
Primary responses of root and leaf elongation to water deficits ...

DOI: 10.1126/scitranslmed.3009278, 256ra135 (2014);6 Sci Transl Med
et al.Hans T. Bjornssondeficits in a mouse model of Kabuki syndromeHistone deacetylase inhibition rescues structural and functional brain
Editor's Summary
new strategy for treating intellectual disability.disorders could be treated by modulating the epigenetic machinery, and that targeting adult neurogenesis may be a deficiency and the hippocampal memory defects were normalized. This suggests that Kabuki syndrome and relatedmouse model. After treatment with AR-42, an agent known to promote open chromatin, both the neurogenesis one of its symptoms. They discover a postnatal deficiency in neurogenesis and hippocampal memory defects in theirutilize a genetically engineered mouse model of Kabuki syndrome, a disorder that includes intellectual disability as
.et alIntellectual disability is common in the population, yet there are few available treatments. Now, Bjornsson
Kabuki Syndrome: A Potentially Reversible Form of Intellectual Disability
http://stm.sciencemag.org/content/6/256/256ra135.full.htmlcan be found at:
and other services, including high-resolution figures,A complete electronic version of this article
http://stm.sciencemag.org/content/suppl/2014/09/29/6.256.256ra135.DC1.html can be found in the online version of this article at: Supplementary Material
http://www.sciencemag.org/about/permissions.dtl in whole or in part can be found at: article
permission to reproduce this of this article or about obtaining reprintsInformation about obtaining
is a registered trademark of AAAS. Science Translational Medicinerights reserved. The title NW, Washington, DC 20005. Copyright 2014 by the American Association for the Advancement of Science; alllast week in December, by the American Association for the Advancement of Science, 1200 New York Avenue
(print ISSN 1946-6234; online ISSN 1946-6242) is published weekly, except theScience Translational Medicine
on
Oct
ober
7, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
o
n O
ctob
er 7
, 201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om

R E S EARCH ART I C L E
NEUROB IOLOGY
Histone deacetylase inhibition rescues structuraland functional brain deficits in a mouse modelof Kabuki syndromeHans T. Bjornsson,1,2*† Joel S. Benjamin,1,3† Li Zhang,1 Jacqueline Weissman,1,4
Elizabeth E. Gerber,1 Yi-Chun Chen,1 Rebecca G. Vaurio,4 Michelle C. Potter,5
Kasper D. Hansen,1,6 Harry C. Dietz1,7,8
ctob
er 7
, 201
4
Kabuki syndrome is caused by haploinsufficiency for either of two genes that promote the opening of chromatin. If animbalance between open and closed chromatin is central to the pathogenesis of Kabuki syndrome, agents that pro-mote chromatin opening might have therapeutic potential. We have characterized a mouse model of Kabuki syn-drome with a heterozygous deletion in the gene encoding the lysine-specific methyltransferase 2D (Kmt2d),leading to impairment of methyltransferase function. In vitro reporter alleles demonstrated a reduction in histone4 acetylation and histone 3 lysine 4 trimethylation (H3K4me3) activity in mouse embryonic fibroblasts fromKmt2d+/bGeo mice. These activities were normalized in response to AR-42, a histone deacetylase inhibitor. In vivo,deficiency of H3K4me3 in the dentate gyrus granule cell layer of Kmt2d+/bGeo mice correlated with reduced neuro-genesis and hippocampal memory defects. These abnormalities improved upon postnatal treatment with AR-42.Our work suggests that a reversible deficiency in postnatal neurogenesis underlies intellectual disability in Kabukisyndrome.
O
onst
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
INTRODUCTION
Kabuki syndrome is an autosomal dominant condition caused by het-erozygous loss-of-function mutations in either of two genes (1–3) withcomplementary functions: lysine-specific methyltransferase 2D (KMT2D)on human chromosome 12 or lysine-specific demethylase 6A (KDM6A)on human chromosome X. KMT2D is a methyltransferase that adds a tri-methylation mark to H3K4 (H3K4me3, an open chromatin mark),whereas KDM6A is a demethylase that removes trimethylation from his-tone 3 lysine 27 (H3K27me3, a closed chromatin mark). Both genes facil-itate the opening of chromatin and promote gene expression (1–3). It istherefore likely that the observed gene dosage sensitivity in Kabuki syn-drome, despite the apparent redundancy of the H3K4 trimethylation ma-chinery, involves a relative imbalance between open and closed chromatinstates for critical target genes. If this is the case, it may be possible to restorethis balance with drugs that promote open chromatin states, such as his-tonedeacetylase inhibitors (HDACi). To test this hypothesis, wehave char-acterized a mouse model of Kabuki syndrome with a heterozygousmutation inKmt2d that results in replacement of the SET (suvar, enhancerof zeste, trithorax) methyltransferase domain (Kmt2d+/bGeo) by a b-Geocassette. Kmt2d+/bGeomice have hippocampal memory defects that corre-late with multiple abnormalities in the granule cell layer of the dentategyrus, a prominent site of adult neurogenesis (4, 5). Guided by the resultsof in vitro analyses using reporter alleles that monitored histone 4 (H4)
1McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School ofMedicine, Baltimore, MD 21205, USA. 2Department of Pediatrics, Johns Hopkins UniversitySchool of Medicine, Baltimore, MD 21205, USA. 3Predoctoral Training Program in HumanGenetics, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins UniversitySchool of Medicine, Baltimore, MD 21205, USA. 4Kennedy Krieger Institute, Baltimore, MD21205, USA. 5Brain Science Institute, Neurology Department, Johns Hopkins UniversitySchool of Medicine, Baltimore, MD 21205, USA. 6Department of Biostatistics, BloombergSchool of Public Health, Baltimore, MD 21205, USA. 7Division of Pediatric Cardiology,Department of Pediatrics, Johns Hopkins University School of Medicine, Baltimore, MD21205, USA. 8Howard Hughes Medical Institute, Baltimore, MD 21205, USA.*Corresponding author. E-mail: [email protected]†These authors contributed equally to this work.
www.Scienc
acetylation and H3K4 trimethylation in cells derived from Kabuki syn-drome mice, we find that oral administration of the HDACi AR-42 to ei-ther young (1- to 2-month-old) or adult (5- to 6-month-old)Kmt2d+/bGeo
micenormalizedboth structural and functional deficits in thedentate gyrusin association with restoration of H3K4 trimethylation.
RESULTS
Kmt2d +/bGeo miceKMT2D is a member of the mixed lineage leukemia (MLL) family ofDrosophila trithorax orthologs that is encoded on human chromosome12 and mouse chromosome 15. An alternative name for KMT2D ismixed lineage leukemia 2 (MLL2). All members of this family containa SET domain, which confers the H3K4 methyltransferase activity, aswell as other domains (6) that delineate individual functions (Fig. 1A).A mouse model harboring a loss-of-function allele for Kmt2b, encodedon human chromosome 19 and mouse chromosome 7, has been char-acterized previously (7), demonstrating hippocampal memory defects.This gene has been alternatively designatedMll4 orMll2, leading to con-fusion in the literature regarding nomenclature for this particular genefamily, as discussed in a recent publication by Bögershausen et al. (8).To specifically assess the underlying pathogenesis of Kabuki syndrome,we have characterized a mouse model with insertion of an expressioncassette encoding a b-galactosidase neomycin resistance fusion protein(b-Geo) into intron 50 of Kmt2d (Mll2) on mouse chromosome 15. In-clusion of a splice acceptor sequence and a 3′ end cleavage and polyad-enylation signal at the 5′ and 3′ ends of the b-Geo cassette, respectively,is predicted to generate a truncated KMT2D protein with peptidesequence corresponding to the first 50 exons of Kmt2d fused to b-Geo,but lacking the SET domain and therefore methyltransferase activity(Fig. 1B and fig. S1A). As predicted from this targeting event, quantita-tive real-time polymerase chain reaction (PCR) analysis of Kmt2dmes-senger RNA in Kmt2d+/bGeo mice demonstrated normal abundance of
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 1

R E S EARCH ART I C L E
sequence corresponding to exon 20 but a 50% reduction for exon 52,when compared to Kmt2d+/+ littermates (Fig. 1C). Expression of aKMT2D–b-galactosidase fusion protein in Kmt2d+/bGeo animals dem-onstrated transcription and translation of the targeted allele (fig. S1B).Furthermore, chromatin immunoprecipitation followed by next-generation
www.Scienc
on
Oct
ober
7, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
sequencing (ChIP-seq) on splenic cells fromKmt2d+/bGeomice andKmt2d+/+
littermates using an antibody against H3K4me3 revealed an overall genome-wide decrease in H3K4me3 in Kmt2d+/bGeo mice (Fig. 1D), supporting thepredicted functional consequences of the mutant allele. Finally, Kmt2d+/bGeo
mice demonstrated facial features that are consistent with Kabuki syn-drome including flattened snout (fig. S2A) and downward rotation ofthe ear canal (fig. S2B). Blinded analysis of x-rays of Kmt2d+/bGeo micerevealed a significantly shorter maxilla (P < 0.005) when compared toKmt2d+/+ littermates (fig. S2, B and C), as judged by the extent of pro-trusion beyond the mandible (fig. S2C).
Kmt2d +/bGeo mice demonstrate hippocampalmemory defectsDisruption of several histone-modifying enzyme genes has been shownto lead to hippocampal memory defects in mice, illustrating a criticalrole for epigenetic homeostasis in memory acquisition (9–11). Kmt2d+/bGeo
miceshowedsignificantdeficits innovelobject recognition(P<0.05;Fig. 1E),Morris water maze probe trial (P < 0.005; Fig. 1F), and contextual fearconditioning (P < 0.05; fig. S3) when compared to Kmt2d+/+ littermates,all consistent with hippocampal memory dysfunction. When performedbefore the hidden platform stage of training, the flag-training phase of theMorris water maze did not reveal significant differences betweenKmt2d+/bGeo and Kmt2d+/+ littermates (fig. S4). Kmt2d+/bGeo mice didnot show decreased activity (fig. S5A), reduced grip strength (fig. S5B),or slower swim speeds (fig. S5C), any of which would be indicative of amore generalized limitation of performance potential in these assays.There were no significant differences in the time that it took Kmt2d+/bGeo
mice to identify the platform (escape latency) compared to Kmt2d+/+ miceduring the training phase (fig. S6).
Decreased dentate gyrus volume and defectiveneurogenesis in Kmt2d+/bGeo miceImmunofluorescence analyses revealed particularly high levels of ex-pression of KMT2D protein in the dentate gyrus granule cell layer ofthe hippocampus in Kmt2d+/+ mice (Fig. 2A) and a marked deficiencyofH3K4me3 in the dentate gyrus granule cell layer ofKmt2d+/bGeomicecompared to Kmt2d+/+ littermates (P < 0.05; Fig. 2, B and C). We alsosaw a similar deficiency of H3K4me3 in the pyramidal layer of the hip-pocampus (P<0.01; fig. S7). The amount ofH3K4me3 showed substan-tial cell-to-cell variability in Kmt2d+/bGeo animals (Fig. 2B), suggestingthat variation in cell state or identity within the granule cell layer or den-tate gyrus may influence vulnerability to the consequences of heterozy-gousKmt2d disruption.Kmt2d+/bGeomice showed a significant decreasein body weight but not brain weight (P < 0.05; fig. S8), and had reduceddentate gyrus granule cell layer volume when standardized to brainweight (P < 0.05; Fig. 2, D and E). This correlated with reduced neuro-genesis in the granule cell layer of Kmt2d+/bGeo mice, as evidenced bysignificantly reduced expression of both doublecortin (DCX) (12) (P <0.001; Fig. 2, F and G) and 5-ethynyl-2′-deoxyuridine (EdU) staining, amarker of both neurogenesis in the granule cell layer and a marker ofneuronal survival when monitored 30 days after labeling (P < 0.01;fig. S9). Confocalmicroscopy revealed an apparent decrease in dendriticbranching complexity of DCX-positive (DCX+) cells in the granule celllayer of Kmt2d+/bGeo mice (fig. S10). However, given the decreasedamounts ofDCX+ cells in thesemice, furtherwork is needed todeterminewhether this is a true or primary manifestation of Kmt2d deficiency.Staining with an antibody against activated caspase 3 did not reveal evi-dence for enhanced cell death in thedentate gyrus inheterozygous-targeted
Fig. 1. Hippocampalmemory defects in a Kmt2d+/bGeomousemodel ofKabuki syndrome. (A) Domain organization of KMT2D in human and
mouse, with the relative position of the H3K4methyltransferase SET domainindicated in red and other domains by additional colors. The human andmurine chromosomal assignment (Chr) is shown. (B) The Kmt2dbGeo target-ing event introduced a b-Geo cassette including a strong splice acceptor (SA)sequence and a 3′ cleavage and polyadenylation signal (pA) into intron 50 ofKmt2d onmouse chromosome 15 (fig. S1A). (C) Real-time PCR using primersspecific for exons 20 or 52 of Kmt2d (arrows) confirmed a substantial reduc-tion (∼50%) in mRNA corresponding to sequences distal to the b-Geoinsertion site when compared to proximal sequences in Kmt2d+/bGeo mice,in comparison to Kmt2d+/+ littermates. Results reflected three technical re-plicates for each of three Kmt2d+/+ and two Kmt2d+/bGeo mice. (D) ChIP-seqrevealed a genome-wide deficiency of H3K4me3 in cells from Kmt2d+/bGeomice when compared to cells from Kmt2d+/+ littermates. A positive valueindicates a higher locus-specific peak in Kmt2d+/bGeo mice. Each pointcorresponds to a genomic location with a peak in at least one sample.Significantly differentially bound loci are red, whereas others are gray.(E) There was no difference in positional preference between genotypesduring the habituation phase [identical objects (L/R)] of the novel objectrecognition test. Kmt2d+/bGeo mice spent less time with a novel object (L)and more with a habituated object (R) compared to Kmt2d+/+ littermates.Kmt2d+/+ littermates also demonstrated significant improvement fromhabituation phase [novel object (L)], whereas Kmt2d+/bGeo mice did not.n = 13 (+/+), n = 10 (+/bGeo). (F) Kmt2d+/bGeo mice showed a reducedfrequency in platform zone crossings during the probe trial phase of Morriswater maze testing. n = 48 (+/+), n = 32 (+/bGeo). *P < 0.05; †P < 0.005; ††P <0.001, t test.
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 2

R E S EARCH ART I C L E
animals (fig. S11). To explore whether there are hippocampal memorydefects in patients with Kabuki syndrome, we analyzed comprehensiveneuropsychological testing performed on three patients with knowndisease-causing mutations in KMT2D (Table 1). Although not all defi-ciencies observed can be explained by hippocampal dysfunction, pa-tients consistently had abnormalities in tasks known to be associatedwith dentate gyrus function (13–15). Other functions linked to otherregions of the hippocampus (16) were also abnormal in some patients,as were some tasks not linked to hippocampus, indicating that other cellpopulations in the central nervous systemmay also play a role. These data
www.Scienc
r 7,
201
4
support the hypothesis that observations inKmt2d+/bGeomice are, at leastin part, reminiscent of findings in Kabuki syndrome in human patients.
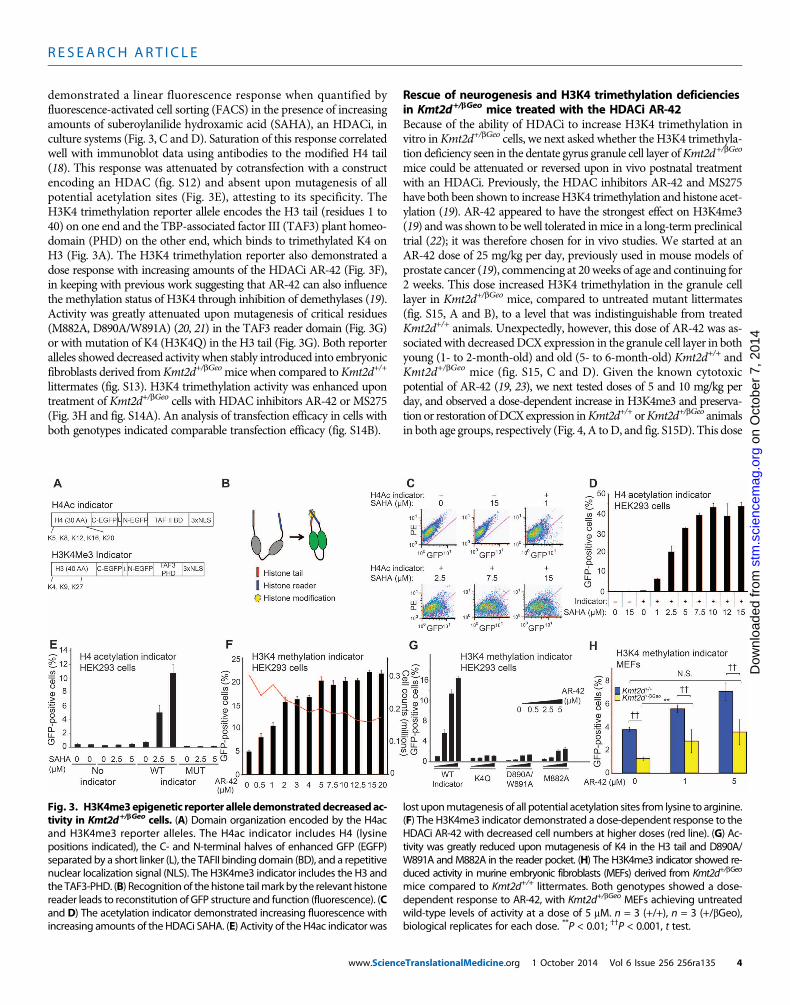
Reporter alleles for epigenetic modifications in embryonicfibroblasts from Kmt2d+/bGeo miceWe created epigenetic reporter systems that monitored either H4 acet-ylation or H3K4 trimethylationmachinery activity in an effort to deter-mine whether there was an ongoing activity deficiency in cells fromKmt2d+/bGeomice (Fig. 3A). Both reporter alleles encode halves of greenfluorescent protein (GFP) separated by a flexible linker region (17) witha histone tail and a histone reader at the N and C termini, respectively.When the histone tail corresponding to either H4 or H3 is modified byacetylation ormethylation, respectively, GFP structure and function arereconstituted, as detected by a fluorescent readout (Fig. 3B). The acetylreporter protein quantifies the activity of the acetylation machinery(acetylation of H4 specifically at sites K5, K8, K12, and K16) and com-prises anH4 tail (residues 1 to 30) on one end and aTATAbox–bindingprotein (TBP)–associated factor II (TAFII) bromodomain on the otherend (Fig. 3A). The TAFII bromodomain only recognizes and binds tothe acetylated H4 tail. This acetylation-dependent reporter protein
on
Oct
obe
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
Fig. 2. A global deficiency of H3K4me3 in the dentate gyrus associatedwith reducedgranule cell layer volumeandneurogenesis inKmt2d+/bGeo
mice. (A) Immunofluorescence revealed intense expression of KMT2D (redsignal) in the dentate gyrus granule cell and pyramidal layers of Kmt2d+/+
mice. (B) Immunofluorescence for H3K4me3 (red) and4′,6-diamidino-2-pheny-lindole (DAPI) (blue) in the granule cell layer of Kmt2d+/bGeomice and Kmt2d+/+
littermates. (C) Quantification revealed a reduced H3K4me3/DAPI signal in-tensity ratio within the granule cell layer of Kmt2d+/bGeo mice comparedto Kmt2d+/+ littermates. n = 9 (+/+), n = 5 (+/bGeo). (D and E) Calculation ofgranule cell layer area (red outline) in every sixth brain slice allowed thedemonstration of reduced granule cell layer volume in Kmt2d+/bGeomice com-pared to Kmt2d+/+ littermates. n = 4 (+/+), n = 5 (+/bGeo). (F and G) Immu-nofluorescence revealed reduced representation of cells positive for DCX, amarker for neurogenesis, in the granule cell layer of Kmt2d+/bGeomice com-pared to Kmt2d+/+ littermates. n = 4 (+/+), n = 4 (+/bGeo). *P < 0.05; ††P <0.001, t test.
Table 1. Neuropsychological findings in patients with Kabuki syn-drome. A retrospective analysis of neuropsychological testing on threepatients with mutations in KMT2D revealed consistent abnormalities offunctions that have been associated with the dentate gyrus. N/A, notadequately tested with used testing regimen; ↓, deficient area (definedas >1 SD below the mean and lower than full-scale IQ or, if unavailable,highest individual test score); metrics linked to the dentate gyrus are inbold font (13–15); metrics more broadly linked to the hippocampus areindicated with an asterisk (16).
Neuropsychologic process/function
eTranslationalMedicine.org 1 October 20
Patient 1
14 Vol 6 Is
Patient 2
sue 256 256r
Patient 3
28 years
15 years 14 yearsFemale
Female MaleAffected gene
KMT2D KMT2D KMT2DFull-scale IQ
87 84 66Perceptual or nonverbal reasoning*
↓ ↓ ↓Verbal reasoning/comprehension
Normal Normal ↓Verbal fluency*
↓ Normal N/ANaming*
Normal Normal NormalVocabulary/reading
Normal Normal N/AProcessing speed
↓ ↓ ↓Basic math calculation
Normal ↓ N/AVisual selective attention*
↓ ↓ N/AVisual working memory*
↓ ↓ ↓Verbal working memory*
Normal Normal ↓Visual delayed memory*
↓ ↓ ↓Verbal delayed memory*
↓ ↓ NormalSwitching/inhibition
↓ ↓ N/AVerbal organization
Normal Normal N/AVisual organization*
↓ ↓ ↓Fine motor
↓ ↓ ↓a135 3
R E S EARCH ART I C L E
n O
ctob
er 7
, 201
4
demonstrated a linear fluorescence response when quantified byfluorescence-activated cell sorting (FACS) in the presence of increasingamounts of suberoylanilide hydroxamic acid (SAHA), an HDACi, inculture systems (Fig. 3, C and D). Saturation of this response correlatedwell with immunoblot data using antibodies to the modified H4 tail(18). This response was attenuated by cotransfection with a constructencoding an HDAC (fig. S12) and absent upon mutagenesis of allpotential acetylation sites (Fig. 3E), attesting to its specificity. TheH3K4 trimethylation reporter allele encodes the H3 tail (residues 1 to40) on one end and the TBP-associated factor III (TAF3) plant homeo-domain (PHD) on the other end, which binds to trimethylated K4 onH3 (Fig. 3A). The H3K4 trimethylation reporter also demonstrated adose response with increasing amounts of the HDACi AR-42 (Fig. 3F),in keeping with previous work suggesting that AR-42 can also influencethe methylation status of H3K4 through inhibition of demethylases (19).Activity was greatly attenuated upon mutagenesis of critical residues(M882A, D890A/W891A) (20, 21) in the TAF3 reader domain (Fig. 3G)or with mutation of K4 (H3K4Q) in the H3 tail (Fig. 3G). Both reporteralleles showed decreased activity when stably introduced into embryonicfibroblasts derived fromKmt2d+/bGeomice when compared toKmt2d+/+
littermates (fig. S13). H3K4 trimethylation activity was enhanced upontreatment of Kmt2d+/bGeo cells with HDAC inhibitors AR-42 or MS275(Fig. 3H and fig. S14A). An analysis of transfection efficacy in cells withboth genotypes indicated comparable transfection efficacy (fig. S14B).
www.Scienc
Rescue of neurogenesis and H3K4 trimethylation deficienciesin Kmt2d+/bGeo mice treated with the HDACi AR-42Because of the ability of HDACi to increase H3K4 trimethylation invitro inKmt2d+/bGeo cells, we next asked whether the H3K4 trimethyla-tion deficiency seen in the dentate gyrus granule cell layer ofKmt2d+/bGeo
mice could be attenuated or reversed upon in vivo postnatal treatmentwith an HDACi. Previously, the HDAC inhibitors AR-42 and MS275have both been shown to increaseH3K4 trimethylation and histone acet-ylation (19). AR-42 appeared to have the strongest effect on H3K4me3(19) andwas shown to bewell tolerated inmice in a long-termpreclinicaltrial (22); it was therefore chosen for in vivo studies. We started at anAR-42 dose of 25 mg/kg per day, previously used in mouse models ofprostate cancer (19), commencing at 20weeks of age and continuing for2 weeks. This dose increased H3K4 trimethylation in the granule celllayer in Kmt2d+/bGeo mice, compared to untreated mutant littermates(fig. S15, A and B), to a level that was indistinguishable from treatedKmt2d+/+ animals. Unexpectedly, however, this dose of AR-42 was as-sociatedwith decreasedDCX expression in the granule cell layer in bothyoung (1- to 2-month-old) and old (5- to 6-month-old) Kmt2d+/+ andKmt2d+/bGeo mice (fig. S15, C and D). Given the known cytotoxicpotential of AR-42 (19, 23), we next tested doses of 5 and 10 mg/kg perday, and observed a dose-dependent increase in H3K4me3 and preserva-tion or restoration ofDCXexpression inKmt2d+/+ orKmt2d+/bGeo animalsin both age groups, respectively (Fig. 4, A toD, and fig. S15D). This dose
ost
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
Fig. 3. H3K4me3epigenetic reporter alleledemonstrateddecreasedac-tivity in Kmt2d+/bGeo cells. (A) Domain organization encoded by the H4ac
lost uponmutagenesis of all potential acetylation sites from lysine to arginine.(F) The H3K4me3 indicator demonstrated a dose-dependent response to the
and H3K4me3 reporter alleles. The H4ac indicator includes H4 (lysinepositions indicated), the C- and N-terminal halves of enhanced GFP (EGFP)separated by a short linker (L), the TAFII binding domain (BD), and a repetitivenuclear localization signal (NLS). The H3K4me3 indicator includes the H3 andthe TAF3-PHD. (B) Recognition of thehistone tailmark by the relevant histonereader leads to reconstitution of GFP structure and function (fluorescence). (Cand D) The acetylation indicator demonstrated increasing fluorescence withincreasing amounts of the HDACi SAHA. (E) Activity of the H4ac indicator was
HDACi AR-42 with decreased cell numbers at higher doses (red line). (G) Ac-tivity was greatly reduced upon mutagenesis of K4 in the H3 tail and D890A/W891A andM882A in the reader pocket. (H) The H3K4me3 indicator showed re-duced activity in murine embryonic fibroblasts (MEFs) derived from Kmt2d+/bGeo
mice compared to Kmt2d+/+ littermates. Both genotypes showed a dose-dependent response to AR-42, with Kmt2d+/bGeo MEFs achieving untreatedwild-type levels of activity at a dose of 5 mM. n = 3 (+/+), n = 3 (+/bGeo),biological replicates for each dose. **P < 0.01; ††P < 0.001, t test.
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 4

R E S EARCH ART I C L E
on
Oct
ober
7, 2
014
mag
.org
also led to a genome-wide increase in H3K4me3 in spleen cells fromKmt2d+/bGeo mice when compared to Kmt2d+/+ littermates on vehicle(Fig. 4E) in associationwith normalization of expression ofKlf10 (fig. S16),a known KMT2D target gene (24). This dose appeared to overcorrect thedeficiency (Fig. 4E), which could be observed when representing data asMA plots (the relationship between log ratios and mean averages, fig.S17) or visualizing the shifts in balance among the two states (fig. S18).We also compared other state combinationswith the same representations,showing a relative normalization of genome-wideH3K4me3 inKmt2d+/bGeo
mice treated with AR-42 when compared to Kmt2d+/+ littermates thatdid (fig. S17E) or did not (fig. S17B) receive drug. The bigger effect atlower intensity log2 counts per million fits with data from ablation ofthe Rubinstein-Taybi gene encoding CREB (cyclic adenosine 3′,5′-monophosphate response element–binding protein)–binding protein(CBP), which has dose-dependent effects on gene expression thoughtto depend on the strength of recruitment for a particular site (25).
Improvement of hippocampal memory defects in Kmt2d+/bGeo
mice treated with AR-42In keeping with the hypothesis that abnormal granule cell layer neuro-genesis contributes to functional deficits, we found that performance inhippocampal memory testing correlated with AR-42 dose-dependenteffects on DCX expression. Specifically, both Kmt2d+/+ and Kmt2d+/bGeo
mice showed improved performance on Morris water maze platformcrossing during probe trial (26) in response to AR-42 (10 mg/kg perday) (P < 0.001), with a greater response in Kmt2d+/bGeo animals and nosignificant difference between genotypes in the treatment group (P = 0.27;Fig. 4F).
stm
.sci
ence
Dow
nloa
ded
from
DISCUSSION
Previous studies have associated structural abnormalities of the dentategyrus with impaired neurogenesis and hippocampal memory defects(27, 28). In accordance with the previously observed phenotype inKmt2b-targeted mice (7), we found that heterozygosity for a loss-of-functionKmt2d allele associates a deficiency ofH3K4me3 in the dentategyrus granule cell layer with hippocampal memory defects in a mousemodel of Kabuki syndrome. Support for a causal relationship is nowincreased by our observation that memory deficits in a mouse modelof Kabuki syndrome can be prevented or even reversed through sys-temic delivery of drugs that directly influence the histone modificationevents that favor chromatin opening.
Our data support the hypothesis that the neurodevelopmental defi-ciency in Kabuki syndrome is maintained by an impairment of adultneurogenesis because of an imbalance between open and closed chro-matin states for critical target genes. In this light, other Mendelian dis-orders involving the histone modification machinery, now numberingmore than 40 (29), might be amenable to therapeutic intervention withHDAC inhibitors (30–32). In keeping with this concept, neurologicalphenotypes in mouse models of Rubinstein-Taybi syndrome with hap-loinsufficiency for the gene encoding the histone acetyltransferase Cbprespond to intracerebroventricular or intraperitoneal administration ofthe HDACi SAHA or trichostatin A, respectively (33, 34); however, nocellular mechanism was described. The specific correlation betweenH3K4me3 and neurogenesis within the dentate gyrus of Kabuki syn-drome mice offers a potential unifying mechanism for hippocampalmemory defects seen in inherited defects of the histone modification
www.Scienc
machinery (10, 11, 33, 34). The further positive correlation of theseevents with functional outcome supports the hypothesis that the fateof the granule cell layer in the dentate gyrus is a critical determinantof both disease pathogenesis and treatment. More work is needed todetermine the relative contribution of precursor cell recruitment, differ-entiation, proliferation, and survival (35, 36). Future studies using lineage-specificKmt2d targetingwill help to elucidate the contributionof individualcell populations (granule cell layer, pyramidal layer, andmolecular layerof the cerebellum) to specific neurodevelopmental phenotypes.
Although there is an overall decrease inH3K4me3 in the dentate gyrusgranule cell layer of Kmt2d+/bGeo mice, we note substantial cell-to-cell
Fig. 4. In vivo effects of AR-42. (A toD) One- to 2-month-oldmice of bothgenotypes showed an increase in H3K4me3 (A and B) (n = 5 to 6 per group)
associated with a dose-dependent increase in neurogenesis in Kmt2d+/bGeomice (C and D) (monitored by normalized DCX expression) (n = 4 to 6 pergroup) upon treatment with the HDACi AR-42. There was no difference ineither H3K4me3 or neurogenesis between Kmt2d+/bGeo and Kmt2d+/+
animals at a dose of 10 mg/kg per day. (E) The genome-wide deficiency ofH3K4me3 seen in Kmt2d+/bGeomice was improved upon treatment with AR-42 (10 mg/kg per day). (F) The reduced frequency of platform crossing seenduring Morris water maze testing of Kmt2d+/bGeomice was normalized upontreatment with AR-42 (10 mg/kg per day) [n = 48 (+/+, no treatment), n = 32(+/bGeo, no treatment), n= 14 (+/+, 10mg/kg per day AR-42), n= 9 (+/bGeo,10 mg/kg per day AR-42)]. *P < 0.05; **P < 0.01; †P < 0.005; ††P < 0.001, t test.
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 5

R E S EARCH ART I C L E
on
Oct
ober
7, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
variation. This might reflect redundancy of enzymes capable of addingthe H3K4 trimethylation mark (7) that could vary in their expression(and therefore compensation capacity) in a cell type–dependent (for ex-ample, differentiation state) or cell state–dependent (for example,electrochemical environment) manner. Alternatively, this could indi-cate that stochastic events thought to contribute to epigenetic individ-uality (37) play a role.
There is precedent that HDACi increases not only histone acetyla-tion but also H3K4 trimethylation (19). Our indicators nicely illus-trate coupling between H4 acetylation and H3K4 trimethylation, withKmt2d+/bGeomice having defects in both systems. Although AR-42 wasused here for this proof of principle study, such a generalized HDACimay have an unacceptable toxicity profile in patients. The reporter al-leles described here have the potential for application in small-moleculescreens to identify drugs with greater potency, specificity, and tolerance.There are also many U.S. Food and Drug Administration–approvedmedications, some that have had longstanding clinical use, that influ-ence epigenetic modifications in addition to their originally establishedfunctions. An example is the anti-epileptic agent valproic acid, whichwas recently shown to be a potent HDACi (38). Several widely usedsupplements or dietary substances, such as folic acid, genistein, and cur-cumin, are known to influence epigenetic modifications (39). A recentpublication suggests that ketosis, as achieved in a ketogenic diet, mightfavor chromatin opening through b-hydroxybutyrate, an endogenousHDACi (40). This indicates the potential that dietary manipulations,such as a ketogenic diet, might have another therapeutic avenue fortreatment in disorders with a deficiency of open chromatin, such asKabuki syndrome. These observationsmay inform the question of po-tential toxicity of interventions that have broad effects on pervasiveepigenetic events. The apparent tolerance to chronic use of such agentsduring postnatal life likely reflects, at least in part, the complex contextwithinwhich gene transcription andultimate function are achieved. Con-tributing factors include DNAmodifications, a repertoire of both posi-tive and negative effectors of transcription, and feedback mechanismsthat titrate both gene expression and protein function. In this light, thepredominant influence of agents such asHDACi as therapiesmayprovepermissive for correction of pathologic alterations in physiologic geneexpression and function rather than obligate and therefore less condu-cive to homeostasis.
Although we were able to demonstrate a beneficial effect of AR-42treatment on neurogenesis at two different ages (1 to 2months and 5 to6months), suggesting that this subphenotype of Kabuki syndromemaybe treatable even in adulthood, it is well established that neurogenesispotential is age-restricted (41). It will be essential to further refine thewindow of opportunity to influence neurogenesis in the granule celllayer in both mouse models and patients. Although our data suggestthat neurogenesis and hippocampal memory can be recovered after2 weeks of treatment with AR-42, it is unclear if this response can besustained even in the presence of chronic treatment. It is also possible,but as yet unproven, that brief treatment in early postnatal stages willresult in the expansion of a stable population of cells within the granulecell layer (despite an ongoing relative deficiency of methyltransferasefunction) and hence achieve long-term recovery of neurologic function.Finally, our ChIP-seq experiments suggest that AR-42 at a dose of10 mg/kg per day led to the most improvement in functional studies(Fig. 4D) but overcorrection of genome-wide H3K4me3 (Fig. 4E). Giventhe favorable tolerance profile of high-doseHDACiwhen used for cancertreatment, this may not be a limiting factor. However, new challenges
www.Scienc
may arise when HDACis are used chronically for Kabuki syndromeor other neurodevelopmental disorders. The combination of in vivoChIP-seq analyses and in vitro reporter allele performance with regardto H3K4me3 status may ultimately allow optimization in the selectionof agent and dose for therapeutic purposes.
Potential limitations of this study include the use of a single hetero-zygous Kmt2d targeted allele that introduces a b-Geo expressioncassette into the open reading frame. Although this strategy should re-capitulate haploinsufficiency for KMT2D, as seen in Kabuki syndromepatients heterozygous for nonsense alleles, a gain-of-function contri-bution of the fusion protein cannot be formally excluded. There arealso inherent limitations in the use of mouse models in the study ofhuman neurocognitive disorders. Finally, lineage-specific cell target-ing will be required to mechanistically validate the correlative link be-tween performance deficits and defects in adult neurogenesis in thedentate gyrus.
In conclusion, our work suggests that a postnatally ongoing and re-versible deficiency of granule cell layer H3K4me3, in association withalterations in adult neurogenesis, underlies intellectual disability in amouse model of Kabuki syndrome. This work adds to the emergingview that multiple genetic etiologies of intellectual disability may beamenable to postnatal therapies (42–44).
MATERIALS AND METHODS
Study designThe purposes of this study were to explore the pathophysiologicalsequence in Kabuki syndrome, a Mendelian disorder of the epigeneticmachinery, and to seek robust disease-associated phenotypes, whichcould be used to monitor therapeutic response. We hypothesized thatbecause both causes of Kabuki syndrome involve the transition fromclosed to open chromatin, this disorder might be caused by a generalimbalance between open and closed chromatin states (favoring closedchromatin) and this ongoing deficiency might be ameliorated withagents that favor chromatin opening such as HDACi. At least 3 to 4biological replicates were used for each biochemical analysis, whereasa sample size of at least 8 to 10 per groupwas used for behavioral testing.Data collection occurred for a predetermined period of time, as dictatedby literature- or core facility–based standards, and no exclusion criteriawere applied. All analyses were performed by examiners blinded togenotype and/or treatment arm. For drug treatments, animals were ran-domly assigned to treatment arms with approximately equivalentnumbers in each group. Box andwhisker plots identify RStudio-definedoutliers (shown as circles), but all data points were used in statisticalanalyses.
Epigenetic reporter allelesEpigenetic reporter alleles were synthesized (OriGene) using publishedsequences for component elements (17, 45). Single-nucleotide muta-tions were created using the QuikChange Lightening kit (Agilent Tech-nologies Inc.). For H4ac indicator, we introduced K5R, K8R, K12R,K16R, and K20R (MUT indicator). For H3K4me3 indicator, we intro-duced K4Q and D890A/W891A and M882A (three separate con-structs). For transient transfections, mouse embryonic fibroblasts (seebelow) were transfected with FuGENE HD (Promega) 48 hours beforeFACS. Transfection efficiency of reporter alleles was comparable intransiently transfected MEFs derived from mice of both genotypes
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 6

R E S EARCH ART I C L E
on
Oct
ober
7, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
(Kmt2d+/bGeo andKmt2d+/+), asmeasured by real-timePCRof genomicDNA. For drug stimulation, drug was added to the medium 24 hoursbefore FACS. For stable transfections in T293 (American Type CultureCollection) cells, blasticidin (10 mg/ml) (Life Technologies) was addedto the medium for several weeks. For stable transfection in MEFs, thereporter was transferred to a ViraPower Lentiviral Expression System(Life Technologies). After selection with blasticidin, the drug of interestwas added 24 hours before FACS. SAHA, AR-42, andMS275were pur-chased from Selleck Chemicals. FACS was performed using either aFACSCalibur (BD Biosciences) or a FACSVerse (BD Biosciences) sys-tem with comparable results. FACS data were analyzed using FlowJo(Tree Star Inc.). A plasmid expressing HDAC3 was acquired fromAddgene (plasmid 13819) and transfected into a stable cell line carryingthe H4 acetyl reporter allele.
AnimalsOur mouse model, Kmt2d+/bGeo, also named Mll2Gt(RRt024)Byg, wasacquired fromBayGenomics (University of California). All experimen-tal mice were on a mixed C57BL/6J and 129/SvEv background.Expected Mendelian ratios were observed when heterozygous animalswere bred to wild type. In heterozygous crosses, however, there wasuniform embryonic lethality of homozygotes by ED12, the earliest de-velopmental stage assayed. For treatment with AR-42, mice were orallygavaged daily with drug (Selleck Chemicals) solubilized in vehicle (0.5%methylcellulose, 0.1% Tween-80, water) or with vehicle alone. Drug de-livery informationwas provided byC. S. Chen and S.K.Kulp fromOhioStateUniversity (19). Drugwas administered for 14 days, andmiceweresacrificed on day 15. Morris water maze testing was initiated at day 7,and a dose of 10 mg/kg per day was used for these studies. For quan-tification of DCX+ cells, doses of 0, 5, 10, and 25 mg/kg per day were used.Genotyping was performed using primers B-GeoF-(CAAATGGCGAT-TACCGTTGA) and B-GeoR-(TGCCCAGTCATAGCCGAATA),which are specific for the targeted allele, and TcrdF-(CAAATGTTGC-TTGTCTGGTG) andTcrdR-(GTCAGTCGAGTGCACAGTTT), whichcontrol for sufficientDNAconcentration. Real-timePCRusing the sameprimers allows discrimination between the heterozygous and homozy-gous state for the targeted allele. Thymuswas dissected from awild-typeneonatal mouse and flash-frozen in optimal cutting temperature(OCT). Slides were fixed with 4% paraformaldehyde for 30min. All ex-periments were performed using mouse protocols approved by theAnimal Care and Use Committee of Johns Hopkins University Schoolof Medicine. Themouse protocols used for this study are in accordancewith the guidelines used by the National Institutes of Health (NIH) formouse care and handling.
Morris water maze testingMice were placed in a 1.1-m-diameter tank filled with room tempera-ture water dyed with nontoxic white paint. For analysis purposes, thetank was divided into four quadrants, with one quadrant containing asmall platform submerged 1.5 cm beneath the water. On each day oftraining, mice were placed in the tank in a random quadrant facingaway from the center and were allowed to swim until they found theplatform and were left there for 30 s. If they did not reach the platformafter 60 s, they were placed on it for 30 s. Each mouse was given fourtrials per day (for 5 days) with no intertrial interval and subsequentlyreturned to its home cage. Latency to reach the platform was measuredduring each trial. The day after the final day of training, the platformwas removed for a probe trial wheremicewere placed in the tank for 90 s.
www.Scienc
The average number of crossings of the platform’s previous location wasrecorded. Visible/flagged platform trainingwas also performed for 3 dayseither before the hidden platform or after the probe trial, where a visibleflag was placed on the submerged platform, and the time for eachmouseto reach the platformwasmeasured for each 60-s trial, four ofwhichwererun in the same way as the hidden platform training. For all training andprobe testing, data were recorded both manually and electronically withANY-maze software (San Diego Instruments) when applicable. Differ-ences in the number of platform crossings during the probe trial werecompared between groups with a Student’s t test with significance valueset at P < 0.05.
Retrospective analysis of neuropsychological testing onpatients with Kabuki syndromeA retrospective chart review was performed using data from patientsthat had clinically indicated neuropsychological testing at the KennedyKrieger Institute (KKI) in years 2004 to 2014. We analyzed test resultsfrom the three individuals with most extensive testing available and aknown disease-associated mutation in KMT2D. All patient data werecollected after consenting patients and stored in secure electronicdatabase at KKI. For this particular analysis per Kennedy Krieger andJohns Hopkins organizational policy, additional Institutional ReviewBoard review was not required (three or fewer patients). We dividedthe individual tasks into 16 categories and used literature to identifytasks known to be associated with dentate gyrus (13–15) or hippocam-pus (nondentate gyrus) (16).
ChIP-seqSpleens were dissected from eightmice, four from each kmt2d genotype(+/bGeo or +/+), where half of each genotype was treated with AR-42and half with vehicle only. Spleens were minced and passed through a40-mm cell strainer to obtain single-cell suspensions. Ten million cellswere used for each ChIP-seq experiment after the native chromatin im-munoprecipitationprotocol, aspreviouslydescribed (46), usingaChIP-gradeantibody against H3K4me3 (9727, Cell Signaling Technology).
ChIP-seq data analysisSequencing was performed using a MiSeq system (Illumina). Paired-end26–base pair reads (4.8 to 9.6 million) were obtained per sample (tableS1). Reads were aligned to the Mus musculus genome, version mm10,using Bowtie 2 (47).We examined each sample with regard to alignmentrate as well as FRIP (fraction of reads in peaks), a measure of the ChIPefficiency (table S1). FRIPwas computed on the basis of peaks called onlyon specific samples using MACS version 1.4.2 (48). For analysis, readsweremerged into onemeta-sample andpeak callingwas performedusingMACS version 1.4.2 (48). This allowed definition of a superset of 33,517peaks in one or more samples. The number of reads overlapping a peakwas computed using BEDTools version 2.17.0 (49) in the following way:each paired-end read was converted to a single interval containing bothmate coordinates (effectively filling in the insert), and these intervals wereexamined for overlaps with the superset of peaks. This created a peak-by-samplematrix of read counts. Differential binding was assessed using theGLM functionality (50) in edgeR version 3.5.27 (51). A single model wasfit, using all eight samples, with tag-wise variance estimation. Differentcontrasts were examined corresponding to the different hypothesesconsidered in the main text, and peaks were considered differentiallybound if they had a Benjamini-Hochberg corrected P values less than5%. Fold change and overall abundance were calculated as per edgeR.
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 7

R E S EARCH ART I C L E
on
Oct
ober
7, 2
014
stm
.sci
ence
mag
.org
ownl
oade
d fr
om
Statistics and plotsFor all box plots generated through RStudio (RStudio Inc.), themarginsof the box show the upper and lower quartiles, the central line shows themedian, and the whiskers show the range. Circles denote outliers asdefined by the RStudio algorithm. For all column, line, and scatterplotgraphs (generated through Microsoft Excel), the error bars representSEM, with the data point representing the mean of each applicablegroup. Unless otherwise stated, significance between two groups wascalculated with a Student’s t test using a significance value of P <0.05. Two-way repeated-measures analyses of variance (ANOVAs)were calculatedwith SPSS (IBM). For every calculatedP value, the statedn represents the number of animals for each group contributing to thatcomparison. For P value nomenclature, *P < 0.05, **P < 0.01, †P < 0.005,††P < 0.001.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/6/256/256ra135/DC1Materials and MethodsFig. S1. Integration site of gene trap in the Kmt2dbGeo allele.Fig. S2. Kmt2d+/bGeo mice show overlapping phenotypic features with patients with Kabukisyndrome.Fig. S3. Kmt2d+/bGeo mice have context-related memory defects.Fig. S4. Kmt2d+/bGeo mice show no deficit in flag trial.Fig. S5. Assessment of motor function in Kmt2d+/bGeo and Kmt2d+/+ mice.Fig. S6. Escape latencies during Morris water maze training.Fig. S7. H3K4me3 is decreased in the pyramidal layer in Kmt2d+/bGeo mice compared to Kmt2d+/+
littermates.Fig. S8. Body and brain size in Kmt2d+/bGeo mice.Fig. S9. EdU incorporation.Fig. S10. Decreased dendrites in DCX+ cells in the granule cell layer of Kmt2d+/bGeo mice.Fig. S11. Staining for activated caspase 3 does not reveal increased apoptosis in the granulecell layer of Kmt2d+/bGeo mice compared to Kmt2d+/+ littermates.Fig. S12. HDAC3 attenuates signal of the H4ac indicator.Fig. S13. Both indicators demonstrate a deficiency in Kmt2d+/bGeo mice.Fig. S14. Improved H3K4 trimethylation activity in Kmt2d+/bGeo cells transiently transfected withH3K4 trimethylation indicator and treated with MS275.Fig. S15. In vivo responses to AR-42.Fig. S16. AR-42–induced expression of a known KMT2D target gene.Fig. S17. MA plots indicate a shift in the balance of H3K4me3 upon treatment with AR-42.Fig. S18. A visualization of shifts in balance between the two states (genotype or AR-42) as afunction of intensity demonstrates an abnormality in Kmt2d+/bGeo that is responsive to AR-42.Fig. S19. Serum control experiments for antibodies used for immunofluorescence.Table S1. A summary of genotypes, drugs, and quality measures of ChIP-seq experiments.References (52, 53)
D
REFERENCES AND NOTES
1. S. B. Ng, A. W. Bigham, K. J. Buckingham, M. C. Hannibal, M. J. McMillin, H. I. Gildersleeve,A. E. Beck, H. K. Tabor, G. M. Cooper, H. C. Mefford, C. Lee, E. H. Turner, J. D. Smith, M. J. Rieder,K. Yoshiura, N. Matsumoto, T. Ohta, N. Niikawa, D. A. Nickerson, M. J. Bamshad, J. Shendure,Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42,790–793 (2010).
2. D. Lederer, B. Grisart, M. C. Digilio, V. Benoit, M. Crespin, S. C. Ghariani, I. Maystadt, B. Dallapiccola,C. Verellen-Dumoulin, Deletion of KDM6A, a histone demethylase interacting with MLL2, in threepatients with Kabuki syndrome. Am. J. Hum. Genet. 90, 119–124 (2012).
3. N. Miyake, S. Mizuno, N. Okamoto, H. Ohashi, M. Shiina, K. Ogata, Y. Tsurusaki, M. Nakashima,H. Saitsu, N. Niikawa, N. Matsumoto, KDM6A point mutations cause Kabuki syndrome. Hum.Mutat. 34, 108–110 (2013).
4. M. Brus, M. Keller, F. Lévy, Temporal features of adult neurogenesis: Differences and simi-larities across mammalian species. Front. Neurosci. 7, 135 (2013).
5. J. Altman, Are new neurons formed in the brains of adult mammals? Science 135, 1127–1128(1962).
6. S. Hunter, P. Jones, A. Mitchell, R. Apweiler, T. K. Attwood, A. Bateman, T. Bernard, D. Binns,P. Bork, S. Burge, E. de Castro, P. Coggill, M. Corbett, U. Das, L. Daugherty, L. Duquenne,
www.Scienc
R. D. Finn, M. Fraser, J. Gough, D. Haft, N. Hulo, D. Kahn, E. Kelly, I. Letunic, D. Lonsdale,R. Lopez, M. Madera, J. Maslen, C. McAnulla, J. McDowall, C. McMenamin, H. Mi, P. Mutowo-Muellenet,N. Mulder, D. Natale, C. Orengo, S. Pesseat, M. Punta, A. F. Quinn, C. Rivoire, A. Sangrador-Vegas,J. D. Selengut, C. J. Sigrist, M. Scheremetjew, J. Tate, M. Thimmajanarthanan, P. D. Thomas,C. H. Wu, C. Yeats, S. Y. Yong, InterPro in 2011: New developments in the family and do-main prediction database. Nucleic Acids Res. 40, D306–D312 (2012).
7. C. Kerimoglu, R. C. Agis-Balboa, A. Kranz, R. Stilling, S. Bahari-Javan, E. Benito-Garagorri, R. Halder,S. Burkhardt, A. F. Stewart, A. Fischer, Histone-methyltransferase MLL2 (KMT2B) is required formemory formation in mice. J. Neurosci. 33, 3452–3464 (2013).
8. N. Bögershausen, E. Bruford, E. B. Wollnik, Skirting the pitfalls: A clear-cut nomenclature forH3K4 methyltransferases. Clin. Genet. 83, 212–214 (2013).
9. Z. Guan, M. Giustetto, S. Lomvardas, J. H. Kim, M. C. Miniaci, J. H. Schwartz, D. Thanos,E. R. Kandel, Integration of long-term-memory-related synaptic plasticity involves bidirectionalregulation of gene expression and chromatin structure. Cell 111, 483–493 (2002).
10. S. Gupta, S. Y. Kim, S. Artis, D. L. Molfese, A. Schumacher, J. D. Sweatt, R. E. Paylor, F. D. Lubin,Histone methylation regulates memory formation. J. Neurosci. 30, 3589–3599 (2010).
11. M. Cohen-Armon, L. Visochek, A. Katzoff, D. Levitan, A. J. Susswein, R. Klein, M. Valbrun,J. H. Schwartz, Long-term memory requires polyADP-ribosylation. Science 304, 1820–1822(2004).
12. M. S. Rao, A. K. Shetty, Efficacy of doublecortin as a marker to analyse the absolute numberand dendritic growth of newly generated neurons in the adult dentate gyrus. Eur. J. Neurosci.19, 234–246 (2004).
13. R. P. Kesner, An analysis of the dentate gyrus function. Behav. Brain Res. 254, 1–7 (2013).14. A. M. Morris, J. C. Churchwell, R. P. Kesner, P. E. Gilbert, Selective lesions of the dentate
gyrus produce disruptions in place learning for adjacent spatial locations. Neurobiol. Learn.Mem. 97, 326–331 (2012).
15. J. R. Epp, A. K. Haack, L. A. Galea, Activation and survival of immature neurons in thedentate gyrus with spatial memory is dependent on time of exposure to spatial learningand age of cells at examination. Neurobiol. Learn. Mem. 95, 316–325 (2011).
16. A. M. Brickman, Y. Stern, S. A. Small, Hippocampal subregions differentially associate withstandardized memory tests. Hippocampus 21, 923–928 (2011).
17. G. S. Baird, D. A. Zacharias, R. Y. Tsien, Circular permutation and receptor insertion withingreen fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 11241–11246 (1999).
18. A. Munshi, T. Tanaka, M. L. Hobbs, S. L. Tucker, V. M. Richon, R. E. Meyn, Vorinostat, ahistone deacetylase inhibitor, enhances the response of human tumor cells to ionizingradiation through prolongation of g-H2AX foci. Mol. Cancer Ther. 5, 1967–1974 (2006).
19. P. H. Huang, C. H. Chen, C. C. Chou, A. M. Sargeant, S. K. Kulp, C. M. Teng, J. C. Byrd, C. S. Chen,Histone deacetylase inhibitors stimulate histone H3 lysine 4 methylation in part via transcrip-tional repression of histone H3 lysine 4 demethylases. Mol. Pharmacol. 79, 197–206 (2011).
20. M. Vermeulen, K. W. Mulder, S. Denissov, V. W. Pijnappel, F. M. van Schaik, R. A. Varier,M. P. Baltissen, H. G. Stunnenberg, M. Mann, H. T. Timmers, Selective anchoring of TFIIDto nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
21. H. van Ingen, F. M. van Schaik, H. Wienk, J. Ballering, H. Rehmann, A. C. Dechesne, J. A. Kruijzer,R. M. Liskamp, H. T. Timmers, R. Boelens, Structural insight into the recognition of theH3K4me3 mark by the TFIID subunit TAF3. Structure 16, 1245–1256 (2008).
22. A. Jacob, J. Oblinger, M. L. Bush, V. Brendel, G. Santarelli, A. R. Chaudhury, S. Kulp, K. M. La Perle,C. S. Chen, L. S. Chang, D. B. Welling, Preclinical validation of AR42, a novel histone deacetylaseinhibitor, as treatment for vestibular schwannomas. Laryngoscope 122, 174–189 (2012).
23. S. Zhang, A. Suvannasankha, C. D. Crean, V. L. White, C. S. Chen, S. S. Farag, The novelhistone deacetylase inhibitor, AR-42, inhibits gp130/Stat3 pathway and induces apoptosisand cell cycle arrest in multiple myeloma cells. Int. J. Cancer 129, 204–213 (2011).
24. C. Guo, C. C. Chang, M. Wortham, L. H. Chen, D. N. Kernagis, X. Qin, Y. W. Cho, J. T. Chi, G. A. Grant,R. E. McLendon, H. Yan, K. Ge, N. Papadopoulos, D. D. Bigner, Y. He, Global identification ofMLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc. Natl. Acad. Sci. U.S.A.109, 17603–17608 (2012).
25. L. H. Kasper, S. Lerach, J. Wang, S. Wu, T. Jeevan, P. K. Brindle, CBP/p300 double null cells revealeffect of coactivator level and diversity on CREB transactivation. EMBO J. 29, 3660–3672(2010).
26. A. Garthe, G. Kempermann, An old test for new neurons: Refining the Morris water maze to studythe functional relevance of adult hippocampal neurogenesis. Front. Neurosci. 7, 63 (2013).
27. A. Ansorg, O. W. Witte, A. Urbach, Age-dependent kinetics of dentate gyrus neurogenesisin the absence of cyclin D2. BMC Neurosci. 13, 46 (2012).
28. S. Denis-Donini, A. Dellarole, P. Crociara, M. T. Francese, V. Bortolotto, G. Quadrato, P. L. Canonico,M. Orsetti, P. Ghi, M. Memo, S. A. Bonini, G. Ferrari-Toninelli, M. Grilli, Impaired adult neurogenesisassociated with short-term memory defects in NF-kB p50-deficient mice. J. Neurosci. 28,3911–3919 (2008).
29. M. Berdasco, M. Esteller, Genetic syndromes caused by mutations in epigenetic genes.Hum. Genet. 132, 359–383 (2013).
30. P. K. Dash, S. A. Orsi, A. N. Moore, Histone deactylase inhibition combined with behavioraltherapy enhances learning and memory following traumatic brain injury. Neuroscience163, 1–8 (2009).
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 8

R E S EARCH ART I C L E
on
Oct
ober
7, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
31. C. G. Vecsey, J. D. Hawk, K. M. Lattal, J. M. Stein, S. A. Fabian, M. A. Attner, S. M. Cabrera,C. B. McDonough, P. K. Brindle, T. Abel, M. A. Wood, Histone deacetylase inhibitorsenhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional acti-vation. J. Neurosci. 27, 6128–6140 (2007).
32. J. Gräff, L. H. Tsai, The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev.Pharmacol. Toxicol. 53, 311–330 (2013).
33. E. Korzus, M. G. Rosenfeld, M. Mayford, CBP histone acetyltransferase activity is a criticalcomponent of memory consolidation. Neuron 42, 961–972 (2004).
34. J. M. Alarcón, G. Malleret, K. Touzani, S. Vronskaya, S. Ishii, E. R. Kandel, A. Barco, Chromatinacetylation, memory, and LTP are impaired in CBP+/– mice: A model for the cognitivedeficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42, 947–959 (2004).
35. P. Yang, L. Guo, Z. J. Duan, C. G. Tepper, L. Xue, X. Chen, H. J. Kung, A. C. Gao, J. X. Zou, H. W. Chen,Histone methyltransferase NSD2/MMSET mediates constitutive NF-kB signaling for cancer cellproliferation, survival, and tumor growth via a feed-forward loop. Mol. Cell. Biol. 32, 3121–3131(2012).
36. S. Lubitz, S. Glaser, J. Schaft, A. F. Stewart, K. Anastassiadis, Increased apoptosis and skeweddifferentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2.Mol.Biol. Cell 18, 2356–2366 (2007).
37. H. T. Bjornsson, M. D. Fallin, A. P. Feinberg, An integrated epigenetic and genetic approachto common human disease. Trends Genet. 20, 350–358 (2004).
38. C. J. Phiel, F. Zhang, E. Y. Huang, M. G. Guenther, M. A. Lazar, P. S. Klein, Histone deacetylaseis a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen.J. Biol. Chem. 276, 36734–36741 (2001).
39. S. M. Meeran, A. Ahmed, T. O. Tollefsbol, Epigenetic targets of bioactive dietary compo-nents for cancer prevention and therapy. Clin. Epigenetics 1, 101–116 (2010).
40. T. Shimazu, M. D. Hirschey, J. Newman, W. He, K. Shirakawa, N. Le Moan, C. A. Grueter, H. Lim,L. R. Saunders, R. D. Stevens, C. B. Newgard, R. V. Farese Jr., R. de Cabo, S. Ulrich, K. Akassoglou,E. Verdin, Suppression of oxidative stress by b-hydroxybutyrate, an endogenous histonedeacetylase inhibitor. Science 339, 211–214 (2013).
41. A. Martinez-Canabal, K. G. Akers, S. A. Josselyn, P. W. Frankland, Age-dependent effects ofhippocampal neurogenesis suppression on spatial learning. Hippocampus 23, 66–74(2013).
42. J. Guy, J. Gan, J. Selfridge, S. Cobb, A. Bird, Reversal of neurological defects in a mousemodel of Rett syndrome. Science 315, 1143–1147 (2007).
43. I. Das, J. M. Park, J. H. Shin, S. K. Jeon, H. Lorenzi, D. J. Linden, P. F. Worley, R. H. Reeves,Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndromemouse model. Sci. Transl. Med. 5, 201ra120 (2013).
44. C. Henderson, L. Wijetunge, M. N. Kinoshita, M. Shumway, R. S. Hammond, F. R. Postma,C. Brynczka, R. Rush, A. Thomas, R. Paylor, S. T. Warren, P. W. Vanderklish, P. C. Kind, R. L. Carpenter,M. F. Bear, A. M. Healy, Reversal of disease-related pathologies in the fragile X mouse model byselective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4, 152ra128 (2012).
45. E. A. Souslova, V. V. Belousov, J. G. Lock, S. Strömblad, S. Kasparov, A. P. Bolshakov, V. G. Pinelis,Y. A. Labas, S. Lukyanov, L. M. Mayr, D. M. Chudakov, Single fluorescent protein-based Ca2+
sensors with increased dynamic range. BMC Biotechnol. 7, 37 (2007).46. G. D. Gilfillan, T. Hughes, Y. Sheng, H. S. Hjorthaug, T. Straub, K. Gervin, J. R. Harris, D. E. Undlien,
R. Lyle, Limitations and possibilities of low cell number ChIP-seq. BMC Genomics 13, 645(2012).
47. B. Langmead, S. L. Salzberg, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9,357–359 (2012).
48. Y. Zhang, T. Liu, C. A. Meyer, J. Eeckhoute, D. S. Johnson, B. E. Bernstein, C. Nusbaum, R. M. Myers,M. Brown, W. Li, X. S. Liu, Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
www.Scienc
49. A. R. Quinlan, I. M. Hall, BEDTools: A flexible suite of utilities for comparing genomicfeatures. Bioinformatics 26, 841–842 (2010).
50. D. J. McCarthy, Y. Chen, G. K. Smyth, Differential expression analysis of multifactor RNA-Seqexperiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
51. M. D. Robinson, D. J. McCarthy, G. K. Smyth, edgeR: A Bioconductor package for differentialexpression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
52. B. L. Loeys, E. E. Gerber, D. Riegert-Johnson, S. Iqbal, P. Whiteman, V. McConnell, C. R. Chillakuri,D. Macaya, P. J. Coucke, A. De Paepe, D. P. Judge, F. Wigley, E. C. Davis, H. J. Mardon, P. Handford,D. R. Keene, L. Y. Sakai, H. C. Dietz, Mutations in fibrillin-1 cause congenital scleroderma: Stiff skinsyndrome. Sci. Transl. Med. 23, 23ra20 (2010).
53. A. Adamczyk, R. Mejias, K. Takamiya, J. Yocum, I. N. Krasnova, J. Calderon, J. L. Cadet, R. L. Huganir,M. V. Pletnikov, T. Wang, GluA3-deficiency in mice is associated with increased social andaggressive behavior and elevated dopamine in striatum. Behav. Brain Res. 229, 265–272(2012).
Acknowledgments: We would like to acknowledge M. Pletnikov, Director of The BehavioralCore at the School of Medicine, for recommendations regarding behavioral test selection andC. S. Chen and S. K. Kulp from the Ohio State University for information regarding drug prep-aration protocols. We would like to thank S. Fontana for advice on culturing MEFs, M. Swaimand E. Gallo for advice on FACS, and M. Zeledon for advice on EdU staining. We would like tothank K. Smith and A. Doyle for reading the manuscript. Funding: This work was supportedby grants to H.C.D. by the Smilow Center for Marfan Syndrome Research and the HowardHughes Medical Institute and to H.T.B. by the NIH Director’s Early Independence Award(DP5OD017877) and a Young Investigator Research Grant from the American Academy ofPediatrics Section on Genetics and Birth Defects. Author contributions: This project wasconceived and designed by H.T.B. and H.C.D. Illustrations were designed by H.T.B. and H.C.D.Genotyping assay and real-time PCR and analysis were performed by H.T.B. Immunoblot andmouse perfusions were run by L.Z. and J.S.B. Gene trap integration site was mapped by L.Z.Cryosectioning, immunofluorescence, granule cell layer area, and EdU were performed by J.S.B.Confocal microscopy was done by J.S.B., E.E.G., and H.T.B. Epigenetic indicator alleles were con-ceived by H.T.B. and H.C.D. and created by H.T.B., and all cellular and flow cytometry work wasdone by L.Z. and H.T.B. Drug gavaging was performed by Y.-C.C. Morris water maze, novel objectrecognition, and open field behavioral tests were run by J.S.B., with fear conditioning and gripstrength tests run by H.T.B. Behavioral tests were selected and analyzed by M.C.P., H.T.B., and J.S.B.Drug trials and mouse colony were managed by J.S.B. ChIP-seq was performed by L.Z. and ana-lyzed by K.D.H. Neuropsychological analyses were performed by J.W. and R.G.V. The paper was writtenand formatted by H.T.B., H.C.D., and J.S.B. with input from M.C.P. Competing interests: H.T.B. andH.C.D. have two provisional patents relevant to this work: “Genetically encoded histone reporterallele constructs,” JHU Ref. No. C12259, and “Methods for treating Mendelian disorders of theepigenetic machinery,” JHU Ref. No. C13033. The other authors declare they have no com-peting interests.
Submitted 10 April 2014Accepted 11 September 2014Published 1 October 201410.1126/scitranslmed.3009278
Citation: H. T. Bjornsson, J. S. Benjamin, L. Zhang, J. Weissman, E. E. Gerber, Y.-C. Chen,R. G. Vaurio, M. C. Potter, K. D. Hansen, H. C. Dietz, Histone deacetylase inhibition rescuesstructural and functional brain deficits in a mouse model of Kabuki syndrome. Sci. Transl.Med. 6, 256ra135 (2014).
eTranslationalMedicine.org 1 October 2014 Vol 6 Issue 256 256ra135 9
Copyright © 2022 FDOKUMEN




















