
Dispersal potential in plant communities depends on environmental conditions
Upload
independentCategory
view
0download
0
of February 6, 2015.This information is current as
Depends on Polyphosphoinositides in T Cellsby the Protein Tyrosine Phosphatase MEG2 Homotypic Secretory Vesicle Fusion Induced
Ishihara, Adam Godzik and Tomas MustelinBottini, Scott Williams, Konstantina Nika, Hisamitsu Huong Huynh, Xiaodong Wang, Weizhong Li, Nunzio
http://www.jimmunol.org/content/171/12/6661doi: 10.4049/jimmunol.171.12.6661
2003; 171:6661-6671; ;J Immunol
Referenceshttp://www.jimmunol.org/content/171/12/6661.full#ref-list-1
, 20 of which you can access for free at: cites 47 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2003 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 6, 2015
http://ww
w.jim
munol.org/
Dow
nloaded from

Homotypic Secretory Vesicle Fusion Induced by theProtein Tyrosine Phosphatase MEG2 Depends onPolyphosphoinositides in T Cells1
Huong Huynh,* Xiaodong Wang,* Weizhong Li,† Nunzio Bottini,* Scott Williams,*Konstantina Nika,* Hisamitsu Ishihara,‡ Adam Godzik,† and Tomas Mustelin2*
Sec14p homology domains are found in a large number of proteins from plants, yeast, invertebrates, and higher eukaryotes. Wereport that the N-terminal Sec14p homology domain of the human protein tyrosine phosphatase PTP-MEG2 binds phosphati-dylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) in vitro and colocalizes with this lipid on secretory vesicle membranes in intactcells. Point mutations that prevented PtdIns(3,4,5)P3 binding abrogated the capacity of PTP-MEG2 to induce homotypic secretoryvesicle fusion in cells. Inhibition of cellular PtdIns(3,4,5)P3 synthesis also rapidly reversed the effect of PTP-MEG2 on secretoryvesicles. Finally, we show that several different phosphoinositide kinases colocalize with PTP-MEG2, thus allowing for localsynthesis of PtdIns(3,4,5)P3 in secretory vesicle membranes. We suggest that PTP-MEG2 through its Sec14p homology domaincouples inositide phosphorylation to tyrosine dephosphorylation and the regulation of intracellular traffic of the secretory pathwayin T cells. The Journal of Immunology, 2003, 171: 6661–6671.
P roteins destined for secretion are synthesized on mem-brane-bound ribosomes and are transported through theendoplasmic reticulum and Golgi to the trans-Golgi net-
work, where they are sorted into small transport vesicles of eitherthe constitutive or the regulated secretory pathways. Homotypicfusion of such transport vesicles results in condensing vacuolesthat subsequently mature into secretory vesicles through an os-motic concentration process. Morphologically, most mature secre-tory vesicles have a dense core of amorphous material. In neuronalcells, they are also larger than the clear synaptic vesicles that con-tain small-molecule neurotransmitters.
Hemopoietic cells use the regulated secretory pathway for a va-riety of functions (1, 2). Platelets contain numerous vesicles withblood clotting factors, vasoactive agents, and growth factors. Mastcells are specialized to degranulate large secretory vesicles thatcontain histamine, vasoactive molecules, and leukotrienes, uponstimulation by Ag-IgE complexes. CTLs contain specialized gran-ules with granzyme, perforin, and other lytic enzymes, which arereleased in a receptor-triggered and directional manner to kill tar-get cells. In many cases, it appears that secretory vesicles havemuch in common with lysosomes and could perhaps be viewed asspecialized lysosomes (2).
A hallmark of the regulated secretory pathway is its response toextracellular stimuli. In both endocrine and hemopoietic cells, theuse of this pathway can be up-regulated severalfold, and in some
instances, the contents of the vesicles are acutely expelled in re-sponse to receptor triggering. The main function of Th cells in theinitiation of an immune response is to actively secrete a number ofcytokines, which recruit and activate other cell types involved ininflammation and the ensuing innate and adaptive immune re-sponse (3). Depending on the subtype of Th cell, these polypep-tides include numerous interleukins (e.g., IL-2, -4, -5, and -12),IFNs, and growth factors (e.g., for granulocytes and monocytes).Despite their importance in the immune system, secretory vesicleshave been relatively little studied in lymphoid cells.
We have reported (4) that the tyrosine-specific protein phospha-tase PTP-MEG2 is involved in regulation of vesicle traffic alongthe regulated secretory pathway in T cells and mast cells. Thisphosphatase contains a unique 250-aa, putative phospholipid-bind-ing domain in its N terminus (5). This region has 28% identity withcellular retinaldehyde-binding protein and 24% identity withSec14p, a yeast protein with phosphatidylinositol (PtdIns)3 transferactivity. A similar Sec14p homology domain is also found in twoprotein tyrosine phosphatases from Xenopus laevis, PTPX1 andPTPX10 (6), and numerous other proteins (7, 8), including manyregulators of Rho, Rac, and Ras proteins (9).
We have studied the Sec14p homology domain of PTP-MEG2and found that it binds a phospholipid with high affinity. Thisbinding is not involved in targeting of PTP-MEG2 to the secretoryvesicle compartment, but appears to be important for the functionof PTP-MEG2 on these vesicles in intact cells. This represents anovel mechanism for cross talk between tyrosine phosphorylationand signaling by phosphoinositides.
Materials and MethodsComputer modeling
A three-dimensional model of aa 8–250 of PTP-MEG2 was built by com-parative modeling, using the crystal structure of Sec14p (Brookhaven Pro-tein Data Bank code 1aua) as a template. Several different alignments were
*Program of Signal Transduction, †Bioinformatics Group, Cancer Research Center,The Burnham Institute, La Jolla, CA 92037; and ‡Division of Molecular Metabolismand Diabetes, Department of Internal Medicine, Tohoku University Graduate Schoolof Medicine, Sendai, Japan
Received for publication April 17, 2003. Accepted for publication October 10, 2003.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This study was supported by Grants AG00252 (to H.H.), GM60049 (to A.G.),CA96949, AI35603, AI48032, and AI53585 (to T.M.) from the National Institutes ofHealth.2 Address correspondence and reprint requests to Dr. Tomas Mustelin, Program ofSignal Transduction, The Burnham Institute, 10901 North Torrey Pines Road, LaJolla, CA 92037. E-mail address: [email protected]
3 Abbreviations used in this paper: PtdIns, phosphatidylinositol; Ins, inositol; PtdSer,phosphatidylserine; PtdIns(3,4,5)P3, PtdIns-3,4,5-trisphosphate; HA, hemagglutinin;TRITC, rhodamine isothiocyanate; GFP, green fluorescent protein; PH, pleckstrinhomology; EEA1, early endosomal Ag 1; h, human.
The Journal of Immunology
Copyright © 2003 by The American Association of Immunologists, Inc. 0022-1767/03/$02.00
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

generated using various alignment methods, including PSI-BLAST (10),FFAS (11), and SAM-99 (12). Despite the relatively low sequence identitybetween the Sec14p homology domain and Sec14p, the statistical signifi-cance of the alignment was high, ranging from an e value of 2 � e�24 forPSI-BLAST alignment to a Z-score of 65 for the FFAS alignment. Thesevalues suggested that the two proteins are certainly homologous, and thatthe alignments were very reliable. Each alignment was used to build athree-dimensional model using a MODELLER automated modeling pro-gram (13), and the resulting models were analyzed for quality using pro-grams ProCheck (14), MatchMaker (Tripos, St. Louis, MO), and Profile3D(15). The resulting models had very high quality with a well-packed inte-rior and a consistent pattern of interactions, again suggesting that the mod-els were reliable and could be used for functional analysis of the Sec14phomology domain. The only differences between models obtained withdifferent alignments concerned the exact length of the alignment, with thePSI-BLAST alignment resulting in a shorter model, with several residuesclipped from the N and C termini of the PTP-MEG2 sequence.
Abs and reagents
The anti-influenza hemagglutinin (HA) tag epitope mAb 12CA5 conju-gated to rhodamine isothiocyanate (TRITC) were from Roche MolecularBiochemicals (Indianapolis, IN). The 16B12 anti-HA from Berkeley An-tibody (Richmond, CA) was used for immunoblotting. Anti-carboxypepti-dase E was from Transduction Laboratories (Lexington, KY). Goat anti-rabbit IgG (F(ab)2)-Alexa 488 was from Molecular Probes (Eugene, OR).Anti-GST mAb was from Santa Cruz Biotechnology (Santa Cruz, CA). Allphospholipids and the anti-PtdIns-3,4,5-trisphosphate (PtdIns(3,4,5)P3)mAb were from Echelon (Salt Lake City, UT). Anti-PtdIns-3-kinase p85Abs were from Upstate Biotechnology (Lake Placid, NY).
Plasmid construction
The cDNA for PTP-MEG2 (5) was kindly provided by P. Majerus (St.Louis, MO). It was subcloned into the pEF/HA vector (16), which adds anHA epitope to the N terminus of the cloned insert. The catalytically inac-tive mutant of PTP-MEG2, PTP-MEG2-C515S, the K55M, and K184Mmutants were generated using the QuickChange site-directed mutagenesiskit (Stratagene, La Jolla, CA). The mutations were verified by nucleotidesequencing. The nucleotides encoding the Sec14p homology domain, aa1–261, were amplified by PCR from the PTP-MEG2 plasmid cDNA. Theamplification product was subcloned into the prokaryotic expression vectorpGEX-4T-1. The fusion protein was expressed in Escherichia coli strainDH5� and purified using glutathione-agarose as described previously (17).
Dot blot assay for phospholipid binding (fat Westerns)
The phospholipid binding specificity of the Sec14p homology domain ofPTP-MEG2 was assayed as described (18). Nitrocellulose filters spottedwith 100 pmol of phospholipids (PIP strips; Echelon) were blocked in 3%fatty acid-free BSA in 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 0.1%Tween 20 for 1 h and incubated with 0.1–1 �g/ml GST-fusion proteinovernight at 4°C. The membrane was washed over 1 h and then incubatedfor 1 h with a 1/1000-diluted anti-GST mAb. The membrane was washedas before, and incubated for 1 h with 1/3000-diluted anti-mouse-HRP con-jugate. Finally, the membrane was washed, and the GST fusion proteinbound to the membrane by virtue of its interaction with phospholipid wasdetected by ECL (ECL kit; Amersham, Arlington Heights, IL).
In vitro transcription and translation of full-length PTP-MEG2for filter binding assay
The cDNA for PTP-MEG2 was cloned into the pET30c vector (Novagen,Madison, WI), and the full-length S-tagged protein was generated using theTNT in vitro coupled transcription-translation system (Promega, Madison,WI). The reaction was first optimized using cold methionine, and the pro-duced PTP-MEG2 protein was detected and quantified using an S-protein-HRP conjugate (Novagen) after SDS-PAGE and transfer of the reactionmixture to nitrocellulose filters. For the lipid overlay assays, PIP stripswere blocked for 1 h as described above and then incubated for 1 h with 25�l of the 35S-labeled in vitro translation reaction mixture diluted in 5 ml ofblocking buffer. After extensive washing with blocking buffer, the stripswere exposed to film.
Cells and transient transfections
Jurkat T leukemia cells were kept at logarithmic growth in RPMI 1640supplemented with 10% heat-inactivated FCS, L-glutamine, and antibiotics.Transient transfections were conducted by electroporation as described be-fore (19–23). Electroporation conditions typically contained 20 � 106 cells
and a total of 5–20 �g of plasmid DNA, and in each transfection, the DNAamount was kept constant by the addition of empty vector. Cells were usedfor experiments 48 h after transfection.
Immunoblots, immunoprecipitation, cell fractionation, andPtdIns 3-kinase assays
Immunoprecipitation was performed as before (19–23). All immunoblotswere developed by the ECL technique (ECL kit; Amersham) according tothe manufacturer’s instructions. Cell fractionation was done as before (24),and PtdIns 3-kinase assays were done as before (16, 21).
Confocal microscopy
Double immunofluorescence staining was done as before (4, 24). Briefly,cells were washed in PBS and fixed in freshly made 3.7% formaldehyde.Fixed cells were permeabilized with 0.1% saponin in PBS, then blocked in2.5% normal goat serum in 0.1% saponin in PBS for 30 min at roomtemperature, and then incubated with primary and secondary Ab diluted inthe same buffer for 1 h each at room temperature. After three washes withPBS, the cells were mounted onto glass slides and viewed under a confocallaser scanning microscope (MRC-1024; Bio-Rad, Hercules, CA). A dif-ferential interference contrast image was also taken of most cells.
ResultsModeling of the Sec14p homology domain of PTP-MEG2
As a rational approach to elucidating the function of the N-termi-nal Sec14p homology domain of PTP-MEG2, we built a three-dimensional model of aa 8–250 of this protein (see Materials andMethods). A comparison of this model with the crystal structure ofSec14p (25) shows that the two display a number of interestingdifferences despite being quite similar overall (Fig. 1, A–D). No-tably, the phospholipid-binding pocket of Sec14p is well con-served in PTP-MEG2, but is shorter and has a cluster of basicamino acid residues in one end (Fig. 1E). This cluster contains Argand Lys residues from the conserved Ala-Arg-Lys-Phe-Asp motif(aa 53–57 in PTP-MEG2), as well as Lys184, Lys209, Arg211, andArg213, which are found in PTP-MEG2, but not in Sec14p (Fig.1F). This model suggested that the PTP-MEG2 Sec14p homologydomain may bind phospholipids that have a more acidic headgroup than the unphosphorylated inositol (Ins) of PtdIns bound bySec14p.
The Sec14p homology domain of PTP-MEG2 bindsPtdIns(3,4,5)P3
To directly test the computer model prediction, we generated aGST-fusion protein containing the Sec14p homology domain ofPTP-MEG2 and measured its capacity to bind phospholipids usinga filter binding assay (18), in which the phospholipids are immo-bilized on nitrocellulose, while the protein is in solution and issubsequently detected by anti-GST or anti-PTP-MEG2 Abs. In thisassay, 3–10 nM Sec14p homology domain of PTP-MEG2 boundbest to PtdIns(3,4,5)P3 and, a bit weaker, to PtdIns(3,5)P2 (Fig. 2).At 10 and 30 nM, some binding was also seen to PtdIns(3,4)P2,PtdIns(4,5)P2, PtdIns(4)P, and PtdIns(5)P. Other phospholipids didnot bind at all to the GST-MEG2 protein. Control GST (Fig. 2F)or GST-hemopoietic protein tyrosine phosphatase (Fig. 4F) did notbind any phospholipids.
To verify that full-length PTP-MEG2 also binds the same phos-pholipids as the GST-Sec14p homology domain, we generated the68-kDa PTP-MEG2 by in vitro transcription and translation of abacterial expression plasmid encoding an N-terminally S-taggedPTP-MEG2 in the presence of [35S]methionine. The phospholipidfilters were then incubated with the translation products, washedextensively, and exposed to film. In these experiments, full-lengthPTP-MEG bound the same phospholipids as the isolated Sec14phomology domain, namely PtdIns(3,4,5)P3 and PtdIns(3,5)P2 andless to other phosphoinositides (Fig. 3A). A control transcription/translation reaction without the DNA, but with [35S]methionine
6662 PTP-MEG2 REGULATION BY PHOSPHOINOSITIDES
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

FIGURE 1. Computer model of theSec14p homology domain of PTP-MEG2. A–D, Comparison of the crys-tal structure of Sec14p with the com-puter model of the Sec14p homologydomain of PTP-MEG2, shown as aribbon graphs (A and B) and asGRASP representation (C and D) withthe surface topology of acidic (red)and basic (blue) charges. The two oc-tylglucoside molecules found in thecrystal of Sec14p are included asspace-fill models. E, A close-up viewof the putative binding pocket for anacidic head group of a ligand phospho-lipid in the Sec14p homology domainof PTP-MEG2. F, Amino acid se-quence alignment of the Sec14p ho-mology domain of PTP-MEG2 andSec14p from Candida albicans (albi)and Saccharomyces cerevisiae (cerev).The basic residues pointed out in E arehighlighted in pale blue.
6663The Journal of Immunology
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

and all other reagents, did not show any binding of phospholipids(Fig. 3B). The presence of full-length S-tagged PTP-MEG2 wasalso confirmed by anti-S immunoblotting (Fig. 3C).
Because yeast Sec14p binds more than just the polar head groupof PtdIns (25, 26), we decided to test how well the polar headgroup of PtdIns(3,4,5)P3, Ins(1,3,4,5)P4, was able to compete withPtdIns(3,4,5)P3 for binding to the Sec14p homology domain ofPTP-MEG2. Although addition of Ins(1,3,4,5)P4 to the bindingassay had a dose-dependent inhibitory effect, this effect was rela-tively minor even at 1 �M Ins(1,3,4,5)P4 (Fig. 4). These data sug-gest (but do not prove) that PTP-MEG2 requires more than just thepolar head group of PtdIns(3,4,5)P3 for high-affinity binding.
Colocalization of PTP-MEG2 with PtdIns(3,4,5)P3 in intact cells
PTP-MEG2 is normally found on the membrane of secretory ves-icles in mast and T cells (4). Expression of catalytically activePTP-MEG2 results in a homotypic fusion of these normally�300-nm vesicles into giant vesicles with diameters of 1–2 �m.Catalytically inactive PTP-MEG2-C515S does not cause vesiclefusion (4). When cells expressing PTP-MEG2 were stained with amAb specific for PtdIns(3,4,5)P3, the resulting fluorescence wasfound to colocalize with PTP-MEG2 on the enlarged vesicles (Fig.5, B and C). A weak plasma membrane staining was also seen inboth untransfected and transfected Jurkat cells (Fig. 5A). As a con-trol, we treated the cells with wortmannin, a fungal inhibitor ofPtdIns 3-kinases, and as expected, PtdIns(3,4,5)P3 was not de-tected in these cells (Fig. 5, D–F) (see below for effects of wort-mannin in PTP-MEG2).
To verify these results with another technique, we used a fusionprotein between green fluorescent protein (GFP) and the Btk ki-nase pleckstrin homology (PH) domain, which is highly specificfor PtdIns(3,4,5)P3 (27). In control Jurkat cells expressing the
FIGURE 4. Polar head group competition assay. A–D, Filter bindingassays for phospholipid selection using 5 nM GST-Sec14p homology do-main of PTP-MEG2 and in the presence of the indicated concentration ofIns(1,3,4,5)P4. E, Graph showing the densitometric quantitation ofPtdIns(3,4,5)P3 binding in A–D. F, A filter incubated with control GST-hemopoietic protein tyrosine phosphatase (GST-HePTP). All filters wereexposed to film for the exact same time.
FIGURE 2. Phospholipid selection by the Sec14p homology domain ofPTP-MEG2. A–D, Filter binding assays for phospholipid selection usingthe indicated concentrations of the GST-Sec14p homology domain of PTP-MEG2. E, Graph showing the densitometric quantitation of PtdIns(3,4,5)P3
binding in A–D. F, A filter incubated with control GST. All filters wereexposed to film for the exact same time.
FIGURE 3. Phospholipid binding by full-length PTP-MEG2. A, Filterbinding assay for phospholipid selection using in vitro-transcribed and-translated [35S]methionine-labeled S-tagged full-length PTP-MEG2. B,Control incubation with same in vitro transcription and translation reactionmixture, but lacking PTP-MEG2 plasmid. C, Anti-S immunoblot of the invitro-transcribed and -translated material.
6664 PTP-MEG2 REGULATION BY PHOSPHOINOSITIDES
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
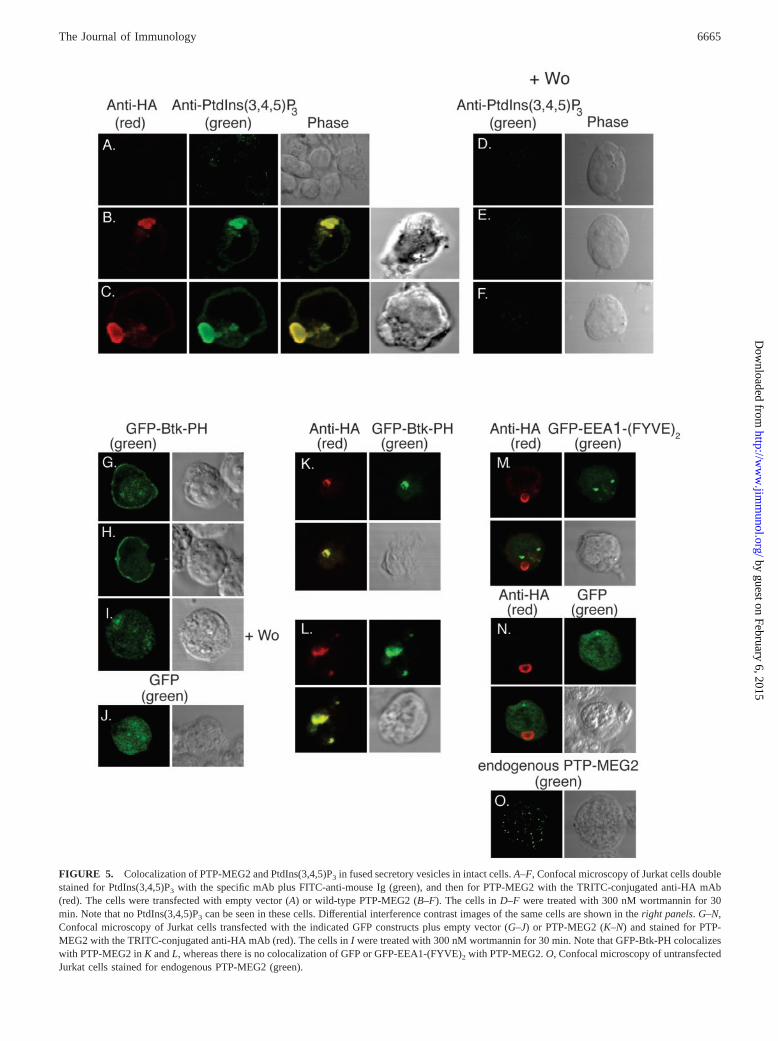
FIGURE 5. Colocalization of PTP-MEG2 and PtdIns(3,4,5)P3 in fused secretory vesicles in intact cells. A–F, Confocal microscopy of Jurkat cells doublestained for PtdIns(3,4,5)P3 with the specific mAb plus FITC-anti-mouse Ig (green), and then for PTP-MEG2 with the TRITC-conjugated anti-HA mAb(red). The cells were transfected with empty vector (A) or wild-type PTP-MEG2 (B–F). The cells in D–F were treated with 300 nM wortmannin for 30min. Note that no PtdIns(3,4,5)P3 can be seen in these cells. Differential interference contrast images of the same cells are shown in the right panels. G–N,Confocal microscopy of Jurkat cells transfected with the indicated GFP constructs plus empty vector (G–J) or PTP-MEG2 (K–N) and stained for PTP-MEG2 with the TRITC-conjugated anti-HA mAb (red). The cells in I were treated with 300 nM wortmannin for 30 min. Note that GFP-Btk-PH colocalizeswith PTP-MEG2 in K and L, whereas there is no colocalization of GFP or GFP-EEA1-(FYVE)2 with PTP-MEG2. O, Confocal microscopy of untransfectedJurkat cells stained for endogenous PTP-MEG2 (green).
6665The Journal of Immunology
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

GFP-Btk-PH construct, fluorescence was diffusely cytoplasmicwith a clear enrichment under the plasma membrane (Fig. 5, G andH), which disappeared upon treatment of the cells with wortman-nin (I). GFP alone was also diffusely distributed (J). In cells co-expressing PTP-MEG2, the GFP-Btk-PH protein accumulated inan intracellular vesicle structure that also stained for PTP-MEG2(Fig. 5, K and L). In contrast, another fusion protein, consisting ofGFP plus two tandem FYVE domains (from early endosomal Ag1 (EEA1); GFP-EEA1-(FYVE)2), which is specific for PtdIns(3)P(28–30), did not colocalize with PTP-MEG2 (Fig. 5M), as was thecase also for GFP alone (N). Taken together, these experimentsshow that PTP-MEG2 colocalizes with PtdIns(3,4,5)P3 in intactcells.
Binding of PtdIns(3,4,5)P3 is required for PTP-MEG2 function
To further test whether binding of PtdIns(3,4,5)P3 to the Sec14phomology domain of PTP-MEG2 is important for the function ofPTP-MEG2, we created three mutants with reduced ability to bindthis phospholipid. Aided by the computer model prediction that anumber of basic amino acid residues in the Sec14p homology do-main may be involved in binding to the acidic head group ofPtdIns(3,4,5)P3 (Fig. 1), we chose two residues, Lys55 and Lys184,and mutated them individually or in combination to methionineresidues. As assessed by dot blots (Fig. 6), the K55M- and
K184M-mutated Sec14p homology domains both had a reducedability to bind PtdIns(3,4,5)P3, whereas the double mutant did notbind at all. When expressed in cells, PTP-MEG2-K55M stillcaused fusion of secretory vesicles, although they tended to remainsmaller, more numerous, and surrounded by small vesicles (Fig. 6,E–G). PTP-MEG2-K184M also caused incomplete fusion with nu-merous small vesicles (Fig. 6, I–K), whereas the double mutant,PTP-MEG2-K55M/K184M, was unable to cause any fusion (M–P). Instead, the fluorescence was mostly seen as small granules inthe region of the Golgi apparatus and trans-Golgi network. Be-cause this pattern is also seen with catalytically inactive PTP-MEG2-C515S (4), these results imply that the Sec14p homologydomain of PTP-MEG2 needs to bind PtdIns(3,4,5)P3 for the en-zyme to cause secretory vesicle fusion in intact cells.
PTP-MEG2 mutants do not colocalize with PtdIns(3,4,5)P3
To further verify that the K55M, K184M, and K55M/K184M mu-tants do not bind PtdIns(3,4,5)P3 in the intact cells, we used theGFP fusion proteins and costained the cells for PTP-MEG2. Theseexperiments showed that only PTP-MEG2 with an intact Sec14phomology domain colocalized with the GFP-Btk-PH domain onthe enlarged vesicles (Fig. 7A). In contrast, the three mutants didnot colocalize with either GFP-Btk-PH or GFP-EEA1-(FYVE)2
(Fig. 7, B–H). Control blots showed that the mutant PTP-MEG2
FIGURE 6. Sec14p homology domain mu-tants of PTP-MEG2 with reduced ligand binding.A, D, H, and L, Filter binding assays for phos-pholipid selection using 5 nM GST-Sec14p ho-mology domain of PTP-MEG2 (A), or its K55M(D), K184M (H), or double K55M/K184M (L)point mutants. B and C, Confocal microscopy ofcells transfected with PTP-MEG2 and stained forPTP-MEG2 with the anti-HA mAb. E–G, Con-focal microscopy of cells transfected with PTP-MEG2-K55M and stained with the anti-HAmAb. I–K, Confocal microscopy of cells trans-fected with PTP-MEG2-K184M and stained withthe anti-HA mAb. M–P, Confocal microscopy ofcells transfected with PTP-MEG2-K55M/K184M and stained with the anti-HA mAb. Theshown cells are representative of the majority ofstained cells.
6666 PTP-MEG2 REGULATION BY PHOSPHOINOSITIDES
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

proteins were expressed at levels similar to intact PTP-MEG2 (Fig.7I), indicating that they were stable. Control blots also showed thatthe two GFP fusion proteins and GFP alone were expressed as theexpected-size proteins (Fig. 7J). A close-up of H shows that thelocation of PTP-MEG2-K55M/K184M remains granular and ve-sicular, although the size of these vesicles are much smaller.
Inhibition of PtdIns(3,4,5)P3 synthesis blocks PTP-MEG2function
The source of PtdIns(3,4,5)P3 in the secretory vesicle compartmentis not clear. In addition to the classical route of synthesis by phos-phorylation of PtdIns(4,5)P2 by type I PtdIns 3-kinases (31), thereis a Golgi- and post-Golgi-localized synthetic (secretory/endo-cytic) pathway that is initiated by type III PtdIns 3-kinase (31, 32),which produces PtdIns(3)P. This lipid acts as a substrate for theD5-specific kinase PIKfyve (33, 34), which phosphorylatesPtdIns(3)P to PtdIns(3,5)P2. Both type III PtdIns 3-kinase andPIKfyve, as well as their products, are known to play regulatoryroles in the secretory pathway in yeast (35). Thus, we favor thissynthetic pathway over the classical route as the source ofPtdIns(3,4,5)P3 in the secretory vesicles. In support of this possi-
bility, the Golgi apparatus and other intracellular membranes alsocontain D4-specific kinases (31), which could phosphorylatePtdIns(3,5)P2 to PtdIns(3,4,5)P3. However, it is interesting to notethat PTP-MEG2 also bound PtdIns(3,5)P2.
Regardless of the route by which PtdIns(3,4,5)P3 is produced forPTP-MEG2 regulation, we reasoned that inhibition of D3-specific(and some D4-specific) kinases by treatment of cells with wort-mannin should prevent its synthesis in the PTP-MEG2-containingvesicles. Indeed, as already shown in Fig. 5, wortmannin causedPtdIns(3,4,5)P3 to disappear from the cells as assessed with thespecific mAb (D–F) or the GFP-Btk-PH construct (I). Further-more, when PTP-MEG2-containing cells were treated with 300nM wortmannin (sufficient to also inhibit type III kinases (31)), thefused secretory vesicle compartment began to fragment and shrinkin a time-dependent manner (Fig. 8, A–K). Within 15–30 min afteraddition of wortmannin, only a multitude of small PTP-MEG2-containing vesicles were seen (Fig. 8, E–H). A quantitation ofvesicles �1 �m in diameter (Fig. 8K) also demonstrates the fastkinetics of the response. A lower dose of wortmannin (100 nM)was partly effective, whereas 50 nM had a minimal effect (notshown). Similarly, another inhibitor of PtdIns 3-kinases,
FIGURE 7. Lack of colocalization of ligandbinding mutants of PTP-MEG2 with PtdIns(3,4,5)P3
or PtdIns(3)P in intact cells. A–H, Confocal micros-copy of Jurkat cells transfected with the indicated GFPconstructs plus PTP-MEG2 (A), PTP-MEG2-K55M(B–D), PTP-MEG2-K184M (E and F), or PTP-MEG2-K55M/K184M (G and H) and stained with theanti-HA mAb. Note that GFP-Btk-PH colocalizeswith PTP-MEG2 in A, whereas there is no colocaliza-tion of GFP-Btk-PH or GFP-EEA1-(FYVE)2 withany of the mutants. I, Immunoblot with anti-HAmAbs of total lysates of Jurkat cells transfected withthe indicated PTP-MEG2 constructs. Note that theyare all expressed at similar levels. J, Control immuno-blot with anti-GFP Abs of total lysates of Jurkat cellstransfected with the indicated GFP fusion proteins. K,Close-up of the lower half of the cell in H, with some-what enhanced contrast, to show the granular/vesicu-lar nature of the distribution of PTP-MEG2-K55M/K184M.
6667The Journal of Immunology
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

LY294002, also caused vesicle shrinking and fission at 50 �M, butwas only partially effective at 25 �M (Fig. 8, L–N). These resultssupport the notion that PtdIns(3,4,5)P3 synthesis is required for thefunction of PTP-MEG2 in intact cells. The relatively high doses ofinhibitors required for the effect may indicate that type III PtdIns
3-kinases, rather than type I, are involved. However, this issueremains to be addressed.
To distinguish between a real change in the morphology of thesecretory vesicle compartment and a possible dissociation of PTP-MEG2 from the vesicle membrane, we also costained the cells forthe secretory vesicle marker carboxypeptidase E, which recapitu-lated the result seen with PTP-MEG2 staining (Fig. 8, A–J). Wealso performed a subcellular fractionation experiment (Fig. 9),which showed that �90% of PTP-MEG2 was sedimented by ul-tracentrifugation at 100,000 � g of a postnuclear supernatant ofsonicated cells, and the remaining fraction was pelleted at 300,000 �g. Following treatment of the cells with wortmannin, there was asmall increase in the latter fraction, but no PTP-MEG2 appeared in thesupernatant (Fig. 9, lane 3). Thus, PTP-MEG2 is membrane boundand remains so upon treatment of the cells with wortmannin. Thisnotion is also supported by the close-up shown in Fig. 7K.
Intracellular location of D3-, D4-, and D5-specificphosphoinositide kinases in T cells
To further support the notion that PtdIns(3,4,5)P3 is present onsecretory vesicle membranes, where it can regulate PTP-MEG2,we stained control cells or PTP-MEG2-expressing cells with Absagainst type I PtdIns 3-kinase p85 (Fig. 10, A–D), the human (h)type III PtdIns 3-kinase hVPS34 (E–H), PI4K� (I and J), or the68-kDa PIP5K� (K–N). These experiments showed that all four
FIGURE 8. Reversal of PTP-MEG2-inducedsecretory vesicle fusion by PtdIns 3-kinase inhibi-tors. A–J, Time course of fragmentation of thefused secretory vesicles after addition of wortman-nin. The cells were stained with anti-carboxypep-tidase E mAb plus FITC-anti-mouse Ig (green) andthen for PTP-MEG2 with the TRITC-conjugatedanti-HA mAb (red). The shown cells were un-treated (A and B), or incubated at 37°C for 5 min (Cand D), 15 min (E and F), 30 min (G and H), or 60min (I and J) with 300 nM wortmannin. The showncells are representative of the majority of stainedcells. K, Histogram showing the response as thefraction of cells with vesicles �1 �m in diameter.L–N, Confocal microscopy of cells transfected withPTP-MEG2 and treated with medium alone (L), or 25�M (M) or 50 �M (N) LY294002 for 30 min.
FIGURE 9. Subcellular fractionation of cells expressing PTP-MEG2 orPTP-MEG2 mutants. Immunoblot with anti-HA mAbs of material pelleted byultracentrifugation at 100,000 � g (top panel), 300,000 � g (middle panel), orremaining in the supernatant after the last centrifugation (bottom panel). Thecells were transfected with the indicated construct, and the cells in lane 3 weretreated with 300 nM wortmannin for 15 min before lysis.
6668 PTP-MEG2 REGULATION BY PHOSPHOINOSITIDES
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
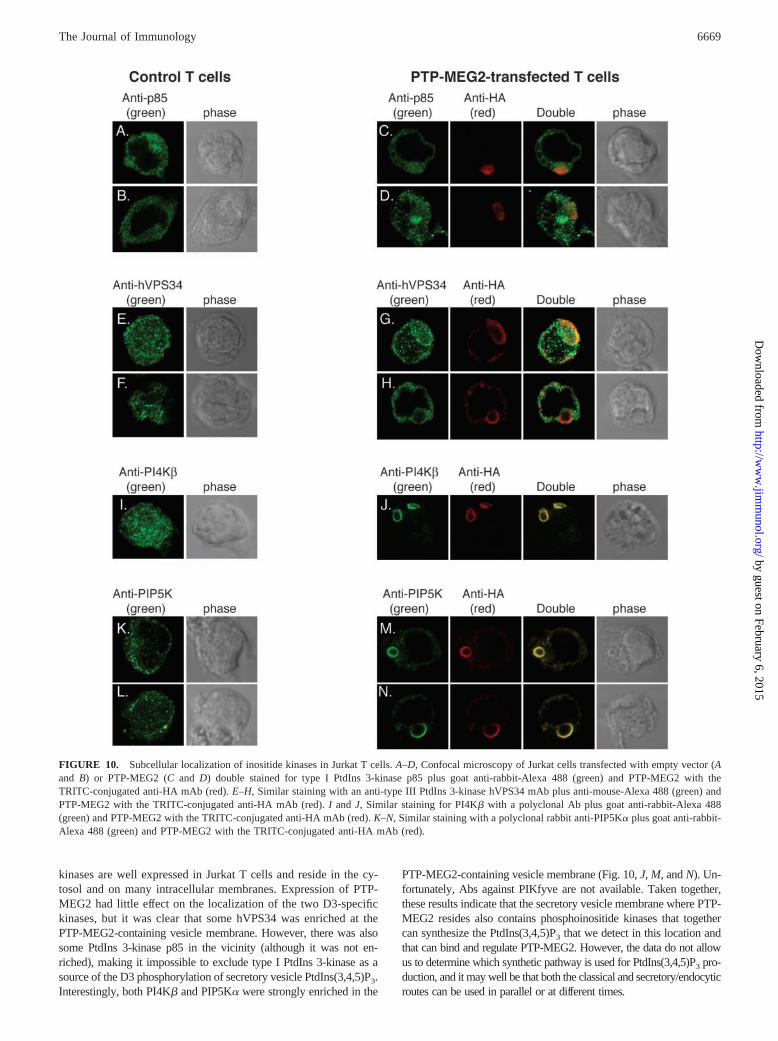
kinases are well expressed in Jurkat T cells and reside in the cy-tosol and on many intracellular membranes. Expression of PTP-MEG2 had little effect on the localization of the two D3-specifickinases, but it was clear that some hVPS34 was enriched at thePTP-MEG2-containing vesicle membrane. However, there was alsosome PtdIns 3-kinase p85 in the vicinity (although it was not en-riched), making it impossible to exclude type I PtdIns 3-kinase as asource of the D3 phosphorylation of secretory vesicle PtdIns(3,4,5)P3.Interestingly, both PI4K� and PIP5K� were strongly enriched in the
PTP-MEG2-containing vesicle membrane (Fig. 10, J, M, and N). Un-fortunately, Abs against PIKfyve are not available. Taken together,these results indicate that the secretory vesicle membrane where PTP-MEG2 resides also contains phosphoinositide kinases that togethercan synthesize the PtdIns(3,4,5)P3 that we detect in this location andthat can bind and regulate PTP-MEG2. However, the data do not allowus to determine which synthetic pathway is used for PtdIns(3,4,5)P3 pro-duction, and it may well be that both the classical and secretory/endocyticroutes can be used in parallel or at different times.
FIGURE 10. Subcellular localization of inositide kinases in Jurkat T cells. A–D, Confocal microscopy of Jurkat cells transfected with empty vector (Aand B) or PTP-MEG2 (C and D) double stained for type I PtdIns 3-kinase p85 plus goat anti-rabbit-Alexa 488 (green) and PTP-MEG2 with theTRITC-conjugated anti-HA mAb (red). E–H, Similar staining with an anti-type III PtdIns 3-kinase hVPS34 mAb plus anti-mouse-Alexa 488 (green) andPTP-MEG2 with the TRITC-conjugated anti-HA mAb (red). I and J, Similar staining for PI4K� with a polyclonal Ab plus goat anti-rabbit-Alexa 488(green) and PTP-MEG2 with the TRITC-conjugated anti-HA mAb (red). K–N, Similar staining with a polyclonal rabbit anti-PIP5K� plus goat anti-rabbit-Alexa 488 (green) and PTP-MEG2 with the TRITC-conjugated anti-HA mAb (red).
6669The Journal of Immunology
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

DiscussionThe high-affinity binding of PtdIns(3,4,5)P3 to the Sec14p homol-ogy domain of PTP-MEG2 represents a novel finding that intro-duces a new domain into the growing list of phosphoinositide-binding protein modules. Phosphoinositides are important secondmessengers in transmembrane signal transduction and intracellularvesicle traffic (36–38). Successive phosphorylation of the D3, D4,and D5 positions of the inositol head group by specific kinases (31)results in up to seven different phosphoinositides, many of whichfunction as docking molecules for protein modules that haveevolved to specifically interact with them (37). Such protein mod-ules include the PH domains (27, 39), FYVE domains (28–30, 35),and C2 domains (40), and are found in protein and lipid kinases,phospholipases, adapter molecules, regulators of small GTP-bind-ing proteins, cytoskeletal proteins, and components of endocyticpathways (28, 35, 37, 40, 41).
The yeast Sec14p protein binds PtdIns and phosphatidylcholineand functions to transport these lipids between cellular mem-branes. Phospholipid binding occurs through a deep elongatedpocket with a lid-like structure that closes over the bound phos-pholipid (25), allowing the protein to extract the lipid from a bi-layer and transport it to another location. In higher eukaryotes, thistransport function has been taken over by a distinct class of PtdInstransport proteins, which play important roles in phosphoinositide-mediated signaling (26). Instead, Sec14p-like proteins have beenretained in a variety of other functions.
Compared with Sec14p, the domain in PTP-MEG2 contains sev-eral basic residues in the putative phospholipid head-bindinggroove (Fig. 1E). Our point mutants suggest that these residues areimportant for phospholipid selection (Fig. 6). This region is highlyvariable among Sec14p homology domains both in topology andcharge, some having a mix of basic and acidic or predominantlyacidic residues, suggesting that the Sec14p homology domain isquite versatile and can evolve to bind different ligands that havecomplementary charges and shapes. However, charged residuesmay not alone account for selectivity, and a crystal structure of theSec14p homology domain of PTP-MEG2 with bound phospholipidwill probably be required for a complete understanding of thestructural basis for ligand selection.
At present, it remains unclear whether the Sec14p homologydomain of PTP-MEG2 binds only the head group ofPtdIns(3,4,5)P3, as do most other protein modules that bind phos-pholipids (e.g., FYVE, PH, and C2 domains), or the entire phos-pholipid, as yeast Sec14p does (25). The high doses of solubleIns(1,3,4,5)P4 required for competition with PtdIns(3,4,5)P3 bind-ing may argue for binding of more than just the polar head group,but this does not necessarily mean that the fatty acid moieties areinvolved. However, unlike Sec14p, which transports PtdIns betweencellular membranes (26), the Sec14p homology domain of PTP-MEG2 seems to remain membrane bound. Thus, we have no reasonto suspect that PTP-MEG2 is involved in phospholipid transport.
Our findings indicate that the high-affinity binding ofPtdIns(3,4,5)P3 to the Sec14p homology domain of PTP-MEG2 isimportant for the activity and function of PTP-MEG2 in intactcells. In the absence of PtdIns(3,4,5)P3, wild-type PTP-MEG2 be-haved like the catalytically inactive C515S mutant, as did theSec14p homology domain mutant K55M/K184M. However, allthese proteins were still associated with the particulate fraction andappeared to localize to the same vesicle compartment, although itsmorphology was changed. Thus, the Sec14p homology domaindoes not mediate targeting of PTP-MEG2 to the phospholipid bi-layer. Rather, there must be another targeting motif in PTP-MEG2that directs the protein to the enclosing membrane of secretory
vesicles. This targeting motif appears to be present within aa 11–261, because a protein with this sequence still is in the particulatefraction (not shown). We suggest that PTP-MEG2 is a permanentresident of the secretory vesicle membrane and there acts as asensor for PtdIns(3,4,5)P3 (and perhaps also PtdIns(3,5)P2). Whenthe lipid is present, PTP-MEG2 dephosphorylates one or severalsubstrates involved in secretory vesicle fusion and/or homeostasis.This model is supported by the recent finding that PTP-MEG2lacking its Sec14p homology domain has a considerably higheractivity than the full-length enzyme (42). Thus, the Sec14p ho-mology domain appears to be involved in intramolecular regula-tion of the catalytic activity of PTP-MEG2.
A curious finding in our studies is that intracellular PtdIns(3,4,5)P3
clearly colocalized only with wild-type PTP-MEG2, but not detect-ably with either PTP-MEG2-C515S (which still binds the lipid) or toPTP-MEG2-K55M/K184M (Fig. 7). Although the differences couldbe due to the detection limit of the GFP-Btk-PH related to the absenceof fusion into a more concentrated compartment, it may also be thatactive PTP-MEG2 has an effect on the amount of PtdIns(3,4,5)P3 inthe secretory vesicle membrane. Such an effect could be due to stim-ulated PtdIns(3,4,5)P3 synthesis, an inhibitory effect on the enzyme(s)that dephosphorylate PtdIns(3,4,5)P3, sequestration of the inositolhead group from such phosphatases, or a combination of these. Re-solving these issues will require identification of the phosphoinositidekinases and phosphatases responsible for the synthesis ofPtdIns(3,4,5)P3 in the post-Golgi vesicles of the secretory pathway.
A recent study (43) disagrees with our findings and instead re-ports that the Sec14p homology domain of PTP-MEG2 bindsphosphatidylserine (PtdSer). We do not understand the basis forthis discrepancy, but we note that binding of phosphoinositides hasbeen reported very recently by two other laboratories (44, 45). Itmay also be significant that the Sec14p domain from PTPX1, theXenopus laevis ortholog of PTP-MEG2, in our hands bound PtdSerand PtdIns, but not phosphoinositides, in experiments conducted inparallel with those shown in Figs. 2 and 3. In contrast, PTP-MEG2never bound any PtdSer. Although we cannot categorically ex-clude the possibility that PTP-MEG2 is more versatile or has amodifiable specificity, the findings we report in this study are notcompatible with binding of PtdSer.
The Sec14p homology domain is apparently an ancient proteinmodule that has evolved to fulfill numerous physiological func-tions in signal transduction and intracellular transport in plants,yeast, invertebrates, and vertebrates (9). A comprehensive (46) da-tabase search (A. Godzik and T. Mustelin, unpublished data) re-vealed many more Sec14p homology domains from several organ-isms, including 19 from Caenorhabditis elegans and 23 fromArabidopsis thaliana, a polyphosphoinositide transfer protein(Ssh1p) from a plant (Glycine max), �100 protein fragments in theDrosophila melanogaster genomic database, numerous proteinsfrom different strains of yeast, and over 20 different mammalianproteins. Like FYVE, PH, and C2 domains, Sec14p homology do-mains are often found in proteins involved in signal transduction,intracellular traffic, and the regulation of small GTP-binding proteinsof the Ras superfamily. The presence of an N-terminal Sec14p ho-mology domain in several GTP/GDP exchange factors for Ras familyproteins is particularly intriguing. The conversion of the GTP/GDPexchange factor Dbl into a transforming oncogene occurred by loss ofan N-terminal fragment that coincides with its Sec14p homology do-main (47). It will be interesting to see what phospholipids or phos-pholipid-like molecules these proteins bind.
AcknowledgmentsWe are grateful to Philip Majerus for the kind gift of the PTP-MEG2cDNA and to Lewis C. Cantley for GFP constructs and advice.
6670 PTP-MEG2 REGULATION BY PHOSPHOINOSITIDES
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

References1. Roche, P. A. 1999. Intracellular protein traffic in lymphocytes: “How do I get
THERE from HERE”? Immunity 11:391.2. Page, L. J., A. J. Darmon, R. Uellner, and G. M. Griffiths. 1998. L is for lytic
granules: lysosomes that kill. Biochim. Biophys. Acta 1401:146.3. Altman, A., K. M. Coggeshall, and T. Mustelin. 1990. Molecular events medi-
ating T cell activation. Adv. Immunol. 48:227.4. Wang, X., H. Huynh, A. Gjorloff-Wingren, E. Monosov, M. Stridsberg,
M. Fukuda, and T. Mustelin. 2002. Enlargement of secretory vesicles by proteintyrosine phosphatase PTP-MEG2 in RBL mast cells and Jurkat T cells. J. Im-munol. 168:4612.
5. Gu, M., I. Warshawsky, and P. W. Majerus. 1992. Cloning and expression of acytosolic megakaryocyte protein-tyrosine-phosphatase with sequence homologyto retinaldehyde-binding protein and yeast SEC14p. Proc. Natl. Acad. Sci. USA89:2980.
6. Del Vecchio, R. L., and N. K. Tonks. 1994. Characterization of two structurallyrelated Xenopus laevis protein tyrosine phosphatases with homology to lipid-binding proteins. J. Biol. Chem. 269:19639.
7. Merkulova, M. I., S. G. Andreeva, T. M. Shuvaeva, S. V. Novoselov,I. V. Peshenko, M. F. Bystrova, V. I. Novoselov, E. E. Fesenko, andV. M. Lipkin. 1999. A novel 45 kDa secretory protein from rat olfactory epithe-lium: primary structure and localisation. FEBS Lett. 450:126.
8. Sato, Y., H. Arai, A. Miyata, S. Tokita, K. Yamamoto, T. Tanabe, and K. Inoue.1993. Primary structure of �-tocopherol transfer protein from rat liver: homologywith cellular retinaldehyde-binding protein. J. Biol. Chem. 268:17705.
9. Aravind, L., A. F. Neuwald, and C. P. Ponting. 1999. Sec14p-like domains inNF1 and Dbl-like proteins indicate lipid regulation of Ras and Rho signaling.Curr. Biol. 9:R195.
10. Altschul, S. F., T. L. Madden, A. A. Schaeffer, J. Zhang, Z. Zhang, W. Miller, andD. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of pro-tein database search programs. Nucleic Acids Res. 25:3389.
11. Rychlewski, L., L. Jaroszewski, W. Li, and A. Godzik. 2000. Comparison ofsequence profiles: strategies for structural predictions using sequence informa-tion. Protein Sci. 9:232.
12. Godzik, A., A. Kolinski, and J. Skolnick. 1992. A topology fingerprint approachto the inverse folding problem. J. Mol. Biol. 227:227.
13. Sali, A., and T. L. Blundell. 1993. Comparative protein modeling by satisfactionof spatial restraints. J. Mol. Biol. 234:779.
14. Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993.PROCHECK: a program to check the stereochemical quality of protein struc-tures. J. Appl. Crystallogr. 26:283.
15. Bowie, J. U., R. Luethy, and D. Eisenberg. 1991. A method to identify proteinsequences that fold into a known three dimensional structure. Science 253:164.
16. von Willebrand, M., T. Jascur, N. Bonnefoy-Berard, H. Yano, A. Altman,Y. Matsuda, and T. Mustelin. 1996. Inhibition of phosphatidylinositol 3-kinaseblocks TCR/CD3-induced activation of the mitogen-activated kinase Erk2. Eur.J. Biochem. 235:828.
17. Saxena, M., S. Williams, J. Gilman, and T. Mustelin. 1998. Negative regulationof T cell antigen receptor signaling by hematopoietic tyrosine phosphatase(HePTP). J. Biol. Chem. 273:15340.
18. Deaka, M., A. Casamayora, R. A. Currieb, C. P. Downes, and D. R. Alessi. 1999.Characterisation of a plant 3-phosphoinositide-depdendent protein kinase-1 ho-mologue which contains a pleckstrin homology domain. FEBS Lett. 451:220.
19. Alonso, A., M. Saxena, S. William, and T. Mustelin. 2001. Inhibitory role fordual-specificity phosphatase VHR in T cell antigen receptor and CD28-inducedErk and Jnk activation. J. Biol. Chem. 276:4766.
20. Couture, C., Z. Songyang, T. Jascur, S. Williams, P. Tailor, L. C. Cantley, andT. Mustelin. 1996. Regulation of the Lck SH2 domain by tyrosine phosphoryla-tion. J. Biol. Chem. 271:24880.
21. Jascur, T., J. Gilman, and T. Mustelin. 1997. Involvement of phosphatidylinositol3-kinase in NFAT activation in T cells. J. Biol. Chem. 272:14483.
22. Saxena, M., S. Williams, J. Brockdorff, J. Gilman, and T. Mustelin. 1999. Inhi-bition of T cell signaling by MAP kinase-targeted hematopoietic tyrosine phos-phatase (HePTP). J. Biol. Chem. 274:11693.
23. Saxena, M., S. Williams, K. Tasken, and T. Mustelin. 1999. Crosstalk betweencAMP-dependent kinase and MAP kinase through hematopoietic protein tyrosinephosphatase (HePTP). Nat. Cell Biol. 1:305.
24. Gjorloff-Wingren, A., M. Saxena, S. Han, X. Wang, A. Alonso, M. Renedo,P. Oh, S. Williams, J. Schnitzer, and T. Mustelin. 2000. Subcellular localizationof intracellular protein tyrosine phosphatases in T cells. Eur. J. Immunol.30:2412.
25. Sha, B., S. E. Phillips, V. A. Bankaitis, and M. Luo. 1998. Crystal structure of theSaccharomyces cerevisiae phosphatidylinositol-transfer protein. Nature 391:506.
26. Cockcroft, S. 1998. Phosphatidylinositol transfer proteins: a requirement in signaltransduction and vesicle traffic. BioEssays 20:423.
27. Salim, K., M. J. Bottomley, E. Querfurth, M. J. Zvelebil, I. Gout, R. Scaife,R. L. Margolis, R. Gigg, C. I. Smith, P. C. Driscoll, et al. 1996. Distinct speci-ficity in the recognition of phosphoinositides by the pleckstrin homology domainsof dynamin and Bruton’s tyrosine kinase. EMBO J. 15:6241.
28. Burd, C. G., and S. D. Emr. 1998. Phosphatidylinositol(3)-phosphate signalingmediated by specific binding to RING FYVE domains. Mol. Cell 2:157.
29. Gaullier, J.-M., A. Simonsen, A. D’Arrigo, B. Bremnes, H. Stenmark, andR. Aasland. 1998. FYVE fingers bind PtdIns(3)P. Nature 394:432.
30. Patki, V., D. C. Lawe, S. Corvera, J. V. Virbasius, and A. Chawla. 1998. Afunctional PtdIns(3)P-binding motif. Nature 394:433.
31. Fruman, D. A., R. E. Meyers, and L. C. Cantley. 1998. Phosphoinositide kinases.Annu. Rev. Biochem. 67:481.
32. Volinia, S., R. Dhand, B. Vanhaesebroeck, L. K. MacDougall, R. Stein,M. J. Zvelebil, J. Domin, C. Panaretou, and M. D. Waterfield. 1995. A humanphosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p pro-tein sorting system. EMBO J. 14:3339.
33. Sbrissa, D., O. C. Ikonomov, and A. Shisheva. 1999. PIKfyve, a mammalianortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. J. Biol.Chem. 274:21589.
34. Shisheva, A., D. Sbrissa, and O. C. Ikonomov. 1999. Cloning, characterization,and expression of a novel Zn2�-binding FYVE finger-containing phosphoinosi-tide kinase in insulin-sensitive cells. Mol. Cell. Biol. 19:623.
35. Wurmser, A. E., J. D. Gary, and S. D. Emr. 1999. Phosphoinositide 3-kinases andtheir FYVE domain-containing effectors as regulators of vacuolar/lysosomalmembrane trafficking pathways. J. Biol. Chem. 274:9129.
36. Corvera, S., A. D’Arrigo, and H. Stenmark. 1999. Phosphoinositides in mem-brane traffic. Curr. Opin. Cell Biol. 11:460.
37. Martin, T. F. 1997. Phosphoinositides as spatial regulators of membrane traffic.Curr. Opin. Neurobiol. 7:331.
38. Rameh, L. E., and L. C. Cantley. 1999. The role of phosphoinositide 3-kinaselipid products in cell function. J. Biol. Chem. 274:8347.
39. Klarlund, J. K., A. Guilherme, J. J. Holik, J. V. Virbasius, A. Chawla, andM. P. Czech. 1997. Signaling by phosphoinositide-3,4,5-trisphosphate throughproteins containing pleckstrin and Sec7 homology domains. Science 275:1927.
40. Chung, S. H., W. J. Song, K. Kim, J. J. Bednarski, J. Chen, G. D. Prestwich, andR. W. Holz. 1998. The C2 domains of Rabphilin3A specifically bind phospha-tidylinositol 4,5-bisphosphate containing vesicles in a Ca2�-dependent manner:in vitro characteristics and possible significance. J. Biol. Chem. 273:10240.
41. Sudhof, T. C., and J. Rizo. 1996. Synaptotagmins: C2-domain proteins that reg-ulate membrane traffic. Neuron 17:379.
42. Qi, Y., R. Zhao, H. Cao, X. Sui, S. B. Krantz, and Z. J. Zhao. 2002. Purificationand characterization of protein tyrosine phosphatase PTP-MEG2. J. Cell. Bio-chem. 86:79.
43. Zhao, R., X. Fu, Q. Li, S. B. Krantz, and Z. J. Zhao. 2003. Specific interactionof tyrosine phosphatase PTP-MEG2 with phosphatidylserine. J. Biol. Chem. 278:22609.
44. Krugmann, S., K. E. Anderson, S. H. Ridley, N. Risso, A. McGregor,J. Coadwell, K. Davidson, A. Eguinoa, C. D. Ellson, P. Lipp, et al. 2002. Iden-tification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases,by selective capture on phosphoinositide affinity matrices. Mol. Cell 9:95.
45. Kruger, J. M., T. Fukushima, V. Cherepanov, N. Borregaard, C. Loeve, C. Shek,K. Sharma, A. K. Tanswell, C.-W. Chow, and G. P. Downey. 2002. Protein-tyrosine phosphatase MEG2 is expressed by human neutrophils: localization tothe phagosome and activation by polyphosphoinositides. J. Biol. Chem.277:2620.
46. Li, W., F. Pio, K. Pawlowski, and A. Godzik. 2000. Saturated Blast: detectingdistant homology using automated multiple intermediate sequence Blast search.Bioinformatics 16:1105.
47. Eva, A., G. Vecchio, M. Diamond, S. R. Tronick, D. Ron, G. M. Cooper, andS. A. Aaronson. 1987. Independently activated dbl oncogenes exhibit similar yetdistinct structural alterations. Oncogene 1:355.
6671The Journal of Immunology
by guest on February 6, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















