
Immunity Mediated by B Cells and Antibodies
46
The production of antibodies is the sole function of the B-cell arm of the immune system. Antibodies are useful in the defense against any pathogen that is present in the extracellular spaces of the body’s tissues. Some human pathogens, such as many species of bacteria, live and reproduce entirely within the extracellular spaces, whereas others, such as viruses, replicate inside cells but are carried through the extracellular spaces as they spread from one cell to the next. Antibodies secreted by plasma cells in secondary lymphoid tissues and bone marrow find their way into the fluids filling the extracellular spaces. Antibodies are not in themselves toxic or destructive to pathogens; their role is simply to bind tightly to them. This can have several consequences. One way in which antibodies reduce infection is by covering up the sites on a pathogen’s surface that are necessary for growth or replication, for example the viral glycoproteins that viruses use to bind to the surface of human cells and initiate infection. Such antibodies are said to neutralize the pathogen. In the development of a vaccine against an infectious agent or its toxic products, the gold standard that a company aims for is the induction of a neutralizing antibody. Antibodies also act as molecular adaptors that bind to pathogens with their antigen-binding arms and to receptors on phagocytic cells with their Fc regions. Thus, opsonization, or coating of a pathogen with antibody, promotes its phagocytosis. The antibody-directed destruction of pathogens caused by opsonization is enhanced by the actions of a set of proteins that do not discriminate between antigens and are present in blood and lymph. These proteins are collectively known as complement because their func- tions complement the antigen-binding function of the antibody. The structure, specificity, and other properties of antibodies were discussed in Chapter 2, and the development of B cells from their origin in bone mar- row to differentiation into antibody-secreting plasma cells was the subject of Chapter 4. This chapter will focus on how antibodies clear infection by tar- geting destructive but nonspecific components of the immune system to an infecting pathogen. In the first part of the chapter we consider the antigens that provoke a B-cell response, how the response develops, and the genera- tion of the different antibody isotypes. The structural differences between antibody isotypes provide a variety of adaptor functions that can target anti- body-bound pathogen to different types of nonspecific effector cell; these aspects of the antibody-mediated immune response will be discussed in the second part of this chapter. In the last part of the chapter, we shall look at the Chapter 7 Immunity Mediated by B Cells and Antibodies 181
Transcript of Immunity Mediated by B Cells and Antibodies

The production of antibodies is the sole function of the B-cell arm of theimmune system. Antibodies are useful in the defense against any pathogenthat is present in the extracellular spaces of the body’s tissues. Some humanpathogens, such as many species of bacteria, live and reproduce entirelywithin the extracellular spaces, whereas others, such as viruses, replicateinside cells but are carried through the extracellular spaces as they spreadfrom one cell to the next. Antibodies secreted by plasma cells in secondarylymphoid tissues and bone marrow find their way into the fluids filling theextracellular spaces.
Antibodies are not in themselves toxic or destructive to pathogens; their roleis simply to bind tightly to them. This can have several consequences. Oneway in which antibodies reduce infection is by covering up the sites on apathogen’s surface that are necessary for growth or replication, for examplethe viral glycoproteins that viruses use to bind to the surface of human cellsand initiate infection. Such antibodies are said to neutralize the pathogen. Inthe development of a vaccine against an infectious agent or its toxic products,the gold standard that a company aims for is the induction of a neutralizingantibody. Antibodies also act as molecular adaptors that bind to pathogenswith their antigen-binding arms and to receptors on phagocytic cells withtheir Fc regions. Thus, opsonization, or coating of a pathogen with antibody,promotes its phagocytosis. The antibody-directed destruction of pathogenscaused by opsonization is enhanced by the actions of a set of proteins that donot discriminate between antigens and are present in blood and lymph.These proteins are collectively known as complement because their func-tions complement the antigen-binding function of the antibody.
The structure, specificity, and other properties of antibodies were discussedin Chapter 2, and the development of B cells from their origin in bone mar-row to differentiation into antibody-secreting plasma cells was the subject ofChapter 4. This chapter will focus on how antibodies clear infection by tar-geting destructive but nonspecific components of the immune system to aninfecting pathogen. In the first part of the chapter we consider the antigensthat provoke a B-cell response, how the response develops, and the genera-tion of the different antibody isotypes. The structural differences betweenantibody isotypes provide a variety of adaptor functions that can target anti-body-bound pathogen to different types of nonspecific effector cell; theseaspects of the antibody-mediated immune response will be discussed in thesecond part of this chapter. In the last part of the chapter, we shall look at the
Chapter 7
Immunity Mediated by B Cells and
Antibodies
181

Chapter 7: Immunity Mediated by B Cells and Antibodies182
functions of the complement system, one of the principal mechanisms oftargeting extracellular pathogens for destruction, and how it is activated byantibody.
Antibody production by B lymphocytesThe antibodies most effective at combating infection are those that are madeearly in an infection and bind strongly to the pathogen. On first exposure toan infectious agent, these two goals make competing demands on theimmune system. As we saw in Chapters 4 and 6, B cells generally require helpfrom activated T cells to mature into antibody-secreting plasma cells; thisdelays the onset of antibody production until around a week after infection.In addition, B cells take time to switch isotype and undergo affinity matura-tion, processes that are necessary for the production of the high-affinity anti-bodies that are most effective at dealing with pathogens. Thus, during thecourse of an infection, the effectiveness of the antibodies produced improvessteadily. This experience is retained in the form of memory B cells and high-affinity antibodies, which provide long-term immunity to reinfection.
A faster primary response is made to certain bacterial antigens that are ableto activate B cells without the need for T-cell help. However, the antibodiesproduced in such a response are predominantly of the IgM isotype and ofgenerally low affinity. They do, however, provide an early defense, helping tokeep the infection at a relatively low level until a better antibody response canbe developed.
7-1 B-cell activation requires cross-linking of surfaceimmunoglobulin
On binding to protein or carbohydrate epitopes on the surface of a microor-ganism, the surface IgM molecules of a naive, mature B cell become physicallycross-linked to each other by the antigen and are drawn into the localized areaof contact with the microbe. This clustering and aggregation of B-cell recep-tors sends signals from the receptor complex to the inside of the cell (Figure7.1). Signal transduction from the B-cell receptor complex resembles, inmany ways, the signaling from the T-cell receptor complex discussed in Sec-tion 6-6, p. 155. Both types of receptor are associated with cytoplasmic pro-tein tyrosine kinases that are activated by receptor clustering, and bothreceptors activate similar intracellular signaling pathways.
Interaction of antigen with surface immunoglobulin is communicated to theinterior of the B cell by the proteins Iga and Igb, which are associated withIgM in the B-cell membrane to form the functional B-cell receptor. Like theCD3 polypeptides of the T-cell receptor complex, the cytoplasmic tails of Igaand Igb each contain two immunoreceptor tyrosine-based activation motifs(ITAMs) with which the Blk, Fyn, and Lyn tyrosine kinases associate. TheITAMs become phosphorylated on tyrosine residues, which allows the Syktyrosine kinase to bind to Igb tails that are doubly phosphorylated. Interac-tion between bound Syk molecules initiates intracellular signaling pathwaysthat lead to changes in gene expression in the nucleus (Figure 7.2).
Figure 7.1 Cross-linking of antigen receptors is the first step inB-cell activation. The B-cell receptors (BCR) on B cells are physicallycross-linked by the repetitive epitopes of antigens (Ag) on the surfaceof a bacterial cell. The B-cell receptor on a mature, naive B cell iscomposed of surface IgM, which binds antigen, and associated Iga
and Igb chains, which provide the signaling capacity.
B-cell receptors are activated bycross-linking with antigens
Ag
BCRIgM
Igα, Igβ
B cell
signals
bacterial cell

183Antibody production by B lymphocytes
Cross-linking of the B-cell receptor by antigen generates a signal that is nec-essary but not sufficient to activate a naive B cell. The additional signalsrequired are delivered in several ways. One set of signals is delivered when theB-cell receptor becomes closely associated with another protein complex onthe B-cell surface known as the B-cell co-receptor. The B-cell co-receptor is acomplex of three proteins: the first is the complement receptor 2 (CR2 orCD21), which binds to complement components deposited on a pathogen;the second is the protein CD19, which acts as the signaling chain of the recep-tor; and the third is the protein CD81 (TAPA-1), whose function is not yetknown. It does act, however, as a cell-surface receptor for the hepatitis C virus.
Certain antigens at pathogen surfaces catalyze a series of enymatic reactionsthat leads to the deposition of several fragments of complement proteins onthe pathogen’s surface close to the antigen molecule. One of these fragments,called C3d, is a ligand for the CR2 component of the B-cell co-receptor. Whenthe receptor of a B cell that is specific for the antigen binds to the antigen theCR2 component of the B-cell co-receptor complex can bind to an adjacentC3d, which serves to bring the B-cell receptor and co-receptor into juxtapo-sition. This co-ligation of the B-cell receptor and the co-receptor brings theIga-bound tyrosine kinase Lyn into close proximity with the CD19 cytoplas-mic tail, which it phosphorylates. The phosphorylated CD19 can then bindintracellular signaling molecules that generate signals that synergize withthose generated by the B-cell receptor complex (Figure 7.3). Simultaneousligation of the B-cell receptor and co-receptor increases the signals by 1000-to 10,000-fold.
Even the combined effects of the B-cell receptor and co-receptor signals aregenerally insufficient to activate a naive B cell. This requires additional signalsprovided by CD4 helper T cells, the effector cells produced upon antigen acti-vation of naive CD4 T cells. Whether a B cell needs T-cell help or not dependson the nature of the antigen, as we shall see in the next section.
The final outcome of B-cell activation is the proliferation and differentiationof the B cell into plasma cells and the secretion of antibodies. The morpho-logical effects of activation are striking: the small resting B cell, which inappearance is all nucleus and no cytoplasm, gives rise to plasma cells com-mitted to antibody secretion and whose large active cytoplasm packed withrough endoplasmic reticulum is testimony to this function (Figure 7.4).
7-2 The antibody response to certain antigens does not requireT-cell help
In their cell walls and capsules, bacterial pathogens possess complex polysac-charides, lipopolysaccharides, and peptidoglycans that are chemically andantigenically distinct from those of mammalian cells and are characterized by
Figure 7.2 Signals from the B-cell receptor initiate a cascade ofintracellular signals. On clustering of the receptors, the receptor-associated tyrosine kinases Blk, Fyn, and Lyn phosphorylate theimmunoreceptor tyrosine-based activation motifs (ITAMS) on thecytoplasmic tails of Iga and Igb (shown in blue and orange,respectively). Subsequently, Syk binds to the phosphorylated ITAMs ofthe Igb chain. Because there are at least two receptor complexes ineach cluster, Syk molecules become bound in close proximity and canactivate each other by transphosphorylation, thus initiating furthersignaling. Therefore, the signals produced are ultimately relayed tothe B-cell nucleus where they induce changes in gene expressionrequired for B-cell activation.
SykBlk, Fyn, or Lyn
Clustering of antigen receptorsallows receptor-associated kinases
to phosphorylate the ITAMs
B cell signals
changes in gene expression in nucleus
bacterial cell
Ag
Syk binds to doubly phosphorylatedITAMs and is activated on binding

Chapter 7: Immunity Mediated by B Cells and Antibodies184
repetitive epitopes. These cell-surface molecules are a major target of theantibody response against extracellular bacterial pathogens, and some ofthese antigens can activate naive B cells without help from CD4 T cells. Suchantigens are known as thymus-independent antigens (TI antigens) becauseimmunodeficient patients born without a thymus are able to make an anti-body response against them. However, they cannot respond to other antigensbecause that requires help from an antigen-activated helper T cell (see Sec-tion 6-17, p. 172). Antigens that need T-cell help are known as thymus-depen-dent antigens (TD antigens). For thymus-independent antigens, the need forT-cell help is overcome in two different ways.
As well as binding to the B-cell receptor, certain thymus-independent anti-gens (called TI-1 antigens) bind to other receptors on B cells, which, in com-bination, induce the B cells to proliferate and differentiate. An example of aTI-1 antigen is the lipopolysaccharide (LPS) of Gram-negative bacteria. LPSbinds to LPS-binding protein and CD14, which then associate with anotherreceptor, called a Toll-like receptor, to produce activating signals. When Bcells are triggered by TI-1 antigens they produce only IgM antibodies, becausecytokines produced by activated T-helper cells are needed for a B cell toswitch its antibody isotype. A surface-associated TI-1 antigen like LPS notonly causes T-cell-independent activation of B cells specific for epitopes ofLPS (Figure 7.5, left panels) but also B cells specific for other antigens of thebacterial cell surface (Figure 7.5, right panels).
The second type of thymus-independent antigen are the TI-2 antigens, whichare typically composed of repetitive carbohydrate or protein epitopes presentat high density on the surface of a microorganism. TI-2 antigens only stimu-late B cells specific for the antigen and probably act by cross-linking B-cellreceptors and co-receptors so extensively that the need for additional signalsis overridden. Responses to TI-2 antigens are usually seen around 48 hoursafter antigen was encountered. Typical antigens of this kind are bacterial cellwall polysaccharides and the responding B cells are often of the B-1 subpop-ulation (see Section 4-6, p. 108). Human B-1 cells develop their full functiononly when a person is about 5 years old, perhaps explaining why infants makerelatively poor antibody responses to polysaccharide antigens. IgM and IgGantibodies are both induced by TI-2 antigens and they are likely to be animportant part of the early B-cell response to some common bacterial infec-tions. Examples of TD, TI-1, and TI-2 antigens and the responses to them arepresented in Figure 7.6.
Although the TI-2 antigens on some bacteria induce an early antibodyresponse that helps to contain the infection, this response has limitations.There is little isotype switching and so the antibodies are predominantly IgM.
The B-cell receptor and co-receptorcooperate in B-cell activation
B cell
bacterial cell
CD19
CD81
C3d
activation signals
CR2
changes in gene expression in nucleus
Ag
Figure 7.3 Signals generated from the B-cell receptor and co-receptor synergize in B-cell activation. Binding of complementreceptor 2 (CR2) to complement fragments (C3d) deposited on thesurface of a pathogen cross-links the B-cell co-receptor complex withthe B-cell receptor. This causes them to cluster together on the B-cellsurface. The cytoplasmic tail of CD19 is then phosphorylated bytyrosine kinases associated with the B-cell receptor. PhosphorylatedCD19 binds intracellular signaling molecules whose signals synergizewith those generated by the B-cell receptor.
Figure 7.4 Plasma cell. Electron micrograph of a plasma cell. Notethe characteristic ‘clockface’ pattern in the nucleus (N), whichresembles the hands and face of a clock, as well as the extensiveendoplasmic reticulum (ER). Photograph courtesy of C. Grossi.
outstanding electronic
permission for thisfigure

185Antibody production by B lymphocytes
Neither is there somatic hypermutation, so there is no possibility for increas-ing the affinity for antigen of the antibodies produced. Lastly, TI-2 antigens donot induce long-term immunological memory and so provide no long-lastingimmunity against reinfection. The development of all these attributesrequires T-cell help, as we shall see next.
7-3 B cells needing T-cell help are activated in secondarylymphoid tissues where they form germinal centers
Although the antibody response to a pathogen may be initiated by thymus-independent antigens, the bulk of the pathogen-specific antibody is eventu-ally produced by B cells stimulated by thymus-dependent antigens. Activa-tion of these B cells occurs in the secondary lymphoid tissues where B cells,specific antigen, and helper CD4 T cells are all brought together. We willdescribe these processes using the lymph node as an example.
Antigens arrive at a node in the lymph draining the infected tissue, whereasantigen-specific lymphocytes enter the node from the blood (see Section 4-9,p. 113). The antigens are transported by dendritic cells that mature and takeup residence in the T-cell area of the lymph node (see Section 6-1, p. 146) orare passively carried along in the lymph to be phagocytosed by macrophagesresident in the lymph node. Either way, the antigens are trapped in the node
Activation of B cells by TI-1 antigens
B cell
bacterial cell
CD19
CD14
LBP
LPS
LPS
CD81
C3d
activation signals
CR2
activation, proliferation, differentiation
LPS-specific, B-cell activation.Production of only LPS-specific IgM
B cell
bacterial cell
CD19
CD14
LBP
LPS
CD81
C3d
activation signals
CR2
activation, proliferation, differentiation
LPS helps activate B cells specific foranother antigen on the bacterial surface
Figure 7.5 Thymus-independent (TI)-1 antigens can activate B cellswithout T-cell help. Certain antigens,such as the lipopolysaccharide (LPS) ofGram-negative bacteria, can on theirown activate B cells to becomeantibody-producing plasma cells. In theleft-hand panels, LPS binds to itsreceptor CD14 on the B-cell surface andalso to an LPS-specific, B-cell receptor.Signaling through both CD14, the B-cell receptor and the associated B-cell co-receptor complex is sufficientto activate the B cell, which gives rise toplasma cells producing anti-LPSantibodies. In the right-hand panels, LPS binding to CD14 provides a co-activating signal for another antigenon the bacterium that binds to itsspecific B-cell receptor. This B cell goeson to produce antibodies specific for thebacterial antigen, not LPS.
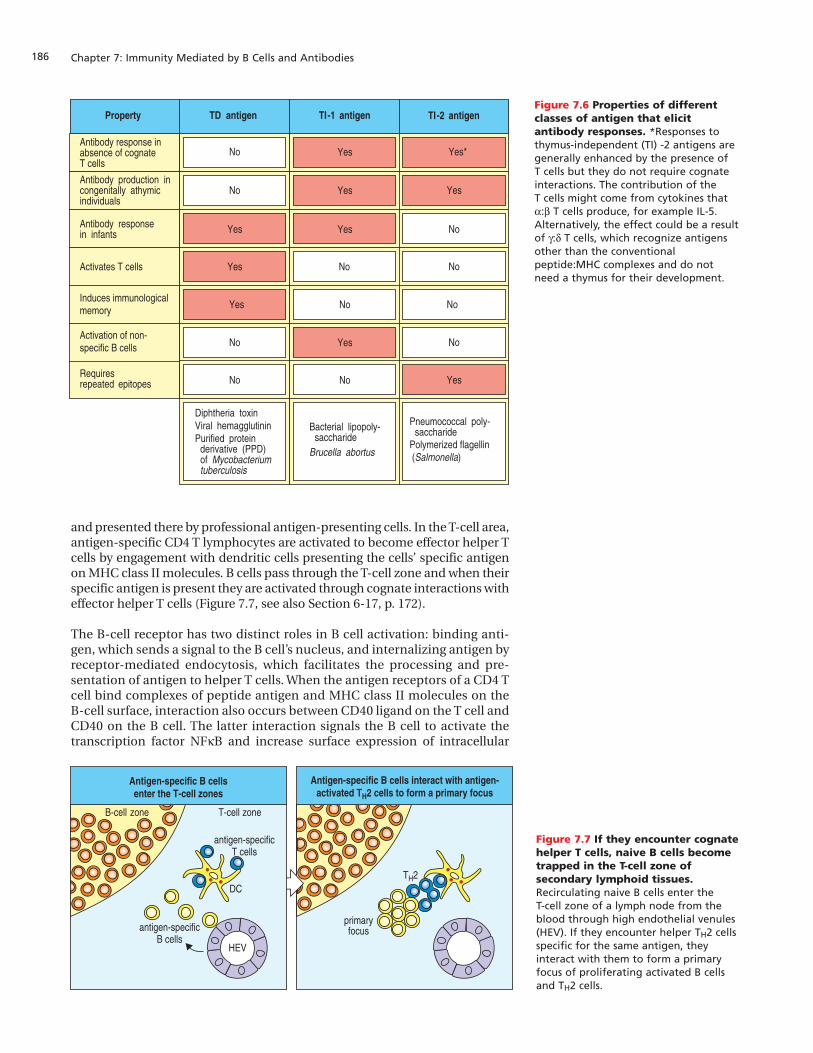
and presented there by professional antigen-presenting cells. In the T-cell area,antigen-specific CD4 T lymphocytes are activated to become effector helper Tcells by engagement with dendritic cells presenting the cells’ specific antigenon MHC class II molecules. B cells pass through the T-cell zone and when theirspecific antigen is present they are activated through cognate interactions witheffector helper T cells (Figure 7.7, see also Section 6-17, p. 172).
The B-cell receptor has two distinct roles in B cell activation: binding anti-gen, which sends a signal to the B cell’s nucleus, and internalizing antigen byreceptor-mediated endocytosis, which facilitates the processing and pre-sentation of antigen to helper T cells. When the antigen receptors of a CD4 Tcell bind complexes of peptide antigen and MHC class II molecules on theB-cell surface, interaction also occurs between CD40 ligand on the T cell andCD40 on the B cell. The latter interaction signals the B cell to activate thetranscription factor NFkB and increase surface expression of intracellular
Chapter 7: Immunity Mediated by B Cells and Antibodies186
Antibody production incongenitally athymicindividuals
Antibody response inabsence of cognate T cells
Antibody responsein infants
TI-1 antigen
Yes
Yes
Yes
Yes
Bacterial lipopoly-saccharide
Brucella abortus
No
TI-2 antigen
Yes
No
Pneumococcal poly-saccharide
Polymerized flagellin (Salmonella)
No
No
Yes*
Activates T cells
TD antigenProperty
No
Yes
Yes
No
No
Induces immunologicalmemory No NoYes
Diphtheria toxinViral hemagglutininPurified protein
derivative (PPD)of Mycobacteriumtuberculosis
Activation of non-specific B cells
Requiresrepeated epitopes No YesNo
Figure 7.6 Properties of differentclasses of antigen that elicitantibody responses. *Responses tothymus-independent (TI) -2 antigens aregenerally enhanced by the presence of T cells but they do not require cognateinteractions. The contribution of the T cells might come from cytokines thata:b T cells produce, for example IL-5.Alternatively, the effect could be a resultof g:d T cells, which recognize antigensother than the conventionalpeptide:MHC complexes and do notneed a thymus for their development.
primaryfocus
Antigen-specific B cells interact with antigen-activated TH2 cells to form a primary focus
T-cell zone
DC
antigen-specificT cells
antigen-specificB cells
B-cell zone
HEV
Antigen-specific B cellsenter the T-cell zones
TH2
Figure 7.7 If they encounter cognatehelper T cells, naive B cells becometrapped in the T-cell zone ofsecondary lymphoid tissues.Recirculating naive B cells enter the T-cell zone of a lymph node from theblood through high endothelial venules(HEV). If they encounter helper TH2 cellsspecific for the same antigen, theyinteract with them to form a primaryfocus of proliferating activated B cellsand TH2 cells.

187Antibody production by B lymphocytes
adhesion molecule 1 (ICAM-1). This strengthens the cognate interactionbetween the B cell and the helper CD4 T cell. A signal produced in the T cellcauses it to secrete cytokines. Of these, interleukin-4 (IL-4) is characteristic ofTH2 cells and essential for B-cell proliferation. Cytokines binding to receptorson the B-cell surface drive the B cell to proliferate and differentiate intoplasma cells (Figure 7.8).
B cells activated by interactions with cognate helper T cells in the T-cell areasof a lymph node form a primary focus of dividing B lymphoblasts that lasts fora few days (see Figure 7.7). Some of these B lymphoblasts move to themedullary cords and differentiate directly into plasma cells under the influ-ence of the cytokines IL-5 and IL-6, which are secreted by TH2 cells. Theysecrete predominantly IgM antibody but some isotype switching can alsooccur in the primary focus. For infections in which the pathogen carries nothymus-independent antigens, this will be the first antibody produced. It willstart to appear several days after the onset of infection.
Other B lymphoblasts from the primary focus move into primary follicles stillattached to their cognate helper T cells. Their rate of division increases toabout once every 6 hours and they become large, metabolically active cellscalled centroblasts (see Section 4-9, p. 113). With the increase in the numberof centroblasts, the morphology of a follicle changes and it becomes domi-nated by the germinal center that contains the newly formed B cells (Figure7.9). Germinal centers appear in secondary lymphoid tissues about 1 weekafter the start of infection and they cause the characteristic swelling of lymphnodes draining an infection.
As they divide, the centroblasts become increasingly closely packed and forma region that is darkly staining in histological sections and is called the darkzone of the germinal center. The centroblasts give rise to nondividing centro-cytes, which leave the close-packed lymphocytes to interact with folliculardendritic cells (FDCs) in the light zone of the germinal center (see Figure7.9). Follicular dendritic cells are the characteristic stromal cell of primarylymphoid follicles. They pick up antigen but do not internalize it, and itremains bound to their surface for long periods of time. They interact with Bcells through a dense network of antigen-loaded dendrites. The folliculardendritic cells are quite distinct from the dendritic cells that present antigento naive T cells and activate them (see Section 6-5, p. 151); they do not derivefrom a hematopoietic stem cell and do not express MHC class II molecules.
Those helper T cells that migrated to the primary follicle together with theactivated B cells also proliferate in the light zone and are intermingled withthe centrocytes. With time, the vast majority of lymphocytes present in agerminal center are clones derived from one or a few founder pairs of anti-gen-activated B and T cells. B cells that were present in the primary folliclebefore entry of the activated B cell–T cell conjugates, and are not specific for
B cell proliferates and differentiatesinto plasma cells
cytokines
CD40CD40L
helper T cell
Helper TH2 cell delivers the second signalvia CD40 ligand and cytokines
Antigen binding to B-cell receptordelivers the first signal to the B cell
B cell
Figure 7.8 B-cell activation inresponse to thymus-dependentantigens requires cognate T-cellhelp. The first signal required for B-cellactivation is delivered through theantigen receptor (left panel). Withthymus-dependent antigens, the secondsignal is delivered by a cognate helper T cell that recognizes a peptidefragment of the antigen bound to MHC class II molecules on the B-cellsurface (center panel). The two signalstogether drive B-cell proliferation anddifferentiation into plasma cells (rightpanel).

the antigen, are pushed to the outside of the germinal center, forming themantle zone (see Figure 7.9).
7-4 Activated B cells undergo somatic hypermutation andaffinity maturation in the specialized microenvironment of thegerminal center
As we have seen in Chapters 4–6, a common theme in lymphocyte develop-ment is for a phase of activation and proliferation to be followed by one ofselection. This is precisely what happens to the B cells maturing in a germinalcenter. Somatic hypermutation, initiated by T-cell cytokines, takes place incentroblasts dividing within the germinal center and gives rise to nondividingcentrocytes with mutated surface immunoglobulin. After hypermutation, thesurface immunoglobulin expressed by an individual centrocyte can have anaffinity for its specific antigen that is higher, lower, or the same as that of theunmutated immunoglobulin. Thus, the population of centrocytes in a germi-nal center expresses immunoglobulins with a range of affinities for the spe-cific antigen.
Centrocytes are programmed to die by apoptosis within a short period unlesstheir surface immunoglobulin is bound by antigen and they are subsequentlycontacted by a helper T cell bearing CD40 ligand. To engage such a helper Tcell, the centrocyte must first bind and process antigen, then present antigenicpeptides at its surface in association with MHC class II molecules. The
Chapter 7: Immunity Mediated by B Cells and Antibodies188
Figure 7.9 Germinal centers are formed when activated B cells enter lymphoid follicles. The germinal center is aspecialized microenvironment in which B-cell proliferation,somatic hypermutation, and selection for antigen binding alloccur. Rapidly proliferating B cells in germinal centers are calledcentroblasts. Closely packed centroblasts form the so-called ‘darkzone’ of the germinal center. This can be seen in the lower partof the center panel, which shows a light micrograph of a sectionthrough a germinal center, and in the accompanying diagram(left panel). As these cells mature, they stop dividing andbecome small centrocytes, moving out into an area of thegerminal center called the ‘light zone’ (in the upper part of the
center panel), where the centrocytes make contact with a densenetwork of follicular dendritic cell processes. The folliculardendritic cells are not stained in the center panel but can beseen clearly in the right panel, in which both follicular dendriticcells (stained blue with antibody against Bu10, a marker offollicular dendritic cells) in the germinal center, as well as themature B cells in the mantle zone (stained brown with anantibody against IgD) can be seen. The plane of this sectionreveals mostly the dense network of follicular dendritic cells inthe light zone, although the less dense network in the dark zonecan just be seen at the bottom of the figure. Photographscourtesy of I. MacLennan.
centrocytes
T cellsH
mantlezone
centroblasts
darkzone
lightzone
folliculardendriticcells
Germinal center (low power) stainedto show follicular dendritic cells
Light micrograph of germinal center(high power)Schematic representation of a germinal center
FDCs
189Antibody production by B lymphocytes
mutated centrocytes now compete with each other, first for access to antigenon follicular dendritic cells and then for antigen-specific helper T cells.
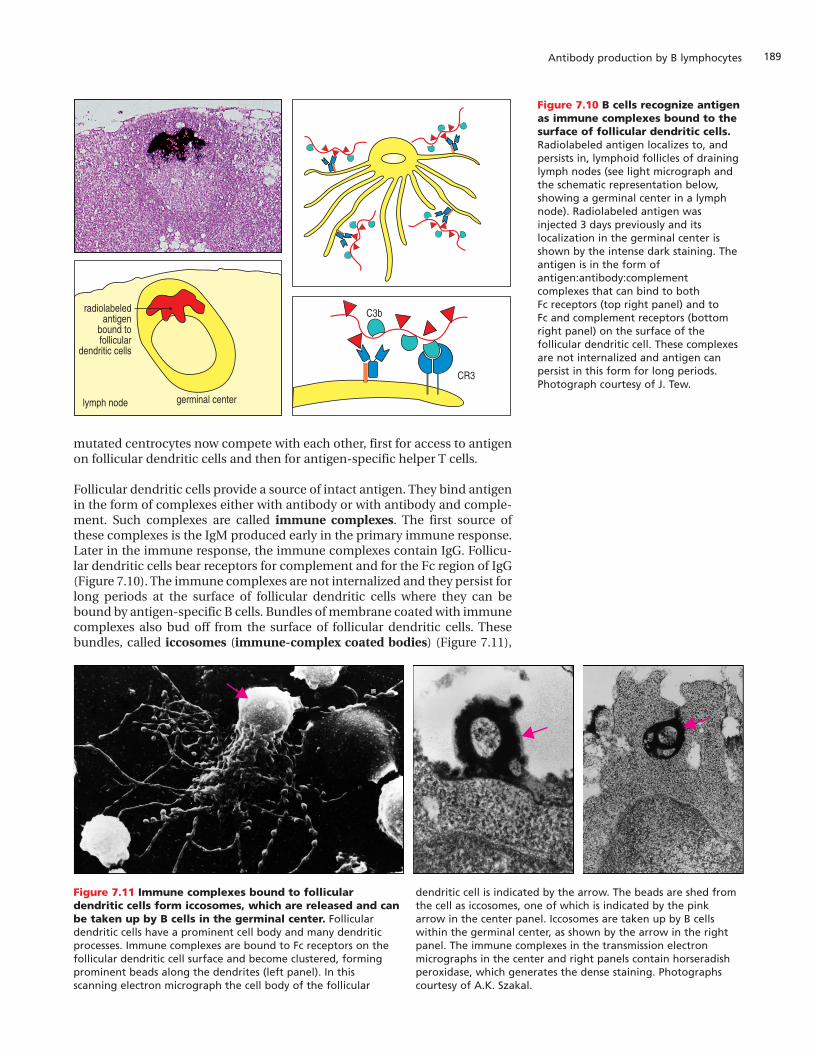
Follicular dendritic cells provide a source of intact antigen. They bind antigenin the form of complexes either with antibody or with antibody and comple-ment. Such complexes are called immune complexes. The first source ofthese complexes is the IgM produced early in the primary immune response.Later in the immune response, the immune complexes contain IgG. Follicu-lar dendritic cells bear receptors for complement and for the Fc region of IgG(Figure 7.10). The immune complexes are not internalized and they persist forlong periods at the surface of follicular dendritic cells where they can bebound by antigen-specific B cells. Bundles of membrane coated with immunecomplexes also bud off from the surface of follicular dendritic cells. Thesebundles, called iccosomes (immune-complex coated bodies) (Figure 7.11),
germinal centerlymph node
radiolabeledantigen
bound tofollicular
dendritic cells
C3b
CR3
Figure 7.10 B cells recognize antigenas immune complexes bound to thesurface of follicular dendritic cells.Radiolabeled antigen localizes to, andpersists in, lymphoid follicles of draininglymph nodes (see light micrograph andthe schematic representation below,showing a germinal center in a lymphnode). Radiolabeled antigen wasinjected 3 days previously and itslocalization in the germinal center isshown by the intense dark staining. Theantigen is in the form ofantigen:antibody:complementcomplexes that can bind to both Fc receptors (top right panel) and to Fc and complement receptors (bottomright panel) on the surface of thefollicular dendritic cell. These complexesare not internalized and antigen canpersist in this form for long periods.Photograph courtesy of J. Tew.
Figure 7.11 Immune complexes bound to folliculardendritic cells form iccosomes, which are released and canbe taken up by B cells in the germinal center. Folliculardendritic cells have a prominent cell body and many dendriticprocesses. Immune complexes are bound to Fc receptors on thefollicular dendritic cell surface and become clustered, formingprominent beads along the dendrites (left panel). In thisscanning electron micrograph the cell body of the follicular
dendritic cell is indicated by the arrow. The beads are shed fromthe cell as iccosomes, one of which is indicated by the pinkarrow in the center panel. Iccosomes are taken up by B cellswithin the germinal center, as shown by the arrow in the rightpanel. The immune complexes in the transmission electronmicrographs in the center and right panels contain horseradishperoxidase, which generates the dense staining. Photographscourtesy of A.K. Szakal.

are bound and taken up by antigen-specific B cells, which then process andpresent the antigen.
Newly formed centrocytes move from the dark zone of the germinal center tocontact follicular dendritic cells in the light zone. If a centrocyte capturessufficient antigen from the follicular dendritic cells or iccosomes, it thenmoves to the outer regions of the light zone where helper T cells are concen-trated. Engagement of peptide:MHC class II by the T-cell receptor complex,and of CD40 on the centrocyte by CD40 ligand on the T cell, induces the cen-trocyte to express the Bcl-xL protein, which prevents its death by apoptosis(Figure 7.12).
Thus, centrocytes with the highest-affinity antigen receptors are selected forsurvival and further differentiation into antibody-producing plasma cells or
Chapter 7: Immunity Mediated by B Cells and Antibodies190
B
Germinal center B cell with low-affinitysurface immunoglobulin
Somatic hypermutation of immunoglobulinV regions in rapidly proliferating
germinal center B cells
Activated B cell
cytokines
BHelperT cell
HelperT cell
Germinal center B cell with high-affinitysurface immunoglobulin
B cell dies by apoptosis Memory B cell
IgG
Plasma cell
CD40 CD40L
HelperT cell
BCR cross-linking
B-cell receptor is not cross-linked and B cellcannot present antigen to T cell
T-cell help and B-cell receptor cross-linkingsustain B cell proliferation and differentiation
Figure 7.12 After somatichypermutation, B cells with high-affinity receptors for antigen arerescued from apoptosis. In thegerminal center, helper T cells induce B cells to undergo somatichypermutation (top panel). B cells thathave undergone somatic hypermutationinteract with follicular dendritic cells(FDCs) that display immune complexeson their surface. B cells whose receptorsbind antigen poorly, or do not bindantigen at all because they havemutated beyond recognition, cannotcompete for access to the FDCs and dieby apoptosis (left panel). B cells withreceptors that bind well receive signalsfrom the FDCs and are induced toexpress Bcl-xL, which prevents apoptosis;these cells survive (right panels). BCR, B-cell receptor.

191Antibody production by B lymphocytes
into long-lived memory cells. In this way, the affinity of antibodies for thespecific antigen increases during the course of an immune response and insubsequent exposures to the same antigen. This process is known as affinitymaturation.
Under the influence of T cells, isotype switching also takes place in B cellswithin the germinal center. Thus, not only does the affinity of the antibod-ies produced increase but also antibodies of different isotypes are made,principally IgG in the case of B cells that differentiate in the lymph nodesand spleen.
For mutated centrocytes that survive selection, the interaction with an anti-gen-specific helper T cell serves several purposes. The mutual engagement ofligands and receptors on the two cells generates an exchange of signals thatinduces the further proliferation of both B and T cells. This serves to expandthe population of selected high-affinity, isotype-switched B cells. Individual Bcells are also directed along pathways of differentiation leading either toplasma cells or to memory B cells. At the height of the adaptive immuneresponse, when the main need is for large quantities of antibodies to fightinfection, the centrocytes that win in this selection leave the germinal centerand differentiate into antibody-producing plasma cells. The differencesbetween resting B cells and plasma cells are summarized in Figure 7.13. In thelater stages of a successful immune response, as the infection subsides, cen-trocytes are thought to differentiate into long-lived, memory B cells, whichnow possess isotype-switched, high-affinity antigen receptors. Plasma cellsare the effector B cells that provide antibody for dealing with today’s infec-tion, whereas the memory B cells represent an investment in the preventionof future infection with the same pathogen should the current infection besuccessfully resolved.
If a centrocyte fails to obtain, internalize, and present antigen it dies by apop-tosis and is phagocytosed by macrophages in the germinal center.Macrophages that have recently engulfed apoptotic centrocytes are a charac-teristic feature of germinal centers and, because of their contents, are calledtingible body macrophages. Somatic hypermutation can produce centro-cytes bearing immunoglobulin that reacts with a self-antigen on the surfaceof cells in the germinal center. When this happens, contact with helper T cellsor other cells in the germinal center will render such centrocytes inactive oranergic—a mechanism similar to the one whereby self-reactive, immature Bcells are inactivated in the bone marrow (see Section 4-7, p. 111).
Figure 7.13 Comparison of resting B cells and plasma cells. The resting B cell expresses an antigen receptor inthe form of surface immunoglobulin,and can also take up protein antigenand present it as a peptide:MHC class IIcomplex. Thus, it can activate helper T cells. Its immunoglobulin genes canalso undergo somatic hypermutation,giving rise to progeny with alteredimmunoglobulin specificity. The plasmacell, in contrast, is a terminallydifferentiated B cell that is dedicated tothe synthesis and secretion of solubleantibody. It no longer divides and itsantibody specificity cannot be changed.
Yes
No
Yes
No
Yes
No
Yes
No
High-rateIg secretion
Isotypeswitch
Somatichyper-
mutationB-lineage cell
No
Resting B cell
Plasma cell
Yes
Property
Inducible
GrowthSurfaceMHC class IISurface Ig
Yes
No
Intrinsic

7-5 Interactions with T cells are required for isotype switchingin B cells
In Chapter 2 we saw how the first immunoglobulins made by B cells are of theIgM and IgD classes, but, after activation by antigen B cells can switch theirheavy-chain isotype to produce IgG, IgA, or IgE. Isotype switching takes placein activated B cells mainly within the germinal center, and the isotype towhich an individual B cell switches is determined by cognate interactionswith helper T cells. The particular isotype to which a switch is made dependson the cytokines secreted by the helper T cell. The roles of individualcytokines in switching the isotype of mouse immunoglobulin heavy chainsare summarized in Figure 7.14. Cytokines secreted by TH2 cells—IL-4, IL-5,and TGF-b—are the predominant players. They initiate the antibody responseby activating naive B cells to differentiate into plasma cells secreting IgM, andalso induce the production of other antibody isotypes including, in humans,the weakly opsonizing antibodies IgG2 and IgG4, as well as IgA and IgE. How-ever, interferon (IFN)-g, the characteristic cytokine produced by TH1 cells,switches B cells to making the IgG2a and IgG3 classes of immunoglobulin (inmice) and the strongly opsonizing antibody IgG1 in humans.
T-cell cytokines induce isotype switching by stimulating transcription fromthe switch regions that lie 5¢ to each heavy-chain C gene. For example, whenactivated B cells are exposed to IL-4, transcription from a site upstream of theswitch regions of Cg1 and Ce can be detected a day or two before switchingoccurs. As with the low-level transcription that occurs in immunoglobulinloci before rearrangement (see Section 4-2, p. 102), this transcription could beopening up the chromatin and making the switch regions accessible to thesomatic recombination machinery that will place a new C gene in juxtaposi-tion to the V-region sequence.
The induction of isotype switching by cognate helper T cells also requires theligation of CD40 on the B-cell surface by CD40 ligand on the T cells. Theimportance of helper T cells and the CD40–CD40 ligand interaction for iso-type switching is apparent from the immunodeficiency of patients who lackCD40 ligand. These patients have abnormally high levels of IgM in their bloodserum, which gives the name hyper-IgM syndrome to their condition, butalmost no IgG and IgA because of the inability of their B cells to switch iso-type. They cannot make antibody responses to thymus-dependent antigensand their secondary lymphoid tissues contain no germinal centers (Figure7.15), showing the general importance of the CD40–CD40 ligand interactionsin T-cell help. Aspects of cell-mediated immunity are also impaired in thesepatients, who are mostly male because the gene for CD40 ligand is on the Xchromosome.
Chapter 7: Immunity Mediated by B Cells and Antibodies192
InducesInhibitsInhibits
Induces
Induces
Induces
Induces
Induces
Inhibits Inhibits
Inhibits
Inhibits
Inhibits
Inhibits
IgEIgG2aIgG3IgM IgG1 IgG2b IgACytokine
IL-4
IL-5
IFN-�
TGF-�
Influence of cytokines on antibody isotype switching in mice
Augmentsproduction
Figure 7.15 Comparison of normaland hyper-IgM syndrome lymphnodes. Bottom panel photographcourtesy of Dr Antonio Perez-Atayde.
Lymph node from patient with hyper-IgM syndrome (no germinal centers)
Lymph node with germinal centers
Figure 7.14 Different cytokinesinduce B cells to switch to differentimmunoglobulin isotypes. Individualcytokines can either induce (green),augment (bright yellow), or inhibit (red)the switching of immunoglobulinsynthesis to a particular isotype. Theinhibitory effects are largely due to thepositive effect of the cytokine onswitching to another isotype. Thiscompilation is drawn from experimentson mouse B cells. There are differencesin humans, but they are not yet as wellworked out. For example, switching toIgA in humans involves TGF-b and IL-10,not IL-5.

193Antibody production by B lymphocytes
Summary
B cells respond to specific antigen with activation, proliferation, and differen-tiation. They then become plasma cells that synthesize and secrete massiveamounts of antibody. Activation of a mature but naive B cell requires signalsdelivered through its antigen receptor and most B cells also need additionalsignals that are delivered only on cognate interaction with an antigen-specific,helper T cell. Activating signals are also delivered through the B-cell co-recep-tor when this is simultaneously ligated with the antigen receptor. The signal-ing pathways used to activate B cells are summarized in Figure 7.16; they aresimilar to those used to activate T cells. The first antibodies made are alwaysIgM; further contact with effector helper T cells is required for activated B cellsto undergo isotype switching, somatic hypermutation, and affinity maturation
CD45
Igb Igk
Syk
tyrosine kinase(Blk, Fyn, or Lyn)
IgM cross-linked by antigen
Receptor cross-linking activatestyrosine kinases Blk, Fyn,and Lyn
Activated kinases phosphorylate theB-cell receptor cytoplasmic domains
Activated kinases activate CD19, BLNK,phospholipase C-γ (PLC-γ), GEFs, and Tec kinases
Syk tyrosine kinasebinds to phosphorylated
Ig and becomesactivated
β
PLC- cleaves phosphatidylinositol bisphosphate (PIP )to yield diacylglycerol (DAG) and inositol trisphosphate (IP )
γ 2
3
The CD45 tyrosinephosphatase allows
activation of receptor-associated kinases
Small G proteins activateMAP kinase cascades
GEFs activate small Gproteins (Ras and Rac)
IP3 increases intracellularCa2+ concentration,
activating a phosphatase,calcineurin
Calcineurin activates a transcription factor, NFAT(nuclear factor of activated
T cells)
The transcription factors NFκB, NFAT, and AP-1 act to induce specific gene transcription,leading to cell proliferation and differentiation
The Ras-induced kinasecascade induces and activatesFos, a component of the AP-1
transcription factor
Protein kinase C activatesa transcription factor, NFκB
DAG and Ca2+ activateprotein kinase C
bacterial cell
Figure 7.16 Simplified outline of theintracellular signaling pathwaysinitiated by cross-linking of B-cellreceptors by antigen. These pathwaysare similar to those that act in naive T cells exposed to antigen (see Figure6.17), but some of the components, suchas the kinase Blk, are specific to B cells.

within the germinal centers of secondary lymphoid organs. All this takes time,during which the pathogen can multiply, spread from the focus of infection,and cause disease. However, if the host survives, there will remain in the cir-culation expanded populations of high-affinity antibodies and memory Bcells programmed to make them again should the need arise. Some antigens,notably certain components of bacterial cell walls and capsules, are capableof inducing a rapid antibody response that does not require T-cell help. Thesethymus-independent antigens are of two types. TI-1 antigens bind to a sec-ond receptor that contribute signals for mitosis and differentiation in addi-tion to those generated through the B-cell antigen receptor. TI-2 antigens aregenerally microbial cell-surface macromolecules with repetitive epitopes thatare present at high density on microbial surfaces and extensively cross-linkthe antigen receptors and co-receptors on the B-cell surface. Antibodies pro-duced against TI antigens are predominantly IgM and the cells producingthem are often of the B-1 lineage. Responses to TI antigens neither induceimmunological memory nor long-lasting immunity.
Antibody effector functionsAs the B-cell response to an infection gets under way, isotype switching diver-sifies the functional properties of the antibody Fc region, which containsbinding sites for other proteins and cells of the immune system. Fc regionsserve two distinct functions: they deliver antibody to anatomical sites thatwould otherwise be inaccessible and they link bound antigen to moleculesor cells that will effect its destruction. Such cells carry receptors called Fcreceptors, which bind to the Fc regions of antibodies of a particular class orsubclass, irrespective of the antibody’s antigen specificity. In this part of thechapter we shall consider how antibodies of different isotypes recruit non-specific effector cells, such as macrophages and neutrophils, into theimmune response by interaction with their Fc receptors.
7-6 IgM, IgG, and IgA antibodies protect the blood andextracellular fluids
In any antibody response, IgM is the first antibody to be produced. It issecreted as a pentamer by plasma cells in the bone marrow, spleen, andmedullary cords of lymph nodes. IgM enters the blood and is carried to sitesof tissue damage and infection throughout the body. The pentameric natureof IgM enables it to bind strongly to microorganisms and particulate antigens,but its large size decreases the extent to which this antibody isotype can pas-sively leave the blood and penetrate infected tissues. There are no receptorsfor the IgM Fc region on phagocytic cells or other leukocytes, so IgM cannotdirectly recruit the destructive capabilities of these cells into the immuneresponse. The Fc region of IgM can, however, bind complement and activatethe complement system, with consequences that we shall consider in the lastpart of this chapter.
Later in an immune response, the dominant blood-borne antibody is thesmaller IgG molecule. An important function of circulating IgM and IgG is toprevent blood-borne infection—septicemia—and the spread of microorgan-isms by neutralizing those that enter the blood. Because the blood circulationis so effective in distributing cells and molecules to all parts of the body, infec-tions of the blood itself can have grave consequences.
IgA is synthesized by plasma cells in secondary lymphoid tissues. MonomericIgA is made by plasma cells derived from B cells that switched their antibodyisotype in the lymph nodes or spleen. In contrast, dimeric IgA is made in the
Chapter 7: Immunity Mediated by B Cells and Antibodies194

195Antibody effector functions
secondary lymphoid tissues underlying mucosal surfaces, as we shall see inthe next section. Like IgG, monomeric IgA enters the extracellular spaces andhelps IgG to protect them against infection by bacteria and virus particles.
7-7 IgA and IgG are transported across epithelial barriers byspecific receptor proteins
Whereas IgM, IgG, and monomeric IgA provide antigen-binding functionswithin the fluids and tissues of the body, dimeric IgA protects the surfaces ofthe epithelia that communicate with the external environment and are par-ticularly vulnerable to infection. These epithelia include the linings of the gas-trointestinal tract, the eyes, nose, throat, the respiratory, urinary, and genitaltracts, and the mammary glands. Dimeric IgA is made in patches of mucosal-associated lymphoid tissues in the lamina propria, the connective tissue thatunderlies the basement membrane of the mucosal epithelium. In these tis-sues, antigen-specific B-cell and T-cell responses to local infections are devel-oped. However, the IgA-secreting plasma cells are on one side of the epithe-lium and their target pathogens are on the other. To reach their targets,dimeric IgA molecules are transported individually across the epithelium bymeans of a receptor on the basolateral surface of the epithelial cells.
The dimeric form of IgA, but not the monomer, binds to a cell-surface recep-tor on the basolateral surface of epithelial cells that is called the poly-Igreceptor because of its specificity for IgA polymers and, to a lesser extent, forpentameric IgM (Figure 7.17). The poly-Ig receptor itself is made up of a seriesof immunoglobulin-like domains. On being bound, the IgA dimer is takeninto the cell by receptor-mediated endocytosis and the antibody:receptorcomplex is carried across the cell to the apical surface in endocytic vesicles.Receptor-mediated transport of a macromolecule from one side of a cell tothe other is known as transcytosis. Once receptor-bound IgA appears on the
Figure 7.17 Transcytosis of dimericIgA antibody across epithelia ismediated by the poly-Ig receptor.Dimeric IgA is made mostly by plasmacells lying just beneath the epithelialbasement membranes of the gut,respiratory tract, tear glands, andsalivary glands. The IgA dimer bound tothe J chain diffuses across the basementmembrane and is bound by the poly-Igreceptor on the basolateral surface ofan epithelial cell. Binding to thereceptor is via the CH3 constant domainsof the IgA heavy chains. The boundcomplex undergoes transcytosis acrossthe cell in a membrane vesicle and isfinally released onto the apical surface.There the poly-Ig receptor is cleaved,releasing the IgA from the epithelial cellmembrane while still being bound to afragment of the receptor called thesecretory component or secretory piece.Carbohydrate (blue hexagon) on thepoly-Ig receptor forms the secretorypiece that binds to mucus at theepithelial surface, thus preventing IgAfrom being washed away into the gutlumen. The residual membrane-boundfragment of the poly-Ig receptor isnonfunctional and is degraded.
Receptor is cleaved,IgA is bound to mucusthrough the secretory
piece
IgA dimer+ secretory component
Receptor-mediatedendocytosis of IgA
basement membrane
lamina propria
tight junction
lumen
Binding of IgA toreceptor on basolateral
face of epithelial cell
epithelial cell
poly-Igreceptor
dimericIgA
IgA-secreting cell
Transport of IgAto apical face of
epithelial cell

apical surface, a protease cleaves the poly-Ig receptor at sites between themembrane-anchoring region and the IgA-binding site. Dimeric IgA isreleased from the membrane still bound to a small fragment of the poly-Igreceptor, which is called the secretory component, or secretory piece, of IgA.The IgA is then held at the mucosal surface, being bound to mucins in mucusby the carbohydrate of the secretory piece.
IgG is actively transported from the blood into the extracellular spaces withintissues by an Fc receptor present on the endothelial cells (Figure 7.18). Thisreceptor is sometimes called the Brambell receptor (FcRB) after the scientistwho first described its function. FcRB is similar in structure to an MHC classI molecule, with the a1 and a2 domains forming a site that binds to the Fcregion of the antibody. In the antibody:receptor complex, two molecules ofFcRB bind to the Fc region of one IgG molecule. The delivery of IgG to theextracellular spaces in connective tissue helps to protect tissues against infec-tion and also protects IgG from the degradation pathways to which serumproteins are subject. As a consequence, IgG molecules have a relatively longhalf-life in relation to most other plasma proteins.
During pregnancy, the fetus is physically protected by the mother from themicroorganisms that inhabit the external environment. At birth, the baby issuddenly exposed to numerous pathogens. Because of their lack of activelyacquired immunity, newborn infants are particularly vulnerable to infectionarising from the microbial colonization of epithelia. To help the infantcounter such attack, it receives IgA from its mother. The IgA is first secretedinto breast milk and then transferred on breast-feeding into the baby’s gut.Within this transferred IgA are antibodies against microorganisms to whichthe mother has previously mounted an IgA response. Within the infant’s gut,the IgA molecules bind to microorganisms and their products, preventingtheir attachment to the gut epithelium and facilitating their expulsion infeces. The transfer of preformed IgA from mother to child in breast milk is anexample of the passive transfer of immunity.
IgA is not the only immunoglobulin isotype that mothers donate to their chil-dren. During pregnancy, IgG from the maternal circulation is transportedacross the placenta and is delivered directly into the fetal bloodstream. Theefficiency of this mechanism is such that at birth human babies have as higha level of IgG in their plasma as their mothers, and as wide a range of antigenspecificities. Transport of IgG across the placenta is performed by FcRB (seeFigure 7.18).
Mice and rats express a homologue of FcRB called FcRn, but its function issomewhat different. In rodents, the receptor is expressed in the intestine fora short period after birth. During this time, the newborn rodent ingestsmaternal IgG in colostrum, the protein-rich fluid in the postnatal mammarygland, which is then transported across the intestinal epithelium into the tis-sues by FcRn.
By means of these specialized transport systems mammals are supplied frombirth with antibodies against common pathogens in their environment. Asthe young mature and make their own antibodies of all the isotypes, these areeach distributed to selected sites in the body (Figure 7.19). Thus, throughout
Chapter 7: Immunity Mediated by B Cells and Antibodies196
FcRB carries IgG across endotheliuminto extracellular spaces
extracellularspaces
endothelialcell
lumen ofcapillary
FcRB
IgG
Figure 7.18 The Brambell receptor (FcRB) transports IgG fromthe bloodstream into the extracellular spaces. An IgG moleculebinds to two FcRB molecules at the apical (luminal) side of theendothelial cell. After receptor-mediated endocytosis, the IgGmolecule is carried in a vesicle across the endothelial cell to the basalside of the cell, where it is released into the extracellular space.
IgG andmonomeric IgA IgM Dimeric
IgA IgE
Figure 7.19 Immunoglobulinisotypes are selectively distributedin the body. IgG and IgM predominatein plasma, whereas IgG and monomericIgA are the major isotypes in theextracellular fluid within the body.Dimeric IgA predominates in secretionsacross epithelia, including breast milk.The fetus receives IgG from the motherby transplacental transport. IgE isassociated mainly with mast cell surfacesand is, therefore, found beneathepithelial surfaces (especially therespiratory tract, gastrointestinal tract,and skin). The brain is normally devoidof immunoglobulin.

197Antibody effector functions
life, the production of different isotypes provides protection against infectionin the extracellular spaces throughout the body.
7-8 Antibody production is deficient in very young infants
During the first year of life there is a window of time when all infants are rel-atively deficient in antibodies and specially vulnerable to infection. Duringpregnancy, maternal IgG antibodies are transported across the placenta intothe fetal circulation, providing newborn infants with antibody levels compa-rable to those of their mothers. As the maternally derived IgG is catabolized,the antibody level gradually decreases until the infant’s immune systembegins to produce its own antibody at about 6 months of age (Figure 7.20).
Consequently, IgG levels are lowest in infants aged 3–12 months and this iswhen they are most susceptible to infection. This problem is particularlyacute in babies born prematurely, who begin life with lower levels of maternalIgG and take longer to attain immunocompetence after birth than babiesborn at term.
7-9 High-affinity IgG and IgA antibodies are used to neutralizemicrobial toxins and animal venoms
Many bacteria secrete protein toxins that cause disease by disrupting the nor-mal function of human cells (Figure 7.21). To have this effect, a bacterial toxinmust first bind to a specific receptor molecule on the surface of the humancell. In some toxins, for example those of diphtheria and tetanus, the recep-tor-binding activity is carried by one polypeptide chain and the toxic functionby another. Antibodies that bind to the receptor-binding polypeptide can besufficient to neutralize a toxin (Figure 7.22). The vaccines for diphtheria andtetanus work on this principle. They are modified toxin molecules, called tox-oids, in which the toxic chain has been denatured to remove its toxicity. Onimmunization, protective neutralizing antibodies are made against the recep-tor-binding chain.
Bacterial toxins are potent at low concentrations, a single molecule of diph-theria toxin is sufficient to kill a cell. To neutralize a bacterial toxin, an anti-body must be of high affinity and essentially irreversible in its binding to thetoxin. It must also be able to penetrate tissues and reach the sites where tox-ins are being released. High-affinity IgG is the main source of neutralizingantibodies for the tissues of the human body, whereas high-affinity IgA servesa similar purpose for the mucosal surfaces.
Poisonous snakes, scorpions, and other animals introduce venoms contain-ing toxic polypeptides into humans through a bite or sting. For some venoms,
Figure 7.20 In the first year of lifeinfants have a transient decrease inlevels of IgG. Before birth, high levelsof IgG are provided by the mother, butafter birth maternally derived IgGdeclines. Although infants produce IgMsoon after birth, the secretion of IgGdoes not begin for about 6 months. Theoverall level of IgG reaches a minimumwithin the first year and then graduallyincreases until adulthood.
conception
0
100
2–6 4 5–3 1 3963birth
fraction of adultlevel of serum
immunoglobulinspassively
transferredmaternal IgG
transientlow IgGlevels
newlysynthesized
IgG
newly synthesizedIgM
newlysynthesized IgA
adult
months years
Chapter 7: Immunity Mediated by B Cells and Antibodies198
a single exposure is sufficient to cause severe tissue damage or even death,and in such situations the primary response of the immune system is too slowto help. As exposure to such venoms is rare, protective vaccines against themhave not been developed. For patients who have been bitten by poisonoussnakes or other venomous creatures, the preferred therapy is to infuse them
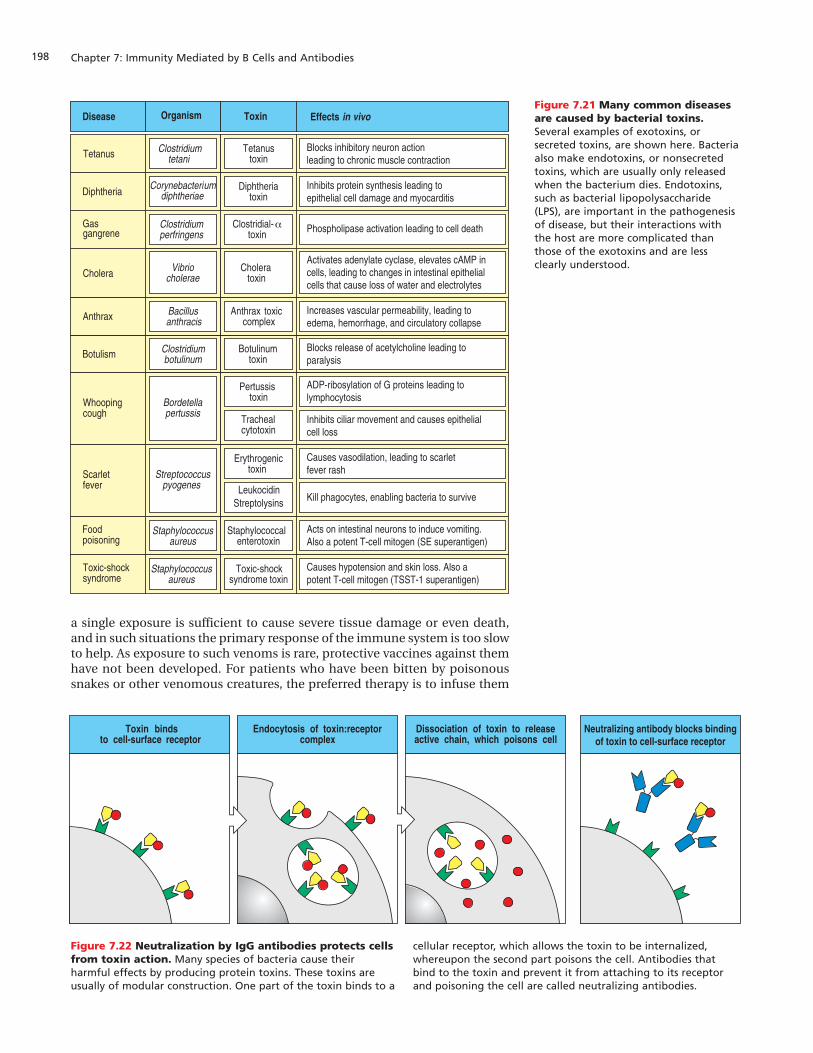
Figure 7.21 Many common diseasesare caused by bacterial toxins.Several examples of exotoxins, orsecreted toxins, are shown here. Bacteriaalso make endotoxins, or nonsecretedtoxins, which are usually only releasedwhen the bacterium dies. Endotoxins,such as bacterial lipopolysaccharide(LPS), are important in the pathogenesisof disease, but their interactions withthe host are more complicated thanthose of the exotoxins and are lessclearly understood.
Dissociation of toxin to releaseactive chain, which poisons cell
Neutralizing antibody blocks bindingof toxin to cell-surface receptor
Toxin bindsto cell-surface receptor
Endocytosis of toxin:receptorcomplex
Disease
Botulism
Foodpoisoning
Toxic-shocksyndrome
Whoopingcough
Scarletfever
Diphtheria
Anthrax
Gasgangrene
Cholera
Organism Toxin Effects in vivo
Clostridiumbotulinum
Staphylococcusaureus
Staphylococcusaureus
Bordetellapertussis
Streptococcuspyogenes
Bacillusanthracis
Clostridiumperfringens
Vibriocholerae
Botulinumtoxin
Staphylococcalenterotoxin
Toxic-shocksyndrome toxin
Pertussistoxin
Erythrogenictoxin
Trachealcytotoxin
LeukocidinStreptolysins
Diphtheriatoxin
Anthrax toxiccomplex
Clostridial-toxin
α
Choleratoxin
Tetanus Clostridiumtetani
Corynebacteriumdiphtheriae
Tetanustoxin
Blocks inhibitory neuron actionleading to chronic muscle contraction
Inhibits protein synthesis leading toepithelial cell damage and myocarditis
Increases vascular permeability, leading toedema, hemorrhage, and circulatory collapse
Blocks release of acetylcholine leading toparalysis
ADP-ribosylation of G proteins leading tolymphocytosis
Inhibits ciliar movement and causes epithelialcell loss
Causes vasodilation, leading to scarlet fever rash
Kill phagocytes, enabling bacteria to survive
Acts on intestinal neurons to induce vomiting.Also a potent T-cell mitogen (SE superantigen)
Causes hypotension and skin loss. Also apotent T-cell mitogen (TSST-1 superantigen)
Phospholipase activation leading to cell death
Activates adenylate cyclase, elevates cAMP incells, leading to changes in intestinal epithelialcells that cause loss of water and electrolytes
Figure 7.22 Neutralization by IgG antibodies protects cellsfrom toxin action. Many species of bacteria cause theirharmful effects by producing protein toxins. These toxins areusually of modular construction. One part of the toxin binds to a
cellular receptor, which allows the toxin to be internalized,whereupon the second part poisons the cell. Antibodies thatbind to the toxin and prevent it from attaching to its receptorand poisoning the cell are called neutralizing antibodies.

199Antibody effector functions
with antibodies specific for the venom. These antibodies are produced byimmunizing large domestic animals—such as horses—with the venom.Transfer of protective antibodies in this manner is known as passive immu-nization and is analogous to the way in which newborn babies acquire pas-sive immunity from their mothers.
7-10 High-affinity neutralizing antibodies prevent viruses andbacteria from infecting cells
The first step in the infection of a human cell by a virus is its attachment tothe cell by means of a cell-surface protein, which is used as the virus receptor.The influenza virus, for example, binds to oligosaccharides on cell-surfaceglycoproteins on epithelial cells of the respiratory tract. The virus bindsthrough a protein in its outer envelope, which is known as the influenzahemagglutinin because the protein can agglutinate, or clump together, redblood cells by binding to oligosaccharides on the red cell surface. Neutraliz-ing antibodies that have been developed during primary immune responsesto influenza and other viruses are the most important aspect of subsequentimmunity to these viruses. Such antibodies coat the virus, inhibit its attach-ment to human cells, and prevent infection (Figure 7.23, upper panels).
Some bacteria that exploit mucosal surfaces maintain their populations bybinding to and colonizing the surface of the epithelial cells, as does the
Virus binds to receptoron cell surface
Receptor-mediatedendocytosis of virus
Acidification of endosome afterendocytosis triggers fusion of viruswith cell and entry of viral DNA
Antibody blocks bindingto virus receptor
Some species of bacteria becomeinternalized and propagate in internal vesicles
Bacteria colonize human cell surfacesby using bacterial adhesins
Antibodies against adhesins blockcolonization and uptake
Figure 7.23 Viral and bacterial infection of cells can beblocked by neutralizing antibodies. Upper panels: for a virusto infect a cell, it must gain entry to the cytoplasm. This requiresbinding of the virus to the cell surface, internalization in anendosome, and fusion of viral and cell membranes to releaseviral nucleic acid into the cytoplasm. Antibodies binding to viralsurface proteins can inhibit either the initial binding of virus or
its subsequent entry into the cell. Lower panels: many bacterialinfections require an interaction between the bacterium and thesurface of a human cell. This is particularly true for infections ofmucosal surfaces. The attachment process involves very specificmolecular interactions between bacterial adhesins and theirligands on human cells. Antibodies specific for epitopes of thebacterial adhesins can, therefore, block infection.

Chapter 7: Immunity Mediated by B Cells and Antibodies200
bacterium Neisseria gonorrhoeae, which causes gonorrhea. Others enterepithelial cells, as do the species of Salmonella that cause food-borne gas-trointestinal infections. IgA antibodies against the adhesion proteins(adhesins) responsible for binding to epithelial cells limit bacterial popula-tions within the gastrointestinal, respiratory, urinary, and reproductive tractsand prevent disease-causing infections in these tissues (see Figure 7.23, lowerpanels).
7-11 The Fc receptors of hematopoietic cells are signalingreceptors that bind the Fc regions of antibodies
Although the binding of a neutralizing antibody to a pathogen or toxin pre-vents further infection, it does not in itself remove the antigen from the body.This is accomplished by phagocytic effector cells, principally neutrophils,blood monocytes, and tissue macrophages. These cells express various recep-tors that bind to the Fc regions of antibodies of different isotypes and areknown generally as Fc receptors.
The Fc receptors of phagocytes and other hematopoietic cells are function-ally and structurally distinct from the FcRB of endothelial cells. They eachconsist of several polypeptide chains, the most important of which is an achain made up of immunoglobulin-like domains (Figure 7.24). This chainbinds to the Fc region of the antibody and determines the isotype specificityof the receptor. Associated with the a chain are other polypeptide chains that
Receptor Fc RI Fc RI(CD89)
Cell type
Relative bindingstrength
Structure 72kDa
MacrophagesNeutrophils Eosinophils
Dendritic cells
IgG1
200 4 4 4 1 20,000 20
MacrophagesNeutrophilsEosinophils
Platelets
MacrophagesNeutrophils Mast cellsEosinophils
Langerhans'cells
B cells
IgG1 IgG1 IgG1
40kDa
-likedomain
ITIMITIM
50–70 kDa
NK cellsEosinophils
MacrophagesNeutrophilsMast cells
FDCs
IgG1
or
Mast cellsEosinophilsBasophils FDCs
IgE IgA1, IgA2
45kDa
9 kDa33 kDa
55–75 kDa
9 kDa
Fc RII-A(CD32) (CD32) (CD32)
Fc RIII(CD16)
Fc RI(CD64)
Fc RII-B2 Fc RII-B1
or
Effect of ligation UptakeStimulationActivation of
respiratory burstInduction of killing
UptakeGranulerelease
(eosinophils)
Uptake No uptakeInhibition
ofstimulation
Inhibitionof
stimulation
Induction UptakeInduction of
killingof killing
(NK cells)
Secretionof granules
Eosinophils†
MacrophagesNeutrophils
�
� �
�
�
� �
�
�
�
�
�
� �� �� � �
� ��
Figure 7.24 Receptors for the Fc regions of immuno-globulins are present on a variety of immune-systemcells. The subunit structure, relative binding strength, andcellular distribution of the Fc receptors are shown. The completemultimolecular structure of most receptors is not yet known butthey may all be multichain molecular complexes similar to theFce receptor I (FceRI). Receptor structure can vary slightly fromone cell type to another. For example, FcgRIII in neutrophils isexpressed as a protein with a glycophosphatidylinositolmembrane anchor and has no associated g chains, whereas innatural killer (NK) cells it is a transmembrane protein associated
with g chains as shown. The information in the figure is based onthe Fc receptors of mouse cells, with the exception of therelative binding strengths that pertain to the human receptors.The Fcg receptors also bind the other subclasses of IgG. FcgRIIIbinds IgG1 and IgG3 with equal strength. For the other FcgRs,IgG1 binds most strongly, IgG2 least strongly, and IgG3 and IgG4with intermediate affinity. FDCs, follicular dendritic cells. *Inthese cases, Fc receptor expression is inducible rather thanconstitutive. †In eosinophils, the molecular weight of CD89a
is 70–100 kDa.

201Antibody effector functions
function either in the folding of the Fc receptor and its movement to the cellsurface or signal the cell once the receptor has bound its ligand. One of thesignaling components, the g chain, is closely related in amino-acid sequenceto the z chain of the T-cell receptor complex.
FcgRII-B1 and -B2 are inhibitory receptors that help to control the activationof naive B cells, mast cells, macrophages, and neutrophils. These receptorsbear immunoreceptor tyrosine-based inhibition motifs (ITIMs) in theircytoplasmic tails, which associate with intracellular proteins that developinhibitory signals.
7-12 Phagocyte Fc receptors facilitate the recognition, uptake,and destruction of antibody-coated pathogens
As we saw in Chapter 6, phagocytic cells can recognize, ingest, and destroybacteria in the absence of specific antibody. This capacity is of paramountimportance in containing infection during the period before an antigen-spe-cific immune response has been made and in enabling macrophages to takeup, process, and present antigen to T cells in the early phases of an adaptiveimmune response. However, the speed with which pathogens can be boundand engulfed by phagocytes greatly increases when the pathogens are coatedwith antibodies, or opsonized. This is because the principal phagocytic cellsof the body—the macrophages and neutrophils—express Fc receptors, calledFcgg receptors, which are specific for the Fc regions of IgG antibodies, partic-ularly that of IgG1 (see Figure 7.24).
When IgG molecules specific for the surface components of a pathogen bindto the pathogen with their Fab arms, the Fc regions are left exposed on theoutside of the antibody-coated particle. The pathogen becomes coated withmany IgG molecules, presenting multiple Fc regions to the Fc receptors on aphagocyte. On contact with a phagocyte, multiple ligand–receptor interac-tions are made, producing a stable and strong binding from interactions thatare individually of low affinity and short-lived. The low affinity of Fcg recep-tors for individual IgG molecules means that they bind only transiently to freeIgG molecules in the absence of antigen. This property enables high concen-trations of IgG of diverse antigenic specificities to circulate in the body’s flu-ids and not clog up the Fc receptors of phagocytes in the absence of antigen.
After the pathogen has been bound to the phagocyte, interactions betweenantibody Fc regions and their receptors facilitate the engulfment of the anti-body-coated pathogen (Figure 7.25). The surface of the phagocyte graduallyextends around the surface of the opsonized pathogen through cycles ofbinding and release between the Fc receptors of the phagocyte and the Fc
macrophage
Antibody binding tobacterium
Antibody-coated bacteriumbinds to Fc receptors
on cell surface
Macrophage membranesurrounds bacterium
Macrophage membranesfuse, creating a membrane-
bounded vesicle, thephagosome
Lysosomes fuse with thephagosome, creatingthe phagolysosome
lysosome
Fc receptors
bacterium
Figure 7.25 Fc receptors onphagocytes trigger the uptake andbreakdown of antibody-coatedpathogens. Specific IgG molecules coatthe pathogen surface, here a bacterium,and tether the bacterium to the surfaceof the phagocyte by binding to the Fc receptors. Signals from the Fcreceptors enhance phagocytosis of thebacterium and the fusion of lysosomescontaining degradative enzymes withthe phagosome.

Chapter 7: Immunity Mediated by B Cells and Antibodies202
regions projecting from the pathogen surface. The engulfment is an activeprocess triggered by signals from the Fc receptors, which is similar, in someways, to walking.
A coating of antibodies makes different sorts of microorganisms appear sim-ilar to the macrophage, and, thus, enables it to deal with them all by using asingle effector mechanism. Encapsulated bacteria such as Streptococcuspneumoniae have evolved cell-surface structures that are resistant to directphagocytosis; for these species a coating with antibody that masks their sur-face is essential if they are to be phagocytosed.
Once an opsonized bacterium has been endocytosed, it becomes enclosed inan acidified vesicle called a phagolysosome, formed from the fusion of thephagosome with lysosomes and neutrophil granules that contain hydrolyticenzymes and microbicidal peptides. Activated neutrophils and macrophagesalso produce oxygen radicals, nitric oxide, and other oxidizing agents withpowerful microbicidal actions. The engulfed bacteria are killed by the com-bined effects of these substances.
As well as destroying microorganisms intracellularly, activated macrophagesalso attack larger antibody-coated parasites, such as worms, that they havebound via their Fc receptors but are too big for them to engulf. In this case,the toxic contents of the lysosomes and diffusible metabolites, such as nitricoxide, are secreted by the macrophage and poured onto the parasite.
7-13 IgE binds to high-affinity Fc receptors on mast cells,basophils, and activated eosinophils
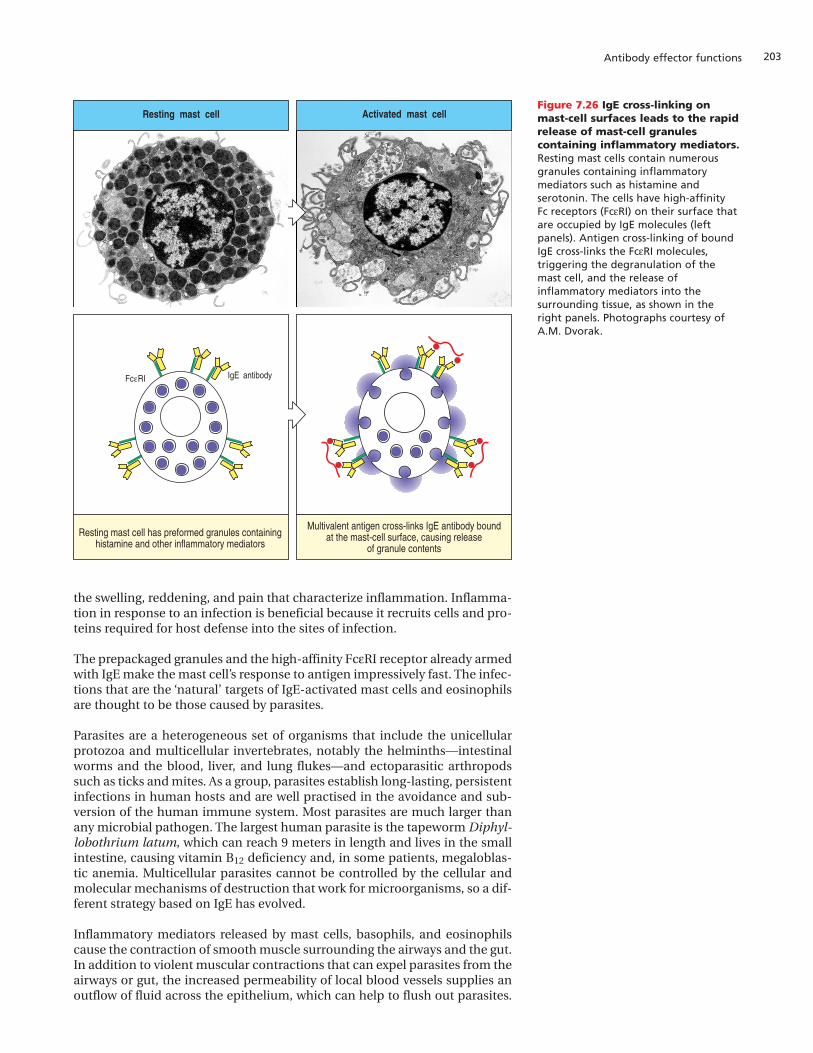
IgE antibodies against a wide variety of different antigens are normally pre-sent in small amounts in all humans. They are produced in responses domi-nated by CD4 TH2 cells, in which the cytokines produced favor switching tothe IgE isotype. A consequence of the low affinity of Fc receptors for IgG isthat free IgG molecules do not form stable interactions with cells expressingthese receptors. The Fc receptor for IgE on mast cells, basophils, and acti-vated eosinophils has quite the opposite properties. This receptor, calledFceRI, has such a high affinity (~1010 M–1) for the Fc region of IgE that IgE mol-ecules are tightly bound in the absence of antigen and the cells are almostalways coated with antibody. In the absence of allergy or parasitic infection, asingle mast cell carries IgE molecules specific for many different antigens.
Mast cells are sentinels posted throughout the body’s tissues, particularly inthe connective tissues lying in the mucosa of the gastrointestinal and respira-tory tracts and in connective tissues along blood vessels—especially those inthe dermis of the skin. The cytoplasm of the resting mast cell is filled withlarge granules containing histamine and other molecules that contribute toinflammation, which are known generally as inflammatory mediators. Mastcells become activated to release their granules when antigen binds to the IgEmolecules bound to FceRI on the mast-cell surface (Figure 7.26). To activatethe cell, the antigen must cross-link at least two IgE molecules and their asso-ciated receptors, which means that the antigen must have at least two topo-graphically separate epitopes recognized by the cell-bound IgE. Cross-linkingof FceRI generates the signal that initiates the release of the mast cell gran-ules. After degranulation the mast cell synthesizes and packages a new set ofgranules.
Inflammatory mediators secreted into the tissues by activated mast cells,basophils, and eosinophils increase the permeability of the local blood ves-sels, enabling other cells and molecules of the immune system to move out ofthe bloodstream and into tissues. This causes local accumulation of fluid and
203Antibody effector functions
the swelling, reddening, and pain that characterize inflammation. Inflamma-tion in response to an infection is beneficial because it recruits cells and pro-teins required for host defense into the sites of infection.
The prepackaged granules and the high-affinity FceRI receptor already armedwith IgE make the mast cell’s response to antigen impressively fast. The infec-tions that are the ‘natural’ targets of IgE-activated mast cells and eosinophilsare thought to be those caused by parasites.
Parasites are a heterogeneous set of organisms that include the unicellularprotozoa and multicellular invertebrates, notably the helminths—intestinalworms and the blood, liver, and lung flukes—and ectoparasitic arthropodssuch as ticks and mites. As a group, parasites establish long-lasting, persistentinfections in human hosts and are well practised in the avoidance and sub-version of the human immune system. Most parasites are much larger thanany microbial pathogen. The largest human parasite is the tapeworm Diphyl-lobothrium latum, which can reach 9 meters in length and lives in the smallintestine, causing vitamin B12 deficiency and, in some patients, megaloblas-tic anemia. Multicellular parasites cannot be controlled by the cellular andmolecular mechanisms of destruction that work for microorganisms, so a dif-ferent strategy based on IgE has evolved.
Inflammatory mediators released by mast cells, basophils, and eosinophilscause the contraction of smooth muscle surrounding the airways and the gut.In addition to violent muscular contractions that can expel parasites from theairways or gut, the increased permeability of local blood vessels supplies anoutflow of fluid across the epithelium, which can help to flush out parasites.
Figure 7.26 IgE cross-linking onmast-cell surfaces leads to the rapidrelease of mast-cell granulescontaining inflammatory mediators.Resting mast cells contain numerousgranules containing inflammatorymediators such as histamine andserotonin. The cells have high-affinity Fc receptors (FceRI) on their surface thatare occupied by IgE molecules (leftpanels). Antigen cross-linking of boundIgE cross-links the FceRI molecules,triggering the degranulation of themast cell, and the release ofinflammatory mediators into thesurrounding tissue, as shown in the right panels. Photographs courtesy ofA.M. Dvorak.
Activated mast cell
Resting mast cell has preformed granules containinghistamine and other inflammatory mediators
Multivalent antigen cross-links IgE antibody boundat the mast-cell surface, causing release
of granule contents
Resting mast cell
FcεRI IgE antibody

Chapter 7: Immunity Mediated by B Cells and Antibodies204
In summary, the combined actions of IgE, mast cells, basophils, andeosinophils serve to physically remove parasite pathogens and other materialfrom the body.
Eosinophils can also use their Fce receptors to act directly against multicellu-lar parasites. Such organisms, even small ones such as the blood fluke Schis-tosoma mansoni, which causes schistosomiasis, cannot be ingested byphagocytes. However, if the parasite induces an antibody response andbecomes coated with IgE, activated eosinophils will bind to it through FceRIand then pour the toxic contents of their granules directly onto its surface(Figure 7.27).
For human populations in developed countries where parasite infections arerare, the mast cell’s response is most frequently seen as a detriment becauseits actions are the cause of allergy and asthma. People with these conditionsmake IgE in response to relatively innocuous substances, for example grasspollens or shellfish, which are often either airborne or eaten. Such substancesare known as allergens. Having made specific IgE, any subsequent encounterwith the allergen leads to massive mast-cell degranulation and a damagingresponse that is quite inappropriate to the threat posed by the antigen or itssource. In extreme cases, the ingestion of an allergen can lead to a systemiclife-threatening inflammatory response called anaphylaxis.
Parasite infections that invoke a protective IgE response are not major healthproblems in the developed world, whereas allergy and asthma do not seem tobe prevalent in the developing countries where infection with parasites isendemic. This is one of various pieces of circumstantial evidence suggestingthat if elements of the immune system are left unstimulated by infection, theycan respond in ways that are frankly unhelpful.
7-14 Fc receptors activate natural killer cells to destroyantibody-coated human cells
Natural killer cells (NK cells) are large effector lymphocytes that circulate inthe blood (see Figure 1.9, p. 10) and whose chief role is in innate immunity.However, they also express an Fc receptor called FcgRIII, or CD16, which isspecific for IgG1 and IgG3. In experimental situations, NK cells have beenshown to recognize and kill human cells coated with antibody against cell-surface components (Figure 7.28). This antibody-dependent cell-mediatedcytotoxicity (ADCC) requires the presence of preformed antibody. This is notavailable during a primary immune response, but NK-cell cytotoxicity might
Figure 7.27 Eosinophils (E) attackinga schistosome larva (SL) in thepresence of serum from an infectedpatient. Large parasites, such as worms,cannot be ingested by phagocytes;however, when the worm is coated withantibody, especially IgE, eosinophils canattack it by using their high-affinity Fc receptors (FceRI). Similar attacks canbe mounted by other Fc-receptor-bearing cells on various large targets.Photograph courtesy of A. Butterworth.
E
E
SL
E
E
SL
Figure 7.28 Antibody-coated targetcells can be killed by natural killercells (NK cells) in antibody-dependent cell-mediatedcytotoxicity (ADCC). NK cells are largegranular lymphocytes that are distinctfrom B and T cells and have FcgRIIIreceptors (CD16) on their surface. Whenthese cells encounter cells coated withIgG antibody, they rapidly kill the targetcell. The importance of ADCC in hostdefense or tissue damage is uncertain.
NK cell
Antibody binds antigens on thesurface of target cells
Fc receptors on NK cellsrecognize bound antibody
Cross-linking of Fc receptors signalsthe NK cell to kill the target cell Target cell dies by apoptosis
Fc RIII(CD16)γ
activatedNK cell
target cell target cell

205The antigen–antibody mediated pathway of complement activation
have a role in secondary responses where antibody is already present.Another situation in which preformed antibody is present is in newborninfants, who have passively acquired IgG against many pathogens to whichthey have yet to be exposed.
Summary
Secreted antibodies are the only effector molecules produced by B cells; theirprincipal function is as adaptor molecules that neutralize the pathogen andbring together pathogens or their products with the effector cells that destroythem. Antibodies can become bound through their Fc regions to Fc receptorson various types of effector cell. These interactions with Fc receptors are spe-cific for immunoglobulin isotype and are used by the immune system for twomain purposes. The first is to deliver antibodies to sites where they would notbe carried by the circulation of the blood and lymph. To this end, the poly-Igreceptor of epithelium provides the lumen of the intestines and othermucosal surfaces with a continual supply of IgA that binds to the microor-ganisms that inhabit and infect those tissues. FcRB delivers IgG from plasmainto the extracellular fluid in tissues and also, during pregnancy, deliversmaternal IgG to the fetal circulation, which is useful after birth against manytypes of infection. The second purpose of the Fc receptors is to attachpathogens or antigens, which have bound to specific antibody, to effectorcells that will respond in ways that eliminate infection. Fc receptors for IgGcan deliver antibody-bound bacteria to phagocytes. In these cases, antibodybinds first to the antigen and then to the Fc receptor. In contrast, the IgE anti-body binds first to the Fc receptor of mast cells, basophils, and activatedeosinophils and then awaits its antigen.
The antigen–antibody mediated pathwayof complement activation The binding of antibodies to antigens involves noncovalent bonds. Interac-tions between them are potentially reversible, depending on local concentra-tions of antibody and antigen, and are, therefore, susceptible to disruption,especially where low-affinity antibodies are concerned. Once a pathogen orantigen has been identified as foreign, it becomes advantageous to mark it fordestruction in a more permanent manner. This is accomplished by a systemof blood proteins known collectively as the complement system, or just com-plement. The name complement was given because the effector functionsprovided by these proteins ‘complement’ the antigen-binding function ofantibodies in the defense against pathogens. Activation of the complementsystem initiates a series of enzymatic reactions in which the proteolytic cleav-age and activation of successive complement components leads to the cova-lent bonding, or ‘fixation’, of particular complement fragments to thepathogen surface. Phagocytes bear surface receptors that recognize thesefragments and this recognition facilitates the uptake and destruction of com-plement-coated microbes by neutrophils and macrophages.
Complement fixed on bacterial surfaces also nucleates a complex of proteinsthat attacks pathogens by poking holes in their cell membranes. Antibodybound to a pathogen triggers complement activation that proceeds by aseries of enzymatic reactions called the classical pathway of complementactivation. Pathogens also trigger complement activation by two other path-ways that do not involve antibody and are considered part of innate immu-nity. They are the lectin pathway of complement activation, which is acti-vated by binding of a plasma protein to mannose-containing peptidoglycans

Chapter 7: Immunity Mediated by B Cells and Antibodies206
on microbial surfaces, and the alternative pathway of complement activa-tion, which is triggered by direct environmental influence of the microbialsurface. The antibody-mediated classical pathway will be discussed here; theother two pathways are covered in Chapter 8.
7-15 Complement components are plasma proteins with variousfunctions
All complement components are made in the liver and circulate in theplasma. They comprise more than 30 proteins with a variety of biochemicalfunctions. Many are enzymes and these are secreted and circulate in an inac-tive form known as a zymogen. Activation of complement takes place in thetissues, into which plasma leaks from the blood, and also in the blood itself.The complement components can be grouped on the basis of their functions;the members of each group are often structurally similar.
The three pathways of complement activation differ in the way they are trig-gered and in their first few reactions (Figure 7.29). However, all three path-ways converge on the same reaction, the cleavage of complement compo-nent C3 into fragments C3b and C3a and the covalent binding of C3b to thepathogen’s surface. This binding of C3b to the surface of pathogens is themost important function of the complement system and is called comple-ment fixation. The bound C3b tags the pathogen for destruction by phago-cytosis and also nucleates protein complexes that damage the pathogen’smembrane. In addition, the soluble C3a fragment recruits inflammatorycells to the site of infection. These three effector mechanisms of complementare common to all three pathways of complement activation. Thus, the clas-sical pathway of complement activation involves some components that arespecific to the classical pathway and others that also function in the otherpathways.
Death of pathogen
Opsonization of pathogens,facilitating uptake and killing
by phagocytes
Recruitment ofinflammatory cells
Perforation ofpathogen cell membrane
C3b covalently bound to surfacecomponents of pathogen
CLASSICAL PATHWAY ALTERNATIVE PATHWAYLECTIN PATHWAY
Antibody binds to specificantigen on pathogen surface
Mannose-binding lectin bindsto pathogen surface
Pathogen surface creates localenvironment conducive to
complement activation
COMPLEMENT ACTIVATION
Figure 7.29 The three pathways ofcomplement activation. The classicalpathway is initiated by the binding ofeither IgM or IgG antibodies to amicrobial surface, whereas the lectin-mediated pathway is initiated by themannose-binding lectin of plasma,which binds to carbohydrates found onbacterial cells. The alternative pathwayis triggered by the local physicochemicalenvironment created by the constituentsof some bacterial surfaces.
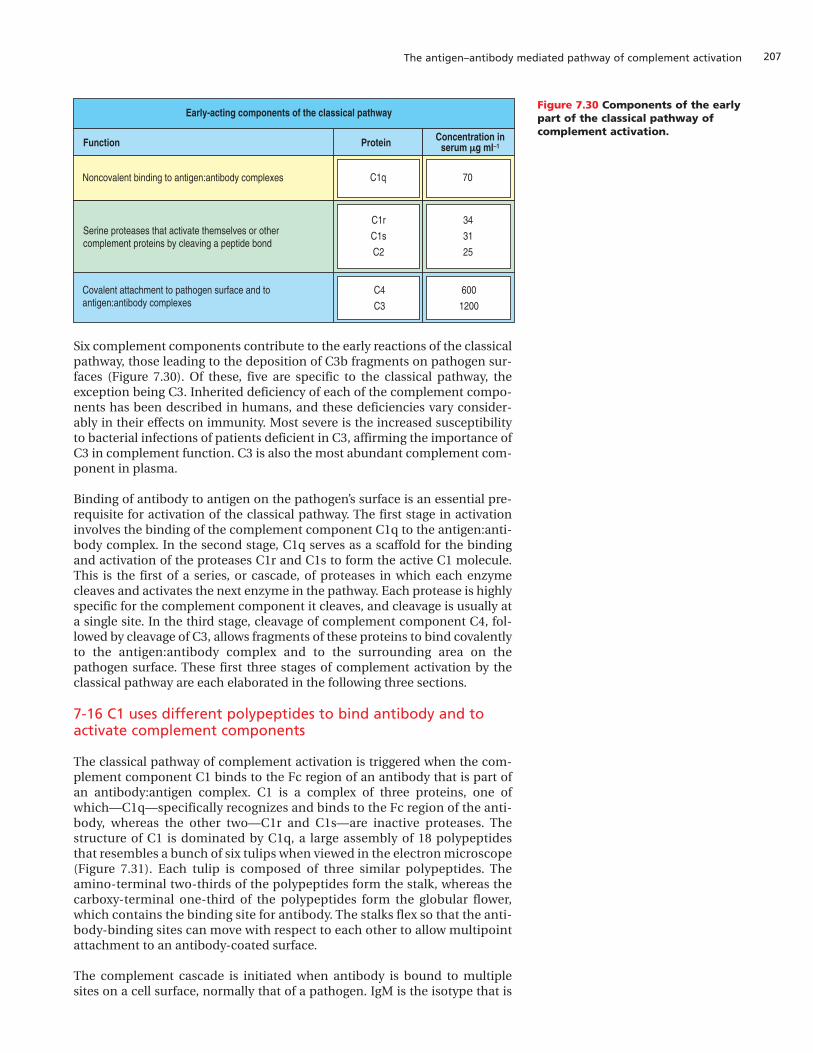
207The antigen–antibody mediated pathway of complement activation
Six complement components contribute to the early reactions of the classicalpathway, those leading to the deposition of C3b fragments on pathogen sur-faces (Figure 7.30). Of these, five are specific to the classical pathway, theexception being C3. Inherited deficiency of each of the complement compo-nents has been described in humans, and these deficiencies vary consider-ably in their effects on immunity. Most severe is the increased susceptibilityto bacterial infections of patients deficient in C3, affirming the importance ofC3 in complement function. C3 is also the most abundant complement com-ponent in plasma.
Binding of antibody to antigen on the pathogen’s surface is an essential pre-requisite for activation of the classical pathway. The first stage in activationinvolves the binding of the complement component C1q to the antigen:anti-body complex. In the second stage, C1q serves as a scaffold for the bindingand activation of the proteases C1r and C1s to form the active C1 molecule.This is the first of a series, or cascade, of proteases in which each enzymecleaves and activates the next enzyme in the pathway. Each protease is highlyspecific for the complement component it cleaves, and cleavage is usually ata single site. In the third stage, cleavage of complement component C4, fol-lowed by cleavage of C3, allows fragments of these proteins to bind covalentlyto the antigen:antibody complex and to the surrounding area on thepathogen surface. These first three stages of complement activation by theclassical pathway are each elaborated in the following three sections.
7-16 C1 uses different polypeptides to bind antibody and toactivate complement components
The classical pathway of complement activation is triggered when the com-plement component C1 binds to the Fc region of an antibody that is part ofan antibody:antigen complex. C1 is a complex of three proteins, one ofwhich—C1q—specifically recognizes and binds to the Fc region of the anti-body, whereas the other two—C1r and C1s—are inactive proteases. Thestructure of C1 is dominated by C1q, a large assembly of 18 polypeptidesthat resembles a bunch of six tulips when viewed in the electron microscope(Figure 7.31). Each tulip is composed of three similar polypeptides. Theamino-terminal two-thirds of the polypeptides form the stalk, whereas thecarboxy-terminal one-third of the polypeptides form the globular flower,which contains the binding site for antibody. The stalks flex so that the anti-body-binding sites can move with respect to each other to allow multipointattachment to an antibody-coated surface.
The complement cascade is initiated when antibody is bound to multiplesites on a cell surface, normally that of a pathogen. IgM is the isotype that is
C4
C3
Function Protein
Early-acting components of the classical pathway
Noncovalent binding to antigen:antibody complexes
Covalent attachment to pathogen surface and toantigen:antibody complexes
C1r
C1s
C2
C1q
600
1200
Concentration inserum µµg ml–1
34
31
25
70
Serine proteases that activate themselves or othercomplement proteins by cleaving a peptide bond
Figure 7.30 Components of the earlypart of the classical pathway ofcomplement activation.

Chapter 7: Immunity Mediated by B Cells and Antibodies208
most efficient at activating complement. The other human isotypes that acti-vate the complement system are IgG1 and IgG3 and, to a lesser extent, IgG2.Pentameric IgM has five Fc regions, each of which can provide a binding sitefor one of the six binding sites of C1q. Multipoint attachment of C1q to IgM isrequired for a stable interaction; this can readily be satisfied by a single mol-ecule of each type. IgG can also bind C1q, but it has only one Fc region; con-sequently, at least two molecules of IgG bound to a microbial surface within30–40 nm of each other are required to bind one molecule of C1q (Figure7.32). This difference explains why IgM activates complement much moreeffectively than IgG, and why free IgG cannot activate complement in solu-tion. Free IgM cannot activate complement because it has a conformationthat does not bind C1q. On binding to specific antigen, IgM changes its con-formation to one that can bind C1q.
C1r and C1s are serine proteases that are activated when C1q binds to an anti-body Fc region. Serine proteases are proteolytic enzymes that have a serineresidue at the active site. They are typically synthesized in an inactive form,the zymogen, and become enzymatically active only after proteolytic cleav-age by another protease. On binding to antibody, one molecule of C1r isinduced to cut itself, thereby becoming enzymatically active. It then cuts and
C1q
C1r C1s
Figure 7.31 The complementcomponent C1. The C1 moleculeconsists of a complex of C1q, C1r, andC1s. The C1q component consists of sixidentical subunits, each with onebinding site for the Fc region of IgM orIgG and extended amino-terminal stalkregions that interact with each otherand with two molecules each of theproteases C1r and C1s. The electronmicrograph on the right contains imagesof three C1q molecules. Photographcourtesy of K.B.M. Reid.
Figure 7.32 The classical pathway ofcomplement activation is initiatedby binding of C1q to antibody on abacterial surface. The binding of C1q to a molecule of pentameric IgM isshown in the left panels. On establishingmultipoint binding to bacterial cell-surface antigens, the IgM moleculeadopts a less planar conformation, theso-called staple conformation (upperpanel). This distortion allows the C1q molecule to establish multipointattachment to the Fc regions of a singleIgM molecule, using the hinges in theC1q stalks to position the globular Fc-binding sites (lower panel). It alsoexposes binding sites for the C1q heads.The binding of C1q to IgG is shown inthe right panels. The C1q moleculeneeds to find pathogen-bound IgGmolecules that are close enough to each other for the C1q molecule to spanbetween them. As a consequence, theactivation of complement by IgGdepends more on the amount anddensity of antibodies bound to apathogen surface than doescomplement activation by IgM.
‘planar’ formof IgM
C1q binds to two or more IgG moleculesC1 binds to a single IgM molecule
‘staple’ formof IgM
IgG molecules bind to antigenson bacterial surface
Pentameric IgM molecule binds to antigenson bacterial surface and adopts ‘staple’ form

209The antigen–antibody mediated pathway of complement activation
activates the second C1r molecule and both C1s molecules. Activated C1s isthe protease that binds, cleaves, and activates the next two components of theclassical pathway, C4 and C2.
7-17 Fragments of C2 and C4 associate on the pathogen surfaceto form the classical C3 convertase
When a C4 molecule interacts with the activated C1s protease, it is cleavedinto a large fragment called C4b and a small fragment called C4a. This cleav-age exposes a high-energy thioester bond, which, in uncut C4, is sequesteredand stabilized within the hydrophobic interior of the protein. On exposure toplasma the thioester is rapidly subjected to nucleophilic attack. The vastmajority of C4b thioesters are hydrolyzed and remain in solution, but somereact with the amino and hydroxyl groups of proteins and carbohydrates atthe pathogen surface, and when this happens C4b becomes covalentlybonded to the pathogen. The overall result is that C4b fragments are cova-lently attached to the pathogen’s surface in the vicinity of the antigen:anti-body complex that started the complement activation. During this reaction,C4b also becomes covalently attached to the antibodies bound to thepathogen and to any associated complement components (Figure 7.33).
Figure 7.33 Cleavage of C4 exposesa reactive thioester bond thatcovalently attaches the C4bfragment to the pathogen surface.Circulating C4 is an inactive serineprotease consisting of a, b, and gpolypeptide chains in which a thioesterbond in the a chain is protected fromhydrolysis within the hydrophobicinterior of the protein. The thioesterbond is denoted in the top two panelsby the circled letters S, C, and O. The C4 molecule is activated by cleavage ofthe a chain by C1s to give fragmentsC4a and C4b. This exposes the thioesterbond of C4b to the environment. Thethioester bonds of most of the C4b fragments will be spontaneouslyhydrolyzed by water as shown in thebottom left panel, but a minority willreact with the hydroxyl and aminogroups on molecules on the pathogen’ssurface, bonding C4b to the pathogensurface as shown in the bottom right panel.
Rcell surface
Attack by H2O Attack by R–OH or R–NH2
C4
C1s
Cleavage by C1s exposes thioester bond
Nucleophilic attack on the thioester bond
Soluble C4b C4b bound to pathogen surface
C4b
nucleophile
C4a
C4bC4b
SH COOH

Chapter 7: Immunity Mediated by B Cells and Antibodies210
Complement component C2 is the second substrate for the activated C1s pro-tease. C2 is a serine protease zymogen that upon cleavage by C1s, produces alarge C2a fragment with protease activity and a small C2b fragment. (For his-torical reasons, the small cleavage product of C2 is called C2b and the largerproduct is called C2a, whereas for other complement proteins, the larger frag-ment is called ‘b’ and the smaller ‘a’.) On release from C1, C2a binds to a C4bfragment bonded to the pathogen surface. This C4b2a complex, called theclassical C3 convertase, is a surface-associated serine protease whose func-tion is to cleave complement component C3 and ‘convert’ it from an inactiveto an active form.
7-18 Cleavage of C3 yields C3b covalently bound to pathogensurfaces
C3 is structurally very similar to C4 and, like C4, it contains a sequesteredthioester bond that becomes activated on cleavage of the molecule. So thereaction that occurs when C3 binds to and is cleaved by C4b2a is just like theC1s-mediated cleavage of C4. Upon cleavage, the smaller C3a fragment isremoved and the thioester bond of the larger C3b fragment becomes exposedand susceptible to attack. Consequently, some of the C3b fragments becomecovalently bonded to the pathogen’s surface around each site where a C4b2acomplex is active (Figure 7.34). Although each C4b2a molecule is only activefor a few minutes it can cleave up to 1000 molecules of C3, many of whichbecome bonded to the microbial surface. So the number of C3b moleculesdeposited on the surface at a site of complement fixation is much larger thanthe number of C4b molecules.
Although the complement system has many components, and can seem hor-ribly complicated, its workings are based on a few simple principles that areelaborated in different ways. Of these principles the most important is the useof thioester bonds in C3 and C4 to covalently tag microbial surfaces. Althoughmany enzymes transiently form thioester bonds during catalysis, for a stableprotein to contain a thioester bond is truly exceptional. Apart from C3 and C4,only one other protein has been found to have this property, the structurallyrelated protease inhibitor of plasma, a2-macroglobulin.
7-19 Partial lack of C4 is the most common immune proteindeficiency in humans
The two types of C4—C4A and C4B— have different properties. The thioesterof the C4A form is preferentially attacked by the amino groups of macromol-ecules, whereas that of C4B is preferentially attacked by the hydroxyl groups.This increases the efficiency of C4 deposition and coverage of the whole
C4b2b binds C3 and cleavesit to C3a and C3b. C3b bindscovalently to the microbial
surface
C2a binds to surface C4bforming the classical C3
convertase, C4b2a
Activated C1s also cleavesC2 to C2a and C2b
Activated C1s cleaves C4 toC4a and C4b. Some C4b binds
covalently to the microbial surface
C4C3
microbial surface
C4a
C4b
C2
C2b
C2a
C2aC4b
C4b
C2a
C4bC3b
C3a
Figure 7.34 Activated C1s cleaves C4and C2 to produce C4b and C2a,which associate to form the classicalC3 convertase. The steps in thereaction are outlined here and detailedin Section 7-19.

211The antigen–antibody mediated pathway of complement activation
pathogen surface. The two genes encoding C4A and C4B are closely linkedand situated in the class III region of the MHC, where, through gene duplica-tion and deletion, further diversification of the C4 genes has evolved (Figure7.35). In humans, 13% of chromosomes lack a functional C4A gene and 18%of chromosomes lack a functional C4B gene; thus, more than 30% of thehuman population is deficient for one or other form of C4, and a partial lackof C4 is the most common human immunodeficiency. Reflecting the comple-mentary functions of the two forms of C4, deficiency in C4A is associated withsusceptibility to the autoimmune disease systemic lupus erythematosus(SLE), whereas deficiency in C4B is associated with lowered resistance toinfection. As well as the simple presence or absence of the C4A and C4Bgenes, there are more than 40 different alleles of the C4 genes, which could beassociated with further differences in C4 function.
7-20 C3b produced by the classical C3 convertase permits theformation of a more powerful alternative C3 convertase
The cleavage of C3 by the classical C3 convertase can be seen as an amplifi-cation step that increases the number of complement fragments attached tothe pathogen surface over that accomplished by cleavage of C4 alone. Thisprocess of amplification is now taken one step further. C4 and C3 are closelyrelated proteins and C3b, like C4b, can assemble a C3 convertase. The othercomponent of this alternative convertase is derived from the plasma proteinfactor B that is closely related in structure and function to C2, which furnishesthe second subunit of the classical C3 convertase (Figure 7.36). When factor Bbinds to C3b on the pathogen surface it becomes susceptible to cleavage byfactor D, another serine protease in plasma, that cleaves off a small Ba frag-ment leaving the larger, and now proteolytically active, Bb fragment associ-ated with C3b. The C3bBb convertase is homologous in both structure andfunction to C4b2a (Figure 7.37): it cleaves C3 molecules, exposing theirthioester bonds to attack by water, plasma proteins, and components of themicrobial surface. The process by which the alternative convertase cleavesand activates C3 is homologous to that followed by the classical convertase
C4 genes
BA
AA
BB
A
B
BAA
BBA
Figure 7.35 Humans differ in the number and type of genes forcomplement component C4. C4A and C4B exhibit differences in theway they bond to pathogen surfaces. The genes for C4A and C4B arelocated in the central part of the MHC, between the class I region andthe class II region (see Figure 3.25, p. 88). Although a majority of MHChaplotypes have one gene for C4A and one for C4B, a considerableminority have other arrangements involving loss or duplication of oneof the genes. These differences lead to variation in C4 function withinthe population and to immunodeficiency in some individuals.
Formation of the alternative C3 convertase C3bBb
microbial surface
C3bC3b C3b C3b
DD
DBB B Bb Ba
+
Figure 7.36 Formation of thealternative C3 convertase. The C3convertase of the alternative pathway isassembled from C3b and the active Bbfragment of factor B. Because C3b, theproduct of the alternative C3convertase, actually makes moreconvertase, this means that thealternative convertase is inherently moreactive than the classical C3 convertase indepositing C3b on pathogen surfaces.

Chapter 7: Immunity Mediated by B Cells and Antibodies212
(Figure 7.38). What distinguishes the convertase C3bBb from C4b2a is that theproduct of the enzymatic reaction, C3b, can assemble more enzyme. This dis-tinctive property of C3bBb produces exponential amplification of the C3 con-version reaction started by C4b2a, an accelerating reaction that would rapidlyconsume the supplies of C3 were there not control mechanisms that dampenthe response, as we shall see later. By the end of the complement-fixing reac-tion, most of the C3b fragments that cover the pathogen surface around theinitiating antigen:antibody complex are as a result of the action of the C3bBbconvertase (Figure 7.39).
In the alternative pathway of complement activation, where antibody is notinvolved, C3bBb is the only participating C3 convertase. For this, and histori-cal reasons, C3bBb is called the C3 convertase of the alternative pathway orthe alternative C3 convertase. Likewise, factor B and factor D are designatedas components of the alternative pathway of complement activation. How-ever, these designations can be misleading because formation and catalysis ofthe C3bBb convertase makes an essential contribution to the antibody-drivenclassical pathway of complement activation, in which C3bBb serves in addi-tion to the classical convertase rather than as an alternative.
7-21 Fragments of C3 and C4 on pathogen surfaces arerecognized by receptors on various cell types
Several types of immune-system cell have surface receptors that bind to theC3 and C4 fragments deposited on the pathogen surface and stimulate a cel-lular response to the pathogen. There are four types of complement recep-tors, falling into two structural groups: CR1 and CR2 in one group and CR3
Two types of C3 convertase
Classical Alternative
C4b
C2a
C3b
Bb
microbial surface
Figure 7.37 The two types of C3 convertase have similarstructures and functions. In the C3 convertase produced by theclassical pathway, C4bC2a, the activated protease C2a cleaves C3 toC3b and C3a (not shown). In the analogous C3 convertase of thealternative pathway, C3bBb, the activated protease Bb carries outexactly the same reaction.
Figure 7.38 C3 activation by thealternative C3 convertase is aprocess analogous to C3 activationby the classical C3 convertase.
C3 activation by the alternative C3 convertase
microbial surface
Bb BbBb
+
C3 activation by the classical C3 convertase
microbial surface
C2a C2a C2a
+
C3
C3C3
C3
C3b
C4b C4b C4b
C3b C3b
C3a
C3a
C3b C3b
C3b C3b

213The antigen–antibody mediated pathway of complement activation
and CR4 in the other. All four receptors differ in their cellular distribution(Figure 7.40). Macrophages and neutrophils (polymorphonuclear leukocytes)express complement receptor 1 (CR1), which on binding to C3b or C4b on apathogen surface facilitates uptake and destruction of the pathogen by thesephagocytic cells (Figure 7.41). In this case, C3b, and to a lesser extent C4b, areacting as opsonins in what is the principal function of complement, todestroy microorganisms. By itself, the interaction of C3b with CR1 does notstimulate phagocytosis and the intracellular killing of pathogens, but itenhances these functions once they have been initiated by the binding of IgGto an Fcg receptor or by the cytokine IFN-g, which is produced by activated Tcells. The interaction of C3b with CR1 is more significant than with C4bbecause complement activation deposits much more C3b than C4b onpathogen surfaces.
Complement receptor 2 (CR2) has a different tissue distribution from CR1,CR3, and CR4 because it is expressed on B cells and follicular dendritic cells.As we saw in Section 7-1, CR2 (also called CD21) is a component of the B-cellco-receptor. It binds to complement fragments iC3b, C3d, and C3dg, whichare formed by degradation of C3b at the pathogen surface. When a B-cell
Figure 7.39 A bird’s-eye view of the fixation of C4b andC3b fragments on a pathogen surface around anantigen:antibody complex. Antibody bound to an antigen ona microbial surface binds C1 (first panel), which leads to thedeposition of C4b (pink circles) around the antigen:antibodycomplex (second panel). When C4b binds C2a to form the
classical C3 convertase, a limited number of C3b molecules(green rectangles) are produced (third panel). These can bind Bbto form the alternative convertase, C3bBb (yellow rectangles),which leads to deposition of many more C3b fragments on themicrobial surface (green rectangles, fourth panel).
C1 binds toantigen:antibody complex Deposition of C4b by C1 Deposition of C3b by C4b2a Deposition of C3b by C3bBb
microbial surface
Receptor
CR2
CR3
CR4
Ligand Functions Cell types
CR1
C3d, C3dg,iC3b
iC3b
Part of B-cell co-receptor
Stimulates phagocytosis
B cells,FDCs
Macrophages, monocytes,polymorphonuclear leukocytes,
FDCs
Macrophages, monocytes,polymorphonuclear leukocytes
C3b, C4b
Promotes C3b and C4b decayStimulates phagocytosis
Erythrocyte transportof immune complexes
Erythrocytes, macrophages,monocytes, polymorphonuclear
leukocytes, B cells, FDCs,podocytes in kidney glomeruli
iC3b Stimulates phagocytosis
Figure 7.40 Distribution andfunction of receptors forcomplement proteins. There areseveral different complement receptorsthat are specific for differentcomplement components or theirfragments. CR1 and CR3 are especiallyimportant in the phagocytosis ofcomplement-coated bacteria bymacrophages and neutrophils. CR1 onerythrocytes clears immune complexesfrom the circulation and CR2 is mainlypresent on B cells, where it is also partof the B-cell co-receptor complex. C3d,C3dg, and iC3b are cleavage products of C3b. FDC, follicular dendritic cell.

Chapter 7: Immunity Mediated by B Cells and Antibodies214
receptor interacts with its specific antigen on a pathogen, the signal deliveredto the B cell is strengthened if the CR2 component of the B-cell co-receptoralso interacts with a nearby iC3b, C3d, or C3dg fragment on the pathogen sur-face (see Figure 7.3). CR2 on follicular dendritic cells in the lymphoid folliclesenables these cells to bind antigens that have been tagged with C3b or itsproducts (see Figure 7.10) and to retain them for long-term stimulation of Bcells. The Epstein–Barr virus (EBV), which causes infectious mononucleosisand certain lymphomas, also binds to CR2 and exploits this interaction toinfect B cells. In this context, CR2 is known as the EBV receptor of human Bcells. CR1 and CR2 are both elongated molecules consisting of a string ofsmall compact structural modules.
In contrast, complement receptors 3 and 4 (CR3 and CR4) are b-integrins.They bind to iC3b on pathogen surfaces. CR3 and CR4 are expressed onphagocytes, where they augment the activities of Fc receptors and CR1 inactivating phagocytosis. Unlike the interaction between C3b and CR1, thebinding of iC3b to CR3 is sufficient in itself to stimulate phagocytosis. b-Inte-grins also function as cell adhesion molecules, and CR3 and CR4 are involvedin the adherence of leukocytes to endothelial cells in sites of inflammation.
7-22 Complement receptors remove immune complexes fromthe circulation
In the previous sections we have been principally concerned with the bindingof antibody and complement to pathogenic microorganisms, which are largeparticles. High-affinity antibodies also bind to soluble protein antigens suchas bacterial toxins, forming complexes that cannot be engulfed by phagocytesbecause they contain too few molecules of IgG to form a stable interactionwith Fcg receptors. Such soluble immune complexes are present in the circu-lation after the immune response to most infections and they are removedthrough the action of complement. The number of IgG molecules in animmune complex is sufficient to bind C1 and activate the enzymes that cleavefirst C4 and then C3, so that the antigen and antibody molecules within thecomplex become covalently tagged with C4b and C3b. Having been tagged inthis way, the complex can now be bound by circulating cells that express CR1.Of these, the most numerous is the erythrocyte, and the vast majority ofimmune complexes become bound to the surface of red blood cells. Duringtheir circulation in the blood, erythrocytes pass through areas of the liver andthe spleen where tissue macrophages remove and degrade the complexes ofcomplement, antibody, and antigen from the erythrocyte surface while leav-ing the erythrocyte unscathed (Figure 7.42).
Granules fuse with phagosomes,releasing toxic oxygen
metabolites that kill bacteria
Encapsulated bacteria cannotbe engulfed by neutrophils
Engulfment of bacteria byneutrophils is mediated by Fc
receptors and complement receptors
Antibody bound to bacteriaactivates complement and bonding
of C3b to bacteria
granules
Fc receptor
complement receptor
C3b
Figure 7.41 Encapsulated bacteriaare more efficiently engulfed byphagocytes when the bacteria arecoated with antibody and C3b.Encapsulated bacteria are naturallyresistant to uptake by phagocytes, hererepresented by a neutrophil. When suchbacteria are coated with antibody andC3b they become susceptible tophagocytosis mediated by Fc receptorsand C3b receptors. Fc receptors bindIgG, whereas complement receptorsbind C3b, inducing efficientphagocytosis and also the activation ofthe neutrophil. When the neutrophil isactivated, its granules fuse with thephagosome containing the bacteria,releasing bactericidal metabolites.Macrophages also phagocytose and kill encapsulated bacteria in the same manner.

215The antigen–antibody mediated pathway of complement activation
If immune complexes are not removed, they have a tendency to enlarge byaggregation and to precipitate at the basement membrane of small blood ves-sels, most notably those of the kidney glomeruli, where blood is filtered toform urine and is under particularly high pressure. Immune complexes thatpass through the basement membrane bind to CR1 receptors expressed bypodocytes, specialized epithelial cells that cover the capillaries. Deposition ofimmune complexes within the kidney probably occurs at some level all thetime, and mesangial cells within the glomerulus are specialized in the elimi-nation of immune complexes and in stimulating the repair of the tissue dam-age they cause.
A feature of the autoimmune disease SLE is a level of immune complexes inthe blood sufficient to cause massive deposition of antigen, antibody, andcomplement on the renal podocytes. These deposits damage the glomeruli,and kidney failure is the principal danger for patients with this disease. A sim-ilar deposition of immune complexes can also be a major problem for patientswho have inherited deficiencies in the early components of the complementpathway and cannot tag their immune complexes with C4b or C3b. Suchpatients cannot clear immune complexes; these accumulate with successiveantibody responses to infection and inflict increasing damage on the kidneys.
7-23 The terminal complement proteins lyse pathogens byforming a membrane pore
As we have seen, the most important product of complement activation isC3b bonded to pathogen surfaces and soluble antigens. However, the cascadeof complement reactions does extend beyond this stage, involving five addi-tional complement components (Figure 7.43). C3b can bind to either of theC3 convertases to produce enzymes that act on the C5 component of com-plement and are called C5 convertases. The C5 convertase of the classicalpathway, the classical C5 convertase, consists of C4b, C2a, and C3b and isdesignated C4b2a3b, whereas the C5 convertase of the alternative pathway,the alternative C5 convertase, consists of Bb plus two C3b fragments and isdesignated C3b2Bb (Figure 7.44).
Complement component C5 is structurally similar to C3 and C4 but lacks thethioester bond and has a different function. It is cleaved by one or other C5convertase into a smaller C5a fragment and a larger C5b fragment. The func-tion of C5b is to initiate the formation of a membrane-attack complex, whichcan make holes in the membranes of bacterial pathogens and eukaryoticcells. In succession, C6 and C7 bind to C5b—interactions that expose ahydrophobic site in C7, which inserts into the lipid bilayer. When C8 binds toC5b a hydrophobic site in C8 is exposed, and on insertion into the membranethis part of C8 initiates polymerization of C9, the component that forms thetransmembrane pores. C9 is structurally similar to perforin, present in thelytic granules of cytotoxic T cells, but forms pores of about 100 Å diametercompared to the 160 Å pores formed by perforin (Figure 7.45). The compo-nents of the membrane-attack complex are listed and their activities summa-rized in Figure 7.43.
Although in the laboratory the perforation of membranes by the membrane-attack complex appears dramatic, clinical evidence demonstrating the
Figure 7.42 Erythrocyte CR1 helps to clear immune complexesfrom the circulation. Immune complexes bind to CR1 onerythrocytes, which transport them to the liver and spleen. Here theyare removed by macrophages expressing receptors for Fc regions andfor bound complement components.
In the spleen and liver, phagocyticcells remove the immune complexes
Small antigen:antibody complexes form inthe circulation and activate complement
Immune complex is coated withcovalently bound C3b
Bound C3b binds to the receptor CR1on erythrocyte surfaces
C1q
C4b2a3b
C3b
macrophage
CR1
erythrocyte

Chapter 7: Immunity Mediated by B Cells and Antibodies216
importance of the C5–C9 components remains limited. The clearest effect ofdeficiency in any of these components is to increase susceptibility to bacteriaof the genus Neisseria, different species of which cause the sexually transmit-ted disease gonorrhea and a common form of bacterial meningitis.
7-24 Small peptides released during complement activationinduce local inflammation
During complement activation, C3, C4, and C5 are each cleaved into two frag-ments of which the larger fragments (C3b, C4b, C5b) continue the pathway ofcomplement activation. The smaller soluble C3a, C4a, and C5a fragments are
Protein
The terminal complement components that form the membrane-attack complex
Concentrationin serum (µg ml-1) Function
C5 85 On activation the soluble C4b fragment initiates assembly of the membrane-attack complex in solution
C6 60 Binds to and stabilizes C5b. Forms a binding site for C7
C7 55 Binds to C5b,6 and exposes a hydrophobic region that permitsattachment to the cell membrane
C8 55
C9 60 Polymerization on the C5b,6,7,8 complex to form a membrane-spanningchannel that disrupts the cell’s integrity and can result in cell death
Binds to C5b,6,7 and exposes a hydrophobic region that insertsinto the cell membrane
Figure 7.43 The terminalcomponents of the complementpathway.
C5 activation by the classical C5 convertase
microbial surface
C2a
+
C5
C5
C4b C5aC3b
C5b
C2a
C4b
C2a
C4bC3b
C5 activation by the alternative C5 convertase
microbial surface
Bb
+
C5
C5
C3b C5aC3b
C5b
Bb
C3b
Bb
C3bC3b
Figure 7.44 Complement componentC5 is cleaved by a C5 convertase togive a soluble active C5b fragment.C5 convertases are formed when C3bbinds to either of the two C3convertases. The C5 convertase of theclassical pathway (top panel) consists ofa complex of C3b, C4b, and C2a,whereas the C5 convertase of thealternative pathway (bottom panel)consists of two molecules of C3b andone of Bb. In both cases, C5 binds to theC3b component of the convertase and iscleaved into fragments C5a and C5b, ofwhich C5b initiates the assembly of theterminal complement components toform the membrane-attack complex.

217The antigen–antibody mediated pathway of complement activation
also physiologically active, increasing inflammation at the site of comple-ment activation through binding to receptors on several cell types. In somecircumstances, these fragments induce anaphylaxis, an acute systemicinflammatory response, and they are therefore referred to as anaphylatoxins.Of the anaphylatoxins, C5a is the most stable and the most potent, followedby C3a and then C4a. Phagocytes, endothelial cells, and mast cells havereceptors specific for C5a and C3a. The two receptors are related and are of atype that is embedded in the cell membrane and signals through the activa-tion of a guanine nucleotide binding protein.
All three anaphylatoxins induce smooth muscle contraction and the degran-ulation of mast cells and basophils, with the consequent release of histamineand other vasoactive substances that increase capillary permeability. Theyalso have direct vasoactive effects on local blood vessels, increasing bloodflow and vascular permeability. These changes make it easier for antibody,complement proteins, phagocytic cells, and lymphocytes to pass out of theblood into the site of an infection (Figure 7.46). Meanwhile, the increasedfluid in the tissues hastens the passage of pathogen-containing antigen-pre-senting cells to the draining lymph nodes and the initiation of B and T lym-phocyte responses.
C5a also acts directly on neutrophils and monocytes to increase their adher-ence to vessel walls and, as a chemoattractant, to direct their migrationtowards sites of antigen deposition. It also increases their capacity for phago-cytosis, as well as raising the expression of CR1 and CR3 on their surfaces. Inthese ways, the anaphylatoxins act in concert with other complement com-ponents to speed the destruction of pathogens by phagocytes.
7-25 Regulatory proteins in plasma limit the extent ofcomplement activation
As we have seen in the previous three sections, complement activation sum-mons powerful cellular and molecular mechanisms of destruction to sites ofinfection. To control the destructive potential of complement there exists abattery of complement control proteins that fall into two broad categories:
Figure 7.45 The membrane-attack complex assembles togenerate a pore in the lipid bilayer membrane. Thesequence of steps and their approximate appearance is shownhere in schematic form. C5b is generated by the cleavage of C5by both the classical C5 convertase C4b2a3b and the alternativeC5 convertase C3b2Bb. C5b then forms a complex by thesuccessive binding of one molecule each of C6, C7, and C8. Informing the complex, C7 and C8 undergo a conformationalchange that exposes hydrophobic sites, which insert into themembrane. This complex causes some membrane damage and
also induces the polymerization of C9. As each molecule of C9 isadded to the polymer, it exposes a hydrophobic site and insertsinto the membrane. Up to 16 molecules of C9 can be added togenerate a transmembrane channel of 100 Å diameter. Thechannel disrupts the bacterial outer membrane, killing thebacterium. In the laboratory, the erythrocyte is a convenient cellwith which to measure complement-mediated lysis. The electronmicrograph shows erythrocyte membranes with membrane-attack complexes seen end on. Photograph courtesy of S. Bhakdiand J. Tranum-Jensen.
outstanding electronic permission for this figure

Chapter 7: Immunity Mediated by B Cells and Antibodies218
plasma proteins that disrupt enzymes formed at particular stages in comple-ment activation and membrane proteins that disrupt the fixation of comple-ment on human cell surfaces.
In the first category is plasma protein C1 inhibitor (C1INH), the major con-troller of the classical pathway of complement activation and also aninhibitor of the clotting and kinin systems. It irreversibly inhibits activatedC1r and C1s by subverting their active sites to form a covalent bond that irre-versibly cross-links the enzyme and its inhibitor. Both molecules are inacti-vated by the process. In effect, C1INH limits the time that C1 moleculesremain active, which, in turn, restricts the supply of C4b and C2a componentsfor making classical C3 convertase (Figure 7.47).
Migration of macrophages, neutrophils,and lymphocytes from blood into tissue
is increased. Microbicidal activity ofmacrophages and neutrophils is also increased
Increased permeability allows increasedfluid leakage from blood vessels
and extravasation of antibodies andcomplement at the site of infection
complementcomponents
IgM
IgG
Anaphylatoxins act on blood vesselsto increase vascular permeability
C4aC3aC5a
Figure 7.46 Local inflammatoryresponses can be induced by thesmall complement fragments C3a,C4a, and especially C5a. These smallanaphylatoxic peptides are produced bycomplement cleavage at the site ofinfection and cause local inflammatoryresponses by acting on local bloodvessels. They cause increased blood flow,increased binding of phagocytes toendothelial cells, and increased vascularpermeability, leading to theaccumulation of fluid, plasma proteins,and cells in the local tissues. Theantibodies, complement, and cellsrecruited by this inflammatory stimulusremove pathogen by enhancing theactivity of phagocytes, which arethemselves also directly stimulated bythe anaphylatoxins.
Figure 7.47 CI inhibitor (C1INH)inhibits the first stages of theclassical pathway of complementactivation.
Each C1INH molecule dissociates one C1ror C1q, becoming covalently bonded to it
Activated Clr and C1s are susceptible toinhibition by C1INH
C1q C1q
C1s
microbial surface
C1r
C1s
C1r
C1INH

219The antigen–antibody mediated pathway of complement activation
As well as its role in complement activation C1INH also inhibits serine pro-teases of the clotting and kinin systems. The importance of C1INH is revealedby the extent of the disease, hereditary angioneurotic edema (HANE), suf-fered by people who have one normal and one defective copy of the C1INHgene. These patients suffer bouts of swelling, called edema in the skin, gut,and airways, usually triggered by some form of physical or mental stress.When edema occurs in the vicinity of the trachea it can lead to suffocation(Figure 7.48). The reduced levels of C1INH in patients’ plasma leads to gener-ally higher levels of active C1 and the fragments it cleaves from C4 and C2,which, during states of stress, can become excessive. A cleavage product ofC2b called C2 kinin causes fluid to enter tissues from blood and produce theedema. Another vasoactive peptide contributing to edema is bradykinin, themain product of the kinin system and overproduced in the absence of C1INH.The symptoms of hereditary angioneurotic edema can be relieved by treat-ment either with stanozol, a steroid for which the curative mechanism isunknown, or by replacement therapy using C1INH purified from the blood ofhealthy donors.
The production and stability of the classical C3 convertase is controlled by theC4-binding protein (C4BP), which binds to C4b and renders it susceptible toinactivation by the plasma serine protease, factor I. C4BP competes with C2afor binding to C4b, so on binding to the C4b component of a C3 convertasemolecule it displaces the C2a component. The stability of C3b is similarly reg-ulated by plasma protein factor H that binds to C3b, facilitating its cleavageto iC3b by factor I (Figure 7.49). In patients who completely lack factor I theformation of C3b and of C3bBb, the alternative C3 convertase, runs awayunchecked until it depletes the reservoir of C3 in the blood and plasma. Whenfaced with bacterial infections, people with factor I deficiency fix abonor-mally low amounts of C3b on bacterial surfaces, making for less efficient bac-terial clearance by phagocytes; consequently, these people are more suscep-tible than usual to ear infections and abscesses caused by pyogenic bacteria.
Figure 7.48 Hereditary angioneuroticedema. Transient localized swelling that occurs in this condition oftenaffects the face.
Figure 7.49 C4-binding protein andfactor H inactivate classical C3 andC5 convertases respectively. Upperpanel: the C4-binding protein (C4BP)binds the C4b component of the classicalC3 convertase (C4b2a), causingdisplacement of the C2a component.When bound to C4BP, C4b is madesusceptible to attack by factor I, whichcleaves it into the inactive fragments,C4c and C4d. Lower panel: factor Hbinds to the C3b component of theclassical C5 convertase (C4b2a3b) anddisplaces it from the complex with C4band C2a. When bound to factor H, C3bis made susceptible to attack by factor I,which degrades it to the inactivefragment iC3b.
Inactivation of the classical C3 convertase by C4-binding protein and factor I
microbial surface
C2a
C4b
C2a
C4b
I
IIC4BP
C4BP C4BP
C4BP
Inactivation of C3b by factor H and factor I to give fragment iC3b
microbial surface
C3b C3b
I
IIH
H H
C3b iC3b
H

Chapter 7: Immunity Mediated by B Cells and Antibodies220
7-26 Regulatory proteins on human cell surfaces protect themfrom the effects of complement activation
The second category of complement control proteins comprises membraneproteins of human cells that interfere with complement activation at humancell surfaces. Several regulators work by preventing bound C4b and C3b frag-ments from becoming active C3 convertases. The decay-accelerating factor(DAF) binds to C4b and C3b components of C3 convertases, causing their dis-sociation and inactivation. Membrane co-factor protein (MCP) also has thisfunction, but, in addition, its binding to C4b and C3b makes them susceptibleto cleavage and inactivation by factor I (Figure 7.50). The functions of MCP aresimilar to those of the soluble complement regulator, factor H, which, itself,can be membrane associated through the use of its binding site for sialic acid,a component of human cell surface carbohydrates but absent from most bac-teria. CR1 also serves to protect the surface of cells on which it is expressed.Like MCP and factor H, CR1 disrupts C3 convertases and makes C3b and C4bsusceptible to cleavage by factor I. During phagocytosis, for example, some ofthe phagocytes’ CR1 molecules will play this protective role whereas otherswill engage the C3b and C4b fragments deposited on the pathogen’s surface.
The activity of the terminal complement components on human cells is alsoregulated by both soluble and surface-associated proteins. The soluble pro-teins called S protein, clusterin, and factor J prevent the soluble complex ofC5b with C6 and C7 from associating with cell membranes. At the human cellsurface, proteins called homologous restriction factor (HRF) and CD59 (alsocalled protectin) prevent recruitment of C9 by the complex of C5b, C6, C7,and C8 (Figure 7.51). DAF, HRF, and CD59 are all linked to the plasma mem-brane by glycosylphosphatidylinositol lipid tails. Impaired synthesis of thistail is the common cause of paroxysmal nocturnal hemoglobinuria, a dis-ease characterized by episodes of complement-mediated lysis of red bloodcells that lack cell-surface DAF, HRF, or CD59.
Many of the diverse proteins that regulate complement are elongated struc-tures built from varying numbers of structurally similar modules known ascomplement control protein (CCP) modules. Each module consists of about60 amino acids that fold into a compact sandwich formed from two slices ofb-pleated sheet stabilized by two conserved disulfide bonds. Proteins madeup of CCP modules are called regulators of complement activation (RCA)and include C4BP, DAF, MCP, and factor H, as well as the complement recep-tors CR1 and CR2. The CCP domains superficially resemble smallimmunoglobulin-like domains and, like the immunoglobulin domain, theCCP module has been used to evolve a diversity of functionally distinctiveimmune-system proteins.
DAF dissociates C3 convertases athuman cell surfaces
human cell surface
Bb
Bb
C3b
DAF
MCP dissociates C3 convertases at humancell surfaces and makes them susceptible
to cleavage by factor I
human cell surface
Bb
C3b
I
MCP
Bb
On human cells CD59 binds to theC5b,6,7,8 complex and prevents
recruitment of C9 to form the pore
On microbial cells complement componentsC5–C9 assemble a complex that
perforates the cell membrane
C6
C5b
C8 C7 C9
C9
CD59
C5b,6,7,8
microbial cell human cell
Figure 7.51 CD59 prevents assemblyof terminal complementcomponents into a membrane pore.By binding to the C5b678 complex, CD59prevents the polymerization of C9 in themembrane to form a pore. Homologousrestriction factor (HRF, not shown) worksin the same way.
Figure 7.50 Regulatory proteins onhuman cells protect them fromcomplement-mediated attack. Upperpanel: decay-accelerating factor (DAF)binds to C3b and C4b, causing thedissociation of existing C3 convertasesand preventing the assembly of newones. Lower panel: membrane co-factorprotein (MCP) binds to C3b and C4b,making them susceptible to proteolyticcleavage by factor I. CR1 (not shown)also acts as an inhibitor in this way.

221The antigen–antibody mediated pathway of complement activation
Summary
The complement system is a major defense mechanism against microbialinfection, particularly extracellular bacteria. Complement components areplasma proteins of several functional groups that become activated by infec-tion in three different ways. In the classical pathway summarized in Figure 7.52,
Death of bacterium
C6,7,8and 9
Phagocytosis ofbacterium
I+B
I+Ba
C3b recognized by CR1receptor of phagocytes
Bb associated with C3bcovalently linked to bacterium
Alternative C3 convertase
C3b and C2a associated withC4b covalently linked
to bacterium
Classical C5 convertase
C3b covalently linkedto bacterium
Amplificationloop
C3
C3a
C5
C5a
C3
C3a
C2
C2b
C4
C4a
C5b in solution
C2a associated with C4bcovalently linked to
bacterium
Activated C1:antigen:antibodycomplex on bacterial surface
Antigen:antibody complexon bacterial surface
Classical C3 convertase
Perforation ofbacterial membrane
C1q,r,s
Bacterium Bacterium-specific IgMand IgG
Figure 7.52 Overview of the classicalpathway of complement activationand action. The function of the earlypart of the pathway is to produce theenzyme C3 convertase, which cleaves C3into C3a and C3b fragments. The C3bfragment binds covalently to thepathogen’s surface. C3b bound tomicrobial surfaces can either bind tocomplement receptors on phagocyticcells, which facilitates the phagocytosisof the pathogen, or can activate theterminal components of complement,which attack the integrity of thepathogen’s cell membrane. The smallerC3a fragment, together with similarfragments cleaved from C4 and C5,induces inflammation by recruitinginflammatory cells into the area ofcomplement activation. C3b onpathogen surfaces can also bind thealternative pathway component Bb toform an alternative C3 convertase,which amplifies the formation of C3b.

Chapter 7: Immunity Mediated by B Cells and Antibodies222
activation occurs when complement component C1 binds to IgM and IgGantibodies that have bound to the cell surface antigens of a pathogen or toimmune complexes. On binding to antibody the C1 molecule becomes anactive protease, which, in turn, cleaves the C4 and C2 components into enzy-matically active fragments. The C4b fragment uses a short-lived thioesterbond to form a covalent bond with a component on the pathogen’s surfaceclose to the antigen:antibody complex. The C2a fragment has proteolyticactivity and associates with surface-bound C4b to form the classical C3 con-vertase enzyme, which cleaves and activates C3. The C3 molecule is related toC4 and its C3b fragment also uses a thioester bond to become covalentlyattached to the pathogen surface. Deposition of C3b on the pathogen surfaceis the central reaction of all three pathways of complement activation. In theclassical pathway, the action of the classical C3 convertase is amplified by theformation and participation of the alternative C3 convertase. Together, theireffect is to cover the pathogen surface with covalently bonded C3b in thevicinity of the antigen: antibody complexes.
C3b fragments at the pathogen cell surface are directly recognized by the CR1receptor of phagocytes, which facilitates the engulfment and destruction ofthe pathogen. Alternatively, C3b in association with a C3 convertase formsanother enzyme, a C5 convertase, that continues the pathway of complementactivation. Cleavage of C5 yields the soluble C5b fragment, which nucleatesassembly of the terminal complement components, C6–9. After initiation insolution, this complex transfers to the cell membrane where it makes a porethat damages the cell membrane and can kill the pathogen. Small fragmentscleaved from C3, C4, and C5 in the sequence of activating reactions summoninflammatory cells to the site of complement activation where they too con-tribute to pathogen death. During complement activation, its capacity foramplification is valuable for developing a rapid and forceful attack on aninfecting microorganism. This power carries with it an inherent tendency torun out of control, which is countered by soluble proteins that dampen theresponse and membrane proteins that impede complement deposition onthe surface of healthy human cells. Figure 7.53 summarizes the functions andfragmentation of the complement components and regulators mentioned inthis part of the chapter.
Summary to Chapter 7The response of B lymphocytes to infection is the secretion of antibodies.These molecular adaptors bind and link the pathogen to effector molecules orcells that will destroy it. In developing an antibody response, the populationof responding B cells can combine a quick but less than optimal response inthe short term with a more effective response that takes time to develop. B-1cells, which do not require cognate helper T cells, and IgM molecules, whichare not dependent on high-affinity binding sites, both represent short-termstrategies. In contrast, B-cell activation driven by cognate T-cell help, withresultant isotype switching and somatic hypermutation, is the long-termstrategy that provides effective protection from subsequent infection by thepathogen. One function of the isotypic diversification of immunoglobulins isto provide antibody responses in different compartments of the human body.IgM, IgG, and monomeric IgA work in the blood, lymph, and connective tis-sues, providing antibody responses to infections within the body’s tissues. Incontrast, dimeric IgA is transported to the luminal side of the gut wall andother mucosal surfaces, where it provides antibody responses against themicroorganisms that colonize these surfaces. A second function of antibodyisotype is to recruit different effector functions into the immune response:receptors for IgG on neutrophils and macrophages deliver pathogens for
223Summary to chapter 7
Function
Binds to antibody that has bound antigen: activates C1r
Early-acting proteins of the classical pathway of complement activation
Activeform
C1q
Cleaves C1s to activate its proteaseC1r
Cleaves C4 and C2C1s
Covalently binds antigens, forms classical C3 and C5 convertasesC4b
Inflammatory mediator (weak)C4a
Active enzyme of classical pathway C3 and C5 convertasesC2b
Precursor of vasoactive C2 kininC2a
Covalently bind antigens. Forms classical C5 convertaseC3b
Inflammatory mediator (moderate)C3a
On activation the soluble C4b fragmentinitiates assembly of the membrane-attackcomplex in solution
C5
Binds to and stabilizes C5b.Forms a binding site for C7C4
Binds to C5b,6 and attaches the complexto the cell membraneC7
Binds to C5b,6,7 and inserts a hydrophobic region into the cell membraneC8
Polymerizes on the C5,6,7,8 complex to form a membrane poreC9
Initiates assembly in solution of membrane-attack complexC5b
Inflammatory mediator (strong)C5a
Inhibits C1C1INH
Disrupts classical C3 convertaseC4BP
Inhibits alternative C3 convertaseFactor H (H)
Cleaves C3b and C4bFactor I (I)
Disrupts C3 convertasesDAF
Disrupts C3 convertasesMCP
Inhibits MAC formationC59
Stabilizes alternative C3 convertaseFactor P (P)
Componentin serum
C1
C4
C2
C3
Function
Covalently binds antigens. Opsonizes pathogensForms C3 and C5 alternative convertases
Activeform
C3b
No known functionBa
Active enzyme of the alternative C3 and C5 convertasesBb
Cleaves B to Ba and Bb when it is bound to C3bD
Componentin serum
C3
Factor B (B)
Factor D (D)
Receptor
CR2
C1qreceptor
CR3
CR4
Ligand Functions Cell types
CR1
C3d, C3dg,iC3b
C1q stalk
iC3b
Part of B-cell co-receptor
Binding of immunecomplexes to phagocytes
Stimulates phagocytosis
B cells, FDCs
B cells,macrophages, monocytes,
platelets,endothelial cells
Macrophages, monocytes,polymorphonuclear leukocytes,
FDCs
Macrophages, monocytes,polymorphonuclear leukocytes
C3b, C4b
Promotes C3b and C4b decayStimulates phagocytosis
Erythrocyte transportof immune complexes
Erythrocytes,macrophages, monocytes,
polymorphonuclear leukocytes,B cells, FDCs
iC3b Stimulates phagocytosis
Early-acting proteins of the alternative pathway of complement activation
Cellular receptors for complement components and fragments
FunctionProtein
Terminal complement components formingthe membrane-attack complex
FunctionName
Control proteins of complement activation
Figure 7.53 Summary of the names andfunctions of the complement proteins,complement receptors, andcomplement-regulatory proteinsassociated with the classical andalternative pathways. DAF, decay-accelerating factor; FDC, follicular dendriticcell; MCP, membrane co-factor protein;MAC, membrane-attack complex.

Chapter 7: Immunity Mediated by B Cells and Antibodies224
phagocytosis, whereas the high-affinity receptor for IgE on mast cells, neu-trophils, and eosinophils ensures that antigens binding IgE provoke aninflammatory response. Proteins of the complement system work synergisti-cally with IgM and IgG to modify pathogens and make them more susceptibleto phagocytosis. Activation of the complement system covers the pathogensurface with covalently bonded complement components—C3b and C4b—that are bound by specific receptors on phagocytic cells, facilitating theuptake and destruction of the pathogen. Other complement fragments pro-duced in the course of activation help induce an inflammatory response atthe site of infection. The activation of phagocytes and inflammatory reactionsby complement provides protection both before and after the antibodyresponse develops. The defense mechanism of complement-mediatedphagocytosis of pathogens evolved long before the existence of antibodies; inmolecular history it was the antibodies that actually provided the comple-mentary function, rather than the complement.
Question 7–1Cross-linking of immunoglobulin by antigen is essentialbut not always sufficient to initiate the signal cascade forB-cell activation. Stimulation of additional receptors isalso necessary for full activation and differentiation ofnaive B cells. Describe these receptors and their ligands,and outline how they help activate the B cell. (Refer toFigures 7.3 and 7.8.)
Question 7–2A. Explain the difference between thymus-dependent
and thymus-independent antigens. B. Describe the characteristics of thymus-independent
antigen-1 (TI-1) and provide an example.C. Repeat this for TI-2 antigen.D. How do these antigens by-pass the requirement for
T-cell help? (Refer to Figures 7.5 and 7.6.)
Question 7–3Which of the following statements is true or false? If astatement is false, explain why.
A. Plasma cells produce secreted antibody, proliferateand undergo somatic hypermutation to produceantibodies with higher affinity for antigen.
B. An immunodeficiency called hyper IgM syndrome ischaracterized by the lack of CD40 ligand expressionon T cells.
C. Antibody-dependent cell-mediated cytotoxicity(ADCC) is mediated by NK cells which bind to anti-body-coated target cells using Fc receptors and killthe target through apoptosis.
D. TI-2 polysaccharide antigens are commonly used invaccines administered to infants because they stim-ulate strong antibody responses.
Question 7–4Explain why CD40 ligand expression on T cells is impor-tant in the T-cell zone of secondary lymphoid tissue andhow this contributes to the formation of a primary focus.(Refer to Figures 7.7 and 7.8.)
Question 7–5
Follicular dendritic cells (FDCs) bear the receptors FcgRand CD2, which bind IgG and complement components(C3b and C3d), respectively, when bound to antigen inthe form of immune complexes.
A. How would an immune complex containing IgMbind to FDCs?
B. Give reasons why FcgR and complement receptorsof FDCs are not internalized by receptor-mediatedendocytosis, as observed in macrophages, whenoccupied by immune complexes. (Refer to Figures7.10, 7.11, and 7.12.)
Question 7–6
A. Discuss the effector functions of IgM antibody.B. Why is IgM so efficient in (i) preventing blood-borne
infections and (ii) fixing complement, but (iii) ineffi-cient in engaging phagocytosis of immune com-plexes? (Refer to Figures 2.29 and 7.24.)
Question 7–7
A. Explain how the poly-Ig receptor transports IgAantibodies across cell barriers and specify the typeof cell barrier involved.
B. What are the final locations of the transported mate-rial? (Refer to Figures 7.17 and 7.19.)
Question 7–8
A. Explain how the Brambell receptor transports IgGantibodies across cell barriers, and specify the typeof cell barrier involved.
B. What is the final location of transported material?(Refer to Figures 7.18 and 7.19.)
Question 7–9
Discuss (a) the similarities and (b) the differencesbetween FceRI and FcgRIII cross-linking and its effects inmast cells and natural killer (NK) cells, respectively. Bespecific. (Refer to Figures 7.24, 7.26 and 7.28.)
Questions

225Questions
Question 7–10 A. Review the differences between the three pathways
of complement (classical, lectin and alternative) interms of how they are activated.
B. Which pathway(s) are considered acquired immuneresponses and which pathway(s) are consideredinnate immune responses and why? (Refer to Fig-ures 7.29 and 7.52.)
Question 7–11Although activation of the three different pathways ofcomplement involves different components, the threepathways converge on a common enzymatic reactionreferred to as complement fixation.
A. Describe this reaction.B. Describe the enzyme(s) responsible.C. Identify the three effector mechanisms of comple-
ment armed by this common pathway. (Refer to Fig-ures 7.34, 7.36, 7.41, 7.44, 7.45, 7.46, and 7.52.)
Question 7–12Describe the course of events that results in the swollenlymph nodes characteristic of many infections. Use thefollowing terms in your answer: T-cell area, primary focus,primary follicle, centroblasts, germinal center, somatichypermutation, centrocytes, follicular dendritic cells, andtingible body macrophages. (Refer to Figure 7.9.)
Question 7–13A. What is meant by the term “passive transfer of
immunity”, and how is it achieved: give examples?B. Define the isotype of the antibody involved in (i)
placental transfer and (ii) breast milk and explainwhy these antibodies are important.
C. Do you think it is possible for a pregnant motherwho has an autoimmune disease to transfer autore-active antibodies to the developing fetus? (Refer toFigures 7.19 and 7.20.)
Question 7–14 Genetic deficiency of complement components affectsimmunity differently depending on which component isabsent or partially lacking. Give two examples of inher-ited deficiencies of complement components and theirconsequences.





















