
Identification of novel proteolytic forms of osteocalcin in human urine
8
Identification of novel proteolytic forms of osteocalcin in human urine Kaisa K. Ivaska, a, * Jukka Hellman, b Johanna Likoj€ arvi, a Sanna-Maria K€ ak€ onen, b Paul Gerdhem, c Kristina Akesson, c Karl J. Obrant, c Kim Pettersson, b and H. Kalervo V€ a€ an€ anen a a Department of Anatomy, Institute of Biomedicine, University of Turku, Kiinamyllynkatu 10, FIN-20520 Turku, Finland b Department of Biotechnology, University of Turku, Tykist € okatu 6A, FIN-20520 Turku, Finland c Department of Orthopaedics, Malm€ o University Hospital, SE-20502 Malm€ o, Sweden Received 20 May 2003 Abstract In this study, we report the isolation and characterization of osteocalcin in human urine using mass spectrometry and N-terminal sequencing. Multiple proteolytic forms of osteocalcin were found, which consisted of 16–27 residues from the middle region of the molecule. Several fragments had residue Gly7 at the N-terminus and the most predominant was fragment 7–31. Additional frag- ments starting from residue Asp14 were detected in the samples of children and young adults. Immunochemical detection of urine osteocalcin fragments had a statistically significant negative correlation to bone mineral density in evaluation of urine samples from 75-year-old women. Thus, the measurement of osteocalcin fragments in urine may have potential applications in diagnostics related to disorders of bone metabolism. Ó 2003 Elsevier Science (USA). All rights reserved. Keywords: Bone; Urine; Osteocalcin; Turnover; Marker; Fragment; Mass spectrometry; Bone mineral density Human osteocalcin (hOC), also designated bone Gla protein, is the most abundant non-collagenous protein in bone matrix. It consists of 49 amino acid residues, three of which are c-carboxylated glutamic acids (Gla). [1]. Although most of the synthesized hOC is adsorbed to bone hydroxyapatite by Gla residues, a small part of it leaks into blood stream where it can be detected [2]. Part of the hOC found in blood is also thought to originate from the resorption, when hOC inside the bone matrix is released during bone degradation [3]. Levels of circulating hOC have been widely used in clinical in- vestigations as a marker of bone formation [4], but the discordant results obtained from different hOC assays have hindered the widespread usage of serum hOC in clinical applications [5–7]. The diversity of the hOC molecule itself has an evi- dent contribution to its immunoreactivity. Multiple proteolytic forms of hOC have been found in circulation [8,9] and urine [8,10]. The hOC fragments might be produced by osteoblasts or released from bone resorp- tion or they might result from degradation in the cir- culation or peripheral organs, such as kidneys, liver, and lungs [2,3,8,9,11–13]. Several proteases, e.g., plas- min, trypsin, and cathepsins, have been reported to cleave hOC into smaller fragments in vitro [14,15]. Also, the carboxylation of glutamic acid residues contributes to the heterogeneity of hOC. When Ca 2þ binds to Gla residues an a-helical structure is known to form [16], while the conformation of non-carboxylated hOC lies somewhere between the random coil and helical form [17]. Thus, a single conformation cannot be defined for the hOC peptide in solution. Furthermore, impaired c- carboxylation of glutamic acid residues has been de- scribed [18]. The half-life of intact hOC in vivo is short and hOC is mainly removed from circulation by glomerular filtration into urine [2]. There is evidence that the production of some of the fragments found in urine occurs before renal clearance and is not a result of it [8]. Because of the rapid Biochemical and Biophysical Research Communications 306 (2003) 973–980 www.elsevier.com/locate/ybbrc BBRC * Corresponding author. Fax: +358-2-333-7352. E-mail address: kaisa.ivaska@utu.fi (K.K. Ivaska). 0006-291X/03/$ - see front matter Ó 2003 Elsevier Science (USA). All rights reserved. doi:10.1016/S0006-291X(03)01093-3
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Identification of novel proteolytic forms of osteocalcin in human urine

Biochemical and Biophysical Research Communications 306 (2003) 973–980
www.elsevier.com/locate/ybbrc
BBRC
Identification of novel proteolytic forms of osteocalcin in human urine
Kaisa K. Ivaska,a,* Jukka Hellman,b Johanna Likoj€aarvi,a Sanna-Maria K€aak€oonen,b
Paul Gerdhem,c Kristina �AAkesson,c Karl J. Obrant,c
Kim Pettersson,b and H. Kalervo V€aa€aan€aanena
a Department of Anatomy, Institute of Biomedicine, University of Turku, Kiinamyllynkatu 10, FIN-20520 Turku, Finlandb Department of Biotechnology, University of Turku, Tykist€okatu 6A, FIN-20520 Turku, Finland
c Department of Orthopaedics, Malm€o University Hospital, SE-20502 Malm€o, Sweden
Received 20 May 2003
Abstract
In this study, we report the isolation and characterization of osteocalcin in human urine using mass spectrometry and N-terminal
sequencing. Multiple proteolytic forms of osteocalcin were found, which consisted of 16–27 residues from the middle region of the
molecule. Several fragments had residue Gly7 at the N-terminus and the most predominant was fragment 7–31. Additional frag-
ments starting from residue Asp14 were detected in the samples of children and young adults. Immunochemical detection of urine
osteocalcin fragments had a statistically significant negative correlation to bone mineral density in evaluation of urine samples from
75-year-old women. Thus, the measurement of osteocalcin fragments in urine may have potential applications in diagnostics related
to disorders of bone metabolism.
� 2003 Elsevier Science (USA). All rights reserved.
Keywords: Bone; Urine; Osteocalcin; Turnover; Marker; Fragment; Mass spectrometry; Bone mineral density
Human osteocalcin (hOC), also designated bone Gla
protein, is the most abundant non-collagenous protein
in bone matrix. It consists of 49 amino acid residues,three of which are c-carboxylated glutamic acids (Gla).[1]. Although most of the synthesized hOC is adsorbed
to bone hydroxyapatite by Gla residues, a small part of
it leaks into blood stream where it can be detected [2].
Part of the hOC found in blood is also thought to
originate from the resorption, when hOC inside the bone
matrix is released during bone degradation [3]. Levels of
circulating hOC have been widely used in clinical in-vestigations as a marker of bone formation [4], but the
discordant results obtained from different hOC assays
have hindered the widespread usage of serum hOC in
clinical applications [5–7].
The diversity of the hOC molecule itself has an evi-
dent contribution to its immunoreactivity. Multiple
proteolytic forms of hOC have been found in circulation
* Corresponding author. Fax: +358-2-333-7352.
E-mail address: [email protected] (K.K. Ivaska).
0006-291X/03/$ - see front matter � 2003 Elsevier Science (USA). All rights
doi:10.1016/S0006-291X(03)01093-3
[8,9] and urine [8,10]. The hOC fragments might be
produced by osteoblasts or released from bone resorp-
tion or they might result from degradation in the cir-culation or peripheral organs, such as kidneys, liver,
and lungs [2,3,8,9,11–13]. Several proteases, e.g., plas-
min, trypsin, and cathepsins, have been reported to
cleave hOC into smaller fragments in vitro [14,15]. Also,
the carboxylation of glutamic acid residues contributes
to the heterogeneity of hOC. When Ca2þ binds to Gla
residues an a-helical structure is known to form [16],
while the conformation of non-carboxylated hOC liessomewhere between the random coil and helical form
[17]. Thus, a single conformation cannot be defined for
the hOC peptide in solution. Furthermore, impaired c-carboxylation of glutamic acid residues has been de-
scribed [18].
The half-life of intact hOC in vivo is short and hOC is
mainly removed from circulation by glomerular filtration
into urine [2]. There is evidence that the production ofsome of the fragments found in urine occurs before renal
clearance and is not a result of it [8]. Because of the rapid
reserved.

974 K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980
clearance from the circulation, serum hOC could reflectthe acute changes in bone metabolism, while some of
urine hOC fragments might serve as an index of long-
term changes [19]. Thus, analysis of urine hOC could
offer an alternative method to monitor bone metabolism.
Due to the presence of multiple hOC fragments, which
reflect potentially diverse degradation cascades, it is es-
sential to apply discriminative methods for the mea-
surement of the hOC fragments of various origins. Inorder to achieve this goal, the exact primary structure of
the fragments has to be known, since it dictates which
epitopes are represented within the fragments. The epi-
tope may often depend on the presence or absence of a
single amino acid residue, which in the case of proteolytic
fragments requires knowledge on the terminal (N- and
C-terminal residue, respectively) residues. Methodologi-
cally, this information can be obtained by mass spec-trometry and/or chemical sequencing methods. Over the
past decade it has become feasible to determine the
masses of biomolecules with high accuracy, which relies
on the ability to discriminate naturally occurring isotopic
forms of the molecules by mass spectrometry. This en-
ables accuracies as high as 10–30 ppm, such as those re-
ported in this study. Together with N-terminal sequence
information obtainable with Edman degradation, it ispossible to deduce the exact primary structure of the
peptide under investigation.
The object of the present study was to identify the
proteolytic fragments of urine hOC from individuals of
different ages. Furthermore, the aim of the study was to
provide evidence for novel information on bone me-
tabolism obtained by the detection of fragmented hOC
in urine samples by simple and convenient immuno-chemical methods.
Experimental
Subjects and sample collection
Urine samples were obtained as the first morning void and stored at
)70 �C until analyzed. Samples were collected from five healthy vol-
unteers, two males aged 4 and 8 years and three females aged 24, 33,
and 75 years. Also, a urine pool was obtained from four females aged
12–16 years (equal amount of hOC from each individual was used).
Subjects did not receive any medication affecting bone metabolism.
Thirty-seven women, all 75 years of age, were a subset of the members
enrolled in the Osteoporosis Prospective Risk Assessment study in the
city of Malm€oo, Sweden [20]. Informed consent was obtained from all
subjects in the study, which was also approved by the local ethics
committee and performed in accordance with the Declaration of
Helsinki.
Reagents for immunochemical detection
Monoclonal antibodies (MAbs) used in this study have been de-
scribed in detail previously [21]. Briefly, MAb 6F9 binds to region 7–19
in hOC molecule and MAbs 2H9 and 3H8 have an epitope on region
20–43. In addition, MAb 3H8 favors Gla-containing forms with 8%
cross-reactivity towards non-carboxylated hOC. MAbs were biotiny-
lated with 50-fold molar excess of biotin-isothiocyanate [22] or labeled
with 200-fold molar excess of europium(III) chelate [23] as described
previously [21]. Synthetic peptide of hOC 1–49 (Gla at positions 17, 21,
and 24) was purchased from Advanced Chemtech (Louisville, USA)
and biotinylated with 30-fold excess of biotin-isothiocyanate. The re-
action was carried out in 50mM NaHCO3–Na2CO3 buffer, pH 9.8, at
+4 �C for 5 h. Buffer exchange was performed with NAP-5 column
(Amersham Biosciences) equilibrated with 50mM Tris–HCl, 150mM
NaCl, and 15mM NaN3, pH 7.75. Streptavidin-coated microtiter
plates, all Delfia reagents and Victor Multilabel Counter, were from
Perkin–Elmer Life Sciences/Wallac, Turku, Finland.
Immunochemical detection of urine hOC
Two-site 6F9/3H8. Four hundred nanograms of biotinylated 6F9 in
50 ll Assay Buffer was added to the wells of streptavidin-coated mi-crotiter plates, incubated with continuous shaking at room tempera-
ture (22 �C) for 30min and washed twice with Delfia Wash Solution.The samples or calibrators (10ll) were added, followed by 100 ng Eu-labeled 3H8 in 50ll Assay Buffer containing 5mmol/L EDTA. After2 h shaking, the plates were washed six times. Detection was carried
out by adding 200ll Delfia Enhancement Solution to each well andmeasuring time-resolved fluorescence after 30min. Synthetic peptide of
hOC 1–43 (Gla at positions 17, 21, and 24, Advanced Chemtech,
Louisville, USA) was used as a calibrator and the standard curve
covered the range from 0.4 to 100ng/ml. The analytical detection limit,
defined as the concentration corresponding to the mean value of 12
determinations of the zero calibrator+ 2SD, was set as 0.07 ng/ml. The
within-assay CVs were less than 5% (N ¼ 12) and the between-assayCVs less than 8% (N ¼ 12).
Two-site 2H9/6F9. The measurement was carried out as above with
400 ng biotinylated 2H9 and 200 ng Eu-labeled 6F9.
Competitive hOC/3H8. Biotinylated hOC (7.5 ng) in 50 ll AssayBuffer was incubated on streptavidin-coated plates for 1 h and washed
six times. Thirty microliters of samples or calibrators and tracer so-
lution with 2 ng Eu-labeled 3H8 in 50 ll Assay Buffer were added,incubated for 1 h and washed six times. The detection was carried out
as above. Pubertal urine diluted in Assay Buffer was used as a cali-
brator and the standard curve covered the range from 1.4 to 46 ng/ml
(hOC in dilutions determined with 6F9/3H8). The analytical detection
limit, defined as the concentration corresponding to the mean value of
24 determinations of the zero calibrator+ 2SD, was set as 1.2 ng/ml.
The within-assay CV was less than 7% (N ¼ 24) and the between assayCV less than 10% (N ¼ 6).
Isolation and fractionation of urine hOC
Urine samples from 4, 8, 12–16, 24, 33, and 75-year-old individuals
were centrifuged for 30min at 10,000g, filtrated (Sterivex-HV 0.45lm,Millipore, USA), and extracted using solid phase extraction cartridges
(Sep-Pak Plus C18, Millipore, USA) primed with acetonitrile. Aceto-
nitrile (40%) was used to elute adsorbed material, fractions containing
hOC were pooled, and acetonitrile was removed by evaporation
(HetoVac, Heto, Denmark). hOC fragments in the extracted material
were isolated by immunoaffinity chromatography. The gel matrix was
prepared according to the instructions of the Affi-Gel Hz Immunoaf-
finity Kit (Bio-Rad Laboratories, USA) and equal amounts of MAbs
6F9, 2H9, and 3H8 were used (1mg MAb mixture per 1ml matrix).
Extracted material was loaded onto columns (2ml) and the gel was
washed with 0.1M Na-phosphate buffer, pH 7, and 0.5M NaCl, fol-
lowed by 0.1M Na-phosphate buffer, pH 7, and 0.3M NaCl, and
0.1M Na-phosphate buffer, pH 7. Finally, 0.1% trifluoroacetic acid
(TFA) was used for elution. 1.0–3.7lg isolated hOC was fractionatedon a Vydac C4 reverse phase high performance liquid chromatography
(RP-HPLC) column (2.1mm� 150mm) equipped with a Vydac C4guard column (both from The Sep/a/ra/tions Group, USA) at a flow

K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980 975
rate of 150ll/min using acetonitrile gradient for elution as describedpreviously [10]. The detection wavelength was 276 nm and peak frac-
tions were collected manually for further characterization. Alterna-
tively, 50ll fractions were collected for immunochemical detection. Acontrol sample was prepared by adding synthetic peptide of hOC 1-49
(50lg, from Advanced Chemtech) into urine (100ml) depleted of hOCwith immunoaffinity chromatography and the sample was subjected to
isolation and fractionation steps. All isolation steps were performed at
room temperature (22 �C).
Matrix-assisted laser desorption ionization time of flight mass spectro-metry and N-terminal sequencing
The HPLC fractions were evaporated to dryness in HetoVac and
reconstituted with 30 ll of 60% acetonitrile in 0.1% TFA. One mi-
croliter samples were mixed with an equal volume of a-cyano-4-hy-droxycinnamic acid (10mg/ml in 60% acetonitrile and 0.1% TFA)
containing calibration mixture 2 from Sequazyme Peptide Mass
Standards Kit (Perseptive Biosystems, USA). Samples were analyzed
in the positive ion reflector mode by matrix-assisted laser desorption
ionization time-of-flight mass spectrometry instrument (MALDI-TOF,
Voyager-DE Pro, Perseptive Biosystems, USA) according to the
manufacturer�s instructions. Linear mode was used for control samplewith spiked synthetic hOC 1-49. Amino-terminal amino acid sequence
analyses of selected fractions were performed with an Applied Bio-
systems (USA) model 477A protein sequencer equipped with an on-
line Applied Biosystems model 120 A phenylthiohydantoin amino acid
analyzer.
Fig. 1. HPLC elution profile of urine hOC fractionation. Fractions
Clinical evaluation and statistics
Urine hOC in 37 75-year-old women was measured with 6F9/3H8
and hOC/3H8. Creatinine measurements were performed by the Jaffe
method and the creatinine-corrected hOC concentrations were used in
all analyses. Bone mineral density (BMD) was assessed at femoral neck
with a Lunar DPX-L-equipment (Lunar, Madison, USA) and T-scores
were calculated according to the manufacturer�s instructions. Thecorrelation between urine hOC and femoral neck T-score was assessed
by Spearman�s rank correlation.
were measured with competitive hOC/3H8 (filled squares) and two-site6F9/3H8 (open squares). Immunoreactivity of fractions is displayed as
percentage of total immunoreactivity and percentages are calculated
for both assays separately. (A) Sample of the 8-year-old subject. The
elution profile was similar in subjects aged 12–16 and 24 years. (B)
Sample of the 75-year-old subject. The elution profile was similar in
subjects aged 4 and 33 years.
Results
Two distinct profiles in the fractionation of urine hOC
HPLC fractionation of isolated urine hOC revealed
several immunoreactive fractions and two distinct elu-
tion profiles. In the fractionation of three urine samples,
two major peaks were observed when fractionation wasmonitored with competitive method hOC/3H8 (Fig. 1A).
These samples were the pool of 12–16-year-old females
and the individual samples of 8 and 24-year-old subjects.
The peak which eluted earlier was undetectable in the
elution profiles of three other samples, namely individ-
ual samples of 4, 33, and 75-year-old subjects (Fig. 1B).
In contrast, the peak which appeared later in the elution
profile and was detected also with two-site method 6F9/3H8 was evident in all samples. This peak represents
main urine hOC which is large enough for two-site im-
munodetection. Fractions eluting at the end of the sec-
ond peak were also weakly positive for the two-site
method 2H9/6F9 (data not shown).
Mass spectrometric characterization reveals several pro-
teolytic forms of hOC
The monoisotopic masses of the six prominent ions
detected in all samples were between 2564 and 2992Da.
The HPLC fractions producing these ions were positive
for two-site method 6F9/3H8. The peak with highest
intensity was seen at 2808Da (Fig. 2A). Three addi-
tional ions of molecular weight 2127, 2240, and 2733Da
were present in three urine samples. The most pro-
nounced of these additional ions was the one at 2127Da(Fig. 2B). The two smallest ones (2127 and 2240Da)
eluted in the area where an additional peak of compet-
itive hOC/3H8 was observed, but the largest ion
(2733Da) was observed in the two-site detectable region
with the ions from 2564 to 2992Da (Fig. 2C). Also, an
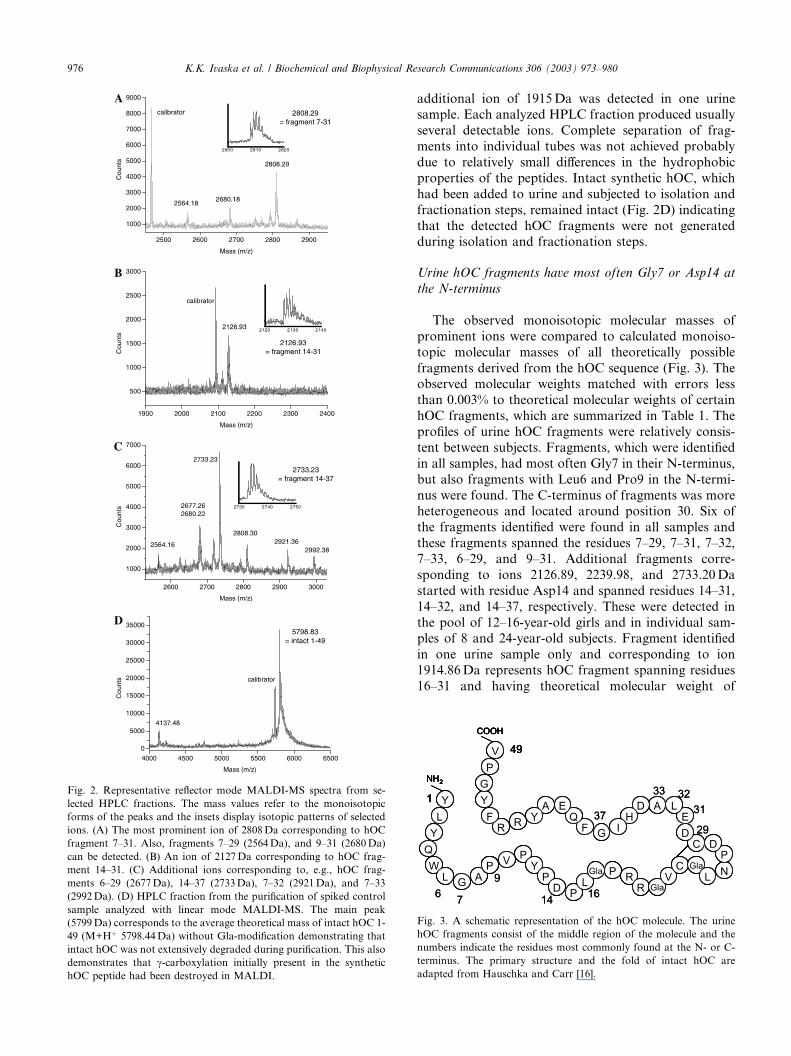
Fig. 2. Representative reflector mode MALDI-MS spectra from se-
lected HPLC fractions. The mass values refer to the monoisotopic
forms of the peaks and the insets display isotopic patterns of selected
ions. (A) The most prominent ion of 2808Da corresponding to hOC
fragment 7–31. Also, fragments 7–29 (2564Da), and 9–31 (2680Da)
can be detected. (B) An ion of 2127Da corresponding to hOC frag-
ment 14–31. (C) Additional ions corresponding to, e.g., hOC frag-
ments 6–29 (2677Da), 14–37 (2733Da), 7–32 (2921Da), and 7–33
(2992Da). (D) HPLC fraction from the purification of spiked control
sample analyzed with linear mode MALDI-MS. The main peak
(5799Da) corresponds to the average theoretical mass of intact hOC 1-
49 (M+Hþ 5798.44Da) without Gla-modification demonstrating that
intact hOC was not extensively degraded during purification. This also
demonstrates that c-carboxylation initially present in the synthetichOC peptide had been destroyed in MALDI.
976 K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980
additional ion of 1915Da was detected in one urinesample. Each analyzed HPLC fraction produced usually
several detectable ions. Complete separation of frag-
ments into individual tubes was not achieved probably
due to relatively small differences in the hydrophobic
properties of the peptides. Intact synthetic hOC, which
had been added to urine and subjected to isolation and
fractionation steps, remained intact (Fig. 2D) indicating
that the detected hOC fragments were not generatedduring isolation and fractionation steps.
Urine hOC fragments have most often Gly7 or Asp14 at
the N-terminus
The observed monoisotopic molecular masses of
prominent ions were compared to calculated monoiso-
topic molecular masses of all theoretically possible
fragments derived from the hOC sequence (Fig. 3). Theobserved molecular weights matched with errors less
than 0.003% to theoretical molecular weights of certain
hOC fragments, which are summarized in Table 1. The
profiles of urine hOC fragments were relatively consis-
tent between subjects. Fragments, which were identified
in all samples, had most often Gly7 in their N-terminus,
but also fragments with Leu6 and Pro9 in the N-termi-
nus were found. The C-terminus of fragments was moreheterogeneous and located around position 30. Six of
the fragments identified were found in all samples and
these fragments spanned the residues 7–29, 7–31, 7–32,
7–33, 6–29, and 9–31. Additional fragments corre-
sponding to ions 2126.89, 2239.98, and 2733.20Da
started with residue Asp14 and spanned residues 14–31,
14–32, and 14–37, respectively. These were detected in
the pool of 12–16-year-old girls and in individual sam-ples of 8 and 24-year-old subjects. Fragment identified
in one urine sample only and corresponding to ion
1914.86Da represents hOC fragment spanning residues
16–31 and having theoretical molecular weight of
Fig. 3. A schematic representation of the hOC molecule. The urine
hOC fragments consist of the middle region of the molecule and the
numbers indicate the residues most commonly found at the N- or C-
terminus. The primary structure and the fold of intact hOC are
adapted from Hauschka and Carr [16].

Table 1
MALDI-MS analysis of urine hOC fragments in urine samples obtained from individuals of different ages
hOC fragment Theoretical mass
(M+Hþ)aIons observed in individual urine samples (M+Hþ)b N-terminal
sequencing4 years 8 years 12–16 years 24 years 33 years 75 years
6–29 2677.26 2677.22 nd 2677.22 2677.26 2677.29 2677.31 LGAPVPY
7–29 2564.17 2564.15 2564.14 2564.12 2564.16 2564.18 2564.18
7–31 2808.24 2808.27 2808.20 2808.25 2808.30 2808.30 2808.29 GAPVPYPD
7–32 2921.33 2921.34 2921.37 2921.32 2921.36 2921.36 2921.34 GAPVPYPDPL
7–33 2992.36 2992.44 2992.32 2992.39 2992.38 2992.41 2992.36
9–31 2680.18 2680.18 2680.18 2680.20 2680.22 2680.23 2680.18 XVPYPXP
14–31 2126.89 nd 2126.93 nd 2126.89 nd nd DPLXPRR
14–32 2239.98 nd 2239.92 2239.94 2239.97 nd nd
14–37 2733.20 nd 2733.20 2733.21 2733.23 nd nd
16–31 1914.81 nd 1914.86 nd nd nd nd
nd, not detected.a The monoisotopic mass values without c-carboxylation are shown.b If the ions were observed in several HPLC fractions, the mean value of individual observations is shown.
Fig. 4. Linear regression for T-score for femoral neck BMD and hOC
fragments detected with competitive hOC/3H8 (A) and two-site 6F9/
3H8 (B), respectively. The Spearman�s rank correlation coefficient forBMD and competitive hOC/3H8 was r ¼ �0:68 (p < 0:0001) and for
BMD and two-site 6F9/3H8 r ¼ �0:37 (p ¼ 0:0259). The urine hOCmeasurements have been corrected for the creatinine excretion.
K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980 977
1914.81Da. All theoretical molecular masses were cal-
culated without Gla-modification, since it has been re-
ported that c-carboxylation is destroyed in MALDI-MSand is not included in the molecular masses of observed
ions [24,25]. The identification of fragments was con-
firmed using N-terminal sequencing, also shown in Ta-
ble 1. Urine may also contain shorter OC fragments,
which remain to be characterized.
Fragments are differentially recognized by immunochem-
ical methods
The most predominant of hOC fragments detected in
all samples was the one of residues 7–31. This fragment
was detected in fractions positive for two-site method
6F9/3H8 and it appears to be the main fragment de-
tected with two-site approach. hOC fragments of resi-dues 7–32 and 7–33 were present in very small amounts
and were identified from fractions positive for two-site
2H9/6F9 and these were the only urine hOC fragments
recognized by this method. Fragments, which started
with residue Asp14, were not detected by the two-site
methods. They were, however, clearly positive for im-
munodetection by the competitive method hOC/3H8,
which inevitably also detected fragments with Gly7 atthe N-terminus. We were unable to separate fragments
6–29, 7–29, and 9–31 to individual fractions but instead,
some of the main fragment 7–31 immunoreactivity co-
eluted with them. One might assume that at least the
competitive method should be able to pick these frag-
ments also for measurement.
Urine hOC reflects the balance of bone turnover
Linear regression for T-score of femoral neck BMD
and urine hOC in the 75-year-old females is shown in
Fig. 4. A statistically significant negative correlation
was observed for bone mass and total urine hOC as

978 K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980
measured by competitive method hOC/3H8, withSpearman�s correlation coefficient r ¼ �0:68, p <0:0001 (Fig. 4A). A statistically significant, but less
pronounced, correlation was obtained for bone mass
and longer hOC fragments starting from Gly7 and de-
tected by two-site method (r ¼ �0:37, p ¼ 0:0259,Fig. 4B).
Taken together, hOC in urine is a complex mixture
of several midmolecule peptide fragments which differfrom each other by only a few amino acid residues at
either N- or C-terminus. The proteolytic forms of
hOC can be divided into two main categories de-
pending on their N-terminus, namely fragments start-
ing from Gly7 (or Leu6, or Pro9) and more truncated
fragments with Asp14 (or Leu16) at the N-terminus,
and the immunochemical detection of identified frag-
ments may provide information on the balance ofbone turnover.
Discussion
Taylor et al. [8] were the first to report the presence of
hOC in urine with a competitive midmolecule hOC ra-
dioimmunoassay. Later, Matikainen et al. [10] were ableto define the cleavage sites of two fragments of hOC
from a pool of urine collected from one pubertal indi-
vidual. The isolated fragments spanned residues 6–30
and 7–30 as determined by N-terminal sequencing and a
linear MALDI-MS. More recently, Srivastava et al. [26]
utilized surface-enhanced laser desorption ionization
time of flight (SELDI-MS) technique for the character-
ization of urine hOC and found a fragment spanningresidues 14–28 of hOC. With the competitive immuno-
assay based on polycolonal antibodies they were able to
detect short-term changes in bone metabolism allowing
a conclusion that the assay detects hOC fragments of
resorptive origin. Also, �AAkesson et al. [27] have reportedthat urine hOC reflects bone resorption rather than bone
formation after tibia osteotomy. These findings make
urine hOC as a bone turnover marker intriguing andemphasize the importance of further clarifying the
nature of urine hOC fragments.
Urine hOC fragments identified in this study con-
sisted of 16–27 amino acid residues from the middle
region of the molecule which allowed the detection of
fragments by immunochemical methods developed
against the midmolecule epitopes. The epitopes in the
middle region of the molecule have been suggested to bethe most stable ones [8,28,29] and they seem to be the
only ones resistant to glomerular filtration and pro-
cessing of urine. The middle region of hOC adopts an
a-helical conformation [16] and this compact structuremay be important for surviving the proteolytic ma-
chinery. The disulphide bond between Cys23 and Cys29
probably further stabilizes these fragments and these
residues were included in all the fragments we identified.The length of urine hOC fragments was highly consis-
tent between different samples and fragments of amino
acid residues 7–29, 7–31, 7–32, 7–33, and 9–31 were
found in all the samples we studied. Also, fragment 6–29
is probably common to all samples, although we failed
to detect it in one subject by MALDI after the purifi-
cation process. This similarity between different samples
suggests that the fragments are not results of randomdegradation processes, but rather generated in a similar
manner in all these individuals in vivo, reflecting the
stability of the middle region of the hOC molecule.
Most of the urine hOC fragments common to all
samples had Gly7 in their N-terminus and the main
hOC fragment detected in all urine samples consisted of
residues 7–31. Matikainen et al. [10] reported that the
main urine hOC fragment detected by two-site immu-noassays consists of residues 7–30, but this discrepancy
is probably due to differences in the accuracy of MALDI
instruments (reflector vs. linear mode analysis) and dif-
ferent interpretation of the results concerning the c-carboxylation. An interesting difference between urine
samples was observed in regard to the presence of
fragments starting from Asp14. The fragments 14–31,
14–32, and 14–37 (and 16–31) were detected in threesamples, in the individual samples from 8 to 24-year-old
individuals and in the pool of 12–16-year-olds. This may
suggest that the Asp14-fragments are produced in high
quantities especially during, e.g., period of high bone
turnover and gaining of bone mass, which might be
typical to these subjects. The cleavage site between
Pro13 and Asp14 has recently been found also by Sri-
vastava and co-workers [26] who were able to charac-terize one hOC fragment corresponding to amino acids
14–28 from a urine sample of a patient with Paget�sdisease. We failed to identify this particular hOC frag-
ment but instead, we identified several similar frag-
ments, namely 14–31, 14–32, 14–37, and 16–31. The
information on c-carboxylation of urine hOC was notobtained due to decarboxylation of samples in MALDI-
MS. However, it has been previously reported that MAb3H8 used in immunochemical detection prefers binding
to c-carboxylated hOC forms [28] and thus, the majorityof urine hOC must be at least partially c-carboxylated.Also, Gla-residues are not extracted in N-terminal se-
quencing which results in blank chromatograms at po-
sitions occupying c-carboxylation [30]. A blank
chromatogram was obtained in sequencing of Asp14
fragments at putative Gla position 17 (data not shown),which suggests c-carboxylation at this site. Also otherGla-containing proteins, e.g., prothrombin have been
reported to exist in urine almost entirely as c-carboxyl-ated forms [31].
The proteases involved in generation of hOC frag-
ments are unclear. The origin of the N-terminus Gly7 is
interesting, since there are no reports on proteinases

K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980 979
cleaving hOC between residues Leu6 and Gly7. Instead,cathepsins B, L, H, and S [14,29] and recently also ca-
thepsinK [32], the main proteolytic enzyme in bone
matrix degradation [33], have been shown to cleave hOC
between residues Gly7 and Ala8. This suggests that ur-
ine hOC fragments starting from Gly7 are not derived
from a cathepsin-like action on hOC and these frag-
ments are not directly derived from breakdown of bone
matrix by cysteine proteases. The protease involved inthe generation of the Asp14 terminus is also unclear. In
contrast to fragments with Gly7 at the N-terminus,
fragments starting from Asp14 can be derived from
peptides generated by cathepsins. The putative resorp-
tive origin of the these fragments is supported by the
preliminary observation by Srivastava et al. [26] that the
measurement of urine hOC 14-28 fragment in alendro-
nate treated patients correlated to the rate of bone re-sorption, defined with urinary and circulating levels of
cross-linked N- or C-terminal telopeptides of type I
collagen.
It has been demonstrated that changes of bone
turnover markers correlate to changes of BMD, but the
correlation of single measurements is usually weaker
[34–37]. Urine hOC as measured by either competitive
hOC/3H8 or two-site 6F9/3H8 immunodetection had asignificant negative correlation to femoral neck BMD,
which is currently used in diagnostics of osteoporosis
and has been proposed to predict hip fractures [38]. The
Spearman�s correlation coefficient was higher for com-petitive method, indicating that the measurement of
both Gly7- and Asp14-fragments of hOC instead of
Gly7-fragments alone might be more informative. To
our knowledge, this is the first report describing therelationship between bone mass and urine hOC. The
development of methods specific for short, putatively
resorptive, fragments only might further improve the
clinical utility of urine hOC.
In conclusion, we have described the identification
of 10 novel proteolytic forms of hOC from urine sam-
ples of healthy volunteers. Fragments consisted of less
than 30 amino acid residues from the middle region ofthe hOC molecule and were abundant enough to
be detected with simple and rapid immunochemical
methods. The knowledge on the detailed peptide
structure of urine hOC makes it feasible to develop
specific immunoassays for their measurement and for
future studies on the biological significance of urine
hOC. The presence of detectable amounts of Asp14-
fragments in individuals characterized by high boneturnover is intriguing, but still needs further evaluation.
Furthermore, the measurement of urine hOC, especially
both Gly7- and Asp14-fragments detected by the com-
petitive immunodetection, was associated with BMD
assessed at femoral neck, indicating potential applica-
tions in osteoporosis diagnostics and prediction of
fractures.
Acknowledgments
This study was supported by the State Technology Development
Center of Finland (TEKES) and National Graduate School for
Musculoskeletal Diseases.
References
[1] J.W. Poser, F.S. Esch, N.C. Ling, P.A. Price, Isolation and
sequence of the vitamin K-dependent protein from human bone.
Undercarboxylation of the first glutamic acid residue, J. Biol.
Chem. 255 (1980) 8685–8691.
[2] P.A. Price, M.K. Williamson, J.W. Lothringer, Origin of the
vitamin K-dependent bone protein found in plasma and its
clearance by kidney and bone, J. Biol. Chem. 256 (1981) 12760–
12766.
[3] C.M. Gundberg, R.S. Weinstein, Multiple immunoreactive forms
of osteocalcin in uremic serum, J. Clin. Invest. 77 (1986) 1762–
1767.
[4] M.J. Power, P.F. Fottrell, Osteocalcin: diagnostic methods and
clinical applications, Crit. Rev. Clin. Lab. Sci. 28 (1991) 287–335.
[5] P.W. Masters, R.G. Jones, D.A. Purves, E.H. Cooper, J.M.
Cooney, Commercial assays for serum osteocalcin give clinically
discordant results, Clin. Chem. 40 (1994) 358–363.
[6] L.J. Deftos, R.L. Wolfert, C.S. Hill, D.W. Burton, Two-site
assays of bone gla protein (osteocalcin) demonstrate immuno-
chemical heterogeneity of the intact molecule, Clin. Chem. 38
(1992) 2318–2321.
[7] P.D. Delmas, C. Christiansen, K.G. Mann, P.A. Price, Bone Gla
protein (osteocalcin) assay standardization report, J. Bone Miner.
Res. 5 (1990) 5–11.
[8] A.K. Taylor, S. Linkhart, S. Mohan, R.A. Christenson, F.R.
Singer, D.J. Baylink, Multiple osteocalcin fragments in human
urine and serum as detected by a midmolecule osteocalcin
radioimmunoassay, J. Clin. Endocrinol. Metab. 70 (1990) 467–
472.
[9] P. Garnero, M. Grimaux, P. Seguin, P.D. Delmas, Characteriza-
tion of immunoreactive forms of human osteocalcin generated in
vivo and in vitro, J. Bone Miner. Res. 9 (1994) 255–264.
[10] T. Matikainen, S.M. K€aak€oonen, K. Pettersson, M. Karp, T.L€oovgren, H.K. V€aa€aan€aanen, J. Hellman, Demonstration of the
predominant urine osteocalcin fragments detectable by two-site
immunoassays, J. Bone Miner. Res. 14 (1999) 431–438.
[11] G. Aliberti, I. Pulignano, M. Proietta, L. Tritapepe, L. Cigognetti,
A. Menichetti, A. Russo, L.V. de Michele, P. Corvisieri, S.
Minisola, Osteocalcin metabolism in the pulmonary circulation,
Clin. Physiol. 20 (2000) 122–125.
[12] W. Farrugia, R.A. Melick, Metabolism of osteocalcin, Calcif.
Tissue Int. 39 (1986) 234–238.
[13] J. Salo, P. Lehenkari, M. Mulari, K. Metsikk€oo, H.K. V€aa€aan€aanen,
Removal of osteoclast bone resorption products by transcytosis,
Science 276 (1997) 270–273.
[14] R. Baumgrass, M.K. Williamson, P.A. Price, Identification of
peptide fragments generated by digestion of bovine and human
osteocalcin with the lysosomal proteinases cathepsin B, D, L, H,
and S, J. Bone Miner. Res. 12 (1997) 447–455.
[15] J.F. Novak, J.D. Hayes, S.K. Nishimoto, Plasmin-mediated
proteolysis of osteocalcin, J. Bone Miner. Res. 12 (1997) 1035–
1042.
[16] P.V. Hauschka, S.A. Carr, Calcium-dependent alpha-helical
structure in osteocalcin, Biochemistry 21 (1982) 2538–2547.
[17] R.A. Atkinson, J.S. Evans, P.V. Hauschka, B.A. Levine, R.
Meats, J.T. Triffitt, A.S. Virdi, R.J. Williams, Conformational
studies of osteocalcin in solution, Eur. J. Biochem. 232 (1995)
515–521.

980 K.K. Ivaska et al. / Biochemical and Biophysical Research Communications 306 (2003) 973–980
[18] J.R. Cairns, P.A. Price, Direct demonstration that the vitamin K-
dependent bone Gla protein is incompletely gamma-carboxylated
in humans, J. Bone Miner. Res. 9 (1994) 1989–1997.
[19] P.A. Price, S.K. Nishimoto, Radioimmunoassay for the vitamin
K-dependent protein of bone and its discovery in plasma, Proc.
Natl. Acad. Sci. USA 77 (1980) 2234–2238.
[20] P. Gerdhem, H. Magnusson, M.K. Karlsson, K. �AAkesson,
Ultrasound of the phalanges is not related to a previous fracture.
A comparison between ultrasound of the phalanges, calcaneus,
and DXA of the spine and hip in 75-year-old women, J. Clin.
Densitom. 5 (2002) 159–166.
[21] J. Hellman, S.M. K€aak€oonen, M.T. Matikainen, M. Karp, T.L€oovgren, H.K. V€aa€aan€aanen, K. Pettersson, Epitope mapping of nine
monoclonal antibodies against osteocalcin: combinations into
two-site assays affect both assay specificity and sample stability, J.
Bone Miner. Res. 11 (1996) 1165–1175.
[22] V.M. Mukkala, E. H€aanninen, I. Hemmil€aa, in: Proceedings of 8th
European Symposium on Organic Chemistry, Sitges, Spain 86,
1993.
[23] H. Takalo, V.M. Mukkala, H. Mikola, P. Liitti, I. Hemmil€aa,Synthesis of europium(III) chelates suitable for labeling of
bioactive molecules, Bioconjug. Chem. 5 (1994) 278–282.
[24] M. Prorok, S.E. Warder, T. Blandl, F.J. Castellino, Calcium
binding properties of synthetic gamma-carboxyglutamic acid-
containing marine cone snail sleeper peptides, conantokin-G and
conantokin-T, Biochemistry 35 (1996) 16528–16534.
[25] D.E. Kalume, J. Stenflo, E. Czerwiec, B. Hambe, B.C. Furie, B.
Furie, P. Roepstorff, Structure determination of two conotoxins
from Conus textile by a combination of matrix-assisted laser
desorption/ionization time-of-flight and electrospray ionization
mass spectrometry and biochemical methods, J. Mass. Spectrom.
35 (2000) 145–156.
[26] A.K. Srivastava, S. Mohan, F.R. Singer, D.J. Baylink, A urine
midmolecule osteocalcin assay shows higher discriminatory power
than a serum midmolecule osteocalcin assay during short-term
alendronate treatmentofosteoporoticpatients,Bone31 (2002) 62–69.
[27] K. �AAkesson, S.-M. K€aakonen, M. Karlsson, T. L€oovgren, K.
Pettersson, K.J. Obrant, Fracture induces long-term effects on
bone turnover in humans, J. Bone Miner. Res. 14 (1999) S272.
[28] S.M. K€aak€oonen, J. Hellman, M. Karp, P. Laaksonen, K.J. Obrant,
H.K. V€aa€aan€aanen, T. L€oovgren, K. Pettersson, Development and
evaluation of three immunofluorometric assays that measure
different forms of osteocalcin in serum, Clin. Chem. 46 (2000)
332–337.
[29] Y. Koboyashi, H. Sakai, S. Ikeda, K. Koboyashi, Y. Kato, S.
Mataki, Processing of NH2- and COOH-terminal peptides of rat
osteocalcin by cathepsin B and L, J. Bone Miner. Metab. 16 (1998)
72–80.
[30] J.R. Cairns, M.K. Williamson, P.A. Price, Direct identification of
gamma-carboxyglutamic acid in the sequencing of vitamin K-
dependent proteins, Anal. Biochem. 199 (1991) 93–97.
[31] N.P. Buchholz, D.S. Kim, P.K. Grover, C.J. Dawson, R.L.
Ryall, The effect of warfarin therapy on the charge properties of
urinary prothrombin fragment 1 and crystallization of calcium
oxalate in undiluted human urine, J. Bone Miner. Res. 14 (1999)
1003–1012.
[32] C.M. Gundberg, M. Clough, J.S. Mort, Proteolysis of human
osteocalcin by MMP�s and cathepsin K, J. Bone Miner. Res. 17(2002) S406.
[33] T. Inui, O. Ishibashi, T. Inaoka, Y. Origane, M. Kumegawa, T.
Kokubo, T. Yamamura, Cathepsin K antisense oligodeoxynucle-
otide inhibits osteoclastic bone resorption, J. Biol. Chem. 272
(1997) 8109–8112.
[34] B.J. Riis, K. Overgaard, C. Christiansen, Biochemical markers of
bone turnover to monitor the bone response to postmenopausal
hormone replacement therapy, Osteoporos. Int. 5 (1995) 276–280.
[35] H. Rosenbrock, V. Seifert-Klauss, S. Kaspar, R. Busch, P.B.
Luppa, Changes of biochemical bone markers during the meno-
pausal transition, Clin. Chem. Lab. Med. 40 (2002) 143–151.
[36] A. Rogers, R.A. Hannon, R. Eastell, Biochemical markers as
predictors of rates of bone loss after menopause, J. Bone Miner.
Res. 15 (2000) 1398–1404.
[37] J.M. Halleen, H. Ylipahkala, S.L. Alatalo, A.J. Janckila, J.E.
Heikkinen, H. Suominen, S. Cheng, H.K. V€aa€aan€aanen, Serum
tartrate-resistant acid phosphatase 5b, but not 5a, correlates with
other markers of bone turnover and bone mineral density, Calcif.
Tissue Int. 71 (2002) 20–25.
[38] S.R. Cummings, D.M. Black, M.C. Nevitt, W. Browner, J.
Cauley, K. Ensrud, H.K. Genant, L. Palermo, J. Scott, T.M.
Vogt, Bone density at various sites for prediction of hip fractures.
The study of osteoporotic fractures research group, Lancet 341
(1993) 72–75.




















