
Understanding the Invitation Acceptance in Agent-initiated ...
Upload
independentCategory
view
3download
0
Human breast carcinoma desmoplasia is PDGF initiated
Zhi-Ming Shao1, Mai Nguyen2,3 and Sanford H Barsky*,1,3
1Department of Pathology, UCLA School of Medicine, Los Angeles, California, CA 90024, USA; 2Department of Surgery UCLASchool of Medicine, Los Angeles, California, CA 90024, USA; 3UCLA/Revlon Breast Center, UCLA School of Medicine, LosAngeles, California, CA 90024, USA
The desmoplastic response to human breast carcinoma isa host myo®broblast-mediated collagenous responseexhibiting synergistic e�ects on tumor progression.Although many paracrine interactions between breastcarcinoma cells and myo®broblasts have been character-ized, the event(s) which initiate desmoplasia haveremained unde®ned. Our studies utilized c-rasH trans-fected MCF-7 cells which overexpress ras p2l and whichare weakly tumorigenic in ovariectomized nude mice.The xenografts are desmoplastic and comprised of 30%myo®broblasts and 60 mg/g of interstitial collagen. Insitu hybridization studies of these xenografts reveal astromal gene expression pattern (stromelysin-3, IGF-IIand TIMP-1) identical to that observed in human tumordesmoplasia. 17-b estradiol increases c-rasH MCF-7growth but abolishes desmoplasia. c-rasH MCF-7 in vitroconstitutively produce myo®broblast mitogenic activitywhich competes with PDGF in a receptor binding assay.This myo®broblast mitogenic activity is unaltered by 17-b estradiol/tamoxifen pretreatment in vitro. Transfectionof c-rasH MCF-7 with a PDGF-A dominant negativemutant, 1308, produced by site-directed mutagenesis(serine?cysteine129) reduces both homo- and heterodimersecretion of PDGF by as much as 90% but does notinterfere with the secretion of other growth factors.Clones with low PDGF, though tumorigenic, are non-desmoplastic. Our results suggest that breast carcinoma-secreted PDGF is the major initiator of tumordesmoplasia. Oncogene (2000) 19, 4337 ± 4345.
Keywords: PDGF; desmoplasia; breast carcinoma;paracrine growth factors; dominant-negative mutants;c-rasH
Introduction
The desmoplastic response to tumor invasion is apoorly understood host mediated collagenous responseresponsible for the `hard lump' appearance of manyhuman cancers, most notable of which is human breastcancer (Barsky and Gopalakrishna, 1987b). Thedesmoplastic response is neither a pathognomonic signnor a sine quan non of malignancy being present inmany benign neoplasms as well as absent in manymalignant tumors. The desmoplastic response is nearlyan exclusively human occurrence, however, being quiterare among spontaneous and transplantable animaltumors and absent in most human tumoral xenograftsin athymic `nude' mice (Povlsen, 1980). During the past
two decades, several mechanistic hypotheses for humantumor desmoplasia have been advanced which includean immune cytokine mechanism, a blood and tissuecoagulation mechanism, and a paracrine growth factormechanism (Dvorak et al., 1981a,b; Peres et al., 1987).The di�culties inherent in proving any of theseproposed mechanisms involve the lack of a goodexperimental model where the desmoplastic responseoccurs and can be successfully perturbated. Of allhuman cancers, the desmoplastic response is mostcommon in carcinomas of the breast and yet well-established ER positive and ER negative human breastcarcinoma cell lines uniformly produce no appreciabledesmoplastic response in athymic mice (Povlsen, 1980).This latter observation is especially surprising since themajority of these cell lines were derived from pleurale�usions whose primary tumor of origin was desmo-plastic (Barsky and Gopalakrishna, 1987a). Since onefundamental di�erence between autochthonous humantumors and human tumoral xenografts in immuno-compromised mice is the comparatively rapid growthof the latter, we hypothesized that one explanation forthe absence of desmoplasia in human xenografts wasthe rapid growth of the tumor cells which mightoverwhelm any host stromal response. We alsoconsidered another possible explanation: that thebreast carcinoma cell lines had lost their desmoplasia-inducing phenotype with increasing in vitro passagethrough the loss of a critical gene product(s) such as aparacrine growth factor(s). It had been demonstratedthat v-rasH and c-rasH, when transfected into MCF-7cells, resulted in estrogen-independent tumorigenicity(Kasid et al., 1985), augmented autocrine and para-crine growth factor production (Dickson et al., 1987),and slow tumoral growth in the absence of exogenousestrogen (Sommers et al., 1990). We reasoned that ifdesmoplasia were indeed both paracrine growth factordependent and dependent on a slow tumoral growthrate, that utilizing these rasH recombinant MCF-7 cellsmight prove e�ective at establishing a human tumorxenograft model of desmoplasia which could besubsequently studied and perturbated to dissect outthe molecular mechanisms involved.
Results
Cell lines
None of the cell lines (W9, W7, neo MCF-7) examinedexhibited a desmoplastic response in the presence ofexogenous estrogen. The tumors which emerged in thepresence of estrogen were very cellular and devoid of atumor stroma. In the absence of estrogen, tumorigeni-
Oncogene (2000) 19, 4337 ± 4345ã 2000 Macmillan Publishers Ltd All rights reserved 0950 ± 9232/00 $15.00
www.nature.com/onc
*Correspondence: SH BarskyReceived 13 May 2000; revised 15 June 2000; accepted 4 July 2000

city and growth were both markedly decreased in all ofthe lines (Table 1). In the absence of estrogen, the W9line however exhibited marked tumor desmoplasia. Itstumor nodules consisted of a striking myo®broblast/®broblast stromal collagenous component (Table 1 andFigure 1a,b). Stromal cells comprised 30% of thetumor (Figure 1c) and collagen was 60 mg/g byhydroxyproline determination (Table 1). These valuesfar exceeded those of the other xenografts (P50.001).In situ hybridization studies of the desmoplastic W9xenograft revealed strong signals of stromal geneexpression (3 ± 10-fold increase over sense background)of stromelysin-3, TIMP-1 and IGF-II (P50.01). Thepattem of stromal gene expression for stromelysin-3and TIMP-1 was zonal, being highest in the stromalregions immediately around the carcinoma cells (Figure1d). For IGF-II, a more di�use signal was observedwithin the stroma. ER negative lines such as the MDA-MB-231 and the MDA-MB-468 grew as very cellulartumors in the absence of estrogen. All other breastcarcinoma xenografts then except for the W9 grew ascellular tumors with little or no stroma formation andno desmoplasia (Figure 1e,f). Estrogen treatment of theW9 stimulated its growth and tumorigenicity butabolished its desmoplasia (Table 1) (P50.01). TheW9, W7 and neo MCF-7 lines exhibited c-rasH p21stability over multiple passages and multiple in vitro/invivo experiments. The W9 line continued to express 40-fold increased levels of c-rasH p21 whereas the W7 andneo MCF-7 lines continued to express normal or lowlevels of c-rasH p21 (Figure 2a).
Myofibroblast mitogenic activity
All of the cell lines examined including both ER positive(MCF-7, T47D), ER negative (MDA-MB-231 andMDA-MB-468) and c-rasH recombinant lines (W9, W7and neo MCF-7) expressed signi®cant myo®broblastmitogenic activity (Figure 2b). The only exception wasthe MDA-MB-157 line, a line derived from a non-desmoplastic human medullary carcinoma (P50.01).The W9 line exhibited signi®cant mitogenic activity butwas not the highest among the lines examined (Figure2b). There was no correlation between c-rasH p21 levelsand myo®broblast mitogenic activity (P40.1). Themyo®broblast mitogenic activity of the W9 line was notaltered by either 17b-estradiol or tamoxifen pretreatment(P40.1) (Figure 2c). The myo®broblast mitogenicfraction on gel ®ltration revealed a major peak (peak 2)
of activity at MW 30 000 (Figure 2d). This peakcompeted with [125I]PDGF in a PDGF receptor bindingassay (Figure 2e). Western blot of both W9 CM (Figure3a) and this fraction con®rmed the presence of PDGF(PDGF-AB, PDGF-AA, PDGF-BB).
Transfection studies
Transfection of c-rasH MCF-7 with a PDGF-A dominantnegative mutant, 1308, produced by site-directedmutagenesis (serine?cysteine129) reduced both homo-and heterodimer secretion of PDGF by as much as 90%(clone 2) (P50.01) (Figures 3a and 4a,b) but did notinterfere with the secretion of other growth factors(P40.1). After transfection of the clones we analysed themRNA by RNase protection analysis by ®rst hybridizingwith a probe from wild-type PDGF-A spanning theregion mutated in the 1308 construct (bases 345 ± 518 ofthe wild-type PDGF-A cDNA). Evidence of the presenceof the 1308 (serine?cysteine129) mRNA was based on thepresence of additional smaller molecular weight bandsproduced by RNase cleavage. Substitution of serine forcysteine129 is thought to destabilize PDGF-A subunitswithin the cell. Suppression of wild-type PDGF geneexpression occurs because the mutant PDGF subunitsdimerize with wild-type subunits (both PDGF-A andPDGF-B) to form inactive or unstable heterodimers.These heterodimers are di�cult to demonstrate withinthe cell because of their instability. The important pointis that these heterodimers are not able to be secreted andthe wild-type homodimerization products (PDGF-AA,PDGF-BB) and the wild-type heterodimerization pro-ducts (PDGF-AB) which the mutant PDGF-A subunitblocks in a trans-dominant fashion are also not secreted.So the best assay for expression of the mutant proteins isthe demonstration that there is no or very low levels ofsecreted PDGF in conditioned media. That is what wehave demonstrated for clone 2 (Figures 3a and 4). Thesecretion of TGFa, TFG-b1, IGF-I and IGF-II whichcharacterized the W9 parent was unaltered, however, inthe PDGF-A transfectants including clone 2 (Figure4c,d,e,f). The PDGF-A W9 clone (clone 2) maintainedboth its estrogen independence and estrogen responsive-ness and exhibited similar degrees of tumorigenicity andgrowth as its W9 parent (P40.1) (Table 1). This clonewith low PDGF, though tumorigenic, was nondesmo-plastic (P50.01) (Figure 3b,c). These results suggest thatbreast carcinoma-secreted PDGF and not host PDGF(derived from platelets or stromal cells) nor any other
Table 1 The desmoplastic response in recombinant MCF-7 lines and the e�ects of estrogena
Tumor Size (diameter) MyofibroblastLine 17b Estradiol incidence (%) (8 weeks) (mm) composition (%) Collagen (mg/g)
PDGFb-W9 + 100 12+4c 5+5d 55e
PDGFb-W9 7 20 3+2 5+4 55W9 + 100 15+3 5+5 5W9 7 20 3+2 30+10 60W7 + 100 14+3 3+2 2.2W7 7 0 ± ± ±Neo + 100 14+4 3+2 1.8Neo 7 0 ± ± ±
aResults of groups of 10 mice are depicted. The W9 line in the absence of exogenous estrogen produced only a 20% incidence of tumors butthese were consistently desmoplastic. With exogenous estrogen, tumorigenicity and growth rate signi®cantly increased but desmoplasiadramatically decreased. No other transfected lines gave signi®cant desmoplasia. bPDGF-A dominant negative mutant (clone 2) of W9 parent.cValues represent mean+standard deviation. dValues represent mean+standard deviation. eValues represent pooled cases of each group
Desmoplasia initiation by PDGFZ-M Shao et al
4338
Oncogene
growth factor elaborated by breast carcinoma cells(TGFa, TFG-b1, IGF-I or IGF-II) is the initiator oftumor desmoplasia.
Discussion
These ®ndings support a non-immune constitutiveparacrine growth factor mechanism for tumor desmo-plasia and support a hypothesis to predict the
occurrence of tumor desmoplasia. Tumors that donot secrete signi®cant PDGF would not be expected toexhibit tumor desmoplasia. Tumors that do secretePDGF but have PDGF receptors may utilize PDGF asan autocrine growth factor and not have any morePDGF available to act in a paracrine fashion. Finallytumors that secrete PDGF and lack PDGF receptorsbut which grow very rapidly may outgrow the nascent®broblast and myo®broblast response that their PDGFelicits and exhibit no desmoplasia. This hypothesis
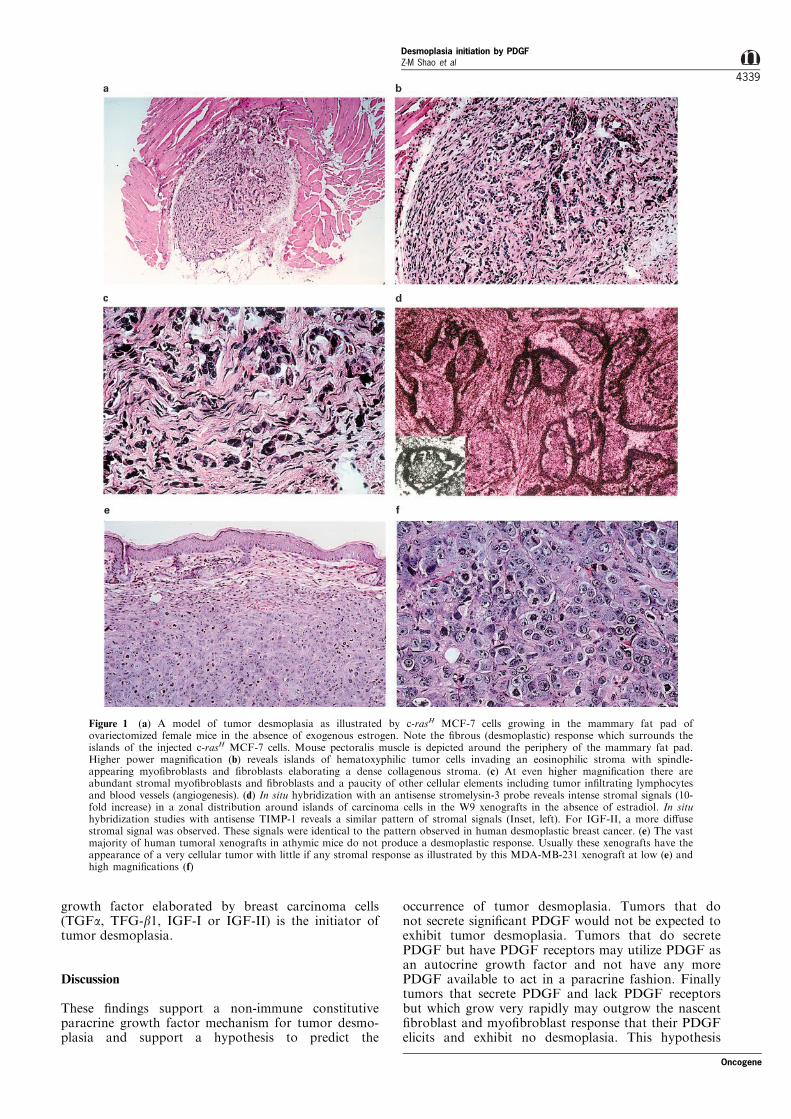
Figure 1 (a) A model of tumor desmoplasia as illustrated by c-rasH MCF-7 cells growing in the mammary fat pad ofovariectomized female mice in the absence of exogenous estrogen. Note the ®brous (desmoplastic) response which surrounds theislands of the injected c-rasH MCF-7 cells. Mouse pectoralis muscle is depicted around the periphery of the mammary fat pad.Higher power magni®cation (b) reveals islands of hematoxyphilic tumor cells invading an eosinophilic stroma with spindle-appearing myo®broblasts and ®broblasts elaborating a dense collagenous stroma. (c) At even higher magni®cation there areabundant stromal myo®broblasts and ®broblasts and a paucity of other cellular elements including tumor in®ltrating lymphocytesand blood vessels (angiogenesis). (d) In situ hybridization with an antisense stromelysin-3 probe reveals intense stromal signals (10-fold increase) in a zonal distribution around islands of carcinoma cells in the W9 xenografts in the absence of estradiol. In situhybridization studies with antisense TIMP-1 reveals a similar pattern of stromal signals (Inset, left). For IGF-II, a more di�usestromal signal was observed. These signals were identical to the pattern observed in human desmoplastic breast cancer. (e) The vastmajority of human tumoral xenografts in athymic mice do not produce a desmoplastic response. Usually these xenografts have theappearance of a very cellular tumor with little if any stromal response as illustrated by this MDA-MB-231 xenograft at low (e) andhigh magni®cations (f)
Oncogene
Desmoplasia initiation by PDGFZ-M Shao et al
4339
a
b
c d
e
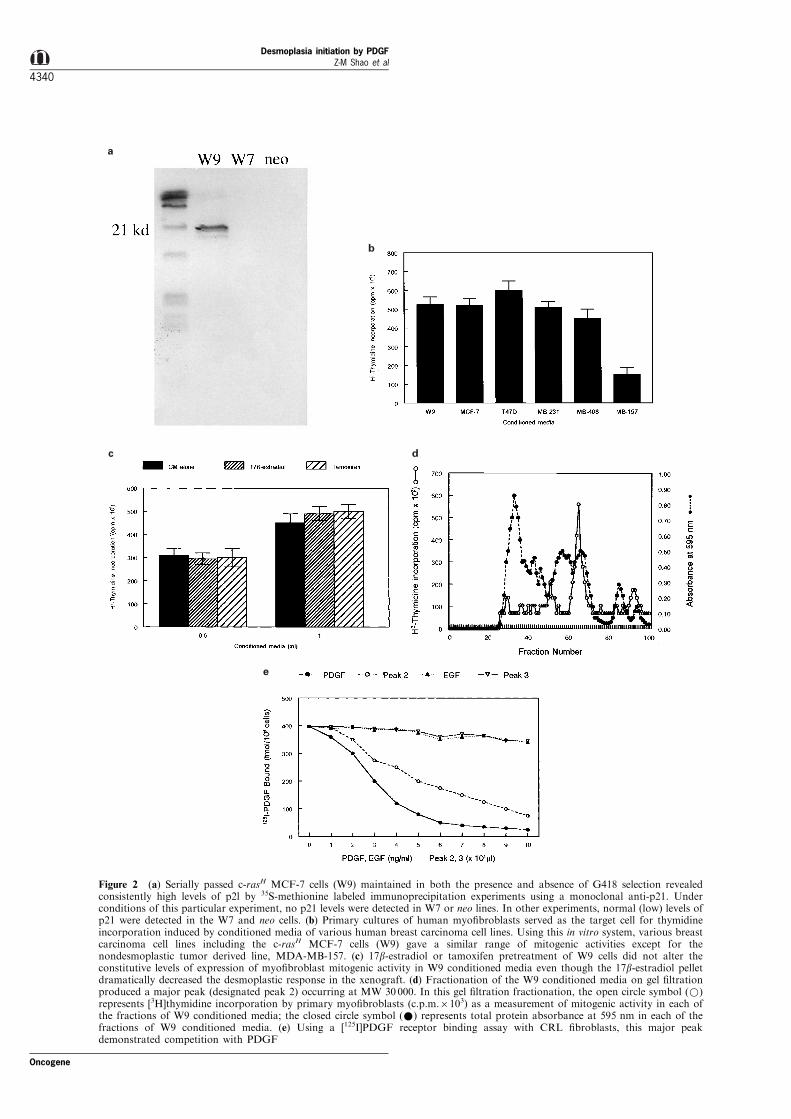
Figure 2 (a) Serially passed c-rasH MCF-7 cells (W9) maintained in both the presence and absence of G418 selection revealedconsistently high levels of p2l by 35S-methionine labeled immunoprecipitation experiments using a monoclonal anti-p21. Underconditions of this particular experiment, no p21 levels were detected in W7 or neo lines. In other experiments, normal (low) levels ofp21 were detected in the W7 and neo cells. (b) Primary cultures of human myo®broblasts served as the target cell for thymidineincorporation induced by conditioned media of various human breast carcinoma cell lines. Using this in vitro system, various breastcarcinoma cell lines including the c-rasH MCF-7 cells (W9) gave a similar range of mitogenic activities except for thenondesmoplastic tumor derived line, MDA-MB-157. (c) 17b-estradiol or tamoxifen pretreatment of W9 cells did not alter theconstitutive levels of expression of myo®broblast mitogenic activity in W9 conditioned media even though the 17b-estradiol pelletdramatically decreased the desmoplastic response in the xenograft. (d) Fractionation of the W9 conditioned media on gel ®ltrationproduced a major peak (designated peak 2) occurring at MW 30000. In this gel ®ltration fractionation, the open circle symbol (*)represents [3H]thymidine incorporation by primary myo®broblasts (c.p.m.6103) as a measurement of mitogenic activity in each ofthe fractions of W9 conditioned media; the closed circle symbol (*) represents total protein absorbance at 595 nm in each of thefractions of W9 conditioned media. (e) Using a [125I]PDGF receptor binding assay with CRL ®broblasts, this major peakdemonstrated competition with PDGF
Desmoplasia initiation by PDGFZ-M Shao et al
4340
Oncogene
would explain why tumor desmoplasia is rare in animaltumors or human tumoral xenografts because all ofthese grow very rapidly; why tumor desmoplasia isuncommon in human sarcomas because these expressPDGF receptors (Malik et al., 1991); and whydesmoplasia is absent in medullary breast cancerbecause these have low levels of PDGF myo®broblastmitogenic activity as exhibited, for example, by theMDA-MB-157. Since our desmoplastic model of tumordesmoplasia occurs in an immunocompromised mouse,immune e�ector cells and their cytokines do not seemnecessary for the initiation or maintenance of thedesmoplastic response.
As has been stated, desmoplasia is an uncommonoccurrence in animal tumors and in most humantumoral xenografts, in part, because these tumors growrapidly. Desmoplasia has however been observed intwo sets of animal tumors, bile duct carcinomas (lines 1and 10) in guinea pigs (Dvorak et al., 1979) and ratcolonic carcinomas (Lieubeau et al., 1994). Interest-ingly, in each of these sets, there was both a verymalignant and a less malignant counterpart. In theguinea pig bile duct carcinomas (Dvorak et al., 1979),the less malignant line, line 1, was desmoplastic. In therat colonic carcinoma lines, the PROb line, whichformed progressive tumors was non-desmoplasticwhereas the REGb line which developed regressivetumors was desmoplastic (Lieubeau et al., 1994). Inboth of these examples, the inverse relationshipbetween growth and desmoplasia was observed. Ourmodel further illustrates this inverse relationship in theslightly di�erent setting of a human tumoral xenograft.
The ®ndings of our study certainly do not exclude arole for other paracrine growth factors (tumor cell orhost derived) or a role for host in¯ammatory cells orhematopoietic cells and their growth factors, ie.,platelet-derived PDGF-AB, PDGF-AA or PDGF-BBin contributing to tumor desmoplasia but excludesthese mediators as major initiators of the response. Inprevious studies we and others (Barsky and Gopalak-rishna, 1987a; Peres et al., 1987), have shown thatdiverse ER positive and ER negative breast carcinomacell lines express PDGF ®broblast/myo®broblast mito-genic activity. In the present study we go the next stepand show that by reducing only tumor-derived PDGF(and not circulating PDGF from platelets) by adominant negative approach, we can abolish tumordesmoplasia. Other investigators have used thisdominant negative PDGF approach to suppresstransformation and growth of human astrocytomacells dependent on PDGF as an autocrine growthfactor (Shamah et al., 1993). Here we interfere withPDGF's role as a paracrine growth factor to suppresstumor desmoplasia. No doubt other paracrine growthfactors produced by the recruited ®broblasts/myo®bro-blasts such as IGF-II and other tumor-derived growthfactors such as TGFa and TGF-b1 exert mutualactions on their respective targets (Cullen et al.,1991). This is in evidence particularly in direct in situhybridization studies of human tumor desmoplasia(Singer et al., 1995) and in in vitro subculturingexperiments of human desmoplastic ®broblasts (Cullenet al., 1991). Without tumor-derived PDGF to initiatethe response, however, the response does not occur inour human xenograft model. Equally important is thedemonstration that growth stimulation of the c-rasH
W9 cells by 17b-estradiol abolishes the desmoplasticresponse not by decreasing the production of PDGF(since 17b-estradiol exerts no e�ect on W9 CMmyo®broblast mitogenic activity in vitro) but byincreasing the growth rate of the W9 cells themselveswhich then outgrow the murine ®broblasts (Table 1).Since the W9 cells express ER-a whereas the murinestromal cells do not, it would be anticipated that theW9 cells would respond to 17b-estradiol by increasedgrowth and decreased desmoplasia. If the Nottingham,Bloom and Richardson grading system is applied toour c-rasH MCF-7 xenografts, the desmoplastic xeno-grafts (W9 in the absence of estrogen) (Figure 1a,b,c)
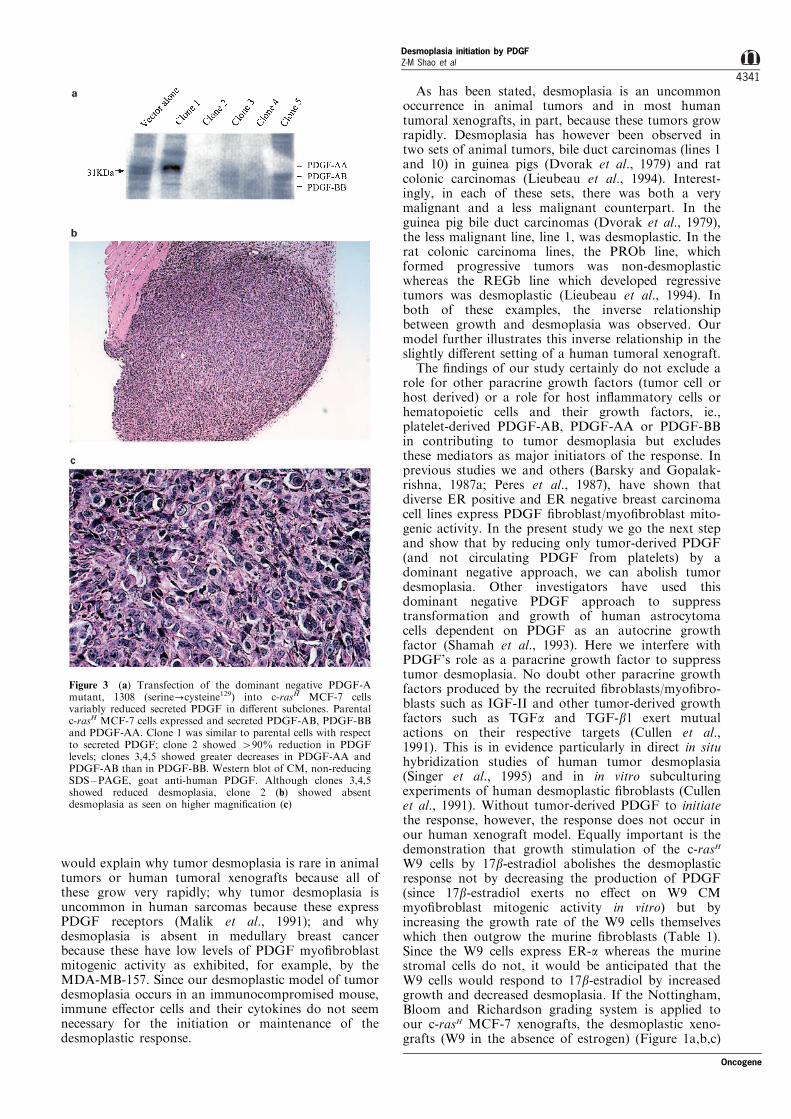
Figure 3 (a) Transfection of the dominant negative PDGF-Amutant, 1308 (serine?cysteine129) into c-rasH MCF-7 cellsvariably reduced secreted PDGF in di�erent subclones. Parentalc-rasH MCF-7 cells expressed and secreted PDGF-AB, PDGF-BBand PDGF-AA. Clone 1 was similar to parental cells with respectto secreted PDGF; clone 2 showed 490% reduction in PDGFlevels; clones 3,4,5 showed greater decreases in PDGF-AA andPDGF-AB than in PDGF-BB. Western blot of CM, non-reducingSDS±PAGE, goat anti-human PDGF. Although clones 3,4,5showed reduced desmoplasia, clone 2 (b) showed absentdesmoplasia as seen on higher magni®cation (c)
Oncogene
Desmoplasia initiation by PDGFZ-M Shao et al
4341

exhibit low numerical histological grade whereas theestrogen-treated non-desmoplastic xenografts exhibithigh numerical histological grade (Figure 1e,f). Thisprogression in histological grade induced by estrogenhas been observed previously in another human breastcarcinoma xenograft (Naundorf et al., 1992).
A line of indirect evidence provided by our study,supporting a tumor-derived paracrine growth factormechanism of desmoplasia and speci®cally a carcino-ma-secreted PDGF initiating mechanism is the patternof gene expression within the stromal cells surroundingthe tumor cells. In situ hybridization studies revealed a3 ± 10-fold increase in select transcripts in the stromal®broblasts/myo®broblasts immediately surrounding theinvasive breast carcinoma cells. This increase was zonalfor stromelysin-3 and TIMP-1 with a dramatic decreasein signal with increasing distance from the tumor cells.This zonal phenomenon present in the c-rasH W9xenografts suggest that the invasive carcinoma cellsthemselves are secreting a di�usable factor which eitherenhances speci®c stromal gene expression or inducesclonal selection of stromal cells with a certain geneexpression pro®le. In either case stromal ®broblasts/myo®broblasts at increasing distances from thecarcinoma cells would be less a�ected. This zonal
e�ect would only be manifest if the source of factorwere the tumor cells themselves and not circulatinghematopoietic elements such as platelets. We andothers have observed the same zonal pattern ofstromelysin-3 and TIMP-1 in actual human cases ofdesmoplastic breast carcinomas (Basset et al., 1990;Tomlinson et al., 1999).
The role of autocrine and paracrine growth factorsin cancer has been appreciated and studied for overtwo decades (Goustin et al., 1986). Carcinoma cells areknown to secrete a number of di�erent factors, bothautocrine and paracrine. Similarly stromal cells secreteboth autocrine and paracrine factors. Looking athuman tumor desmoplasia there are a number ofautocrine and paracrine loops in play, some involvinggrowth stimulation and some involving growth inhibi-tion (Horgan et al., 1987; Adams et al., 1988; Osborneet al., 1989; Yee et al., 1989). PDGF, however, is theonly factor out of the many growth factors secreted bybreast carcinoma cells to have a sole paracrine actionon neighboring stromal cells. All other factors (TGFa,TFG-b1, IGF-I or IGF-II) have autocrine actions aswell (Osborne and Arteaga, 1990; Yee et al., 1988;Prager et al., 1994; Arteaga and Osborne, 1989).Furthermore PDGF receptors are present on stromal
Figure 4 Representative clones (clones 1,2,3) of those depicted in Figure 3a are shown in their respective lanes. Although thesecretion of PDGF-AA (a), PDGF-BB (b) and PDGF-AB (a,b) was reduced in most of the PDGF-MCF-7 clones, especially clone 2,the secretion of other growth factors including TGFa (c), TGF-b1 (d), IGF-I (e) and IGF-II (f) was not altered. Western blot, non-reducing SDS±PAGE, (a) anti-human PDGF-AA; (b) anti-human PDGF-BB; (c) anti-human TGFa; (d) anti-human TGF-b1; (e)anti-human IGF-I; (f) anti-human IGF-II
Desmoplasia initiation by PDGFZ-M Shao et al
4342
Oncogene

cells but are lacking on epithelial (carcinoma) cells aswell as endothelial cells (Malik et al., 1991). PDGF caninduce stromal mitogenesis and stromal downstreamgenes such as IGF-I and IGF-II in vitro (Cullen et al.,1991; Miura et al., 1994). Whereas these secondarygenes can be hormonally regulated (Manni et al., 1994;Noguchi et al., 1993), PDGF secretion does not seemto be under hormonal control (Dickson et al., 1987).These ®ndings and the ®ndings in our present studycollectively suggest that PDGF is an independent highlevel paracrine factor whose primary target is stromalcells and that carcinoma-secreted PDGF speci®cally isthe primary initiator of tumor desmoplasia, a paracrinephenomenon distinct from angiogenesis.
The desmoplastic response in human breast carcino-ma is a host myo®broblast-mediated collagenousresponse which comprises, to a large extent, thetumor's microenvironment and which exhibits syner-gistic e�ects on breast carcinoma progression. Thedesmoplastic response likely represents a substantialbarrier to future gene and immunotherapies because itscollagenous matrix is relatively impervious to immunee�ector cells and its ®broblast/myo®broblast cellularcomposition is adsorptive to locally injected orsystemically administered gene therapy vectors suchas amphotropic retroviruses or CAR-1 receptor-speci®cadenoviruses. Therefore it might prove e�cacious froma therapeutic standpoint in the future if we couldinhibit the desmoplastic response itself.
Materials and methods
Cell lines
Standard ER positive (MCF-7, T47D, MDA-MB-157) andER negative (MDA-MB-231 and MDA-MB-468) humanbreast carcinoma cell lines were used in these studies. Inaddition recombinant MCF-7 lines (W9, W7 or neo) whichhad been transfected with c-rasH or the neo selectablemarker in previous studies (Sommers et al., 1990) were a giftof Ed Gelmann (Lombardi Cancer Center, GeorgetownUniversity). These lines were grown in the presence of0.8 mg/ml Geneticin (G418). In these previous studies(Sommers et al., 1990), it had been demonstrated that theW9 clone expressed a 40-fold increase in rasH p21 whereasthe W7 and neo clones demonstrated only normal levels ofp21. It had also been shown in previous studies that rasH
transfection reduced the dependency of MCF-7 on exogen-ous estrogen by increasing the secretion of a number ofdi�erent growth factors including IGF-I, IGF-II, TGFa andTGF-b1 (Dickson et al., 1987). Adult human ®broblasts,CRL 1477 (ATCC, Rockville, MD, USA) were used inPDGF binding studies.
Anti-rasH p21 immunoprecipitation studies
We sought to con®rm the p21 data by immunoprecipitationstudies to verify the stability of the lines. W9, W7 and neoMCF-7 lines were subjected to metabolic labeling with 35S-methionine and anti-rasH p21 immunoprecipitation studiesaccording to standard protocols (Muschel et al., 1985). 10 l(1 mg) of a monoclonal rat anti-rasH p21 (clone Y13-259)(Oncogene Science, Manhasset, NY, USA) and a goat anti-rat IgG/Protein-A agarose complex were used. The immuno-precipitate was collected by centrifugation at 2500 r.p.m. for15 min. Pelleted samples (20 ml) were analysed by SDS±PAGE.
Transfection studies
The W9 line was subjected to additional transfection with adominant negative mutant of PDGF-A, 1308 (serine?cys-teine129), a gift of Charles Stiles (Dana-Farber CancerInstitute, Boston, MA, USA). This dominant negativemutant had been produced by site directed mutagenesis(serine?cysteine129) of the murine PDGF-A chain andcloned into the pLNCX expression vector, termedpLNCXO8. This dominant negative mutant had beenfound to exert dominant negative e�ects on both the Aand B chains of PDGF, cross phylogenetic lines rangingfrom Xenopus to man, and dramatically reduce PDGFsecretion (Mercola et al., 1990; Shamah et al., 1993). 105
W9 cells were cotransfected with a 10-fold molar excess ofpLNCX08 to pHyg (a gift of Dr Bill Sugden, McArdleLaboratory for Cancer Research, University of WisconsinWI, USA) using Lipofectin reagent (Gibco ±BRL) andstandard transfection protocols (Barsky et al., 1997). Aftertransfection, selection was accomplished with 0.1 mg/mlhygromycin B (Calbiochem, La Jolla, CA, USA). Emergingclones were removed with cloning rings, cultured in thepresence of both 0.8 mg/ml Geneticin (G418) and 0.1 mg/ml hygromycin B and studied for the secretion of PDGFand other growth factors. Western blots of CM wereanalysed with speci®c antibodies which could distinguishPDGF-AA from PDGF-BB (Hart et al., 1990) and otherspeci®c antibodies against PDGF-AB/AA/BB, TGFa, TGF-b1, IGF-I and IGF-II: polyclonal rabbit anti-humanPDGF-AA and anti-human PDGF-BB (Genzyme Diagnos-tics, Cambridge, MA, USA); polyclonal goat anti-humanPDGF (R&D Systems, Minneapolis, MN, USA); murinemonoclonal anti-human TGFa, clone 134A-2B3 (OncogeneResearch Products, Cambridge, MA, USA); murine mono-clonal anti-human TGF-b1, clone 9016.2 (R&D Systems);murine monoclonal anti-human IGF-1, clone 82-9A(Oncogene Research Products); and polyclonal rabbit anti-human IGF-II (Pepro Tech Inc., Rocky Hill, NJ, USA).Antibodies were used according to manufacturers' dilutionswith standard Western blot protocols (Sternlicht et al.,1997). Selected clones were injected into nude mice andgrown as xenografts.
Screening and fractionation of CM by [3H]thymidine uptakestudies
Myo®broblasts were cultured from desmoplastic humanbreast carcinoma explants according to previous methods(Barsky et al., 1984). 56105 myo®broblasts were seeded in60 mm culture dishes containing serum-free media. After24 h, the cells were treated with CM from the human breastcarcinoma cell lines being studied and myo®broblast[3H]thymidine incorporation was measured by standardmethods (Peres et al., 1987). Speci®cally, W9 CM wascollected over 24 h, concentrated 10-fold using Centriprep-10concentrators (Amicon, Beverly, MA, USA) and lyophilized.Lyophilized CM was then dissolved in 2 ml of 1 M aceticacid and applied to an Ultrogel AcA-44 column(1.56100 cm) initially calibrated with various molecularweight marker proteins and equilibrated with 1 M aceticacid. The column was eluted at a ¯ow rate of 15 ml/h andfractions of 2 ml were collected. 0.2 ml aliquots wereassayed for myo®broblast [3H]thymidine incorporationstudies.
PDGF receptor binding assay
PDGF was labeled with 125I (Heldin et al., 1981) and PDGFreceptor competing activity in fractionated W9 CM wasmeasured as inhibition of binding of [125I]PDGF to con¯uentcultures of CRL 1477 ®broblasts as described previously(Peres et al., 1987; Bronzert et al., 1987). Puri®ed PDGF andEGF served as competition controls.
Oncogene
Desmoplasia initiation by PDGFZ-M Shao et al
4343

Xenograft studies
56106 cells of the W9, W7, neo MCF-7, and selected clonesof the PDGF-A (serine?cysteine129)-W9 transfectants wereinjected into the mammary fat pads of 6 week oldovariectomized female nude mice (BALB/c nu/nu) (HarlanSprague-Dawley, Madison, WI, USA) both in the presenceand absence of a 0.7 mg 60 day release 17b-estradiol pellet(Innovative Research, Sarasota, FL, USA). After 8 weeks,tumorigenicity and size were noted and extirpated tumornodules were subjected to detailed histological analysis,immunocytochemistry, in situ hybridization and collagenextraction studies. All studies utilized groups of 10 mice.
Measurement of hydroxyproline (total collagen)
Pooled tumoral xenografts from each group were homo-genized in 5 ml of 10% trichloroacetic acid at 48C andcentrifuged at 40006g for 10 min. The pellet was washedtwice successively with 10% trichloroacetic acid, ethanol :ether (3 : 1), and ether. The washed pellet was dried. Collagenwas measured after acid hydrolyis (6 M HCI for 18 h) ashydroxyproline (Bergman and Loxley, 1963).
Histological, immunocytochemical and in situ hybridizationstudies
Extirpated xenografts were examined histologically for thepresence of desmoplasia. In immunocytochemical studies,carcinoma cells were readily identi®ed by anti-cytokeratinantibodies, ®broblasts by antivimentin antibodies and myo®-broblasts by anti-smooth muscle actin antibodies and thepercentage of each cell type recorded as demonstrated inprevious studies (Barsky et al., 1984; Barsky and Gopalak-rishna, 1987b). In situ hybridization studies were conductedwith riboprobes made from murine stromelysin-3, humanstromelysin-3, human IGF-II and human TIMP-1 cDNAs,provided by ATCC (IMAGE Consortium), (Rockville, MD,USA). The pBluescript SK plasmid (Stratagene, La Jolla,CA, USA) containing EcoRI ±NotI fragments of therespective cDNAs was linearized with XbaI for antisense
strand preparation from the T7 promoter and with HindIIIfor sense strand preparation from the T3 promoter. [35S]-UTP-labeled RNA transcripts were synthesized at concentra-tions of 2 ± 56105 c.p.m./ml. Fresh frozen sections of thedi�erent xenografts were studied with in situ hybridizationprotocols (O'Connell et al., 1998). Antisense signals werecompared to sense background and recorded with digitalimage analysis.
Effects of 17b-estradiol/tamoxifen
The e�ects of 17b-estradiol/tamoxifen on W9 CM myo®-broblast [3H]thymidine incorporation in vitro were deter-mined. W9 cells were grown in DMEM phenol red-freemedia with 5% dextran-charcoal and sulfatase treated FBS((CSS) (GIBCO/BRL), Grand Island, NY, USA) and treatedwith doses of 1079 to 1077 M 17b-estradiol and doses of1076 to 1074 M tamoxifen for 48 h prior to washing and a24 h collection of CM. The CM was assayed formyo®broblast [3H]thymidine incorporation as before. Thee�ects of 17b-estradiol (0.7 mg 60 day release 17b-estradiolpellet) on tumorigenicity, size and histology (desmoplasia) ofneo MCF-7, W7, W9 and the PDGF-A-W9 clones weredetermined.
Statistical analysis
All in vitro experiments were performed in triplicate. Allanimal experiments were performed with groups of 10 mice.Results were analysed with standard tests of statisticalsigni®cance, including the 2-tailed Student's t-test and aone-way analysis of variance (ANOVA).
AcknowledgmentsThis work was supported by USPHS grants CA40225,CA01351, CA71195 and CA83111, funds from the Cali-fornia Institute for Cancer Research (CICR), the CancerResearch Co-Ordinating Committee (CRCC), and theJonsson Comprehensive Cancer Center (JCCC).
References
Adams EF, Newton CJ, Braunsberg N, Shaikh N, GhilchikM and James VHT. (1988). Breast Cancer Res. Treat., 11,165 ± 172.
Arteaga CL and Osborne CK. (1989). Cancer Res., 49,6237 ± 6241.
Barsky SH and Gopalakrishna R. (1987a). Biochem.Biophys. Res. Commun., 149, 1125 ± 1131.
Barsky SH and Gopalakrishna R. (1987b). Cancer Res., 47,1663 ± 1667.
Barsky SH, Green WR, Grotendorst GR and Liotta LA.(1984). Am. J. Pathol., 115, 329 ± 333.
Barsky SH, Sternlicht MD, Safarians S, Nguyen M, Chin K,Stewart SD, Hiti AL and Gray JW. (1997). Oncogene, 15,2077 ± 2091.
Basset P, Bellocq JP, Wolf C, Stoll I, Hutin P, Limacher JM,Podhajcer OL, Chenard MP, Rio MC and Chambon P.(1990). Nature, 348, 699 ± 704.
Bergman I and Loxley R. (1963). Anal. Chem., 35, 1961 ±1965.
Bronzert DA, Pantazis P, Antoniades N, Kasid A, DavidsonN, Dickson RB and Lippman ME. (1987). Proc. Natl.Acad. Sci. USA, 84, 5763 ± 5767.
Cullen KJ, Smith HS, Hill S, Rosen N and Lippman ME.(1991). Cancer Res., 51, 4978 ± 4985.
Dickson RB, Kasid A, Hu� KK, Bates SE, Knabbe C,Bronzert D, Gelmann EP and Lippman ME. (1987). Proc.Natl. Acad. Sci. USA, 84, 837 ± 841.
Dvorak HF, Dvorak AM, Manseau EJ, Wiberg L andChurchill WH. (1979). J. Natl. Cancer Inst., 62, 1459 ±1472.
Dvorak HF, Dickersin GR, Dvorak AM, Manseau EJ andPyne K. (1981a). J. Natl. Cancer Inst., 67, 335 ± 345.
Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn Pand Bitzer AM. (1981b). Science, 212, 923 ± 924.
Goustin AS, Leof EB, Shipley GD and Moses HL. (1986).Cancer Res., 46, 1015 ± 1029.
Hart CE, Bailey M, Curtis DA, Osborn S, Raines E, Ross Rand Forstrom JW. (1990) Biochemistry, 29, 166 ± 172.
Heldin CH, Westermark B and Wasteson A. (1981). Proc.Natl. Acad. Sci. USA, 78, 3664 ± 3668.
Horgan K, Jones DL and Mansel RE. (1987) Br. J. Surg., 79,227 ± 229.
Kasid A, Lippman ME, Papageorge AG, Lowry DR andGelmann EP. (1985) Science, 228, 725 ± 728.
Lieubeau B, Garrigue L, Barbieux I, Me¯ah K and GregoireM. (1994). Cancer Res., 54, 6526 ± 6532.
Malik R, Gope M, Womer RB, Nagashunmugam T,Margolin JF, Basu A and Scher CD. (1991). CancerRes., 51, 5626 ± 5631.
Manni A, Badger B, Wei L, Zaenglein A, Grove R, Khin S,Heitjan D, Shimasaki S and Ling N. (1994). Cancer Res.,54, 2934 ± 2942.
Mercola M, Deininger PL, Shamah SM, Porter J, Wang Cand Stiles CD. (1990). Genes Dev., 4, 2333 ± 2341.
Desmoplasia initiation by PDGFZ-M Shao et al
4344
Oncogene

Miura M, Li SW, Dumenil G and Baserga R. (1994). CancerRes., 54, 2472 ± 2477.
Muschel RJ, Williams JE, Lowy DR and Liotta LA. (1985).Am. J. Pathol., 121, 1 ± 8.
Naundorf H, Fichtner I, Buttner B and Frege J. (1992) J.Cancer Res. Clin. Oncol., 119, 35 ± 40.
Noguchi M, Thomas M, Koyasaki N, Ohta N, Taniya T,Kitagawa H, Miyazaki I and Mizukami Y. (1993). Mol.Cell. Endocrinol., 92, 69 ± 76.
O'Connell JT, Shao ZM, Drori E, Basbaum CB and BarskySH. (1998). Hum. Pathol., 29, 1517 ± 1523.
Osborne CK and Arteaga CL. (1990). Breast Cancer Res.Treat., 15, 3 ± 11.
Osborne CK, Coronado EB, Kitten LJ, Arteaga CI, FuquaSAW, Ramasharma K, Marshall M and Li CH. (1989).Mol. Endocrinol., 3, 1701 ± 1709.
Peres R, Betsholtz C, Westermark B and Heldin CH. (1987).Cancer Res., 47, 3425 ± 3429.
Povlsen CO. (1980). Antibiot. Chemother., 28, 15 ± 28.
Prager D, Li HL, Asa S and Melmed S. (1994). Proc. Natl.Acad. Sci. USA, 91, 2181 ± 2185.
Shamah SM, Stiles CD and Guha A. (1993). Mol. Cell. Biol.,13, 7203 ± 7212.
Singer C, Rasmussen A, Smith HS, Lippman ME, Lynch HTand Cullen KJ. (1995). Cancer Res., 55, 2448 ± 2454.
Sommers CL, Papageorge A, Wilding G and Gelmann EP.(1990). Cancer Res., 50, 67 ± 71.
Sternlicht MD, Kedeshian P, Shao ZM, Safarians S andBarsky SH. (1997). Clin. Cancer Res., 3, 1949 ± 1958.
Tomlinson J, Barsky SH, Nelson S, Singer S, Pezeshki B, LeeMC, Eilber F and Nguyen M. (1999). Clin. Cancer Res., 5,3516 ± 3522.
Yee D, Cullen KJ, Paik S, Perdue JF, Hampton B, SchwartzA, Lippman ME and Rosen N. (1988). Cancer Res., 48,6691 ± 6696.
Yee D, Paik S, Lebovic GS, Marcus RR, Favoni RE, CullenKJ, Lippman ME and Rosen N. (1989). Mol. Endocrinol.,3, 509 ± 517.
Oncogene
Desmoplasia initiation by PDGFZ-M Shao et al
4345
Copyright © 2022 FDOKUMEN




















