
Improving the prognosis for patients with glioblastoma: the rationale for targeting Src
13
TOPIC REVIEW Improving the prognosis for patients with glioblastoma: the rationale for targeting Src John de Groot Vanessa Milano Received: 9 January 2009 / Accepted: 30 April 2009 / Published online: 13 May 2009 Ó Springer Science+Business Media, LLC. 2009 Abstract Glioblastoma is the most common and aggressive form of primary brain tumor. The prognosis for patients diagnosed with glioblastoma is poor, with a median survival of 12–14 months and a 5-year survival rate of \ 5%. The upfront standard treatment for patients with newly diagnosed glioblastoma, consisting of surgery fol- lowed by chemotherapy combined with radiotherapy, provides only short-term survival benefits. Recurrent glio- blastoma is an extremely challenging therapeutic setting because of the aggressive and resistant nature of the tumor. A set of key molecular targets in oncology is the Src family of non-receptor protein kinases. Dysregulated signaling via the Src kinases has been shown to underlie glioma-related proliferation, angiogenesis, migration, and survival. Here we review the biologic role of Src in malignant glioma and discuss key preclinical studies demonstrating the potential utility of inhibiting Src in glioma. Proof of clinical benefit is forthcoming from the first clinical studies involving the newest generation of small molecule Src inhibitors cur- rently in clinical trials for recurrent glioblastoma. Blocking Src alone will likely not translate into a significant clinical benefit; thus, strategies for combining Src inhibitors with potential synergistic therapeutic modalities will be dis- cussed. This review will focus on dasatinib, the most advanced Src inhibitor being tested in glioblastoma, which is currently in phase I/II trials in this setting. Keywords Glioblastoma Á Src family of tyrosine kinases Á Src inhibitors Á Dasatinib Introduction Glioblastoma is designated by the World Health Organi- zation (WHO) as a grade IV astrocytoma [1] and may arise de novo or from a pre-existing lower-grade glioma. Glio- blastomas are characterized pathologically by high cellu- larity and mitotic activity, nuclear atypia, psuedopalisating necrosis, and microvascular proliferation (angiogenesis). Angiogenesis is driven by the secretion of vascular endo- thelial growth factor (VEGF; originally called vascular permeability factor or VPF), placental growth factor (PlGF), basic fibroblast growth factor (bFGF), and other pro-angiogenic factors which also increase vascular per- meability. This increased vascular permeability results in the deterioration of the blood brain barrier (BBB) and underlies the tumor enhancement observed with T1-post gadolinium magnetic resonance imaging (MRI) scans. In addition to abnormal blood vessel proliferation, glioblas- toma tumor cells are highly motile. Tumor infiltration of the brain can be both local (confined to regions close to the primary tumor) and more diffuse: glioblastoma cells often eventually spread throughout the brain [2]. Those tumor cells that migrate into healthy regions of the brain are difficult to treat due to the inherent limitations to drug delivery imposed by an intact BBB. J. de Groot (&) The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box 431, Houston, TX 77030-4009, USA e-mail: [email protected] V. Milano Department of Biological Sciences, The University of Notre Dame, 107 Galvin Life Science Center, Notre Dame, IN 46556- 5645, USA e-mail: [email protected] 123 J Neurooncol (2009) 95:151–163 DOI 10.1007/s11060-009-9916-2
Transcript of Improving the prognosis for patients with glioblastoma: the rationale for targeting Src

TOPIC REVIEW
Improving the prognosis for patients with glioblastoma:the rationale for targeting Src
John de Groot Æ Vanessa Milano
Received: 9 January 2009 / Accepted: 30 April 2009 / Published online: 13 May 2009
� Springer Science+Business Media, LLC. 2009
Abstract Glioblastoma is the most common and
aggressive form of primary brain tumor. The prognosis for
patients diagnosed with glioblastoma is poor, with a
median survival of 12–14 months and a 5-year survival rate
of \5%. The upfront standard treatment for patients with
newly diagnosed glioblastoma, consisting of surgery fol-
lowed by chemotherapy combined with radiotherapy,
provides only short-term survival benefits. Recurrent glio-
blastoma is an extremely challenging therapeutic setting
because of the aggressive and resistant nature of the tumor.
A set of key molecular targets in oncology is the Src family
of non-receptor protein kinases. Dysregulated signaling via
the Src kinases has been shown to underlie glioma-related
proliferation, angiogenesis, migration, and survival. Here
we review the biologic role of Src in malignant glioma and
discuss key preclinical studies demonstrating the potential
utility of inhibiting Src in glioma. Proof of clinical benefit
is forthcoming from the first clinical studies involving the
newest generation of small molecule Src inhibitors cur-
rently in clinical trials for recurrent glioblastoma. Blocking
Src alone will likely not translate into a significant clinical
benefit; thus, strategies for combining Src inhibitors with
potential synergistic therapeutic modalities will be dis-
cussed. This review will focus on dasatinib, the most
advanced Src inhibitor being tested in glioblastoma, which
is currently in phase I/II trials in this setting.
Keywords Glioblastoma �Src family of tyrosine kinases � Src inhibitors � Dasatinib
Introduction
Glioblastoma is designated by the World Health Organi-
zation (WHO) as a grade IV astrocytoma [1] and may arise
de novo or from a pre-existing lower-grade glioma. Glio-
blastomas are characterized pathologically by high cellu-
larity and mitotic activity, nuclear atypia, psuedopalisating
necrosis, and microvascular proliferation (angiogenesis).
Angiogenesis is driven by the secretion of vascular endo-
thelial growth factor (VEGF; originally called vascular
permeability factor or VPF), placental growth factor
(PlGF), basic fibroblast growth factor (bFGF), and other
pro-angiogenic factors which also increase vascular per-
meability. This increased vascular permeability results in
the deterioration of the blood brain barrier (BBB) and
underlies the tumor enhancement observed with T1-post
gadolinium magnetic resonance imaging (MRI) scans. In
addition to abnormal blood vessel proliferation, glioblas-
toma tumor cells are highly motile. Tumor infiltration of
the brain can be both local (confined to regions close to the
primary tumor) and more diffuse: glioblastoma cells often
eventually spread throughout the brain [2]. Those tumor
cells that migrate into healthy regions of the brain are
difficult to treat due to the inherent limitations to drug
delivery imposed by an intact BBB.
J. de Groot (&)
The University of Texas M. D. Anderson Cancer Center, 1515
Holcombe Boulevard, Box 431, Houston, TX 77030-4009, USA
e-mail: [email protected]
V. Milano
Department of Biological Sciences, The University of Notre
Dame, 107 Galvin Life Science Center, Notre Dame, IN 46556-
5645, USA
e-mail: [email protected]
123
J Neurooncol (2009) 95:151–163
DOI 10.1007/s11060-009-9916-2

The oncogenic properties of Src signaling
Since the original discovery of viral Src almost a century
ago, the oncoprotein Src (c-Src) has become widely studied
[3]. The Src family of tyrosine kinases (SFKs), of which
there are 9 members (Src, YES, FYN, LYN, LCK, HCK,
FGR, YRK, and BLK), are all non-receptor, membrane-
associated proteins. Src is linked with the development and
progression of multiple cancer types [4]. The causal rela-
tionship between Src dysregulation and the cancer pheno-
type can be explained, in part, by downstream activation of
the phosphatidylinositol 3-kinase (PI3K) and mitogen
activated protein kinase (MAPK) pathways. These key
pathways stimulate proliferation, invasion, tumorigenesis,
angiogenesis, and impairment of apoptosis [5]. Src acti-
vation can be effected by receptor over-expression and less
commonly by mutation. High levels of activated Src have
been described in breast cancer, non-small cell lung cancer,
leukemia, colon cancer, gliomas, and other solid tumors
[4, 6–9].
Although the mechanisms by which Src promotes can-
cer are not completely understood, it is likely to be a
central regulator at the interface between extracellular
signals and intracellular pathway activation. Src can be
activated by integrin engagement or by the activation of
cell surface receptors including insulin-like growth factor-1
receptor (IGF-1R), epidermal growth factor receptor
(EGFR), and platelet-derived growth factor receptor
(PDGFR). In turn, Src mediates the phosphorylation of
multiple intracellular substrates (EGFR, focal adhesion
kinase [FAK], proline-rich tyrosine kinase 2 [PYK2],
paxillin, signal transducer and activator of transcription-3
[STAT3], and cyclin D) [10]. In addition to promoting
tumor growth during tumorigenesis, Src also mediates
critical functions such as cell adhesion, invasion [11],
angiogenesis [12], and the inhibition of apoptosis [13].
Furthermore, Src can cooperate with EGFR-mediated sig-
naling to enhance tumor biology [14]. Another important
pathway is the VEGF-induced signaling in endothelial cells
which is dependent on Src-mediated FAK activation. This
suggests Src plays an important role in angiogenesis
through its regulation of endothelial cell migration and any
subsequent invasion of tissues [13].
The collective evidence suggests Src plays a central role
in promoting tumorigenesis and disease progression. It is
therefore not surprising that the potential clinical benefits
of targeted therapy versus Src have garnered significant
interest in several oncology settings. There is a particularly
strong rationale for targeting Src in glioblastoma. In the
majority of these tumors, Src expression is evident and is
likely to contribute to the malignant and aggressive phe-
notype of the disease. This review provides an update on
the use of therapeutic approaches currently in development
for the treatment of glioblastoma with an emphasis on
targeting Src kinase.
Current therapeutic options for glioblastoma
Despite recent advances in the molecular classification,
technologic improvements in surgery and radiotherapy, and
the integration of novel molecular targeted therapies, the
clinical outcome of patients with malignant glioma remains
poor. The general standard of care is surgery and radio-
therapy plus temozolamide, followed by adjuvant temo-
zolamide [15]. Although malignant gliomas are diffusely
infiltrative tumors, maximal safe surgical resection of
[98%, although involving a high degree of intervention, is
associated with improved survival [16]. Following surgery,
radiotherapy is the mainstay of treatment. Adjuvant
radiotherapy of 50–60 Gy is associated with an increase in
survival by 14–36 weeks [17, 18]. Historically, chemo-
therapy was generally considered to only provide a mar-
ginal improvement in survival in this setting [19]. More
recently, however, the role of chemotherapy in the man-
agement of malignant gliomas has been further clarified. In
a landmark study, Stupp and colleagues demonstrated that
the addition of temozolomide to radiotherapy followed by
adjuvant temozolomide for 6 months improved median
overall survival by 2.5 months [20]. A greater improve-
ment was also reported in patients with a methylated
methyl guanine methyl DNA transferase (MGMT) pro-
moter [21]. Patients treated with chemoradiation and
adjuvant temozolomide had a median overall survival of
14.6 months with a 2-year survival of 26.5%, double that
of the non-temozolomide group. Ongoing trials (RTOG
0525) will determine if MGMT status is a predictive
marker of response to temozolomide and other alkylating
agents or if it is a prognostic marker of outcome.
Despite these encouraging data, almost all patients
recur. Because of the highly infiltrative nature of these
tumors, recurrence is usually within 2 cm of the original
tumor location [22]. Conventional chemotherapy has been
the mainstay therapy for recurrent malignant gliomas until
the recent development of molecularly-targeted therapy.
Nitrosoureas such as carmustine (CCNU) or lomustine
(BCNU) have been extensively evaluated in patients with
recurrent malignant gliomas prior to the introduction of
temozolomide in the last decade. Regimens showing some
activity in recurrent glioblastoma include carmustine [23],
irinotecan alone [24] or in combination with carmustine
[25] or bevacizumab [26, 27], celecoxib [28], carboplatin
[29], and procarbazine as part of the PCV regimen (pro-
carbazine, carmustine and vincristine) [30, 31]. Results
from recent clinical trials using the combination of the
angiogenesis inhibitor bevacizumab (humanized anti-VEGF
152 J Neurooncol (2009) 95:151–163
123

antibody) and irinotecan are encouraging and may herald
the approval of this regimen for the treatment of recurrent
glioblastoma [27, 32], and a phase III study of bev-
acizumab in the first-line treatment of glioblastoma is
expected to be initiated soon.
At the time of tumor recurrence, life expectancy is
severely reduced. Patients with glioblastoma have 6-month
progression-free survival (PFS) rates of 9–15%, a median
PFS of 9 weeks and overall survival of only 3–4 months.
Patients with anaplastic astrocytoma have 6-month PFS
rates of 31% and median PFS rates of 13 weeks [33].
However, the dramatic improvement in our understanding
of the molecular and genetic alterations that drive gli-
omagenesis and tumor progression heralds the develop-
ment of new targets and potentially effective therapies.
Molecular targeting of glioblastoma
Like most cancers, gliomas arise through the sequential
accumulation of genetic abnormalities such as the activa-
tion of oncogenes and loss of tumor suppressor genes that
ultimately lead to increased cell proliferation, resistance to
apoptosis, increased invasion and sustained angiogenesis.
At the molecular level, these genetic changes result from
the loss of normal control of intracellular signaling path-
ways and lead to the activation of signal transduction
cascades such as the PI3K pathway. Identification of early
or critical molecular events that promote cellular trans-
formation and drive malignant cell growth is central to the
development of new molecular therapies. As our under-
standing of specific signal aberrations improves, novel
anticancer therapeutics that inhibit one or more molecular
targets can be used to block these key signals. The dramatic
clinical success seen with imatinib for the treatment of
chronic myeloid leukemia (CML) demonstrates the
potential efficacy of targeted therapy in cancer [34]. This
approach is now being enthusiastically pursued in patients
with malignant glioma.
Identifying key molecular targets in glioblastoma
The molecular profiling of glioblastoma tumors has iden-
tified numerous genes that are important for promoting
tumor proliferation and survival. Detailed analysis of
patient tumor samples has identified numerous genes in
malignant gliomas that are important for the regulation of
signaling networks responsible for sustained cellular pro-
liferation. Glioblastomas are traditionally classified into
two subgroups depending on genetic classification and the
clinical presentation of the tumor. Primary or de novo
glioblastomas are thought to arise spontaneously and
are the most common subtype of glioblastoma. They are
characterized by overexpression or amplification of EGFR,
inactivation of the tumor suppressor gene phosphatase and
tensin homolog deleted on chromosome 10 (PTEN) and
inactivation of p16INK4A or p14ARF. Secondary glioblas-
tomas are less commonly encountered. These arise from
WHO grade II tumors (low grade gliomas) and are char-
acterized by inactivating mutations or loss of TP53, loss of
heterozygosity (LOH) of chromosome 17p, and overex-
pression of PDGFR. Further progression occurs to the more
aggressive anaplastic astrocytoma (WHO grade III) tumors
which demonstrate LOH of 19q, mutation of retinoblas-
toma susceptibility locus 1 (Rb1), and overexpression or
amplification of CDK4 and HDM2. Final progression to
glioblastoma occurs with the loss of PTEN.
Two main mediators of the malignant phenotype have
been identified as potential targets for the treatment of
glioma: the cell cycle machinery that drives uncontrolled
cellular proliferation and the key growth factor receptors
can drive signaling through the MAPK and PI3K path-
ways. Dysregulation of the cell cycle leads to uncon-
trolled cellular proliferation and is typically mediated by
alterations in retinoblastoma (Rb), p53, and CDK4/6 sig-
naling [35, 36]. These molecular signals all offer potential
targets for therapy. In addition to loss of cell cycle con-
trol, extracellular growth factors can promote cell prolif-
eration, survival, and migration. The net effect of the
activation of EGFR, IGF-1R, PDGFR, and other receptor
tyrosine kinases together with loss of pathway-controlling
tumor suppressor genes is the sustained downstream
activation of multiple signal transduction networks. The
Ras-MAPK pathway, which regulates cellular prolifera-
tion [37], and the PI3K-Akt pathway, which coordinately
controls cell division, tumor growth, angiogenesis, apop-
tosis, invasion and cellular metabolism [38] are the best
characterized and most important signaling transduction
cascades in gliomas. Confirmation of decades of molec-
ular analyses of glioma tumors is supported by the pre-
liminary report recently published by the Cancer Genome
Atlas project [39]. Laboratory and clinical research efforts
have led to the development of novel, mechanism-based
strategies to identify and ‘‘target’’ specific molecular
alterations. These strategies offer exciting opportunities to
inhibit important pathways mediating cancer cell growth
and survival.
One recent study using bead-based methodology for
detecting phosphorylation of multiple tyrosine kinases
identified SRC kinase as being frequently activated in
glioblastoma cell lines and primary glioblastoma patient
samples [40]. At present, a number of targeted agents
blocking pivotal signaling pathways in glioblastoma are
under evaluation for the treatment of malignant gliomas
(Table 1). Several new agents entering into clinical trials,
J Neurooncol (2009) 95:151–163 153
123

like dasatinib and sorafenib, inhibit multiple targets, which
may allow for the simultaneous inhibition of several criti-
cal targets with the same drug. Currently there are
numerous clinical trials using combinations of agents to
inhibit multiple targets using both combinations of targeted
therapeutics as well as combining targeted agents with
chemotherapy and/or radiation (reviewed in [41, 42]).
The role of Src in glioblastoma
The SFKs are a family of homologous members that are
located within the cytosol and act as intermediate intra-
cellular signal transducing proteins under physiologic
conditions [43, 44]. Dysregulation of the ratio of activated
to inactivated SFKs has been implicated in the develop-
ment of a number of primary tumors including colon
cancer, breast cancer, non-small cell lung cancer, pancre-
atic cancer, and glioma [4, 6–9, 45]. Such dysregulation
may be a consequence of a mutation in Src itself or over-
expression of Src-regulating proteins.
Transgenic mice which spontaneously express v-Src
develop glioblastoma tumors, the molecular biology of
which closely matches that of the human form [46]. This
finding points to the central role of Src in glioma devel-
opment and progression. Src is a key component of many
pathways important for the development and progression
of glioblastoma in that it is linked to proteins that are
overexpressed or exhibit constitutively active mutant forms
in glioblastoma cells (like EGFR, PDGFR, VEGFR, and
c-KIT).
Src plays an important role in tumor cell proliferation
via its activation of growth factor activated receptor tyro-
sine kinases, including EGFR, PDFGR, fibroblast growth
factor (FGF), and hepatocyte growth factor (HGF) [14, 47–
50]. Cyclic phosphorylation of EGFR, for example, acti-
vates the AKT pathway, a downstream effecter of PI3K,
and has been implicated in the stimulation of matrix
metalloproteinase-2 (MMP-2) secretion [51]. Src kinase
activity has also been proposed as necessary for tumor cells
to enter the cell cycle upon exposure to PDGF [10]. It is
thought that the activation of MAPK p42/44 by PDGF is
mediated through Src and Grb-2/PI3K function [52] and
Src-mediated activation of c-Abl by PDGF has also been
demonstrated to promote mitogenesis [53]. Src has also
been linked with STAT3 phosphorylation and extracellular
signal-regulated kinase (ERK) activation, resulting in the
inhibition of apoptosis in response to stress [54].
In addition to its role in regulating cell proliferation, Src
also affects cell adhesion, migration and invasion. Src
interaction with FAK leads to the formation of key com-
plexes with p130Cas, integrin avb3 and paxillin [55] and
activates PI3K [56]. Tumor cell migration is a key issue for
the diffuse nature (and unfavourable prognosis) of glio-
blastoma multiforme (GBM) in the clinic. Src also plays an
important role in focal adhesion disassembly since its
expression results in disruption of focal adhesions and
stress fibres leading to the loss of adhesion to the extra-
cellular matrix (ECM) [57]. This Src-mediated disruption
of focal adhesions leads to a decrease in cell-cell and cell-
ECM adhesion and is an important process central to cell
migration and invasion. In addition to its effects on
motility, Src may enhance cellular invasion by regulating
the expression of MMPs and tissue inhibitors of metallo-
proteinases (TIMPs) [58]. For example, FAK, a key sub-
strate of Src, can activate c-JUN kinase which results in
expression of MMP-2 and MMP-9 [59]. MMPs are known
to degrade the ECM, releasing angiogenic factors and
promoting cellular invasion.
Diffuse tumor infiltration into normal brain is one of the
key elements responsible for the unfavourable prognosis of
glioblastoma. Upregulated Src signaling may contribute to
this phenomenon via several different signaling pathways.
In human glioblastoma cells, LYN kinase activity is
exceptionally high and accounts for the majority of pan-Src
activity [60]. Additionally, LYN has been shown to aid the
localization of integrin avb3 to focal adhesion sites after
activation by PDGFR [61], thus promoting cell migration.
The promotion of the migration of glioblastoma cells by
phorbol 12-myristate 13-acetate (PMA)-activated PKC is
also regulated by Src via the Cas/Crk/Rak1 pathway [62].
Table 1 Selected multi-kinase inhibitors
Agent Company Target(s)
AEE-778 Novartis EGFR, VEGFR
AZD-2171 Astra Zeneca pan-VEGFR
Imatinib Novartis Abl, c-Kit, PDGFR
Lapatinib GSK EGFR, HER2
MLN-518 Millinium PDGFR, Flt-3, c-Kit
NVP-TAE226 Novartis FAK, IGF-1R
Pazopanib GSK VEGFR, PDGFR, c-Kit
Sorafenib Bayer Raf, VEGFR-2,-3, PDGFR,
Flt-3
Sunitinib Pfizer VEGFR, PDGFR, c-Kit, Flt-3
Vandetanib Astra Zeneca EGFR, VEGFR
Vatalinib Novartis VEGFR, PDGFR
XL-880 Exelixis c-met, VEGFR, PDGFR,
Tie-2
Bribvanib BMS VEGFR, FGFR
XL418 Exelixis AKT/p70S6K
XL765 Exelixis PI3K, mTOR
BEZ235 Novartis PI3K, mTOR
XL184 Exelixis c-met, VEGFR
OSI-906 OSI pharmaceuticals IGF-1R
154 J Neurooncol (2009) 95:151–163
123

Finally, we recently demonstrated a significant decrease in
tumor cell invasion in vitro following the pharmacologic
blockade of Src activity with dasatinib. Treated glioma
cells changed morphologically to became spindle shaped
with a loss of focal adhesion complex formation. Predict-
ably, the loss of focal adhesions was accompanied by a
decrease in FAK autophosphorylation [63].
The tumor suppressor PTEN controls the activity of the
SFK member FYN in glioblastoma cells, controlling
glioblastoma cell migration via PI3K [64]. However, de
novo glioblastoma neoplasms typically exhibit a high
frequency of PTEN mutation or dysregulation [65]. The
relationship between SFKs and PTEN is reciprocal, as Src
may phosphorylate PTEN and decrease its activity, lead-
ing to increased PI3K activity and cell growth [66].
Furthermore, CD95-induced recruitment of the SFK
member YES and the p85 subunit of PI3K have been
shown to signal invasion of glioblastoma via the glycogen
synthase kinase 3-b pathway and expression of MMPs in
a glioblastoma cell line [67]. Further evidence is provided
by Src-deficient transgenic mice, which develop consid-
erably less infiltrative glioblastomas compared with wild
type animals after orthotopic implantation of glioblastoma
cells in the brain [68]. These compelling data implicate a
role for Src in blood vessel growth and permeability
which has a reciprocal role in limiting tumor invasion into
normal brain.
Glioblastomas are highly vascularised and exhibit a high
degree of infiltration into surrounding brain tissue. EGFR is
well known as an activator of angiogenesis in normal
health and as a necessary component of tumor survival
[69]. Notably, a high frequency of glioblastomas are his-
tologically characterized by endothelial cell proliferation.
Endothelial cell proliferation is stimulated by multiple
pro-angiogenic growth factors, most notably VEGF.
VEGF-induced signaling in endothelial cells is dependent
on Src-mediated FAK activation, suggesting that Src plays
an important role in angiogenesis through its regulation of
endothelial cell migration and invasion [13]. Paugh et al.,
have shown that Src is involved in mediating the overex-
pression of plasminogen activator inhibitor-1 (PAI-1),
which is involved in glioblastoma angiogenesis, via an
EGFR-driven pathway [70]. Tumor growth may also be
driven at least in part by Src upregulation in conjunction
with cross-phosphorylation between both EGFR and FAK
[71]. Increased FAK signaling, which can be induced by
Src phosphorylation, promotes glioblastoma cell prolifer-
ation in animal models [72]. Also, Src-dependent phos-
phorylation of FAK is responsible for focal adhesion
formation and migration, and is also a promoter of survival
signaling via actin assembly and calpain activity [73].
Furthermore, hypoxia is an important stimulus for the
secretion of VEGF and Src mediates this process [74].
Finally, confirmation of the importance of Src in angio-
genesis comes from studies that block Src activity. Inac-
tivating Src [75] or blocking Src activity with small
molecule inhibitors [76] has been shown to inhibit angio-
genesis in vivo and in vitro.
Taken together, these factors suggest that targeted
inhibition of Src and its family members may be a rational
approach to treating glioblastoma. Src signaling pathways
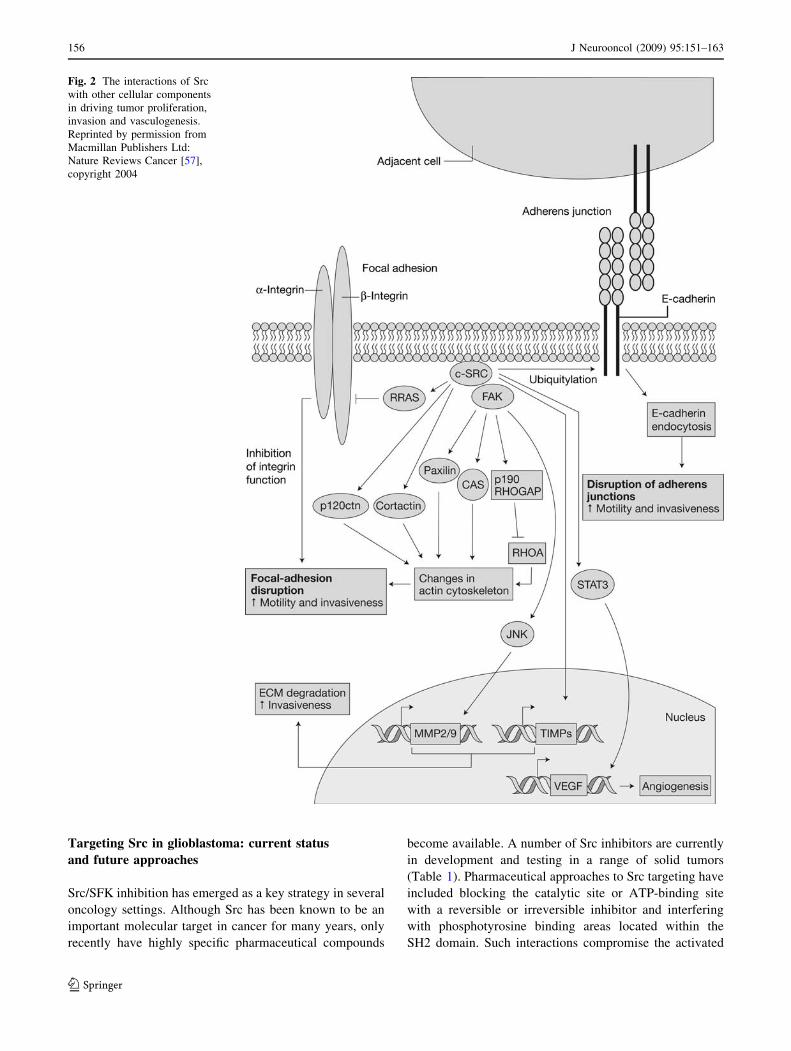
and interactions are presented in Figs. 1 [3] and 2 [57].
Fig. 1 Overview of the
downstream multiple molecular
pathways activated by Src.
Reprinted by permission from
Macmillan Publishers Ltd:
Nature Reviews Molecular Cell
Biology [3], copyright 2001
J Neurooncol (2009) 95:151–163 155
123
Targeting Src in glioblastoma: current status
and future approaches
Src/SFK inhibition has emerged as a key strategy in several
oncology settings. Although Src has been known to be an
important molecular target in cancer for many years, only
recently have highly specific pharmaceutical compounds
become available. A number of Src inhibitors are currently
in development and testing in a range of solid tumors
(Table 1). Pharmaceutical approaches to Src targeting have
included blocking the catalytic site or ATP-binding site
with a reversible or irreversible inhibitor and interfering
with phosphotyrosine binding areas located within the
SH2 domain. Such interactions compromise the activated
Fig. 2 The interactions of Src
with other cellular components
in driving tumor proliferation,
invasion and vasculogenesis.
Reprinted by permission from
Macmillan Publishers Ltd:
Nature Reviews Cancer [57],
copyright 2004
156 J Neurooncol (2009) 95:151–163
123

tertiary structure of Src and the protein scaffolding that
associates with the SH2 domain of Src. The development
of Src inhibitors has been most successful through a
combination of structure-based and screening-based
approaches. These agents may have anti-tumor activity
when used alone or augment the effects of chemotherapy or
radiotherapy. Currently, dasatinib (BMS-354825), sunitinib
(SU11248), PP2, and SU6656 have been evaluated to
varying degrees as SFK inhibitors vis-a-vis glioblastoma
[77, 78]. Of these agents, development of dasatinib is the
most advanced.
Dasatinib is an oral inhibitor of Src tyrosine kinase, as
well as Bcr-Abl, Kit, PDGFRb, and Eph receptors [79–82].
Dasatinib has potent in vitro anti-proliferative and anti-
metastatic activity mediated by Src kinase inhibition,
which has translated into preliminary evidence of clinical
activity in some patients with cancer. Dasatinib is FDA
approved for the treatment of CML and Philadelphia-
chromosome-positive (Ph?) acute lymphocytic leukemia
and is currently under investigation in a number of solid
tumors. Multiple phase I and II clinical trials of dasatinib
alone or in combination with cytotoxic chemotherapy are
ongoing in patients with solid tumors, including hormone-
refractory prostate cancer, breast cancer, metastatic colo-
rectal cancer, and non-small cell lung cancer. Preliminary
reports suggest that some patients with heavily pretreated,
chemotherapy-refractory disease respond to dasatinib.
Radiographic and prostate-specific antigen responses were
documented in a phase II study of patients with castration-
resistant prostate cancer [83] and confirmed responses have
also been reported in a phase I study of dasatinib in com-
bination with 5-fluoruracil, leucovorin, oxaliplatin (FOL-
FOX) and cetuximab in patients with metastatic colorectal
cancer [84]. These encouraging preliminary data support
the further investigation of dasatinib in patients with solid
tumors.
Dasatinib also appears to be well tolerated in patients
with solid tumors. In a phase I multiple ascending-dose
trial of dasatinib in patients with advanced solid tumors, of
11 patients treated with 140 mg once daily, only one
patient required a dose interruption and none required dose
reduction. No grade 3–4 hematologic toxicities were
observed [85]. In a separate phase I study in heavily pre-
treated patients with advanced solid tumors, of 67 subjects
treated with dasatinib twice daily, only four patients had
grade 3–4 hematologic toxicities at any time on treatment,
with no grade 3–4 thrombocytopenia reported [86].
Hematologic toxicity has also been uncommon in ongoing
phase II studies in patients with breast or prostate cancer.
Among 44 patients with advanced breast cancer treated
with 140–200 mg dasatinib twice daily, only three instan-
ces of grade 3–4 neutropenia were reported with no grade
3–4 anemia or thrombocytopenia observed [87]. These data
would suggest that dasatinib- associated myelosuppression
is rare in patients with solid tumors and that patients with
leukemia may develop myelotoxicity as a result of potent
inhibition of the disease process and not a specific toxicity
of dasatinib to normal hematopoiesis. Common nonhema-
tologic toxicities with dasatinib include fatigue, nausea/
vomiting or diarrhea, fluid retention, headache, and mus-
culoskeletal pain.
In the ongoing phase II trial of dasatinib in patients with
glioblastoma (RTOG0627), patients with glioblastoma
appeared to tolerate dasatinib better than patients with
other solid tumors. There are many potential reasons for
the improved tolerability of dasatinib in this patient pop-
ulation, including the use of high doses of corticosteroids
and the potential concurrent use of agents that activate the
CYP450 system, including proton pump inhibitors (fre-
quently given in conjunction with steroids). To evaluate
potential drug-drug interactions that might alter the
metabolism of dasatinib, an extension of this study is
planned that will evaluate dasatinib pharmacokinetics
during intrapatient dose escalation.
There is strong support for the use of dasatinib in glio-
blastoma from several preclinical studies. Dasatinib has
been shown to reduce levels of phosphorylated Src, AKT,
and ribosomal protein S6 in glioblastoma cell lines, par-
ticularly in cells with activated PTEN [63]. In addition,
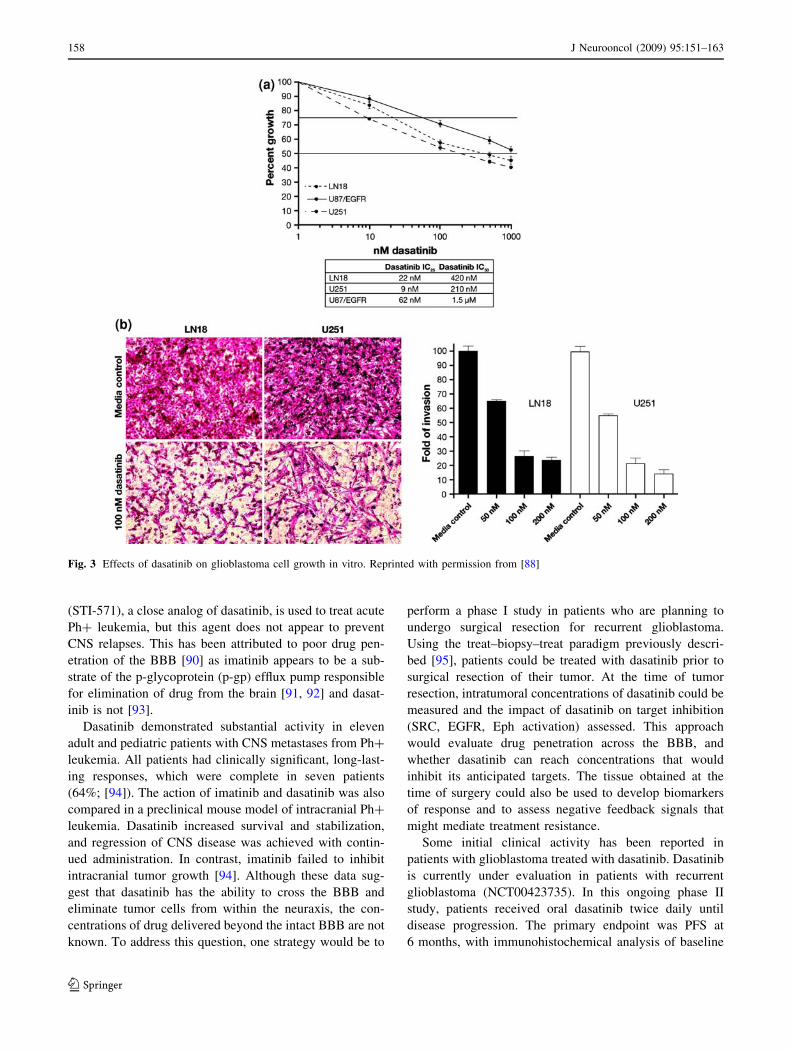
dasatinib reduces glioblastoma cell growth and invasion
(Fig. 3) [88]. At a mechanistic level, dasatinib has been
suggested to reduce the invasive potential of glioma cells
by disrupting paxillin localization to focal adhesions,
decreasing autophosphoylation of FAK, and decreasing
phosphorylation of FAK by Src. Combining dasatinib with
cytotoxic chemotherapy results in an additive or synergistic
increase in cell growth inhibition, most notably combined
with the alkylating agent temozolomide. The combination
of dasatinib and temozolomide has been reported to effect
additive increases in cell cycle disruption and autophagic
cell death when compared with the effect of temozolomide
alone. Preliminary evidence also advocates the combina-
tion of radiotherapy with Src inhibition [51, 89].
Penetration of the BBB is a key issue when treating
primary and metastatic brain tumors. There may be less of
an impediment to drug delivery to the primary, enhancing
glioblastoma as the tumor may disrupt the BBB and allow
greater access for anticancer agents. However, the more
diffuse GBM becomes within the central nervous system
(CNS), the more of an issue the intact BBB becomes. For
example, in a highly infiltrative glioblastoma, distant sites
of the tumor within the normal brain may remain unaf-
fected by drugs. As a result, the disease is likely to progress
in distant areas of the brain. Insight into the ability of Src
inhibitors to penetrate into the CNS comes from the
treatment of metastases to the brain. For example, imatinib
J Neurooncol (2009) 95:151–163 157
123
(STI-571), a close analog of dasatinib, is used to treat acute
Ph? leukemia, but this agent does not appear to prevent
CNS relapses. This has been attributed to poor drug pen-
etration of the BBB [90] as imatinib appears to be a sub-
strate of the p-glycoprotein (p-gp) efflux pump responsible
for elimination of drug from the brain [91, 92] and dasat-
inib is not [93].
Dasatinib demonstrated substantial activity in eleven
adult and pediatric patients with CNS metastases from Ph?
leukemia. All patients had clinically significant, long-last-
ing responses, which were complete in seven patients
(64%; [94]). The action of imatinib and dasatinib was also
compared in a preclinical mouse model of intracranial Ph?
leukemia. Dasatinib increased survival and stabilization,
and regression of CNS disease was achieved with contin-
ued administration. In contrast, imatinib failed to inhibit
intracranial tumor growth [94]. Although these data sug-
gest that dasatinib has the ability to cross the BBB and
eliminate tumor cells from within the neuraxis, the con-
centrations of drug delivered beyond the intact BBB are not
known. To address this question, one strategy would be to
perform a phase I study in patients who are planning to
undergo surgical resection for recurrent glioblastoma.
Using the treat–biopsy–treat paradigm previously descri-
bed [95], patients could be treated with dasatinib prior to
surgical resection of their tumor. At the time of tumor
resection, intratumoral concentrations of dasatinib could be
measured and the impact of dasatinib on target inhibition
(SRC, EGFR, Eph activation) assessed. This approach
would evaluate drug penetration across the BBB, and
whether dasatinib can reach concentrations that would
inhibit its anticipated targets. The tissue obtained at the
time of surgery could also be used to develop biomarkers
of response and to assess negative feedback signals that
might mediate treatment resistance.
Some initial clinical activity has been reported in
patients with glioblastoma treated with dasatinib. Dasatinib
is currently under evaluation in patients with recurrent
glioblastoma (NCT00423735). In this ongoing phase II
study, patients received oral dasatinib twice daily until
disease progression. The primary endpoint was PFS at
6 months, with immunohistochemical analysis of baseline
Fig. 3 Effects of dasatinib on glioblastoma cell growth in vitro. Reprinted with permission from [88]
158 J Neurooncol (2009) 95:151–163
123

tumor tissue to determine if a molecular signature of da-
satinib targets (e.g. the presence of Src, PDGFR, EPHA2,
and c-KIT) predicts sensitivity to or clinical outcome to
dasatinib treatment. An interim analysis of this study was
recently presented at the Society for Neuro-Oncology
annual meeting. A total of 78% of patients had 2 or more of
the presumptive molecular targets of dasatinib described
above. Although efficacy data are not available, dasatinib
at a dose of 100 mg/day was reasonably well tolerated with
no patients having grade 4 or 5 toxicities [96]. Although
other SFK member (such as the highly expressed LYN and
FYN) protein expression or phosphorylation status may
need to be evaluated in this study, the expression of other
SFK-related proteins may prove more predictive of
response. Among a larger panel of genes identified, Huang
et al., identified a six gene expression profile that corre-
lated with breast cancer cell line response to dasatinib. The
six genes that correlated with response included EPHA2,
CAV1, CAV2, ANXA1, PTRF, and IGFBP2 [97], all of
which are either targets of dasatinib, substrates for SFK or
are part of downstream signaling pathways mediated by
SFK. Although Src expression itself did not correlate with
response, the overall activation status of the Src pathway
was important. Ultimately, the use of a biomarker panel to
aid in the selection of patients most likely to respond to
dasatinib has enormous clinical utility and is the first step
in individualizing cancer treatment.
Like most targets in glioblastoma, however, single-agent
activity is likely to be limited and SFK inhibition will be
most effective in combination with other therapies. Src
inhibitors combined with radiation or cytotoxic chemo-
therapy or other novel agents could effectively inhibit
tumor growth and overcome intrinsic resistance to single-
agent therapy [98]. An alternate approach is to use agents
like dasatinib which have promiscuous inhibitory activity
against multiple tyrosine kinases (dasatinib also inhibits
PDGFR, c-KIT, and EPHA2). Currently, dasatinib is being
investigated in combination with erlotinib (an EGFR
inhibitor) in patients with malignant glioblastoma in a
phase I study (NCT00609999). Src is thought to potentiate
EGFR signaling [99] and thus targeting both EGFR and Src
may have synergistic benefit to patients with glioblastoma.
There has recently been an interest in developing combi-
nation strategies that may block the pro-invasive phenotype
driven by the use of anti-VEGF therapies in glioblastoma
[26, 100]. Given its ability to block in vitro tumor migra-
tion [63], dasatinib may be effective in inhibiting tumor
invasion when used in combination with anti-angiogenic
agents. Dasatinib, characterized previously as an anti-
invasive but cytostatic agent in glioblastoma, may also be
successfully combined with radiation therapy or anti-
angiogenic therapy, which has been recently shown to
increase tumor invasion [26]. We are opening a phase I/II
clinical of dasatinib in combination with radiation and
temozolomide followed by adjuvant dasatinib and tem-
ozolomide for patients with newly diagnosed glioblastoma.
In addition to evaluating the impact of Src inhibition on
progression free survival and outcome, we will be inter-
rogating tumor tissue for potential biomarkers of response.
These studies will improve our understanding of the role of
Src in glioma development and the impact of inhibiting Src
on patient outcome.
Summary and conclusions
Malignant gliomas are diffusely infiltrative tumors that
migrate and invade extensive areas of the brain; this
invasive phenotype often mediates tumor recurrence.
Glioblastoma is a disease with a poor prognosis; most
patients will progress within six to nine months despite
treatment with surgery, radiation and adjuvant chemo-
therapy. Clearly, new therapeutic options are needed.
Agents that prevent the invasion of tumor cells throughout
the brain are likely to provide meaningful improvements in
survival for patients with glioblastoma. The prominent role
of Src in activating downstream signaling through the Ras/
MAPK and PI3K pathways in promoting tumor prolifera-
tion and invasion makes it an attractive molecular target in
glioblastoma. Pharmacologic inhibition of Src/SFK repre-
sents a promising strategy for improving glioblastoma
treatment by limiting tumor invasion into normal brain, a
rationale supported by preclinical data.
Although Src inhibitors are poised to deliver promising
results, the optimal therapeutic strategy will involve
rationale combination of Src inhibitors with agents that
exert synergistic anti-glioma activity. Because the biology
of malignant gliomas involves a complex network of inter-
connected signaling pathways, careful preclinical interro-
gation is necessary to determine the optimal treatment
combinations. Evaluation of tumor tissue before and after
treatment with Src inhibitors will be necessary to fully
realize the impact of this therapy on the tumor and to
inform the next generation of clinical trials involving
treatments that block mechanisms of escape from Src
inhibition. As more clinical information about Src/SFK
inhibitors emerges, a clearer picture of the molecular
determinants of response will define who will benefit from
this strategy and how it integrates into current glioblastoma
therapy. In the future, molecular profiles will be used to
determine which molecular targets predict response
allowing for individualization of cancer therapy.
Integrating SFK inhibitors into the treatment of primary
brain tumors provides an invaluable opportunity to advance
the field of neuro-oncology. Realistically, single-agent
studies might be expected to lead to clinical responses in
J Neurooncol (2009) 95:151–163 159
123

recurrent glioblastoma but have little impact on progres-
sion free survival due to the activation of multiple redun-
dant signaling pathways in these tumors [101]. However,
when combined in synergistic combinations with chemo-
therapy and/or radiation, these agents may significantly
improve 6-month progression free and overall survival
rates compared with historical controls. Given the highly
invasive phenotype of malignant gliomas, Src inhibitors
may improve overall survival by inhibiting tumor invasion
into normal brain hidden from the effects of cytotoxic
therapies. Novel endpoints could be used to measure a
decrease in tumor cell invasion using noninvasive MR
imaging biomarker endpoints in these studies. There is also
a potential role for Src/SFK inhibitors in the treatment of
brain metastases from other primary tumors. Also, given
the elevated incidence of glioblastoma in children, the
inclusion of pediatric patients should be strongly encour-
aged in clinical trials. A first step toward this may be to
lower the inclusion age for recruitment to trials to include
younger adults/older children.
In summary, Src inhibitors hold great promise for the
treatment of glioblastoma. In the near future, the treatment
of glioblastoma will include combining the old SRC
(Surgery, Radiotherapy, Chemotherapy) with the new Src
(sarcoma tyrosine kinase inhibitors).
Acknowledgments Editorial and writing support was provided by
Gardiner-Caldwell US, funded by Bristol-Myers Squibb.
References
1. Louis DN, Ohgaki H, Wiestler OD et al (2007) The 2007 WHO
classification of tumours of the central nervous system. Acta
Neuropathol 114:97–109. doi:10.1007/s00401-007-0243-4
2. Giese A, Bjerkvig R, Berens ME et al (2003) Cost of migration:
invasion of malignant gliomas and implications for treatment. J
Clin Oncol 21:1624–1636. doi:10.1200/JCO.2003.05.063
3. Martin GS (2001) The hunting of the Src. Nat Rev Mol Cell Biol
2:467–475. doi:10.1038/35073094
4. Summy JM, Gallick GE (2003) Src family kinases in tumor
progression and metastasis. Cancer Metastasis Rev 22:337–358.
doi:10.1023/A:1023772912750
5. Thomas SM, Brugge JS (1997) Cellular functions regulated by
Src family kinases. Annu Rev Cell Dev Biol 13:513–609. doi:
10.1146/annurev.cellbio.13.1.513
6. Brabek J, Constancio SS, Siesser PF et al (2005) Crk-associated
substrate tyrosine phosphorylation sites are critical for invasion
and metastasis of SRC-transformed cells. Mol Cancer Res
3:307–315. doi:10.1158/1541-7786.MCR-05-0015
7. Dehm S, Senger MA, Bonham K (2001) SRC transcriptional
activation in a subset of human colon cancer cell lines. FEBS
Lett 487:367–371. doi:10.1016/S0014-5793(00)02354-1
8. Dehm SM, Bonham K (2004) SRC gene expression in human
cancer: the role of transcriptional activation. Biochem Cell Biol
82:263–274. doi:10.1139/o03-077
9. Irby RB, Yeatman TJ (2000) Role of Src expression and acti-
vation in human cancer. Oncogene 19:5636–5642. doi:10.1038/
sj.onc.1203912
10. Courtneidge SA (2002) Role of Src in signal transduction
pathways. The Jubilee Lecture. Biochem Soc Trans 30:11–17.
doi:10.1042/BST0300011
11. Frame MC (2002) Src in cancer: deregulation and consequences
for cell behaviour. Biochim Biophys Acta 1602:114–130
12. Niu G, Wright KL, Huang M et al (2002) Constitutive Stat3
activity up-regulates VEGF expression and tumor angiogenesis.
Oncogene 21:2000–2008. doi:10.1038/sj.onc.1205260
13. Abu-Ghazaleh R, Kabir J, Jia H et al (2001) Src mediates
stimulation by vascular endothelial growth factor of the phos-
phorylation of focal adhesion kinase at tyrosine 861, and
migration and anti-apoptosis in endothelial cells. Biochem J
360:255–264. doi:10.1042/0264-6021:3600255
14. Tice DA, Biscardi JS, Nickles AL et al (1999) Mechanism of
biological synergy between cellular Src and epidermal growth
factor receptor. Proc Natl Acad Sci USA 96:1415–1420. doi:
10.1073/pnas.96.4.1415
15. National Comprehensive Cancer Network (NCCN) (2008) Clin-
ical practice guidelines in oncology: central nervous system
cancers, V.1.2008. Available at http://www.nccn.org/professionals/
physician_gls/PDF/cns.pdf. Accessed Sept 2008
16. Hentschel SJ, Sawaya R (2003) Optimizing outcomes with
maximal surgical resection of malignant gliomas. Cancer Con-
trol 10:109–114
17. Walker MD, Alexander E Jr, Hunt WE et al (1978) Evaluation
of BCNU and/or radiotherapy in the treatment of anaplastic
gliomas. A cooperative clinical trial. J Neurosurg 49:333–343
18. Walker MD, Green SB, Byar DP et al (1980) Randomized
comparisons of radiotherapy and nitrosoureas for the treatment
of malignant glioma after surgery. N Engl J Med 303:1323–
1329
19. Stewart LA (2002) Chemotherapy in adult high-grade glioma: a
systematic review and meta-analysis of individual patient data
from 12 randomised trials. Lancet 359:1011–1018. doi:10.1016/
S0140-6736(02)08091-1
20. Stupp R, Mason WP, Van den Bent MJ et al (2005) Radio-
therapy plus concomitant and adjuvant temozolomide for glio-
blastoma. N Engl J Med 352:987–996. doi:10.1056/NEJMoa
043330
21. Hegi ME, Diserens AC, Gorlia T et al (2005) MGMT gene
silencing and benefit from temozolomide in glioblastoma. N
Engl J Med 352:997–1003. doi:10.1056/NEJMoa043331
22. Stupp R, Hegi ME, Van den Bent MJ et al (2006) Chang-
ing paradigms–an update on the multidisciplinary management
of malignant glioma. Oncologist 11:165–180. doi:10.1634/the
oncologist.11-2-165
23. Hart MG, Grant R, Garside R et al (2008) Chemotherapeutic
wafers for high grade glioma. Cochrane Database Syst Rev
CD007294
24. Prados MD, Lamborn K, Yung WK et al (2006) A phase 2 trial
of irinotecan (CPT-11) in patients with recurrent malignant
glioma: a North American brain tumor consortium study. Neuro
Oncol 8:189–193. doi:10.1215/15228517-2005-010
25. Brandes AA, Tosoni A, Basso U et al (2004) Second-line che-
motherapy with irinotecan plus carmustine in glioblastoma
recurrent or progressive after first-line temozolomide chemo-
therapy: a phase II study of the Gruppo Italiano Cooperativo di
Neuro-Oncologia (GICNO). J Clin Oncol 22:4779–4786. doi:
10.1200/JCO.2004.06.181
26. Norden AD, Young GS, Setayesh K et al (2008) Bevacizumab
for recurrent malignant gliomas: efficacy, toxicity, and patterns
of recurrence. Neurology 70:779–787. doi:10.1212/01.wnl.0000
304121.57857.38
27. Vredenburgh JJ, Desjardins A, Herndon JE et al (2007) Bev-
acizumab plus irinotecan in recurrent glioblastoma multiforme.
J Clin Oncol 25:4722–4729. doi:10.1200/JCO.2007.12.2440
160 J Neurooncol (2009) 95:151–163
123

28. Reardon DA, Quinn JA, Vredenburgh J et al (2005) Phase II trial
of irinotecan plus celecoxib in adults with recurrent malignant
glioma. Cancer 103:329–338. doi:10.1002/cncr.20776
29. Yung WK, Mechtler L, Gleason MJ (1991) Intravenous carbo-
platin for recurrent malignant glioma: a phase II study. J Clin
Oncol 9:860–864
30. Newton HB, Junck L, Bromberg J et al (1990) Procarbazine
chemotherapy in the treatment of recurrent malignant astrocy-
tomas after radiation and nitrosourea failure. Neurology 40:
1743–1746
31. Rodriguez LA, Prados M, Silver P et al (1989) Reevaluation of
procarbazine for the treatment of recurrent malignant central
nervous system tumors. Cancer 64:2420–2423. doi:10.1002/
1097-0142(19891215)64:12\2420::AID-
CNCR2820641204[3.0.CO;2-B
32. Cloughesy T, Prados M, Wen PY et al. (2008) A phase II,
randomized, non-comparative clinical trial of the effect of
bevacizumab (BV) alone or in combination with irinotecan
(CPT) on 6-month progression free survival (PFS6) in recurrent,
treatment-refractory glioblastoma (GBM). J Clin Oncol 26:
2010b
33. Wong ET, Hess KR, Gleason MJ et al (1999) Outcomes and
prognostic factors in recurrent glioma patients enrolled onto
phase II clinical trials. J Clin Oncol 17:2572–2578
34. O’Brien SG, Guilhot F, Larson RA et al (2003) Imatinib com-
pared with interferon and low-dose cytarabine for newly diag-
nosed chronic-phase chronic myeloid leukemia. N Engl J Med
348:994–1004. doi:10.1056/NEJMoa022457
35. Louis DN (2006) Molecular pathology of malignant gliomas.
Annu Rev Pathol 1:97–117. doi:10.1146/annurev.pathol.1.110
304.100043
36. Sanson M, Thillet J, Hoang-Xuan K (2004) Molecular changes
in gliomas. Curr Opin Oncol 16:607–613. doi:10.1097/01.cco.
0000142485.81849.cc
37. Sebolt-Leopold JS, Herrera R (2004) Targeting the mitogen-
activated protein kinase cascade to treat cancer. Nat Rev Cancer
4:937–947. doi:10.1038/nrc1503
38. Castellino RC, Durden DL (2007) Mechanisms of disease: the
PI3K-Akt-PTEN signaling node–an intercept point for the con-
trol of angiogenesis in brain tumors. Nat Clin Pract Neurol
3:682–693. doi:10.1038/ncpneuro0661
39. Cancer Genome Atlas (2008) Comprehensive genomic charac-
terization defines human glioblastoma genes and core pathways.
Nature 455:1061–1068. doi:10.1038/nature07385
40. Du J, Bernasconi P, Clauser KR et al (2009) Bead-based pro-
filing of tyrosine kinase phosphorylation identifies SRC as a
potential target for glioblastoma therapy. Nat Biotechnol 27:77–
83. doi:10.1038/nbt.1513
41. Chi AS, Wen PY (2007) Inhibiting kinases in malignant glio-
mas. Expert Opin Ther Targets 11:473–496. doi:10.1517/14728
222.11.4.473
42. Gonzalez J, de GJ (2008) Combination therapy for malignant
glioma based on PTEN status. Expert Rev Anticancer Ther
8:1767–1779. doi:10.1586/14737140.8.11.1767
43. Lutz MP, Esser IB, Flossmann-Kast BB et al (1998) Overex-
pression and activation of the tyrosine kinase Src in human
pancreatic carcinoma. Biochem Biophys Res Commun 243:
503–508. doi:10.1006/bbrc.1997.8043
44. Zeng L, Si X, Yu WP et al (2003) PTP alpha regulates integrin-
stimulated FAK autophosphorylation and cytoskeletal rear-
rangement in cell spreading and migration. J Cell Biol 160:137–
146. doi:10.1083/jcb.200206049
45. Irby RB, Mao W, Coppola D et al (1999) Activating SRC
mutation in a subset of advanced human colon cancers. Nat
Genet 21:187–190. doi:10.1038/5971
46. Weissenberger J, Steinbach JP, Malin G et al (1997) Develop-
ment and malignant progression of astrocytomas in GFAP-v-src
transgenic mice. Oncogene 14:2005–2013. doi:10.1038/sj.onc.
1201168
47. Bowman T, Broome MA, Sinibaldi D et al (2001) Stat3-medi-
ated Myc expression is required for Src transformation and
PDGF-induced mitogenesis. Proc Natl Acad Sci USA 98:7319–
7324. doi:10.1073/pnas.131568898
48. De Mali KA, Godwin SL, Soltoff SP et al (1999) Multiple roles
for Src in a PDGF-stimulated cell. Exp Cell Res 253:271–279.
doi:10.1006/excr.1999.4669
49. Landgren E, Blume-Jensen P, Courtneidge SA et al (1995)
Fibroblast growth factor receptor-1 regulation of Src family
kinases. Oncogene 10:2027–2035
50. Mao W, Irby R, Coppola D et al (1997) Activation of c-Src by
receptor tyrosine kinases in human colon cancer cells with high
metastatic potential. Oncogene 15:3083–3090. doi:10.1038/sj.
onc.1201496
51. Park CM, Park MJ, Kwak HJ et al (2006) Ionizing radiation
enhances matrix metalloproteinase-2 secretion and invasion of
glioma cells through Src/epidermal growth factor receptor-
mediated p38/Akt and phosphatidylinositol 3-kinase/Akt sig-
naling pathways. Cancer Res 66:8511–8519. doi:10.1158/0008-5472.CAN-05-4340
52. Conway AM, Rakhit S, Pyne S et al (1999) Platelet-derived-
growth-factor stimulation of the p42/p44 mitogen-activated
protein kinase pathway in airway smooth muscle: role of per-
tussis-toxin-sensitive G-proteins, c-Src tyrosine kinases and
phosphoinositide 3-kinase. Biochem J 337(Pt 2):171–177. doi:
10.1042/0264-6021:3370171
53. Furstoss O, Dorey K, Simon V et al (2002) c-Abl is an effector
of Src for growth factor-induced c-myc expression and DNA
synthesis. EMBO J 21:514–524. doi:10.1093/emboj/21.4.514
54. Kitagawa D, Tanemura S, Ohata S et al (2002) Activation of
extracellular signal-regulated kinase by ultraviolet is mediated
through Src-dependent epidermal growth factor receptor phos-
phorylation. Its implication in an anti-apoptotic function. J Biol
Chem 277:366–371. doi:10.1074/jbc.M107110200
55. Cox BD, Natarajan M, Stettner MR et al (2006) New con-
cepts regarding focal adhesion kinase promotion of cell migra-
tion and proliferation. J Cell Biochem 99:35–52. doi:10.1002/
jcb.20956
56. Band CJ, Mounier C, Posner BI (1999) Epidermal growth factor
and insulin-induced deoxyribonucleic acid synthesis in primary
rat hepatocytes is phosphatidylinositol 3-kinase dependent and
dissociated from protooncogene induction. Endocrinology
140:5626–5634. doi:10.1210/en.140.12.5626
57. Yeatman TJ (2004) A renaissance for SRC. Nat Rev Cancer
4:470–480. doi:10.1038/nrc1366
58. Noritake H, Miyamori H, Goto C et al (1999) Overexpression of
tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) in
metastatic MDCK cells transformed by v-src. Clin Exp Metas-
tasis 17:105–110. doi:10.1023/A:1006596620406
59. Hsia DA, Mitra SK, Hauck CR et al (2003) Differential regu-
lation of cell motility and invasion by FAK. J Cell Biol
160:753–767. doi:10.1083/jcb.200212114
60. Stettner MR, Wang W, Nabors LB et al (2005) Lyn kinase
activity is the predominant cellular SRC kinase activity in
glioblastoma tumor cells. Cancer Res 65:5535–5543. doi:
10.1158/0008-5472.CAN-04-3688
61. Ding Q, Stewart J Jr, Olman MA et al (2003) The pattern of
enhancement of Src kinase activity on platelet-derived growth
factor stimulation of glioblastoma cells is affected by the inte-
grin engaged. J Biol Chem 278:39882–39891. doi:10.1074/
jbc.M304685200
J Neurooncol (2009) 95:151–163 161
123

62. Nomura N, Nomura M, Sugiyama K et al (2007) Src regulates
phorbol 12-myristate 13-acetate-activated PKC-induced migra-
tion via Cas/Crk/Rac1 signaling pathway in glioblastoma cells.
Int J Mol Med 20:511–519
63. Milano V, LaFortune T, de Groot JF (2008) Dasatinib-induced
authophagy is synergistically enhanced in combination with
temozolomide and is further augmented in PTEN-functional
glioma. In: Proceedings of the 99th annual meeting of the
American association for cancer research, Apr 12–16, 2008; San
Diego, CA
64. Dey N, Crosswell HE, De P et al. (2008) The protein phos-
phatase activity of PTEN regulates SRC family kinases and
controls glioma migration. Cancer Res 68:1862–1871. doi:
10.1158/0008-5472.CAN-07-1182
65. Simpson L, Parsons R (2001) PTEN: life as a tumor suppressor.
Exp Cell Res 264:29–41. doi:10.1006/excr.2000.5130
66. Edwards J, Krishna NS, Witton CJ et al (2003) Gene amplifi-
cations associated with the development of hormone-resistant
prostate cancer. Clin Cancer Res 9:5271–5281
67. Kleber S, Sancho-Martinez I, Wiestler B et al (2008) Yes and
PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell
13:235–248. doi:10.1016/j.ccr.2008.02.003
68. Lund CV, Nguyen MT, Owens GC et al (2006) Reduced glioma
infiltration in Src-deficient mice. J Neurooncol 78:19–29. doi:
10.1007/s11060-005-9068-y
69. Dancey JE, Freidlin B (2003) Targeting epidermal growth factor
receptor–are we missing the mark? Lancet 362:62–64. doi:
10.1016/S0140-6736(03)13810-X
70. Paugh BS, Paugh SW, Bryan L et al (2008) EGF regulates plas-
minogen activator inhibitor-1 (PAI-1) by a pathway involving
c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells.
FASEB J 22:455–465. doi:10.1096/fj.07-8276com
71. Schaller MD, Hildebrand JD, Shannon JD et al (1994) Auto-
phosphorylation of the focal adhesion kinase, pp125FAK,
directs SH2-dependent binding of pp60src. Mol Cell Biol
14:1680–1688
72. Natarajan M, Hecker TP, Gladson CL (2003) FAK signaling in
anaplastic astrocytoma and glioblastoma tumors. Cancer J 9:
126–133. doi:10.1097/00130404-200303000-00008
73. Westhoff MA, Serrels B, Fincham VJ et al (2004) SRC-medi-
ated phosphorylation of focal adhesion kinase couples actin and
adhesion dynamics to survival signaling. Mol Cell Biol 24:
8113–8133. doi:10.1128/MCB.24.18.8113-8133.2004
74. Mukhopadhyay D, Tsiokas L, Zhou XM et al (1995) Hypoxic
induction of human vascular endothelial growth factor expres-
sion through c-Src activation. Nature 375:577–581. doi:
10.1038/375577a0
75. Kilarski WW, Jura N, Gerwins P (2003) Inactivation of Src
family kinases inhibits angiogenesis in vivo: implications for a
mechanism involving organization of the actin cytoskeleton.
Exp Cell Res 291:70–82. doi:10.1016/S0014-4827(03)00374-4
76. Laird AD, Li G, Moss KG et al. (2003) Src family kinase
activity is required for signal tranducer and activator of tran-
scription 3 and focal adhesion kinase phosphorylation and vas-
cular endothelial growth factor signaling in vivo and for
anchorage-dependent and -independent growth of human tumor
cells. Mol Cancer Ther 2:461–469
77. Angers-Loustau A, Hering R, Werbowetski TE et al (2004) SRC
regulates actin dynamics and invasion of malignant glial cells in
three dimensions. Mol Cancer Res 2:595–605
78. de Bouard S, Herlin P, Christensen JG et al (2007) Antiangio-
genic and anti-invasive effects of sunitinib on experimental
human glioblastoma. Neuro Oncol 9:412–423. doi:10.1215/152
28517-2007-024
79. Lee FY, Wen ML, Bhide R et al (2005) Dasatinib (BMS-
354825) overcomes multiple mechanisms of imatinib resistance
in chronic myeloid leukemia. Blood (ASH Annual Meeting
Abstracts) 106:Abstract 1994
80. Lombardo LJ, Lee FY, Chen P et al (2004) Discovery of N-(2-
chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-
1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide
(BMS-354825), a dual Src/Abl kinase inhibitor with potent
antitumor activity in preclinical assays. J Med Chem 47:6658–
6661. doi:10.1021/jm049486a
81. Schittenhelm MM, Shiraga S, Schroeder A et al (2006) Dasat-
inib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits
the kinase activity of wild-type, juxtamembrane, and activation
loop mutant KIT isoforms associated with human malignancies.
Cancer Res 66:473–481. doi:10.1158/0008-5472.CAN-05-2050
82. Shah NP, Lee FY, Luo R et al (2006) Dasatinib (BMS-354825)
inhibits KITD816V, an imatinib-resistant activating mutation
that triggers neoplastic growth in the majority of patients with
systemic mastocytosis. Blood 108:286–291. doi:10.1182/blood-
2005-10-3969
83. Yu EY, Massard C, Gross M et al (2009) A phase II study of
once-daily dasatinib for patients with castration-resistant pros-
tate cancer (CA180085). J Clin Oncol (ASCO Genitourinary
Cancers Symposium Abstracts) (abstract 165)
84. Kopetz S, Wolff RA, Glover K et al (2008) Phase I study of Src
inhibition with dasatinib in combination with 5-fluoruracil,
leucovorin, oxaliplatin (FOLFOX) and cetuximab in metastatic
colorectal cancer. J Clin Oncol (ASCO Gastrointestinal Cancers
Symposium Abstracts) (abstract 325)
85. Bristol-Myers Squibb (2009) Synopsis: Final Clinical Study
Report for CA180021. Available at http://ctr.bms.com/pdf//
CA180021.pdf. Accessed Mar 2009
86. Bristol-Myers Squibb (2009) Synopsis: Final Clinical Study
Report for CA180003. Available at http://ctr.bms.com/pdf/CA
180003.pdf. Accessed Mar 2009
87. Finn RS, Bengala C, Ibrahim N et al (2008) Phase II trial of
dasatinib in triple-negative breast cancer: results of study
CA180059. 31st Annual San Antonio Breast Cancer Sympo-
sium, 10–14 Dec, 2008 (abstract 3118)
88. Milano V, Piao Y, La Fortune T et al (2009) Dasatinib-induced
autophagy is enhanced in combination with temozolomide in
glioma. Mol Cancer Ther 8:394–406. doi:10.1158/1535-7163.
MCT-08-0669
89. Cuneo KC, Geng L, Tan J et al (2006) SRC family kinase
inhibitor SU6656 enhances antiangiogenic effect of irradiation.
Int J Radiat Oncol Biol Phys 64:1197–1203. doi:10.1016/j.
ijrobp.2005.11.014
90. Takayama N, Sato N, O’Brien SG et al (2002) Imatinib mesylate
has limited activity against the central nervous system
involvement of Philadelphia chromosome-positive acute lym-
phoblastic leukaemia due to poor penetration into cerebrospinal
fluid. Br J Haematol 119:106–108. doi:10.1046/j.1365-2141.
2002.03881.x
91. Bihorel S, Camenisch G, Lemaire M et al (2007) Influence of
breast cancer resistance protein (Abcg2) and p-glycoprotein
(Abcb1a) on the transport of imatinib mesylate (Gleevec) across
the mouse blood-brain barrier. J Neurochem 102:1749–1757.
doi:10.1111/j.1471-4159.2007.04808.x
92. Decleves X, Bihorel S, Debray M et al (2008) ABC transporters
and the accumulation of imatinib and its active metabolite
CGP74588 in rat C6 glioma cells. Pharmacol Res 57:214–222.
doi:10.1016/j.phrs.2008.01.006
93. Mahon FX, Hayette S, Lagarde V et al (2008) Evidence that
resistance to nilotinib may be due to BCR-ABL, Pgp, or Src
kinase overexpression. Cancer Res 68:9809–9816. doi:10.1158/
0008-5472.CAN-08-1008
94. Porkka K, Koskenvesa P, Lundan T et al (2008) Dasatinib
crosses the blood-brain barrier and is an efficient therapy for
162 J Neurooncol (2009) 95:151–163
123

central nervous system Philadelphia chromosome-positive leu-
kemia. Blood 112:1005–1012. doi:10.1182/blood-2008-02-14
0665
95. Cloughesy TF, Yoshimoto K, Nghiemphu P et al (2008) Anti-
tumor activity of rapamycin in a Phase I trial for patients with
recurrent PTEN-deficient glioblastoma. PLoS Med 5:e8. doi:
10.1371/journal.pmed.0050008
96. Lassman A, Wang M, Glibert M et al (2008) Phase ii trial of
dasatinib for recurrent glioblastoma (rtog 0627). Neuro Oncol
10:824
97. Huang F, Reeves K, Han X et al (2007) Identification of can-
didate molecular markers predicting sensitivity in solid tumors
to dasatinib: rationale for patient selection. Cancer Res 67:2226–
2238. doi:10.1158/0008-5472.CAN-06-3633
98. Dancey JE, Chen HX (2006) Strategies for optimizing combi-
nations of molecularly targeted anticancer agents. Nat Rev Drug
Discov 5:649–659. doi:10.1038/nrd2089
99. Maa MC, Leu TH, McCarley DJ et al (1995) Potentiation
of epidermal growth factor receptor-mediated oncogenesis by
c-Src: implications for the etiology of multiple human cancers.
Proc Natl Acad Sci USA 92:6981–6985. doi:10.1073/pnas.92.
15.6981
100. Bergers G, Hanahan D (2008) Modes of resistance to anti-
angiogenic therapy. Nat Rev Cancer 8:592–603. doi:10.1038/
nrc2442
101. Stommel JM, Kimmelman AC, Ying H et al (2007) Coactivation
of receptor tyrosine kinases affects the response of tumor cells to
targeted therapies. Science 318:287–290. doi:10.1126/science.
1142946
J Neurooncol (2009) 95:151–163 163
123




















