
Genetic contribution of the HLA region to the familial clustering of coeliac disease
11
Ann. Hum. Genet. (1997), 61, 307–317 Printed in Great Britain 307 Genetic contribution of the HLA region to the familial clustering of coeliac disease F. PETRONZELLI", M. BONAMICO#, P. FERRANTE", R. GRILLO", B. MORA", P. MARIANI#, I. APOLLONIO", G. GEMME$ M. C. MAZZILLI" " Dipartimento di Medicina Sperimentale, Universita [ ‘ La Sapienza ’, Viale Regina Elena, 324–00161 Roma, Italy # Dipartimento di Pediatria, Universita [ ‘ La Sapienza ’, Roma, Italy $ Istituto Gaslini, Universita [ di Genova, Italy (Received 22.12.95. Accepted 14.4.97) In order to assess the effect of the HLA region on familiality of coeliac disease (CD), we carried out a study on 121 CD index cases and 325 first degree relatives. The transmission disequilibrium test confirmed the importance of the HLA-DR3 haplotype in CD susceptibility. However, the different distortion found in affected children inheriting maternal or paternal DR3 alleles suggested that the sex of the parent might influence the risk conferred by this haplotype. The increase in risk to siblings of affected individuals relative to the risk in the general population (λ s ) and the contribution of the HLA genes to this clustering (λ sHLA ) have also been estimated. Non-overlapping data from the literature have been collected and combined with our sample to extend such analysis. Then, the percentage contribution of the HLA region to the development of CD among siblings was 36–2%. This result confirms that the HLA genotypes are an important genetic background to CD development but shows that additional susceptibility factors remain to be identified. Coeliac disease (CD), also known as gluten sensitive enteropathy (GSE), is a condition in which the immune response to ingested gluten is accompanied by intestinal mucosal damage (Trier, 1991). The resulting atrophy of the villi of the proximal small intestine leads to mal- absorption of nutrients and the clinical features of CD. The symptoms and the epithelial damage are reversed by the exclusion of gluten from the diet. The pathogenesis of CD is believed to involve interaction between genetic and environ- mental factors (Polanco, 1981). The reported prevalence of clinically manifest CD is around 1:1000. However, because of the increasing recognition of subclinical or silent cases, the true prevalence is much higher than supposed, and it probably approaches 1 : 300 (Catassi et al. 1994 ; Troncone et al. 1992). The familial clustering of the disease is documented by the increased prevalence in relatives ; in a multicentred study, undertaken to investigate the prevalence of CD among first-degree relatives of patients, an estimated risk of 8–7 % was found (Auricchio et al. 1988). The genetic contribution to this increased risk is supported by the high rate of concordance for disease (71 %) among monozygotic twins (Polanco et al. 1981). Among sib pairs in which both siblings are affected with CD, the HLA haplotype sharing is significantly more than expected as reviewed by Scholz & Albert (1983). Furthermore, several studies have reported that CD more frequently occurs in HLA-DR3, DQ2 positive subjects. Indeed, a significant part of the genetic sus- ceptibility to CD seems associated with products of the major histocompatibility complex (MHC)
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Genetic contribution of the HLA region to the familial clustering of coeliac disease

Ann. Hum. Genet. (1997), 61, 307–317
Printed in Great Britain
307
Genetic contribution of the HLA region to the familial clustering
of coeliac disease
F. PETRONZELLI", M. BONAMICO#, P. FERRANTE", R. GRILLO", B. MORA",
P. MARIANI#, I. APOLLONIO", G. GEMME$ M. C. MAZZILLI"
"Dipartimento di Medicina Sperimentale, Universita[ ‘La Sapienza’, Viale Regina Elena,
324–00161 Roma, Italy#Dipartimento di Pediatria, Universita[ ‘La Sapienza’, Roma, Italy
$ Istituto Gaslini, Universita[ di Genova, Italy
(Received 22.12.95. Accepted 14.4.97)
In order to assess the effect of the HLA region on familiality of coeliac disease (CD), we carried out
a study on 121 CD index cases and 325 first degree relatives. The transmission disequilibrium test
confirmed the importance of the HLA-DR3 haplotype in CD susceptibility. However, the different
distortion found in affected children inheriting maternal or paternal DR3 alleles suggested that the
sex of the parent might influence the risk conferred by this haplotype. The increase in risk to siblings
of affected individuals relative to the risk in the general population (λs) and the contribution of the
HLA genes to this clustering (λsHLA
) have also been estimated. Non-overlapping data from the
literature have been collected and combined with our sample to extend such analysis. Then, the
percentage contribution of the HLA region to the development of CD among siblings was 36±2%.
This result confirms that the HLA genotypes are an important genetic background to CD
development but shows that additional susceptibility factors remain to be identified.
Coeliac disease (CD), also known as gluten
sensitive enteropathy (GSE), is a condition in
which the immune response to ingested gluten is
accompanied by intestinal mucosal damage
(Trier, 1991). The resulting atrophy of the villi of
the proximal small intestine leads to mal-
absorption of nutrients and the clinical features
of CD. The symptoms and the epithelial damage
are reversed by the exclusion of gluten from the
diet. The pathogenesis of CD is believed to
involve interaction between genetic and environ-
mental factors (Polanco, 1981).
The reported prevalence of clinically manifest
CD is around 1:1000. However, because of the
increasing recognition of subclinical or silent
cases, the true prevalence is much higher than
supposed, and it probably approaches 1:300
(Catassi et al. 1994; Troncone et al. 1992). The
familial clustering of the disease is documented
by the increased prevalence in relatives; in a
multicentred study, undertaken to investigate
the prevalence of CD among first-degree relatives
of patients, an estimated risk of 8±7% was found
(Auricchio et al. 1988). The genetic contribution
to this increased risk is supported by the high
rate of concordance for disease (71%) among
monozygotic twins (Polanco et al. 1981).
Among sib pairs in which both siblings are
affected with CD, the HLA haplotype sharing is
significantly more than expected as reviewed by
Scholz & Albert (1983). Furthermore, several
studies have reported that CD more frequently
occurs in HLA-DR3, DQ2 positive subjects.
Indeed, a significant part of the genetic sus-
ceptibility to CD seems associated with products
of the major histocompatibility complex (MHC)

308 F. P
encoded within the class II region (Sollid &
Thorsby, 1993).
Genes in the class II region are clustered into
three major subregions named HLA-DP, -DQ
and -DR. Each class II molecule on the cell
surface occurs as a heterodimer consisting of an
α chain encoded by an A gene and a β chain
encoded by a B gene. A hallmark of the HLA
gene complex is linkage disequilibrium between
alleles at different loci, which greatly complicates
any attempt to pinpoint disease susceptibility to
a specific locus. In this regard, the HLA typing of
patients and controls from different populations
could be of great relevance. Concerning CD,
Spanish (Mearin et al. 1983) and Italian studies
(Trabace et al. 1984; Morellini et al. 1988)
revealed a lower association of the disease to the
DR3 haplotype than reported in the North
Europe populations while a new association to
DR5,7 heterozygous combination was found.
This observation led to the exclusion of a direct
involvement of the DR3 molecule in the patho-
genesis of CD. More recently, different studies
(Morellini et al. 1986; Sollid et al. 1989; Hall et al.
1992) have provided evidence for a primary HLA
association of CD to the DQ(α1*0501, β1*0201)
heterodimer, encoded in cis on the DR3 haplo-
type or in trans in the DR5,7 heterozygous
individuals. The immunopathogenic importance
of this association is supported by the finding
that, in the small intestine, the CD-associated
DQ heterodimer represents the main gluten-
presenting molecule (Lundin et al. 1990).
In a group of Italian paediatric patients we
have reported (Mazzilli et al. 1992) that 92% of
CD patients (50% DR3 and 42% DR5,7) carried
the high-risk DQα}β dimer. In accordance with
previous studies (Tosi et al. 1986), most of the
dimer negative patients typed as DR4,DQ8. A
few DR3 and DR5,7 negative cases were also
described (Grillo et al. 1996).
The aim of the present study was to evaluate
the risk of coeliac disease among first-degree
relatives of affected children in relation to the
presence of susceptible HLA molecules. The data
presented here are discussed together with those
reported in the literature in an attempt to
contribute in defining the relative role of the
HLA region and other genes in the familial
clustering of the disease.
Families
The whole sample consisted of 447 subjects :
121 unrelated children with coeliac disease and
325 first-degree relatives (212 parents, 113 sib-
lings) and one twin. Altogether, 144 patients
were observed (121 index cases, 18 siblings and 5
parents). In the 18 families with 2 affected
siblings we considered as ‘ index case’ the first
being diagnosed. The diagnosis remained un-
certain in 2 parents and 4 siblings. The twin pair
included two HLA identical affected males but
not enough data were available to conclude if
they were monozygous.
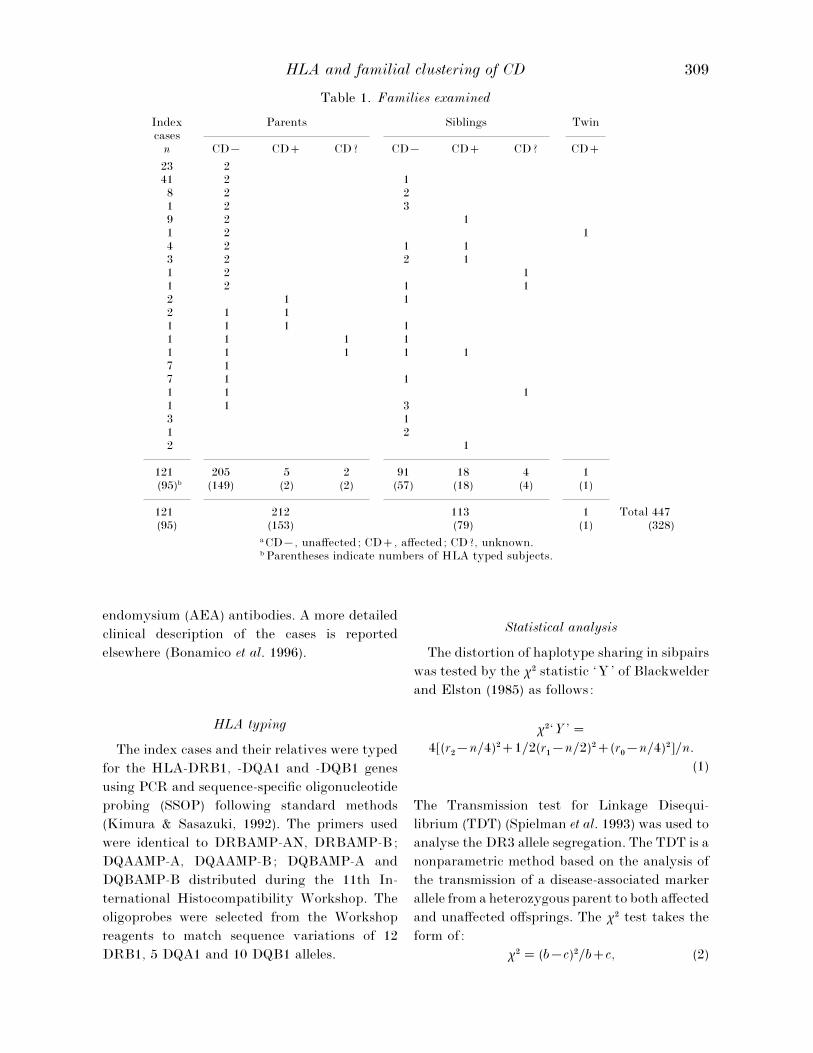
The composition of the families is reported in
Table 1. HLA typing was performed in 153
parents, 79 siblings and one twin of 81 index
cases. In total, 95 index cases were HLA typed
including 28 from earlier studies (Ferrante et al.
1992; Mazzilli et al. 1992). The numbers of typed
subjects in the different groups are indicated in
brackets in the table. In order to complete the
HLA haplotypic segregation the typing was
carried out in twelve more parents but they were
not considered in any of the analyses reported in
the following paragraphs because of lack of
clinical information.
Diagnosis
The diagnosis of CD was based on small
intestinal biopsies, according to the new Euro-
pean Society for Paediatric Gastroenterology
and Nutrition (ESPGAN) criteria (Walker-Smith
et al. 1990). While all the index cases were
biopsied, as far as the relatives are concerned
only ‘suspected’ cases were requested to undergo
the biopsies. The suspicion of CD was based on
clinical examination and on the presence of
antigliadin (AGA), IgA and IgG, and}or anti-
HLA and familial clustering of CD 309
Table 1. Families examined
Indexcases
Parents Siblings Twin
n CD® CD CD? CD® CD CD? CD
23 241 2 18 2 21 2 39 2 11 2 14 2 1 13 2 2 11 2 11 2 1 12 1 12 1 11 1 1 11 1 1 11 1 1 1 17 17 1 11 1 11 1 33 11 22 1
121 205 5 2 91 18 4 1(95)b (149) (2) (2) (57) (18) (4) (1)
121 212 113 1 Total 447(95) (153) (79) (1) (328)
a CD®, unaffected; CD, affected; CD?, unknown.b Parentheses indicate numbers of HLA typed subjects.
endomysium (AEA) antibodies. A more detailed
clinical description of the cases is reported
elsewhere (Bonamico et al. 1996).
HLA typing
The index cases and their relatives were typed
for the HLA-DRB1, -DQA1 and -DQB1 genes
using PCR and sequence-specific oligonucleotide
probing (SSOP) following standard methods
(Kimura & Sasazuki, 1992). The primers used
were identical to DRBAMP-AN, DRBAMP-B;
DQAAMP-A, DQAAMP-B; DQBAMP-A and
DQBAMP-B distributed during the 11th In-
ternational Histocompatibility Workshop. The
oligoprobes were selected from the Workshop
reagents to match sequence variations of 12
DRB1, 5 DQA1 and 10 DQB1 alleles.
Statistical analysis
The distortion of haplotype sharing in sibpairs
was tested by the χ# statistic ‘Y’ of Blackwelder
and Elston (1985) as follows:
χ#‘Y ’¯4[(r
#®n}4)#1}2(r
"®n}2)#(r
!®n}4)#]}n.
(1)
The Transmission test for Linkage Disequi-
librium (TDT) (Spielman et al. 1993) was used to
analyse the DR3 allele segregation. The TDT is a
nonparametric method based on the analysis of
the transmission of a disease-associated marker
allele from a heterozygous parent to both affected
and unaffected offsprings. The χ# test takes the
form of:
χ#¯ (b®c)#}bc, (2)

310 F. P
where b is the number of transmitted alleles and c
the number of transmissions of other alleles.
The λsparameter (Risch, 1987) has been used
to define the relationship between the risk in
siblings (Ks) and the population prevalence (K)
as follows:
λs¯K
s}K. (3)
The contribution of the HLA region (λsHLA
) has
been estimated from the expected proportion of
affected sib pairs sharing zero haplotypes identi-
cal-by-descent (IBD) (0±25) divided by the ob-
served proportion.
Assuming a multiplicative model as suggested
by Risch (1987), the HLA contribution to the λs
can be expressed as percentage by the following
formula:
% λsHLA
¯ 100(log λsHLA
}log λs). (4)
Nomenclature
The latest recommendation from the WHO
HLA Nomenclature Committee was used
(Bodmer et al. 1995).
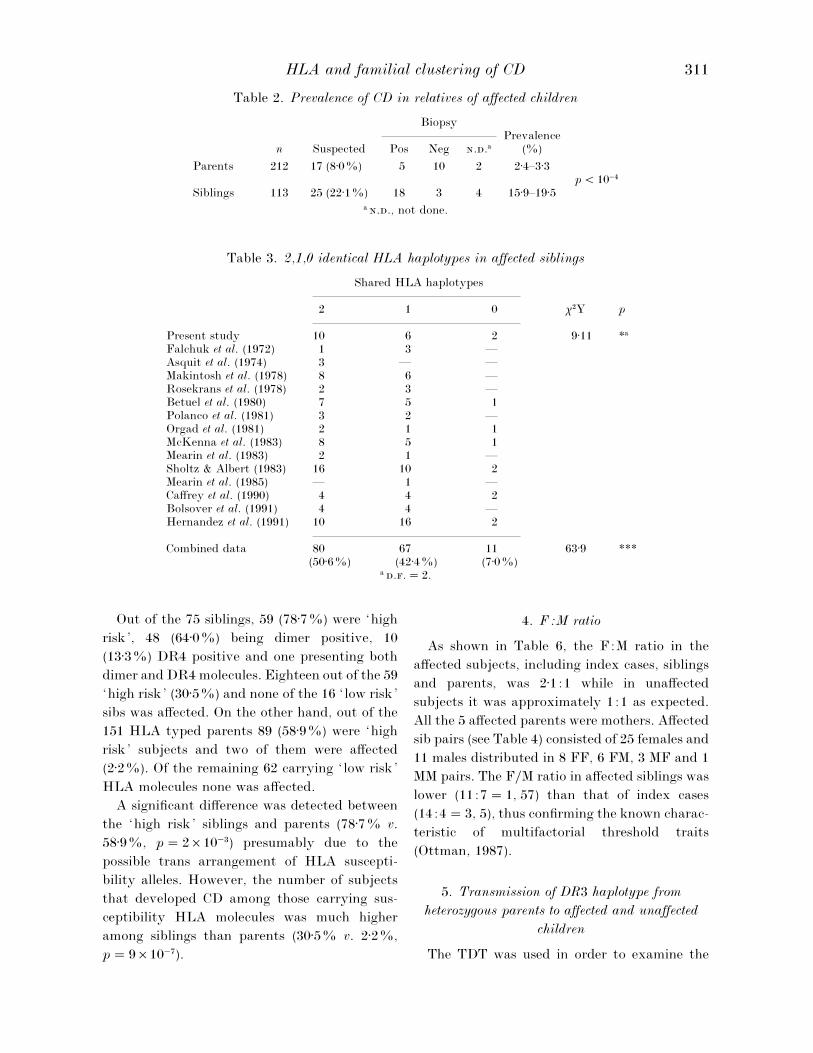
1. Prevalence of CD in first degree relatives
On the basis of clinical symptoms and presence
of antigliadin (IgA and IgG) and}or anti-
endomysium antibodies, a diagnosis of suspected
CD was made for 17}212 parents (8±0%), 25}113
siblings (22±1%) and for the twin. All the cases
identified by the first screening were asked to
undergo the biopsy}ies. Five parents, 18 siblings
and the twin were found to be affected on the
basis of intestinal mucosa atrophy. Two parents
and 4 siblings have not completed the diagnostic
procedures yet or refused to undergo the biopsy;
they have been classified as unknown and not
considered in the analysis reported in the fol-
lowing paragraphs. Thus the prevalence of the
disease in the parents of affected children was
2±4% (5}212) if only the biopsied cases were
considered or 3±3% (7}212) taking into account
also the unknown subjects (Table 2). Similarly,
the prevalence in the siblings of the index cases
was evaluated in a range from 15±9% to 19±5%.
Using the chi-square test of a 2¬2 contingency
table, there was a significant difference between
the two groups even comparing the two closest
values (p! 10−%).
2. HLA haplotype sharing in sib pairs
The sharing of HLA haplotypes IBD has been
analysed in the sib pairs collected in the present
study together with those available from the
literature (Table 3). The frequencies of sharing
two, one or zero HLA haplotypes in affected
siblings were obviously distorted in our sample
as well as in the combined data, as shown by the
χ# ‘Y’ test statistic. Taken together, the results
on 158 affected sib pairs gave frequencies of
50±6%, 42±4% and 7±0% for sharing 2,1 and 0
HLA haplotypes respectively. On the other hand,
non-affected siblings did not differ significantly
from the expected proportion, both in our sample
(22±8%, 56±1%, 21±1%) and in the combined one
(21±4%, 51±1%, 27±5%).
In Table 4 is reported some information
concerning the 18 CD sib pairs of the present
study together with the 4 cases with ‘unknown’
siblings and the pair of twins. Regarding the
clinical presentation of the disease, all the index
cases showed a manifest form, typical or atypical,
while most siblings were clinically silent. All
dimer negative index cases and affected siblings
typed as DR4. Three out of 4 siblings classified as
CD ‘unknown’ showed HLA typing at risk of
CD. The twins were both DR5}7 positive.
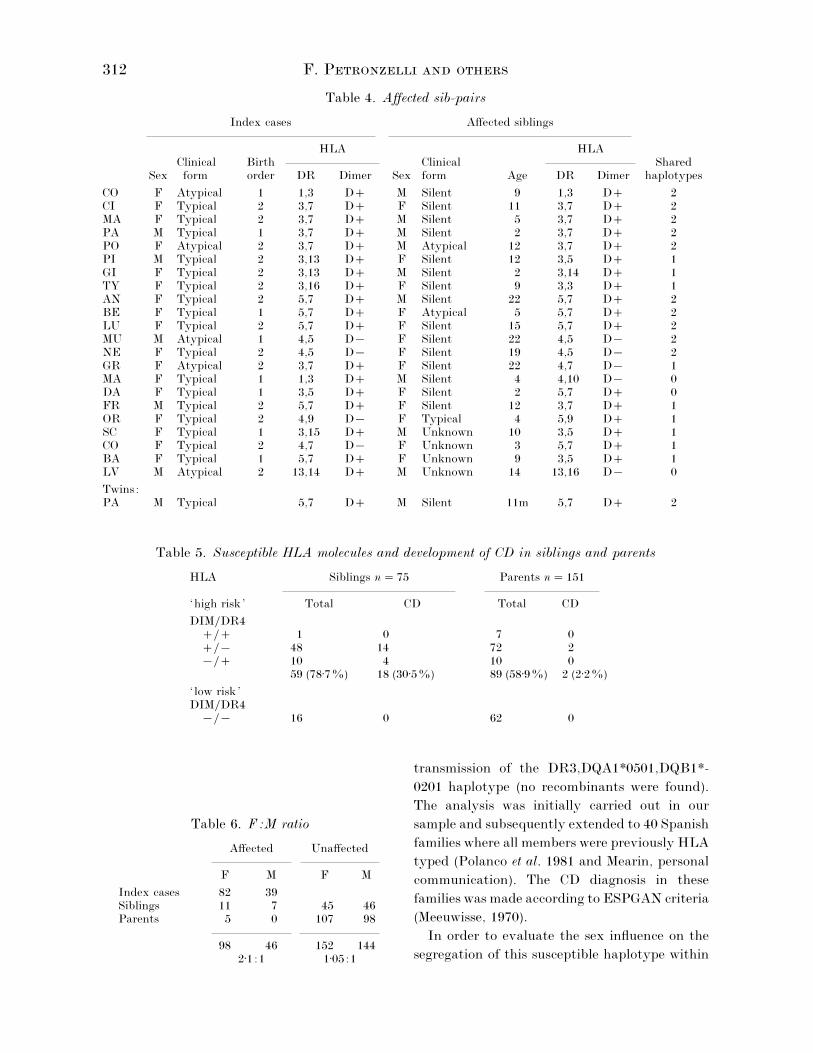
3. Recurrence of CD in relatives and HLA typing
Seventy-five siblings and 151 parents were
HLA typed, including all the 18 siblings and only
2 of the five parents identified as affected. The
subjects carrying the DQ(α1*0501, β1*0201)
dimer and}or DR4 molecule were classified as
‘high risk’, while the absence of these antigens
characterised the ‘ low risk’ group (Table 5).
HLA and familial clustering of CD 311
Table 2. Prevalence of CD in relatives of affected children
BiopsyPrevalence
n Suspected Pos Neg ..a (%)
Parents 212 17 (8±0%) 5 10 2 2±4–3±3p! 10−%
Siblings 113 25 (22±1%) 18 3 4 15±9–19±5a .., not done.
Table 3. 2,1,0 identical HLA haplotypes in affected siblings
Shared HLA haplotypes
2 1 0 χ#Y p
Present study 10 6 2 9±11 *a
Falchuk et al. (1972) 1 3 —Asquit et al. (1974) 3 — —Makintosh et al. (1978) 8 6 —Rosekrans et al. (1978) 2 3 —Betuel et al. (1980) 7 5 1Polanco et al. (1981) 3 2 —Orgad et al. (1981) 2 1 1McKenna et al. (1983) 8 5 1Mearin et al. (1983) 2 1 —Sholtz & Albert (1983) 16 10 2Mearin et al. (1985) — 1 —Caffrey et al. (1990) 4 4 2Bolsover et al. (1991) 4 4 —Hernandez et al. (1991) 10 16 2
Combined data 80 67 11 63±9 ***(50±6%) (42±4%) (7±0%)
a ..¯ 2.
Out of the 75 siblings, 59 (78±7%) were ‘high
risk’, 48 (64±0%) being dimer positive, 10
(13±3%) DR4 positive and one presenting both
dimer and DR4 molecules. Eighteen out of the 59
‘high risk’ (30±5%) and none of the 16 ‘ low risk’
sibs was affected. On the other hand, out of the
151 HLA typed parents 89 (58±9%) were ‘high
risk’ subjects and two of them were affected
(2±2%). Of the remaining 62 carrying ‘ low risk’
HLA molecules none was affected.
A significant difference was detected between
the ‘high risk’ siblings and parents (78±7% v.
58±9%, p¯ 2¬10−$) presumably due to the
possible trans arrangement of HLA suscepti-
bility alleles. However, the number of subjects
that developed CD among those carrying sus-
ceptibility HLA molecules was much higher
among siblings than parents (30±5% v. 2±2%,
p¯ 9¬10−().
4. F:M ratio
As shown in Table 6, the F:M ratio in the
affected subjects, including index cases, siblings
and parents, was 2±1:1 while in unaffected
subjects it was approximately 1:1 as expected.
All the 5 affected parents were mothers. Affected
sib pairs (see Table 4) consisted of 25 females and
11 males distributed in 8 FF, 6 FM, 3 MF and 1
MM pairs. The F}M ratio in affected siblings was
lower (11:7¯ 1, 57) than that of index cases
(14:4¯ 3, 5), thus confirming the known charac-
teristic of multifactorial threshold traits
(Ottman, 1987).
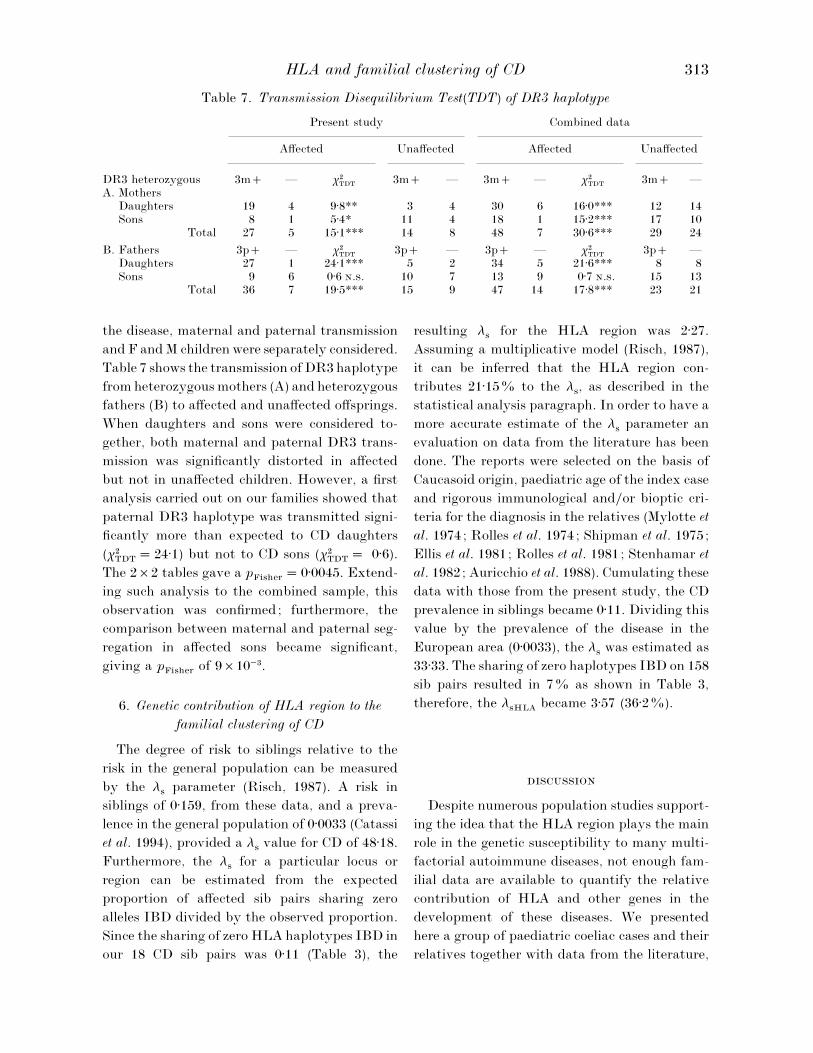
5. Transmission of DR3 haplotype from
heterozygous parents to affected and unaffected
children
The TDT was used in order to examine the
312 F. P
Table 4. Affected sib-pairs
Index cases Affected siblings
HLA HLAClinical Birth Clinical Shared
Sex form order DR Dimer Sex form Age DR Dimer haplotypes
CO F Atypical 1 1,3 D M Silent 9 1,3 D 2CI F Typical 2 3,7 D F Silent 11 3,7 D 2MA F Typical 2 3,7 D M Silent 5 3,7 D 2PA M Typical 1 3,7 D M Silent 2 3,7 D 2PO F Atypical 2 3,7 D M Atypical 12 3,7 D 2PI M Typical 2 3,13 D F Silent 12 3,5 D 1GI F Typical 2 3,13 D M Silent 2 3,14 D 1TY F Typical 2 3,16 D F Silent 9 3,3 D 1AN F Typical 2 5,7 D M Silent 22 5,7 D 2BE F Typical 1 5,7 D F Atypical 5 5,7 D 2LU F Typical 2 5,7 D F Silent 15 5,7 D 2MU M Atypical 1 4,5 D® F Silent 22 4,5 D® 2NE F Typical 2 4,5 D® F Silent 19 4,5 D® 2GR F Atypical 2 3,7 D F Silent 22 4,7 D® 1MA F Typical 1 1,3 D M Silent 4 4,10 D® 0DA F Typical 1 3,5 D F Silent 2 5,7 D 0FR M Typical 2 5,7 D F Silent 12 3,7 D 1OR F Typical 2 4,9 D® F Typical 4 5,9 D 1SC F Typical 1 3,15 D M Unknown 10 3,5 D 1CO F Typical 2 4,7 D® F Unknown 3 5,7 D 1BA F Typical 1 5,7 D F Unknown 9 3,5 D 1LV M Atypical 2 13,14 D M Unknown 14 13,16 D® 0
Twins:PA M Typical 5,7 D M Silent 11m 5,7 D 2
Table 5. Susceptible HLA molecules and development of CD in siblings and parents
HLA Siblings n¯ 75 Parents n¯ 151
‘high risk’ Total CD Total CD
DIM}DR4} 1 0 7 0}® 48 14 72 2®} 10 4 10 0
59 (78±7%) 18 (30±5%) 89 (58±9%) 2 (2±2%)
‘ low risk’DIM}DR4
®}® 16 0 62 0
Table 6. F:M ratio
Affected Unaffected
F M F M
Index cases 82 39Siblings 11 7 45 46Parents 5 0 107 98
98 46 152 1442±1:1 1±05:1
transmission of the DR3,DQA1*0501,DQB1*-
0201 haplotype (no recombinants were found).
The analysis was initially carried out in our
sample and subsequently extended to 40 Spanish
families where all members were previously HLA
typed (Polanco et al. 1981 and Mearin, personal
communication). The CD diagnosis in these
families was made according to ESPGAN criteria
(Meeuwisse, 1970).
In order to evaluate the sex influence on the
segregation of this susceptible haplotype within
HLA and familial clustering of CD 313
Table 7. Transmission Disequilibrium Test(TDT) of DR3 haplotype
Present study Combined data
Affected Unaffected Affected Unaffected
DR3 heterozygousA. Mothers
3m — χ#TDT
3m — 3m — χ#TDT
3m —
Daughters 19 4 9±8** 3 4 30 6 16±0*** 12 14Sons 8 1 5±4* 11 4 18 1 15±2*** 17 10
Total 27 5 15±1*** 14 8 48 7 30±6*** 29 24
B. Fathers 3p — χ#TDT
3p — 3p — χ#TDT
3p —Daughters 27 1 24±1*** 5 2 34 5 21±6*** 8 8Sons 9 6 0±6 .. 10 7 13 9 0±7 .. 15 13
Total 36 7 19±5*** 15 9 47 14 17±8*** 23 21
the disease, maternal and paternal transmission
and F and M children were separately considered.
Table 7 shows the transmission of DR3 haplotype
from heterozygous mothers (A) and heterozygous
fathers (B) to affected and unaffected offsprings.
When daughters and sons were considered to-
gether, both maternal and paternal DR3 trans-
mission was significantly distorted in affected
but not in unaffected children. However, a first
analysis carried out on our families showed that
paternal DR3 haplotype was transmitted signi-
ficantly more than expected to CD daughters
(χ#TDT
¯ 24±1) but not to CD sons (χ#TDT
¯ 0±6).
The 2¬2 tables gave a pFisher
¯ 0±0045. Extend-
ing such analysis to the combined sample, this
observation was confirmed; furthermore, the
comparison between maternal and paternal seg-
regation in affected sons became significant,
giving a pFisher
of 9¬10−$.
6. Genetic contribution of HLA region to the
familial clustering of CD
The degree of risk to siblings relative to the
risk in the general population can be measured
by the λs
parameter (Risch, 1987). A risk in
siblings of 0±159, from these data, and a preva-
lence in the general population of 0±0033 (Catassi
et al. 1994), provided a λsvalue for CD of 48±18.
Furthermore, the λs
for a particular locus or
region can be estimated from the expected
proportion of affected sib pairs sharing zero
alleles IBD divided by the observed proportion.
Since the sharing of zero HLA haplotypes IBD in
our 18 CD sib pairs was 0±11 (Table 3), the
resulting λs
for the HLA region was 2±27.
Assuming a multiplicative model (Risch, 1987),
it can be inferred that the HLA region con-
tributes 21±15% to the λs, as described in the
statistical analysis paragraph. In order to have a
more accurate estimate of the λs
parameter an
evaluation on data from the literature has been
done. The reports were selected on the basis of
Caucasoid origin, paediatric age of the index case
and rigorous immunological and}or bioptic cri-
teria for the diagnosis in the relatives (Mylotte et
al. 1974; Rolles et al. 1974; Shipman et al. 1975;
Ellis et al. 1981; Rolles et al. 1981; Stenhamar et
al. 1982; Auricchio et al. 1988). Cumulating these
data with those from the present study, the CD
prevalence in siblings became 0±11. Dividing this
value by the prevalence of the disease in the
European area (0±0033), the λswas estimated as
33±33. The sharing of zero haplotypes IBD on 158
sib pairs resulted in 7% as shown in Table 3,
therefore, the λsHLA
became 3±57 (36±2%).
Despite numerous population studies support-
ing the idea that the HLA region plays the main
role in the genetic susceptibility to many multi-
factorial autoimmune diseases, not enough fam-
ilial data are available to quantify the relative
contribution of HLA and other genes in the
development of these diseases. We presented
here a group of paediatric coeliac cases and their
relatives together with data from the literature,

314 F. P
with the double aim to contribute in evaluating
the prevalence of CD in relatives of affected
children and in assessing the HLA role on the
increased risks of disease in the siblings.
The screening procedure used in this study
consisted of two steps. All first-degree relatives
of the index cases were invited to take part in a
first level of study that included clinical and
immunological examinations and the ‘suspected’
subjects were asked to undergo biopsy}ies. The
diagnosis was confirmed in 86% of siblings and
33% of parents. This discrepancy could be
explained by the fact that with age AGA results
tend to become worse predictors of CD (Picarelli
et al. 1996). Furthermore, adults often manifest
clinical symptoms suggestive of CD which in fact
are due to different gastroenterological disorders.
Then the prevalence of CD in the 325 first-
degree relatives studied was 7% if only subjects
that had undergone biopsies were considered, or
8±9% including cases positive at the first screen-
ing but that have not concluded the diagnostic
iter, yet. This result is in agreement with those
previously reported in relatives of paediatric CD
patients where the average prevalence of 8±2%
can be calculated (Mylotte et al. 1974; Rolles et
al. 1974; Shipman et al. 1975; Ellis et al. 1981;
Rolles et al. 1981; Stenhamar et al. 1982;
Auricchio et al. 1988). Since the biopsy screening
procedure has been used in most of these cases,
our data attest the reliability of non-invasive
pre-screening protocol in detecting manifest as
well as subclinical cases, confirming previous
observations (Catassi et al. 1994).
When parents and siblings were separately
considered, the risks in the two groups were
significantly different, being in a range of 2±4–
3±3% and 15±9–19±5%, respectively. These re-
sults are in agreement with the suggested
approximate 5:1 ratio of children}adult patients
(Magazzu' et al. 1994). The prevalence of CD in
siblings here estimated is one of the higher
reported. However, we would like to emphasise
that most of them were asymptomatic.
The mode of inheritance of CD susceptibility
has not been established but it probably involves
several interacting loci. The association of CD
with the HLA region is the only consistently
observed genetic feature in this multifactorial
disorder. What is now largely accepted is the
direct role of the DQ(α1*0501, β1*0201) dimer in
the pathogenesis of the disease. The proposed
mechanisms may be the involvement in the
thymic selection of TCR repertoire and}or the
preferential binding and presentation of relevant
gliadin peptides. Although HLA typing is not
diagnostically significant, we propose it as a pre-
screening test in CD relatives to restrict the
number of subjects that must undergo further
investigations. Unlike the anti-gliadin and anti-
endomysium antibodies that occur when disease
occurs, the HLA alleles are predisposing factors
detectable at birth and therefore might have a
prospective value.
All affected relatives found in our sample
carried high risk HLA alleles, but in some familial
groups the disease was inherited with different
haplotypes. These families formally confirm that
HLA genes are not markers of a linked disease
gene but may themselves work as predisposing
factors. Really, the HLA susceptibility appears
primarily conferred by the concurrent presence
of DQA1*0501 and DQB1*0201 alleles, coding in
cis or in trans the DQα}β heterodimer. In the
majority of the patients a single copy of these
high risk alleles is sufficient for the development
of the disease (Petronzelli et al., 1993). This
observation leads us to consider the dominant
model as the most appropriate to explain the
HLA contribution to CD susceptibility. A gene
dosage effect of the DQB1*0201 allele in dimer
positive individuals has been recently reported
by Ploski et al. (1993). In our opinion, this result
is not in contrast with this model of inheritance
since a homozygosity influence on the patho-
logical phenotype has been observed for several
dominant genes. Furthermore, the high risk
conferred by the double dose of DQB1*0201
allele and the fact that in many families the
dimer is carried in trans could justify the
observed increase in frequency of sibling pairs
sharing two haplotypes that previously sug-
gested a recessive model of inheritance (Green-
berg et al. 1982).

HLA and familial clustering of CD 315
Since DR3,DQA1*0501,DQB1*0201 is the
most frequent haplotype in CD patients, TDT
has been used to analyse its segregation in the
families. Looking at the whole affected progeny,
a high distortion of DR3 transmission from both
parents was found, as expected on the basis of
the strong association of DR3 with the disease.
However, the segregation from heterozygous
fathers became highly distorted in female but
not in male children. Our data combined with 40
Spanish families, that we regard as very similar
to our own sample, not only confirmed the trend
observed but also allowed the comparison of
DR3 maternal and paternal segregation in
affected sons. These data support the idea that
the sex of the parent transmitting the DR3
together with that of the child inheriting it may
influence the susceptibility conferred by this
haplotype. A similar model, even if in a single
gene disease, has been proposed for Myotonic
Distrophy (Gennarelli et al. 1994).
An attempt to quantify the HLA contribution
to the familial clustering of the disease was done
using the λsparameter. The contribution of the
HLA region evaluated on the affected sib pairs
from the present study resulted as 21±15% of the
increased risk to siblings. Indeed, the presence of
high risk HLA molecules seems a necessary but
not sufficient genetic condition since the disease
occurs in approximately 1 out of 3 sibs and 1 out
of 50 parents carrying predisposing HLA factors.
A further evaluation of these parameters was
done on a larger sample consisting of our families
and data available from the literature. A λsHLA
of
3±57, was calculated using haplotype sharing
frequency of 158 sib pairs and possibly represents
a more precise estimate of the HLA role. Hence,
the contribution of the HLA susceptible alleles
to the development of CD has been quantified as
36±2% of the increase in risk to siblings leaving a
large proportion to unidentified factors.
In the absence of logical candidate loci, a
genome-wide screening that makes use of a large
set of well spaced highly polymorphic markers
has proven to be a very powerful method for
searching for genomic regions which might
contain loci involved in genetically complex
diseases (Weeks & Lathrop, 1995). The search for
genes predisposing to CD has already been
undertaken by Zhong et al. (1996) that recently
published the results of a first screening carried
out on 45 affected sibling pairs where some
candidate regions have been identified. Even if
these results need to be confirmed in a larger
sample, they represent the first indication of a
chromosomal location of other predisposing
genes.
We are grateful to the patients and their families forparticipating in the study. We would like to thank Prof.I. Polanco and Dr M. L. Mearin for kindly providingsome unpublished pedigrees and Prof. L. Terrenato forcritically reading the manuscript. This work was partiallysupported by CNR, ‘Progetto Finalizzato IngegneriaGenetica’ and by MURST 40% ‘Immunoregolazione’.
A, P. (1974). Family studies in celiac disease. InCeliac disease (eds. W. Th. Hekkens & A. S. Pena),pp. 322–325. Leiden: Krose.
A, S., M, G., T, R., V, J.,M, M. & P, I. (1988). Celiac disease as afamilial condition: identification of asymptomaticceliac patients within family groups. GastroenterologyInternational, 1, 25–31.
B, H., G, L., D, L., B, J.,F, F. & L, J. C. (1980). Celiac diseaseand its association with HLA markers. In Hysto-compatibility testing 1980, vol. 1 (ed. P. I. Terasaki),pp. 668–672. UCLA Tissue Typing Laboratory Pub-lisher.
B, W. C. & E, R. C. (1985). A com-parison of sib-pair linkage tests for disease suscep-tibility loci. Genet. Epidemiol. 2, 85–97.
B, J. G., M, G. E., A, E. D., B,W. F., B, R. E., C, D., D, B.,E, H. A., M, B., M, W. R., P, P.,S, T., S, G. M. T., S, J.L., S, A. & T, P. I. (1995). No-menclature for factors of the HLA system, 1995.Tissue Antigens 46, 1–18.
B, W. J., H, M. A., V, R. W.,W, K. I. & C, P. J. (1991). A family studyconfirms that the HLA-DP associations with CeliacDisease are the result of an extended HLA-DR3haplotype. Hum. Immunol. 31, 100–108.
B, M., M, P., M, M. C., T,P., L, P., F, P., P, A.,M, A., G, G. & I, C. (1996).Prevalence and clinical pattern of celiac disease amongsiblings of celiac children. J. Ped. Gastroenterol. Nutr.23: 159–163.
C, C., H, G. A., N, M. J., C, P.G., K, P., F, L., M, P., G,R., F, C., W, D., S, J. A. (1990).HLA-DP and celiac disease: family and populationstudies. Gut 31, 663–667.

316 F. P
C, C., R, I. M., F, E., R, M.,B, F., C, F., C, G. V. & G,P. L. (1994). Celiac disease in the year 2000: exploringthe iceberg. The Lancet 343, 200–203.
E, A. (1981). Celiac disease: previous family studies.In The genetics of celiac disease (ed. R. B. McConnell),pp. 197–200. Lancaster : MTP Press.
F, Z. M., R, G. N. & S, W. (1972).Predominance of histocompatibility antigen HL-A8 inpatients with gluten-sensitive enteropathy. J. Clin.Invest. 51, 1602–1606.
F, P., P, F., M, P., B-, M., M, M. C. (1992). Oligotyping of Italianceliac patients with the 11th International Histo-compatibility Workshop reagents. Tissue antigens 39,38–39.
G, M., D, B., B, M., M-, I. & N, G. (1994). Meiotic drive at themyotonic dystrophy locus. J. Med. Genet. 31, 980.
G, D. A., H, S. E. & R, J. I. (1982).Evidence for recessive and against dominant in-heritance at the HLA-linked locus in coeliac disease.Am. J. Hum. Genet. 34, 263–277.
G, R., P, F., F, P., M, B.,B, M., M, M. C. (1996). Unusual HLAtyping in Celiac disease. Dis. Markers. 13, 61–64.
H, M., M, M. C., S, M. L., B, F.,B, A., B, G., C, P. J., C-, G. R., F, P., G, W., H, M.,K, A., K, E., L, J. S., M-, V., M, K., P, F., R,E., T, G., V, B. A., W, K. I., W,T. & A, E. (1992). Celiac disease study XIthworkshop report. In HLA 1991, vol. 1 (eds. K. Tsuji,M. Aizawa, T. Sasazuki), pp. 722–729. Oxford: Uni-versity Press.
H, J. L., M, J. P., MC, C. C.,MC, C. F., S, F. M. & E, R. C.(1991). Evidence for a dominant gene mechanismunderlying coeliac disease in West of Ireland. Genet.Epidemiol. 8, 13–27.
K, A. & S, T. (1992). Eleventh Inter-national Histocompatibility Workshop reference pro-tocol for the HLA DNA-typing technique. In HLA1991, vol. 1 (eds. K. Tsuji, M. Aizawa, T. Sasazuki), pp.397–419. Oxford: University Press.
L, K. E. A., S, L. M., Q, E., M-, G., G, H. A., E, J. & T, E.(1990). T lymphocyte recognition of celiac diseaseassociated cis or trans encoded HLA-DQ α,β hetero-dimer. J. Immunol. 145, 136–139.
M' , G., B, G., C, F., I, G., DD, F., P, R., C, F., M, I.,R, C., A, A., R, N., B, E.,T, G. & G, L. (1994). Increasing incidenceof childhood celiac disease in Sicily: results of amulticenter study. Acta Paediatr. 83, 1065–1069.
M, P. & A, P. (1978). HLA and coeliacdisease. Brit. Med. Bull 34, 291–294.
M, M. C., F, P., M, P., M,E., P, F., T, P. & B, M.(1992). A study of Italian paediatric celiac diseasepatients confirms that the primary HLA association isto the DQ(α1*0501, β1*0201). heterodimer. Hum.Immunol. 33, 133–139.
MK, R., S, F. M., MN, B., S,S., A, E. & MC, C. F. (1983). Family andpopulation studies of HLA and coeliac disease in theWest of Ireland. Tissue Antigens 22, 175–181.
M, M. L., B, I., P4 , A. S., P, I.,V, C., S, G.T. M., D V, R. R.P. & V R, J. J. (1983). HLA-DR phenotypes inSpanish coeliac children: their contribution to theunderstanding of the genetics of the disease. Gut 24,532–537.
M, M. L., B, J., M, N., S,E., S, M., B, I., S, G.M.T., P, A. S., V G, H. H. & V
R, J. J. (1985). HLA-DR antigens and phenotypesin Dutch coeliac children and their families. ClinicalGenetics 27, 45–50.
M, G. W. (1970). Diagnostic criteria in coeliacdisease. Acta Paediatr. Scand. 59, 461–463.
M, M., B, M., O, C., L, P.,T, S., C, S., M, I. & M-, M. C. (1986). Are DQ dimers by transassociationthe molecular basis of coeliac disease susceptibility?7th International Congress of Human Genetics, Berlin.
M, M., T, S., M, M. C., L, P.,C, S., B, M., M, I. &G, E. (1988). A study of HLA class II antigensin an Italian paediatric population with coeliac disease.Disease Markers 6, 23–28.
M, M., E-M, B., F, P. F.,MN, B. & MC, C. F. (1974). Familystudies in coeliac disease. Q. J. Med. 171, 359–369.
O, S., A, S., J, A., M, J. & G,E. (1981). Immunogenetics of childhood celiac disease:the association with HLA DR3 and DR7 in unrelatedpatients and multiply affected families. Israel J. Med.Sci. 17, 1041–1044.
O, R. (1987). Simple test of the multifactorial-polygenic model with sex dependent threshold. J.Chron. Dis. 40, 165–170.
P, F., M, G., F, P., B-, M., R, G., C, L. & M, M. C.(1993). Different dose effect of HLA DQαβ hetero-dimers in insulin dependent diabetes mellitus andceliac disease susceptibility. Human Immunol. 36,156–162.
P, A., T, P., M, P., D G-, F., G, M., G, M., P, P.,B, M., B, G. (1996). The use of athreshold serum level of antigliadin antibodies im-proves diagnostic efficiency of the test in adult coeliacdisease but is not successful as a screening test. TheItalian Journal of astroenterology 28, 70–75.
P, R., T, J. E. E. & S, L. M. (1993).On the HLA-DQ(1*0501,1*0201). associated suscep-tibility in celiac disease: a possible gene dosage effectof DQB1*0201. Tissue Antigens 41, 173–177.
P, I., B, I., V L, A., S-, I., M K, P., G, J., D’A, J.,V, C., V R, J. J. & P, A. S. (1981).Gluten sensitive enteropathy in Spain: genetic andenvironmental factors. In The genetics of coeliac disease(ed. R. B. McConnell), pp. 211–231. Lancaster : MTPPress.

HLA and familial clustering of CD 317
R, N. (1987). Assessing the role of HLA-linked andunlinked determinants of disease. Am. J. Hum. Genet.40, 1–14.
R, C. J., K-M, T. O. & S, W. K. (1974).Family study of coeliac disease. In Coeliac disease (eds.W. T. J. M. Hekkens & A. S. Pena), pp. 320–321.Leiden: Stenfort Kroese).
R, C. J., K-M, T. B., W-K, S. &A, C. M. (1981). The familial incidence ofasymptomatic coeliac disease. In The genetics of coeliacdisease (ed. R. B. McConnell), pp 235–243. Lancaster :MTP Press.
R, P. C. M., P, A. S., H, W.T. J.M., D V, R. R. P. & H, A. J. (1978). Coeliacdisease. A family study in the Netherlands. InPerspectives in coeliac disease (ed. B. McNicholl, C. F.McCarthy & P. F. Fottrell), pp. 146–154. Lancaster :MTP Press.
S, R. T., W, A. L., K, R. & T,R. R. (1975). A family study of coeliac disease. Aust.N.Z.J. Med. 5, 250–255.
S, S. & A, E. (1983). HLA and diseases :involvement of more than one HLA-linked deter-minant of disease susceptibility. Immunological Rev.70, 77–88.
S, M. L., M, G., E, J., G, H.,V, F. & T, E. (1989). Evidence for aprimary association of celiac disease to a particularHLA-DQ α,β heterodimer. J. Exp. Med. 169, 345–350.
S, L. M. & T, E. (1993). HLA susceptibilitygenes in celiac disease: genetic mapping and role inpathogenesis. Gastroenterology 105, 910–922.
S, R. S., MG, R. E. & E, W. J.(1993). Transmission test for linkage disequilibrium:
the insulin gene region and insulin-dependent DiabetesMellitus (IDDM). Am. J. Hum. Genet. 52, 506–516.
S, L., B, A. & W, J. (1982).A family study of coeliac disease. Acta Paediatr. Scand.71, 625–628.
T, R., T, N., P, J., D M, M.,W, J. C. & H, P. H. (1986). A radio-immunoassay typing study of non-DQw2-associatedceliac disease. Clin. Immunol. Immunopathol. 39,168–172.
T, S., G, A., R, M., M, D.,C, I., T, A., M, M. C. &G, E. (1984). HLA-ABC and DR antigens inceliac disease. Vox Sanguinis 46, 102–106.
T, J. S. (1991). Celiac sprue. N. Engl. J. Med. 325,1709–1719.
T, R., C, N., Z, N., M,G., M, L. & A, S. (1992). Coeliac disease:a common food intolerance on an immunological basis.In Common food intolerances 1: Epidemiology of coeliacdisease, vol. 2 (eds. S. Auricchio & J. K. Visakorpi), pp.1–11. Basel : Karger.
W-S, J. A., G, S., S, J.,S, D. H. & V, J. K. (1990). Revisedcriteria for diagnosis of coeliac disease. Archives ofDisease in Childhood 65, 909–911.
W, D. E. & L, M. (1995). Polygenic disease:methods for mapping complex disease traits. TIG 11(12), 513–519.
Z, F., MC, C. C., O, J. M., E, R. C.,S, F. M., MC, C. F. & M, J. P.(1996). An autosomal screen for genes that predisposeto celiac disease in the western counties of Ireland.Nature Genet. 14, 329–333.




















