
Human papillomavirus-16 associated squamous cell carcinoma of the head and neck (SCCHN): A natural...
9
Human papillomavirus-16 associated squamous cell carcinoma of the head and neck (SCCHN): A natural disease model provides insights into viral carcinogenesis Robert L. Ferris a,b, * , Ivan Martinez d , Nicky Sirianni b , Jun Wang b , Andre ´s Lo ´ pez-Albaitero b , Susanne M. Gollin c , Jonas T. Johnson b , Saleem Khan d a UPCI Research Pavilion, The Hillman Cancer Center, 5117 Centre Avenue, Room 1.19d, Pittsburgh, PA 15213-1863, USA b Departments of Otolaryngology and Immunology, University of Pittsburgh Medical Center and Cancer Institute, Pittsburgh, PA, USA c Department of Human Genetics, University of Pittsburgh Graduate School of Public Health and University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA d Department of Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA Received 11 October 2004; accepted 23 November 2004 Abstract Uncertainty regarding the causality of human papillomaviruses (HPVs) in squamous cell carcinoma of the head and neck (SCCHN) necessitates better in vitro models. We carried out molecular analyses of a novel, naturally HPV-16-transformed SCCHN cell line (UPCI:SCC090) and show high copy number of HPV-16 DNA, present in a head to tail, tandemly repeated integrated state. Sequence analysis of the HPV-16 long control region (LCR) in UPCI:SCC090 revealed a deletion of 163 bp, removing a portion of the enhancer sequence, including the binding sites for the transcription factors YY1 and NF1. The E6 and E7 oncogenes of HPV-16 are expressed at high levels in this cell lines, as determined by quantitative reverse transcriptase-polymerase chain reaction (RT- PCR). UPCI:SCC090 contains wild-type tumour suppressor TP53 gene, and undetectable p53 protein, except after treatment with cisplatin, specific proteasome inhibitors or by E6 RNA interference, suggesting E6-dependent degradation of p53 in this cell line. The results of our studies are consistent with a causative role of HPV-16 in the pathogenesis of SCCHN. Ó 2005 Elsevier Ltd. All rights reserved. Keywords: Human papillomavirus; Head and neck cancer 1. Introduction Human papillomaviruses (HPVs) are small, double- stranded DNA viruses that are associated with upper and lower genital tract neoplasias [1,2]. HPV types 16 and 18 are associated with a majority of cases of HPV-induced cervical cancers [3–5]. In most benign and preneoplastic cervical lesions, the HPV DNA is present as a nuclear plasmid. However, in carcinomas the HPV DNA is usually found to be integrated into one or more human chromosomes [6], leading to upreg- ulation of the viral E6 and E7 genes and tumour pro- gression. Although there has been an increase in the number of studies dealing with the role of HPVs in squa- mous cell carcinoma of the head and neck (SCCHN) during carcinogenesis, such studies have been hampered by the inability to study the disease in vitro. Such studies require a well-characterised cellular model of HPV-asso- ciated SCCHN for molecular analyses similar to those used in the study of cervical carcinomas [7]. There were approximately 500 000 new cases of SCCHN worldwide in 2001 [8], usually associated with such risk factors as heavy consumption of alcohol and/ 0959-8049/$ - see front matter Ó 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.ejca.2004.11.023 * Corresponding author. Tel.: +1 412 623 1416; fax: +1 412 623 1415. E-mail address: [email protected] (R.L. Ferris). www.ejconline.com European Journal of Cancer 41 (2005) 807–815 European Journal of Cancer
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Human papillomavirus-16 associated squamous cell carcinoma of the head and neck (SCCHN): A natural...

European
www.ejconline.com
European Journal of Cancer 41 (2005) 807–815
Journal of
Cancer
Human papillomavirus-16 associated squamous cell carcinoma ofthe head and neck (SCCHN): A natural disease model
provides insights into viral carcinogenesis
Robert L. Ferris a,b,*, Ivan Martinez d, Nicky Sirianni b, Jun Wang b,Andres Lopez-Albaitero b, Susanne M. Gollin c, Jonas T. Johnson b, Saleem Khan d
a UPCI Research Pavilion, The Hillman Cancer Center, 5117 Centre Avenue, Room 1.19d, Pittsburgh, PA 15213-1863, USAb Departments of Otolaryngology and Immunology, University of Pittsburgh Medical Center and Cancer Institute, Pittsburgh, PA, USA
c Department of Human Genetics, University of Pittsburgh Graduate School of Public Health and University of Pittsburgh Cancer Institute,
Pittsburgh, PA, USAd Department of Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Received 11 October 2004; accepted 23 November 2004
Abstract
Uncertainty regarding the causality of human papillomaviruses (HPVs) in squamous cell carcinoma of the head and neck
(SCCHN) necessitates better in vitro models. We carried out molecular analyses of a novel, naturally HPV-16-transformed SCCHN
cell line (UPCI:SCC090) and show high copy number of HPV-16 DNA, present in a head to tail, tandemly repeated integrated state.
Sequence analysis of the HPV-16 long control region (LCR) in UPCI:SCC090 revealed a deletion of 163 bp, removing a portion of
the enhancer sequence, including the binding sites for the transcription factors YY1 and NF1. The E6 and E7 oncogenes of HPV-16
are expressed at high levels in this cell lines, as determined by quantitative reverse transcriptase-polymerase chain reaction (RT-
PCR). UPCI:SCC090 contains wild-type tumour suppressor TP53 gene, and undetectable p53 protein, except after treatment with
cisplatin, specific proteasome inhibitors or by E6 RNA interference, suggesting E6-dependent degradation of p53 in this cell line.
The results of our studies are consistent with a causative role of HPV-16 in the pathogenesis of SCCHN.
� 2005 Elsevier Ltd. All rights reserved.
Keywords: Human papillomavirus; Head and neck cancer
1. Introduction
Human papillomaviruses (HPVs) are small, double-
stranded DNA viruses that are associated with upper
and lower genital tract neoplasias [1,2]. HPV types 16and 18 are associated with a majority of cases of
HPV-induced cervical cancers [3–5]. In most benign
and preneoplastic cervical lesions, the HPV DNA is
present as a nuclear plasmid. However, in carcinomas
the HPV DNA is usually found to be integrated into
0959-8049/$ - see front matter � 2005 Elsevier Ltd. All rights reserved.
doi:10.1016/j.ejca.2004.11.023
* Corresponding author. Tel.: +1 412 623 1416; fax: +1 412 623 1415.
E-mail address: [email protected] (R.L. Ferris).
one or more human chromosomes [6], leading to upreg-
ulation of the viral E6 and E7 genes and tumour pro-
gression. Although there has been an increase in the
number of studies dealing with the role of HPVs in squa-
mous cell carcinoma of the head and neck (SCCHN)during carcinogenesis, such studies have been hampered
by the inability to study the disease in vitro. Such studies
require a well-characterised cellular model of HPV-asso-
ciated SCCHN for molecular analyses similar to those
used in the study of cervical carcinomas [7].
There were approximately 500000 new cases of
SCCHN worldwide in 2001 [8], usually associated with
such risk factors as heavy consumption of alcohol and/

808 R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815
or tobacco. Although the distribution of episomal and
integrated HPV forms in both precancerous and cancer-
ous lesions of the head and neck has not been deter-
mined, limited evidence suggests similar mechanisms to
those observed in cervical carcinomas [9–13]. Epidemio-
logical and molecular studies have shown that morethan 90% of cervical tumours exclusively harbour
integrated viral sequences [14]. Molecular studies of
HPV-associated SCCHN are necessary for a better
understanding of the physical state and potential role
of this virus in carcinogenesis, and for the development
of new, more targeted therapeutic strategies.
The carcinogenic mechanism of HPV-induced
SCCHN may differ from that of anogenital cancers.Although well-characterised in vitro cellular model sys-
tems exist for cervical carcinoma, including the CaSki,
HeLa and SiHa cell lines, no clearly defined model for
HPV-associated SCCHN exists, despite occasional re-
ports of HPV DNA in head and neck cancer cell lines.
Furthermore, the SCCHN field is complicated by the
reporting of oral or gingival keratinocyte cultures that
are transiently or stably transfected with genes encodingE6 and E7, without the presence of the other HPV genes
[15]. Thus, the ideal in vitro model for HPV-associated
SCCHN would be a naturally HPV-16-transformed cell
line derived from a de novo tumour from the oropharynx
of a SCCHN patient. Here, we report the molecular
characterisation of a recently identified HPV-16+ oro-
pharyngeal cell line (UPCI:SCC090).
2. Materials and methods
2.1. Cell lines
Cell lines were cultured as described in Ref. [16].
SiHa, C-33A, PCI-30, PCI-13 (gifts from Dr. Theresa
Whiteside, UPCI), L-18 ([17], a gift from Lou Laimins),and UPCI:SCC090: DMEM + 10% FBS (fetal bovine
serum) + 2% L-glutamine + 1% Penicillin/Streptomycin
(Invitrogen). For CaSki cells, Roswell Park Memorial
Institute (RPMI) 1640 was used.
The UPCI:SCC090 cell line was derived by the ex-
plant method from a 44-year old male (now deceased)
smoker with an oropharyngeal SCCHN arising in the
base of tongue. His tumour was staged as T2N1M0according to the 4th Edition American Joint Committee
on Cancer (AJCC) guidelines, and the histology was
moderately to poorly differentiated invasive squamous
cell carcinoma with basaloid features.
2.2. HPV-positivity, Southern blotting and the
identification of integrated HPV-16 DNA
DNA from the following cell lines was isolated by
phenol–chloroform–isoamyl alcohol (25:24:1) extraction
and ethanol-precipitation [18]: UPCI:SCC90; the cervi-
cal carcinoma cell line, CaSki, containing tandemly inte-
grated copies of HPV-16 DNA; and L-18, a human
keratinocyte cell line containing episomal copies of
HPV-18 DNA [17]. HPV-positivity in the UP-
CI:SCC090 cell line was tested using M09/M11 PCRprimers which amplify an approximately 450-bp con-
served region of the L1 gene of HPVs [19]. HPV-16-spe-
cific polymerase chain reaction (PCR) was done by
amplifying a 477-bp region of the E6 gene of this virus
using 5 0-ATGCACCAAAAGAGAACTGC-3 0 as the
forward primer, and 5 0-TTACAGCTGGGTTTCTC-
TAC-3 0 as the reverse primer. The glyceraldehyde-3-
phosphate dehydrogenase (G3PDH) gene was amplifiedas a control using the forward primer 5 0-
ACCACAGTCCATGCCATCAC-3 0 and the reverse
primer 5 0-TCCACCACCCTGTTGCTGTA-3 0 which
amplify a 556-bp DNA region. PCR for the L1, E6
and G3PDH were performed in a 50 ll volume contain-
ing 50 mM KCl, 10 mM Tris (pH 8.3), 1.5 mM MgCl2,
0.01% gelatin, 200 lM deoxynucleoside triphosphate
(dNTP) mix, 0.4 lM of each primer and 2.5 units ofthe Taq DNA polymerase. The DNA was denatured
at 94 �C for 5 min, followed by 40 PCR amplification cy-
cles that consisted of denaturation (94 �C, 1 min),
annealing (55–60 �C, 1 min) and extension (72 �C, 2
min). An additional extension step of 72 �C for 5 min
was included at the end of the reaction. The PCR-ampli-
fied DNA was analysed by agarose gel electrophoresis
[20]. For Southern blot analysis, 5 lg of CaSki and UP-CI:SCC090 DNA was digested with three different
restriction endonucleases. BglII does not cleave the
HPV-16 genome, BamHI cleaves it once and KpnI
cleaves it at two sites. For the L-18 cell line, the DNA
was treated with BglII which does not cleave the HPV-
18 DNA, EcoRV (which cleaves it once) and BamHI
that cleaves the HPV-18 DNA twice. The digested
DNA was electrophoresed on a 0.7% agarose gel andthe Southern blots were probed with a 32P-labelled plas-
mid containing the complete HPV-16 genome as de-
scribed in Ref. [18]. The blots were subjected to
autoradiography at �80 �C. The identity of HPV-16 in
UPCI:SCC90 was confirmed by PCR amplification of
portions of the E2, E6, E7 and L1 genes followed by
automated DNA sequencing of the amplified products.
The following HPV-16-specific primers were used forthe PCR amplification. E2 (1,228 bp product) forward
primer 5 0-GGAAATCCAGTGTATGAGCTTAATG-
3 0 and reverse primer 5 0-GTAATGTTGTGGATG-
CAGTATCAAG-3 0; E6/E7 (735 bp product) forward
primer 5 0-ATGCACCAAAAGAGAACTGC-3 0 and re-
verse primer 5 0-TGCCCATTAACAGGTCTTCC-3 0.
The MY09/MY11 primers [19] were used for the ampli-
fication of the L1 gene and primers described above forthe amplification of the E6 region of HPV-16. The PCR
conditions were the same as described above, except for

R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815 809
the E2 gene for which extension reactions were carried
out for 2.5 min. DNA sequencing was carried out using
an Automated Applied Biosystems PRISM 3100 Genet-
ic Analyzer.
2.3. Identification of the region deleted in the HPV-16
DNA in the UPCI:SCC090 cell line
The long control region (LCR) of the HPV-16 DNA
was amplified by PCR. The sequences of the primers
used were: 5 0-TTTTGGCACAAAATGTGTTTTT-3 0
for the forward primer (HPV-16 positions 7470–7491)
and 5 0-GCACAGAGCTG CAAACAACTAT for the
downstream primer (positions 150–171). The reactionmixtures contained 200 lM of each deoxynucleoside tri-
phosphate (dNTP), 200 ng of UPCI:SCC090 DNA, 1
lM of each primer, and 5 units of the Pfu polymerase
(Stratagene, La Jolla, CA). The conditions of amplifica-
tion were as follows: 94 �C for 5 min; 94 �C for 1 min, 60
�C for 1 min, and 72 �C for 2 min for 40 cycles; and 72
�C for 7 min. The PCR-amplified DNA was isolated by
gel electrophoresis and subjected to automated DNAsequencing.
2.4. Quantitative real-time PCR (qPCR)
Relative HPV-16 E6 and E7 DNA copy number in
the UPCI:SCC090 cells was determined by type-specific
primers/probe and conditions [21]. Control DNA quan-
tification was performed, amplifying a series of micro-satellite repeats (QuMA) [22], and using a serially
diluted HPV-16 E6-encoding plasmid. Comparison
was made with CaSki and SiHa (see Table 1). Input
copy numbers were determined using HPV-16 E6-con-
taining plasmid DNA, and unknown samples norma-
lised to E6 input plasmid amounts. Relative expression
of E6 and E7 was calculated using the delta CT method
described previously in Ref. [22]: (Relative expres-sion = 2�DCT; where DCT = CT(Target gene) � CT(QuMA)).
While this equation is exactly accurate only for quanti-
tative real-time RT-PCR (qRT-PCR) reactions that
are 100% efficient, it provides an estimate, particularly
when compared with internal known reagents, such as
the E6-encoding plasmid and the CaSki cell line, which
Table 1
HPV-16 qPCR in SCC90 cells
Cell line [E6]a [E7]a
SCC90 91 171
CaSki 105 249
SiHa 0.46 1.0
qPCR, quantitative real-time PCR.a Relative to control QuMa DNA amplification, using 2�DCT
method.
has been quantified in terms of the integrated HPV
DNA present.
2.5. Isolation of RNA and RT-PCR analysis
Expression of the HPV-16 E2 and E6 genes wasinvestigated by reverse transcriptase (RT)-PCR analy-
sis. RNA was isolated using the ULTRASPEC RNA
isolation system (Biotecx) according to the manufac-
turer�s protocol. Before DNA synthesis, RNA was
treated with DNaseI, amplification grade (Invitrogen)
for 15 min at room temperature to avoid DNA con-
tamination. DNaseI was then inactivated by the addi-
tion of 25 mM ethylene diamine tetraacetic acid(EDTA) followed by incubation at 65 �C for 10
min. The lack of contaminating DNA was confirmed
by the failure of PCR amplification in reactions con-
taining the Taq polymerase, but lacking the reverse
transcriptase. The cDNA synthesis was performed at
37 �C for 1 h in a final volume of 20 ll using 1 lgof total RNA template, 0.5 lg of oligo (dT)15, 10
mM dNTPs, 30 U of RNase inhibitor and 200 unitsof MMLV reverse transcriptase. Expression of the
HPV-16 E6 gene was studied using the primer pair
described above. To detect expression of the HPV-16
E2 gene, PCR amplification was done using 5 0-
AAAGTGGACATTACAAGACGTTAGC-3 0 for the
forward primer and 5 0-GTGAGCTGTTAAATG-
CAGTGAGG-3 0 for the reverse primer that are ex-
pected to generate a 554-bp product. The expressionof the cellular G3PDH gene was used as a control.
PCR was performed in a 50 ll volume containing
50 mM KCl, 10 mM Tris (pH 8.3), 1.5 mM MgCl2,
0.01% gelatin, 200 lM dNTP mix, 0.4 lM of each pri-
mer and 2.5 units of the Taq DNA polymerase. The
DNA was denatured at 94 �C for 5 min, followed
by 40 PCR amplification cycles that consisted of dena-
turation (94 �C, 1 min), annealing (55–60 �C, 1 min)and extension (72 �C, 2 min). An additional extension
step of 72 �C for 5 min was included at the end of the
reaction. The PCR products were analysed by electro-
phoresis on 1% agarose gels.
2.6. Quantitative real-time RT-PCR
Reverse transcription was performed with randomhexamer primers and Superscript � (Invitrogen Corp.)
as described previously in Ref. [9]. As described in
Ref. [21], qRT-PCR was then carried out on the Applied
Biosystems 7700 Sequence Detection Instrument at 95
�C for 12 min, PCR was performed at 95 �C for 15 s,
60 �C for 60 s. Relative expression of the target gene:
endogenous control gene, b-glucuronidase (GUS), was
calculated using the delta CT method described previ-ously: (Relative expression = 2�DCT; where DCT =
CT(Target gene) � CT(GUS)) [20].

Fig. 1. Polymerase chain reaction (PCR) analysis of UPCI:SCC090
and CaSki DNA. Amplification of human papillomavirus (HPV)-16
genes L1, detected by consensus L1 primers, and E6, amplified using
type-specific HPV-16 primers as described in Section 2.
810 R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815
2.7. RNA interference (siRNA) for HPV-16 E6
UPCI:SCC090 cells were plated in 24-well plates in
media, as above. At 50% confluence, cells were transfec-
ted with Oligofectamine (Invitrogen) following the man-
ufacturer�s protocol. siRNA specific for E6, or irrelevant(scrambled sequence) were ordered pre-duplexed and
ready for transfection from Dharmacon. Two HPV-16
E6 primers were mixed together using a total concentra-
tion of 60 pmol/well. siRNA target sequences are as fol-
lows: E6-1 5 0-AAGAGCUGAAACAACUAUAC-3 0,
E6-2 5 0-AACUGCGACGUGAGGUAUAUG-3 0, IR-
REL 5 0-AAGCACACACGUAGACAUUCG-3 0. Cells
were assayed for HPV16-E6 gene activity 48 h aftertransfection.
2.8. Determination of p53 expression and stability
Cells were treated with the indicated drugs and con-
centrations (described below) before lysis using l% Non-
idet P-40 (NP-40) and protease inhibitor cocktail
(Promega). Cisplatin was used at a final concentrationof 40 lM. Lactacystin [23] was used at a final concentra-
tion of 50 lM; MG-132 was used at a final concentra-
tion of 50 lM. Drug treatments were carried out for 6
h prior to cell lysis, sodium dodecyl sulphate-polyacryl-
amide gel electrophoresis (SDS-PAGE) electrophoresis
and Western blotting. Cellular extracts (5–10 ll totalprotein) were electrophoresed and immunoblotted using
anti-p53 DO-7 mAb (BD Pharmingen) and secondaryAb linked to horse radish peroxidase (HRP) or fluoro-
chromes (Amersham Pharmacia). Equal protein loading
was determined using the Pierce bicinchoninic acid
(BCA) protein quantitation reagent kit and confirmed
by blotting the polyvinylidene fluoride (PVDF) mem-
brane with anti-b-actin Ab (Sigma). The intensity of
protein bands was determined by densitometric analysis
of the immunoblots.
3. Results
3.1. UPCI:SCC090 cells contain integrated HPV-16
DNA
PCR analysis using the consensus M09/M11 primers
for the L1 gene showed the presence of a 450-bp band,
indicating the presence of HPV DNA in the UP-
CI:SCC090 cell line (Fig. 1). DNAs from CaSki (HPV-
16-positive) and C-33A (HPV-negative) were used as po-sitive and negative controls, respectively. In addition,
the G3PDH gene was amplified as a positive control
for all the cell lines. Since HPV-16 is the most common
type found in SCCHN, HPV-16-specific PCR reactions
were performed using DNA from this cell line as the
template. PCR analysis using primers for the E6 gene
showed that this cell line contained HPV-16 DNA
(Fig. 1). Primers specific for HPV-18 E6 gene failed to
amplify any DNA from the UPCI:SCC090 cell line(Fig. 1). To confirm that this cell line contains HPV-16
DNA, specific PCR primers were used to amplify por-
tions of the HPV-16 E2, E6, E7 and L1 genes. Auto-
mated DNA sequencing of 400–500 bp regions of the
above PCR products showed that the DNA corre-
sponded to that of HPV-16 (data not shown). Further-
more, the amplified DNA sequence resembled the
European E-G131G variant of HPV-16 [24]. These re-sults confirmed that the UPCI:SCC090 cell line contains
HPV-16 DNA.
We then carried out Southern blot analysis to deter-
mine the physical state of the HPV-16 DNA in the UP-
CI:SCC090 cell line. In these studies, we used DNA
from CaSki which contains tandem head to tail repeats
of integrated HPV-16 DNA [25] and the L-18 cell line
that contains episomal HPV-16 DNA [17] as controls.When the Southern blot was hybridised to a HPV-16
probe, uncut UPCI:SCC090 DNA and DNA cleaved
with BglII that does not cleave HPV-16 generated a sin-
gle, slow-migrating band similar to that observed with
the CaSki DNA (Fig. 2). By contrast, uncut L-18
DNA contained two major bands corresponding to the
supercoiled (SC) and open-circular (OC) forms of the
HPV-18 DNA which cross-hybridises to the HPV-16probe (Fig. 2). These results demonstrated that as is
the case with CaSki cells, the UPCI:SCC090 cell line
contains integrated HPV-16 DNA. When the UP-
CI:SCC090 DNA was treated with BamHI that cleaves
the HPV-16 DNA once, a major 7.8 kb band was ob-
served (Fig. 2). Since the HPV-16 DNA is present in
an integrated state in this cell line, the 7.8 kb band is pre-
sumably generated from the release of unit-length DNA
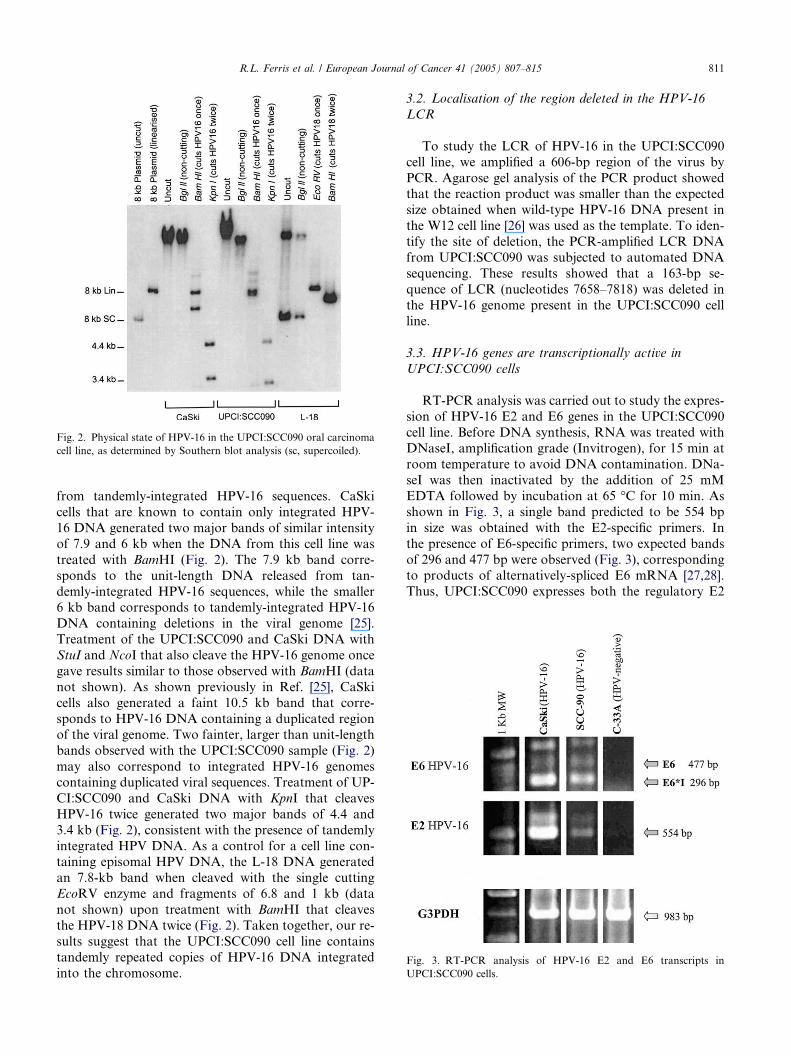
Fig. 2. Physical state of HPV-16 in the UPCI:SCC090 oral carcinoma
cell line, as determined by Southern blot analysis (sc, supercoiled).
Fig. 3. RT-PCR analysis of HPV-16 E2 and E6 transcripts in
UPCI:SCC090 cells.
R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815 811
from tandemly-integrated HPV-16 sequences. CaSki
cells that are known to contain only integrated HPV-
16 DNA generated two major bands of similar intensity
of 7.9 and 6 kb when the DNA from this cell line was
treated with BamHI (Fig. 2). The 7.9 kb band corre-
sponds to the unit-length DNA released from tan-
demly-integrated HPV-16 sequences, while the smaller6 kb band corresponds to tandemly-integrated HPV-16
DNA containing deletions in the viral genome [25].
Treatment of the UPCI:SCC090 and CaSki DNA with
StuI and NcoI that also cleave the HPV-16 genome once
gave results similar to those observed with BamHI (data
not shown). As shown previously in Ref. [25], CaSki
cells also generated a faint 10.5 kb band that corre-
sponds to HPV-16 DNA containing a duplicated regionof the viral genome. Two fainter, larger than unit-length
bands observed with the UPCI:SCC090 sample (Fig. 2)
may also correspond to integrated HPV-16 genomes
containing duplicated viral sequences. Treatment of UP-
CI:SCC090 and CaSki DNA with KpnI that cleaves
HPV-16 twice generated two major bands of 4.4 and
3.4 kb (Fig. 2), consistent with the presence of tandemly
integrated HPV DNA. As a control for a cell line con-taining episomal HPV DNA, the L-18 DNA generated
an 7.8-kb band when cleaved with the single cutting
EcoRV enzyme and fragments of 6.8 and 1 kb (data
not shown) upon treatment with BamHI that cleaves
the HPV-18 DNA twice (Fig. 2). Taken together, our re-
sults suggest that the UPCI:SCC090 cell line contains
tandemly repeated copies of HPV-16 DNA integrated
into the chromosome.
3.2. Localisation of the region deleted in the HPV-16
LCR
To study the LCR of HPV-16 in the UPCI:SCC090
cell line, we amplified a 606-bp region of the virus by
PCR. Agarose gel analysis of the PCR product showedthat the reaction product was smaller than the expected
size obtained when wild-type HPV-16 DNA present in
the W12 cell line [26] was used as the template. To iden-
tify the site of deletion, the PCR-amplified LCR DNA
from UPCI:SCC090 was subjected to automated DNA
sequencing. These results showed that a 163-bp se-
quence of LCR (nucleotides 7658–7818) was deleted in
the HPV-16 genome present in the UPCI:SCC090 cellline.
3.3. HPV-16 genes are transcriptionally active in
UPCI:SCC090 cells
RT-PCR analysis was carried out to study the expres-
sion of HPV-16 E2 and E6 genes in the UPCI:SCC090
cell line. Before DNA synthesis, RNA was treated withDNaseI, amplification grade (Invitrogen), for 15 min at
room temperature to avoid DNA contamination. DNa-
seI was then inactivated by the addition of 25 mM
EDTA followed by incubation at 65 �C for 10 min. As
shown in Fig. 3, a single band predicted to be 554 bp
in size was obtained with the E2-specific primers. In
the presence of E6-specific primers, two expected bands
of 296 and 477 bp were observed (Fig. 3), correspondingto products of alternatively-spliced E6 mRNA [27,28].
Thus, UPCI:SCC090 expresses both the regulatory E2

812 R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815
gene and the E6 oncogene of HPV-16, consistent with
the presence of tandemly integrated copies of the virus
in this cell line. Further, these results show that integra-
tion of the HPV-16 DNA in this cell line does not inter-
rupt the viral E2 gene.
3.4. Quantitative real-time PCR to quantitate relative
copies of HPV-16 E6 DNA
We performed qPCR for HPV-16 E6 and E7 gene to
quantify viral DNA copy numbers present in UP-
CI:SCC090 cells, compared with known high (CaSki)
and low (SiHa) copy number cervical carcinoma cells.
The cycle threshold (Ct) for UPCI:SCC090 cells was sig-nificantly lower than SiHa cells, which are known to
contain 1–2 copies of integrated HPV-16 genomic cop-
ies. Back-calculation using quantitatively amplified ser-
ies of microsatellite repeats as described in Ref. [22],
and developing a standard curve based on a serially di-
luted HPV-16 E6 encoding plasmid, enabled the relative
determination of HPV-16 copy numbers in UP-
CI:SCC090 cells. Thus, UPCI:SCC090 contains approx-imately 100–150 copies of HPV-16 DNA (Table 1),
similar to CaSki cells (known to contain several hundred
copies per host cellular genome).
3.5. Real-time quantitative reverse transcription PCR
demonstrates high expression of HPV-16 E6 and E7
oncogenes
We measured E6 and E7 mRNA levels in UP-
CI:SCC090 cells to evaluate the likelihood that HPV-
16 contributed to carcinogenesis in these cells. As shown
in Fig. 4, high-level E6 and E7 expression was observed.
Consistently higher (P < 0.005) levels of E6 and E7
expression were observed in UPCI:SCC090 cells com-
pared with CaSki and SiHa cells, indicating the likely
importance of these gene products in UPCI:SCC090carcinogenesis.
Fig. 4. Expression of HPV-16 E6 and E7 in UPCI:SCC090 cells by
quantitative reverse-transcription RT-PCR (qRT-PCR) relative to the
control gene, b-glucuronidase (GUS). *Relative to control QuMa
DNA amplication, using 2�DCT method.
3.6. Genotype of TP53 in UPCI:SCC090 cells
Although TP53 is mutated in some HPV-16 associ-
ated carcinomas, the mutation frequency is lower than
in non-HPV-associated tumours, and this correlates
with viral gene expression [29]. Consistent with this no-tion, we performed cDNA sequence analysis for the
TP53 gene in UPCI:SCC090 cells (data not shown). Di-
rect fluorogenic sequence analysis of both coding and
non-coding strands, with subsequent BLAST alignment
demonstrated that UPCI:SCC090 cells contain wild-
type TP53 in exons 2 through 11. Using differential
restriction digestion [30], we confirmed that UP-
CI:SCC090 cells are heterozygous for proline and argi-nine at codon 72.
3.7. Reversal of p53 degradation by anti-neoplastic and
anti-viral drug treatment
Based on the wild-type sequence of TP53 in UP-
CI:SCC090 cells, and E6-induced, ubiquitin-mediated
proteolysis of p53, we postulated that antineoplasticchemotherapeutic effects might be mediated through res-
toration of p53 expression. Fig. 5(a) shows an immuno-
blot of UPCI:SCC090 lysates, indicating that p53 is
essentially undetectable in these cells, likely due to the
enhanced ubiquitination and proteasomal degradation
induced by HPV-16 E6 protein associating with the
E6-AP (ubiquitin E3 ligase). After cisplatin treatment
(40 lM for 6 h), reversal of this E6 effect and p53 expres-sion was clearly observed in UPCI:SCC090 and CaSki
cells (data not shown). A similar effect was also seen
with proteasome inhibitors lactacystin [23] and leuci-
nal–leucinal–norleucinal (referred to as LLnL or MG-
132).
3.8. siRNA knockdown of HPV-16 E6 results in
accumulation of p53
To clarify the carcinogenic effect of HPV-16 E6 on
p53 expression, UPCI:SCC090 cells were transfected
with siRNA against HPV-16 E6. E6 RNA levels were re-
duced up to 70% as shown by qRT-PCR (data not
shown). We found that repression of HPV-16 E6 expres-
sion led to the accumulation of p53 in these cells (Fig.
5(b)). The level of p53 accumulation observed in siR-NA-treated UPCI:SCC090 cells was lower than that
seen after treatment with potent proteasome inhibitors
(Fig. 5(a)), likely due to incomplete knockdown of E6
expression by siRNA.
4. Discussion
Epidemiological correlation between mucosatropic
HPV infection and a subgroup of SCCHN is most com-

Fig. 5. Recovery of p53 expression by treatment of UPCI:SCC090
cells with the chemotherapeutic agent cisplatin or by proteasome
inhibitors (a) or by downregulation of HPV-16 E6 expression by RNA
interference (b). (a) Recovery of p53 from HPV-l6-induced degrada-
tion. Western blot analysis of the p53 protein in UPCI:SCC090 and
CaSki cells using DO-7 anti-p53 monoclonal antibodies after treat-
ment with cisplatin (40 lM for 6 h; lanes 2 and 6) and after treatment
with specific proteasome inhibitors lactacystin (10 lM; lanes 3 and 7)
or MG-132 (50 lM, lanes 4 and 8). (b) Western blot analysis of the p53
protein after transient transfection of UPCI:SCC090 cells with a 21-
mer oligonucleotides designed for short hairpin RNA interference of
the E6 gene (siRNA), see Section 2 (1FN-c, interferon-c).
R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815 813
pelling for the oropharyngeal site (tonsil or base of ton-
gue). In general, tobacco exposure is inversely associated
with the presence of HPV-infection in SCCHN, but
these features are not absolute. The determinants of
each factor in the aetiology of SCCHN has been difficult
to identify due to a lack of natural in vitro cellular mod-
els of this disease. Cell lines such as we have described in
this report are critical to our understanding of this dis-ease and comparing features between HPV-associated
SCCHN and cervical carcinoma are limited. Reports
of propagation of naturally occurring, HPV-trans-
formed SCCHN cell lines have been infrequent, with
few cell lines maintaining their HPV DNA [31,32]. Epi-
somal HPV DNA, by itself, is generally expected to be
an unlikely aetiological factor for SCCHN, due to the
low-level expression of the transforming E6 and E7 pro-teins. However, the presence of mutations in HPV LCR
could result in an increased expression of these onco-
genes [25,31,33–35] and this may lead to cellular trans-
formation. Integrated viral DNA that leads to high
levels of E6/E7 expression is much more likely to be
present in naturally transformed, HPV-16-infected
SCCHN cell lines. Because a causal association of
HPV-induced carcinogenesis is uncertain, the need forin vitro cellular models is paramount to enable the char-
acterisation of HPV-associated SCCHN.
Recently, it has been shown by fluorescent in situ
hybridisation (FISH) and restriction-site PCR that the
UPCI:SCC090 cell line contains integrated HPV-16
DNA in chromosomes 3, 6, 9q and 13q [33]. Further-
more, the integration sites were found to be generally lo-
cated in common fragile sites. Our quantitative PCRstudies demonstrate that the HPV-16 DNA is present
at approximately 100–150 copies per cell in UP-
CI:SCC090 (Table 1). Southern blot analysis of uncut
UPCI:SCC090 DNA with an HPV-specific probe
showed the presence of a high molecular weight DNA,
but no signal corresponding to the SC or OC forms of
HPV-16 DNA was observed (Fig. 2). Since treatment
of UPCI:SCC090 DNA with BamHI that cleavesHPV-16 DNA only once generated a single band of
7.8 kb (Fig. 2), our results suggest that the viral genome
is integrated in tandem copies in a head to tail orienta-
tion in this cell line. Such an arrangement of integrated
sequences would release unit length HPV-16 DNA upon
treatment with a single-cutting enzyme. This interpreta-
tion is consistent with the observations with the CaSki
cell line which also generates an 8 kb band upon treat-ment with BamHI (Fig. 2). However, CaSki cells also
generated a second major band of approximately 6 kb,
which represents tandem integration of a second, deleted
form of HPV-16 DNA in this cell line [25]. Deletions
and rearrangements of HPV DNA are frequently ob-
served in cervical carcinoma cell lines containing inte-
grated HPV DNA [33–35]. We used DNA from the L-
18 cell line containing episomal copies of HPV-18DNA as a control. The results obtained with the L-18
DNA were consistent with the presence of episomal
HPV-18 DNA in this cell line (Fig. 2).
UPCI:SCC090 cells contain integrated HPV-16 DNA
and express high levels of E6, which is important for cel-
lular transformation [36,37]. These observations are
consistent with a causative role for HPV-16 in the carci-
nogenesis of SCCHN. Interestingly, UPCI:SCC090 alsoexpressed the viral E2 gene (Fig. 3). This suggests that,
as is the case in CaSki cells, the E2 gene is not disrupted
or deleted in the UPCI:SCC090 cell line. Previous stud-
ies have identified several mutations in the LCR of
HPVs in cases of cervical carcinoma, especially those
in which the HPV genome is present in an episomal
form [1,34]. PCR amplification of the HPV-16 LCR in
the UPCI:SCC090 cell line followed by DNA sequenc-ing showed that a 163-bp sequence (nucleotides 7656–
7818) was deleted in the HPV-16 genome present in
the UPCI:SCC090 cell line. This region includes a por-
tion of the HPV-16 enhancer sequence, two YY1 bind-
ing sites and one NF1 binding site [34,35]. Regions
within the HPV LCR are known to negatively regulate
HPV-16 E6 and E7 expression. For example, binding
of the YY1 factor to its binding site has been suggestedto interfere with the formation of the transcription initi-
ation complex at the P97 promoter for the E6/E7 genes

814 R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815
[35]. Since the HPV-16 genome in the UPCI:SCC090 cell
line contains a deletion of the binding sites for the YY1
and NFI proteins, our results suggest that this may re-
sult in the upregulation of E6/E7 expression, even in
the presence of viral E2 protein. The additional se-
quences missing in the HPV-16 LCR in UPCI:SCC090cells may also play a negative role in E6/E7 oncogene
expression. Integrated copies of HPV-16 are also present
in the tumour from which the UPCI:SCC090 cell line
was derived (data not shown), suggesting that expres-
sion of the E6/E7 oncogenes from the integrated state
may have contributed to the development of the tumour
from which this cell line was derived.
Phenotypic and genotypic analysis shows that UP-CI:SCC090 represents an interesting and valid model
of HPV-16-associated SCCHN. It is derived from a de
novo oropharyngeal tumour, a site with roughly 50%
prevalence of HPV-16 DNA in numerous studies
[38,39]. This cell line contains wild-type p53 which is
rapidly degraded in untreated cells, but is recoverable
by the action of specific proteasome inhibitors and E6
knockdown by siRNA (Fig. 5), consistent with the func-tional p53 inactivation by the HPV-16 E6 protein. Bio-
logically active concentrations of cisplatin, a commonly
used chemotherapeutic drug in SCCHN, showed similar
recovery of p53 expression (Fig. 5(a)). Although cervical
cancer may not be as sensitive to this drug as SCCHN,
this fact raises the possibility that these diseases
although generally associated with the same viral sub-
types, may develop through distinct aetiological mecha-nisms. Such issues may now be tested in vitro comparing
features of cell lines derived from individuals with each
disease.
Because of the mounting molecular epidemiological
data in favour of the presence an HPV-16-associated
subtype of SCCHN, particularly in the oropharynx,
the UPCI:SCC090 cell line promises to be quite valuable
as an in vitro model system to test similarities betweenHPV-associated SCCHN, HPV-negative SCCHN, and
HPV-induced cervical carcinoma. Further studies are
expected to clarify the aetiological role of HPV-16 in a
clinically significant subset of SCCHN.
Conflict of interest statement
None declared.
Acknowledgements
This work was supported by the Hillman Founda-
tion, American Head and Neck Society, The American
Academy of Otolaryngology/Head and Neck Surgery,the Stout Family fund for Clinical Research, a pilot
grant from the Oral Cancer Center at the University
of Pittsburgh and the University of Pittsburgh Eye
and Ear Foundation.
References
1. zur Hausen H. Papillomaviruses and cancer: from basic studies to
clinical application. Nat Rev Cancer 2002, 2(5), 342–350.
2. Herrero R, Castellsague X, Pawlita M, et al. Human papilloma-
virus and oral cancer: The International Agency for Research on
Cancer multicenter study. J Natl Cancer Inst 2003, 95(23),
1772–1783.
3. Scheffner M, Romanczuk H, Munger K, et al. Functions of
human papillomavirus proteins. Curr Top Microbiol Immunol
1994, 186, 83–99.
4. Huibregtse JM, Scheffner M, Howley PM. E6-AP directs the HPV
E6-dependent inactivation of p53 and is representative of a family
of structurally and functionally related proteins. Cold Spring Harb
Symp Quant Biol 1994, 59, 237–245.
5. Braakhuis BJ, Snijders PJ, Keune WJ, et al. Genetic patterns in
head and neck cancers that contain or lack transcriptionally
active human papillomavirus. J Natl Cancer Inst 2004, 96(13),
998–1006.
6. Park JS, Hwang ES, Park SN, et al. Physical status and
expression of HPV genes in cervical cancers. Gynecol Oncol
1997, 65(l), 121–129.
7. Ke LD, Adler-Storthz K, Mitchell MF, et al. Expression of
human papillomavirus E7 mRNA in human oral and cervical
neoplasia and cell lines. Oral Oncol 1999, 35(4), 415–420.
8. Greenlee RT, Hill-Harmon MB, Murray T, et al. Cancer statis-
tics, 2001. CA Cancer J Clin 2001, 51(l), 15–36.
9. Bercovich JA, Centeno CR, Aguilar OG, et al. Presence and
integration of human papillomavirus type 6 in a tonsillar
carcinoma. J Gen Virol 1991, 72(Pt 10), 2569–2572.
10. Scheurlen W, Stremlau A, Gissmann L, et al. Rearranged HPV 16
molecules in an anal and in a laryngeal carcinoma. Int J Cancer
1986, 38(5), 671–676.
11. Steenbergen RD, Hermsen MA, Walboomers JM, et al. Inte-
grated human papillomavirus type 16 and loss of heterozygosity at
11q22 and 18q21 in an oral carcinoma and its derivative cell line.
Cancer Res 1995, 55(22), 5465–5471.
12. Venuti A, Manni V, Morello R, et al. Physical state and
expression of human papillomavirus in laryngeal carcinoma
and surrounding normal mucosa. J Med Virol 2000, 60(4),
396–402.
13. Maitland NJ, Cox MF, Lynas C, et al. Detection of human
papillomavirus DNA in biopsies of human oral tissue. Brit J
Cancer 1987, 56(3), 245–250.
14. Vernon SD, Unger ER, Miller DL, et al. Association of human
papillomavirus type 16 integration in the E2 gene with poor
disease-free survival from cervical cancer. Int J Cancer 1997, 74(l),
50–56.
15. Yoo GH, Washington J, Oliver J, et al. The effects of exogenous
p53 overexpression on HPV-immortalized and carcinogen trans-
formed oral keratinocytes. Cancer 2002, 94(1), 159–166.
16. Heo DS, Snyderman C, Gollin SM, et al. Biology, cytogenetics,
and sensitivity to immunological effector cells of new head and
neck squamous cell carcinoma lines. Cancer Res 1989, 49(18),
5167–5175.
17. Frattini MG, Lim HB, Doorbar J, et al. Induction of human
papillomavirus type 18 late gene expression and genomic ampli-
fication in organotypic cultures from transfected DNA templates.
J Virol 1997, 71(9), 7068–7072.
18. Maniatis T, Fritsch EF, Sambrook J. Molecular cloning : a
laboratory manual. 2nd ed. Cold Spring Harbor, NY, Cold
Spring Harbor Laboratory, 1989.

R.L. Ferris et al. / European Journal of Cancer 41 (2005) 807–815 815
19. Manos MM, Ting Y, Wright DK, et al. Use of polymerase chain
reaction amplification for the detection of genital human papil-
lomavirusesCancer cells. pp. 209–214.
20. Wang J, Xi L, Hunt JL, et al. Expression pattern of chemokine
receptor 6 (CCR6) and CCR7 in squamous cell carcinoma of the
head and neck identifies a novel metastatic phenotype. Cancer Res
2004, 64(5), 1861–1866.
21. Wang-Johanning F, Lu DW, Wang Y, et al. Quantitation of
human papillomavirus 16 E6 and E7 DNA and RNA in residual
material from ThinPrep Papanicolaou tests using real-time poly-
merase chain reaction analysis. Cancer 2002, 94(8), 2199–2210.
22. Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of
FMR1 mRNA in carrier males: a new mechanism of involvement
in the fragile-X syndrome. Am J Hum Genet 2000, 66(1), 6–15.
23. Fenteany G, Standaert RF, Reichard GA, et al. A beta-lactone
related to lactacystin induces neurite outgrowth in a neuroblas-
toma cell line and inhibits cell cycle progression in an osteosar-
coma cell line. Proc Natl Acad Sci USA 1994, 91(8), 3358–3362.
24. Yamada TMM, Peto J, Greer CE, et al. Human papillomavirus
type 16 sequence variation in cervical cancers: a worldwide
perspective. J Virol 1997, 71(3), 2463–2472.
25. Meissner JD. Nucleotide sequences and further characterization
of human papillomavirus DNA present in the CaSki, SiHa and
HeLa cervical carcinoma cell lines. J Gen Virol 1999, 80(Pt 7),
1725–1733.
26. Stanley MA, Browne HM, Appleby M, et al. Properties of a non-
tumorigenic human cervical keratinocyte cell line. Int J Cancer
1989, 43(4), 672–676.
27. Fuchs PG, Pfister H. Transcription of papillomavirus genomes.
Intervirology 1994, 37(3–4), 159–167.
28. Mansur CP, Androphy EJ. Cellular transformation by papillo-
mavirus oncoproteins. Biochim Biophys Acta 1993, 1155(3),
323–345.
29. Dai M, Clifford GM, le Calvez F, et al. Human papillomavirus
type 16 and TP53 mutation in oral cancer: matched analysis of the
IARC multicenter study. Cancer Res 2004, 468–471.
30. Shen H, Zheng Y, Sturgis EM, et al. P53 codon 72 polymorphism
and risk of squamous cell carcinoma of the head and neck: a case-
control study. Cancer Lett 2002, 183(2), 123–130.
31. Shillitoe EJ, Noonan S. Strength and specificity of different gene
promoters in oral cancer cells. Oral Oncol 2000, 36(2), 214–220.
32. Sacks PG. Cell, tissue and organ culture as in vitro models to
study the biology of squamous cell carcinomas of the head and
neck. Cancer Metast Rev 1996, 15(1), 27–51.
33. Ragin CC, Reshmi SC, Gollin SM. Mapping and analysis of
HPV16 integration sites in a head and neck cancer cell line. Int J
Cancer 2004, 110(5), 701–709.
34. Rose BR, Thompson CH, Zhang J, et al. Sequence variation in
the upstream regulatory region of HPV 18 isolates from cervical
cancers. Gynecol Oncol 1997, 66(2), 282–289.
35. O�Connor MJ, Tan SH, Tan CH, et al. YY1 represses human
papillomavirus type 16 transcription by quenching AP-1 activity. J
Virol 1996, 70(10), 6529–6539.
36. Banks L, Crawford L. Analysis of human papillomavirus type 16
polypeptides in transformed primary cells. Virology 1988, 165(l),
326–328.
37. Crook T, Morgenstern JP, Crawford L, et al. Continued expres-
sion of HPV-16 E7 protein is required for maintenance of the
transformed phenotype of cells co-transformed by HPV-16 plus
EJ-ras. EMBO J 1989, 8(2), 513–519.
38. Mork J, Lie AK, Glattre E, et al. Human papillomavirus
infection as a risk factor for squamous-cell carcinoma of the head
and neck. N Engl J Med 2001, 344(15), 1125–1131.
39. Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal
association between human papillomavirus and a subset of head
and neck cancers. J Natl Cancer Inst 2000, 92(9), 709–720.




















