
Flow carbonylation of sterically hindered ortho-substituted ...

Applied Catalysis A: General 190 (2000) 117–135
Heat-treatment of isomorphously substituted ZSM-5 (MFI) zeolitesAn ESR and Mössbauer spectroscopy and kinetic study
Pál Fejesa,∗, János B. Nagyb, Károly Lázárc, János Halászaa József Attila University, Applied Chemistry Department, 6720 Szeged, Hungary
b Laboratoire de RMN, Department de Chimie, FUNDP, Namur, Belgiumc Institute of Isotope and Surface Chemistry, 1525 Budapest, P.O. Box 77, Hungary
Accepted 28 June 1999
Abstract
Fe-ZSM-5 zeolites comprise of three types (I, II and III) of iron (Fe3+) sites in both as synthesized (AS) and heat-treatedstates. Previous evidence could be confirmed by the assignment of ESR lines, registered in the X-band, stating: (i) type Iframework Fe3+ sites (in Th oxygen coordination), where charge compensation occurs mainly by Na+ ions and the crystalfield (cf) approximates cubic symmetry, produce resonances at near tog= 2.00; (ii) type II and probably type III binuclearFe...Fe dioxo- and oxo-bridges manifest themselves in producing slightly distorted surroundings of axial symmetry, and therelevant powder-averaged subspectrum is superimposed on the previous one in theg∼= 2.45–1.98 interval. As the temperatureof the heat-treatment (HT) is raised, type II and III sites will be annihilated with preference, under concomitant production ofamorphous Fe2O3 possessing molecular dispersity. (iii) The weak ESR signal (2–4%) atg∼= 4.27 is attributed to Th coordinatedframework sites in the surface layers (≈ 1 u.c. thick) under the influence of solid surface tension, giving rise to fully rhombiccf symmetry.
The size of the ejected Fe2O3 particles, as estimated from Mössbauer spectra (no hyperfine structure at 77 K) is less than2.9 nm. The extremely large electron affinity of Fe3+ ions manifests itself in decreased ionicity (and unexpectedly dominantcubic symmetry), activity in biomimetic oxidations, autoreduction and spontaneous reoxidation etc.
In the liquid phase oxidation by hydrogen peroxide ofn-hexane and cyclohexane the heat-treated Ti-ZSM-5 samplesexhibited both activity- and selectivity dependence on HT. On the basis of experiences drawn from the heat-treatmentsproposal is made for the structures of the defect site (producing the 960 cm−1 IR signal) and the active centre of selectiveoxidation. The proposal seems to be in accord with the experimental observations (isotopic exchange, MAS-NMR behaviouretc.) published so far. ©2000 Elsevier Science B.V. All rights reserved.
Keywords:Heat-treatment; ESR and Mössbauer spectroscopy; As synthesized; ZSM-5 zeolites
1. Introduction
In part I [1] focus was on the way how and to whatdegree intentional and accidental overheating, which
∗ Corresponding author.E-mail address:[email protected] (P. Fejes)
may take place for example during the removal bycalcination of the organic template in isomorphouslysubstituted zeolites, may influence lattice stabilityand thereby the distribution of the substituting ionsin framework and extra-framework positions. X-raydiffractometry and X-ray photoelectron spectroscopy(XPS) revealed a linear/quasi-linear decrease of
0926-860X/00/$ – see front matter ©2000 Elsevier Science B.V. All rights reserved.PII: S0926-860X(99)00298-7

118 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
framework Fe3+ and Ti4+ species, respectively, withFe-ZSM-5 (Si/Fe = 15.0) and Ti-ZSM-5 (Si/Ti = 29.8)samples in the temperature interval: 773–1073 K.The ejected materials (Fe2O3 and TiO2) seem tobe amorphous and have extremely high (probably:‘molecular’) dispersity. The heat-treatments increasedthe Si/T ratio and concomitantly lattice shrinkageand a slight (≤ 9 cm−1) blue-shift of the respectiveIR spectra has taken place. This framework con-traction was extraordinary in that it occurred almostexclusively along the sinusoidal channels (i.e. verti-cal to the [1 0 0] crystallographic plane) of the MFIstructure.
In the case of the Fe-ZSM-5 sample the fate ofthe three types of framework sites below has beenfollowed more closely:
Both type II and III sites are produced during thehydrothermal crystallization of Fe-silicate gel. Ac-cording to probability theory the amount of type IIIsites is proportional to (Si/Fe)−2. The existence oftype II, edge-sharing tetrahedral (framework) andoctahedral (extra-framework) sites is hypothetic andbased mainly on the observation of Mulay et al. [2]that under the conditions of the preparation of the pre-cursor Fe-silicate gel (pH≈ 2.0) the concentration ofa dimeric [Fe2(OH)2(H2O)8]4+ ion, exhibiting a sim-ilar structure as type II sites, is measurable and it mayreach even 90% of total iron content at still higher pH.From among the three types of Fe3+ environments inthe zeolitic structure less stable against overheatingseem to be those sites where charge compensation oc-curs with (hydrated octahedral) Fe(OH)+2 ions (typeIII sites); type II sites are supposed to exhibit mediumstability and most stable are (also in agreementwith literature data; cf. [3]) those Fe3+ frameworksites where the charge compensating ions are Na+ions.
The Ti4+ framework sites in our samples, synthe-sized also by the ‘sol–gel technique’ to provide possi-bility for comparison, appear to exhibit lesser heat re-sistance than those prepared from tetraethyl orthosil-icate and titanium(IV) butoxide as raw-materials. (It
remains to be seen whether the greater stability (asclaimed in the respective patents) is due to higher ini-tial Ti4+ incorporation or other causes). A detailedX-ray diffractometry, XPS and IR study of heat-treatedTi-ZSM-5 specimens has been published in the 1stpart of this paper [1].
Fe-ZSM-5, as test-substance, was selected becausehigh-spin (3d5, 6S5/2) Fe ions are both ESR and Möss-bauer active. There is no doubt, much more informa-tion could have been obtained from the ESR spectraif it were possible to study single crystals rather thanpolycrystalline powders, because it is known (see e.g.[4–8]) that the stereotype spectra cannot be resolvedby any means, thus they are only to a limited extentsuitable to provide information on the symmetry con-ditions of the substituting and already ejected Fe3+ions. As ESR spectroscopy does not permit distinc-tion between framework- and extra-framework Fe3+sites located in identical/nearly identical crystal fieldsymmetries, it was obligatory to investigate the sameheat-treated Fe-ZSM-5 samples by Mössbauer spec-troscopy as well, to acquire information on the oxi-dation state (under the conditions of in situ high vac-uum Mössbauer studies autoreduction of Fe3+ is pos-sible), ferric ions’ environments, possible growth oreven crystallization of the dislodged Fe2O3 clustersor particles, their approximate size and disposition toform magnetically ordered domains.
This paper reports on the ESR and Mössbauer spec-troscopy studies of heat-treated Fe-ZSM-5 samples. Inorder to see and follow the changes in catalytic activ-ity and selectivity, the heat-treated Ti-ZSM-5 samplesmentioned above were tested in the liquid phase se-lective oxidation ofn-hexane and cyclohexane usingH2O2 as oxidant.
2. Experimental part
2.1. Brief summary of the synthesis method andheat-treatment
The iron (Fe3+) and titanium containing ZSM-5(MFI) samples were prepared according to the ‘sol–geltechnique’ described in detail elsewhere [9]. In thecase of Fe-ZSM-5 this was a necessity [10], on theother hand it was only one possibility from among a

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 119
wider variety of recipes when synthetizing Ti-ZSM-5,however, only identical synthesis methods provide apotential for comparison. The main points of the syn-theses can be summarized as follows1. Formation of the respective metal orthosilicate,
as sol, in a strongly acidic (pH< 1.0) aqueoussolution of the metal salt by admixing a sodiummetasilicate solution (Si/Na< 1.8) drop by dropunder intensive mixing.
2. Gelation at pH = 4.0–4.3, and 313 K.3. Washing of the gel particles by percolation in or-
der to remove the salts using distilled water. Thispurification is especially important in the case ofthe titanium precursor gel (Si/Na > 1000 should bereached for the dry xerogel), because even tracesof alkali cations might prevent the incorporationof titanium [11,12].
4. Adjustment of the composition of the slurries; re-duction of the particle size of the gel by milling,followed by crystallization at 443 K, applyingrocking agitation.
Dealing with consequences of ‘caring’ calcinationand heat-treatments thereafter, we give a brief sum-mary about these techniques.
Unlike from ‘standard’ calcination where the as syn-thesized zeolite samples are heated at∼773 K in air,‘caring’ calcination means heating (3◦/min) in pure N2up to 773 K in order to decompose and remove about90–92% of the organics, followed by cooling down to673 K while maintaining the N2 flow. Thereafter, dis-connecting the N2 from the oven, the sample is heatedup under the conditions of increasing O2 concentra-tion (from the air) to remove the carbonaceous restof the template. Further heat-treatments (when neces-sary) were carried out in ‘shallow bed’ using Pt cru-cibles in the preheated oven, lasting for 3 h.
The calcined/heat-treated samples were stored in adesiccator over cc. aqueous ammonium chloride solu-tion. The samples for registering the ESR spectra wereconditioned as such; for accumulating the Mössbauerspectra and for the kinetic studies evacuated samples(pressure< 1.3× 10−2 Pa) were used.
2.2. ESR spectra
The ESR spectra were recorded on a BrukerB.E.R.-420 spectrometer at room temperature (r.t.)
and liquid nitrogen temperature (l.n.t). The spec-trometer was operated at X-band frequency and amicrowave power of 13 mW and using 100 KHz fieldmodulation. The evaluation of the spectra was facil-itated by a set of pitch-signals (Mn2+ in crystallineMgO). Approximate line intensities were computedaccording to the formula of Wertz et al. [13]
Irel = Amp(1Hpp)2
whereAmp is the peak-to-peak amplitude, and1Hpprepresents the peak-to-peak width.
It should be emphasized that theIrel values so com-puted do not permit to draw far-reaching consequencesbecause bothAmp and1Hpp are resultants of a verycomplex superposition of spectral lines (vide infra),nevertheless are useful for qualitative purposes.
2.3. Mössbauer spectra
Amounts of 400–600 mg samples were pressedinto wafers and at 77 and 300 K in situ Mössbauerspectra were recorded after caring calcination andheat treatments, respectively, performed at 773, 873,973 and 1073 K. All isomer shift data are given rela-tive to metallica-iron, the accuracy of the positionaldata is ca.±0.03 mm/s. The measuring cell and therelated further experimental details are described in[14]. First, spectra were recorded on the ‘as received’samples which (during the transportation) might havetaken up water vapour from the atmosphere. The ad-sorbed water was removed by evacuation (for 2 h, toa final pressure of 10−1 Pa) at 573 K. Then, in orderto facilitate distinction among the various species ofiron, a reducing treatment was applied at 570 K inflowing purified hydrogen (0.3 l/h, 100 kPa) for 1 h.Finally, the evacuation was repeated at 573 K, tocheck whether the distribution of iron species waschanged by reduction. The corresponding spectra wererecorded after each step of the subsequent treatments.
The data extracted from the fits are displayed inTable 1. Isomer shift (IS, relative toa-iron), quad-rupole splitting (QS) and relative intensity (RI, spec-tral area, %) values are reported. The assignmentsof various coordination states shown in the Tableare primarily based on data taken from previous re-ports [15–17]. For illustration, a series of spectraobtained on the sample calcined at 973 K is shown in

120 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
Table 1Mössbauer parameters extracted from the fits of the in situ spectraa
Measurement at 300 K 77 K
Sample calcined at Treatment tb Component IS QS RI IS QS RI
773 K A.R. Fe3+oh 0.31 0.77 100 0.43 0.88 100
573 K Fe3+Td, a 0.21 1.75 20
10−1 Pa Fe3+Td, b 0.25 1.23 59
Fe2+ 1.10 2.01 21
573 K Fe3+Td/oh 0.33 1.40 31
H2 Fe3+oh 0.30 0.86 94 0.36 0.73 37
1 bar Fe2+a 0.94 2.73 6 1.08 2.48 17
Fe2+b 1.36 3.10 14
873 K A.R. Fe3+oh 0.31 0.81 100 0.40 0.87 100
573 K Fe3+Td, a 0.22 1.86 25
10−1 Pa Fe3+Td, b 0.27 1.20 56
Fe2+a 0.97 1.53 18
573 K Fe3+Td, a 0.32 1.74 9
H2 Fe3+Td, b 0.29 1.14 78 0.33 1.29 65
1 bar Fe3+Td/oh 0.28 0.60 17
Fe2+a 1.17 2.38 22
Fe2+b 1.17 2.17 6 1.45 2.88 4
573 K Fe3+Td, a 0.22 1.72 27
10−1 Pa Fe3+Td/oh 0.28 1.03 42
Fe2+a 0.84 2.29 31
973 K A.R. Fe3+Td/oh 0.33 0.85 100 0.41 1.05 68
Fe3+oh 0.42 0.66 32
570 K Fe3+Td, a 0.24 1.94 19
10−1 Pa Fe3+Td, b 0.26 1.51 34
Fe3+Td, oh 0.31 0.90 33
Fe2+ 1.10 1.88 14
570 K Fe3+Td/oh 0.33 1.14 58 0.44 1.03 44
H2 Fe3+oh 0.31 0.67 36 0.40 0.60 19
1 bar Fe2+a 1.09 2.45 20
Fe2+b 1.09 2.92 6 1.22 3.05 17
Fe3+Td/oh 0.40c 1.20 48
Fe3+oh 0.39 0.73 46
Fe2+b 1.27 3.09 7
570 K Fe3+Td/oh 0.33 0.98 73 0.37 1.13 18
H2 Fe3+oh 0.30 0.54 21 0.34 0.77 39
1 bar Fe2+a 0.89 2.36 6 1.13 2.23 14
(repeat) Fe2+b 1.38 2.70 29
570 K Fe3+Td, a 0.21 1.89 4

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 121
Table 1 (Continued)
Measurement at 300 K 77 K
Sample calcined at Treatment tb Component IS QS RI IS QS RI
10−1 Pa Fe3+Td, b 0.24 1.53 31
Fe3+Td/oh 0.29 0.91 37
Fe2+ 1.05 1.96 27
1073 K A.R. Fe3+Td/oh 0.33 1.05 50 0.42 0.96 72
Fe3+oh 0.32 0.65 50 0.41 0.56 28
573 K Fe3+Td/oh 0.31 1.11 65
10−1 Pa Fe3+oh 0.34 0.65 35
573 K Fe3+Td/oh 0.34 1.09 45 0.45 1.02 37
H2 Fe3+oh 0.33 0.66 55 0.45 0.61 45
1 bar Fe2+a 0.97 1.98 9
Fe2+b 1.14 2.67 8
573 K Fe3+Td/oh 0.33 1.08 48
10−1 Pa Fe3+oh 0.35 0.62 44
Fe2+ 1.09 1.72 9
a IS: Isomer shift, related toa-iron, mm/s, QS: quadrupole splitting, mm/s, RI: relative spectral area, %. Remarks: after the 573 Khydrogen treatments always the 77 K spectrum was recorded first.
b A.R.: as received.c Repetition of the measurement again at 77 K (after the 77 and 300 K one in H2).
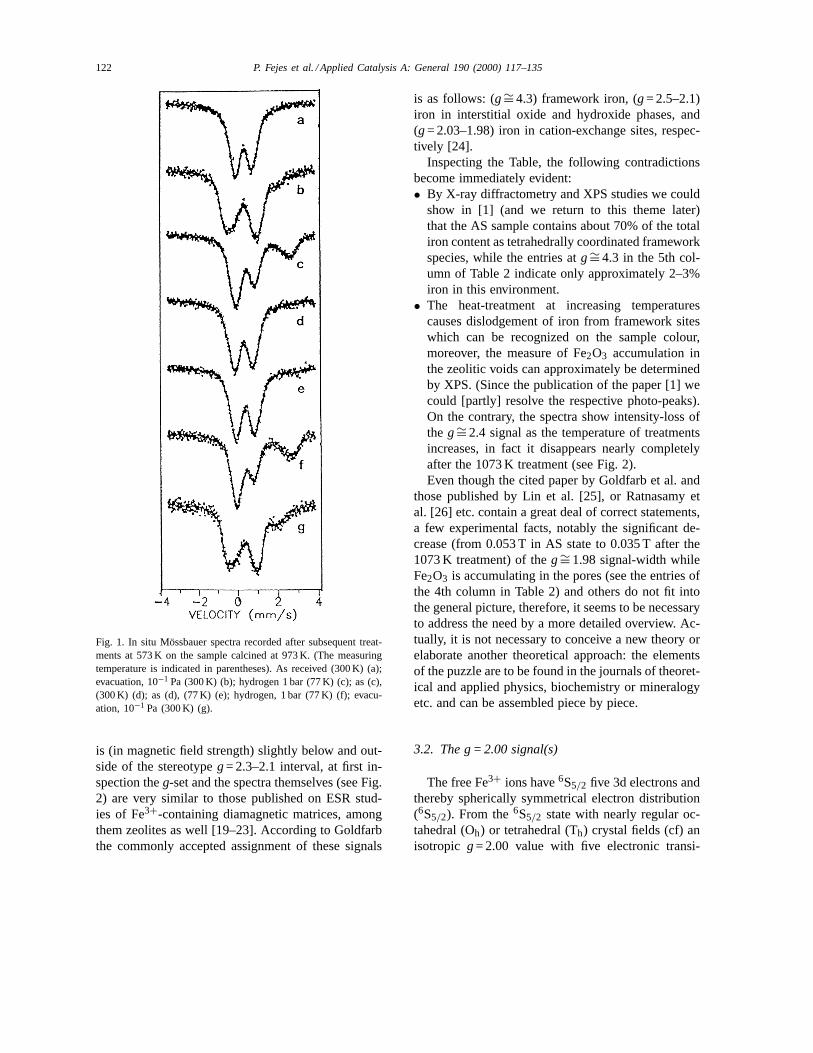
Fig. 1. The values of absorption (A, spectral area re-lated to the base line) were also calculated and the1A/1T slopes were estimated from the 77 and 300 Kspectra. Considering full spectra (without making anydistinction between the various Fe3+ components)and assuming linear relationship betweenA and Tthe corresponding1A/1T slopes were calculated; thevalues are situated between−10−3 and −4× 10−4.From these slopes the average value of the Debyetemperature: 300 K< 2D < 450 K can be estimated[18]. These2D values indicate fairly strong bonds.Consequently, in a first approximation, the RI valuesare proportional to the quantities of the given species,thus, they can be used for relative comparisons toestimate the changes in the amounts of the samecomponent after different treatments.
2.4. Experimental in selective oxidation
The experimental arrangement was similar to thosegenerally used when carrying out such catalytic re-actions: thermostated and stirred teflon autoclaves
(∼200 ml); temperature between 323 and 373 K underautogenous pressure, using different mole ratios ofH2O2 to n-hexane and cyclohexane in acetone as in-termediary solvent, to avoid phase separation. About200 mg catalyst was used in each case. For productanalysis a GC (type: Shimadzu 16A) was utilized,equipped with capillary column (length: 30 m; filling:SPB-1) and FI-detector.
Neither the Ti-silicate precursor gel, nor the ASTi-ZSM-5 sample, nor the specimen after the 1073 Kheat-treatment contained anatase phase; in their laserRaman spectra the most intense band of anatase (at145 cm−1) was completely missing (not shown).
3. Results and discussion
3.1. ESR studies
The ESR parameters of the AS and heat-treatedsamples are compiled in Table 2. Three characteris-tic features can be recognized atg∼= 4.27, g∼= 2.46and g∼= 1.98 and even when the mid valueg∼= 2.46
122 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
Fig. 1. In situ Mössbauer spectra recorded after subsequent treat-ments at 573 K on the sample calcined at 973 K. (The measuringtemperature is indicated in parentheses). As received (300 K) (a);evacuation, 10−1 Pa (300 K) (b); hydrogen 1 bar (77 K) (c); as (c),(300 K) (d); as (d), (77 K) (e); hydrogen, 1 bar (77 K) (f); evacu-ation, 10−1 Pa (300 K) (g).
is (in magnetic field strength) slightly below and out-side of the stereotypeg= 2.3–2.1 interval, at first in-spection theg-set and the spectra themselves (see Fig.2) are very similar to those published on ESR stud-ies of Fe3+-containing diamagnetic matrices, amongthem zeolites as well [19–23]. According to Goldfarbthe commonly accepted assignment of these signals
is as follows: (g∼= 4.3) framework iron, (g= 2.5–2.1)iron in interstitial oxide and hydroxide phases, and(g= 2.03–1.98) iron in cation-exchange sites, respec-tively [24].
Inspecting the Table, the following contradictionsbecome immediately evident:• By X-ray diffractometry and XPS studies we could
show in [1] (and we return to this theme later)that the AS sample contains about 70% of the totaliron content as tetrahedrally coordinated frameworkspecies, while the entries atg∼= 4.3 in the 5th col-umn of Table 2 indicate only approximately 2–3%iron in this environment.
• The heat-treatment at increasing temperaturescauses dislodgement of iron from framework siteswhich can be recognized on the sample colour,moreover, the measure of Fe2O3 accumulation inthe zeolitic voids can approximately be determinedby XPS. (Since the publication of the paper [1] wecould [partly] resolve the respective photo-peaks).On the contrary, the spectra show intensity-loss ofthe g∼= 2.4 signal as the temperature of treatmentsincreases, in fact it disappears nearly completelyafter the 1073 K treatment (see Fig. 2).Even though the cited paper by Goldfarb et al. and
those published by Lin et al. [25], or Ratnasamy etal. [26] etc. contain a great deal of correct statements,a few experimental facts, notably the significant de-crease (from 0.053 T in AS state to 0.035 T after the1073 K treatment) of theg∼= 1.98 signal-width whileFe2O3 is accumulating in the pores (see the entries ofthe 4th column in Table 2) and others do not fit intothe general picture, therefore, it seems to be necessaryto address the need by a more detailed overview. Ac-tually, it is not necessary to conceive a new theory orelaborate another theoretical approach: the elementsof the puzzle are to be found in the journals of theoret-ical and applied physics, biochemistry or mineralogyetc. and can be assembled piece by piece.
3.2. The g = 2.00 signal(s)
The free Fe3+ ions have6S5/2 five 3d electrons andthereby spherically symmetrical electron distribution(6S5/2). From the6S5/2 state with nearly regular oc-tahedral (Oh) or tetrahedral (Th) crystal fields (cf) anisotropic g= 2.00 value with five electronic transi-

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 123
Table 2ESR parameters for different types of Fe3+ sites in heat-treated Fe-ZSM-5 zeolites
Sample ESR spectrum g-value 1Hpp (width) Irel (relative intensity) Gaina
registered at (in T) (arbitrary units)
AS 4.09 0.029 14.7 (3.2%)2.48 – – 5× 104
1.98 0.053 439.7Caring calcination, temperature≤773 K 4.27 0.028 6.7 (2.4%)
2.49 – – 1× 105
1.98 0.060 278.0Heat-treated at 873 K r. t. 4.29 0.028 7.0 (2.4%)
2.51 – – 1× 105
1.98 0.062 283.8Heat-treated at 973 K 4.27 0.018 1.1 (0.4%)
2.46 – – 5× 104
1.98 0.040 274.5Heat-treated at 1073 K 4.27 0.018 1.6 (0.5%)
2.48 – – 3.2× 104
1.98 0.035 307.2AS 4.09 0.023 238.6 (5.8%)
2.83 ? – – 8× 103
1.97 0.056 4084.3Heat-treated at 1073 K l. n. t. 4.27 0.017 120.0 (1.0%)
– – – 4× 103
1.97 0.053 11907.9
a The ESR spectra of Fig. 2 are strictly comparable if the respective gains are considered.
tions can be expected. The lines are badly (in strictsense: not) separated. This approximation is more orless valid in our case too: theg= 2.00 (overlapping)lines in Fig. 2. are partly due to type I (and maybetype III) sites of framework Fe3+ ions in tetrahedralcoordination, with Na+ (and to a lesser extent TPA+)and hydrated [Fe(OH)2]+ cations as charge com-pensating ions. Those with Na+ are the most stableFe-substituted lattice sites, exhibiting unexpected heatresistance (incidentally in excellent agreement withthe observations of McNicol and Pott [3]), which isconvincingly shown by the fact that their ‘as syn-thesized’ (AS) contribution of 40% (equivalent to3.95× 10−4 mol Fe3+/g zeolite) to all by Fe3+ ionssubstituted framework sites in the Fe-ZSM-5 zeolitehad been reduced only to∼35% after a severe 3 hheat-treatment at 1073 K.
In (intact) type II sites the distance between(Th coordinated) framework and (Oh coordinated)extra-framework Fe3+ ions is about 0.27 nm, i.e.less than the critical 0.9 nm where dipole–dipole andspin exchange interactions begin to give rise to fairlystrong (>100 cm−1) magnetic coupling. This causes
distortion of the cubic crystal-field, therefore thesesites exhibit different ESR habits (vide infra).
The preservation of the undistorted, cubic symme-try of type I and type III framework Fe3+ sites beingin permanent coulomb intraction with charge compen-sating Na+ and [Fe(OH)2]+ ions need a word of com-ment. The situation is that even in a perfect Th or Ohcrystal field there will be some distortion: the chargecloud will be slightly ellipsoidal instead of a perfectsphere [27]. The distortion of high-spin Fe3+ ions iscaused by the charge distribution of other ions sitedin the surroundings (modified by the Sternheimer an-tishielding factor), thereby any effect of whatsoeverorigin that may contribute to the reduction of the ef-fective charges or increases the separation may help tomaintain cubic symmetry or at least reduce large dis-tortion. According to Beran et al. [28] calculations, us-ing the CNDO/2 method extended for transition met-als, show that the negative charge densities localizedon the oxygen atoms coordinated to Fe3+ ions are sig-nificantly lower than the charge on the remaining oxy-gen atoms. This can be explained by donation of elec-tron density to Fe3+ ions, which exhibit the strongest

124 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
Fig. 2. ESR spectra of the heat-treated Fe-ZSM-5 zeolites. From (a) to (e): spectra registered at room temperature: as synthetised sample(a); after caring calcination (b); heat-treatments at 873 K (c); at 973 (d); at 1073 K (e). Spectra A and E were registered at liquid nitrogentemperature: as synthetised sample (A); heat-treated at 1073 K (E). (Magnetic scales to the spectra can be computed from the entries ofcolumn 4. in Table 2).
electron acceptor ability of all the transition metalions studied so far. Thereby the negative charge onthe {FeO4/2}− tetrahedra is reduced substantially. Inthe previous chapter we mentioned that except XPS,Mössbauer spectroscopic and Kinetic studies no pre-caution was taken against atmospheric moisture, thusthe exchange ions, Na+ and [Fe(OH)2]+ in our ze-olitic samples were certainly hydrated. The hydrationshell contributed to electrostatic shielding and the in-creased size to larger charge separation.
3.3. The g = 4.3 signal(s)
Examination of the spectra in Fig. 2. shows thatthe g= 4.27 signal is in our case very weak and itsintensity is decreasing by the heat-treatments.
Strong rhombic crystal-fields giving rise to theg= 4.27 signal can be produced by a number ofchemical and physical influences:• Meads et al. have found good correlation between
the fraction of total (triplet) intensity in the 0.1558 T
resonance (corresponding tog∼= 4.3 in the X-band)and the Hinckley crystallinity index, indicating thatimperfect crystallisation (stacking faults etc.) maycreate rhombic crystal fields [29]. As Fig. 1 in thefirst part of this paper [1] and a wealth of unpub-lished and published (see e.g. [9]) SEM picturesprove, the isomorphously substituted ZSM-5 zeo-lites synthesized by the ‘sol–gel technique’ are per-fectly crystallized, thus this explanation would beat variance with the experimental facts.
• It has been shown in [9] that the29Si MAS NMRspectra of the zeolite samples synthesized with the‘sol–gel technique’ registered without and withcross-polarisation (CP) are identical, hence, it canbe stated that these zeolite samples are practicallyfree of defect sites in their AS states. Although theheat-treatments by (partial) dislodgement of theincorporated ions produce a great deal of vacancies(in this case hydrated nests) and every Fe3+ (orTi4+) site, irrespective of its coordination, shouldbe regarded liable to distortion if it is adjacent to

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 125
such a defect, the mathematical probability prac-tically excludes such a coincidence. Without CPthese defects produced in the calcination and fol-lowing heat-treatments are ‘invisible’ in the MASNMR spectra.
• As concerns the weak, low-symmetry ESR sig-nal at g∼= 4.27, the experimental spectra of ourFe-ZSM-5 specimen can be interpreted on the as-sumption that this feature is due to framework (Th)and extra-framework (Oh) Fe3+ sites residing in the0.5–1.0 nm thick surface layers of the crystallitesand the serious reduction in symmetry is caused bythe strain induced by lattice contraction throughoutthe crystallites under the influence of the surfacetension (‘positive pressure’) of the solid. In thecase of zeolites the estimated order of magnitudeis 0.8–1.2 J/m2 (800–1200 erg/cm2), cf. [30]. Itis supposed that the effect consists mainly in theunidirectional compression of the surface layers.The Fe-ZSM-5 zeolites (at not too high Si/Fe ra-
tios) crystallize in spherulitic conglomerates of littleplatelet-like crystals, less than 0.5mm each, overgrownepitaxially fairly well in the direction of crystal axis‘c’ (vertical to [0 0 1]), but the stacking is imperfect inthe other two directions. The number of sites whichare under strain in the about 1 u.c. thick outer layersof the crystallites is estimated to be about 2–4% (oreven less) of all sites substituted by Fe3+ ions whichis in agreement with the spectra. The decrease of sig-nal intensity by heat-treatments is due to relaxationand reduction of framework Fe3+ sites.
This interpretation of the fully rhombic ESR signal(and no doubt, the approach is not generally valid) issupported by the following experimental facts. Parkerobserved in 1969 that theg= 4.27 ESR resonanceof high-spin Fe3+ depends sensitively on the parti-cle size and the aspect ratio (length/width) of kaoli-nite crystals [31]. Boldú et al. observed [32] that theoccurrence-probability of low-symmetry cf sites in-creases as the external surface increases. Hall dealtwith this theme in 1980 [5]. Ione observed that theincrease in the crystallinity of ZSM-5 phase, underotherwise identical conditions, was significantly cor-related with theg= 4.27 ESR signal intensity [33]; asimilar observation was published by Sweeney et al.[34] and others.When reading the literature on syn-theses of transition metal substituted zeolites one getsthe impression in the light of the previous thoughts
that the degree of incorporation had been far better inmany instances than the authors thought solely on thebasis of theg= 4.27 ESR line intensity, which is inreality not a reliable measure of substitution.
3.4. The signals attributed to ‘oxo’-bridged speciesand iron(III)-oxide
Common element for both type II and III sites thatthey comprise of a Fe3+-ion surrounded by four oxy-gens in Th coordinationand a Fe3+-ion having sixoxygen ligands in Oh symmetry. While in the case oftype II sites the tetrahedron and octahedron share com-mon edge (with an approximate Fe. . . Fe distance of0.27 nm), for type III sites the coordination is uncer-tain: the crystallization of the gel may lead to the for-mation of real Fe–O–Fe oxobridges (the experimentalobservation that Fe3+ cannot be exchanged by NH+
4ions [35] supports this view) with a Fe. . . Fe distanceof about 2× 0.19 = 0.38 nm, or the structure assumesother than an apex-sharing configuration, character-ized mainly by ionically bonded complex ions havingcomplete Th and Oh ligand spheres. (The Oh Fe3+is most likely a dihydroxo species, fully hydrated:[Fe(OH)2·(H2O)4]+). Both types, in particular type IIsites, where by the very small Fe. . . Fe distance strongmagnetic exchange interactions can be contemplated,are capable to distort the cubic cf symmetry. At slightdistortion the symmetry will be axial, although in thecase of type II sites the dipole-dipole and spin-spin ex-change interactions may cause an additional wideningof the ESR signal(s).
The contributions by type II and III sites to the totaliron content of the Fe-ZSM-5 sample investigated herecan be seen on the entries of Table 3 for the AS stateand for that corresponding to the specimen after thelast heat-treatment at 1073 K.
The total iron content is based on EDX spectra.The distribution of iron between framework andextra-framework sites was computed from XPS spec-tra [1] and are uncertain up to about±4 (relative) %.Reliable analytical methods permitting unequivocaldistinction between the various framework sites arenot available yet. The data given here are estimatesbased on K+ ion-exchange and presumed stabilities.These studies should contribute to this lacuna ofknowledge.

126 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
Table 3Distribution of Fe3+ between different types of sites in AS stateand after heat-treatment at 1073 K
Type I Type II Type III Sum
AS statein % of the total iron content40 14–21 46–39 100in mmol/g zeolite0.395 0.138–0.207 0.454–0.385 0.988
Type I Type II Type III Fe2O3 Sum
After heat-treatment at 1073 Kin % of the total iron content∼35 ∼5 0 ∼60 100in mmol/g zeolite∼0.346 ∼0.049 0 ∼0.593 0.988
Fig. 3. The general appearance of a powder-averaged ESR spec-trum produced by high-spin Fe3+ species in axially symmetricenvironments.
Several approaches with various types of zero field(zf) splittings and accordingly with unlike degree ofsophistication in the mathematical description havebeen investigated [36,37] to arrive eventually at ahalf-forgotten paper by Lunsford [38], who couldshow already in 1965 that the asymmetric Fe3+powder-lines nearg= 2.00, exhibiting a clearly vis-ible ‘hump’ (see Fig. 3) at slightly lower magneticfields are characteristic for powder-averaged spectraof axially symmetric Fe3+ species.
The averaged polycrystalline spectrum can be eas-ily computed. Fitting our experimental spectrum to thecomputed one by applying a software for spectrumsimulation and a non-linear parameter estimation pro-gram in order to obtain the ‘a’ values seems to bepossible. (We report on these results elsewhere).
There is no doubt that a part of our spectra (fromthe first AS sample to the last one) between a slightlylarger magnetic field interval than indicated previouslybelongs to this class of cf symmetry; in other wordsthe hump at about 0.27 T has nothing to do with ironoxide/hydroxide debris in the zeolitic voids as is gen-erally believed even today [39–41]. (It is, of course,another question whether iron oxide/hydroxide finelydispersed in amorphous alumina matrices is capableto produce resonance absorption atg= 2.3–2.1?).
The slight down-field shift, i.e. the lower limit ofspherical integration (g11) when computing the pow-der spectra may be due to the very specific behaviourof type II sites. As their heat-resistance does notexceed much that of type III sites, their destructioncauses a marked decrease of magnetic exchange lead-ing to a narrower line-shape (1Hpp = 0.06→0.035 T)aroundg= 1.98 (see Table 2). This is amazing, indeed,for the amount of dislodged Fe2O3 (which shouldcause an increase of theg= 2.3 ESR line intensityaccording accepted assignments) increases substan-tially. This means in other words that the magneti-cally coupled dioxo-bridged Fe...Fe site interactions,in type II sites with energies exceeding 100 cm−1
(8.36 J/mol) or even attaining the magnitude of 300cm−1 (25.08 J/mol) found by Bleany and Bowersand others in copper acetate [42], are replaced bythe weaker (|J|≤ 100 cm−1, vide infra) interactionsof Fe2O3 clusters/particles ejected from the latticeduring the treatments. In spite of its ‘weakness’,magnetic coupling is clearly seen from the fact thatCurie’s law is not obeyed (in Table 2Irel (l.n.t.)/Irel(r.t.)� 3.87). This observation supports the existenceof type II sites in our Fe-ZSM-5 sample.
The faulty assignment of the ESR feature atg= 2.3–2.1 is still more clearly seen when we knowthat Fe2O3 produced during the heat-treatment mainlyfrom both type II and III sites in ultrafine (‘molec-ular’) particle size produces symmetrical ESR linescentered aroundg= 2.00 [21,43–45]. The gradual in-crease of the particle size (if it is actually occurring)brings about a slow development of asymmetry and

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 127
Fig. 4. Ferromagnetic resonance absorption atg= 2.00, after Itonet al. [21].
broadening of the line-shape, then reaching a criticaldiameter (dcr) at about
dcr ∼= 2 + 10−7 T 1/3 (wheredcr is in cm, T in K)
for a-Fe2O3 (1)
anddcr ∼= 2.5 × 10−7 T 1/3 for a-FeOOH
above the blocking temperature (TB) the previoussuperantiferromagnetic/superparamagnetic behaviourturns to 100% ferro-/ferrimagnetism [46] and in paral-lel a great deal of physical properties (from saturationmagnetization to ESR-, or more precisely: ferro-magnetic resonance, FMR, -absorption etc.) changeprofoundly. If crystalline, this magnetically orderedphase presents itself by an abrupt and sharp FMRabsorption atg= 2.00, as shown in Fig. 4 after Iton etal. [21], and at the same time in the57Fe Mössbauerspectrum a six-line magnetic hyperfine splitting canbe observed.
Looking at the measured/estimated material bal-ance (vide supra) in the final state, the ejected amountof Fe2O3 is 0.593 mmol/g zeolite. A silicalite ofMFI structure possessing ideal composition and rep-resented by the formula: Si96O192 might consistof 6.81× 1019 unit cells/g, thus, after the 1073 Kheat-treatment it contains four, maximum five Fe2O3units/u.c. four Fe2O3 units are just enough to buildup two units cells ofa-Fe2O3, exhibiting rhombic(corundum) structure with the space-group D6
3d − R3c(a= 0.542 nm,α = 55
◦17), still, we are convinced that
the ejected Fe2O3 in the zeolitic voids is amorphous,
irrespective of whether it originates from type II ortype III sites. This belief is based among others onthe well-known fact that even when the magneticexchange interactions throughout the whole latticeof the diamagnetic host are not too large for Fe2O3of extremely low dispersity, they are strong enoughto impede any movement of the ejected Fe2O3 units,keeping them separated. The situation changes dra-matically when the samples are concurrently steamed:the hydrated Fe2O3 becomes extraneous in the fairlyhydrophobic SiO2 lattice and migrates onto the sur-face of the zeolite thereby creating possibility forparticle growth. Similar observations, concerning dis-persity in particular, are reported e.g. in [19,47] andelsewhere.
Speculating on the role Fe complexes, in particu-lar dioxo- or oxobridged Fe...Fe pairs in zeolites mayplay in biomimetic oxidations or in denox reactions,one cannot get rid of the idea that the weakening ofthe Fe–O bonds, which results eventually in the for-mation of the so calleda-oxygen [48], or even in Fe3+autoreduction, is mainly due to the extreme large elec-tron affinity of Fe3+ ions. No prophetic capabilities areneeded to predict an increased interest for ferric activesites in zeolitic matrices in the near future.It is worth-while now to see what kind of additional knowledgecan be obtained from the57Fe Mössbauer spectra.
3.5. Mössbauer studies
3.5.1. Changes in the coordination states — speciesof iron distinguished by evacuation
The parameters extracted from spectra recorded onthe as received samples are similar; no characteris-tic differences are found. In general, the isomer shiftvalues are above 0.3 mm/s the QS values are below1.0 mm/s (r.t. data), thus, based on these values oc-tahedral coordination might be assigned for all ironspecies, hardly any effect of the different calcinationtemperatures can be revealed (for the 973 K calcinedsample see Fig. 1a).
It might also be mentioned that for the fitting ofthese spectra different line widths were allowed forthe lines of doublets, but the intensities (areas) in thetwo corresponding lines were kept still at the samevalue. (At all the other fits both the intensities and linewidths were constrained to the same value for the cor-

128 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
responding two lines of a doublet). As an explanationfor the different line widths it might be suggested thatin the dinuclear iron-iron centers the two iron ions arebridged through (hydr)oxo bridges, which may trans-fer a certain magnetic coupling and the difference inthe relaxation times for the two|1/2 >↔ |1/2 > andfor the |3/2>↔ |1/2> transitions may result in differ-ent line widths (but retaining the same intensity forthe two lines [49]).
In contrast, expressed differences can be observedin the spectra recorded after evacuation at 573 K. Thefirst important feature is the appearance of compo-nents with IS< 0.3 mm/s and QS > 1.2 mm/s. Thesepairs of IS and QS values clearly indicate the presenceof tetrahedral Fe3+. In analogy to previous studies[17,50] the component of larger QS (1.8–1.9) can beattributed to the H-form (type I/a) structure (denotedas Fe3+
Td,a in Table 1). The other tetrahedral componentwith slightly larger isomer shift (0.25< IS< 0.27) andsmaller QS (1.2–1.5) can also be assigned to frame-work substituted iron ions, present in structures oftypes I, II and III (Fe3+
Td.b).The second important feature is the appearance of
Fe2+ components. Since the autoreduction is char-acteristic also for extra-framework ions, this com-ponent can probably be attributed to the counterpartextra-framework ions in structures of type III. How-ever, the presence of type II structure cannot beexcluded either; the ISFe2+ < 1.0 mm/s value may in-dicate intervalent states in dinuclear Fe–O–Fe pairsof structure II [49]. (For illustration the spectrum ofthe 973 K calcined sample is shown in Fig. 1b).
The samples exhibit distinct differences after evac-uation depending on the calcination temperature.Practically the 773 and 873 K calcinations result inthe same effects, the relative contributions of ironspecies are similar in these two spectra; the predom-inant species is Fe3+
Td . Performing a low-temperature(140 K) and a room temperature measurement invacuum the Debye temperature can be estimatedfor Fe3+
Td . The (A300 K − A140 K)/1T slope is ca.−5× 10−4 K−1, corresponding to2D,Td ≈ 450 K,indicative of a strong, ionic bond in Th environment.
The 973 K treatment results in large extent of ironremoval and appearence of octahedral Fe3+
Oh.a, proba-bly (at least partly) in oxidic environment (1/3 spec-tral area). This component becomes prevailing afterthe 1073 K calcination. In correspondence, as for the
framework substituted part, the contribution of typeI/a structure (H-form) hardly changes in samples cal-cined at 773, 873 and 973 K. Instead, upon calcina-tion at 973 K the contribution of Fe3+
Td.b component de-creases considerably, indicating ejection of substantialamounts of Fe3+ residing in type I and II environmentsthereby greatly reducing iron–iron interactions.
The calcination at 1073 K resulted in the removal ofiron from the framework in the largest extent. ThoughXPS data substantiate the presence of about 40% ofthe total iron content in Th coordination further on[1], the presence of Fe3+
Td components could not be de-tected any more in the Mössbauer spectrum. Instead,the overwhelming part of iron is located in Oh envi-ronment. Besides the Fe3+
Oh.a another octahedral com-
ponent Fe3+Oh,b also appears with a small QS value
(0.65 mm/s). The removed iron is probably accomo-dated still in the ZSM-5 framework, as high-dispersionoxide, similarly as reported for Y zeolite [47]. Thissuggestion is verified by the lack of magnetic split-ting in our spectra; i.e. small magnetic particles mayexhibit paramagnetic resonance spectra provided theircharacteristic size exceeds a threshold value. At l.n.temperature the threshold value for the appearance ofthe magnetic splitting in the spectra for iron oxide is∼2.9 nm [51,52]. Thus, the actual size of the removediron in oxidic particles did not attain this thresholdvalue in detectable amounts in any of our samples.
In the sample calcined at 1073 an approximate valuecan also be obtained for the Debye temperature ofthe Fe3+
Oh sites, since the prevailing coordination statein this sample is octahedral. The (A300 K − A77 K)/1Tslope is−8.7× 10−4 K−1, corresponding to a Debyetemperature:2D,Oh = 320 K. This value is consider-ably smaller than that found for the Fe3+
Td sites. Thisis in full agreement with the considerations above,suggesting that the octahedral iron oxide is locatedin the voids in form of highly dispersed particles andis not attached strongly to the framework. (For largea-Fe2O3 and a-FeOOH particles2D ∼= 500 K is re-ported [18]).
3.6. Reduction/oxidation processes — relations tocatalytic properties
The next step in the series of treatments followingthe evacuation was a reducing treatment in hydrogenat 573 K. After the treatment the samples were cooled

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 129
down immediately to 77 K (within ca. 1/2 h) and thein situ spectra were recorded. In the sample, calcinedat the highest temperature, at 1073 K, only a smallamount of Fe2+ (RI = 17%) was detected. Thus, theoverwhelming part of iron cannot be reduced to Fe2+state at 1073 K, the high temperature calcination sta-bilized this component in form of a highly dispersedoxidic phase. However, dinuclear Fe–O–Fe pairs areprobably still present in a minor amount, the com-ponent characterized by IS = 0.97 and QS = 1.98 mm/scan probably be attributed to intervalence oxidationstates [53].
In the other three samples (calcined at 773, 873 and973 K) the effect of reduction was more pronounced,the amount of Fe2+ component increased consider-ably (to RI values 31, 26 and 37%, in the 77 K spectra,respectively). Surprisingly, the presence of Fe2+ com-ponent in a large portion is temporary; by increasingthe temperature of measurement from 77 to 300 K thecontribution of Fe2+ decreased considerably, even be-low the RI values recorded after evacuation. (Thus, asa net effect of the hydrogen treatment, an increase ofthe Fe3+ contribution, i.e. not reduction but even theopposite process, a slight degree of oxidation couldbe established when comparing only the room temper-ature spectra of the evacuated and hydrogen treatedsamples; cf. Fig. 1c, d).
In order to obtain further information on this net‘oxidation’ process, additional measurements wereperformed on the sample calcined at 973 K. First, thesample was measured again at 77 K; cooling alone(in hydrogen) was insufficient to restore the reducedstate: almost the same low percentages were foundat both 77 and 300 K (6% and 7% RI; Fig. 1e). Torevert the oxidation process the hydrogen treatmentwas repeated at 573 K, and the sample was measuredagain at 77 K (Fig. 1f).
This treatment resulted in the restoration of the pre-vious states, the corresponding pairs of spectra anddata are almost the same (cf. Fig. 1c, f and Table 1).At the end, all the samples were evacuated again, (ex-cept the sample calcined at 773 K) and found that thespectra were similar to those recorded after the firstevacuation, i.e. the distribution of iron did not changesignificantly among the various species, proving thatthe samples are stable, no considerable removal of ironhas taken place from the framework during the appliedevacuation and reduction treatments; (Fig. 1g and b).
The most noticeable feature is the spontaneous oxi-dation of Fe2+ species while maintaining the hydrogenatmosphere by the simple raise of the measuring tem-perature from 77 to 300 K. A similar phenomenon wasobserved in Fe-ferrierite zeolite recently; this ferrisil-icate exhibited excellent activity for mild oxidations(oxidation ofn-hexane and hydroxylation of phenol).For explanation simultaneous development of variousredox processes was suggested taking place on bothFe3+ ions of the Feframework–O–Feextra-framework pairs[54].
The present series of papers may help to further elu-cidate these very interesting phenomena which mightbe of great importance from the point of view ofbiomimetic oxidations and automotive emission con-trol. As mentioned previously, the (partial) loss of neg-ative charge on the oxygen(s) between dioxo- or oxo-bridged Feframework... Feextra-framework pairs is a pre-requisite to the formation ofa-oxygen, being activein selective oxidations. The possibility of reoxidation(mainly) of type II Fe2+ sites is also necessary to re-peat the cycle.
The Mössbauer spectra reveal the presence oftwo types of Fe2+ sites: Fe2+
a and Fe2+b after re-
duction. The former one has small IS and QS val-ues. The IS77 K ∼= 1.1 mm/s is a relatively smallvalue, it may indicate the existence of an interva-lence state (+2< Fe2+
a < +3). The symmetry aroundthis component is more distorted than around Fe2+
b(QSFe2+
a< QSFe2+
b; in the case of high-spin 3d6 Fe2+
ions, the greater is the symmetry the larger is theQS - opposite to conditions of high-spin Fe3+ [55]).The Fe2+
b component is most probably the counte-rion in type III structure since a more symmetric(oxy)hydrate sphere of ligands can be formed aroundit. The signal of the Fe2+
a component is probablyoriginated from the extra-framework component oftype II structure bonded to the framework ferric ionby a di(hydr)oxo bridge exhibiting the smaller sym-metry and possibly the intervalence oxidation state,too. It can be surmized that this type II site Fe3+
a(or Fe2+
a ) ion is the genuine participant in the re-versible redox process mentioned previously: whenin Fe2+
a state, it will be reoxidized easily upon in-creasing the (measuring) temperature from 77 to300 K (see Table 1). The Fe3+
Td framework compo-nent(s) of the Fe...Fe twin(s) exhibit larger symme-tries.

130 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
As a further proof of this concept it should be men-tioned here that between corner- or edge sharing tetra-hedra thermally activited electron delocalization (ED)may take place amidst Fe3+. . . Fe2+ pairs at about300–400 K provided the distances are short. The crit-ical Fe. . . Fe distance seems to be 0.27–0.33 nm [53],thus in the ED only type II sites may be involved (ofcourse, after partial reduction). Burns explicitly men-tions the requirement that ED needs ferromagneticcoupling between adjacent Fe sites, which is again anindirect proof for the existence of type II sites.
The strange redox behaviour mentioned previouslyis due to the reaction step
Fe3+–O2−–Fe3+ + H2
reduction(heating)�
oxidation(cooling)2Fe2+ + H2O
(2)
since the1Hr ∼= 96 kJ/mol increase of the reaction en-thalpy is overcompensated by the negative (−T1Sr)product (1Sr ∼= 0.188 kJ/mol deg.) already at about510 K.
At ambient temperature1Gr (300 K)∼= 39.6 kJ/mol,thus for the reversed reaction:1Gr =−39.6 kJ/mol,i.e. it becomes preferred at room temperature. Thefast cooling to 77 K after reduction is equivalent to‘freezing in’ the reversed step of Eq. (2).
The autoreduction
Fe3+–O2−–Fe3+autoreduction(heating)�
reoxidation(cooling)2Fe2+ + O (3)
seems to be similar to reaction (2). Actually it is, ifatomic (and unfortunately not molecular) or ‘weakly’bonded oxygen is available, like in H2O, N2Oand other momentarily unknown reactants. In thebiomimetic selective oxidations the virtually ‘atomic’oxygen (denoted ‘a-oxygen’; vide supra) can be uti-lized according to Eq. (3) when the reactions (2) and(3) are coupled in cycle. Instead of H2O the N2Oseems to be a better oxidant requiring a lower tem-perature where the1Gr Gibbs’ free energies for bothreactions, or for the overall reaction, including theoxidation step too, become(s) negative.
Eventually the two coupled steps are very sim-ilar (as shown, also from thermodynamic point ofview) to the Mars/van Krevelen mechanism of selec-tive oxidation, the main difference being that thesetwin Fe3+. . . Fe3+ sites, whoseexact identification
fails yet, do not belong to a continuous bulk oxi-dic phase. It should be mentioned, however, that the(Cr2O3-promoted) bulk-phase catalysts (based onFe2O3 a few decades before) are capable to reproduceexactly the same steps in the water-gas shift reactioncycle, with H2O and CO as reactants, at about 520 K.It is also true, that the Fe2O3/MoO3 mixed oxidecatalysts, as bulk phase materials, are well knownand utilized in the selective oxidation of methanolto formaldehyde. Here the molecular oxygen will beactivated by the Mo component.
At the end of this chapter mention should be madeabout another important consequence of the extremeelectron affinity of ferric ions. The Mössbauer iso-mer shift, IS, depends on the (virtual) electron den-sity at the atomic nucleus. The shorter Fe3+–O dis-tance in Th, or the larger in Oh coordination mani-fest themselves in the respective ISTh < ISOh values.Shorter distance means reduced nominal charge (cf.Beran, [28]) on the ferric ion, which brings about anupward shift (in energy) of the electronic core levels.In other words, in XPS spectroscopy the core levelphoto-electrons from Th framework ferric ions will beemitted at lower energy than those originating fromOh extra-framework iron ions, proving that the assign-ments with respect of the order of the Th and Oh pho-toelectron peaks have been done correctly in the pub-lication [1].
As the Ti4+–O bond length in Th coordination isshorter than that in Oh coordination (∼0.171 versus∼0.210 nm), the inevitable result is a decrease of thetetrahedral Ti 2p electron binding energies. The se-quence of the respective Ti4+ XPS peaks is also anal-ogous to Fe3+.
3.7. Kinetic studies
As discussed copiously in the 1st part of this pa-per [1], the Si/Ti = 29.8 ratio of the AS Ti-ZSM-5zeolite sample means 3.12 Ti4+ ions per u. c. of thezeolite on the average. From these only 53.5%, i. e.1.67/u. c. are siting in the framework, 1.42/u. c. arein extra-framework positions from the very begin-ning. The reason of the incomplete substitution canbe explained in similar terms as discussed there inrelation to Fe3+. Though the experimental data for Tiare relatively scarce, it is agreed upon that in ‘not tooconcentrated’ sulfuric acid solutions –Ti–O–Ti–O–

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 131
Table 4Conversion and cyclohexanone selectivity in the oxidation of cyclohexane on Ti-ZSM-5 in dependence of the temperature and heat-treatment
Catalyst Solvent Time of Conversion (%) Cyclohexanonereaction (h) selectivity (%)
Ti-ZSM-5 873 K Acetone 1.5 2.6 30.44 8.4 41.96 11.0 51.08 15.7 56.0
12 40.0 61.0
Ti-ZSM-5 1073 K Acetone 1.5 9.1 25.04 12.5 32.06 18.0 43.18 24.7 47.0
12 49.1 59.6
chains of variable lengths exist, where each Ti iscoordinated approximately octahedrally to two bridg-ing oxygen atoms and a sulfate anion, or watermolecule(s) [56]. There is good reason to believe thatin the weakly alkaline milieu of the synthesis slurryonly one Ti4+ will be incorporated in the lattice, theremaining part of the chain (which may consist of 1–2additional elements) is protruding into the zeoliticvoid(s).
The heat-treatment brings about dislodgement offramework Ti, here too, however, as FT-IR spectra,taken in the structure sensitive domain, attest, the localdegradation of the zeolitic lattice at the framework sitetakes place in sharp contrast to what we have seen inthe case of Fe-ZSM-5.
Let us mention first a few experimental facts1. Whereas no indication of any ‘finger-print’ band
could be found for the heat-treated Fe-ZSM-5samples in the mid-IR region at about 960 cm−1,the Ti-ZSM-5 samples exhibited the well-knownfeature at 965.4 cm−1 , the intensity of whichseemingly increased with the temperature of treat-ments (cf. Fig. 11 in [1]).
2. According to Fig. 10 in [1] the amount of ejectedTi (in Oh coordination) increases in a nearly linearfashion as the temperature of treatments increases.
3. Comparing the catalytic activities of twoTi-ZSM-5 samples heat-treated at 873 (sample I)and 1073 K (sample II) in the selective oxidationof cyclohexane using H2O2 as oxidant, Table 4convincingly shows that under otherwise identi-cal conditions the conversion versus time curvefor sample II is situated above that for sample I.
The products of oxidation are cyclohexanol, cy-clohexanone, epoxide and acetylcyclohexane.
4. Similarly, at a pre-set time of reaction (4 h) then-hexane conversion increases with the tempera-ture of treatments.
Here the catalytic reaction leads to the forma-tion of alcohols and ketones (2- and 3-hexanol and2- and 3-hexanone).
From these facts a very important information canimmediately be retrieved: the selective oxidation ac-tivity is due to a Ti species which is not part, or is notfully part of the Ti-ZSM-5 lattice. This species will begenerated during the template removal by calcination(and further intentional heat-treatments).
The parallelism observable between ejected amountof Ti, catalytic activity and intensity of the≈960 cm−1
‘finger-print’ band can be rationalized in terms of theejection process itself, producing an extra-frameworkTi species and leaving behind a specific defect site.
This paper deals with the structural, catalytic andother experimentally perceptible consequences ofheat-treatments, thus, by various reasons, it is not jus-tified to give a detailed overview here about ‘the stateof the art’, published since 1983 on various experi-mental and theoretical aspects of the theme. Nonethe-less, we, too, elaborated a working hypothesis whichseems to be interesting enough and so it is worth tocite just a few additional arguments, which maybesupport the hypothesis, or are at variance with it.
We think so that, contrary to Fe-ZSM-5, duringheat-treatments the dislodgement of the substituting Tiis not complete: one Ti–O–Si link to the lattice remains(certainly, this link will be disrupted fully at higher

132 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
temperatures, leading to the formation of inactive ti-tanium(IV) oxide), concomitantly a characteristic de-fect site is produced through formation of a distortedsquare planar arrangement of four Si–O–Si– bonds, asshown below on a detail of the Ti-ZSM-5 structure:
The ‘square-planar’ nests are actually ‘emptynests’, observed first by McDaniel and Maher inthe process of ultrastabilisation [57]. We think, wehave observed first their intensive lattice vibration at930 cm−1 during dealumination of mordenite by nitro-syl chloride and other dealuminating agents [58–61].In the case of Ti-ZSM-5 the IR band they produce ap-pears near to 960 cm−1. Later, extending the studiesof a long row of very capable authors commencingwith Barrer [62] and ending by Karge [63], using1H NMR spectroscopy and derivatography we couldshow that empty nests are produced from thermallyunstable hydrated nests by dehydroxylation [64]. Alltogether we published about 10–12 papers on thistopic.
Others, Bellussi, Esposito, Taramasso etc. in Mi-lan and Corma, Camblor, Martinez etc. in Valencia(Spain) approached the theme from the direction ofsynthesis and application of Ti zeolites in the liquidphase selective oxidation. Camblor et al. state in [65]correctly that the 960 cm−1 ‘finger-print’ IR band can-not be associated with a metal-oxygen bond, becausewith slight modification the same feature appears inthe spectra of Ti, V and other zeolites or even in those
of crushed silica. They recognized also that the inten-sity of the respective band will be enhanced after cal-cination. To our opinion the reason is dehydroxylationof the hydrated nests.
It has been found by Bellussi et al. [66] and Cambloret al. [67] that the 960 cm−1 band shows no hydrogenisotopic effect (the empty nests are ‘empty’, indeed,without H), but are sensitive to H217O and H2
18Oexchange.
We know that the ‘empty nests’ can with some re-versibility be dehydrated and even rehydrated:
Si–16O–Si+ H218O → Si–16OH + Si–18OH
thus, in one step only 50% exchange can be at-tained, provided the loss of water possessing differentlabeling takes place with equal probability duringdehydroxylation. The formation of new -Si-18O-Si-linkages generate additional chemical shifts in the29Si MAS NMR spectrum, as Notary refers to it in[68].
As concerns active sites, our working hypothesisdeviates in some details from that Corma et al. putforward in [69]. They keep the Ti in the framework(in Th coordination) and assume the formation of astable five-membered cyclic structure (species I), inwhich a protic solvent molecule, ROH, coordinates tothe Ti center and stabilizes the Ti-peroxo complex via

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 133
hydrogen bonding:
If the catalytically active sites are ‘half-ejected’Si–O–Ti groups, they should be capable to produce‘a-oxygen’ (O) from H2O2, and the role protic sol-vents may play in the oxidation activity of thesecatalysts, as Corma et al. stipulate in [69], can beexpressed by the following formalism:
The ‘a-oxygen’ enters the molecule to be oxidized,and the active site regenerates itself by virtue of push-ing off the water by the hydrogen peroxide.
The two approaches, when we disregard the bond-ing order of Ti, differ only in subtle respects and itwill not be an easy task to prove the validity of eithersuggestion. The more is this true, if we consider thehuge amount of experimental data and the conscious-ness accumulated since the first publications on thistopic. Currently we are not prepared to the task of anykind of jurisdiction, as the prime consideration is hereto pursue the tenet advanced in the Introduction: toshow that uncontrolled calcination and overheating ofTi-ZSM-5 or other catalysts might seriously influenceboth quality and quantity of active sites responsible forthe selective oxidation of a great variety of reactants.
We hope that we could convincingly demonstrate in[1] and in this paper that as synthesized Ti-ZSM-5 cat-alysts should be calcined by the greatest care: 773 Kis above the temperature limit of (the least stable) Tiframework-site stability, thus, calcination is always ac-companied by ejection of Ti from the framework. If thecatalytic activity is due to the number of ‘half-ejected’Ti species, as we believe, then per unit mass the num-ber of these active sites reaches maximum as the tem-perature of the treatments increases. At still higher
temperatures the ‘half-ejection’ turns to ‘full-ejection’causing loss of activity. From all this follows thatunimpaired tetrahedral Ti (and other) framework sitesare too stable under the conditions of liquid phase se-lective oxidation to be able to play the role of activesites.
4. Conclusions
1. X-band ESR spectra of isomorphously substitutedFe-ZSM-5 zeolite specimens have proven the ex-istence of type I framework sites in cubic sym-metry (g values near to 2.00; Debye tempera-ture2D ∼= 450 K); type II and III binuclear Fe...Fedi(hydr)oxo-bridged Fe species in both Th and Ohoxygen environments, exhibiting axial symmetry(g values of the powder averaged subspectra ap-pear in the 2.45–1.98 interval), partly in strongmagnetic coupling; and fully rhombic Th and Ohcoordinated Fe3+ sites in the surface layers of thecrystallites, strained under the influence of solidsurface tension (signal atg= 4.27).
Increasingly severe heat-treatments cause dis-lodgement of Fe2O3 (Fe...Fe distance∼0.38 nm;2D ∼= 320 K) of high dispersity with concomitantannihilation of the strong magnetic coupling oftype II sites (Fe...Fe distance∼0.27 nm); instead,the weaker magnetic interaction of ejected Fe2O3can be observed in the spectra.
2. The lack of magnetic hyperfine splitting in theMössbauer spectra, even at 77 K, sets the upperlimit of particle size of ejected Fe2O3 to about 3nm. The observed redox behaviour validates thepresence of type II and III sites. Type III sitescan be made responsible for the Fe3+→Fe2+ au-toreduction, observable after heating in neutralmedium. Development and temporary stabiliza-tion of intervalent Fe states are also detected inthe Mössbauer spectra and were ascribed to typeII dimeric sites.
3. Even when the as synthesized (AS) forms of Fe-and Ti-ZSM-5 show a few similarities (only par-tial substitution of Si by Fe and Ti, respectively,due to the analogous ‘solution chemistry’, dur-ing the synthesis; ejection in very similar fashionof Fe2O3 and TiO2 when burning off the tem-plate etc.), the mechanism of lattice damage and

134 P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135
thereby the development of catalytically activesites upon heating seems to be completely differ-ent: while the ejected Fe2O3 loses the link to thelattice, the Ti is (at not too high temperatures)only ‘half-ejected’ and retains some connection tothe lattice via a Ti–O–Si bond. Parallelism seemsto be existing between the number of active sites,these ‘half-ejected’ species and the intensity ofthe∼960 cm−1 IR band.
The results of ESR and Mössbauer studies confirmour previous observation (see in [1]) that (intentionalor accidental) heat treatment exerts major influence onboth the number and chemical quality of active sites,therefore, this operation should be carried out with thegreatest care.
Acknowledgements
The authors: P. Fejes, K. Lázár and H. Halász ac-knowledge herewith the grants by the OTKA Com-mission under No.-s T 029463, T 021131, T 016338,respectively. Thanks are due also to Mrs. Barna forthe synthesis work and precise typing.
References
[1] P. Fejes, J.B. Nagy, J. Halász, A. Oszkó, J. Appl. Catal. A:General 175 (1998) 89.
[2] L.N. Mulay, P.W. Selwood, J. Am. Chem. Soc. 77 (1955)2693.
[3] B.D. Mc Nicol, G.T. Pott, J. Catal. 25 (1972) 223.[4] K. Lázár, R. Fricke, H. Kosslick, J. Cejka, G. Vorbeck, A.M.
-Szeleczky, Stud. Surf. Sci. Catal. 94 (1995) 219.[5] P.L. Hall, Clay Miner. 15 (1980) 321.[6] N.P. Evmiridis, Inorg. Chem. 25 (1986) 4362.[7] A.V. Kucherov, A.A. Slinkin, Zeolites 8 (1988) 110.[8] H. Karge, Stud. Surf. Sci. Catal. 105 (1997) 1901.[9] P. Fejes, J.B. Nagy, K. Kovács, Gy. Vankó, Appl. Catal. A:
General 145 (1996) 155.[10] H. Robson, Microporous and Mesoporous Mater. 22 (1998)
632.[11] G. Bellussi, A. Carati, M.G. Clerici, A. Esposito, Proc. 5th
Symp. on Sci. Bases for Prep. of Heter. Catal., Louvain laNeuve, Belgium, 1990, p. 201.
[12] G. Bellussi, G. Maddinelli, A. Carati, A. Gevasini, R. Millini,9th Zeolite Conf., Montreal, Canada, 1992, Poster A-8.
[13] J.E. Wertz, J.R. Bolton, Electron Spin Resonance: ElementaryTheory and Practical Application, McGraw-Hill, New York,1972.
[14] K. Lázár, K. Matusek, J. Mink, S. Dobos, L. Guczi, A.Vizi-Orosz, L. Markó, W.M. Reiff, J. Catal. 87 (1984) 163.
[15] R.G. Burns, Hyperfine Interactions 91 (1994) 739.[16] A. Meagher, V. Nair, R. Szostak, Zeolites 8 (1988) 3.[17] S. Sivasanker, K. Lázár, J. Catal. 147 (1994) 207.[18] J.W. Niemantsverdriet, A.M. van der Kraan, W.N. Delgass,
J. Catal. 89 (1984) 138.[19] B.A. Goodman, Clay Miner. 13 (1978) 351.[20] E.G. Derouane, M. Mestdagh, L. Vielvoye, J. Catal. 33 (1974)
169.[21] L.E. Iton, R.B. Beal, D.T. Hodul, J. Mol. Catal. 21 (1983)
151.[22] W.J. Ball, J. Dwyer, A.A. Garforth, W.J. Smith, Proc. 7th Int.
Conf. on Zeolites, Tokyo, 1986, p. 137.[23] M.L.S. Correa, M. Wallau, U. Schuchardt, Stud. Surf. Sci.
Catal. 105 (1997) 277.[24] D. Goldfarb, M. Bernardo, K.G. Strohmaier, D.E.W. Vaughan,
H. Thomann, J. Am. Chem. Soc. 116 (1994) 6344.[25] D.H. Lin, G. Coudurier, J.C. Vedrine, in: P.A. Jacobs, R.A.
van Santen (Eds.), Zeolites: Facts, Figures, Future, Elsevier,Amsterdam, 1984, p. 1431.
[26] P. Ratnasamy, R. Kumar, Catal. Today 9 (1991) 328.[27] B. Bleany, K.W.H. Stevens, Progr. Phys. 16 (1953) 137.[28] S. Beran, P. Jiru, B. Wichterlova, Zeolites 2 (1982) 252.[29] R.E. Meads, P.J. Malden, Clay Miner. 10 (1975) 313.[30] G.A. Somorjai, Introduction to Surface Chemistry and
Catalysis, Wiley, New York, 1994, p. 271.[31] T.W. Parker, Clay Miner. 8 (1969) 135.[32] J.L. Boldu, E. Munoz, Y. Chen, M.M. Abraham, J. Chem.
Phys. 80 (1984) 574.[33] K.G. Ione, L.A. Vostrikova, V.M. Mastikhin, J. Mol. Catal.
31 (1985) 355.[34] W.V. Sweeney, D. Coucouvanis, R.E. Coffman, J. Chem.
Phys. 59 (1973) 369.[35] S. Kaliaguine, G. Lemay, A. Adnot, S. Burelle, Zeolites 10
(1990) 559.[36] B. Wichterlova, J. Nováková, L. Kubelková, P. Mikusik,
Zeolites 5 (1985) 21.[37] G. Calis, P. Treriken, E. de Boer, A. Swofs, M.A. Hefni,
Zeolites 7 (1987) 319.[38] J.H. Lunsford, J. Chem. Phys. 42 (1965) 2617.[39] B. Wichterlova, Zeolites 1 (1981) 181.[40] L.M. Kustov, V.B. Kazansky, P. Ratnasamy, Zeolites 7 (1987)
79.[41] P.N. Joshi, S.V. Avate, V.P. Shiralkar, J. Phys. Chem. 97
(1993) 9749.[42] B. Bleaney, K. Stevens, Progr. Phys. 16 (1953) 153.[43] L.E. Iton, R.B. Beal, P.J. Hanort, J. Mol. Catal. 27 (1984)
95.[44] H. M. Ziethen, A.X. Trautwein, H. Winkler, F. Schmidt, in:
P.A. Jacobs, R.A. van Santen (Eds.), Zeolites: Facts, Figures,Future, Elsevier, Amsterdam, 1989, p. 1043.
[45] T. Inui, H. Matsuda, O. Yamase, H. Nagata, K. Fukuda, T.Ukawa, A. Miyamoto, J. Catal. 98 (1986) 491.
[46] I.P. Suzdalev, Soviet Phys. Solid State 12 (1970) 775.[47] Y. Okamoto, H. Kihuta, Y. Ohto, S. Nasu, O. Terasaki, Stud.
Surf. Sci. Catal. 105 (1997) 2051.[48] G.I. Panov, V.I. Sobolev, K.A. Dubkov, A.S. Kharitonov, Stud.
Surf. Sci. Catal. 101 (1996) 493.

P. Fejes et al. / Applied Catalysis A: General 190 (2000) 117–135 135
[49] M. Blume, Phys. Rev. Lett. 14 (1965) 96.[50] K. Lázár, G. Borbély, H. Beyer, Zeolites 11 (1991) 214.[51] H. Ziethen, A.X. Trautwein, Stud. Surf. Sci. Catal. 46 (1989)
789.[52] J.L. Dormann, Rev. Phys. Appl. 16 (1981) 275.[53] R.G. Burns, in: K. Prassides (Ed.), Mixed Valency Systems:
Applications in Chemistry, Physics and Biology, KluwerAcademic Publishres, Dordrecht, 1991, p. 175.
[54] K. Lázár, G. Lejeune, R.K. Ahedi, S.S. Shevade, A.N.Kotasthane, J. Phys. Chem. B, in press.
[55] N.N. Greenwood, T.G. Gibb, Mössbauer Spectroscopy,Chapman & Hall, London, 1971.
[56] F.A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry,Wiley, New York, 1988, p. 656.
[57] C.V. Mc Daniel, P.K. Maher, in: J.A. Rabo (Ed.), ZeoliteChemistry and Catalysis, American Chemical Society,Washington, DC, 1976, p. 312.
[58] P. Fejes, I. Hannus, I. Kiricsi, Zeolites 4 (1984) 73.[59] P. Fejes, I. Kiricsi, I. Hannus, Gy. Schöbel, Stud. Surf. Sci.
Catal. 24 (1985) 263.
[60] P. Fejes, I. Kiricsi, I. Hannus, Gy. Schöbel, in: D. Kalló, Kh.M. Minachev (Eds.), Catalysis on Zeolites, Akadémiai Kiadó,Budapest, 1988, pp. 205–229.
[61] P. Fejes, I. Kiricsi, I. Hannus, I.andT. Székely, Gy. Tasi,Zeolites 9 (1989) 392.
[62] R.M. Barrer, Zeolites and Clay Minerals as Sorbentsand Molecular Sieves, Academic Press, London, 1978,Chap. 7.
[63] H.G. Karge, Z. Phys. Chem. N. F. 122 (1980) 103.[64] P. Fejes, I. Hannus, I. Kiricsi, H. Pfeifer, D. Freude, W. Oeme,
Zeolites 5 (1985) 45.[65] M.A. Camblor, A. Corma, J. Pérez-Pariente, J. Chem. Soc.,
Chem. Commun. (1993) 557.[66] G. Bellussi, A. Carati, M.G. Clerici, G. Maddinelli, R. Millini,
J. Catal. 133 (1992) 220.[67] M.A. Camblor, A. Corma, A. Martinez, J. Pérez-Pariente, J.
Chem. Soc., Chem. Commun. (1992) 589.[68] B. Notari, Adv. Catal. 41 (1996) 272–278.[69] A. Corma, P. Esteve, A. Martinez, J. Catal. 161 (1996) 11.
Copyright © 2022 FDOKUMEN




















