
Genetically proxied therapeutic inhibition of antihypertensive ...

1
HDAC inhibition overcomes acute resistance to MEK inhibition in BRAF mutant
colorectal cancer by down-regulation of c-FLIPL.
Robbie Carson1,†, Basak Celtikci1, †, Cathy Fenning1, Arman Javadi1, Nyree Crawford1, Lucia
Perez Carbonell1, Mark Lawler1, Daniel B. Longley1, Patrick G. Johnston1, †, Sandra Van
Schaeybroeck1,2, †
1. Centre for Cancer Research and Cell Biology, School of Medicine, Dentistry and
Biomedical Science, Queen’s University Belfast, 97 Lisburn Road, Belfast, BT9 7AE, UK
† equal contribution
Running title: Regulation of STAT3 and c-FLIPL in BRAF mutant colon cancer.
Keywords: BRAF, colorectal cancer, MEK, c-MET-JAK-STAT3, c-FLIP
Financial support from Cancer Research UK (C212/A7402); Cancer Research UK fellowship
(C13749/A7261).
2. Corresponding author: Dr. Sandra Van Schaeybroeck, Centre for Cancer Research and
Cell Biology, Queen’s University Belfast, 97 Lisburn Road, Belfast, Northern Ireland, BT9
7AE. [email protected]; Telephone: +44 2890 972954; Fax: +44 2890972776.
Conflicts of interest: P.G. Johnston has an ownership interest in both Almac Diagnostics and
Fusion Antibodies. He is a consultant/advisor for, and has received honoraria from, Chugai
pharmaceuticals, Sanofi-Aventis and Pfizer. All other authors have no conflicts of interest to
declare.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

2
TRANSLATIONAL RELEVANCE
Oncogenic V600E BRAF mutations are associated with poor clinical prognosis in colorectal
cancer (CRC). Inhibitors of the BRAF-MAPK pathway are effective therapies for BRAF
mutant (MT) melanoma, but are ineffective in the majority of BRAFMT CRCs. Understanding
MAPK inhibitor resistance is therefore an important unmet clinical need in BRAFMT CRC. In
this study, we have identified a novel resistance mechanism to MEK1/2 inhibition that is
mediated via STAT3 and the anti-apoptotic protein c-FLIPL and which is dependent on c-
MET activity. Furthermore, inhibiting c-MET or down-regulating c-FLIP using RNAi or HDAC
inhibitors significantly increased MEK inhibitor-induced apoptosis in BRAFMT CRC in vitro
and in xenograft models. Taken together, our results indicate that combining MEK1/2
inhibitors with c-MET or HDAC inhibitors may be promising therapeutic strategies for this
poor prognostic CRC subgroup and should be explored in future clinical trials.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

3
ABSTRACT
Purpose: Activating mutations in the BRAF oncogene are found in 8-15% of colorectal
cancer (CRC) patients and have been associated with poor survival. In contrast to BRAF
mutant (MT) melanoma, inhibition of the MAPK pathway is ineffective in the majority of
BRAFMT CRC patients. Therefore, identification of novel therapies for BRAFMT CRC is
urgently needed.
Experimental Design: BRAFMT and WT CRC models were assessed in vitro and in vivo.
Small molecule inhibitors of MEK1/2, MET and HDAC were employed, over-expression and
siRNA approaches were applied, and cell death was assessed by flow cytometry, Western
blotting, cell viability and caspase activity assays.
Results: Increased c-MET-STAT3 signalling was identified as a novel adaptive resistance
mechanism to MEK inhibitors (MEKi) in BRAFMT CRC models in vitro and in vivo. Moreover,
MEKi treatment resulted in acute increases in transcription of the endogenous caspase-8
inhibitor c-FLIPL in BRAFMT cells, but not in BRAFWT cells, and inhibition of STAT3 activity
abrogated MEKi-induced c-FLIPL expression. In addition, treatment with c-FLIP-specific
siRNA or HDAC inhibitors abrogated MEKi-induced upregulation of c-FLIPL expression and
resulted in significant increases in MEKi-induced cell death in BRAFMT CRC cells. Notably,
combined HDAC inhibitor/MEKi treatment resulted in dramatically attenuated tumor growth
in BRAFMT xenografts.
Conclusions: Our findings indicate that c-MET/STAT3-dependent upregulation of c-FLIPL
expression is an important escape mechanism following MEKi treatment in BRAFMT CRC.
Thus, combinations of MEKi with inhibitors of c-MET or c-FLIP (eg. HDAC inhibitors) could
be potential novel treatment strategies for BRAFMT CRC.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

4
INTRODUCTION
BRAF is mutated in ∼8% of all cancers, and approximately 10% of colorectal cancer tumors
harbour a BRAFT1799A transversion, encoding the constitutively active V600E-BRAF
oncoprotein (1, 2). BRAF belongs to the RAF protein-serine/threonine kinase family, which
also comprises RAF-1/c-RAF and A-RAF (3). In normal signalling conditions, RAF proteins
phosphorylate and activate the mitogen-activated protein kinase (MAPK) signalling axis in
response to active GTP-bound RAS. BRAFV600E mutations lead to constitutively active
BRAF, resulting in sustained MAPK signalling, independent of upstream kinase activity (4).
Recent phase III studies have shown that patients with BRAF mutant (MT) tumors have the
worst overall survival compared to patients with RASMT (KRAS or NRAS) or KRAS/BRAF
wild type (WT) CRC tumors (5-7). Furthermore, some studies have suggested that the
BRAFV600E mutation should be included as marker of resistance to epidermal growth factor
receptor (EGFR)-targeted therapies, such as cetuximab and panitumumab (8-10). Therefore,
novel therapeutic strategies are urgently needed for BRAFMT CRC patients.
Tumors carrying a mutationally activated oncogene frequently show “addiction” to the
oncoprotein or activated signalling pathway upon which the cancer cells became dependent
(11). Recently, the Food and Drug Administration (FDA) approved the selective BRAF
inhibitor Vemurafenib for the treatment of patients with unresectable or metastatic melanoma
with the BRAFV600E mutation. Although the BRAF inhibitors Vemurafenib or Dabrafenib,
MEK1/2 inhibition (Trametinib) or a BRAF/MEKi combination resulted in dramatic responses
between 60%-80% in BRAFMT melanoma (12-14), disappointing responses of 5% and 12%
were seen for Vemurafenib alone or Dabrafenib/Trametinib combination respectively in
BRAFMT CRC (15, 16). Understanding mechanisms of resistance to MAPK inhibition in
BRAFMT CRC may lead to new therapeutic strategies for this poor prognostic CRC
subgroup.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

5
Herein, we report a novel resistance mechanism to MEK1/2 inhibition in BRAFMT CRC,
which is mediated via STAT3 and the anti-apoptotic protein c-FLIPL. This survival response
is dependent on c-MET and JAK1/2 activity and is abrogated using c-MET and JAK1/2-
STAT3 inhibitors. We also show that concomitant treatment with inhibitors of c-MET or c-
FLIP (using HDAC inhibitors) and MEK1/2 inhibition leads to marked increases in
therapeutic efficacy in BRAFMT in vitro and xenograft models. Taken together, our results
indicate that combining MEK1/2i with inhibitors of c-MET or c-FLIP (eg. HDAC inhibitors)
could be potential novel treatment strategies for BRAFMT CRC.
MATERIALS AND METHODS
Materials
AZD6244, AZD1480 and AZD9150 were obtained from AstraZeneca (Macclesfield, UK),
Crizotinib from Pfizer (Peapack, New Jersey, USA), TG101348 from Axon Medchem
(Groningen, The Netherlands), Trametinib, AZD8931, Vorinostat and Entinostat (MS-275)
from Selleck Chemicals LLC (Suffolk, UK), Stattic from Sigma-Aldrich (Dorset, UK), UO126
from Promega (Southampton, UK) and PD98059 from Cell Signaling (Beverly, MA, USA).
siRNAs targeting STAT3 (STAT3_7), JAK1 (JAK1_1), JAK2 (JAK2_7) and caspase 9
(CASP9_5) were purchased from Qiagen (Crawley, UK); the c-FLIP siRNA sequence used
was 5’ AAG CAG TCT GTT CAA GGA GCA; the caspase 8 sequence used was 5’ GAG
UCU GUG CCC AAA UCA ATT (17).
Cell culture.
Authentication and culture of LIM2405, LIM1215, HT-29, VACO432/VT1, RKO F6-8 and
CACO-2 CRC cells have previously been described (18-20). All cells were passaged for a
maximum of 2 months, after which new seed stocks were thawed for experimental use.
LIM2405 and LIM1215 cell lines, established in 1992, were a gift from Dr. Whitehead
(Ludwig Institute of Melbourne and Vanderbilt University, Nashville, TN) in 2010. These cell
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

6
lines were tested for morphology/growth rate/response to mitogens/xenograft
growth/expression of brush-border and mucin-related antigens/mutational analysis (21, 22).
VACO432, VT1 and RKO F6-8 were kindly provided by Prof B. Vogelstein (Johns Hopkins
University School of Medicine, Baltimore) in 2012. HT-29 (2001), CACO2 (2005), COLO205
(2012) and COLO320 (2012) cells were obtained from the American Type Culture Collection
(Authentication by short tandem repeat (STR) profiling/karyotyping/isoenzyme analysis).
Generation of stable c-FLIPL-overexpressing cells.
c-FLIPL containing pBABE puro vector was transfected into Phoenix 293T cells along with a
VSVG (vesicular stomatitis virus (vsv) glycoprotein) coat protein plasmid. Following 24h
transfection, filtered medium with polybrene (1:1000) was added to HT-29 cells for 48 hours,
and thereafter 1μg/mL puromycin (Life Technologies, Inc.) added for selection of c-FLIPL
overexpressing HT-29 cells. Overexpression of c-FLIPL was confirmed by Western blotting.
Cell Viability assays
Cell viability assays were done as previously described (20). Representative results of at
least 3 independent experiments are shown.
Flow cytometric analysis and cell death measurement.
Apoptosis was determined using propidium iodide (PI) staining to evaluate the percentage of
cells with DNA content <2N as previously described (18). For annexin/PI analysis, cells were
harvested and analysed according to the manufacturer’s instructions (BD Biosciences).
Representative results of at least 3 independent experiments are shown.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

7
Western blotting.
Western blot analysis was carried out as previously described (20). Anti-FLIP (NF6; Alexis,
San Diego, CA, USA), anti-caspase 8 (12F5; Alexis) and anti-poly(ADP-ribose) polymerase
(PARP; eBioscience; San Diego, CA, USA) mouse monoclonal antibodies were used in
conjunction with a horseradish peroxidase–conjugated anti-mouse secondary antibody
(Amersham). Anti-cleaved caspase 3 (Cell Signaling Technology, Beverly, MA, USA), anti-
hyperacetylated Hystone H4 (Millipore, Watford, UK) were used in conjunction with a HRP-
conjugated anti-rabbit secondary antibody (Amersham, Buckinghamshire, UK). Equal
loading was assessed using β-actin (Sigma) mouse monoclonal primary antibody.
Caspase-activity assays
Caspase-8 or caspase-3/7-GLO reagents (25μl) (Promega, Southampton, UK) were
incubated with 5μg of protein lysate diluted in cell culture medium in a total volume of 50μl
for 45 minutes at room temperature. Luciferase activity was measured using a luminometer.
STAT3 reporter assay
The STAT3 reporter, negative and positive control constructs were obtained from Qiagen
(Crawley, UK). Luminescence was measured following addition of 50μl of luciferase assay
reagent into each well.
siRNA transfections
siRNA transfections were carried out using Hiperfect (Qiagen) as previously described (20).
ELISA
Soluble c-MET and HGF ELISA assays were carried out as previously described (20).
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

8
Real-time reverse transcription-PCR analysis
RNA was isolated using the GeneJET RNA purification kit (Thermo Scientific, Leicestershire,
UK) and reverse transcribed using the Moloney murine leukemia virus-based reverse
transcriptase kit (Invitrogen). Q-PCR analysis was performed using the LightCycler® 480
probes master mix or the Roche SYBR Green method (LightCycler® 480II, Roche).
In vivo study. In vivo studies were conducted as previously described using BALB/c nude
mice (18). AZD6244 10mg/kg/bid p.o, Entinostat (MS-275) 10mg/kg/qd p.o and Crizotinib
25mg/kg/qd were administered by oral gavage. The control groups received vehicle
(methocel/polysorbate buffer). In the combination groups, AZD6244 was administered
together with MS-275 or Crizotinib. Each treatment group contained 8 animals. Two hours
following final dosing, mice were sacrificed and tumor tissue was excised and snap-frozen in
liquid nitrogen for immunoblotting (PPL2704). All animal experiments were carried out
according to UKCCCR guidelines.
Statistical analysis
Student’s t-tests and 2-way ANOVA were calculated using the GraphPad software (Prism4).
2-way ANOVA test was used to determine the significance of change in levels of apoptosis
between different treatment groups. All changes in levels of apoptosis that are described as
significant had p values that were <0.05 (* denotes p<0.05; ** denotes p<0.01; *** denotes
p<0.001; ns denotes not significant compared with control).
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

9
RESULTS
MEK1/2 inhibition decreases cell viability but does not induce apoptosis in BRAFV600E
CRC. To explore the sensitivity of BRAFMT CRC cells to MEK1/2 inhibition, we evaluated
the effects of the clinically employed MEK1/2 inhibitors AZD6244 and Trametinib
(GSK1120212) on viability of the parental VACO432 CRC cell line, which carries the V600E
mutation in BRAF (BRAFV600E/+), its isogenic VT1 clone with a disrupted BRAFV600E
allele (23), and a panel of BRAFMT (LIM2405, HT-29, COLO205, RKO F6-8) and WT
(CACO-2, LIM1215 and COLO320) CRC cells (Supplementary Fig. S1A, upper panel).
BRAFMT CRC cells showed a higher sensitivity to MEK1/2 inhibition with IC50 values
between 20nM-3.7μM and 0.1nM-9.4nM for AZD6244 and Trametinib respectively,
compared with IC50 values of >6.8μM and >13nM for BRAFWT cells (p<0.001 for both
AZD6244 and Trametinib). Although BRAFMT CRC cells were more sensitive to MEKi than
BRAFWT cells, apoptosis induction 24h after treatment with MEKi was relatively inefficient
as assessed by activation of caspase-3 (Supplementary Fig. S1A, lower panel) and flow
cytometry (Supplementary Fig. S1B).
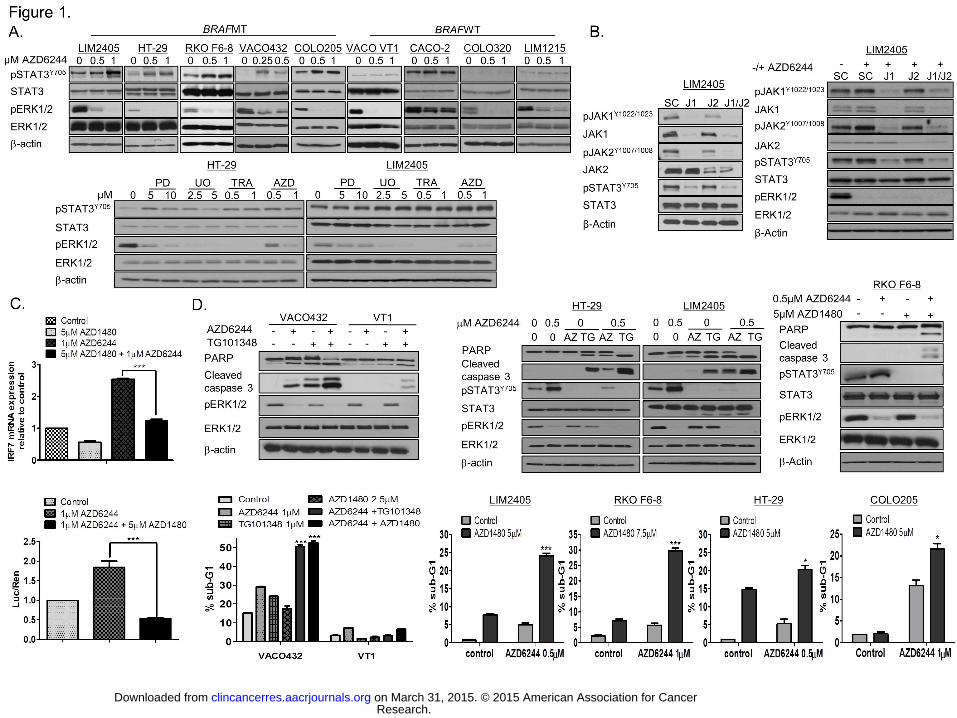
STAT3 pathway activation and JAK1/JAK2 are key mediators of resistance to MEK
inhibitors in BRAFMT colorectal cancer cells. We have recently shown that KRAS mutant
cells respond to MEK1/2 inhibitors with increased STAT3Y705 phosphorylation (20). Hence,
we investigated the effect of AZD6244, PD98059, UO126 and Trametinib on STAT3
activation in our panel of BRAFMT and WT CRC cell lines and found that STAT3
phosphorylation (Y705) was acutely increased in the BRAFMT cell lines in response to each
agent, but not in the BRAFWT/KRASWT cells (Fig. 1A). In addition, we also found acute
increases in STAT3 transcriptional activity and expression of the STAT3 target genes IRF-7,
RPS18 and SOCS3 following AZD6244 treatment in the BRAFMT CRC cells
(Supplementary Fig. S1C). Constitutive pSTAT3 levels were higher in BRAFMT VACO432
cells compared to its isogenic WT clone; however there was no clear pattern of pSTAT3
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

10
expression across a panel of non-matched BRAFMT and WT CRC cells (Supplementary Fig.
S1D). STAT3 is activated in the context of MEKi in BRAFMT but not BRAFWT cells, a
phenomenon known as an adaptive, acutely induced resistance mechanism (24). We also
assessed the effect of STAT3 silencing on apoptosis induction by flow cytometry and
Western blotting for PARP cleavage and activation of caspase 3. A significant increase in
apoptosis was observed when BRAFMT cells were co-treated with STAT3 siRNA and
AZD6244 (Supplementary Fig. S1B, E). Collectively, these data indicate that STAT3 is an
important survival pathway following MEK1/2 inhibition in BRAFMT CRC.
Next, we investigated the involvement of the upstream kinases JAK1 and JAK2 in regulating
STAT3 activity and survival of BRAFMT and WT CRC cells. MEK inhibitor-induced STAT3
phosphorylation was associated with increased JAK1 and JAK2 phosphorylation
(Supplementary Fig. S2A). In addition, RNAi against JAK1 decreased basal and MEKi-
induced pJAK2 levels and resulted in more potent inhibition of both basal and MEKi-induced
STAT3 activity compared to the effects of JAK2 silencing (Fig. 1B). Moreover, treatment with
the JAK1/2 inhibitor AZD1480 (25) resulted in potent inhibition of MEKi-induced STAT3
transcriptional activity, as indicated by decreased STAT3 reporter activity and mRNA
expression of IRF-7 (Fig. 1C, upper and lower panels). These results indicate that JAK1 and
JAK2 cooperate to regulate basal and MEKi-induced STAT3 survival response in BRAFMT
CRC, which is consistent with our previous data in KRASMT CRC cells (20).
We next investigated the involvement of JAK1/2 in regulating survival of BRAFMT and WT
CRC cells. Combined treatment of AZD1480 or TG101348 (20, 26) with AZD6244 resulted in
potent increases in apoptosis as indicated by flow cytometry and increased PARP cleavage
and caspase 3 processing in the BRAFMT VACO432 cell line but not the WT VT1 clone (Fig.
1D, upper and lower panels). Calculation of combination index (CI) values confirmed strong
synergy between AZD1480 or TG101348 and AZD6244 in the BRAFMT VACO432 cell line
(Supplementary Fig. S2B). Importantly, similar results were obtained in a wider panel of
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

11
BRAFMT (HT-29, LIM2405, RKO F6-8, COLO205), but not BRAFWT CRC cell line models
(Fig. 1D; Supplementary Fig. S2B-D).
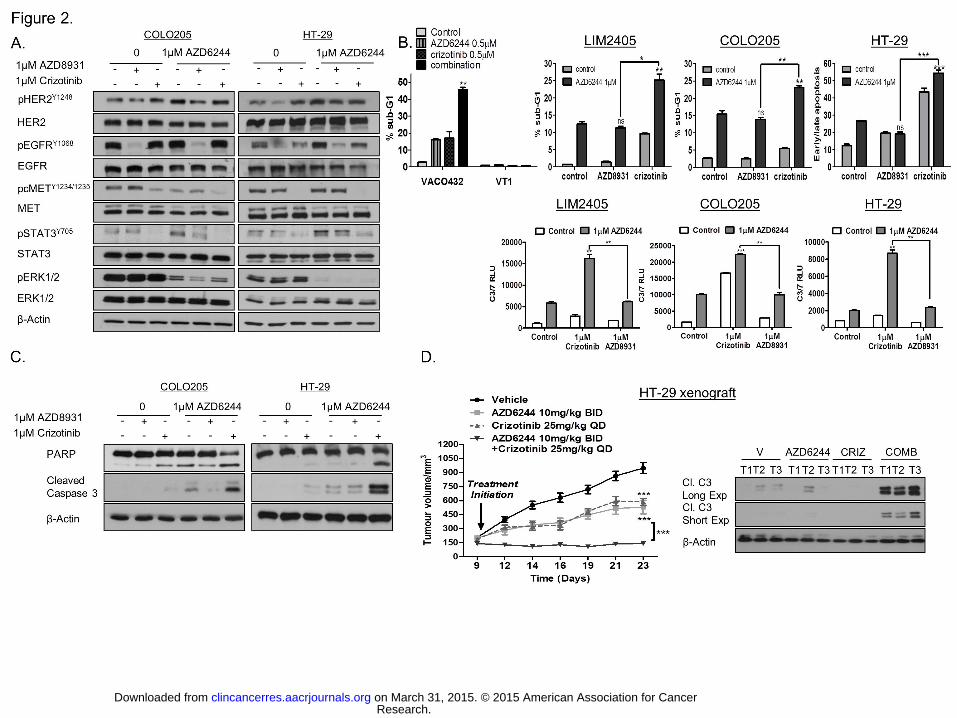
c-MET regulates MEKi-induced STAT3 activation and survival in BRAFMT CRC cells.
Previous studies have shown the role of the epidermal growth factor receptor family in
resistance to BRAF inhibitors in BRAFMT cancers, including CRC (27, 28). Secretion of the
hepatocyte growth factor (HGF), the ligand for c-MET, has also been shown to confer
resistance to BRAF inhibition in BRAFMT melanoma (29). Based on these studies and our
previous data in KRASMT CRC (20), we assessed the effect of MEK inhibition on
EGFRY1068, HER2Y1248 and c-METY1234/1235 phosphorylation and found acute increases in
phosphorylation of these receptors, as early as 3 hours following treatment with AZD6244 in
BRAFMT cells (Supplementary Fig. S3A). Basal and AZD6244-induced STAT3
phosphorylation were inhibited following treatment with the c-MET/ALK inhibitor Crizotinib,
but unaffected by treatment with the dual EGFR/HER2 inhibitor AZD8931 in a panel of
BRAFMT CRC cells, indicating that activation of STAT3 following MEK inhibition is not
driven by EGFR or HER2 in BRAFMT CRC (Fig. 2A; Supplementary Fig. S3B).
We next assessed the effect of c-MET inhibition on MEK-induced apoptosis in BRAFMT
CRC cells. Co-treatment with Crizotinib and AZD6244 resulted in significant increases in
apoptosis in the BRAFMT VACO432 cell line, but not in the BRAFWT daughter cell line (Fig.
2B). Similar results were obtained when apoptosis was assessed by Western blotting for
PARP and caspase 3 cleavage, flow cytometry and caspase-8 and -3/7 activity assays
following combined AZD6244/Crizotinib treatment in BRAFMT LIM2405, COLO205, HT-29
and RKO cells, but not in BRAFWT cells (Fig. 2B, C; Supplementary Fig. S3C-E).
Importantly, although both AZD8931/AZD6244 and Crizotinib/AZD6244 combinations
resulted in significant decreases in cell viability in BRAFMT cells, only Crizotinib enhanced
apoptotic cell death when combined with AZD6244 in the panel of BRAFMT cells (Fig. 2B, C;
Supplementary Fig. S3D and F). In order to define the relative importance of the extrinsic
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

12
and intrinsic apoptotic pathways in mediating AZD6244/Crizotinib-induced apoptosis, we
used siRNA specifically directed against caspase 8 (extrinsic pathway) or caspase 9
(intrinsic pathway) (Supplementary Fig. S3G). Notably, silencing of either caspase
significantly decreased apoptosis following AZD6244/Crizotinib treatment in BRAFMT
LIM2405 cells. This suggests that the cell death induced by this combination proceeds via a
caspase-8-mediated activation of the caspase-9-dependent intrinsic apoptosis pathway.
Taken together, these data would indicate that the c-MET/STAT3 signalling is an important
survival response induced by MEK1/2 inhibition in BRAFMT CRC.
Based on the compelling in vitro evidence, we next assessed the therapeutic efficacy of
combined c-MET and MEK inhibition in a BRAFMT HT-29 xenograft model using AZD6244
and Crizotinib (Fig. 2D). We detected significantly increased pSTAT3 levels in xenografts
treated with AZD6244; moreover, Crizotinib resulted in decreased basal and AZD6244-
induced pSTAT3 levels, consistent with our in vitro observation (Supplementary Fig. S3H).
Importantly, the METi/MEKi combination led to a marked reduction in growth of BRAFMT
HT-29 xenografts compared with treatment with each agent individually (Fig. 2D, left panel).
Furthermore, caspase 3 cleavage was observed in the HT-29 tumors following treatment
with AZD6244 in combination with Crizotinib, indicative of apoptosis induction (Fig. 2D, right
panel). These results indicate that agents directed against c-MET may be highly effective
combination partners for MEK1/2-targeted therapies in BRAFMT CRC.
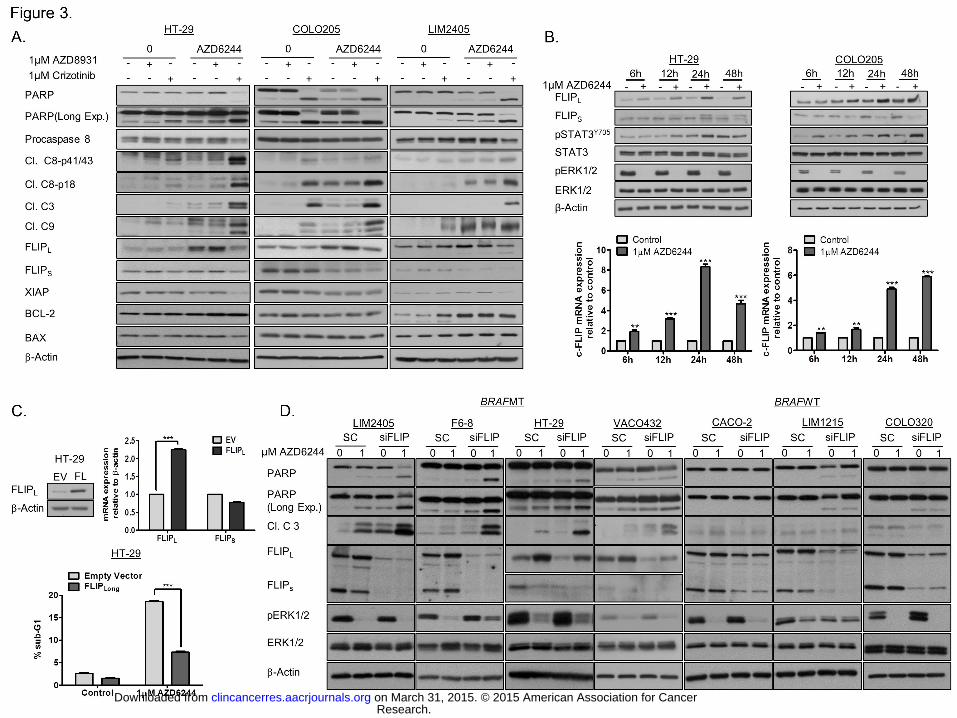
Inhibitors of MEK1/2 upregulate c-FLIPL expression in BRAFMT CRC. To investigate the
mechanism of apoptosis further, we assessed expression levels of pro- and anti-apoptotic
proteins following AZD6244/Crizotinib treatment in BRAFMT HT-29, COLO205 and LIM2405
CRC cells (Fig. 3A). Notably, expression of c-FLIPL, but not c-FLIPS, was acutely increased
following AZD6244 treatment in all three BRAFMT cell lines (Fig. 3A). In contrast, AZD6244
treatment had little impact on c-FLIPL levels in BRAFWT CRC models (Supplementary Fig.
S4A). We also found that the increases in c-FLIPL levels in BRAFMT CRC cells was a
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

13
transcriptional event, as AZD6244 increased c-FLIPL mRNA levels as early as 6h post-
treatment (Fig. 3B; Supplementary Fig. S4B). Importantly, co-treatment with AZD6244 and
Crizotinib, but not AZD8931, abrogated AZD6244-induced c-FLIPL expression, which was
associated with processing of pro-caspase 8 and potent caspase-3, -9 and PARP cleavage
(Fig. 3A).
c-FLIPL is important in regulating sensitivity to MEKi in BRAFMT CRC cells
To assess further the importance of c-FLIPL in regulating response to MEK1/2 inhibition, we
developed a BRAFMT HT-29 cell line model that stably overexpresses c-FLIPL (Fig. 3C). c-
FLIPL expression in HT-29-FL cell line was approximately 2-2.5-fold higher compared with EV
(Empty vector) (Fig. 3C, upper panel). Overexpression of c-FLIPL in this cell line correlated
with reduced levels of AZD6244-induced apoptosis compared with the EV cell line (Fig. 3C,
lower panel). These results suggested a role for c-FLIPL in mediating resistance to MEK-
targeted agents in BRAFMT CRC cells. To investigate this, we assessed the effect of c-FLIP
silencing on apoptosis induction using Western blotting for PARP cleavage and activation of
caspase-3. Co-treatment with c-FLIP siRNA and AZD6244 resulted in significant increases
in apoptosis in the BRAFMT LIM2405, RKO F6-8, VACO432 and HT-29 cell lines, but not in
the BRAFWT CACO-2, LIM1215, COLO320 and VT1 cells (Fig. 3D; Supplementary Fig.
S4C). These results are the first to demonstrate the importance of c-FLIPL in regulating
sensitivity to MEK inhibition in BRAFMT CRC.
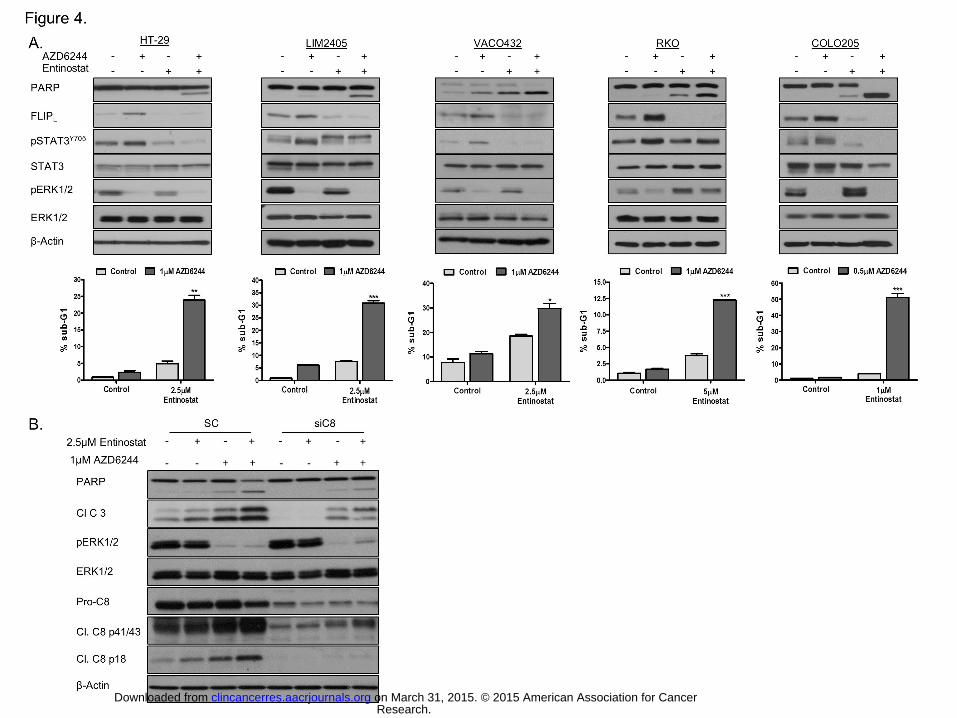
Co-inhibition of HDAC 1-3 and MEK1/2 results in cell death and inhibits growth of
BRAFMT xenograft models. Our previous studies and those of others have shown that
HDAC inhibitors with anti-HDAC1-3 activity are efficient post-transcriptional suppressors of
FLIP expression (30, 31). Treatment with the pan-HDAC inhibitor Vorinostat or the HDAC 1-
3 inhibitor Entinostat inhibited both basal and AZD6244-induced c-FLIPL expression, and this
was associated with marked increases in apoptosis in the BRAFMT cell line panel (Fig. 4A;
Supplementary Fig. S5A and B). We also determined that the apoptosis induced by
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

14
Entinostat/AZD6244 was highly dependent on caspase 8 (Fig. 4B), consistent with a FLIP-
dependent mechanism of cell death.
To extend these in vitro findings, we next assessed the therapeutic efficacy of combined
Entinostat and MEK inhibition in the BRAFMT HT-29 xenograft model. Although both
Entinostat and AZD6244 slowed tumor growth, the Entinostat/MEKi combination led to a
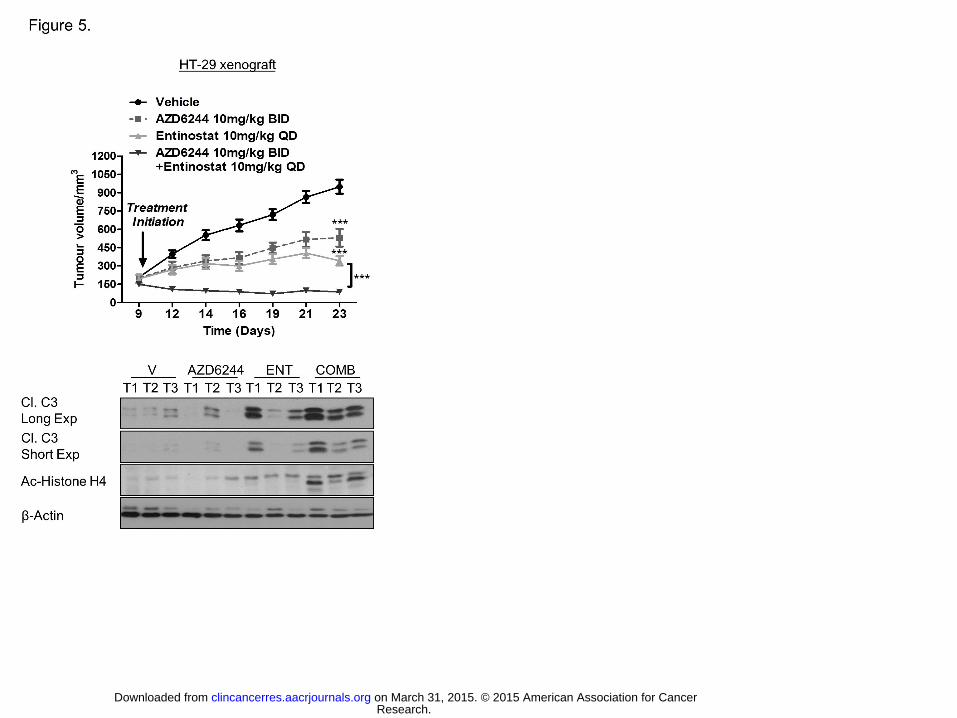
supra-additive reduction in growth in HT-29 BRAFMT xenograft model (Fig. 5). Treatment
with Entinostat resulted in increased acetylation of H4, a marker of HDAC inhibition and
decreased basal and AZD6244-induced STAT3 activation (Fig. 5; Supplementary Fig. S5C).
In addition, combined Entinostat/AZD6244 treatment resulted in high levels of caspase 3
activation in HT-29 tumors (Fig. 5). These findings indicate that HDAC1-3-targeted agents
may be highly effective when used in conjunction with MEK1/2 targeted therapies to treat
BRAFMT CRC.
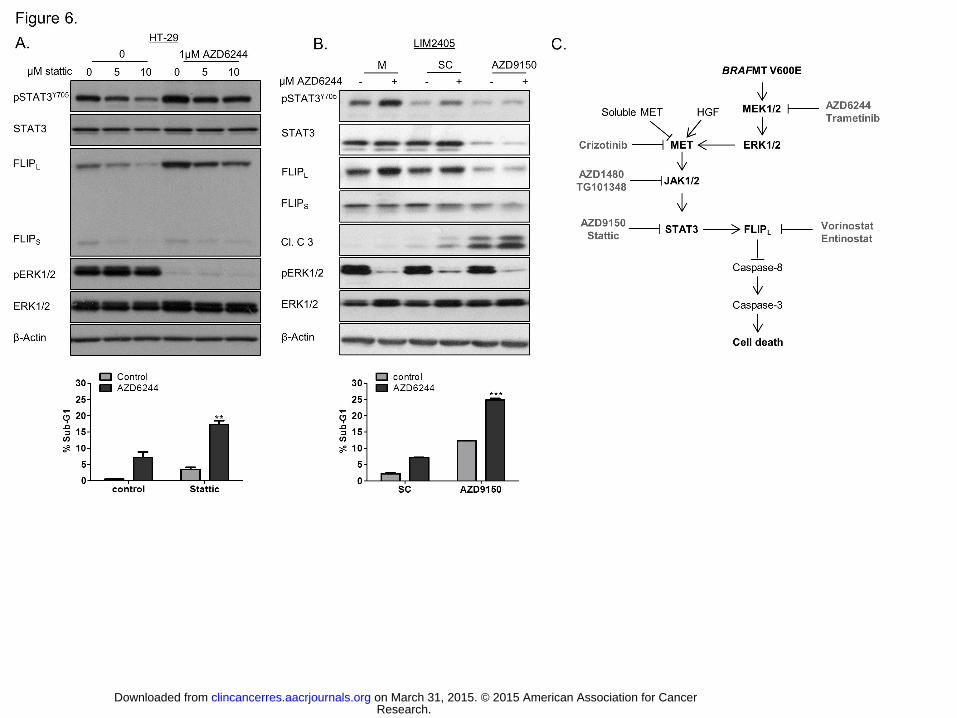
STAT3 regulates MEK1/2 inhibitor induced increases in c-FLIP expression. To
investigate the relationship between MEK1/2 inhibitor-induced increases in STAT3 activity
and c-FLIPL expression levels, we used the STAT3 small molecule inhibitor Stattic, which
targets the STAT3 SH2 domain and prevents dimerization (32), RNAi and antisense against
STAT3. Treatment with Stattic resulted in decreases in basal and AZD6244-induced
STAT3Y705 phosphorylation levels, and this was associated with decreased AZD6244-
induced c-FLIPL mRNA and protein levels (Fig. 6A; Supplementary Fig. S6A). To exclude
any off-target effects from Stattic that may affect c-FLIPL expression, we analysed the effect
of STAT3 siRNA and AZD9150, a 16 oligonucleotide antisense molecule targeting the 3’
untranslated region of the STAT3 mRNA which is in clinical development (33). Both
AZD9150 and STAT3 silencing decreased basal and AZD6244-induced STAT3Y705
phosphorylation levels and c-FLIPL expression levels (Fig. 6B; Supplementary Fig. S6B). In
addition, co-treatment with AZD6244 and Stattic or AZD9150 resulted in significant
increases in apoptosis (Fig. 6), similar to those observed with HDACi/MEKi combination (Fig.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

15
4). Taken together, these results would suggest that STAT3 directly regulates c-FLIPL
survival response in response to MEK inhibition in BRAFMT CRC.
DISCUSSION
Activating mutations in the BRAF oncogene occur in 8-15% of CRC and are associated with
the poor clinical outcome (34). The studies showing a lack of clinical benefit from EGFR-
targeted agents in BRAFMT CRC, remain controversial (9, 35). Although selective BRAF
and MEK1/2 inhibitors such as Vemurafenib and Trametinib have resulted in dramatic
responses in BRAFMTV600E melanoma, BRAFMT CRC with the identical missense mutation
has shown very poor responses to these treatments (15, 16). In both our in vitro and in vivo
data, we corroborate the findings seen in human patients. Understanding of the intrinsic
resistance mechanisms to MAPK inhibition in BRAFMT CRC has the potential to identify
novel therapeutic strategies for this poor prognostic genetic subgroup. Recently, there have
been a number of studies that have investigated resistance mechanisms to the BRAF
inhibitor Vemurafenib in BRAFMT CRC. Indeed, two studies have observed increased
activity of EGFR following treatment with Vemurafenib in BRAFMT CRC cells (27, 36).
Moreover, BRAFMT melanoma cells were found to have low levels of EGFR, which might
explain the favorable response of BRAFMT melanoma to BRAFi. Other studies reported the
role of the PI3K/AKT and mTOR pathways in mediating resistance to Vemurafenib in
BRAFMT CRC cells (37-39). Our study provides evidence that STAT3-dependent c-FLIPL
upregulation is a major resistance mechanism to MEK inhibitors in BRAFMT CRC models.
Constitutive activation of STAT3 has been reported in a broad range of tumors and tumor-
derived cell lines, including melanoma and colon cancer (40). A number of studies have
shown that deregulated STAT3 not only enhances tumor cell proliferation and survival, but
also can promote oncogenesis by bridging chronic inflammation to tumor formation (41-43).
Furthermore, recent data from our lab and other studies have shown a role for STAT3 in
regulating resistance to EGFR and MEK1/2 targeted therapies in lung and colon cancer,
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

16
driven by activated EGFR, HER2, ALK, MET and mutant KRAS (20, 44). In this study, we
have found that treatment with different MEK inhibitors resulted in acute increases in STAT3
phosphorylation in BRAFMT CRC cells.
Although recent studies have identified mutations within the SH2 dimerization and activation
domain of STAT3 in large granular lymphocytic leukaemia (45) and inflammatory
hepatocellular adenomas (IHCAs) (46), STAT3 activation is usually dependent on growth
factor receptors signalling and their associated janus kinases. Here, we found that basal and
MEKi-induced STAT3 activation in BRAFMT CRC cells was mediated through JAK1 and
JAK2. Using selective inhibitors of JAK1/2, we further demonstrated the differential
dependency of BRAFMT and BRAFWT cells on STAT3 for survival, particularly in the
context of co-treatment with MEK inhibitors. Collectively, these results indicate that JAK
activation in response to inhibition of MEK1/2 in BRAFMT CRC cells contributes to
resistance to MEK inhibitors in these cancer cells.
In addition, we found that STAT3 was activated by upstream activation of c-MET. Soluble
HGF, the ligand for c-MET, was not detected in the culture medium of BRAFMT cell line
models; however, consistent with our data in KRASMT cell lines (20), we found that MEK
regulated the levels of soluble decoy MET, the natural antagonist of c-MET, in BRAFMT
CRC models in vitro and in vivo (Supplementary Fig. S7). Based on their findings of a
Vemurafenib-induced feedback activation of EGFR and their in vivo data, previous studies
have suggested that the addition of an EGFR inhibitor to Vemurafenib could be a novel
treatment approach for BRAFMT CRC (27). Interestingly, our studies showed that the c-
MET-STAT3 signalling axis appears to have a more significant role in survival and MEKi-
induced resistance in BRAFMT CRC cells, as the Crizotinib/AZD6244 combination was a
more effective inducer of apoptosis compared to AZD6244 combined with gefitinib (EGFR
inhibitor) or AZD8931 (EGFR/HER2 inhibitor). The importance of c-MET as mediator of
acute resistance to MEK1/2 inhibitors was demonstrated in vivo, where co-treatment of
BRAFMT CRC xenografts with Crizotinib blocked AZD6244-induced STAT3 activation and
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

17
resulted in supra-additive reductions in tumor growth and marked induction of apoptosis
(24). Collectively, these results indicate that inhibitors of the c-MET pathway in conjunction
with MEK inhibitors could be a novel treatment strategy for BRAFMT CRC tumors.
Mechanistically, we found that STAT3 activation protects BRAFMT cancer cells from
apoptosis following MEK inhibition, by acutely upregulating expression of the anti-apoptotic
protein c-FLIPL. Notably, treatment with AZD6244 resulted in acute increases in c-FLIPL
mRNA and protein levels only in BRAFMT cells, and inhibition of STAT3 using stattic or
AZD9150 attenuated MEKi-induced upregulation of c-FLIPL expression and resulted in
significant increases in MEKi-induced apoptosis. Moreover, we also demonstrate for the first
time that FLIPL overexpression abrogates the effect of MEKi in BRAFMT CRC cells. We
previously have shown that c-FLIP is an important regulator of apoptosis and drug
resistance and a poor prognostic marker in CRC (17, 47, 48). Using siRNA against c-FLIP
and multiple cell line models, we now show that c-FLIP is also a critical mediator of
resistance to MEKi in BRAFMT CRC. Our previous studies and those of others have shown
that pan-HDAC inhibitors and more specific HDAC1-3 inhibitors act as efficient post-
transcriptional suppressors of FLIP expression (30, 49). Combined treatment of BRAFMT
CRC models with the HDAC1-3 inhibitor Entinostat and AZD6244 blocked AZD6244-induced
c-FLIPL upregulation and resulted in enhanced levels of apoptosis induction. In addition, we
also demonstrated that MEK inhibition in conjunction with Entinostat was highly effective at
blocking the growth of BRAFMT CRC xenografts. This is the first study showing that
combined HDAC/MEK inhibition could be a promising treatment strategy for BRAFMT CRC
patients. Before carrying forward into a clinical trial, this combination study needs to be
confirmed in additional BRAFMT models.
In conclusion, we have identified STAT3-mediated c-FLIP upregulation as an important
resistance mechanism to MEKi in BRAFMT CRC that is acutely induced as a consequence
of increases in c-MET and JAK1/2 activation (Fig. 6C). From a cancer therapeutics
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

18
perspective, the substantial tumor growth inhibition observed in our xenograft studies
support the evaluation of c-MET/MEK co-inhibition or HDAC1-3/MEK co-inhibition in clinical
trials for patients with metastatic BRAFMT colorectal cancer.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

19
REFERENCES
1. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54.
2. Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61-70.
3. Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, et al. Raf family kinases: old dogs have learned new tricks. Genes & cancer 2011;2:232-60.
4. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116:855-67.
5. Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet 2011;377:2103-14.
6. Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol 2009;27:5931-7.
7. Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 2011;29:2011-9.
8. Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 2009;101:715-21.
9. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008;26:5705-12.
10. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753-62.
11. Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes & development 2007;21:3214-31.
12. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809-19.
13. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107-14.
14. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012;367:1694-703.
15. Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Lee RJ, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol 2010;28: 15s (suppl; abstr 3534).
16. Corcoran RB, Atreya CE, Falchook GS, Infante JR, Hamid O, Messersmith WA, et al. Phase 1-2 trial of the BRAF inhibitor dabrafenib (D) plus MEK inhibitor trametinib (T) in BRAF V600 mutant colorectal cancer (CRC): Updated efficacy and biomarker analysis. J Clin Oncol 2014;32: 5s (suppl; abstr 3517).
17. Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, Galligan L, et al. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 2006;25:838-48.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

20
18. Kyula JN, Van Schaeybroeck S, Doherty J, Fenning CS, Longley DB, Johnston PG. Chemotherapy-Induced Activation of ADAM-17: A Novel Mechanism of Drug Resistance in Colorectal Cancer. Clin Cancer Res 2010;16:3378-89.
19. Van Schaeybroeck S, Kyula JN, Fenton A, Fenning CS, Sasazuki T, Shirasawa S, et al. Oncogenic Kras promotes chemotherapy-induced growth factor shedding via ADAM17. Cancer Res 2011;71:1071-80.
20. Van Schaeybroeck S, Kalimutho M, Dunne PD, Carson R, Allen W, Jithesh PV, et al. ADAM17-Dependent c-MET-STAT3 Signaling Mediates Resistance to MEK Inhibitors in KRAS Mutant Colorectal Cancer. Cell reports 2014;7:1940-55.
21. Whitehead RH, Zhang HH, Hayward IP. Retention of tissue-specific phenotype in a panel of colon carcinoma cell lines: relationship to clinical correlates. Immunol Cell Biol 1992;70 ( Pt 4):227-36.
22. Zhang HH, Walker F, Kiflemariam S, Whitehead RH, Williams D, Phillips WA, et al. Selective inhibition of proliferation in colorectal carcinoma cell lines expressing mutant APC or activated B-Raf. Int J Cancer 2009;125:297-307.
23. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009;325:1555-9.
24. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714-26.
25. Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009;16:487-97.
26. Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 2008;13:311-20.
27. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100-3.
28. Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer discovery 2013;3:520-33.
29. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500-4.
30. Kerr E, Holohan C, McLaughlin KM, Majkut J, Dolan S, Redmond K, et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ 2012;19:1317-27.
31. Riley JS, Hutchinson R, McArt DG, Crawford N, Holohan C, Paul I, et al. Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer. Cell death & disease 2013;4:e951.
32. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chemistry & biology 2006;13:1235-42.
33. Hong DS, Younes A, Fayad L, Fowler NH, Hagemeister FG, Mistry R, et al. A phase I study of ISIS 481464 (AZD9150), a first-in-human, first-in-class, antisense oligonucleotide inhibitor of STAT3, in patients with advanced cancers. J Clin Oncol 31, 2013 (suppl; abstr 8523) 2013.
34. Yaeger R, Cercek A, Chou JF, Sylvester BE, Kemeny NE, Hechtman JF, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer. Cancer 2014;120:2316-24.
35. Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

21
metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer 2012;48:1466-75.
36. Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery 2012;2:227-35.
37. Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Jr., Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 2013;19:657-67.
38. Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 2012;72:779-89.
39. Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res 2013;19:2688-98.
40. Bromberg J. Stat proteins and oncogenesis. J Clin Invest 2002;109:1139-42. 41. Li Y, Du H, Qin Y, Roberts J, Cummings OW, Yan C. Activation of the signal
transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res 2007;67:8494-503.
42. Judd LM, Bredin K, Kalantzis A, Jenkins BJ, Ernst M, Giraud AS. STAT3 activation regulates growth, inflammation, and vascularization in a mouse model of gastric tumorigenesis. Gastroenterology 2006;131:1073-85.
43. Ogata H, Kobayashi T, Chinen T, Takaki H, Sanada T, Minoda Y, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology 2006;131:179-93.
44. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug Resistance via Feedback Activation of Stat3 in Oncogene-Addicted Cancer Cells. Cancer Cell 2014;26:207-21.
45. Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012;366:1905-13.
46. Pilati C, Amessou M, Bihl MP, Balabaud C, Nhieu JT, Paradis V, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. The Journal of experimental medicine 2011;208:1359-66.
47. Wilson TR, McLaughlin KM, McEwan M, Sakai H, Rogers KM, Redmond KM, et al. c-FLIP: a key regulator of colorectal cancer cell death. Cancer Res 2007;67:5754-62.
48. McLornan DP, Barrett HL, Cummins R, McDermott U, McDowell C, Conlon SJ, et al. Prognostic significance of TRAIL signaling molecules in stage II and III colorectal cancer. Clin Cancer Res 2010;16:3442-51.
49. Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, Anthony DA, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci U S A 2008;105:11317-22.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

22
FIGURE LEGENDS
Figure 1. JAK1/2 regulate MEK inhibitor-induced STAT3 survival response in BRAFMT
CRC cells. A. Upper panel: BRAFMT and WT CRC cells were treated with AZD6244 for
24h and STAT3 and ERK1/2 expression/activity levels determined by Western blotting.
Lower panel: HT-29 and LIM2405 cells were treated with indicated doses of PD98059 (PD),
UO126 (UO), Trametinib (TRA) or AZD6244 (AZD) for 24h and STAT3 and ERK1/2
expression and activity levels determined by Western blotting. B. Left panel: LIM2405 cells
were transfected with 10nM JAK1 siRNA, JAK2 siRNA or co-transfected with 10nM JAK1
and JAK2 siRNA for 24h. Right panel: LIM2405 cells were transfected with 10nM JAK1
siRNA (J1), JAK2 siRNA (J2) or co-transfected with 10nM JAK1 and JAK2 siRNA (J1/2) for
24 hours, followed by treatment with AZD6244 for 6h. JAK1, pJAK1Y1022/1023, JAK2,
pJAK2Y1007/1008, pSTAT3Y705, STAT3, pERK1/2 and ERK1/2 levels were determined by
Western blotting. C. Upper panel: HT-29 cells were pre-incubated with AZD1480 for 3 hours
and thereafter treated with AZD6244 for 24 hours and IRF7 mRNA levels determined by
real-time PCR. Relative mRNA expression was calculated using the DDCt method with
normalisation to β-actin and GAPDH. Lower panel: HT-29 cells were co-treated with
AZD1480 and AZD6244 for 24 hours and a luciferase construct STAT3 reporter assay was
used to measure STAT3 activity. D. BRAFMT and WT cells were pre-incubated with
TG101348 (TG) or AZD1480 (AZ) for 4 hours, followed by treatment with AZD6244 for 24h.
Upper panel: PARP, cleaved caspase 3, pERK1/2 and ERK1/2 levels were determined by
Western blotting; Lower panel: apoptosis was assessed by flow cytometric analysis with
Propidium Iodide (PI).
Figure 2. Combined c-MET and MEK inhibition results in apoptosis in BRAFMT in vitro
and in vivo models. A. COLO205 and HT-29 cells were pre-treated with Crizotinib or
AZD8931 for 3h, and thereafter treated with AZD6244 for 24 hours and pHER2Y1248, HER2,
pEGFRY1068, EGFR, pcMETY1234/1235, MET, pSTAT3Y705, STAT3, pERK1/2 and ERK1/2 levels
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

23
determined by Western blotting. B. Upper panel: VACO432, VT1, LIM2405, COLO205 and
HT-29 cells were pre-treated with Crizotinib or AZD8931 for 3h, and thereafter treated with
AZD6244 for 48 hours and apoptosis was assessed by flow cytometric analysis with PI.
Lower panel: Caspase 3/7 activity levels were measured in HT-29, LIM2405 and COLO205
cells, following pre-incubation with Crizotinib or AZD8931 for 3h, and treatment with
AZD6244 for 24 hours. C. Expression levels of PARP and cleaved C3 in COLO205 and HT-
29 cells following treatment with Crizotinib or AZD8931 and AZD6244 for 24h. D. Left panel:
Growth rate of HT-29 xenografts in BALB/c Nude mice treated with vehicle, AZD6244,
Crizotinib or combined AZD6244/Crizotinib treatment. Differences in growth were
determined using Student’s t test and by calculating subsequent P values. ***, P < 0.001.
Right panel: WB analysis of cleaved C3 in HT-29 vehicle- (Vh), AZD6244-, Crizotinib-
(CRIZ) and AZD6244/Crizotinib (COMB)-treated xenografts.
Figure 3. c-FLIPL regulates sensitivity of BRAFMT CRC cells to MEK inhibition. A. HT-
29, COLO205, and LIM2405 cells were pre-treated with Crizotinib or AZD8931 for 3h, and
thereafter treated with AZD6244 for 24 hours and PARP, pro-caspase 8, caspase 8 p41/43,
caspase 8 p18, cleaved caspase 3, cleaved caspase 9, FLIPL, FLIPS, XIAP, BCL-2 and BAX
levels determined by Western blotting. B. HT-29 and COLO205 cells were treated with
AZD6244 for the indicated time. Upper panel: FLIPL, FLIPS, pSTAT3Y705, STAT3, pERK1/2
and ERK1/2 levels were determined by Western blotting. Lower panel: c-FLIP mRNA levels
were determined by real-time PCR. Relative mRNA expression was calculated using the
DDCt method with normalisation to β-actin and GAPDH. C. Upper panel: expression levels
of FLIPL and FLIPS in HT-29 empty vector (EV) and FLIPL overexpressing cells, determined
by Western blotting and Real-time PCR. Lower panel: HT-29 empty vector (EV) and FLIPL
overexpressing cells were treated with AZD6244 for 48 hours and apoptosis assessed by
flow cytometric analysis. D. BRAFMT LIM2405, RKO F6-8, HT-29 and VACO432 cells and
BRAFWT CACO-2, LIM-1215 and COLO320 cells were transfected with c-FLIP siRNA
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

24
(10nM) for 24 hours and thereafter treated with AZD6244 for 24h. PARP, cleaved caspase 3,
FLIPL, FLIPS, pERK1/2 and ERK1/2 levels were determined by Western blotting.
Figure 4. Combined HDAC1-3-MEK1/2-inhibition results in marked apoptosis in
BRAFMT CRC cells. A. HT-29, LIM2405, VACO432, RKO F6-8 and COLO205 cells were
pre-incubated with Entinostat for 12 hours (2.5μM in HT-29, LIM2405 and VACO432; 5μM in
RKO and 1μM in COLO205), followed by treatment with AZD6244 for 24h. Upper panel:
PARP, FLIPL, pSTAT3Y705, STAT3, pERK1/2 and ERK1/2 levels were determined by
Western blotting. Lower panel: Apoptosis was assessed by flow cytometric analysis. B.
LIM2405 cells were transfected with caspase 8 siRNA (10nM) for 24 hours and thereafter
treated with Entinostat and AZD6244 for 24h. PARP, cleaved caspase 3, pERK1/2, ERK1/2,
pro-caspase 8, cleaved C8 p41/43, cleaved C8 p18 levels were determined by Western
blotting.
Figure 5. Combined HDAC and MEK inhibition results in synergistic reduction in
growth of BRAFMT CRC in vivo. Upper panel: Growth rate of HT-29 xenografts in BALB/c
Nude mice treated with vehicle, AZD6244, Entinostat or combined AZD6244/Entinostat
treatment. Differences in growth were determined using Student’s t test and by calculating
subsequent P values. ***, P < 0.001. Lower panel: WB analysis of cleaved C3 and Ac-
Histone H4 in HT-29 vehicle- (Vh), AZD6244-, Entinostat- (ENT) and AZD6244/Entinostat
(COMB)-treated xenografts.
Figure 6. STAT3 regulates MEKi-induced FLIPL expression levels. A. Upper panel: HT-
29 cells were co-treated with Stattic and AZD6244 for 24 hours and pSTAT3Y705, STAT3,
FLIPL, FLIPS, pERK1/2 and ERK1/2 levels determined by Western blotting. Lower panel:
Flow cytometric analysis of HT-29 following treatment with 5μM Stattic and 1μM AZD6244
for 48 hours. B. Upper panel: pSTAT3Y705, STAT3, FLIPL, FLIPS, cleaved C3, pERK1/2 and
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

25
ERK1/2 levels in LIM2405 cells following transfection with 10nM AZD9150 for 48 hours and
treatment with AZD6244 for 24 hours. Lower panel: Flow cytometric analysis of LIM2405
following transfection 10nM AZD9150 for 48 hours and treatment with 1μM AZD6244 for 24
hours. C. Schematic overview of mechanism of STAT3 mediated c-FLIPL expression
following MEKi treatment in BRAFMT CRC.
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701

Published OnlineFirst March 26, 2015.Clin Cancer Res Robbie Carson, Basak Celtikci, Catherine S. Fenning, et al. c-FLIPL.in BRAF mutant colorectal cancer by down-regulation of HDAC inhibition overcomes acute resistance to MEK inhibition
Updated version
10.1158/1078-0432.CCR-14-2701doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2015/03/27/1078-0432.CCR-14-2701.DC1.html
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on March 31, 2015. © 2015 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2015; DOI: 10.1158/1078-0432.CCR-14-2701
Copyright © 2022 FDOKUMEN




















