
Heterogeneous photocatalytic transformations of s-triazine derivatives
20
Res. Chem. Intermed., Vol. 23, No. 4, pp. 291 310(1997) VSP 1997 HETEROGENEOUS PHOTOCATALYTIC TRANSFORMATIONS OF s-TRIAZINE DERIVATIVES C. MINERO, V. MAURINO, and E. PELIZZETTI* Dipartimento di Chimica Analitica, Universith di Torino, 10125 Torino, Italy. Received 3 September t 996; accepted 27 December 1996 Abstract-Photocatalytic processesarising from irradiated semiconductor oxide suspensionscontaining 1,3,5-triazine (s-triazine) derivatives are described. Whereas unsubstituted 1,3,5-triazine undergoes hydrolysis, irrespective of the presence of the photocatalyst, other chloro-, amino-, mercapto-, allyloxo-, carboxy-derivativesgive rise, in the presence of band-gap excited semiconductoroxide, to nearly stoichiometric formation of 2,4,6-trihydroxy-1,3,5-triazine (cyanuricacid). This last compound is stable toward photocatalytic conditions as well as to "OH chemically generated in homogeneous solution (H202/UV or Fenton's reagent). Only partial conversionin cyanuric acid is observedfor 2,4,6- tris(2-pyridyl)-l,3,5-triazine and possible explanation is given. The formation of inorganic species (nitrate, chloride, sulfate and ammonium ions) is reported and the mechanism of their evolution presented. INTRODUCTION Various derivatives of 1,3,5-triazine [1] are of considerable commercial importance, e.g. 1,3,5-triazine herbicides (atrazine, simazine etc.) [2], melamine derivatives [3] and isocyanuril compounds [1]. Transformations of these compounds are of significant interest either for their persistence and stability and for gaining information on kinetic parameters and intermediates identification. Heterogeneous photocatalysis over irradiated semiconductor oxide has been proven to efficiently degrade organic compounds through a series of oxidative/reductive pathways [4-5]. This behavior is of interest to gain insight in the mechanisms of transformation and for efficient detoxification of waters [6-8]. Several intermediates have been identified and cyanuric acid has been recognized as the final product in the photocatalytic degradation of herbicides [9,10] and melamine [11]. This result represents a peculiar property since in previously reported studies on other organic compounds, complete mineralization to CO2 has been demonstrated [7]. This prompted us to extend the investigation on photocatalytic behavior of a series of 1,3,5-triazine derivatives, including the unsubstituted 1,3,5-triazine (Chart 1). Some new and characteristic features in the degradation patterns have been observed.
Transcript of Heterogeneous photocatalytic transformations of s-triazine derivatives

Res. Chem. Intermed., Vol. 23, No. 4, pp. 291 310(1997) �9 VSP 1997
H E T E R O G E N E O U S P H O T O C A T A L Y T I C TRANSFORMATIONS OF s-TRIAZINE DERIVATIVES
C. MINERO, V. MAURINO, and E. PELIZZETTI* Dipartimento di Chimica Analitica, Universith di Torino, 10125 Torino, Italy.
Received 3 September t 996; accepted 27 December 1996
Abstract-Photocatalytic processes arising from irradiated semiconductor oxide suspensions containing 1,3,5-triazine (s-triazine) derivatives are described. Whereas unsubstituted 1,3,5-triazine undergoes hydrolysis, irrespective of the presence of the photocatalyst, other chloro-, amino-, mercapto-, allyloxo-, carboxy- derivatives give rise, in the presence of band-gap excited semiconductor oxide, to nearly stoichiometric formation of 2,4,6-trihydroxy-1,3,5-triazine (cyanuric acid). This last compound is stable toward photocatalytic conditions as well as to "OH chemically generated in homogeneous solution (H202/UV or Fenton's reagent). Only partial conversion in cyanuric acid is observed for 2,4,6- tris(2-pyridyl)-l,3,5-triazine and possible explanation is given. The formation of inorganic species (nitrate, chloride, sulfate and ammonium ions) is reported and the mechanism of their evolution presented.
INTRODUCTION
Various derivatives of 1,3,5-triazine [1] are of considerable commercial importance,
e.g. 1,3,5-triazine herbicides (atrazine, simazine etc.) [2], melamine derivatives [3]
and isocyanuril compounds [1]. Transformations of these compounds are of
significant interest either for their persistence and stability and for gaining
information on kinetic parameters and intermediates identification.
Heterogeneous photocatalysis over irradiated semiconductor oxide has been
proven to efficiently degrade organic compounds through a series of
oxidative/reductive pathways [4-5]. This behavior is of interest to gain insight in the
mechanisms of transformation and for efficient detoxification of waters [6-8].
Several intermediates have been identified and cyanuric acid has been recognized
as the final product in the photocatalytic degradation of herbicides [9,10] and
melamine [11]. This result represents a peculiar property since in previously
reported studies on other organic compounds, complete mineralization to CO2 has
been demonstrated [7]. This prompted us to extend the investigation on
photocatalytic behavior of a series of 1,3,5-triazine derivatives, including the
unsubstituted 1,3,5-triazine (Chart 1). Some new and characteristic features in the
degradation patterns have been observed.
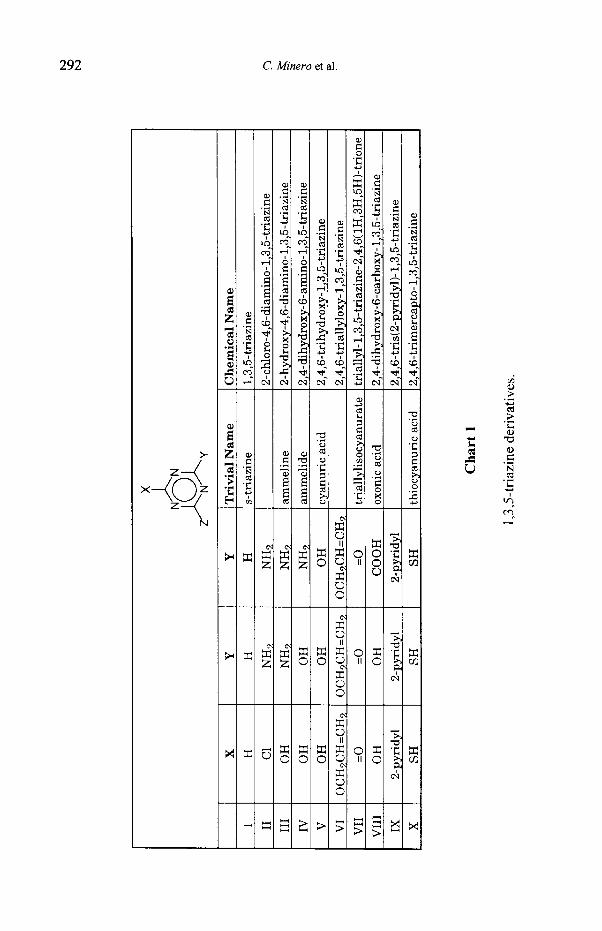
ix.)
bo
I II
III
IV
V
VI
VII
V
III
IX
X
X
X
Y
Y
Tri
vial
Nam
e C
hem
ical
N
ame
H
H
H
s-tr
iazi
ne
1,3,
5-tr
iazi
ne
Cl
OH
NH2
NH
~.
OH
O
H
NH
~.
NH
2
NH
2 O
H
OH
O
H
OC
H2C
H=C
HT
, O
CH
2CH
=C
H 2
OC
H2C
H=C
H~.
=O
=0
=0
OH
O
H
CO
OH
2-
pyri
dyl
SH
2-py
ridy
l
SH
2-py
ridy
l
SH
amm
elin
e
amm
elid
e cy
anur
ic a
cid
tria
llyl
isoc
yanu
rate
oxon
ic a
cid
thio
cyan
uric
aci
d
2-ch
loro
-4,6
-dia
min
o- 1
,3,5
-tri
azin
e
2-hy
drox
y-4~
6-di
amin
o- l~
3,5-
tria
zine
2,4-
dihy
drox
y-6-
amin
o- 1,
3,5-
tria
zine
2,
4,6-
trih
ydro
xy- l
~3~5
-tri
azin
e
2~4,
6-tr
iall
ylox
y- 1 f
l,5-
tria
zine
tr
iall
yl- l
t3t5
-tri
azin
e-2~
4~6(
1Hfl
H~5
H)-
trio
ne
2,4-
dihy
drox
y-6-
carb
oxy-
l~3f
i-tr
iazi
ne
2,4,
6-tr
is(2
-pyr
idyl
)- lf
lfi-
tria
zine
2,4,
6-tr
imer
capt
o- 1
~3,5
-tri
azin
e
r
Ch
art
1
1,3,
5-tr
iazi
ne d
eriv
ativ
es.

Photocatalytic Transformations of s-Triazine Derivatives 293
EXPERIMENTAL SECTION
Materials and Reagents 1,3,5-triazine (Aldrich 98%), cyanuric acid (Aldrich 98%), 2-chloro-4,6-diamino- 1,3,5-triazine (Aldrich 95%), 2,4,6-triallyloxy-1,3,5-triazine (Aldrich 97%), triallyl- 1,3,5-triazine-2,4,6(1H,3H,5H)-trione (Aldrich 98%), oxonic acid, potassium salt (Aldrich 97%), trithiocyanuric acid (Aldrich 95%), 2,4,6-tris(2-pyridyl)-l,3,5- triazine (Aldrich, 98%), didodecyldimethylammonium bromide (DDDMAB) (Eastman Kodak Co., Rochester, N.Y.), acetonitrile, methanol and ethanol (LiChrosolv, Merck) were used as received. Ammeline and ammelide were synthesized following the procedure described elsewhere [9]. Other reagents used were at least analytical grade. Titania TiO2 P25 (BET area ca 55 mJg-', Degussa A.G., Frankfurt, Germany) and Zirconia Z r O 2 (Aldrich, 99%) were irradiated in aerated aqueous suspension for at least 12 hours and washed with deionized-doubly distilled water in order to avoid interference from organic impurities and inorganic ions adsorbed on the photocatalyst.
Degradation Experiments The slurries were prepared by suspending with sonication the required amount of photocatalyst powder and addition of the needed amount of the aqueous stock solution of substrate. The pH of the slurries before irradiation was adjusted by adding drops of 1M solutions of NaOH or HCIO4 as required, pH 5.5 is the natural pH of a TiOz suspension (500 mg L~).
The irradiation experiments were carried out in Pyrex glass (with cut-off at 295 nm) or quartz cylindrical cells (4.0 cm diameter, 2.3 cm height), containing 5 mL of the aqueous suspension of the photocatalyst powder and substrate, using a 1500 watt Xenon lamp (Solarbox, CO.FO.MEGRA, Milan, Italy) equipped with a 340 nm cut-off filter and simulating AM1 solar light. Homogeneous degradation with the HzOz/UV system were carried out in the quartz cell, using the Xenon lamp without any cut-off filter. The irradianee spectrum and the cells were described elsewhere [12,13]. The total photonic fluxes in the cells were: 3.45 �9 10 7 and 1.92 �9 10 7 Einstein s" in the range 280-400 nm and 340-400 nm, respectively, as determined by ferrioxalate actinometry. The cell temperature during irradiation was 60 ~ During irradiation the slurries were magnetically stirred.
Blank runs (cut-off at 340 nm) on all compounds in the absence of photocatalyst showed no detectable degradation of the substrate after 100 hours of irradiation.
Fenton's reagent runs were carried out on the substrates solution containing H202 1.0 �9 10 .5 M and FeSO4 1.0 �9 10 .3 M at pH 3 (H2SO4). After the desired time

294 c Minero et al.
the reaction was quenched by adding methanol.
Analytical Determinations
The HPLC determinations were carried out with a Hitachi mod. L6200 pump, equipped with a Rheodine injector (20 p.L loop) and UV detector (Hitachi mod. L4200), on the filtered (Millex HV 0.45 gm, Millipore) irradiated aqueous samples. Ammeline was quantified by ion pair chromatography with a bonded phase octadecylsilica column (LiChrocart 100 RP18 Merck, 125 mm length, 4 mm i.d., 5 gm packing); the mobile phase was 0.01 M sodium hexanesulfonate (Aldrich 98%), 0.0t M H3PO4 in water/acetonitrile 95/5 at 1 mL rain l . The detection was carried
out at 220 nm. Ammelide and cyanuric acid were detected by ion interaction chromatography with the immobilized reagent technique (IIIR) [14]. An octadecylsilica column (LiChrospher 100 RP- 18 endcapped Merck, 5 gm packing, 250 mm length, 4 mmid.) was loaded with DDDMAB 0.01 M in acetonitrile/water 25/75 with the procedure described elsewhere [9]. The sample elutions were carried out with a 5 ~ 10 .3 M phosphate buffer solution (pH 7.7) at 1 mL min -I, with UV detection at 220 nm. In these conditions ammeline, ammelide and cyanuric acid showed retention times of 2.6, 6.2, 7.8 min respectively. Oxonic acid was quantified by ion-pair chromatography employing a bonded phase octadecylsilica column (LiChrocart 100 RP18 Merck, 125 mm length, 4 mm i.d., 5 gm packing); the mobile
phase was 0.01 M tetrabutylammonium chloride, 0.01 M Na2HPO4, 0.005 M NaH2PO4 in water/methanol 90/10 at 1 mL min 1. The detection was carried out at 250 nm. 2,4,6-triallyloxy-l,3,5-triazine andtriallyl-l,3,5-triazine-2,4,6(1H,3H,5H)- trione analysis were performed on a bonded phase octadecylsilica column (same as above) with water/acetonitrile 50/50 as mobile phase. The detection was carried out at 205 nm.
The quantitative determination of 2,4,6-tris(2-pyridyl)-l,3,5-triazine was performed on a spectrophotometer (Uvikon mod 200, Kontron, Milan, Italy), measuring the absorbance of the Fe 2+ complex at 596 nm. 4 mL of the filtered irradiated sample were mixed with 1 mL of 0.001 M FeSO4 solution and 1 mL of acetate buffer (2 M, pH 5); the resultant solution was thoroughly mixed and allowed to stand 5 minutes in the dark before absorbance measurement.
The formation of nitrate, chloride and sulfate was monitored by suppressed ion chromatography, utilizing a Biotronik IC 5000 apparatus equipped with a Dionex AS4-SC separation column and an alkaline buffer eluant containing Na2CO3 (1.8 raM) and NaHCO3 (1.7 mM) at the flow rate of 1.5 mL rain -I.
Nitrite and ammonium ion analysis were performed using the Griess- Horvath reagent (Merck) and the Berthellot reagent (Merck) respectively, following
the procedures suggested by the vendor.

Photocatalytic Transformations of s-Triazine Derivatives 295
Total organic carbon (TOC) determinations were carried out by using a Shimadzu model TOC-5000 total carbon analyzer on the filtered (0.45 m) irradiated samples.
RESULT AND DISCUSSION
Primary Events and Reactive Species in the Photocatalysis on 7702
Band gap irradiation of titanium dioxide generates electron/hole pairs
- - +
7 ~ 0 2 + hv "* ece + hv~ (1)
After charge separation (Eq. (1)), other reactions can compete with electron/hole recombination. The charge carriers reaching the surface can be trapped by intrinsic traps (sub-surface energy traps for the hole and surface traps for the electrons) and by extrinsic surface traps via interfacial electron transfer with adsorbed electron donors (Redz)a~s and acceptors (OX~)ads, respectively, as depicted in Eqs. (2) and (3)
+
hvB + (Red2)aa., --* (Oxz)aa.,. (2)
eCB + (OXl)ad.,. -'* (Redl)aa " (3)
For hydrated and hydroxylated TiO 2 anatase surfaces [15], as in the present case, hole trapping by interfacial electron transfer yields surface-bound "OH radicals
+
hvB + H20 d " -* "OHm,. + H + (4)
+
hvB + OHS~,, --* "OHm,,. (5)

296 C Minero et al.
In aerated solutions trapped electrons may be scavenged oxygen to give the superoxide anion
by adsorbed
e~ B + 02~d,. "-* O~ ~d.,. (6)
which can be further reduced to peroxide dianion. Surface peroxo-species can be formed [15]. Other reactions and species, depending on solution pH, can occur on the titanium dioxide particle and involve the solvent. They have been discussed in detail'elsewhere [6,7,16]. Heterogeneous photocatalytic processes involve reactions at the surface/solution interfaces where (i) the oxidizing species can be holes (more oxidizing than aqueous hydroxyl radical) or trapped holes (i.e. ~ less oxidizing than aqueous hydroxyl radical); (ii) the reducing species can be conduction band electrons or trapped electrons, both being much less reducing than aqueous hydrated electrons.
Both direct observation with magnetic resonance techniques and indirect kinetic evidence point to the presence of a surface bound hydroxyl radical on irradiated aqueous TiOz suspensions. Many times, the same intermediates formed from ~ radicals generating systems, like pulse radiolysis or Fenton's reagent, are also involved in heterogeneous photocatalytic processes [7]. Nonetheless, time resolved diffused reflectance spectroscopic measurements indicate the transient formation of singly oxidized (or reduced) intermediates derived from a range of organic and inorganic reagents on irradiated aqueous TiO2 suspensions [17]. Substrates not reactive toward hydroxyl radicals are degraded employing TiO2 photocatalysis, with rates of decay highly influenced by the semiconductor valence band edge position [ 11 ]. Experiments with 2,4-dichlorophenoxyacetic acid also led to conclude that in the photocatalytic degradation of this substrate a dual radical/hole mechanism is operating [18]. These evidences suggest that the exact mechanism is influenced by several factors, and the organic substrates (initial and intermediates), depending on their nature and on the experimental conditions, can react through direct interaction with the carriers (Eq. (2) and (3)) and/or with the trapped holes (hereafter called ~ radicals). Besides the solvent, other species are present at the interface or in solution (e.g. HO2*/O2* , 02, H202) which can participate to the complex degradation scheme leading to the transformations of the organics presented here.
The loss of the substrates considered in this work obeyed (pseudo) first- order kinetics over at least two half-lives. This behavior is explained in the framework of a detailed kinetic analysis of photocatalytic processes [19]. Table 1 shows the rate constants (kobs) measured under the present photocatalytic conditions.

Photocatalytic Transformations of s-Triazine Derivatives
Table 1
Observed pseudo-first-order rate constants for the photocatalytic degradation of the selected s-triazine derivatives.
297
Compound Initial Kinetic
Concentration Constant (h "~) (10 "4 M)
2,4-amino-6-chloro-s-triazine 0.35 0.15 + 0.02
Ammeline 0.80 0.40 • 0.05
Ammelide 0.40 0.11 • 0.02
Triallylcyanurate 2.00 10.5 �9 0.5
Triallylisocyanurate 2.00 13.5 • 0.7
Oxonic acid 1.25 5.0 + 0.5
2,4,6-tris(2-pyridil)-s-triazine 0.80 7.2 • 0.5
It is worth noting that kob s for substrates not oxidized by Fenton's reagent (ammeline, ammelide and 2,4-diamino-6-chloro-s-triazine), thus presumably degraded with TiOz/light through direct hole oxidation, are 10-100 times lower than substrates degraded by homogeneous "OH generating systems
1, 3, 5- Triazine 1,3,5-Triazine is highly soluble in water and is reported to be very sensitive to
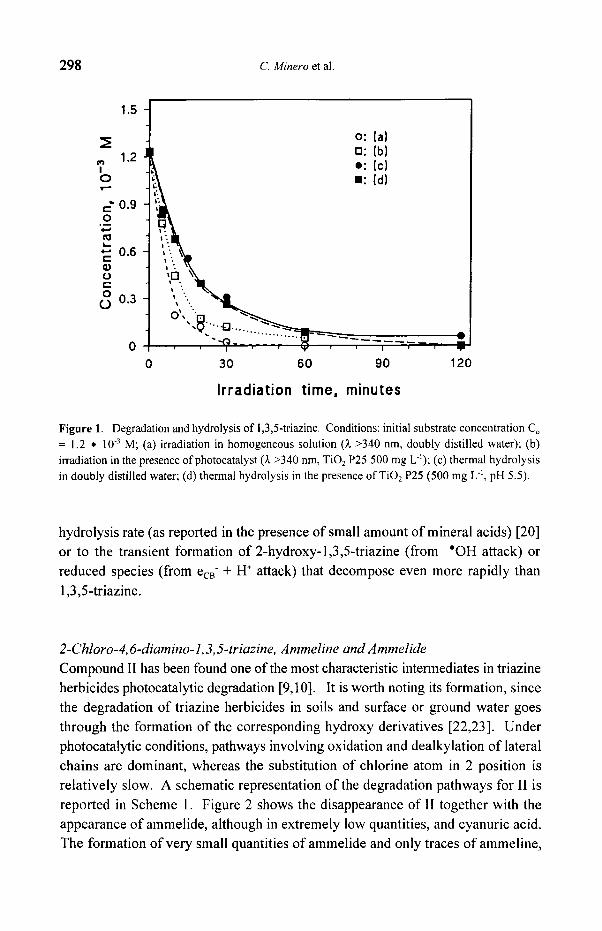
solvolysis in water and other solvents containing hydroxyl groups [1]. In an aqueous solution containing 1.2 �9 103 M kept in the dark, less than 6 ~ 10 -5 M of
1,3,5-triazine can be detected after 2 hours at 60 ~ being ammonium formate the main decomposition product (ca. 50% of the stoichiometric amount for a complete hydrolysis) [20]. As shown in Figure 1, irradiation in the presence of TiO2 influences the rate of 1,3,5-triazine disappearance, which is reduced to less than 6 * 10 .5 M in ca. 30 minutes. Again ca. 50% of the stoichiometric amount of ammonium formate
has been recovered, and no cyanuric acid has been detected. The absence of any product originating from "OH addition to the 1,3,5-triazine ring structure, further confirmed by the absence of cyanuric acid also when 1,3,5-triazine is treated with
the Fenton's reagent, arises probably from the instability of 2-hydroxy-l,3,5- triazine. In fact this last compound is not reported in the literature [1] and attempts to prepare it have been unsuccessful [21 ]. Therefore the rate increase observed in Figure 1 in the presence of TiO2 and light can be due either to an increase in
298 c. Minero et al.
1.5
z ;
fn 1.2 I o
c = 0.9 0
�9 - ' 0.6 c
o r
0 0 0.3
0
o: (a) o: (b) o: (c) m: ( d )
. . . .
0 30 60 90 120
I r r a d i a t i o n t i m e , minutes
Figure 1. Degradation and hydrolysis of i,3,5-triazine. Conditions: initial substrate concentration Co = 1.2 �9 10 .3 M; (a) irradiation in homogeneous solution (t >340 nm, doubly distilled water); (b) irradiation in the presence of photocatalyst (t >340 nm, TiO 2 P25 500 mg L-~); (c) thermal hydrolysis in doubly distilled water; (d) thermal hydrolysis in the presence of TiO2 P25 (500 mg L ~, pH 5.5).
hydrolysis rate (as reported in the presence of small amount of mineral acids) [20]
or to the transient formation of 2-hydroxy-l,3,5-triazine (from "OH attack) or
reduced species (from ecB + H § attack) that decompose even more rapidly than
1,3 ,5- tr iaz ine .
2-Chloro-4, 6-diamino-1, 3, 5-triazine, Ammeline and Ammelide
Compound II has been found one of the most characteristic intermediates in triazine
herbicides photocatalytic degradation [9,10]. It is worth noting its formation, since
the degradation of triazine herbicides in soils and surface or ground water goes
through the formation of the corresponding hydroxy derivatives [22,23]. Under
photocatalytic conditions, pathways involving oxidation and dealkylation of lateral
chains are dominant, whereas the substitution of chlorine atom in 2 position is
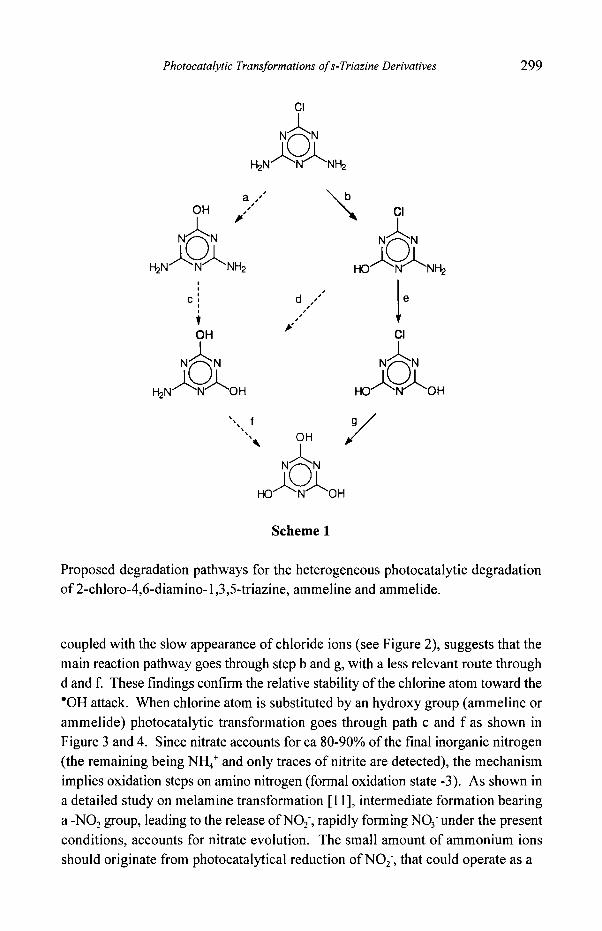
relatively slow. A schematic representation of the degradation pathways for II is
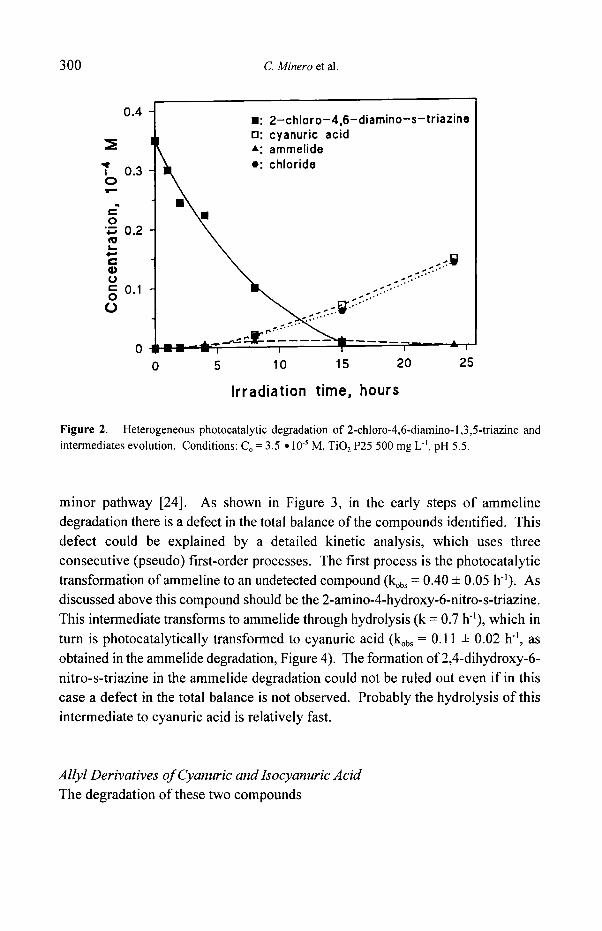
reported in Scheme 1. Figure 2 shows the disappearance of II together with the
appearance of ammelide, although in extremely low quantities, and cyanuric acid.
The formation of very small quantities of ammelide and only traces of ammeline,
Photocatalytic Transformations of s-Triazine Derivatives 299
CI N..-/..N
a pl
OH "" N N
H2N NH2
c
V OH
1-12NL~NLO H
d / ,/
• CI
HOL~NLNH2
Cl
H O L @ ~ O H
",, f y "', OH
N)' N
Scheme 1
Proposed degradation pathways for the heterogeneous photocatalytic degradation of 2-chloro-4,6-diamino- 1,3,5-triazine, ammeline and ammelide.
coupled with the slow appearance of chloride ions (see Figure 2), suggests that the main reaction pathway goes through step b and g, with a less relevant route through d and f. These findings confirm the relative stability of the chlorine atom toward the "OH attack. When chlorine atom is substituted by an hydroxy group (ammeline or ammelide) photocatalytic transformation goes through path c and f as shown in Figure 3 and 4. Since nitrate accounts for ca 80-90% of the final inorganic nitrogen (the remaining being NH4 + and only traces of nitrite are detected), the mechanism implies oxidation steps on amino nitrogen (formal oxidation state -3). As shown in a detailed study on melamine transformation [11], intermediate formation bearing a -NO2 group, leading to the release of NO2, rapidly forming NO3 under the present conditions, accounts for nitrate evolution. The small amount of ammonium ions should originate from photocatalytical reduction of NOz, that could operate as a
300 C. Minero et al.
'qr I 0
0.4
0.3
=" 0
' ~ o.2
e,,
~- 0.1 0
r
n: 2-chloro-4 ,6-diamino-s- t r iaz ine . n: cyanuric acid
::
, .,. 555. ' ." '" '
0 5 10 15 20 25
I r r a d i a t i o n t i m e , hours
F i g u r e 2. Heterogeneous photocatalytic degradation o f 2-chloro-4,6-diamino-l ,3 ,5- t r iazine and
intermediates evolution. Conditions: C o = 3.5 �9 10 -~ M, TiO 2 P25 500 mg L j , pH 5.5.
minor pathway [24]. As shown in Figure 3, in the early steps of ammeline degradation there is a defect in the total balance of the compounds identified. This defect could be explained by a detailed kinetic analysis, which uses three consecutive (pseudo) first-order processes. The first process is the photocatalytic
transformation ofammeline to an undetected compound (kob s = 0.40 4- 0.05 h'l). AS discussed above this compound should be the 2-amino-4-hydroxy-6-nitro-s-triazine. This intermediate transforms to ammelide through hydrolysis (k = 0.7 hl), which in
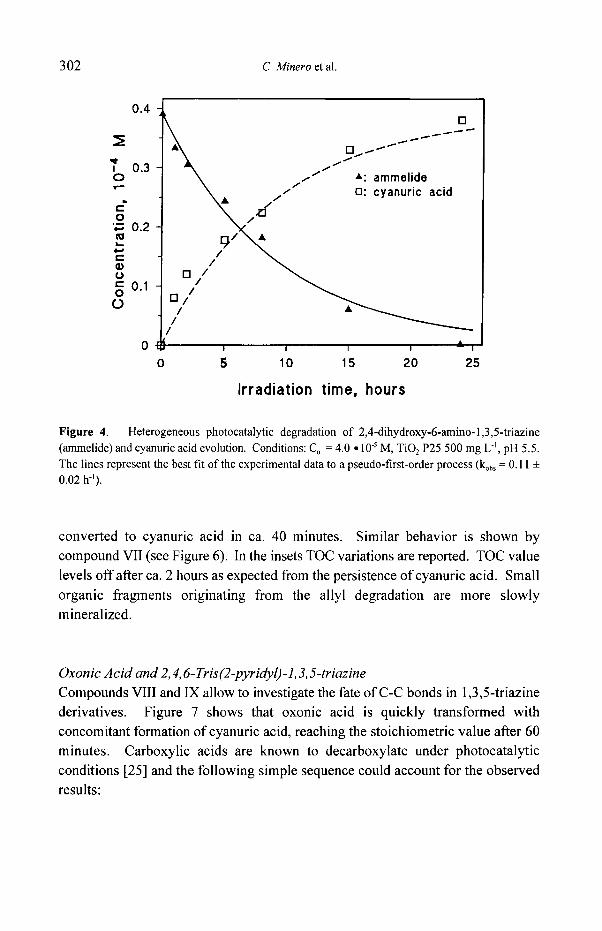
turn is photocatalytically transformed to cyanuric acid ( k o b s = 0.11 + 0.02 h l , as obtained in the ammelide degradation, Figure 4). The formation of 2,4-dihydroxy-6- nitro-s-triazine in the ammelide degradation could not be ruled out even if in this case a defect in the total balance is not observed. Probably the hydrolysis of this intermediate to cyanuric acid is relatively fast.
Allyl Derivatives of Cyanuric and Isocyanuric Acid The degradation of these two compounds
Photocatalytic Transformations of s-Triazine Derivatives 3 01
H2C=HCH2CO N OCH2CH=-CH2
0
H2C=HCH2CO -~NI~N/OCH2CH=CH2
I OCH2CH=CH 2
VI VII
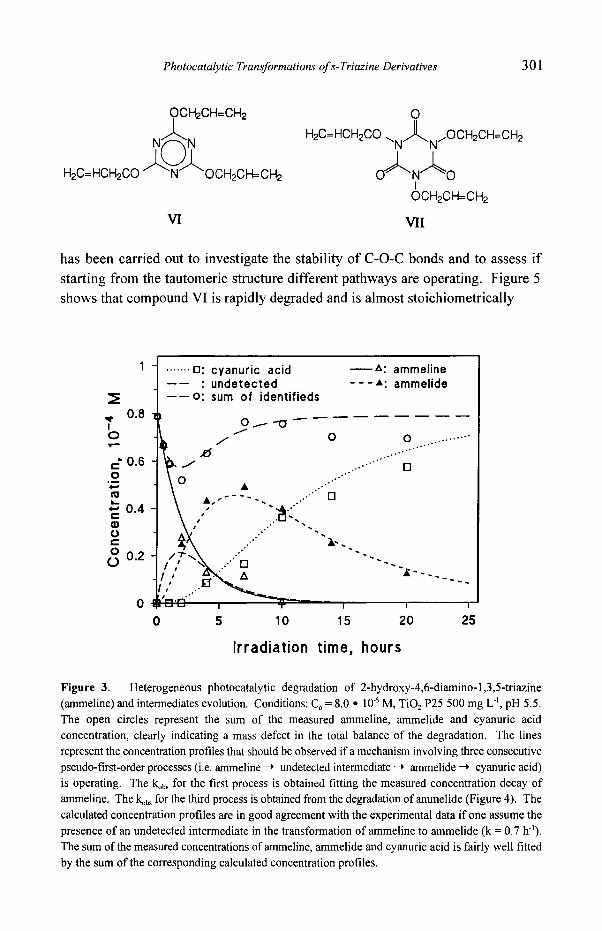
has been carried out to investigate the stability of C-O-C bonds and to assess if starting from the tautomeric structure different pathways are operating. Figure 5 shows that compound VI is rapidly degraded and is almost stoichiometrically
1 t ....... 13: cyanuric acid A: arnmeline -I - - - - : undetected - - - A : ammelide I - - - - ~ sum of identifieds
,~ 0.8 "1[ 0 . ~ - " ~ . . . . . . . . . . I
O t l b f f " O 0 .............. T ' - -
........... ........ ~" 0.6 j ......... []
.R
0.4
0
0 =~= L . i L . I I =1" I I I
0 5 10 15 20 25
Irradiation time, hours
Figure 3. Heterogeneous photocatalytic degradation of 2-hydroxy-4,6-diamino-l,3,5-triazine (ammeline) and intermediates evolution. Conditions: Co = 8.0 �9 10 .5 M, TiO 2 P25 500 mg L 4, pH 5.5. The open circles represent the sum of the measured ammeline, ammelide and cyanuric acid concentration, clearly indicating a mass defect in the total balance of the degradation. The lines
represent the concentration profiles that should be observed if a mechanism involving three consecutive pseudo-first-order processes (i.e. ammeline -~ undetected intermediate -* ammelide ~ cyanuric acid) is operating. The kob ~ for the first process is obtained fitting the measured concentration decay of ammeline. The kob, for the third process is obtained from the degradation ofammelide (Figure 4). The calculated concentration profiles are in good agreement with the experimental data if one assume the presence of an undetected intermediate in the transformation of ammeline to ammelide (k = 0.7 h4). The sum of the measured concentrations of ammeline, ammelide and cyanuric acid is fairly well fitted by the sum of the corresponding calculated concentration profiles.
302 C. Minero et al.
0.4
i 0.3 0 T-"
c" 0
�9 ~, 0.2 Oil
(J ~" 0.1 0
0
/ / / A: ammelide - ~ / z ] / / / rl: cyanuric acid
/ / [] ii i i ]/11 �9
I I I I - - I
0 5 10 15 20 25
Irradiation time, hours
Figure 4. Heterogeneous photocatalytic degradation of 2,4-dihydroxy-6-amino-l,3,5-triazine (ammelide) and cyanuric acid evolution. Conditions: C o = 4.0 �9 10 .5 M, TiO 2 P25 500 mg L -~, pH 5.5. The lines represent the best fit of the experimental data to a pseudo-first-order process (kob s = 0.11 + 0.02 h'l).
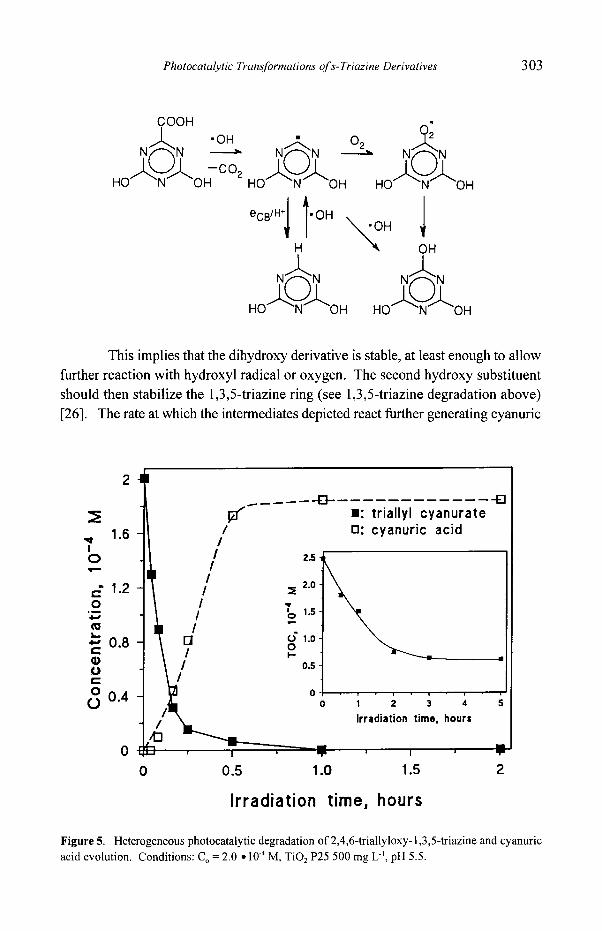
converted to cyanuric acid in ca. 40 minutes. Similar behavior is shown by
compound VII (see Figure 6). In the insets TOC variations are reported. TOC value
levels of faf ter ca. 2 hours as expected from the persistence ofcyanuric acid. Small
organic fragments originating from the allyl degradation are more slowly
mineralized.
Oxonic Ac id and 2, 4, 6- Tris (2-pyridyl)- l, 3, 5-triazine
Compounds VIII and IX allow to investigate the fate of C-C bonds in 1,3,5-triazine
derivatives. Figure 7 shows that oxonic acid is quickly transformed with
concomitant formation ofcyanur ic acid, reaching the stoichiometric value after 60
minutes. Carboxylic acids are known to decarboxylate under photocatalytic
conditions [25] and the following simple sequence could account for the observed
results:
Photocatalytic Transformations of s-Triazine Derivatives 303
COOH ~ .o~. ~.~ o~ L~.. ~ " ~ ,,
eo.'"" t"o" ,,~o. 1 H OH
This implies that the dihydroxy derivative is stable, at least enough to allow
further reaction with hydroxyl radical or oxygen. The second hydroxy substituent
should then stabilize the 1,3,5-triazine ring (see 1,3,5-triazine degradation above)
[26]. The rate at which the intermediates depicted react further generating cyanuric
2-1
1.6 I 0
~ 1.2 o
~ 0 . 8 c
0 c o 0 . 4
o
0 0.5 1.0
. . . . . --El- [ ] i ~ ": triallyl cyanurate
/ n: cyanuric acid /
/ z.s
! / .5 2.0
/ 7 1.s / o /
[ ] (.)" 1.0 0
I r o.s 0 �9 i . . . = . i �9
1 2 3 4
k k . . , . . . . , , l ~ ~ Irradiation time, houri
1.5 2
Irradiation time, hours
Figure 5. Heterogeneous photocatalytic degradation of 2,4,6-triallyloxy-i,3,5-triazine and cyanuric acid evolution. Conditions: C O = 2.0 �9 10 -4 M, TiO: P25 500 mg L ~, pH 5.5.
3 04 C. Minero et al.
I
0
2
.,1
1.6 ,,O" /
/ /
~" 1.2 / o / :E
�9 ~ I
=- / ,- ' 0.8 /3 o
e.- / 0
0 0.4 /
0 ~ , ~. ,
f . , , . " [ ~
2 . 5
2 . 0
~o l_s
(,3" 1 . 0
0 . 5
0 0.5
E] =': t r i a l l y l is0cyanurate m: cyanuric acid
, , , �9 ,
2 3 4
Irradiation time. hour s
I ' I ' I
1.0 1.5 2
Irradiation time, hours
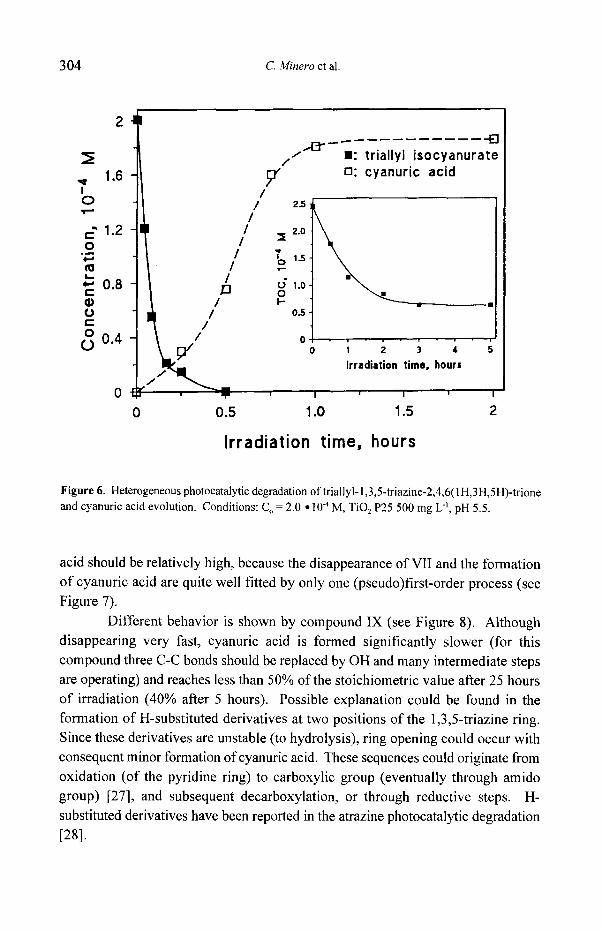
Figure 6. Heterogeneous photocatalytic degradation of triallyl-l,3,5-triazine-2,4,6(IH,3H,5H)-trione and cyanuric acid evolution. Conditions: C O = 2.0 ~ 10 -4 M, TiO 2 P25 500 mg L "l, pH 5.5.
acid should be relatively high, because the disappearance of VlI and the formation of cyanuric acid are quite well fitted by only one (pseudo)first-order process (see Figure 7).
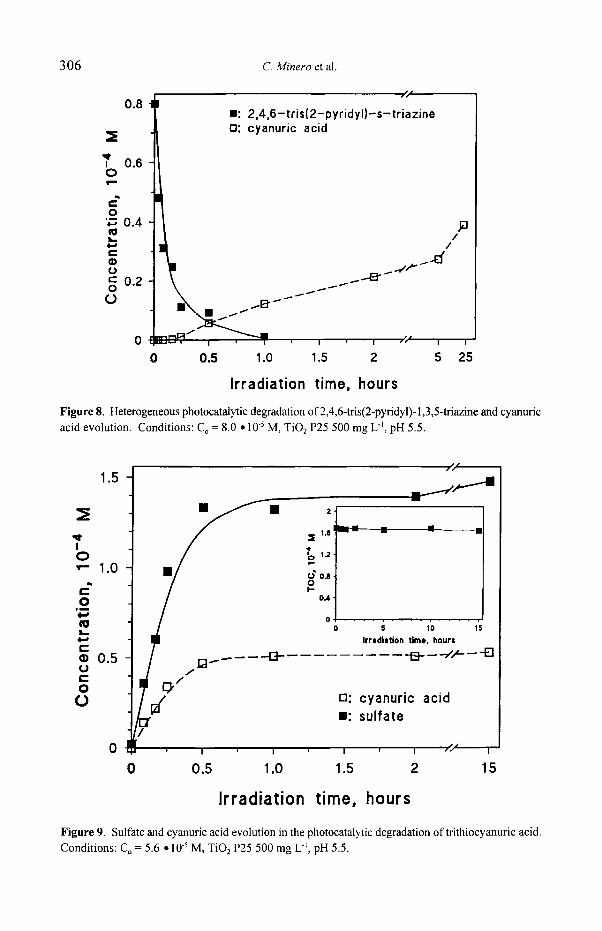
Different behavior is shown by compound IX (see Figure 8). Although
disappearing very fast, cyanuric acid is formed significantly slower (for this compound three C-C bonds should be replaced by OH and many intermediate steps are operating) and reaches less than 50% of the stoichiometric value after 25 hours of irradiation (40% after 5 hours). Possible explanation could be found in the formation of H-substituted derivatives at two positions of the 1,3,5-triazine ring. Since these derivatives are unstable (to hydrolysis), ring opening could occur with consequent minor formation ofcyanuric acid. These sequences could originate from
oxidation (of the pyridine ring) to carboxylic group (eventually through amido group) [27], and subsequent decarboxylation, or through reductive steps. H- substituted derivatives have been reported in the atrazine photocatalytic degradation [28].
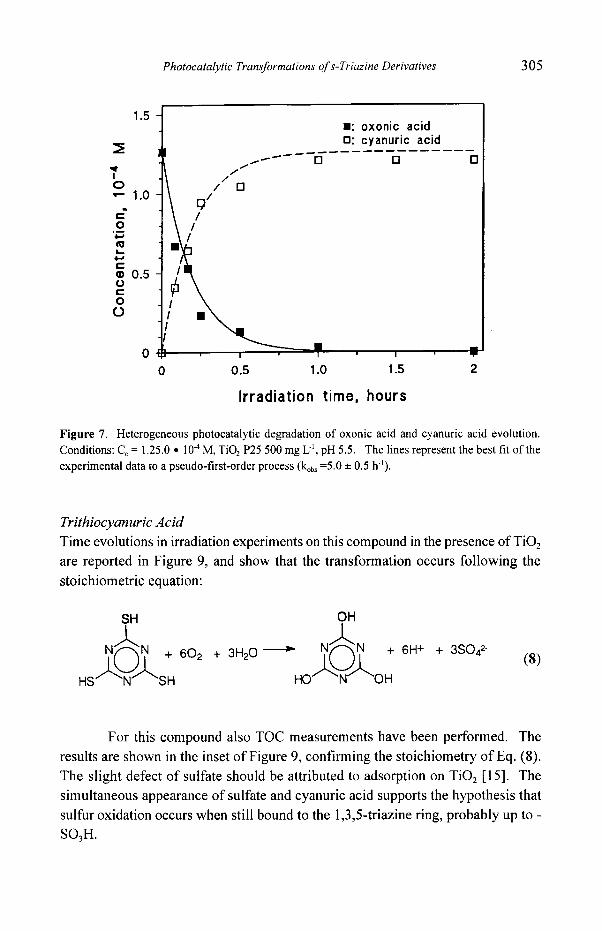
Photocatalytic Transformations of s- Triazine Derivatives 3 0 5
I 0
=- o , i
I,. w C q) o r o O
1.5
1.0
0.5
0
m: oxonic acid o: cyanuric acid
J / \f~
r I ~ ~ , ~ i
0 0.5 1 o0 1.5 2
I r rad ia t ion t ime, hours
Figure 7. Heterogeneous photocatalytic degradation of oxonic acid and cyanuric acid evolution. Conditions: Co = 1.25.0 �9 10 4 M, TiO 2 P25 500 mg L 4, pH 5.5. The lines represent the best fit of the
experimental data to a pseudo-first-order process (kob, =5.0 • 0.5 h").
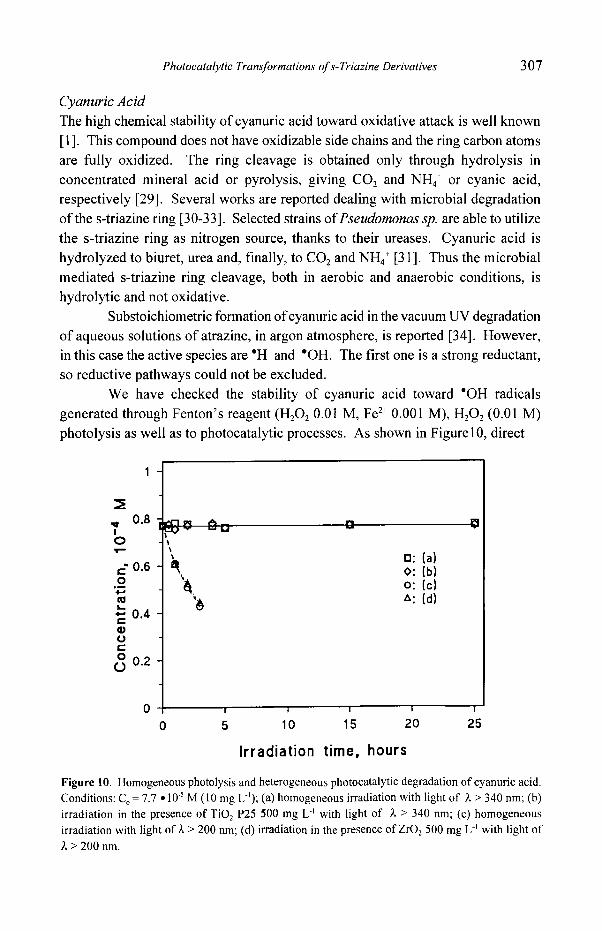
Trithiocyanuric Acid Time evolutions in irradiation experiments on this compound in the presence of TiOz are reported in Figure 9, and show that the transformation occurs following the
stoichiometric equation:
SH OH
N~J~N + + ~ N~--/~N + 6H+ HsJL~k~N'~SH 602 3 H 2 0 HO~.~N~.O H
+ 3SO4 2 (8)
For this compound also TOC measurements have been performed. The results are shown in the inset of Figure 9, confirming the stoichiometry of Eq. (8). The slight defect of sulfate should be attributed to adsorption on TiO2 [15]. The simultaneous appearance of sulfate and cyanuric acid supports the hypothesis that sulfur oxidation occurs when still bound to the 1,3,5-triazine ring, probably up to -
SO3H.
306 C. Minero et al.
e t
0.8 ,l_
T '=: 2,4,6-tris(2-pyridyl)-s-triazine
D: cyanuric acid //El ~.0 0"6
' ~ 0 .4
=o 0.2 ~ i ..e.... .-- -'E~'~''''d
0 /
0 0 .5 1.0 1.5 2 5 25
Irradiation time, hours
Figure 8. Heterogeneous photocatalytic degradation of 2,4,6-tris(2-pyridyl)-l,3,5-triazine and cyanuric acid evolution. Conditions: Co = 8.0 �9 10 5 M, TiO2 P25 500 mg L l , pH 5.5.
I C) 1 - -
=- 0
o i 4 ~
t-
o c 0 0
1.5
1.0
0.5
0 4 j i I ' I
0 0.5 1.0
Id " f J
2 .
1.o ! rl~'~-
d oJ- O I--
0 . 4 .
0 �9 . 0
- - m - - - - n
�9 �9 , . . . . , . . . . ,
5 10 15
I r r o d i e t i o n t i m e , h o u r s
t~ . _ -//--- - - -El
a: cyanuric acid =': sulfate
I ' I J~t I
1.5 2 15
Irradiation time, hours Figure 9. Sulfate and cyanuric acid evolution in the photocatalytic degradation of trithiocyanuric acid. Conditions: Co = 5.6 �9 l0 -5 M, TiO2 P25 500 mg L L, pH 5.5.
Photocatalytic Transformations of s-Triazine Derivatives 307
Cyanuric Acid
The high chemical stability ofcyanuric acid toward oxidative attack is well known [ 1 ]. This compound does not have oxidizable side chains and the ring carbon atoms are fully oxidized. The ring cleavage is obtained only through hydrolysis in concentrated mineral acid or pyrolysis, giving CO2 and NH4 § or cyanic acid, respectively [29]. Several works are reported dealing with microbial degradation of the s-triazine ring [30-33]. Selected strains ofPseudomonas sp. are able to utilize the s-triazine ring as nitrogen source, thanks to their ureases. Cyanuric acid is hydrolyzed to biuret, urea and, finally, to CO2 and NH4 + [31]. Thus the microbial mediated s-triazine ring cleavage, both in aerobic and anaerobic conditions, is hydrolytic and not oxidative.
Substoichiometric formation ofcyanuric acid in the vacuum UV degradation of aqueous solutions of atrazine, in argon atmosphere, is reported [34]. However, in this case the active species are ~ and ~ The first one is a strong reductant, so reductive pathways could not be excluded.
We have checked the stability of cyanuric acid toward "OH radicals generated through Fenton's reagent (H202 0.01 M, Fe 2§ 0.001 M), H202 (0.01 M) photolysis as well as to photocatalytic processes. As shown in Figurel0, direct
0.8 I 0 T'-
c" 0.6 o
i . _
0.4 e -
u c o 0.2
0
a'-I ~, a ,_ 0 171
~, o: (a) �9 o: (b) '~. o: (c)
,~) A: {d)
I I I I I
0 5 10 15 20 25
I r r a d i a t i o n t i m e , h o u r s
Figure 10. Homogeneous photolysis and heterogeneous photocatalytic degradation of cyanuric acid.
Conditions: Co = 7.7 �9 10 .5 M (10 mg L-'); (a) homogeneous irradiation with light of ~ > 340 nm; (b)
irradiation in the presence of TiO 2 P25 500 mg L" with light of ~, > 340 nm; (c) homogeneous
irradiation with light o f ~ > 200 nm; (d) irradiation in the presence ofZrO 2 500 mg L ~ with light of
> 200 nm.

308 C. Minero et al.
photolysis at > 340 nm or through band-gap excitation of TiO2 does not affect cyanuric acid stability. TOC measurements confirm that cyanuric acid under these conditions is not mineralized. Disappearance of cyanuric acid is observed when
irradiated with light > 200 nm, and the rate of disappearance is slightly higher at basic pH. The presence of TiO2 does not improve the degradation rate. Even the use of ZrO2, a semiconductor with the redox potential of valence band higher than TiO2 [35], does not improve the degradation rate significantly. Under these conditions, the substrate disappearance is to be attributed to direct photolysis of cyanuric acid
or its dissociated forms.
CONCLUSIONS
The photocatalytic behavior of 1,3,5-triazine derivatives has shown some characteristic features, which can be summarized as follows: (i) 1,3,5-Triazine decomposes through thermal hydrolysis and irradiated semiconductors may accelerate this decomposition without leading to the formation of stable OH adducts, like cyanuric acid. (ii) Cyanuric acid is stable toward photocatalytic processes involving band-gap
excited TiO2. This is a rather unusual behavior, since other organics have been shown to be efficiently mineralized to CO2 and inorganic ions. As far as water purification treatments based on photocatalytic processes are concerned, it must be concluded that compounds having 1,3,5-triazine ring in their structure are likely to form cyanuric acid as final product. However, it has been demonstrated that this product exhibits a very low toxicity [36] and can be biologically degraded [31]. (iii) Amino and hydroxy-amino derivatives of 1,3,5-triazine lead, through fairly well
established pathways, to cyanuric acid formation in nearly stoichiometric ratio. (iv) Chlorine atom on 1,3,5-triazine structure bearing two other amino substituent is only slowly displaced in respect to the amino substituent itself. This contrasts with other aromatic derivatives undergoing photocatalytic degradation, in which chloride evolves in the early part of the degradation. (v) Alkyloxy-1,3,5-triazine and alkyl derivatives of the isocyanuric acid are both rapidly and stoichiometrically converted into cyanuric acid. (vi) Trithiocyanuric acid is also converted into cyanuric acid releasing sulfate ions. The oxidation of sulfur is likely to occur when linked to the ring. (vii) Degradation of compounds with C-C bonds shows that for a single carboxy substituent quantitative conversion to cyanuric acid occurs. When three C-C bonds are present only 50% of the stoichiometric amount of cyanuric acid is recovered. The formation of unstable two H-substituted triazine is presumably responsible for this finding.

Photocatalytic Transformations of s-Triazine Derivatives 309
Acknowledgment This research has been received generous support from the following agencies: CNR, MURST, CIEMAT and Progetto Sistema Lagunare Veneziano.
R E F E R E N C E S
1 ,
2. 3.
4.
5. 6. 7.
~
9.
10.
11.
12.
13. 14.
15. 16.
17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28.
E.M. Smolin and L. Rapoport. In: The Chemistry of Heterocyclic Compounds, Vol. 13., A. Weissenberg (Ed.), Interscience, New York, 1959. G.L. Bery, Farm Chemical Handbook, Meister Publ., Willonghby, Ohio, 1985. Kirk-Othmer, The Encyclopedia of Chemical Technology, Wiley Interscience, New York, 1984, 3rd Ed. N. Serpone and E. Pelizzetti (Eds.), Photocatalysis. Fundamentals and Applications, Wiley, New York, 1989. M.R. Hoffmann, S.T. Martin, W. Choi, and D.W. Bahnemann, Chem. Rev. 95, 69 (1995). D.F. Ollis, E. Pelizzetti, and N. Serpone, Environ. Sci. Technol. 25, 1522 (1991). D.W. Bahnemann, J. Cunningham, M.A. Fox, E. Pelizzetti, P. Pichat, and N. Serpone. In: Aquatic andSurface Chemistry, G.R. Helz, R.G. Zepp, and D.G. Crosby (Eds.), Lewis, Boca Raton, 1994, p. 261. E. Legrini, E. Oliveros, and A.M. Braun, Chem. Rev. 93, 671 (1993). E. Pelizzetti, V. Maurino, C. Minero, V. Carlin, E. Pramauro, O. Zerbinati, and M.L. Tosato, Environ. Sci. Technol. 24, 1559 (1990). E. Pelizzetti, C. Minero, M. Vincenti, E. Pramauro, V. Carlin, and M. Dolci, Chemosphere 24, 891 (1992). E. Pelizzetti, C. Minero, V. Maurino, and M. Vincenti, Melamine Transformation through Advanced Redox Processes, submitted. E. Pelizzetti, C. Minero, V. Maurino, A. Sclafani, H. Hidaka, and N. Serpone, Environ. Sci. Techno123, 1380 (1989). C. Minero, E. Pelizzetti, S. Malato, and J. Blanco, Chemosphere 26, 2103 (1993). P.K. Dasgupta. In: Ion Chromatography, J.G. Tarter (Ed.), Marcel Dekker, New York, 1987, p. 257. J. Augustynski, Struct. Bonding (Berlin)69, 1 (1988). R.W. Matthews. In: Photochemical Conversion and Storage of Solar Energy, E. Pelizzetti and M Schiavello (Eds.), Kluwer, Dordrecht, 1991, p. 427. R.B. Draper and M.A. Fox, Langmuir 6, 1396 (1990). Y. Sun and J.J. Pignatello, Environ. Sci. Technol. 29, 2065 (1995). C. Minero, Sol. Energy Mat. Sol. Cells38, 421 (1995). C. Grundman and A. Kreutzberger, Z Am. Chem. Soc. 76, 5646 (1954). A. Burger and E.D. Hornbaker, J. Am. Chem. Soc. 75, 4579 (1953). G.G. Choudry, Curt. Top. Environ. Toxicol. Chem. 7, 143 (1984). C.I. Harris, J. Agric. FoodChem. 15, 157 (1967). Y. Osawa, J. Phys. Chem. 88, 3069 (1984). T. Sakata, T. Kawai, and K. Hashimoto, J. Phys. Chem. 88, 2344 (1984). Ref. 1, p.202. C. Maillard, C. Guillard, and P. Pichat, Chemosphere 24, 1085 (1992). P.L. Yue and D. Allen. In: Photocatalytic Purification and Treatment of Water and Air, D.F.OIIis and H. AI-Ekabi (Eds.), Elsevier, Amsterdam, 1993, p.607.

310 C. Minero et al.
29. 30. 31, 32.
33.
34.
35,
36.
Ref. 1, pag 44. A.M. Cook, P. Beilstein, H. Grossenbacher, and R. Hutter, Biochem. J. 231, 25 (1985). A.M. Cook, FEMS Microbiol. Rev. 46, 93 (1987). P.C.Kearney, M.Y.Muldoon, C.J.Somich, J.M.Ruth, and D.V.Voaden, ~ Agric. Food. Chem. 36, 1301 (1988). M. Radosevich, S.J. Traina, Y.-L. Hao, and O.H. Tuovinen, Appl. Environ. Microbiol. 61, 297 (1995). M.C. Gonzales, A.M. Braun, A. Bianco Prevot, and E. Pelizzetti, Chemosphere 28, 2121 (1994). S.R. Morrison, Electrochemistry at Semiconductor and Oxidized Metal Electrodes, Plenum, New York, 1980. E. Canelli, Am. ~ Public Health 64, 155 (1974).




















