
Highly sensitive and selective determination of cupric ions by using...
7
Highly sensitive and selective determination of uranium in natural waters through a novel solidified floating organic drop microextraction method coupled with spectrophotometric determination† Ali Reza Ghiasvand, * Nahid Heidari and Payman Hashemi Ultra trace amounts of uranyl ions were extracted using a reliable, simple and selective solidified floating organic drop microextraction (SFODME) method, and determined by UV-Vis spectrophotometry. Di-2- ethylhexylphosphoric acid (HDEHP) was applied as the complexing ligand during the microextraction process, and Arsenazo III (AIII) was used as the chromogenic reagent through spectrophotometric determination. The effects of important experimental parameters on the formation of UO 2 –AIII complex and color development were studied. Other important experimental parameters affecting microextraction recovery such as type and volume of extracting organic solvent, acid concentration of sample solution, ligand concentration, stirring rate, extraction time and temperature, and salt addition were also evaluated and optimized. Under the optimized conditions, a linear response was obtained over the range of 0.8–75 ng mL 1 for uranyl ions. The interfering effect of some foreign ions on the extraction and determination of uranyl ions was also investigated. Limit of detection, relative standard deviation and enrichment factor of the proposed SFODME-UV method were obtained as 0.1 ng mL 1 , 3.7% and 125, respectively, for uranyl ions. The proposed method was applied successfully for the extraction and determination of uranyl ions in natural water samples. The results of the proposed SFODME method and those reported by official standard method were in good agreement. 1. Introduction Uranium is known to have both chemical and radioactive toxicity towards the human body. Its compounds are carcino- genic and have irreversible effects on some tissues such as kidney. 1,2 Due to elevated levels of uranyl ions in natural waters and the requirement for proper storage and disposal of uranium nuclear fuel wastes, its extraction and determination techniques need to be improved. In recent years, several tech- niques have been used to extract and measure uranyl ions from aqueous samples, including solvent extraction (SE), 3 ultral- tration, 4 solid phase extraction (SPE), 5–8 supercritical uid extraction (SFE), 9 cloud-point extraction (CPE), 10–12 homoge- neous liquid–liquid extraction (HLLE), 13 dispersive liquid– liquid microextraction (DLLME) 14 and membrane methods consisting of bulk liquid membrane (BLM), 15 emulsion liquid membrane (ELM) 16 and supported liquid membrane (SLM). 17 However, there are still difficulties due to increasing uranium in natural waters. Hence, the development of reliable, simple, inexpensive and practical methods for the determination of uranyl ions in water samples is of paramount importance. Neutron activation analysis (NAA), 18 energy dispersive X-ray uorescence (ED-XRF), 19 inductively coupled mass spectrometry (ICP-MS) 20 and radiochemical methods, such as alpha spec- trometry, 21 gamma spectrometry 22 and liquid scintillation counting, 23 are suitable for the sensitive determination of uranyl ions. However, these methods are expensive and are available mostly in equipped and specialized laboratories. 24 Spectrophotometric methods, which can provide sufficient sensitivity, are cost-effective, common in laboratories and are the easiest techniques for the determination of uranyl ions. 25,26 Several organic and inorganic reagents have been used for the spectrophotometric determination of uranyl ions. Among them, dibenzoylmethane (DBM) and Arsenazo III (AIII) have the ability to selectively measure very low concentrations of uranyl ions. 27,28 However, AIII is the most sensitive reagent for the spectrophotometric determination of uranyl ions. 29,30 Due to problems associated with classical extraction methods, liquid and solid-phase microextraction procedures have been developed to eliminate or minimize these limita- tions. Each of these new microextraction methods also has limitations and disadvantages. Solidied oating organic drop Department of Chemistry, Lorestan University, 5 th km Khoramabad-Tehran Hwy, P.O. Box: 465, Khoramabad, Iran, 67138-17133. E-mail: [email protected]; [email protected]; [email protected]; Fax: +98 661 6200610-12; Tel: +98 661 6200610-12 † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ay00981a Cite this: Anal. Methods, 2014, 6, 5992 Received 24th April 2014 Accepted 3rd June 2014 DOI: 10.1039/c4ay00981a www.rsc.org/methods 5992 | Anal. Methods, 2014, 6, 5992–5998 This journal is © The Royal Society of Chemistry 2014 Analytical Methods PAPER
Transcript of Highly sensitive and selective determination of cupric ions by using...

AnalyticalMethods
PAPER
Department of Chemistry, Lorestan Universi
Box: 465, Khoramabad, Iran, 67138-17
[email protected]; payman_hashemi@
Tel: +98 661 6200610-12
† Electronic supplementary informa10.1039/c4ay00981a
Cite this: Anal. Methods, 2014, 6, 5992
Received 24th April 2014Accepted 3rd June 2014
DOI: 10.1039/c4ay00981a
www.rsc.org/methods
5992 | Anal. Methods, 2014, 6, 5992–5
Highly sensitive and selective determination ofuranium in natural waters through a novel solidifiedfloating organic drop microextraction methodcoupled with spectrophotometric determination†
Ali Reza Ghiasvand,* Nahid Heidari and Payman Hashemi
Ultra trace amounts of uranyl ions were extracted using a reliable, simple and selective solidified floating
organic drop microextraction (SFODME) method, and determined by UV-Vis spectrophotometry. Di-2-
ethylhexylphosphoric acid (HDEHP) was applied as the complexing ligand during the microextraction
process, and Arsenazo III (AIII) was used as the chromogenic reagent through spectrophotometric
determination. The effects of important experimental parameters on the formation of UO2–AIII complex
and color development were studied. Other important experimental parameters affecting
microextraction recovery such as type and volume of extracting organic solvent, acid concentration of
sample solution, ligand concentration, stirring rate, extraction time and temperature, and salt addition
were also evaluated and optimized. Under the optimized conditions, a linear response was obtained over
the range of 0.8–75 ng mL�1 for uranyl ions. The interfering effect of some foreign ions on the
extraction and determination of uranyl ions was also investigated. Limit of detection, relative standard
deviation and enrichment factor of the proposed SFODME-UV method were obtained as 0.1 ng mL�1,
3.7% and 125, respectively, for uranyl ions. The proposed method was applied successfully for the
extraction and determination of uranyl ions in natural water samples. The results of the proposed
SFODME method and those reported by official standard method were in good agreement.
1. Introduction
Uranium is known to have both chemical and radioactivetoxicity towards the human body. Its compounds are carcino-genic and have irreversible effects on some tissues such askidney.1,2 Due to elevated levels of uranyl ions in natural watersand the requirement for proper storage and disposal ofuranium nuclear fuel wastes, its extraction and determinationtechniques need to be improved. In recent years, several tech-niques have been used to extract and measure uranyl ions fromaqueous samples, including solvent extraction (SE),3 ultral-tration,4 solid phase extraction (SPE),5–8 supercritical uidextraction (SFE),9 cloud-point extraction (CPE),10–12 homoge-neous liquid–liquid extraction (HLLE),13 dispersive liquid–liquid microextraction (DLLME)14 and membrane methodsconsisting of bulk liquid membrane (BLM),15 emulsion liquidmembrane (ELM)16 and supported liquid membrane (SLM).17
However, there are still difficulties due to increasing uranium in
ty, 5th km Khoramabad-Tehran Hwy, P.O.
133. E-mail: [email protected];
yahoo.com; Fax: +98 661 6200610-12;
tion (ESI) available. See DOI:
998
natural waters. Hence, the development of reliable, simple,inexpensive and practical methods for the determination ofuranyl ions in water samples is of paramount importance.Neutron activation analysis (NAA),18 energy dispersive X-rayuorescence (ED-XRF),19 inductively coupledmass spectrometry(ICP-MS)20 and radiochemical methods, such as alpha spec-trometry,21 gamma spectrometry22 and liquid scintillationcounting,23 are suitable for the sensitive determination ofuranyl ions. However, these methods are expensive and areavailable mostly in equipped and specialized laboratories.24
Spectrophotometric methods, which can provide sufficientsensitivity, are cost-effective, common in laboratories and arethe easiest techniques for the determination of uranyl ions.25,26
Several organic and inorganic reagents have been used for thespectrophotometric determination of uranyl ions. Among them,dibenzoylmethane (DBM) and Arsenazo III (AIII) have the abilityto selectively measure very low concentrations of uranylions.27,28 However, AIII is the most sensitive reagent for thespectrophotometric determination of uranyl ions.29,30
Due to problems associated with classical extractionmethods, liquid and solid-phase microextraction procedureshave been developed to eliminate or minimize these limita-tions. Each of these new microextraction methods also haslimitations and disadvantages. Solidied oating organic drop
This journal is © The Royal Society of Chemistry 2014

Paper Analytical Methods
microextraction (SFODME) method is a miniaturized form ofliquid–liquid extraction (LLE), which was started in 2007 as aproper alternative to other microextraction procedures.31,32 Inthis method, a microdroplet of a proper organic solvent, withmelting point close to room temperature, is oated on thesurface of an aqueous sample solution under agitation. Aerthe completion of the extraction, the aqueous sample is cooledto solidify the organic drop. Then, the solidied organicmicrodroplet is transferred into a conical micro-vial, andallowed to melt at room temperature. Finally, the analyteconcentrated into the organic microdroplet is determined byintroducing it to a proper analytical instrument.
SFODME coupled to different analytical instruments, espe-cially UV-Vis spectrophotometry,33,34 ame atomic absorptionspectrometry (F-AAS),35 electrothermal atomic absorptionspectrometry (ET-AAS),36 high performance liquid chromatog-raphy (HPLC),37 and gas chromatography (GC),38 has beenapplied for the extraction of organic33,36,37,39–42 and inorganicanalytes.35,38,43–54
In this research, a simple, sensitive and reliable SFODMEmethod was established for the selective preconcentration ofuranyl ions using HDEHP, which was followed by UV-Vis spec-trophotometric determination using AIII. To the best of ourknowledge, there has been no report on the application ofHDEHP as a commercial, low cost and selective extractingreagent for the extraction of uranyl ions by the SFODMEmethodcoupled with spectrophotometry.
2. Experimental2.1. Materials
Organic solvents of 1-undecanol (98%), 1-dodecanol (98%), 2-dodecanol (95%), and n-hexadecane (98%) were purchasedfromMerck Chemical Co (Darmstadt, Germany). Reagent grademethanol, ethanol, sodium chloride, sodium nitrate and highpurity nitric acid (HNO3), hydrochloric acid (HCl), andperchloric acid (HClO4) were supplied by Merck (Darmstadt,Germany) or Fluka (Buchs, Switzerland). Double distilled waterwas used throughout the experiments. Di-2-ethylhexyl phos-phoric acid (HDEHP) was obtained from Merck (97% pure).Analar grade disodium salt of Arsenazo III (1,8-dihydroxy-2,7-bis(2-arsonophenylazo)naphthalene-3,6-disulphonic acid) wasobtained from Merck. A 0.2% (w/v) stock solution of AIII wasprepared by dissolving 0.2 g of its disodium salt in 100 mLmethanol. Analytical grade uranyl nitrate supplied by Merckwas of the highest purity available, and was dried in a vacuumover P2O5. A stock solution of uranyl ions (1000 mg mL�1) wasprepared by dissolving an appropriate amount ofUO2(NO3)2$6H2O in 0.5 M HNO3. Working solutions of uranylions were prepared daily by proper dilution of its stock solutionwith double distilled water.
Scheme 1 The simple planar structure of Arsenazo III (1,8-dihydroxy-2,7-bis(2-arsonophenylazo)naphthalene-3,6-disulphonic acid).
2.2. Instruments
A double-beam spectrophotometer (UV-160, Shimadzu, Kyoto,Japan) was used for absorbance measurements. A pure glass Fi-streem double distiller (Fisons Scientic Apparatus,
This journal is © The Royal Society of Chemistry 2014
Loughborough, England) was used for the preparation ofdouble distilled water. Weight measurements were performedusing a Shimadzu AX-120 digital balance (Shimadzu, Japan).20 mL extraction vials obtained from Supelco were used for theSFODME experiments. Stirring of solutions was carried out on aHeidolph MR 3001 K heater magnetic-stirrer (Kelheim, Ger-many) using a 8 mm � 1.5 mm PTFE coated stirring bar. A100 mL microsyringe (Hamilton Bonaduz AG, Switzerland) wasemployed for the placing of the HDEHP solution on the top-center surface of the sample solution. A stainless steel mini-spatula was applied to transfer the solidied organic drop. Forthe easy and clean removal of solidied organic drop (SFO), thepan of the mini-spatula was slightly tilted, like that of a table-spoon, and a hole was created in the center.
2.3. Methods
15 mL portion of the sample solution (0.1 M HClO4) containing50 ng mL�1 of uranyl ion was placed into an extraction vial.Using a 100 mL microsyringe, 50 mL of HDEHP solution (5% w/vin 1-dodecanol) was lightly placed on the top-center surface ofthe sample solution. The cap of the extraction vial was closedand it was kept in a water bath (35 �C) and stirred at 900 rpm.Aer 40 min of stirring to achieve equilibration, the extractionvial was immersed in an ice-bath for 5 min. The solidiedorganic drop was then transferred to a 2 mL vial using a mini-spatula and mixed with 500 mL of AIII solution (0.2% in meth-anol) as the chromogenic reagent. Aer a 10 min rest time forthe color to stabilize, the absorbance of the solution wasrecorded at 620 nm (lmax) against a reagent blank. Theconcentration of uranyl ions was then determined using anexternal calibration curve (0.1–10 mg mL�1; R2 ¼ 0.9993).
3. Result and discussion3.1. The study of important parameters affecting theformation of uranyl–AIII complex
Arsenazo III (AIII) has different active sites (Scheme 1) and itscomplex formation with metal ions is strongly dependent onpH, ionic strength and its concentration.27,55 Therefore, it wasnecessary to qualify its reaction with uranyl ions for theconditions of the proposed SFODME procedure. Thus, someimportant experimental parameters affecting the formation ofUO2
2+–AIII complex were investigated.For the study of the time required for complete complex
formation, different concentrations of uranyl ion were mixedwith 0.2% AIII solution (both dissolved in methanol) and
Anal. Methods, 2014, 6, 5992–5998 | 5993
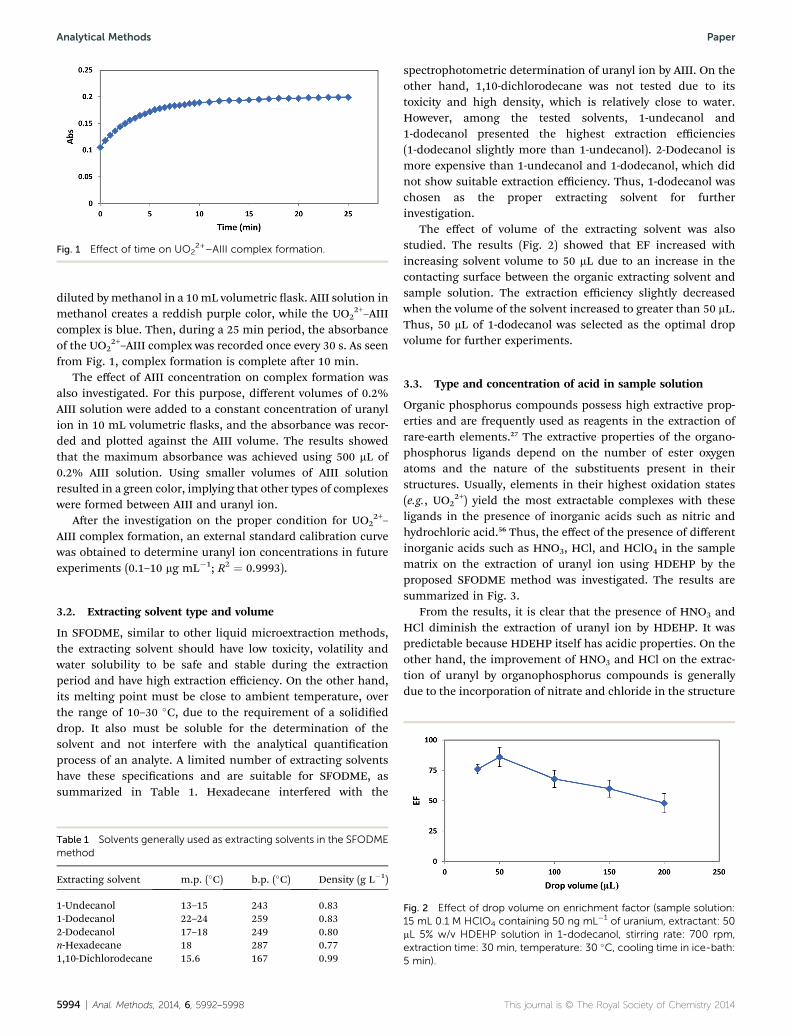
Fig. 1 Effect of time on UO22+–AIII complex formation.
Analytical Methods Paper
diluted bymethanol in a 10mL volumetric ask. AIII solution inmethanol creates a reddish purple color, while the UO2
2+–AIIIcomplex is blue. Then, during a 25 min period, the absorbanceof the UO2
2+–AIII complex was recorded once every 30 s. As seenfrom Fig. 1, complex formation is complete aer 10 min.
The effect of AIII concentration on complex formation wasalso investigated. For this purpose, different volumes of 0.2%AIII solution were added to a constant concentration of uranylion in 10 mL volumetric asks, and the absorbance was recor-ded and plotted against the AIII volume. The results showedthat the maximum absorbance was achieved using 500 mL of0.2% AIII solution. Using smaller volumes of AIII solutionresulted in a green color, implying that other types of complexeswere formed between AIII and uranyl ion.
Aer the investigation on the proper condition for UO22+–
AIII complex formation, an external standard calibration curvewas obtained to determine uranyl ion concentrations in futureexperiments (0.1–10 mg mL�1; R2 ¼ 0.9993).
3.2. Extracting solvent type and volume
In SFODME, similar to other liquid microextraction methods,the extracting solvent should have low toxicity, volatility andwater solubility to be safe and stable during the extractionperiod and have high extraction efficiency. On the other hand,its melting point must be close to ambient temperature, overthe range of 10–30 �C, due to the requirement of a solidieddrop. It also must be soluble for the determination of thesolvent and not interfere with the analytical quanticationprocess of an analyte. A limited number of extracting solventshave these specications and are suitable for SFODME, assummarized in Table 1. Hexadecane interfered with the
Table 1 Solvents generally used as extracting solvents in the SFODMEmethod
Extracting solvent m.p. (�C) b.p. (�C) Density (g L�1)
1-Undecanol 13–15 243 0.831-Dodecanol 22–24 259 0.832-Dodecanol 17–18 249 0.80n-Hexadecane 18 287 0.771,10-Dichlorodecane 15.6 167 0.99
5994 | Anal. Methods, 2014, 6, 5992–5998
spectrophotometric determination of uranyl ion by AIII. On theother hand, 1,10-dichlorodecane was not tested due to itstoxicity and high density, which is relatively close to water.However, among the tested solvents, 1-undecanol and1-dodecanol presented the highest extraction efficiencies(1-dodecanol slightly more than 1-undecanol). 2-Dodecanol ismore expensive than 1-undecanol and 1-dodecanol, which didnot show suitable extraction efficiency. Thus, 1-dodecanol waschosen as the proper extracting solvent for furtherinvestigation.
The effect of volume of the extracting solvent was alsostudied. The results (Fig. 2) showed that EF increased withincreasing solvent volume to 50 mL due to an increase in thecontacting surface between the organic extracting solvent andsample solution. The extraction efficiency slightly decreasedwhen the volume of the solvent increased to greater than 50 mL.Thus, 50 mL of 1-dodecanol was selected as the optimal dropvolume for further experiments.
3.3. Type and concentration of acid in sample solution
Organic phosphorus compounds possess high extractive prop-erties and are frequently used as reagents in the extraction ofrare-earth elements.27 The extractive properties of the organo-phosphorus ligands depend on the number of ester oxygenatoms and the nature of the substituents present in theirstructures. Usually, elements in their highest oxidation states(e.g., UO2
2+) yield the most extractable complexes with theseligands in the presence of inorganic acids such as nitric andhydrochloric acid.56 Thus, the effect of the presence of differentinorganic acids such as HNO3, HCl, and HClO4 in the samplematrix on the extraction of uranyl ion using HDEHP by theproposed SFODME method was investigated. The results aresummarized in Fig. 3.
From the results, it is clear that the presence of HNO3 andHCl diminish the extraction of uranyl ion by HDEHP. It waspredictable because HDEHP itself has acidic properties. On theother hand, the improvement of HNO3 and HCl on the extrac-tion of uranyl by organophosphorus compounds is generallydue to the incorporation of nitrate and chloride in the structure
Fig. 2 Effect of drop volume on enrichment factor (sample solution:15 mL 0.1 M HClO4 containing 50 ng mL�1 of uranium, extractant: 50mL 5% w/v HDEHP solution in 1-dodecanol, stirring rate: 700 rpm,extraction time: 30 min, temperature: 30 �C, cooling time in ice-bath:5 min).
This journal is © The Royal Society of Chemistry 2014
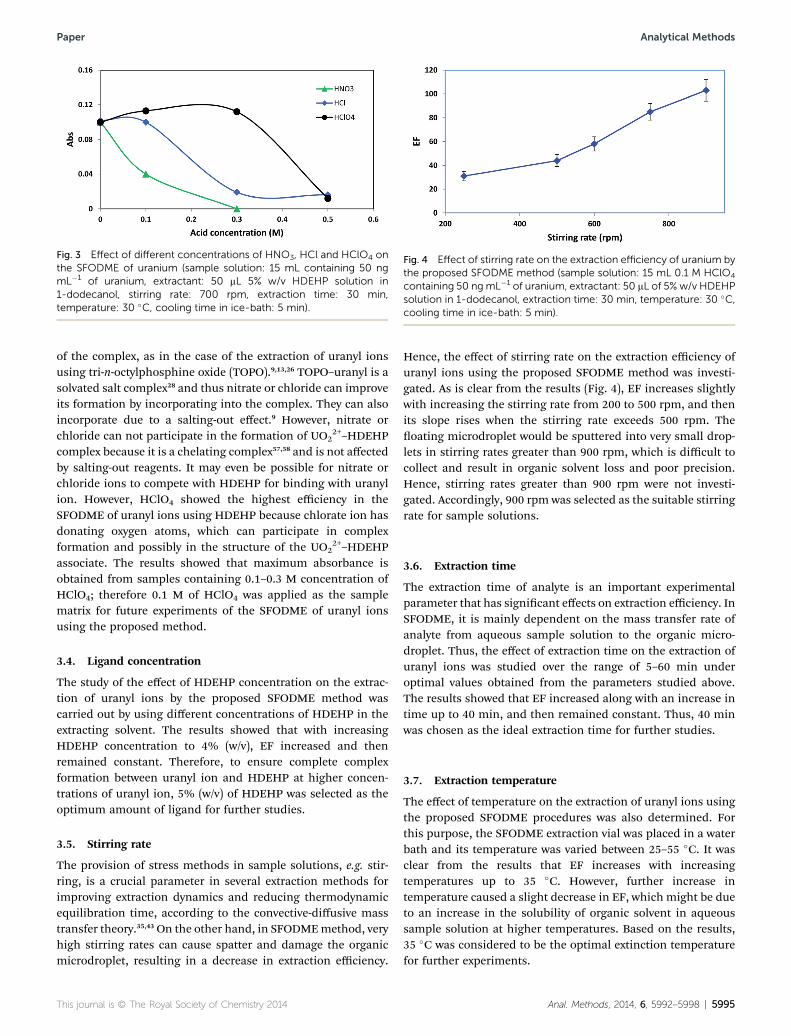
Fig. 3 Effect of different concentrations of HNO3, HCl and HClO4 onthe SFODME of uranium (sample solution: 15 mL containing 50 ngmL�1 of uranium, extractant: 50 mL 5% w/v HDEHP solution in1-dodecanol, stirring rate: 700 rpm, extraction time: 30 min,temperature: 30 �C, cooling time in ice-bath: 5 min).
Fig. 4 Effect of stirring rate on the extraction efficiency of uranium bythe proposed SFODME method (sample solution: 15 mL 0.1 M HClO4
containing 50 ngmL�1 of uranium, extractant: 50 mL of 5% w/v HDEHPsolution in 1-dodecanol, extraction time: 30 min, temperature: 30 �C,cooling time in ice-bath: 5 min).
Paper Analytical Methods
of the complex, as in the case of the extraction of uranyl ionsusing tri-n-octylphosphine oxide (TOPO).9,13,26 TOPO–uranyl is asolvated salt complex28 and thus nitrate or chloride can improveits formation by incorporating into the complex. They can alsoincorporate due to a salting-out effect.9 However, nitrate orchloride can not participate in the formation of UO2
2+–HDEHPcomplex because it is a chelating complex57,58 and is not affectedby salting-out reagents. It may even be possible for nitrate orchloride ions to compete with HDEHP for binding with uranylion. However, HClO4 showed the highest efficiency in theSFODME of uranyl ions using HDEHP because chlorate ion hasdonating oxygen atoms, which can participate in complexformation and possibly in the structure of the UO2
2+–HDEHPassociate. The results showed that maximum absorbance isobtained from samples containing 0.1–0.3 M concentration ofHClO4; therefore 0.1 M of HClO4 was applied as the samplematrix for future experiments of the SFODME of uranyl ionsusing the proposed method.
3.4. Ligand concentration
The study of the effect of HDEHP concentration on the extrac-tion of uranyl ions by the proposed SFODME method wascarried out by using different concentrations of HDEHP in theextracting solvent. The results showed that with increasingHDEHP concentration to 4% (w/v), EF increased and thenremained constant. Therefore, to ensure complete complexformation between uranyl ion and HDEHP at higher concen-trations of uranyl ion, 5% (w/v) of HDEHP was selected as theoptimum amount of ligand for further studies.
3.5. Stirring rate
The provision of stress methods in sample solutions, e.g. stir-ring, is a crucial parameter in several extraction methods forimproving extraction dynamics and reducing thermodynamicequilibration time, according to the convective-diffusive masstransfer theory.35,43On the other hand, in SFODMEmethod, veryhigh stirring rates can cause spatter and damage the organicmicrodroplet, resulting in a decrease in extraction efficiency.
This journal is © The Royal Society of Chemistry 2014
Hence, the effect of stirring rate on the extraction efficiency ofuranyl ions using the proposed SFODME method was investi-gated. As is clear from the results (Fig. 4), EF increases slightlywith increasing the stirring rate from 200 to 500 rpm, and thenits slope rises when the stirring rate exceeds 500 rpm. Theoating microdroplet would be sputtered into very small drop-lets in stirring rates greater than 900 rpm, which is difficult tocollect and result in organic solvent loss and poor precision.Hence, stirring rates greater than 900 rpm were not investi-gated. Accordingly, 900 rpm was selected as the suitable stirringrate for sample solutions.
3.6. Extraction time
The extraction time of analyte is an important experimentalparameter that has signicant effects on extraction efficiency. InSFODME, it is mainly dependent on the mass transfer rate ofanalyte from aqueous sample solution to the organic micro-droplet. Thus, the effect of extraction time on the extraction ofuranyl ions was studied over the range of 5–60 min underoptimal values obtained from the parameters studied above.The results showed that EF increased along with an increase intime up to 40 min, and then remained constant. Thus, 40 minwas chosen as the ideal extraction time for further studies.
3.7. Extraction temperature
The effect of temperature on the extraction of uranyl ions usingthe proposed SFODME procedures was also determined. Forthis purpose, the SFODME extraction vial was placed in a waterbath and its temperature was varied between 25–55 �C. It wasclear from the results that EF increases with increasingtemperatures up to 35 �C. However, further increase intemperature caused a slight decrease in EF, which might be dueto an increase in the solubility of organic solvent in aqueoussample solution at higher temperatures. Based on the results,35 �C was considered to be the optimal extinction temperaturefor further experiments.
Anal. Methods, 2014, 6, 5992–5998 | 5995
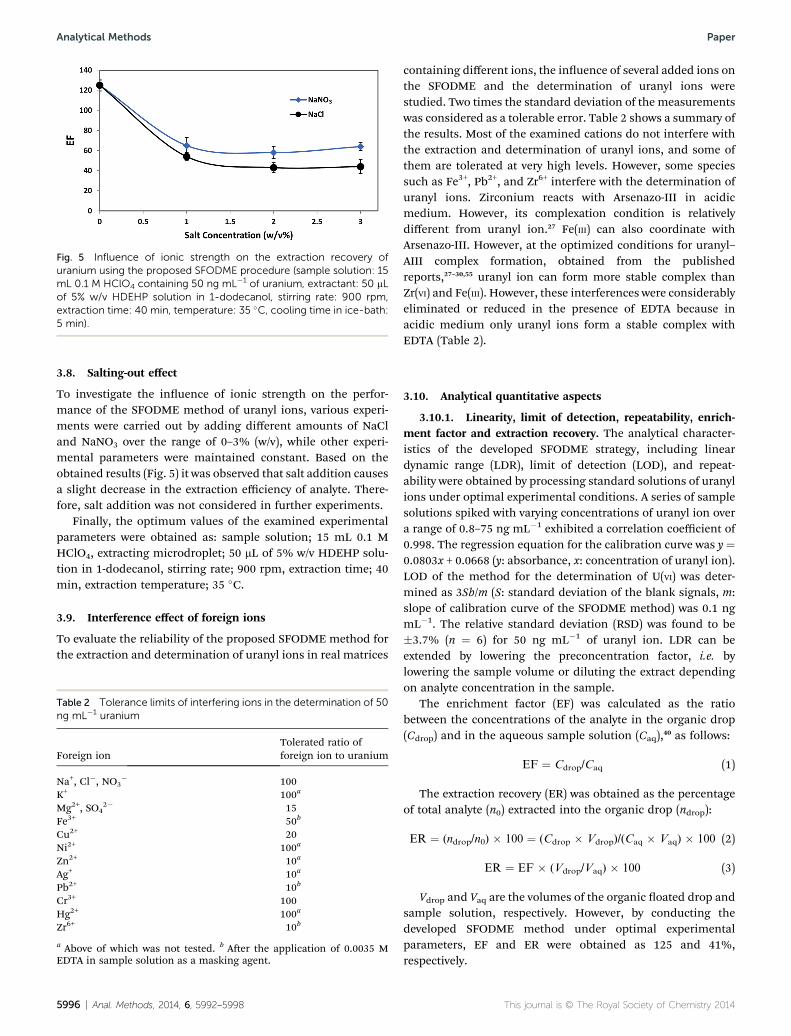
Fig. 5 Influence of ionic strength on the extraction recovery ofuranium using the proposed SFODME procedure (sample solution: 15mL 0.1 M HClO4 containing 50 ng mL�1 of uranium, extractant: 50 mLof 5% w/v HDEHP solution in 1-dodecanol, stirring rate: 900 rpm,extraction time: 40 min, temperature: 35 �C, cooling time in ice-bath:5 min).
Analytical Methods Paper
3.8. Salting-out effect
To investigate the inuence of ionic strength on the perfor-mance of the SFODME method of uranyl ions, various experi-ments were carried out by adding different amounts of NaCland NaNO3 over the range of 0–3% (w/v), while other experi-mental parameters were maintained constant. Based on theobtained results (Fig. 5) it was observed that salt addition causesa slight decrease in the extraction efficiency of analyte. There-fore, salt addition was not considered in further experiments.
Finally, the optimum values of the examined experimentalparameters were obtained as: sample solution; 15 mL 0.1 MHClO4, extracting microdroplet; 50 mL of 5% w/v HDEHP solu-tion in 1-dodecanol, stirring rate; 900 rpm, extraction time; 40min, extraction temperature; 35 �C.
3.9. Interference effect of foreign ions
To evaluate the reliability of the proposed SFODME method forthe extraction and determination of uranyl ions in real matrices
Table 2 Tolerance limits of interfering ions in the determination of 50ng mL�1 uranium
Foreign ionTolerated ratio offoreign ion to uranium
Na+, Cl�, NO3� 100
K+ 100a
Mg2+, SO42� 15
Fe3+ 50b
Cu2+ 20Ni2+ 100a
Zn2+ 10a
Ag+ 10a
Pb2+ 10b
Cr3+ 100Hg2+ 100a
Zr6+ 10b
a Above of which was not tested. b Aer the application of 0.0035 MEDTA in sample solution as a masking agent.
5996 | Anal. Methods, 2014, 6, 5992–5998
containing different ions, the inuence of several added ions onthe SFODME and the determination of uranyl ions werestudied. Two times the standard deviation of the measurementswas considered as a tolerable error. Table 2 shows a summary ofthe results. Most of the examined cations do not interfere withthe extraction and determination of uranyl ions, and some ofthem are tolerated at very high levels. However, some speciessuch as Fe3+, Pb2+, and Zr6+ interfere with the determination ofuranyl ions. Zirconium reacts with Arsenazo-III in acidicmedium. However, its complexation condition is relativelydifferent from uranyl ion.27 Fe(III) can also coordinate withArsenazo-III. However, at the optimized conditions for uranyl–AIII complex formation, obtained from the publishedreports,27–30,55 uranyl ion can form more stable complex thanZr(VI) and Fe(III). However, these interferences were considerablyeliminated or reduced in the presence of EDTA because inacidic medium only uranyl ions form a stable complex withEDTA (Table 2).
3.10. Analytical quantitative aspects
3.10.1. Linearity, limit of detection, repeatability, enrich-ment factor and extraction recovery. The analytical character-istics of the developed SFODME strategy, including lineardynamic range (LDR), limit of detection (LOD), and repeat-ability were obtained by processing standard solutions of uranylions under optimal experimental conditions. A series of samplesolutions spiked with varying concentrations of uranyl ion overa range of 0.8–75 ng mL�1 exhibited a correlation coefficient of0.998. The regression equation for the calibration curve was y ¼0.0803x + 0.0668 (y: absorbance, x: concentration of uranyl ion).LOD of the method for the determination of U(VI) was deter-mined as 3Sb/m (S: standard deviation of the blank signals, m:slope of calibration curve of the SFODME method) was 0.1 ngmL�1. The relative standard deviation (RSD) was found to be�3.7% (n ¼ 6) for 50 ng mL�1 of uranyl ion. LDR can beextended by lowering the preconcentration factor, i.e. bylowering the sample volume or diluting the extract dependingon analyte concentration in the sample.
The enrichment factor (EF) was calculated as the ratiobetween the concentrations of the analyte in the organic drop(Cdrop) and in the aqueous sample solution (Caq),40 as follows:
EF ¼ Cdrop/Caq (1)
The extraction recovery (ER) was obtained as the percentageof total analyte (n0) extracted into the organic drop (ndrop):
ER ¼ (ndrop/n0) � 100 ¼ (Cdrop � Vdrop)/(Caq � Vaq) � 100 (2)
ER ¼ EF � (Vdrop/Vaq) � 100 (3)
Vdrop and Vaq are the volumes of the organic oated drop andsample solution, respectively. However, by conducting thedeveloped SFODME method under optimal experimentalparameters, EF and ER were obtained as 125 and 41%,respectively.
This journal is © The Royal Society of Chemistry 2014

Table 3 Extraction and determination of uranium in water samples using the proposed SFODME method
Water sample Uranium added (ng mL�1)
Uranium determined (ng mL�1)
SFODME method ICP-MSa
Tap water (Lorestan University, Khoramabad) 50 49.2 (3.5)b —River water (Bisheh waterfall, Khoramabad) 50 48.5 (4.1) —Spring water 1 (Gachin mine, Bandar Abbass) 0 65.2 (2.8) 63.7 (2.2)Spring water 2 (Gachin mine, Bandar Abbass) 0 52.8 (3.1) 51.4 (3.8)
a Results reported by the Applied Geological Research Center of Geological Survey of Iran. b RSD of three replicate experiments.
Table 4 Comparison of the proposed SFODME-UV method to somepreconcentration procedures for the extraction and determination ofuranium
Method LOD (ng mL�1) RSD EF Ref.
SFODME-UV 0.1 3.7 125 Present workSPE-UV 0.5 4 — 34SPE-UV 2.7 <10 100 5CPE-UV (Triton X-114) 0.5 2.3 — 59SPE-ICP-AES 6.14 — 143 60SPE-DPP 20 1.6 100 7SPE-UV 0.1 3 — 26CPE-ICP-OES 1 6.1 43.7 11
Paper Analytical Methods
3.10.2. Analysis of real samples. To evaluate the accuracyand applicability of the proposed SFODME method to realsamples, it was applied in the recovery and determination ofuranyl ions from two water samples. A tap water sample (fromLorestan University) and a river water sample (from Bishehwaterfall located 65 km from Khoramabad) were analyzed usingthe added-found method. Uranyl ion was not detected in realsamples. Therefore, these real samples were spiked with thetarget analyte at a concentration of 50 ng mL�1. The proposedSFODME method was also applied to extract and determine thecontents of uranyl ions in two water samples collected from twodifferent springs around Gachin uranium mine near BandarAbbass city. The results showed satisfactory agreement betweenthe concentration obtained by the proposed SFODME methodand those obtained by ICP-MS. The results summarized in Table3 indicate that the developed method can be successfullyapplied for the determination of uranyl ions in real watersamples.
4. Conclusion
In this study, a sensitive, effective and reliable method for thepreconcentration and determination of uranyl ions in naturalwater samples was developed. UV-Vis spectrophotometry, asimple, low cost and commonly used method, was applied forthe detection of uranyl ions. The developed SFODME-UVmethod largely minimizes the consumption of toxic organicsolvents and signicantly increases the sensitivity for thedetermination of uranyl ions. Ease of operation, sensitivity,selectivity, low sample volume, low cost and high enrichmentfactors are some advantages of the proposed strategy compared
This journal is © The Royal Society of Chemistry 2014
to the other reported methods (Table 4) in the litera-ture.5,7,11,26,34,59 The proposed SFODME-UV method wassuccessfully conducted for the recovery and determination ofuranyl ions in natural water samples.
References
1 H. M. Hartmann, F. A. Monette and H. I. Avci,Hum. Ecol. RiskAssess., 2000, 6, 851–874.
2 E. S. Cra, A. W. Abu-Qare, M. M. Flaherty, M. C. Garofolo,H. L. Rincavage and M. B. Abou-Donia, J. Toxicol. Environ.Health, Part B, 2004, 7, 297–317.
3 S. A. El-Reefy, N. S. Awwad and H. F. Aly, J. Chem. Technol.Biotechnol., 1997, 69, 271–275.
4 J. D. Roach and J. H. Zapien,Water Res., 2009, 43, 4751–4759.5 S. Ozdemir and E. Kilinc,Microchim. Acta, 2012, 178, 389–397.6 M. E. Mahmoud, I. M. M. Kenawy, E. M. Soliman,M. A. Hafez, M. A. A. Akl and R. R. A. Lashein, Anal. Sci.,2008, 24, 381–387.
7 D. Dojozan, M. H. Pournaghi-Azar and J. Toutounchi-Asr,Talanta, 1998, 46, 123–128.
8 G. Daneshvar, A. Jabbari, Y. Yamini and D. Paki, J. Anal.Chem., 2009, 64, 602–608.
9 M. Shamsipur, A. R. Ghiasvand and Y. Yamini, J. Supercrit.Fluids, 2001, 20, 163–169.
10 T. Madrakian, A. Ahami and A. Mousavi, Talanta, 2007, 71,610–614.
11 S. Shariati, Y. Yamini and M. K. Zanjani, J. Hazard. Mater.,2008, 156, 583–590.
12 D. Totur, S. Bozkurt and M. Merdivan, J. Radioanal. Nucl.Chem., 2012, 292, 321–327.
13 A. R. Ghiasvand and E. Mohagheghzadeh, Anal. Sci., 2004,20, 917–919.
14 Y. Liu, E. Zhao, W. Zhu, H. Gao and Z. Zhou, J. Chromatogr. A,2009, 1216, 885–891.
15 R. Davarkhah, F. Khanramaki, M. Asgari, B. Salimi,P. Ashtari and M. Shamsipur, J. Radioanal. Nucl. Chem.,2013, 298, 1–8.
16 P. S. Kulkarni, S. Mukhopadhyay, M. P. Bellary andS. K. Ghosh, Hydrometallurgy, 2002, 64, 49–58.
17 S. Biswas, P. N. Pathak and S. B. Roy, Desalination, 2012, 290,74–82.
18 E. Montoya, P. Mendoza, P. Bedregal, O. Baltuano andI. Cohen, J. Radioanal. Nucl. Chem., 2012, 291, 175–178.
Anal. Methods, 2014, 6, 5992–5998 | 5997

Analytical Methods Paper
19 V. Natarajan, N. K. Porwal and S. V. Godbole, IJCT, 2012, 19,399–402.
20 J. Avivar, L. Ferrer, M. Casas and V. Cerda, J. Anal. At.Spectrom., 2012, 27, 327–334.
21 G. Andreou, M. Efstathiou and I. Pashalidis, J. Radioanal.Nucl. Chem., 2012, 291, 865–867.
22 L. Dragounova and P. Rulık, Appl. Radiat. Isot., 2013, 81, 123–127.
23 Y. Yoon, S. Cho, K. Lee, K. Ko and K. Ha, J. Radioanal. Nucl.Chem., 2013, 296, 397–402.
24 P. A. Dimovasilis and M. I. Prodromidis, Sens. Actuators, B,2011, 156, 689–694.
25 M. N. A. Lutfullah, N. Rahman and S. N. H. Azmi, J. Hazard.Mater., 2008, 155, 261–268.
26 M. Shamsipur, A. R. Ghiasvand and Y. Yamini, Anal. Chem.,1999, 71, 4892–4895.
27 Z. Marczenko, Separation and spectrophotometricdetermination of elements, Ellis Harwood, London, 1986.
28 J. Minczewski, J. Chwastowska and R. Dybczynski,Separation and preconcentration methods in inorganic traceanalysis, Ellis Harwood, London, 1982.
29 A. Niazi, J. Braz. Chem. Soc., 2006, 17, 1020–1026.30 A. Safavi and M. Bagheri, Anal. Chim. Acta, 2005, 530, 55–60.31 M. R. Khalili Zanjani, Y. Yamini, S. Shariati and J. A. Jonsson,
Anal. Chim. Acta, 2007, 585, 286–293.32 I. Durukan, Ç. Arpa Sahin and S. Bektas,Microchem. J., 2011,
98, 215–219.33 H. Xu, Z. Ding, L. Lv, D. Song and Y.-Q. Feng, Anal. Chim.
Acta, 2009, 636, 28–33.34 J. B. Ghasemi and E. Zolfonoun, Talanta, 2010, 80, 1191–
1197.35 Ç. A. Sahin and I. Tokgoz, Anal. Chim. Acta, 2010, 667, 83–87.36 M.-I. Leong and S.-D. Huang, J. Chromatogr. A, 2008, 1211, 8–
12.37 C.-C. Chang and S.-D. Huang, Anal. Chim. Acta, 2010, 662,
39–43.38 J. Ma, J. Zhang, X. Du, X. Lei and J. Li, Microchim. Acta, 2010,
168, 153–159.39 V. Vickackaite and E. Pusvaskiene, J. Sep. Sci., 2009, 32,
3512–3520.
5998 | Anal. Methods, 2014, 6, 5992–5998
40 M.-I. Leong and S.-D. Huang, J. Chromatogr. A, 2009, 1216,7645–7650.
41 Y.-H. Jian, Y. Hu, T. Wang, J.-L. Liu, C. Zhang and Y. Li, Chin.J. Anal. Chem., 2010, 38, 62–66.
42 C. Wu, H. Liu, W. Liu, Q. Wu, C. Wang and Z. Wang, Anal.Bioanal. Chem., 2010, 397, 2543–2549.
43 M. S. Bidabadi, S. Dadfarnia and A. M. H. Shabani, J. Hazard.Mater., 2009, 166, 291–296.
44 I. Lopez-Garcıa, R. E. Rivas and M. Hernandez-Cordoba,Anal. Bioanal. Chem., 2010, 396, 3097–3102.
45 Y.-Y. Wang, G.-Y. Zhao, Q.-Y. Chang, X.-H. Zang, C. Wangand Z. Wang, Chin. J. Anal. Chem., 2010, 38, 1517–1522.
46 R. E. Rivas, I. Lopez-Garcia and M. Hernandez-Cordoba,Anal. Methods, 2010, 2, 225–230.
47 M. Mohamadi and A. Mostafavi, Talanta, 2010, 81, 309–313.48 M. R. Moghadam, A. M. H. Shabani and S. Dadfarnia, J.
Hazard. Mater., 2011, 197, 176–182.49 T. Asadollahi, S. Dadfarnia and A. M. H. Shabani, Talanta,
2010, 82, 208–212.50 K. Shrivas and D. K. Patel, J. Hazard. Mater., 2010, 176, 414–
417.51 C. Mahugo-Santana, Z. Sosa-Ferrera, M. E. Torres-Padron
and J. J. Santana-Rodrıguez, TrAC, Trends Anal. Chem.,2011, 30, 731–748.
52 M. Ghambarian, M. R. Khalili-Zanjani, Y. Yamini, A. Esraliand N. Yazdanfar, Talanta, 2010, 81, 197–201.
53 H. R. Sobhi, Y. Yamini, A. Esrali and R. H. H. B. Abadi, J.Chromatogr. A, 2008, 1196, 28–32.
54 S. Dadfarnia, A. M. Salmanzadeh and A. M. H. Shabani, Anal.Chim. Acta, 2008, 623, 163–167.
55 H. Brown and B. Rydqvist, Biophys. J., 1981, 36, 117–137.56 A. E. Lemire, A. F. Janzen and K. Marat, J. Inorg. Nucl. Chem.,
1963, 25, 109–115.57 T. Sato, J. Inorg. Nucl. Chem., 1965, 27, 1853–1860.58 H. S. Ferreira, M. d. A. Bezerra and S. L. Costa Ferreira,
Microchim. Acta, 2006, 154, 163–167.59 V. K. Jain, R. A. Pandya, S. G. Pillai and P. S. Shrivastav,
Talanta, 2006, 70, 257–266.
This journal is © The Royal Society of Chemistry 2014










![N -[4-( N -Cyclohexylsulfamoyl)phenyl]acetamide](https://static.fdokumen.com/doc/165x107/632f4f4de68feab59a0210b7/n-4-n-cyclohexylsulfamoylphenylacetamide.jpg)









