
Hydrogeochemical modeling of a thermal system and lessons learned for CO 2 geologic storage
13
Hydrogeochemical modeling of a thermal system and lessons learned for CO 2 geologic storage L.F. Auqué ⁎, P. Acero, M.J. Gimeno, J.B. Gómez, M.P. Asta Geochemical Modeling Group, Petrology and Geochemistry Area, Earth Sciences Department, University of Zaragoza, Spain Faculty of Geology, C/ Pedro Cerbuna 12, 50009 Zaragoza, Spain abstract article info Article history: Received 14 July 2009 Received in revised form 10 September 2009 Accepted 10 September 2009 Editor: J. Fein Keywords: Natural analogues Dedolomitization Thermal waters Geochemical modeling Geological storage of carbon dioxide is presently considered to be one of the main strategies to mitigate the impact of the emissions of this gas on global warming. Among the various alternatives considered for CO 2 geological storage, one of the main geological candidates for hosting injected CO 2 in the long term are deep porous reservoir rock formations saturated with brackish or saline solutions. Although valuable information on the expected hydrogeochemical processes involved in the CO 2 storage in such deep saline aquifers can be obtained in laboratory or modeling studies, the only direct source of information about the long-term behavior of geological storages for CO 2 in deep aquifers is natural analogues. In this work, a classical and simple geochemical methodology is successfully applied to the study of the features and hydrogeochemical processes determining the evolution of a Spanish thermal system (the Alhama–Jaraba complex), which can be considered as a natural analogue for deep geological CO 2 storage in carbonate rocks. The geology, structure, depth and hydrogeochemistry of the Alhama–Jaraba thermal system are very similar to the expected features of a potential CO 2 reservoir in carbonate materials. The processes determining the hydrogeochemical evolution in the Alhama–Jaraba thermal system have been successfully identified and quantified with the assistance of ion–ion plots, speciation–solubility calculations and mass-balance calculations. Furthermore, the feasibility of the proposed conceptual hydrogeochemical model for this system has been verified by using reaction-path calculations. Mass-balance calculation results have indicated that the observed hydrogeochemical evolution between springs is mainly due to halite dissolution and dedolomitization triggered by gypsum or anhydrite dissolution. CO 2 (g) mass transfer has been estimated to be negligible, which suggests that the main processes responsible for the variation in the TIC and the CO 2 (g) pressure during deep circulation are dissolution and precipitation reactions for carbonate minerals. All the processes identified in the Alhama–Jaraba thermal system are relevant for the long-term evolution of a deep CO 2 storage site hosted by carbonate rocks. As shown in this study, the application of classical geochemical tools provides an excellent starting point for understanding the behavior of prospective storage systems. Moreover, the existence of dedolomitization is very relevant for the hydraulic properties of carbonate aquifers potentially used for CO 2 geological storage because of the effects on porosity and, therefore, permeability during the long-term evolution of such systems. Furthermore, dedolomitization may represent a mechanism of mineral trapping for CO 2 sequestration under certain conditions. © 2009 Elsevier B.V. All rights reserved. 1. Introduction Geological disposal and storage of carbon dioxide is at present considered to be one of the main strategies to mitigate the impact of the emissions of this gas on global warming (Metz et al., 2005 and references therein). Among the various alternatives considered for CO 2 geological storage, the main geological candidates for hosting injected CO 2 are: oil and gas reservoirs, deep saline aquifers, coal beds, caverns and mines (Hitchon et al., 1999; Holloway et al., 1999; Bachu, 2000; Metz et al., 2005; Wildenborg and Lokhorst, 2005). Owing to their larger storage capacity at a global scale, deep porous reservoir rock formations saturated with brackish or saline solutions are generally regarded as the most effective geological reservoirs for the long-term storage of CO 2 (Metz et al., 2005 and references therein). Although other types of hosting rock formations have also been considered (evaporites, basalts, etc.), most of the earlier studies have focused on the geological storage of CO 2 in saline aquifers hosted by sedimentary rocks, especially in carbonate (Gunter et al., 2000; Shiraki and Dunn, 2000; Christensen and Holloway, 2004; Chemical Geology 268 (2009) 324–336 ⁎ Corresponding author. Geochemical Modeling Group, Petrology and Geochemistry Area, Earth Sciences Department, University of Zaragoza, Spain. Tel.: +34 976761067; fax: +34 976761106. E-mail address: [email protected] (L.F. Auqué). 0009-2541/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.chemgeo.2009.09.011 Contents lists available at ScienceDirect Chemical Geology journal homepage: www.elsevier.com/locate/chemgeo
Transcript of Hydrogeochemical modeling of a thermal system and lessons learned for CO 2 geologic storage

Chemical Geology 268 (2009) 324–336
Contents lists available at ScienceDirect
Chemical Geology
j ourna l homepage: www.e lsev ie r.com/ locate /chemgeo
Hydrogeochemical modeling of a thermal system and lessons learned for CO2
geologic storage
L.F. Auqué ⁎, P. Acero, M.J. Gimeno, J.B. Gómez, M.P. AstaGeochemical Modeling Group, Petrology and Geochemistry Area, Earth Sciences Department, University of Zaragoza, SpainFaculty of Geology, C/ Pedro Cerbuna 12, 50009 Zaragoza, Spain
⁎ Corresponding author. Geochemical Modeling GrouArea, Earth Sciences Department, University of Zaragozafax: +34 976761106.
E-mail address: [email protected] (L.F. Auqué).
0009-2541/$ – see front matter © 2009 Elsevier B.V. Adoi:10.1016/j.chemgeo.2009.09.011
a b s t r a c t
a r t i c l e i n f oArticle history:Received 14 July 2009Received in revised form 10 September 2009Accepted 10 September 2009
Editor: J. Fein
Keywords:Natural analoguesDedolomitizationThermal watersGeochemical modeling
Geological storage of carbon dioxide is presently considered to be one of the main strategies to mitigate theimpact of the emissions of this gas on global warming. Among the various alternatives considered for CO2
geological storage, one of the main geological candidates for hosting injected CO2 in the long term are deepporous reservoir rock formations saturated with brackish or saline solutions. Although valuable informationon the expected hydrogeochemical processes involved in the CO2 storage in such deep saline aquifers can beobtained in laboratory or modeling studies, the only direct source of information about the long-termbehavior of geological storages for CO2 in deep aquifers is natural analogues.In this work, a classical and simple geochemical methodology is successfully applied to the study of thefeatures and hydrogeochemical processes determining the evolution of a Spanish thermal system (theAlhama–Jaraba complex), which can be considered as a natural analogue for deep geological CO2 storage incarbonate rocks. The geology, structure, depth and hydrogeochemistry of the Alhama–Jaraba thermal systemare very similar to the expected features of a potential CO2 reservoir in carbonate materials.The processes determining the hydrogeochemical evolution in the Alhama–Jaraba thermal system have beensuccessfully identified and quantified with the assistance of ion–ion plots, speciation–solubility calculationsand mass-balance calculations. Furthermore, the feasibility of the proposed conceptual hydrogeochemicalmodel for this system has been verified by using reaction-path calculations.Mass-balance calculation results have indicated that the observed hydrogeochemical evolution betweensprings is mainly due to halite dissolution and dedolomitization triggered by gypsum or anhydritedissolution. CO2(g) mass transfer has been estimated to be negligible, which suggests that the mainprocesses responsible for the variation in the TIC and the CO2(g) pressure during deep circulation aredissolution and precipitation reactions for carbonate minerals.All the processes identified in the Alhama–Jaraba thermal system are relevant for the long-term evolution ofa deep CO2 storage site hosted by carbonate rocks. As shown in this study, the application of classicalgeochemical tools provides an excellent starting point for understanding the behavior of prospective storagesystems. Moreover, the existence of dedolomitization is very relevant for the hydraulic properties ofcarbonate aquifers potentially used for CO2 geological storage because of the effects on porosity and,therefore, permeability during the long-term evolution of such systems. Furthermore, dedolomitization mayrepresent a mechanism of mineral trapping for CO2 sequestration under certain conditions.
p, Petrology and Geochemistry, Spain. Tel.: +34 976761067;
ll rights reserved.
© 2009 Elsevier B.V. All rights reserved.
1. Introduction
Geological disposal and storage of carbon dioxide is at presentconsidered to be one of the main strategies to mitigate the impact ofthe emissions of this gas on global warming (Metz et al., 2005 andreferences therein). Among the various alternatives considered forCO2 geological storage, the main geological candidates for hosting
injected CO2 are: oil and gas reservoirs, deep saline aquifers, coal beds,caverns and mines (Hitchon et al., 1999; Holloway et al., 1999; Bachu,2000; Metz et al., 2005; Wildenborg and Lokhorst, 2005).
Owing to their larger storage capacity at a global scale, deepporous reservoir rock formations saturated with brackish or salinesolutions are generally regarded as the most effective geologicalreservoirs for the long-term storage of CO2 (Metz et al., 2005 andreferences therein). Although other types of hosting rock formationshave also been considered (evaporites, basalts, etc.), most of theearlier studies have focused on the geological storage of CO2 in salineaquifers hosted by sedimentary rocks, especially in carbonate (Gunteret al., 2000; Shiraki and Dunn, 2000; Christensen and Holloway, 2004;

325L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
Brosse et al., 2005; Lagneau et al., 2005; Noiriel et al., 2005, andreferences therein; André et al., 2007) and sandstone formations(Pearce et al., 1996; Czernichowski et al., 1996; Gunter et al., 1997;Johnson et al., 2001, 2004; Rochelle et al., 2002; Savage et al., 2002;Kaszuba et al., 2003, 2005; Kirste et al., 2004; Xu et al., 2000, 2004,2005; Allen et al. 2005; Bateman et al., 2005; Gaus et al., 2005; Knausset al., 2005; Palandri et al., 2005; White et al., 2005; Bertier et al.,2006; Chadwick et al., 2006; Audigane et al., 2007).
In order to appropriately select the adequate sedimentary formationto dispose and store CO2 (storage capacity estimation) and to guaranteethe safety of the deep geological reservoirs (safety assessment), theinteractions between the CO2-enriched solutions and the hosting rocksmust be identified and characterised. Even though valuable informationon the hydrogeochemical processes expected throughout these inter-actions can be obtained from modeling exercises (Gunter et al., 1997,2000; Cipolli et al., 2004; Johnson et al., 2001, 2004; Pruess et al., 2004;Gaus et al., 2005; Lagneau et al., 2005; Moore et al., 2005; White et al.,2005; André et al., 2007; Audigane et al., 2007; Xu et al., 2004, 2005,2007; Gherardi et al., 2007) and experimental works both in the fieldand in the laboratory (Gunter et al., 1997, 2000; Shiraki andDunn, 2000;Rochelle et al., 2002; Kaszuba et al., 2003, 2005; Soong et al., 2004;Bateman et al., 2005; Emberley et al., 2005; Knauss et al., 2005; Noirielet al., 2005; Bertier et al., 2006), the only direct source of informationabout the long-term behavior of geological storage for CO2 in deepaquifers is natural analogues. Two types of systems are currently beingstudied as natural analogues of geologic CO2 storage; (1) natural trapsthat have effectively stored CO2 at geological timescales (e.g. gas fields),and (2) systems inwhich a naturalmigration of CO2 towards the surfacetakes place (e.g. hydrothermal systems). While natural CO2 traps andsystems with high CO2 pressures (e.g. from deep sources) provide veryvaluable information about the processes and interactions expected inthe reservoir sections closer to the injection wells, hydrothermalsystemswith lowCO2 pressures are probably themain natural analoguefor studying the expectedwater–rock interactions in the long termor inthe reservoir parts more distant from the injected supercritical CO2.
Apart from the possibility of studying the structural characteristicsand geochemical processes expected in geologic storage media, naturalanalogues offer the opportunity to test and refine the methodologiesneeded for the selection and safety assessmentof sites considered for theunderground disposal of CO2 (Pearce et al., 1996; Stevens et al., 2001).One of the first tasks during the selection and design of CO2 geologicalstorages will be the characterisation of the prospective sites and theknowledge of their pristine conditions before CO2 injection. In line withthese goals, the study of natural systems similar to CO2 storage sites (fortheir structure or controllingprocesses) represents a uniqueopportunityto test and optimize themethodologies for sampling, analysis, modelingand interpretation that will be applied in future CO2 reservoirs.
In this work, a classical and simple geochemical methodology issuccessfully applied to the study of the features and hydrogeochemicalprocesses determining the evolution of a Spanish thermal system (theAlhama–Jaraba complex) which can be considered as a naturalanalogue for many of the processes expected in a deep geologicalCO2 storage placed in carbonate rocks. The Alhama–Jaraba complex(NE Spain) is a thermal system in a deep (around 1000 m) carbonateaquifer confined by low-permeability caprocks. The Alhama–Jarabathermal groundwaters emerge at around30 °C inmore than 30 springsand there is evidence of CO2-degassing processes associated with theshallowest part of the upflow. Owing to this geological structure andgeneral hydrogeochemical behavior, this system represents a goodopportunity to explore the long-term water–rock interactions and totest the methodology for the geochemical characterisation of pros-pective or selected CO2 storage sites.
With these goals, the results of ion–ion plots, speciation–solubilitycalculations, mass-balance and reaction-path calculations have beencombined to interpret the hydrogeological and hydrogeochemicalbehavior of this system and to identify and quantify some of the
controlling processes expected throughout long-term CO2 geologicalstorage or in the reservoir sectionsmore distant from the CO2 injectionpoints. Similar methodologies have been successfully applied to thecharacterisation of hydrogeochemical processes in other carbonateaquifers and to constrain the possible regional or local flow paths(Plummer et al., 1990; Sacks et al., 1995; Lopez-Chicano et al., 2001;Uliana and Sharp, 2001; Frondini, 2008; Leybourne et al., 2009).
2. Geological and hydrogeological setting
The Alhama and Jaraba springs (NE Spain, Fig. 1) belong to one of themain thermal systems in Spain. There are several thermal resorts andwater bottling plants in the area at present. The Jaraba thermal complex,close to the Mesa River, consists of 14 catalogued springs flowing at anelevation of 737 m.a.s.l. with an approximate flow rate around 600 L/s(IGME, 1980; De Toledo and Arqued, 1990; Sanchez et al., 2004). TheAlhama thermal complex, close to the Jalon River, is formed by a dozencatalogued springs with a combined flow rate around 550 L/s andflowing at an elevation of 660 m.a.s.l. Apart from these well-knownspring complexes, there are other minor hot springs in the area nearby(Tena et al. 1995; Sanz and Yelamos, 1998; Sanchez et al., 2004), such asthe Embid spring (19 L/s) and the two Deza springs (20 L/s) (Fig. 1).
Geologically, the thermal system is located at the edge with theTertiary Almazan Basin (W Iberian Range; Fig. 1). Most of the thermalsprings are associated with Upper Cretaceous Carbonate rocks and arestructurally controlled along NW–SE and NE–SW directions (IGME,1982). The lithologies represented in the area range from Palaeozoicto Tertiary and Quaternary materials. The Paleozoic rocks (quartzites,sandstones, marls, limestones and dolostones) follow a NW–SEdirection and act as a low-permeability barrier termed as the “Atecathreshold”. The Jurassic and Cretaceous rocks comprise mainly high-permeability carbonates (dolostones, limestones, etc.) and low-permeability sandstones, marls and evaporitic rocks and appearmainly in the south part of the area (between Alhama and Mochales,Fig. 1). The Tertiary rocks are generally marls, sands, evaporites andcarbonates belonging to the Calatayud and Almazan Basins. Finally,there are scattered river deposits corresponding to terraces and tufas,covering a good part of the studied area.
Structurally, the area is formed by a series of anticlinal andsynclinal structures following NW–SE trends and clearly vergingtowards the SW. The structural and hydrologic configuration of thiszone is also determined by NW–SE faults created during the Hercinianorogeny and later reactivated by Alpine tectonics, changing progres-sively to W–E directions.
From the hydrogeologic point of view, twomain aquifers have beendescribed in earlier works as being related to the existence of thermalsprings (IGME, 1980; IGME, 1987; De Toledo and Arqued, 1990; Sanzand Yelamos, 1998; Sanchez et al., 2004); (1) Solorio aquifer, formedmainly by Jurassic carbonate rocks, and (2) Alhama aquifer, cor-responding to Upper Cretaceous carbonate rocks. Althoughmost of theearlier hydrogeologic studies consider that themain recharge areas arelocated in the Solorio Range and in the surroundings of Deza (for theSolorio and Alhama aquifers, respectively) there is no generalagreement on the general hydrogeologic model for this system.Nevertheless, themost recent studies have suggested that both aquiferlevels are connected and that the flow directions of thermal ground-waters are structurally controlled and may correspond to two maincirculation directions (Fig. 1); (1) SW–NE, from the Solorio Range andflowing underneath the Tertiary rocks of the Almazan Basin towardsJaraba and Alhama (Sanchez et al., 2004), and (2) NW–SE, from Dezatowards Embid and Alhama (Sanz and Yelamos, 1998). Groundwaterflow and thermal springs are generally associated with Cretaceousrocks in both circulation directions. However, the rocks in the rechargearea for the Mochales–Alhama flow path (i.e. Solorio Range) aremainly Jurassic, which indicates that some hydraulic connectionbetween the Jurassic and Cretaceous rocks must exist, consistently

Fig. 1. Geological map and location of the groups of springs included in this study.
326 L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
with the hypothesis of connection between the twomain aquifer unitsin the studied area.
In any case, the petrological and mineralogical features of theJurassic and Cretaceous rocks hosting the two described aquifers(Solorio and Alhama) are very similar. The Cretaceous carbonate rocksconsist mainly of massive and bedded limestones, dolomitic lime-stones and dolostones. Interbedded or dispersed evaporitic minerals(mainly gypsum, CaSO4·2H2O, and/or anhydrite, CaSO4) also appear inthe carbonate sequences associatedwith the development of localizedhypersaline lagoon or coastal sabkha environments (Floquet et al.,1981;Meléndez et al., 1985; Alonso et al., 1993). Someof the carbonatesedimentation episodes in these Upper Cretaceous materials show amore terrigenous character with intercalations of sandstones, clays-tones, siltstones, dolomitic siltstones, dolomitic marls, limestones anddolomitic limestones. From a mineralogical point of view, thesesiliciclasticmaterials are formed by quartz, K-feldspar, lithic fragmentsand white mica in a fine matrix consisting of illite and kaolinite. Withregard to the Jurassic rocks of the area, they correspond to Liassicmaterials with lithologies very similar to the Cretaceous rocks. Theyare mainly formed by massive, bedded and brecciated dolostones andlimestones with intercalations of evaporitic rocks (formed mainly bygypsum/anhydrite) and marls (Aurell et al., 2002).
3. Materials and methods
The study of the possible hydrogeochemical evolution paths in theAlhama–Jaraba thermal system will allow not only quantifying thegeochemical processes controlling the evolution of thermal waters, butalso checking the hydrological hypotheses proposed for this area. For
these reasons, the geochemical patterns along the two main proposedflow directions associated with thermal springs in this area (Mochales–Jaraba–Alhama and Deza–Embid–Alhama) will be examined.
The water sampling location is displayed in Table 1 and in Fig. 1.There are samples representative of thermal springs (Jaraba, Alhamaand Embid) and other non-thermal samples representing the hypo-thetical recharge areas or, at least, less evolved waters from theseareas (Mochales and Deza).
At each sampling point, separated samples for anion and cationanalyses were taken in bottles pre-washedwith HCl and subsequentlyrinsed three times with double-distilled water. Samples for cationanalyses were filtered at 0.1 μm and acidified to pH less than 1 withultrapure HNO3. Temperature wasmeasured in situ by a thermometer(accuracy better than 0.1 °C). pHwas determined by a combined glasselectrode with automated temperature correction, calibrated on aregular basis with standard buffer solutions of pH 4 and 7 at similartemperature to the sampled springs (from 25 to 30 °C). The reportedaccuracy for pH measurements is better than ±0.03 pH units.Electrical conductivity was measured by using a conductivity meterwith a K=1 cell and with automated temperature correction in the10–70 °C temperature range.
Total alkalinity and chloride contents were determined by titrationwith a Mettler titrator with end-point electrode. For the hydrogeo-chemical conditions corresponding to our samples, total and carbonatealkalinity can be considered as equivalent. Fluoride content wasmeasured by using a selective electrode. Sulfate and nitrate concentra-tions were determined by colorimetry using modifications of theNemethmethod (Nemeth, 1963) and the Brucine method (Jenkins andMedsken, 1964), respectively. Inductively Coupled Plasma-Atomic
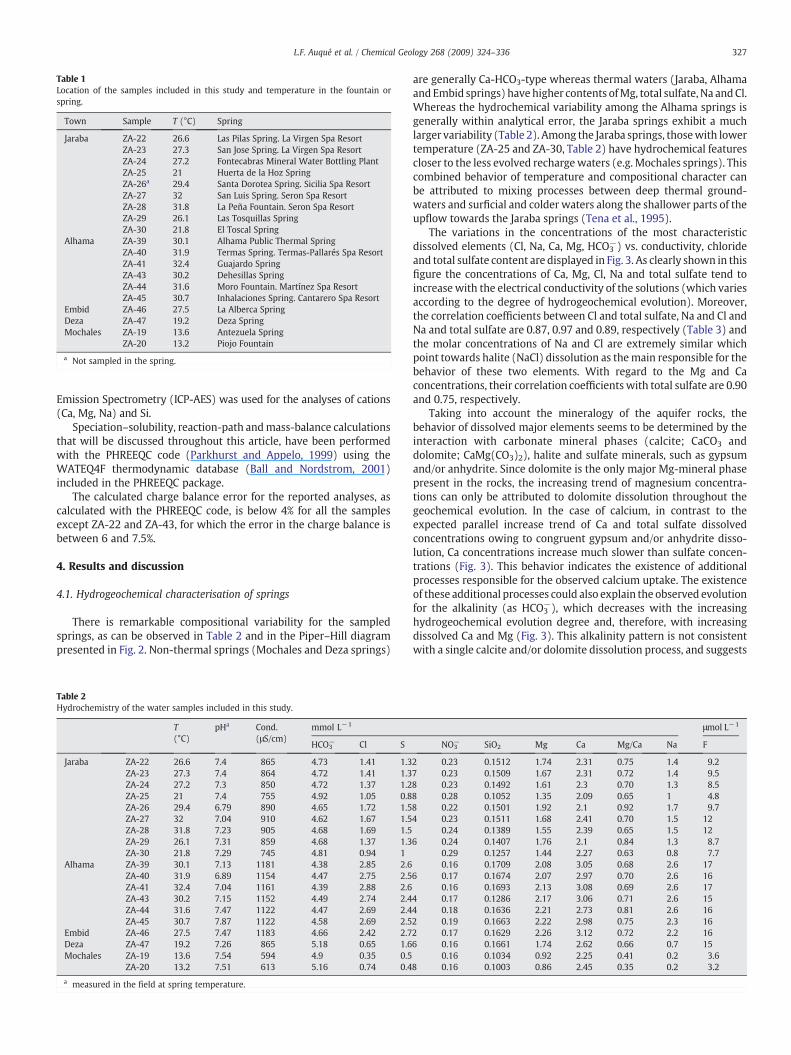
Table 1Location of the samples included in this study and temperature in the fountain orspring.
Town Sample T (°C) Spring
Jaraba ZA-22 26.6 Las Pilas Spring. La Virgen Spa ResortZA-23 27.3 San Jose Spring. La Virgen Spa ResortZA-24 27.2 Fontecabras Mineral Water Bottling PlantZA-25 21 Huerta de la Hoz SpringZA-26a 29.4 Santa Dorotea Spring. Sicilia Spa ResortZA-27 32 San Luis Spring. Seron Spa ResortZA-28 31.8 La Peña Fountain. Seron Spa ResortZA-29 26.1 Las Tosquillas SpringZA-30 21.8 El Toscal Spring
Alhama ZA-39 30.1 Alhama Public Thermal SpringZA-40 31.9 Termas Spring. Termas-Pallarés Spa ResortZA-41 32.4 Guajardo SpringZA-43 30.2 Dehesillas SpringZA-44 31.6 Moro Fountain. Martínez Spa ResortZA-45 30.7 Inhalaciones Spring. Cantarero Spa Resort
Embid ZA-46 27.5 La Alberca SpringDeza ZA-47 19.2 Deza SpringMochales ZA-19 13.6 Antezuela Spring
ZA-20 13.2 Piojo Fountain
a Not sampled in the spring.
327L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
Emission Spectrometry (ICP-AES) was used for the analyses of cations(Ca, Mg, Na) and Si.
Speciation–solubility, reaction-path andmass-balance calculationsthat will be discussed throughout this article, have been performedwith the PHREEQC code (Parkhurst and Appelo, 1999) using theWATEQ4F thermodynamic database (Ball and Nordstrom, 2001)included in the PHREEQC package.
The calculated charge balance error for the reported analyses, ascalculated with the PHREEQC code, is below 4% for all the samplesexcept ZA-22 and ZA-43, for which the error in the charge balance isbetween 6 and 7.5%.
4. Results and discussion
4.1. Hydrogeochemical characterisation of springs
There is remarkable compositional variability for the sampledsprings, as can be observed in Table 2 and in the Piper–Hill diagrampresented in Fig. 2. Non-thermal springs (Mochales and Deza springs)
Table 2Hydrochemistry of the water samples included in this study.
T(°C)
pHa Cond.(μS/cm)
mmol L−1
HCO3− Cl S
Jaraba ZA-22 26.6 7.4 865 4.73 1.41 1.3ZA-23 27.3 7.4 864 4.72 1.41 1.3ZA-24 27.2 7.3 850 4.72 1.37 1.2ZA-25 21 7.4 755 4.92 1.05 0.8ZA-26 29.4 6.79 890 4.65 1.72 1.5ZA-27 32 7.04 910 4.62 1.67 1.5ZA-28 31.8 7.23 905 4.68 1.69 1.5ZA-29 26.1 7.31 859 4.68 1.37 1.3ZA-30 21.8 7.29 745 4.81 0.94 1
Alhama ZA-39 30.1 7.13 1181 4.38 2.85 2.6ZA-40 31.9 6.89 1154 4.47 2.75 2.5ZA-41 32.4 7.04 1161 4.39 2.88 2.6ZA-43 30.2 7.15 1152 4.49 2.74 2.4ZA-44 31.6 7.47 1122 4.47 2.69 2.4ZA-45 30.7 7.87 1122 4.58 2.69 2.5
Embid ZA-46 27.5 7.47 1183 4.66 2.42 2.7Deza ZA-47 19.2 7.26 865 5.18 0.65 1.6Mochales ZA-19 13.6 7.54 594 4.9 0.35 0.5
ZA-20 13.2 7.51 613 5.16 0.74 0.4
a measured in the field at spring temperature.
are generally Ca-HCO3-type whereas thermal waters (Jaraba, AlhamaandEmbid springs) have higher contents ofMg, total sulfate, Na andCl.Whereas the hydrochemical variability among the Alhama springs isgenerally within analytical error, the Jaraba springs exhibit a muchlarger variability (Table 2). Among the Jaraba springs, thosewith lowertemperature (ZA-25 and ZA-30, Table 2) have hydrochemical featurescloser to the less evolved rechargewaters (e.g. Mochales springs). Thiscombined behavior of temperature and compositional character canbe attributed to mixing processes between deep thermal ground-waters and surficial and colder waters along the shallower parts of theupflow towards the Jaraba springs (Tena et al., 1995).
The variations in the concentrations of the most characteristicdissolved elements (Cl, Na, Ca, Mg, HCO3
−) vs. conductivity, chlorideand total sulfate content are displayed in Fig. 3. As clearly shown in thisfigure the concentrations of Ca, Mg, Cl, Na and total sulfate tend toincrease with the electrical conductivity of the solutions (which variesaccording to the degree of hydrogeochemical evolution). Moreover,the correlation coefficients between Cl and total sulfate, Na and Cl andNa and total sulfate are 0.87, 0.97 and 0.89, respectively (Table 3) andthe molar concentrations of Na and Cl are extremely similar whichpoint towards halite (NaCl) dissolution as themain responsible for thebehavior of these two elements. With regard to the Mg and Caconcentrations, their correlation coefficients with total sulfate are 0.90and 0.75, respectively.
Taking into account the mineralogy of the aquifer rocks, thebehavior of dissolved major elements seems to be determined by theinteraction with carbonate mineral phases (calcite; CaCO3 anddolomite; CaMg(CO3)2), halite and sulfate minerals, such as gypsumand/or anhydrite. Since dolomite is the only major Mg-mineral phasepresent in the rocks, the increasing trend of magnesium concentra-tions can only be attributed to dolomite dissolution throughout thegeochemical evolution. In the case of calcium, in contrast to theexpected parallel increase trend of Ca and total sulfate dissolvedconcentrations owing to congruent gypsum and/or anhydrite disso-lution, Ca concentrations increase much slower than sulfate concen-trations (Fig. 3). This behavior indicates the existence of additionalprocesses responsible for the observed calcium uptake. The existenceof these additional processes could also explain the observed evolutionfor the alkalinity (as HCO3
−), which decreases with the increasinghydrogeochemical evolution degree and, therefore, with increasingdissolved Ca and Mg (Fig. 3). This alkalinity pattern is not consistentwith a single calcite and/or dolomite dissolution process, and suggests
μmol L−1
NO3− SiO2 Mg Ca Mg/Ca Na F
2 0.23 0.1512 1.74 2.31 0.75 1.4 9.27 0.23 0.1509 1.67 2.31 0.72 1.4 9.58 0.23 0.1492 1.61 2.3 0.70 1.3 8.58 0.28 0.1052 1.35 2.09 0.65 1 4.88 0.22 0.1501 1.92 2.1 0.92 1.7 9.74 0.23 0.1511 1.68 2.41 0.70 1.5 12
0.24 0.1389 1.55 2.39 0.65 1.5 126 0.24 0.1407 1.76 2.1 0.84 1.3 8.7
0.29 0.1257 1.44 2.27 0.63 0.8 7.70.16 0.1709 2.08 3.05 0.68 2.6 17
6 0.17 0.1674 2.07 2.97 0.70 2.6 160.16 0.1693 2.13 3.08 0.69 2.6 17
4 0.17 0.1286 2.17 3.06 0.71 2.6 154 0.18 0.1636 2.21 2.73 0.81 2.6 162 0.19 0.1663 2.22 2.98 0.75 2.3 162 0.17 0.1629 2.26 3.12 0.72 2.2 166 0.16 0.1661 1.74 2.62 0.66 0.7 15
0.16 0.1034 0.92 2.25 0.41 0.2 3.68 0.16 0.1003 0.86 2.45 0.35 0.2 3.2

Fig. 2. Representation of the composition of the water samples obtained in this study in a Piper–Hill diagram.
328 L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
that the water–carbonate interaction processes are controlled, at leastin part, by the precipitation of a secondary phase uptaking a fraction ofthe dissolved Ca and part of the alkalinity.
As a general trend, the compositional evolution for all the studiedsprings seems to be consistent with the existence of the two mainflow directions proposed in earlier hydrogeological studies (i.e. a flowdirection from Mochales to Jaraba and then to Alhama and a secondflow direction from Deza to Embid and then also to Alhama, wherethe two flow directions seem to join). For the hypothetical flowdirection from Mochales towards Alhama, Mg/Ca ratio initiallyincreases between Mochales and Jaraba and then remains almostconstant between Jaraba and Alhama (ratio Mg/Ca between 0.63 and0.84 in all the samples but ZA-26, from Jaraba), together with aprogressive increase in sodium, sulfate and chloride concentrations(Table 2 and Fig. 2). Globally, this hydrochemical evolution is similarto the evolution pattern already described for many other carbonateaquifers (Back et al., 1983; Plummer et al., 1990; Lopez-Chicano et al.,2001).
For the hypothetical flow direction Deza–Embid–Alhama, thegeochemical evolution seems to be very similar to the one describedfor the flow line Mochales–Jaraba–Alhama but the variations are lessevident because the initial concentrations (i.e. Deza waters) arelarger, especially for total sulfate concentrations, and because thehydrochemistry of the thermal waters from Embid and Alhama is toosimilar to recognize any sharp variation trend (Table 2). As describedfor the flow path Mochales–Jaraba–Alhama, the ratio Mg/Ca is almostconstant in all these samples (Table 2).
With regard to the water temperature of the springs, the highesttemperatures in the Alhama and Jaraba samples are very similar(Table 2). Nonetheless, the temperature variability is much largerfor the Jaraba (between 21 and 32 °C) than for the Alhama springs(temperature always higher than 30 °C and temperature variabilitysmaller than 2.3 °C). In spite of these differences, the estimation of
the temperature conditions at depth, based on the combination ofclassical chemical geothermometers (e.g. quartz geothermometer;Fournier and Potter, 1982) and geochemical modeling calculationsbased on the multiple mineral equilibrium approach described byReed and Spycher (1984), shows the existence of similar temper-ature (ranging between 40 and 50 °C) in the deep reservoir for allthe thermal springs included in this study (Tena et al., 1995). Takinginto account that the average surface temperature in the study areais 14 °C and the geothermal gradient in this zone is around 30 mK/m(Fernández et al., 1989), this suggests a maximum depth betweenapproximately 850 and 1200 m for these carbonate aquifers, whichis very consistent with the estimated thickness by geophysicalmethods of Tertiary materials in this area of the Almazan Basin(Sanchez et al., 2004) under which the groundwaters are thought tocirculate. Moreover, this depth matches very closely the depthrange currently proposed for deep geologic storage of supercriticalCO2 under average geothermic and lithostatic conditions (Metzet al., 2005).
4.2. Hydrogeochemical modeling: speciation–solubility calculations andprocesses controlling the geochemical behavior of the system
Speciation–solubility calculations were carried out with the assis-tance of the PHREEQC code (Parkhurst and Appelo, 1999) and using thethermodynamic database WATEQ4F.dat (Ball and Nordstrom, 2001),distributed with the code. The main results of these calculations,including the saturation indices for carbonate and sulfate mineralphases and the values obtained for TIC (Total Inorganic Carbon) and forthepartial pressureof CO2 inequilibriumwith the samples, are shown inTable 4.
The saturation indices for gypsum, anhydrite, halite and fluorite(CaF2) in the thermal springs of Alhama, Jaraba and Embid indicatethat all the studied waters are clearly subsaturated with respect
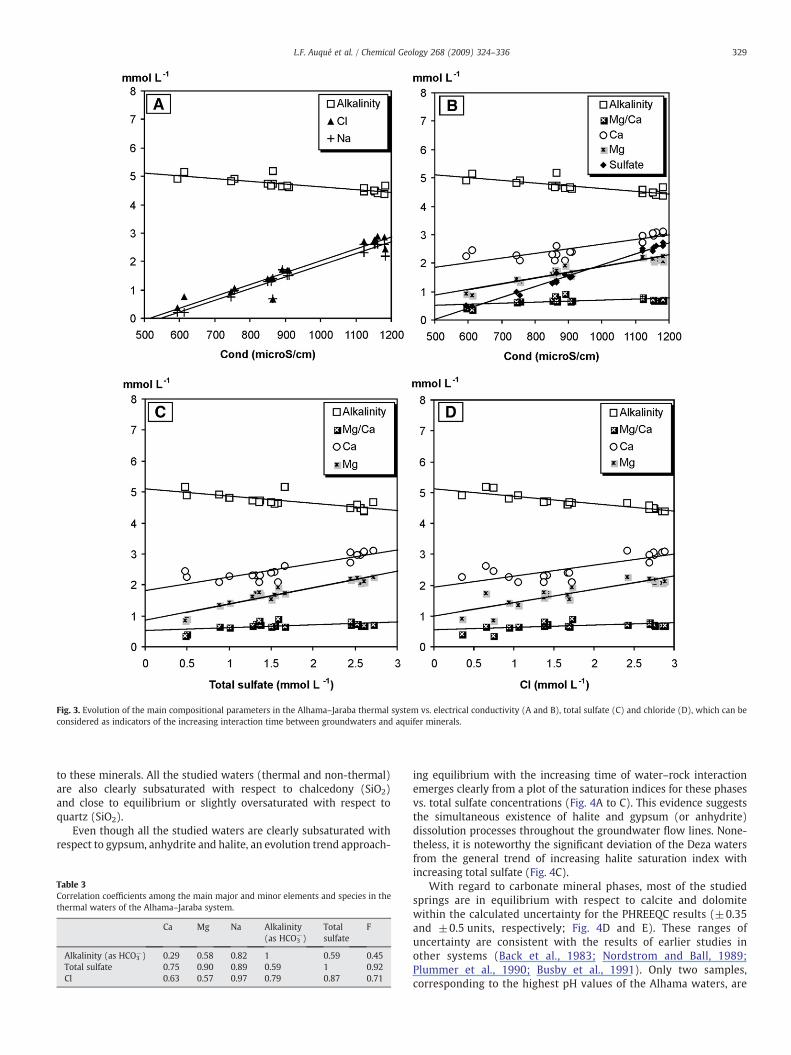
Fig. 3. Evolution of the main compositional parameters in the Alhama–Jaraba thermal system vs. electrical conductivity (A and B), total sulfate (C) and chloride (D), which can beconsidered as indicators of the increasing interaction time between groundwaters and aquifer minerals.
329L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
to these minerals. All the studied waters (thermal and non-thermal)are also clearly subsaturated with respect to chalcedony (SiO2)and close to equilibrium or slightly oversaturated with respect toquartz (SiO2).
Even though all the studied waters are clearly subsaturated withrespect to gypsum, anhydrite and halite, an evolution trend approach-
Table 3Correlation coefficients among the main major and minor elements and species in thethermal waters of the Alhama–Jaraba system.
Ca Mg Na Alkalinity(as HCO3
−)Totalsulfate
F
Alkalinity (as HCO3−) 0.29 0.58 0.82 1 0.59 0.45
Total sulfate 0.75 0.90 0.89 0.59 1 0.92Cl 0.63 0.57 0.97 0.79 0.87 0.71
ing equilibrium with the increasing time of water–rock interactionemerges clearly from a plot of the saturation indices for these phasesvs. total sulfate concentrations (Fig. 4A to C). This evidence suggeststhe simultaneous existence of halite and gypsum (or anhydrite)dissolution processes throughout the groundwater flow lines. None-theless, it is noteworthy the significant deviation of the Deza watersfrom the general trend of increasing halite saturation index withincreasing total sulfate (Fig. 4C).
With regard to carbonate mineral phases, most of the studiedsprings are in equilibrium with respect to calcite and dolomitewithin the calculated uncertainty for the PHREEQC results (±0.35and ±0.5 units, respectively; Fig. 4D and E). These ranges ofuncertainty are consistent with the results of earlier studies inother systems (Back et al., 1983; Nordstrom and Ball, 1989;Plummer et al., 1990; Busby et al., 1991). Only two samples,corresponding to the highest pH values of the Alhama waters, are
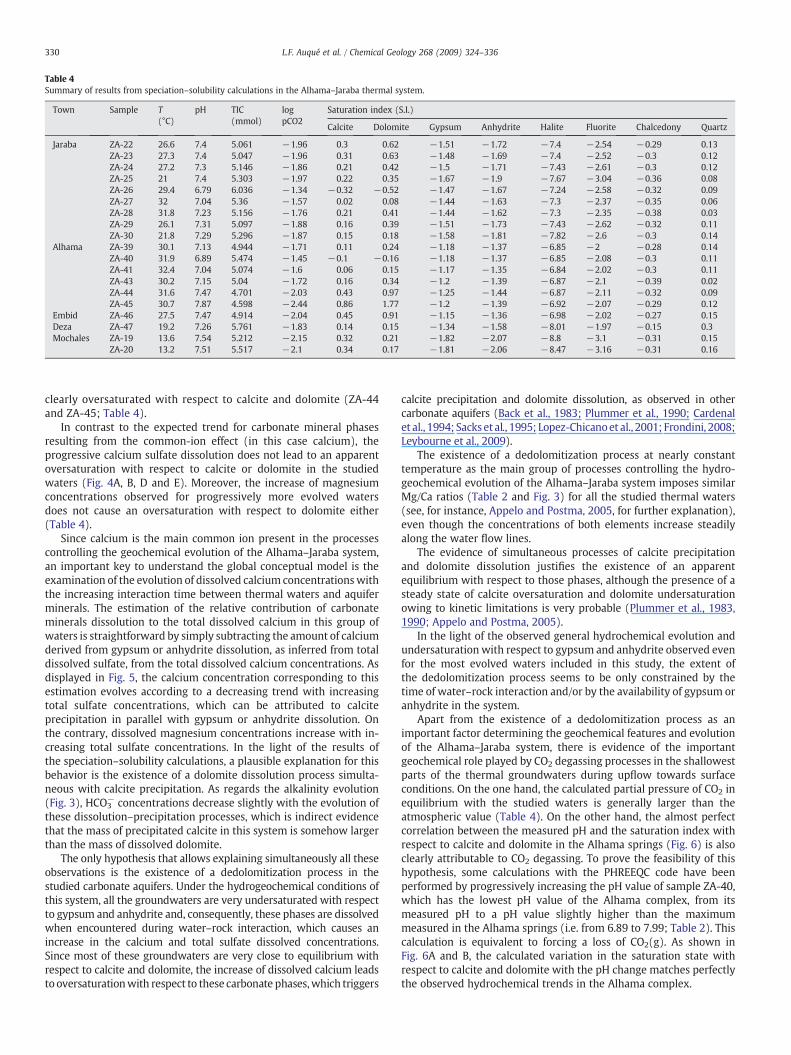
Table 4Summary of results from speciation–solubility calculations in the Alhama–Jaraba thermal system.
Town Sample T(°C)
pH TIC(mmol)
logpCO2
Saturation index (S.I.)
Calcite Dolomite Gypsum Anhydrite Halite Fluorite Chalcedony Quartz
Jaraba ZA-22 26.6 7.4 5.061 −1.96 0.3 0.62 −1.51 −1.72 −7.4 −2.54 −0.29 0.13ZA-23 27.3 7.4 5.047 −1.96 0.31 0.63 −1.48 −1.69 −7.4 −2.52 −0.3 0.12ZA-24 27.2 7.3 5.146 −1.86 0.21 0.42 −1.5 −1.71 −7.43 −2.61 −0.3 0.12ZA-25 21 7.4 5.303 −1.97 0.22 0.35 −1.67 −1.9 −7.67 −3.04 −0.36 0.08ZA-26 29.4 6.79 6.036 −1.34 −0.32 −0.52 −1.47 −1.67 −7.24 −2.58 −0.32 0.09ZA-27 32 7.04 5.36 −1.57 0.02 0.08 −1.44 −1.63 −7.3 −2.37 −0.35 0.06ZA-28 31.8 7.23 5.156 −1.76 0.21 0.41 −1.44 −1.62 −7.3 −2.35 −0.38 0.03ZA-29 26.1 7.31 5.097 −1.88 0.16 0.39 −1.51 −1.73 −7.43 −2.62 −0.32 0.11ZA-30 21.8 7.29 5.296 −1.87 0.15 0.18 −1.58 −1.81 −7.82 −2.6 −0.3 0.14
Alhama ZA-39 30.1 7.13 4.944 −1.71 0.11 0.24 −1.18 −1.37 −6.85 −2 −0.28 0.14ZA-40 31.9 6.89 5.474 −1.45 −0.1 −0.16 −1.18 −1.37 −6.85 −2.08 −0.3 0.11ZA-41 32.4 7.04 5.074 −1.6 0.06 0.15 −1.17 −1.35 −6.84 −2.02 −0.3 0.11ZA-43 30.2 7.15 5.04 −1.72 0.16 0.34 −1.2 −1.39 −6.87 −2.1 −0.39 0.02ZA-44 31.6 7.47 4.701 −2.03 0.43 0.97 −1.25 −1.44 −6.87 −2.11 −0.32 0.09ZA-45 30.7 7.87 4.598 −2.44 0.86 1.77 −1.2 −1.39 −6.92 −2.07 −0.29 0.12
Embid ZA-46 27.5 7.47 4.914 −2.04 0.45 0.91 −1.15 −1.36 −6.98 −2.02 −0.27 0.15Deza ZA-47 19.2 7.26 5.761 −1.83 0.14 0.15 −1.34 −1.58 −8.01 −1.97 −0.15 0.3Mochales ZA-19 13.6 7.54 5.212 −2.15 0.32 0.21 −1.82 −2.07 −8.8 −3.1 −0.31 0.15
ZA-20 13.2 7.51 5.517 −2.1 0.34 0.17 −1.81 −2.06 −8.47 −3.16 −0.31 0.16
330 L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
clearly oversaturated with respect to calcite and dolomite (ZA-44and ZA-45; Table 4).
In contrast to the expected trend for carbonate mineral phasesresulting from the common-ion effect (in this case calcium), theprogressive calcium sulfate dissolution does not lead to an apparentoversaturation with respect to calcite or dolomite in the studiedwaters (Fig. 4A, B, D and E). Moreover, the increase of magnesiumconcentrations observed for progressively more evolved watersdoes not cause an oversaturation with respect to dolomite either(Table 4).
Since calcium is the main common ion present in the processescontrolling the geochemical evolution of the Alhama–Jaraba system,an important key to understand the global conceptual model is theexamination of the evolution of dissolved calcium concentrationswiththe increasing interaction time between thermal waters and aquiferminerals. The estimation of the relative contribution of carbonateminerals dissolution to the total dissolved calcium in this group ofwaters is straightforward by simply subtracting the amount of calciumderived from gypsum or anhydrite dissolution, as inferred from totaldissolved sulfate, from the total dissolved calcium concentrations. Asdisplayed in Fig. 5, the calcium concentration corresponding to thisestimation evolves according to a decreasing trend with increasingtotal sulfate concentrations, which can be attributed to calciteprecipitation in parallel with gypsum or anhydrite dissolution. Onthe contrary, dissolved magnesium concentrations increase with in-creasing total sulfate concentrations. In the light of the results ofthe speciation–solubility calculations, a plausible explanation for thisbehavior is the existence of a dolomite dissolution process simulta-neous with calcite precipitation. As regards the alkalinity evolution(Fig. 3), HCO3
− concentrations decrease slightly with the evolution ofthese dissolution–precipitation processes, which is indirect evidencethat the mass of precipitated calcite in this system is somehow largerthan the mass of dissolved dolomite.
The only hypothesis that allows explaining simultaneously all theseobservations is the existence of a dedolomitization process in thestudied carbonate aquifers. Under the hydrogeochemical conditions ofthis system, all the groundwaters are very undersaturated with respectto gypsum and anhydrite and, consequently, these phases are dissolvedwhen encountered during water–rock interaction, which causes anincrease in the calcium and total sulfate dissolved concentrations.Since most of these groundwaters are very close to equilibrium withrespect to calcite and dolomite, the increase of dissolved calcium leadsto oversaturationwith respect to these carbonate phases,which triggers
calcite precipitation and dolomite dissolution, as observed in othercarbonate aquifers (Back et al., 1983; Plummer et al., 1990; Cardenalet al., 1994; Sackset al., 1995; Lopez-Chicanoet al., 2001; Frondini, 2008;Leybourne et al., 2009).
The existence of a dedolomitization process at nearly constanttemperature as the main group of processes controlling the hydro-geochemical evolution of the Alhama–Jaraba system imposes similarMg/Ca ratios (Table 2 and Fig. 3) for all the studied thermal waters(see, for instance, Appelo and Postma, 2005, for further explanation),even though the concentrations of both elements increase steadilyalong the water flow lines.
The evidence of simultaneous processes of calcite precipitationand dolomite dissolution justifies the existence of an apparentequilibrium with respect to those phases, although the presence of asteady state of calcite oversaturation and dolomite undersaturationowing to kinetic limitations is very probable (Plummer et al., 1983,1990; Appelo and Postma, 2005).
In the light of the observed general hydrochemical evolution andundersaturation with respect to gypsum and anhydrite observed evenfor the most evolved waters included in this study, the extent ofthe dedolomitization process seems to be only constrained by thetime of water–rock interaction and/or by the availability of gypsum oranhydrite in the system.
Apart from the existence of a dedolomitization process as animportant factor determining the geochemical features and evolutionof the Alhama–Jaraba system, there is evidence of the importantgeochemical role played by CO2 degassing processes in the shallowestparts of the thermal groundwaters during upflow towards surfaceconditions. On the one hand, the calculated partial pressure of CO2 inequilibrium with the studied waters is generally larger than theatmospheric value (Table 4). On the other hand, the almost perfectcorrelation between the measured pH and the saturation index withrespect to calcite and dolomite in the Alhama springs (Fig. 6) is alsoclearly attributable to CO2 degassing. To prove the feasibility of thishypothesis, some calculations with the PHREEQC code have beenperformed by progressively increasing the pH value of sample ZA-40,which has the lowest pH value of the Alhama complex, from itsmeasured pH to a pH value slightly higher than the maximummeasured in the Alhama springs (i.e. from 6.89 to 7.99; Table 2). Thiscalculation is equivalent to forcing a loss of CO2(g). As shown inFig. 6A and B, the calculated variation in the saturation state withrespect to calcite and dolomite with the pH change matches perfectlythe observed hydrochemical trends in the Alhama complex.
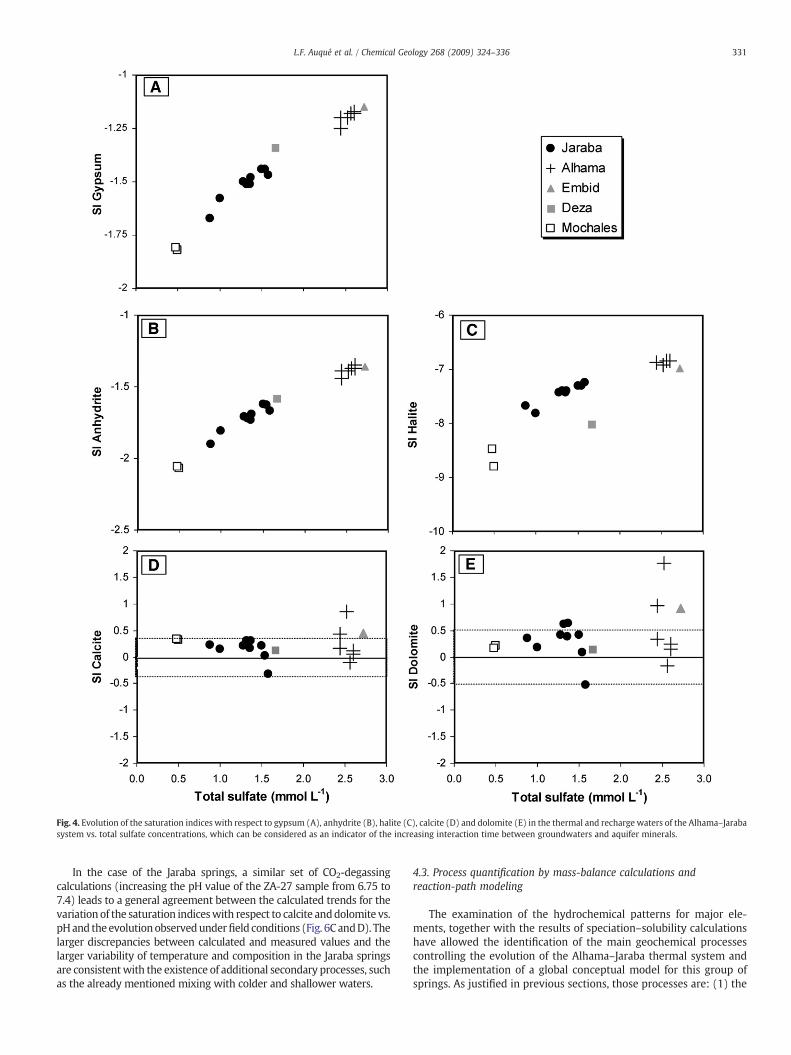
Fig. 4. Evolution of the saturation indices with respect to gypsum (A), anhydrite (B), halite (C), calcite (D) and dolomite (E) in the thermal and recharge waters of the Alhama–Jarabasystem vs. total sulfate concentrations, which can be considered as an indicator of the increasing interaction time between groundwaters and aquifer minerals.
331L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
In the case of the Jaraba springs, a similar set of CO2-degassingcalculations (increasing the pH value of the ZA-27 sample from 6.75 to7.4) leads to a general agreement between the calculated trends for thevariation of the saturation indiceswith respect to calcite anddolomite vs.pHand the evolutionobservedunderfield conditions (Fig. 6CandD). Thelarger discrepancies between calculated and measured values and thelarger variability of temperature and composition in the Jaraba springsare consistentwith the existence of additional secondary processes, suchas the already mentioned mixing with colder and shallower waters.
4.3. Process quantification by mass-balance calculations andreaction-path modeling
The examination of the hydrochemical patterns for major ele-ments, together with the results of speciation–solubility calculationshave allowed the identification of the main geochemical processescontrolling the evolution of the Alhama–Jaraba thermal system andthe implementation of a global conceptual model for this group ofsprings. As justified in previous sections, those processes are: (1) the
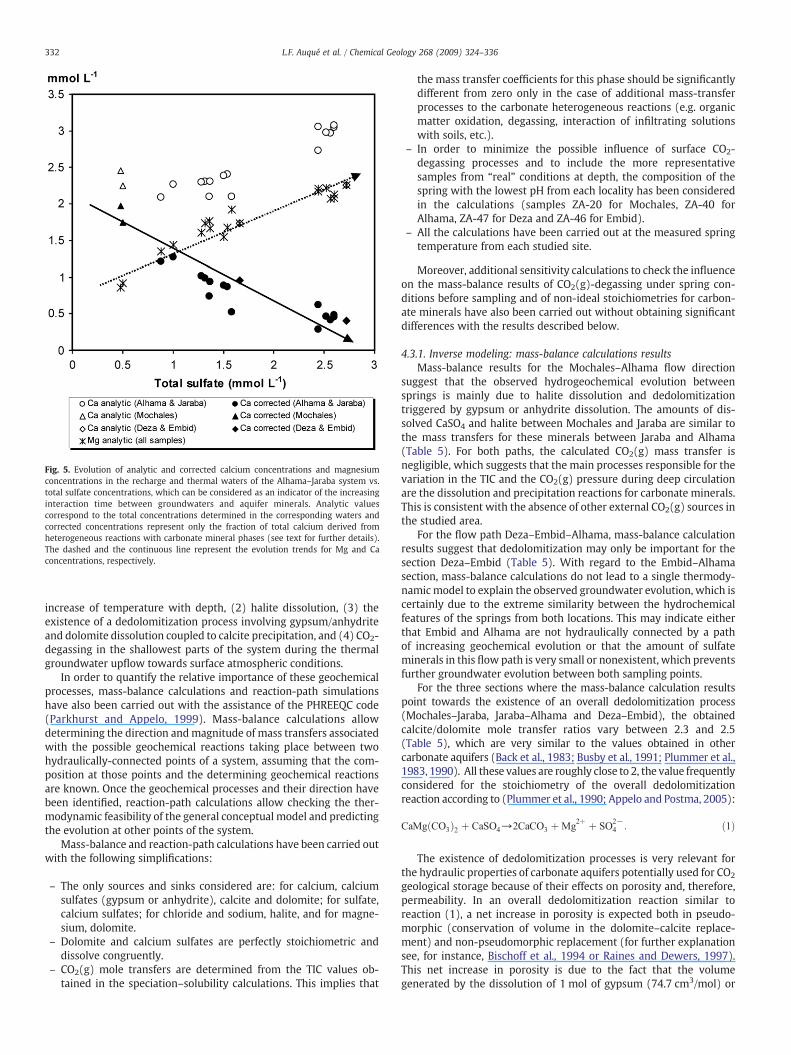
Fig. 5. Evolution of analytic and corrected calcium concentrations and magnesiumconcentrations in the recharge and thermal waters of the Alhama–Jaraba system vs.total sulfate concentrations, which can be considered as an indicator of the increasinginteraction time between groundwaters and aquifer minerals. Analytic valuescorrespond to the total concentrations determined in the corresponding waters andcorrected concentrations represent only the fraction of total calcium derived fromheterogeneous reactions with carbonate mineral phases (see text for further details).The dashed and the continuous line represent the evolution trends for Mg and Caconcentrations, respectively.
332 L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
increase of temperature with depth, (2) halite dissolution, (3) theexistence of a dedolomitization process involving gypsum/anhydriteand dolomite dissolution coupled to calcite precipitation, and (4) CO2-degassing in the shallowest parts of the system during the thermalgroundwater upflow towards surface atmospheric conditions.
In order to quantify the relative importance of these geochemicalprocesses, mass-balance calculations and reaction-path simulationshave also been carried out with the assistance of the PHREEQC code(Parkhurst and Appelo, 1999). Mass-balance calculations allowdetermining the direction andmagnitude of mass transfers associatedwith the possible geochemical reactions taking place between twohydraulically-connected points of a system, assuming that the com-position at those points and the determining geochemical reactionsare known. Once the geochemical processes and their direction havebeen identified, reaction-path calculations allow checking the ther-modynamic feasibility of the general conceptual model and predictingthe evolution at other points of the system.
Mass-balance and reaction-path calculations have been carried outwith the following simplifications:
– The only sources and sinks considered are: for calcium, calciumsulfates (gypsum or anhydrite), calcite and dolomite; for sulfate,calcium sulfates; for chloride and sodium, halite, and for magne-sium, dolomite.
– Dolomite and calcium sulfates are perfectly stoichiometric anddissolve congruently.
– CO2(g) mole transfers are determined from the TIC values ob-tained in the speciation–solubility calculations. This implies that
the mass transfer coefficients for this phase should be significantlydifferent from zero only in the case of additional mass-transferprocesses to the carbonate heterogeneous reactions (e.g. organicmatter oxidation, degassing, interaction of infiltrating solutionswith soils, etc.).
– In order to minimize the possible influence of surface CO2-degassing processes and to include the more representativesamples from “real” conditions at depth, the composition of thespring with the lowest pH from each locality has been consideredin the calculations (samples ZA-20 for Mochales, ZA-40 forAlhama, ZA-47 for Deza and ZA-46 for Embid).
– All the calculations have been carried out at the measured springtemperature from each studied site.
Moreover, additional sensitivity calculations to check the influenceon the mass-balance results of CO2(g)-degassing under spring con-ditions before sampling and of non-ideal stoichiometries for carbon-ate minerals have also been carried out without obtaining significantdifferences with the results described below.
4.3.1. Inverse modeling: mass-balance calculations resultsMass-balance results for the Mochales–Alhama flow direction
suggest that the observed hydrogeochemical evolution betweensprings is mainly due to halite dissolution and dedolomitizationtriggered by gypsum or anhydrite dissolution. The amounts of dis-solved CaSO4 and halite between Mochales and Jaraba are similar tothe mass transfers for these minerals between Jaraba and Alhama(Table 5). For both paths, the calculated CO2(g) mass transfer isnegligible, which suggests that the main processes responsible for thevariation in the TIC and the CO2(g) pressure during deep circulationare the dissolution and precipitation reactions for carbonate minerals.This is consistent with the absence of other external CO2(g) sources inthe studied area.
For the flow path Deza–Embid–Alhama, mass-balance calculationresults suggest that dedolomitization may only be important for thesection Deza–Embid (Table 5). With regard to the Embid–Alhamasection, mass-balance calculations do not lead to a single thermody-namicmodel to explain the observed groundwater evolution, which iscertainly due to the extreme similarity between the hydrochemicalfeatures of the springs from both locations. This may indicate eitherthat Embid and Alhama are not hydraulically connected by a pathof increasing geochemical evolution or that the amount of sulfateminerals in this flow path is very small or nonexistent, which preventsfurther groundwater evolution between both sampling points.
For the three sections where the mass-balance calculation resultspoint towards the existence of an overall dedolomitization process(Mochales–Jaraba, Jaraba–Alhama and Deza–Embid), the obtainedcalcite/dolomite mole transfer ratios vary between 2.3 and 2.5(Table 5), which are very similar to the values obtained in othercarbonate aquifers (Back et al., 1983; Busby et al., 1991; Plummer et al.,1983, 1990). All these values are roughly close to 2, the value frequentlyconsidered for the stoichiometry of the overall dedolomitizationreaction according to (Plummer et al., 1990; Appelo and Postma, 2005):
CaMgðCO3Þ2 þ CaSO4→2CaCO3 þMg2þ þ SO
2−4 : ð1Þ
The existence of dedolomitization processes is very relevant forthe hydraulic properties of carbonate aquifers potentially used for CO2
geological storage because of their effects on porosity and, therefore,permeability. In an overall dedolomitization reaction similar toreaction (1), a net increase in porosity is expected both in pseudo-morphic (conservation of volume in the dolomite–calcite replace-ment) and non-pseudomorphic replacement (for further explanationsee, for instance, Bischoff et al., 1994 or Raines and Dewers, 1997).This net increase in porosity is due to the fact that the volumegenerated by the dissolution of 1 mol of gypsum (74.7 cm3/mol) or
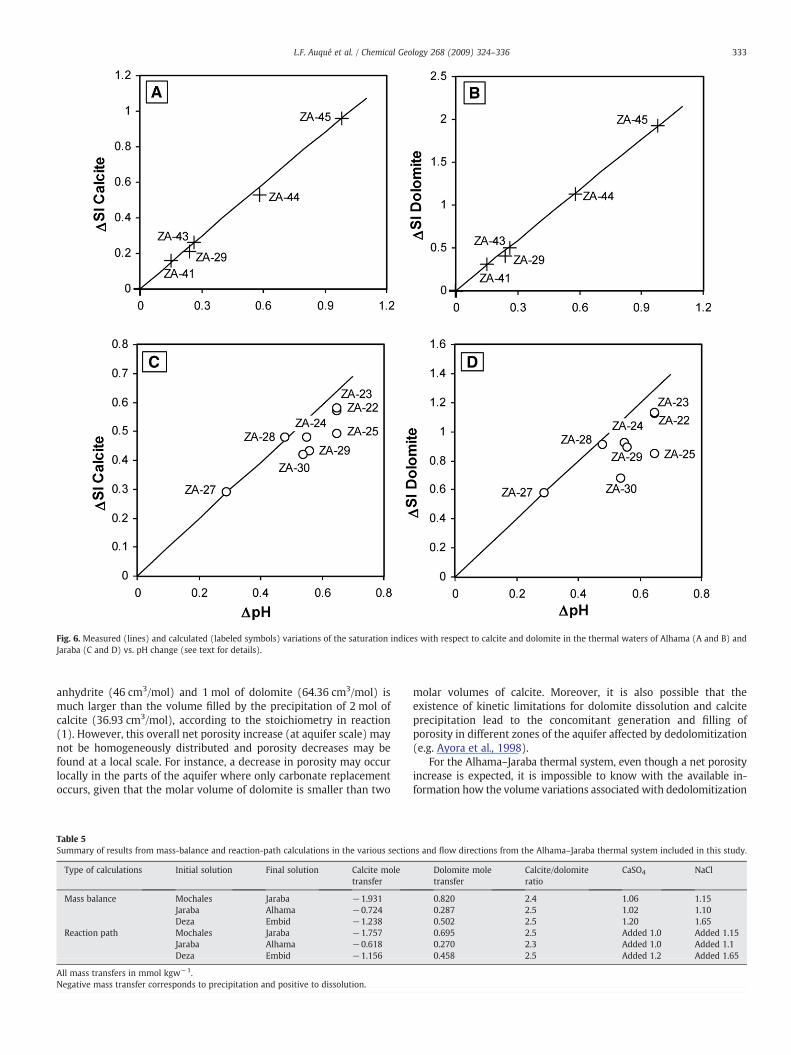
Fig. 6. Measured (lines) and calculated (labeled symbols) variations of the saturation indices with respect to calcite and dolomite in the thermal waters of Alhama (A and B) andJaraba (C and D) vs. pH change (see text for details).
333L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
anhydrite (46 cm3/mol) and 1 mol of dolomite (64.36 cm3/mol) ismuch larger than the volume filled by the precipitation of 2 mol ofcalcite (36.93 cm3/mol), according to the stoichiometry in reaction(1). However, this overall net porosity increase (at aquifer scale) maynot be homogeneously distributed and porosity decreases may befound at a local scale. For instance, a decrease in porosity may occurlocally in the parts of the aquifer where only carbonate replacementoccurs, given that the molar volume of dolomite is smaller than two
Table 5Summary of results from mass-balance and reaction-path calculations in the various sectio
Type of calculations Initial solution Final solution Calcite moletransfer
Mass balance Mochales Jaraba −1.931Jaraba Alhama −0.724Deza Embid −1.238
Reaction path Mochales Jaraba −1.757Jaraba Alhama −0.618Deza Embid −1.156
All mass transfers in mmol kgw−1.Negative mass transfer corresponds to precipitation and positive to dissolution.
molar volumes of calcite. Moreover, it is also possible that theexistence of kinetic limitations for dolomite dissolution and calciteprecipitation lead to the concomitant generation and filling ofporosity in different zones of the aquifer affected by dedolomitization(e.g. Ayora et al., 1998).
For the Alhama–Jaraba thermal system, even though a net porosityincrease is expected, it is impossible to know with the available in-formation how the volume variations associated with dedolomitization
ns and flow directions from the Alhama–Jaraba thermal system included in this study.
Dolomite moletransfer
Calcite/dolomiteratio
CaSO4 NaCl
0.820 2.4 1.06 1.150.287 2.5 1.02 1.100.502 2.5 1.20 1.650.695 2.5 Added 1.0 Added 1.150.270 2.3 Added 1.0 Added 1.10.458 2.5 Added 1.2 Added 1.65

334 L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
are distributed locally throughout the studiedflowpaths and, therefore,the evaluation of the possible permeability variations would be merelyspeculative. However, this would be an important point in anycarbonate aquifer considered for hosting a CO2 storage, since porosityvariations may modify the hydraulic conductivity of the carbonateaquifer and affect its ability to store CO2 in the long term. This highlightsthe importance of a detailed mineralogical and geochemical character-isation of the rocks and sediments considered for hosting CO2 reservoirs,which is the only way to accurately predict the evolution of suchsystems in the long term. Moreover, even though mass-balance resultsrepresent a first step in the understanding of this problem, they provideonly net porosity variations along flow paths and more sophisticatedmodeling approaches (e.g. coupled flow and reactive transportincluding kineticmodeling)would be needed to dealwith local porositychanges.
4.3.2. Direct modeling: reaction-path calculationsAs explained above, mass-balance calculations have not allowed
unequivocally interpreting and justifying the geochemical processescontrolling the hydrochemical evolution for the Embid–Alhama flowdirection. Nevertheless, inverse calculations have indicated that theevolution of the thermal solutions in the flow direction Mochales–Jaraba–Alhama and in the section Deza–Embid seem to be mainlydetermined by a dedolomitization process. Consequently, simplifiedreaction-path calculations have been carried out as a direct modelingcheck of the conceptual model feasibility for these flow sections of thestudied thermal system.
With this aim, calculations in which calcium sulfate and halitewere added to the Mochales, Jaraba and Deza solutions were carriedout. In these calculations, calcite and dolomite equilibria are imposedin order to obtain the calcite and dolomite mole transfer amountspromoted by the irreversible gypsum or anhydrite dissolution in thesections Mochales–Jaraba, Jaraba–Alhama and Deza–Embid. No otherCO2 sources or sinks are considered in the calculations, in agreementwith the results of mass-balance calculations. The amounts of addedphases were those obtained in the mass-balance calculations for eachsection (Table 5).
The results of the reaction-path calculations are summarized inTables 5 and 6. As displayed in Table 5, the obtained calcite anddolomite mole transfers for both sections after addition of calciumsulfate closely match the results from mass-balance calculations.Moreover, the calculated composition after evolution by dedolomiti-zation for the spring waters of Jaraba (from the Mochales solutioncomposition), Alhama (from the Jaraba solution compositions) andEmbid (from the Deza solution compositions) is the same (within 10%uncertainty) as observed in the corresponding thermal solutions(Table 6). The good general agreement obtained for both types ofresults (mass transfers and solution compositions) prove that theproposed dedolomitization model is qualitatively and quantitativelyadequate to describe the behavior of the Mochales–Jaraba–Alhamaflow direction and the section Deza–Embid of the Deza–Alhama flowpath.
Table 6Comparison between the measured hydrochemistry of the Jaraba and Alhama thermalwaters and the calculated compositions obtained for both sampling locations by meansof reaction-path calculations (see text for details).
mmol kgw−1
pH Alk. Ca Mg S Na Cl
Jaraba Measured 7.04 4.62 2.40 1.70 1.50 1.50 1.70Calculated 7.03 4.43 2.45 1.55 1.54 1.35 1.89
Alhama Measured 6.89 4.47 2.97 2.07 2.56 2.60 2.80Calculated 6.98 4.46 3.07 1.97 2.52 2.60 2.80
Embid Measured 7.02 4.66 3.12 2.26 2.72 2.20 2.42Calculated 7.02 4.70 3.12 2.20 2.86 2.35 2.30
The largest differences withmeasured compositions correspond tothe concentrations of Na and Cl of the Jaraba springs calculated fromthe composition of the Mochales spring waters (Table 6). This slightdiscrepancy could be attributed to the existence in this system sectionof other processes not included in the geochemical model, such ascation exchange. In fact, the calculated compositions in the sectionMochales–Jaraba match more closely the measured hydrochemistrywhen cation exchange processes involving Na and Ca are included inthe conceptual model (not shown). Nonetheless, we have preferrednot to include additional processes in the model for the sake ofsimplicity and robustness of the proposed conceptual model.
Finally, an additional result arises from the performed reaction-path simulations. During the simulated dedolomitization trend, TICconcentrations progressively decrease whereas CO2 partial pressuresincrease. These results are perfectly consistent with the mass-balanceresults, which indicate that the estimated mass of precipitated calciteis more than double the mass of dissolved dolomite.
These trends are obscured in the studied thermal waters by theoutgassing processes suffered during their ascent off. However,similar evolution trends for TIC and pCO2 have been observed inother carbonate aquifers where dedolomitization occurs under closedsystem conditions with respect to CO2 (e.g. Back et al., 1983; Plummeret al., 1990) and in purely theoretical thermodynamic simulations ofthe dedolomitization processes under closed system conditions(Parkhurst et al., 1990).
This result would imply that, under certain conditions, dedolomi-tization may produce a net mineral trapping of CO2, which is one ofthe most desirable mechanisms for CO2 sequestration. However, thispossibility must be further studied as the TIC balance can be affectedand/or modified by, for example, the carbonate replacement modality(pseudomorphic vs. non-pseudomorphic) or by the occurrence ofprocesses additional to dedolomitization (e.g. Plummer et al., 1990;Raines and Dewers, 1997).
5. Conclusions
The processes determining the hydrogeochemical evolution in theAlhama–Jaraba thermal system have been successfully identified andquantified with the assistance of classical geochemical tools (ion–ionplots, speciation–solubility calculations) and inverse (mass-balancecalculations) and direct (reaction-path calculations) geochemicalmodeling. Even in the sections of the system where those controllinggeochemical processes have not been completely defined, the appliedmethodology has contributed to identify the main uncertainties andto constrain the possible recharge areas and flow directions. Theimplementation of mass-balance calculations has provided supportfor the hydrogeological hypotheses and has allowed quantifying themass transfers between sampling locations. Furthermore, the feasi-bility of the proposed conceptual hydrogeochemical model for thestudied system has been verified by using reaction-path calculations.
All the processes identified in the Alhama–Jaraba thermal systemare relevant for the long-term expected evolution of CO2 storage indeep carbonate aquifers and/or for the behavior of the CO2 reservoirsections more distant from the CO2 injection wells.
As also feasible under certain evolution scenarios in the springsplaced in the surroundings of a CO2 deep geological storage incarbonate rocks, the observed hydrochemistry in the Alhama–Jarabasprings is influenced by the existence of CO2(g)-degassing processesduring the upflow of thermal groundwaters towards surface condi-tions. Apart from these processes, the hydrogeochemical evolution ofthe studied groundwaters is determined by the temperature varia-tions with depth, by halite dissolution and, mainly, by dedolomitiza-tion triggered by gypsum/anhydrite dissolution.
Dedolomitization has already proven in recent studies to beeffective in determining the evolution of carbonate aquifers affectedby deep CO2 influx. In some of these aquifers dedolomitization may

335L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
occur over a wide pCO2 values from 0.1 to 10 bar (e.g. Frondini, 2008).A detailed comparison between themass transfers and their effects onthe hydrogeochemistry for such groundwater systems and for othersystems not affected by CO2 deep sources (like the Alhama–Jarabasystem) may provide valuable information for the understanding ofthe expected processes in a CO2 storage in carbonate aquifers.
Moreover, the existence of dedolomitization is very relevant forthe hydraulic properties of carbonate aquifers potentially used for CO2
geological storage because of their effects on porosity and, therefore,permeability. Porosity variations may modify the hydraulic conduc-tivity of the carbonate aquifer and affect its ability to store CO2 inthe long term. As explained in this study, even though the overalldedolomitization process (as described in reaction (1)) should cause anet increase of the aquifer porosity, it may also produce decreases ofporosity (depending on the kinetic–spatial-relations between thesequence of the involved processes, gypsum/anhydrite dissolution,calcite precipitation and dolomite dissolution) at a local scale. Thisconclusion highlights the importance of a detailed mineralogical andgeochemical characterisation of the rocks and sediments consideredfor hosting CO2 reservoirs, which is the only way to accurately predictthe evolution of such systems in the long term.
Furthermore, mass balance and reaction-path modeling indicatethat dedolomitization processes may also represent a sink of CO2
through mineral sequestration, at least under some circumstances,and this fact merits further studies.
As shown in this study, the application of classical geochemicaltools provides an excellent starting point for understanding the long-term behavior of prospective CO2 storage systems. Hydrothermalsystems with relatively low CO2 pressures, like the Alhama–Jarabasystem, are probably the main natural analogue for studying theexpected water–rock interactions in the long term or in the reservoirparts more distant from the injected supercritical CO2 and provide aunique opportunity to test and optimize the site characterisationmethodologies that will be applied in future CO2 reservoirs.
Acknowledgements
This study has been partially carried out with the support of theR+D Contracts PSE-120000-2007-6 and PSE-120000-2008-6 withthe Spanish Government and of the Program for CO2 GeologicalStorage of the “Ciudad de la Energia” Foundation.We thank the Editor,Dr. Jeremy B. Fein, for his help and comments. The constructivecriticisms from Dr. I. Hutcheon and an anonymous reviewer havesignificantly improved the original manuscript.
References
Allen, D.E., Strazisar, B.R., Soong, Y., Hedges, S.W., 2005. Modeling carbon dioxidesequestration in saline aquifers: significance of elevated pressures and salinities.Fuel Process. Technol. 86 (14–15), 1569–1580.
Alonso, A., Floquet, M., Mas, R., Melendez, A., 1993. Late Cretaceous carbonateplatforms: origin and evolution, Iberian Range, Spain. In: Masse, J.P. (Ed.), AAPGSpec. Publ., 56, pp. 297–316.
André, L., Audigane, P., Azaroual, M., Menjoz, A., 2007. Numerical modeling of fluid–rock chemical interactions at the supercritical CO2–liquid interface during CO2
injection into a carbonate reservoir, the Dogger aquifer (Paris Basin, France).Energy Convers. Manage. 48 (6), 1782–1797.
Appelo, C.A.J., Postma, D., 2005. Geochemistry, Groundwater and Pollution, second ed.A.A. Balkema, Rotterdam.
Audigane, P., Gaus, I., Czernichowski-Lauriol, I., Pruess, K., Xu, T., 2007. Two-dimensional reactive transport modeling of CO2 injection in a saline aquifer atthe Sleipner site, North Sea. Am. J. Sci. 307, 974–1008.
Aurell, M., Melendez, G., Oloriz, F., Badenas, B., Caracuel, J.E., Garcia-Ramos, J.C., Goy, A.,Linares, A., Quesada, S., Robles, S., Rodríguez-Tovar, F.J., Rosales, I., Sandoval, J.,Suarez de Centi, C., Tavera, J.M., Valenzuela, M., 2002. Jurassic. In: Gibbons, W.,Moreno, T. (Eds.), The Geology of Spain. Geological Society of London, London, UK.
Ayora, C., Taberner, C., Saaltink, M.W., Carrera, J., 1998. The genesis of dedolomites: adiscussion based on reactive transport modelling. J. Hydrol. 209, 346–365.
Back, W., Hanshaw, B.B., Plummer, L.N., Rahn, P.H., Rightmire, C.T., Rubin, M., 1983.Process and rate of dedolomitization: mass transfer and 14C dating in a regionalcarbonate aquifer. Geol. Soc. Am. Bull. 94, 1415–1429.
Ball, J.W., Nordstrom, D.K., 2001. User's manual for WATEQ4F, with revisedthermodynamic data base and test cases for calculating speciation of major,trace, and redox elements in natural waters. US Geological Survey Open File Report91-183, USA.
Bachu, S., 2000. Sequestration of CO2 in geological media: criteria and approach for siteselection in response to climate change. Energy Convers. Manage. 41 (9), 953–970.
Bateman, K., Turner, G., Pearce, J.M., Noy, D.J., Birchall, D.J., Rochelle, C.A., 2005. Large-scale column experiment: study of CO2, porewater, rock reactions and model testcase. Oil Gas Sci. Technol. 60, 161–175.
Bertier, P., Swennen, R., Laenen, B., Lagrou, D., Dreesen, R., 2006. Experimentalidentification of CO2–water–rock interactions caused by sequestration of CO2 inWestphalian and Buntsandstein sandstones of the Campine Basin (NE-Belgium).J. Geochem. Explor. 89, 10–14.
Bischoff, J.L., Juliá, R., Shanks, W.C., Rosenbauer, R.J., 1994. Karstification withoutcarbonic acid: bedrock dissolution gypsum-driven dedolomitization. Geology 22,995–998.
Brosse, E., Magnier, C., Vincent, B., 2005. Modelling fluid–rock interaction induced bythe percolation of CO2-enriched solutions in core samples: the role of reactivesurface area. Oil Gas Sci. Technol. 60, 287–305.
Busby, J.F., Plummer, L.N., Lee, R.W., Hanshaw, B.B., 1991. Geochemical evolution ofwater in the Madison aquifer in parts of Montana, South Dakota andWyoming. U.S.Geol. Surv. Prof. Pap., 1273-F. 89 pp.
Cardenal, J., Benavente, J., Cruz-Sanjulian, J., 1994. Chemical evolution of groundwaterin Triassic gypsum-bearing carbonate aquifers (Las Alpujarras, southern Spain).J. Hydrol. 161, 3–20.
Chadwick, R.A., Bernstone, C., May, F., Thibeau, S., Zweigel, P. (Eds.), 2006. Best Practicefor the Storage of CO2 in Saline Aquifers. Observations and Guidelines from theSACS and CO2STORE Projects.
Christensen, N.P., Holloway, S., 2004. Geological Storage of CO2 from combustion offossil fuel, The GESTCO Project. Summary Report, 2nd Edition. 32 pp.
Cipolli, F., Gambardella, B., Marini, L., Ottonello, G., Zuccolini, M.V., 2004. Geochemistryof high-pH waters from serpentinites of the Gruppo di Voltri (Genova, Italy)and reaction path modeling of CO2 sequestration in serpentinite aquifers. Appl.Geochem. 19 (5), 787–802.
Czernichowski, L.I., Sanjuan, B., Rochelle, C., Bateman, K., Pearce, J., Blackwell, P., 1996.Inorganic geochemistry. The Underground Disposal of Carbon Dioxide, Final Reportof Joule II Project No. CT92-0031, pp. 183–276. Chapter 7.
De Toledo, F.O., Arqued, V., 1990. Estudio de los recursos hidraúlicos subterráneos de losacuíferos relacionados con la provincia de Zaragoza. Unidad Hidrogeológica 43,Sierra del Solorio. MOPU, 09.803.183/0411. 224 pp. (in Spanish).
Emberley, S., Hutcheon, I., Shevalier, M., Durocher, K., Mayer, B., Gunter, W.D.,Perkins, E.H., 2005. Monitoring of fluid–rock interaction and CO2 storage throughproduced fluid sampling at the Weyburn CO2-injection enhanced oil recovery site,Saskatchewan, Canada. Appl. Geochem. 20, 1131–1157.
Fernández, M., Marzán, I., Correia, A., Ramalho, C., 1989. Heat flow, heat production,and lithospheric thermal regime in the Iberian Peninsula. Tectonophysics 291,29–53.
Floquet, M., Meléndez, A., Pedauye, R., Pardo, G., Villena, J., 1981. El Cretácico del bordeseptentrional de la Rama Castellana de la Cordillera Ibérica (Sector Central). ElCretácico de la Cordillera Ibérica (Sector Central). Libro Guía de las Jornadas deCampo, Grupo Español del Mesozoico, pp. 157–207. in Spanish.
Fournier, R.O., Potter II, R.W., 1982. A revised and expanded silica (quartz) geo-thermometer. Geotherm. Res. Coun. Bull. 11, 3–12.
Frondini, F., 2008. Geochemistry of regional aquifers hosted by carbonate-evaporiteformations in Umbria and southern Tuscany (central Italy). Appl. Geochem. 23,2091–2104. doi:10.1016/j.apgeochem.2008.05.001.
Gaus, I., Azaroual, M., Czernichowski-Lauriol, I., 2005. Reactive transport modelling ofthe impact of CO2 injection on the clayey cap rock at Sleipner (North Sea). Chem.Geol. 217 (3–4), 319–337.
Gherardi, F., Xu, T.F., Pruess, K., 2007. Numerical modeling of self-limiting and self-enhancing caprock alteration induced by CO2 storage in a depleted gas reservoir.Chem. Geol. 244, 103–129.
Gunter, W.D., Wiwchar, B., Perkins, E.H., 1997. Aquifer disposal of CO2-rich greenhousegases: extension of the time scale of experiment for CO2-sequestering reactions bygeochemical modelling. Mineral. Petrol. 59, 121–140.
Gunter, W.D., Perkins, E.H., Hutcheon, I., 2000. Aquifer disposal of acid gases: modellingof water–rock reactions for trapping of acid wastes. Appl. Geochem. 15, 1085–1095.
Hitchon, B., Gunter, W.D., Gentzis, T., Bailey, R.T., 1999. Sedimentary basins andgreenhouse gases: a serendipitous association. Energy Convers. Manage. 40 (8),825–843.
Holloway, S., Rochelle, C.A., Pearce, J.M., 1999. Geological sequestration of carbon dioxide:implications for the coal industry. Trans. Inst. Min. Metall., Sect. B 108, B19–B22.
IGME, 1980. Informe hidrogeológico del subsistema acuífero Sierra del Solorio (Sistemaacuífero 57). in Spanish.
IGME, 1982. Estudio de las manifestaciones termales de Extremadura, Salamanca,Aragón y Rioja, orientados a su posible explotación como recursos geotérmicos.Memoria. Tomo II (in Spanish).
IGME, 1987. Estudio de detalle del borde septentrional de la Sierra del Solorio (Sistemaacuífero 57). in Spanish.
Jenkins, D., Medsken, L., 1964. A brucine method for the determination of nitrate inocean, estuarine, and fresh waters. Anal. Chem. 36, 610.
Johnson, J., Nitao, J., Steefel, C., Knauss, K., 2001. Reactive transport modeling of geologicCO2 sequestration in saline aquifers: the influence of intra-aquifer shales and therelative effectiveness of structural, solubility, andmineral trapping during progradeand retrograde sequestration. First National Conference on Carbon Sequestration,Washington, DC, May 14–17, 2001.

336 L.F. Auqué et al. / Chemical Geology 268 (2009) 324–336
Johnson, J., Nitao, J., Morris, J., 2004. Reactive transport modeling of cap rock integrityduring natural and engineered CO2 storage. Carbon Dioxide Capture for Storage inDeep Geologic Formations. Elsevier.
Kaszuba, J.P., Janecky, D.R., Snow, M.G., 2003. Carbon dioxide reaction processes in amodel brine aquifer at 200 °C and 200 bars: implications for geologic sequestrationof carbon. Appl. Geochem. 18, 1065–1080.
Kaszuba, J.P., Janecky, D.R., Snow, M.G., 2005. Experimental evaluation of mixed fluidreactions between supercritical carbon dioxide and NaCl brine: relevance to theintegrity of a geologic carbon repository. Chem. Geol. 217, 277–293.
Kirste, D.M., Watson, N.M., Tingate, P.R., 2004. Geochemical modelling of CO2–water–rock interaction in the Pretty Hill Formation, Otway Basin. PESA EasternAustralasian Basins Symposium, Adelaide, 19–22 September, 2004, pp. 403–411.
Knauss, K.G., Johnson, J.W., Steefel, C.I., 2005. Evaluation of the impact of CO2, co-contaminant gas, aqueous fluid and reservoir rock interactions on the geologicsequestration of CO2. Chem. Geol. 217, 339–350.
Lagneau, V., Pipart, A., Catalette, H., 2005. Reactive transport modelling of CO2
sequestration in deep saline aquifers. Oil Gas Sci. Technol. 60 (2), 231–247.Leybourne, M.I., Betcher, R.N., McRitchie, W.D., Kaszycki, C.A., Boyle, D.R., 2009.
Geochemistry and stable isotopic composition of tufa waters and precipitates fromthe Interlake Region, Manitoba. Chem. Geol. 260 (3), 221–233.
Lopez-Chicano, M., Bouamama, M., Vallejos, A., Pulido-Bosch, A., 2001. Factors whichdetermine the hydrogeochemical behaviour of karstic springs. A case study fromthe Betic Cordilleras. Appl. Geochem. 16 (9), 1179–1192.
Meléndez, A., Meléndez, F., Portero, J., Ramírez del Pozo, J., 1985. Stratigraphy,sedimentology and paleogeography of Upper Cretaceous evaporiric-carbonateplatform in the central part of the Sierra Iberica. In: Mila, M.D., Rosel, J. (Eds.),Excursion Guidebook. International Association of Sedimentologists, 6th EuropeanRegional Meeting, Lleida, Spain, pp. 189–211.
Metz, B.; Davidson, O.; de Coninck, H; Loos, M. and Meyer, L. (eds.), 2005. IPCC SpecialReport on Carbon Dioxide Capture and Storage. Prepared by Working Group III ofthe Intergovernmental Panel on Climate Change. Cambridge University Press,Cambridge, United Kingdom and New York, NY, USA, 442 pp.
Moore, J., Adams, M., Allis, R., Lutz, S., Rauzi, S., 2005. Mineralogical and geochemicalconsequences of the long-term presence of CO2 in natural reservoirs: an examplefrom the Springerville-St. Johns field, Arizona, and New Mexico, USA. Chem. Geol.217 (3–4), 365–385.
Nemeth, K., 1963. Photometric determination of sulphate in soil extracts. Z. PflErnahr.Dung. 103, 193–196.
Noiriel, C., Bernard, D., Gouze, Ph., Thibault, X., 2005. Hydraulic properties andmicrogeometry evolution accompanying limestone dissolution by acidic water. OilGas Sci. Technol. 60, 177–192.
Nordstrom, D.K., Ball, J.W., 1989. Mineral saturation states in natural waters and theirsensitivity to thermodynamic and analytic errors. Sci. Geol. Bull. 42, 269–280.
Palandri, J.L., Rosenbauer, R.J., Kharaka, Y.K., 2005. Ferric iron in sediments as a novelCO2 mineral trap: CO2–SO2 reaction with hematite. Appl. Geochem. 20, 2038–2048.
Parkhurst, D.L., Appelo, C.A.J., 1999. User's guide to PHREEQC (Version 2), a computerprogram for speciation, batch-reaction, one-dimensional transport, and inversegeochemical calculations. Water Resources Research Investigations Report 99-4259.312 pp.
Parkhurst, D.L., Thorstenson, D.C. and Plumer, L.N. 1990. PHREEQE, a computer programfor geochemical calculations. Revised by J.V. Tisaranni & P.D. Glynn. U.S. Geol. Surv.Water Res. Inv., 80–96, 193 pp.
Pearce, J.M., Holloway, S., Wacker, H., Nelis, M.K., Rochelle, C., Bateman, K., 1996.Natural occurrences as analogues for the geological disposal of carbon dioxide.Energy Convers. Manage. 37, 1123–1128.
Plummer, L.N., Parkhurst, D.L., Thorstenson, D.C., 1983. Development of reactionmodels for ground-water systems. Geochim. Cosmochim. Acta 47, 665–686.
Plummer, L.N., Busby, J.F., Lee, R.W., Hanshaw, B.B., 1990. Geochemical modeling of theMadison aquifer in parts of Montana, Wyoming and South Dakota. Water Resour.Res. 26, 1981–2014.
Pruess, K., Garcia, J., Kovscek, T., Oldenburg, C., Rutqvist, J., Steefel, C., Xu, T.F., 2004.Code intercomparison builds confidence in numerical simulation models forgeologic disposal of CO2. Energy 29 (9–10), 1431–1444.
Reed, M., Spycher, N., 1984. Calculation of pH and mineral equilibria in hydrothermalwaters with application to geothermometry and studies of boiling and dilution.Geochim. Cosmochim. Acta 48, 1479–1492.
Raines, M.A., Dewers, T.A., 1997. Dedolomitization as driving mechanism for karstgeneration in Permian Blaine Formation, Southwestern Oklahoma. USA. Carbonate.Evaporite. 12, 24–31.
Rochelle, C.A., Bateman, K., Pearce, J.M., 2002. Geochemical interactions betweensupercritical CO2 and the Utsira Formation: an experimental study. CommissionedReport CR/02/060. British Geological Survey. 62 pp.
Sacks, L.A., Herman, J.S., Kauffman, S.J., 1995. Controls on high sulfate concentrations inthe upper Floridan aquifer in southwest Florida. Water Resour. Res. 31 (10),2541–2551.
Sanchez, J.A., Coloma, P., Perez-Garcia, A., 2004. Evaluation of geothermal flow at thesprings in Aragon (Spain), and its relation to geologic structure. Hydrol. J. 12,601–609.
Sanz, E., Yelamos, J.G., 1998. Methodology for the study of unexploited aquifers withthermal waters: application to the aquifer of the Alhama de Aragon Hot Spring.Ground Water 6, 913–923.
Savage, D., Noy, D., Miharac, M., 2002. Modelling the interaction of bentonite withhyperalkaline fluids. Appl. Geochem. 17, 207–223.
Shiraki, R., Dunn, T.L., 2000. Experimental study on water–rock interactions during CO2
flooding in the Tensleep Formation, Wyoming, USA. Appl. Geochem. 15, 265–279.Soong, Y., Goodman, A.L., McCarthy-Jones, J.R., Baltrus, J.P., 2004. Experimental and
simulation studies on mineral trapping of CO2 with brine. Energy Convers. Manage.45, 1845–1859.
Stevens, S.H., Pearce, J.M., Riggs, A.A.J, 2001. Natural analogs for geologic storage of CO2:an integrated global research program. First National Conference on CarbonSequestration. Washington D.C., May 14–17, 2001.
Tena, J.M., Auque, L.F., Gimeno, M.J., Mandado, J., 1995. Evolucion fisicoquimica ygeotermometria del sistema hidrotermal de Alhama–Jaraba (Provincia de Zar-agoza). Institucion Fernando el Catolico, Zaragoza, Spain. 178 pp. (in Spanish).
Uliana,M.N., Sharp, J.M., 2001. Tracing regional flow paths tomajor springs in Trans-PecosTexas using geochemical data and geochemical models. Chem. Geol. 179 (1–4),53–72.
Wildenborg, T., Lokhorst, A., 2005. Introduction on CO2 geological storage. Classificationof storage options. Oil Gas Sci. Technol. 60 (3), 513–515.
White, S.P., Allis, T.R.G., Moore, J., Chidsey, T., Morgan, C., Gwynn, W., Adams, M., 2005.Simulation of reactive transport of injected CO2 on the Colorado Plateau, Utah. USA.Chem. Geol. 217, 387–405.
Xu, T., Apps, J., Pruess, K., 2000. Analysis of mineral trapping for CO2 disposal in deepaquifers. Lawrence Berkeley National Laboratory Report LBNL-46992. Berkeley,California.
Xu, T.F., Apps, J.A., Pruess, K., 2004. Numerical simulation of CO2 disposal by mineraltrapping in deep aquifers. Appl. Geochem. 19 (6), 917–936.
Xu, T., Apps, J.A., Pruess, K., 2005. Mineral sequestration of carbon dioxide in asandstone–shale system. Chem. Geol. 217, 295–318.
Xu, T., Apps, J.A., Pruess, K., Yamamoto, H., 2007. Numerical modeling of injection andmineral trapping of CO2 with H2S and SO2 in a sandstone formation. Chem. Geol.242, 319–334.




















