
Implications for Alzheimer's disease - CiteSeerX
11
Neurobiology of Aging 27 (2006) 1239–1249 Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human A(1–42): Implications for Alzheimer’s disease Debra Boyd-Kimball a , H. Fai Poon a , Bert C. Lynn a,b , Jian Cai d , William M. Pierce Jr. d , Jon B. Klein e , Jmil Ferguson f , Christopher D. Link f , D. Allan Butterfield a,c,∗ a Department of Chemistry, Center of Membrane Sciences, and Sanders-Brown Center on Aging, 121 Chemistry-Physics Building, University of Kentucky, Lexington, KY 40506-0055, USA b Core Proteomics Laboratory, University of Kentucky, Lexington, KY, USA c Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA d Department of Pharmacology and Toxicology, University of Louisville School of Medicine and VAMC, Louisville, KY, USA e Kidney Disease Program and Proteomics Core Laboratory, University of Louisville School of Medicine and VAMC, Louisville, KY, USA f Institute for Behavioral Genetics, University of Colorado, Boulder, CO, USA Received 20 December 2004; received in revised form 17 May 2005; accepted 1 July 2005 Available online 11 August 2005 Abstract Protein oxidation has been shown to lead to loss of protein function, increased protein aggregation, decreased protein turnover, decreased membrane fluidity, altered cellular redox poteintial, loss of Ca 2+ homeostaisis, and cell death. There is increasing evidence that protein oxidation is involved in the pathogenesis of Alzheimer’s disease and amyloid beta-peptide (1–42) has been implicated as a mediator of oxidative stress in AD. However, the specific implications of the oxidation induced by A(1–42) on the neurodegeneration evident in AD are unknown. In this study, we used proteomic techniques to identify specific targets of oxidation in transgenic Caenorhabditis elegans (C. elegans) expressing human A(1–42). We identified 16 oxidized proteins involved in energy metabolism, proteasome function, and scavenging of oxidants that are more oxidized compared to control lines. These results are discussed with reference to Alzheimer’s disease. © 2005 Elsevier Inc. All rights reserved. Keywords: Alzheimer’s disease; Amyloid -peptide; Protein oxidation; Proteomics; C. elegans 1. Introduction Protein oxidation [13] is extensive in Alzheimer’s dis- ease (AD) [14,26,30,39]. Recent proteomics studies in our laboratory have identified specific targets of protein oxida- tion in AD brain, assessed by increased carbonyls, such as, creatine kinase BB, glutamine synthase, ubiquitin carboxy- terminal hydrolase L-1, dihydropyrimidase-related protein 2, -enolase, and heat shock cognate 71, indicating that a number of cellular mechanisms are affected including energy metabolism, protein turnover, and neuronal communication ∗ Corresponding author. Tel.: +1 859 257 3184; fax: +1 859 257 5876. E-mail address: [email protected] (D.A. Butterfield). [16,17]. Alzheimer’s disease brain is characterized patholog- ically by synapse loss and by the presence of senile plaques, neurofibrillary tangles, and neuropil threads. Extracellular senile plaques are composed primarily of fibrilar deposits of amyloid beta peptide (1–42) [A(1–42)], a product of proteolytic cleavage of the transmembrane protein, amyloid precursor protein (APP). On the other hand, intracellular neu- rofibrillary tangles are composed of paired helical filaments formed from hyperphosphorylated tau, a microtubule associ- ated protein [25,33,57]. A(1–42) has been proposed to play a central role in the pathogenesis of AD as a mediator of oxidative stress [11,12,15]. However, the mechanism by which A(1–42)- induced protein oxidation occurs is unknown. Several 0197-4580/$ – see front matter © 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2005.07.001
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Implications for Alzheimer's disease - CiteSeerX

Neurobiology of Aging 27 (2006) 1239–1249
Proteomic identification of proteins specifically oxidized inCaenorhabditis elegans expressing human A�(1–42):
Implications for Alzheimer’s disease
Debra Boyd-Kimball a, H. Fai Poon a, Bert C. Lynn a,b, Jian Cai d, William M. Pierce Jr. d,Jon B. Klein e, Jmil Ferguson f, Christopher D. Link f, D. Allan Butterfield a,c,∗
a Department of Chemistry, Center of Membrane Sciences, and Sanders-Brown Center on Aging, 121 Chemistry-Physics Building,University of Kentucky, Lexington, KY 40506-0055, USA
b Core Proteomics Laboratory, University of Kentucky, Lexington, KY, USAc Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA
d Department of Pharmacology and Toxicology, University of Louisville School of Medicine and VAMC, Louisville, KY, USAe Kidney Disease Program and Proteomics Core Laboratory, University of Louisville School of Medicine and VAMC, Louisville, KY, USA
f Institute for Behavioral Genetics, University of Colorado, Boulder, CO, USA
Received 20 December 2004; received in revised form 17 May 2005; accepted 1 July 2005Available online 11 August 2005
A
miitha©
K
1
eltct2nm
0d
bstract
Protein oxidation has been shown to lead to loss of protein function, increased protein aggregation, decreased protein turnover, decreasedembrane fluidity, altered cellular redox poteintial, loss of Ca2+ homeostaisis, and cell death. There is increasing evidence that protein oxidation
s involved in the pathogenesis of Alzheimer’s disease and amyloid beta-peptide (1–42) has been implicated as a mediator of oxidative stressn AD. However, the specific implications of the oxidation induced by A�(1–42) on the neurodegeneration evident in AD are unknown. Inhis study, we used proteomic techniques to identify specific targets of oxidation in transgenic Caenorhabditis elegans (C. elegans) expressinguman A�(1–42). We identified 16 oxidized proteins involved in energy metabolism, proteasome function, and scavenging of oxidants thatre more oxidized compared to control lines. These results are discussed with reference to Alzheimer’s disease.
2005 Elsevier Inc. All rights reserved.
eywords: Alzheimer’s disease; Amyloid �-peptide; Protein oxidation; Proteomics; C. elegans
. Introduction
Protein oxidation [13] is extensive in Alzheimer’s dis-ase (AD) [14,26,30,39]. Recent proteomics studies in ouraboratory have identified specific targets of protein oxida-ion in AD brain, assessed by increased carbonyls, such as,reatine kinase BB, glutamine synthase, ubiquitin carboxy-erminal hydrolase L-1, dihydropyrimidase-related protein, �-enolase, and heat shock cognate 71, indicating that aumber of cellular mechanisms are affected including energyetabolism, protein turnover, and neuronal communication
∗ Corresponding author. Tel.: +1 859 257 3184; fax: +1 859 257 5876.E-mail address: [email protected] (D.A. Butterfield).
[16,17]. Alzheimer’s disease brain is characterized patholog-ically by synapse loss and by the presence of senile plaques,neurofibrillary tangles, and neuropil threads. Extracellularsenile plaques are composed primarily of fibrilar depositsof amyloid beta peptide (1–42) [A�(1–42)], a product ofproteolytic cleavage of the transmembrane protein, amyloidprecursor protein (APP). On the other hand, intracellular neu-rofibrillary tangles are composed of paired helical filamentsformed from hyperphosphorylated tau, a microtubule associ-ated protein [25,33,57].
A�(1–42) has been proposed to play a central role inthe pathogenesis of AD as a mediator of oxidative stress[11,12,15]. However, the mechanism by which A�(1–42)-induced protein oxidation occurs is unknown. Several
197-4580/$ – see front matter © 2005 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2005.07.001

1240 D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249
hypotheses have been proposed including the aggregationstate of the peptide. Early studies demonstrated an associationbetween fibrillar peptide aggregates and toxicity [36,46].More recently, however, it has been shown in C. elegansexpressing A�(1–42) that protein oxidation precedes fibril-lar deposition of the peptide suggesting that small, solubleoligomers of the peptide are the toxic species [21].
In this study, we utilized proteomic techniques to iden-tify proteins that are specifically oxidized in a transgenic C.elegans expressing human A�(1–42) in body wall muscle(CL4176). Moreover, in order to evaluate the role of pro-tein aggregation per se in oxidative stress, we analyzed atransgenic C. elegans expressing a green fluorescent protein(GFP) fusion protein, which forms rapid aggregates of GFPin the worms (CL2337). Finally, to control for non-specificprotein oxidation resulting from muscle dysfunction itself,we examined the oxidative effects in transgenic C. elegansexpressing the ypkA subunit of Yersinia pseudotuberculosis,a serine/threonine kinase known to affect the cytoskeleton(XA1440) [28]. We identified 16 proteins to be oxidativelymodified in C. elegans expressing A�(1–42), five proteins tobe oxidatively modified in C. elegans expressing aggregatedGFP, and 1 protein was oxidatively modified in C. elegansexpressing ypkA. The proteins identified in this study areinvolved in a variety of cellular functions including cytoskele-tal integrity, scavenging of oxidants, signal transduction, lipidmPtiwod
2
2
fTf
2p
sc[ae1mY
three of these strains become paralyzed after temperatureupshift induction of the transgene. Transgenic strain CL1234,which contains the same smg-1ts mutation and dominant rol-6 transgene marker as the strains above, but does not expresstransgenic protein in muscle, was used as a control. This straindid not become paralyzed after temperature upshift.
Developmentally staged C. elegans populations werepropagated at 16 ◦C, then upshifted to 25 ◦C as third stage lar-vae. Upshifted populations were harvested after 24 h, washedfree of bacteria, resuspended in H2O, quick frozen in a liquidN2 bath, and stored at −80 ◦C. For each strain, populationsof animals from multiple growth plates were pooled, thenaliquoted before freezing. Individual aliquots were then sep-arately processed.
2.3. Sample preparation
Samples were homogenized by sonication in lysis buffer[10 mM HEPES pH 7.4 containing 137 mM NaCl, 4.6 mMKCl, 1.1 mM KH2PO4, 0.6 mM MgSO4, and proteaseinhibitors: leupeptin (0.5 �g/mL), pepstatin (0.7 �g/mL),type IIS soybean trypsin inhibitor (0.5 �g/mL), and PMSF(40 �g/mL)] and protein concentration was estimated bythe Pierce BCA method. One hundred micrograms of pro-tein was aliquoted from each sample and were incubatedat room temperature for 30 min in four volumes of 10 mM2caooctatb(nt
2b
pilrgdv318fT
etabolism, proteasome function, and energy metabolism.rotein oxidation has been shown to alter protein conforma-
ion leading to loss of function [1,16,26,31,41,49]. Thus, its likely that oxidation of the proteins identified in this studyill lead to loss of function. The implications of oxidationf these proteins are discussed with respect to Alzheimer’sisease.
. Methods
.1. Chemicals
All chemicals were of the highest purity and were obtainedrom Sigma (St. Louis, MO, USA) unless otherwise noted.he oxyblot protein oxidation detection kit was purchased
rom Chemicon International (Temecula, CA, USA).
.2. Transgenic strain construction and samplereparation.
Stable (chromosomally integrated) transgenic C. eleganstrains with temperature-inducible transgene expression wereonstructed using the smg-1ts system as previously described35]. Induction of strain CL4176 results in intramuscularccumulation of human A�(1–42). Strain CL2337 induciblyxpresses an aggregating variant of GFP with a C-terminal6 residue addition (“degron peptide”) [34] in body walluscle. Similarly, strain XA1440 inducibly expresses the
. pseudotuberculosis ypkA toxin in body wall muscle. All
,4-dinitrophenylhydrazine (DNPH) in 2 M HCl for proteinarbonyl derivatization/oxyblots or 2 M HCl for gel mapsnd mass spectrometry analysis, according to the methodf Levine et al. [32]. Proteins were precipitated by additionf ice-cold 100% trichloroacetic acid (TCA) to a final con-entration of 15% for 10 min on ice. Precipitates were cen-rifuged for 2 min at 14,000 × g 4 ◦C. The pellet was retainednd washed with 500 �L of 1:1 (v/v) ethyl acetate/ethanolhree times. The final pellet was dissolved in rehydrationuffer containing 8 M urea, 2 M thiourea, 2% CHAPS, 0.2%v/v) biolytes, 50 mM dithiothreitol (DTT), and bromophe-ol blue. Samples were sonicated in rehydration buffer on icehree times for 20 s intervals.
.4. Two-dimensional gel electrophoresis and Westernlotting
Two-dimensional polyacrylamide gel electrophoresis waserformed with a Bio-Rad system using 110-mm pH 3–10mmobilized pH gradients (IPG) strips and Criterion 8–16%inear gradient resolving gels. IPG strips were activelyehydrated at 50 V 20 ◦C overnight. One hundred micro-rams of protein was loaded per strip during active rehy-ration. Isoelectric focusing of strips loaded with proteinia active rehydration was performed at 20 ◦C as follows:00 V for 2 h linear gradient, 500 V 2 h linear gradient,000 V 2 h linear gradient, 8000 V 8 h linear gradient, and000 V 10 h rapid gradient. Gel strips were equilibratedor 10 min prior to second-dimension separation in 0.375 Mris–HCl pH 8.8 containing 6 M urea (Bio-Rad, Hercules,

D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249 1241
CA, USA), 2 % (w/v) sodium dodecyl sulfate (SDS), 20%(v/v) glycerol, and 0.5% DTT (Bio-Rad) followed by re-equilibration for 10 min in the same buffer containing 4.5%iodoacetamide (IA) (Bio-Rad) in place of DTT. Controland A� strips were placed on the Criterion gels, prestainedmolecular standards were applied, and electrophoresis wasperformed at 200 V for 65 min. It is conceivable that somevery large proteins (>100 kDa) may not transfer well to themembrane, and if this were the case in this study, thenwhat is reported would be a partial basis set of oxida-tively modified proteins in C. elegans that express humanA�(1–42).
2.5. Coomassie blue staining
Gels were fixed in a solution containing 40% (v/v)methanol, 7% (v/v) acetic acid for 20 min and stained for 2 hat room temperature with agitation in 50 mL of Coomassieblue gel stain (Bio-Rad). Gels were destained overnight byseveral washes with 50 mL dIH2O.
2.6. Oxyblot immunochemical detection
For immunoblotting analysis, electrophoresis was per-formed as stated previously and gels were transferred toa nitrocellulose membrane. The membranes were blockedwsTicpaipat5ss
2
bcgcw(wtw2fw
in the dark at room temperature. The liquid was drawn offand the gel pieces were incubated with 50 mM NH4HCO3at room temperature for 15 min. Acetonitrile was added tothe gel pieces for 15 min at room temperature. All solventswere removed and the gel pieces were allowed to dry for30 min. The gel pieces were rehydrated with addition ofa minimal volume of 20 ng/�L modified trypsin in 50mMNH4HCO3. The gel pieces were chopped and incubated inthe presence of trypsin with shaking overnight (∼18 h) at37 ◦C.
2.8. Analysis of gel images
The analyses of the gel maps and membranes to compareprotein expression and carbonyl immunoreactivity contentbetween control and A� treated samples were performed withPDQuest software (Bio-Rad). Images from SYPRO Rubystained gels, used to measure protein content, were obtainedusing a UV transilluminator (λex = 470 nm, λem = 618 nm,Molecular Dynamics, Sunnyvale, CA, USA). Oxyblots, usedto measure carbonyl immunoreactivity, were scanned with aMicrotek Scanmaker 4900.
2.9. Mass spectrometry
For this study, mass spectra were recorded at both the Uni-vaUUitrpl(io1hrpsbi1Rcncm
Ln�5
ith 3% bovine serum albumin (BSA) in phosphate bufferedaline containing 0.01% (w/v) sodium azide and 0.2% (v/v)ween 20 (PBST) overnight at 4 ◦C. The membranes were
ncubated with anti-2,4-dinitrophenylhydrazone (DNP) poly-lonal antibody diluted 1:100 in PBST for 2 h at room tem-erature with rocking. Following completion of the primaryntibody incubation, the membranes were washed three timesn PBST for 5 min each. An anti-rabbit IgG alkaline phos-hatase secondary antibody was diluted 1:3000 in PBSTnd incubated with the membranes for 2 h at room tempera-ure. The membranes were washed in PBST three times formin and developed using Sigmafast Tablets (BCIP/NBT
ubstrate). Blots were dried and scanned with Adobe Photo-hop.
.7. In-gel digestion
Samples were prepared according to the method describedy Thongboonkerd et al. [54]. Briefly, the protein spots wereut and removed from the gel with a clean razor blade. Theel pieces were placed into individual, clean 1.5 mL micro-entrifuge tubes and kept overnight at −20 ◦C. The gel piecesere thawed and washed with 0.1 M ammonium bicarbonate
NH4HCO3) for 15 min at room temperature. Acetonitrileas added to the gel pieces and incubated for an addi-
ional 15 min. The liquid was removed and the gel piecesere allowed to dry. The gel pieces were rehydrated with0 mM DTT (Bio-Rad) in 0.1 M NH4HCO3 and incubatedor 45 min at 56 ◦C. The DTT was removed and replacedith 55 mM IA (Bio-Rad) in 0.1 M NH4HCO3 for 30 min
ersity of Kentucky Mass Spectrometry Facility (UKMSF)nd the Department of Pharmacology and Toxicology at theniverisity of Louisville. For all mass spectra obtained atKMSF a Bruker Autoflex matrix assisted laser desorption
onization-time of flight (MALDI-TOF) mass spectrome-er (Bruker Daltonic, Billerica, MA, USA) operated in theeflection mode was used to generate peptide mass finger-rints. Peptides resulting form in-gel digestion were ana-yzed on a 384 position, 600 �m Anchor-ChipTM TargetBruker Daltonics, Bremen, Germany) and prepared accord-ng to AnchorChip recommendations (AnchorChip Technol-gy, Rev. 2, Bruker Daltonics, Bremen Germany). Briefly,�L of digestate was mixed with 1 �L of alpha-cyano-4-ydroxycinnamic acid (0.3 mg/mL in ethanol:acetone, 2:1atio) directly on the target and allowed to dry at room tem-erature. The sample spot was washed with 1 �L of 1% TFAolution for approximately 60 s. The TFA droplet was gentlylown off the sample spot with compressed air. The result-ng diffuse sample spot was recrystallized (refocused) using�L of a solution of ethanol:acetone: 0.1% TFA (6:3:1 ratio).eported spectra are a summation of 100 laser shots. Externalalibration of the mass axis was used for acquisition and inter-al calibration using either trypsin autolysis ions or matrixlusters was applied post acquisition for accurate mass deter-ination.For all mass spectra obtained at the University of
ouisville, a nitrocellulose solution was made by dissolving aitrocellulose membrane in 1:1 acetone:isopropanol solvent.-cyano-4-hydroxycinnamic acid (�-CN) was washed with0 �L of acetone and acetone phase was discarded. The �-

1242 D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249
CN was dissolved in acetone to a concentration of 10 mg/mLand the nitrocellulose and �-CN solutions were mixed to 1:4ratio and 1 �L of this mixture was deposited onto the 96 wellMALDI target plate. The samples were prepared for addi-tion to the plate by mixing 2 �L of sample with 2 �L of10 mg/mL �-CN solution in 0.1% trifluoroacetic acid in 1:1H2O/acetonitrile. The sample mixtures (1 �L) were loadedonto each thin film. After the sample mixtures were dried,1.5 mL of 2% formic acid in 18 M� water was added to eachspot. The formic solution was removed by gentle blotting.This washing step was performed twice. The samples werethen dried at room temperature. Fragment size was deter-mined by MALDI-TOF mass spectrometry.
Mass spectral data were obtained using a Micromass TOF-Spec 2E instrument with a 337 nm N2 laser at 20–35%power in the positive ion reflectron mode. Spectral data wereobtained by averaging 10 spectra, each of which was the com-posite of 10 laser firings. The mass axis was calibrated usingknown peaks from tryptic autolysis.
2.10. Analysis of peptide sequences
Peptide mass fingerprinting was used to identify pro-teins from tryptic peptide fragments by ultilzing the MAS-COT search engine (www.matrixscience.com) based on theestwcoise−
cation of the protein is not correct. MOWSE scores greaterthan 57 were considered to be significant (p < 0.05). All pro-tein identifications were in the expected size and pI rangebased on position in the gel.
2.11. Statistical analysis
Statistical comparison of carbonyl levels of proteins,matched with anti-DNP positive spots on 2D-oxyblots fromC. elegans expressing A�(1–42) and control C. elegans sam-ples, were performed using both ANOVA, and Student’st-tests. Statistical comparisions were also conducted betweenC. elegans expressing GFP:degron fusion protein and con-trol and between C. elegans expressing ypkA and control.p-Values of <0.05 were considered to be significant. For allanalyses, the carbonyl level of each spot was normalized tothe protein level of the corresponding spot in the gel. Onlythose spots found to be significantly oxidized compared tocontrol carbonyl levels were subjected to proteomic analysis.As discussed by Maurer et al. [43], there are no statistical testscurrently applicable to proteomics data. However, due to therelatively small number of proteins analyzed in this study,compared to the thousands of genes that are analyzed bymicroarrays, ANOVA and Student’s t-test were used [8,43].
3
e(ptab
F trol C.r
ntire NCBI and SwissProt protein databases. Databaseearches were conducted allowing for up to one missedrypsin cleavage and using the assumption that the peptidesere monoisotopic, oxidized at methionine residues, and
arbamidomethylated at cysteine residues. Mass tolerancef 150 ppm/g was the window of error allowed for match-ng the peptide mass values. Probability-based MOWSEcores were estimated by comparison of search results againststimated random match population and were reported as10 × log10(p), where p is the probability that the identifi-
ig. 1. Coomassie blue stained 2D gels (A) and 2D-oxyblots (B) from conepresent the area enlarged in Fig. 2.
. Results
Comparison of protein oxidation levels in C. elegansxpressing A�(1–42) (CL4176) and control C. elegansCL1234) was carried out by first identifying carbonylatedroteins via anti-DNP immunochemical development of pro-eins transferred to a nitrocellulose membrane, or 2D-oyxblotnalysis (Fig. 1B). Individual protein spots were matchedetween the 2D-PAGE maps and the 2D-oxyblots and the
elegans CL1234 and C. elegans expressing A�(1–42) CL4176. The boxes

D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249 1243
Fig. 2. Enlargements of 2D gel (A) and 2D-oxyblot (B) images show the position of protein spots and carbonyl immunoreactivity, respectively. The 2D-oxyblotfrom C. elegans expressing A�(1–42) is labeled with the proteins identified in this study.
carbonyl immunoreactivity of each spot was normalized tothe protein content in the 2D-PAGE (Fig. 1A). Enlargementsof the boxed areas are shown in Fig. 2. Additionally, com-parisons were made in the same manner between C. elegansexpressing GFP::degron (CL2337) and control C. elegans(CL1234) and between C. elegans expressing the Y. pseudo-tuberculosis toxin ypkA (XA1440) and control C. elegans(CL1234). As previously described for strain CL4176 [35],the CL2337 and XA1440 control strains also become par-alyzed 24 h after induction of expression of the transgenicprotein. Under these conditions, the GFP::degron forms intra-cellular aggregates, while the ypkA protein remains soluble
in the cytoplasm (neither protein forms detectable amyloid).These strains therefore control for the cellular effects ofprotein aggregation and general muscle dysfunction. In thisstudy, we confirm previous reports that many, but not all, indi-vidual proteins exhibit carbonyl immunoreactivity [16–19](Figs. 2–6).
Mass spectrometry analysis allowed for the identifica-tion of protein spots found to be increasingly carbonylatedby A�(1–42) expression (Table 1). These oxidized pro-teins include proteins involved in lipid metabolism, energyproduction, protein degradation, and muscle-specific func-tions. In C. elegans expressing aggregating GFP, eight
F trol C.b
ig. 3. Coomassie blue stained 2D gels (A) and 2D-oxyblots (B) from conoxes represent the area enlarged in Fig. 4.
elegans CL1234 and C. elegans expressing aggregated GFP, CL2337. The
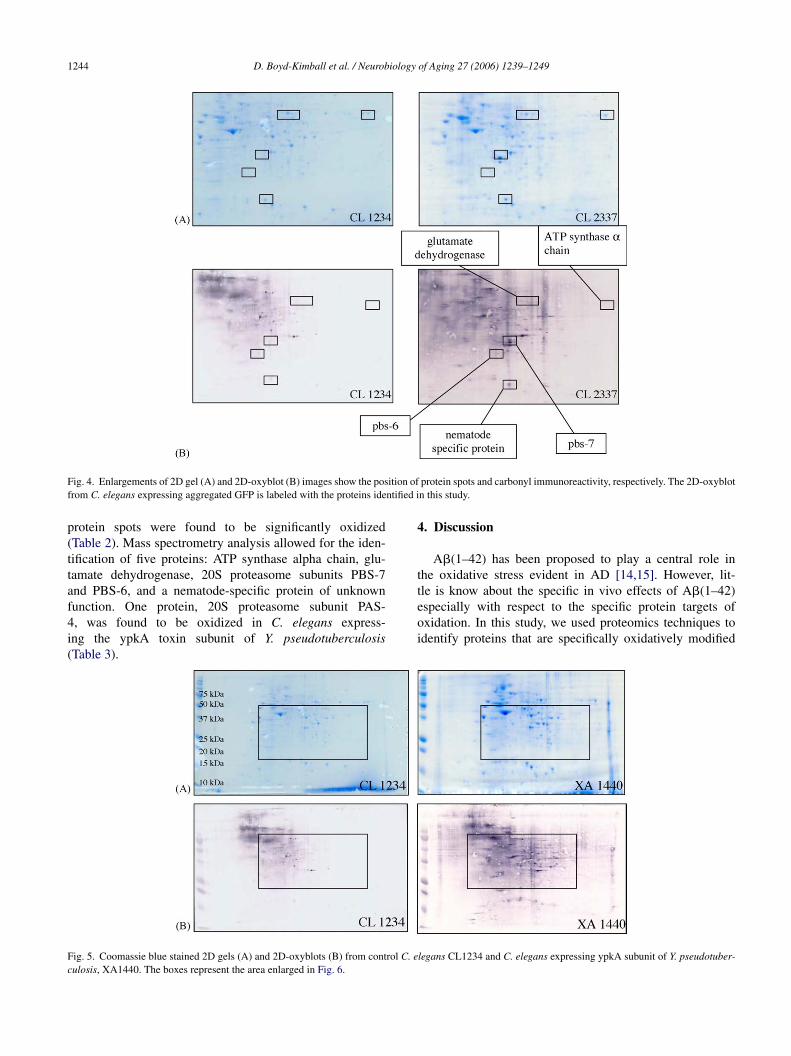
1244 D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249
Fig. 4. Enlargements of 2D gel (A) and 2D-oxyblot (B) images show the position of protein spots and carbonyl immunoreactivity, respectively. The 2D-oxyblotfrom C. elegans expressing aggregated GFP is labeled with the proteins identified in this study.
protein spots were found to be significantly oxidized(Table 2). Mass spectrometry analysis allowed for the iden-tification of five proteins: ATP synthase alpha chain, glu-tamate dehydrogenase, 20S proteasome subunits PBS-7and PBS-6, and a nematode-specific protein of unknownfunction. One protein, 20S proteasome subunit PAS-4, was found to be oxidized in C. elegans express-ing the ypkA toxin subunit of Y. pseudotuberculosis(Table 3).
4. Discussion
A�(1–42) has been proposed to play a central role inthe oxidative stress evident in AD [14,15]. However, lit-tle is know about the specific in vivo effects of A�(1–42)especially with respect to the specific protein targets ofoxidation. In this study, we used proteomics techniques toidentify proteins that are specifically oxidatively modified
F rol C. ec
ig. 5. Coomassie blue stained 2D gels (A) and 2D-oxyblots (B) from contulosis, XA1440. The boxes represent the area enlarged in Fig. 6.
legans CL1234 and C. elegans expressing ypkA subunit of Y. pseudotuber-

D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249 1245
Fig. 6. Enlargements of 2D gel (A) and 2D-oxyblot (B) images show the position of protein spots and carbonyl immunoreactivity, respectively. The 2D-oxyblotfrom C. elegans expressing the ypkA subunit of Y. pseudotuberculosis is labeled with the protein identified in this study.
in C. elegans expressing A�(1–42) (CL4176). This modelexpresses intracellular A�(1–42), and thus we have specif-ically identified intracellular targets of A� oxidation. Ourstudy cannot resolve whether the observed protein oxida-tion results from direct interaction of A� with the oxidizedprotein, or from secondary effects (e.g., general cellularoxidative stress resulting from interference with mitochon-drial function). However, our analysis of transgenic controlstrains demonstrates that the specific protein oxidation wehave observed is not a byproduct of general cellular dys-
function or the accumulation of protein aggregates. Sixteenoxidized proteins were identified in C. elegans expressingA�(1–42). These proteins are involved in a variety of cellu-lar function including energy metabolism, proteasome func-tion, cellular structure, lipid transport, and signal transduc-tion. Five proteins were oxidized in C. elegans expressingaggregated GFP, and only one protein was found to be oxi-dized in C. elegans expressing ypkA. The only commonproteins between the three groups were proteosomal sub-units, suggesting that oxidative damage to the proteasome
Table 1Summary of proteomics-identified proteins in CL4176, the C. elegans model that expresses human A�(1–42)a
Gene, protein name Mowse score Peptidesmatched
Coverage (%) Oxidation (%) MW (Da) pI p-Value
T08G2.3, medium-chain acyl-CoA dehydrogenase 51 5 14 540 ± 86 45132 8.37 <0.037K06A5.6, short-chain acyl-CoA dehydrogenase 82 8 16 540 ± 86 46033 7.63 <0.037F17C11.9, translation elongation factor EF-1 � 124 12 29 540 ± 86 44474 6.29 <0.037F46E10.10, malate dehydrogenase 125 10 30 540 ± 86 36012 6.6 <0.037F46H5.3, arginine kinase 187 20 57 44636 ± 7494 40365 6.17 <0.007K04D7.1, RACK1 ortholog 76 9 26 540 ± 85 36150 6.44 <0.037C36E6.5 (mlc-2), myosin regulatory light chain 130 11 71 1777 ± 460 18648 5.06 <0.04C36E6.3 (mlc-1), myosin regulatory light chain 68 5 35 539 ± 86 18605 5.06 <0.037M03F4.2 (act-4), actinb 44 5 17 539 ± 85 42093 5.30 <0.037R07H5.8, adenosine kinase 65 6 21 539 ± 85 37404 5.67 <0.037F15E11.1, nematode specific protein 119 10 64 539 ± 85 17418 6.04 <0.037W02D3.5 (lbp-6), lipid binding protein 97 8 66 539 ± 86 15631 6.77 <0.037F01G10.1, transketolase 166 16 25 540 ± 86 65970 6.18 <0.037Z 7F 6R 6
rmaticsi essed as t).
he mole
K945.2 (pas-7), proteasome alpha subunit 7239H11.5 (pbs-7), proteasome beta subunit 6607B1.4 (gst-36), glutathionine S-transferase sigma class 77a MALDI-TOF MS analysis of tryptic peptides coupled to protein info
mmunoreactivity/protein expression values were averaged (n = 3) and exprignificance of elevated protein carbonyls relative to control worms (see texb MOWSE score for identification of actin was not significant; however, t
25 9436 ± 2859 27735 5 <0.0521 298451 ± 79992 26951 5.61 <0.03327 540 ± 86 23918 5.88 <0.037
led to the identity of each protein listed. For each protein the carbonyls percent oxidation compared to control ± S.E.M. The p-value listed is the
cular weight and pI correspond with the protein spot of interest.

1246 D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249
Table 2Summary of proteomics-identified proteins in CL2337, the C. elegans model that expresses GFP::degron fusion proteina
Gene, protein name Mowse score Peptides matched Coverage (%) Oxidation (%) MW (Da) pI p-Value
H28016.1, ATP synthase � chain 132 12 24 867 ± 86 57752 8.98 <0.05F15E11.1, nematode specific protein 119 10 64 551 ± 108 17418 6.40 <0.05ZK829.4, glutamate dehydrogenase 245 23 49 657 ± 151 59214 6.40 <0.05ZK829.4, glutamate dehydrogenase 100 10 18 362 ± 74 58758 6.9 <0.09F39H11.5 (pbs-7), proteasome beta subunit 66 6 21 3527 ± 1137 26951 5.61 <0.07C02F5.9 (pbs-6), proteasome beta sununit 61 6 25 425 ± 62 29140 5.63 <0.05
a MALDI-TOF MS analysis of tryptic peptides coupled to protein informatics led to the identity of each protein listed. For each protein the carbonylimmunoreactivity/protein expression values were averaged (n = 3) and expressed as percent oxidation compared to control ± S.E.M. The p-value listed is thesignificance of elevated protein carbonyls relative to control worms (see text).
may be a general result of cellular toxicity leading to muscledysfunction.
A set of proteins associated with energy and metabolismwere identified exclusively in the C. elegans express-ing A�(1–42), a finding consistent with altered energymetabolism in AD [44,48,55]. Acyl-CoA dehydrogenase,malate dehydrogenase, transketolase, arginine kinase, andadenosine kinase were all found to be oxidized by A�(1–42),but not in the other two C. elegans models, suggesting thatoxidation of proteins involved in energy metabolism is adirect effect of exposure to A�(1–42) independent of pro-tein aggregation and paralysis. Our laboratory reported usingproteomics that several energy and metabolism-related pro-teins were oxidatively modified in AD brain [9,10,16–18].
Acyl-CoA dehydrogenase is an enzyme involved in fattyacid metabolism that catalyzes the conversion of acyl-CoAto trans-�2-enoyl-CoA coupled to the reduction of FAD toFADH2. This is the first reaction in the conversion of acyl-CoA to acetyl-CoA, which can then pass into the citric acidcycle to lead to the generation of ATP from fatty acids. Asnoted, prior studies suggest that oxidative modification leadsto loss of protein function [26,30]. Thus, loss of functionof acyl-CoA dehydrogenase would inhibit the conversion ofacyl-CoA to acetyl-CoA and thus prevent the production ofATP from fatty acid catabolism, conceivably related to theknown energy alteration and to fatty acid alterations in ADb
TSt
GMPCOMpp
mbelw
Malate dehydrogenase is an enzyme that catalyzes thelast step of the citric acid cycle, the reversible oxidation ofmalate to oxaloacetate coupled with the reduction of NAD+ toNADH. Malate dehydrogenase is located in both the cytosoland the mitochondrial matrix and participates in the malate-aspartate shuttle that passively feeds electrons from cytosolicNADH into the electron transport chain. Loss of function ofmalate dehydrogenase due to oxidative modification wouldsignificantly decrease the efficiency of the citric acid cycleas well as the transport of electrons from cytosolic NADHinto the mitochondrial matrix, and consequently, decreasethe production of ATP.
Transketolase is an enzyme that catalyzes two independentreactions of the pentose phosphate pathway, which generatesNADPH and ribose-5-phosphate. NADPH is necessary fora variety of synthetic pathways, as well as, the reduction ofoxidized glutathione by glutathione reductase, while ribose-5-phosphate is required for synthesis of nucleic acids. Thereactions catalyzed by transketolase generate products thatcan directly enter glycolysis to produce ATP: glyceraldehyde-3-phosphate and fructose-6-phosphate. Expression of trans-ketolase was found to be significantly reduced in C. elegansexpressing A�(1–42) [35], and the activity of transketolasehas been shown to be decreased in AD brain [24]. Lossof function of transkeltolase would prevent the generationof NADPH, as well as the synthesis of intermediates oftcLtlda
dcpttdkaah
rain [47].
able 3ummary of proteomics-identified proteins in XA1440, the C. elegans model
hat expresses the ypkA subunit of Yersinia pseudotuberculoisis toxina
ene, protein name C36B1.4 (pas-4), proteasome alpha subunitowse score 84
eptides matched 6overage (%) 18xidation (%) 26835 ± 7399W (Da) 28336
I 5.93-Value <0.04a MALDI-TOF MS analysis of tryptic peptides coupled to protein infor-atics led to the identity of each protein listed. For each protein the car-
onyl immunoreactivity/protein expression values were averaged (n = 3) andxpressed as percent oxidation compared to control ± S.E.M. The p-valueisted is the significance of elevated protein carbonyls relative to controlorms (see text).
he pentose phosphate pathway that could feed into gly-olysis, and the synthesis of nucleic acids for DNA repair.ack of NADPH would lead to decreased activity of syn-
hetic pathways resulting in decrease protein turnover andoss of activity of glutathione reductase, and consequently,ecreased availability of reduced glutathione, an importantntioxidant in the brain [4].
Arginine kinase is a member of the phosphagen (guani-ino) kinase family of highly conserved enzymes thatatalyze the reversible transfer of phosphate from ahophorylated guanidino (∼NH-CN2H4
+) substrate to ADPo satisfy short-term ATP requirements. Through this reac-ion cells can support nerve activity that would otherwiserain ATP from other essential functions [5,52,58]. Arginineinase has significant sequence similarity to creatine kinase,nd is likely to serve the same function in C. elegans muscles creatine kinase does in mammalian cells. Creatine kinaseas been shown to be oxidized in AD brain [3,16].

D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249 1247
Adenosine kinase catalyzes the reversible phosphoryla-tion of adenosine by ATP to form ADP and AMP to reg-ulate intra- and extracellular levels of adenosine. Loss offunction of arginine kinase and adenosine kinase wouldresult in altered cellular phosphate stores for the quickresponse generation of ATP production. In the brain, highlevels of ATP production are necessary to maintain neu-ronal membrane potential via the Na+/K+-ATPase. Lackof ATP could lead to loss of membrane potential, loss ofimpulse transmission, altered long term potentiation, influxof Ca2+, and neuronal apoptosis all of which could lead tothe neurodegeneration evident in AD. This could be espe-cially important for the synaptic region of neurons, thoughtby many to be the site of initial attack in AD neurons[42].
The proteasome is a multi-subunit protein complex thatplays a role in the proteolytic degradation of intracellularproteins and consists of seven alpha subunits and sevenbeta subunits. The proteasome is believed to be particu-larly important for the turn over of misfolded and aggre-gated proteins [27]. A significant decrease in proteasomeactivity has been reported in AD [29], and inhibition ofthe proteasome has been shown to lead to increased mito-chondrial reactive oxygen species production and decreasemitochondrial turnover [50]. Oxidation of proteasomal sub-units was detected in all three C. elegans models. However,tseimA
oiohplpamtfibsadtgimcdtn
elegans model expressing ypkA, which controlled for paral-ysis. This finding suggests that the oxidation of myosin inresponse to A�(1–42) was a direct effect of the proxim-ity of the protein to the sight of reactive oxygen speciesproduction.
A�(1–42) has been shown to induce the formation ofthe lipid peroxidation product 4-hydroxynonenal (HNE)[14,30,38]. HNE is a reactive alkenal, found to be increased inAD brain [40], that reacts by Michael addition with protein-bound cysteine, lysine, and histidine to add carbonyl func-tionality [13,23]. Glutathione S-transferases (GSTs) catalyzethe enzymatic reaction of reactive alkenals, such as HNE,with glutathione, a cysteine containing antioxidant that isabundant in the brain [20,23,31,56]. In our study, oxidation ofone GST was found in C. elegans expressing A�(1–42) anddecreased GST activity has been reported in AD [37]. Loss offunction of GSTs would result in an increased vulnerabilityof neurons to reactive alkenals, and consequently, oxida-tive damage. Our laboratory recently reported both increasedHNE-binding to GST and increased expression of this proteinin AD brain [51].
Lipid binding protein 6 (LBP-6) is a fatty-acid bind-ing protein associated with lipid metabolism and fatty acidtransport. Oxidation of LBP-6 was found to be induced byA�(1–42). This finding is consistent with altered cholesterolhomeostasis and decreased membrane fluidity in AD. Addi-tetiCmttbo
rtcitwaasrwdwRfvicc
he only specific overlap in proteins occurred with protea-ome beta subunit 7, which was oxidized in both the C.legans model expressing A�(1–42) and the model express-ng aggregated GFP, suggesting that aggregated A�(1–42)
ay play a role in the loss of proteasome activity reported inD.Cytoskeletal alterations in AD also include the significant
xidation of �-actin and the trend toward oxidative mod-ficantion of �-tubulin in AD [2]. Actin was significantlyxidized in C. elegan expressing A�(1–42). It must be noted,owever, that the MOWSE score for the identification of thisrotein did not reach significance; nevertheless, the molecu-ar weight and isoelectric point of actin corresponds to therotein spot of interest [7]. Actin, found in both neuronsnd glial cells, is a core subunit of microfilaments. Actinicrofilaments are particularly concentrated in presynaptic
erminals, dendritic spines, and growth cones. Actin micro-laments play a role in the neuronal membrane cytoskeletony maintaining the distribution of membrane proteins, andegregating axonal and dendritic proteins [6]. Oxidation ofctin can lead to loss of membrane cytoskeletal structure,ecreased membrane fluidity, and trafficking of synaptic pro-eins. Moreover, actin is involved in the elongation of therowth cone and loss of function of actin could play a rolen the loss of synapse and neuronal communication docu-
ented in AD [42]. Oxidation of myosin regulatory lighthain and myosin light chain 1 were also found to be oxi-ized in response to A�(1–42). This is most likely is due tohe expression of A�(1–42) within the muscular wall of theematode. Oxidation of myosin was not detected in the C.
ionally, it has been found that cholesterol modulates theffects of A�(1–42) on neuronal membranes [22], althoughhe exact role of cholesterol in AD is not clear. It is interest-ng that acyl-CoA is also oxidized in A�(1–42)-expressing. elegans. Conceivably, mitochondrial resident fatty acidetabolism is altered by A�(1–42) and in AD brain. Taken
ogether, the loss of function of LBP-6 could play a role inhe altered cholesterol homeostasis in AD suggesting a linketween altered membrane fluidity and A�(1–42)-inducedxidative stress.
Guanine nucleotide-binding proteins (G proteins) areesponsible for signal transduction from hormones, neuro-ransmitters, and chemokines. G proteins normally are asso-iated with seven TM receptors that bind ligands resultingn the activation of the G proteins, subsequently leading tohe activation of cellular signaling pathways. These path-ays include regulation of metabolic enzymes, ion channels,
nd transporters [45]. Oxidation of G proteins can lead toltered Ca2+ homeostasis, loss of activation of second mes-angers such as cAMP, altered cell cycle, neurotransmitterelease, and ultimately cell death. Based on the gene fromhich the particular protein identified in this study is pro-uced it is believed that the protein is an ortholog of RACK1,hich shows similarities to the � subunit of G proteins.ACK1 has been shown to be significantly decreased in AD
rontal cortexc compared to control [6]. Receptors for acti-ated C kinase (RACK) are a family of proteins involvedn anchoring activated protein kinase C (PKC) to subcellularompartments. PKC activation has been shown to regulate ionhannels, receptor desensitization, transcription, neurotrans-

1248 D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249
mitter release, and synaptic efficacy [53]. Loss of function ofRACK1 would lead to loss of interaction with activated PKCand consequently loss of subcellular compartmentalizationpossibly affecting the interaction of activated PKC with itsprotein substrates.
Only aggregated GFP induced oxidation of glutamatedehydrogenase (GDH) suggesting that aggregation, but notA�(1–42)-induced oxidative stress is responsible for the oxi-dation of this protein in this model system. GDH is anenzyme located in the mitochondrial matix that can act ineither a metabolic or a catabolic direction. In the biosyn-thetic direction, GDH catalyzes the reductive amination of �-ketoglutarate with NADPH to yield glutamate. Alternatively,GDH can catalyze the formation of �-ketoglutarate fromglutamate with NAD+ and ammonium ion. The catabolicactivity of GDH is particularly important for the eliminationof excitotoxic glutamate. The activity of glutamine synthetase(GS), an enzyme which catalyzes the conversion of glutamateto glutamine, has been shown to be significantly decreasedin AD brain. Moreover, it is likely that the loss of activ-ity of GS is due to the oxidative modified of GS in ADbrain [16,26]. Additionally, it has been shown that A�(1–42)-induces oxidative modification of the glutamate transporterEAAT2 by 4-hydroxynonenal (HNE) in synaptosomes [30].Likewise, EAAT2 activity is decreased in AD brain [41] and ithas been shown that EAAT2 is modified by HNE in AD brain[iimialqcoci
fiAapssipAAA
A
[
We are indebted to Creg Darby for providing the XA1440strain.
References
[1] Aksenov MY, Aksenova MV, Butterfield DA, Markesbery WR.Oxidative modification of creatine kinase BB in Alzheimer’s dis-ease brain. J Neurochem 2000;74:2520–7.
[2] Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markes-bery WR. Protein oxidation in the brain in Alzheimer’s disease.Neuroscience 2001;103:373–83.
[3] Aksenov MY, Tucker HM, Nair P, Askenova MV, Butterfield DA,Estus S, et al. The expression of several mitochondrial and nucleargenes encoding the subunits of electron transport chain enzymecomplexes, cytochrome c oxidase, and NADH dehydrogenase in dif-ferent brain regions in Alzheimer’s disease brain. Neurochem Res1999;24:767–74.
[4] Aksenov MY, Markesbery WR. Changes in thiol content and expres-sion of glutathione redox system genes in the hippocampus andcerebellum in Alzheimer’s disease. Neurosci Lett 2001;302:141–5.
[5] Azzi A, Clark SA, Ellington WR, Chapman MS. The roleof phosphagen specificity loops in arginine kinase. Protein Sci2004;13:575–85.
[6] Battaini F, Pascale A, Lucchi L, Pasinetti GM, Govoni S. Proteinkinase C anchoring deficit in postmortem brains of Alzheimer’s dis-ease patients. Exp Neurol 1999;159:559–64.
[7] Beck KA, Nelson J. The spectrin-based membrane skele-ton as a membrane protein-sorting machine. Am J Physiol1996;270:C1263–70.
[
[
[
[
[
[
[
[
[
30]. Loss of function of GDH, GS, and EAAT2 would resultn a decreased conversion or uptake of glutamate, resultingn accumulation of extracellular glutamate. The excess gluta-
ate would stimulate NMDA receptors leading to an increasen Ca2+ influx. Altered calcium homeostasis would lead tolteration in long term potentiation (LTP) and consequently,earning and memory, mitochondrial swelling with conse-uent ROS leakage and release of pro-apoptotic cytochrome, stress in the endoplasmic reticulum, as well as, activationf calcium-sensitive proteases such as calpain. Clearly, suchhanges would lead to neuronal death and may be importantn AD.
In summary, utilizing proteomics techniques we identi-ed proteins significantly oxidized in C. elegans expressing�(1–42). The proteins identified in this study are involved invariety of cellular functions including energy metabolism,roteasome function, signal transduction, lipid transport,cavenging of oxidants, and cytoskeletal maintenance, con-istent with functions found to be altered in AD brain. Moremportantly, however, the proteins identified in this studyrovide evidence of the in vivo protein oxidation effects of�(1–42) and insight into the neurodegenerative effects of�(1–42)-induced protein oxidation in the pathogenesis ofD.
cknowledgements
This work was supported in part by NIH grants to D.A.B.AG-05119; AG-10836] and C.D.L [AG12423, AG21037].
[8] Boguski MS, McIntosh MW. Biomedical informatics for proteomics.Nature 2003;422:233–7.
[9] Butterfield DA. Proteomics: a new approach to investigate oxidativestress in Alzheimer’s disease brain. Brain Res 2004;1000:1–7.
10] Butterfield DA, Castegna A. Energy metabolism in Alzheimer’s dis-ease brain: insights from proteomics. Appl Proteomics Genomics2003;2:67–70.
11] Butterfield DA. Amyloid beta-peptide (1–42)-induced oxida-tive stress and neurotoxicity: implications for neurodegenerationin Alzheimer’s disease brain. Free Radic Res 2002;36:1307–13.
12] Butterfield DA. Amyloid beta-peptide [1–42]-associated free radical-induced oxidative stress and neurodegeneration in Alzheimer’sdisease brain: mechanisms and consequences. Curr Med Chem2003;10:2651–9.
13] Butterfield DA, Stadtman ER. Protein oxidation processes in agingbrain. Adv Cell Aging Gerontol 1997;2:161–91.
14] Butterfield DA, Lauderback CM. Lipid peroxidation and proteinoxidation in Alzheimer’s disease brain: potential causes and con-sequences involving amyloid beta-peptide-associated free radicaloxidative stress. Free Radic Biol Med 2002;32:1050–60.
15] Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence ofoxidative damage in Alzheimer’s disease brain: central role for amy-loid beta-peptide. Trends Mol Med 2001;7:548–54.
16] Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB,Pierce WM, et al. Proteomic identification of oxidatively modifiedproteins in Alzheimer’s disease brain part I: creatine kinase BB,glutamine synthetase, and ubiquitin carboxy-terminal hydrolase L-1.Free Radic Biol Med 2002;33:562–71.
17] Castegna A, Aksenov M, Thongboonkerd, Klein JB, Pierce WM,Booze R, et al. Proteomic identification of oxidatively modifiedproteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, �-enolase, and heat shock cognate 71. J Neurochem2002;82:1524–32.
18] Castegna A, Thongboonkerd V, Klein J, Lynn BC, Wang YL, OsakaH, et al. Proteomic analysis of brain proteins in the gracile axonal

D. Boyd-Kimball et al. / Neurobiology of Aging 27 (2006) 1239–1249 1249
dystrophy (gad) mouse, a syndrome that emanates from dysfunc-tional ubiquitin carboxyl-terminal hydrolase L-1, reveals oxidationof key proteins. J Neurochem 2004;88:1540–6.
[19] Castegna A, Thongboonkerd V, Klein JB, Lynn B, MarkesberyWR, Butterfield DA. Proteomic identification of nitrated proteinsin Alzheimer’s disease brain. J Neurochem 2003;85:1394–401.
[20] Cooper AJL. Glutathione in the brain: disorder of glutathionemetabolism. In: The molecular and genetic basis of neurologicaldisease. Newton: Butterworth-Heinemann; 1997. pp. 1195–230.
[21] Drake J, Link CD, Butterfield DA. Oxidative stress precedes fib-rillar deposition of Alzheimer’s disease amyloid �-peptide (1–42)in a transgenic Caenorhabditis elegans model. Neurobiol Aging2003;24:415–20.
[22] Eckert GP, Kirsch C, Leutz S, Wood WG, Muller WE. Cholesterolmodulates amyloid beta-peptide’s membrane interactions. Pharma-copsychiatry 2003;36:S136–43.
[23] Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of4-hydroxynonenal, malonaldehyde and related aldehydes. Free RadicBiol Med 1991;11:81–128.
[24] Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, etal. Reduced activities of thiamine-dependent enzymes in the brainsand peripheral tissues of patients with Alzheimer’s disease. ArchNeurol 1988;45:836–40.
[25] Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibril-lary tangles in P301L tau transgenic mice induced by A�42 fibrils.Science 2001;293:1491–5.
[26] Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M,et al. Brain regional correspondence between Alzheimer’s diseasehistopathology and biomarkers of protein oxidation. J Neurochem1995;65:2146–56.
[27] Jayarapu K, Griffin TA. Protein-protein interactions among human
[
[
[
[
[
[
[
[
[
[
[38] Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP.A role for 4-hydroxynonenal, an aldehydic product of lipid per-oxidation, in disruption of ion homeostasis and neuronal deathinduced by amyloid beta-peptide. J Neurochem 1997;68:255–64.
[39] Markesbery MR. Oxidative Stress Hypothesis in Alzheimer’s Dis-ease. Free Radic Biol Med 1997;23:134–47.
[40] Markesbery WR, Lovell MA. Four-hydroxynonenal, a product oflipid peroxidation, is increased in the brain in Alzheimer’s disease.Neurobiol Aging 1998;19:33–6.
[41] Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Defi-cient glutamate transport is associated with neurodegeneration inAlzheimer’s disease. Ann Neurol 1996;40:759–66.
[42] Masliah E, Mallory M, Hansen L, De Teresa R, Alford M, TerryR. Synaptic and neuritic alterations during the progression ofAlzheimer’s disease. Neurosci Lett 1994;174:67–72.
[43] Maurer MH, Feldmann Jr RE, Bromme JO, Kalenka A. Compari-son of statistical approaches for the analysis of proteome expressiondata of differentiating neural stem cells. J Proteome Res 2005;4:96–100.
[44] Messier C, Gagnon M. Glucose regulation and brain aging. J NutrHealth Aging 2000;4:208–13.
[45] Neves SR, Ram PT, Iyengar R. G protein pathways. Science2002:1636–9.
[46] Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic �-amyloid protein in hippocampal cul-tures. Eur J Pharmacol—Mol Pharmacol Section 1991;207:367–8.
[47] Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR.Regional membrane phospholipid alterations in Alzheimer’s disease.Neurochem Res 1998;23:81–8.
[
[
[
[
[
[
[
[
[
[
[
20S proteasome subunits and proteassemblin. Biochem Biophys ResCommun 2004;314:523–8.
28] Juris SJ, Rudolph AE, Huddler D, Orth K, Dixon JE. A dis-tinctive role for the Yersinia protein kinase: actin binding, kinaseactivation, and cytoskeleton disruption. Proc Nat Acad Sci USA2000;97:9431–6.
29] Keller JN, Hanni KB, Markesbery WM. Impaired proteasome func-tion in Alzheimer’s disease. J Neurochem 2000;75:436–9.
30] Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI,Markesbery WR, et al. The glial glutamate transporter, GLT-1, isoxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s dis-ease brain: the role of Abeta 1–42. J Neurochem 2001;78:413–6.
31] Leiers B, Kampkotter A, Grevelding CG, Link CD, Johnson TE,Henkle-Duhrsen K. A stress-responsive glutathione S-transferaseconfers resistance to oxidative stress in Caenorhabditis elegans. FreeRadic Biol Med 2003;34:1405–15.
32] Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assaysfor determination of oxidatively modified proteins. Meth Enzymol1994;233:346–57.
33] Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al.Enhanced neurofibrillary degeneration in transgenic mice expressingmutant tau and APP. Science 2001;293:1487–91.
34] Link CD. Invertebrate models of Alzheimer’s disease. Genes BrainBehav 2005;4:147–56.
35] Link CD, Taft A, Kapulkin V, Duke K, Kim S, Fei Q, et al.Gene expression analysis in a transgenic Caenorhabditis eleganAlzheimer’s disease model. Neurobiol Aging 2003;24:397–413.
36] Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibrilformation and is inhibited by congo red. Proc Natl Acad Sci USA1994;91:12243–7.
37] Lovell MA, Xie C, Markesbery WR. Decreased glutathione trans-ferase activity in brain and ventricular fluid in Alzheimer’s disease.Neurology 1998;51:1562–6.
48] Scheltens P, Korf ESC. Contribution of neuroimaging in the diagno-sis of Alzheimer’s disease and other dementias. Curr Opin Neurol2000;13:391–6.
49] Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, WaegG, et al. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal,alters the conformation of cortical synaptosomal membrane protein.J Neurochem 1997;69:1161–9.
50] Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q,et al. Proteasome inhibition alters neural mitochondrial homeostasisand mitochondria turnover. J Biol Chem 2004;279:20699–707.
51] Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 inAlzheimer’s disease brain: Implications for accumulation of reactivelipid peroxidation products. Neurochem Res 2004;29:2215–20.
52] Suzuki T, Kamidochi M, Inoue N, Kawamichi H, Yazawa Y,Furukohri T, et al. Arginine kinase evolved twice: evidence thatechinoderm arginine kinase originated from creatine kinase. BiochemJ 1999;340:671–5.
53] Tanka C, Nishizuka Y. The protein kinase C family for neuronalsignaling. Annu Rev Neurosci 1994;17:551–67.
54] Thongboonkerd V, Luengpailin J, Cao J, Pierce QM, Cai J, KleinJB, et al. Fluoride exposure attenuates expression of Streptococcuspyrogenes virulence factors. JBC 2002;277:16599–605.
55] Vanhanen M, Soininen H. Glucose intolerance, cognitive impairmentand Alzheimer’s disease. Curr Opin Neurol 1998;11:673–7.
56] Xie C, Lovell MA, Markesberry WR. Glutathione transferase pro-tects neuronal cultures against four hydroxynonenal toxicity. FreeRadic Biol Med 1998;25:979–88.
57] Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid �
peptide induced tau phosphorylation and loss of cholinergic neuronsin rat primary septal cultures. Neuroscience 2002;115:201–11.
58] Zhou G, Somasundaram T, Blanc E, Parthasarathy G, Ellington WR,Chapman MS. Transition state structure of arginine kinase: Implica-tions for catalysis of bimolecular reactions. PNAS 1998;95:8449–54.




















