
Hydrogen production by oxidative steam reforming of ethanol over an Ir/CeO 2 catalyst
7
Hydrogen production by oxidative steam reforming of ethanol over an Ir/CeO 2 catalyst Weijie Cai, Baocai Zhang, Yong Li, Yide Xu, Wenjie Shen * State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China Received 5 September 2006; received in revised form 16 December 2006; accepted 15 January 2007 Available online 24 January 2007 Abstract Oxidative steam reforming of ethanol over an Ir/CeO 2 catalyst was investigated. Ethanol dehydrogenation to acetaldehyde and decomposition to methane and carbon monoxide were the primary reactions at low temperatures, and complete reforming of ethanol was achieved at 773 K with hydrogen, carbon oxides and methane as the only products in the outlet stream. More importantly, stability test revealed that the Ir/CeO 2 catalyst could show rather stable catalytic performance for 60 h time-on-stream without deactivation. The improvement was attributed to the effective prevention of the sintering of the highly dispersed Ir particles through the strong interaction between Ir and CeO 2 and to the significant resistance to coke deposition due to the higher oxygen storage-release capacity of ceria. Ó 2007 Elsevier B.V. All rights reserved. Keywords: Ethanol; Hydrogen; Oxidative steam reforming; Ir/CeO 2 ; Catalytic stability 1. Introduction As addressed by the greenhouse effect and the energy efficiency, ethanol which is readily obtained from the fer- mentation of renewable biomass is regarded as one the most promising fuels for the production of hydrogen for fuel cell applications [1–3]. Steam reforming (SR), partial oxidation (POX), and oxidative steam reforming (OSR) or autothermal reforming (ATR) were proposed to be effective routes for producing hydrogen. Steam reforming of ethanol has been extensively studied over Ni, Co, Pt and Rh catalysts supported by metal oxides [1,4–7]. How- ever, this process is highly endothermic and requires pro- viding huge amount of energy, and thus it is inconvenient for the onboard production of hydrogen. Partial oxidation of ethanol was also reported to be effective for H 2 produc- tion over ceria-supported Pt and Rh catalysts [8–10]. The exothermic nature of this process provides the advantages of being a fast reaction, and is suitable for rapid speed vari- ations. But the theoretical yield of hydrogen through POX is much lower than that of SR. Consequently, oxidative steam reforming of ethanol, which is a combination of SR and POX reactions, was proposed as the most effective process by considering the energy efficiency and the hydro- gen yield. In this case, the exothermic partial oxidation of ethanol would supply the heat necessary for the endother- mic steam reforming of ethanol. By feeding ethanol, water, and oxygen in a molar ratio of 1/1.8/0.6, the overall reac- tion can be thermally neutral and the hydrogen concentra- tion could approach to about 70% in the outlet stream. CH 3 CH 2 OH þ 1:8H 2 O þ 0:6O 2 ! 4:8H 2 þ 2CO 2 DH o 298 ¼þ4:4 kJ mol 1 : Supported Ni, Pt and Rh catalysts were recently reported to be effective for this reaction [11–13]. We have recently reported that ceria-supported Ir cata- lysts could provide excellent stability and activity for steam reforming of ethanol at 650 °C, and no catalytic deactiva- tion was observed within 300 h time-on-stream with approximately stoichiometric H 2 O/ethanol ratio of 3.2 1566-7367/$ - see front matter Ó 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.catcom.2007.01.017 * Corresponding author. Tel.: +86 411 84379085; fax: +86 411 84694447. E-mail address: [email protected] (W. Shen). www.elsevier.com/locate/catcom Catalysis Communications 8 (2007) 1588–1594
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Hydrogen production by oxidative steam reforming of ethanol over an Ir/CeO 2 catalyst

www.elsevier.com/locate/catcom
Catalysis Communications 8 (2007) 1588–1594
Hydrogen production by oxidative steam reformingof ethanol over an Ir/CeO2 catalyst
Weijie Cai, Baocai Zhang, Yong Li, Yide Xu, Wenjie Shen *
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
Received 5 September 2006; received in revised form 16 December 2006; accepted 15 January 2007Available online 24 January 2007
Abstract
Oxidative steam reforming of ethanol over an Ir/CeO2 catalyst was investigated. Ethanol dehydrogenation to acetaldehyde anddecomposition to methane and carbon monoxide were the primary reactions at low temperatures, and complete reforming of ethanolwas achieved at 773 K with hydrogen, carbon oxides and methane as the only products in the outlet stream. More importantly, stabilitytest revealed that the Ir/CeO2 catalyst could show rather stable catalytic performance for 60 h time-on-stream without deactivation. Theimprovement was attributed to the effective prevention of the sintering of the highly dispersed Ir particles through the strong interactionbetween Ir and CeO2 and to the significant resistance to coke deposition due to the higher oxygen storage-release capacity of ceria.� 2007 Elsevier B.V. All rights reserved.
Keywords: Ethanol; Hydrogen; Oxidative steam reforming; Ir/CeO2; Catalytic stability
1. Introduction
As addressed by the greenhouse effect and the energyefficiency, ethanol which is readily obtained from the fer-mentation of renewable biomass is regarded as one themost promising fuels for the production of hydrogen forfuel cell applications [1–3]. Steam reforming (SR), partialoxidation (POX), and oxidative steam reforming (OSR)or autothermal reforming (ATR) were proposed to beeffective routes for producing hydrogen. Steam reformingof ethanol has been extensively studied over Ni, Co, Ptand Rh catalysts supported by metal oxides [1,4–7]. How-ever, this process is highly endothermic and requires pro-viding huge amount of energy, and thus it is inconvenientfor the onboard production of hydrogen. Partial oxidationof ethanol was also reported to be effective for H2 produc-tion over ceria-supported Pt and Rh catalysts [8–10]. Theexothermic nature of this process provides the advantages
1566-7367/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.catcom.2007.01.017
* Corresponding author. Tel.: +86 411 84379085; fax: +86 41184694447.
E-mail address: [email protected] (W. Shen).
of being a fast reaction, and is suitable for rapid speed vari-ations. But the theoretical yield of hydrogen through POXis much lower than that of SR. Consequently, oxidativesteam reforming of ethanol, which is a combination ofSR and POX reactions, was proposed as the most effectiveprocess by considering the energy efficiency and the hydro-gen yield. In this case, the exothermic partial oxidation ofethanol would supply the heat necessary for the endother-mic steam reforming of ethanol. By feeding ethanol, water,and oxygen in a molar ratio of 1/1.8/0.6, the overall reac-tion can be thermally neutral and the hydrogen concentra-tion could approach to about 70% in the outlet stream.
CH3CH2OHþ 1:8H2Oþ 0:6O2 ! 4:8H2 þ 2CO2
DH o298 ¼ þ4:4 kJ mol�1:
Supported Ni, Pt and Rh catalysts were recently reportedto be effective for this reaction [11–13].
We have recently reported that ceria-supported Ir cata-lysts could provide excellent stability and activity for steamreforming of ethanol at 650 �C, and no catalytic deactiva-tion was observed within 300 h time-on-stream withapproximately stoichiometric H2O/ethanol ratio of 3.2

W. Cai et al. / Catalysis Communications 8 (2007) 1588–1594 1589
[5]. In this work, we extend to study the performance of theIr/CeO2 catalyst for oxidative steam reforming of ethanol.
2. Experimental
2.1. Catalyst preparation
CeO2 was prepared by precipitation of cerium salt withurea in aqueous solution. 60 g (NH4)2Ce(NO3)6 and 200 gurea was dissolved into 2000 ml water and the mixturewas gradually heated to 363 K under stirring and kept atthis temperature for 27 h. After filtration and being thor-oughly washed with distilled water, the obtained solidwas dried at 373 K for 12 h, and calcined at 673 K for5 h in air. The Ir/CeO2 catalyst with Ir loading of 2 wt.%was prepared by the deposition–precipitation method.The CeO2 powder was first suspended into the aqueoussolution containing Ir precursor (H2IrCl6 Æ 6H2O) andheated to 348 K under stirring. 0.25 M Na2CO3 aqueoussolutions were then gradually added until the pH value ofthe mixture reached 9.0 and further aged for 1 h at348 K. After filtration and washing with hot water, theresulting solid was dried at 373 K overnight and finally cal-cined at 673 K for 5 h in air. Inductively coupled plasmaatomic emission spectrometer (ICP-AES) analysis revealedthat the actual amount of Ir was 2.1 wt.%, as designed.
2.2. Catalyst characterization
BET surface area measurements were conducted using aNova 4200e (Quantachrome) instrument. Prior to the mea-surements, the samples were degassed in vacuum at 573 Kfor 2 h.
Power X-ray diffraction (XRD) patterns were recordedon a Rigaku D/MAX-RB diffractor with Cu Ka radiationoperated at 40 kV and 100 mA. The mean crystallite sizesof CeO2 were calculated from the Scherrer equation.
HRTEM images were taken on a Philips Tecnai G220transmission electron microscope. Specimens were pre-pared by ultrasonically suspending the sample in ethanol.A drop of the suspension was deposited on a thin carbonfilm supported on a standard copper grid and dried in air.
Hydrogen temperature-programmed reduction (H2-TPR) measurement was performed with a conventionalsetup equipped with TCD detector. Samples (100 mg) wereloaded and pretreated at 773 K for 1 h under He flow(40 ml/min) to remove the adsorbed carbonates andhydrates. After cooling down to room temperature andintroducing the reduction agent of 5% H2/He(40 ml/min),the temperature was then programmed to rise at a rampof 10 K/min.
Oxygen storage capacity measurements were carried outin a micro-reactor coupled to a quadrupole mass spectrom-eter (Omnistar, Balzers). Prior to analysis, the samples(100 mg) were reduced with 5% H2/Ar (50 ml/min) at1273 K. Then the sample was switched to Ar flow for15 min at 1273 K, and then slowly cooled to 723 K. A
5% O2/Ar mixture (50 ml/min) was passed through thesample. The mass spectrometer was used to measure thecomposition of the effluent. Oxygen consumption was cal-culated from the curve corresponding to m/e = 32.
2.3. Temperature-programmed desorption and surfacereaction
Temperature-programmed desorption (TPD) and sur-face reaction (TPSR) experiments were carried out with amicro-reactor equipped with a quadrupole mass spectrom-eter (Omnistar, Balzers). Before analysis, the samples werereduced with 5% H2/He (40 ml/min) at 673 K for 1 h. Aftercooling down to room temperature, adsorption of ethanolwas then performed by passing He through a saturatorcontaining pure ethanol at 273 K for 1 h. The sample wasflushed with He for 30 mim, and then the catalyst washeated with a rate of 15 K/min under the flowing of He(for TPD) and 3.0% H2O/1.0% O2/He (for TPSR) with aflowrate of 40 ml/min.
2.4. Catalytic measurements
Oxidative steam reforming of ethanol was conductedwith a continuous flow fixed-bed quartz micro-reactorunder atmospheric pressure. Catalysts (300 mg) (grain sizeof 40–60 mesh) were loaded and sandwiched by two quartzwool layers. Prior to reaction, the catalyst was reducedwith 5.0% H2/He (30 ml/min) at 673 K for 1 h. Ethanolaqueous solution with a molar ratio of H2O/EtOH = 1.8:1was injected by a micro-pump into a vaporizer where it washeated to 473 K and mixed with the oxygen stream comingfrom the mass-flow controller, resulting in a typical feedgas composition of CH3CH2OH/H2O/O2 = 1.0/1.8/0.6(molar ratio). The effluent was analyzed on-line by gaschromatography equipped with TCD and FID detectors.
3. Results and discussion
3.1. Physical and chemical characteristics of the Ir/CeO2
catalyst
The BET surface areas of the CeO2 support and the Ir/CeO2 catalyst were measured to be 158 m2/g and 153 m2/g,respectively, indicating that the loading of Ir only slightlydecreased the surface area of ceria.
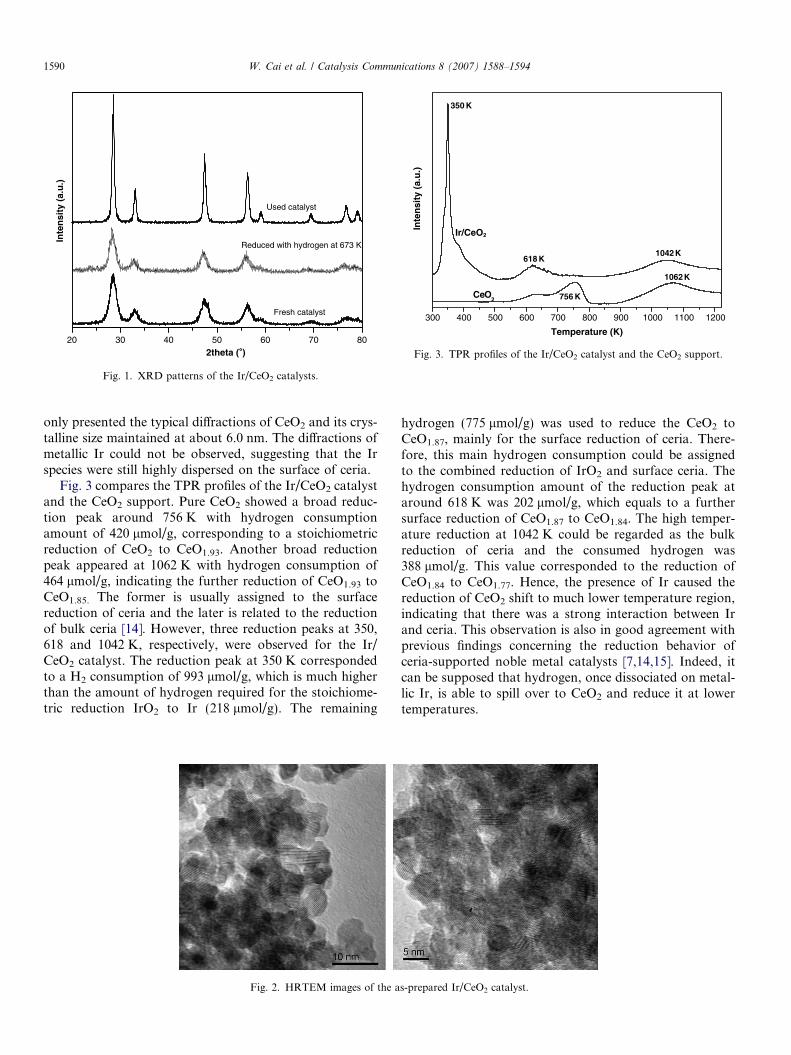
Fig. 1 shows the XRD patterns of the Ir/CeO2 catalysts.For the as-prepared catalyst, only the diffraction peaks ofCeO2 with fluorite structure was observed, and the averagecrystalline size was about 6.0 nm. No diffraction peaks dueto Ir species could be detected, indicating that Ir particlesare highly dispersed on CeO2 and they are too small tobe detected. Fig. 2 shows the HRTEM images of the Ir/CeO2 catalyst. It is clear that the Ir particles with sizes of2–3 nm were well dispersed on the surface of ceria, andthe EDX spectroscopy confirmed the presence of Ir. Whenthe Ir/CeO2 was reduced with hydrogen at 673 K, there still
20 30 40 50 60 70 80
Used catalyst
Inte
nsi
ty (
a.u
.)
2theta (°)
Fresh catalyst
Reduced with hydrogen at 673 K
Fig. 1. XRD patterns of the Ir/CeO2 catalysts.
300 400 500 600 700 800 900 1000 1100 1200
350 K
618 K
756 K
1062 K
1042 K
Ir/CeO2
CeO2
Temperature (K)
Inte
nsi
ty (
a.u
.)
Fig. 3. TPR profiles of the Ir/CeO2 catalyst and the CeO2 support.
1590 W. Cai et al. / Catalysis Communications 8 (2007) 1588–1594
only presented the typical diffractions of CeO2 and its crys-talline size maintained at about 6.0 nm. The diffractions ofmetallic Ir could not be observed, suggesting that the Irspecies were still highly dispersed on the surface of ceria.
Fig. 3 compares the TPR profiles of the Ir/CeO2 catalystand the CeO2 support. Pure CeO2 showed a broad reduc-tion peak around 756 K with hydrogen consumptionamount of 420 lmol/g, corresponding to a stoichiometricreduction of CeO2 to CeO1.93. Another broad reductionpeak appeared at 1062 K with hydrogen consumption of464 lmol/g, indicating the further reduction of CeO1.93 toCeO1.85. The former is usually assigned to the surfacereduction of ceria and the later is related to the reductionof bulk ceria [14]. However, three reduction peaks at 350,618 and 1042 K, respectively, were observed for the Ir/CeO2 catalyst. The reduction peak at 350 K correspondedto a H2 consumption of 993 lmol/g, which is much higherthan the amount of hydrogen required for the stoichiome-tric reduction IrO2 to Ir (218 lmol/g). The remaining
Fig. 2. HRTEM images of the a
hydrogen (775 lmol/g) was used to reduce the CeO2 toCeO1.87, mainly for the surface reduction of ceria. There-fore, this main hydrogen consumption could be assignedto the combined reduction of IrO2 and surface ceria. Thehydrogen consumption amount of the reduction peak ataround 618 K was 202 lmol/g, which equals to a furthersurface reduction of CeO1.87 to CeO1.84. The high temper-ature reduction at 1042 K could be regarded as the bulkreduction of ceria and the consumed hydrogen was388 lmol/g. This value corresponded to the reduction ofCeO1.84 to CeO1.77. Hence, the presence of Ir caused thereduction of CeO2 shift to much lower temperature region,indicating that there was a strong interaction between Irand ceria. This observation is also in good agreement withprevious findings concerning the reduction behavior ofceria-supported noble metal catalysts [7,14,15]. Indeed, itcan be supposed that hydrogen, once dissociated on metal-lic Ir, is able to spill over to CeO2 and reduce it at lowertemperatures.
s-prepared Ir/CeO2 catalyst.

W. Cai et al. / Catalysis Communications 8 (2007) 1588–1594 1591
3.2. Temperature-programmed desorption and surface
reaction of ethanol
Fig. 4 shows the TPD and TPSR profiles of adsorbedethanol on the Ir/CeO2 catalyst. Upon adsorption of etha-nol at room temperature, ethoxy species are readily formedon CeO2, Pt/CeO2 and Rh/CeO2 [8,16–18]. Meanwhile,partial oxidation of ethoxy species by surface oxygenanions of ceria results in the formation of acetate species.Simultaneous desorption of acetaldehyde and acetonewas observed at around 552 K in the TPD profile ofadsorbed ethanol, probably due to the dehydrogenationof ethanol to acetaldehyde and the concurrently occurreddecarbonylation of acetaldehyde to acetone. The formationof CO2 at 606 K and the appearance of methane indicatedthat the decomposition of the ethoxy species unreacted.The desorption of CH4, CO and H2 at 640-670 K couldbe attributed to the decomposition of the acetate speciesformed by the oxidation of the ethoxy species with ceria,as previously proposed [8,16].
Interestingly, the addition of water and oxygen to thecarrier gas significantly influenced the TPD features, asdemonstrated by the TPSR profile. In the presence of waterand oxygen, acetaldehyde formation was observed ataround 350 K with the simultaneous desorption of ethanol.This clearly indicated that oxidative dehydrogenation ofethanol to acetaldehyde took place using oxygen probablyfrom the ceria. Small amounts of hydrogen formation wasobserved at 370 K together with the consumption of oxy-gen in the feed. With further increasing temperature, eth-oxy and acetate species formed on the surface underwentdecomposition into methane and CO. The significant con-
400 500 600 700 800
Inte
nsi
ty (
a.u
.)
H2
CO
CH4X5
CH3COCH3X10
CO2
CH3CHO
CH3CH2OHX30
CH3CH2OHX30
660K
670K
648K
606K
552K
607K
552K
He
300 350 400 450 500 550 600
350K
350K
370K
507K
H2
CH3CHO
CO
CH4
O2
CO2
Inte
nsi
ty (
a.u
.)
Temperature (K)Temperature (K)
504K
370K
H2O/O
2/He
Fig. 4. TPD and TPSR profiles of adsorbed ethanol on the Ir/CeO2
catalyst.
sumption of oxygen at 504 K was accompanied by the for-mation of methane, CO, CO2. The large production CO2
was mainly caused by the further oxidation of carbon mon-oxide. In addition, the acetate species formed at room tem-perature and those remained unreacted are oxidized, alsoproducing more CO2. With further increasing the tempera-ture, there was no products could be detected, suggestingthat all the species adsorbed on the catalyst surface werereacted with water and oxygen in the flow gas. Comparedwith the TPD feature where only inert He was used as car-rier gas, the presence of water and oxygen greatly shiftedthe desorption and surface reaction of adsorbed ethanolto much lower temperatures. Both the oxidative dehydro-genation of adsorbed ethanol to acetaldehyde and thedecomposition of ethoxy and acetate species occurred atmuch lower temperatures.
Previous TPD and TPSR studies of adsorbed ethanol onceria-support metals revealed that two peaks correspond-ing to the formation of CH4, CO and H2 were observedat around 480 K and at high temperature region (above500 K) [8,16,17]. The first peak was attributed to the ethoxydecomposition, whereas the second was due to the acetatedecomposition. The production of CO2 stemmed from thecarbonate decomposition. In this work, only one peak ofH2, CO and CH4 were formed. The redox properties ofthe catalysts could possibly explain the differencesobserved. It was noted that the previous works used ceriawith much lower surface areas (9–63 m2/g) [8,16,17], whilewe used a high surface area ceria (158 m2/g) as support. Itwas readily confirmed that ceria with high surface areascould show excellent redox properties than these with lowsurface areas [19]. This can be further confirmed by theoxygen storage capacities (OSC) of the current catalysts.The oxygen uptake of the pure ceria support was measuredto be 421 lmol/g and the corresponding value of the Ir/CeO2 catalyst was 1054 lmol/g. As expected, these valuesare much higher than the previously reported results usingceria with lower surface areas [8,16,17]. Since the redoxability of the Ir/ceria system was greatly improved by usingceria with high surface area, the promoted adsorption andsurface reactions of ethoxy and acetate species on the cur-rent catalyst is reasonable.
3.3. Influence of reaction temperature and contact time
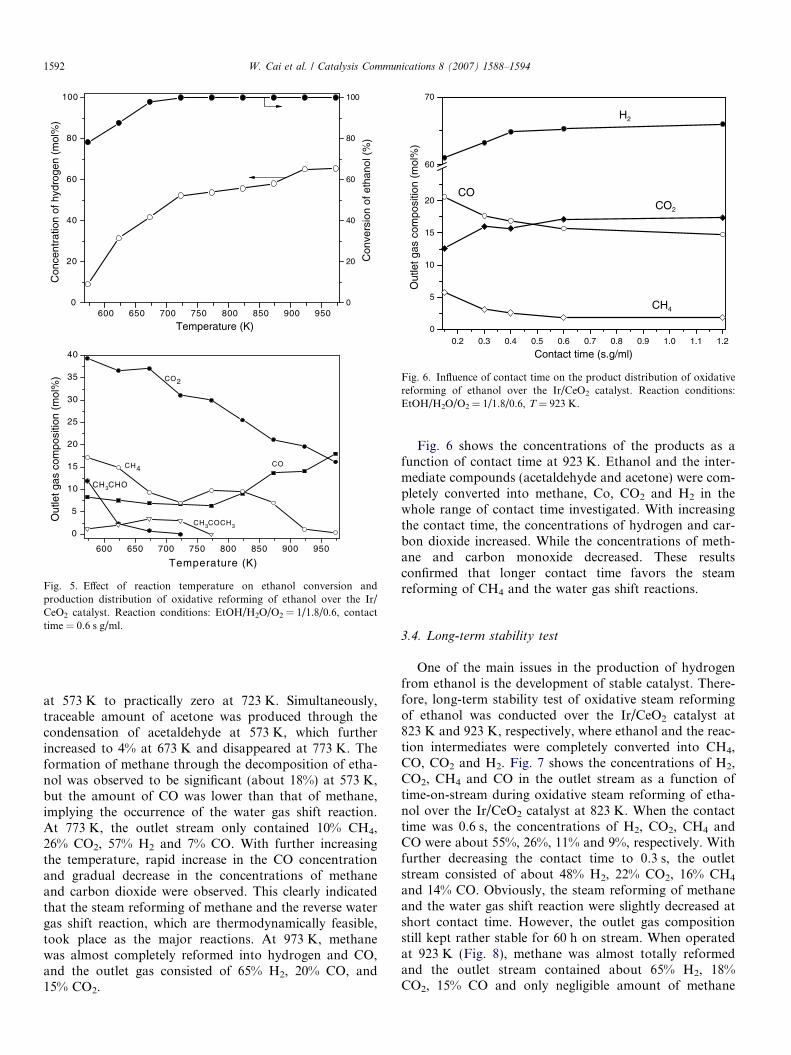
Fig. 5 shows the effect of reaction temperature on theproduct distribution of oxidative steam reforming of etha-nol over the Ir/CeO2 catalyst. It can be seen that both theconversion of ethanol and the concentration of H2
increased progressively with the temperature. With furtherincreasing the temperature up to 973 K, the hydrogen con-centration in the effluent approached to 65%. Concerningthe distribution of C-containing products, significantamount of CO2 were formed at lower temperatures, sug-gesting that ethanol was mainly oxidized into CO2. Mean-while, the concentration of acetaldehyde, formed throughdehydrogenation of ethanol, decreased rapidly from 12%
600 650 700 750 800 850 900 9500
20
40
60
80
100
0
20
40
60
80
100
Temperature (K)
Con
vers
ion
of e
than
ol (
%)
Con
cent
ratio
n of
hyd
roge
n (m
ol%
)
600 650 700 750 800 850 900 950
0
5
10
15
20
25
30
35
40
CO2
CO
CH3COCH3
CH3CHO
CH4
Temperature (K)
Out
let g
as c
ompo
sitio
n (m
ol%
)
Fig. 5. Effect of reaction temperature on ethanol conversion andproduction distribution of oxidative reforming of ethanol over the Ir/CeO2 catalyst. Reaction conditions: EtOH/H2O/O2 = 1/1.8/0.6, contacttime = 0.6 s g/ml.
0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 1.1 1.20
5
10
15
20
60
70
Out
let g
as c
ompo
sitio
n (m
ol%
)
Contact time (s.g/ml)
H2
COCO2
CH4
Fig. 6. Influence of contact time on the product distribution of oxidativereforming of ethanol over the Ir/CeO2 catalyst. Reaction conditions:EtOH/H2O/O2 = 1/1.8/0.6, T = 923 K.
1592 W. Cai et al. / Catalysis Communications 8 (2007) 1588–1594
at 573 K to practically zero at 723 K. Simultaneously,traceable amount of acetone was produced through thecondensation of acetaldehyde at 573 K, which furtherincreased to 4% at 673 K and disappeared at 773 K. Theformation of methane through the decomposition of etha-nol was observed to be significant (about 18%) at 573 K,but the amount of CO was lower than that of methane,implying the occurrence of the water gas shift reaction.At 773 K, the outlet stream only contained 10% CH4,26% CO2, 57% H2 and 7% CO. With further increasingthe temperature, rapid increase in the CO concentrationand gradual decrease in the concentrations of methaneand carbon dioxide were observed. This clearly indicatedthat the steam reforming of methane and the reverse watergas shift reaction, which are thermodynamically feasible,took place as the major reactions. At 973 K, methanewas almost completely reformed into hydrogen and CO,and the outlet gas consisted of 65% H2, 20% CO, and15% CO2.
Fig. 6 shows the concentrations of the products as afunction of contact time at 923 K. Ethanol and the inter-mediate compounds (acetaldehyde and acetone) were com-pletely converted into methane, Co, CO2 and H2 in thewhole range of contact time investigated. With increasingthe contact time, the concentrations of hydrogen and car-bon dioxide increased. While the concentrations of meth-ane and carbon monoxide decreased. These resultsconfirmed that longer contact time favors the steamreforming of CH4 and the water gas shift reactions.
3.4. Long-term stability test
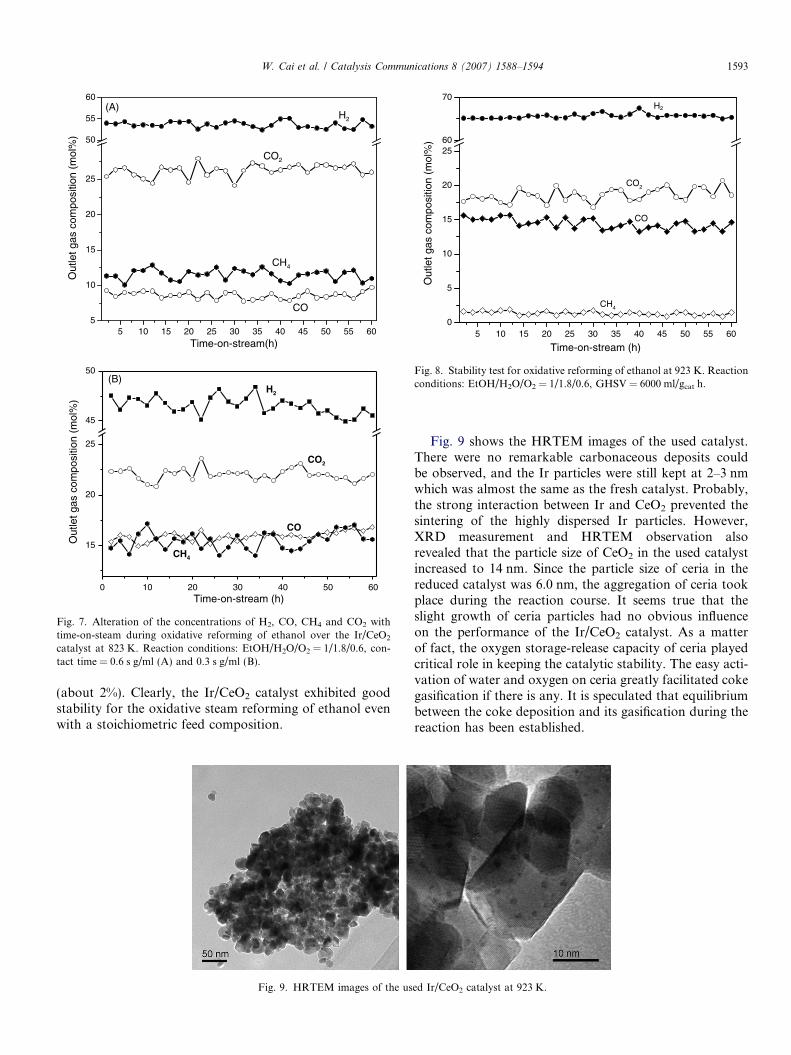
One of the main issues in the production of hydrogenfrom ethanol is the development of stable catalyst. There-fore, long-term stability test of oxidative steam reformingof ethanol was conducted over the Ir/CeO2 catalyst at823 K and 923 K, respectively, where ethanol and the reac-tion intermediates were completely converted into CH4,CO, CO2 and H2. Fig. 7 shows the concentrations of H2,CO2, CH4 and CO in the outlet stream as a function oftime-on-stream during oxidative steam reforming of etha-nol over the Ir/CeO2 catalyst at 823 K. When the contacttime was 0.6 s, the concentrations of H2, CO2, CH4 andCO were about 55%, 26%, 11% and 9%, respectively. Withfurther decreasing the contact time to 0.3 s, the outletstream consisted of about 48% H2, 22% CO2, 16% CH4
and 14% CO. Obviously, the steam reforming of methaneand the water gas shift reaction were slightly decreased atshort contact time. However, the outlet gas compositionstill kept rather stable for 60 h on stream. When operatedat 923 K (Fig. 8), methane was almost totally reformedand the outlet stream contained about 65% H2, 18%CO2, 15% CO and only negligible amount of methane
5 10 15 20 25 30 35 40 45 50 55 605
10
15
20
25
50
55
60
CO2
H2
CH4
CO
Out
let g
as c
ompo
sitio
n (m
ol%
)
Time-on-stream(h)
(A)
0 10 20 30 40 50 60
15
20
25
45
50
CH4
CO
CO2
H2
Out
let g
as c
ompo
sitio
n (m
ol%
)
Time-on-stream (h)
(B)
Fig. 7. Alteration of the concentrations of H2, CO, CH4 and CO2 withtime-on-steam during oxidative reforming of ethanol over the Ir/CeO2
catalyst at 823 K. Reaction conditions: EtOH/H2O/O2 = 1/1.8/0.6, con-tact time = 0.6 s g/ml (A) and 0.3 s g/ml (B).
5 10 15 20 25 30 35 40 45 50 55 600
5
10
15
20
2560
70
CH4
CO
H2
CO2
Out
let g
as c
ompo
sitio
n (m
ol%
)
Time-on-stream (h)
Fig. 8. Stability test for oxidative reforming of ethanol at 923 K. Reactionconditions: EtOH/H2O/O2 = 1/1.8/0.6, GHSV = 6000 ml/gcat h.
W. Cai et al. / Catalysis Communications 8 (2007) 1588–1594 1593
(about 2%). Clearly, the Ir/CeO2 catalyst exhibited goodstability for the oxidative steam reforming of ethanol evenwith a stoichiometric feed composition.
Fig. 9. HRTEM images of the us
Fig. 9 shows the HRTEM images of the used catalyst.There were no remarkable carbonaceous deposits couldbe observed, and the Ir particles were still kept at 2–3 nmwhich was almost the same as the fresh catalyst. Probably,the strong interaction between Ir and CeO2 prevented thesintering of the highly dispersed Ir particles. However,XRD measurement and HRTEM observation alsorevealed that the particle size of CeO2 in the used catalystincreased to 14 nm. Since the particle size of ceria in thereduced catalyst was 6.0 nm, the aggregation of ceria tookplace during the reaction course. It seems true that theslight growth of ceria particles had no obvious influenceon the performance of the Ir/CeO2 catalyst. As a matterof fact, the oxygen storage-release capacity of ceria playedcritical role in keeping the catalytic stability. The easy acti-vation of water and oxygen on ceria greatly facilitated cokegasification if there is any. It is speculated that equilibriumbetween the coke deposition and its gasification during thereaction has been established.
ed Ir/CeO2 catalyst at 923 K.

1594 W. Cai et al. / Catalysis Communications 8 (2007) 1588–1594
4. Conclusions
The Ir/CeO2 catalyst is significantly active and selectivefor hydrogen production by oxidative steam reforming ofethanol. The complete conversion of ethanol was achievedat 773 K with hydrogen, carbon oxides and methane as theonly products in the outlet stream. With further increasingthe reaction temperature, reforming of methane and watergas shift became the major reactions. Stability test for 60 htime-on-stream revealed that the catalyst did not show anydeactivation, and no remarkable carbon deposition wasobserved. The promotion effect of CeO2 was attributed tothe effective prevention of the sintering of the highly dis-persed Ir particles and to the strong resistance to cokedeposition based on its higher reducibility and the oxygenstorage-release capacity, which facilitates coke gasificationif there is any.
References
[1] A.N. Fatsikostas, X.E. Verykios, J. Catal. 225 (2004) 439–452.[2] D.K. Liguras, D.I. Kondarides, X.E. Verykios, Appl. Catal. B 43
(2003) 345–354.[3] F. Auoretre, C. Descorme, J. Catal. 233 (2005) 464–477.
[4] F. Frusteri, S. Freni, L. Spadaro, V. Chiodo, G. Bonura, S. Donato,S. Cavallaro, Catal. Commun. 5 (2004) 611–661.
[5] B. Zhang, X. Tang, Y. Li, W. Cai, Y. Xu, W. Shen, Catal. Commun.7 (2006) 367–372.
[6] H.V. Fajardo, L.F.D. Probst, Appl. Catal. A 306 (2006)134–141.
[7] R.M. Navarro, M.C. Alvarez-Galvan, M.C. Sanchez, F. Rosa, J.L.G.Fierro, Appl. Catal. B 55 (2005) 229–241.
[8] L.V. Mattos, F.B. Nornha, J. Catal. 233 (2005) 453–463.[9] L.V. Mattos, F.B. Nornha, J. Power Sources 145 (2005) 10–15.
[10] J.R. Salge, G.A. Deluga, L.D. Schmidt, J. Catal. 235 (2005)69–78.
[11] J. Kugai, S. Velu, C. Song, M.H. Engehard, Y.H. Chin, J. Catal. 238(2006) 430–440.
[12] F. Frusteri, S. Freni, V. Chiodo, S. Donato, G. Bonura, S. Cavallaro,Int. J. Hydrogen Energ. 31 (2006) 2193–2199.
[13] V. Fierro, O. Akdim, H. Provendier, C. Mirodatos, J. Power Sources145 (2005) 659–666.
[14] A. Trovarelli, Catal. Rev. Sci. Eng. 38 (1996) 439–445.[15] F. Sadi, D. Duprez, F. Gerard, A. Miloudi, J. Catal. 213 (2003) 226–
234.[16] A. Yee, S.J. Morrison, H. Idriss, J. Catal. 186 (1999) 279–295.[17] P.Y. Sheng, A. Yee, G.A. Bowmaker, H. Idriss, J. Catal. 208 (2002)
393–403.[18] A. Erdohelyi, J. Rasko, T. Kecskes, M. Toth, M. Domok, K. Baan,
Catal. Today 116 (2006) 367–376.[19] F. Giordano, A. Trovarelli, C. Leitenburg, M. Giona, J. Catal. 193
(2000) 273–282.




















