
i Dipped Natural Rubber Latex Thin Films - OhioLINK ETD ...
214
Dipped Natural Rubber Latex Thin Films: Hypoallergenic Accelerator Formulations for Crosslinking, and Composites with Waste-Derived Fillers DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Jessica Lauren Slutzky Graduate Program in Food, Agricultural and Biological Engineering The Ohio State University 2019 Dissertation Committee: Advisor: Dr. Katrina Cornish Dr. John Lannutti Dr. Frederick Michel Jr. Dr. Alfred Soboyejo
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of i Dipped Natural Rubber Latex Thin Films - OhioLINK ETD ...

i
Dipped Natural Rubber Latex Thin Films: Hypoallergenic Accelerator Formulations for
Crosslinking, and Composites with Waste-Derived Fillers
DISSERTATION
Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy
in the Graduate School of The Ohio State University
By
Jessica Lauren Slutzky
Graduate Program in Food, Agricultural and Biological Engineering
The Ohio State University
2019
Dissertation Committee:
Advisor: Dr. Katrina Cornish
Dr. John Lannutti
Dr. Frederick Michel Jr.
Dr. Alfred Soboyejo

ii
Copyrighted by
Jessica Lauren Slutzky
2019

ii
Abstract
Bio-based polymeric materials are of great commercial interest to attain
environmental sustainability. Natural rubber (cis-1,4 polyisoprene) (NR) is a commodity
that is extensively used in industrial, consumer, and medical industries. Over 5,000
plants produce natural rubber, but over 90% of the world’s supply of natural rubber is
extracted from one plant species: the Brazilian rubber tree, Hevea brasiliensis. Hevea
natural rubber (NR) has a high molecular weight, and can be produced in yields sufficient
to meet market demands. NR contains a high amount of allergic proteins that can cause
severe allergic reactions. One alternative sources of NR can be derived from the shrub
Parthenium argentatum, commonly known as guayule. Guayule natural rubber (GNR)
has a high molecular weight and does not contain proteins associated with allergic
reactions, rendering it circumallergenic. However, previous work has shown that GNR
and NR have differences in various properties, preventing GNR from being a direct
substitute for NR in many applications and giving it advantages in others. Therefore, the
proposed work focuses on creating bio-based elastomeric materials by optimizing
vulcanization chemistries, as well as creating composites using fillers derived from waste
streams.
Vulcanization chemistries of circumallergenic GNR and hypoallergenic NR
(made by removal of soluble proteins which reduce allergic potential) for thin film
applications was optimized using the chemical accelerators diisopropyl xanthogen
polysulphide (DIXP) and zinc diisononyl dithiocarbamate (ZDNC). DIXP and ZDNC
do not induce Type IV allergic reactions such as contact dermatitis, a unique benefit in

iii
comparison to other rubber chemical accelerators. The thin films were manufactured
from natural rubber latex using traditional coagulation dipping methods onto stainless
steel formers. The effects of chemical accelerator concentration on mechanical,
rheological, morphological, and thermal properties of circumallergenic GNR and
hypoallergenic NR thin films were investigated. Many of the GNR and NR thin films
possessed mechanical properties superior to ASTM standards for surgical gloves and
condoms. Multivariate models of mechanical properties as a function of film thickness
and chemical accelerator concentration were generated to quantify differences between
GNR and NR thin films. Data analytic methods such as canonical correlation analysis
further quantified differences in mechanical properties between GNR and NR thin films,
with GNR having greater elongation at break than NR, but NR having a higher Young’s
Modulus and strength at break than GNR vulcanized films. Thicker films for both NR
and GNR showed an increase in Young’s Modulus and strength at break. Scanning
electron microscopic (SEM) analysis of GNR and NR thin films showed that smooth thin
films without void spaces or defects were created. The glass transition temperatures and
thermal degradation curves of GNR and NR thin films were determined to quantify
differences in vulcanization, with GNR having a lower glass transition temperature than
NR vulcanized thin films. By comparing vulcanization chemistries of GNR and NR thin
films, differences in thin film properties attributed to species origin can be quantified.
Commercial thin film products made from NR often contain fillers from non-
renewable resources to improve mechanical properties and thermal stability. Fillers from
agricultural and industrial waste streams were compounded into thin films, using
traditional coagulation dipping methods. Fillers included guayule bark bagasse, carbon

iv
fly ash, and calcium carbonate derived from eggshells, utilized at various particle sizes
and loadings. The chemical accelerators used in these composites include zinc
diethyldithiocarbamate (ZDEC), diphenyl guanidine (DPG), and dipentamethylene
thiuram polysulfide (DPTT), which are traditional chemical accelerators associated with
increased contact dermatitis risk, but create vulcanized thin films with superior stability
compared to DIXP and ZDNC accelerators. The vulcanization chemistries in these films
were not optimized in order to determine the sole effect of fillers on GNR and NR thin
film properties. In addition, NR latex with and without soluble protein was utilized to
determine how protein content impacts film properties. Due to the increased allergic
potential of these films in comparison to those manufactured in the first method, these
films have applications as industrial coatings and should not be implemented in medical
or consumer applications.
Reinforcement of NR and GNR compounds were achieved using fillers that were
nano sized, especially at loadings below 2 parts per hundred rubber (phr) of carbon fly
ash. Adding fillers to GNR typically caused increased elongation at break, whereas NR
had a decreased elongation at break. The differences in bulk mechanical properties of
NR and GNR compounds with fillers can be attributed to variances in the polymer-filler
interaction; non-rubber components such as proteins and phospholipids vary between
GNR and NR and can affect surface activity of a filler. Variances in bulk mechanical
properties of GNR due to different fillers are attributed to properties of the filler,
including particle structure, size, bulk density, alkalinity, and surface activity. Particles
of larger sizes, such as 300 microns, can provide texture to NR and GNR thin films,
which could be utilized for the commercialization of industrial non-slip surfaces. These

v
results can assist in successful commercialization of GNR, and create more sustainable
NR and GNR composites.

vi
Dedicated to my family.

vii
Acknowledgements
I would like to thank my committee members Drs. Katrina Cornish, John
Lannutti, Frederick Michel, and Alfred Soboyejo. Their guidance and support made this
dissertation possible. I would like to express my utmost gratitude for Dr. Katrina Cornish
and the department of Food, Agricultural and Biological Engineering at Ohio State.
Thank you to my high school math teacher, Ferd Schneider, for encouraging me to pursue
engineering. In addition, I would like to thank Dr. Louis Chicoine of Nationwide
Children’s Hospital for my first research job. I would also like to thank John Shepherd,
who funded my undergraduate scholarship. Above all, I want to thank my family.
I would like to thank Ohio Agricultural Research and Development Center
(OARDC) and the Institute for Materials Research (IMR) for the funding of this project.
I would also like to thank the University of Akron Research Foundation for their
mentorship in entrepreneurship.

viii
Vita
June 2006……………………………………Graduated from Walnut Hills High School
Cincinnati, Ohio
June 2011……………………………………B.S., Food, Agricultural & Biological
Engineering, The Ohio State University
June 2011……………………………………B.S., Psychology, The Ohio State University
September 2011-August 2014………………Ohio Agricultural Research and
Development Center (OARDC), Graduate
Research Associate, Doctoral Student, The
Ohio State University
August 2014-May 2015…………………….OARDC, Graduate Research Associate,
Charles Thorne Memorial Associateship,
Doctoral Candidate, The Ohio State
University
May 2015- March 2018……………………..Research Scientist, Battelle Memorial
Institute, Columbus, Ohio
July 2018- present…………………………..Research Scientist, Checkerspot, Berkeley,
California

ix
Publications
Cornish, K., Bates, G.M., Slutzky J.L., Meleshcuk A., Xie W., Sellers K., Mathias R., Boyd M., Castaneda
R., Wright M., Borel L., 2019. Extractable protein levels in latex products, and their associated
risks, emphasizing American dentistry. Biology and Medicine. 11:2 (7 pages). DOI:
10.4172/0974-8369.1000456.
Slutzky J.L., Baral N., Shah A., Ezeji T., Cornish K., Christy A., 2016. Acetone-Butanol-Ethanol
Fermentation of Corn Stover: Current Production Methods, Economic Viability, and Commercial
Use, FEMS Microbiology Letters. 363:6.
Chen B., Xue J., Meng X., Slutzky J.L., Calvert A.E., Chicoine L.G., 2014. Resveratrol prevents hypoxia-
induced arginase II expression and proliferation of human pulmonary artery smooth muscle cells
via Akt-dependent signaling, American Journal of Physiology - Lung Cellular and Molecular
Physiology. 307:L317-L325.
Fields of Study
Major Field: Food, Agricultural & Biological Engineering

x
Table of Contents
Abstract………………………………………………………………………………........ii
Acknowledgments…………………………………………………………………....….vii
Vita……………………………………………………………………………………...viii
List of Tables……………………………………………………………………..…….xiii
List of Figures……………………………………………………………………………xv
Chapter 1: Introduction…………………………………………………………...……….1
Chapter 2: Statement of Problem………………………………………………………….3
Chapter 3: Literature Review…………………………………………………………...…6
3.1 Introduction to Polymers…………………………………………………..6
3.2 Introduction to Elastomers……………………………………………....15
3.3 Specific Elastomer Structure and Properties……………………………31
3.4 Elastomer Compounding………………………………………….……..61
3.5 Manufacturing Methods for Latex……………………………………….79
3.6 Manufacturing Methods for Rubber……………………………………..82
Chapter 4: Mechanical properties of Type I circumallergenic & Type IV hypoallergenic
guayule natural rubber latex thin films…………………………………………………..86
4.1 Introduction………………………………………………………………87
4.2 Experimental…………………………………………………………….91
4.3 Results and Discussion…………………………………………….…….94
4.4 Conclusion…………………..………………………………………….103
Chapter 5: Mechanical and thermal properties of type I & type IV hypoallergenic Hevea
natural rubber latex thin films.………………………………………………………..107

xi
5.1 Introduction……………………………………………………………..108
5.2 Experimental……………………………………………………….…..112
5.3 Results and Discussion…………………………………………………116
5.4 Conclusions……………………………………………………………..126
Chapter 6: Canonical correlation analysis of type I and type IV circumallergenic guayule
natural rubber thin films and type I and type IV hypoallergenic ultra-low protein Hevea
natural rubber thin films.………………………………………………………………..128
6.1 Introduction……………………………………………………………..129
6.2 Experimental………………………………………………………..….133
6.3 Results and Discussion………………………………………….…..….139
6.4 Conclusions……………………………………………………………..143
Chapter 7: Mechanical properties of guayule natural rubber latex thin film composites
with biobased fillers.………………………………………………………………...….145
7.1 Introduction………………………………………………………….….146
7.2 Experimental…………………………………………………………...150
7.3 Results and Discussion……………………………………………...….154
7.4 Conclusions…………………………………………………………….161
Chapter 8: Mechanical properties of Hevea natural rubber latex thin film composites with
biobased fillers.………………………………………………………………………...162
8.1 Introduction………………………………………………………….…163
8.2 Experimental………………………………………………………..…165
8.3 Results and Discussion…………………………………………………171
8.4 Conclusions……………………………………………………………180

xii
Chapter 9: Conclusion.…………………………………………………………………181
Chapter 10: Future Studies…………………………………………………………..…183
References………………………………………………………………………………184

xiii
List of Tables
Table 3. 1. Common elastomers……………..……….……………………………...….26
Table 4. 1. Latex Compound Recipe……………………………………………………91
Table 4. 2. Dwell time and average thin film thickness, SEM < 0.01 mm……………....94
Table 4. 3. Tensile data for GNRL thin films, which will be used for subsequent thermal
analysis via DMA, DSC, and TGA……………………………………………………101
Table 4. 4. Glass transition temperatures obtained from DMA for GNRL thin films....100
Table 4. 5. Glass transition temperatures obtained from DSC for GNRL thin films.....101
Table 4. 6. Thermal degradation temperatures for GNRL thin films……………..…...102
Table 5. 1. Latex Compound Recipe…………………………………………………..112
Table 5. 2. Dwell time and average thin film thickness……………………………..…116
Table 5. 3. Tensile data for ultra-low protein Hevea NRL thin films, which will be used
for subsequent thermal analysis via DMA, DSC, and TGA……………………..……..120
Table 5. 4. Glass transition temperatures obtained from DMA for ultra-low protein
Hevea NRL thin films………………………………………………………………..…122
Table 5. 5. Glass transition temperatures obtained from DSC for ultra-low protein Hevea
NRL thin films…………………………………………………………………………123
Table 5. 6. Thermogravimetric data for ultra-low protein Hevea NRL thin films……125
Table 6. 1. Latex Compound Recipe for Guayule Natural Rubber Latex………….….133
Table 6. 2. Latex Compounding Recipe for Ultra-low Protein Hevea NRL…….…….133
Table 6. 3. Dwell time and average thin film thickness……………………………..…135
Table 6. 4. Notation of different experimental data used for statistical analysis……....136

xiv
Table 6. 5. Mixed linear and non-linear stochastic multivariate regression models using
film thickness (mm) as the significant predictor…………………………….………….139
Table 6. 6. Mixed linear and non-linear stochastic multivariate regression models using
DIXP (phr) as the significant predictor…………………………………………….…...140
Table 6. 7. Mixed linear and non-linear stochastic multivariate regression models using
ZDNC (PHR) as the significant predictor………………………………………………140
Table 6. 8. Canonical correlation values for multivariate models of tensile properties.142
Table 7. 1. Latex Compound Recipe…………..………………………………………151
Table 8. 1. Filler bulk densities………………………………..………………………167
Table 8. 2. Latex Compound Recipe……………………………...…………………..168

xv
List of Figures
Fig. 3. 1: Macromolecular structure of amorphous (A), crystalline (B), and semi-
crystalline polymers (C)………………………………………………………………..….7
Fig. 3. 2: Free volume of a polymer as a function of temperature………….………..…...8
Fig. 3. 3: Stress-strain curve of polymeric materials……..………………………..…….12
Fig. 3. 4: Polymer crazing…..…………………………………………………………...12
Fig. 3. 5: SEM of fracture surfaces for a brittle fracture of natural rubber (A), and ductile
fracture in a natural rubber-thermoplastic blend (B) …………………………...……….14
Fig. 3. 6: Stress-strain curve of elastomer thermosets with increasing crosslink density.16
Fig. 3. 7: Ideally elastic polymers represented by Hooke’s Law……………..................18
Fig. 3. 8: Kelvin-Voight model of viscoelastic materials……….……………………....21
Fig. 3. 9: Creep deformation of polymers………………………………………...….….21
Fig. 3. 10: Loading curve and associated hysteresis……………………..……………...23
Fig. 3. 11: Typical stress relaxation curve of viscoelastic materials………...………..…24
Fig. 3. 12: Polymer chemical structure, consisting of carbon backbone and pendant
groups, such as methyl and aromatic rings………………………………………………25
Fig. 3. 13: Polymerization of polybutadiene rubber……………………………..……....33
Fig. 3. 14: Polymerization of polyisobutylene rubber………………………………..….31
Fig. 3. 15: Structure of isobutylene-isoprene rubber, or butyl rubber…………………...35
Fig. 3. 16: Structure of ethylene propylene diene terpolymer (EPDM)………….….…..36
Fig. 3. 17: Structure of styrene butadiene rubber (SBR)……………………………..….37
Fig. 3. 18: Polybutadiene linked through the 1-and 4-carbon atoms, and through the 1-
and 2-carbon atoms…………………………………………………………………..…..39

xvi
Fig. 3. 19: Structure of polychloroprene…………………………….………………..…40
Fig. 3. 20: Structure of acrylonitrile butadiene rubber (NBR)…………………………..42
Fig. 3. 21: Structure of polysulfide Thiokol A…………………………………………..44
Fig. 3. 22: Structure of poly(vinylidene fluoride-co-hexafluoropropylene)……….. …..46
Fig. 3. 23: Structure of polydimethylsiloxane (PDMS)…………………………………47
Fig. 3. 24: Structure of styrenic block copolymer elastomers………………………...…49
Fig. 3. 25: Structure of polyamide thermoplastic elastomers……………………………51
Fig. 3. 26: Chemical reaction for polyurethane synthesis…………………………...…..54
Fig. 3. 27: The mevalonate pathway, which produces isopentenyl pyrophosphate (IPP),
the monomer for cis-1,4-polyisoprene……………………………………………….…..57
Fig. 4. 1. 3D graphs of mechanical properties for guayule thin films…………………..97
Fig. 4. 2. Glass transition temperatures obtained from DMA for GNRL thin films…...100
Fig. 4.3. Differential scanning calorimetry of GNRL formulations……………………101
Fig. 4. 4. Thermogravimetric analysis of GNRL formulations………………………...102
Fig. 5. 1. 3D graphs of mechanical properties for ultra-low protein Hevea NRL thin
films…………………………………………………………………………………….119
Fig. 5. 2. Glass transition temperatures obtained from DMA for ultra-low protein Hevea
NRL thin films…………………………………………………………………………122
Fig. 5. 3. Differential scanning calorimetry of ultra-low protein Hevea NRL
formulations…………………………………………………………………………….124
Fig. 5. 4. Thermogravimetric analysis of ultra-low protein Hevea formulations………125
Fig. 7.1. Mechanical Properties of GNRL with eggshells…………………………...…155
Fig. 7.2. Mechanical Properties of GNRL with guayule bagasse………………………157

xvii
Fig. 7.3. Mechanical Properties of GNRL with carbon fly ash………………………...158
Fig. 8. 1. Macro (solid line) and micro sized (dashed line) fillers’ particle size
distribution……………………………………………………………………………...166
Fig. 8. 2. Particle size distribution of nano sized fillers………………………………..171
Fig. 8. 3. Transmission electron micrographs of nano sized fillers……..……………...172
Fig. 8. 4. Tensile Properties of Eggshell-NRL composites……………………...……..174
Fig. 8. 5. Tensile Properties of Carbon fly ash-NRL composites……………………...176
Fig. 8. 6. Tensile Properties of guayule bagasse-NRL composites…………………….178

1
Chapter 1: Introduction
Thin film elastomers are extensively used in a variety of applications including
industrial coatings, consumer coatings, and medical products such as surgical balloons,
gloves, and condoms (Cornish et al., 2007). Natural rubber (NR) is predominately used
in thin film applications due to its superior stretch and softness compared to synthetic
elastomers such as nitrile. However the proteins found in NR derived from the Brazilian
rubber tree, Hevea brasiliensis, are severely allergenic and associated with Type I IgE-
mediated allergies (Cornish, 2012; Hamilton and Cornish, 2010; Siler et al., 1996). As a
result, synthetic elastomers became widely used in thin film elastomer applications
despite their inferior mechanical properties (Cornish, 2012; Cornish et al., 2007). NR
from other plant sources, such as the shrub Parthenium argentatum, commonly known as
guayule, is of commercial interest partly due to its lack of allergic proteins (Hamilton and
Cornish, 2010; Siler et al., 1996). However, a key step to increasing the commercial
potential of guayule natural rubber (GNR) includes rigorous material characterization to
determine its differences to NR and define niche applications where GNR is the most
ideal material to use.
Key differences between NR and GNR include polymer macromolecular structure
and biochemical composition (Monadjemi et al., 2016). Macromolecular structural
differences between NR and GNR are attributed to differences in plant species
metabolism and biosynthesis of isoprene units, which are still undefined (Puskas et al.,
2014). Biochemical components entrapped during the extraction of GNR and NR include
proteins, polyphenols, alkaloids, fatty acids, and phospholipids, which vary among plant
species (McMahan et al., 2015). As a result, despite natural elastomers being comprised

2
of cis-1,4-polyisoprene, there are vast differences in structure and chemical composition
which have a subsequent impact upon polymer compounding, manufacture, and final
product properties (Monadjemi et al., 2016).
Therefore, the engineering and characterization of GNR must be compared to NR
in order to define material differences attributed to species origin. GNR is known to have
a more linear polymer structure compared to NR (Hager et al., 1979). The lack of
polymer branching in GNR decreases molecular entanglements compared to NR. This
allows for greater chain slippage and higher ultimate elongations. Since GNR has fewer
chain entanglements than NR, crosslinking optimization is different between GNR and
NR to avoid brittle fracture. In addition, biochemical components entrapped in aqueous
natural rubber can interfere, as well as improve crosslink formation (Cornish et al., 2007).
For example, proteins act as a natural surfactant and can improve the incorporation of
hydrophilic compounding chemicals into the hydrophobic, hydrocarbon structure of cis-
1,4-polyisoprene (McMahan et al, 2015). Other biochemical components such as
phospholipids can initiate stress-induced crystallization behavior of GNR and NR, which
subsequently impacts tensile strength (Cornish, 2001; Steinbuchel, 2003). Therefore, it is
imperative to optimize GNR and NR independently for a given manufacturing process
due to their vast differences, despite both being comprised of high molecular weight cis-
1,4-polyisoprene.

3
Chapter 2: Statement of the Problem
Current global demand for elastomers exceeds the natural rubber supply, which is
supplemented by synthetic elastomers derived from petroleum (Cornish, 2014). Global
demand for elastomers will continue to grow, especially in emerging markets such as
India and China as consumers in those markets gain purchasing power (Cornish, 2014).
Currently, over 90% of the global natural rubber supply is derived from Hevea
brasiliensis, the Brazilian rubber tree, which is predominately grown in tropical regions
of Southeast Asia, thus requiring extensive exports for rubber products in North America
and Europe (van Beilen and Poirier, 2007). In addition, Hevea natural rubber (NR) and
natural rubber latex (NRL) is capable of eliciting severe type I allergic responses,
attributed to allergic proteins entrapped during the extraction process (Cornish et al.,
1999). Type I allergies to NR and NRL proteins have caused great concern in the
medical industry, prompting bans of NR and NRL products in many hospitals. Current
commercial alternatives to NR and NRL include nitrile and other synthetic elastomers,
which often have inferior mechanical properties compared to NR and NRL (van Jole,
2007). As a result, alternative, renewable materials to alleviate type I sensitization and
subsequent allergic reaction need to be developed to promote consumer safety and
environmental sustainability.
The hypothesis of this work focuses on finding applications of guayule natural
rubber latex (GNRL) and hypoallergenic NRL for consumer and medical industries by
optimizing chemical compounding and manufacturing techniques, while providing a
comparative analysis to NRL products. Understanding the differences between GNRL,
hypoallergenic NRL, and traditional NRL will provide insight into differences in

4
intermolecular interactions between rubber molecules and cross-linking chemicals, as
well as solid fillers. Once these latex chemical formulations are optimized, they can be
processed via coagulated dipping to create thin film elastomer finished products.
Examples of finished products include surgical balloons, medical gloves, condoms,
catheters, dental dams, and biocompatible coatings.
Fillers are often used in compounding of natural rubber latex, to make natural
rubber latex composites. Fillers can provide a reinforcing effect, improving mechanical
properties. This work will also focus on evaluating waste-derived agro-industrial
residues for natural rubber latex composites, for both guayule and Hevea.
Characterization of mechanical properties of NRL films attributed to type of filler,
particle size, and loading will be assessed in different natural rubber lattices.
The overall goal of this work is to promote natural rubber latex products, through
improved mechanical properties attributed to cross-link optimization, and the use of new
fillers for natural rubber latex composites. This goal will be accomplished via the
following objectives:
• Objective 1: Understand the physico-chemical behavior changes of vulcanized
GNRL, hypoallergenic NRL, and stabilized NRL containing soluble protein,
using the chemical accelerators diisopropyl xanthogen polysulfide (DIXP) and
diisonyl dithiocarbamate (ZDNC), therefore eliminating a type IV, contact
dermatitis allergy.
• Objective 2: Optimize cross-linking and natural rubber latex mechanical
properties using mathematical modeling.

5
• Objective 3: Engineer new natural rubber latex composites using low-cost,
waste-derived material.

6
Chapter 3: Literature review
3.1. Introduction to Polymers
A polymer is molecule containing chemically bonded repetitive units, termed
monomers. Polymeric materials have molar masses in excess of 103 g/mol, and are larger
than 1 nm in size (Young and Lovell, 1991). The macromolecular morphology of
polymers varies due to chemical composition and secondary bonding, with the resulting
three-dimensional aggregate macromolecular structure dictating physical properties
(Young and Lovell, 1991). Polymer molecules generally pack together in a non-uniform
fashion, creating a combination of ordered, crystalline regions mixed with disordered,
amorphous regions (Figure 3.1). Crystallinity occurs when linear polymer chains are
structurally oriented in a uniform three-dimensional matrix, with polymer chains
extending out from crystalline domains into amorphous regions where they are coiled and
tangled (Young and Lovell, 1991). The extent, or degree, of crystallinity in a polymer
depends on polymer chain length, branching, and secondary bonding (such as hydrogen
bonding, and dipole-dipole interactions) (Young and Lovell, 1991). Some polymers lack
crystalline regions, and are completely amorphous.

7
Fig. 3. 1: Macromolecular structure of amorphous (A), crystalline (B), and semi-
crystalline polymers (C)
3.1.1. Thermal Transitions of Polymers
Material specific thermal transitions such as the glass transition temperature (Tg)
and the melting temperature (Tm) provide insight into polymer morphology. Tm is the
temperature at which crystalline domains lose their structure and melt (Young and
Lovell, 1991). Tm is indicative of the degree of crystallinity, as well as the type of crystal
structure in a polymer (Young and Lovell, 1991). Tg is the temperature above which
amorphous domains of a polymer are capable of segmental chain motion; below the Tg a
polymer is capable of translational motion only and behaves like a brittle, rigid glass
(Young and Lovell, 1991). At temperatures above the Tg, the polymer is capable of
segmental and translational motion, resulting in viscoelastic rubbery behavior (Young
and Lovell, 1991). Tg varies among polymers due to differences in the small amount of
unfilled volume associated with the end of a polymer chain, termed free volume (Young
8
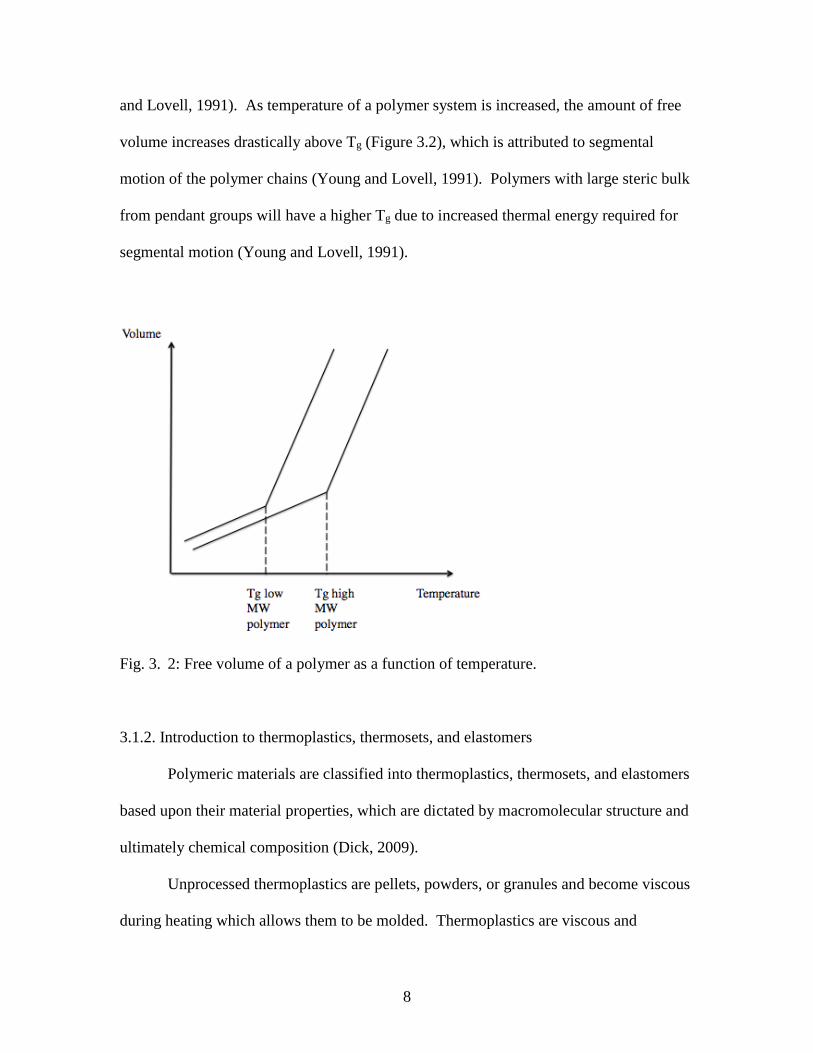
and Lovell, 1991). As temperature of a polymer system is increased, the amount of free
volume increases drastically above Tg (Figure 3.2), which is attributed to segmental
motion of the polymer chains (Young and Lovell, 1991). Polymers with large steric bulk
from pendant groups will have a higher Tg due to increased thermal energy required for
segmental motion (Young and Lovell, 1991).
Fig. 3. 2: Free volume of a polymer as a function of temperature.
3.1.2. Introduction to thermoplastics, thermosets, and elastomers
Polymeric materials are classified into thermoplastics, thermosets, and elastomers
based upon their material properties, which are dictated by macromolecular structure and
ultimately chemical composition (Dick, 2009).
Unprocessed thermoplastics are pellets, powders, or granules and become viscous
during heating which allows them to be molded. Thermoplastics are viscous and

9
moldable above a specified Tm, and solidify when cooled below the Tm. Thermoplastics
have a Tm and a Tg that are attributed to the properties of their crystalline and amorphous
regions, respectively (Young and Lovell, 1991). For example, thermoplastics with a
higher degree of crystallinity have a higher Tm, requiring more thermal energy to melt
their crystalline regions (Young and Lovell, 1991). Thermoplastics with longer chain
lengths, more extensive chain branching, and stronger secondary bonding have a higher
Tg due to segmental chain motion being hindered, requiring more energy (Young and
Lovell, 1991).
Thermosets are polymers that are chemically crosslinked to create intermolecular
and intramolecular bonds, and therefore lack the ability to be reshaped at any temperature
(Dick, 2009). The crosslinks in thermosets prevent chain slippage, which improve bulk
mechanical properties (Dick, 2009). The type of crosslinker used in a thermoset depends
on the type of active sites found in a polymer, such as unsaturated carbons or epoxy
groups (Dick, 2009). Differences in chemical composition among thermosets can provide
differences in morphology as well: some thermosets have crystalline regions, or are
completely amorphous in structure. Thermosets that have crystalline regions have
distinct Tm and Tg, whereas amorphous thermosets only have a Tg (Young and Lovell,
1991). Thermosets with crystalline regions, termed thermoset resins, are typically liquid
chemicals with a low molecular weight and low viscosity that are polymerized into long
chains and high molecular weight molecules with a high viscosity which are subsequently
crosslinked to stiffen the thermoset (Young and Lovell, 1991). Thermosets that are
completely amorphous in structure are typically large molecular weight molecules
previously polymerized, and then subsequently crosslinked into a thermoset (Young and

10
Lovell, 1991). In both thermoset resins and amorphous thermosets, the finished
thermoset product has a three-dimensional network of chemical crosslinks with better
mechanical properties than thermoplastics (Dick, 2009).
Elastomers are characterized by their ability to be stretched and return to its
original shape without permanent deformation. Elastomers can be thermoplastics, or
lightly crosslinked thermosets. Elastomers are typically able to undergo reversible strain
at temperatures above Tg due to external forces producing intramolecular conformational
changes, or long-range segmental motion, in amorphous regions (Dick, 2009).
3.1.3. Mechanical Properties of Polymers
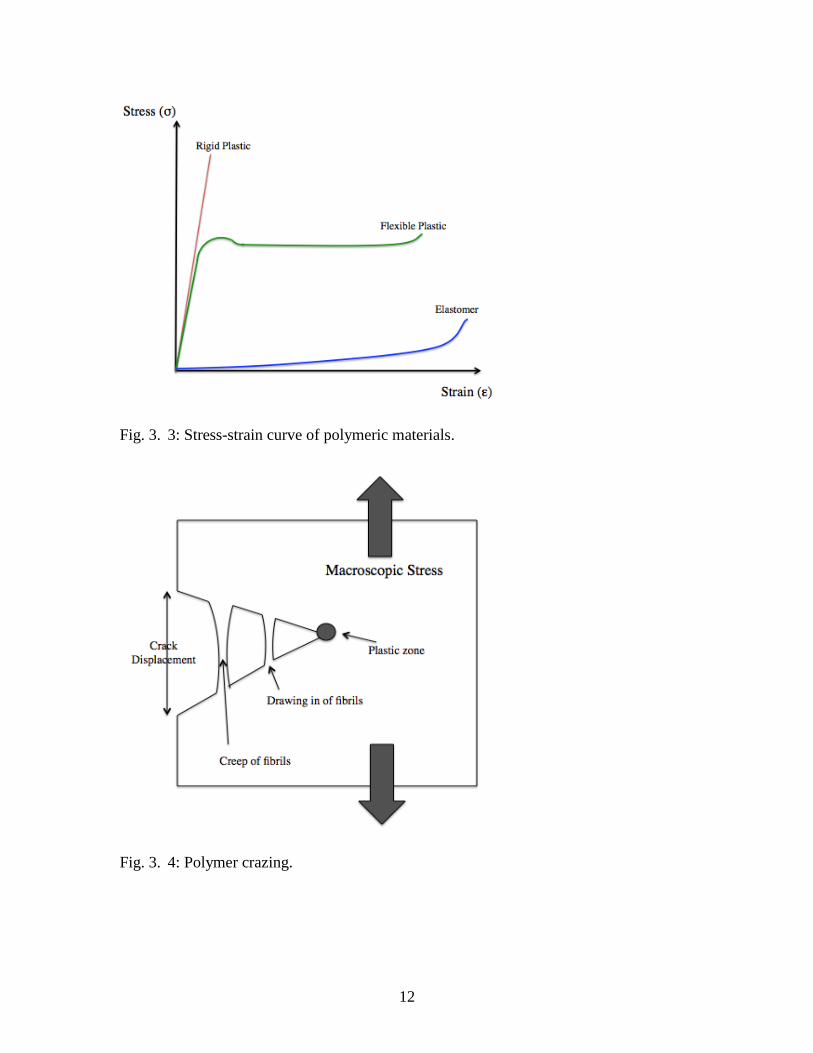
Stress-strain curves of macromolecular materials further distinguish
thermoplastics, thermosets, and elastomers (Fig. 3.3). When subjected to a uniaxial load,
thermoplastics deform reversibly (elastically) until a maximum stress, the yield point, is
reached. Thermoplastics stressed beyond the yield point deform irreversibly, until the
material ultimately fractures (Young and Lovell, 1991). Thermoplastics are capable of
undergoing both ductile and brittle fracture, depending on the conditions of the
mechanical stress (Young and Lovell, 1991). Thermoplastics stressed in reduced
temperatures, with increased strain rates, sharp notches, and increased thickness are more
susceptible to brittle fracture (Young and Lovell, 1991).
Polymers can undergo yielding mechanisms, such as shear yielding and crazing
while undergoing mechanical stresses (Young and Lovell, 1991). Crazing is
characterized by polymer chains rearranging in highly stressed regions under uniaxial
loads, creating localized plastic deformations with microvoids connected by polymer
fibrilliar bridges (Fig. 3.4) (Young and Lovell, 1991). These polymer fibrilliar bridges

11
eventually coalesce to form a crack, which can ultimately lead to mechanical failure.
Shear yielding is a yielding mechanism that occurs parallel to the direction of the force,
whereas crazing occurs normal to the direction of the force.
Crazing in thermoplastics is attributed to changes in physical and chemical
bonding (Calhoun and Peacock, 2006). Mechanical stress can cause the weak physical
Van der Waals forces between polymer chains to separate, creating a microscopic void
space (Calhoun and Peacock, 2006). The covalent bonds in the polymer backbone chain
compensate for the mechanical stress, reducing the polymer chain length and increase the
void space initiated by disruption in the Van der Waals forces (Young and Lovell, 1991).
These void spaces are bridged by fibrils a few nanometers in diameter, which are
molecules of the stretched polymer chain. Crazing occurs internally in a polymeric
material, absorbing fracture energy and therefore improving the fracture toughness of the
material (Young and Lovell, 1991). Crazes typically form in regions associated with
flaws, and most frequently occur in amorphous, brittle polymers (Young and Lovell,
1991). Crazing can ultimately lead to crack propagation, and these cracks may grow until
bulk mechanical failure occurs (Grellman and Seidler, 2001).
12
Fig. 3. 3: Stress-strain curve of polymeric materials.
Fig. 3. 4: Polymer crazing.

13
Polymers can undergo brittle or ductile fracture, depending on the failure
conditions. Polymers favor brittle fracture with reduced temperatures, increased strain
rates, geometric notches, and increased thickness (Grellman and Seidler, 2001). Brittle
fracture in polymers occurs during high strain rate impacts, and is characterized by a
smooth fracture surface (Grellman and Seidler, 2001). Polymers that undergo ductile
failure display necking and permanent deformation, which is irreversible (Grellman and
Seidler, 2001). Necking in ductile fracture is attributed to polymeric molecular chains
unfolding and re-aligning in the direction of the applied stress (Grellman and Seidler,
2001). Amorphous polymers undergo molecular uncoiling, followed by polymer chains
alignment, causing the neck to propagate until polymer chain scission(Grellman and
Seidler, 2001). Crystalline polymers undergo chain unfolding in the amorphous regions
between the lamellae of the crystals, followed by crystal fracture and re-alignment of
crystalline regions(Grellman and Seidler, 2001). Amorphous polymers that are
crosslinked into thermosets ultimately undergo brittle fracture, attributed to the covalent
bonds of crosslinks being severed (Grellman and Seidler, 2001). The fracture
mechanisms vary between elastomers depending on the respective microarchitecture of
the material, which is a result of the aggregate structures based on chemical composition.

14
Fig. 3. 5: SEM of fracture surfaces for a brittle fracture of natural rubber (A), and ductile
fracture in a natural rubber-thermoplastic blend (B) (Grellman and Seidler, 2001).

15
3.2. Introduction to Elastomers
Elastomers are materials that can exhibit a rapid and large reversible strain in
response to mechanical stress, and therefore have high resiliency (Dick, 2009). Elastic
strain can be attributed to chemical bond stretching, bond angle deformation, or crystal
structure deformation (Dick, 2009). Elastomers are classified as thermoplastics with
semi-crystalline regions, or amorphous polymers that utilize crosslinking to become
thermosets (Dick, 2009). Amorphous regions of elastomers contain predominately
amorphous structures oriented into random coils. However, there are crystalline regions
in thermoplastic semi-crystalline polymers, and amorphous regions of elastomers can
phase transform into strain-induced crystals (i.e., strain-induced crystallization) (Dick,
2009). Within amorphous regions, unstrained elastomers exist in a random, coiled
structure. As strain is applied, mechanical energy is dissipated by the re-orientation of
molecular chains into an uncoiled, aligned, crystalline structures within amorphous
regions (Grellman and Seidler, 2001). Mechanical failure of elastomers occurs when
chains are completely uncoiled and chemical bonds begin to break typically in very high
strains of 750%-2000%, dependent upon the chemical composition and three-dimensional
structure of the elastomer (Dick, 2009). Crosslinking of elastomers can hinder polymer
chain uncoiling under strain by restricting macromolecular translational movement, thus
improving mechanical properties and elastomer utility (Figure 3.6) (Dick, 2009).
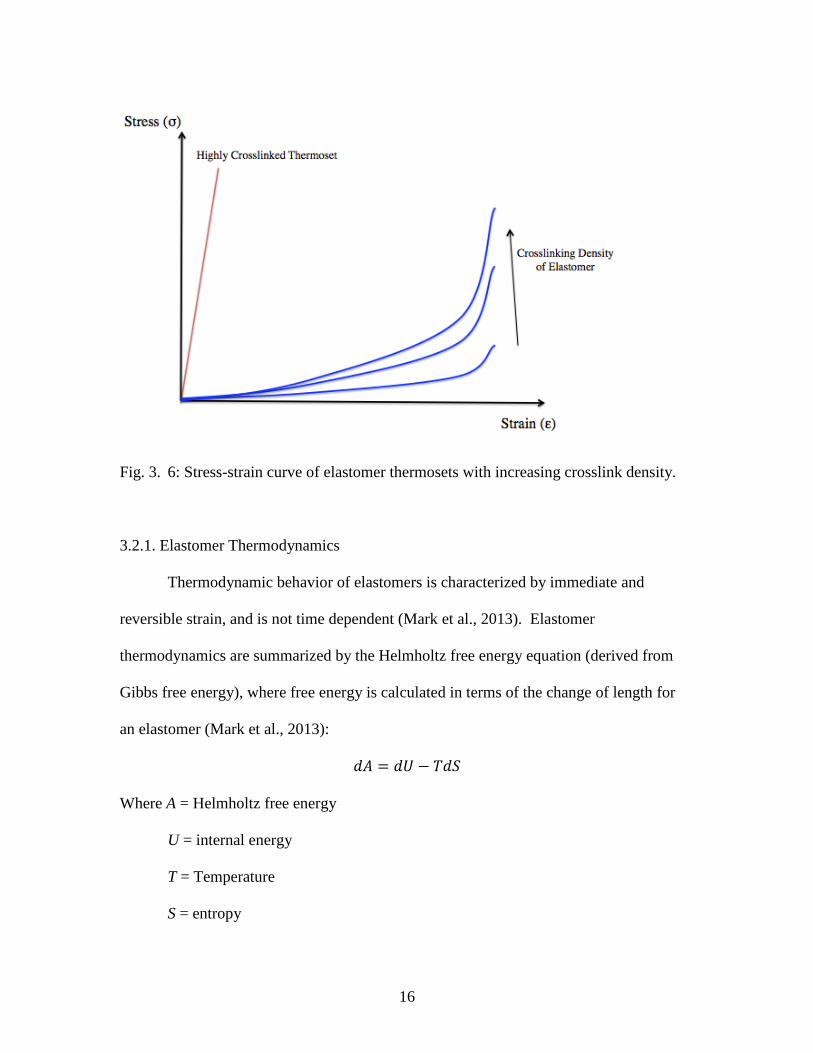
16
Fig. 3. 6: Stress-strain curve of elastomer thermosets with increasing crosslink density.
3.2.1. Elastomer Thermodynamics
Thermodynamic behavior of elastomers is characterized by immediate and
reversible strain, and is not time dependent (Mark et al., 2013). Elastomer
thermodynamics are summarized by the Helmholtz free energy equation (derived from
Gibbs free energy), where free energy is calculated in terms of the change of length for
an elastomer (Mark et al., 2013):
𝑑𝐴 = 𝑑𝑈 − 𝑇𝑑𝑆
Where A = Helmholtz free energy
U = internal energy
T = Temperature
S = entropy

17
Entropy of an elastomeric system is determined by macromolecular
conformations. Entropy is maximized when the elastomer is contracted, due to the
infinite macromolecular conformations due to dynamic flexibility (changes in spatial
orientation while in equilibrium) (Mark et al., 2013). In contrast, when an elastomer is
elongated, the entropy of the system is minimized due to the macromolecular structure
becoming more linear, and thus losing conformational freedom (Mark et al., 2013).
When an elastomer mechanically fails, the elastomer has minimized the entropy of the
system (Mark et al., 2013).
Enthalpy, or the internal energy, is dictated by the thermal energy introduced into
a system. If a stretched elastomer is heated, its length will decrease as the system
dissipates thermal energy by increasing coiled macromolecular conformations (Mark et
al., 2013). Similarly, if a stretched elastomer is allowed to contract, its temperature will
decrease (Mark et al., 2013). In contrast, if an elastomer is stretched, its temperature will
increase (Mark et al., 2013).
Ideal, purely elastic polymers obey Hooke’s law (Figure 3.7), equilibrating their
macromolecular structure proportionally to the amount of strain applied (Mark et al.,
2013). Elastic deformations are attributed to polymer chain segmental motions,
dependent upon free volume and are a thermodynamic phenomenon (Mark et al., 2013).
At low strains, elastomers more closely follow Hooke’s law due to impeding uncoiling of
polymer chains attributed to macromolecular entanglement (Mark et al., 2013).
However, elastomers deviate from Hooke’s law at high strain due to the elongation of
polymer chains, crosslinks’ bonds becoming strained, and the phase transformation of

18
amorphous regions into crystalline structures (Mark et al., 2013). Elastomers also deviate
from Hooke’s law under severe compression, attributed to limited free volume (Mark et
al., 2013).
Fig. 3. 7: Ideally elastic polymers represented by Hooke’s Law
The non-ideal behavior of elastomers is attributed to their viscous component
which, in turn, is attributed to permanent or non-reversible deformation caused by
translational motion of the polymer structure while under strain (Mark et al., 2013). This
translational, segmental motion of polymers is dependent upon kinetics and causes
deviations from ideal thermodynamics associated with a polymeric system (Mark et al.,
2013).
3.2.2. Elastomer Kinetics
Viscoelastic materials such as elastomers are non-Newtonian (non-ideal) and
exhibit a temperature dependent, non-linear response to a strain rate (Mark et al., 2013).
Segmental motion of macromolecules is the cause of non-ideal behavior associated with

19
the viscous component of an elastomer (Mark et al., 2013). The Tg of a polymer
determines the thermal energy required for segmental motions in macromolecular
systems, defining the boundary between elastic and visco-elastic behavior (Mark et al.,
2013). When a polymer is below its Tg, segmental motions are not possible and the
polymer is glassy and behaves elastically like an ideal (Bingham) plastic (Mark et al.,
2013). When the temperature of a polymer is above its Tg, long-range segmental motion
occurs and the polymer behaves as an elastomer (Mark et al., 2013). Phenomena such as
creep, hysteresis, and stress relaxation are attributed to the viscous component in
polymers.
3.2.2.1. Creep
Creep is a time dependent increase in deformation under constant stress (Mark et
al., 2013). The amount of creep is dependent upon material properties, exposure time and
temperature, as well as the applied structural load (Mark et al., 2013). In general, if the
stress is removed within a certain time (specified by TA in Fig. 3.9), the strain recovers
partially very rapidly (specified by B in Fig. 3.9) and is termed elastic recovery (Mark et
al., 2013). When a material is exposed to a stress below the material’s elastic limit
beyond a certain time (specified by C in Fig. 3.9), permanent deformation occurs which
is due to the slow viscous component of the polymer (Mark et al., 2013). Creep of
polymers is an important mechanical characteristic to consider when characterizing
dimensional stability.
Creep of polymers can be modeled using the Kelvin-Voight model, represented
by a Hookean spring and a Newtonian dashpot in parallel (Figure 3.8). The creep strain
is given by the following convolution integral (Mark et al., 2013):

20
𝜀(𝑡) = 𝜎𝐶𝑜 + 𝜎𝐶 ∫ 𝑓(𝜏) (1 − 𝑒(−
𝑡𝜏
)) 𝑑𝜏
∞
0
Where:
σ = applied stress
Co = instantaneous creep compliance
C = creep compliance coefficient
τ = retardation time
f(τ) = distribution of retardation times
Viscoelastic materials experience a time-dependent increase in strain when
subjected to a step constant stress. The time-dependent increase in strain varies with
different amounts of stress. The transition from a linear to non-linear viscoelasticity is
characterized by a material specific amount of stress, termed the critical stress (Mark et
al., 2013). Below the critical stress, the viscoelastic material has linear viscoelasticity
whereas above the critical stress the polymer’s creep rate increases non-linearly (Mark et
al., 2013). As constant stress is applied to the system, the material undergoes strain until
the material fails. If the stress is maintained on the material for a short amount of time,
the material behaves elastically. If the stress is held on the material above its critical
value of applied stress, the creep modulus is dependent upon the stress applied (Mark et
al., 2013). A viscoelastic creep modulus-time curve can represent multiple strain versus
time responses for various stress loads that are under the material’s critical stress value.
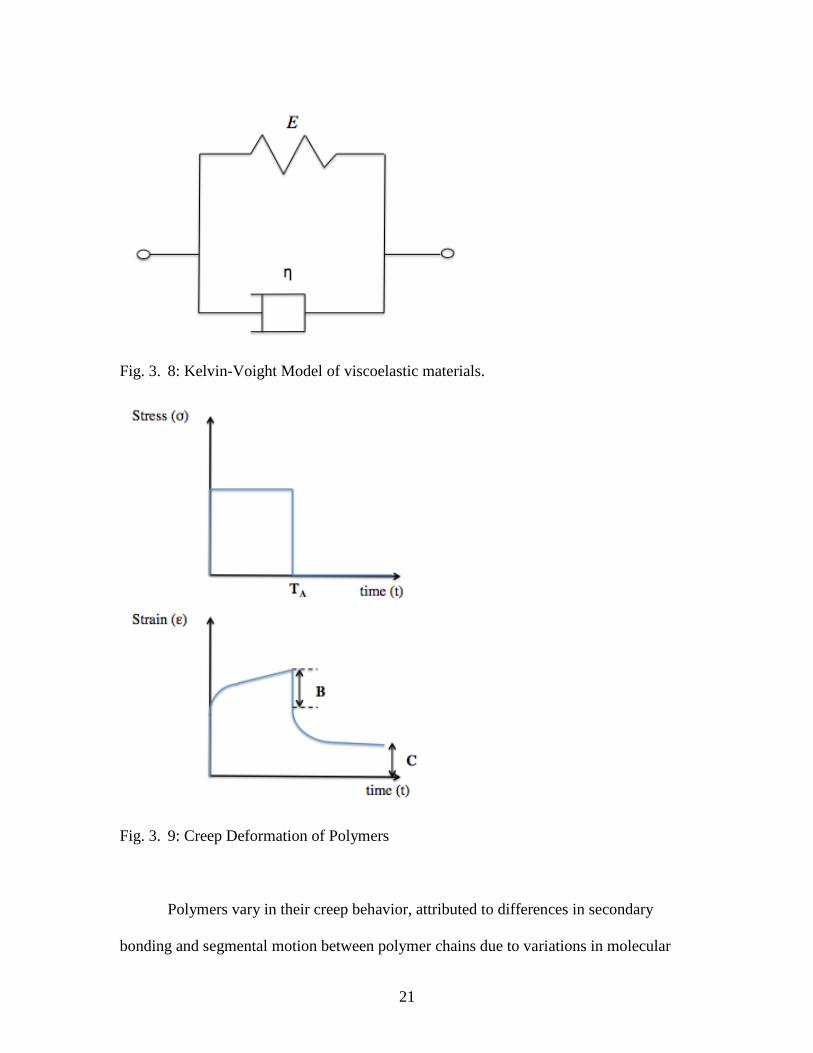
21
Fig. 3. 8: Kelvin-Voight Model of viscoelastic materials.
Fig. 3. 9: Creep Deformation of Polymers
Polymers vary in their creep behavior, attributed to differences in secondary
bonding and segmental motion between polymer chains due to variations in molecular

22
weight and chemical composition. In general, more thermally stable polymers, such as
those with a higher molecular weight or containing aromatic rings, are more creep
resistant (Mark et al., 2013).
3.2.2.2. Elastic hysteresis
Elastic hysteresis is characterized by the deformation of a material due to current
stress and past stresses (Mark et al., 2013). In context of viscoelastic mechanics, force
loading and subsequent unloading has a time-dependent behavior deviant from the ideal
elastic behavior described by Hooke’s law. As forces are loaded onto an elastomer,
energy is dissipated by extension of the elastomer. However, when that same force is
unloaded, less energy is required by the elastomer to retain its original shape and the
excess energy is dissipated as heat (Mark et al., 2013). The thermal energy dissipated by
the system is due to internal macromolecular friction (Mark et al., 2013). As a result,
elastomers are commonly used as stress dampeners due to an elastomer’s ability to
absorb mechanical compression energy and dissipate this energy as heat (Mark et al.,
2013). The large area contained within the hysteresis loop (Figure 3.10) shows that an
elastomer dissipates energy by internal macromolecular friction, compared to stiff
materials that would have a smaller hysteresis loop (Mark et al., 2013).
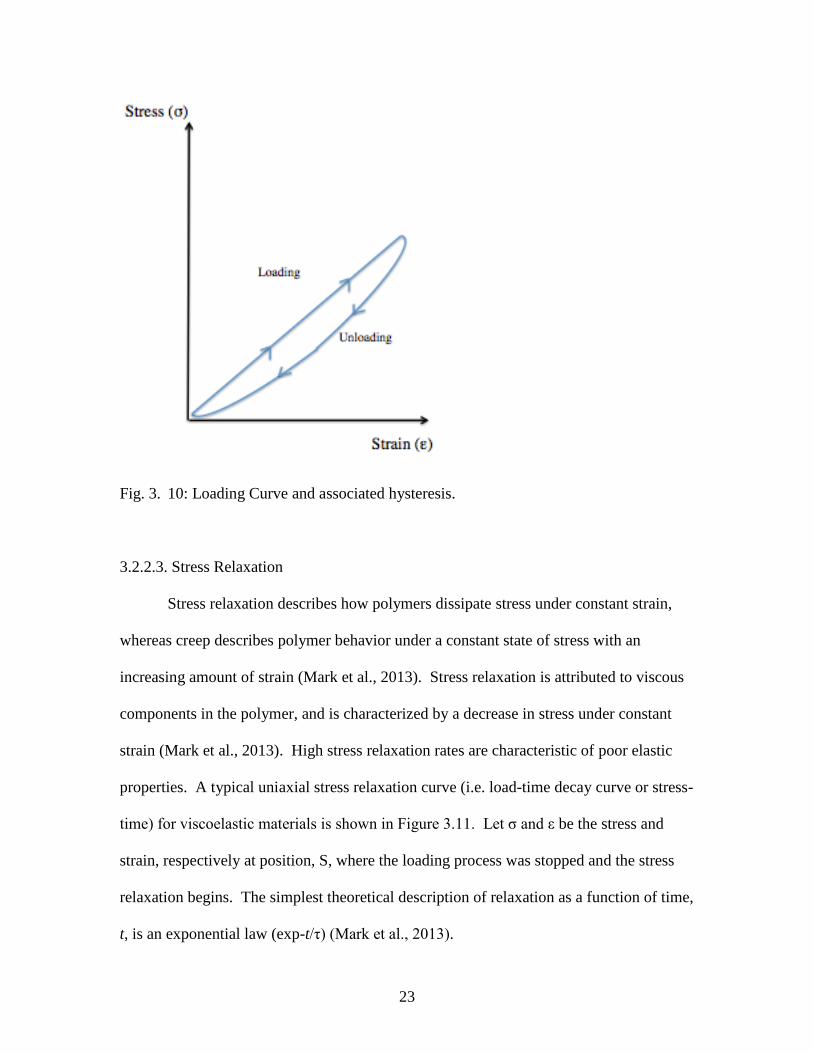
23
Fig. 3. 10: Loading Curve and associated hysteresis.
3.2.2.3. Stress Relaxation
Stress relaxation describes how polymers dissipate stress under constant strain,
whereas creep describes polymer behavior under a constant state of stress with an
increasing amount of strain (Mark et al., 2013). Stress relaxation is attributed to viscous
components in the polymer, and is characterized by a decrease in stress under constant
strain (Mark et al., 2013). High stress relaxation rates are characteristic of poor elastic
properties. A typical uniaxial stress relaxation curve (i.e. load-time decay curve or stress-
time) for viscoelastic materials is shown in Figure 3.11. Let σ and ε be the stress and
strain, respectively at position, S, where the loading process was stopped and the stress
relaxation begins. The simplest theoretical description of relaxation as a function of time,
t, is an exponential law (exp-t/τ) (Mark et al., 2013).
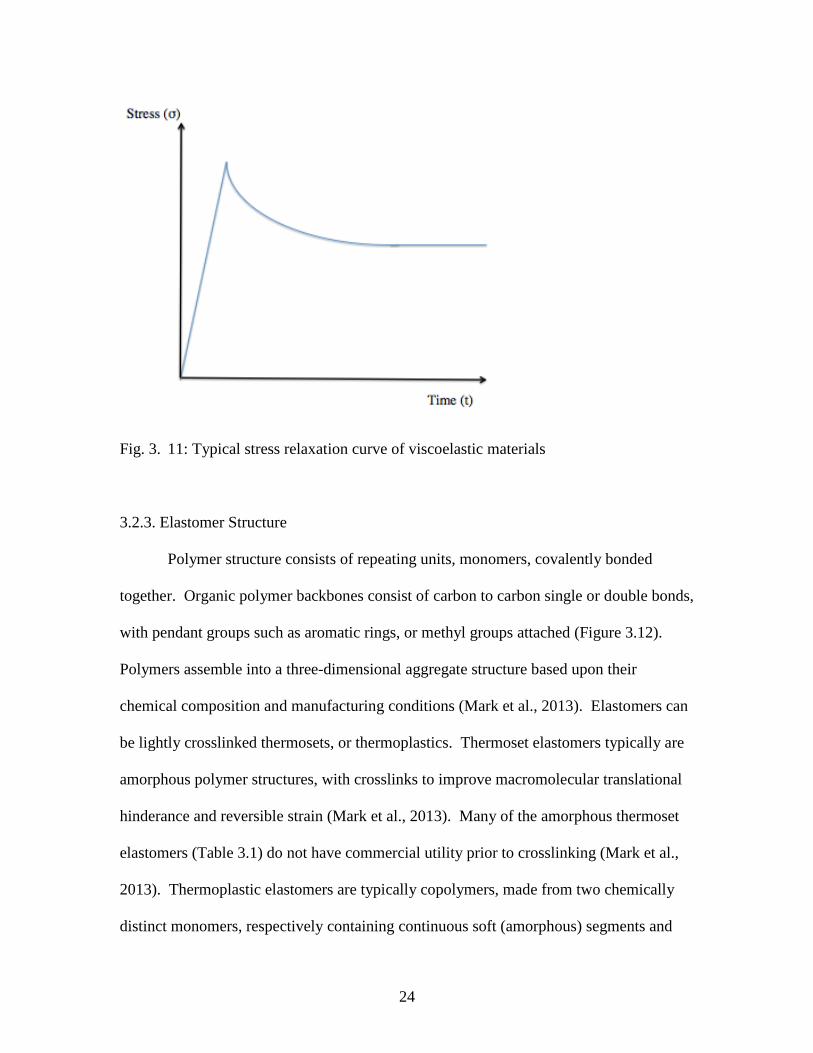
24
Fig. 3. 11: Typical stress relaxation curve of viscoelastic materials
3.2.3. Elastomer Structure
Polymer structure consists of repeating units, monomers, covalently bonded
together. Organic polymer backbones consist of carbon to carbon single or double bonds,
with pendant groups such as aromatic rings, or methyl groups attached (Figure 3.12).
Polymers assemble into a three-dimensional aggregate structure based upon their
chemical composition and manufacturing conditions (Mark et al., 2013). Elastomers can
be lightly crosslinked thermosets, or thermoplastics. Thermoset elastomers typically are
amorphous polymer structures, with crosslinks to improve macromolecular translational
hinderance and reversible strain (Mark et al., 2013). Many of the amorphous thermoset
elastomers (Table 3.1) do not have commercial utility prior to crosslinking (Mark et al.,
2013). Thermoplastic elastomers are typically copolymers, made from two chemically
distinct monomers, respectively containing continuous soft (amorphous) segments and

25
hard (crystalline) segments (Table 3.2). Thermoplastic elastomers typically have ultimate
strains less than thermoset elastomers, due to their crystalline regions not being able to
uncoil to dissipate stress like amorphous regions (Mark et al., 2013). However, unlike
some thermoset elastomers, thermoplastic elastomers can be used in injection and other
molding processes. Thermoplastic elastomers can also be engineered to optimize their
ratio of soft and hard copolymer segments, enabling precise tailoring of properties for
given applications (Mark et al., 2013).
Fig. 3. 12: Polymer chemical structure, consisting of carbon backbone and pendant
groups, such as methyl and aromatic rings.

26
Table 3. 1: Common elastomers (Mark et al., 2013)

27
Elastomers are predominately hydrocarbon molecules that lack polar groups and
do not possess strong intermolecular forces such as hydrogen bonding and dipole-dipole
interactions (Mark et al., 2013). The chemical structure of the amorphous regions in
elastomers is important due to its ability to provide reversible strain through polymer
chain segmental motion under mechanical stress, typical of elastomers (Mark et al.,
2013). The minimal intermolecular forces among polymer chains allow for chain
uncoiling (Mark et al., 2013). Rapid strain of elastomers requires minimal steric
hindrance within the macromolecular structure (Mark et al., 2013). As a result, elastomer
structures often lack large pendant groups in order to allow for rapid uncoiling of
amorphous polymer structures when under strain (Mark et al., 2013). In addition,
crosslinking is required for reversible strain in amorphous elastomers. Crosslinking
inhibits translational motion, contributing to strain reversibility. Ideally, crosslinking to
the extent of a percolation network (a random pathway of crosslinks throughout the bulk
polymer material) provides reversible strain, whereas crosslinking beyond a percolation
network (multiple, dense pathways of crosslinks) can decrease elastomeric performance
and result in brittle fracture (Mark et al., 2013).
3.2.4. Elastomer Polymerization Processes
Natural rubber (NR) is enzymatically synthesized in vivo by rubber producing
plants such as Hevea brasiliensis Muell. Arg. and Parthenium argentatum Gray (Cornish,
2014). All other elastomers are synthesized from petrochemicals that require the
polymerization of monomers, a combination of monomers, or further modification of an
existing polymer (Mark et al., 2013). Synthetic elastomers are homopolymers when
derived from one type of monomer, such as polyisoprene or polybutadiene (Young and

28
Lovell, 1991). Copolymers are comprised of two or more different monomers, and
include elastomers such as styrene-butadiene rubber (SBR) (Mark et al., 2013). There
are four methods for elastomer polymerization: bulk, solution, suspension, and emulsion
(Mark et al., 2013).
3.2.4.1. Bulk Polymerization
Bulk polymerization utilizes a single monomer and a suitable catalyst, and is
heated or pressurized to initiate polymerization. Bulk polymerization produces solid
polymer through polymerization mechanisms such as polycondensation, free radical, or
coordination (Mark et al., 2013). Solid polyurethane elastomers utilize a
polycondensation mechanism, whereas ethylene acrylic elastomers use the free radical
polymerization mechanism (Mark et al., 2013). Ethylene-propylene-diene rubber
(EPDM) uses a coordination polymerization method to create an ethylene-propylene
(EPM) copolymer that is subsequently terpolymerized to create EPDM (Mark et al.,
2013). In general, temperature and molecular weight control is difficult in bulk
polymerization due to the solid state of the polymer and uncontrollable auto acceleration
of the polymerization reaction (Mark et al., 2013).
3.2.4.2. Solution Polymerization
Solution polymerization uses an inert solvent as the medium for the monomers
and catalysts. Catalysts in solution polymerization are soluble, or are finely suspended in
solution. Solution polymerization allows for precise control of the polymerization
reaction, with improved temperature and viscosity control compared to bulk
polymerization (Mark et al., 2013). Catalyst type and concentration can dictate the
molecular weight and structure of the final polymer, and allow some control of the auto

29
acceleration of the polymerization reaction (Mark et al., 2013). Solution polymerization
uses polymerization methods such as polycondensation, free radical, cationic, anionic,
and coordination (Mark et al., 2013). In addition to bulk polymerization methods,
polyurethanes can be synthesized by solution polymerization using a condensation
mechanism (Mark et al., 2013). Ethylene-vinyl acetate rubber (EVM) uses a free radical
solution polymerization method (Mark et al., 2013). Epicholorohydrin rubber (ECO),
and butyl rubber (IIR) use a cationic polymerization method, whereas SBR,
polybutadiene rubber (BR), and polyisoprene rubber (IR) use an anionic polymerization
method (Mark et al., 2013). Coordination mechanisms are used to create BR, IR, EPM,
and EPDM in solution (Mark et al., 2013). Disadvantages of solution polymerization
include removal of excess solvent from the finished polymer (Mark et al., 2013).
3.2.4.3. Suspension Polymerization
Suspension polymerization uses monomers and a monomer soluble catalyst
suspended as monomer/catalyst droplets in water. Suspending agents are used in the
aqueous medium, to prevent coalescence during polymerization. Polymerization occurs
within the monomer droplets, creating larger polymer beads. The final polymer beads are
insoluble in water, and can be retrieved by filtration (Vivaldo-Lima, et al., 1997).
Polymer mechanisms used in suspension polymerization include free radical, cationic,
and coordination (Vivaldo-Lima, et al., 1997). EVM is made by a free radical
mechanism, in solution and suspension (Vivaldo-Lima, et al., 1997). EPM is made by a
coordination reaction in solution and suspension polymerization (Vivaldo-Lima, et al.,
1997). Disadvantages of suspension polymerization include auto acceleration, and

30
difficulties associated with filtering and isolating the polymer product beads (Vivaldo-
Lima, et al., 1997).
3.2.4.4. Emulsion Polymerization
Emulsion polymerization occurs in an aqueous medium, in micelles of monomers.
Surfactants are used to create an emulsion of monomers, catalysts, and modifiers.
Catalysts initiate the polymerization reaction, whereas modifiers can control polymer
structure and molecular weight (Lovell and El-Asser, 1997). Stabilizers such as
antioxidants are added to the emulsion, and unreacted monomers are recovered from the
emulsion. This creates a final emulsion polymer product that is stable, and can be
directly used in manufacturing of thin films and coatings or coagulated into solid state
(Lovell and El-Asser, 1997). Free radical polymerization is the method primarily used in
emulsion polymerization, and is used to create rubbers such as SBR, chloroprene rubber
(CR), acrylonitrile-butadiene rubber (NBR), EVM, and fluorocarbon rubber (FPM)
(Lovell and El-Asser, 1997). Disadvantages associated with emulsion polymerization
include difficulty in isolation of the polymer product, and removal of surfactants to
generate pure polymer products (Lovell and El-Asser, 1997).

31
3.3. Specific Elastomer Structure and Properties
Natural rubber (NR), cis-1,4-polyisoprene, is the only naturally produced
elastomer with high molecular weights sufficient for industrial use and has a completely
amorphous structure, when not under strain (Tanaka, 2001). All other elastomers are
synthetic, and can have an amorphous or semi-crystalline structure (Mark et al., 2013).
Synthetic elastomers are polymerized from petroleum byproducts, or alternatively are
modified polymerized synthetic materials. Elastomers are further classified according to
their utility: general purpose, solvent resistant, or temperature resistant (Mark et al.,
2013). General-purpose elastomers are predominately aliphatic and aromatic
hydrocarbons with amorphous structures and include natural and synthetic elastomers
(Mark et al., 2013). General-purpose elastomers have an unsaturated polymer backbone
and can be crosslinked with sulfur, with the exception of ethylene propylene rubber that
is saturated and therefore is cured using peroxides (Mark et al., 2013). Petroleum and
solvent resistant elastomers generally incorporate nitrile, amide, or chloride groups into
their structures, and include rubbers such as nitrile, polychloroprene, epichlorohydrin,
polyurethane, and chlorinated polyethylene (Mark et al., 2013). Temperature resistant
elastomers incorporate molecules such as fluorine, sulfur, or silicon into their polymer
structure to produce elastomers resistant to degradation at high temperatures (Mark et al.,
2013). Temperature resistant elastomers include polyacrylate, fluorocarbon,
chlorosulfonated polyethylene, and silicone (Mark et al., 2013). Since solvent and
temperature resistant elastomers lack unsaturated sites, these polymers must utilize non-
sulfur cures, which will be detailed in subsequent sections.

32
The following sections go into further detail about various types of elastomers,
their respective structures and unique properties.
3.3.1. Synthetic Elastomer Structures and Properties
Synthetic elastomers account for more than half of the world’s annual 27.5
million metric tons consumption of rubbers (Mark et al., 2013) . Synthetic elastomers are
polymerized from petroleum-derived monomers. Synthetic rubbers have temperature and
solvent resistance compared to natural rubber (NR) and are used predominately in
applications such as tire treads, hoses, belts, flooring, dampeners, and medical devices
(Mark et al., 2013). Synthetic elastomers are thermosets or thermoplastics.
3.3.1.1. Aliphatic and Aromatic Hydrocarbon Elastomers
3.3.1.1.1. Aliphatic Hydrocarbon Elastomers
Synthetic hydrocarbon elastomers are general-purpose elastomers, as they have
poorer temperature and solvent resistance than other elastomers that contain halogens,
nitrogen, esters, or ethers. Hydrocarbons elastomers are divided into two classes:
aliphatic (non-aromatic), and aromatic elastomers (Mark et al., 2013).
Synthetic non-aromatic hydrocarbon elastomers include polybutadiene rubber
(PBR), polyisobutylene rubber (PIB), isobutylene isoprene rubber (IIR), and ethylene
propylene diene monomer rubber (EPDM) (Mark et al., 2013). Bulk differences between
synthetic non-aromatic hydrocarbon elastomers are attributed to variations in chemical
structure, and are specified in detail below.
PBR is a synthetic rubber comprised from the monomer 1,3-butadiene (Figure
3.13) (Mark et al., 2013). PBR can be polymerized in three forms: cis, trans, and vinyl.
Butadiene monomers polymerized from end-to-end, create the cis and trans forms (Mark

33
et al., 2013). The ratio of trans vs. cis isomers in PBR is controlled during synthesis, and
impacts both microarchitecture and bulk properties (Mark et al., 2013). Trans double
bonds formed during polymerization cause rigidity in the polymer chain, allowing for
formation of microcrystalline regions (Mark et al., 2013). The cis double bonds formed
during polymerization generate a bend in the polymer chain, preventing efficient polymer
chain packing and therefore increasing amorphous regions in the polymer. PBR with a
high percentage of cis double bond configurations (over 92%) is an elastomeric material,
and is manufactured by using a Ziegler-Natta catalyst during synthesis (Mark et al.,
2013).
PBR accounted for about 25% of all synthetic rubbers consumed in 2012 (Mark et
al., 2013). PBR has a high resistance to wear, and is commonly used in tires or as an
additive to improve the mechanical strength of plastics (Mark et al., 2013). PBR also has
high electrical resistivity, and is used in electrical component coatings (Mark et al.,
2013). In addition, PBR has high resilience and forms the elastic cores of golf balls.
Other applications for PBR include inner tubing for hoses, railway pads, and bridge
blocks (Mark et al., 2013).
Fig. 3. 13: Polymerization of polybutadiene rubber.

34
Polyisobutylene (PIB) is a homopolymer of isobutylene, consisting of a saturated
hydrocarbon backbone with two pendant methyl groups attached to alternating carbons
on the polymer backbone (Fig. 3.14) (Mark et al., 2013). PIB is polymerized using
cationic addition (Mark et al., 2013). PIB has good flex properties and is colorless to a
light-yellow color (Mark et al., 2013). PIB is gas impermeable, and is primarily utilized
as an inner liner in tires and other inflatable items (Mark et al., 2013).
Fig. 3. 14: Polymerization of polyisobutylene rubber.
Butyl rubber (IIR) is a copolymer, comprised of 98% isobutylene and 2%
isoprene (Mark et al., 2013). The small amounts of isoprene allows IIR to be crosslinked
with sulfur, improving bulk mechanical properties (Mark et al., 2013). Butyl rubber is
used for shock absorption applications, and has low gas and moisture permeability (Mark
et al., 2013). In addition, butyl rubber has outstanding resistance to heat, aging, weather,
ozone, chemicals, flexing, abrasion, and tearing (Mark et al., 2013). Applications that
use butyl rubber materials include shock mounts, sealant for rubber roof repair, tubeless
tire liners, inner tubes, stoppers, sealants, adhesives, and liners (Mark et al., 2013).
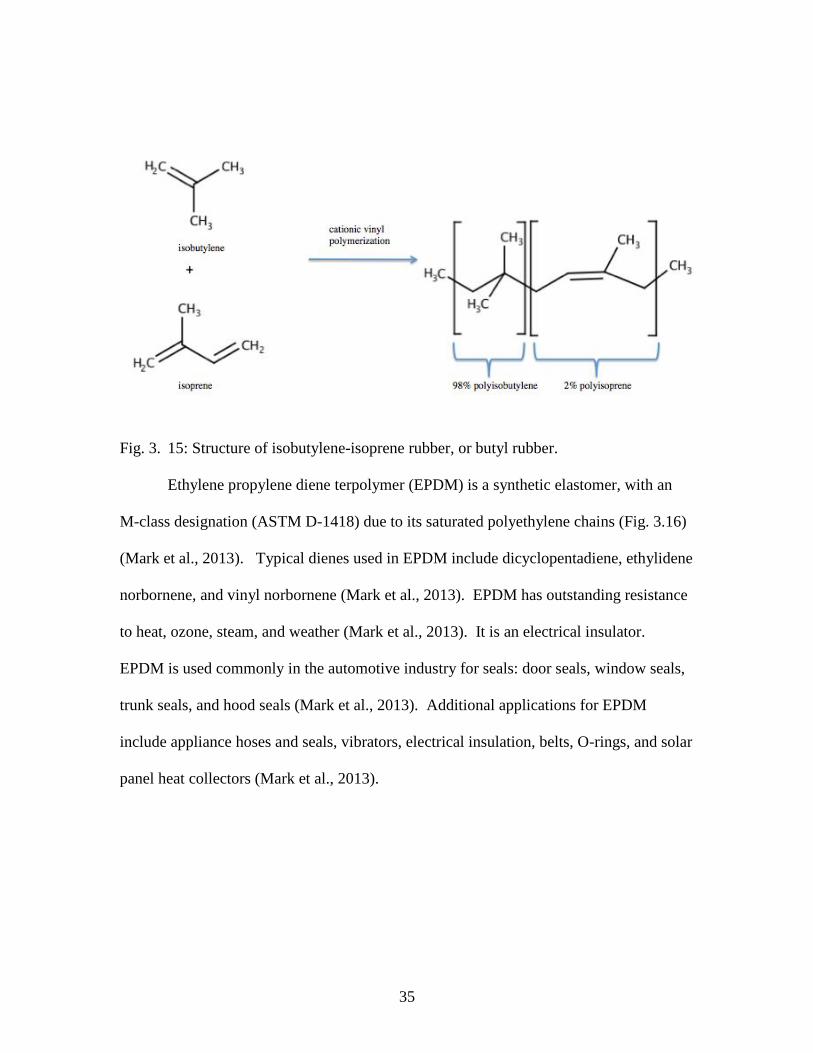
35
Fig. 3. 15: Structure of isobutylene-isoprene rubber, or butyl rubber.
Ethylene propylene diene terpolymer (EPDM) is a synthetic elastomer, with an
M-class designation (ASTM D-1418) due to its saturated polyethylene chains (Fig. 3.16)
(Mark et al., 2013). Typical dienes used in EPDM include dicyclopentadiene, ethylidene
norbornene, and vinyl norbornene (Mark et al., 2013). EPDM has outstanding resistance
to heat, ozone, steam, and weather (Mark et al., 2013). It is an electrical insulator.
EPDM is used commonly in the automotive industry for seals: door seals, window seals,
trunk seals, and hood seals (Mark et al., 2013). Additional applications for EPDM
include appliance hoses and seals, vibrators, electrical insulation, belts, O-rings, and solar
panel heat collectors (Mark et al., 2013).
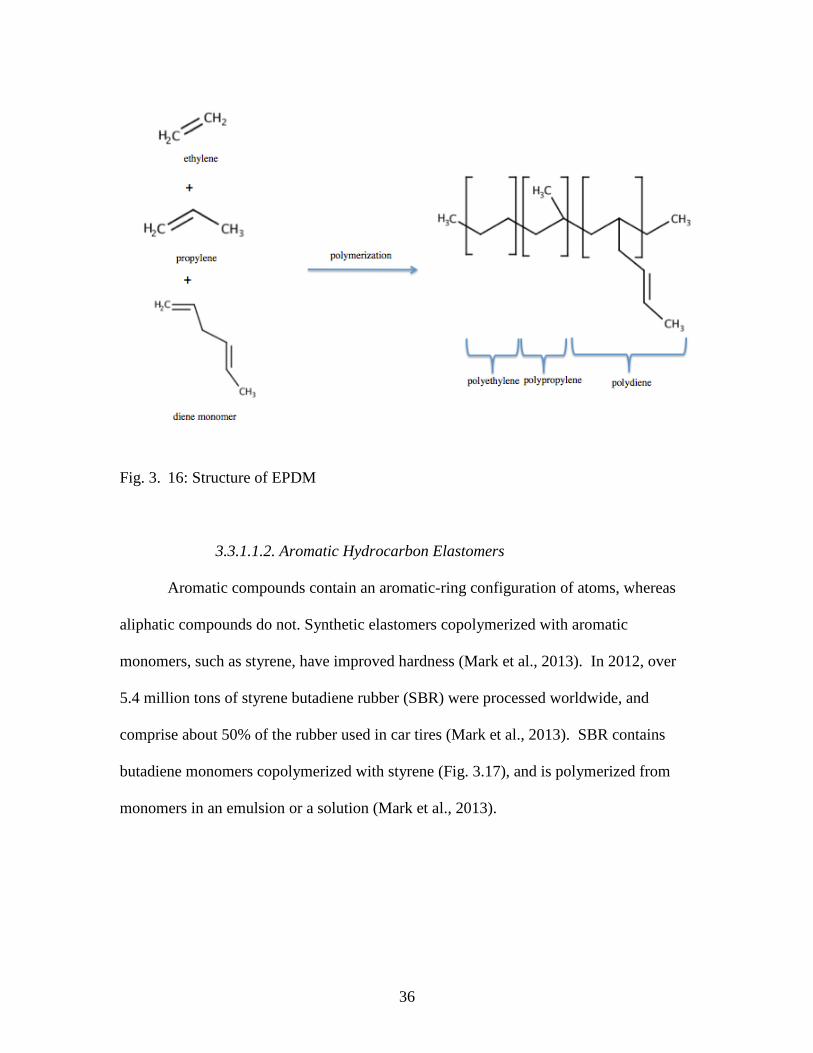
36
Fig. 3. 16: Structure of EPDM
3.3.1.1.2. Aromatic Hydrocarbon Elastomers
Aromatic compounds contain an aromatic-ring configuration of atoms, whereas
aliphatic compounds do not. Synthetic elastomers copolymerized with aromatic
monomers, such as styrene, have improved hardness (Mark et al., 2013). In 2012, over
5.4 million tons of styrene butadiene rubber (SBR) were processed worldwide, and
comprise about 50% of the rubber used in car tires (Mark et al., 2013). SBR contains
butadiene monomers copolymerized with styrene (Fig. 3.17), and is polymerized from
monomers in an emulsion or a solution (Mark et al., 2013).
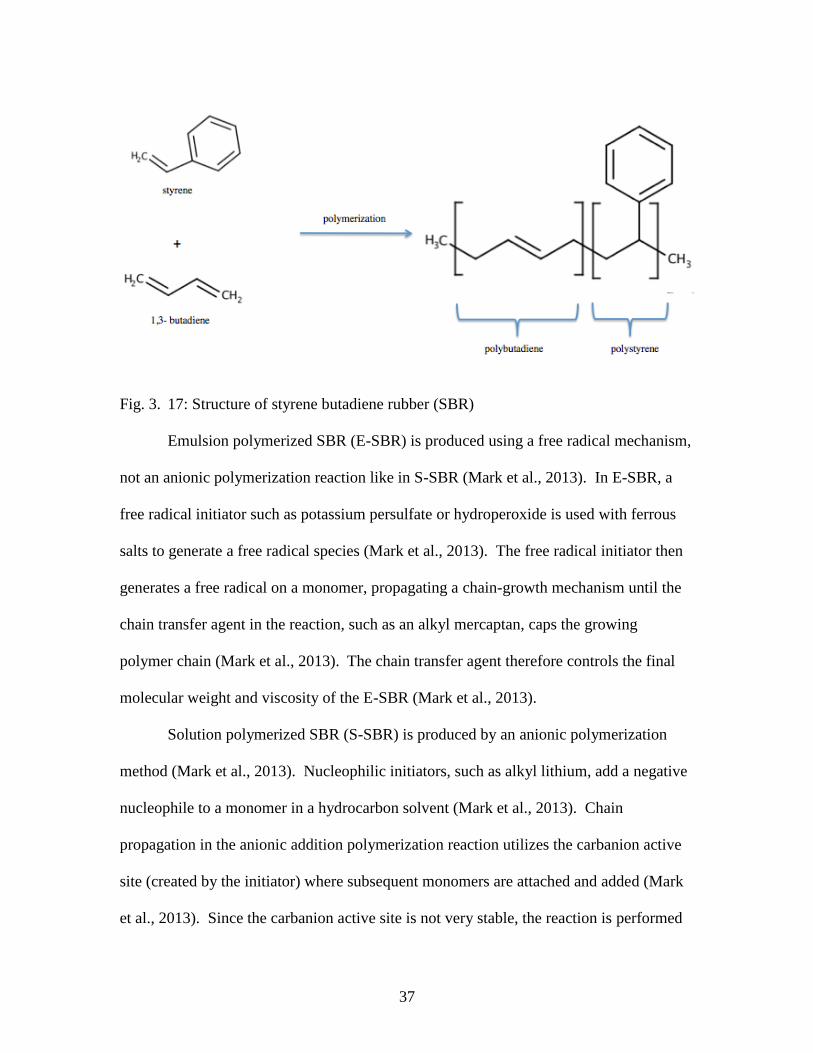
37
Fig. 3. 17: Structure of styrene butadiene rubber (SBR)
Emulsion polymerized SBR (E-SBR) is produced using a free radical mechanism,
not an anionic polymerization reaction like in S-SBR (Mark et al., 2013). In E-SBR, a
free radical initiator such as potassium persulfate or hydroperoxide is used with ferrous
salts to generate a free radical species (Mark et al., 2013). The free radical initiator then
generates a free radical on a monomer, propagating a chain-growth mechanism until the
chain transfer agent in the reaction, such as an alkyl mercaptan, caps the growing
polymer chain (Mark et al., 2013). The chain transfer agent therefore controls the final
molecular weight and viscosity of the E-SBR (Mark et al., 2013).
Solution polymerized SBR (S-SBR) is produced by an anionic polymerization
method (Mark et al., 2013). Nucleophilic initiators, such as alkyl lithium, add a negative
nucleophile to a monomer in a hydrocarbon solvent (Mark et al., 2013). Chain
propagation in the anionic addition polymerization reaction utilizes the carbanion active
site (created by the initiator) where subsequent monomers are attached and added (Mark
et al., 2013). Since the carbanion active site is not very stable, the reaction is performed

38
at low temperatures close to 0oC (Mark et al., 2013). The reaction has no formal
termination mechanism, but the carbanionic active site is often quenched by trace
impurities such as oxygen, carbon dioxide, or water (Mark et al., 2013). Spontaneous
termination occurs as well due to carbanion decay, resulting in hydride elimination (Mark
et al., 2013). The spontaneous termination of the polymerization reaction can create
issues with S-SBR processability, specifically with respect to molecular weight
distributions and long chain branching (Mark et al., 2013). To improve processability,
coupling agents such as SiCl4 and SnCl4 are used during the carbanionic polymerization
to broaden the molecular weight distributions and create branched polymer structures
(Mark et al., 2013). S-SBR is typically used in extruded and molded rubber goods
including specialty tire applications due to its better wet grip and rolling resistance than
E-SBR (Mark et al., 2013).
Additional variations in SBR polymerization include the ratio of styrene to
butadiene monomers and the type of butadiene isomer used (Mark et al., 2013). Higher
concentrations of styrene create a rubber that is harder, less elastic, and has better
abrasion resistance than BR (Mark et al., 2013). Butadiene monomers can be added in
1,4- (including cis-1,4 and trans-1,4 isomers) or in 1,2- units to the growing polymer
backbone (Fig. 3.18) (Mark et al., 2013). The relative concentration of 1,2 vs. 1,4-
addition during polymerization is dependent upon the type of polymerization reaction.

39
Fig. 3. 18: Polybutadiene linked through the 1-and 4-carbon atoms, and through the 1-
and 2-carbon atoms.
As a result, SBR is predominately used in tire treads, cables, and footwear, among
other applications where abrasion resistance is needed (Mark et al., 2013). Increasing the
styrene content in SBR also increases the Tg of the material and therefore can impact
physical properties of the material.
SBR has good chemical resistance to weak organic acids, alcohols, moderate
chemicals, and ketones (Mark et al., 2013). SBR has poor resistance to ozone, strong
acids, fats, oils, and other hydrocarbons (Mark et al., 2013). SBR has poor heat
resistance compared to most other elastomers, and is used in temperatures ranging from -
60oF to 250oF (Mark et al., 2013).
3.3.1.2. Halogen and Nitrile Substituted Elastomers
Halogen and nitrile substituted elastomers have superior oil and organic solvent
resistance compared to hydrocarbon elastomers. The most popular halogen substituted
elastomer is polychloroprene (CR) (Fig. 3.19) (Mark et al., 2013). In addition to superior
resistance to organics, CR has low flammability, good toughness, and good ozone and
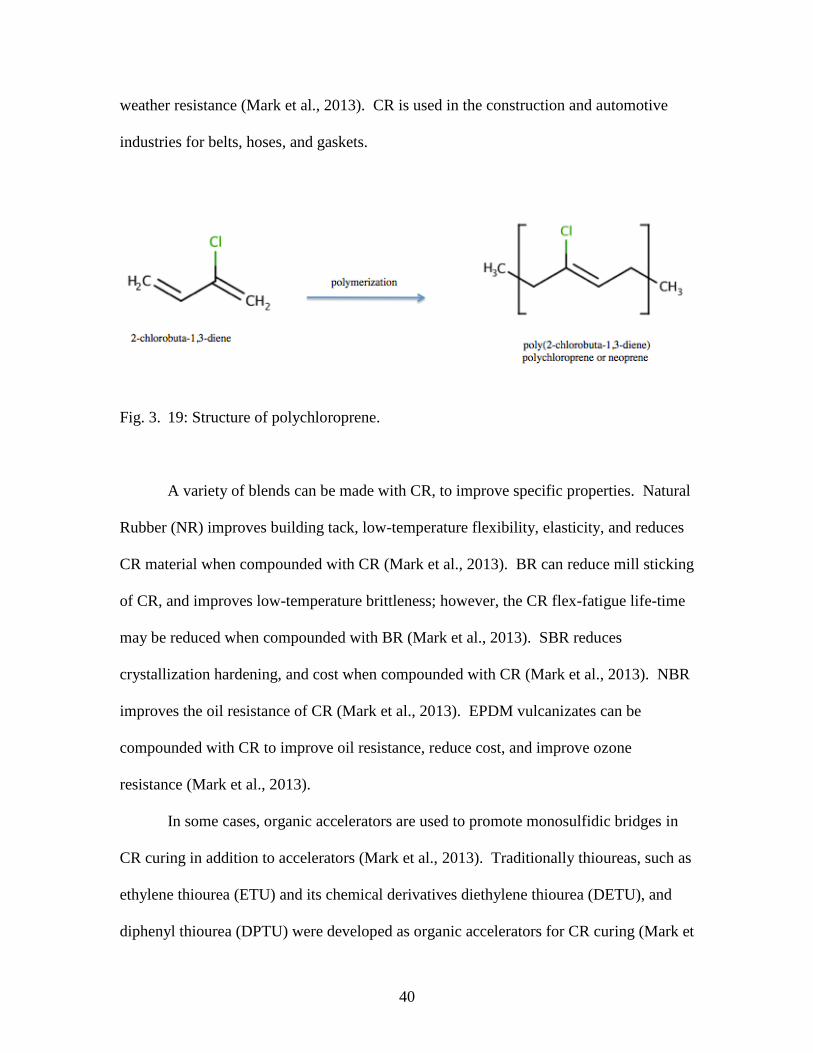
40
weather resistance (Mark et al., 2013). CR is used in the construction and automotive
industries for belts, hoses, and gaskets.
Fig. 3. 19: Structure of polychloroprene.
A variety of blends can be made with CR, to improve specific properties. Natural
Rubber (NR) improves building tack, low-temperature flexibility, elasticity, and reduces
CR material when compounded with CR (Mark et al., 2013). BR can reduce mill sticking
of CR, and improves low-temperature brittleness; however, the CR flex-fatigue life-time
may be reduced when compounded with BR (Mark et al., 2013). SBR reduces
crystallization hardening, and cost when compounded with CR (Mark et al., 2013). NBR
improves the oil resistance of CR (Mark et al., 2013). EPDM vulcanizates can be
compounded with CR to improve oil resistance, reduce cost, and improve ozone
resistance (Mark et al., 2013).
In some cases, organic accelerators are used to promote monosulfidic bridges in
CR curing in addition to accelerators (Mark et al., 2013). Traditionally thioureas, such as
ethylene thiourea (ETU) and its chemical derivatives diethylene thiourea (DETU), and
diphenyl thiourea (DPTU) were developed as organic accelerators for CR curing (Mark et

41
al., 2013). However, ETUs have been linked to cancer in laboratory animals, and
alternatives to thioureas have been developed, such as N-methyl-thiazolidine-2-thione
(Mark et al., 2013). Removing thioureas from curing systems can result in a slower cure,
and vulcanizates with higher set properties and lower heat resistance (Mark et al., 2013).
CR can be crosslinked by metal oxides, and do not need accelerators such as other
diene rubbers. Zinc oxide (ZnO) and magnesium oxide (MgO) are the most frequently
used metal oxides in CR crosslinking; a combination of ZnO and MgO is used to cure CR
to optimize mechanical properties (Mark et al., 2013). Typically 5 phr ZnO and 4 phr
MgO are used (Mark et al., 2013).
Nitrile substituted elastomers include acrylonitrile butadiene rubber (NBR), a
copolymer of butadiene and acrylonitrile (Fig. 3.20). NBR is used commonly in the
automotive non-tire and industrial rubber business due to its oil and heat resistance (Mark
et al., 2013). The composition of acrylonitrile in NBR impacts the physical and chemical
properties. There are over 188 different grades of NBR manufactured internationally,
varying due to polymerization method (batch, continuous, hot, cold), and molecular
weight distribution (Mark et al., 2013). Modifications to NBR include carboxylated,
precrosslinked, ACN/isoprene/butadiene, liquid, carbon black masterbatches, plasticizer
extended, and nitrile/pvc blends (Mark et al., 2013). When compounding NBR, the
acrylonitrile (ACN) content and the viscosity of the NBR grade should be taken into
consideration first (Mark et al., 2013). NBR grades with a broad molecular weight
distribution are used for extrusion and calendaring processes, while narrow molecular
weight distributions are used for molding (Mark et al., 2013).

42
Reinforcing fillers are commonly compounded with NBR, to improve tensile and
tear strength, abrasion resistance, chemical resistance, resilience and low compression set
(Mark et al., 2013). Carbon black is commonly used in NBR, as well as non-black fillers
such as silica, silicate, clays, talc, and calcium carbonate (Mark et al., 2013). Plasticizers
are used with NBR to reduce costs, and utilize polar plasticizers such as highly aromatic
mineral oils, and esterized oils (Mark et al., 2013).
NBR is crosslinked using sulfur and peroxide cures, and zinc oxide/peroxide for
carboxylated nitriles (Mark et al., 2013). Sulfur cures with NBR are best for dynamic
applications at moderate temperatures, where heat resistance and low compression set are
not major factors. Sulfur-free cure systems are limited in application (Mark et al., 2013).
Peroxide cure systems provide the best NBR heat and compression set resistance, its
ability to return to its original geometry after a prolonged compressive stress at elevated
temperatures (Mark et al., 2013).
Fig. 3. 20: Structure of acrylonitrile butadiene rubber (NBR).

43
3.3.1.3. Sulfide Elastomers
Polysulfide rubber (PSR) has superior chemical resistance towards hydrocarbons.
PSR is used in sealant applications due to its superior dimensional stability, flexibility,
low moisture vapor transmission, low gas transmission and weatherability (Mark et al.,
2013). Commercial brands of PSR include Thiokol FA, Thiokol ST, and Thiokol LP,
which are synthesized via nucleophilic substitution with sodium polysulfide and
dichlorides (Mark et al., 2013). Thiokol FA is made from di-2-chloroethyl formal and
ethylene dichloride, and is used in specialty roller applications requiring resistance to
ketones, aromatic solvents, and some chlorinated solvents (Fig. 3.21) (Mark et al., 2013).
Thiokol ST is a branched polysufide formed from di-2-chloroethyl formal with about 2%
1,2,3-trichloropropane as trifunctional branching units, used for mechanical goods.
Thiokol LP is formed from the cleavage of Thiokol FA and Thiokol ST (mercaptan-
terminated polymers) (Mark et al., 2013). Thiokol LP is liquid and is primarily used as a
sealant, coating, and binder (Mark et al., 2013).
Solid polysulfides (Thiokol ST and Thiokol FA) are typically vulcanized to
provide excellent low temperature properties (Mark et al., 2013). Using an ester type
plasticizer can further improve low-temperature properties (Mark et al., 2013).
Polysulfides provide good impermeability to solvents and gases (Mark et al., 2013).
Thiokol FA has excellent weathering and ozone resistance, making it ideal for weather
strips and sealants (Mark et al., 2013).
Compounding of polysulfide includes vulcanization agents, fillers, and
plasticizers (Mark et al., 2013). Zinc oxide is the predominant vulcanization agent used
in Thiokol FA (Mark et al., 2013). Thiokol ST is optimally cured using zinc peroxide.

44
Carbon black, and other elastomers are good reinforcing agents for polysulfides (Mark et
al., 2013). Non-black fillers are not as effective in reinforcement of polysulfide
elastomers (Mark et al., 2013). Low pH fillers, such as clay, should be avoided and will
slow the rate of vulcanization (Mark et al., 2013). Polysulfides are blended with other
elastomers such as nitrile rubber, NBR, or neoprene to improve their physical properties
and processability (Mark et al., 2013). Thiokol FA is typically blended with neoprene to
improve strength and processing, but reduces solvent resistance (Mark et al., 2013).
Fig. 3. 21: Structure of polysulfide Thiokol A.
3.3.1.4. Fluorocarbon Elastomers
Fluorocarbon elastomers (FKM) are typically used in harsh applications where
chemical and heat resistance are needed, in the automotive and aerospace industries

45
(Mark et al., 2013). A common type of FKM is the dipolymer of poly(vinylidene
fluoride-co-hexafluoropropylene), synthesized from monomers of hexafluoropropylene
(HFP) and vinylidene fluoride (VDF or VF2), shown in Figure 3.22. An additional
monomer tetrafluoroethylene (TFE) can be used instead of VF2, to increase the fluorine
level in the FKM (Mark et al., 2013). FKM are typically prepared by high-pressure, free-
radical emulsion polymerization (Mark et al., 2013). Organic or inorganic peroxy-
compounds, such as ammonium persulfate are used as initiators (Mark et al., 2013).
Compounding FKM is different from most other elastomer compounding;
plasticizers are not tolerated, and most chemicals used in rubber compounding are not
recommended due to FKM’s extreme resistance to chemicals and heat. FKM is typically
cured using Bisphenol AF and accelerator salts (Mark et al., 2013). Amines can also be
used to cure FKM, but confer an inferior scorch safety and compression set resistance
(Mark et al., 2013). Peroxide curing systems can only be used with FKM that contain a
peroxide cure site. All cure systems for FKM need metal oxides, which act as acid
acceptors and capture HF formed during vulcanization (Mark et al., 2013). Magnesium
oxide (3 phr) and calcium hydroxide (6 phr) are typically used for bisphenol cures,
whereas lower levels of zinc oxides (1 to 3 phr) are used for peroxide cures (Mark et al.,
2013).
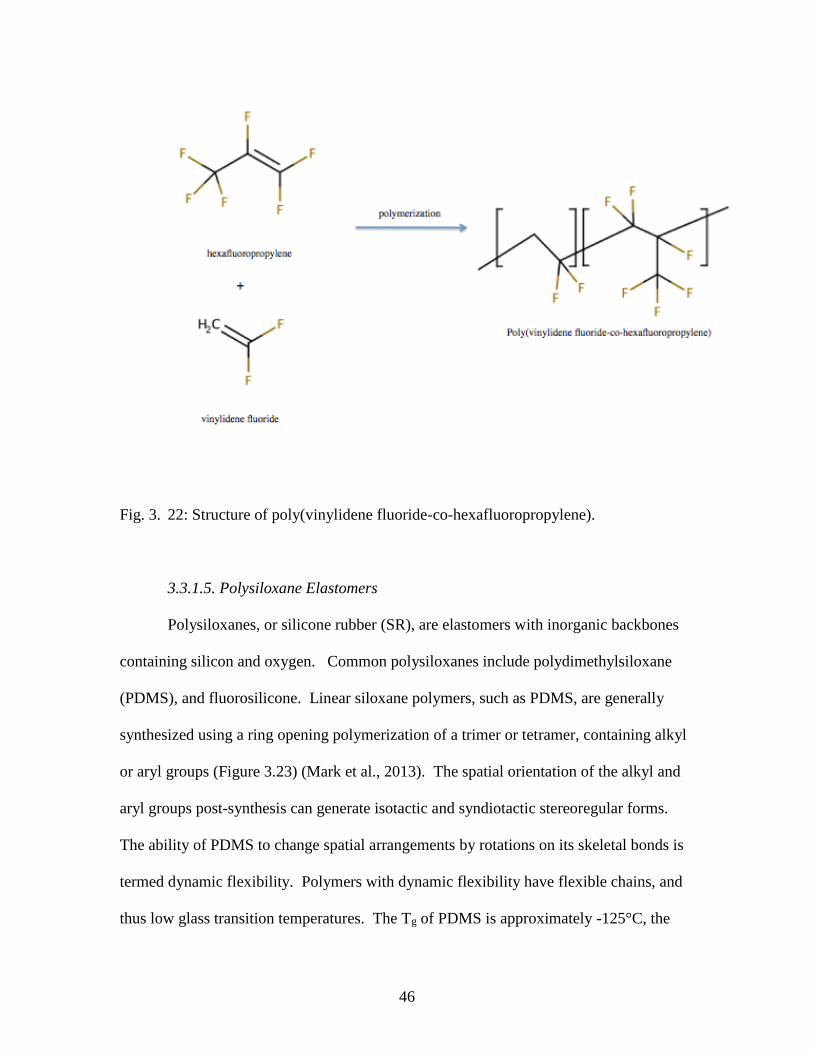
46
Fig. 3. 22: Structure of poly(vinylidene fluoride-co-hexafluoropropylene).
3.3.1.5. Polysiloxane Elastomers
Polysiloxanes, or silicone rubber (SR), are elastomers with inorganic backbones
containing silicon and oxygen. Common polysiloxanes include polydimethylsiloxane
(PDMS), and fluorosilicone. Linear siloxane polymers, such as PDMS, are generally
synthesized using a ring opening polymerization of a trimer or tetramer, containing alkyl
or aryl groups (Figure 3.23) (Mark et al., 2013). The spatial orientation of the alkyl and
aryl groups post-synthesis can generate isotactic and syndiotactic stereoregular forms.
The ability of PDMS to change spatial arrangements by rotations on its skeletal bonds is
termed dynamic flexibility. Polymers with dynamic flexibility have flexible chains, and
thus low glass transition temperatures. The Tg of PDMS is approximately -125°C, the

47
lowest of all polymers (Wypych, 2012). In addition to dynamic flexibility, PDMS has
chain flexibility along its backbone; the oxygen skeletal atoms (Si-O-Si) are the smallest
atoms with multi-valency required for a polymer chain structure and allows for rotational
movement along the polymer backbone (Wypych, 2012). The bond angle of Si-O-Si is
readily deformable, due to its large bond angle of 143°, compared to a stable tetrahedral
bond angle of 109.5° (Wypych, 2012). The ability for chain rotations and chain
deformity in PDMS results in an ability of the polymer chain to be compact while in a
random coil; therefore PDMS has a very low melting temperature (40°C) due to the
resulting equilibrium flexibility (Mark et al., 2013). Therefore, strain induced
crystallization rarely occurs in PDMS (Wypych, 2012). To improve stiffness in
polysiloxanes, large side groups are added to the backbone, or rigid units are polymerized
into the backbone (Wypych, 2012).
In addition to a low glass temperature, other properties of siloxane polymers
include high permeability to gases. As a result, siloxanes are frequently used in
applications such as gas separation membranes and soft contact lenses (Wypych, 2012).
Fig. 3.23: Structure of polydimethylsiloxane.

48
3.3.1.6. Thermoplastic Elastomers
Thermoplastic elastomers are typically segmented block copolymers that are
phase separated into amorphous and semi-crystalline regions. The hard segments are
semi-crystalline regions acting as physical crosslinks, reducing chain slippage (Mark et
al., 2013). The soft segments are the amorphous regions that create a matrix, which
contributes to the flexibility and resiliency of the material. The properties of
thermoplastic elastomers vary due to the proportion of hard and soft segments, molecular
weight distribution, method of preparation, and thermal history that affects the degree of
phase separation and domain formation (Mark et al., 2013). Thermoplastic elastomers
lack chemical crosslinks, and therefore utilize physical crosslinks to make sufficient
molecular entanglements (Mark et al., 2013).
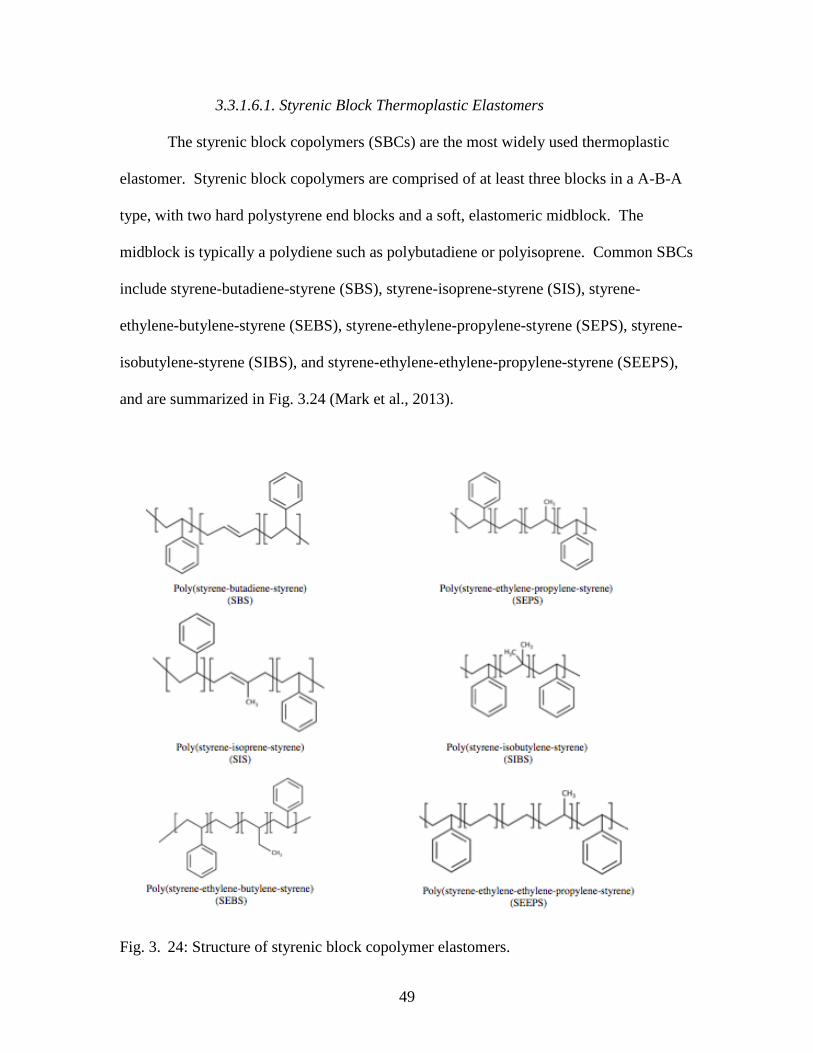
49
3.3.1.6.1. Styrenic Block Thermoplastic Elastomers
The styrenic block copolymers (SBCs) are the most widely used thermoplastic
elastomer. Styrenic block copolymers are comprised of at least three blocks in a A-B-A
type, with two hard polystyrene end blocks and a soft, elastomeric midblock. The
midblock is typically a polydiene such as polybutadiene or polyisoprene. Common SBCs
include styrene-butadiene-styrene (SBS), styrene-isoprene-styrene (SIS), styrene-
ethylene-butylene-styrene (SEBS), styrene-ethylene-propylene-styrene (SEPS), styrene-
isobutylene-styrene (SIBS), and styrene-ethylene-ethylene-propylene-styrene (SEEPS),
and are summarized in Fig. 3.24 (Mark et al., 2013).
Fig. 3. 24: Structure of styrenic block copolymer elastomers.

50
SBCs are used in footwear, and adhesives. SBCs are also used as a hardener in
asphalt to improve rut resistance. SBCs are also compounded to create grips and
cosmetic finishes for consumer goods, automotive parts, and packaging (Mark et al.,
2013).
3.3.1.7.2. Polyamide Thermoplastic Elastomers
The polyamide elastomers are segmented block copolymers that include
poly(esteramides) (PEA), poly(etheresteramides) (PEEA), poly(carbonateesteramides)
(PCEA), and polyether-block-amides (PE-b-A) (Mark et al., 2013). Typical of other
segmented coblock polymers, polyamides are phase separated into hard and soft
segments. The hard segments of polyamides are aliphatic polyamides, and the soft
segments are aliphatic polyethers and/or polyesters. Hard and soft segments are linked
by ester or amide groups.
Polyamides have superior temperature resistance compared to other thermoplastic
elastomers. Polyamides also have superior abrasion resistance, with a hardness in the
range of Shore 80A to Shore 70D (Mark et al., 2013). Polyamides are also used for
insulation materials. Additional applications include industrial applications, consumer
goods, automotive applications, electronics, hot melt adhesives, powder coatings for
metals, and impact modifiers in thermoplastics (Mark et al., 2013). Since polyamides are
biodegradable, they are also being developed for biomedical applications including drug
delivery systems, hydrogels, and tissue engineering (Mark et al., 2013).

51
Fig. 3. 25: Structure of polyamide thermoplastic elastomers.
3.3.1.6.3. Polyether Ester Thermoplastic Elastomers
Polyether ester elastomers are coblock polymers with hard polyester segments
embedded in a soft polyether matrix. The polyester crystalline regions act as physical
crosslinks, which provide polyether ester elastomers with superior heat resistance with a
wide useful temperature range. Polyether ester elastomers have good resistance to
hydrocarbon based greases and oils, but has limited resistance to halogenated solvents
and aqueous acids and bases (Mark et al., 2013). Polyether esters have excellent wear
properties and high tensile strength, but have a higher and more limited hardness range
(30 to 82 Shore D) compared to other thermoplastic elastomers (Mark et al., 2013).
3.3.1.6.4. Polyolefin Thermoplastic Elastomers
Polyolefins are hydrocarbon thermoplastics formed from the polymerization of
olefins including propylene, ethylene, isoprenes, and butenes (Mark et al., 2013).
Polyolefin thermoplastic elastomers (TPEs) depend on the crystallization of polymer
chains to produce elastomeric characteristics. Properties of polyolefins depend on the
type of monomers and route of polymerization, resulting in distinct types of polyolefin
TPEs such as: blends or thermoplastic polyolefins (TPOs), dynamically vulcanized
blends (TPVs) of ethylene-propylene random copolymer (EPM) or ethylene-propylene
diene monomer (EPDM) with an olefin, random block copolymers (e.g., ethylene α-

52
olefin copolymers), block copolymers (e.g., hydrogenate polybutadiene-isoprene-
butadiene block copolymer), stereo polymers (e.g., stereoblock polypropylene) and graft
copolymers (e.g., polyisobutylene-g-polystyrene) (Mark et al., 2013).
Polyolefin TPEs have different types of crystallization due to structural
differences. Random block copolymers polyolefin TPEs contain long ethylene segments
that crystallize and form physical crosslinks for amorphous chain segments (Mark et al.,
2013). Stereoblock copolymers utilize tacticity to create crystalline regions (Mark et al.,
2013). Polyolefin graft copolymers contain crystalline polyolefin chains grafted to an
amorphous polyolefin backbone. Most graft and block copolymers are elastic due to
reversible, physical crosslinks and rubber chain entanglements, utilizing an entropic
retractive force (Mark et al., 2013). TPOs are mechanical blends, and therefore are co-
continuous with both an elastomeric and crystalline polyolefin phase (Mark et al., 2013).
TPVs are crosslinked, and therefore have a continuous polyolefin phase that surrounds
the discontinuous elastomer regions.
Polyolefin blend TPEs (TPOs) are predominately made from elastomeric
ethylene-propylene random copolymer (EPM), and crystalline isotactic polypropylene
(iPP). EPM-iPP blends are typically shear mixed in the range of 100-1000 s-1 (Mark et al.,
2013). The viscosity ratios of EPM and iPP can be adjusted to provide continuous phases
over a wide range of volume fraction of the blend (e.g., 80/20 to 20/80) (Mark et al.,
2013). TPOs are compounded with additional additives such as fillers, reinforcing
agents, plasticizers, lubricants, processing aids, flow modifiers, antioxidants, heat
stabilizers, etc. (Mark et al., 2013). Most additives are contained in the rubber phase of
TPOs, softening and extending the rubber (Mark et al., 2013).

53
3.3.1.6.5. Polyurethane Elastomers
Polyurethanes have superior abrasion resistance and tear strength, better oxygen
resistance, but are susceptible to rapid breakdown from heat and water. Polyurethanes
are produced by the polyaddition reaction of a diisocyanate or a polymeric isocyanate
with a polyol, with appropriate catalysts and additives (Mark et al., 2013). Urethane
groups are formed along the polymer backbone, typically in a reaction between
isocyanate and hydroxyl groups.
Polyurethane elastomers (PUs) are formed by reacting a diisocyanate (aromatic or
aliphatic), a long-chain diol, and a small molecule chain-extender diol or diamine (Fig.
3.26). The resulting polyurethane consists of hard diisocyanate-chain extender segments
in the soft macrodiol matrix (Mark et al., 2013). The microphase separation between the
hard and soft segments vary between types of PUs due to differences in chemical
composition and processing.
PUs utilize physical crosslinks from its hard domains, but can be chemically
crosslinked as well using a tri-or multifunctional constituents (Mark et al., 2013). Once
PUs are crosslinked, they cannot be remolded and become a semi-crystalline thermoset.
As a result, thermoplastic PUs with only physical crosslinks are of interest due its ability
to be remolded and thus recycled (Mark et al., 2013).

54
Fig. 3. 26: Chemical reaction for polyurethane synthesis.
3.3.2. Natural Rubber Latex and Natural Rubber
Over 2,500 species of plants produce cis-1,4- polyisoprene (Mooibroek and
Cornish, 2000). Natural rubber latex (NRL) is an aqueous emulsion comprised of rubber
particles comprised of high molecular weight cis-1,4-polyisoprene macromolecules, and
other biochemical metabolites such as proteins, fatty acids, and antioxidants that are all
synthesized in vivo (Puskas et al., 2014). NRL can be coagulated to form solid natural
rubber (NR), which entraps non-rubber particle latex biochemical metabolites within the
rubber matrix (Cornish et al., 2008). NRL is used to make thin film products such as
gloves, balloons, and coatings whereas NR is used to make bulk rubber items such as
tires and gaskets (Cornish et al., 2008).
The only commercial sources of NRL and NR are available from Hevea
brasiliensis, the Brazilian or Para rubber tree. Other plant species which make high
molecular weight rubber include Parthenium argentatum (commonly called guayule),
and Taraxacum koksaghyz (rubber dandelion) (Cornish et al., 2008). It is currently

55
unknown why plants produce rubber, and some of the underlying mechanisms of NR
biosynthesis remain unclear (Puskas et al., 2014).
3.3.1.1. Extraction of Natural Rubber Latex
NRL is an aqueous emulsion of rubber particles present in laticiferous vessels
(ducts) or parenchymal (single) cells of rubber producing plants (Cornish et al., 2005).
The NRL from Hevea (HNRL) is extracted by “tapping” the rubber tree, characterized by
making incisions along the trunk and collecting the latex as it bleeds from the laticifers.
The latex vessels are comprised of an anastomosed cell system, located in the secondary
phloem in the trunk and arranged as a paracirculatory system (Cornish et al., 2005).
Sieve tubes create a series of circular rings in the plant by fusing to one another, and the
incisions of the “tapping” process typically penetrate the contact points between sieve
tubes. Rubber particles, as well as cytoplasmic components, are released from the cut
laticifers, which retain most larger cellular components such as organelles and nuclei.
In the guayule shrub, rubber particles accumulate in the parenchymal cells of the
shoot and root bark, and so tapping is not possible (Cornish et al., 1999). Thus, guayule
NRL (GNRL) is produced by homogenizing fresh plants in aqueous medium to release
rubber particles from individual cells, and the particles are then purified while
maintaining them in aqueous suspension (Cornish et al., 1999). Solvent extraction also
can be used to extract rubber from dried guayule shrub. However, solvent extraction of
rubber from guayule has many technical issues, including difficult separation of the
viscous extractant from finely dispersed solids, accumulation of terpenes in recycled
solvents, and the separation of a low molecular weight rubber and resin fractions from
high quality GNR (Cornish et al., 1999).

56
GRNL contains terpene resins, but otherwise does not contain a low molecular-
weight rubber fraction (unlike GNR produced by solvent extraction) (Cornish et al.,
2008). However, highly purified GNRL must be stabilized with emulsion additives such
as ammonia, potassium hydroxide, and/or amine and phenolic antioxidants, and
surfactants (Cornish et al., 2008). Stabilized GNRL in sealed containers has an
impressive shelf life of at least 10 years (Cornish et al, 2008).
3.5.2. Rubber Bio-synthesis
Rubber particles are comprised of a monolayer proteo-phospholipid membrane
encasing hydrophobic cis-1,4-polyisoprene chains (Puskas et al., 2014). The monolayer
proteo-phospholipid membrane stabilizes the rubber particles, preventing aggregation in
the aqueous environment of the plant cell or laticifer (Puskas et al., 2014). The
biosynthesis of NR is catalyzed by the rubber transferase enzyme complex (cis-prenyl
transferase, RT-ase), which is bound to the rubber particle’s membrane. The
polymerization of rubber occurs at the active sites of the amphiphilic RT-ase which
contains glycosylated hydrophilic regions that regulate the placement of subunits in the
rubber particle membrane (Puskas et al., 2014).
The monomer of cis-1,4-polyisoprene is isopentenyl pyrophosphate (IPP) which
is synthesized from acetyl-CoA via the cytoplasmic mevalonate pathway (Fig. 3.27.). IPP
is isomerized to 1,1-dimethylallyl pyrophosphate (DMAPP) by the enzyme
pyrophosphate isomerase (Kumar et al., 2012). DMAPP is then catalyzed by specific
trans-prenyl transferases, adding 1-3 IPP monomers to form oligomeric allylic
pyrophosphates (APPs), creating the initiator for the polymerization of IPP monomers

57
(Kumar et al., 2012). The RT-ase requires divalent cation co-factors such as Mg2+, and
Mn2+ (Kumar et al., 2012).
Rubber transferase, an amphiphilic enzyme, is located at the interface between the
rubber particle core and the aqueous phase of the cytoplasm (Puskas et al., 2014).
Hypothesized polymerization mechanisms for natural rubber include a combination of
chain growth and polycondensation mechanisms (Puskas et al., 2014).
The molecular weight and molecular weight distributions of cis-1,4-polyisoprene
are species dependent and are impacted by plant age, genotype, environment, and
extraction process (Cornish, 2001). NR also contains a gel phase, which is comprised of
insoluble rubber that has been naturally crosslinked (Cornish, 2001). There are two
different components to gel: a hard and a soft gel. The hard gel is formed by radical
reactions between sulfur containing proteins and the dimethyl allyl double bond in the
head group of NR (McMahan et al, 2015). The soft gel is produced by hydrogen bonding
between phosphates and phospholipids at the end group of NR (McMahan et al, 2015).
The NR gel phase varies between plant species and environmental conditions. NR and
TNR (NR from T. kok-saghyz) are very similar in gel content and composition, whereas
GNR is different, having little protein or gel (McMahan et al, 2015).
Fig. 3. 27: The mevalonate pathway, which produces isopentenyl pyrophosphate (IPP),
the monomer for cis-1,4-polyisoprene.

58
3.3.1.2 Hevea brasiliensis Natural Rubber Latex
Hevea rubber is obtained as latex (HRNL) which is about 35% by weight rubber
particles. It also includes about 0.5% proteins, 0.6% phospholipids, and 0.09%
tocotrienols as non-rubber components. Solid rubber from coagulated latex contains
about 2.8% acetone-soluble fraction (tocotrienols, fatty acids, sterols, etc.), 2.5% protein
fraction, 0.2% ash fraction, and 95-98% of hydrocarbon rubber (Amnuaypronsri et al.,
2008; McMahan and Lhamo, 2015; Tanaka, 2001).
Lutoids are the most abundant non-rubber particle in Hevea latex (Tanaka, 2001).
Lutoids are spherical membrane-bound vacuoles ranging in diameter from 0.5-5 um and
have been completely characterized (Wititsuwannakul and Wititsuwannakul, 2005).
Within the lutoid membrane, there are dissolved metabolites such as acids, minerals,
proteins, and sugars. Lutoids have been found to contain acid phosphatase, lysozyme,
and acid hydrolases, which are characteristic of lysosomes. Lutoids regulate the
homeostasis of the laticiferous system, and contribute to latex coagulation. Lutoid
membranes contain high levels of phophatidic acid, and therefore have a strong negative
charge that promotes their stabilization in aqueous media. Contents of lutoids include
anionic and cationic protein Hevein is an anionic protein that is 70% of the total proteins
found in lutoids (Gidrol et al., 1994). Hevein causes latex to coagulate by agglutination
with the 23 kDA protein in rubber particles (rubber transferase). Lutoids also contain
numerous acid hydrolases and peroxidases, which are utilized during cell homeostasis
(Gidrol et al., 1994).

59
3.3.1.3. Guayule Natural Rubber Latex
Guayule is a non-laticiferous plant, the rubber particles are biosynthesized and
accumulate in the parenchyma cells of the stems and roots (Cornish, 2014). Guayule,
however, does have resin canals within its parenchyma tissue, which produce resins that
contain sesquiterpene ethers, triterpenoids, and fatty acid triglycerides but not rubber
particles (Cornish, 2014). Histochemical staining and fluorescent microscopy has shown
that rubber particles are localized in epithelial cells in parenchyma tissue and in pith, as
well as around resin canals (Cornish and Backhaus, 1990).
3.3.1.4. Other sources of Natural Rubber
Taraxacum kok-saghyz (rubber dandelion)
Natural rubber is also produced by Taraxacum kok-saghyz, which is a
diploid (n=16) sexually reproducing dandelion species native to Kazakhstan. The rubber
particles produced by 4-month and 12- month old T. kok-saghyz has been characterized as
having a unimodal distribution, with particle sizes ranging from 0.2 to 0.7 μm with an
average particle size of 320 nm, and an average molecular weight of 4,750 kDa (Schmidt
et al., 2010). NMR analysis has confirmed that the purity of poly(cis-1,4-isoprene) in T.
kok-saghyz is >95% (Schmidt et al., 2010). The yield of natural rubber in T. kok-saghyz
during plant growth increases rapidly during the first 8 months of growth; after 8 months
the yield plateaus at 130-150 mg dry rubber/mL latex. Rubber particles from T. kok-
saghyz contain cis-1,4-polyprenylcistransferase (rubber transferase), which is the same
enzyme identified in the biosynthesis of H. brasiliensis rubber (Schmidt et al., 2010), and
it has been hypothesized that biosynthesis of natural rubber in T. kok-saghyz is analogous
to H. brasiliensis biosynthesis.

60
Other proteins found in natural rubber latex from T. kok-saghyz have been
characterized, and show significant cross-reactivity between T. kok-saghyz proteins and
anti-H. brasiliensis latex antibodies (Cornish et al., 2015). For instance, the H.
brasiliensis rubber particle membrane bound protein and severe allergen Hev b1, has
specific antibodies that cross-react with at least seven proteins found in T. kok-saghyz
latex. Therefore, it can be inferred that the natural rubber from T. kok-saghyz has
potential to trigger allergic reactions, and perhaps even sensitize people to Type I latex
allergy. Therefore, commercial applications utilizing T. kok-saghyz should follow similar
precautions to those currently in place for H. brasiliensis with regards to allergenic
potential and safety.

61
3.4. Elastomer Compounding
Compounding is the mixing of a base polymer with additives or other polymers to
make the base polymer perform better, cost less, improve processability, and/or improve
its physical appearance (Donnet and Custodero, 2013; Rattanasom et al., 2007).
Thermoplastics and thermoset elastomers are compounded with additives such as fillers,
antidegradants, blowing agents, desiccants, flame retardants, odorants and deodorants,
peptisers, pigments, processing aids and plasticizers, retarders, and tackifiers (Dick,
2009). Elastomers are compounded to optimize polymer properties for specific
applications, and variations in elastomer compounding are due to polymer structure and
specific additives. Thermosets are compounded to generate crosslinks, which, like the
elastomers, require chemical activators and accelerators in addition to the crosslinking
agent.
3.4.1. Crosslinking
Thermoset elastomers require curing to create a three-dimensional network of
chemically bonded intramolecular and intermolecular crosslinks, forming a continuous
polymer network (Donnet and Custodero, 2013). The type of cure used to crosslink a
thermoset depends on the chemical structure and intended application of the polymer.
Types of cures include peroxide, sulfur, metal oxides, phenolic resins, quinones, etc. and
have different reaction chemistries, resulting in varying structure and quality of crosslinks
(Kruzelak et al., 2016). Sulfur and peroxide cures are the most commonly used for
crosslinking rubber materials.
3.4.1.1. Peroxide cures

62
Peroxide cures are often used in fully saturated elastomers, such as silicones
(Kruzelak et al., 2016). Organic peroxides are thermally decomposed to create free
radicals, which can create an active site on the polymer carbon backbone. The
subsequent reaction between two active sites creates a carbon-carbon crosslink.
Polymers cured with peroxides have good heat-aging stability and low compression set,
which is attributed to the carbon-carbon crosslinks (Kruzelak et al., 2016). Organic
peroxides can be used in a wide variety of elastomers including saturated and unsaturated
hydrocarbons (EPDM), fluoroelastomers, nitrile rubbers, and silicones (Kruzelak et al.,
2016).
Coagents (activators) are compounded with the polymer to optimize the peroxide
cure. Coagents increase the crosslinking efficiency of the peroxide, typically not
exceeding 0.3 phr (Dick, 2009). Coagents generally increase the hardness and tensile
strength, while decreasing the elongation at break. Coagents are classified based on their
contribution to the cure. Type I coagents increase both the rate and the state of the cure.
Type I coagents are typically polar, multifunctional low molecular weight compounds
that form very reactive radicals, which are subsequently homopolymerized or grafted to
polymer chains (Dick, 2009). Type II coagents form less reactive radicals and only
contribute to the state of the cure (Dick, 2009). Type I coagents homopolymerize and
form crosslinks through radical addition reactions, whereas Type II coagents typically
contain an extractable allylic hydrogen and form crosslinks through intramolecular
cyclization and intermolecular propagation reactions (Dick, 2009). Crosslinking at
higher temperatures in the range of 100°C-160°C can be done with peroxides, with
formulations typically containing a small amount of vinyl groups (Dick, 2009).

63
3.4.1.2. Sulfur cures
Sulfur is the most common crosslinking agent used with unsaturated rubbers to
produced vulcanized rubber. The rhombic form of sulfur is typically used for
vulcanization; it exists as an eight member- sulfur ring structure (Dick, 2009). The
amorphous form of sulfur is a metastable high polymer, with a molecular weight of
100,000 to 300,000 (Dick, 2009). Amorphous sulfur is insoluble in most solvents and
rubber, and therefore is typically only used prevent surface blooming on uncured rubber
surfaces where tack is desired (Dick, 2009).
Typically, 1 to 3 phr of sulfur is used for most rubber products (Dick, 2009). For
dry rubber crosslinking, the dispersion of sulfur for is improved using sulfur treated with
carbon black, magnesium carbonate, or oils (Dick, 2009). In addition, masterbatches of
sulfur with rubbers or other grafted polymers are used to improved dispersion. For latex
crosslinking, emulsions of sulfur are used, therefore incorporating oils for improved
dispersion.
Vulcanization with sulfur alone is an inefficient process, and requires the use of
chemical activators and accelerators. The chemical reaction between sulfur and the
double bonds of various polymers requires 40 to 55 sulfur atoms, without a chemical
accelerator, and can take up to 6 hours to complete the reaction at 140°C (Dick, 2009).
The crosslinks of vulcanizates made without chemical accelerators are extremely prone to
oxidative degradation and have inadequate mechanical properties for rubber applications
(Dick, 2009).
Chemical accelerators are used in rubber compounding to increase the speed of
vulcanization, and permit vulcanization to proceed at lower temperature and with greater

64
efficiency. There are two categories of accelerators: primary and/or secondary
accelerators. Primary accelerators are used at a concentration of 0.5 to 1.5 phr in most
rubber compounds, and most are from six chemical classes: thiazoles, sulfenamides,
thiurams, guanidines, dithiocarbamates, xanthates, and thioureas, with sulfonamides and
thiazoles being the main classes of accelerators used (Dick, 2009). Primary accelerators
usually have a long scorch time and cure quickly during vulcanization. Secondary
accelerators are typically used at 10-40% loading of the primary accelerator, and boost
the cure and increase the crosslink density (Dick, 2009). The most common secondary
accelerators include guanidines, thiurams, and dithiocarbamates (Dick, 2009).
Thiazoles are a commonly used class of primary accelerators. Thiazoles have
improved scorch safety and allow for high temperature cures with a short cure time and
broad vulcanization plateau (Dick, 2009). The most common commercial thiazoles are
bis(2-benzothiazole) disulfide (MBTS), 2-mercaptobenzothiazole (MBT), and the zinc
salt of mercaptobenzothiazole (ZMBT) (Dick, 2009). The thiazoles typically need
activation from zinc oxide or stearic acid, and require the use of a second accelerator to
expedite the cure speed (Dick, 2009). Secondary accelerators are typically used at 10-
40% loading of the primary accelerator, and include diphenyl guanidine (DPG) or
diothrotolyl guanidine (DOTG) (Dick, 2009).
Sulfenamides are another commonly used class of primary accelerators.
Sulfenamides are relatively safe, and are known for their scorch delay (Dick, 2009).
Sulfenamides allow for easy processing and molding of rubber compounds, providing a
broad vulcanization plateau and good aging resistance (Dick, 2009). However,
sulfenamides lack good storage ability, and will decompose when exposed to high

65
humidity and heat, as well as acids (Dick, 2009). Sulfenamides are typically used with
secondary accelerators such as DPG, DOTG, or tetramethylthuiram mono or disulfide
(TMTM, TMTD) (Dick, 2009). These co-accelerators increase the cure rate but also
reduce the scorch safety (Dick, 2009).
Thuirams are effective sulfur cure accelerators that contain two or more sulfur
atoms. As a result, these compounds not only function as accelerators but also act as
sulfur donors, and can allow for a “sulfur-less” cure therefore not needing elemental
sulfur in the rubber compound (Dick, 2009). Rubbers with little to no sulfur content, or
low unsaturation such as IIR and EPDM, typically use thuirams (Dick, 2009). The most
popular thiurams are tetramethyl thiuram monosulfide (TMTM), tetramethyl thiuram
disulfide (TMTD), and dipentamethylene thiuram tetrasulfide (DPTT) (Dick, 2009).
Rubbers cured with thuirams typically have excellent heat and water vapor resistance
(Dick, 2009). However, thiurams are expensive compared to other rubber accelerators,
and also have a tendency to bloom to the surface (Dick, 2009).
Guanidines are condensation products of aromatic amines (aniline) and
carbondisulfide with subsequent substitution of the thione functionality (>C=S) for a
primary ketimine group (>C=NH) (Dick, 2009). Guanidines have a slow cure rate, and
require the use of zinc oxide for activation (Dick, 2009). Guanidines are commonly used
in thick walled rubber products, but are most commonly used as secondary accelerators
with thiazoles (Dick, 2009). The most common guanidines used are diphenyl guanidine
(DPG), and N,N’-diorthotolyl guanidine (DOTG) (Dick, 2009). Guanidines provide a
high crosslink density, and good mechanical properties including a high modulus and
good compression set (Dick, 2009). Guanidines can cause a brown discoloration of

66
rubber goods, and therefore are not recommended for lightly colored objects (Dick,
2009).
Dithiocarbamates are ultra-fast accelerators that have a minimal induction time,
and therefore require a retarder to avoid scorch when dithiocarbamates are used as a
primary accelerator (Dick, 2009). Dithiocarbamates also require activators such as zinc
oxide or fatty acids (Dick, 2009). Common dithiocarbamates include zinc dimethyl
dithiocarbamate (ZDMC), zinc diethyl dithiocarbamate (ZDEC), and zinc diburyl
dithiocarbamate (ZDBC) (Dick, 2009). As the alkyl group of the dithiocarbamates are
lengthened, the scorch safety of the compound increases, with ZDBC having the highest
scorch safety (Dick, 2009). However, the zinc salt of the dithiocarbamates decreases
solubility in the non-polar rubber matrix, and therefore have a tendency to bloom to the
surface at high concentrations (Dick, 2009). Dithiocarbamates are typically used in low
sulfur cures, low temperature cures, and for white, transparent, or brightly colored rubber
goods (Dick, 2009). Dithiocarbamates are also used as secondary accelerators to speed
cure (Dick, 2009).
Xanthates are ultra-fast primary accelerators, predominately used for the
vulcanization of rubber latex and rubber in solution at low temperatures (Dick, 2009).
Xanthates are polar, and some are even soluble in water (Dick, 2009). The most
common xanthates are zinc xanthate (ZIX), and sodium isopropyl xanthate (NaIX) (Dick,
2009).
Thioureas are ultra-fast primary or secondary accelerators. Thioureas are
predominately used for the vulcanization of polychloroprene rubbers. Commonly used

67
thioureas include ethylene thiourea (ETU), dipentamethylene thiourea (DPTU), and
dibutyl thiourea (DBTU) (Dick, 2009).
3.4.1.3. Polyurethane crosslinking
Flexible and semi-flexible foams, cast elastomers, and coating systems are
crosslinked segmented polyurethanes, consisting of soft and hard segments.
Polyurethanes are prepared using polyurethane prepolymer, which is combined with a
chain extender such as a short chain glycol and/or a crosslinking agent having a
functionality of three or more (Mark et al., 2013). Isocyanurates of different
diisocyanates, polyamides, and polyols are used as crosslinking agents (Mark et al, 2013).
Polyurethane systems can also be crosslinked without the addition of crosslinking agents,
attributed to allophanate and biuret bonds made during polyurethane synthesis (Mark et
al., 2013).

68
3.4.1.4. Metallic oxides
Carboxylated nitrile, butadiene, and styrene-butadiene rubbers can be crosslinked
via the reaction of zinc oxide with the carboxylated groups on the polymer chains,
forming a zinc salt with the carboxylate groups (Dick, 2009). Polychloroprene
(neoprenes) and chlorosulfonated polyethylene are also vulcanized using metal oxides,
zinc oxide being most common (Dick, 2009).
3.4.1.5. Addition cure
Silicone rubber can use addition or condensation cures for room temperature
vulcanization for molding and casting applications (Dick, 2009). Addition cures are
catalyzed using platinum, and are favored for their toughness and use in high temperature
applications (Dick, 2009). Product types that use addition curing include: solid silicone
rubber, liquid silicones, silicone gels, 2-part silicone rubber, and UV-curable silicone
rubbers (Dick, 2009). The platinum catalyst creates a three-dimensional polymer
network by reacting the crosslinker’s Si-H groups with the vinyl groups of the silicone
polymer (Dick, 2009). Platinum catalysts used in silicone addition cures can be
deactivated, or poisoned with chemicals such as with nitrogen, sulfur, phosphorus, sulfur
vulcanized rubbers, polyurethanes, and tin associated with condensation cured silicone
rubbers (Dick, 2009).
3.4.1.6. Condensation-based cures
Silicone rubber is cured by condensation reactions for general mold making and
prototype applications. In condensation curing, terminal hydroxyl groups of silicone
polymers react with a siloxane curing agent in presence of a tin or organotitanium
catalyst and a small amount of water (Dick, 2009). This reaction releases volatile

69
compounds such as alcohols, acetic acid, and amines (Dick, 2009). Thus condensation
silicones undergo slight shrinkage during cure from release of volatiles, whereas
shrinkage is negligible for addition cures where there are no volatiles (Dick, 2009).
Product types that utilize condensation curing include 2-part silicone rubber, and 1-
component silicone rubbers (Dick, 2009). Tin-catalyzed or organotitanium condensation
cures are typically cheaper than platinum-catalyzed addition cures, and therefore
condensation cures are used more widely in economic manufacturing (Dick, 2009).
Condensation cured silicones are more tear resistant than addition cured silicones.
3.4.2. Fillers
Addition of inert materials dispersed in the bulk of an elastomer prior to curing,
can improve strength and hardness but typically at the expense of elasticity and resilience
(Leblanc, 2002). Size and shape of the fillers, as well as degree of dispersion are
important factors. Some fillers will improve properties of an elastomer; providing a
reinforcing effect that is characterized by improved stiffness, high resistance to tearing
and abrasion, and enhanced tensile strength (Donnet and Custodero, 2013). Other fillers
are diluents, and typically are used to reduce cost of the bulk product, typically without
any improvements on polymer performance (Rattanasom et al., 2007).
Particle size is the most fundamental property of a filler which affects
reinforcement of the elastomer the most. Particle sizes ranging from 1000-5000nm
provide a small reinforcement; particles less than 1000nm provide a medium
reinforcement; particles smaller than 100nm provide the strongest reinforcement (Hamed,
2000).

70
Fillers are typically hydrophilic, whereas most polymer matrices are more
hydrophobic due to the large quantity of hydrocarbons. To strengthen the reinforcing
effect, the interfacial bonding between a filler and its polymer matrix needs to be
improved (Kohls and Beaucage, 2002). To improve interactions between filler and
matrix, fillers are often surface modified. Modification of filler surface to improve
hydrophobicity is most commonly achieved through silane coupling agents, via the
hydrolysis of hydroxyl groups found on a filler’s surface.
3.4.2.1. Carbon Black/ Carbon Fly Ash
Carbon black is useful in increasing strength and hardness for elastomers, and is
the most common filler used in the rubber industry. Carbon black is a colloidal form of
elemental carbon, typically produced by the combustion of oil or natural gases. The
average particle sizes of carbon blacks for industrial use typically range from 10nm to
500nm (Kausar, 2017). Aggregate size also varies with particle size, and can consist of
four shape categories: spheroidal, ellipsoidal, linear, and branched (Kauser, 2017).
Particle size and shape can affect the filler-rubber interface and hence the reinforcement
of carbon black in elastomer systems; Van Der Waals forces between the carbon black
surface and the rubber matrix, mechanical interlocking of the rubber onto the filler
surface, and rubber chains grafted to carbon black surfaces via free radical reactions
between carbon atoms all provide a strong reinforcing effect (Dick, 2009). Smaller sized
carbon blacks have a greater adhesion to the rubber matrix and therefore provide a
stronger reinforcing effect, compared to larger sized carbon blacks with weaker adhesion
to the rubber matrix (Dick, 2009).

71
3.4.2.2. Clays/Silicas
Silica is the second most common filler used in the rubber industry, and is
commonly called a “white” filler. However, since silica is not quite as reactive with
rubber as carbon black, silane coupling agents are used to modify the surface of silica
particles. Clay is typically used as a cheap filler to reduce costs, and has a poor
reinforcing ability because of its large particle size and low surface activity (Tohsan and
Ikeda, 2014; Dick, 2009). To improve the reinforcing effect of clays in polymers, the
clays need to be intercalated or exfoliated. Successful intercalation or exfoliation of clays
improve mechanical, barrier, and thermal properties (Dick, 2009). This can be done via
solution blending, latex compounding, direct intercalation of molten polymer, and in-situ
polymerization (Dick, 2009).
3.4.2.3. Calcium Carbonate and Whitening Agents
Calcium carbonate is used in its most common form, calcite, as a filler in
elastomers. Calcite is produced from three different mineral sources: chalk, limestone,
and marble (Dick, 2009). Calcite produced from chalk is typically the purest form of
calcite, and consists of loosely bonded, uniform crystals that are about 3 microns in
diameter. Milling breaks bulk chalk deposits into its basic, 3 microns crystal size (Dick,
2009).
To achieve calcium carbonate particle sizes smaller than 3 microns, calcium
carbonate is precipitated by carbonation of a calcium hydroxide slurry (Dick, 2009). The
crystal phase, particle size, and shape can all be controlled by the reaction conditions and
additives (Dick, 2009). Most commonly, rhombic calcite crystals with a size of 50 – 100

72
nm, coated with fatty acids for improved compatibility with polymers are used for nano
fillers (Dick, 2009).
3.4.2.4. Natural Fibers
Natural or lignocellulosic fibers consist of helically wound cellulose micro fibrils,
embedded in amorphous lignin matrix. Cellulose (α -cellulose), lignin, pectins,
hemicellulose, and waxes are the major components of natural fibers. Lignocellulosic
fibers have been used extensively in composites, because of their low cost, low density
per unit volume, and sometimes acceptable specific strength (Abraham et al., 2013b).
Natural fibers have poor fiber/matrix interactions, water resistance and lower durability
(Abrahamn et al., 2013a). This is attributed to the weak interfacial bonds between
hydrophilic natural fibers and non-polar organophilic polymer matrices (Abraham et al.,
2011; Angellier et al., 2005). Commonly, natural fibers are surface treated to improve
interfacial adhesion with polymer matrices using physical, mechanical and/or chemical
approaches (Liu et al., 2008). Thermal stability of fibers is important when considering
their application as fillers. The manufacturing and processing of composites at high
temperatures can lead to degradation of the natural fiber, which can result in unfavorable
bulk properties.
3.4.3. Anti-degradants
Polymers are subject to degradation on exposure to environments such as: storage,
oxygen, heat, UV light and weathering, ozone, catalytic degradation from heavy metal
ions (Cu, Mn, Fe, etc.), and dynamic flex-fatigue (Dick, 2009). Aging due to heat results
in loss of elasticity and tensile strength, whereas oxygen-mediated failures (such as
caused by ozone) can result in extensive cracking as well as loss in elasticity and tensile

73
strength (Dick, 2009). Most anti-degradants include antioxidants and antiozonants,
typically classified into amine type anti-degradants and phenolic type anti-degradants
(Dick, 2009).
3.4.4. Blowing Agents
Blowing agents are used to create a cellular structure via a foaming process in
materials that undergo hardening or a phase transition, including polymers. A blowing
agent is typically added to a polymer in a liquid stage, undergoing a reaction that foams
and subsequently hardens to create a porous structure. The cellular structure in a matrix
reduces density while increasing thermal and acoustic insulation, with an overall increase
in relative stiffness compared to the original polymer (Dick, 2009).
There are typically two classes of blowing agents: physical, and chemical.
Physical blowing agents are endothermic, requiring heat to volatilize the liquid blowing
agent. Physical blowing agents in industrial applications include hydrocarbons such as
pentane, isopentane, and cyclopentane, and liquid carbon dioxide (Dick, 2009).
Chemical blowing agents create a cellular structure via the release of gaseous products
and typically undergo an exothermic reaction. Common chemical blowing agents include
isocyanate and water for polyurethanes, azodicarbonamide for vinyls, hydrazine and
other nitrogen-based materials for thermoplastic and elastomer foams, and sodium
bicarbonate for thermoplastic foams (Dick, 2009). Mixtures of physical and chemical
blowing agents are used in the production of flexible polyurethane foams with very low
densities. Using both physical and chemical blowing agents minimizes temperature
increases during synthesis, mitigating thermal degradation or damage (Dick, 2009). For

74
example, polyurethane foams utilize chemical blowing agents, isocyanate and water, in
combination with the physical blowing agent of liquid carbon dioxide.
3.4.5. Bonding promoters
Adhesion between two surfaces is the result of interatomic and intermolecular
interactions at its respective surfaces. Silane coupling agents are most commonly used to
improve adhesion between rubber and inorganic fillers or substrates (Liu et al., 2008).
Primers or other additives are used typically in coating formulations, and are most
commonly silicone-based (Dick, 2009). Other types of bonding promoters are used in
complex composites such as rubber reinforced with steel wires, or nylon fibers. Bonding
promoters used to improve adhesion of steel and nylon consist of resorcinol and a
formaldehyde donor (Dick, 2009). The type of formaldehyde donor impacts the adhesion
strength, and the kinetics of vulcanization. Therefore, it is important to optimize the type
of bonding promoter and loading in a formulation for an individual application.
3.4.6. Desiccants
Desiccants are used in rubber compounding to remove water that may be from
fillers, the bulk material, or a result of the vulcanization reaction. Having water present
in a rubber compounds can lead to porosity in structure, and therefore desiccants are
added. Calcium oxide is the most commonly used desiccant in rubber compounds, and
can be used across a wide variety of rubber chemistries (Dick, 2009). Calcium oxide
does influence the kinetics of vulcanization, and formulations need to be adjusted to
maintain desired physical properties (Dick, 2009).

75
3.4.7. Flame retardants
Transition metal materials, including oxides, salts, organometallics or metal
chelate complexes, are used as flame retardants for polymers. Metal halides such as
antimony oxide, zinc borate, aluminum hydroxide, and chlorinated paraffins are
commonly used as flame retardants (Dick, 2009).
3.4.8. Odorants and deodorants
Odorants and deodorants are often used to mask the distinct aromas associated
with natural and synthetic elastomers, often associated with sulfur based crosslinkers or
stabilizers (Dick, 2009). Odorants and deodorants are used to make the final product
acceptable to the user. Since fragrances are volatile at higher temperatures, most are used
in low temperature thermoplastics such as olefins, for extrusion and molding
applications.
3.4.9. Peptisers
Peptisers are added in the earliest stage of natural rubber manufacturing, helping
to break down the rubber rapidly during mastication to create a homogenous masticate
(Dick, 2009). Peptisers reduce the viscosity of natural rubber and latex by catalyzing
rubber mastication. Reducing the viscosity of natural rubber to a workable level can
reduce mixing time and thus provides time, energy, cost, and environmental benefits
(Dick, 2009). Traditionally, pentachloro triophenol based peptisers were used, but are
replaced by dibenzoamido diphenyl disulfide chemistries to reduce environmental health
risks (Dick, 2009).

76
3.4.10. Pigments
Pigments are used to improve heat and light resistance, as well as to add color to
elastomeric products. The selection of a pigment depends on the application
requirements for light stability, heat stability, resistance to bleed and migration, and
shade (Dick, 2009). Pigments are classified as organic, inorganic, or hybrid pigments.
Pigments are available in a variety of forms, including dry powders, color concentrates,
and liquids.
Organic pigments commonly used in plastics include quinacridones (red, violet,
orange), dioxazines (violet), isoindolines (yellow, orange, red), perylenes, flavanthrones,
and anthraquinones (Dick, 2009). However, organic dyes are sensitive to heat and
chemicals, and their color can fade with long-term sun exposure.
The most common inorganic pigments include oxides, sulfides, hydroxides,
chromates, and other metal complexes such as cadmium, zinc, titanium, lead, and
molybdenum (Dick, 2009). Inorganic pigments are more thermally stable than organic
pigments, and are opaquer and more resistant to migration, chemicals, and fading. The
most widely used white pigment is titanium oxide, which is used alone or with other
colorants to produce pastel shades. Inorganic pigment powders are insoluble in the
rubber matrix and therefore cannot phase separate and do not bloom to the surface of the
elastomer with use.
Hybrid pigments are formed from a stable dye sphere that surrounds an inorganic
substrate such as silica, titanium dioxide, or aluminosilicates (Dick, 2009). Silica is
commonly used as a filler in polymers, but has poor dispersion due to its innate
hydrophilic properties and preference to agglomerate (Liu et al, 2008). Dyes have been

77
used to modify silica to improve dispersion while providing visually appealing end
product colors.
3.4.11. Plasticizers/processing aids
Plasticizers and processing aids have the function of reducing the energy required
for processing while assisting in the dispersion of rubber additives (Bergman and
Trimbach, 2014). In general, plasticizers reduce hardness, stiffness, and mechanical
properties, but increase elongation at break and improve flexibility at low temperatures
(Petrovic et al., 2013). Plasticizers are often used to reduce the viscosity of the polymer
compound, allowing for the addition of fillers (Dick, 2009). Process aids which do not
modify the viscosity of the compound are commonly a combination of waxes, and/or
fatty acid salts at a concentration of 1-3 phr (Bergman and Trimbach, 2014).
The most common plasticizers are mineral oils, consisting of aromatic, napthenic,
and paraffinic oils (Petrovic et al., 2013). When selecting a plasticizer, compatibility
between the type of oil and the polymer matrix needs to be considered (Petrovic et al.,
2013).
3.4.12. Retarders
Retarders, also known as pre-vulcanization inhibitors, readily react with
accelerators and slowly release them for vulcanization. Ideal retarders increase the scorch
time at processing temperatures, without delaying the time to reach 90% cure and without
significantly changing the maximum rheometer torque (Dick, 2009). The most common
use of retarders is to increase the scorch time, therefore gaining flow time in a mold or

78
calendaring process. This is useful in applications where the polymer is underfilling the
mold, and increasing the preform weight results in thick flashes and flow lines.
3.4.13. Tackifiers
Tackifiers are chemical compounds used to increase tack, the stickiness of the
surface of an adhesive. Tackifiers are amorphous, glassy, low molecular weight
hydrocarbon polymers. Tackifiers can influence the color, odor, and stability of an
adhesive. Tackifers are used in applications where adhesion between two surfaces is
required. Resins, polymerized materials with a molecular weight less than 10,000, are
used with rubber to improve flow properties, in addition to increasing tack (Dick, 2009).
However, not all resins are tackifiers. Resins that are tackifiers as well include aromatic
resins (phenolic), petroleum-based resins, and plant based resins (wood rosin and
terpenes) (Dick, 2009).
3.4.14. Type IV allergy associated with elastomer compounding
Some elastomer compounding chemicals can cause a type IV allergic reaction,
clinically presented as non-immunological irritant contact dermatitis. Rubber additives
associated with a clinical type IV allergic response include over 30 chemicals, associated
with the chemical classifications of thiurams, benzothiazoles, mercaptos, ureas,
thiocarbamates, diamines, and others (Nettis et al., 2002). A type IV allergy is clinically
identified using a skin patch test, with readings taken 2 to 4 days post-exposure. Contact
dermatitis, characterized by erthematosquamous eczema, is characteristic of a type IV
allergic reaction.

79
3.5. Manufacturing Methods for Latex
Latex is an aqueous emulsion that contains a dispersion of polymer particles.
Elastomer latex systems include natural polyisoprene, and synthetic polyisoprene, nitrile,
polybutadiene-styrene, and acrylics (Dick, 2009). Latex systems need to be compounded
prior to manufacture, and vary in formulations due to polymer chemistry and structure, as
well as the presence of other chemical additives. In the case of natural rubber latex,
biochemical metabolites (such as proteins) entrapped in the latex during removal from
feedstock can affect compounding and therefore manufacturing.
Latex systems will need an emulsifying agent to maintain the emulsion, and
prevent phase separation of the polymer and water phases. Proteins and other
biochemical metabolites are emulsifying agents that stabilize natural latex, whereas
anionic and non-ionic soaps or detergents are typically used as emulsifying agents for
synthetic latex systems. Natural latex is typically buffered at a basic pH, whereas
synthetic latex requires a more neutral pH; this is attributed to the isoelectronic point and
colloidal stability of the emulsions. As a result, variations in polymer latex chemistry can
vary greatly and should be taken into consideration when developing a manufacturing
technique.
3.5.1. Dipping
Latex dipping is the most commonly used process for a variety of thin film
products. The dipping process consists of the following steps: insertion of a former into
compounded latex, removing the former, drying to remove liquids, followed by
vulcanization and finally stripping the product from the former. Formers are typically
made from aluminum, stainless steel, ceramics, or glass. Automated dippers are

80
commonly used for products such as gloves, balloons, and condoms, generating several
thousand per hour.
There are a few variations in the dipping process, depending on how thick the
final product needs to be. Straight dipping is used for the thinnest products, typically for
items 0.6 mm and thinner. Condoms are typically made using straight dipping, where a
glass former is typically dipped in latex, dried, a second layer is dipped, dried, followed
by the bead rolling, vulcanization, washing and stripping of the former using water spray.
Products should then be leached in water to remove excess compounding ingredients, and
then surface treated via chlorination to reduce stickness and tack. Condoms are then
tested according to ASTM and ISO standards, and then packaged for consumer use.
Coagulant dipping is used for products that have a thickness up to 1.5 mm, such
as gloves. Coagulant dipping varies from straight dipping in that the former is dipped
into an alcoholic solution of calcium salt, dried, and then dipped into latex. Coagulants
such as positively charged calcium ions, neutralize the negative electrical charge on
natural rubber particles and synthetic latex with anionic emulsifiers, which destabilize the
forces keeping colloids apart. Coagulant dipping is less effective on synthetic latex
systems that use non-ionic emulsifiers.
Heat sensitive dipping is used for products with a thickness up to 5 mm, including
baby nipples and some industrial products. To achieve such a thick dipped item, latex is
compounded with a heat sensitivity compound, such as polyvinylmethyl ether.

81
3.5.2. Open Cell Foams (Dunlop, Talalay)
Open cell elastomer foams, foams with interconnected pores, are typically
formulated from NRL or synthetic polyisoprene latex. There are two methods that are
used, the Dunlop and the Talalay methods which vary in how the foam is gelled prior to
vulcanization. The Dunlop process utilizes a chemical gelling agent, such as zinc oxide
whereas the Talalay process utilizes carbon dioxide gas and the formation of carbonic
acid as the gelling agent. The morphology of the open cell architecture also varies
between the methods, with the Talalay method having a more uniform cell structure.
The Dunlop method compounded latex with a gelling agent such as zinc oxide,
and is whipped to incorporate air into the compounded latex. After the latex is foamed,
poured into a mold and closed, and subsequently vulcanized. The foam is washed post-
vulcanization, and then dried.
In the Talalay method, latex is compounded and foamed without a gelling agent,
and then partially fills a large mold. The mold is closed, and the latex is expanded by
vacuum to fill the mold, followed by cooling of the mold to freeze the foam structure, and
introduction of carbon dioxide gas, with subsequent heating for vulcanization. The
carbon dioxide causes gelation in the Talalay method, via formation of carbonic acid and
lowering of the pH.

82
3.6. Manufacturing Methods for Rubber
Common dry rubber manufacturing techniques include closed cell foams,
extrusion, and molding. The chemical composition of rubber needs to be taken into
account during manufacturing, as the process of manufacture varies among the types of
elastomer used. Thermoplastic elastomers are more readily molded due to their thermal
transitions around their respective melting points, whereas thermosets do not have a
melting point. Thermoset elastomers typically are cured or vulcanized while being
manufactured, a major distinction from thermoplastics which do not need a cure cycle.
Natural rubber has multiple biochemical metabolites, such as proteins and phospholipids,
entrapped in its dry rubber matrix, which can affect manufacturing and need to be taken
into account during compounding.
3.6.1. Closed cell foams (semi-open cell foams)
Closed cell foams, foams with pores that are not connected, are dense foam
products. Closed cell foams are used in the automotive industry, construction for
insulation and other thermal management applications, and for consumer products such
as wetsuits, gloves, and orthopedic braces. EPDM, silicones, natural rubber, and
neoprene are all commonly used rubbers for closed cell foams.
Closed cells foams are made using a one or two step process with a gas, typically
either nitrogen or supercritical carbon dioxide. In one-step foaming, carbon dioxide
dissolution, expansion and cooling of samples takes place inside a high pressure mold.
The two-step foaming process, expansion of the sample into a foam can be done inside

83
the mold used to initially dissolve the carbon dioxide, or outside of the initial mold by
heating carbon dioxide-saturated samples using an oil-bath or hot press.
3.6.2. Extrusion
Extrusion is a process in which a polymer is forced through a die of the required
cross section under pressure. Typically, rubber products are extruded and then cross-
linked or vulcanized. There are two types of extrusion processes: ram extrusion and
screw extrusion. Both extrusion processes begin with unvulcanized rubber compound
being fed into an extruder. In screw extrusion, the revolving screw within the extruder
will begin to bring rubber into the die, increasing the pressure and temperature of the
material as it approaches the die. In ram extrusion, a batch approach is used where a
piston pushes a given volume of material through the orifice of the extrusion die. When
the material reaches the die in both processes, the pressure forces the material through the
die orifice, causing swelling of the materials. Since this swelling occurs, extruded parts
typically have tolerances on their cross sections. After extrusion, the material is
vulcanized, and may shrink. Extrusion dies are precise and specific tools, often custom
made for a given product. Commonly extruded rubber materials include EPDM,
neoprene, SBR, nitrile and silicone.
3.6.3. Molding
3.6.3.1. Compression Molding
Compression molding is the most common type of mold used in the rubber
industry. Typically, compression molding consists of placing a precut or shaped polymer
or composite into a two-piece mold that is closed. The pressure applied by the press

84
forces the material to fit the shape of the mold. Any excess material flows out of the
mold and is termed flash. Tire treads are typically cured in a compression mold.
3.6.3.3. Transfer Molding
Transfer molding is used to mold complicated shapes, not possible using
conventional compression molding. Transfer molding has a more complicated mold than
those used in compression molding; transfer molds have an area of the mold which holds
uncured polymer and then distributes this uncured polymer into the mold cavity where
curing occurs.
3.6.3.3. Injection Molding
Injection molding traditionally were used for thermoplastics, but has been
developed so that rubber compounds can be molded and vulcanized by this method.
Injection molding follows a cycle of feed, injection, and demolding, with low rejection
rates and lower finishing costs.
3.6.4. Blown Film Extrusion
Blown film extrusion is the most common method to make plastic films,
especially for the packaging industry. Elastomers are typically not processed using
blown film extrusion, due to the lack of crystallization from thermal cooling by most
elastomers which is required for successful blown film extrusion. Elastomers such as
natural rubber latex can be used in a blend with highly crystalline thermoplastics, such as
low-density polyethylene (Mahapram and Poompradub, 2011). In general, blown film
extrusion involves the extrusion of a tube of molten polymer through a die, and inflating
it to several times its initial diameter to form a thin film bubble. This bubble is then
collapsed and used a lay-flat film or can be made into bags.

85
Blown film extrusion consists of four main steps:
1. Thermoplastic material starts in a pellet form, where it is compacted and melted to
form a continuous, viscous liquid. The molten thermoplastic is then extruded
through an annular die.
2. Air is then injected through a hole in the center of the die, and the resulting
constant and even pressure gradient causes the extruded melt to expand into a
bubble. Air leaving the bubble is replaced as a constant rate, to ensure uniform
thickness of the film.
3. The bubble is cooled via convective air, and begins to solidify. The line at which
the polymer transitions from molten to solid is termed the frost line, due to the
change in polymer opaqueness attributed to the diffraction of light by crystallites
in the polymer that are formed during cooling.
4. After solidification, the film moves into a set of nip rollers that collapse the
bubble and flatten it into two flat film layers.

86
Chapter 4: Mechanical properties of type I circumallergenic & type IV
hypoallergenic guayule natural rubber latex thin films.
J. Lauren Slutzky a, and Katrina Cornish a,b
aOhio State University, Department of Food, Agricultural and Biological Engineering,
1680 Madison Avenue, Wooster, Ohio 44691 USA
bOhio State University, Department of Horticulture and Crop Science, 1680 Madison
Avenue, Wooster, Ohio 44691 USA
*Corresponding author: Katrina Cornish, Williams Hall, 1680 Madison Avenue,
Wooster, Ohio 44691, USA. Tel: 330-263-3982 Fax: 330-263-3887 Email:
Abstract
Type I latex allergy sensitization and subsequent allergic reactions to Hevea
natural rubber latex proteins have created an industry demand for thin film barriers that
circumvent the allergic response (i.e. are circumallergenic). Other allergens associated
with natural rubber and synthetic polymer thin films are attributed to residual thiazole,
thiuram, and carbamate accelerators that can cause type IV allergies, characterized by
contact dermatitis or delayed contact hypersensitivity. Type I circumallergenic and type
IV hypoallergenic thin films were created utilizing natural rubber latex from the plant
species Parthenium argentatum Gray (guayule), cured with the accelerators diisopropyl
xanthogen polysulphide (DIXP) and zinc diisononyl dithiocarbamate (ZDNC). Guayule
latex is circumallergenic with respect to type I latex allergy, because its proteins do not
cross-react with Hevea associated allergic proteins. Type IV allergies are diminished
because DIXP is consumed during the vulcanization process, and skin tests have shown

87
that ZDNC does not cause dermal reactions or delayed contact hypersensitivity, because
it does not bloom to the surface. These type I circumallergenic and type IV
hypoallergenic thin films have mechanical properties superior to those described in
American Standard for Testing Materials D 3577, the standard for rubber surgical gloves,
and could be used to make commercial medical thin film barriers, such as medical gloves,
condoms, balloons and dental dams.
Keywords: Guayule, natural rubber latex, type I latex allergy, type IV latex allergy,
elastomer thin films
4.1. Introduction
Elastomeric medical products, such as examination and surgical gloves, catheters,
masks, dental dams, orthodontic rubber bands, and condoms often are made from natural
rubber latex (NRL). NRL is derived from the plant species, Hevea brasiliensis, Muell
Arg. commonly known as the Brazilian or Para Rubber Tree. However, NRL contains
numerous allergens capable of eliciting a spectrum of type I allergic responses, mediated
by IgE, ranging in severity from contact dermatitis, contact urticaria, and delayed
hypersensitivity through systemic reactions, such as hives and edema, to life-threatening
anaphylaxis and death. Estimates of healthcare workers with immunogenic NRL
immediate hypersensitivity have ranged from 3% to 17.9%, depending on populations
studied and criteria on what defines an allergic response (Hamann et al., 2001; Haman et
al., 1998; Nabavizadeh et al., 2009; Liss and Sussman, 1999). Direct skin contact by
vulcanized elastomer products also can induce type IV delayed hypersensitivity reactions
mediated by antigen specific sensitized T lymphocytes, and are attributed to residual
accelerators used to crosslink natural, as well as synthetic, diene elastomers (Pak et al.,

88
2012). Type IV contact dermatitis to rubber products is one of the most common causes
of occupational contact dermatitis (Meyer et al., 2000). Also, eczema associated with a
type IV allergic response can increase susceptibility to a type I sensitization and
immunogenic response for individuals with high exposure to medical NRL products, such
as gloves (Miri et al., 2007).
Natural rubber latex derived from guayule is a circumallergenic (circumvents the
allergic response) alternative to Hevea derived NRL. Purified guayule NRL contains less
than 1% of the proteins in Hevea NRL, and 90% of the trace proteins are a single protein,
cytochrome P450 oxidase, an allene oxide synthase (Cornish et al., 2008; Cornish et al.,
2006; Pan et al., 1995). This protein is known to be poorly immunogenic, and the P450-
protein family has not been associated with allergic reactions in humans (personal
communication, Dr. H.P.Rihs, BGFA, Ruhr-University-Bochum, Germany). There are
no detailed studies of the remaining 10% of guayule NRL proteins because the amount of
these proteins is too small to either evoke an immunogenic response or to concentrate the
individual proteins sufficiently to permit an effective study. As a group, concentrated
total guayule latex proteins were found to be less than normally immunogenic in rabbits
(Siler et al., 1996). Anti-guayule protein murine polyclonal IgG did not cross-react with
Hevea latex proteins (Siler et al., 1996). Furthermore, in vitro studies have shown that
Hevea latex-specific human IgE antibodies do not bind to guayule latex proteins, and in
vivo studies confirmed guayule latex proteins to not cause reactions in Hevea-sensitized
healthcare workers (Siler et al., 1996; Carey et al., 1995).
Type IV allergens in medical natural and synthetic elastomer products are
attributed to residual accelerators used to increase the rate and efficacy of sulfur cross-

89
links formation and, in some cases, to anti-oxidants. The commonly used rubber
accelerators, thiazoles, thiurams, and carbamates, are recognized by the U.S. Food and
Drug Administration as sensitizing agents capable of eliciting type IV allergic reactions
(van Jole, 2008). Other health hazards associated with thiuram and carbamate rubber
accelerators include formation of fugitive N-nitrosamines, known carcinogens and
teratogens. Current alternatives for diene elastomer crosslinking include gamma and UV
irradiation, organic peroxide cures, zinc oxide activators without accelerators (utilizing
the carboxyl-zinc ionic bond), and placement of functional groups onto the polymer
backbone that can form crosslinks post product fabrication (van Jole, 2008). However,
since gamma and UV irradiation, and organic peroxides create carbon and not sulfur
crosslinks, they generally possess inferior mechanical properties compared to vulcanized
rubbers (van Jole, 2008).
The rubber accelerators, zinc diisononyl dithiocarbamate (ZDNC) and diisopropyl
xanthogen (DIXP), utilize sulfur in crosslinks, and have diminished residual chemicals
associated with type IV allergens (Chakraborty and Couchman, 2006). ZDNC has a
lower allergenic potential than conventional dithiocarbamates due to its high molecular
weight (Chakraborty and Couchman, 2006). This limits its ability to bloom to the
surface of latex films, and less ZDNC can be extracted from finished rubber articles
compared to common industry accelerators such as ZBEC (Chakraborty and Couchman,
2006). DIXP is a fugitive xanthate accelerator which, unlike other fugitive accelerators,
is consumed completely into sulfur crosslinks, and the byproducts of this reaction are
volatile isopropanol and carbon disulfide (Chakraborty and Couchman, 2006). DIXP
contains no nitrogen, and so cannot form carcinogenic and teratogenic N-nitrosamines.

90
The objective of this study was to optimally vulcanize guayule NRL (GNRL) with
the accelerators DIXP and ZDNC to create type I circumallergenic and type IV
hypoallergenic thin film elastomers with mechanical properties suitable for surgical
gloves, and other thin film elastomer products, such as catheters and condoms. The
materials made in this study are the first type I circumallergenic and type IV
hypoallergenic NRL thin films reported with mechanical properties superior those
specified in American Standard for Testing Materials (ASTM) D 3577, the standard for
rubber surgical gloves.

91
4.2. Experimental
4.2.1. Materials and sample preparation
4.2.1.1. Emulsion chemistry/compounding
GNRL was made using a base compounding recipe (Table 4.1) while varying the
amount of the accelerators DIXP and ZDNC (Robinson Brothers Ltd, UK). The amount
of accelerators and other materials were added to GNRL at specified concentrations
based on parts per hundred dry rubber (phr). The sulfur emulsion, antioxidant dispersion,
and zinc oxide dispersion were supplied by Akron Dispersions (Akron, OH, USA) and
the ammonium hydroxide was supplied by W.W. Grainger, Inc (Salt Lake City, UT,
USA). Deionized water was added to the compounded emulsion until 48% solids by
volume was achieved, and was prevulcanized for 2.5 hours while stirring with a 30 rpm
hand mixer.
Table 4.1. Latex Compound Recipe.
Quantity (phr)
Guayule NRL 100
Sulfur 2
Ammonium Hydroxide 1
ZnO 0.3
Antioxidant 2
ZDNC 0.2, 0.4, 0.6, 0.8, 1.0, 1.2 or 1.4
DIXP 1, 1.2, 1.4, 1.6, 1.8, 2.0, 2.1 or 2.2
4.2.1.2. Thin film manufacture by dipping
Thin film elastomer products are manufactured by dipping formers into emulsions
with subsequent heating to remove liquids and vulcanize the NRL. Initially, a stainless

92
steel former was heated to 70 ºC and dipped (10 second dwell) into a coagulant solution
(25% aqueous calcium nitrate in 70% isopropyl alcohol). After the solvent evaporated,
the coagulant-coated former was reheated to 70 ºC then dipped into a compounded latex
emulsion and held there for different dwell times during which a thin film of coagulated
latex was deposited (dwell times varied from 5, 15, 30, 45, and 60 sec). Once the latex
gelled via heating (15 min at 100 ºC), it was leached in water (30 min at 55 ºC), followed
by stripping of the former and subsequent vulcanization of the rubber article (20 min at
105 ºC). The rubber article was then placed in a tumble dryer post-vulcanization (60 min
at 60 ºC). All guayule natural rubber latex (GNRL) thin films in this study were made
using a Diplomat automated dipper (DipTech Systems Inc., Kent, OH, USA).
4.2.2. Tensile Properties
Four dumbbell samples of each compound at each dwell time were cut using Die
C according to ASTM D 412 (ASTM International, 2013a). Evaluation of the tensile
mechanical properties followed ASTM D 412 and was determined using an Instron 3366
with Bluehill v. 2.17 software package (Instron, Norwood, MA, USA) [15]. Samples
were tested using a crosshead speed of 500 mm/min at room temperature (26°C). The
reported mean ± the standard error from the mean (SE) values are averages of at least 4
samples. In addition, statistical analysis was performed using Minitab, version 16.0
(State College, PA). Significant differences (P-values < 0.05) in tensile data amongst
GNRL compounds for each film thickness were determined using one-way analysis of
variance (ANOVA) and the Fisher method.
4.2.3. Dynamic Mechanical Analyzer (DMA)

93
The glass transition temperatures of the GNRL thin films were studied using a TA
Instrument Q800 (New Castle, DE), as previously described (Monadjemi et al., 2016).
The samples were equilibrated at -110°C and subsequently heated to 150°C with a
heating rate of 5°C/min. The sample dimensions were approximately 25 x 3.0 x 0.7 mm3
with an amplitude of 15 μm, preload of 1N, and frequency of 1 Hz using the tension film
clamps. The values presented are averages of four samples.
4.2.4. Differential Scanning Calorimetry (DSC)
TA Instruments Q100 (New Castle, DE) differential scanning calorimetry (DSC)
was used to determine the state of the crosslinked elastomers as a function of
temperature, using a previously described procedure (Musto et al., 2016). The average
glass transition temperatures were determined from thermograms obtained by heating 5
to 10 mg samples from -20 to 200 ºC at a rate of 10 ºC/min. The samples were allowed
to anneal at 200 ºC for 3 to 5 min to remove the thermal history of the polymers, and
subsequently cooled to -85 ºC, held for 3 to 5 min, and reheated from -85 to 200 ºC with
a heating rate of 10 ºC/min. The reported values are averages of four samples.
4.2.5. Thermogravimetric Analysis (TGA)
Thermogravimetric analysis (TGA) was used to study thermal decomposition
properties using a TA Instruments TGA 5000 (New Castle, DE). The samples were
heated under nitrogen from room temperature to 500 ºC with a heating rate of 20 ºC/min.
The reported values are averages of four samples.

94
4.3. Results and Discussion
4.3.1. Dwell time and film thickness
When the film thickness for each respective dwell time was measured for four
replicate samples, each set had a standard error less than 0.01 mm (Table 4.2). The
relationship between dwell time and average thin film thickness showed a linear
relationship (R2=0.98, df = 3).
Table 4. 2. Dwell time and average thin film thickness, SEM < 0.01 mm
Dwell Time (s) Average Film
Thickness (mm)
5 0.15
15 0.19
30 0.21
45 0.23
60 0.26
4.3.2. Tensile properties
3D surface graphs were created using mesh points based on an average of four
samples, to show how modulus at 500% elongation (MPa), tensile strength (MPa), and
ultimate elongation (%) varied with different levels of DIXP and ZDNC for each film
thickness (Fig. 4.1).
The modulus at 500% elongation typically was not significantly different across
film thicknesses for each formulation. Exceptions are films made with 1.0 phr DIXP and
0.4 phr ZDNC, which showed significant differences in modulus at 500% elongation for
the 150 and 190 μm thinner films and the thicker films of 210, 230, and 260 μm. The

95
modulus was statistically higher for the thinnest films of 150 μm compared to the thickest
films of 260 μm for the following formulations: 1.4 phr DIXP for all ZDNC loadings
above 0.6 phr, and 2 phr DIXP for ZDNC loadings of 1 phr, 1.2 phr. There was no
statistical difference in modulus for film thicknesses for the formulations of 2 phr DIXP
with 0.6 phr ZDNC, and 2 phr DIXP with 0.8 phr ZDNC, nor at comparative film
thicknesses.
The modulus at 500% elongation in the thinnest films was particularly sensitive to
the ratio and concentration of the two accelerators (Figure 4.1(a) and (d)). The thinnest
film (Figure 4.1(a)) has a modulus made with 1.4 phr DIXP was statistically significantly
higher than for films made with 2.0 phr DIXP, with ZDNC concentrations of 0.4, 0.6, and
0.8 phr. However, this difference was not observed in treatments containing higher
concentrations of ZDNC, such as 1.4 phr DIXP with 1.0 phr and 1.2 phr ZDNC
compared to 2.0 phr DIXP with 1.0 phr and 1.2 phr ZDNC. In the thicker films (Figure
4.1 (g), (j) and (m)) modulus at 500% elongation became quite uniform over the different
accelerator concentrations and ratios, with the exception of the 1 phr DIXP, with 0.4 phr
ZDNC film was being statistically lower than films from all other formulations at the
same film thicknesses.
Tensile strength varied considerably among all film thicknesses in the tested
accelerator treatments. Tensile strength (Figure 4.1 (b), (e), (h), (k) and (n)) was
strongest in the films made with 2.0 phr DIXP/0.6 phr ZDNC and 2.0 phr DIXP/0.8 phr
ZDNC for all film thicknesses, and these had similar strength to each other. The films
made with 2.0 phr DIXP/0.6 phr ZDNC, and 2.0 DIXP/0.8 phr ZDNC, were significantly
stronger than films from the other formulations and across the different thicknesses.

96
Relative minima in tensile strength were observed in 1.4 phr DIXP/1.2 phr ZDNC films
(Figure 4.1 (b) (e) and (k)), which were significant lower than films made with 2.0 phr
DIXP/0.8 phr ZDNC, as well as 2.0 phr DIXP/0.6 phr ZDNC.
The largest ultimate elongations were obtained in 2.0 phr DIXP/0.8 phr ZDNC
(Figure 4.1 (c), (f) and (i)) across most thicknesses. The thinnest 2.0 phr DIXP/0.8 phr
ZDNC films had an elongation at break of 1888 ± 78.68 % (Figure 4.1(c)), significantly
higher than all other films except those made with 2.0 phr DIXP/0.6 phr ZDNC. The
lowest ultimate elongations for most films occurred in 1.4 phr DIXP/1.2 phr ZDNC, and
1.6 phr DIXP/1.2 phr ZDNC (Figure 4.1 (c), (f), (i) and (l)), and these were statistically
lower than 2.0 phr DIXP/0.8 phr ZDNC and the 2.0 phr DIXP/0.6 phr ZDNC
formulations. In contrast, the thickest films (Figure 4.1(o)) had maximum ultimate
elongations in 1.4 phr DIXP regardless of the level of ZDNC, and were statistically
significantly higher than the minimum ultimate elongations in 2.0 phr DIXP/0.6 phr
ZDNC.

97
Fig. 4. 1. 3D graphs of mechanical properties for guayule thin films: modulus at 500%
elongation (MPa), tensile strength (MPa), and ultimate elongation (%), while varying the
amount of rubber accelerators DIXP (phr) and ZDNC (phr) for different film thicknesses.
DIXP: diisopropyl xanthogen polysulphide; phr: parts per hundred dry rubber; ZDNC:
zinc diisononyl dithiocarbamate.
Film
Thickness
(µm)
Modulus at
500% Elongation (MPa)
Tensile Strength (MPa)
Ultimate Elongation (%)
150
190
210
230
260
(a) (b) (c)
(d) (e) (f)
(l)
(i) (h) (g)
(m)
(k) (j)
(n) (o)

98
Three formulations in Fig. 4.1 were selected for thermal analysis (Table 4.3). The
formulation with the best tensile properties (DIXP 2.0 phr/ ZDNC 0.8 phr) was selected,
as well as two formulations that had statistically inferior mechanical properties (Table
4.3) and a total accelerator concentration higher (DIXP 2.2 phr/ZDNC 0.4 phr) or lower
(DIXP 1.0 phr/ ZDNC 0.4 phr) than the optimal formulation (Table 4.3). This allows for
thermal analysis across formulations with different crosslinking densities, including
optimally cured films, an under-crosslinked material that undergoes ductile fracture, and
an over-crosslinked material that undergoes brittle fracture.
Table 4.3. Tensile data for GNRL thin films, which will be used for subsequent thermal
analysis via DMA, DSC, and TGA.
Sample Tensile Properties:
Modulus at
500% Elongation
(MPa)
Tensile
Strength (MPa)
Ultimate
Elongation (%)
Guayule NRL:
DIXP 1.0; ZDNC 0.4 0.58 ± 0.04 0.76 ± 0.06 884 ± 41
DIXP 2.0; ZDNC 0.8 1.58 ± 0.02 28.96 ± 2.57 1888 ± 78
DIXP 2.2; ZDNC 0.4 1.68 ± 0.07 18.70 ± 5.64 961± 334
4.3.3. Dynamic Mechanical Analysis (DMA)
The glass transition temperature (Tg) is the temperature at which polymers
transition from glassy behavior, where segmental motion is restricted, to rubbery
behavior that is characterized by molecular relaxations involving cooperative segmental
motion. The segmental motion of polymers allows for reversible strain in a material, the
defining behavior of an elastomer. As a result, characterization of the Tg can provide
insight into the physio-chemical interactions of materials, and how molecular motion can

99
become hindered due to vulcanization. The Tg typically increases in polymers that
undergo more extensive crosslinking, as molecular segmental motion becomes hindered
by inter- and intra-molecular crosslinks.
A low frequency of 1 Hz was used to test film samples, since higher frequencies
can increase the Tg due to reduced molecular relaxation of the polymeric system. Storage
moduli transitions were observed between -54 ºC and -49 ºC for all GNRL films (Table
4.4 and Fig. 4.2) but the initial temperature transition occurred at a lower temperature in
the optimal film than in the others. Storage modulus decreased with increasing
temperature above Tg (Fig. 4.2). The two suboptimal films, which had different tensile
properties (Table 4.3, p <0.05) did not have statistically significant different storage
moduli at respective temperatures (Fig. 4.2). However, the film with the optimal
performance (DIXP 2.0 phr/ ZDNC 0.8 phr) maintained much higher storage modulus
over temperatures above its Tg than the other two films.
Tan delta (δ) is the ratio of loss modulus (E”) to storage modulus (E’), and
reflects the energy dissipated as heat during a heating cycle (Fig. 4.2). The damping peak
occurred between -39 °C and -37 °C for all three films, higher than the glass transition
values derived from the storage modulus (Table 4.4), which is very common in the
analysis of elastomers. The loss modulus peak is another way to determine the glass
transition: an increase in the loss modulus represents an increase in viscous behavior due
to increased cooperative segmental motion. The loss modulus peak occurred between -49
°C and -47 °C (Table 4.4). The storage modulus (Fig. 4.2) of the optimized film (DIXP
2.0 phr/ZDNC 0.8 phr) is significantly stronger above Tg compared to the suboptimial
films.
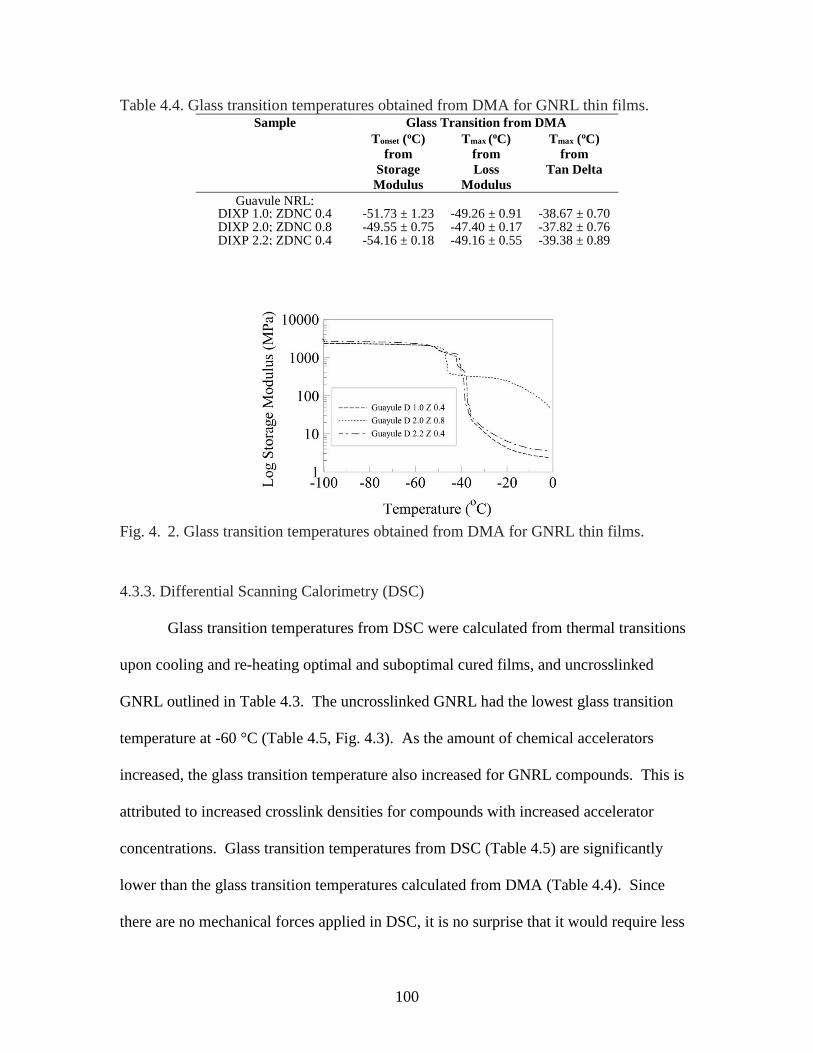
100
Table 4.4. Glass transition temperatures obtained from DMA for GNRL thin films. Sample Glass Transition from DMA
Tonset (oC)
from
Storage
Modulus
Tmax (oC)
from
Loss
Modulus
Tmax (oC)
from
Tan Delta
Guayule NRL: DIXP 1.0; ZDNC 0.4 -51.73 ± 1.23 -49.26 ± 0.91 -38.67 ± 0.70 DIXP 2.0; ZDNC 0.8 -49.55 ± 0.75 -47.40 ± 0.17 -37.82 ± 0.76 DIXP 2.2; ZDNC 0.4 -54.16 ± 0.18 -49.16 ± 0.55 -39.38 ± 0.89
Fig. 4. 2. Glass transition temperatures obtained from DMA for GNRL thin films.
4.3.3. Differential Scanning Calorimetry (DSC)
Glass transition temperatures from DSC were calculated from thermal transitions
upon cooling and re-heating optimal and suboptimal cured films, and uncrosslinked
GNRL outlined in Table 4.3. The uncrosslinked GNRL had the lowest glass transition
temperature at -60 °C (Table 4.5, Fig. 4.3). As the amount of chemical accelerators
increased, the glass transition temperature also increased for GNRL compounds. This is
attributed to increased crosslink densities for compounds with increased accelerator
concentrations. Glass transition temperatures from DSC (Table 4.5) are significantly
lower than the glass transition temperatures calculated from DMA (Table 4.4). Since
there are no mechanical forces applied in DSC, it is no surprise that it would require less

101
energy to undergo cooperative segmental motion, resulting in a lower glass transition
temperature when compared to DMA data.
Table 4.5. Glass transition temperatures obtained from DSC for GNRL thin films.
Average of four samples.
Sample Glass Transition (Second Melting)
Teig (oC) Tmg (oC)
Guayule NRL:
Uncrosslinked -60.39 ± 1.02 -60.25 ± 1.95
DIXP 1.0; ZDNC 0.4 -56.75 ± 2.58 -54.42 ± 2.14
DIXP 2.0; ZDNC 0.8 -54.01 ± 2.12 -49.96 ± 0.74
DIXP 2.2; ZDNC 0.4 -52.45 ± 1.18 -48.25 ± 0.86
Teig = extrapolated onset temperature, Tmg = midpoint temperature
Fig. 4. 3. Differential scanning calorimetry of GNRL formulations. Average of four
samples.
4.3.3. Thermogravimetric Analysis (TGA)
Thermal degradation was observed from TGA curves depicting the weight loss of
GNRL (Fig. 4.4) outlined in Table 4. There were no distinctive differences between the
three different films’ peak temperature for major weight loss, which was 371 ºC. (Fig.
4.4). The uncrosslinked GNRL had a significantly lower percent residue remaining,
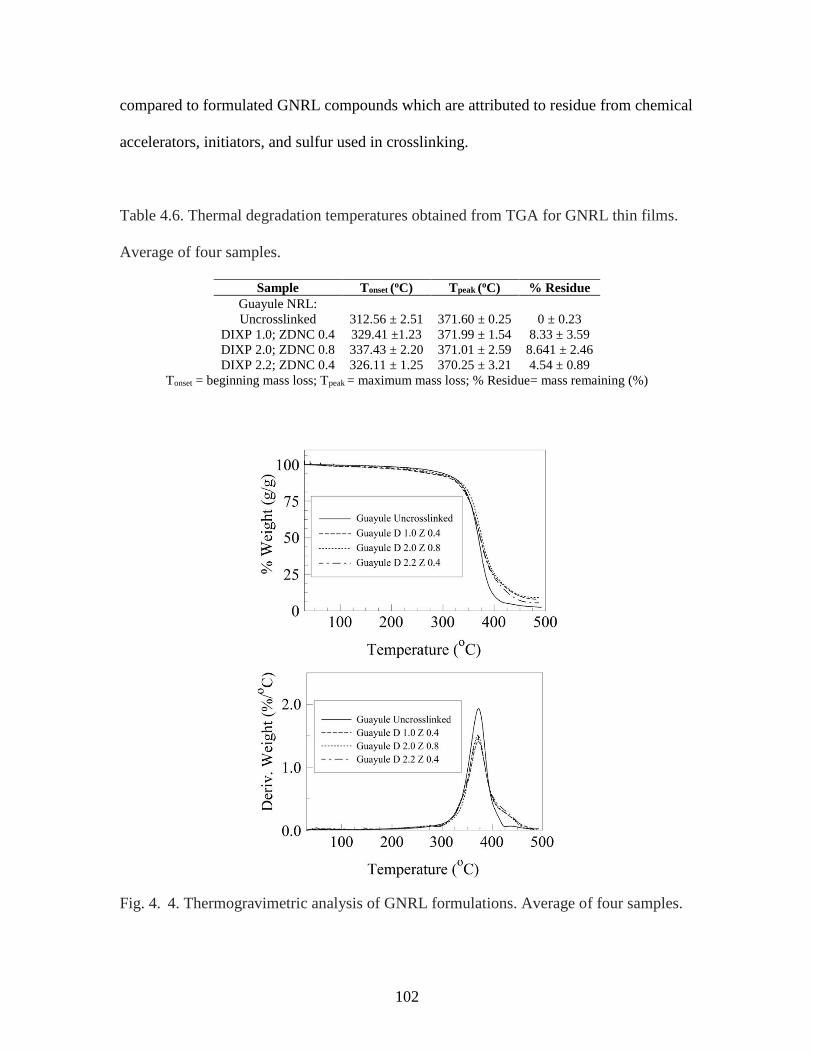
102
compared to formulated GNRL compounds which are attributed to residue from chemical
accelerators, initiators, and sulfur used in crosslinking.
Table 4.6. Thermal degradation temperatures obtained from TGA for GNRL thin films.
Average of four samples.
Sample Tonset (oC) Tpeak (oC) % Residue
Guayule NRL:
Uncrosslinked 312.56 ± 2.51 371.60 ± 0.25 0 ± 0.23
DIXP 1.0; ZDNC 0.4 329.41 ±1.23 371.99 ± 1.54 8.33 ± 3.59
DIXP 2.0; ZDNC 0.8 337.43 ± 2.20 371.01 ± 2.59 8.641 ± 2.46
DIXP 2.2; ZDNC 0.4 326.11 ± 1.25 370.25 ± 3.21 4.54 ± 0.89
Tonset = beginning mass loss; Tpeak = maximum mass loss; % Residue= mass remaining (%)
Fig. 4. 4. Thermogravimetric analysis of GNRL formulations. Average of four samples.

103
4.4. Conclusion
Dipped thin film elastomers are employed in medical applications such as surgical
balloons, catheters, dental dams, condoms and gloves. One of the most taxing
specifications is for surgical gloves, which must protect patients and surgeons during
hours of surgery without failing. The ASTM D3577 standard specification for natural
rubber surgical gloves demands tensile strength greater than 24 MPa, modulus at 500%
elongation of less than 5.5 MPa and ultimate elongation greater than 750% (ASTM
International, 2013b). All three performance parameters are exceeded by many of the
thin films formulated and made in this study (Figure 4.1(a) to (o)).
Crosslinking GNRL to confer good physical properties requires a synergistic
amount of primary and secondary accelerator and this has to be in balance with the added
sulfur crosslinker (Gao et al., 2013, Vennemann et al, 2013). The strongest GNRL films
had an accelerator/sulfur ratio of 1.4, which indicates that the accelerator package of 2
phr DIXP/0.8 phr ZDNC is semi-efficient. Semi-efficient accelerator systems are able to
promote a higher rate of crosslinking compared to conventional systems, incorporating
polysulfidic, di and monosulfidic bonds (Nocil Ltd., 2010).
Vulcanization temperature also significantly impacts crosslink structure and tensile
properties. Optimum crosslinks are formed using the lowest temperature possible,
regardless of the type of accelerator used (Nocil Ltd., 2010). In our accelerator package,
the primary accelerator is DIXP and it has a very short vulcanization plateau which
occurs from 80-100 °C (Chakraborty and Couchman, 2006). Therefore, the use of ultra-
fast accelerators such as ZDNC increases the rate of cure despite the relatively low cure
temperature required. ZDNC, a dithiocarbmate, induces a faster cure rate and higher

104
crosslink density, which can be attributed to higher crosslink density and improved
tensile strength of films (Fig. 1B, E, H, K and N) compared to using DIXP as a sole
accelerator (Nocil Ltd., 2010).
There are over 50 different types of vulcanization accelerators commercially
available, with various accelerator classes utilizing different crosslinking mechanisms
(Nocil Ltd., 2010). Within accelerator classes, reaction kinetics can vary due to
differences in molecular weight and solubility in the polymer emulsion (Nocil Ltd.,
2010). Therefore, this study of DIXP and ZDNC is unique to GNRL with its emulsion
chemistry containing high amounts of potassium hydroxide. A different stabilization
package for GNRL may change accelerator reaction kinetics, crosslink density, and,
ultimately, tensile properties.
The GNRL thin films in this study have mechanical properties that exceed other
glove films of natural or synthetic origin (Krutzer et al., 2014). Studies of commercially
available polychloroprene, anionically polymerized polyisoprene, and zinc polymerized
polyisoprene thin films reported lower tensile strengths and higher ultimate elongations
than NRL thin films (Krutzer et al., 2014). The crosslinking mechanisms for the
commercial films tested were unspecified, and so it is not known whether these films
contain accelerators capable of causing a type IV allergic response (Krutzer et al., 2014),
although it seems likely.
Type I circumallergenic elastomeric thin films have been created using synthetic
polymers such as polychloroprene, and polyisoprene, yet all have poorer tensile
properties than the GNRL films in this study (Chen et al., 2011; Tao, 2002).
Polychloroprene films have been reported utilizing DIXP and ZDNC, in addition to the

105
accelerator DPG (a known type IV allergen) but these films contain residual DPG,
increasing their type IV allergic potential, and did not even meet the considerably lower
performance requirements set by the ASTM standards for synthetic surgical gloves (van
Jole, 2006). Polychloroprene was also used to make type I circumallergenic and type IV
hypoallergenic thin films using only DIXP as accelerator, but did not meet ASTM
standards for synthetic surgical gloves (Krutzer et al., 2014). However, the GNRL films
created in our study have superior ultimate elongations, and are softer than the
polychloroprene films (Chen et al., 2011). Softer materials require less force for material
deformation, minimizing hand fatigue for surgeons during hours of surgery.
Other type I circumallergenic and type IV hypoallergenic thin film elastomers
have been made from nitrile butadiene rubber. Carboxyl functional side groups were
added to nitrile monomer units to induce crosslinking without sulfur or accelerators (Tao,
2002). However, these nitrile films have inferior ultimate elongations, and are 2.5x stiffer
than the GNRL thin films made in our study (Tao, 2002).
Overall, GNRL thin films have superior properties for glove applications than
other type I circumallergenic and type IV hypoallergenic elastomers (Cornish et al., 2007;
Krutzer et al., 2014; Chen et al., 2011; Tao, 2002), creating a unique combination of
strength, high elongation and softness (as shown by its low modulus at 500% elongation).
Thus, gloves made from GNRK filkms combine outstanding disease protection with
consumer comfort unmatched by other elastomeric materials.
Future research to optimize DIXP and ZDNC in other polymer lattices will
determine the extent to which emulsion chemistry and polymer origin impact
vulcanization reaction kinetics and ultimately tensile properties. Future quantification of

106
crosslink density, and the type of crosslink (polysulifidic, monosulfidic, or disulfidic) in
GNRL thin films can distinguish the underlying mechanisms of how accelerator
concentrations impact tensile properties.

107
Chapter 5: Mechanical and thermal properties of type I & type IV hypoallergenic
Hevea natural rubber latex thin films.
J. Lauren Slutzky a*, and Katrina Cornish a,b
aOhio State University, Department of Food, Agricultural and Biological Engineering,
1680 Madison Avenue, Wooster, Ohio 44691 USA
bOhio State University, Department of Horticulture and Crop Science, 1680 Madison
Avenue, Wooster, Ohio 44691 USA
*Corresponding author: Katrina Cornish, Williams Hall, 1680 Madison Avenue,
Wooster, Ohio 44691, USA. Tel: 330-263-3982 Fax: 330-263-3887 Email:
Abstract:
Type I latex allergy sensitization and subsequent allergic reactions to Hevea
natural rubber latex proteins have created an industry demand for thin film barriers that
are hypoallergenic. Other allergens associated with natural rubber and synthetic polymer
thin films are attributed to residual thiazole, thiuram, and carbamate accelerators that are
prone to cause type IV allergies, characterized by contact dermatitis or delayed contact
hypersensitivity. This work focuses on the manufacturing of type I and type IV
hypoallergenic thin films utilizing natural rubber latex from the plant species Hevea
brasiliensis, cured with the accelerators diisopropyl xanthogen polysulphide (DIXP) and
zinc diisononyl dithiocarbamate (ZDNC). Ultra- low protein Hevea latex is
hypoallergenic with respect to type I latex allergy, because its extractable protein and
rubber bound proteins have been precipitated and discarded prior to compounding and
subsequent manufacture. Type IV allergies are diminished because DIXP is consumed

108
during the vulcanization process, and skin tests have shown that ZDNC does not cause
dermal reactions or delayed contact hypersensitivity. Fabrication of the type I and type
IV hypoallergenic thin films was carried out using a coagulant dipping method, and the
effects of the accelerator loading on the mechanical and thermal properties of cured ultra-
low protein Hevea thin films were characterized. Many of the formulated films have
mechanical properties superior to those described in American Standard for Testing
Materials D 3577, the standard for rubber surgical gloves, and could be used to make
commercial medical thin film barriers such as medical gloves, condoms, balloons and
dental dams. Thermal analysis of films also confirmed that accelerator loading affected
crosslinking.
Keywords: ultra-low protein Hevea, natural rubber latex, type I latex allergy, type IV
latex allergy, elastomer thin films
5.1. Introduction
Over 40,000 consumer products are made from natural rubber, including tires,
gloves, catheters, condoms, and clothing elastic (Kelly, 1995). NRL is derived from the
plant species, Hevea brasiliensis, commonly known as the Brazilian or Para Rubber Tree.
However, NRL contains over 200 different proteins, over 60 of which are human
allergens, capable of eliciting a spectrum of type I allergic responses, ranging in severity
from contact dermatitis, contact urticarial, and delayed hypersensitivity through systemic
reactions, such as hives and edema, to life-threatening anaphylaxis and death (Spaner et
al., 1989).
Repeated exposure to Hevea NRL allergenic proteins can sensitize individuals,
inducing IgE antibodies. With increased exposure to Hevea NRL products, an individual

109
is more likely to create IgE antibodies associated with Hevea NRL allergenic proteins: At
the height of the crisis 68% of children with spina bifida were sensitized due to frequent
spinal catheterization with Hevea NRL products, 5-15% of healthcare workers were
sensitized from Hevea NRL gloves, 10% of industrial rubber workers were sensitized,
whereas 0.2%-8.2% of the general population was sensitized (Grzybowski et al., 1996;
Alenius et al., 2002).
Direct skin contact by vulcanized elastomer products also can induce type IV
delayed hypersensitivity reactions mediated by antigen specific sensitized T lymphocytes,
and are attributed to residual accelerators used to crosslink natural as well as synthetic
diene elastomers (Pak et al., 2012). Type IV contact dermatitis to rubber products is one
of the most common causes of occupational contact dermatitis (Sasseville, 2008). In
addition, 50% of individuals with type IV allergy will develop a type I allergy to Hevea
NRL proteins, since contact dermatitis has compromised their dermal barrier and
therefore is more susceptible to allergenic protein absorption (Hayes et al., 2000).
Ultra-low protein Hevea NRL is type I hypoallergenic, due to an aluminum
hydroxide treatment which removes proteins from the NRL. A slurry of aluminum
hydroxide is introduced at the processing stage, binding to the non-rubber particles and
soluble protein in NRL (Swason, 2008). These aluminum bound protein particles are
subsequently removed by centrifugation (Swason, 2008). In addition to removing
antigenic proteins, this process also removes other non-rubber biochemical metabolites
and impurities, creating a cleaner and stable ultra-low protein Hevea NRL (Swason,
2008).

110
Type IV allergens in medical natural and synthetic elastomer products are
attributed to residual accelerators used to increase the rate and efficacy of sulfur cross-
links formation and in some cases, to anti-oxidants. The commonly used rubber
accelerators, thiazoles, thiurams, and carbamates, are recognized by the U.S. Food and
Drug Administration as sensitizing agents capable of eliciting type IV allergic reactions
(Taylor and Leow, 2000). Additional concerns with sulfur vulcanization accelerators
include the potential formation of the carcinogenic and teratogenic N-nitrosamines.
Nitrosamines form when nitrites from the environment react with nitrosatable substances,
such as secondary amines found in many vulcanization chemistries (Mutsuga, 2013).
Current alternatives for diene elastomer crosslinking include gamma and UV
irradiation, organic peroxide cures, zinc oxide activators without accelerators (utilizing
the carboxyl-zinc ionic bond), and placement of functional groups onto the polymer
backbone that can form crosslinks post product fabrication. However, since gamma and
UV irradiation, and organic peroxides create carbon and not sulfur crosslinks, they
generally possess inferior mechanical properties compared to vulcanized rubbers (van
Jole, 2008).
Novel sulfur vulcanization chemistries continue to be developed by chemists,
with aims of reducing allergenic potential, mitigating the formation of nitrosamines, and
improving vulcanization efficiency. The new chemical accelerators diisopropyl
xanthogen polysulfide (DIXP) and zinc diisononyl dithiocarbamate (ZDNC) (developed
by Robinson Bros. Ltd., U.K.) have reduced allergenic potential, lack secondary amines,
and can be used in tandem for the efficient vulcanization of NRL (Chakraborty, 2005).
DIXP is a fugitive xanthate accelerator which is completely consumed into sulfur

111
crosslinks, and its byproducts are volatile isopropanol and carbon disulfide (Chakraborty,
2005). ZDNC has a lower allergic potential than conventional dithiocarbamates because
its high molecular weight renders it soluble in the rubber matrix, and therefore less
ZDNC can be extracted from finished rubber articles compared to common industry
dithiocarbamates (Chakraborty, 2005).
The objective of this study was to vulcanize ultra-low protein Hevea NRL with
the accelerators DIXP and ZDNC to create type I and type IV hypoallergenic thin film
elastomers with mechanical properties suitable for surgical gloves, and other thin film
elastomer products, such as catheters and condoms. The materials made in this study are
type I and type IV hypoallergenic NRL thin films with mechanical properties superior
those specified in American Standard for Testing Materials (ASTM) D 3577, the standard
for rubber surgical gloves.

112
5.2. Experimental
5.2.1. Materials and sample preparation
5.2.1.1. Emulsion chemistry/compounding
Ultra-low Hevea NRL (Vystar, U.S.A) was formulated using a base compounding
recipe (Table 5.1) while varying the amount of the accelerators DIXP and ZDNC. The
amount of accelerators and other materials were added to ultra-low Hevea NRL at
specified concentrations based on parts per hundred dry rubber (phr). The sulfur
emulsion, antioxidant dispersion, and zinc oxide dispersion were supplied by Akron
Dispersions (Akron, OH, USA) and the ammonium hydroxide was supplied by W.W.
Grainger, Inc (Salt Lake City, UT, USA). Deionized water was added to the
compounded emulsion until 48% solids by volume was achieved, and was prevulcanized
for 2.5 hours while stirring with a 30 rpm hand mixer.
Table 5.1. Latex Compound Recipe.
Quantity (phr)
Ultra-low protein Hevea NRL 100
Sulfur 2
Ammonium Hydroxide 1
ZnO 0.3
Antioxidant 2
ZDNC 0.2, 0.4, or 0.6
DIXP 0.2, 0.4, 0.6, 0.8, or 1.0

113
5.2.1.2. Thin film manufacture by dipping
Thin film elastomer products are manufactured by dipping formers into emulsions
with subsequent heating to remove liquids and vulcanize the NRL. Initially, a stainless
steel former was heated to 70 ºC and dipped (10 second dwell) into a coagulant solution
(25% aqueous calcium nitrate in 70% isopropyl alcohol). After the solvent evaporated,
the coagulant-coated former was reheated to 70 ºC then dipped into a compounded latex
emulsion and held there for different dwell times during which a thin film of coagulated
latex was deposited (dwell times varied from 5, 15, 30, 45, and 60 sec). Once the latex
gelled via heating (15 min at 100 ºC), it was leached in water (30 min at 55 ºC), followed
by stripping of the former and subsequent vulcanization of the rubber article (20 min at
105 ºC). The rubber article was then placed in a tumble dryer post-vulcanization (60 min
at 60 ºC). All ultra-low protein Hevea NRL thin films in this study were made using a
Diplomat automated dipper (DipTech Systems Inc., Kent, OH, USA).
5.2.2. Tensile Properties
Four dumbbell samples of each compound at each dwell time were cut using Die
C according to ASTM D 412 (ASTM International, 2013a). Mechanical properties were
tested following ASTM D 412 and were determined using an Instron 3366 with Bluehill
v. 2.17 software package (Instron, Norwood, MA, USA) (ASTM International, 2013a).
Samples were tested using a crosshead speed of 500 mm/min at room temperature
(26°C). The reported mean ± the standard error from the mean (SE) values are averages
of at least 4 samples. In addition, statistical analysis was performed using Minitab,
version 16.0 (State College, PA). Significant differences (p-values < 0.05) in tensile data

114
amongst ultra-low protein Hevea NRL compounds for each film thickness were
determined using one-way analysis of variance (ANOVA).
5.2.3. Dynamic Mechanical Analyzer (DMA)
The mechanically induced glass transition temperatures of the cured films were
studied using a TA Instrument Q800 (New Castle, DE), as previously described
(Monadjemi et al., 2016). The samples were equilibrated at -110°C and subsequently
heated to 150 °C with a heating rate of 5 °C/min. The sample dimensions were
approximately 25 x 3.0 x 0.7 mm3 with an amplitude of 15 μm, preload of 1N, and
frequency of 1 Hz using the tension film clamps. The values presented are averages of
four samples.

115
5.2.4. Differential Scanning Calorimetry (DSC)
TA Instruments Q100 (New Castle, DE) differential scanning calorimetry (DSC)
was used to determine the state of the crosslinked elastomers as a function of
temperature, using a previously described procedure (Musto et al., 2016). The average
glass transition temperatures were determined from thermograms obtained by heating 5
to 10 mg samples from -20 to 200 °C at a rate of 10 °C/min. The samples were allowed
to anneal at 200 °C for 3 to 5 min to remove the thermal history of the polymers, and
subsequently cooled to -85 °C, held for 3 to 5 min, and reheated from -85 to 200 °C with
a heating rate of 10 °C/min. The reported values are averages of four samples.
5.2.5. Thermogravimetric Analysis (TGA)
Thermogravimetric analysis (TGA) was used to study thermal decomposition
using a TA Instruments TGA 5000 (New Castle, DE). The samples were heated under
nitrogen from room temperature to 500 °C with a heating rate of 20° C/min. The
reported values are averages of four samples.

116
5.3. Results and Discussion
5.3.1. Dwell time and film thickness
When the film thickness for each respective dwell time was measured for four
samples, each set had a standard error less than 0.01 mm (Table 5.2). The relationship
between dwell time and average thin film thickness showed a linear relationship
(R2=0.98, df=3).
Table 5.2. Dwell time and average thin film thickness, standard error from mean <10 μm Dwell Time (s) Average Film
Thickness (μm)
5 150
15 190
30 210
45 230
60 260
5.3.2: Tensile properties
3D surface graphs were created using mesh points based on an average of four
samples, to show how modulus at 500% elongation (MPa), tensile strength (MPa), and
ultimate elongation (%) varied with different levels of DIXP (phr) and ZDNC (phr) for
each film thickness (Fig. 5.1).
The modulus at 500% elongation in the thinnest films was particularly sensitive to
the ratio and concentration of the two accelerators (Figure 5.1(a) and (d)). The thinnest
films (Figure 5.1(a)) had statistically significant lower modulus for formulations
containing 0.2 phr DIXP with ZDNC concentrations of 0.0 and 0.2 phr, in comparison to
films containing 0.4 phr DIXP with ZDNC concentrations of 0.2, 0.4, and 0.6 phr. There

117
are no statistical differences in modulus at 500% elongation for the thinnest films (Figure
5.1(a)) between formulations containing higher concentrations of DIXP, such as 0.6 phr
DIXP with 0.4 or 0.6 phr ZDNC, compared to 0.8 phr DIXP with 0.2, 0.4, and 0.6 phr
ZDNC. In the thickest films (Figure 5.1 (e)), modulus at 500% elongation became
uniform for formulations containing 1.0 phr DIXP with 0.2, 0.4, and 0.6 phr ZDNC
(Figure 5.1 (e)), whereas in thinner films (Figure 5.1 (b), (c), (d)) the 1.0 phr DIXP/0.2
phr ZDNC formulations are statistically significantly softer for modulus at 500%
elongation compared to the formulations containing 1.0 phr DIXP/0.6 phr ZDNC.
Tensile strength varied considerably among all film thicknesses in the tested
accelerator treatments. Tensile strength (Figure 5.1 (b), (e), and (h)) was strongest, and
similar, in films made with 0.4 phr DIXP/0.4 phr ZDNC, and 0.6 phr DIXP/0.4 phr
ZDNC for the film thicknesses of 150, 190, and 210 μm. The thinnest films of the 0.4 phr
DIXP/0.4 phr ZDNC polymer compound were 150 μm thick and had a tensile strength of
31.38 ± 1.29 MPa (Figure 5.1(b)). Films made with 0.4 phr DIXP/0.4 phr ZDNC, and 0.6
phr DIXP/0.4 phr ZDNC were weaker than films from other formulations at film
thicknesses of 150, 190, and 210 μm. Films made with higher concentrations of DIXP in
the thicker 230 and 260 μm films had tensile properties similar to films from 0.4 phr
DIXP/0.4 phr ZDNC and 0.6 phr DIXP/0.4 phr ZDNC. Relative minima in tensile
strength were observed in 0.2 phr DIXP/0.0 phr ZDNC (Figure 5.1 (b) (e) and (k)), which
were statistically significantlky weaker than films made with 0.4 phr DIXP/0.4 phr
ZDNC and 0.6 phr DIXP/0.4 phr ZDNC.
There was little statistical significance among formulations for ultimate
elongation. The largest ultimate elongations, in the thinnest films of 150 μm, were

118
obtained in 0.8 phr DIXP/0.4 phr ZDNC (Figure 5.1 (c)), and had an elongation at break
of 1686 ± 64%. This film was only significantly different from the group of films with
the lowest ultimate elongations, i.e., 0.2 phr DIXP/0.0 phr ZDNC, 0.2 phr DIXP/0.2 phr
ZDNC, and 0.4 phr DIXP/0.6 phr ZDNC. For the films 190 μm thick, the largest
ultimate elongations were in films made with 0.6 phr DIXP/0.2 phr ZDNC formulation
(Figure 5.1 (f)), and had an elongation at break of 2107 ± 42%, and was significantly
higher than the other films of this thickness. The highest ultimate elongation in thickest
films (260 μm) were also obtained from this fomulation, (Figure 5.1 (o)), and had an
elongation at break of 2204 ± 299%. However, only the films with the ultimate
elongation minimum, (made with 0.2 phr DIXP/0.0 phr ZDNC) had significantly lower
elgonation at break.
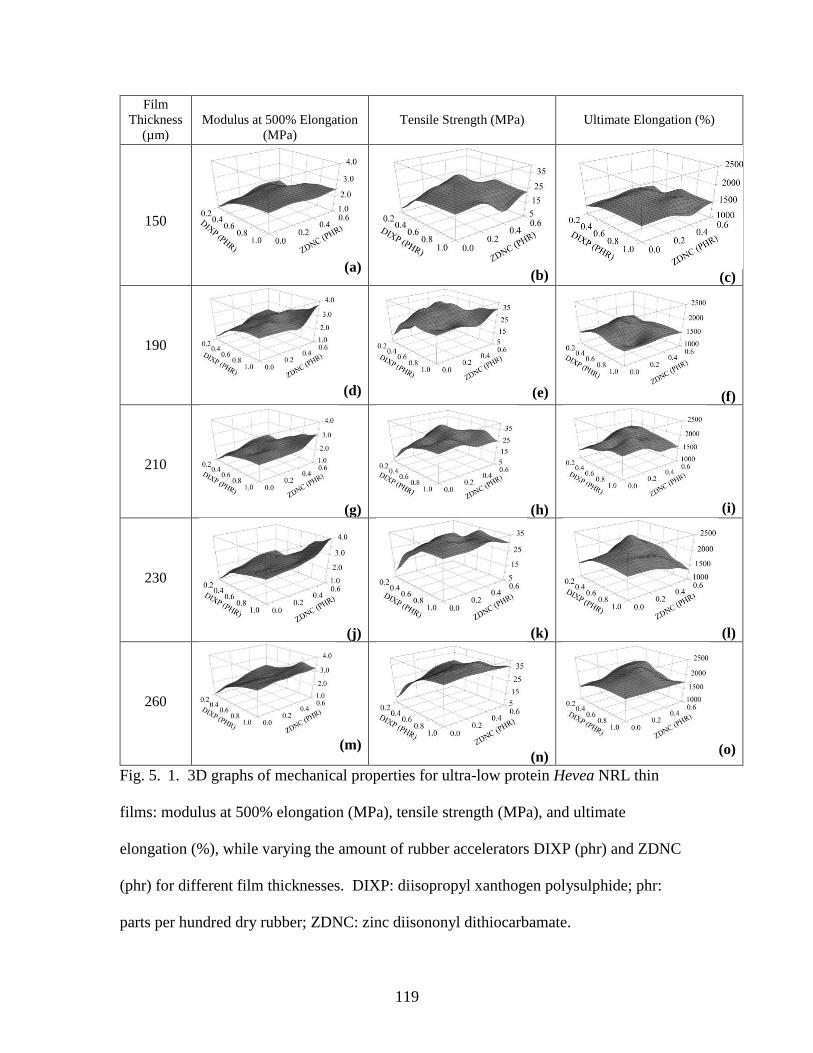
119
Film
Thickness
(µm)
Modulus at 500% Elongation
(MPa)
Tensile Strength (MPa)
Ultimate Elongation (%)
150
(a)
(b)
(c)
190
(d)
(e)
(f)
210
(g)
(h)
(i)
230
(j)
(k)
(l)
260
(m)
(n)
(o)
Fig. 5. 1. 3D graphs of mechanical properties for ultra-low protein Hevea NRL thin
films: modulus at 500% elongation (MPa), tensile strength (MPa), and ultimate
elongation (%), while varying the amount of rubber accelerators DIXP (phr) and ZDNC
(phr) for different film thicknesses. DIXP: diisopropyl xanthogen polysulphide; phr:
parts per hundred dry rubber; ZDNC: zinc diisononyl dithiocarbamate.

120
Three formulations in Figure 5.1 were selected for thermal analysis (Table 5.3).
The formulation with the best tensile properties (DIXP 0.4 phr/ZDNC 0.4 phr) was
selected, as well as two formulations that had statistically inferior mechanical properties
and a total accelerator concentration higher or lower than the optimal formulation (Table
5.3). This allows for thermal analysis across formulations with different crosslinking
densities, including optimally-cured films (DIXP 0.4 phr /ZDNC 0.4 phr), an under-
crosslinked material (DIXP 0.2 phr /ZDNC 0.2 phr) that undergoes ductile fracture, and
an over-crosslinked material (DIXP 1.0 phr/ZDNC 0.2 phr) that undergoes brittle
fracture.
Table 5.3. Tensile data for ultra-low protein Hevea NRL thin films, which will be used
for subsequent thermal analysis via DMA, DSC, and TGA.
Sample Tensile Properties:
Modulus at
500% Elongation
(MPa)
Tensile
Strength (MPa)
Ultimate
Elongation (%)
Ultra-low Protein Hevea
NRL:
DIXP 0.2; ZDNC 0.2 1.19 ± 0.08 6.30 ± 0.44 1238 ± 36
DIXP 0.4; ZDNC 0.4 2.14 ± 0.04 31.38 ± 1.29 1590 ± 31
DIXP 1.0; ZDNC 0.6 2.51 ± 0.08 23.35 ± 2.52 1496 ± 45

121
5.3.3. Dynamic Mechanical Analysis (DMA)
The glass transition temperature (Tg) is the temperature at which polymers
transition from glassy behavior, where segmental motion is restricted, to rubbery
behavior that is characterized by molecular relaxations involving cooperative segmental
motion. The segmental motion of polymers allows for reversible strain in a material, the
defining behavior of an elastomer. As a result, characterization of the Tg can provide
insight into the physio-chemical interactions of materials, and how molecular motion can
become hindered due to vulcanization. The Tg typically increases in polymers that
undergo more extensive crosslinking, as molecular segmental motion becomes hindered
by inter- and intra-molecular crosslinks.
A low frequency of 1 Hz was used for the testing of samples, since higher
frequencies can increase the Tg due to reduced molecular relaxation of the polymeric
system. The storage moduli transitions were observed between -56ºC and -53ºC for all
ultra-low protein Hevea NRL (Table 5.4 and Figure 5.2). Ultra-low protein Hevea NRL
showed a significant decrease in storage modulus with increasing temperature (Figure
5.2). However, ultra-low protein Hevea NRL formulations with statistically significant
tensile properties (Table 5.3, p <0.05) did not have statistically significant different
storage moduli at respective temperatures (Table 5.4).
Tan delta (δ) is the ratio of loss modulus (E”) to storage modulus (E’), and
reflects the energy dissipated as heat during a heating cycle (Figure 5.2). The dampening
peak for ultra-low protein Hevea NRL formulations occurred between -44 °C and -37 °C,
and tan delta peak values are higher than the glass transition values derived from the
storage modulus (Table 5.4), which is very common in the analysis of elastomers. The

122
loss modulus peak is another way to determine the glass transition: an increase in the loss
modulus represents an increase in viscous behavior due to increased cooperative
segmental motion. The loss modulus peak for ultra-low protein Hevea NRL formulations
occurred between -54 °C and -50 °C (Table 5.4).
Table 5.4. Glass transition temperatures obtained from DMA for ultra-low protein Hevea
NRL thin films.
Sample Glass Transition from DMA
Tonset (oC)
from
Storage
Modulus
Tmax (oC)
from
Loss
Modulus
Tmax (oC)
from
Tan Delta
Ultra-low Protein Hevea
NRL:
DIXP 0.2; ZDNC 0.2 -56.65 ± 0.23 -54.37 ± 1.88 -41.74 ± 3.16 DIXP 0.4; ZDNC 0.4 -53.20 ± 0.77 -50.30 ±0.34 -36.95 ± 0.59 DIXP 1.0; ZDNC 0.6 -56.60 ± 0.37 -54.00 ± 0.51 -44.05 ± 1.53
Fig. 5. 2. Glass transition temperatures obtained from DMA for ultra-low protein Hevea
NRL thin films.

123
5.3.3. Differential Scanning Calorimetry (DSC)
Glass transition temperatures from DSC were calculated from thermal transitions
upon cooling and re-heating of ultra-low protein Hevea samples outlined in Table 5.3.
Uncrosslinked ultra-low protein Hevea NRL was included for DSC analysis; the
uncrosslinked material is ultra-low protein Hevea NRL latex dried at ambient
temperatures for 1 week, and does not contain any formulation additives. The
uncrosslinked ultra-low protein Hevea NRL had the lowest glass transition temperature at
-67.5°C (Figure 5.3, Table 5.5). As the amount of chemical accelerators increased, the
glass transition temperature also increased for ultra-low protein Hevea NRL compounds.
This is attributed to increased crosslink densities for compounds with increased
accelerator concentrations. Glass transition temperatures from DSC (Table 5.5) are
significantly lower than the glass transition temperatures calculated from DMA (Table
5.4). Since there are no mechanical forces applied in DSC, it is no surprise that it would
require less energy to undergo cooperative segmental motion, resulting in a lower glass
transition temperature than estimated by DMA data.
Table 5.5. Glass transition temperatures obtained from DSC for ultra-low protein Hevea
NRL thin films. Average of four samples.
Sample Glass Transition (Second Melting)
Teig (oC) Tmg (oC)
Ultra-low Protein Hevea NRL:
Uncrosslinked -67.50 ± 1.25 -64.81 ± 0.69
DIXP 0.2; ZDNC 0.2 -59.86 ± 0.23 -58.30 ± 1.68
DIXP 0.4; ZDNC 0.4 -59.26 ± 0.84 -57.31 ± 1.48
DIXP 1.0; ZDNC 0.6 -57.35 ± 1.54 -56.51 ± 0.87
Teig = extrapolated onset temperature, Tmg = midpoint temperature
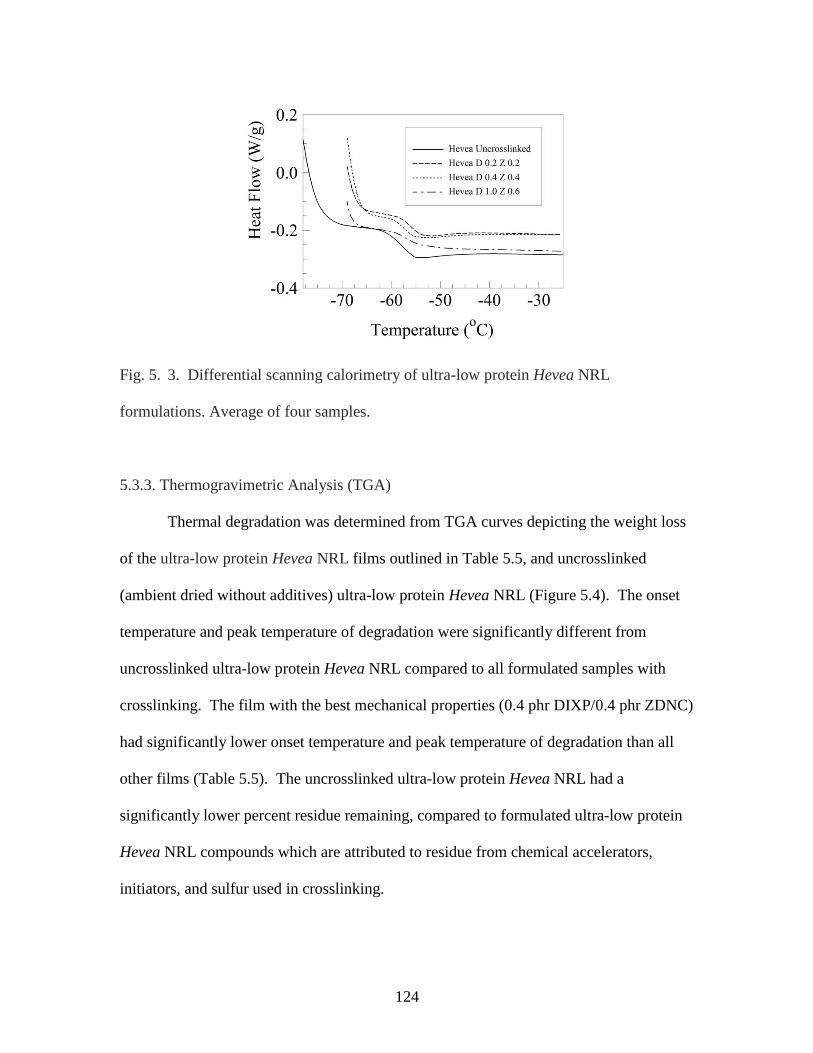
124
Fig. 5. 3. Differential scanning calorimetry of ultra-low protein Hevea NRL
formulations. Average of four samples.
5.3.3. Thermogravimetric Analysis (TGA)
Thermal degradation was determined from TGA curves depicting the weight loss
of the ultra-low protein Hevea NRL films outlined in Table 5.5, and uncrosslinked
(ambient dried without additives) ultra-low protein Hevea NRL (Figure 5.4). The onset
temperature and peak temperature of degradation were significantly different from
uncrosslinked ultra-low protein Hevea NRL compared to all formulated samples with
crosslinking. The film with the best mechanical properties (0.4 phr DIXP/0.4 phr ZDNC)
had significantly lower onset temperature and peak temperature of degradation than all
other films (Table 5.5). The uncrosslinked ultra-low protein Hevea NRL had a
significantly lower percent residue remaining, compared to formulated ultra-low protein
Hevea NRL compounds which are attributed to residue from chemical accelerators,
initiators, and sulfur used in crosslinking.
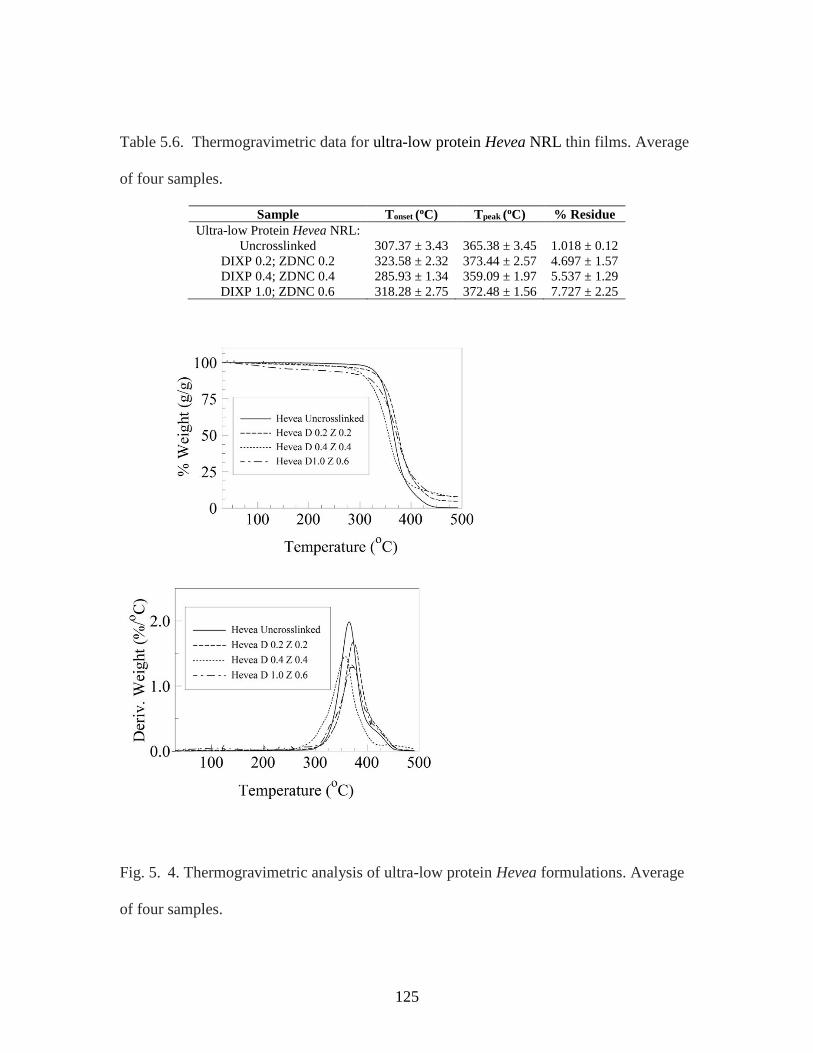
125
Table 5.6. Thermogravimetric data for ultra-low protein Hevea NRL thin films. Average
of four samples.
Sample Tonset (oC) Tpeak (oC) % Residue
Ultra-low Protein Hevea NRL:
Uncrosslinked 307.37 ± 3.43 365.38 ± 3.45 1.018 ± 0.12
DIXP 0.2; ZDNC 0.2 323.58 ± 2.32 373.44 ± 2.57 4.697 ± 1.57
DIXP 0.4; ZDNC 0.4 285.93 ± 1.34 359.09 ± 1.97 5.537 ± 1.29
DIXP 1.0; ZDNC 0.6 318.28 ± 2.75 372.48 ± 1.56 7.727 ± 2.25
Fig. 5. 4. Thermogravimetric analysis of ultra-low protein Hevea formulations. Average
of four samples.

126
5.4. Conclusions
Type I and type IV hypoallergenic thin film elastomers can be used in
applications such as surgical gloves. The specifications for surgical gloves is detailed in
ASTM D3577: natural rubber surgical gloves demands tensile strength greater than 24
MPa, modulus at 500% elongation of less than 5.5 MPa and ultimate elongation greater
than 750% [16]. All three performance parameters are exceeded by many of the thin
films formulated in this study (Figure 5.1).
The films had significantly different tensile properties based on formulation.
Thermal differences between films were not statistically significant; however all films
had a higher glass transition temperature compared to uncrosslinked ultra-low protein
Hevea NRL. Therefore accelerator ratio and concentration affects tensile properties;
however, the accelerator ratio and concentration does not affect thermal properties of
ultra-low protein Hevea NRL films, such as glass transition temperature. Thermal
measurements such as glass transition temperature cannot provide the resolution of
crosslink density and quality, but do provide significant changes compared to
uncrosslinked, ambient dried ultra-low protein Hevea NRL.
To quantify significant property differences between ultra-low protein Hevea
NRL films, mechanical properties from tensile testing should be prioritized as a result to
determine the effect of crosslinking on film properties. The storage modulus of ultra-low
protein Hevea NRL films is not significantly different between films, and therefore only
tensile properties should used to assess films to determine optimal formulation and
properties.

127
The strongest ultra-low protein Hevea NRL films (DIXP 0.4 phr/ ZDNC 0.4 phr)
has an accelerator ratio of 1:1; whereas the strongest guayule NRL films in Chapter 4
(DIXP 2.0 phr/ ZDNC 0.8 phr) has an accelerator ratio of 2.5:1. This vast difference in
chemical accelerator loadings and ratios between optimized formulations can be
attributed to differences in the NRL chemistry. All NRL emulsions are buffered to a
alkaline pH to extend shelf-life; Ultra-low protein Hevea NRL is stabilized with
ammonium hydroxide, whereas GNRL is stabilized with potassium hydroxide. The
differences in the stabilization packages of NRL needs to be considered when
determining an ideal accelerator concentration and ratio, specifically due to the role of
ammonium hydroxide and zinc in creating efficient carbon-sulfur bonds. NRL stabilized
with ammonium hydroxide such as ultra-low protein Hevea NRL have excess ammonium
hydroxide; ammonium hydroxide is an activator for zinc-based accelerators (such as
ZDNC) and can promote carbon-sulfur bond formation. Future work could focus on
understanding how ammonium hydroxide concentration in NRL affects accelerator
concentration and ratio.

128
Chapter 6: Canonical correlation analysis of type I and type IV circumallergenic
guayule natural rubber thin films and type I and type IV hypoallergenic ultra-low
protein Hevea natural rubber thin films.
J. Lauren Slutzky a, Alfred Soboyejo a , and Katrina Cornish a,b*
aOhio State University, Department of Food, Agricultural and Biological Engineering,
1680 Madison Avenue, Wooster, Ohio 44691 USA
bOhio State University, Department of Horticulture and Crop Science, 1680 Madison
Avenue, Wooster, Ohio 44691 USA
*Corresponding author: Katrina Cornish, Williams Hall, 1680 Madison Avenue,
Wooster, Ohio 44691, USA. Tel: 330-263-3982 Fax: 330-263-3887 Email:
Abstract:
Parthenium argentatum Gray, commonly called guayule, is an industrial crop that
produces low protein laex containing proteins that do not cross-react with antibodies
raised against Hevea brasiliensis Muell. Arg. associated allergic proteins. In Chapter 4,
guayule natural rubber latex (GNRL) (Chapter 4) and hypoallergenic ultra-low protein
Hevea natural rubber latex (NRL) (Chapter 5) thin films were cured with the accelerators
diisopropyl xanthogen polysulphide (DIXP) and alkyldithiocarbamate (ZDNC), and the
tensile properties were assessed according to the American Standard for Testing
Materials (ASTM) 3577, which defines surgical glove specifications. The aim of this
chapter was to develop multivariate stochastic regression models using ASTM 3577
specifications such as modulus at 500% elongation (MPa), tensile strength (MPa), and
ultimate elongation (%) as a function of the concentration of chemical accelerators DIXP

129
and ZDNC, as well as film thickness, for GNRL and ultra-low protein Hevea NRL. In
addition canonical correlation analysis (CCA) was used to demonstrate the individual
effect of DIXP, ZDNC, and film thickness on the tensile properties. CCA was used to
determine Pearson correlation (R2) relationships within and between the input variables
of DIXP, ZDNC, and film thickness and the output variables of the tensile properties.
CCA was also used in this study to maximize the correlation between input variables and
output variables by calculating a singular value decomposition correlation (R2), which
was used to compare the linear, multiplicative, and predictor transformed multivariate
regression methods. The abstraction and integration of correlations from multivariate
stochastic regression analysis and CCA can reduce the time needed to improve product
development by reducing the range of input variable which must be used to test
performance.
Keywords: Guayule, natural rubber latex, multivariate stochastic regression, canonical
correlation analysis
6.1. Introduction
Over 2,500 plants produce rubber, but only a few plants produce rubber with high
molecular weight capable of macromolecular entanglement for commercial applications
(Perumal et al., 2013). Currently, Hevea brasiliensis Muell. Arg. accounts for over 90%
of the global supply of natural rubber (Perumal et al., 2013). However, latex from Hevea
contains proteins that are capable of sensitizing and eventually eliciting a type I allergic
reaction, characterized by skin inflammation, nausea, and perhaps anaphylaxis and
ultimately death (Cornish and Siler, 1996). There are over a dozen serious human
allergens found in Hevea latex (Carey et al., 1995; Miri et al., 2007).

130
Natural rubber latex derived from the guayule shrub circumvents the Hevea type I
latex allergy; guayule does not contain any of the Hevea associated allergens (Cornish et
al., 2006; Cornish, 2012). Also, GNRL contains very little protein. Therefore, guayule
natural rubber latex (GNRL) does not pose a threat to consumers with regards to
developing a type I latex allergy, and is safe to use by consumers with type I latex
allergies (Cornish et al., 2007).
Natural rubber is typically crosslinked in order to have mechanical properties
suitable for commercial applications. Natural rubber latex thin film elastomers utilize the
diene bonds in the cis-1,4-polyisoprene structure for crosslinking with sulfur, which
hinders macromolecular translational movement and improves bulk mechanical
properties. Activators and accelerators are used to improve the rate and efficiency of
sulfur crosslink formation. Commonly used accelerators such as thiurams, thiazoles, and
carbamates are acknowledged by the U.S. Food and Drug Administration as capable of
eliciting type IV allergies, characterized by contact dermatitis and compromise of the
dermal barrier (Miri et al., 2007).
As a result, alternative accelerators have been developed by Robinson Brothers
Ltd (West Bromwich, U.K.), namely zinc diisononyl dithiocarbamate (ZDNC) and
diisopropyl xanthogen (DIXP). ZDNC has a higher molecular weight compared to other
thiocarbamates, which hinders ZDNC from blooming to the finished rubber article’s
surface (Chakraborty et al., 2006). DIXP leaves no residual chemicals, generating the
volatile byproducts of isopropanol and carbon disulfide (Chakraborty et al., 2006; van
Jole, 2007). In Chapter 4, DIXP in conjunction with ZDNC was used to create type I
circumallergenic and type IV hypoallergenic guayule natural rubber latex (GNRL)

131
elastomeric thin films; In Chapter 5, type I and type IV hypoallergenic ultra-low protein
Hevea natural rubber latex (NRL) elastomeric thin films were created.
This chapter uses the data from Chapter 4 and 5, in multivariate stochastic
regression analysis, to establish quantitative structure-property relationships (QSRPs),
between the predictor variables of accelerator concentration and film thicknesses, and the
corresponding response variables of the tensile properties outlined in ASTM 3577.
Multivariate models using parametric regression, via the linear least squares method are
used. Both linear and nonlinear regression models were used for the simulation, as well
as models developed from a predictor variable transformation. To compare models,
coefficient analysis to quantify the effects of each independent variable on individual
tensile properties, and the Pearson correlation coefficient (R2), were compared among
models to determine the best fit.
This study used multivariate ordination methods to reduce dimensionality as well
as to recognize patterns in these multivariate data sets, specifically by using canonical
correlation analysis (CCA). Ordinations were divided into two groups: unconstrained
and constrained methods. Unconstrained methods included principle component
analysis, and reduced dimensions on the basis of minimizing residual variance (Soboyejo,
2011). Unconstrained ordination methods were used to recognize relative dispersions
among groups of data (Soboyejo, 2011). In contrast, constrained ordination methods
relate a matrix of response variables with a matrix of quantitative predictor variables, and
are used in this study (Soboyejo, 2011). Specifically, CCA utilizes ordination axes that
maximize their correlation with linear combinations of some quantitative predictor
variables, producing a singular value decomposition correlation (R2) (Rickman et al.,

132
2017). Constrained ordination methods have been termed a “direct” gradient response
(Rickman et al., 2017).
For each type of multivariate stochastic regression modeling technique used, the
transformed data was organized into matrices of predictor variables and response
variables for CCA. CCA is widely used in parametric multivariate data analysis to
analyze correlations within and between multiple independent (predictor) variables and
multiple dependent (response) variables. In this study, the predictor variables are the
accelerator concentrations of DIXP and ZDNC, and film thickness; the response variables
are the tensile properties of modulus at 500% (MPa), tensile strength (MPa), and ultimate
elongation (%). CCA was used in the regression analysis to comprehensively evaluate
the reliability of the proposed models, to determine if one type of regression modeling
could be used for all of the response variables by comparative analysis of singular value
decomposition correlation (R2) for each type of data transformation.

133
6.2. Experimental
6.2.1. Materials and sample preparation
6.2.1.1. Emulsion chemistry/compounding
GNRL and ultra-low protein Hevea NRL were made using a base compounding
recipe (Table 6.1 and Table 6.2) while varying the amount of the accelerators DIXP and
ZDNC (Robinson Brothers Ltd, UK). The amount of accelerators and other materials
were added to GNRL at specified concentrations based on parts per hundred dry rubber
(phr). The sulfur emulsion, antioxidant dispersion, and zinc oxide dispersion were
supplied by Akron Dispersions (Akron, OH, USA) and the ammonium hydroxide was
supplied by W.W. Grainger, Inc (Salt Lake City, UT, USA). Deionized water was added
to the compounded emulsion until 48% solids by volume was achieved, and was
prevulcanized for 2.5 hours while stirring with a 30 rpm hand mixer.
Table 6.1. Latex Compound Recipe for Guayule Natural Rubber Latex
Dry Weight
Guayule NRL 100 Sulfur 2
Ammonium Hydroxide 1
ZnO 0.3
Antioxidant 2
ZDNC 0.2, 0.4, 0.6, 0.8, 1.0, 1.2 or 1.4
DIXP 1, 1.2, 1.4, 1.6, 1.8, 2.0, 2.1 or 2.2
Table 6.2. Latex Compounding Recipe for Ultra-low Protein Hevea NRL
Dry Weight
Ultra-low Protein Hevea NRL 100 Sulfur 2
Ammonium Hydroxide 1
ZnO 0.3
Antioxidant 2
ZDNC 0.2, 0.4, or 0.6
DIXP 0.2, 0.4, 0.6, 0.8, or 1.0

134
6.2.1.2. Thin film manufacture by dipping
Thin film elastomer products are manufactured by dipping formers into emulsions
with subsequent heating to remove liquids and vulcanize the NRL. Initially, a stainless
steel former was heated to 70 ºC and dipped (10 second dwell) into a coagulant solution
(25% aqueous calcium nitrate in 70% isopropyl alcohol). After the solvent evaporated,
the coagulant-coated former was reheated to 70 ºC then dipped into a compounded latex
emulsion and held there for different dwell times during which a thin film of coagulated
latex was deposited (dwell times varied from 5, 15, 30, 45, and 60 sec). Once the latex
gelled via heating (15 min at 100 ºC), it was leached in water (30 min at 55 ºC), followed
by stripping of the former and subsequent vulcanization of the rubber article (20 min at
105 ºC). The rubber article was then placed in a tumble dryer post-vulcanization (60 min
at 60 ºC). All thin films in this study were made using a Diplomat automated dipper
(DipTech Systems Inc., Kent, OH, USA).
6.2.2. Tensile Properties
Four dumbbell samples of each compound at each dwell time were cut using Die
C according to ASTM D 412 (ASTM International, 2013a). Tensile mechanical
properties were determined followed ASTM D 412 using a tensiometer (Model 3366,
Instron, Norwood, MA, USA). As outlined in ASTM 3577, the surgical glove standard,
tensile properties of modulus at 500% (MPa), tensile strength (MPa), and ultimate
elongation (%) were calculated for each sample (ASTM International, 2013b). The
reported mean ± the standard error from the mean (SE) values are averages of at least 4
samples. In addition, statistical analysis was performed using Minitab, version 16.0
(State College, PA). Significant differences (P-values < 0.05) in tensile data amongst

135
GNRL compounds for each film thickness were determined in Chapter 4 using one-way
analysis of variance (ANOVA), and ultra-low protein Hevea NRL compounds were
assessed for significant differences (P-values < 0.05) in tensile data in Chapter 5.
6.2.3. Statistical Analysis
6.2.3.1. Data
6.2.3.1.1 Dwell Time and Film Thickness
When the film thickness for each respective dwell time was measured for four
samples, each has a standard error less than 0.01mm (Table 6.3). The relationship
between dwell time and average thin film thickness showed a linear relationship
(R2=0.98, df=3). This relationship was consistent between GNRL and ultra-low protein
Hevea NRL thin films.
Table 6.3. Dwell time and average thin film thickness, SEM < 0.01mm.
Dwell Time (s) Average Film Thickness
(mm)
5 0.15
15 0.19
30 0.21
45 0.23
60 0.26
6.2.3.1.2 Tensile Data
Mechanical properties for GNRL and ultra-low protein Hevea NRL thin films
used in this study were determined in Chapters 4 and 5. The data used includes
mechanical properties of vulcanized thin films of varying thicknesses, using the type IV
hypoallergenic accelerators, DIXP and ZDNC.

136
The variables were categorized into response variables (Yi) which represent the
different mechanical properties of the vulcanized thin films, and explanatory variables
(Xi), which represent the film thicknesses, and loading of chemical accelerators DIXP
and ZDNC (Table 6.4).
Table 6.4. Notation of different experimental data used for statistical analysis.
Variable Notation Experimental Data for Model
X1 Film Thickness (mm)
X2 DIXP (PHR)
X3 ZDNC (PHR)
Y1 Stress at 500% Elongation (MPa)
Y2 Tensile Strength (MPa)
Y3 Ultimate Elongation (%)
6.2.3.2. Multivariate stochastic regression models
Multivariate stochastic regression models were made using JMP 11 Statistical
Analysis Software (SAS Institute Inc., Cary, NC). The approach used film thickness and
concentration of DIXP and ZDNC accelerators as explanatory variables (Table 6.4) to
predict the thin films’ mechanical properties as designated by ASTM 3577.
The first multivariate stochastic regression models were developed using a linear
model, summarized by the notation:
𝑌 = 𝑎 + ∑ 𝑏𝑖𝑥𝑖
𝑘
𝑖=1
where the predictors X1, X2, X3 are the same for each independent model of Y1, Y2, and
Y3 (Soboyejo, 2011).
Next, multiplicative non-linear models were created. The notation for a
multiplicative non-linear model is summarized by the following:

137
𝑌 = 𝑎 ∏ 𝑥𝑖𝑏𝑖
𝑘
𝑖=1
where the predictors X1, X2, X3 are the same for each independent model of Y1, Y2, and
Y3 (Soboyejo, 2011).
The mixed models of non-linear and linear regression models are summarized by the
following notation:
𝑌 = 𝑋𝑖
𝑎 + ∑ 𝑏𝑖𝑥𝑖𝑘𝑖=1
ln 𝑌 = 𝑙𝑛𝑋𝑖
𝑎 + ∑ 𝑏𝑖𝑙𝑛𝑥𝑖𝑘𝑖=1
𝑍𝑖 =𝑋𝑖
𝑌= 𝑎 + ∑ 𝑏𝑖𝑥𝑖
𝑘
𝑖=1
𝑙𝑛𝑥𝑖
𝑙𝑛𝑦𝑖= 𝑎 + ∑ 𝑏𝑖𝑥𝑖
𝑘
𝑖=1
For i=1, 2…k
Using this notation, mixed model regressions were made with each correlated predictor
value (termed the significant predictor variable) (Soboyejo, 2011).
Modeling techniques for the multivariate regression analysis were assessed using
the square of multiple correlation (R2) for a multivariate regression model for each tensile
property assessed. The R2 value for each mechanical property was calculated for a series
of regression models including linear, nonlinear, and mixed significant predictor
modeling.

138
6.2.3.3. Canonical correlation analysis (CCA)
The relationships between the sets of processing variables (DIXP concentration,
ZDNC concentration, and film thickness) and tensile property variables (modulus at 500%
elongation, tensile strength, and ultimate elongation) were investigated to create linear
composites of the respective variable sets. In CCA, the correlation of interest was
between the linear variates created for thin film manufacturing and tensile sets of data
(Rickman et al., 2017). CCA initially calculates a canonical loading for each individual
data set (i.e., the manufacturing data, and tensile data), and then calculates a canonical
correlation from the canonical variates for each respective data set (Rickman et al., 2017).
CCA can determine correlations between manufacturing data and tensile data, as well as
show correlations within tensile data (Rickman et al., 2017).

139
6.3 Results and Discussion
6.3.1. Multivariate stochastic regression models
The R2 values for the linear regression for all three models in both types of latex
were below statistical significance of 0.95 and therefore, the data was modeled using a
multiplicative model. The R2 values for all multiplicative non-linear models were below
statistical significance of 0.95, and so the mixing of linear and non-linear models was used
next to model the tensile data. Mixed model (linear and non-linear) regressions were made
with each correlated predictor value (termed the significant predictor variable), as
summarized in Table 6.5, Table 6.6 and Table 6.7.
Table 6.5. Mixed linear and non-linear stochastic multivariate regression models using
film thickness (mm) as the significant predictor.
Regression Equations R2
Guayule NRL Formulations:
[ln(X1)/ln(Y1 )]= -7.27 – 2.03(ln(X1)) + 1.16(ln(X2)) – 0.21(ln(X3))
0.014
[ln(X1)/ln(Y2)] = -.60 -0.438(ln(X1)) – 1.71(ln(X2)) – 0.459(ln(X3))
0.138
[ln(X1)/ln(Y3)] = 0.019 + 0.152(ln(X1)) + 0.0175(ln(X2)) +
0.00925(ln(X3))
0.886
Ultra-low Protein Hevea NRL Formulations:
[ln(X1)/ln(Y1 )]= 1.47 + 3.26(ln(X1)) + 4.04(ln(X2)) – 3.21(ln(X3))
0.957
[ln(X1)/ln(Y2)] = 0.358 + 0.507(ln(X1)) + 0.0887(ln(X2)) +
0.00862(ln(X3))
0.726
[ln(X1)/ln(Y3)] = 0.0214 + 0.148(ln(X1)) + 0.00093(ln(X2)) +
0.000149(ln(X3))
0.977

140
Table 6.6. Mixed linear and non-linear stochastic multivariate regression models using
DIXP (phr) as the significant predictor.
Regression Equations R2
Guayule NRL Formulations:
[ln(X2)/ln(Y1 )]= 1.14 + 0.621(ln(X1)) + 1.96(ln(X2)) +0.175(ln(X3))
0.396
[ln(X2)/ln(Y2)] = -0.119 – 0.0792(ln(X1)) +0.337(ln(X2)) -0.0158(ln(X3))
0.680
[ln(X2)/ln(Y3)] = -0.00611-0.00344ln(X1)) + 0.137(ln(X2)) – 0.00161(ln(X3))
0.996
Ultra-low Protein Hevea NRL Formulations:
[ln(X2)/ln(Y1 )]= 0.41 + 1.29(ln(X1)) + 4.69(ln(X2)) - 2.72(ln(X3))
0.944
[ln(X2)/ln(Y2)] = 0.149 + 0.0740(ln(X1)) +0.382(ln(X2)) + 0.00831(ln(X3))
0.947
[ln(X2)/ln(Y3)] = 0.00837+ 0.00459ln(X1)) + 0.137(ln(X2)) – 0.00161(ln(X3))
0.999
Table 6.7. Mixed linear and non-linear stochastic multivariate regression models using
ZDNC (PHR) as the significant predictor.
Regression Equations R2
Guayule NRL Formulations:
[ln(X3)/ln(Y1 )]= -2.74 -1.45(ln(X1)) + 0.97(ln(X2)) + 2.15(ln(X3))
0.146
[ln(X3)/ln(Y2)] = -0.346 – 0.451(ln(X1)) -0.893(ln(X2)) +0.150(ln(X3))
0.176
[ln(X3)/ln(Y3)] = 0.00267+0.00376ln(X1)) + 0.00899(ln(X2)) + 0.141(ln(X3))
0.992
Ultra-low Protein Hevea NRL Formulations:
[ln(X3)/ln(Y1 )]= -50.6 -2.2(ln(X1)) + 14.6(ln(X2)) - 51(ln(X3))
0.986
[ln(X3)/ln(Y2)] = 0.442 + 0.120(ln(X1)) +0.0235(ln(X2)) +0.546(ln(X3))
0.997
[ln(X3)/ln(Y3)] = 0.0193 + 0.00849ln(X1)) + 0.00053(ln(X2)) + 0.140(ln(X3))
0.999
The multivariate models created showed that DIXP and ZDNC were both
significant predictor variables for response variables of stress at 500% elongation (MPa),
tensile strength (MPa), and ultimate elongation (%) in GNRL and ultra-low protein
Hevea NRL thin films. Comparison of GNRL and ultra-low protein Hevea NRL
compounding data and multivariate models give insight into differences in their emulsion
chemistry. Multivariate models of GNRL were only significant for response variables of
ultimate elongation when using a mixed-model with DIXP and ZDNC as the significant
predictors. In contrast, similar multivariate models for ultra-low protein Hevea NRL were

141
statistically significant for all three response variables of stress at 500% elongation,
tensile strength, and ultimate elongation. This indicates that latex origin must be
considered during compounding studies.
6.3.2. Canonical Correlation Analysis
A CCA was performed on the data used to make the aforementioned models
(Tables 6.5-6.7). The matrices of predictor variables and response variables for CCA
were the same as used in multivariate analysis (Table 6.4).
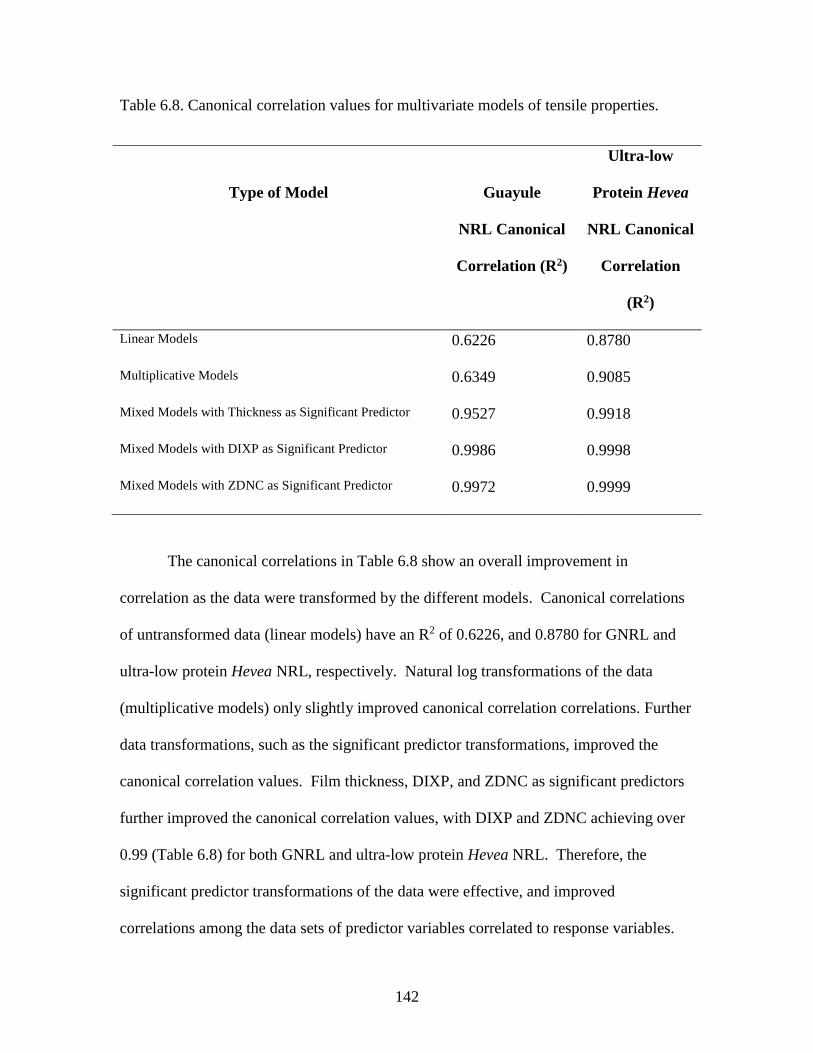
142
Table 6.8. Canonical correlation values for multivariate models of tensile properties.
Type of Model
Guayule
NRL Canonical
Correlation (R2)
Ultra-low
Protein Hevea
NRL Canonical
Correlation
(R2)
Linear Models 0.6226 0.8780
Multiplicative Models 0.6349 0.9085
Mixed Models with Thickness as Significant Predictor 0.9527 0.9918
Mixed Models with DIXP as Significant Predictor 0.9986 0.9998
Mixed Models with ZDNC as Significant Predictor 0.9972 0.9999
The canonical correlations in Table 6.8 show an overall improvement in
correlation as the data were transformed by the different models. Canonical correlations
of untransformed data (linear models) have an R2 of 0.6226, and 0.8780 for GNRL and
ultra-low protein Hevea NRL, respectively. Natural log transformations of the data
(multiplicative models) only slightly improved canonical correlation correlations. Further
data transformations, such as the significant predictor transformations, improved the
canonical correlation values. Film thickness, DIXP, and ZDNC as significant predictors
further improved the canonical correlation values, with DIXP and ZDNC achieving over
0.99 (Table 6.8) for both GNRL and ultra-low protein Hevea NRL. Therefore, the
significant predictor transformations of the data were effective, and improved
correlations among the data sets of predictor variables correlated to response variables.

143
6.4. Conclusions
The statistical modeling approach proved to be an effective way to estimate how
film thickness, and accelerator amount and type individually contribute to the mechanical
properties of both GNRL and ultra-low protein Hevea NRL thin film elastomers. This is
notable due to the complexity of GNRL and ultra-low protein Hevea NRL systems and the
large number of chemical accelerators that can be optimized in these lattices. In addition,
this modeling approach can be used to predict properties in various lattices, compounding
formulations, and film geometry (such as film thickness). Loading of DIXP and ZDNC
were significant for elongation at break in GNRL thin films. However, we were not able to
model the effect of DIXP and ZDNC on mechanical stress, including stress at 300%
elongation, and stress at break. This is most likely due to complexity in the GNRL system,
such as the role of the latex buffering system on crosslink formation, and biochemical
metabolites in the lattice such as phospholipids. Therefore, it is clear that modeling of
GNRL systems is complex and needs to account for additional predictor variables outside
the scope of this study to increase statistical significance.
In contrast, this modeling approach using the ultra-low protein Hevea NRL data set
was effective in estimating the individual contributions of chemical accelerator loading and
film thickness on thin film mechanical properties. The ultra-low protein Hevea NRL
system found that all tensile standards as outlined in ASTM 3577 are effectively modeled,
in contrast to only ultimate elongation in the GNRL data set. This shows that ultra-low
protein Hevea NRL is more predictive lattice in formulation and process conditions
modeling; this may be attributed to a less complex lattice compared to GNRL. Ultra-low
protein Hevea NRL is buffered in ammonia hydroxide, which is ideal for crosslinking of

144
thin film elastomers (and may lead to more predicative performance) compared to
potassium hydroxide stabilized GNRL. Including predictor variables that quantify
buffering systems in lattices may improve the effectiveness of the mixed modeling of these
latex systems and provide insight into what predictor variables dictate mechanical
properties.

145
Chapter 7: Mechanical properties of guayule natural rubber latex thin film
composites with biobased fillers.
J. Lauren Slutzky a*, and Katrina Cornish a,b
aOhio State University, Department of Food, Agricultural and Biological Engineering,
1680 Madison Avenue, Wooster, Ohio 44691 USA
bOhio State University, Department of Horticulture and Crop Science, 1680 Madison
Avenue, Wooster, Ohio 44691 USA
*Corresponding author: Katrina Cornish, Williams Hall, 1680 Madison Avenue,
Wooster, Ohio 44691, USA. Tel: 330-263-3982 Fax: 330-263-3887 Email:
Abstract:
Many natural rubber latex consumer products contain fillers used to diminish
costs, or create new desirable physical properties that aid in processability or utility. Yet
these commonly used fillers are not sustainable industries, and dedicate energy and
natural resources such as petroleum for their production. Thus, sustainable alternatives
for natural rubber latex composites need to be developed. This work focuses on the
manufacturing of natural rubber latex composites from the plant species Parthenium
argentatum, commonly referred to as guayule, compounded with fillers derived from
waste streams: calcium carbonate from eggshells (mostly calcium carbonate), carbon fly
ash, and guayule bark bagasse. Tensile properties of composites were assessed according
to ASTM D 412 to assess how type of sustainable filler, as well as filler loading and
particle size effect mechanical properties.

146
In general, all latex composites had stronger tensile properties with smaller
particle sizes at lower loadings. The tensile strength of latex composites at high loadings
were often inferior compared to latex films without fillers, but a reinforcing effect was
found in low loadings of nano-sized fillers. In general, the elongation at break was
increased in latex composites compared to latex films without fillers. Many of the
composites still exceeded the tensile requirements described in ASTM D 3577, the
surgical glove standard. Latex composites with sustainable fillers can create polymer
films with qualities desirable in many applications. The use of such fillers can create
novel materials and decrease cost of manufacture.
Keywords: Guayule, natural rubber latex composites, biofillers, calcium carbonate,
carbon fly ash, guayule bagasse, elastomer thin films
7.1. Introduction
Guayule natural rubber latex (GNRL) is a colloidal suspension of both rubber and
non-rubber constituents, derived from the rubber producing perennial shrub, Parthenium
argentatum. Guayule stores natural rubber latex in its parenchyma cells, and therefore
the shrub must be homogenized in an aqueous buffer from which the latex is then
purified and stabilized to create a colloidal suspension of both rubber particles and non-
rubber constituents (Cornish et al., 2008). Non-rubber constituents in GNRL include
rubber particle associated proteins and resins (low-molecular weight, acetone-extractable
material) which include saponifiable lipids (Cornish et al., 2008).
Fillers are inert materials that are dispersed into the bulk of a material prior to
manufacture and curing. Fillers can improve properties of a material, providing a

147
reinforcing effect that is characterized by improved stiffness, higher resistance to tearing
and abrasion, and enhanced tensile strength (Donnet, 2003; Kohls and Beaucage, 2002).
Other fillers are diluents, and typically are used to reduce cost of the bulk product,
typically without any improvements in polymer performance (Rothon, 2000). Particle
size is the most fundamental property of a filler, which affects reinforcement of the
elastomer the most (Rothon, 2000; Dick, 2009). Particle sizes ranging from 1000-5000
nm provide a small reinforcement; particles less than 1000 nm provide a medium
reinforcement; particles smaller than 100 nm provide the strongest reinforcement
(Rothon, 2000; Dick, 2009). Other properties can affect the interactions between a
polymer and filler, including surface chemistries, and particle geometry (Rothon, 2000;
Dick, 2009).
The most common reinforcement filler for natural rubber is carbon black
(Frohlich et al., 2005). Carbon black is produced by the thermal decomposition or partial
combustion of petroleum or natural gas, and typically contains more than 95% pure
carbon with minimal impurities of oxygen, hydrogen and nitrogen. Carbon black is
manufactured to create controlled particle sizes that range from 10 nm to 500 nm, which
fuse into chain-like aggregates (Rothon, 2000; Dick, 2009). Carbon fly ash is a coal
combustion product that is composed of fine particulates of burned fuel that are driven
out of coal-fired boilers with flue gases. In the past, carbon fly ash was released in to the
atmosphere, but air pollution standards now require its captured, resulting in a large
global supply. Other researchers have investigated the possibility of replacing carbon
black with the cheaper carbon fly ash in elastomers such as styrene-butadiene rubber, and
dry natural rubber (Barrera and Cornish, 2015).

148
The most common diluent filler for natural rubber is the calcite form of calcium
carbonate. In addition to being a diluent, calcium carbonate is a detacktifying agent, can
increase stiffness, and provides abrasion resistance (Dick, 2009). Calcium carbonate is
typically dry milled or ground to crystallites as small as 1 micron, whereas nano-
precipitated calcium carbonate creates crystallites with sizes ranging from 1- 100 nm.
(Dick, 2009). Ground calcium carbonate is derived from chalk, sediments of crushed
marine shells (Dick, 2009). Alternative natural sources of calcium carbonate, such as
eggshells, have shown promise as reinforcing or diluent for natural rubber when used to
partially or completely replace carbon black (Barrera and Cornish, 2016) or silica (Ren et
al., 2019).
The production of GNRL results in a significant amount of waste fiber, or guayule
bark bagasse (Cornish et al., 2008). Guayule bark bagasse has successfully used in
applications such as termite resistant wood products, chemical derivatives, paper
products, and energy sources (Cornish et al., 2008). Guayule bagasse has been shown to
have higher chemical extracts, especially the amount of resins, compared to other sources
of wood fibers such as maple and milkweed (Cornish et al., 2008). Dry natural rubber
composites have been made with guayule bagasse previously (Barrera and Cornish,
2015).
The utilization of alternative fillers such as carbon fly ash, calcium carbonate-rich
eggshells, and guayule bagasse could reduce the final cost of a manufactured rubber
product. However, mechanical properties may decline as a result of insufficient filler-
rubber interactions, compared to crosslinked GNRL films without any fillers, especially
at high loadings. The aim of this study was to analyze GNRL composite films made with

149
the alternative fillers carbon fly ash, calcium carbonate from eggshells, or guayule
bagasse. The effect of particle size and loading level were evaluated.

150
7.2. Experimental
7.2.1. Materials and sample preparation
GNRL was extracted as described (Cornish et al., 2008), from fresh guayule shrub
grown in Arizona. Compounding chemicals, specifically the accelerators zinc
diethyldithiocarbamate (ZDEC), diphenyl guanidine (DPG), and
dipentamethylenethiuram tetrasulfide (DPTT), activator zinc oxide, sulfur, and
antioxidant were purchased from Akron Dispersions (Akron, OH). The ammonium
hydroxide was purchased from W.W. Grainger, Inc (Salt Lake City, UT, USA).
The waste filler raw materials were generously donated as follows: eggshells (ES)
by Troyer’s Home Pantry (Apple Creek, OH), carbon fly ash (CFA) by Cargill Salt
(Akron, OH), and guayule bagasse (GB) was generated as a co-product of latex
extraction at our facility from guayule shrubs donated by PanAridus (Casa Grande, AZ).
7.2.1.1. Emulsion chemistry/compounding
Fillers were ground using an IKA A11 basic mill (Wilmington, NC). Macro sized
particles were separated using a size 50 and 400 mesh sieve from Fisher Scientific
(Pittsburgh, PA), with resulting particles ranging from 300 µm to 38 µm. Micro sized
particles were separated using a size 400 mesh, isolating particles 38 µm and smaller.
Nano sized fillers were made by dispersing the micro sized particles in distilled water,
and then wet milling to submicron size using a Planetary Ball Mill 100 manufactured by
Glen Mills (Clifton, NJ). The carbon fly ash and guayule bark bagasse were dry milled
as received, whereas the eggshells were washed and membranes were peeled from the
eggshells prior to milling.

151
GNRL was made using a compounding recipe (Table 7.1) while varying the
amount, type, and particle size of filler. The amount of fillers and other materials were
added to GNRL at specified concentrations based on parts per hundred dry rubber (phr).
Fillers were dispersed in deionized water (amount of water needed to achieve 48% solids
by volume was used to disperse fillers), and stirred for 1 hour with a 30 rpm hand mixer.
The latex compound recipe without fillers was prevulcanized for 2.5 hours while stirring
with a 30 rpm hand mixer. The filler-water dispersion was then added to the
prevulcanized latex compound, and stirred for an additional 1 hour with a 30 rpm hand
mixer.
Table 7.1. Latex Compound Recipe.
Dry Weight
Guayule NRL 100
Sulfur 2
Ammonium Hydroxide 1
ZnO 1
Antioxidant 2
Zinc Diethyldiothiocarbamate (ZDEC)
Accelerator
0.5
Diphenyl guandine (DPG) Accelerator 0.4
Dipentamethylenethiuram (DPTT)
Accelerator
0.6
Filler 1, 2, 5 or 10

152
7.2.1.2. Thin film manufacture by dipping
Thin film elastomer products are manufactured by dipping formers into emulsions
with subsequent heating to remove liquids and vulcanize the NRL. Initially, a stainless
steel former was heated to 70 ºC and dipped (10 second dwell) into a coagulant solution
(25% aqueous calcium nitrate in 70% isopropyl alcohol). After the solvent evaporated,
the coagulant-coated former was reheated to 70 ºC then dipped into a compounded latex
emulsion and held there for different dwell times during which a thin film of coagulated
latex was deposited (dwell times varied from 5, 15, 30, 45, and 60 sec). Once the latex
gelled via heating (15 min at 100 ºC), it was leached in water (30 min at 55 ºC), followed
by stripping of the former and subsequent vulcanization of the rubber article (20 min at
105 ºC). The rubber article was then placed in a tumble dryer post-vulcanization (60 min
at 60 ºC). All GNRL thin films in this study were made using a Diplomat automated
dipper (DipTech Systems Inc., Kent, OH, USA).
7.2.2. Tensile Properties
Four dumbbell samples of each compound at each dwell time were cut using Die
C according to ASTM D 412 (ASTM International, 2013b). Film thickness varied from
0.23 mm to 0.24 mm for all composites. Evaluation of the tensile mechanical properties
followed ASTM D 412 and was determined using an Instron 3366 with Bluehill v. 2.17
software package (Instron, Norwood, MA, USA) (ASTM International, 2013b). Samples
were tested using a crosshead speed of 500 mm/min at room temperature (26°C). The
reported mean ± the standard error from the mean (SE) values are averages of at least 4
samples. In addition, statistical analysis was performed using Minitab, version 16.0

153
(State College, PA). Significant differences (P-values < 0.05) in tensile data amongst
GNRL compounds for each film thickness were determined using one-way analysis of
variance (ANOVA) and the Fisher method.
7.2.3. Scanning Electron Microscopy (SEM)
The morphology of the various fillers was investigated using a Hitachi S-3500N
scanning electron microscope (Tokyo, Japan). Samples were sputter-coated with a thin
layer of platinum (0.2 KÅ) by an Anatech Hummer 6.2 Sputtering system prior to
analysis.

154
7.3. Results and Discussion
7.3.1. Tensile properties
Eggshell-GNRL composites showed a range of varied tensile mechanical
properties, compared to unfilled GNRL thin films. Eggshell-GNRL composites showed a
significant stiffening in some composites, namely macrosized eggshell at 1 phr and
microsized eggshell at 2 phr, compared to unfilled GNRL thin films. However other
eggshell-GNRL composites had a significant softening effect compared to unfilled
GNRL films; this included all nanosized eggshell composites, as well as microsized and
macrosized eggshell composites at high loadings, 5 and 10 phr (Fig. 7.1a). Elongation at
break was significantly increased in all nanosized eggshell composites, and in higher
loadings (5 and 10 phr) of microsized eggshell-GNRL composites (Fig. 7.1b).
Macrosized eggshell-GNRL composites, in all loadings, did not significantly change
elongation at break, compared to unfilled GNRL thin films (Fig. 7.1b). There is no
reinforcement effect in any of the eggshell-GNRL composites; tensile strength is not
improved significantly compared unfilled GNRL thin films (Fig. 7.1c). Eggshells at 2
phr loading, in any size, provide a diluent effect; there is no significant change tensile
strength compared to unfilled GNRL thin films (Fig. 7.1c).
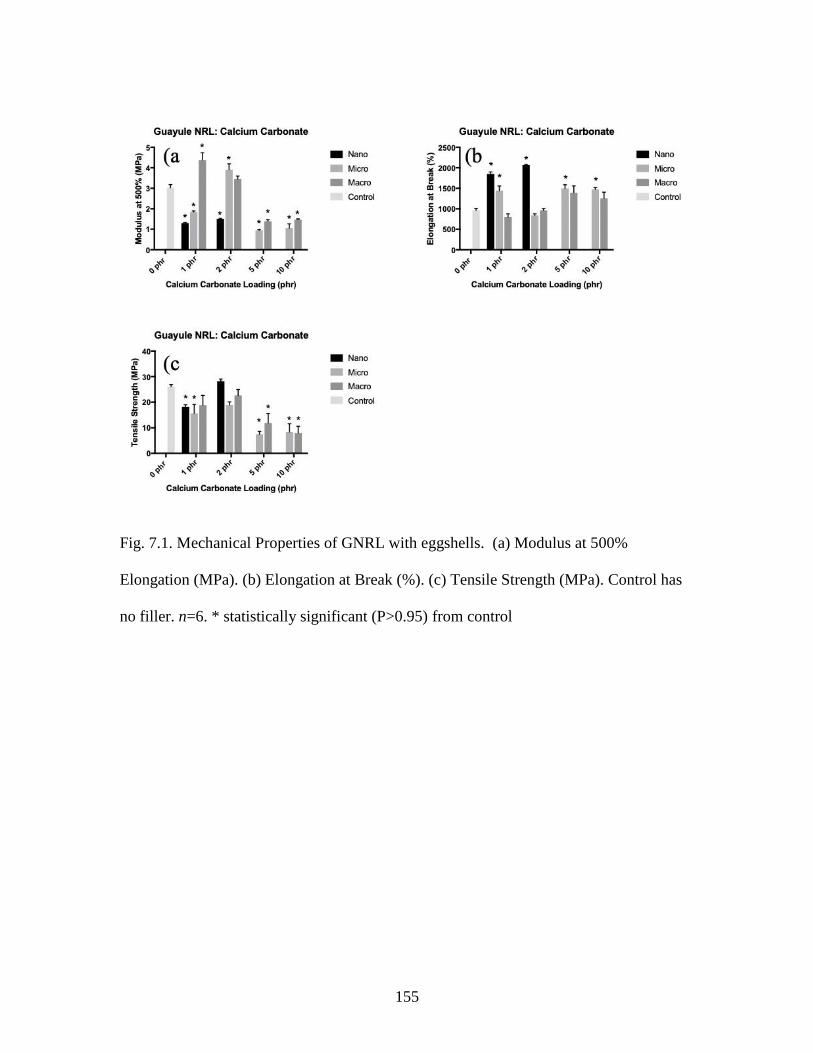
155
Fig. 7.1. Mechanical Properties of GNRL with eggshells. (a) Modulus at 500%
Elongation (MPa). (b) Elongation at Break (%). (c) Tensile Strength (MPa). Control has
no filler. n=6. * statistically significant (P>0.95) from control

156
Guayule bagasse (GB)-GNRL composite films all have decreased tensile strength
compared to unfilled GNRL films (Fig. 7.2c). GB-NRL films are softer in composites
containing high loadings of macro fillers and nanofillers; whereas GB-NRL films are
stiffer in composites with low loadings of macro fillers (Fig. 7.2a). GB-NRL films that
are softer (Fig. 7.2a), also have a higher elongation at break (Fig. 7.2b); this can be
attributed to the fillers interfering in crosslinking and resulting in a softer, more ductile
composite film. The guayule bagasse fillers were not able to provide a reinforcement
effect, but GB-NRL composites such as 1 phr micro are able to produce a diluent effect
for modulus at 500% and elongation at break, however are not able to maintain tensile
strength properties.
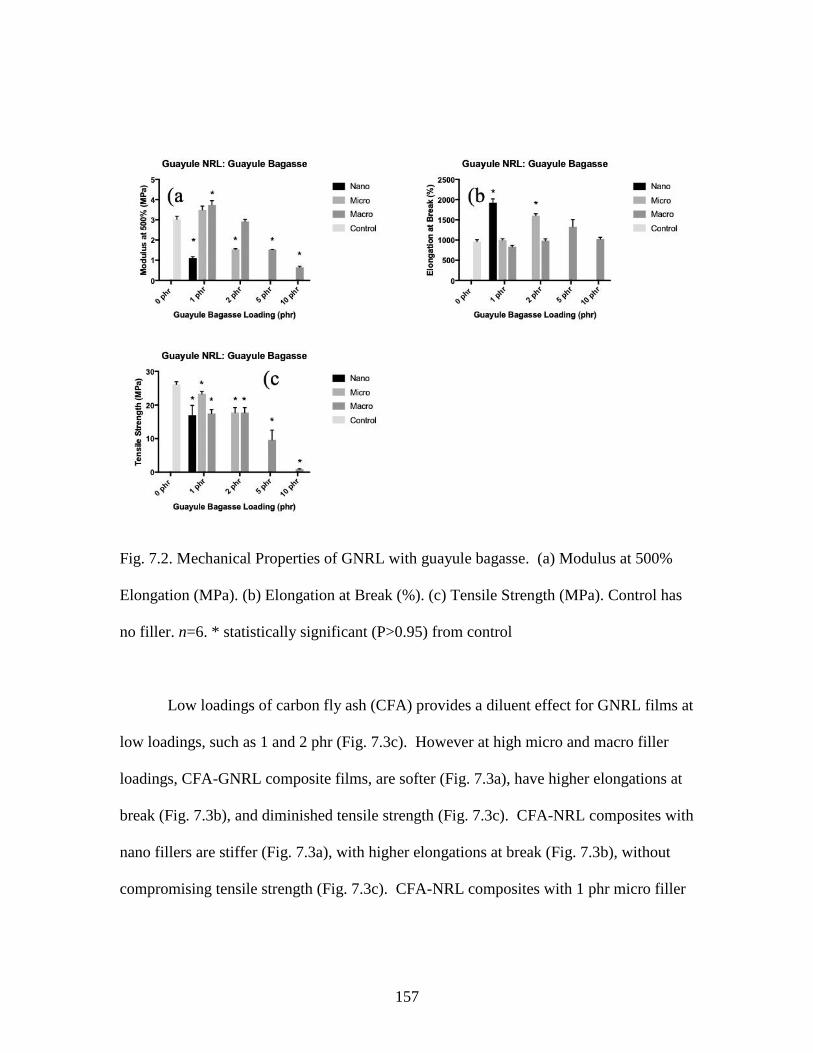
157
Fig. 7.2. Mechanical Properties of GNRL with guayule bagasse. (a) Modulus at 500%
Elongation (MPa). (b) Elongation at Break (%). (c) Tensile Strength (MPa). Control has
no filler. n=6. * statistically significant (P>0.95) from control
Low loadings of carbon fly ash (CFA) provides a diluent effect for GNRL films at
low loadings, such as 1 and 2 phr (Fig. 7.3c). However at high micro and macro filler
loadings, CFA-GNRL composite films, are softer (Fig. 7.3a), have higher elongations at
break (Fig. 7.3b), and diminished tensile strength (Fig. 7.3c). CFA-NRL composites with
nano fillers are stiffer (Fig. 7.3a), with higher elongations at break (Fig. 7.3b), without
compromising tensile strength (Fig. 7.3c). CFA-NRL composites with 1 phr micro filler
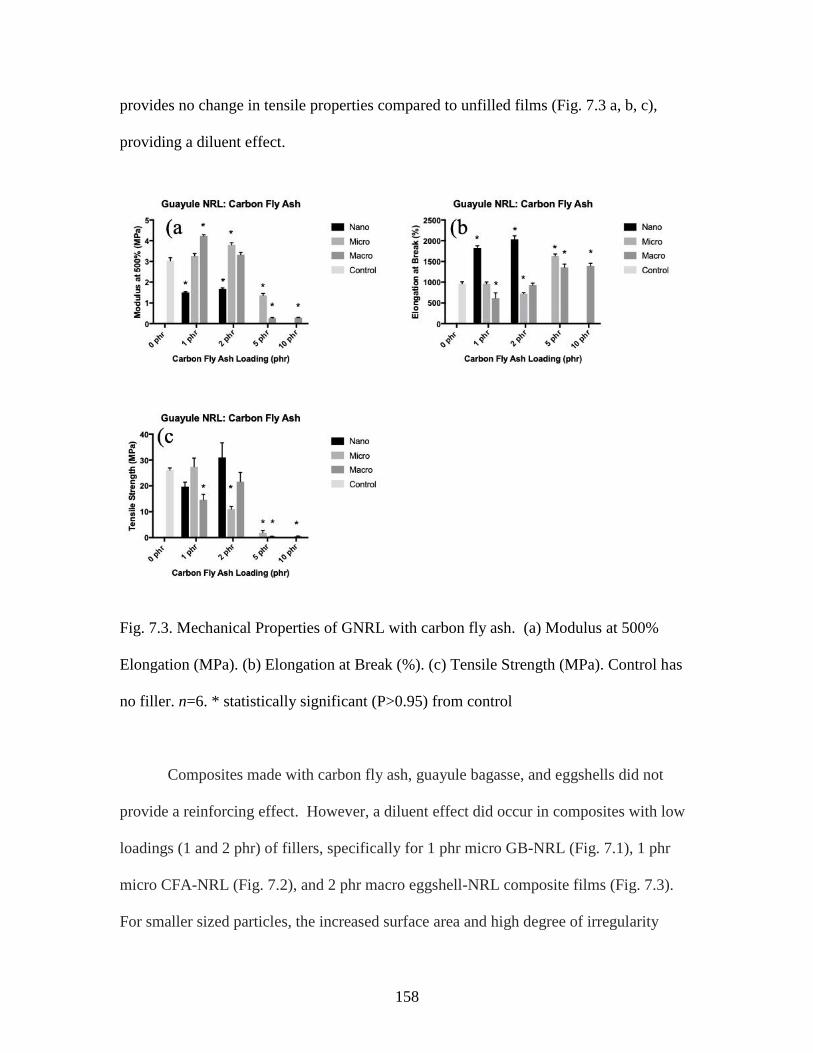
158
provides no change in tensile properties compared to unfilled films (Fig. 7.3 a, b, c),
providing a diluent effect.
Fig. 7.3. Mechanical Properties of GNRL with carbon fly ash. (a) Modulus at 500%
Elongation (MPa). (b) Elongation at Break (%). (c) Tensile Strength (MPa). Control has
no filler. n=6. * statistically significant (P>0.95) from control
Composites made with carbon fly ash, guayule bagasse, and eggshells did not
provide a reinforcing effect. However, a diluent effect did occur in composites with low
loadings (1 and 2 phr) of fillers, specifically for 1 phr micro GB-NRL (Fig. 7.1), 1 phr
micro CFA-NRL (Fig. 7.2), and 2 phr macro eggshell-NRL composite films (Fig. 7.3).
For smaller sized particles, the increased surface area and high degree of irregularity

159
restricts the polymer chain motion under applied strain. This behavior is not observed in
macro sized fillers due to the reduced surface area and more regular, laminar shape. In 5
and 10 phr filler loadings properties were decreased; the NRL acted as a binder material
for the fillers. Nano sized composites with high loadings (5 and 10 phr) could not be
successfully made; the emulsions were unstable and coagulated during mixing due to
insufficient wetting of the fillers.
Composites made with micro waste-derived fillers performed better than
composites made with macro particles (Figs. 7.1-7.3). This is attributed to the larger
particle size of the macro fillers; macro fillers have less surface area per unit weight than
smaller particles, having less interaction with the polymer, and also generating flaws
within the composite. Micro sized particles have a lower bulk density than macro sized
particles, most likely attributed to the broader particle size distribution of the macro sized
particles. The broader particle distribution allows smaller particles to fit between the
void spaces of bigger particles.
Composites that provided a diluent effect contained micro sized fillers.
Interaction between these waste-derived fillers and interactions between the particles and
rubber, help maintain physical properties. Hydrophobicity of the waste-derived fillers
contribute to the filler’s affinity for the rubber.
Filler-filler interactions existing between particles with polar surfaces are
attributed to hydrogen bonding; whereas non-polar fillers have filler-filler interactions
due to van der Waals forces. Filler-filler interactions can promote the formation of a
percolation network, that can contribute to the overall reinforcement of the material by

160
restricting chain mobility. The linear chain structure of GNRL allows it to flow more
easily into a filler network than the more branched HNRL.
Surface chemistry of fillers can effect the efficiency of crosslinking. CFA is
composed of alumino-silicate (>50%); silanol groups can react with rubber compounding
ingredients and lead to slower cures. Eggshells are over 95% calcium carbonate, which
can expedite cure rates due to its alkalinity. Since processing conditions were the same
for all films, slower cure rates may contribute to a lower crosslink density, which results
in lower mechanical properties.

161
7.4. Conclusions
Agro-industrial residues such as eggshells, carbon fly ash, and guayule bagasse
are abundant solid wastes with heterogeneous compositions. These characteristics make
them promising materials for NRL composites, comparable in properties to unfilled NRL
films. In addition, the renewable character of agro-industrial residues can improve the
sustainability of NRL products while adding value to these waste-derived fillers.
Therefore, the results encourage the further study of utilizing renewable waste-derived
materials to reduce the amount of non-renewable fillers in NRL composites. Future work
includes tailoring mechanical properties of NRL composites by modification of fillers
such as surface functionalization with silane coupling agents, and blending particles of
various origins, and sizes to achieve specific requirements for various end applications.

162
Chapter 8: Mechanical properties of Hevea natural rubber latex thin film
composites with biobased fillers.
J. Lauren Slutzky a*, and Katrina Cornish a,b
aOhio State University, Department of Food, Agricultural and Biological Engineering,
1680 Madison Avenue, Wooster, Ohio 44691 USA
bOhio State University, Department of Horticulture and Crop Science, 1680 Madison
Avenue, Wooster, Ohio 44691 USA
*Corresponding author: Katrina Cornish, Williams Hall, 1680 Madison Avenue,
Wooster, Ohio 44691, USA. Tel: 330-263-3982 Fax: 330-263-3887 Email:
Abstract:
Natural rubber latex (NRL) products are often compounded with non-sustainable
reinforcing fillers such as carbon black. This work focuses on the manufacturing of NRL
composites using natural rubber latex from the plant species Hevea brasiliensis with and
without soluble proteins, compounded with sustainable fillers from waste streams:
calcium carbonate-rich eggshells, carbon fly ash, and guayule bark bagasse. The soluble
protein in NRL contains severe allergens, but serves as a naturally occurring surfactant
that may improve filler dispersion in NRL. To make the NRL composite thin films,
fillers were compounded into NRL, followed by coagulant dipping, and vulcanization.
The effect of filler type, size, and loading in ultra-low protein and standard protein NRL
composites on mechanical properties were characterized, according to ASTM D 412.
Ultra-low protein NRL composites were more susceptible to softening compared
than standard protein NRL composites. Nano sized fillers increase elongation at break of

163
ultra-low protein NRL composites across all filler types; whereas nano fillers in standard
NRL composites have no effect on elongation at break compared to unfilled standard
NRL films. Overall, none of the fillers in any loading had a reinforcement effect. Many
composites did not have significantly different properties compared to unfilled NRL thin
films, indicating a diluent effect.
Keywords: Hevea, Natural rubber latex composites, biofillers, calcium carbonate, carbon
fly ash, guayule bagasse, elastomer thin films
8.1. Introduction
Natural rubber latex (NRL) is a colloidal suspension of rubber and non-rubber
constituents, derived from the plant species Hevea brasiliensis. Standard NRL typically
contains 60% dry rubber, with an additional 1.6% dry non-rubber constituents that
includes proteins and phospholipids (Sansatsadeekul et al., 2011). Ultra-low protein
Hevea NRL undergoes an aluminum hydroxide treatment that removes soluble protein
and other impurities, resulting in 0.5% dry non-rubber constituents (Swason, 2008). The
effect of non-rubber constituents in NRL thin films have been shown to have profound
effects on mechanical properties, attributed to protein-polymer interactions (Monadjemi
et al., 2016). However, the effect of soluble protein in NRL composites with biofillers
has not been previously investigated.
Fillers are expected to adhere to rubber particles during thin film dipping and
vulcanization to provide a reinforcing effect. However, many fillers are not able to
adhere to rubber particles without a coupling agent, which can add cost to the final
product. Fillers that are not effectively adhered to rubber particles provide a diluent
effect; diluent fillers are inert and added to polymer materials at levels that don’t cause

164
premature material failure, to reduce the cost of the final product. Diluent fillers such as
calcium carbonate, sodium fluorohectorite, and kaolinite clay are common bulk
cheapeners used in NRL products (Dick, 2009) but frequently reduce product
performance. However, calcium carbonate and kaolinite clay requires extensive mining,
which is dangerous and can ultimately cause detriment to ecological systems (Dick,
2009). Sodium fluorohectorite is synthesized at extremely high temperatures, using glass
precursors, which is energy intensive and a dangerous synthesis (Stoter et al., 2013).
Therefore diluent fillers derived from waste streams are of interest; fillers derived from
waste streams require little energy for preparation, and are essentially negligible in cost.
Waste streams that are applicable as diluent fillers for NRL composites include
calcium carbonate-rich eggshells, carbon fly ash, and guayule bark bagasse. Eggshells,
carbon fly ash, and guayule bark bagasse have successfully been incorporated into natural
rubber composites as diluent fillers, and in NRL and GNRL composites as partial
replacements of carbon black (Barrera and Cornish, 2015; Barrera and Cornish, 2016) or
silica (Ren et al., 2019) when they can also act as reinforcing fillers and also improve the
dispersion of the conventional filler. This study analyzed the effect of NRL soluble
protein and filler type and loading, on mechanical properties of NRL composites made
with the alternative fillers carbon fly ash, calcium carbonate-rich eggshells, or guayule
bagasse. The fillers varied in particle sizes, loading, and type of latex (with or without
soluble protein).

165
8.2. Experimental
8.2.1. Materials
Ultra-low protein NRL (trade name Vytex) and standard protein NRL (trade name
Centex) were purchased from Centrotrade (Chesapeake, VA). Compounding chemicals,
specifically the accelerators zinc diethyldithiocarbamate (ZDEC), diphenyl guanidine
(DPG), and dipentamethylenethiuram tetrasulfide (DPTT), activator zinc oxide, sulfur,
and antioxidant were purchased from Akron Dispersions (Akron, OH). The ammonium
hydroxide was purchased from W.W. Grainger, Inc (Salt Lake City, UT, USA).
The waste filler raw materials were generously donated as follows: eggshells (ES)
by Troyer’s Home Pantry (Apple Creek, OH), carbon fly ash (CFA) by Cargill Salt
(Akron, OH), and guayule bagasse (GB) was generated as a co-product of latex
extraction at our facility from guayule shrubs donated by PanAridus (Casa Grande, AZ).
8.2.2. Preparation of Fillers
Raw materials were dried at 55 oC and then ground using an IKA A11 basic mill
(Wilmington, NC). Macro sized particles were separated using a size 50 and 400 mesh
sieve from Fisher Scientific (Pittsburgh, PA), with resulting particles ranging from 300
µm to 38 µm. Micro sized particles were separated using a size 400 mesh, isolating
particles 38 µm and smaller. Nano sized fillers were made by dispersing the micro sized
particles in distilled water, and then wet milling to submicron size using a Planetary Ball
Mill 100 manufactured by Glen Mills (Clifton, NJ). The carbon fly ash and guayule bark
bagasse were dry milled as received, whereas the eggshells were washed and membranes
were peeled from the eggshells prior to milling. Particle size distributions were verified
using a particle size analyzer LA-950V2, Horiba Scientific (Irvine, CA) (Fig. 8.1). Filler

166
bulk densities were determined by weighing a known volume of each filler and
calculating the bulk density as mass/volume occupied (Table 8.1).
Fig. 8. 1. Macro (solid line) and micro sized (dashed line) fillers’ particle size
distribution. a) Carbon fly ash; macro (median 76.20 μm, mean 89.32 μm, SD 61.94 μm),
micro (median 11.25 μm, mean 12.12 μm, SD 4.93 μm), b) eggshells; macro )median
230.33 μm, mean 241.46 μm, SD 111.01 μm), micro (median 16.07 μm, mean 23.19 μm,
SD 20.79 μm), c) guayule bagasse; macro (median 243.10 μm, mean 279.33 μm, SD
188.56 μm), micro (median 49.09 μm, 54.75 μm, SD 30.63 μm).

167
Table 8.1. Filler bulk densities
Filler Size Density (g/cm3)
Carbon fly ash Macro 0.74 ± 0.02 Carbon fly ash Micro 0.57 ± 0.01
Guayule bagasse Macro 0.53 ± 0.02
Guayule bagasse Micro 0.36 ± 0.07
Eggshells Macro 1.32 ± 0.02
Eggshells Micro 0.79 ± 0.05
8.2.3. Emulsion chemistry/compounding
NRL composites were made using a compounding recipe (Table 8.1) while
varying the amount, type, and particle size of filler. The amount of fillers and other
materials were added to NRL at specified concentrations based on parts per hundred dry
rubber (phr). Fillers were dispersed in deionized water (amount of water needed to
achieve 48% solids by volume was used to disperse fillers), and stirred for 1 hour with a
30-rpm hand mixer. The latex compound recipe without fillers was prevulcanized for 2.5
hours while stirring with a 30-rpm hand mixer. The filler-water dispersion was then
added to the prevulcanized latex compound, and stirred for an additional 1 hour with a
30-rpm hand mixer.

168
Table 8.2. Latex Compound Recipe.
Dry Weight
NRL 100
Sulfur 2
Ammonium Hydroxide 1
ZnO 1
Antioxidant 2
Zinc Diethyldiothiocarbamate (ZDEC)
Accelerator
0.5
Diphenyl guandine (DPG) Accelerator 0.4
Dipentamethylenethiuram (DPTT)
Accelerator
0.6
Filler 1, 2, 5 or 10

169
8.2.4. Thin film manufacture by dipping
Thin film elastomer products are manufactured by dipping formers into emulsions
with subsequent heating to remove liquids and vulcanize the NRL. Initially, a stainless
steel former was heated to 70 ºC and dipped (10 second dwell) into a coagulant solution
(25% aqueous calcium nitrate in 70% isopropyl alcohol). After the solvent evaporated,
the coagulant-coated former was reheated to 70 ºC then dipped into a compounded latex
emulsion and held there for different dwell times during which a thin film of coagulated
latex was deposited (dwell times varied from 5, 15, 30, 45, and 60 sec). Once the latex
gelled via heating (15 min at 100 ºC), it was leached in water (30 min at 55 ºC), followed
by stripping of the former and subsequent vulcanization of the rubber article (20 min at
105 ºC). The rubber article was then placed in a tumble dryer post-vulcanization (60 min
at 60 ºC). All NRL thin films in this study were made using a Diplomat automated dipper
(DipTech Systems Inc., Kent, OH, USA).
8.2.5. Tensile Properties
Four dumbbell samples of each compound at each dwell time were cut using Die
C according to ASTM D 412 (ASTM International, 2013a). Film thickness varied from
0.24 mm to 0.23 mm for all composites. Evaluation of the tensile mechanical properties
followed ASTM D 412 and was determined using an Instron 3366 with Bluehill v. 2.17
software package (Instron, Norwood, MA, USA) (ASTM International, 2013a). Samples
were tested using a crosshead speed of 500 mm/min at room temperature (26 °C). The
reported mean ± the standard error from the mean (SE) values are averages of at least 4
samples. In addition, statistical analysis was performed using Minitab, version 16.0
(State College, PA). Significant differences (P-values < 0.05) in tensile data amongst

170
NRL compounds for each film thickness were determined using one-way analysis of
variance (ANOVA) and the Fisher method.
8.2.6. Scanning Electron Microscopy (SEM)
The morphology of the various fillers was investigated using a Hitachi S-3500N
scanning electron microscope (Tokyo, Japan). Samples were sputter-coated with a thin
layer of platinum (0.2 KÅ) by an Anatech Hummer 6.2 Sputtering system prior to
analysis.
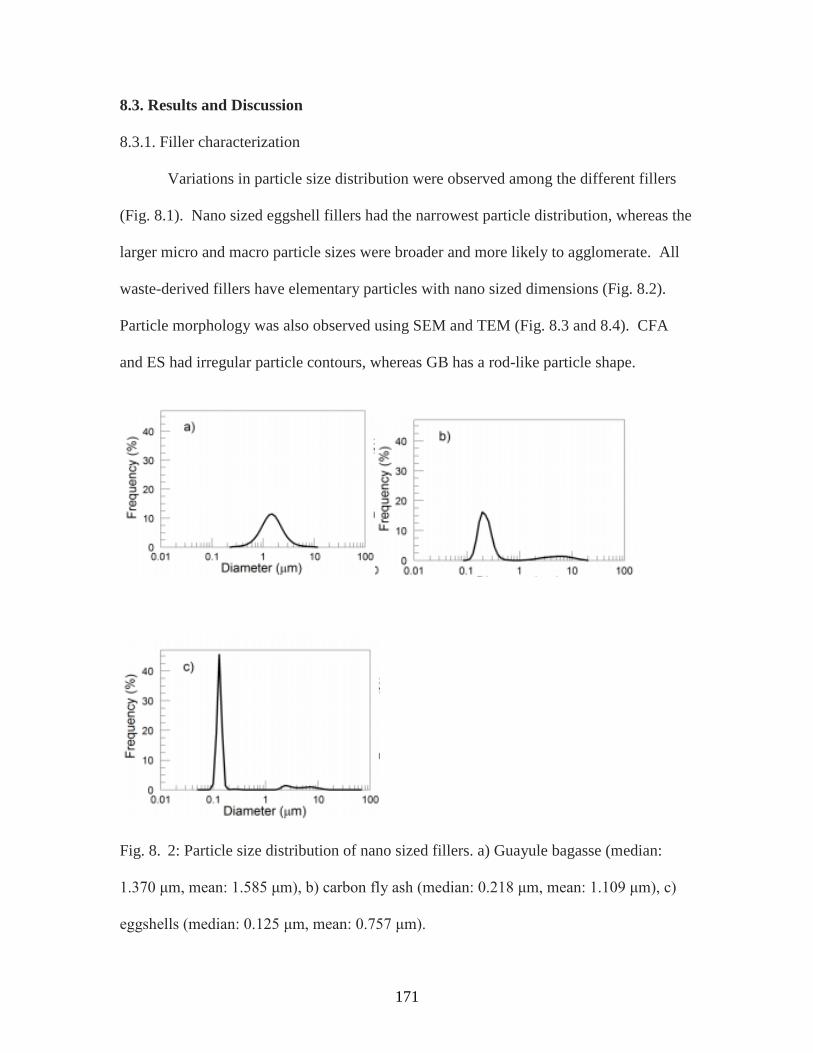
171
8.3. Results and Discussion
8.3.1. Filler characterization
Variations in particle size distribution were observed among the different fillers
(Fig. 8.1). Nano sized eggshell fillers had the narrowest particle distribution, whereas the
larger micro and macro particle sizes were broader and more likely to agglomerate. All
waste-derived fillers have elementary particles with nano sized dimensions (Fig. 8.2).
Particle morphology was also observed using SEM and TEM (Fig. 8.3 and 8.4). CFA
and ES had irregular particle contours, whereas GB has a rod-like particle shape.
Fig. 8. 2: Particle size distribution of nano sized fillers. a) Guayule bagasse (median:
1.370 μm, mean: 1.585 μm), b) carbon fly ash (median: 0.218 μm, mean: 1.109 μm), c)
eggshells (median: 0.125 μm, mean: 0.757 μm).
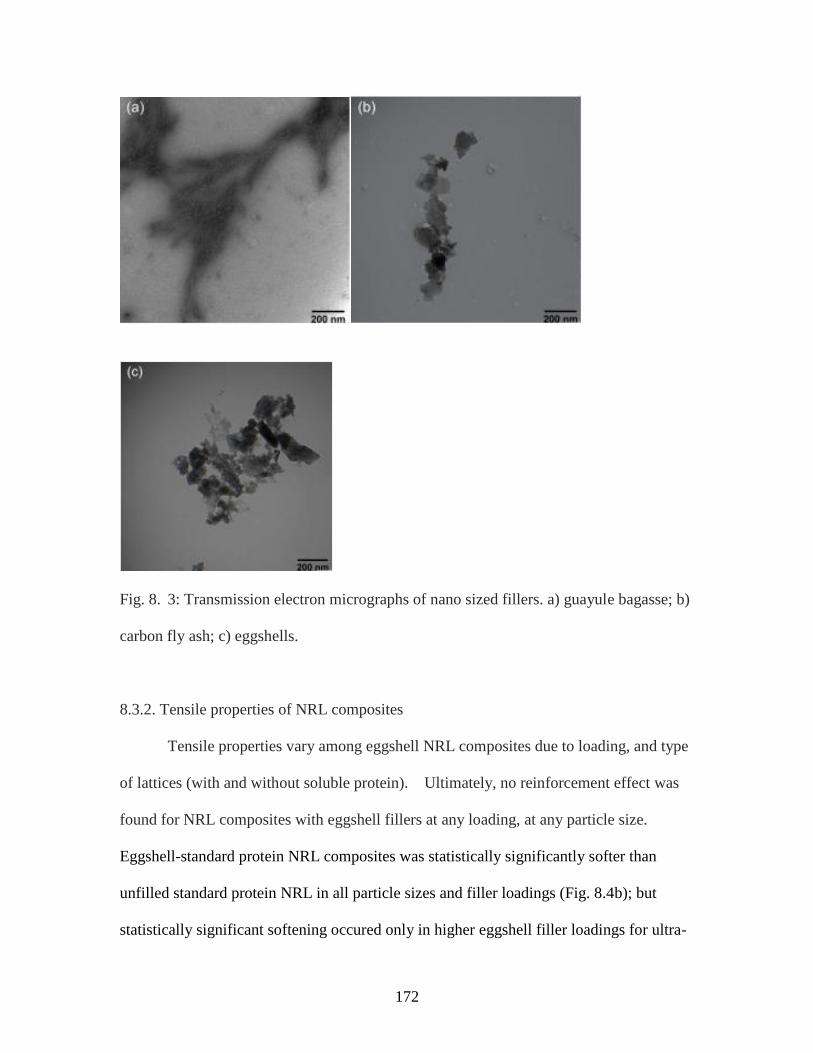
172
Fig. 8. 3: Transmission electron micrographs of nano sized fillers. a) guayule bagasse; b)
carbon fly ash; c) eggshells.
8.3.2. Tensile properties of NRL composites
Tensile properties vary among eggshell NRL composites due to loading, and type
of lattices (with and without soluble protein). Ultimately, no reinforcement effect was
found for NRL composites with eggshell fillers at any loading, at any particle size.
Eggshell-standard protein NRL composites was statistically significantly softer than
unfilled standard protein NRL in all particle sizes and filler loadings (Fig. 8.4b); but
statistically significant softening occured only in higher eggshell filler loadings for ultra-

173
low protein NRL composites (Fig. 8.4a). Eggshell-ultra-low protein NRL composites
showed a significant increase in elongation at break with nanosized 1 phr and 2 phr
loadings, as well as a microsized 5 phr loading, compared to an unfilled ultra-low protein
NRL film (Fig. 8.4c). However, in eggshell-standard protein NRL composites,
elongation at break is improved only with nanosized 2 phr loading (Fig. 8.4d). Eggshell
fillers at any size or any loading do not improve the tensile strength of NRL composite
films, regardless of presence or absence of soluble latex protein (Fig. 8.4f).
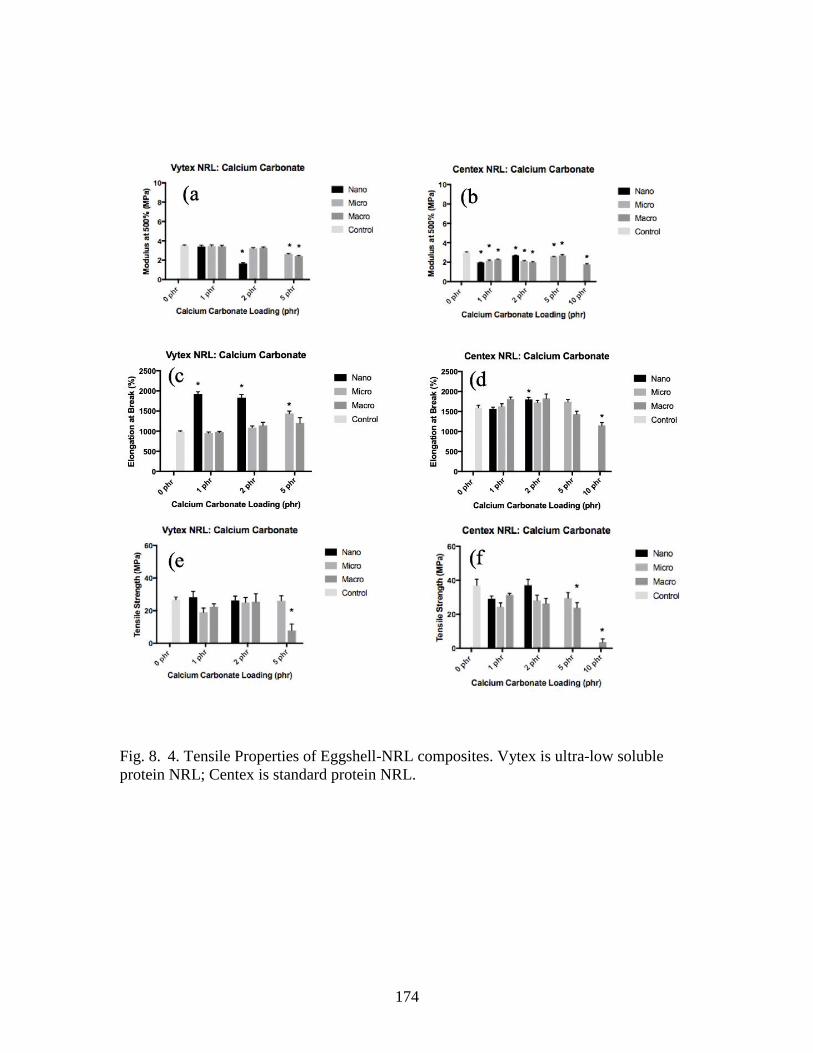
174
Fig. 8. 4. Tensile Properties of Eggshell-NRL composites. Vytex is ultra-low soluble
protein NRL; Centex is standard protein NRL.

175
Tensile properties of carbon fly ash NRL composites varied with filler loading
and amount of protein in the latex. Compared to unfilled NRL thin films, carbon fly ash-
ultra-low protein NRL composites significantly softened, specifically with nanosized
fillers at 1 and 2 phr loadings and in macro-sized fillers at 2, 5 and 10 phr loadings (Fig.
8.5a). Microsized carbon fly ash fillers in ultra-low protein NRL also softened, but
notably only at higher loadings of 5 and 10 phr. However, carbon fly ash composites
with standard protein NRL softened less than the protein films; only carbon fly ash
composites with high loadings (5 and 10 phr) of macrosized fillers caused a significant
softening effect in thin films (Fig. 8.5b). The amount of protein in the NRL also affected
elongation at break of carbon fly ash composites, which significantly increased in 1 and 2
phr nanosizded fillers (Fig. 8.5c). Elongation at break was unaffected by carbon fly ash
fillers of any size or any loading in standard NRL-carbon fly ash composites (Fig. 8.5d).
There was no reinforcing effect for carbon fly ash compared to unfilled NRL thin films,
regardless of the amount of soluble protein in NRL (Fig. 8.5e, Fig. 8.5f). At higher
loadings such as 5 and 10 phr, there is a significant decrease in properties in micro and
macro sized fillers in films made from both ultra-low protein and standard protein NRL.
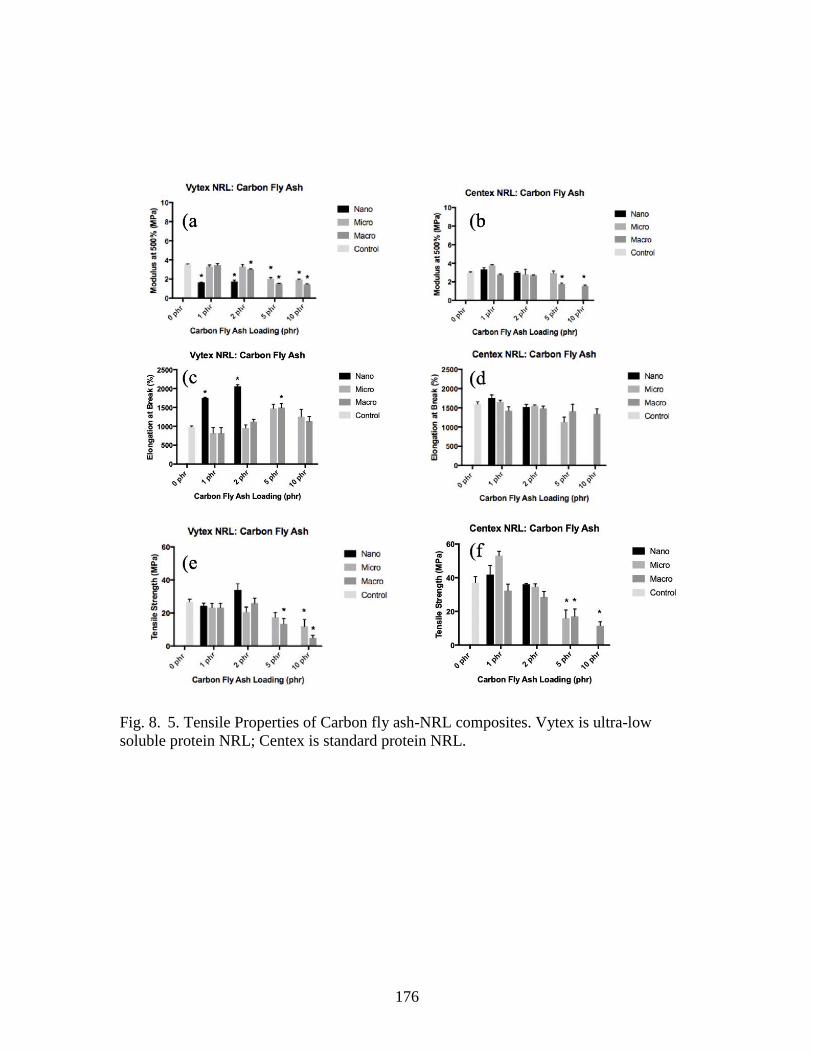
176
Fig. 8. 5. Tensile Properties of Carbon fly ash-NRL composites. Vytex is ultra-low
soluble protein NRL; Centex is standard protein NRL.

177
Guayule bagasse was not capable of providing a reinforcing effect, regardless of
particle size, loading, or amount of soluble protein in NRL (Fig. 8.6). Guayule bagasse
composites softened in nanosized guayule bagasse-ultra-low protein NRL composites, as
well as high loadings of macrosized guayule bagasse (Fig. 8.6a). However, guayule
bagasse did not soften guayule-bagasse-standard soluble protein NRL composites (Fig.
8.6b). Elongation at break is significantly increased in nanosized guayule bagasse-ultra-
low protein NRL composites, compared to unfilled ultra-low protein NRL composites
(Fig. 8.6c). However, elongation at break significantly decreased in 1phr microsized
guayule bagasse-ultra-low protein NRL composites, compared to unfilled ultra-low
protein NRL composites (Fig. 8.6c). In guayule bagasse at nano and micro sizes did not
affect standard-soluble protein NRL composites (Fig. 8.6d), but macro sized guayule
bagasse-standard soluble protein NRL composites had significantly lower elongation at
break. Guayule bagasse fillers decreased tensile strength of composites especially in
those made with standard-protein NRL, most marked with macrosized filled standard-
protein NRL thin films (Fig 6.8f). Guayule bagasse did not reinforce ultra-low protein
NRL composites and 1 phr microsized guayule bagasse filled composites were
significantly weaker than the ultra-low protein NRL thin films without fillers (Fig 6.8e).
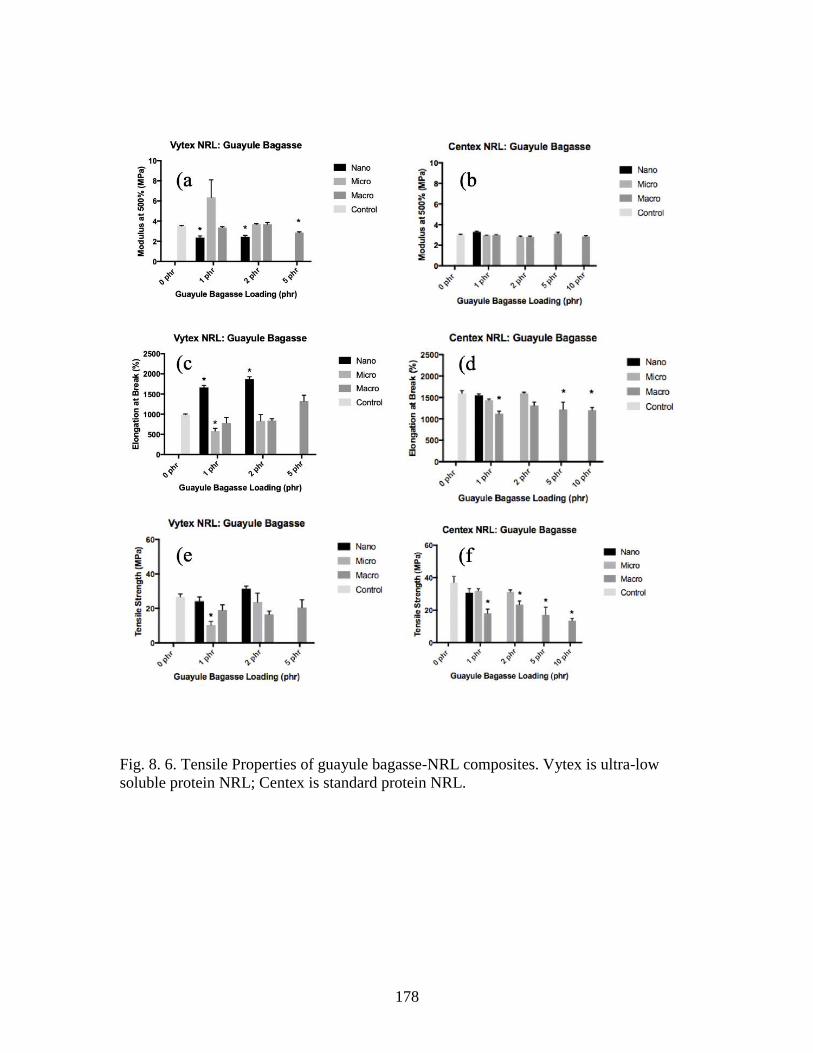
178
Fig. 8. 6. Tensile Properties of guayule bagasse-NRL composites. Vytex is ultra-low
soluble protein NRL; Centex is standard protein NRL.

179
Composites made with carbon fly ash, guayule bagasse, and eggshells did not
provide a reinforcing effect, regardless of type of latex used. Structure of the fillers can
influence reinforcing effect. The eggshells and CFA fillers have a high surface area due
to its platy, rough structure and high porosity (Fig. 8.2). The porosity promotes a wetting
effect, that can improve interfacial adhesion between the polymer and filler. The fillers
in the NRL composite films were not surface treated; surface modification of fillers to
improve surface hydrophobicity makes it more compatible with the rubber polymer.
Hydrophobic surface chemistries in fillers can improve polymer-filler interaction and
contributed to the observed reinforcing effect. Naturally occurring resins in GB can affect
mechanical properties of GB-NRL composites. Resins can potentially provide a
plasticizing effect, increasing ductility and properties such as elongation at break and
modulus at 500% elongation.
Many of the composites made in this study meet the physical properties required
for their application in a range of rubber products. Potential applications include medical
surgical gloves, and textured dipped coatings. The filler-NRL composites that provide a
diluent effect could be used to manufacture NRL products with a low carbon footprint
and are most sustainable than conventional fillers such as mined calcium carbonate, and
carbon black.

180
8.4. Conclusions
Waste-derived fillers can provide a diluent effect for NRL composite films,
regardless of the level of soluble protein in the NRL. Diluent composite NRL films were
achieved by low loadings of micro waste-derived fillers. This is important due to the
renewable character of these materials, and possible applications for NRL composites.

181
Chapter 9: Conclusion
The main goals of this research were to develop thin film applications for type I
circumallergenic GNRL and type I hypoallergenic ultra-low protein Hevea NRL; this
included optimizing a type IV hypoallergenic accelerator package to make medical grade
elastomers, and developing latex thin film composites with agro-industrial residues for
industrial grade elastomers.
The type IV hypoallergenic accelerator package was successfully used to
compound thin films that meet the specifications for surgical gloves (ASTM 3577) for
both type I circumallergenic GNRL, and type I hypoallergenic ultra-low protein Hevea
NRL. Instrinsic differences in the composition of GNRL and ultra-low Hevea NRL
required markedly different accelerator concentrations and ratios to maximize their
mechanical performance and achieve or exceed the tensile properties outlined in ASTM
3577; GNRL required 3x more accelerator than ultra-low protein Hevea NRL
formulations, probably due to its even lower protein content as well as differences in
stabilizing systems. Statistical modeling of GNRL formulations correlated only one type
of accelerator loading and ultimate elongation; whereas ultra-low protein Hevea NRL
statistical modeling correlated both accelerator types, and film thickness with all tensile
mechanical properties modeled. Thus, GNRL must have other significant properties
which would be better predictors of behavior but were not identified or taken in the
current models for statistical modeling. Also, the buffering systems of the GNRL and
ultra-low protein Hevea NRL are drastically different; GNRL was stabilized with
potassium hydroxide to stabilize the emulsion (a strong base) whereas ultra-low protein
Hevea NRL is stabilized by ammonium hydroxide (a weak base). Ammonium hydroxide

182
is known to work as an activator with zinc-based catalyst systems, and therefore
buffering systems of the lattices would have an effect on crosslinking and accelerator
efficiency and may cause the stronger correlations in formulation-property statistical
models seen in this latex.
The composites of GNRL and ultra-low protein Hevea NRL with agro-industrial
wastes did not reinforcement the composite latex films, regardless of latex type, filler
type, or loading. However, several sizes and loadings could be used without damanging
properties until higher loadings were used. Fillers did affect crosslinking in some
composites in GNRL and NRL (with and without soluble protein) significantly
decreasing tensile strength, and softening the modulus at 500% elongation, compared to
unfilled thin films. Soluble protein in latex had an effect on % elongation at break: nano
fillers increased elongation at break of GNRL and ultra-low protein Hevea NRL
composites, but did not occur in standard soluble protein NRL. Thus, latex composition,
including nonrubber components, and polymer-filler interations, are important to the final
performance of composite films.

183
Chapter 10: Future Studies
This research revealed the complexity and variation in types of latex and
their effect on crosslinking, and ultimately tensile mechanical properties. This work
provides a foundation for future research in understanding and predicting how variances
in latex chemistry affect chemical compounding and ultimately, mechanical properties.
Future work should include: investigating the crosslinking efficiency of a GNRL
stabilized in ammonium hydroxide for chemical accelerator packages, optimizing
chemical accelerator packages for the filler composite work, and functionalizing fillers
with a hydrophobic or sulfur surface group to improve the reinforcing effect in the latex
composites. Additional material characterization could be done of the composites
formulated in this work could include degradation studies, oxidation resistance, and
analysis of dynamic properties.

184
References:
Abraham, E., Deepa, B., Pothan, L.A., Jacob, M., Thomas, S., Cvelbar, U., Anandjiwala,
R., 2011. Extraction of nanocellulose fibrils from lignocellulosic fibres: A novel
approach. Carbohydr. Polym. 86, 1468–1475. doi:10.1016/j.carbpol.2011.06.034
Abraham, E., Deepa, B., Pothan, L.A., John, M., Narine, S.S., Thomas, S., Anandjiwala,
R., 2013a. Physicomechanical properties of nanocomposites based on cellulose
nanofibre and natural rubber latex. Cellulose 20, 417–427. doi:10.1007/s10570-
012- 9830-1
Abraham, E., Thomas, M.S., John, C., Pothen, L. a., Shoseyov, O., Thomas, S., 2013b.
Green nanocomposites of natural rubber/nanocellulose: Membrane transport,
rheological and thermal degradation characterisations. Ind. Crops Prod. 51, 415–
424. doi:10.1016/j.indcrop.2013.09.022
Alenius, H., Turjanmaa, K., Palosuo, T., 2002. Natural Rubber latex allergy.
Occupational and Environmental Medicine. 59, 419-424.
Amnuaypornsri, S., Sakdapipanich, J., Toki, S., Hsiao, B.S., Ichikawa, N., Tanaka, Y.,
2008. Strain-Induced Crystallization of Natural Rubber: Effect of Proteins and
Phospholipids. Rubber Chem. Technol. 81, 753–766. doi:10.5254/1.3548230
Angellier, H., Molina-Boisseau, S., Dufresne, A., 2005a. Mechanical properties of waxy
maize starch nanocrystal reinforced natural rubber. Macromolecules 38, 9161–
9170. doi:10.1021/ma0512399

185
ASTM International, 2013a. ASTM standard D412-15a. Standard test methods for
vulcanized rubber and thermoplastic elastomers- tension. Doi:10.1520/D0412-
15A
ASTM International, 2013b. ASTM standard D3577. Standard specification for rubber
surgical gloves.
Barrera, C.S., Cornish, K., 2016. High performance waste-derived filler/carbon black
reinforced guayule natural rubber composites. Ind. Crops Prod. 86, 132–142.
doi:10.1016/j.indcrop.2016.03.021
Barrera, C.S., Cornish, K., 2015. Novel Mineral and Organic Materials from Agro-
Industrial Residues as Fillers for Natural Rubber. J. Polym. Environ. 23, 437–448.
doi:10.1007/s10924-015-0737-4
Bergman, C., Trimbach, J., 2014. Influence of Plasticizers on the Properties of Natural
Rubber Compounds. KGK Rubberpoint. 67, 40-49.
Calhoun, A., Peacock, A., 2006. Polymer Chemistry Properties and Applications. Hanser.
Carey, A.B., Cornish, K., Schrank, P.J., 1995. Cross reactivity of alternative plant sources
of latex in subjects with systemic IgE mediated sensitivity to Hevea brasiliensis
latex. Annuals of Allergy, Asthma, Immunology. 74, 317-320.
Chakraborty, K.B., Couchman, R., 2006. Technological and physico-chemical behavior
of safer accelerators in synthetic polyisoprene latex. 17, 1-12.

186
Chen, S.F., Wont, W.C., Low, C.Y., 2011. Vulcanization composition having reduced
allergic potential. Patent Application CA2802956.
Cornish, K., 2001. Similarities and differences in rubber biochemistry among plant
species. Phytochemistry 57, 1123–1134. doi:10.1016/S0031-9422(01)00097-8
Cornish, K., 2014. Biosynthesis of natural rubber (NR) in different rubber-producing
species, in: Chemistry, Manufacture and Applications of Natural Rubber.
Elsevier, pp. 3–29. doi:10.1533/9780857096913.1.3
Cornish, K., 1996. Hypoallergenic natural rubber products from Parthenium argentatum
(Gray) and other non-Hevea brasiliensis species. U.S. Patent, No. 5,580,942.
Cornish, K., Backhaus, R.A., 1990. Rubber transferase activity in rubber particles of
guayule. Phytochemistry. 29, 3809-3813. Doi: 10.1016/0031-9422(90)85337-F.
Cornish, K., Chapman, M.H., Nakayama, F.S., Vinyard, S.H., Whitehand, L.C., 1999.
Latex quantification in guayule shrub and homogenate. 10, 121-136. Doi:
10.1016/S0926-6690(99)00016-3
Cornish, K., Fishman, B., Nguyen, K., Williams, J., 2007. Guayule natural rubber thin
film articles. Patent Application WO2009078883 A1, USA.
Cornish, K., McMahan, C.M., Xie, W., 2006. ASTM D1076 Category 4 Latex
Quantifying Guayule (NRG) and Hevea (NR) Latex Protein. In: International
Latex Conference, 15-16.

187
Cornish, K., Whitehand, L.C., Van Fleet, J.E., Brichta, J.L., Chapman, M.H., Knuckles,
B.E., 2005. Latex yield and quality during storage of guayule (Parthenium
argentatum Gray) homogenates. Ind. Crops Prod. 22, 75–85.
doi:10.1016/j.indcrop.2004.07.004
Cornish, K., Williams, J., Hall, J.L., McCoy, R.G., 2008. Production and properties of
Yulex® - The Natural Solution to Latex Allergy. Rubber Chem. Technol. 81,
709– 722. doi:10.5254/1.3548227
Cornish, K., Williams, J., Nguyen, K., Fishman, B., 2007. Guayule natural rubber latex
thin film articles. U.S. Patent, No 84316676B2.
Dick, J.S., 2009. Rubber Technology: Compounding and Testing for Performance 2E.
Hanser.
Donnet, J.B., 2003. Nano and microcomposites of polymers elastomers and their
reinforcement. Compos. Sci. Technol. 63, 1085–1088. doi:10.1016/S0266-
3538(03)00028-9
Donnet, J.-B., Custodero, E., 2013. Reinforcement of Elastomers by Particulate Fillers,
in: The Science and Technology of Rubber. Elsevier, pp. 383–416.
doi:10.1016/B978-0-12-394584-6.00008-X
Gao, T., Huang, M., Puwang, L., 2013. Effect of accelerator type on dynamic properties
of natural rubber vulcanizates. Advanced Materials Research. 834, 138-141.

188
Gidrol, X., Chrestin, H., Tan, H.L., Kush, A., 1994. Hevein, a lectin-like protein from
Hevea brasiliensis is involved in the coagulation of latex. 25, 9278-83.
Grellmann, W., Seidler, S., 2001. Deformation and Fracture Behaviour of Polymers.
Springer.
Grzybowski, S., Peyser, P.A., Johnson, C.C., 1996. The prevalence of anti-latex IgE
antibodies among registered nurses. Jour. Allergy and Clinical Immuno. 98, 535-
544.
Hager, T., MacArthur, A., McIntyre, D., Seeger, R., 1979. Chemistry and Structure of
Natural Rubbers. Rubber Chemistry and Technology. 52, 693-709.
Doi:10.5254/1.3535234
Hamann, C.P., Rogers, P.A., Sullivan, K., 2001. Latex allergy update. Infection Control
Today. 5, 36-38.
Hamann, C.P., Turjanmaa, K., Rietschel, R., Siew, C., Owensby, D., Gruninger, S.E.,
1998. Natural rubber latex hypersensitivity: incidence and prevalence of type I
allergy in the dental professional. The Journal of the American Dental
Association. 129, 219-220.
Hamed, G.R., 2000. Reinforcement of Rubber. Rubber Chem. Technol. 73, 524-533.
Doi: 10.5254/1.3547603

189
Hamilton, R.G., Cornish, K., 2010. Immunogenicity studies of guayule and guayule latex
in occupationally exposed workers. Ind. Crops Prod. 31, 197–201.
doi:10.1016/j.indcrop.2009.09.012
Hayes, B.B., Afshari, A., Millecchia, L., Willard, P.A., Povoski, S.P., Meade, B.J., 2000.
Evaluation of percutaneous penetration of natural rubber latex proteins.
Toxological Sciences. 56, 262-270. Doi: 10.1093/toxsci/56.2.262
Kauser, A., 2017. Contemporary applications of carbon black-filled polymer composites
overview of essential aspects. Journ. Plastic Film & Sheeting. Doi:
10.1177/8756087917725773
Kelly, K., 1995. Management of the Latex-Allergic Patient. Immunol Allergy Clinical
Am. 15, 139-156.
Kohls, D.J., Beaucage, G., 2002. Rational design of reinforced rubber. Curr. Opin. Solid
State Mater. Sci. 6, 183-194. Doi: 10.1016/S1359-0286(02)00073-6
Krutzer, B., Ros, M., Smit, J., de Jong, W., 2014, A review of synthetic lattices in
surgical glove use. Report, Kraton Innovation Center.
Kruzelak, J., Sykora, R., Hudec, I., 2016. Sulphur and peroxide vulcanization of rubber
compounds- overview. Chemical Papers. 70, 1533-1555. Doi: 10.1515/chempap-
2016-0093.

190
Kumar, S., Hahn, F.M., Baidoo, E., Kahlon, T.S., Wood, D.F., McMahan, C.M., Cornish,
K., Keasling, J.D., Daniell, H., Whalen, M., 2012. Remodeling the isoprenoid
pathway in tobacco by expressing cytoplasmic mevalonate pathway in
chloroplasts. Metabolic Engineering. 14, 19-28. Doi:
10.1016/j/ymben.2011.11.005
Leblanc, J., 2002. Rubber-filler interactions and rheological properties in filled
compounds. Prog. Polym. Sci. 27, 627-687. Doi: 10.5254/1.3538716
Liss, G.M., Sussman, G.L., 1999. Latex sensitization: occupational versus general
population prevalence rates. American Journal of Industrial Medicine. 35, 196-200.
Lovell, P.A., El-Aasser, M.S., 1997. Emulsion Polymerization and Emulsion Polymers.
John Wiley & Sons Ltd
Mahapram, S., Poompradub, S., 2011. Preparation of natural rubber (NR) latex/low
density polyethylene (LDPE) blown film and its properties. Polymer Testing. 30:
716-725. Doi: 10.1016/j.polymertesting.2011.06.006
Mark, J.E., Erman, B., Roland, C.M., 2013. The Science and Technology of Rubber 4th
edition. Doi: 10.1016/C2011-0-05820-9
McMahan, C., Kostyal, D., Lhamo, D., Cornish, K., 2015. Protein influences on guayule
and Hevea natural rubber sol and gel. J. Appl. Polym. Sci. 42051, n/a-n/a. 185
doi:10.1002/app.42051

191
McMahan, C., Lhamo, D., 2015. Study of Amino Acid Modifiers in Guayule Natural
Rubber. Rubber Chem. Technol. 88, 310–323. doi:10.5254/rct.15.85931
Miri, S., Pourpak, Z., Zarinara, A., 2007. Prevalence of type I allergy to natural rubber
latex and type IV allergy to latex and rubber additives in operating room staff
with glove-related symptoms. Allergy and Asthma Proceedings. 28, 557-563.
Meyer, J.D., Chen, Y., Holt, D.L., 2000. Occupational contact dermatitis in the UK: a
surveillance report from Epiderm & OPRA. Occupational Medicine. 50, 265-273.
Monadjemi, S.M., McMahan, C.M., Cornish, K., 2016. Effect of Non-Rubber
Constituents on Guayule and Hevea Rubber Intrinsic Properties. Journal of
Research Updates in Polymer Science. 5, 87-96. doi:10.6000/1929-
5995.2016.05.03.01
Mooibroek, H., Cornish, K., 2000. Alternative sources of natural rubber. Appl.
Microbiol. Biotechnol. 53, 355–365. doi:10.1007/s002530051627
Musto, S., Barbera, V., Maggio, M., Mauro, M., Guerra, G., Galimberti, M., 2016.
Crystallinity and crystalline phase orientation of poly(1,4-cis-isoprene) from
Hevea brasiliensis and Taraxacum kok-saghyz. Polymer Advanced Technologies.
7, 1082-1089. Doi: 10.1002/pat.3774
Mutsuga, M., 2013. Analysis of N-Nitrosamine Migration from Rubber Soothers. Journal
of Analytical Chemistry. 4, 277-285.

192
Nabavizadeh, S.H., Anushiravani, A., Amin, R., 2009. Natural rubber latex
hypersensitivity with skin prick test in operating room personnel. Iranian Journal
of Allergy, Asthma and Immunology 8, 219-220.
Nettis, E., Assennato, G., Ferrannini, A., Tursi, A., 2002. Type I allergy to natural rubber
latex and type IV allergy to healthcare workers with glove-related skin symptoms.
Clin. Exp All 32, 441-447.
Nocil Ltd., 2010. Vulcanization and Accelerators. Report, Technical Note.
Pak, V.M., Watkins, M., Green-McKenzie, J., 2012. What is the role of thiurams in
allergy to natural rubber latex products? Journal of Occupational and
Environmental Medicine. 54, 649-650.
Pan, Z., Durst, F., Werck-Reichhart, 1995. The major protein of guayule rubber particles
is a cytochrome P450: enzyme and its reaction products. Journal of Biological
Chemistry. 270, 8487-8494.
Perumal, V., Geetha, N., Palanivel, S., Thulaseedharan, A., 2013. Natural rubber
producing plants: An overview. African Journal of Biotechnology. 12,1297-1310.
Doi: 10.5897/AJBX12.016
Petrovic, Z.S., Ionescu, M., Milic, J., Halladay, J.R., 2013. Soybean Oil Plasticizers as
Repleacement of Petroleum in Rubber. Rubber Chem. and Tech. 86, 233-249.

193
Puskas, J.E., Chiang, K., Barkakaty, B., 2014. Natural rubber (NR) biosynthesis:
perspectives from polymer chemistry, in: Chemistry, Manufacture and
Applications of Natural Rubber. Elsevier, pp. 30–67.
doi:10.1533/9780857096913.1.30
Quirk, R.P., 1988. Overview of curing and crosslinking elastomers. Prog. Rubber Plastic
Technology 4, 31.
Rattanasom, N., Saowapark, T., Deeprasertkul, C., 2007. Reinforcement of natural rubber
with silica/carbon black hybrid filler. Polym. Test. 26, 369–377.
doi:10.1016/j.polymertesting.2006.12.003
Ren, X., Geng, Y., Soboyejo, A.B.O., Cornish, K., 2019. Reinforced mechanical
properties of functionalized silica and eggshell filled guayule natural rubber
composites. Rubber Chemistry and Technology. Doi:10.5254/rct.19.81485
Rickman, J.M., Wang, Y., Rollett, A.D., Harmer, M.P., Compson, C., 2017. Data
analytics suing canonical correlation analysis and Monte Carlo simulation.
Computational Materials. Doi:10.1038/s41524-017-0028-9
Rothon, R.N., 2000. Particulate Fillers for Polymers. Rapra. ISBN: 1-85957-310-X
Sansatsadeekul, J., Sakdapipanich, J., Rojurthai, P., 2011. Characterization of associated
proteins and phospholipids in natural rubber latex. J Biosci Bioeng. 111, 628-634.
Doi: 10.1016/j.biosc.2011.01.013

194
Sasseville, D., 2008. Occupational contact dermatitis. Allergy, Asthma, and Clinical
Immunology: Office Journal of Canadian Society of All. and Clin. Immuno. 54,
649-650. doi:10.1097/JOM.0b013e31824be452.
Schmidt, T., Lenders, M., Hillebrand, A., van Deenen, N., Munt, O., Reichelt, R.,
Eisenreich, W., Fischer, R., Prufer, D., Gronover, C.S., 2010. Characterization of
rubber particles and rubber chain elongation in Taraxacum kok-saghyz. BMC
Biochemistry, 11:11. Doi: 10.1186/1471-2091-11-11.
Seymour, B., 1989. Polymer science before and after 1989: notable development in the
lifetime of Maurtis Dekker. J. Macromols. Sci. Chem. 26, 1023-1032.
Siler, D.J., Cornish, K., 1995. Measurement of Protein in Natural Rubber Latex. Anal.
Biochem. 229, 278–281.
Siler, D.J., Cornish, K., 1994. Hypoallergenicity of guayule rubber particle proteins
compared to Hevea latex proteins. Ind. Crops Prod. 2, 307–313.
doi:10.1016/0926- 6690(94)90122-8
Siler, D.J., Cornish, K., Hamilton, R.G., 1996. Absence of cross-reactivity of IgE
antibodies from subjects allergic to Hevea brasiliensis latex with a new source of
natural rubber latex from guayule (Parthenium argentatum). J. Allergy Clin.
Immunol. 98, 895–902. doi:Doi 10.1016/S0091-6749(96)80005-4
Soboyejo, A.B., 2011. Probabilistic methods in engineering and biosystems engineering.

195
Spaner, D., Dolovich, J., Tarlo, S., Sussman, G., Butto, K., 1989. Hypersensitivity to
natural rubber latex. J. Allergy Clin. Immunol. 83, 1135–1137. doi:
10.1016/0091-6749(89)90457-0.
Steinbuchel, A., 2003. Production of rubber-like polymers by microorganisms. Curr.
Opin. Microbiol. 6, 261–270. doi:10.1016/S1369-5274(03)00061-4
Stoter, M., Kunz, D.A., Schmidt, M., Hirsemann, D., Kalo, H., Putz, B., Senker, J., Breu,
J., 2013. Nanoplatelets of Sodium Hectorite Showing Aspect Ratios of 20000 and
superior purity. Langmuir. 29, 1280-1285.
Swason, M., 2008. Vytex Natural Rubber Latex: An Innovative Source for Natural
Rubber Products. International Latex Conference.
Tanaka, Y., 2001. Structural Characterization of Natural Polyisoprenes: Solve the
Mystery of Natural Rubber Based on Structural Study. Rubber Chem. Technol.
74, 355–375. doi:10.5254/1.3547643
Tao, J., 2002. Soft nitrile gloves having improved glove relaxation properties. Patent
Application EP 1352616 A1.
Taylor, J.S., Leow, Y.H., 2000. Cutaneaous Reactions to Rubber. Rubber Chemistry and
Technology: Rubber Reviews. 73, 427-485.
Tohsan, A., Ikeda, Y., 2014. Generating particulate silica fillers in situ to improve the
mechanical propertie4s of natural rubber (NR), in: Chemistry, Manufacture and
Applications of Natural Rubber. Elsevier, pp. 169-192.

196
van Jole, E., 2007. Latex accelerator composition. European Patent, No 1913071B1.
van Beilen, J.B., Poirier, Y., 2007. Establishment of new crops for the production of
natural rubber. Trends Biotechnol. 25, 522–529.
doi:10.1016/j.tibtech.2007.08.009
Vennemann, N., Schwarze, C., Kummerlowe, C., 2013. Determination of crosslink
density and network structure of NR vulcanizates by means of TSSR. Advanced
Materials Research. 844, 482-485.
Vivaldo-Lima, E., Wood, P.E., Hamielec, A.E., 1997. An Updated Review on
Suspension Polymerization. Ind. Eng. Chem. Res. 36, 939-965. Doi:
10.1021/ie960361g
Wititsuwannakul, D., Wititsuwannakul, R., 2005. Natural Rubber and Structure of Latex,
Biochemistry of. Biopolymers Online. Doi: 10.1002/3527600035.bpol2006
Wypych, G., 2012. Handbook of Polymers. ChemTec Publishing.
Yasuyuki, T., 2001. Structural Characterization of Natural Polyisoprenes: Solve the
Mystery of Natural Rubber Study. Rubber Chemistry and Technology. 74, 355-
375.
Young, R.J., Lovell, P.A., 1991. Introduction to Polymers: Second Edition. Springer-
Science. Doi: 10.1007/978-1-4899-3176-4.




















