, pH 6.5, 1 mmol ⁄L ino-sine diphosphate, 1 mmol ⁄L MnCl2, 1 mmol ⁄L dithiothreitol, 0.25 mmol ⁄LNADH, 2 mmol ⁄L phosphoenolpyruvate, 50 mmol ⁄L NaHCO3, 7.2 U malicdehydrogenase and 10 lL hepatic cytosol. Enzyme activity was determined at25�C for 2 min as the decrease in absorbance at 340 nm.
Glucose-6-phosphatase activity was determined in liver samples using aspectrophotometric assay according to the methods of Alegre et al.21 withslight modification. The reaction mixture (final volume 1 mL) contained100 mmol ⁄L sodium HEPES, pH 6.5, 26.5 mmol ⁄L glucose-6-phospate,1.8 mmol ⁄L EDTA, both previously adjusted to pH 6.5, 2 mmol ⁄L NADP+,0.6 kIU ⁄L mutarotase, 10 lL hepatic cytosol and 6 kIU ⁄L glucose dehydro-genase. Enzyme activity is expressed in U ⁄mg protein.
Liver glycogen content was estimated using direct enzymatic digestion byamyloglycosidase22 in the liver of db ⁄ db mice treated with different doses ofcarbenoxolone or vehicle.
Estimation of GK protein levels
After completion of the 10 day treatment with carbenoxolone, liver tissueswere stored at )80�C until analysis. Approximately 100 mg liver tissue washomogenized in 1 mL lysis buffer containing 50 mmol ⁄L Tris–HCl (pH 8.0),150 mmol ⁄L NaCl, 0.02% NaN3, 0.1% sodium dodecyl sulphate (SDS),1 mmol ⁄L EDTA, 0.5% sodium deoxycholate, 100 mg ⁄mL phenylmethyl-sulphonyl fluoride, 1 mg ⁄mL leupeptin and 1% Nonidet P-40 for 30 min at4�C. After lysis, cellular debris were removed by centrifugation at 12 000 gfor 20 min at 4�C. Protein concentrations were quantified using a commer-cially available kit. Protein samples with 6 · SDS loading buffer containing0.75 mol ⁄L Tris–HCl (pH 6.8), 18% SDS, 0.2% bromophenol blue, 20%(v ⁄ v) glycerol and 200 mmol ⁄L DTT were denatured by boiling for 10 min,then separated by 10% SDS–polyacrylamide gel electrophoresis and trans-ferred to nitrocellulose membranes in a cold chamber.
Membranes were blocked with 5% non-fat milk for 1 h and incubatedovernight at 4�C with anti-glucokinase (GCK) polyclonal antibody (mousemonclonal antibody against GCK; sc-55496; Santa Cruz Biotechnology,Santa Cruz, CA, USA). After three washes with Tris-buffered saline Tween-20 (TBST) for 10 min each time, membranes were incubated for 2 h with goatanti-mouse horseradish peroxidase-conjugated secondary antibodies andfinally washed thoroughly with TBST. Protein bands of interest were visual-ized using an enhanced chemiluminescence (ECL) reagent detection systemand quantified using a Gel Doc 2000 densitometer (Bio-Rad, Hercules, CA,USA).
Estimation of mesenteric fat
Mice were killed at the end of the study and mesenteric fat was collected andweighed using a digital balance (PB303; Mettler Toledo, Mumbai, India).
Free fatty acid release in isolated adipocytes
Adipocytes were isolated from mesenteric fat by the collagenase digestionmethod.23 Adipocytes (2.5 · 106 cells ⁄mL) were pre-incubated at 37�C for30 min in Krebs’ buffer containing 5.0 mmol ⁄L glucose. Adrenaline
(10 lmol ⁄L) was then added to the reaction mixtures and the resulting solu-tions were incubated at 37�C for 3 h. The reactions were stopped by soakingthe tube in ice water. The samples were then centrifuged at 1096 g for 10 minat 4�C. The concentrations of FFA in the supernatant were determined usingan FFA kit.
Insulin release in isolated islets
Pancreatic islets were isolated by the collagenase digestion method,23 handpicked from the pancreatic homogenate and cultured overnight in islet cell cul-ture medium (RPMI 1640) containing 11 mmol ⁄L glucose, 5000 U ⁄mL pen-icillin, 5 mg ⁄mL streptomycin and 10% fetal bovine serum at 37�C. Theinsulin release assay24 in islets from chronically treated mice was performedin Kreb’s buffer containing 0.05% bovine serum albumin. Insulin concentra-tions were determined using an insulin ELISA kit.
Statistical analysis
Data are presented as the mean ± SEM. Data were analysed using GRAPHPADPRISM 4 (GraphPad Software, La Jolla, CA, USA) and two-tailed P < 0.05was considered significant. The effects of vehicle or treatment on glucose andinsulin during the OGTT and on glucose during the ITT after 10 days treat-ment were assessed by two-way ANOVA (with time as a repeated measure) andBonferroni’s post hoc test. All groups were compared with each other forBonferroni comparison, with a total of 15 comparisons in the OGTT and ITTfor glucose. The effects of the 11b-HSD1 inhibitor carbenoxolone on percent-age cortisol area (as an index of 11b-HSD1 enzyme inhibition), bodyweight,feed intake, energy expenditure (EE), respiratory quotient (RQ), serum lipidparameters, potassium, glucose, insulin, leptin, enzyme activities, proteinexpression and hepatic glycogen were compared with values in the vehicle-treated group using one-way ANOVA with Dunnett’s test.
RESULTS
Inhibitory effect of carbenoxolone on 11b-HSD1 activityin the liver of db ⁄dbmice
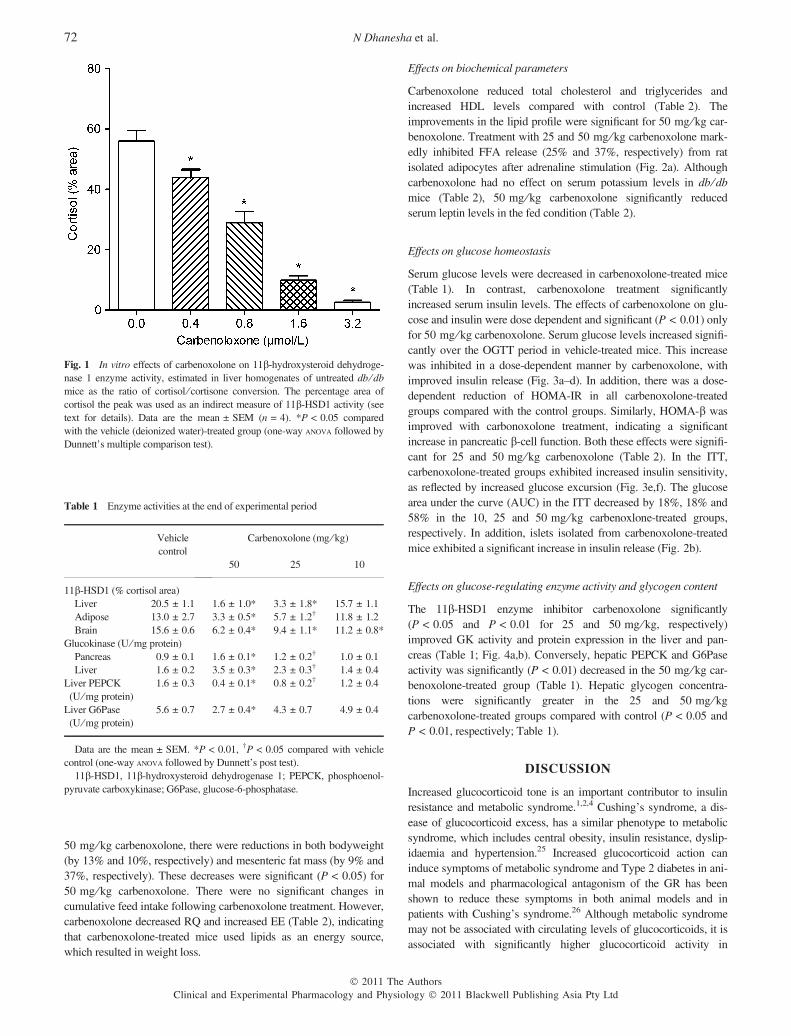
The effect of carbenoxolone on 11b-HSD1 activity was determinedin livers of untreated db ⁄ db mice. There was a trend towards a con-centration-dependent decrease in 11b-HSD1 activity after 2 h incu-bation with carbenoxolone, as reflected by a reduction in thepercentage cortisol area. The conversion of cortisone to cortisol wassignificantly reduced in the presence of 0.4, 0.8, 1.6 and 3.2 lmol ⁄L,carbenoxolone by 21%, 48%, 82% and 95%, respectively (Fig. 1).
Effects of 10 day treatment with carbenoxolone on metabolicparameters
Effects on 11b-HSD1 in the liver, adipose tissue and brain
After 10 days treatment with carbenoxolone, there was a significantdecrease in 11b-HSD1 activity in carbenoxolone-treated db ⁄ db mice.Two hours after the last carbenoxolone dose (either 10, 25 or50 mg ⁄kg), the enzyme activity assay (i.e. percentage cortisol area, ameasure of the conversion of cortisone to cortisol) revealed reduc-tions in 11b-HSD1 activity in the liver (23%, 84% and 92%, respec-tively), adipose (9%, 56% and 75%, respectively) and brain (28%,40% and 60%, respectively; Table 1).
Effects on bodyweight, fat mass, feed intake and EE
Bodyweight did not change significantly over the 10 day treatmentperiod in vehicle-treated mice. However, in mice treated with 10 or
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
Inhibition of 11b-HSD1 in db ⁄ db mice 71
50 mg ⁄kg carbenoxolone, there were reductions in both bodyweight(by 13% and 10%, respectively) and mesenteric fat mass (by 9% and37%, respectively). These decreases were significant (P < 0.05) for50 mg ⁄kg carbenoxolone. There were no significant changes incumulative feed intake following carbenoxolone treatment. However,carbenoxolone decreased RQ and increased EE (Table 2), indicatingthat carbenoxolone-treated mice used lipids as an energy source,which resulted in weight loss.
Effects on biochemical parameters
Carbenoxolone reduced total cholesterol and triglycerides andincreased HDL levels compared with control (Table 2). Theimprovements in the lipid profile were significant for 50 mg ⁄kg car-benoxolone. Treatment with 25 and 50 mg ⁄kg carbenoxolone mark-edly inhibited FFA release (25% and 37%, respectively) from ratisolated adipocytes after adrenaline stimulation (Fig. 2a). Althoughcarbenoxolone had no effect on serum potassium levels in db ⁄ dbmice (Table 2), 50 mg ⁄kg carbenoxolone significantly reducedserum leptin levels in the fed condition (Table 2).
Effects on glucose homeostasis
Serum glucose levels were decreased in carbenoxolone-treated mice(Table 1). In contrast, carbenoxolone treatment significantlyincreased serum insulin levels. The effects of carbenoxolone on glu-cose and insulin were dose dependent and significant (P < 0.01) onlyfor 50 mg ⁄ kg carbenoxolone. Serum glucose levels increased signifi-cantly over the OGTT period in vehicle-treated mice. This increasewas inhibited in a dose-dependent manner by carbenoxolone, withimproved insulin release (Fig. 3a–d). In addition, there was a dose-dependent reduction of HOMA-IR in all carbenoxolone-treatedgroups compared with the control groups. Similarly, HOMA-b wasimproved with carbonoxolone treatment, indicating a significantincrease in pancreatic b-cell function. Both these effects were signifi-cant for 25 and 50 mg ⁄kg carbenoxolone (Table 2). In the ITT,carbenoxolone-treated groups exhibited increased insulin sensitivity,as reflected by increased glucose excursion (Fig. 3e,f). The glucosearea under the curve (AUC) in the ITT decreased by 18%, 18% and58% in the 10, 25 and 50 mg ⁄kg carbenoxlone-treated groups,respectively. In addition, islets isolated from carbenoxolone-treatedmice exhibited a significant increase in insulin release (Fig. 2b).
Effects on glucose-regulating enzyme activity and glycogen content
The 11b-HSD1 enzyme inhibitor carbenoxolone significantly(P < 0.05 and P < 0.01 for 25 and 50 mg ⁄ kg, respectively)improved GK activity and protein expression in the liver and pan-creas (Table 1; Fig. 4a,b). Conversely, hepatic PEPCK and G6Paseactivity was significantly (P < 0.01) decreased in the 50 mg ⁄ kg car-benoxolone-treated group (Table 1). Hepatic glycogen concentra-tions were significantly greater in the 25 and 50 mg ⁄ kgcarbenoxolone-treated groups compared with control (P < 0.05 andP < 0.01, respectively; Table 1).
DISCUSSION
Increased glucocorticoid tone is an important contributor to insulinresistance and metabolic syndrome.1,2,4 Cushing’s syndrome, a dis-ease of glucocorticoid excess, has a similar phenotype to metabolicsyndrome, which includes central obesity, insulin resistance, dyslip-idaemia and hypertension.25 Increased glucocorticoid action caninduce symptoms of metabolic syndrome and Type 2 diabetes in ani-mal models and pharmacological antagonism of the GR has beenshown to reduce these symptoms in both animal models and inpatients with Cushing’s syndrome.26 Although metabolic syndromemay not be associated with circulating levels of glucocorticoids, it isassociated with significantly higher glucocorticoid activity in
Fig. 1 In vitro effects of carbenoxolone on 11b-hydroxysteroid dehydroge-nase 1 enzyme activity, estimated in liver homogenates of untreated db ⁄ dbmice as the ratio of cortisol ⁄ cortisone conversion. The percentage area ofcortisol the peak was used as an indirect measure of 11b-HSD1 activity (seetext for details). Data are the mean ± SEM (n = 4). *P < 0.05 comparedwith the vehicle (deionized water)-treated group (one-way ANOVA followed byDunnett’s multiple comparison test).
Table 1 Enzyme activities at the end of experimental period
Vehiclecontrol
Carbenoxolone (mg ⁄ kg)
50 25 10
11b-HSD1 (% cortisol area)Liver 20.5 ± 1.1 1.6 ± 1.0* 3.3 ± 1.8* 15.7 ± 1.1Adipose 13.0 ± 2.7 3.3 ± 0.5* 5.7 ± 1.2† 11.8 ± 1.2Brain 15.6 ± 0.6 6.2 ± 0.4* 9.4 ± 1.1* 11.2 ± 0.8*
Glucokinase (U ⁄mg protein)Pancreas 0.9 ± 0.1 1.6 ± 0.1* 1.2 ± 0.2† 1.0 ± 0.1Liver 1.6 ± 0.2 3.5 ± 0.3* 2.3 ± 0.3† 1.4 ± 0.4
Liver PEPCK(U ⁄mg protein)
1.6 ± 0.3 0.4 ± 0.1* 0.8 ± 0.2† 1.2 ± 0.4
Liver G6Pase(U ⁄mg protein)
5.6 ± 0.7 2.7 ± 0.4* 4.3 ± 0.7 4.9 ± 0.4
Data are the mean ± SEM. *P < 0.01, †P < 0.05 compared with vehiclecontrol (one-way ANOVA followed by Dunnett’s post test).
11b-HSD1, 11b-hydroxysteroid dehydrogenase 1; PEPCK, phosphoenol-pyruvate carboxykinase; G6Pase, glucose-6-phosphatase.
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
72 N Dhanesha et al.

metabolically active tissues such as the liver and adipose.3,4,25 Highglucocorticoid tone results from the regeneration of active cortisolfrom inactive cortisone by prereceptor metabolism, which is gov-erned by 11b-HSD1.6 Activation of 11b-HSD1 in the liver contrib-utes to the development of Type 2 diabetes in db ⁄db mice.13 Herein,we found that in vivo inhibition of 11b-HSD1 in the liver, adiposeand brain with carbenoxolone, a prototype inhibitor of 11b-HSD1,significantly improved the symptoms of Type 2 diabetes and obesityin db ⁄db mice. In addition to a reduction in bodyweight andadiposity, 11b-HSD1 inhibition had beneficial effects on glycolyticand gluconeogenic enzymes, including increases in GK activity andprotein levels in the pancreas and liver and reductions in PEPCK andG6Pase activity in the liver. The results of the present study supportfurther investigations of 11b-HSD1 inhibitors as important targets inthe treatment of metabolic syndrome in humans.In situ studies have revealed high 11b-HSD1 mRNA expression in
obese individuals in target tissues.27 In addition, 11b-HSD1 enzymeactivity and GR expression are significantly higher in visceral com-pared with subcutaneous fat in obese humans.25 Glucocorticoids havebeen reported to inhibit pre-adipocyte proliferation and to promoteadipocyte differentiation, thereby promoting adipogenesis.28 How-ever, it has been reported that carbenoxolone treatment in obese Zuc-ker fa ⁄ fa rats failed to have any effect on obesity, probably due toinsufficient 11b-HSD1 inhibition in the adipose tissue.29 Therefore,should inhibition of 11b-HSD1 with synthetic inhibitors prove effec-tive at lowering obesity and dyslipidaemia in humans, additional ben-eficial effects on adipose 11b-HSD1 are anticipated. In the presentstudy, carbenoxolone-treated db ⁄db mice had reduced mesenteric fatmass and bodyweight, with lower FFA release from isolated adipo-cytes, whereas feed intake was unaffected. The enzyme activity assayrevealed a dose-dependent reduction in adipose and brain 11b-HSD1following chronic carbenoxolone treatment of these mice. Together,these data suggest that 11b-HSD1 inhibitors are likely to exert major
pharmacological actions in adipose tissue. Indeed, it has beenreported that the 11b-HSD1 inhibitor carbenoxolone decreases body-weight in obese and hyperlipidaemic low-density lipoprotein (LDL)receptor-knockout mice.30 In the present study, carbenoxolone treat-ment significantly reduced serum cholesterol and triglyceride levelsand increased serum HDL–cholesterol in db ⁄ db mice. In addition,carbenoxolone exerted antilipolytic effects, as observed by the inhibi-tion of adrenaline-induced FFA release from adipocytes. This effectwould have decreased FFA flux to the liver. Decreased fatty aciduptake in the liver can result in decreased very low-density lipopro-tein secretion. This could be one of the mechanisms underlying thelipid reductions seen after carbenoxolone treatment. However, themechanism by which 11b-HSD1 inhibition reduces serum lipid con-centrations is not yet fully known, although studies in 11bHSD1-knockout mice reveal increased hepatic expression of enzymesinvolved in fat catabolism and their coordinating transcription factorperoxisome proliferator-activated receptor a.11 Moreover, glucocor-ticoids have been reported to reduce HDL and to increase LDL.4
These effects may not be secondary to reductions in fat mass andbodyweight. Although carbenoxolone treatment did not affect feedintake in the present study, Feng et al.31 have demonstrated signifi-cant reduction in feed intake after 14 days treatment with the selec-tive and potent 11b-HSD1 inhibitor emodin in diet-induced obesemice. Similar appetite-reducing effects of selective 11b-HSD1 inhibi-tors have been reported in the diet-induced obese mouse model.32
These differences may be due to a significant deficit in leptin signal-ling in the db ⁄db model and the selectivity of the inhibitors used inthese studies. For the purposes of our study, the lack of effect of car-benoxolone on feed intake eliminated the need for concern regardingpossible stress effects of 11b-HSD1 inhibition on appetite.Carbenoxolone treatment significantly reduced brain 11b-HSD1
activity. Whereas food intake was similar in control and carbenoxo-lone-treated mice during the experiment, EE was significantly higher
Table 2 Effect of 11b-hydroxysteroid dehydrogenase 1 inhibition on metabolic parameters
Vehicle control Carbenoxolone (mg ⁄ kg)
50 25 10
Bodyweight (g)Day 0 48.4 ± 1.8 48.1 ± 2.7 50.6 ± 1.3 48.4 ± 1.8Day 10 47.8 ± 1.9 41.7 ± 2.6** 48.2 ± 2.0 43.1 ± 3.7
Mesenteric fat mass (g ⁄ kg bodyweight) 1188 ± 48 751 ± 53* 1034 ± 48 1086 ± 50Food consumption (g ⁄ day) 6.5 ± 0.3 5.8 ± 0.4 6.1 ± 0.3 6.8 ± 0.9EE (kcal ⁄ day per kg0.75) 97.0 ± 2.9 125 ± 5** 113 ± 6 108 ± 4RQ 1.01 ± 0.01 0.87 ± 0.01** 0.96 ± 0.01 0.92 ± 0.01Serum glucose (mg ⁄ dL) 443 ± 39 200 ± 34** 284 ± 28** 347 ± 83Serum insulin (ng ⁄mL) 2.5 ± 0.2 4.3 ± 0.7** 3.5 ± 0.2 3.3 ± 0.3Serum leptin (ng ⁄mL) 58.4 ± 8.8 26.8 ± 5.6** 52.0 ± 4.6 60.8 ± 6.0Serum potassium (mmol ⁄L) 6.4 ± 0.8 6.8 ± 1.5 6.1 ± 1.2 5.9 ± 0.9Serum triglycerides (mg ⁄ dL) 213 ± 28 153 ± 12** 189 ± 11 195 ± 42Serum total cholesterol (mg ⁄ dL) 223 ± 15 155 ± 12** 166 ± 27** 180 ± 15Serum HDL-C (mg ⁄ dL) 60.3 ± 5.5 111 ± 16** 87.2 ± 12.7 65.9 ± 8.2Liver glycogen content (mg ⁄ g liver) 49.2 ± 4.8 72 ± 8** 62.5 ± 6.7* 48.9 ± 2.3HOMA-IR 74.4 ± 9.0 43.3 ± 10.0** 55.7 ± 6.0* 63.4 ± 14.4HOMA-b 58.0 ± 6.6 317 ± 133** 145 ± 49* 186 ± 69
Values are the mean ± SEM. *P < 0.05, **P < 0.01 compared with vehicle control (one-way ANOVA followed by Dunnett’s post test).EE, energy expenditure; RQ, respiratory quotient; HDL-C, high-density lipoprotein–cholesterol; HOMA-IR, homeostasis model assessment of insulin
resistance; HOMA-b, homeostasis model assessment of pancreatic b-cell function.
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
Inhibition of 11b-HSD1 in db ⁄ db mice 73

in carbenoxolone-treated mice compared with controls. The increasein EE was associated with a decrease in RQ, without any change inlocomotor activity, indicating that the decrease in bodyweight inobese db ⁄ db mice could be attributed to a reduction in central gluco-corticoid regeneration. Indeed, a reciprocal relationship existsbetween leptin sensitivity and glucocorticoid action.1,33 The presentstudy indicates that increased leptin sensitivity, as indicated by lowerleptin levels, may translate into increased EE and utilization of fatmass.In the present study, chronic 11b-HSD1 inhibition by carbenoxo-
lone significantly improved glucose tolerance in db ⁄db mice. Theimprovement in glucose tolerance was accompanied by a significant
and sustained inhibition of hepatic 11b-HSD1 activity. Evidenceindicates that glycaemic improvement is a hallmark of effective11b-HSD1 inhibition in vivo, as observed in KKAy mice34 anddiet-induced obese mice.35 Although such glycaemic improvementhas not been observed in LDL receptor-knockout mice,30 theimprovement in the present study highlights the underlying patho-logy related to 11b-HSD1 upregulation in the liver of db ⁄db mice.Indeed, improvements in glycaemic control by selective 11b-HSD1inhibitors have been reported in db ⁄dbmice.36
Gluconeogeneis is increased in Type 2 diabetes and contributessignificantly to fasting and post-prandial hyperglycaemia in db ⁄ dbmice.14 Because elevated glucocorticoid tone correlates wellwith high hepatic gluconeogenesis in db ⁄db mice and because anta-gonizing glucocorticoids at the receptor level suppresses the key
(a)
(b)
Fig. 2 Effects of 11b-hydroxysteroid dehydrogenase 1 inhibition on adipo-cytes and islets following chronic (10 day) carbenoxolone treatment of mice.(a) Non-esterified free fatty acid (NEFA) release after adrenaline stimulationof adipocytes. (Control values have been normalized to 100%). (b) Insulinrelease from isolated pancreatic islets after incubation in the presence of16.7 mmol ⁄L glucose. Data are the mean ± SEM (n = 7). *P < 0.05compared with the vehicle (deionized water)-treated group (one-way ANOVA
followed by Dunnett’s multiple comparison test).
(a) (b)
(c) (d)
(e) (f)
Fig. 3 Effects of chronic (10 day) carbenoxolone treatment on glycaemiccontrol in db ⁄ dbmice. (s), vehicle (deionized water) control; (d), 10 mg ⁄ kg,p.o., carbenoxolone; (h), 25 mg ⁄ kg, p.o., carbenoxolone; (n), 50 mg ⁄ kg,p.o., carbenoxolone. (a–d) Results of the oral glucose tolerance test (OGTT)following a 1.5 g ⁄ kg oral glucose load at Time 0 min. (a) Serum glucose lev-els, (b) area under the glucose curve (AUCglucose), (c) serum insulin levels and(d) AUCinsulin during the OGTT. (e,f) Results of the insulin tolerance test(ITT) following injection of 1 l ⁄ kg, i.p. insulin. (e) Serum glucose levels and(f) AUCglucose during the ITT. Data are the mean ± SEM (n = 7). *P < 0.05compared with the vehicle control group (two-way ANOVA with time as therepeated measure, followed by Bonferroni’s post hoc test). There was a totalof 15 comparisons for glucose and 12 for insulin.
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
74 N Dhanesha et al.

gluconeogenic enzymes in db ⁄ db mice, it would be expected thatantagonizing glucocorticoid effects would be beneficial for glycae-mic control in this species.13,33 However, the actions of glucocortic-oids on target tissues, such as the liver and adipose, are notnecessarily dependent on circulating glucocorticoid levels, but ratheron their prereceptor metabolism, which is regulated by 11b-HSD1.6
Hence, inhibition of 11b-HSD1 offers a useful pharmacological inter-vention that is likely to suppress hepatic gluconeogenic enzymes.The key enzymes that control gluconeogenesis and glucose outputfrom the liver are G6Pase and PEPCK and the expression of theseenzymes in the liver is increased in db ⁄ db mice.6,14 In the presentstudy, carbenoxolone treatment significantly reduced the activity ofPEPCK and G6Pase in the liver, which can be strongly correlatedwith 11b-HSD1 inhibition in the liver. These results suggest thatpharmacological inhibition of 11b-HSD1 by carbenoxolone reduceshepatic gluconeogenesis, which may have contributed to thedecreased fasting blood glucose levels.Glucocorticoids play a critical role in glucose metabolism by func-
tionally opposing insulin effects and also inhibiting pancreatic insulinsecretion. Therefore, it was not surprising that the improvement inglucose tolerance produced by 11b-HSD1 inhibition in the presentstudy was accompanied by increases in insulin levels. Pancreaticislets isolated from chronically treated mice exhibited significantlyincreased insulin release, which further supports a direct antihyper-glycaemic action of carbenoxolone. Because pancreatic GK activityand expression were increased in carbenoxolone-treated mice, therecould be a correlation between the increased insulin-secretory capac-ity of islets and 11b-HSD1 inhibition.A reduction in HOMA-IR (a ratio indicating insulin resistance) and
an improvement in HOMA-b (a ratio indicating the insulin-secretarycapacity of islet b-cells) were observed with chronic carbenoxolonetreatment in the present study. In addition, in the ITT of overnight-fasted mice, carbenoxolone-treated mice exhibited improved insulinsensitivity compared with control mice. The glucose–insulin interplayin db ⁄db mice is known to be age dependent.37 The initial adaptationto insulin resistance results in marked hyperinsulinaemia, followed byb-cell necrosis and insulin deficiency when the mice reach 24 weeksof age.37 In the present study, carbenoxolone resulted in a reductionof hepatic 11b-HSD1 activity that was correlated with hypoglycaemiaby the suppression of the key gluconeogenic enzymes PEPCK andG6Pase in db ⁄db mice. However, improved insulin secretion accom-panying the antihyperglycaemic effect indicated improvement in pan-creatic activity. The GK enzyme plays a key role in glucosehomeostasis, regulating insulin secretion in response to glucose avail-ability in b-cells in the pancreas and modulating glucose utilizationand output in the liver.38 In the present study, carbenoxolone treat-ment dose-dependently increased GK activity and expression in boththe liver and pancreas. Suppression of PEPCK and G6Pase by11b-HSD1 inhibition could reduce hepatic gluconeogenesissufficiently to decrease the accumulation of hepatic glycogen andcirculating glucose levels. In addition, the increased glucose utiliza-tion and increased glycogen synthesis following GK activation canreduce hepatic glucose output. Hepatic GK is a potential target forpharmacological treatment of Type 2 diabetes, as evidenced by thefact that liver-specific GK-knockout mice exhibit mild hyperglyca-emia39 and rats overexpressing GK in the liver have reduced bloodglucose levels.40 The increase in hepatic GK can result in increasedutilization of blood glucose for energy production or glycogen storagein the liver.41 Indeed, hepatic glycogen content was significantlyhigher in carbenoxolone-treated mice in the present study.In conclusion, we have found that inhibition of the critical step in
glucocorticoid regeneration in liver, adipose and brain with the proto-type 11b-HSD1 inhibitor carbenoxolone significantly improves thesymptoms of Type 2 diabetes and metabolic syndrome in db ⁄ dbmice. The data suggest that carbenoxolone treatment improves leptin
(a)
(b)
Fig. 4 Effects of chronic (10 day) carbenoxolone treatment on glucokinaseprotein levels in the (a) liver and (b) pancreas, as determined by western blotanalysis. Data are the mean ± SEM (n = 7). *P < 0.05 compared with thevehicle control group (one-way ANOVA followed by Dunnett’s multiplecomparison test).
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
Inhibition of 11b-HSD1 in db ⁄ db mice 75

sensitivity in db ⁄db mice with increases in EE and lipolysis. In addi-tion, 11b-HSD1 inhibition is associated with decreased activity ofthe gluconeogenic enzymes PEPCK and G6Pase in the liver and withimprovements in GK activity and expression in the liver and pan-creas. Together, these results indicate that 11b-HSD1 inhibition hasenormous therapeutic potential to reduce the symptoms of metabolicsyndrome by improving liver, adipose and pancreas function.
ACKNOWLEDGEMENTS
The authors are thankful to Dr Prakash Davadra (Department of Ana-lytical Research, Zydus Research Centre, Ahmedabad, India) for helpwith the HPLC analysis and Mr Pankaj R Patel (Chairman, ZydusCadila) for his encouragement and support. This paper is ZRCcommunication no. 288.
REFERENCES
1. Baxter JD. Glucocorticoid hormone action. Pharmacol. Ther. B 1976; 2:605–69.
2. Dallman MF, Strack AM, Akana SF et al. Feast and famine: Criticalrole of glucocorticoids with insulin in daily energy flow. Front. Neuro-endocrinol. 1994; 14: 303–47.
3. Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies ofCushing’s syndrome: Diagnosis and therapy. Treat. Endocrinol. 2002;1: 79–94.
4. Day C. Metabolic syndrome, or what you will: Definitions and epidemi-ology. Diab. Vasc. Dis. Res. 2007; 4: 32–8.
5. Stahn C, Lowenberg M, Hommes DW, Buttgereit F. Molecular mecha-nisms of glucocorticoid action and selective glucocorticoid receptoragonists.Mol. Cell. Endocrinol. 2007; 275: 71–8.
6. Draper N, Stewart PM. 11Beta-hydroxysteroid dehydrogenase and thepre-receptor regulation of corticosteroid hormone action. J. Endocrinol.2005; 186: 251–71.
7. Seckl JR, Walker BR. Minireview: 11Beta-hydroxysteroid dehydroge-nase type 1. A tissue-specific amplifier of glucocorticoid action. Endo-crinology 2001; 142: 1371–6.
8. Masuzaki H, Paterson J, Shinyama H et al. A transgenic model of vis-ceral obesity and the metabolic syndrome. Science 2001; 294: 2166–70.
9. Paterson JM, Morton NM, Fievet C et al. Metabolic syndrome withoutobesity: Hepatic overexpression of 11beta-hydroxysteroid dehydroge-nase type 1 in transgenic mice. Proc. Natl Acad. Sci. USA 2004; 101:7088–93.
10. Kotelevtsev Y, Holmes MC, Burchell A et al. 11beta-hydroxysteroiddehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc.Natl Acad. Sci. USA 1997; 94: 14924–9.
11. Morton NM, Holmes MC, Fievet C et al. Improved lipid and lipoproteinprofile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 2001;276: 41293–300.
12. Madiehe AM, Hebert S, Mitchell TD, Harris RB. Strain-dependent stim-ulation of growth in leptin-treated obese db ⁄ db mice. Endocrinology2002; 143: 3875–83.
13. Aoki K, Homma M, Hirano T et al. mRNA and enzyme activity ofhepatic 11beta-hydroxysteroid dehydrogenase type 1 are elevated inC57BL ⁄KsJ-db ⁄ db mice. Life Sci. 2001; 69: 2543–9.
14. Friedman JE, Sun Y, Ishizuka T et al. Phosphoenolpyruvate carboxy-kinase (GTP) gene transcription and hyperglycemia are regulated byglucocorticoids in genetically obese db ⁄ db transgenic mice. J. Biol.Chem. 1997; 272: 31475–81.
15. Coppola GM, Kukkola PJ, Stanton JL et al. Perhydroquinolylbenza-mides as novel inhibitors of 11beta-hydroxysteroid dehydrogenase type1. J. Med. Chem. 2005; 20: 6696–712.
16. Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. Car-benoxolone increases hepatic insulin sensitivity in man: A novel role for11-oxosteroid reductase in enhancing glucocorticoid receptor activation.J. Clin. Endocrinol. Metab. 1995; 80: 3155–9.
17. Andrews RC, Rooyackers O, Walker BR. Effects of the 11 beta-hydrox-ysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity inmen with type 2 diabetes. J. Clin. Endocrinol. Metab. 2004; 88: 285–91.
18. Horigome H, HommaM, Hiran T, Oka K, Niitsuma T, Hayashi T. Mag-nolol fromMagnolia officinalis inhibits 11beta-hydroxysteroid dehydro-genase without increases of corticosterone and thymocyte apoptosis inmice. Planta Med. 2001; 67: 33–7.
19. Davidson AL, Arion WJ. Factors underlying significant underestima-tions of glucokinase activity in crude liver extracts: Physiological impli-cations of higher cellular activity. Arch. Biochem. Biophys. 1987; 253:156–67.
20. Bentle LA, Lardy HA. Interaction of anions and divalent metal ions withphosphoenolpyruvate carboxykinase. J. Biol. Chem. 1976; 251: 2916–21.
21. Alegre M, Ciudad CJ, Fillat C, Guinovart JJ. Determination of glucose-6-phosphatase activity using the glucose dehydrogenase-coupled reac-tion. Anal. Biochem. 1988; 173: 185–9.
22. Roehrig KL, Allred JB. Direct enzymatic procedure for the determina-tion of liver glycogen. Anal. Biochem. 1974; 58: 414–21.
23. Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolationfrom mouse pancreas. Nat. Protoc. 2009; 4: 1649–52.
24. Kanitkar M, Galande S, Bhonde R. Curcumin prevents streptozotocin-induced islet damage by scavenging free radicals: A prophylactic andprotective role. Eur. J. Pharmacol. 2007; 577: 183–91.
25. Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect ‘Cush-ing’s disease of the omentum’? Lancet 1997; 349: 1210–13.
26. Livingstone DE, Jones GC, Smith K et al. Understanding the role ofglucocorticoids in obesity: Tissue-specific alterations of corticosteronemetabolism in obese Zucker rats. Endocrinology 2000; 141: 560–3.
27. Stimson RH, Andrew R, McAvoy NC, Tripathi D, Hayes PC, WalkerBR. Increased whole-body and sustained liver cortisol regeneration by11{beta}-hydroxysteroid dehydrogenase type 1 in obese men with type2 diabetes provides a target for enzyme inhibition. Diabetes 2011; 60:720–5.
28. Morton NM. Obesity and corticosteroids. 11Beta-hydroxysteroid type 1as a cause and therapeutic target in metabolic disease. Mol. Cell. Endo-crinol. 2010; 316: 154–64.
29. Livingstone DE, Walker BR. Is 11beta-hydroxysteroid dehydrogenasetype 1 a therapeutic target? Effects of carbenoxolone in lean and obeseZucker rats. J. Pharmacol. Exp. Ther. 2004; 305: 167–72.
30. Nuotio-Antar AM, Hachey DL, Hasty AH. Carbenoxolone treatmentattenuates symptoms of metabolic syndrome and atherogenesis in obese,hyperlipidemic mice. Am. J. Physiol. Endocrinol. Metab. 2007; 293:E1517–28.
31. Feng Y, Huang SL, Dou W et al. Emodin, a natural product, selectivelyinhibits 11beta-hydroxysteroid dehydrogenase type 1 and amelioratesmetabolic disorder in diet-induced obese mice. Br. J. Pharmacol. 2010;161: 113–26.
32. Wang SJ, Birtles S, de Schoolmeester J et al. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 reduces food intake and weightgain but maintains energy expenditure in diet-induced obese mice.Diabetologia 2006; 49: 1333–7.
33. Livingstone DE, Grassick SL, Currie GL, Walker BR, Andrew R. Dys-regulation of glucocorticoid metabolism in murine obesity: Comparableeffects of leptin resistance and deficiency. J. Endocrinol. 2009; 201:211–18.
34. Barf T, Vallgarda J, Emond R et al. Arylsulfonamidothiazoles as a newclass of potential antidiabetic drugs. Discovery of potent and selectiveinhibitors of the 11beta-hydroxysteroid dehydrogenase type 1. J. Med.Chem. 2002; 45: 3813–15.
35. Taylor A, Irwin N, McKillop AM, Flatt PR, Gault VA. Sub-chronicadministration of the 11beta-HSD1 inhibitor, carbenoxolone, improves
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
76 N Dhanesha et al.

glucose tolerance and insulin sensitivity in mice with diet-induced obes-ity. Biol. Chem. 2008; 389: 441–5.
36. Alberts P, Nilsson C, Selen G et al. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivityin hyperglycemic mice strains. Endocrinology 2004; 144: 4755–62.
37. Orland MJ, Permutt MA. Quantitative analysis of pancreatic proinsulinmRNA in genetically diabetic (db ⁄ db) mice. Diabetes 1987; 36: 341–7.
38. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influ-ence stress responses? Integrating permissive, suppressive, stimulatory,and preparative actions. Endocr. Rev. 2000; 21: 55–89.
39. Postic C, Shiota M, Niswender KD et al. Dual roles for glucokinase inglucose homeostasis as determined by liver and pancreatic beta cell-spe-cific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999; 274:305–15.
40. Ferre T, Pujol A, Riu E, Bosch F, Valera A. Correction of diabeticalterations by glucokinase. Proc. Natl Acad. Sci. USA 1996; 93: 7225–30.
41. Iynedjian PB, Gjinovci A, Renold AE. Stimulation by insulin of gluco-kinase gene transcription in liver of diabetic rats. J. Biol. Chem. 1988;263: 740–4.
� 2011 The AuthorsClinical and Experimental Pharmacology and Physiology � 2011 Blackwell Publishing Asia Pty Ltd
Inhibition of 11b-HSD1 in db ⁄ db mice 77




















