
Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death
Upload
independentCategory
view
5download
0
ORIGINAL ARTICLE
High-throughput single-cell sequencing identifiesphotoheterotrophs and chemoautotrophs infreshwater bacterioplankton
Manuel Martinez-Garcia, Brandon K Swan, Nicole J Poulton, Monica Lluesma Gomez,Dashiell Masland, Michael E Sieracki and Ramunas StepanauskasBigelow Laboratory for Ocean Sciences, West Boothbay Harbor, ME, USA
Recent discoveries suggest that photoheterotrophs (rhodopsin-containing bacteria (RBs) andaerobic anoxygenic phototrophs (AAPs)) and chemoautotrophs may be significant for marineand freshwater ecosystem productivity. However, their abundance and taxonomic identitiesremain largely unknown. We used a combination of single-cell and metagenomic DNA sequencingto study the predominant photoheterotrophs and chemoautotrophs inhabiting the euphotic zoneof temperate, physicochemically diverse freshwater lakes. Multi-locus sequencing of 712 singleamplified genomes, generated by fluorescence-activated cell sorting and whole genome multipledisplacement amplification, showed that most of the cosmopolitan freshwater clusters containphotoheterotrophs. These comprised at least 10–23% of bacterioplankton, and RBs were thedominant fraction. Our data demonstrate that Actinobacteria, including clusters acI, Luna andacSTL, are the predominant freshwater RBs. We significantly broaden the known taxonomic range offreshwater RBs, to include Alpha-, Beta-, Gamma- and Deltaproteobacteria, Verrucomicrobia andSphingobacteria. By sequencing single cells, we found evidence for inter-phyla horizontal genetransfer and recombination of rhodopsin genes and identified specific taxonomic groups involved inthese evolutionary processes. Our data suggest that members of the ubiquitous betaproteobacteriaPolynucleobacter spp. are the dominant AAPs in temperate freshwater lakes. Furthermore,the RuBisCO (ribulose 1,5-bisphosphate carboxylase/oxygenase) gene was found in several singlecells of Betaproteobacteria, Bacteroidetes and Gammaproteobacteria, suggesting that chemoauto-trophs may be more prevalent among aerobic bacterioplankton than previously thought. This studydemonstrates the power of single-cell DNA sequencing addressing previously unresolved questionsabout the metabolic potential and evolutionary histories of uncultured microorganisms, whichdominate most natural environments.The ISME Journal (2012) 6, 113–123; doi:10.1038/ismej.2011.84; published online 30 June 2011Subject Category: integrated genomics and post-genomics approaches in microbial ecologyKeywords: photoheterotrophs; rhodopsin; pufM; single cell; RuBisCO; bacterioplankton
Introduction
Photosynthetic reactions performed by algae andcyanobacteria are the primary autochthonoussources of energy and organic carbon in mostaquatic ecosystems. However, recent studies demon-strate that some heterotrophic, planktonic bacteriaharness solar energy to produce ATP, in this waysupplementing their energy requirements but notfixing inorganic carbon (Zubkov, 2009). Such photo-heterotrophs include rhodopsin-containing bacteria(RBs) (Beja et al., 2002) and aerobic anoxygenic
phototrophs (AAPs) (Yurkov and Beatty, 1998;Beja et al., 2002). Both RBs and AAPs are abundantin the ocean (Beja et al., 2000; de la Torre et al.,2003; Sabehi et al., 2005; Jiao et al., 2007, 2010;DeLong and Beja, 2010), potentially contributingsignificantly to the ecosystem productivity. Inmarine ecosystems, members of Proteobacteria,Flavobacteria, Planctomycetes and Euryarchaeahave been found to contain rhodopsins (DeLongand Beja, 2010), whereas AAPs have been identifiedamong Alpha- and Gammaproteobacteria (Allgaieret al., 2003; Cho et al., 2007). In contrast to themarine environments, only a handful of studies onphotoheterotrophy have been conducted in fresh-water ecosystems (Waidner and Kirchman, 2005;Atamna-Ismaeel et al., 2008; Masın et al., 2008;Sharma et al., 2008, 2009; Eiler et al., 2009). So far,only Actinobacteria have been found to possessrhodopsins in freshwater ecosystems, as a result of
Received 10 March 2011; revised 3 May 2011; accepted 4 May2011; published online 30 June 2011
Correspondence: R Stepanauskas, Single Cell Genomics Center,Bigelow Laboratory for Ocean Sciences, PO Box 475, 180McKown Point Road, West Boothbay Harbor, ME 04575-0475,USA.E-mail: [email protected]
The ISME Journal (2012) 6, 113–123& 2012 International Society for Microbial Ecology All rights reserved 1751-7362/12
www.nature.com/ismej

metagenomic- and cultivation-based studies (Sharmaet al., 2009). In the case of freshwater AAPs, severalalpha- and betaproteobacteria strains have beenisolated (Yurkov and Beatty, 1998; Suyama et al.,1999; Page et al., 2004; Gich and Overmann, 2006;Wagner-Dobler and Biebl, 2006) and a few surveyshave been conducted to study the diversity anddistribution of genes involved in aerobic AAP, suchas pufM and BchlY (Waidner and Kirchman, 2005;Yutin et al., 2005; Masın et al., 2008). Thus, the existingdata suggest that RBs and AAPs are present and diversein freshwater environments, but their abundance andtaxonomic identities remain largely unknown.
Chemoautotrophs constitute another potentiallyunderappreciated functional group of freshwaterbacterioplankton. Several recent studies demon-strate significant CO2 fixation in the dark in bothanoxic and oxygenated water column, and some ofthis CO2 fixation appears to be driven by non-pigmented prokaryotes of unknown taxonomicaffiliation (Garcıa-Cantizano et al., 2005; Casamayoret al., 2008). In contrast, the only molecular surveyon the diversity of the ribulose 1,5-bisphosphatecarboxylase/oxygenase (RuBisCO) gene in fresh-water bacterioplankton that we are aware of (Tabitaet al., 2008) concluded (based on RuBisCO phylo-geny) that photosynthetic organisms were the onlyautotrophs in the epilimnion. The paucity ofinformation about the taxonomic identities of im-portant photoheterotrophs and chemoautotrophs isprimarily the result of methodological limitations. Onthe one hand, it is well known that current cultivationtechniques do not recover the vast majority ofthe environmental microbial diversity (Rappe andGiovannoni, 2003). On the other hand, most culture-independent research tools, such as environmentalpolymerase chain reaction (PCR)-based gene surveysor metagenomic shotgun sequencing, are poorlysuited to link metabolic genes to taxonomic markersat the organism level (Rusch et al., 2007).
To circumvent these methodological limitationsand to significantly expand our knowledge of photo-heterotroph and chemoautotroph diversity in fresh-water ecosystems, we employed multi-locus DNAsequencing from individual microbial cells (Raghu-nathan et al., 2005; Zhang et al., 2006), which hasbeen proven suitable for the study of unculturedmicroorganisms (Kvist et al., 2007; Marcy et al., 2007;Stepanauskas and Sieracki, 2007; Woyke et al., 2009).Our approach enabled us to link photoheterotrophicand chemoautotrophic gene markers to specifictaxonomic groups of bacterioplankton inhabiting theepilimnia of temperate freshwater lakes.
Methods
Sample collectionWater samples were collected from 0.5 to 1 m depthof the temperate freshwater lakes Mendota, Damar-iscotta, Sparkling and Trout Bog (Supplementary
Table S1). The same samples were used for meta-genomic shotgun sequencing and for single-cellanalyses. For metagenomics, microbial biomassfrom 0.5 to 4.5 l was collected on 0.2-mm pore sizemembranes (Supor PES filters, Pall Corporation, NY,USA) and stored at �80 1C until DNA extraction.For single-cell analyses, replicate, 1-ml aliquots ofenvironmental samples were cryopreserved with6% glycine betaine (Sigma, St Louis, MO, USA)at �80 1C until used (Cleland et al., 2004).
Metagenomic analysesDNA extractions from Damariscotta samples werecarried out using the PowerWater kit (MoBioLaboratories Inc., Carlsbad, CA, USA) followingthe manufacturer’s protocol. The obtained DNAwas concentrated with Microcon YM-10 columns(Millipore, Bedford, MA, USA) until reaching thedesired concentration for 454 pyrosequencing. ForMendota, Sparkling and Trout Bog samples, DNAextractions were performed using the xanthogenate-sodium dodecyl sulfate protocol slightly modifiedfrom that described by Tillett and Neilan (2000).Briefly, each filter was incubated with 250 ml ofTE buffer (10 mM Tris-Cl, pH 7.5 and 1 mM EDTA)and 25ml of lysozyme (10 mg ml) for 10 min at roomtemperature. Then, 50 ml of 10 mg ml proteinase Kwas added and incubated at 55 1C for 1 h, and thenproceeded as described (Tillett and Neilan, 2000). Atotal of eight shotgun libraries (two per lake: springand summer) were constructed at the KTH GenomeCenter (Stockholm, Sweden) and the Institutefor Genome Sciences (Baltimore, MD, USA) usingthe 454 GS FLX Titanium Sequencing Platform(Roche, Branford, CT, USA) according to themanufacturer’s instructions (Supplementary TableS2). The obtained 454 reads were quality-trimmedand the redundant reads were removed from thedata set (Gomez-Alvarez et al., 2009). The individual454 reads were then annotated using both theRAMMCAP pipeline (Li, 2009) implemented in theCAMERA (https://portal.camera.calit2.net) andthe MG-RAST server (Meyer et al., 2008). In addi-tion, we built a local database with our metagenomicreads and used it for BLASTx similarity searches todetect the rhodopsin, pufLM, BchlY, RuBisCO andrecA/radA sequences present in the metagenomes.We used the standalone BLAST 2.2.22þ package(ftp://ftp.ncbi.nlm.nih.gov/blast/) with an E-valuecutoff of 10�5. Sequences detected by the standaloneBLAST, CAMERA and MG-RAST were used todesign new or to improve existing primers usingPrimer 3 (Untergasser et al., 2007).
Single-cell sorting, whole genome amplification,and PCR screening of SAG librariesBefore cell sorting, environmental samples withprokaryote cell abundances above 5� 105 ml�1 werediluted 10� with sterile-filtered lake water and
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
114
The ISME Journal

pre-screened through a 70-mm mesh-size cell strainer(Becton Dickinson, Franklin Lakes, NJ, USA). Forprokaryote detection, diluted subsamples (1–3 ml)were incubated for 10–120 min with SYTO-9DNA stain (5mM final concentration; Invitrogen,Carlsbad, CA, USA). The high nucleic acid (HNA)and low nucleic acid (LNA) cell fractions weresorted separately. Cell sorting was performed with aMoFlo (Beckman Coulter, Danvers, MA, USA) flowcytometer using a 488 nm argon laser for excitation, a70mm nozzle orifice and a CyClone robotic arm fordroplet deposition into microplates. The cytometerwas triggered on side scatter. The ‘single 1 drop’ modewas used for maximal sort purity, which ensures theabsence of non-target particles within the target celldrop and the drops immediately surrounding the cell.The accuracy of 10mm fluorescent bead depositioninto the 384-well plates was verified by microscopi-cally examining the presence of beads in the platewells. Of the 2–3 plates examined each sort day, o2%wells were found to not contain a bead and onlyo0.5% wells were found to contain more than onebead, indicating thus very high purity of single cells.The latter is most likely caused by co-deposition oftwo beads attached to each other, which at certainorientation may have similar optical properties to asingle bead. Cells were deposited into 384-well platescontaining 0.6ml per well of either (a) 1� TE bufferor (2) prepGEM Bacteria (Zygem, Solana Beach, CA,USA) reaction mix, and stored at �80 1C until furtherprocessing. Of the 384 wells, 315 were dedicatedfor single cells, 66 were used as negative controls (nodroplet deposition) and 3 received 10 cells each(positive controls).
The cells that were sorted into TE buffer (most ofthe single amplified genomes (SAGs)) were lysedand their DNA was denatured using cold KOH(Raghunathan et al., 2005). The cells that weresorted into the prepGEM Bacteria reaction mix (SAGnames starting with AAA041) were first lysedfollowing Zygem instructions and then exposed toKOH treatment as above. There was no statisticallysignificant difference (Po0.05) between the compo-sition of SAGs obtained using a KOH lysis treatmentor a combination of prepGEM enzymes and KOHfor cell lysis. Genomic DNA from the lysed cells wasamplified using multiple displacement amplifica-tion (MDA) to generate enough template for sub-sequent, multiple PCR-based or genomic sequencing(Dean et al., 2002; Raghunathan et al., 2005) in 10 mlfinal volume. The MDA reactions contained 2 U/mlRepliphi polymerase (Epicentre, Madison, WI,USA), 1� reaction buffer (Epicentre), 0.4 mM eachdNTP (Epicentre), 2 mM dithiothreitol (Epicentre),50 mM phosphorylated random hexamers (IDT) and1mM SYTO-9 (Invitrogen) (all final concentration).The MDA reactions were run at 30 1C for 12–16 h,and then inactivated by 15 min incubation at 65 1C.The amplified genomic DNA was stored at �80 1Cuntil further processing. We refer to the MDAproducts originating from individual cells as SAGs.
The instruments and the reagents were deconta-minated for DNA before sorting and MDA setup, asdescribed previously (Stepanauskas and Sieracki,2007). DNA contaminants in MDA reagents werecrosslinked by an UV treatment in Stratalinker(Stratagene, Santa Clara, CA, USA) for 40–90 min,rendering them unamplifiable by MDA. During UVtreatment, reagents were placed on ice to avoidoverheating. An empirical optimization of the UVexposure was performed to ensure the removal ofamplifiable contaminants without inactivatingMDA. Cell sorting and MDA setup were performedin a high-efficiency particulate air-filtered environ-ment. As a quality control, the kinetics of all MDAreactions were monitored by measuring the SYTO-9 fluorescence using either LightCycler 480 (Roche)or FLUOstar Omega (BMG, Cary, NC, USA). Thecritical point (Cp) was determined for each MDAreaction as the time required to produce half of themaximal fluorescence. The Cp is inversely corre-lated to the amount of DNA template (Zhang et al.,2006). Only microplates in which Cp values weresignificantly lower in 1-cell wells compared to0-cell wells (Po0.05; Wilcoxon’s two-sample test)were used in further analysis. Humic Lake TroutBog SAG libraries had very low MDA success rate,probably due to the high concentration of humicacids. Thus, that particular sample was not con-sidered in our estimates of photoheterotrophabundance.
The MDA products were diluted 50-fold insterile TE buffer. Then, 0.5 ml aliquots of the diluteMDA products served as templates in 5 ml real-time PCR screens. The SSU rRNA, pufM, BchlY,rhodopsin and RuBisCO genes were targetedin these PCR using primers and thermal cyclingconditions specified in Supplementary TableS3. Forward (50-GTAAAACGACGGCCAGT-30) orreverse (50-CAGGAAACAGCTATGACC-30) M13sequencing primer was appended to the 50 end ofeach PCR primer to aid direct sequencing of thePCR products. All PCRs were performed usingLightCycler 480 SYBR Green I Master mix (Roche)in a LightCycler 480 II real-time thermal cycler(Roche). The real-time PCR kinetics and theamplicon melting curves served as proxies detect-ing SAG target genes. New 20 ml PCR reactionswere set up for the PCR-positive SAGs andthe amplicons were sequenced from both endsusing M13 targets and Sanger technology by theBeckman Coulter Genomics.
Single-cell sorting, whole genome amplificationand real-time PCR screens were performed at theBigelow Laboratory Single Cell Genomics Center(www.bigelow.org/scgc). Our previous studies andother recent publications using our single-cellsequencing techniques demonstrate the reliabilityof our methodology with insignificant levels of DNAcontamination (Stepanauskas and Sieracki, 2007;Woyke et al., 2009; Fleming et al., 2011; Hess et al.,2011; Heywood et al., 2011).
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
115
The ISME Journal

Phylogenetic analysisThe 16S rRNA gene sequences obtained from SAGswere aligned using the SILVA aligner (Pruesse et al.,2007). Only sequences displaying X80% of thealignment quality score in the SILVA aligner wereconsidered for the analysis (http://www.arb-silva.de/). Phylogenetic analysis based on maximumlikelihood (1000 bootstrap replications) was per-formed with RAxML version 7.0.3 (Stamatakis,2006) implemented in ARB package (Ludwig et al.,2004) using the reference ARB database 102 contain-ing 460 783 high-quality 16S rRNA sequences(http://www.arb-silva.de). The core tree was calcu-lated with the closest reference sequences and thenpartial sequences from SAGs (360–833 nucleotidepositions) were added using the ARB parsimonytool. Sequences of pufM, BchlY, rhodopsin andRuBisCO from SAGs were translated to amino acids,aligned with ClustalW and manually revised. Theresulting protein alignment was used as a scaffoldfor constructing the corresponding nucleotidealignment using RevTrans 1.4 Server (Wernerssonand Pedersen, 2003). Both the protein and thenucleotide alignments were used to infer theevolutionary history of the studied genes based onmaximum likelihood (1000 bootstrap replications)using RAxML version 7.0.4. Recombination detec-tion was performed following the model ofdual multiple change-point on aligned nucleotidesequences with the program DualBrothers (Mininet al., 2005). GenBank accession numbers: 16S rRNA(HQ662961–HQ663702), rhodopsin (HQ663727–HQ663845), pufM (HQ663703–HQ663715), BchlY(HQ663724–HQ663726) and RuBisCO (HQ663716–HQ663723).
Results and discussion
Taxonomic composition of SAGsWater samples to build the SAG libraries werecollected from the euphotic zone of the temperatefreshwater lakes Mendota, Damariscotta, Sparklingand Trout Bog (see Supplementary Table S1 for lakecharacteristics). A total of 3150 SAGs of randomlysorted freshwater planktonic prokaryotes weregenerated and PCR-screened for the 16S rRNA gene(Table 1). We successfully sequenced the 16S rRNA
gene from 712 SAGs, yielding 5–30% successrate, depending on the lake and season (Table 1).Combined, Actinobacteria, Betaproteobacteria andGammaproteobacteria comprised 61–97% of SAGsfrom the studied lakes (Supplementary Figure S1).Each of these groups were dominated by clustersthat were previously found to be abundant infreshwater environments using other methods, suchas the Actinobacteria group acI (SupplementaryFigure S2a) and the betaproteobacteria Polynucleo-bacter spp. (Supplementary Figure S2b) (Warneckeet al., 2005; Allgaier and Grossart, 2006; Rusch et al.,2007; Jezberova et al., 2010). Other ubiquitous albeitless abundant freshwater clusters represented in theSAG libraries included the AlphaproteobacteriaLD12 clade (Zwart et al., 2002), Bacteroidetes,Deltaproteobacteria and Verrucomicrobia (Supple-mentary Figures S2d–g). No archaeal 16S rRNAsequences were detected in the studied SAGlibraries or in the metagenomic shotgun libraries ofthe lakes annotated with CAMERA and MG-RASTpipelines. Overall, bacterial diversity data obtainedby metagenomics and single-cell sequencingshowed similar taxonomic composition, with59–83% of 16S rRNA gene sequences retrievedusing the two techniques displayed 497% similar-ity (Supplementary Figure S3). Furthermore, similarrelative abundances were obtained in the 454shotgun and SAG libraries for the predominantfreshwater groups such as Actinobacteria andBetaproteobacteria, which together comprised45–60% of total SAGs in the studied lakes. There-fore, the diversity and relative abundance of theobtained SAGs are consistent with metagenomic dataand previous studies of similar freshwater environ-ments (Zwart et al., 2002; Warnecke et al., 2005;Allgaier and Grossart, 2006; Rusch et al., 2007;Jezberova et al., 2010), indicating that our single-cellsequencing techniques were suitable to represent thefull spectrum of the most abundant epilimnetic bac-terioplankton groups in temperate freshwater lakes.
In natural aquatic environments, bacteria withHNA and LNA content are commonly observedwith flow cytometry after cell staining with nucleicacid-specific fluorescent dyes (Gasol and DelGiorgio, 2008). We compared the taxonomic compo-sition of SAGs generated from the HNA and LNAbacterioplankton fractions (Figure 1). Results show
Table 1 Summary of SAGs analyzed and genes obtained from single cells
Lake SAGs 16S rRNAa Rhodopsin pufM BchlY Photoheterotrophsb (%) RuBisCO
Damariscotta (spring) 945 200 (21%) 40 4 2 23 1Damariscotta (summer) 945 179 (19%) 34 1 1 20 0Mendota (spring) 630 188 (30%) 33 3 0 20 1Sparkling (spring) 630 145 (23%) 12 5 0 10 6Total 3150 712 (23%) 119 13 3 133 8
Abbreviations: AAP, aerobic anoxygenic phototroph; RB, rhodopsin-containing bacteria; RuBisCO, ribulose 1,5-bisphosphate carboxylase/oxygenase; SAG, single amplified genome.aNumber of SAGs yielding 16S rRNA gene sequences (success rate, %).bA total of 133 photoheterotrophic cells were identified. Frequency of photoheterotrophs (RBs and AAPs) among the SAGs that yielded 16S rRNA gene.
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
116
The ISME Journal

that the taxonomic composition of LNA cellsdiffered from HNA cells in Damariscotta springand Mendota spring samples, whereas no significantdifferences were observed in Damariscotta summerand Sparkling spring samples. This suggests thattaxonomic differences between HNA and LNA cellsare subject to spatial and temporal variation. Somemarine studies suggest that only HNA cells aremetabolically active (Gasol and Del Giorgio, 2008),whereas other reports contradict this simple dichot-omy (Jochem et al., 2004; Zubkov et al., 2004;Longnecker et al., 2005, 2006; Bouvier et al., 2007).Recently, Wang et al. (2009) have shown in fresh-water that LNA bacteria affiliated to the Polynucleo-bacter cluster utilize natural assimilable organiccarbon and show high growth rates. Interestingly,95% of the Polynucleobacter spp. SAGs originatedfrom HNA cells in our study, whereas there was nosuch HNA/LNA separation among ActinobacteriaacI (51% HNA) and Alphaproteobacteria LD12 (45%HNA) SAGs. This contrasts with findings frommarine systems, where SAR11, the sister groupof LD12, is predominantly found in the LNA fraction(Hill et al., 2010).
Abundance of photoheterotrophsSequences of rhodopsin, pufM and BchlY genesrecovered from metagenomic shotgun sequencing ofthe studied freshwater samples (SupplementaryTables S4) were used to design multiple, optimizedprimers to PCR amplify and sequence these genesfrom individual SAGs (Supplementary Table S3).Owing to cost constraints, only two pairs ofrhodopsin primers, representing the most abundantmetagenomic sequences, were used in the SAGscreening (Supplementary Table S3); these primerscovered 50–100% of the forward targets and78–100% of the reverse targets found in the fourmetagenomic data sets (Supplementary Table S4A).The pufM and BchlY primers used in the SAGanalysis covered 100% of the diversity of these
genes found in the studied metagenomes (Supple-mentary Table S4B). In total, we PCR-amplified andsequenced 119 rhodopsin, 13 pufM and 3 BchlYgenes from SAGs. As the 16S rRNA genes were alsosequenced from the same SAGs, this multi-locussequencing analysis of individual cells providedcultivation-unbiased taxonomic identity of 133photoheterotrophic freshwater bacteria (Table 1).
Among the studied environmental samples,rhodopsin genes were detected in 8–20% of theSAGs and either pufM or BchlY or both weredetected in 2–3% of the SAGs (Table 1). This shouldbe considered a conservative estimate of photohe-terotrophic bacterioplankton abundance, due toPCR limitations, such as primer–target mismatches(discussed above) and template secondary struc-tures (Potvin and Lovejoy, 2009). The unevengenome amplification by MDA (Zhang et al., 2006;Woyke et al., 2009) may also lead to some PCRreactions to fail. However, the range of AAPabundance detected here is within the publishedrange for temperate freshwater systems obtained byinfrared epifluorescence microscopy (o1–20% oftotal bacteria) (Masın et al., 2008). Contrary to AAPs,RB abundances cannot be estimated by microscopy.Thus, single-cell sequencing circumvents currentmethodological limitation to study rhodopsin abun-dances in microbial communities (DeLong and Beja,2010). As an alternative way to determine photoheter-otroph abundance, we calculated the ratios of rho-dopsin and pufM genes to the conserved singlecopy gene recA in the metagenomic data sets obtainedfrom the same lake water samples. Assuming thatno more than one copy of these genes occurs in eachcell, rhodopsin and pufM genes were present in37–56% and 3–37% of the studied freshwaterbacterioplankton samples, respectively (Figure 2).
Figure 1 Principal coordinates analysis of weighted UniFracpairwise distances between 16S rRNA gene sequences from thestudied environmental samples and the HNA and LNA cellfractions. A neighbor-joining tree (Jukes–Cantor substitutionmodel) including all 16S rRNA gene sequences from SAGs servedas the input data for the Fast UniFrac analysis. The archaeonNitrosopumilus maritimus (CP000866) was used as an outgroup.
Figure 2 The relative frequency of photoheterotrophs infreshwater bacterioplankton, as determined by single-cellapproach and metagenomic sequencing. The frequency of photo-heterotrophs among SAGs was determined as the fraction of 16SrRNA-positive SAGs from which rhodopsin or pufM gene wasrecovered. The frequency of photoheterotrophs in metagenomeswas determined as the ratio of either rhodopsin or pufM torecAþ radA. M, metagenomics; SCG, single-cell genomics; Dam,Damariscotta Lake.
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
117
The ISME Journal

These metagenomics-based estimates likely betterreflect the true frequencies of phototrophs in thestudied samples, albeit they do not provide informa-tion on photoheterotroph identities, which is a majoradvantage of single-cell sequencing. Furthermore, 454shotgun sequencing is also prone to some biases thatmay distort gene frequency information (Morgan et al.,2010). Despite the existing methodological limitationsfor photoheterotroph quantification, it is clearthat AAPs and RBs constitute a major fraction offreshwater bacterioplankton, at least 8–23% acrossvarious types of lakes.
Identity of photoheterotrophsRhodopsin genes obtained from SAGs formedtwo major clusters (Figure 3a). The first clusterwas composed of 95 actinobacteria and 5 gamma-proteobacteria SAGs and grouped together with thepreviously published rhodopsin sequences fromclones and cultures named as ‘actinorhodopsins’(Atamna-Ismaeel et al., 2008; Sharma et al., 2008,2009) (Figure 3b). The second cluster was composedof rhodopsin sequences from 15 SAGs (Figure 3c)belonging to Alpha-, Beta-, Gamma- and Deltapro-teobacteria, Verrucomicrobia, Sphingobacteria andActinobacteria. The latter SAG sequences groupedtogether with previously published sequencesdesignated as ‘proteorhodopsins’ (Beja et al., 2000)(detailed phylogenetic information from the 16SrRNA gene analysis is provided in SupplementaryFigure S2). Three rhodopsin sequences from theSAGs AAA041-G17 (Betaproteobacteria), AAA278-C16 (Deltaproteobacteria) and AAA487-P23 (Verruco-microbia) were phylogenetically positioned in-betweenthese two major rhodopsin clusters. In addition, thealphaproteobacterium SAG AAA024-J18 carried arhodopsin that was phylogenetically related toxanthorhodopsins (Figure 3a), which are protonpumps that are abundant in hypersaline environ-ments (Balashov et al., 2005). Interestingly, 45%of SAGs that carried the rhodopsin gene wereHNA bacteria, indicating that rhodopsin geneswere equally abundant among HNA and LNAbacterioplankton.
Before our study, only Actinobacteria have beenfound to contain rhodopsins in freshwater environ-ments (Sharma et al., 2009). Our data demonstratesActinobacteria, including clusters acI, Luna andacSTL, as the predominant phylum containingrhodopsin genes in temperate freshwater lakes. Inaddition, we significantly broadened the knowntaxonomic range of rhodopsin-containing freshwaterbacterioplankton, to include Alpha-, Beta-, Gamma-and Deltaproteobacteria, Verrucomicrobia andSphingobacteria. In fact, rhodopsin-containingDeltaproteobacteria, Verrucomicrobia and Sphingo-bacteria have never been previously reported fromany type of environment.
In most cases, phylogenies of the 16S rRNAand genes involved in photoheterotrophy were
congruent (Figure 3). For instance, we demonstratedthat freshwater rhodopsins, related to the marineSAR11 clade, belong to the SAR11 sister groupLD12 (Figure 3c and Supplementary Figure S2c).However, this congruency has exceptions. In onecase, five gammaproteobacteria SAGs, originatingfrom multiple environmental samples, had rhodop-sin sequences clustering with Actinobacteria,suggesting their origin through horizontal genetransfer (HGT). In another case, an actinobacteriaSAG AAA278-O22 contained two rhodopsins, onetypical for this phylum and another closely relatedto sequences from Betaproteobacteria. This impliesthat B5% of the observed rhodopsins may haveevolved from HGT events. Earlier findings ofrhodopsin HGT among phylogenetically distantmicrobes is consistent with our results (McCarrenand DeLong, 2007). Besides HGT, our study providesthe first evidence for recombination betweenactinorhodopsin- and proteorhodopsin-like genes(Figure 3d), resulting in composite rhodopsins, suchas those found in the SAGs AAA278-C16 (Deltapro-teobacteria) and AAA041-G17 (Betaproteobacteria)that are phylogenetically positioned in-betweenthe two major rhodopsin clusters (Figure 3a).
We detected either pufM or BchlY or both in15 SAGs (Table 1). Unexpectedly, most of theseputative AAPs (53%) were Betaproteobacteria,primarily members of the Polynucleobacter cluster(Figure 4). Two alphaproteobacteria and threegammaproteobacteria SAGs related to Roseomonasand Pseudomonas spp. also had pufM genes.Thus, members of the ubiquitous Polynucleobactercluster (Jezberova et al., 2010) may be among thepredominant freshwater AAPs. This is contraryto earlier data obtained from cultures, wherefreshwater AAPs have been primarily detectedamong Alphaproteobacteria (Yurkov and Beatty,1998; Suyama et al., 1999; Page et al., 2004; Gichand Overmann, 2006; Wagner-Dobler and Biebl,2006). Interestingly, genes involved in aerobic AAPare absent in Polynucleobacter strain sp. QLW-P1DMWA-1, the only planktonic freshwater bacter-ium with whole genome information available. Ourstudy demonstrates how single-cell sequencing canprovide more reliable and extensive informationabout the metabolic potential of specific microbialassemblage members compared with other availablemethods.
Potential planktonic chemoautotrophsWe detected RuBisCO genes in several SAGs ofBeta- and Gammaproteobacteria and Bacteroidetes,raising the possibility that they fix inorganic carbon(Figure 4). These SAGs appear to represent aerobic,planktonic organisms, as closely related 16S rRNAgene clones have been obtained from the euphoticand well oxygenated waters of different lakesand continents (Supplementary Figure S2). More-over, none of these SAGs are related to known
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
118
The ISME Journal

chemolitoautotrophic or photoautotrophic anaero-bic bacteria, which would indicate resuspensionfrom sediments or hypolimnion. All the RuBisCO
sequences obtained from these SAGs were form IAor IB, which are typically found in aerobic environ-ments (Tabita et al., 2008). Significantly, PCR
Figure 3 Maximum-likelihood tree of 119 rhodopsin proteins from single cells: (a) general tree; (b) subtree of actinorhodopsin-likeproteins and (c) subtree of proteorhodopsin-like proteins. In all, 120 amino-acid positions were used in the tree construction. Bootstrapvalues X50 are displayed. SAGs obtained from the HNA and LNA cells are indicated in regular and italic fonts, respectively.The taxonomic identity of rhodopsin-containing SAGs, based on their 16S rRNA gene phylogeny, is provided next to the SAG name(for detailed phylogeny of the 16S rRNA genes, see Supplementary Figure S2). (d) Recombination analysis of rhodopsin genes. The dualmultiple change-point model that considers the spatial variation of tree topologies and the substitution process parameters was appliedin a Bayesian framework using reversible jump Markov chain Monte Carlo sampling to approximate the joint posterior distribution of allmodel parameters. Parameters of transition:transversion (k) and expected divergence (m) and spatial variation of tree topologies areindicated. Each one of the breakpoints shown in the tree topologies together with k and m parameters indicate a putative recombinationevent. Recombination was not detected within the actinorhodopsin and proteorhodopsin clusters.
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
119
The ISME Journal

primers used in our SAG screens (SupplementaryTable S3) displayed 6–9 nucleotide mismatcheswith most RuBisCO genes detected by metage-nomics and failed to amplify RuBisCO genes evenfrom some cyanobacterial SAGs. Unfortunately, ourattempts to design broader-range RuBisCO primershave failed so far, due to the high diversity ofthe typical conserved regions of this gene in ourmetagenomic data sets. The ratio of RuBisCO versusrecA genes ranged 6–77% in the metagenomes(Supplementary Figure S4). Both the SAG analysisand the metagenomic sequencing suggested that thehighest frequency of cells with RuBisCO genes werein the oligotrophic Sparkling Lake. It is important tonote that in this study we did not target otherautotrophic carbon fixation pathways than theCalvin–Benson–Bassham cycle. Thus, our analysismay have significantly underestimated the fractionof potential planktonic chemoautotrophs.
In addition to CO2 fixation, RuBisCO may alsobe involved in the central redox cofactor recycling
in AAP bacteria inhabiting reducing environments,such as soils and sediments, sometimes thrivingon organic substrates more reduced than biomass(McKinlay and Harwood, 2010). It remains to bedetermined whether similar mechanisms can besignificant among bacterioplankton inhabiting oxy-genated water column. Chemoautotrophy ratherthan the recycling of redox cofactors appears a morelikely role of the detected SAG RuBisCO genes forthe following reasons: (1) we did not find photo-trophy genes pufM or BchlY in any of the SAGs thatcontained RuBisCO; and (2) RuBisCO was mostabundant in SAGs from the oligotrophic SparklingLake, which is the lake containing the lowestconcentrations of organic substrates (see Supple-mentary Table S1). Thus, our study providestaxonomic identities of potential freshwater che-moautotrophs that may be involved in the aerobicCO2 fixation, a metabolic process that requiresfurther attention to fully understand carbon cyclingin freshwater environments.
Figure 4 Maximum-likelihood phylogenetic analysis of pufM and RuBisCO genes and the corresponding 16S rRNA sequences fromsingle cells (limited to Betaproteobacteria owing to space constrains). The taxonomic identity of the pufM- and RuBisCO-containingSAGs is indicated next to the SAG name. BchlY-containing SAGs are indicated in the phylogenetic tree of 16S rRNA gene (for detailedphylogeny of the 16S rRNA genes, see Supplementary Figure S2). Bootstrap (1000 replicate) values X50 are displayed. In the case ofpufM gene, the analysis was conducted on nucleotide sequence alignment (250 nucleotide positions). For the RuBisCO gene, the analysiswas based on protein sequence alignment (amino-acid positions 100–262).
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
120
The ISME Journal

Concluding remarks
Using a combination of single-cell sequencing andmetagenomics, we vastly expanded the knowledgeof the predominant photoheterotrophs and potentialchemoautotrophs inhabiting the euphotic zoneof temperate freshwater lakes. We found that all ofthe ubiquitous freshwater bacterioplankton clusters,such as Actinobacteria acI, Luna and acSTL, Poly-nucleobacter spp. (Betaproteobacteria) and LD12(Alphaproteobacteria) contain photoheterotrophs,suggesting that photoheterotrophy is an importantcompetitive strategy for freshwater bacterioplank-ton. Our approach enabled us to perform a high-throughput, cost-effective study while circumvent-ing many analytical limitations inherent to earliertechniques, such as cultivation and metagenomics.For example, more than a decade of studiesof marine proteorhodopsins have resulted in directidentification of only 37 bacteria containing rho-dopsins, most of which belong to a few taxonomicgroups (DeLong and Beja, 2010). Here, in a 1-yearstudy we identified 118 predominant freshwaterbacteria containing rhodopsins, with no apparenttaxonomic biases. Furthermore, our single-cellsequencing results indicate HGT and recombinationof rhodopsin genes in freshwater bacterioplanktonand link the gene’s evolutionary history to thetaxonomic identities of specific microbial groupsthat are involved in these evolutionary processes.Finally, SAGs generated here represent the largestand cultivation-unbiased genomic DNA library offreshwater bacterioplankton, opening unprece-dented opportunities for additional analysesof specific loci and whole genome sequencing thatwill provide further insights into the metabolicpotential and evolutionary histories of freshwaterbacterioplankton.
Acknowledgements
We thank Wendy Korjeff-Bellows and Jane Heywood forfieldwork assistance, Stefan Bertilsson and Siv Anderssonfor access to their metagenomic data, Katherine McMahonand Todd Miller for Wisconsin lake sampling andDNA extraction, and Vladimir M Minin for his advice onrecombination event analysis. This research was sup-ported by the NSF Grants DEB-841933 and OCE-821374to RS and by a Maine Technology Institute researchinfrastructure grant to the Bigelow Laboratory.
References
Allgaier M, Grossart H-P. (2006). Diversity and seasonaldynamics of actinobacteria populations in four Lakesin Northeastern Germany. Appl Environ Microbiol 72:3489–3497.
Allgaier M, Uphoff H, Felske A, Wagner-Dobler I. (2003).Aerobic anoxygenic photosynthesis in Roseobacter
clade bacteria from diverse marine habitats. ApplEnviron Microbiol 69: 5051–5059.
Atamna-Ismaeel N, Sabehi G, Sharon I, Witzel K-P,Labrenz M, Jurgens K et al. (2008). Widespreaddistribution of proteorhodopsins in freshwater andbrackish ecosystems. ISME J 2: 656–662.
Balashov SP, Imasheva ES, Boichenko VA, Anton J,Wang JM, Lanyi JK. (2005). Xanthorhodopsin: a protonpump with a light-harvesting carotenoid antenna.Science 309: 2061–2064.
Beja O, Suzuki MT, Heidelberg JF, Nelson WC,Preston CM, Hamada T et al. (2002). Unsuspecteddiversity among marine aerobic anoxygenic photo-trophs. Nature 415: 630–633.
Beja O, Aravind L, Koonin EV, Suzuki MT, Hadd A,Nguyen LP et al. (2000). Bacterial rhodopsin: evidencefor a new type of phototrophy in the Sea. Science 289:1902–1906.
Bouvier T, Del Giorgio PA, Gasol JM. (2007). A compara-tive study of the cytometric characteristics of high andlow nucleic-acid bacterioplankton cells from differentaquatic ecosystems. Environ Microbiol 9: 2050–2066.
Casamayor EO, Garcıa-Cantizano J, Pedros-Alio C. (2008).Carbon dioxide fixation in the dark by photosyntheticbacteria in sulfide-rich stratified lakes with oxic–anoxic interfaces. Limnol Oceanogr 53: 1193–1203.
Cho J-C, Stapels MD, Morris RM, Vergin KL,Schwalbach MS, Givan SA et al. (2007). Polyphyleticphotosynthetic reaction centre genes in oligotrophicmarine Gammaproteobacteria. Environ Microbiol 9:1456–1463.
Cleland D, Krader P, McCree C, Tang J, Emerson D. (2004).Glycine betaine as a cryoprotectant for prokaryotes.J Microbiol Methods 58: 31–38.
de la Torre JR, Christianson LM, Beja O, Suzuki MT,Karl DM, Heidelberg J et al. (2003). Proteorhodopsingenes are distributed among divergent marine bacter-ial taxa. Proc Natl Acad Sci USA 100: 12830–12835.
Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward Pet al. (2002). Comprehensive human genome amplifi-cation using multiple displacement amplification.Proc Natl Acad Sci USA 99: 5261–5266.
DeLong EF, Beja O. (2010). The light-driven proton pumpproteorhodopsin enhances bacterial survival duringtough times. PLoS Biol 8: e1000359.
Eiler A, Beier S, Sawstrom C, Karlsson J, Bertilsson S.(2009). High ratio of bacteriochlorophyll biosynthesisgenes to chlorophyll biosynthesis genes in bacteria ofhumic lakes. Appl Environ Microbiol 75: 7221–7228.
Fleming EJ, Langdon AE, Martinez-Garcia M, Stepanaus-kas R, Poulton N, Masland D et al. (2011). What’s newis old: resolving the identity of Leptothrix ochraceausing single cell genomics, pyrosequencing and FISH.PLoS One 6: e17769.
Garcıa-Cantizano J, Casamayor E, Gasol J, Guerrero R,Pedros-Alio C. (2005). Partitioning of CO2; incorpora-tion among planktonic microbial guilds and estima-tion of in situ specific growth rates. Microb Ecol 50:230–241.
Gasol JM, Del Giorgio PA. (2008). Physiological structureand single-cell activity in marine bacterioplankton.In: Kirchman DL (ed). Microbial Ecology of theOceans. John Wiley & Sons Inc: Hoboken, NJ,pp 243–298.
Gich F, Overmann J. (2006). Sandarakinorhabdus limno-phila gen. nov., sp. nov., a novel bacteriochlorophylla-containing, obligately aerobic bacterium isolated
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
121
The ISME Journal

from freshwater lakes. Int J Syst Evol Microbiol 56:847–854.
Gomez-Alvarez V, Teal TK, Schmidt TM. (2009). Systema-tic artifacts in metagenomes from complex microbialcommunities. ISME J 3: 1314–1317.
Hess M, Sczyrba A, Egan R, Kim T-W, Chokhawala H,Schroth G et al. (2011). Metagenomic discoveryof biomass-degrading genes and genomes from cowrumen. Science 331: 463–467.
Heywood JL, Sieracki ME, Bellows W, Poulton NJ,Stepanauskas R. (2011). Capturing diversity of marineheterotrophic protists: one cell at a time. ISME J 5:674–684.
Hill PG, Zubkov MV, Purdie DA. (2010). Differentialresponses of Prochlorococcus and SAR11-dominatedbacterioplankton groups to atmospheric dust inputs inthe tropical Northeast Atlantic Ocean. FEMS MicrobiolLett 306: 82–89.
Jezberova J, Jezbera J, Brandt U, Lindstrom ES,Langenheder S, Hahn MW. (2010). Ubiquity of Poly-nucleobacter necessarius ssp. asymbioticus in lenticfreshwater habitats of a heterogenous 2000 km2 area.Environ Microbiol 12: 658–669.
Jiao N, Zhang Y, Zeng Y, Hong N, Liu R, Chen F et al.(2007). Distinct distribution pattern of abundance anddiversity of aerobic anoxygenic phototrophic bacteriain the global ocean. Environ Microbiol 9: 3091–3099.
Jiao N, Zhang F, Hong N. (2010). Significant rolesof bacteriochlorophyll a supplemental to chlorophylla in the ocean. ISME J 4: 595–597.
Jochem FJ, Lavrentyev PJ, First MR. (2004). Growth andgrazing rates of bacteria groups with different apparentDNA content in the Gulf of Mexico. Mar Biol 145:1213–1225.
Kvist T, Ahring B, Lasken R, Westermann P. (2007).Specific single-cell isolation and genomic amplifica-tion of uncultured microorganisms. Appl MicrobiolBiotechnol 74: 926–935.
Li W. (2009). Analysis and comparison of very largemetagenomes with fast clustering and functionalannotation. BMC Bioinform 10: 359.
Longnecker K, Homen DS, Sherr EB, Sherr BF. (2006).Similar community structure of biosyntheticallyactive prokaryotes across a range of ecosystem trophicstates. Aquat Microb Ecol 42: 265–276.
Longnecker K, Sherr BF, Sherr EB. (2005). Activity andphylogenetic diversity of bacterial cells with highand low nucleic acid content and electron transportsystem activity in an upwelling ecosystem. ApplEnviron Microbiol 71: 7737–7749.
Ludwig W, Strunk O, Westram R, Richter L, Meier H,Yadhukumar et al. (2004). ARB: a software environ-ment for sequence data. Nucl Acids Res 32:1363–1371.
Marcy Y, Ouverney C, Bik EM, Losekann T, Ivanova N,Martin HG et al. (2007). Dissecting biological ‘darkmatter’ with single-cell genetic analysis of rare anduncultivated TM7 microbes from the human mouth.Proc Natl Acad Sci USA 104: 11889–11894.
Masın M, Nedoma J, Pechar L, Koblızek M. (2008).Distribution of aerobic anoxygenic phototrophsin temperate freshwater systems. Environ Microbiol10: 1988–1996.
McCarren J, DeLong EF. (2007). Proteorhodopsin photo-system gene clusters exhibit co-evolutionary trendsand shared ancestry among diverse marine microbialphyla. Environ Microbiol 9: 846–858.
McKinlay JB, Harwood CS. (2010). Carbon dioxidefixation as a central redox cofactor recycling mec-hanism in bacteria. Proc Natl Acad Sci USA 107:11669–11675.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass E, KubalM et al. (2008). The metagenomics RAST server—apublic resource for the automatic phylogenetic andfunctional analysis of metagenomes. BMC Bioinform 9:386.
Minin VN, Dorman KS, Fang F, Suchard MA. (2005).Dual multiple change-point model leads to moreaccurate recombination detection. Bioinformatics 21:3034–3042.
Morgan JL, Darling AE, Eisen JA. (2010). Metagenomicsequencing of an in vitro-simulated microbialcommunity. PLoS One 5: e10209.
Page KA, Connon SA, Giovannoni SJ. (2004). Representa-tive freshwater bacterioplankton isolated from CraterLake, Oregon. Appl Environ Microbiol 70: 6542–6550.
Potvin M, Lovejoy C. (2009). PCR-based diversityestimates of artificial and environmental 18S rRNAgene libraries. J Eukaryot Microbiol 56: 174–181.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W,Peplies J et al. (2007). SILVA: a comprehensive onlineresource for quality checked and aligned ribosomalRNA sequence data compatible with ARB. Nucl AcidsRes 35: 7188–7196.
Raghunathan A, Ferguson Jr HR, Bornarth CJ, Song W,Driscoll M, Lasken RS. (2005). Genomic DNA ampli-fication from a single bacterium. Appl EnvironMicrobiol 71: 3342–3347.
Rappe MS, Giovannoni SJ. (2003). The uncultured micro-bial majority. Annu Rev Microbiol 57: 369–394.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB,Williamson S, Yooseph S et al. (2007). The SorcererII global ocean sampling expedition: Northwest Atlan-tic through Eastern Tropical Pacific. PLoS Biol 5: e77.
Sabehi G, Loy A, Jung K-H, Partha R, Spudich JL,Isaacson T et al. (2005). New insights into metabolicproperties of marine bacteria encoding proteorhodop-sins. PLoS Biol 3: e273.
Sharma AK, Sommerfeld K, Bullerjahn GS, Matteson AR,Wilhelm SW, Jezbera J et al. (2009). Actinorhodopsingenes discovered in diverse freshwater habitats andamong cultivated freshwater Actinobacteria. ISME J 3:726–737.
Sharma AK, Zhaxybayeva O, Papke RT, Doolittle WF.(2008). Actinorhodopsins: proteorhodopsin-like genesequences found predominantly in non-marine envir-onments. Environ Microbiol 10: 1039–1056.
Stamatakis A. (2006). RAxML-VI-HPC: maximum like-lihood-based phylogenetic analyses with thousandsof taxa and mixed models. Bioinformatics 22:2688–2690.
Stepanauskas R, Sieracki ME. (2007). Matching phylo-geny and metabolism in the uncultured marinebacteria, one cell at a time. Proc Natl Acad Sci USA104: 9052–9057.
Suyama T, Shigematsu T, Takaichi S, Nodasaka Y,Fujikawa S, Hosoya H et al. (1999). Roseatelesdepolymerans gen. nov., sp. nov., a new bacteriochlor-ophyll a-containing obligate aerobe belonging to thebeta-subclass of the Proteobacteria. Int J Syst Bacteriol49: 449–457.
Tabita FR, Hanson TE, Satagopan S, Witte BH, Kreel NE.(2008). Phylogenetic and evolutionary relationshipsof RubisCO and the RubisCO-like proteins and the
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
122
The ISME Journal

functional lessons provided by diverse molecularforms. Philos Trans R Soc Ser B 363: 2629–2640.
Tillett D, Neilan BA. (2000). Xanthogenate nucleic acidisolation from cultured and environmental cyanobac-teria. J Phycol 36: 251–258.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R,Leunissen JAM. (2007). Primer3Plus, an enhanced webinterface to Primer3. Nucl Acids Res 35: W71–W74.
Wagner-Dobler I, Biebl H. (2006). Environmentalbiology of the marine Roseobacter lineage. Annu RevMicrobiol 60: 255–280.
Waidner LA, Kirchman DL. (2005). Aerobic anoxygenicphotosynthesis genes and operons in unculturedbacteria in the Delaware River. Environ Microbiol 7:1896–1908.
Wang Y, Hammes F, Boon N, Chami M, Egli T. (2009).Isolation and characterization of low nucleic acid(LNA)-content bacteria. ISME J 3: 889–902.
Warnecke F, Sommaruga R, Sekar R, Hofer JS, Pernthaler J.(2005). Abundances, identity, and growth state ofActinobacteria in mountain lakes of different UVtransparency. Appl Environ Microbiol 71: 5551–5559.
Wernersson R, Pedersen AG. (2003). RevTrans: multiplealignment of coding DNA from aligned amino acidsequences. Nucl Acids Res 31: 3537–3539.
Woyke T, Xie G, Copeland A, Gonzalez JM, Han C, Kiss Het al. (2009). Assembling the marine metagenome, onecell at a time. PLoS One 4: e5299.
Yurkov VV, Beatty JT. (1998). Aerobic anoxygenicphototrophic bacteria. Microbiol Mol Biol Rev 62:695–724.
Yutin N, Suzuki MT, Beja O. (2005). Novel primersreveal wider diversity among marine aerobic anoxy-genic phototrophs. Appl Environ Microbiol 71:8958–8962.
Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J,Chisholm SW et al. (2006). Sequencing genomes fromsingle cells by polymerase cloning. Nat Biotechnol 24:680–686.
Zubkov MV, Allen JI, Fuchs BM. (2004). Coexistenceof dominant groups in marine bacterioplanktoncommunity; a combination of experimental andmodelling approaches. J Mar Biol Assoc UK 84:519–529.
Zubkov MV. (2009). Photoheterotrophy in marine prokar-yotes. J Plankton Res 31: 933–938.
Zwart G, Crump BC, Agterveld MPK-v, Hagen F, Han S-K.(2002). Typical freshwater bacteria: an analysisof available 16S rRNA gene sequences from planktonof lakes and rivers. Aquat Microb Ecol 28: 141–155.
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Photoheterotrophs and chemoautotrophs in freshwater bacterioplanktonM Martinez-Garcia et al
123
The ISME Journal

Supplementary Information for
High throughput single cell sequencing identifies photoheterotrophs and
chemoautotrophs in freshwater bacterioplankton
Manuel Martinez-Garcia, Brandon K. Swan, Nicole J. Poulton, Monica Lluesma Gomez,
Dashiell Masland, Michael E. Sieracki and Ramunas Stepanauskas*
*To whom correspondence should be addressed. E-mail: [email protected]
This supplementary information file includes: Supplementary Figure Legends (S1-S4) Supplementary Figures (S1-S4) Supplementary Tables (S1-S4) References for Supplementary Information
1

Supplementary Figure Legends Fig. S1. Taxonomic composition of SAGs from the HNA and LNA bacterioplankton
fractions from the Lakes Damariscotta (spring and summer), Mendota (spring), and
Sparkling (spring). SAG taxonomic identities were determined by using the Ribosomal
Database Project Classifier (http://rdp.cme.msu.edu/) and ARB software.
Fig. S2. Best maximum likelihood trees (1000 bootstrap) of the 16S rRNA genes from
SAGs belonging to (a) Actinobacteria, (b) Betaproteobacteria, (c) Alphaproteobacteria,
(d) Gammaproteobacteria, (e) Bacteroidetes (Sphingobacteria), (f) Deltaproteobacteria,
and (g) Verrucomicrobia. Single cells containing rhodopsin (blue), pufM (red), BchlY
(red), and RuBisCO (green) genes is indicated next to the SAG name. Bootstrap values ≥
50 are displayed.
Fig. S3. Bacterial composition comparison between metagenomics and single cell
sequencing. 16S rRNA gene sequences retrieved by metagenomics were quality control
filtered the same as SAGs sequences (see methods). Given the high number of short 16S
rRNA sequences from 454, sequences were also filtered according to length (≥200
nucleotides) and overlapping region (≥150 nucleotides) within the same 16S rRNA gene
nucleotide positions (340-806) analyzed for SAGs. Total number of 454-16S rRNA
sequences from metagenomics considered for the analysis was: 40, 34, and 44 for
Mendota, Damariscotta (spring), and Damariscotta (summer) respectively. 454 shotgun
library from Sparkling Lake was not considered due to the small number of reads.
Pairwise sequence similarity matrix was calculated with ARB software.
Fig. S4. The relative abundance of RuBisCO genes obtained by metagenomics.
recA/radA gene was used for data normalization.
2

Fig S1
3

Fig S2A
4

Fig S2B
5

Fig S2C
6

Fig S2D
7

Fig S2E
8

Fig S2F
Fig S2G
9
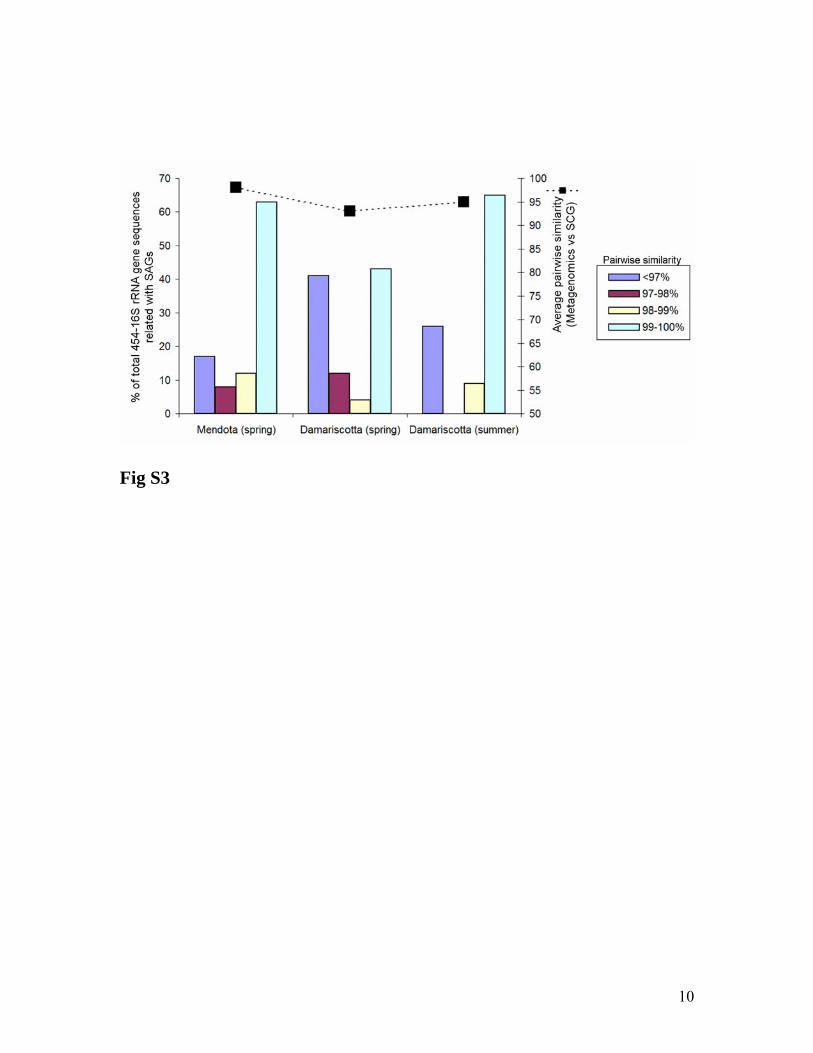
Fig S3
10

Fig S4
11

12
Tab
le S
1. L
ake
char
acte
rist
ics
and
sam
ple
info
rmat
ion
Lak
e T
roph
ic
stat
usa
Loc
atio
n la
titud
e [N
] lo
ngitu
de* [
W]
Mea
n de
pth* (
m)
Mea
na pH
S
ampl
ing
date
(20
09)
Chl
aa
(µg/
L)
Vol
ume
filt
ered
(L
) fo
r D
NA
ext
ract
ion
43°
6'19
.58"
05
/12
2.5
0.5
Men
dota
E
utro
phic
89
°24'
28.7
1"
12.8
8.
4 08
/23
13.3
0.
5 44
°10'
38.3
1"
04/2
8 3.
2 4.
5 D
amar
isco
tta
Mes
otro
phic
69
°29'
12.4
2"
9.0
6.7
08/1
9 3.
1 1.
5 46
° 0'
34.1
3"
05/2
8 1.
5 0.
5 S
park
ling
Olig
otro
phic
89
°42'
2.24
" 10
.9
7.4
08/1
2 0.
2 0.
5 46
° 2'
27.
59"
05/2
8 11
.8
0.5
Tro
ut B
og
Hum
ic
89°
41' 9
.6"
5.6
4.8
08/1
2 72
.9
0.5
a Dat
a ob
tain
ed f
rom
The
Nor
th T
empe
rate
Lak
es L
ong
Ter
m E
colo
gica
l Res
earc
h (h
ttp://
lter.
lim
nolo
gy.w
isc.
edu/
inde
x.ht
ml)
and
D
epar
tmen
t of
Env
iron
men
tal P
rote
ctio
n of
Mai
ne (
http
://w
ww
.mai
ne.g
ov/)
. Mor
e de
taile
d in
form
atio
n ab
out c
hara
cter
istic
s su
ch
as d
isso
lved
org
anic
car
bon
(DO
C)
cont
ent
can
be f
ound
in
thes
e w
eb p
ages
. C
hl a
dat
a fr
om t
he L
ake
Dam
aris
cotta
was
m
easu
red
fluo
rom
etri
call
y at
Big
elow
lab
orat
ory.
Tab
le S
2. S
umm
ary
of t
he
454
met
agen
omic
sh
otgu
n s
equ
enci
ng
Met
agen
ome
No.
of
454
read
s (N
o. o
f nu
cleo
tides
) B
efor
e fi
lter
inga
A
fter
fil
teri
nga
% a
rtif
acts
af
ter
454
filt
erin
g
Ave
rage
se
quen
ce
leng
th
Men
dota
Spr
ing
4843
50 (
1732
9906
5)
3625
60 (
1313
4187
0)
25.2
36
2
Men
dota
Sum
mer
55
4444
(21
0765
835)
49
8550
(19
1375
633)
10
.1
384
Dam
aris
cotta
Spr
ing
33
7219
(13
1058
407)
30
5746
(11
9917
588)
9.
3 39
2
Dam
aris
cotta
Sum
mer
39
4404
(15
2578
395)
35
6323
(13
9131
704)
9.
6 39
0
Spar
klin
g Sp
ring
11
9298
(39
6030
48)
5711
6 (1
9449
905)
52
.1
340
Spar
klin
g Su
mm
er
5262
5 (1
7827
280)
47
202
(161
4869
1)
10.3
34
2
Tro
ut S
prin
g 24
5084
(80
0709
97)
1852
43 (
6135
2381
) 24
.4
331
Tro
ut S
umm
er
1357
49 (
4941
2536
) 12
1361
(44
6798
80)
10.5
36
8 a 45
4 fi
lter
use
d fo
r re
mov
ing
arti
fici
al r
epli
cate
s ge
nera
ted
duri
ng t
he 4
54 p
yros
eque
ncin
g (G
omez
-Alv
arez
et
al.,
2009
)
Su
pp
lem
enta
ry T
able
s

Tab
le S
3. P
rim
ers
and
PC
R c
ycli
ng
con
dit
ion
s ge
ne
Pri
mer
nam
e P
rim
er s
eque
nce
(5' t
o 3'
) a
Ref
eren
ce
PCR
cyc
ling
cond
itio
ns
pufM
_uni
Ffr
esh
(for
war
d)
GG
NA
AY
YT
GT
TY
TA
YA
AC
C
mod
ifie
d fr
om (
Yut
in e
t al.,
20
05)
pufM
_uni
Rfr
esh
(rev
erse
) C
CC
AT
SGT
CC
AN
CK
CC
AR
AA
m
odif
ied
from
(Y
utin
et a
l.,
2005
) pu
fM
pufM
_WA
Wfr
esh
(rev
erse
) A
YN
GC
RA
AC
CA
CC
AN
GC
CC
A
as in
(Y
utin
et a
l., 2
005)
a. D
enat
urin
g: 9
5°C
for
5 m
in
b. 4
0 cy
cles
: 94°
C f
or 2
0 s,
50
°C f
or 1
min
, and
72°
C
for
1 m
in
c. F
inal
ext
ensi
on: 7
2°C
for
10
min
d.
Mel
ting
cur
veb
Bch
YF
(fo
rwar
d)
CC
NC
AR
AG
NA
TG
TG
YC
CN
GC
m
odif
ied
from
(Y
utin
et a
l.,
2009
) B
chlY
B
chY
R (
reve
rse)
G
GR
TC
NR
CB
GG
RA
AV
AT
YT
C
mod
ifie
d fr
om (
Yut
in e
t al.,
20
09)
sam
e as
puf
M g
ene
PR
pf1
(for
war
d)
TA
YC
GY
TA
YG
TN
GA
YT
GG
m
odif
ied
from
(Sh
arm
a et
al.,
20
09)
PR
pf2
(for
war
d)
TA
YM
GW
TA
YA
TT
GA
YT
GG
ne
w p
rim
er
PR
pr1
(rev
erse
) A
TY
GG
RT
AN
AC
RC
CC
CA
m
odif
ied
from
(Sh
arm
a et
al.,
20
09)
Rho
dops
in
PR
pr2
(rev
erse
) G
GR
TA
AA
TN
GC
CC
AW
CC
ne
w p
rim
er
a. D
enat
urin
g: 9
5°C
for
5 m
in
b. 4
0 cy
cles
: 94°
C f
or 2
0 s,
46
°C f
or 1
min
, and
72°
C
for
45 s
. c.
Fin
al e
xten
sion
: 72°
C f
or 1
0 m
in
d. M
elti
ng c
urve
b
cbbL
F (
forw
ard)
G
AC
TT
CA
CC
AA
AG
AC
GA
CG
A
as
in (
Els
aied
and
Nag
anum
a,
2001
)
RuB
isC
O
(cbb
L)
cbbL
R (
reve
rse)
T
CG
AA
CT
TG
AT
TT
CT
TT
CC
A
as in
(E
lsai
ed a
nd N
agan
uma,
20
01)
a. D
enat
urin
g: 9
5°C
for
5 m
in
b. 2
cyc
les:
94°
C f
or 2
0 s,
37
°C f
or 3
0 s,
and
72°
C f
or
3 m
in.
c. 3
8 cy
cles
: 94°
C f
or 2
0 s,
53
°C f
or 3
0 s,
and
72°
C
for
1 m
in.
d. F
inal
ext
ensi
on: 7
2°C
for
7 m
in
e. M
elti
ng c
urve
b
P
rok_
340F
(fo
rwar
d)
C
CT
AY
GG
GR
BG
CA
SC
AG
m
odif
ied
from
(T
akai
and
H
orik
oshi
, 200
0)
Pro
k_80
6R (
reve
rse)
G
GA
CT
AY
NN
GG
GT
AT
CT
AA
T
mod
ifie
d fr
om (
Tak
ai a
nd
Hor
ikos
hi, 2
000)
a. D
enat
urin
g: 9
5°C
for
5 m
in
b. 4
0 cy
cles
: 94°
C f
or 2
0 s,
54
°C f
or 2
0 s,
and
72°
C
for
30 s
. c.
Fin
al e
xten
sion
: 72°
C f
or 1
0 m
in.
d. M
elti
ng c
urve
b
27F
’ (f
orw
ard)
A
GR
GT
TY
GA
TY
MT
GG
CT
CA
G
as in
(L
ane,
199
1)
as in
ref
eren
ce
907R
(re
vers
e)
CC
G T
CA
AT
T C
MT
TT
R A
GT
TT
as
in (
Cas
amay
or e
t al.,
200
0)
as in
ref
eren
ce
Arc
h_34
5F (
forw
ard)
C
CT
AY
G G
GG
YG
C A
SC
AG
as
in (
Gan
tner
et a
l. 20
10)
as
in r
efer
ence
PA
rch_
519F
(fo
rwar
d)
CA
G C
MG
CC
G C
GG
TA
A
as in
(T
eske
and
Sor
ense
n,
2007
) as
in r
efer
ence
Arc
h_10
00R
(re
vers
e)
GG
C C
AT
GC
A C
YW
CY
T C
TC
as
in (
Gan
tner
et a
l. 20
10)
as in
ref
eren
ce
16S
rR
NA
Arc
h_91
5R (
reve
rse)
G
TG
CT
C C
CC
CG
C C
AA
TT
C C
T
as in
(C
asam
ayor
et a
l., 2
000)
as
in r
efer
ence
a A
ll f
orw
ard
and
reve
rse
prim
ers
had
M13
for
war
d (G
TA
AA
AC
GA
CG
GC
CA
GT
) an
d re
vers
e (C
AG
GA
AA
CA
GC
TA
TG
AC
C)
prim
ers
resp
ecti
vely
in 5
', w
hich
was
use
d as
targ
et to
seq
uenc
e th
e re
sult
ing
ampl
icon
s. P
rim
er s
eque
nce
mod
ific
atio
n is
indi
cate
d in
bol
d an
d un
derl
ined
. b M
elti
ng c
urve
for
all
rea
ctio
ns w
as p
erfo
rmed
as
foll
owin
g: 9
5°C
for
5 s
, 52°
C f
or 1
min
, and
a c
onti
nuou
s te
mpe
ratu
re r
amp
(0.1
1°C
/s)
from
52
to 9
7°C
.
13

Tab
le S
4A. C
over
age
of t
he
rhod
opsi
n g
ene
div
ersi
ty, a
s d
eter
min
ed b
y m
etag
enom
ic s
equ
enci
ng,
by
the
PC
R p
rim
ers
use
d in
th
is s
tud
y
No.
of
rhod
opsi
n ge
nes
foun
d in
the
454
libra
ry
No.
of
454
read
s co
ntai
ning
the
forw
ard
targ
et
Per
cent
age
of p
rim
er
cove
rage
for
the
forw
ard
targ
eta
No.
of
454
read
s co
ntai
ning
the
reve
rse
targ
et
Per
cent
age
of p
rim
er
cove
rage
for
the
reve
rse
targ
etb
Men
dota
(sp
ring
) 61
30
71
%
31
78%
D
amar
isco
tta
(spr
ing)
41
22
72
%
10
100%
D
amar
isco
tta
(sum
mer
) 65
34
86
%
31
87%
S
park
ling
(spr
ing)
6
5 50
%
1 10
0%
Tro
ut (
spri
ng)
6 2
100%
3
100%
a F
orw
ard
prim
ers
PR
pf1
and
PR
pf2
used
in th
e pr
esen
t stu
dy
b Rev
erse
pri
mer
s P
Rpr
1 an
d P
Rpr
2 us
ed in
the
pres
ent s
tudy
T
able
S4B
. C
over
age
of t
he
pufM
and
Bch
lY g
ene
div
ersi
ty,
as d
eter
min
ed b
y m
etag
enom
ic
seq
uen
cin
g, b
y th
e P
CR
pri
mer
s u
sed
in
th
is s
tud
y. P
rim
er c
over
age
was
100
% f
or b
oth
gen
es,
forw
ard
an
d re
vers
e
No.
of
pufM
gen
es f
ound
in th
e 45
4 lib
rary
a N
o. o
f B
chlY
gen
es f
ound
in th
e 45
4 li
brar
yb M
endo
ta (
spri
ng)
4 5
Dam
aris
cotta
(sp
ring
) 11
28
D
amar
isco
tta (
sum
mer
) 16
20
S
park
ling
(spr
ing)
4
2 T
rout
(sp
ring
) 5
5 a P
rim
ers
pufM
F, p
ufM
_uni
R, a
nd p
ufM
_WA
W w
ere
used
for
the
pufM
gen
e am
plif
icat
ion
from
SA
Gs
b P
rim
ers
Bch
lYF
and
Bch
lYR
wer
e us
ed f
or th
e B
chlY
gen
e am
plif
icat
ion
from
SA
Gs
14

15
References Casamayor EO, Schafer H, Baneras L, Pedros-Alio C, Muyzer G (2000). Identification of
and spatio-temporal differences between microbial assemblages from two neighboring sulfurous lakes: comparison by microscopy and denaturing gradient gel electrophoresis. Appl Environ Microbiol 66: 499-508.
Elsaied H, Naganuma T (2001). Phylogenetic diversity of ribulose-1,5-bisphosphate carboxylase/oxygenase large-subunit genes from deep-sea microorganisms. Appl Environ Microbiol 67: 1751-1765.
Gantner S, Andersson AF, Alonso-Sáez L, Bertilsson S Novel primers for 16S rRNA-based archaeal community analyses in environmental samples (2010) J Microbiol Methods 84: 12-18.
Gomez-Alvarez V, Teal TK, Schmidt TM (2009). Systematic artifacts in metagenomes from complex microbial communities. ISME J 3: 1314-1317.
Lane DJ (1991). 16S/23S rRNA sequencing. In: Stackebrandt EaG, M. (ed). Nucleic Acid Techniques in Bacterial Systematics John Wiley and Sons: New York. pp 115–175.
Sharma AK, Sommerfeld K, Bullerjahn GS, Matteson AR, Wilhelm SW, Jezbera J et al. (2009). Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. ISME J 3: 726-737.
Takai K, Horikoshi K (2000). Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl Environ Microbiol 66: 5066-5072.
Teske A, Sorensen KB (2007). Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J 2: 3-18.
Yutin N, Suzuki MT, Beja O (2005). Novel primers reveal wider diversity among marine aerobic anoxygenic phototrophs. Appl Environ Microbiol 71: 8958-8962.
Yutin N, Suzuki MT, Rosenberg M, Rotem D, Madigan MT, Suling J et al. (2009). BchY-based degenerate primers target all types of anoxygenic photosynthetic bacteria in a single PCR. Appl Environ Microbiol 75: 7556-7559.
Copyright © 2022 FDOKUMEN




















