
GNE genotype explains 20% of phenotypic variability in GNE ...
11
ARTICLE OPEN ACCESS GNE genotype explains 20% of phenotypic variability in GNE myopathy Oksana Pogoryelova, PhD,* Ian J. Wilson, PhD,* Hank Mansbach, MD, Zohar Argov, MD, Ichizo Nishino, PhD, and Hanns Lochm¨ uller, MD Neurol Genet 2019;5:e308. doi:10.1212/NXG.0000000000000308 Correspondence Dr. Pogoryelova [email protected] Abstract Objective To test the hypothesis that common GNE mutations influence disease severity; using statistical analysis of patient cohorts from different countries. Methods Systematic literature review identified 11 articles reporting 759 patients. GNE registry data were used as a second data set. The relative contributions of the GNE mutations, homozygosity, and country to the age at onset were explored using linear modeling, and relative importance measures were calculated. The rate of ambulation loss for GNE mutations, homozygosity, country, and age at onset was analyzed using Cox proportional hazards models. Results A spectrum of symptoms and large variability of age at onset and nonambulatory status was observed within families and cohorts. We estimated that 20% of variability is explained by GNE mutations. Individuals harboring p.Asp207Val have an expected age at onset 8.0 (s.e1.0) years later than those without and probability of continued ambulation at age 40 of 0.98 (95% confidence interval [CI] 0.96–1). In contrast, p.Leu539Ser results in onset on average 7.2 (s.e.2.7) years earlier than those without this mutation, and p.Val603Leu has a probability of continued ambulance of 0.61 (95% CI 0.50–0.74) at age 40, but has a nonsignificant effect on age at onset. Conclusions GNE myopathy severity significantly varies in all cohorts, with 20% of variability explained by the GNE mutation. Atypical symptoms and clinical presentation suggest that physical and instrumental examination should include additional clinical tests. Proven and measurable effect of GNE mutations on the disease severity should be factored in patient management and clinical research study for a better data interpretation. *Contributed equally to the study. From the Institute of Genetic Medicine (O.P., I.J.W.), Newcastle University, Newcastle upon Tyne, United Kingdom; Ultragenyx Pharmaceutical (H.M.), CA; Department of Neurology (Z.A.), Hadassah-Hebrew University Medical Center, Jerusalem, Israel; Department of Neuromuscular Research (I.N.), National Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, Japan; Department of Neuropediatrics and Muscle Disorders (H.L.), Medical Center—University of Freiburg, Faculty of Medicine, Freiburg, Germany; Centro Nacional de An´ alisis Gen´ omico (CNAG-CRG) (H.L.), Center for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona, Catalonia, Spain; and Children’s Hospital of Eastern Ontario Research Institute (H.L.), University of Ottawa, Ottawa, Canada and Division of Neurology, Department of Medicine, The Ottawa Hospital, Ottawa, Canada. Supported by TREAT-NMD operating grants, FP6 LSHM-CT-2006-036825, 20123307 UNEW FY2013, and AFM 16104. Further support came from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 305444 (RD-Connect) and Medical Research Council, UK (reference G1002274, grant ID 98482). Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG. The Article Processing Charge was funded by the RCUK. This is an open access article distributed under the terms of the Creative Commons Attribution License 4.0 (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Copyright © 2019 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. 1
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of GNE genotype explains 20% of phenotypic variability in GNE ...

ARTICLE OPEN ACCESS
GNE genotype explains 20 of phenotypicvariability in GNE myopathyOksana Pogoryelova PhD Ian J Wilson PhD Hank Mansbach MD Zohar Argov MD Ichizo Nishino PhD
and Hanns Lochmuller MD
Neurol Genet 20195e308 doi101212NXG0000000000000308
Correspondence
Dr Pogoryelova
Oksanapogoryelovanclacuk
AbstractObjectiveTo test the hypothesis that commonGNEmutations influence disease severity using statisticalanalysis of patient cohorts from different countries
MethodsSystematic literature review identified 11 articles reporting 759 patients GNE registry datawere used as a second data set The relative contributions of theGNEmutations homozygosityand country to the age at onset were explored using linear modeling and relative importancemeasures were calculated The rate of ambulation loss for GNE mutations homozygositycountry and age at onset was analyzed using Cox proportional hazards models
ResultsA spectrum of symptoms and large variability of age at onset and nonambulatory status wasobserved within families and cohorts We estimated that 20 of variability is explained by GNEmutations Individuals harboring pAsp207Val have an expected age at onset 80 (se10) yearslater than those without and probability of continued ambulation at age 40 of 098 (95confidence interval [CI] 096ndash1) In contrast pLeu539Ser results in onset on average 72(se27) years earlier than those without this mutation and pVal603Leu has a probability ofcontinued ambulance of 061 (95 CI 050ndash074) at age 40 but has a nonsignificant effect onage at onset
ConclusionsGNE myopathy severity significantly varies in all cohorts with 20 of variability explained bythe GNE mutation Atypical symptoms and clinical presentation suggest that physical andinstrumental examination should include additional clinical tests Proven and measurable effectofGNEmutations on the disease severity should be factored in patient management and clinicalresearch study for a better data interpretation
Contributed equally to the study
From the Institute of Genetic Medicine (OP IJW) Newcastle University Newcastle upon Tyne United Kingdom Ultragenyx Pharmaceutical (HM) CA Department of Neurology(ZA) Hadassah-Hebrew University Medical Center Jerusalem Israel Department of Neuromuscular Research (IN) National Institute of Neuroscience National Center ofNeurology and Psychiatry Tokyo Japan Department of Neuropediatrics and Muscle Disorders (HL) Medical CentermdashUniversity of Freiburg Faculty ofMedicine Freiburg GermanyCentro Nacional de Analisis Genomico (CNAG-CRG) (HL) Center for Genomic Regulation Barcelona Institute of Science and Technology (BIST) Barcelona Catalonia Spain andChildrenrsquosHospital of EasternOntario Research Institute (HL) University ofOttawaOttawa Canada andDivision ofNeurology Department ofMedicine TheOttawaHospital OttawaCanada
Supported by TREAT-NMDoperating grants FP6 LSHM-CT-2006-036825 20123307UNEWFY2013 and AFM16104 Further support came from the EuropeanUnion Seventh FrameworkProgramme (FP72007-2013) under grant agreement No 305444 (RD-Connect) and Medical Research Council UK (reference G1002274 grant ID 98482)
Funding information and disclosures are provided at the end of the article Full disclosure form information provided by the authors is available with the full text of this article atNeurologyorgNG
The Article Processing Charge was funded by the RCUK
This is an open access article distributed under the terms of the Creative Commons Attribution License 40 (CC BY) which permits unrestricted use distribution and reproduction in anymedium provided the original work is properly cited
Copyright copy 2019 The Author(s) Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology 1
GNE myopathy is an autosomal recessive rare distal myopathyIt is a slow progressing disease caused by mutations in the GNEgene which affects sialic acid synthesis Clinical course variesfrom mild to severely debilitating forms leading to end-stageskeletal muscle weakness The genetic basis for GNE myopathywas established in 20011 more than 180mutations are currentlyknown among them several founder or recurrent mutations2ndash7
Wide phenotypic variability is described in the literature evenwithin families carrying the same mutation Some unique fea-tures of GNE myopathy are observed only in certainpopulations89 Population-based studies suggest that GNE ge-notype may influence predisposition to a faster or slower pro-gression of the disease but were often not large enough to reachstatistical significance5610 Molecular mechanisms underlyingmuscle weakness have been studied in cell and enzyme studiessuggesting that different missense mutations may affect enzy-matic activity to a different extent Primary muscle cells withGNE mutations showed a significant reduction in sialic acidlevels11 suggesting that this might be the cause of the muscleweakness A defined link betweenGNE genotype and phenotypewould have a major impact on the clinical trial planning androutine disease management and care The aim of the study is toexplore cohort-based studies to test the hypothesis that GNEgenotype influences GNE myopathy disease severity
MethodsLiterature reviewPubMed database search was conducted on 12 May 2017using search words ldquoGNE myopathyrdquo ldquoHIBMrdquo ldquoquadricepssparing myopathyrdquo ldquodistal myopathy with rimmed vacuolesrdquoIn total 494 records were identified Duplicated recordssingle case reports and scientific publication based on mo-lecular studies cells and animal models were eliminatedOther records were checked for the following cohort size ge15patients availability of demographic and clinical descriptionand description of the GNE mutations in the cohort Elevenarticles were selected for analysis (total 759 cases table 1)Seven articles contained per patient data (UK India BulgariaIran China 1 China 2 and Japan 3) and 4 articles (IsraelJapan 1 Japan 2 and Japan 4) provided cohort summaryinformation In total n = 360 patients had individual leveldata n = 342 with defined age at onset and n = 247 withknown ambulation status
Registry dataGNE myopathy registry (previously described12) was used asanother independent data set The registry data were in-terrogated for number of patients per country (subset A) andfor patients with homozygous mutations regardless of theircountry of residence (subset B) Six countries were presented
by ge14 patients (total n = 135) Four GNE mutations werepresent in the homozygous state ge3 patients (total n = 31)The study received approval from Newcastle and NorthTyneside 1 Research Ethics Committee reference number13NE0123 Informed consent was obtained from allpatients participating in the registry
Statistical modeling of age at onsetIndividuals with known genotype and known age at onset wereselected for this analysis (n = 342) Those variants with at least10 observations in the data set were used for further analysisTen mutations (table 2) were 77 of the total changes in thedata Genetic explanatory variables were constructed for eachmutation under a dominance model where the variable iscoded for the presenceabsence of a mutation and allelic dos-age model that gives the allele count (0 1 or 2) for the 10changes Other explanatory variables used were the geographiclocation of the study and whether an individual was homozy-gous A series of linear models were fitted for the age at onsetregressed against all combinations of location andor homo-zygosity and modeling the genetic contribution as either allelicdose or dominance models using the R statistical package13
Selection of the better fitting models was done using thestepwise regression with model choice made using the AkaikeInformation Criterion (AIC)14 The relative importance ofthe different factors in explaining variation in the age at onsetwas assessed using the relaimpo package within R15 The bestfitting models were then used to predict the age at onset forindividuals in the GNE registry
Age of ambulation lossIndividuals in the onset data that had information aboutambulation status either continued ambulation at assessmentor the age at loss of ambulation (n = 220) were used foranalysis of ambulation status Mutations with more than 10observations in the data set were used for further analysis (n =6 mutations table 2) Cox proportional hazards models16
were used to evaluate the relationship of continued ambula-tion to the genotype data as described above the study andthe age at onset Initially univariate survival models were fittedand a threshold p value of 01 was used to select those vari-ables to consider in a multivariate analysis A multivariatemodel was chosen from the best combination of significantunivariate models Choices between non-nested models weremade using the AIC Model fit was assessed using survivalconcordance for registry individuals
Data availability statementThe study analyzes published data Anonymized GNE registrydata can be shared upon request to the GNE Registry SteeringCommittee
GlossaryCI = confidence interval Ace-ER = N-acetylneuraminic acid extended release AIC = Akaike Information Criterion
2 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Table 1 Demographic description of the GNE cohorts
Countrywhere thestudy wasconducted
Ethnicalbackground
Numberofpatients
Percentageof malefemale ratio(n)
Age atbaselinemean (plusmnSD)
Mean age atonset (plusmnSD)[range]
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD)
GNE geneticsdescription of thecohort
Cardiacinvolvement
Respiratoryinvolvement
Japan 19 Japanese 121 455 (5566) 277 (96) 430 (52) 354 (113)mean 211 yafter the onset
pV603L and pD207Vaccount for 710 ofcases
Normal in all testedpatients (289)
FVC normal (33)decreased in 67patients
Japan 217 Japanese 27 333 (918) 430 (129) 259 (103)[15ndash58]
360 (9) Not available pV603L and pD207Vaccount for 750 of allmutations
Structural andrhythmabnormalities in296 patients
OSASa (n = 2) FVCdrop minus33 in 1 y innonambulant subset
Japan 324 Japanese 212 448 (95117) 416 (141) 284 SD102 [10-61]
234 (154) 368 (113)[range 19ndash78]
pV603L and pD207Vaccount for 887 allmutations
Not available Not available
Japan 425 Japanese 71 338 (2444) 431 (130) 248 SD 83 [12-58]
520 (37)[50-80 ]
349 (117)[range 18ndash70]Mean 170 (21)y after the onset
pV603L and pD207V862 of all mutations
Not available Not available
Bulgaria5 Roma people 50 620 (3119) Not available 244 (631)[10ndash42]
640 (32) Not availablemean 1034(431) y afteronset
pI618T account for 98of all mutations
Structural andrhythmabnormalities in455 patients
FVC [range 60ndash75]
UK7 White BritishIrish IndianPakistan
26 539 (1412) 477 30 (92) [10ndash44] 192 (5) 463 (80) pA662V and pD409Yaccount for 63
Mild systolicdysfunction- 1patient
Normal in all patients
Israel4 Middle EasternJews KaraitesArabs
129 Not available Not available [17ndash48] Not available Not availablegt15 y sinceonset
pM743T accounts for100 of all mutations
None Not available
Iran10 Iranian non-Jewish (MuslimPersian)
18 500 (99) 34 [range24ndash47]
257 [19ndash31] 167 (3) 373 13ndash15 ysince onset
High prevalence ofpM743T
Normal in allpatients
Normal in all patients
China 16 Chinese 35 457 (1619) 373 (102)[range 22ndash66]
306 (73)[20ndash43]
Not available Not available pD207V allelefrequency in the cohortof 357
Not available Not available
China 226 Chinese 53 509 (2726) 315 (84) [range11ndash46]
205 (81) [5-39] Not available Not available No single dominatingmutation in the studycohort
Not available Not available
India8 Indians 17 706 (125) 341 (71) [range25ndash49]
270 (63)[21ndash46]
59 (1) Not available 765 patients harbourpV727M
Not available Not available
a Obstructive sleep apnoea syndrome
Neurolo
gyorgN
GNeurologyG
enetics|
Volume5N
umber
1|
February
20193
ResultsAge at onsetEleven cohorts were selected for analysis Four publicationsfrom Japan met the inclusion criteria and may have over-lapping patients Two publications from China may haveoverlapping patients but this is unlikely The overlap in pa-tient populations between those cohorts cannot be de-termined therefore they were analyzed as individual patientcohorts Bulgaria UK Israel Iran and India are presented bya single publication (table 1)
Number of patients in populations varied between 17 (India)and 212 (Japan) Average age at baseline varied between 315years (China) and 477 years (UK) The earliest onset at 10years was detected in Japan UK and Bulgaria while the latestonset was observed at 61 years in Japan giving Japan thelargest variability of age at onset (figure 1)
Linear models were fitted to the age at onset on those indi-viduals with information about genotype and age at onset (n =341) Genetic variables provided a better fit to the data thanlocation and homozygosity (maximum adjusted r2 was 008without individual mutation explanatory variables) Modelsusing allelic dose models were marginally worse than domi-nance models Statistically significant mutations werepAsp207Val (increase in age at onset of 8 years p = 9e-16)pAla631Val (increase of 48 years p = 0002) andpLeu539Ser (decrease of 72 years p = 0008) Confidenceintervals are given in table 2 The proportion of variabilityexplained by these 3 genetic factors was estimated to be188 using the reliampo package For univariate models theage at onset was a highly significant predictor of continued
ambulance (p lt 2e-16) along with individual homozygosity(p = 7e-16) and the mutations pVal603Leu (p = 0007)pAsp207Val (p = 00002) and pILe618Thr (p = 34e-14)In a multivariate model using all variables homozygositywas only marginally significant (p gt 001) Age at onset anddominance for the 3 mutations mdash the change in hazard forthe 3 mutationsmdashwas used in the final model (table 2) Theeffect of the age at onset and the 3 mutations (figure 2) Wecan show the effects of the 3 mutations by comparingmutations to an individual with onset at age 30 and withoutany of the 3 mutations who has a probability of 085 (95confidence interval [CI] 077ndash094) of continued ambulanceat age 40
Age of ambulationLowest proportion of nonambulant patients was described inIndia (59) and the highest in Bulgaria (640) resulting inalmost every third GNE patient in the world being non-ambulant (325) Authors of the selected manuscripts de-scribed reaching nonambulatory status in 2 ways (1) Age atnonambulatory status (5 publications) and duration(ie years) since the onset to nonambulatory status (3 pub-lications) The former shows that patients reach non-ambulatory status at an average 381 years old with overa decade (114 years) difference between ldquoJapan 4rdquo and UKcohorts The latter shows that the progress since onset to thewheelchair bound state can take between 103 and 211 yearswith an average of 162 years
A mutation at pVal603Leu gives a probability of ambulanceat age 40 of 061 (95 CI 049ndash074) a mutation atpIleThr gives 073 (95 CI 0163ndash083) and a mutation atpAsp207Val has a probability of continued ambulance of 098
Table 2 Genomic positions and amino acid changes for the 10 changes analyzed in the onset data
Genomicposition
Amino acidchange Ref SNP ID
Frequencyin onsetdata
Populationspresent
CountinExAC
Count in1000genomes
Frequencyambulationdata
Estimatedchange inage atonset
Estimatedhazard formodel amb2(95 CI)
36219937 pVal603Leu rs121908632 029 2 3 1 034 31 (15ndash63)
36246117 pAsp207Val rs139425890 017 2 5 2 015 80 (6199) 012 (003ndash05)
36219891 pILe618Thr 016 4 1 0 023 19 (096ndash39)
36218221 pAla631Val rs62541771 0047 3 10 0 (0002) 48 (12 85)
36217396 pMet743Leu rs28937594 0033 2 4 0 0043
36249315 pCys44Ser 0027 2 0 0 (0007)
36236861 pArg277Trp rs121908629 0020 4 0 0 0025
36236887 pLeu268Leu 0018 1 0 0 0027
36227394 pAsp409Tyr rs199877522 0017 1 14 0 (0)
36222884 pLeu539Ser 0015 1 0 0 (0) minus72 (minus127-18)
All genomic positions are on chromosome 9 and based on amino acid changesmapped to transcript ENST00000377902 Changeswith bracketed frequenciesin the ambulation data were not included in the ambulation model
4 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
(95 CI 096ndash1) When considering only the Japanese co-hort we confirm that the presence of pAsp207Val is associ-ated with a muchmilder phenotype (probability of ambulanceat age 40 is 098 95 CI [094ndash10]) compared to individualswith no copies probability 058 [045 073] Homozygotes ofpVal603Leu have a much more severe phenotype withprobability of continued ambulance of 039 95 CI[024ndash061] compared to a heterozygote probability 09095 CI [080ndash10]
GenotypesFounder or high-prevalence mutations were significantly prev-alent in 9 cohorts In 2 populations (Israel and Bulgaria) foundermutation appears in nearly all patients in homozygous stateJapan and UK showed 2 mutations dominating the cohortsChina 1 and China 2 cohorts showed a different spectrum ofmutations with one dominating mutation in former study andmultiple mutations without clear dominance in the latter
Cardiorespiratory functionIt is considered that GNEmyopathy does not increase the risk ofcardiomyopathy and respiratory failure However a 1 yearlongnatural history study conducted in Japan showed that forced vitalcapacity (FVC) declines in nonambulatory patients17 De-scription of the respiratory function was available in 5 of 10studies UK and Iran described it as ldquonormal in all patientsrdquo FVCwas either normal or mildly to moderately decreased in all other
tested patients No severe decline in FVC was reported Car-diomyopathy was not reported nevertheless some structuraland rhythm abnormalities were present in 296 and 455 ofpatients from ldquoJapan 2rdquo and Bulgaria respectively Reportedabnormalities included borderline reduction in ejection fractionand impaired relaxation (in patients who had concomitant dis-eases such as diabetes or hypertension) ECG in a few patientsshowed evidence of abnormal repolarization and left ventricularhypertrophy Rhythm abnormalities such as extrasystolic ar-rhythmia and borderline nonspecific intraventricular delay wereobserved on a few occasions Sudden death of unknown causewas reported in 4 nonambulant patients without a family historyof cardiac rhythm abnormalities5
Atypical featuresMuscle biopsy appearance without rimmed vacuoles wasobserved in the UK and Japan and is likely to be linked to thesite of the biopsy (ie quadriceps as the least affected muscle)or early stage of the disease Thrombocytopenia was reportedin 2 Japanese cohorts (ldquoJapan 1rdquo and ldquoJapan 2rdquo) A high rate ofan early onset presentation (lt20 years) was reported in ldquoJa-pan 4rdquo cohort Asymmetrical weakness was evident in the UKand both Chinese cohorts Quadriceps sparing almost a pa-thognomonic feature of GNEmyopathy was absent in a smallnumber of patients from Israel China 1 and China 2 cohortsFacial and neck weakness and limb-girdle muscle weakness atearly stages of the disease were observed in China and Israel
Figure 1 Forest plot showing data overview by onset on a cohort level
Studies are grouped by source individual level data (bottom panel) summary statistics form articles (middle panel) and by country from the registry (toppanel with dark background) Means are given by large circles the confidence intervals by thick black lines and the minimum and maximum values by xSample sizes for each study are indicated to the right The overall mean (calculated from individual level data) is shown by the vertical grey line Student-tconfidence intervals were calculated for registry countries and individual studies Confidence intervals for summary studies were taken from the articles
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 5
Analysis of the registry cohortAge at onset and nonambulatory status were compared between6 country-specific cohorts with n ge 14 patients Baseline data arepresented in table 3 Using the model and parameter valuesestimated in from section 6 to predict onset times for the registrydata (n = 126)was not fully successful with an r2 of 004 betweenthe self-reported onset values in the registry data and the onsetvalues predicted from this model based on the registry geneticdata So there is a relationship between estimated onset time andpredicted time but this is not as strong as in the meta data set
Registry data with ambulance data had a sample size of 113Survival concordance for registry individuals was calculated usingthe parameters from the best fit survival model This workson pairs of individuals and averages how often the individualwith the lowest ambulance time also has the highest risk scoreThis is then averaged over all pairs of individuals Hence perfectmatches would have a concordance of 1 and a model that wasrandom would have a concordance of 05 The concordance forthe registry data was 078 which shows good agreement
Registry data were also interrogated to identify homozygouspatients irrespectively of their country of residence Four sub-sets with the number of patients ge4 were analyzed Because of
a small number of available data we present it only for de-scriptive analysis Baseline demographic data genotype andclinical parameters are presented in table 3
DiscussionDisease severity and variability in its presentation are factors toconsider in the disease management and planning clinical re-search Patients are keen to know their prognosis and likelytrajectory of progression tomake personal choices Rare diseasestatus leads to scarcity of the data and availability of biologicalsamples to study Therefore an integrated analysis of theavailable data provides an additional insight in understandingthe correlation between GNE genotype and phenotype
Cohort-based studies provided country-specific data Analysisof these cohorts showed the presence of outliers with an earlyor late onset of the first symptoms in every cohort which isnot explained by a particular mutation We find that almost20 of the variability in age at onset is due to the GNEgenotype with the change to pAsp207Val being associatedwith on average an 8-year increase in age at onset In contrastpLeu539Ser results in onset on average 72 years earlier thanthose without this mutation
Figure 2 Plots of probability of loss of ambulation with age for Cox-proportional hazard models
(A) shows fitted values for model 2 withage for an individual withoutpVal603Leu pIle618Thr orpAsp207Val (B C and D) give expectedcurves for an individual with and with-out the given mutation with shadedareas indicating 90 confidenceintervals
6 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

GNE myopathy is an autosomal recessive rare distal myopathyIt is a slow progressing disease caused by mutations in the GNEgene which affects sialic acid synthesis Clinical course variesfrom mild to severely debilitating forms leading to end-stageskeletal muscle weakness The genetic basis for GNE myopathywas established in 20011 more than 180mutations are currentlyknown among them several founder or recurrent mutations2ndash7
Wide phenotypic variability is described in the literature evenwithin families carrying the same mutation Some unique fea-tures of GNE myopathy are observed only in certainpopulations89 Population-based studies suggest that GNE ge-notype may influence predisposition to a faster or slower pro-gression of the disease but were often not large enough to reachstatistical significance5610 Molecular mechanisms underlyingmuscle weakness have been studied in cell and enzyme studiessuggesting that different missense mutations may affect enzy-matic activity to a different extent Primary muscle cells withGNE mutations showed a significant reduction in sialic acidlevels11 suggesting that this might be the cause of the muscleweakness A defined link betweenGNE genotype and phenotypewould have a major impact on the clinical trial planning androutine disease management and care The aim of the study is toexplore cohort-based studies to test the hypothesis that GNEgenotype influences GNE myopathy disease severity
MethodsLiterature reviewPubMed database search was conducted on 12 May 2017using search words ldquoGNE myopathyrdquo ldquoHIBMrdquo ldquoquadricepssparing myopathyrdquo ldquodistal myopathy with rimmed vacuolesrdquoIn total 494 records were identified Duplicated recordssingle case reports and scientific publication based on mo-lecular studies cells and animal models were eliminatedOther records were checked for the following cohort size ge15patients availability of demographic and clinical descriptionand description of the GNE mutations in the cohort Elevenarticles were selected for analysis (total 759 cases table 1)Seven articles contained per patient data (UK India BulgariaIran China 1 China 2 and Japan 3) and 4 articles (IsraelJapan 1 Japan 2 and Japan 4) provided cohort summaryinformation In total n = 360 patients had individual leveldata n = 342 with defined age at onset and n = 247 withknown ambulation status
Registry dataGNE myopathy registry (previously described12) was used asanother independent data set The registry data were in-terrogated for number of patients per country (subset A) andfor patients with homozygous mutations regardless of theircountry of residence (subset B) Six countries were presented
by ge14 patients (total n = 135) Four GNE mutations werepresent in the homozygous state ge3 patients (total n = 31)The study received approval from Newcastle and NorthTyneside 1 Research Ethics Committee reference number13NE0123 Informed consent was obtained from allpatients participating in the registry
Statistical modeling of age at onsetIndividuals with known genotype and known age at onset wereselected for this analysis (n = 342) Those variants with at least10 observations in the data set were used for further analysisTen mutations (table 2) were 77 of the total changes in thedata Genetic explanatory variables were constructed for eachmutation under a dominance model where the variable iscoded for the presenceabsence of a mutation and allelic dos-age model that gives the allele count (0 1 or 2) for the 10changes Other explanatory variables used were the geographiclocation of the study and whether an individual was homozy-gous A series of linear models were fitted for the age at onsetregressed against all combinations of location andor homo-zygosity and modeling the genetic contribution as either allelicdose or dominance models using the R statistical package13
Selection of the better fitting models was done using thestepwise regression with model choice made using the AkaikeInformation Criterion (AIC)14 The relative importance ofthe different factors in explaining variation in the age at onsetwas assessed using the relaimpo package within R15 The bestfitting models were then used to predict the age at onset forindividuals in the GNE registry
Age of ambulation lossIndividuals in the onset data that had information aboutambulation status either continued ambulation at assessmentor the age at loss of ambulation (n = 220) were used foranalysis of ambulation status Mutations with more than 10observations in the data set were used for further analysis (n =6 mutations table 2) Cox proportional hazards models16
were used to evaluate the relationship of continued ambula-tion to the genotype data as described above the study andthe age at onset Initially univariate survival models were fittedand a threshold p value of 01 was used to select those vari-ables to consider in a multivariate analysis A multivariatemodel was chosen from the best combination of significantunivariate models Choices between non-nested models weremade using the AIC Model fit was assessed using survivalconcordance for registry individuals
Data availability statementThe study analyzes published data Anonymized GNE registrydata can be shared upon request to the GNE Registry SteeringCommittee
GlossaryCI = confidence interval Ace-ER = N-acetylneuraminic acid extended release AIC = Akaike Information Criterion
2 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Table 1 Demographic description of the GNE cohorts
Countrywhere thestudy wasconducted
Ethnicalbackground
Numberofpatients
Percentageof malefemale ratio(n)
Age atbaselinemean (plusmnSD)
Mean age atonset (plusmnSD)[range]
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD)
GNE geneticsdescription of thecohort
Cardiacinvolvement
Respiratoryinvolvement
Japan 19 Japanese 121 455 (5566) 277 (96) 430 (52) 354 (113)mean 211 yafter the onset
pV603L and pD207Vaccount for 710 ofcases
Normal in all testedpatients (289)
FVC normal (33)decreased in 67patients
Japan 217 Japanese 27 333 (918) 430 (129) 259 (103)[15ndash58]
360 (9) Not available pV603L and pD207Vaccount for 750 of allmutations
Structural andrhythmabnormalities in296 patients
OSASa (n = 2) FVCdrop minus33 in 1 y innonambulant subset
Japan 324 Japanese 212 448 (95117) 416 (141) 284 SD102 [10-61]
234 (154) 368 (113)[range 19ndash78]
pV603L and pD207Vaccount for 887 allmutations
Not available Not available
Japan 425 Japanese 71 338 (2444) 431 (130) 248 SD 83 [12-58]
520 (37)[50-80 ]
349 (117)[range 18ndash70]Mean 170 (21)y after the onset
pV603L and pD207V862 of all mutations
Not available Not available
Bulgaria5 Roma people 50 620 (3119) Not available 244 (631)[10ndash42]
640 (32) Not availablemean 1034(431) y afteronset
pI618T account for 98of all mutations
Structural andrhythmabnormalities in455 patients
FVC [range 60ndash75]
UK7 White BritishIrish IndianPakistan
26 539 (1412) 477 30 (92) [10ndash44] 192 (5) 463 (80) pA662V and pD409Yaccount for 63
Mild systolicdysfunction- 1patient
Normal in all patients
Israel4 Middle EasternJews KaraitesArabs
129 Not available Not available [17ndash48] Not available Not availablegt15 y sinceonset
pM743T accounts for100 of all mutations
None Not available
Iran10 Iranian non-Jewish (MuslimPersian)
18 500 (99) 34 [range24ndash47]
257 [19ndash31] 167 (3) 373 13ndash15 ysince onset
High prevalence ofpM743T
Normal in allpatients
Normal in all patients
China 16 Chinese 35 457 (1619) 373 (102)[range 22ndash66]
306 (73)[20ndash43]
Not available Not available pD207V allelefrequency in the cohortof 357
Not available Not available
China 226 Chinese 53 509 (2726) 315 (84) [range11ndash46]
205 (81) [5-39] Not available Not available No single dominatingmutation in the studycohort
Not available Not available
India8 Indians 17 706 (125) 341 (71) [range25ndash49]
270 (63)[21ndash46]
59 (1) Not available 765 patients harbourpV727M
Not available Not available
a Obstructive sleep apnoea syndrome
Neurolo
gyorgN
GNeurologyG
enetics|
Volume5N
umber
1|
February
20193
ResultsAge at onsetEleven cohorts were selected for analysis Four publicationsfrom Japan met the inclusion criteria and may have over-lapping patients Two publications from China may haveoverlapping patients but this is unlikely The overlap in pa-tient populations between those cohorts cannot be de-termined therefore they were analyzed as individual patientcohorts Bulgaria UK Israel Iran and India are presented bya single publication (table 1)
Number of patients in populations varied between 17 (India)and 212 (Japan) Average age at baseline varied between 315years (China) and 477 years (UK) The earliest onset at 10years was detected in Japan UK and Bulgaria while the latestonset was observed at 61 years in Japan giving Japan thelargest variability of age at onset (figure 1)
Linear models were fitted to the age at onset on those indi-viduals with information about genotype and age at onset (n =341) Genetic variables provided a better fit to the data thanlocation and homozygosity (maximum adjusted r2 was 008without individual mutation explanatory variables) Modelsusing allelic dose models were marginally worse than domi-nance models Statistically significant mutations werepAsp207Val (increase in age at onset of 8 years p = 9e-16)pAla631Val (increase of 48 years p = 0002) andpLeu539Ser (decrease of 72 years p = 0008) Confidenceintervals are given in table 2 The proportion of variabilityexplained by these 3 genetic factors was estimated to be188 using the reliampo package For univariate models theage at onset was a highly significant predictor of continued
ambulance (p lt 2e-16) along with individual homozygosity(p = 7e-16) and the mutations pVal603Leu (p = 0007)pAsp207Val (p = 00002) and pILe618Thr (p = 34e-14)In a multivariate model using all variables homozygositywas only marginally significant (p gt 001) Age at onset anddominance for the 3 mutations mdash the change in hazard forthe 3 mutationsmdashwas used in the final model (table 2) Theeffect of the age at onset and the 3 mutations (figure 2) Wecan show the effects of the 3 mutations by comparingmutations to an individual with onset at age 30 and withoutany of the 3 mutations who has a probability of 085 (95confidence interval [CI] 077ndash094) of continued ambulanceat age 40
Age of ambulationLowest proportion of nonambulant patients was described inIndia (59) and the highest in Bulgaria (640) resulting inalmost every third GNE patient in the world being non-ambulant (325) Authors of the selected manuscripts de-scribed reaching nonambulatory status in 2 ways (1) Age atnonambulatory status (5 publications) and duration(ie years) since the onset to nonambulatory status (3 pub-lications) The former shows that patients reach non-ambulatory status at an average 381 years old with overa decade (114 years) difference between ldquoJapan 4rdquo and UKcohorts The latter shows that the progress since onset to thewheelchair bound state can take between 103 and 211 yearswith an average of 162 years
A mutation at pVal603Leu gives a probability of ambulanceat age 40 of 061 (95 CI 049ndash074) a mutation atpIleThr gives 073 (95 CI 0163ndash083) and a mutation atpAsp207Val has a probability of continued ambulance of 098
Table 2 Genomic positions and amino acid changes for the 10 changes analyzed in the onset data
Genomicposition
Amino acidchange Ref SNP ID
Frequencyin onsetdata
Populationspresent
CountinExAC
Count in1000genomes
Frequencyambulationdata
Estimatedchange inage atonset
Estimatedhazard formodel amb2(95 CI)
36219937 pVal603Leu rs121908632 029 2 3 1 034 31 (15ndash63)
36246117 pAsp207Val rs139425890 017 2 5 2 015 80 (6199) 012 (003ndash05)
36219891 pILe618Thr 016 4 1 0 023 19 (096ndash39)
36218221 pAla631Val rs62541771 0047 3 10 0 (0002) 48 (12 85)
36217396 pMet743Leu rs28937594 0033 2 4 0 0043
36249315 pCys44Ser 0027 2 0 0 (0007)
36236861 pArg277Trp rs121908629 0020 4 0 0 0025
36236887 pLeu268Leu 0018 1 0 0 0027
36227394 pAsp409Tyr rs199877522 0017 1 14 0 (0)
36222884 pLeu539Ser 0015 1 0 0 (0) minus72 (minus127-18)
All genomic positions are on chromosome 9 and based on amino acid changesmapped to transcript ENST00000377902 Changeswith bracketed frequenciesin the ambulation data were not included in the ambulation model
4 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
(95 CI 096ndash1) When considering only the Japanese co-hort we confirm that the presence of pAsp207Val is associ-ated with a muchmilder phenotype (probability of ambulanceat age 40 is 098 95 CI [094ndash10]) compared to individualswith no copies probability 058 [045 073] Homozygotes ofpVal603Leu have a much more severe phenotype withprobability of continued ambulance of 039 95 CI[024ndash061] compared to a heterozygote probability 09095 CI [080ndash10]
GenotypesFounder or high-prevalence mutations were significantly prev-alent in 9 cohorts In 2 populations (Israel and Bulgaria) foundermutation appears in nearly all patients in homozygous stateJapan and UK showed 2 mutations dominating the cohortsChina 1 and China 2 cohorts showed a different spectrum ofmutations with one dominating mutation in former study andmultiple mutations without clear dominance in the latter
Cardiorespiratory functionIt is considered that GNEmyopathy does not increase the risk ofcardiomyopathy and respiratory failure However a 1 yearlongnatural history study conducted in Japan showed that forced vitalcapacity (FVC) declines in nonambulatory patients17 De-scription of the respiratory function was available in 5 of 10studies UK and Iran described it as ldquonormal in all patientsrdquo FVCwas either normal or mildly to moderately decreased in all other
tested patients No severe decline in FVC was reported Car-diomyopathy was not reported nevertheless some structuraland rhythm abnormalities were present in 296 and 455 ofpatients from ldquoJapan 2rdquo and Bulgaria respectively Reportedabnormalities included borderline reduction in ejection fractionand impaired relaxation (in patients who had concomitant dis-eases such as diabetes or hypertension) ECG in a few patientsshowed evidence of abnormal repolarization and left ventricularhypertrophy Rhythm abnormalities such as extrasystolic ar-rhythmia and borderline nonspecific intraventricular delay wereobserved on a few occasions Sudden death of unknown causewas reported in 4 nonambulant patients without a family historyof cardiac rhythm abnormalities5
Atypical featuresMuscle biopsy appearance without rimmed vacuoles wasobserved in the UK and Japan and is likely to be linked to thesite of the biopsy (ie quadriceps as the least affected muscle)or early stage of the disease Thrombocytopenia was reportedin 2 Japanese cohorts (ldquoJapan 1rdquo and ldquoJapan 2rdquo) A high rate ofan early onset presentation (lt20 years) was reported in ldquoJa-pan 4rdquo cohort Asymmetrical weakness was evident in the UKand both Chinese cohorts Quadriceps sparing almost a pa-thognomonic feature of GNEmyopathy was absent in a smallnumber of patients from Israel China 1 and China 2 cohortsFacial and neck weakness and limb-girdle muscle weakness atearly stages of the disease were observed in China and Israel
Figure 1 Forest plot showing data overview by onset on a cohort level
Studies are grouped by source individual level data (bottom panel) summary statistics form articles (middle panel) and by country from the registry (toppanel with dark background) Means are given by large circles the confidence intervals by thick black lines and the minimum and maximum values by xSample sizes for each study are indicated to the right The overall mean (calculated from individual level data) is shown by the vertical grey line Student-tconfidence intervals were calculated for registry countries and individual studies Confidence intervals for summary studies were taken from the articles
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 5
Analysis of the registry cohortAge at onset and nonambulatory status were compared between6 country-specific cohorts with n ge 14 patients Baseline data arepresented in table 3 Using the model and parameter valuesestimated in from section 6 to predict onset times for the registrydata (n = 126)was not fully successful with an r2 of 004 betweenthe self-reported onset values in the registry data and the onsetvalues predicted from this model based on the registry geneticdata So there is a relationship between estimated onset time andpredicted time but this is not as strong as in the meta data set
Registry data with ambulance data had a sample size of 113Survival concordance for registry individuals was calculated usingthe parameters from the best fit survival model This workson pairs of individuals and averages how often the individualwith the lowest ambulance time also has the highest risk scoreThis is then averaged over all pairs of individuals Hence perfectmatches would have a concordance of 1 and a model that wasrandom would have a concordance of 05 The concordance forthe registry data was 078 which shows good agreement
Registry data were also interrogated to identify homozygouspatients irrespectively of their country of residence Four sub-sets with the number of patients ge4 were analyzed Because of
a small number of available data we present it only for de-scriptive analysis Baseline demographic data genotype andclinical parameters are presented in table 3
DiscussionDisease severity and variability in its presentation are factors toconsider in the disease management and planning clinical re-search Patients are keen to know their prognosis and likelytrajectory of progression tomake personal choices Rare diseasestatus leads to scarcity of the data and availability of biologicalsamples to study Therefore an integrated analysis of theavailable data provides an additional insight in understandingthe correlation between GNE genotype and phenotype
Cohort-based studies provided country-specific data Analysisof these cohorts showed the presence of outliers with an earlyor late onset of the first symptoms in every cohort which isnot explained by a particular mutation We find that almost20 of the variability in age at onset is due to the GNEgenotype with the change to pAsp207Val being associatedwith on average an 8-year increase in age at onset In contrastpLeu539Ser results in onset on average 72 years earlier thanthose without this mutation
Figure 2 Plots of probability of loss of ambulation with age for Cox-proportional hazard models
(A) shows fitted values for model 2 withage for an individual withoutpVal603Leu pIle618Thr orpAsp207Val (B C and D) give expectedcurves for an individual with and with-out the given mutation with shadedareas indicating 90 confidenceintervals
6 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

Table 1 Demographic description of the GNE cohorts
Countrywhere thestudy wasconducted
Ethnicalbackground
Numberofpatients
Percentageof malefemale ratio(n)
Age atbaselinemean (plusmnSD)
Mean age atonset (plusmnSD)[range]
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD)
GNE geneticsdescription of thecohort
Cardiacinvolvement
Respiratoryinvolvement
Japan 19 Japanese 121 455 (5566) 277 (96) 430 (52) 354 (113)mean 211 yafter the onset
pV603L and pD207Vaccount for 710 ofcases
Normal in all testedpatients (289)
FVC normal (33)decreased in 67patients
Japan 217 Japanese 27 333 (918) 430 (129) 259 (103)[15ndash58]
360 (9) Not available pV603L and pD207Vaccount for 750 of allmutations
Structural andrhythmabnormalities in296 patients
OSASa (n = 2) FVCdrop minus33 in 1 y innonambulant subset
Japan 324 Japanese 212 448 (95117) 416 (141) 284 SD102 [10-61]
234 (154) 368 (113)[range 19ndash78]
pV603L and pD207Vaccount for 887 allmutations
Not available Not available
Japan 425 Japanese 71 338 (2444) 431 (130) 248 SD 83 [12-58]
520 (37)[50-80 ]
349 (117)[range 18ndash70]Mean 170 (21)y after the onset
pV603L and pD207V862 of all mutations
Not available Not available
Bulgaria5 Roma people 50 620 (3119) Not available 244 (631)[10ndash42]
640 (32) Not availablemean 1034(431) y afteronset
pI618T account for 98of all mutations
Structural andrhythmabnormalities in455 patients
FVC [range 60ndash75]
UK7 White BritishIrish IndianPakistan
26 539 (1412) 477 30 (92) [10ndash44] 192 (5) 463 (80) pA662V and pD409Yaccount for 63
Mild systolicdysfunction- 1patient
Normal in all patients
Israel4 Middle EasternJews KaraitesArabs
129 Not available Not available [17ndash48] Not available Not availablegt15 y sinceonset
pM743T accounts for100 of all mutations
None Not available
Iran10 Iranian non-Jewish (MuslimPersian)
18 500 (99) 34 [range24ndash47]
257 [19ndash31] 167 (3) 373 13ndash15 ysince onset
High prevalence ofpM743T
Normal in allpatients
Normal in all patients
China 16 Chinese 35 457 (1619) 373 (102)[range 22ndash66]
306 (73)[20ndash43]
Not available Not available pD207V allelefrequency in the cohortof 357
Not available Not available
China 226 Chinese 53 509 (2726) 315 (84) [range11ndash46]
205 (81) [5-39] Not available Not available No single dominatingmutation in the studycohort
Not available Not available
India8 Indians 17 706 (125) 341 (71) [range25ndash49]
270 (63)[21ndash46]
59 (1) Not available 765 patients harbourpV727M
Not available Not available
a Obstructive sleep apnoea syndrome
Neurolo
gyorgN
GNeurologyG
enetics|
Volume5N
umber
1|
February
20193
ResultsAge at onsetEleven cohorts were selected for analysis Four publicationsfrom Japan met the inclusion criteria and may have over-lapping patients Two publications from China may haveoverlapping patients but this is unlikely The overlap in pa-tient populations between those cohorts cannot be de-termined therefore they were analyzed as individual patientcohorts Bulgaria UK Israel Iran and India are presented bya single publication (table 1)
Number of patients in populations varied between 17 (India)and 212 (Japan) Average age at baseline varied between 315years (China) and 477 years (UK) The earliest onset at 10years was detected in Japan UK and Bulgaria while the latestonset was observed at 61 years in Japan giving Japan thelargest variability of age at onset (figure 1)
Linear models were fitted to the age at onset on those indi-viduals with information about genotype and age at onset (n =341) Genetic variables provided a better fit to the data thanlocation and homozygosity (maximum adjusted r2 was 008without individual mutation explanatory variables) Modelsusing allelic dose models were marginally worse than domi-nance models Statistically significant mutations werepAsp207Val (increase in age at onset of 8 years p = 9e-16)pAla631Val (increase of 48 years p = 0002) andpLeu539Ser (decrease of 72 years p = 0008) Confidenceintervals are given in table 2 The proportion of variabilityexplained by these 3 genetic factors was estimated to be188 using the reliampo package For univariate models theage at onset was a highly significant predictor of continued
ambulance (p lt 2e-16) along with individual homozygosity(p = 7e-16) and the mutations pVal603Leu (p = 0007)pAsp207Val (p = 00002) and pILe618Thr (p = 34e-14)In a multivariate model using all variables homozygositywas only marginally significant (p gt 001) Age at onset anddominance for the 3 mutations mdash the change in hazard forthe 3 mutationsmdashwas used in the final model (table 2) Theeffect of the age at onset and the 3 mutations (figure 2) Wecan show the effects of the 3 mutations by comparingmutations to an individual with onset at age 30 and withoutany of the 3 mutations who has a probability of 085 (95confidence interval [CI] 077ndash094) of continued ambulanceat age 40
Age of ambulationLowest proportion of nonambulant patients was described inIndia (59) and the highest in Bulgaria (640) resulting inalmost every third GNE patient in the world being non-ambulant (325) Authors of the selected manuscripts de-scribed reaching nonambulatory status in 2 ways (1) Age atnonambulatory status (5 publications) and duration(ie years) since the onset to nonambulatory status (3 pub-lications) The former shows that patients reach non-ambulatory status at an average 381 years old with overa decade (114 years) difference between ldquoJapan 4rdquo and UKcohorts The latter shows that the progress since onset to thewheelchair bound state can take between 103 and 211 yearswith an average of 162 years
A mutation at pVal603Leu gives a probability of ambulanceat age 40 of 061 (95 CI 049ndash074) a mutation atpIleThr gives 073 (95 CI 0163ndash083) and a mutation atpAsp207Val has a probability of continued ambulance of 098
Table 2 Genomic positions and amino acid changes for the 10 changes analyzed in the onset data
Genomicposition
Amino acidchange Ref SNP ID
Frequencyin onsetdata
Populationspresent
CountinExAC
Count in1000genomes
Frequencyambulationdata
Estimatedchange inage atonset
Estimatedhazard formodel amb2(95 CI)
36219937 pVal603Leu rs121908632 029 2 3 1 034 31 (15ndash63)
36246117 pAsp207Val rs139425890 017 2 5 2 015 80 (6199) 012 (003ndash05)
36219891 pILe618Thr 016 4 1 0 023 19 (096ndash39)
36218221 pAla631Val rs62541771 0047 3 10 0 (0002) 48 (12 85)
36217396 pMet743Leu rs28937594 0033 2 4 0 0043
36249315 pCys44Ser 0027 2 0 0 (0007)
36236861 pArg277Trp rs121908629 0020 4 0 0 0025
36236887 pLeu268Leu 0018 1 0 0 0027
36227394 pAsp409Tyr rs199877522 0017 1 14 0 (0)
36222884 pLeu539Ser 0015 1 0 0 (0) minus72 (minus127-18)
All genomic positions are on chromosome 9 and based on amino acid changesmapped to transcript ENST00000377902 Changeswith bracketed frequenciesin the ambulation data were not included in the ambulation model
4 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
(95 CI 096ndash1) When considering only the Japanese co-hort we confirm that the presence of pAsp207Val is associ-ated with a muchmilder phenotype (probability of ambulanceat age 40 is 098 95 CI [094ndash10]) compared to individualswith no copies probability 058 [045 073] Homozygotes ofpVal603Leu have a much more severe phenotype withprobability of continued ambulance of 039 95 CI[024ndash061] compared to a heterozygote probability 09095 CI [080ndash10]
GenotypesFounder or high-prevalence mutations were significantly prev-alent in 9 cohorts In 2 populations (Israel and Bulgaria) foundermutation appears in nearly all patients in homozygous stateJapan and UK showed 2 mutations dominating the cohortsChina 1 and China 2 cohorts showed a different spectrum ofmutations with one dominating mutation in former study andmultiple mutations without clear dominance in the latter
Cardiorespiratory functionIt is considered that GNEmyopathy does not increase the risk ofcardiomyopathy and respiratory failure However a 1 yearlongnatural history study conducted in Japan showed that forced vitalcapacity (FVC) declines in nonambulatory patients17 De-scription of the respiratory function was available in 5 of 10studies UK and Iran described it as ldquonormal in all patientsrdquo FVCwas either normal or mildly to moderately decreased in all other
tested patients No severe decline in FVC was reported Car-diomyopathy was not reported nevertheless some structuraland rhythm abnormalities were present in 296 and 455 ofpatients from ldquoJapan 2rdquo and Bulgaria respectively Reportedabnormalities included borderline reduction in ejection fractionand impaired relaxation (in patients who had concomitant dis-eases such as diabetes or hypertension) ECG in a few patientsshowed evidence of abnormal repolarization and left ventricularhypertrophy Rhythm abnormalities such as extrasystolic ar-rhythmia and borderline nonspecific intraventricular delay wereobserved on a few occasions Sudden death of unknown causewas reported in 4 nonambulant patients without a family historyof cardiac rhythm abnormalities5
Atypical featuresMuscle biopsy appearance without rimmed vacuoles wasobserved in the UK and Japan and is likely to be linked to thesite of the biopsy (ie quadriceps as the least affected muscle)or early stage of the disease Thrombocytopenia was reportedin 2 Japanese cohorts (ldquoJapan 1rdquo and ldquoJapan 2rdquo) A high rate ofan early onset presentation (lt20 years) was reported in ldquoJa-pan 4rdquo cohort Asymmetrical weakness was evident in the UKand both Chinese cohorts Quadriceps sparing almost a pa-thognomonic feature of GNEmyopathy was absent in a smallnumber of patients from Israel China 1 and China 2 cohortsFacial and neck weakness and limb-girdle muscle weakness atearly stages of the disease were observed in China and Israel
Figure 1 Forest plot showing data overview by onset on a cohort level
Studies are grouped by source individual level data (bottom panel) summary statistics form articles (middle panel) and by country from the registry (toppanel with dark background) Means are given by large circles the confidence intervals by thick black lines and the minimum and maximum values by xSample sizes for each study are indicated to the right The overall mean (calculated from individual level data) is shown by the vertical grey line Student-tconfidence intervals were calculated for registry countries and individual studies Confidence intervals for summary studies were taken from the articles
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 5
Analysis of the registry cohortAge at onset and nonambulatory status were compared between6 country-specific cohorts with n ge 14 patients Baseline data arepresented in table 3 Using the model and parameter valuesestimated in from section 6 to predict onset times for the registrydata (n = 126)was not fully successful with an r2 of 004 betweenthe self-reported onset values in the registry data and the onsetvalues predicted from this model based on the registry geneticdata So there is a relationship between estimated onset time andpredicted time but this is not as strong as in the meta data set
Registry data with ambulance data had a sample size of 113Survival concordance for registry individuals was calculated usingthe parameters from the best fit survival model This workson pairs of individuals and averages how often the individualwith the lowest ambulance time also has the highest risk scoreThis is then averaged over all pairs of individuals Hence perfectmatches would have a concordance of 1 and a model that wasrandom would have a concordance of 05 The concordance forthe registry data was 078 which shows good agreement
Registry data were also interrogated to identify homozygouspatients irrespectively of their country of residence Four sub-sets with the number of patients ge4 were analyzed Because of
a small number of available data we present it only for de-scriptive analysis Baseline demographic data genotype andclinical parameters are presented in table 3
DiscussionDisease severity and variability in its presentation are factors toconsider in the disease management and planning clinical re-search Patients are keen to know their prognosis and likelytrajectory of progression tomake personal choices Rare diseasestatus leads to scarcity of the data and availability of biologicalsamples to study Therefore an integrated analysis of theavailable data provides an additional insight in understandingthe correlation between GNE genotype and phenotype
Cohort-based studies provided country-specific data Analysisof these cohorts showed the presence of outliers with an earlyor late onset of the first symptoms in every cohort which isnot explained by a particular mutation We find that almost20 of the variability in age at onset is due to the GNEgenotype with the change to pAsp207Val being associatedwith on average an 8-year increase in age at onset In contrastpLeu539Ser results in onset on average 72 years earlier thanthose without this mutation
Figure 2 Plots of probability of loss of ambulation with age for Cox-proportional hazard models
(A) shows fitted values for model 2 withage for an individual withoutpVal603Leu pIle618Thr orpAsp207Val (B C and D) give expectedcurves for an individual with and with-out the given mutation with shadedareas indicating 90 confidenceintervals
6 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

ResultsAge at onsetEleven cohorts were selected for analysis Four publicationsfrom Japan met the inclusion criteria and may have over-lapping patients Two publications from China may haveoverlapping patients but this is unlikely The overlap in pa-tient populations between those cohorts cannot be de-termined therefore they were analyzed as individual patientcohorts Bulgaria UK Israel Iran and India are presented bya single publication (table 1)
Number of patients in populations varied between 17 (India)and 212 (Japan) Average age at baseline varied between 315years (China) and 477 years (UK) The earliest onset at 10years was detected in Japan UK and Bulgaria while the latestonset was observed at 61 years in Japan giving Japan thelargest variability of age at onset (figure 1)
Linear models were fitted to the age at onset on those indi-viduals with information about genotype and age at onset (n =341) Genetic variables provided a better fit to the data thanlocation and homozygosity (maximum adjusted r2 was 008without individual mutation explanatory variables) Modelsusing allelic dose models were marginally worse than domi-nance models Statistically significant mutations werepAsp207Val (increase in age at onset of 8 years p = 9e-16)pAla631Val (increase of 48 years p = 0002) andpLeu539Ser (decrease of 72 years p = 0008) Confidenceintervals are given in table 2 The proportion of variabilityexplained by these 3 genetic factors was estimated to be188 using the reliampo package For univariate models theage at onset was a highly significant predictor of continued
ambulance (p lt 2e-16) along with individual homozygosity(p = 7e-16) and the mutations pVal603Leu (p = 0007)pAsp207Val (p = 00002) and pILe618Thr (p = 34e-14)In a multivariate model using all variables homozygositywas only marginally significant (p gt 001) Age at onset anddominance for the 3 mutations mdash the change in hazard forthe 3 mutationsmdashwas used in the final model (table 2) Theeffect of the age at onset and the 3 mutations (figure 2) Wecan show the effects of the 3 mutations by comparingmutations to an individual with onset at age 30 and withoutany of the 3 mutations who has a probability of 085 (95confidence interval [CI] 077ndash094) of continued ambulanceat age 40
Age of ambulationLowest proportion of nonambulant patients was described inIndia (59) and the highest in Bulgaria (640) resulting inalmost every third GNE patient in the world being non-ambulant (325) Authors of the selected manuscripts de-scribed reaching nonambulatory status in 2 ways (1) Age atnonambulatory status (5 publications) and duration(ie years) since the onset to nonambulatory status (3 pub-lications) The former shows that patients reach non-ambulatory status at an average 381 years old with overa decade (114 years) difference between ldquoJapan 4rdquo and UKcohorts The latter shows that the progress since onset to thewheelchair bound state can take between 103 and 211 yearswith an average of 162 years
A mutation at pVal603Leu gives a probability of ambulanceat age 40 of 061 (95 CI 049ndash074) a mutation atpIleThr gives 073 (95 CI 0163ndash083) and a mutation atpAsp207Val has a probability of continued ambulance of 098
Table 2 Genomic positions and amino acid changes for the 10 changes analyzed in the onset data
Genomicposition
Amino acidchange Ref SNP ID
Frequencyin onsetdata
Populationspresent
CountinExAC
Count in1000genomes
Frequencyambulationdata
Estimatedchange inage atonset
Estimatedhazard formodel amb2(95 CI)
36219937 pVal603Leu rs121908632 029 2 3 1 034 31 (15ndash63)
36246117 pAsp207Val rs139425890 017 2 5 2 015 80 (6199) 012 (003ndash05)
36219891 pILe618Thr 016 4 1 0 023 19 (096ndash39)
36218221 pAla631Val rs62541771 0047 3 10 0 (0002) 48 (12 85)
36217396 pMet743Leu rs28937594 0033 2 4 0 0043
36249315 pCys44Ser 0027 2 0 0 (0007)
36236861 pArg277Trp rs121908629 0020 4 0 0 0025
36236887 pLeu268Leu 0018 1 0 0 0027
36227394 pAsp409Tyr rs199877522 0017 1 14 0 (0)
36222884 pLeu539Ser 0015 1 0 0 (0) minus72 (minus127-18)
All genomic positions are on chromosome 9 and based on amino acid changesmapped to transcript ENST00000377902 Changeswith bracketed frequenciesin the ambulation data were not included in the ambulation model
4 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
(95 CI 096ndash1) When considering only the Japanese co-hort we confirm that the presence of pAsp207Val is associ-ated with a muchmilder phenotype (probability of ambulanceat age 40 is 098 95 CI [094ndash10]) compared to individualswith no copies probability 058 [045 073] Homozygotes ofpVal603Leu have a much more severe phenotype withprobability of continued ambulance of 039 95 CI[024ndash061] compared to a heterozygote probability 09095 CI [080ndash10]
GenotypesFounder or high-prevalence mutations were significantly prev-alent in 9 cohorts In 2 populations (Israel and Bulgaria) foundermutation appears in nearly all patients in homozygous stateJapan and UK showed 2 mutations dominating the cohortsChina 1 and China 2 cohorts showed a different spectrum ofmutations with one dominating mutation in former study andmultiple mutations without clear dominance in the latter
Cardiorespiratory functionIt is considered that GNEmyopathy does not increase the risk ofcardiomyopathy and respiratory failure However a 1 yearlongnatural history study conducted in Japan showed that forced vitalcapacity (FVC) declines in nonambulatory patients17 De-scription of the respiratory function was available in 5 of 10studies UK and Iran described it as ldquonormal in all patientsrdquo FVCwas either normal or mildly to moderately decreased in all other
tested patients No severe decline in FVC was reported Car-diomyopathy was not reported nevertheless some structuraland rhythm abnormalities were present in 296 and 455 ofpatients from ldquoJapan 2rdquo and Bulgaria respectively Reportedabnormalities included borderline reduction in ejection fractionand impaired relaxation (in patients who had concomitant dis-eases such as diabetes or hypertension) ECG in a few patientsshowed evidence of abnormal repolarization and left ventricularhypertrophy Rhythm abnormalities such as extrasystolic ar-rhythmia and borderline nonspecific intraventricular delay wereobserved on a few occasions Sudden death of unknown causewas reported in 4 nonambulant patients without a family historyof cardiac rhythm abnormalities5
Atypical featuresMuscle biopsy appearance without rimmed vacuoles wasobserved in the UK and Japan and is likely to be linked to thesite of the biopsy (ie quadriceps as the least affected muscle)or early stage of the disease Thrombocytopenia was reportedin 2 Japanese cohorts (ldquoJapan 1rdquo and ldquoJapan 2rdquo) A high rate ofan early onset presentation (lt20 years) was reported in ldquoJa-pan 4rdquo cohort Asymmetrical weakness was evident in the UKand both Chinese cohorts Quadriceps sparing almost a pa-thognomonic feature of GNEmyopathy was absent in a smallnumber of patients from Israel China 1 and China 2 cohortsFacial and neck weakness and limb-girdle muscle weakness atearly stages of the disease were observed in China and Israel
Figure 1 Forest plot showing data overview by onset on a cohort level
Studies are grouped by source individual level data (bottom panel) summary statistics form articles (middle panel) and by country from the registry (toppanel with dark background) Means are given by large circles the confidence intervals by thick black lines and the minimum and maximum values by xSample sizes for each study are indicated to the right The overall mean (calculated from individual level data) is shown by the vertical grey line Student-tconfidence intervals were calculated for registry countries and individual studies Confidence intervals for summary studies were taken from the articles
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 5
Analysis of the registry cohortAge at onset and nonambulatory status were compared between6 country-specific cohorts with n ge 14 patients Baseline data arepresented in table 3 Using the model and parameter valuesestimated in from section 6 to predict onset times for the registrydata (n = 126)was not fully successful with an r2 of 004 betweenthe self-reported onset values in the registry data and the onsetvalues predicted from this model based on the registry geneticdata So there is a relationship between estimated onset time andpredicted time but this is not as strong as in the meta data set
Registry data with ambulance data had a sample size of 113Survival concordance for registry individuals was calculated usingthe parameters from the best fit survival model This workson pairs of individuals and averages how often the individualwith the lowest ambulance time also has the highest risk scoreThis is then averaged over all pairs of individuals Hence perfectmatches would have a concordance of 1 and a model that wasrandom would have a concordance of 05 The concordance forthe registry data was 078 which shows good agreement
Registry data were also interrogated to identify homozygouspatients irrespectively of their country of residence Four sub-sets with the number of patients ge4 were analyzed Because of
a small number of available data we present it only for de-scriptive analysis Baseline demographic data genotype andclinical parameters are presented in table 3
DiscussionDisease severity and variability in its presentation are factors toconsider in the disease management and planning clinical re-search Patients are keen to know their prognosis and likelytrajectory of progression tomake personal choices Rare diseasestatus leads to scarcity of the data and availability of biologicalsamples to study Therefore an integrated analysis of theavailable data provides an additional insight in understandingthe correlation between GNE genotype and phenotype
Cohort-based studies provided country-specific data Analysisof these cohorts showed the presence of outliers with an earlyor late onset of the first symptoms in every cohort which isnot explained by a particular mutation We find that almost20 of the variability in age at onset is due to the GNEgenotype with the change to pAsp207Val being associatedwith on average an 8-year increase in age at onset In contrastpLeu539Ser results in onset on average 72 years earlier thanthose without this mutation
Figure 2 Plots of probability of loss of ambulation with age for Cox-proportional hazard models
(A) shows fitted values for model 2 withage for an individual withoutpVal603Leu pIle618Thr orpAsp207Val (B C and D) give expectedcurves for an individual with and with-out the given mutation with shadedareas indicating 90 confidenceintervals
6 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

(95 CI 096ndash1) When considering only the Japanese co-hort we confirm that the presence of pAsp207Val is associ-ated with a muchmilder phenotype (probability of ambulanceat age 40 is 098 95 CI [094ndash10]) compared to individualswith no copies probability 058 [045 073] Homozygotes ofpVal603Leu have a much more severe phenotype withprobability of continued ambulance of 039 95 CI[024ndash061] compared to a heterozygote probability 09095 CI [080ndash10]
GenotypesFounder or high-prevalence mutations were significantly prev-alent in 9 cohorts In 2 populations (Israel and Bulgaria) foundermutation appears in nearly all patients in homozygous stateJapan and UK showed 2 mutations dominating the cohortsChina 1 and China 2 cohorts showed a different spectrum ofmutations with one dominating mutation in former study andmultiple mutations without clear dominance in the latter
Cardiorespiratory functionIt is considered that GNEmyopathy does not increase the risk ofcardiomyopathy and respiratory failure However a 1 yearlongnatural history study conducted in Japan showed that forced vitalcapacity (FVC) declines in nonambulatory patients17 De-scription of the respiratory function was available in 5 of 10studies UK and Iran described it as ldquonormal in all patientsrdquo FVCwas either normal or mildly to moderately decreased in all other
tested patients No severe decline in FVC was reported Car-diomyopathy was not reported nevertheless some structuraland rhythm abnormalities were present in 296 and 455 ofpatients from ldquoJapan 2rdquo and Bulgaria respectively Reportedabnormalities included borderline reduction in ejection fractionand impaired relaxation (in patients who had concomitant dis-eases such as diabetes or hypertension) ECG in a few patientsshowed evidence of abnormal repolarization and left ventricularhypertrophy Rhythm abnormalities such as extrasystolic ar-rhythmia and borderline nonspecific intraventricular delay wereobserved on a few occasions Sudden death of unknown causewas reported in 4 nonambulant patients without a family historyof cardiac rhythm abnormalities5
Atypical featuresMuscle biopsy appearance without rimmed vacuoles wasobserved in the UK and Japan and is likely to be linked to thesite of the biopsy (ie quadriceps as the least affected muscle)or early stage of the disease Thrombocytopenia was reportedin 2 Japanese cohorts (ldquoJapan 1rdquo and ldquoJapan 2rdquo) A high rate ofan early onset presentation (lt20 years) was reported in ldquoJa-pan 4rdquo cohort Asymmetrical weakness was evident in the UKand both Chinese cohorts Quadriceps sparing almost a pa-thognomonic feature of GNEmyopathy was absent in a smallnumber of patients from Israel China 1 and China 2 cohortsFacial and neck weakness and limb-girdle muscle weakness atearly stages of the disease were observed in China and Israel
Figure 1 Forest plot showing data overview by onset on a cohort level
Studies are grouped by source individual level data (bottom panel) summary statistics form articles (middle panel) and by country from the registry (toppanel with dark background) Means are given by large circles the confidence intervals by thick black lines and the minimum and maximum values by xSample sizes for each study are indicated to the right The overall mean (calculated from individual level data) is shown by the vertical grey line Student-tconfidence intervals were calculated for registry countries and individual studies Confidence intervals for summary studies were taken from the articles
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 5
Analysis of the registry cohortAge at onset and nonambulatory status were compared between6 country-specific cohorts with n ge 14 patients Baseline data arepresented in table 3 Using the model and parameter valuesestimated in from section 6 to predict onset times for the registrydata (n = 126)was not fully successful with an r2 of 004 betweenthe self-reported onset values in the registry data and the onsetvalues predicted from this model based on the registry geneticdata So there is a relationship between estimated onset time andpredicted time but this is not as strong as in the meta data set
Registry data with ambulance data had a sample size of 113Survival concordance for registry individuals was calculated usingthe parameters from the best fit survival model This workson pairs of individuals and averages how often the individualwith the lowest ambulance time also has the highest risk scoreThis is then averaged over all pairs of individuals Hence perfectmatches would have a concordance of 1 and a model that wasrandom would have a concordance of 05 The concordance forthe registry data was 078 which shows good agreement
Registry data were also interrogated to identify homozygouspatients irrespectively of their country of residence Four sub-sets with the number of patients ge4 were analyzed Because of
a small number of available data we present it only for de-scriptive analysis Baseline demographic data genotype andclinical parameters are presented in table 3
DiscussionDisease severity and variability in its presentation are factors toconsider in the disease management and planning clinical re-search Patients are keen to know their prognosis and likelytrajectory of progression tomake personal choices Rare diseasestatus leads to scarcity of the data and availability of biologicalsamples to study Therefore an integrated analysis of theavailable data provides an additional insight in understandingthe correlation between GNE genotype and phenotype
Cohort-based studies provided country-specific data Analysisof these cohorts showed the presence of outliers with an earlyor late onset of the first symptoms in every cohort which isnot explained by a particular mutation We find that almost20 of the variability in age at onset is due to the GNEgenotype with the change to pAsp207Val being associatedwith on average an 8-year increase in age at onset In contrastpLeu539Ser results in onset on average 72 years earlier thanthose without this mutation
Figure 2 Plots of probability of loss of ambulation with age for Cox-proportional hazard models
(A) shows fitted values for model 2 withage for an individual withoutpVal603Leu pIle618Thr orpAsp207Val (B C and D) give expectedcurves for an individual with and with-out the given mutation with shadedareas indicating 90 confidenceintervals
6 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
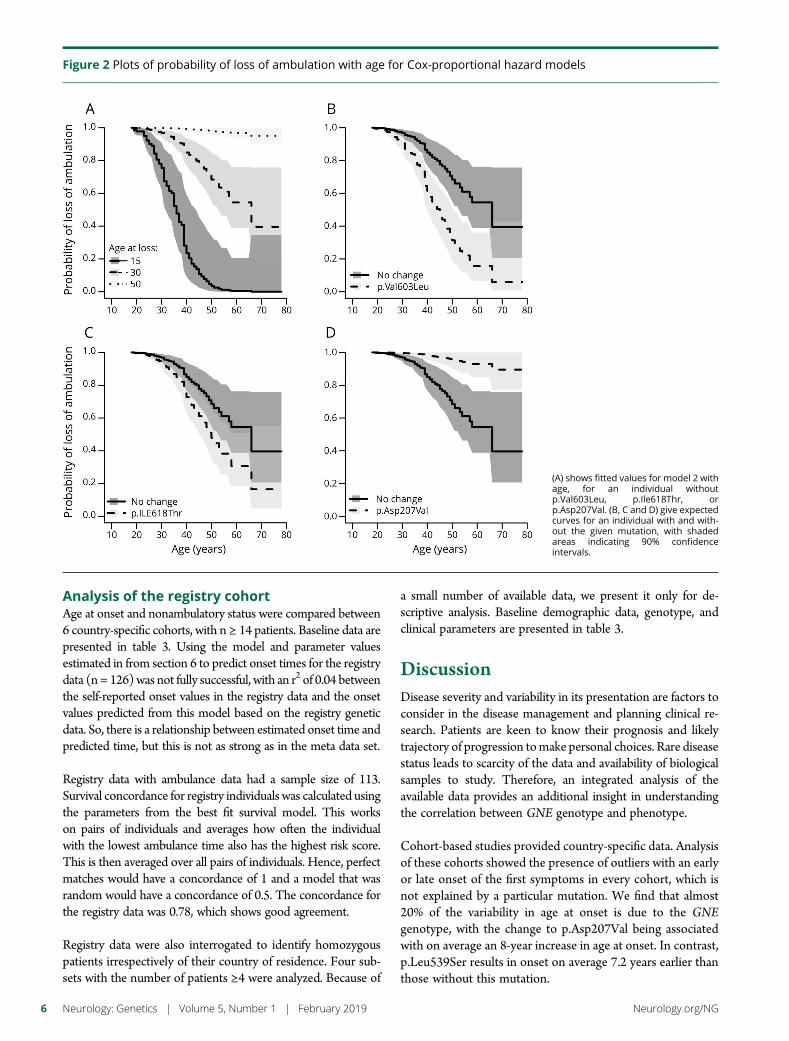
Analysis of the registry cohortAge at onset and nonambulatory status were compared between6 country-specific cohorts with n ge 14 patients Baseline data arepresented in table 3 Using the model and parameter valuesestimated in from section 6 to predict onset times for the registrydata (n = 126)was not fully successful with an r2 of 004 betweenthe self-reported onset values in the registry data and the onsetvalues predicted from this model based on the registry geneticdata So there is a relationship between estimated onset time andpredicted time but this is not as strong as in the meta data set
Registry data with ambulance data had a sample size of 113Survival concordance for registry individuals was calculated usingthe parameters from the best fit survival model This workson pairs of individuals and averages how often the individualwith the lowest ambulance time also has the highest risk scoreThis is then averaged over all pairs of individuals Hence perfectmatches would have a concordance of 1 and a model that wasrandom would have a concordance of 05 The concordance forthe registry data was 078 which shows good agreement
Registry data were also interrogated to identify homozygouspatients irrespectively of their country of residence Four sub-sets with the number of patients ge4 were analyzed Because of
a small number of available data we present it only for de-scriptive analysis Baseline demographic data genotype andclinical parameters are presented in table 3
DiscussionDisease severity and variability in its presentation are factors toconsider in the disease management and planning clinical re-search Patients are keen to know their prognosis and likelytrajectory of progression tomake personal choices Rare diseasestatus leads to scarcity of the data and availability of biologicalsamples to study Therefore an integrated analysis of theavailable data provides an additional insight in understandingthe correlation between GNE genotype and phenotype
Cohort-based studies provided country-specific data Analysisof these cohorts showed the presence of outliers with an earlyor late onset of the first symptoms in every cohort which isnot explained by a particular mutation We find that almost20 of the variability in age at onset is due to the GNEgenotype with the change to pAsp207Val being associatedwith on average an 8-year increase in age at onset In contrastpLeu539Ser results in onset on average 72 years earlier thanthose without this mutation
Figure 2 Plots of probability of loss of ambulation with age for Cox-proportional hazard models
(A) shows fitted values for model 2 withage for an individual withoutpVal603Leu pIle618Thr orpAsp207Val (B C and D) give expectedcurves for an individual with and with-out the given mutation with shadedareas indicating 90 confidenceintervals
6 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

Escherichia coli and insect cell disease models showed thatepimerase and kinase enzymatic activity significantly variedamong selected mutations18 Slow progression of the diseasemay partially be explained by mitochondrial alterations19
However the correlation between the mutations and level ofmembrane sialylation is not consistent which may indicatethat involvement of other factors affecting the cellular andclinical phenotype20 An indirect marker of disease severityndashage of nonambulation status has a marked variability12 Thedifferences between age at nonambulatory status were asso-ciated with the age at onset and 3 founder mutations While itis difficult to disentangle population effects from geneticeffects special fitting models for nonambulatory age werebetter fitted to genetic data than to population source andthese models showed good concordance with those used topredict continued ambulance in the GNE registry Significantdifference between Japan Iran and Bulgaria in age at non-ambulatory status and proportion of nonambulant patientsin the cohorts also point to the fact that the speed at whichthe disease progresses may be attributable to the founder
mutations highly prevalent in those populations Whencomparisons are made only with homozygous Japanesepopulations the differences in survival curves almost disap-pear Roma people founder mutation (pILe618Thr) showsno evidence of association with the age at onset but is asso-ciated with a lower age of loss of ambulation The mutationpLeu539Ser only seen in the Chinese data is associated withan onset 72 years earlier but is not seen in the ambulationdata and we cannot make any inferences about its impact onambulation As these mutations are not shared with otherstudy populations it is difficult to extract the effects of ge-notype The differences in health care approaches can alsopotentially play a role in preserving ambulation
Cohorts with a higher number of nonambulant patients alsohave a higher number of patients with compromised re-spiratory function This confirms previous observation ona multinational level that respiratory function is normally pre-served until the very nonambulatory stages of the disease Wealso did not observe a correlation between cardiomyopathy and
Table 3 Analysis of the registry data
Countryofresidence
Ethnicity ofthe studycohort
Numberofpatients
Malefemaleratio (nn)
Age atbaselinemean(plusmnSD)
Mean ageat onset(plusmnSD)
Percentage ofnonambulantpatients (n)
Mean age atnonambulatorystate (plusmnSD) Mutation description
India Asian 14 571 (86) 341 (82) 256 (56) 77 (1) na 14 different mutations intotal pVal727Met is themost commonaccounting for 423 ofall mutations
Italy White 20 400 (812) 397 (116) 255 (67) 400 (8) 359 (97) 9 mutations in totalpAsn550Ser andpMet743Thr are the mostcommon accounting for214 of all mutationseach
SouthKorea
Asian 22 455 (1012) 342 (86) 257 (72) 333 (7) 334 (80) 5 mutations in total 2most common mutationspVal603Leu andpCys44Ser accounting for50 and 385 of allmutations respectively
USA 85 White15 Asian
21 524 (1110) 473 (125) 293 (91) 200 (4) 433 (75) 11 mutations in total 3most commonpAla662Val pVal727Metand pMet743Thr accountfor 214 143 and143 respectively of allmutations
UK 762White238 Asian
21 429 (912) 479 (103) 312 (79) 238 (5) 426 (116) 9 mutations in total 2most commonpAsp409Tyr andpAla662Val account for250 and 450 of allmutations respectively
Iran Asian 37 486 (1819) 363 (80) 287 (57) 94 (3) 387 (58) 9 mutations in total onemost commonpMet743Thr accounts for536 of all mutations
Grandtotal
135 474 (6471) 394 (110) 278 (72) 215 (28) 389 (92)
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 7
GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

GNE myopathy but a number of mild-to-moderate structuraland rhythm heart abnormalities were observed
Rare symptoms found only in one country (eg thrombo-cytopenia or Beevor sign) could be attributable to the de-scribed ethnic groups only or to observational bias due to thedifferences in medical practices across the world A new GNEmutation has been found to cause a congenital macro-thrombocytopenia without noticeable muscle weakness todate21 It supports the observation that platelet count may becompromised in GNE myopathy and therefore it may berecommended to include platelet count test in the routinemanagement of the disease
Spectrum of the disease presentation severity and specific fea-tures is important in planning clinical trials for adequate patientstratification and reduced bias due to known covariancesN-acetylneuraminic acid extended release (Ace-ER) phase 2 and3 clinical trials showed different results where phase 2 showedthat Ace-ER stabilizes the disease progression and causes sta-tistical significant difference between treatment and placebogroups22 However phase 3 showed no benefit of Ace-ER onmuscle strength23 We speculate that difference in the resultsbetween 2 studies could partially be accounted for differences inthe study cohorts In phase 2 trial sites were located in 2countries the United States and Israel where pMet743Thr ismost common Phase 3 study enrolled a larger number ofpatients from 7 countries with 4 of them being in Europe andone in Canadamdashwhere pMet743Thr is much less prevalentThis genotype difference in the cohorts could have potentiallyhad an impact on the discrepancy in the study results Wetherefore suggest that GNE mutation should be taken in con-sideration as a factor for patient stratification in the clinical trials
This article is a first attempt to look at the genotypemdashphenotype correlation in GNE myopathy through a system-atic review and meta-analysis The analysis shows that GNEgenotype has an impact on the phenotype and accounts for20 of variability in onset of the disease and has a significantinfluence on reaching nonambulatory status The analysisindicated that there are other disease modifying factors thatinfluence phenotype Cohort-specific features expand clinicalunderstanding of the disease and warrant a closer look at theplatelet count in GNE patients across the world Better un-derstanding of the disease progression based on the genotypeis important for counselling and management of GNEpatients Potential impact of genotype on the disease pro-gression is also important for clinical trial design and patientstratification We acknowledge study limitations such assample size and factors (eg differences in environment dietand health care systems) that can potentially influence theanalysis Therefore a replication of the analysis on a larger anddifferent cohort would have to be conducted in the future
AcknowledgmentThe authors would also like to thank GNE myopathyRegistry Steering Committee and NDF for supporting this
research This work was supported by European UnionSeventh Framework Programme (FP72007-2013) undergrant agreement No 305444 (RD-Connect) and MedicalResearch Council UK (reference G1002274 grant ID98482)
Study fundingNo targeted funding reported
DisclosureO Pogoryelova has served as a clinical trial investigator forUltragenyx Pharmaceutical Inc I J Wilson reports no dis-closures Z Argov was a consultant for Ultragenyx Pharma-ceutical and was Special Medical Advisor for CEO of BioBlastPharma The GNE research at Hadassah received grantsfrom AFM NDF ISF and GIF I Nishino was an advisorfor Ultragenyx Pharmaceutical The GNE myopathy researchis supported currently by Intramural Research Grant(29-4 and 29-3) for Neurological and Psychiatric Disorders ofNCNP AMED under grant numbers JP17ek0109285h0001JP17ek0109085h0003 JP17ek0109196h0001 andJP17kk0205001s0202 and Health Labour and Welfare Sci-ences Research Grants (H29-Nanchito (Nan)-Ippan-030)and was previously by Health Labour and Welfare SciencesResearch Grants Patients Association for Distal Myopathiesand Neuromuscular Disease Foundation H Mansbach isemployed and holds stock options with Ultragenyx Pharma-ceutical Inc H Lochmuller has served as a clinical trial in-vestigator for Ultragenyx Pharmaceutical Inc Financialsupport to the Newcastle University and the Newcastle NHSTrust for research projects and clinical trials by AMO Phar-maceuticals Biogen GW Pharma Pfizer PTC TherapeuticsRoche and Ultragenyx Full disclosure form informationprovided by the authors is available with the full text of thisarticle at NeurologyorgNG
Publication historyReceived by Neurology Genetics September 7 2018 Accepted in finalform December 20 2018
Appendix 1 Authors contribution
Name Location Role Contribution
OksanaPogoryelovaMD PhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy dataacquisition analyzedthe data drafted themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Ian WilsonPhD
Institute of GeneticMedicine NewcastleUniversityNewcastle uponTyne UK
Author Design andconceptualizedstudy analyzed thedata statisticalanalysis drafted themanuscript forintellectual content
8 Neurology Genetics | Volume 5 Number 1 | February 2019 NeurologyorgNG
References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

References1 Eisenberg I Avidan N Potikha T et al The UDP-N-acetylglucosamine 2-epimerase
N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion bodymyopathy Nat Genet 20012983ndash87
2 Celeste FV Vilboux T Ciccone C et al Mutation update for GNE gene variantsassociated with GNE myopathy Hum Mutat 201435915ndash926
3 Nishino I Noguchi S Murayama K et al Distal myopathy with rimmed vacuoles isallelic to hereditary inclusion body myopathy Neurology 2002591689ndash1693
4 Argov Z Eisenberg I Grabov-Nardini G et al Hereditary inclusion body myopathythe Middle Eastern genetic cluster Neurology 2003601519ndash1523
5 Chamova T Guergueltcheva V Gospodinova M et al GNE myopathy in Romapatients homozygous for the pI618T founder mutation Neuromuscul Disord 201525713ndash718
6 Zhao J Wang Z Hong D et al Mutational spectrum and clinical features in 35unrelated mainland Chinese patients with GNE myopathy J Neurol Sci 201535421ndash26
7 Chaouch A Brennan KM Hudson J et al Two recurrent mutations are associatedwith GNEmyopathy in the North of Britain J Neurol Neurosurg Psychiatry 2014851359ndash1365
8 Preethish-Kumar V Pogoryelova O Polavarapu K et al Beevorrsquos sign a potentialclinical marker for GNE myopathy Eur J Neurol 201623e46ndash48
9 Mori-Yoshimura M Hayashi YK Yonemoto N et al Nationwide patient registry forGNE myopathy in Japan Orphanet J Rare Dis 20149150
10 Haghighi A Nafissi S Qurashi A et al Genetics of GNE myopathy in the non-JewishPersian population Eur J Hum Genet 201624243ndash251
11 Noguchi S Keira Y Murayama K et al Reduction of UDP-N-acetylglucosamine2-epimeraseN-acetylmannosamine kinase activity and sialylation in distal myopathywith rimmed vacuoles J Biol Chem 200427911402ndash11407
12 Pogoryelova O Cammish P Mansbach H et al Phenotypic stratification andgenotype-phenotype correlation in a heterogeneous international cohort of GNEmyopathy patients first report from the GNE Myopathy Disease Monitoring Pro-gram registry portion Neuromuscul Disord 201828158ndash168
13 Team RC R A Language and Environment for Statistical Computing Austria RFoundation for Statistical Computing Vienna 2018
14 Akaike H A new look at the statistical model identification IEEE Trans AutomaticControl 197419716ndash723
15 Relative UG Importance for linear regression in R the package relaimpo J Stat Softw2006171ndash27
16 Venables WN Ripley BD Modern Applied Statistics with S-Plus Berlin GermanySpringer-Verlag 1994
17 Mori-Yoshimura M Oya Y Yajima H et al GNE myopathy a prospective naturalhistory study of disease progression Neuromuscul Disord 201424380ndash386
18 Penner J Mantey LR Elgavish S et al Influence of UDP-GlcNAc 2-epimeraseManNAc kinase mutant proteins on hereditary inclusion body myopathy Bio-chemistry 2006452968ndash2977
19 Eisenberg I Novershtern N Itzhaki Z et al Mitochondrial processes are impaired inhereditary inclusion body myopathy Hum Mol Genet 2008173663ndash3674
20 Salama I Hinderlich S Shlomai Z et al No overall hyposialylation in hereditaryinclusion body myopathy myoblasts carrying the homozygous M712T GNE muta-tion Biochem Biophys Res Commun 2005328221ndash226
21 Futterer J Dalby A Lowe GC et al Mutation in GNE is associated with a severe formof congenital thrombocytopenia Blood 20181321855ndash1858
22 Argov Z Caraco Y Lau H et al Aceneuramic acid extended release administrationmaintains upper limb muscle strength in a 48-week study of subjects with GNEmyopathy results from a phase 2 randomized controlled study J Neuromuscul Dis2016349ndash66
23 Lochmuller H Behin A Caraco Y et al A phase 3 randomized study evaluating sialicacid extended-release for GNE myopathy Neurology Epub 2019 Jan 25
24 Cho A Hayashi YK Monma K et al Mutation profile of the GNE gene in Japanesepatients with distal myopathy with rimmed vacuoles (GNE myopathy) J NeurolNeurosurg Psychiatry 201485914ndash917
25 Mori-Yoshimura M Monma K Suzuki N et al Heterozygous UDP-GlcNAc2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE generesult in a less severe GNE myopathy phenotype compared to homozygousN-acetylmannosamine kinase domain mutations J Neurol Sci 2012318100ndash105
26 Lu X Pu C Huang X Liu J Mao Y Distal myopathy with rimmed vacuoles clinicaland muscle morphological characteristics and spectrum of GNE gene mutations in 53Chinese patients Neurol Res 2011331025ndash1031
Appendix 1 (continued)
Name Location Role Contribution
HankMansbachMD
UltragenyxPharmaceutical CAUSA
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
Zohar ArgovMD
Department ofNeurologyHadassah-HebrewUniversity MedicalCenter JerusalemIsrael
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
IchizoNishino MDPhD
Department ofNeuromuscularResearch NationalInstitute ofNeuroscienceNational Center ofNeurology andPsychiatry TokyoJapan
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
HannsLochmullerMD PhD
Childrenrsquos Hospital ofEastern OntarioResearch InstituteUniversity of OttawaOttawa Canada andDivision of NeurologyDepartment ofMedicine The OttawaHospital OttawaDepartment ofNeuropediatrics andMuscle DisordersMedical Center ndashUniversity of FreiburgFaculty of MedicineFreiburg GermanyCentro Nacional deAnalisis Genomico(CNAG-CRG) Centerfor GenomicRegulation BarcelonaInstitute of Scienceand Technology(BIST) BarcelonaCatalonia Spain
Author Interpreted the datarevised themanuscript forintellectual contentMember of the GNERegistry SteeringCommittee
NeurologyorgNG Neurology Genetics | Volume 5 Number 1 | February 2019 9
DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

DOI 101212NXG000000000000030820195 Neurol Genet
Oksana Pogoryelova Ian J Wilson Hank Mansbach et al genotype explains 20 of phenotypic variability in GNE myopathyGNE
This information is current as of February 1 2019
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet
ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet

ServicesUpdated Information amp
httpngneurologyorgcontent51e308fullhtmlincluding high resolution figures can be found at
References httpngneurologyorgcontent51e308fullhtmlref-list-1
This article cites 23 articles 4 of which you can access for free at
Citations httpngneurologyorgcontent51e308fullhtmlotherarticles
This article has been cited by 1 HighWire-hosted articles
Subspecialty Collections
ishttpngneurologyorgcgicollectionnatural_history_studies_prognosNatural history studies (prognosis)
httpngneurologyorgcgicollectionmuscle_diseaseMuscle disease
httpngneurologyorgcgicollectioncohort_studiesCohort studies
httpngneurologyorgcgicollectionassociation_studies_in_geneticsAssociation studies in genetics
httpngneurologyorgcgicollectionall_geneticsAll Geneticsfollowing collection(s) This article along with others on similar topics appears in the
Permissions amp Licensing
httpngneurologyorgmiscaboutxhtmlpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpngneurologyorgmiscaddirxhtmlreprintsusInformation about ordering reprints can be found online
reserved Online ISSN 2376-7839Published by Wolters Kluwer Health Inc on behalf of the American Academy of Neurology All rightsan open-access online-only continuous publication journal Copyright Copyright copy 2019 The Author(s)
is an official journal of the American Academy of Neurology Published since April 2015 it isNeurol Genet




















