
Electronic spectra of substituted metal deuteroporphyrins
13
JOURNAL OF MOLECULAR SPECTROSCOPY 16, 451-463 (1965) Electronic Spectra of Substituted Metal Deuteroporphyrins WINSLOW S. CAUGHEY Department of Physiological Chemistry, The Johns Hopkins University, Baltimore, Maryland* and Monadnock Research Institute, Antrim, New Hampshire RALPH M. DEAL? Monadnock Research Institute, Antrim, New Hampshire AND CHARLES WEISS~ AND MARTIN GOUTERMAN Conant Chemical Laboratory, Harvard University, Cambridge, Massachusetts The spectra of 23 metal 2,4-substituted deuteroporphyrins IX in chloro- form solutions are given or reported in the region 650-250 ml*. Four r --f R* bands (&, B, N, L) are identified in the region, along with bands 7 and r which are associated with carbonyl and Zn substitution, respectively. Theoretical dis- cussion centers on the 390-250 rnp region. Over the past eight years one of the authors (W. C.) and his coworkers have been engaged in a systematic study of the spectra of porphyrin molecules.’ In the preceding paper (I), two of the authors (C. W. and M. G.) summarized the development of theory and presented t’he results of extensive calculations based on SCMO-PPP (Self-Consistent Molecular Orbital Pariser-Parr-Pople) theory. In this paper, we have joined to present some unpublished data and some inter- pretations dealing mainly with the region at higher energy than the extremely intense Soret absorption band (E > 100 000 liters/mole cm) that occurs near 400 rnp in all porphyrins. THEORY AS APPLIED TO EXPERIMENT The theory of porphyrin electronic states was developed to interpret the varia- tions in the visible and Soret bands which result from changes in substitution at * Present address. t Present address: Department of Chemistry, Kalamazoo College, Kalamazoo, Michigan. $ Present address: U. S. Army Engineer Research and Development Laboratories, Fort Belvoir, Virginia. 1 Other parts of this study are being prepared for publication: (a) W. S. Caughey, W. Y. Fujimoto, and J. L. York; (b) B. 1). McLees and W. S. Caughey; (c) W. S. Caughey, 451
Transcript of Electronic spectra of substituted metal deuteroporphyrins

JOURNAL OF MOLECULAR SPECTROSCOPY 16, 451-463 (1965)
Electronic Spectra of Substituted Metal Deuteroporphyrins
WINSLOW S. CAUGHEY
Department of Physiological Chemistry, The Johns Hopkins University, Baltimore, Maryland*
and Monadnock Research Institute, Antrim, New Hampshire
RALPH M. DEAL?
Monadnock Research Institute, Antrim, New Hampshire
AND
CHARLES WEISS~ AND MARTIN GOUTERMAN
Conant Chemical Laboratory, Harvard University, Cambridge, Massachusetts
The spectra of 23 metal 2,4-substituted deuteroporphyrins IX in chloro- form solutions are given or reported in the region 650-250 ml*. Four r --f R* bands (&, B, N, L) are identified in the region, along with bands 7 and r which are associated with carbonyl and Zn substitution, respectively. Theoretical dis- cussion centers on the 390-250 rnp region.
Over the past eight years one of the authors (W. C.) and his coworkers have been engaged in a systematic study of the spectra of porphyrin molecules.’ In the preceding paper (I), two of the authors (C. W. and M. G.) summarized the development of theory and presented t’he results of extensive calculations based on SCMO-PPP (Self-Consistent Molecular Orbital Pariser-Parr-Pople) theory. In this paper, we have joined to present some unpublished data and some inter- pretations dealing mainly with the region at higher energy than the extremely intense Soret absorption band (E > 100 000 liters/mole cm) that occurs near 400 rnp in all porphyrins.
THEORY AS APPLIED TO EXPERIMENT
The theory of porphyrin electronic states was developed to interpret the varia- tions in the visible and Soret bands which result from changes in substitution at
* Present address. t Present address: Department of Chemistry, Kalamazoo College, Kalamazoo, Michigan. $ Present address: U. S. Army Engineer Research and Development Laboratories, Fort
Belvoir, Virginia. 1 Other parts of this study are being prepared for publication: (a) W. S. Caughey, W. Y.
Fujimoto, and J. L. York; (b) B. 1). McLees and W. S. Caughey; (c) W. S. Caughey,
451
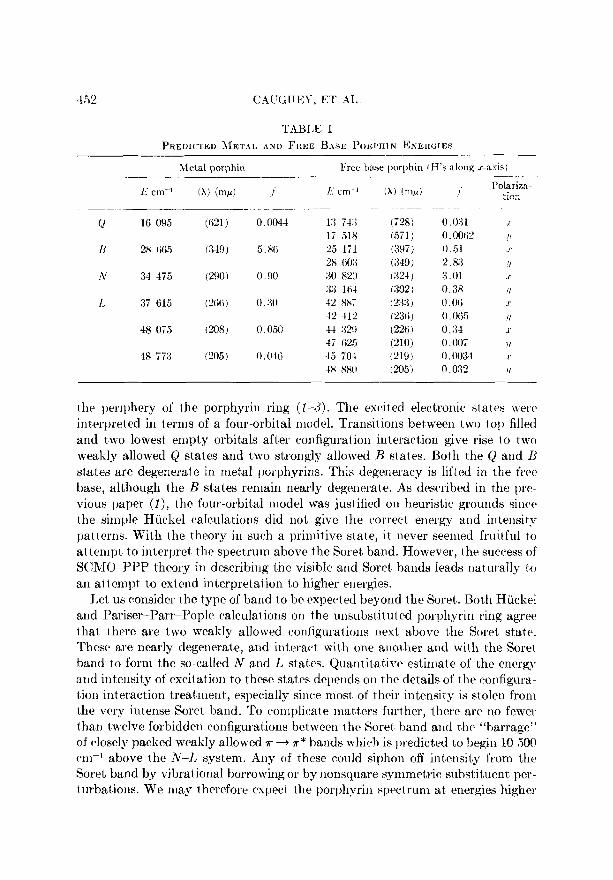
TABLE I
PREDICTED ~IETAL AND FREE BASE POWHIN ENERGIES
0 16 095
N 28 665
IV 34 475
I. 37 615
48 055
48 773
Metal porphin Free IBase porphin (H’s along x-axis)
- (621) 0.004-i 13 743
li 518
(349) 5.86 25 lil 28 603
ma) 0.00 90 821) 33 1li-l
(L’iXii 0.30 12 m; 42 412
(208) 0.050 44 329 47 625
(205) 0.04ti 45 TO-l 48 880
(A) (m/A) I
(728) 0.0:31 (571) 0. ooril’ (397) 0.51 (349) 2.83 ( :x&l ) 3.01 Go21 ) 0.38 I 233 ) o.oci (236~ 0.065 (226 ) 0.34 (210) 0.007 (219) 0.00:34 (205) 0 03” ._
I’olariza- tiorl
.r
I
!/
!I
the peril)hery of Ihe porphyrin ring (I-3). The exc*ited electronic states were interpreted in terms of a four-orbital model. Transitions between two 101) filled and two lowest, empty orbitals after configuration interaction give rise to two weakly allowed Q states and two strongly allowed I? states. Both the Q and B st,ates are degenerate in metal porphyrins. This degeneracy is lifted in t,he free base, although the B states remain nearly degenerate. As described in the pre- vious paper (I), the four-orbital model was justified on heurist,ic grounds since the simljle Hiickcl calculations did not give the correct energy and intellsit\ patterns. With the theory in such a primitivr stat,e, it never seemed fruitful to attempt to interpret the spectrum above the Soret, band. However, the success of SCM&PPP theory in describing the visible and Soret, bands leads naturally to an attempt to cxtcnd interpretation t#o higher energies.
Let us consider the type of band to be expected beyond the Soret,. Both Hiickrl and Pariser-Parr-Pople calculations on the unsubstituted porphyrin ring agree that, there are two weakly allowed configurations next above t’he Soret state. These are nearly degenerate, and interacat with one another and with the Soret band to form the so-called N and L states. Quantitative estimate of the cncrg) and intensity of excitation to these states depends on the details of the configura- tion interaction treatment, especially since most of their intensity is st,oleu from the very intense Soret band. To ~omplicat e matters further, there are 110 few-e1 t)han twelve forbidden configurations bctwccn the Soret band and the “barrage” of closely packed weakly allowed 7r + r* bands which is ljredicted t,o begin 10 500
m-1 above the N-L system. Any of these could siphon off intensit,y from the Soret band by vibrational borrowing or by nonsquare symmetric substituent per- turbations. We may therefore expect the porphyrin spectrum at energies higher

METAL DEUTEROPORPHYRINR 453
&OCH3 C&H,
(0)
kH2 kH2
kH, :H,
>k f 0 0CH3 0 OCH,
(b)
c 8OCH, d&H3
w (dl
FIG. 1. (a) Deuteroporphyrin IX dimethyl ester. (b) 2,4_Diethyldeuteroporphyrin IX (mesoporphyrin IX) dimethyl ester. (c) 2,4-Divinyldeuteroporphyrin IX (protoporphyrin IX) dimethyl ester. (d) 2,4-Diacetyldeuteroporphyrin IX dimethyl ester.
than that of bhe Soret band to be a rather confused melange of weak electroni- cally allowed and electronically forbidden but vibronically allowed transitions. We shall see that t,he experimental spectra are qualitatively consistent with this prediction, a not terribly useful confirmation of the theory.
Table I lists the energies and intensit#ies for t,he dozen lowest allowed excited states for unsubstituted porphin. The calculation made use of “traditional” parameters (1) and cont,ained 29 configurations. The left column shows Ddh porphin, wit,h its systematic degeneracy between x and y bands. It should be noted that this “metal” porphin takes 110 account of the electronegativity of any specific metal. It merely symmetrizes the pyrrole and pyridine nitrogen atoms of the free base. An earlier analysis (3) suggested that Cu most nearly corresponds to this metal calculation.
The right column of Table I shows free base porphin wit’h t’he protons on the m-axis. The .r-y degeneracy of the metal is now lifted, and in general the z polar-

45‘4 CAUGHEY, ET AL
06 -
7
,
/
/
COBALT
B
NICKEL IQ
PALLADIUM
250 300 350 400 450 500 550 600 650
WAVELENGTH-mp
FIG;. 2. Metal deuteroporphyrins IS spectra in chloroform. Top: Cobalt (II) (1.95 X 10-S M); dimethyl ester. Middle: Nickel (II) (4.6 X 10-j M); diethyl ester. Bottom: Pal- ladium (II) (2.5 X lo-” M); dimethyl ester. Path length for visible and ultraviolet regions 1 cm, for Poret region 0.1 cm. Note change in horizontal wale at 350 m+
ized state is predicted to have lower energy. There are two serious discrepancies between this calculation and experiment. One is that Qz is calculated to be stronger than Q2, . It has been shown (1) that correct prediction of this property requires a very exact’ balance of one and two electron terms. The correct result is obtainable with slight adjustment of the Hamiltonian. The second discrepancy arises in that N, is calculated to draw off much of the intensity of B, , a result not shown by the data (4). This error may arise from an overestimation of the dif- ference between pyrrole and pyridine nitrogen at’oms in porphin combined with the general overest’imation of configur&ion interaction found in these SCMO- PPP calculat,ions (1). Because of the t.heoretical errors in the free base of unsub-
I 08 -
04 -
02 -
L I I I
N
w
002 -
_I -
02 -
06 -
N
I93
II MAGNESIUM
B Q
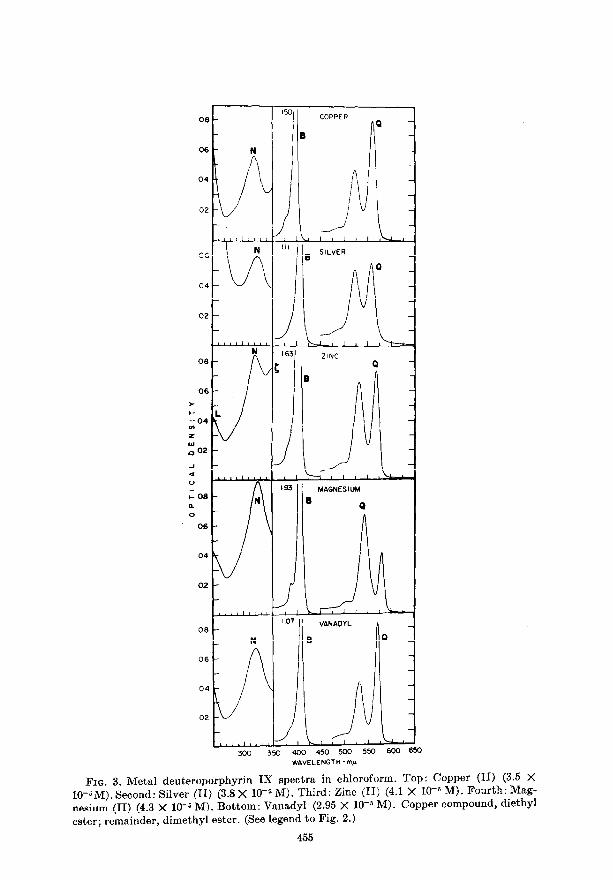
FIG. 3. Metal deuteroporphyrin IX spectra in chloroform. Top: Copper (II) (3.5 X 10-&M). Second: Silver (II) (3.8 X lO-& M). Third: Zinc (II) (4.1 X lo-& M). Fourth: Mag- nesium (II) (4.3 X 10-5 M). Bottom: Vanadyl (2.95 X 1W6 M). Copper compound, diethyl ester; remainder, dimethyl ester. (See legend to Fig. 2.)
455

456 CAUGHEY, ET AI,.
COPPER
i
)_ h!L N
:0 d
MAGNESIUM 06 - N
A i f- B
06
xx) 350 400 450 5cxl 550 600 650 wn\,c, Chl?TY_ _
FIG. 4. Metal protoporphyrin IX dimethyl esters in chlorof~~rm Top: Copper (11) (9.5 X 10-S M). Middle: Zinc (II) (2.7 X 10-S M). Bottom: Magnesium (II) (3.1 X lo-” M). (See
legend to Fig. 2.)
stituted porphin, we would conclude that the metal derivatives of uusubstituted porphin would supply the most meaningful data for roniparison with t,heory. LitUe such data is available. The availabiIit,y of unsubstituted porphin has beeu limited by the difficulty of it.s synthesis. Further, substituents are ofteu necessary t,o raise solubility to experimentally convenient levels. For these reasons, the present paper reports on metal complexes of 2 ,kubstit,uted deuteroporphyrins
IX which were prepared for use in studies of the influence of porphyrin structure OH reactions and properties of natural porphyrins and derivatives. (See Footnote 1 and References 5 and 6.) (See Fig. 1 for structures of porphyrins uanied in this Jtaper. )

METAL DEUTEROPORPHYRINS 457
Before going on to the data, it is important to underline certain other limita- t,ions of the calculations: (i) No account is taken of substituents; we are compar- ing all data to t’he one Oa porphin calculation of Table I. (ii) The calculations only give ?r + ?r* excit,at’ions. In the free base n + ?r* excitations might occur, and in transition metals with open d shells t,here might be charge transfer and d -+ d t’ransitions. (iii) The calculations take no account of the possible nonplanarity of the porphyrin ring. Of these, we can perhaps safely ignore the nonplanarity, because of the overall success of t,he SC&IO-PPP calculations (I), and the d -+ d transitions, which are generally so weak as to be unobservable except to the red of the first visible absorption. The importance of the ot’her limitations can be determined only through comparison with dat,a.
EXPERIMENTAL
Figures 2 and 3 show t’he electronic absorption spectra of various metal com- plexes of deuteroporphyrin IX in the region from 650-250 rnp. (The solutions shown are transparent from 700-650 mp.) In Fig. 4 are displayed the spectra of three met,al complexes of 2,4-diacetyldeuteroporphyrin IX. The ultraviolet spectra of three metal mesoporphyrins (Zn, Pd, Cu) have already been pub- lished in Reference 1. The spect.ra were recorded either on t,he Beckman model DK-2 or on t#he Carey model 11 spectrophotomet,er. The compounds were pre- pared according ho procedures which will be described in separate publications (see Foot8note 1).
All the compounds were carefully purified and characterized by elemental analyses and such spectroscopic measurements as NMR and IR. All spectra shown in Figs. 2-6 are solution spectra taken at room temperature. The solvent used in each was chloroform (Baker reagent), cont’aining about 0.5% ethanol. This was stored over calcium oxide and freshly filtered before use. Concentra- Oions were different for each spect’rum (see legend under each figure) and may be in error by as much as 10% in certain cases where very small quantities of por- phyrin were used.
CLASSIFICATION AND NOMENCLATURE
In discussing molecular absorptions we might dist,inguish three types of names: a r,ommon name, a generic name, and a group theoretical name. Thus the lowest absorption band of benzene is called by the names cr, lL* , lBzu and the strong porphyrin band near 400 mp is called Soret, IB, ‘22, . The common name is merely for convenience. The generic name implies something about the nature of the excited state and differs from the group theoretical name in that it is preserved when molecular symmetry is changed by substitution. Further, it can differ even when the symmet,ry of two states is identical, as with the ‘& and 1B states of porphin. We have tried t.o follow Platt (7) in the choice of generic names.

458 CAUGHEY. ET AL
For the bands of porphyrin above t#he Soret band, the question arises as to whether to refer t#o t,hem by common and/or generic names. This choice depends on the confidence of the investigators in the current theory. We have chosen to give two bands t,he common names 7, { and two bands the generic names N, I,. We now give the empirical characterization of these bands:
(1) q bands appear in carbonyl substituted porphyrins around 370 mp. They are broad and appear as a small hump on the tail of the Soret band.
(2) !: bands appear as a well resolved shoulder in some Zn porphyrins around 350 mp.
(3) N bands have E - 10 000-25 000 liters/mole cm and appear at, 350-320 111~. The band appears in three forms:
(a) In metal alkylporphyrins ,2 it appears as a broad band with maximum between 330-320 rnp, usually with a barely resolved shoulder at 310 1111.1 and sug- gf&ions of further structure.
(b) In free base porphyrins, this band is reduced to a shoulder or infle&on, presumably due t’o the greater width of the Soret band owing to the loss of square symm ct,ry .
(c) In porphyrins with vinyl and carbonyl substituents, t,his band is often in- lensified and may be red shifted as far as 345 mp.
(4) L bands also have E - 10 000-25 000 and appear at 310-255 mu. They are sometimes doubled or have a shoulder. In the present data, this band appears in four forms:
(a) a broad singIe or doubIe peak in cobalt, nickel, and palladium alkyl- porphyrins;
(h) a peak, usually double, in metal and free base diacetyl deuteroporphyrins; (c) a broad shoulder in free base and metal protoporphyrins; (d) a well-resolved mound at 280-265 rnp in other free base porphyrins.
ASSIGNMENTS
hs their names imply, we identify N and L bands with excitation to the N and 1, states predicted by the theory and given in Table I. Both bands have t,he ill- tensity to be expect.ed of a weakly allowed r --+ ?r* t,ransition. The N band ap- pears from 4600-6800 cm-l above t’he Soret band, as compared with 5800 cn--’ predicted by t’heory; the L band appears 83OC-15400 cm-’ above the Soret and 3900-8500 cm-l above the N band, as compared to 8950 and 3100 cm-1 predicted by the Dheory, respectively. The L state is further predicted to have less intrinsic intensity t~han the N, a prediction consistent wil,h the fact that t.he L band does not appear in many symmetrically substituted porphins, or is present as a barely visible blip. The vagaries of the band ident,ified with t,he L stat$e are such that in some cases the identification may be in error and that some “L” bands may be due to higher n -+ P* st,ates, to forbidden bands made allowed by substituent per- t.urbations, or possibly to charge transfer transitions.
2 We apply the term “alkyl porphyrins” to all porphyrins which have saturated substit- uents or hydrogen on the betapyrrole carbon atoms.

METAL DEUTEROPORPH YRINS
Table II
A. Deuteroporphyrin IX estersa.
Qd B N L Other
Frge Base z-1 619(Q,) 53O(Q 1 399 -317 -266
16160 1%37oy 25060 -31550 -37590
Cobalt (II) r,q~ 549 (513) 392 319 263 -1 cm 18210 (19490) 25510 31350 28020 (310 sh)
Nickel (II) w 549.5 (514) 390.5 325 288 -1
cm 18200 (19460) 25610 30770 34720
Palladu"m(II)m!L 544 (509) 393 320 280,268 -1 cm 18380 (19650) 25450 31250 35710.37310 (3lq sh)
copper (II) w-1 559.5 (522) 397.5 318.5 cm 17870 (19160) 25160) 31400 - (311 sh)
Silver (II) rwl 558.5 (525) 408 326 cm 17910 (19050) 24510 30670 - (310 sh)
Zinc (II) nw 569 (531) 402 320 -253 sh 350 (6) -1
cm 17570 (18830) 24880 31250 39530 28570
Magnesium(Il)mw 578.5 (542) 408 326 -1
cm 17290 (18450) 24510 30670
Vanadyl w 570.5 (532) 407.5 321 -1
cm 17530 (188001 24540 31150
B. Mesoporphyrin IX estersa.
Free Base 6lY(Q ) 329 sh 269
16160Y
532(Q 1
1Ez3ooy
399.5
25030 31250 37170
cobalt (II) 553 (517) 392.5 324 266
18080 (19340) 25480 30860 37590
Nickel (II) 550.5 (514) 392 330 290
18170 (19460) 25510 30300 34480
Palladium (II) 548 (511) 393 327 282,271 sh (310 shj
18250 (19570) 25450 30580 35460.36900 sh
Copper (II) 563 (525) 398 327 (311 sh)
17760 (19050) (25130) 30580
Zinc (II) 570.5 (532.5) 401.5 331.5 -258 sh 350 (5)
17530 (18780) 24910 30160 -38760 28570
459
C. Protoporphyrin IX estersa
Qd B N L Other
Free BaSe 630(QJ 54l(Qy) 407.5 274,258 c
15870 18-180 24540 38760.36500 --__-
Nickel (II) 561 (523.5) 400 -2337 511 b
17830 119100) 25000 -29670
Copper (II) 570 (531.5) 405.5 335 -27, sh -322 sh
_______l7xa___~881o___2_r1_6Go___2Ra5o --36630 --_
Zinc (II) 578.5 (541.5) 412.5 -353 m-268 sh -322 sh
_______-_L7ZTT-__ f18-170) 24240 28330____37310_____:31060__
Magnesium (II) 593 (552.5) 419 343 268 sh 322 sh
__ __16~~0____fuu~33~70 29L~L~O___~----

460 CAUGHEY. ET AL.
Q3 B 1 N
Free Base 640 (Qs) 552(Qy) 424.5 375 270-310 c 32260-
-__15_83_1____1813il___aiSG3___2_6_670_____ 37040 __
Cobalt (II) 568 (534) 415 -365 331 285,-266sh
~_____17~~__~_187_23___24100~ .-27400 30210 35090.-37590
Nickel (II) 571.5 (532.51 415.5 -369 335 sll 359,255
______~17~~3_____~8_zS~ 24070 -27100 29850 32360.39220
Palladlw (II) 5 d5 (526) 418 362 327 291.261 sh
_____Luoo____fLd~~aL _ 23920 27621) 30580 34350.38310
copper (II) 5Sl (5421 420 -369 326 284
~_L7LZ&___118-150) 23810 -27100_ 30670 35210
Silver (II) 579 (543) 423.5 373 331.5 -259 sh
17270 (18420) 23283 _ 26810 30165 38610
zinc (II) 592 (555) 430.5 371 330e 268
16893 (1832,JJ 23230 26950 30300 37310 _____M-----
Notes to Table II -__-_ __-
The weak intensity of the q band makes it probable that this band corresponds
to a forbidden transition buried under the O-l overtone of the Soret band in square porphyrins. In diacetyldeuteroporphyrins, this band is intensified by the nonsquare symmetric substituent perturbation and is uncovered by the large red shift of the Soret band. There is also the possibility that the r] band should be identified with the N state, and is obscured in other porphins by the Soret O-l overtone. If this be the case, the “N” bands shouId be identified with the L state and t,he “L” bands with higher energy excited states. It might be possible to settle this quest& with Pariser-Parr t’ype calculations into which the carbonyl
pi orbitals are introduced explicitly. The [ band was early observed in Zn tetraphenyl porphin (8), and was as-
signed by Kobayashi (9) to the state we have called N. The present data now indicate that this band is peculiar to zinc proto- and alkyl porphyrins. It does not appear in t,he spectrum of zinc diacetyl deut,eroporphyrin or zinc mesoporphyrin in pyridine.

METAL 1)EUTEROPORPHYRINS 461
The tenhative assignment.s of the ultraviolet spectrum of 27 free base and metal porphins, along with the experimental energies of their Soret and visible transi- tions, are tabulated in Table II. This interpretation of the ultraviolet spectrum is, of course, not unique and is subject to revision or rejection as more data is accumulated. In particular, we have not really excluded the possibility that one or more of t,he bands listed in Table II as 7r + r* transitions will actually turn out to be n + a* transit.ions involving the ring nitrogens. On the other hand, they are almost certainly too intense to be carbonyl n -+ a* or metal dd (crystal field) tsansitions. In addition, we would expect both from the theory and from the appearance of the experimental spectra that there are a large number of weak transitions which are not resolved in t’he solution spect’ra now available to us. These might be resolvable by low temperature.
Measurements of t.he red and near-IR specka of concentrated solutions of carbonyl substituted free base porphins were made to seek evidence for a car- bony1 to ring n -+ ?r* band. Such a transition had been postulated by Fernandez
I I I I 1 I I I
L B SILVER n
PALLADIUM
L 7)
250 300 350 400 450 500 550 600 650
WAVELENGTH-my
FIG. 5. Metal 2,Gdiacetyldeuteroporphyrin Ix dimethyl esters in chloroform. Top: Copper (11) (5.0 X 10-S M). Second: Silver (II) (5.0 X W5 M). Third: Zinc (II) (5.0 X 1@5 M). Bot,tom: Palladium (II) (5.0 X 10v5 M). (See legend to Fig. 2.)
4@2 CAIi(:HEY, ET .41,.
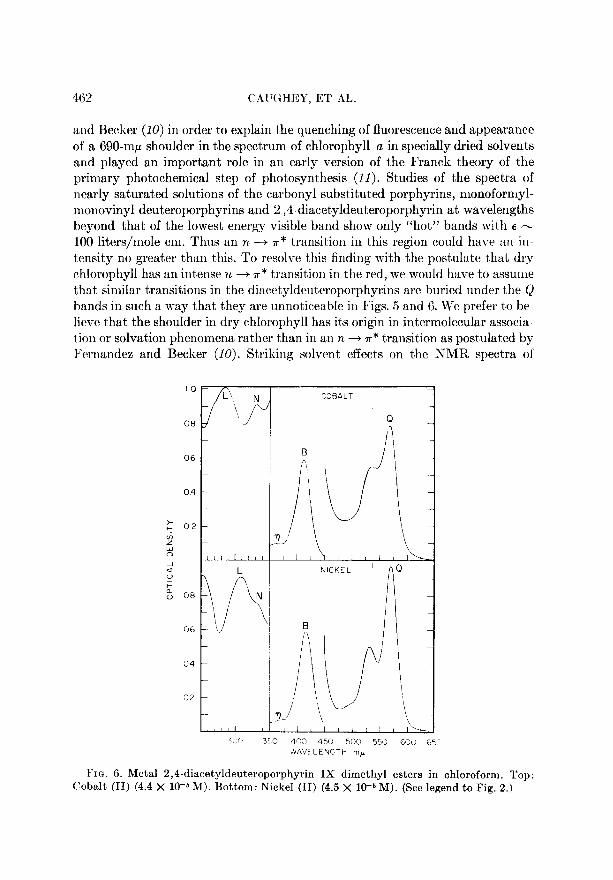
and Becker (10) in order to explain the quenching of fluorescence and appearance
of a 690-rnp shoulder in the spectrum of chlorophyll a in specially dried solvents and played an important role in an early version of the Fran& theory of the primary photochemical step of photosynthesis (II). Studies of the spectra of nearly saturated solutions of the carbonyl substituted porphyrins, monoformyl- monovinyl deuteroporphyrins and 2,4-diacetyldeuteroporphyrin at wavelengths beyond that of the lowest energy visible band show only “hot” bands with a m 100 liters/mole cm. Thus an n -+ T * transit#ion in this region could have an itl-
tensity no greater than this. To resolve this finding with the post’ulate that dry chlorophyll has an intense 1~ --+ T* transition in the red, we would have to assume
t,hat similar transitions in the diacet’yldeuteroporphyrins are buried under the Q bands in such a way that they are unnoticeable in Figs. 5 and 6. We prefer to be- lieve that the shoulder in dry chlorophyll has it,s origin in intermolecular associa- tion or solvation phenomena rather than in an n -+ ?r* transition as postulated by Fernandes and Becker (10). St’riking solvent effect’s on t#he NMR spectra of
IO
06
06
COBALT
1
2 i’l’ 3FO 4m 450 500 550 6Ou 651 NAVELENGTH mp
FIG. 6. Metal 2,4-diacetyldeuteroporphyrin IX dimethyl esters in chloroform. Top: Cobalt (II) (4.4 X lO-& M). Bottom: Nickel (II) (4.5 X W6 M). (See legend to Fig. 2.)

METAL DEUTEROPORPHYRINS 463
chlorophylls and other magnesium porphyrins can be explained in terms of such phenomena (1.2, IS).
ACKNOWLEDGMENT
This work was supported in part by Contract No. SA-43-ph-1914 between Cancer Chemo- therapy National Service Center, National Cancer Institute, National Institutes of Health and Monadnock Research Institute and in part by U. S. Public Health Service Grants No. HE-06079 and GM-10833. One of the authors (W. S. C.) gratefully acknowledges the sup- port of a Lederle Medical Faculty Award. Another author (C. W.) was supported during this research by a National Institutes of Health pre-doctoral fellowship.
RECEIVED March 6, 1965
REFERENCES
1. C. WEISS, H. KOBAYASHI, AND M. GOUTERMAN, J. Mol. Spectry. 16, 415 (1965). 2. J. R. PLATT, in “Radiation Biology,” (A. Hollaender, ed.), Vol. III, Chap. 2. McGraw-
Hill, New York, 1956. 3. M. GOUTERMAN, J. Chem. Phys. 30, 1139 (1959). 4. C. RIMINGTON, S. F. MASON, AND 0. KENNARD, Spectrochim. Acta 12, 65 (1958). 5. W. S. CAUGHEY, R. M. DEAL, B. D. MCLEES, AND J. 0. ALBEN, J. Am. Chem. Sot. 84,
1735 (1962). 6. W. 8. CAUGHEY, J. 0. ALBEN, AND C. A. BEAUDREAU, in “Oxidases and Related Redox
Systems,” (T. E. King, H. S. Mason, and M. Morrison, eds.). Wiley, New York, in press.
7. J. W. PLATT, J. Opt. Sot. Am. 43, 252 (1953). 8. G. R. SEELY AND M. CALVIN, J. Chem. Phys. 23,106s (1955). 9. H. KOBAYASHI, J. Chem. Phys. 30,1362-1373 (1959).
10. J. FERNANDEZ AND R. BECKER, J. Chem. Phys. 31, 467 (1959). 11. J. FRANCK, J. L. ROSENBERG, AND C. WEISS, in L‘Luminescence of Organic and Inor-
ganic Systems, (H. P. Kallman and G. M. Spruch, eds.). Wiley, New York, 1962. See also J. Franck and J. L. Rosenberg, J. Theoret. Biol. 7, 276 (1964).
1~. P. K. IBER, dissertation, The Johns Hopkins University, 1963. 13. G. L. CLOSS, J. J. KATZ, F. C. PENNINGTON, M. R. THOMAS, AND H. H. STRAIN, J. Am.
Chem. Sot., 86. 3809 (1963).




















