
Gene-specific and global methylation patterns predict outcome in patients with acute myeloid...
10
ORIGINAL ARTICLE Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia S Deneberg 1,4 , M Gro ¨vdal 1,4 , M Karimi 1,2 , M Jansson 1 , H Nahi 1 , A Corbacioglu 3 , V Gaidzik 3 , K Do ¨hner 3 , C Paul 1 , TJ Ekstro ¨m 2 , E Hellstro ¨ m-Lindberg 1 and S Lehmann 1 1 Division of Hematology, Department of Medicine, Karolinska Institutet, Huddinge, Stockholm, Sweden; 2 Department of Molecular Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden and 3 Department of Internal Medicine III, University Hospital of Ulm, Ulm, Germany This study was designed to analyze the effect of global and gene-specific DNA methylation patterns on the outcome of patients with acute myeloid leukemia (AML). Methylation of CDKN2B (p15), E-cadherin (CDH) and hypermethylated in cancer 1 (HIC1) promoters and global DNA methylation by luminometric methylation assay (LUMA) was analyzed in 107 AML patients and cytogenetic and molecular mutational analysis was performed. In addition, genome-wide promoter- associated methylation was assessed using the Illumina HumanMethylation27 array in a proportion of the patients. Promoter methylation was discovered in 66, 66 and 51% of the patients for p15, CDH and HIC1, respectively. In multivariate analysis, low global DNA methylation was associated with higher complete remission rate (hazard ratio (HR) 5.9, P ¼ 0.005) and p15 methylation was associated with better overall (HR 0.4, P ¼ 0.001) and disease-free survival (HR 0.4, P ¼ 0.016). CDH and HIC1 methylation were not associated with clinical outcome. Mutational status and karyotype were not significantly asso- ciated with gene-specific methylation or global methylation. Increased genome-wide promoter-associated methylation was associated with better overall and disease-free survival as well as with LUMA hypomethylation. We conclude that global and gene-specific methylation patterns are independently asso- ciated with the clinical outcome in AML patients. Leukemia (2010) 24, 932–941; doi:10.1038/leu.2010.41; published online 18 March 2010 Keywords: acute myeloid leukemia; DNA methylation; outcome; epigenetics Introduction Acute myeloid leukemia (AML) is a hematological malignancy with frequent and nonrandom genetic changes. Chromosomal translocations and gene mutations distinguish subtypes of the disease and have a role in proliferation and differentiation and also affect prognosis in AML. 1,2 Epigenetic modification of DNA is an important biological function regulating gene expression in normal and tumor cells. Decrease in the whole-genome methyl- ation level is widespread in neoplastic diseases. 3,4 Methylation of promoter-specific CpG islands, and hence silencing of tumor suppressor and cell cycle control genes, is considered to be an important step in tumor development and has been shown previously in AML. 5 In one large study estrogen receptor methylation has been shown to correlate with better overall survival (OS) in AML. 6 Although methylation of other genes has been shown to associate with prognosis, the results are somewhat contradictory. 7–9 In high-risk myelodysplastic syndrome (MDS) and in AML after MDS, methylation of CDKN2b (p15), E-cadherin (CDH) and hypermethylated in cancer 1 (HIC1) have recently been shown to be associated with failure to achieve complete remission (CR) after induction chemotherapy. 10 We analyzed gene-specific promoter methylation of p15, CDH and HIC1 as well as global DNA methylation using the luminometric methylation assay (LUMA) technique in a well-characterized cohort of 107 AML patients. Furthermore, we examined genome-wide CpG island-associated promoter methylation of 27 578 promoter CpG sites, corresponding to 14 495 individual genes, in 20 AML patients using the Illumina HumanMethylation27 BeadChip. We found that low global LUMA methylation levels correlated with an increased CR rate and that methylation of p15 correlated with better disease-free survival (DFS) and OS in uni- and multivariate analyses. There was a correlation between less global LUMA methylation and increased genome-wide promoter-associated methylation using the Illumina array. We found no significant associations between the methylation pattern and the cytogenetic risk groups or the mutation status of FLT3 (Fms-like tyrosine kinase 3), NPM1 (nucleophosmin), CEBPA (CCAAT enhancer binding protein-a) and NRAS (neuroblastoma RAS viral oncogene homolog). Materials and methods Patients and sampling A total of 107 previously untreated AML patients treated at the Karolinska University Hospital at Huddinge were included in the study. Patients with AML M3 or a previous history of MDS were excluded. Standard induction chemotherapy including cytarabine and an anthracycline was given aiming at CR. Patients who achieved CR were given standard consolidation therapy according to the applicable national or study protocol including allogeneic stem cell transplantation (n ¼ 23). Of all patients reaching CR, 18 (24%) received allogeneic stem cell transplantation, 41 (54%) received full consolidation therapy and 16 (22%) received reduced chemotherapy consolidation because of treatment toxicities. For diagnosis and classification of CR and progression, World Health Organization criteria were applied. 11 DNA was extracted from the diagnostic bone marrow samples (n ¼ 79) or peripheral blood (n ¼ 28) after mononuclear cell isolation (Lymphoprep; Axis-Shield PoC, Oslo, Norway). Informed consent was obtained before the specimens were collected. The study was approved by the regional ethics committee. Chromosome banding was performed using standard laboratory techniques. Mutation analyses of FLT3 (internal tandem duplications and tyrosine kinase domain Received 19 October 2009; revised 10 December 2009; accepted 21 January 2010; published online 18 March 2010 Correspondence: Dr S Deneberg, Hematology Center, M54, Karolinska University Hospital, Huddinge, 14186 Stockholm, Sweden. E-mail: [email protected] 4 These authors contributed equally to this work. Leukemia (2010) 24, 932–941 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Gene-specific and global methylation patterns predict outcome in patients with acute myeloid...

ORIGINAL ARTICLE
Gene-specific and global methylation patterns predict outcome in patients with acutemyeloid leukemia
S Deneberg1,4, M Grovdal1,4, M Karimi1,2, M Jansson1, H Nahi1, A Corbacioglu3, V Gaidzik3, K Dohner3, C Paul1, TJ Ekstrom2,E Hellstrom-Lindberg1 and S Lehmann1
1Division of Hematology, Department of Medicine, Karolinska Institutet, Huddinge, Stockholm, Sweden; 2Department ofMolecular Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden and 3Department of InternalMedicine III, University Hospital of Ulm, Ulm, Germany
This study was designed to analyze the effect of global andgene-specific DNA methylation patterns on the outcome ofpatients with acute myeloid leukemia (AML). Methylation ofCDKN2B (p15), E-cadherin (CDH) and hypermethylated incancer 1 (HIC1) promoters and global DNA methylation byluminometric methylation assay (LUMA) was analyzed in 107AML patients and cytogenetic and molecular mutationalanalysis was performed. In addition, genome-wide promoter-associated methylation was assessed using the IlluminaHumanMethylation27 array in a proportion of the patients.Promoter methylation was discovered in 66, 66 and 51% of thepatients for p15, CDH and HIC1, respectively. In multivariateanalysis, low global DNA methylation was associated withhigher complete remission rate (hazard ratio (HR) 5.9, P¼ 0.005)and p15 methylation was associated with better overall (HR 0.4,P¼0.001) and disease-free survival (HR 0.4, P¼ 0.016). CDH andHIC1 methylation were not associated with clinical outcome.Mutational status and karyotype were not significantly asso-ciated with gene-specific methylation or global methylation.Increased genome-wide promoter-associated methylation wasassociated with better overall and disease-free survival as wellas with LUMA hypomethylation. We conclude that global andgene-specific methylation patterns are independently asso-ciated with the clinical outcome in AML patients.Leukemia (2010) 24, 932–941; doi:10.1038/leu.2010.41;published online 18 March 2010Keywords: acute myeloid leukemia; DNA methylation; outcome;epigenetics
Introduction
Acute myeloid leukemia (AML) is a hematological malignancywith frequent and nonrandom genetic changes. Chromosomaltranslocations and gene mutations distinguish subtypes of thedisease and have a role in proliferation and differentiation andalso affect prognosis in AML.1,2 Epigenetic modification of DNAis an important biological function regulating gene expression innormal and tumor cells. Decrease in the whole-genome methyl-ation level is widespread in neoplastic diseases.3,4 Methylationof promoter-specific CpG islands, and hence silencing of tumorsuppressor and cell cycle control genes, is considered to be animportant step in tumor development and has been shownpreviously in AML.5 In one large study estrogen receptormethylation has been shown to correlate with better overallsurvival (OS) in AML.6 Although methylation of other geneshas been shown to associate with prognosis, the results are
somewhat contradictory.7–9 In high-risk myelodysplastic syndrome(MDS) and in AML after MDS, methylation of CDKN2b (p15),E-cadherin (CDH) and hypermethylated in cancer 1 (HIC1) haverecently been shown to be associated with failure to achievecomplete remission (CR) after induction chemotherapy.10
We analyzed gene-specific promoter methylation of p15,CDH and HIC1 as well as global DNA methylation usingthe luminometric methylation assay (LUMA) technique in awell-characterized cohort of 107 AML patients. Furthermore,we examined genome-wide CpG island-associated promotermethylation of 27 578 promoter CpG sites, corresponding to14495 individual genes, in 20 AML patients using the IlluminaHumanMethylation27 BeadChip. We found that low globalLUMA methylation levels correlated with an increased CR rateand that methylation of p15 correlated with better disease-freesurvival (DFS) and OS in uni- and multivariate analyses. Therewas a correlation between less global LUMA methylation andincreased genome-wide promoter-associated methylation usingthe Illumina array. We found no significant associations betweenthe methylation pattern and the cytogenetic risk groups or themutation status of FLT3 (Fms-like tyrosine kinase 3), NPM1(nucleophosmin), CEBPA (CCAAT enhancer binding protein-a)and NRAS (neuroblastoma RAS viral oncogene homolog).
Materials and methods
Patients and samplingA total of 107 previously untreated AML patients treated at theKarolinska University Hospital at Huddinge were included inthe study. Patients with AML M3 or a previous history of MDSwere excluded. Standard induction chemotherapy includingcytarabine and an anthracycline was given aiming at CR.Patients who achieved CR were given standard consolidationtherapy according to the applicable national or study protocolincluding allogeneic stem cell transplantation (n¼ 23). Of allpatients reaching CR, 18 (24%) received allogeneic stem celltransplantation, 41 (54%) received full consolidation therapyand 16 (22%) received reduced chemotherapy consolidationbecause of treatment toxicities. For diagnosis and classificationof CR and progression, World Health Organization criteria wereapplied.11 DNA was extracted from the diagnostic bone marrowsamples (n¼ 79) or peripheral blood (n¼ 28) after mononuclearcell isolation (Lymphoprep; Axis-Shield PoC, Oslo, Norway).Informed consent was obtained before the specimens werecollected. The study was approved by the regional ethicscommittee. Chromosome banding was performed usingstandard laboratory techniques. Mutation analyses of FLT3(internal tandem duplications and tyrosine kinase domain
Received 19 October 2009; revised 10 December 2009; accepted 21January 2010; published online 18 March 2010
Correspondence: Dr S Deneberg, Hematology Center, M54, KarolinskaUniversity Hospital, Huddinge, 14186 Stockholm, Sweden.E-mail: [email protected] authors contributed equally to this work.
Leukemia (2010) 24, 932–941& 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00
www.nature.com/leu

mutations at codon D835 and I836), NPM1, CEBPA, and NRASwere performed as previously described.12 Patients weredivided into three age groups before multivariate analysis(o55, 55–75 and 475 years), and into three cytogenetic riskgroups. In all, 5 patients had a low-risk karyotype (t(8;21), inv16), 77 had intermediate-risk karyotype (normal karyotypeand aberrations not defined as high or low risk) and 25 had ahigh-risk karyotype (del 7, del 5q, inv 3, t(6;9), complex (X5aberrations)). The low- and intermediate-risk groups wereanalyzed together as there were too few samples in the low-risk group to allow for meaningful multivariate analysis.
Bisulfite treatment and PCRDNA was modified with sodium bisulfite as previouslydescribed.13 A total of 2.5mg of DNA from each sample wasdenatured in NaOH for 15 min at 37 1C, followed by incubationin sodium bisulfite at 55 1C for 16 h. Thereafter, DNA wasrecovered using the GeneClean II kit (Qbiogene, Montreal, QC,Canada), desulfonated in NaOH and precipitated in ethanol.PCR specific for bisulfite-reacted p15, CDH and HIC1 promoterswas carried out in a final volume of 25ml containing 50–100 ngof bisulfite-modified DNA, 1� TEMPase PCR Buffer (Bie &Berntsen AS, Herlev, Denmark), 0.2 mmol/l deoxyribonucleo-tide triphosphate (ABgene, Epsom, UK), 0.5mmol/l each offorward and reverse primers14 and 0.75 units of TEMPase HotStart DNA polymerase (Bie & Berntsen). PCR was performed in aPX2 Thermal cycler (Thermo Electron Corporation, Waltham,MA, USA). The polymerase was activated by incubation at 95 1Cfor 15 min followed by 40 cycles of 95 1C for 30 s, 55 1C (p15and HIC1) or 48 1C (CDH) for 30 s, 72 1C for 30 s and a finalextension at 72 1C for 5 min. PCR results were examined byelectrophoresis in a 2.5% agarose gel. Amplification of non-bisulfite-treated DNA yielded no products for either primer pair.The p15, CDH and HIC1 amplicons contained 13, 15 and 9CpG residues, respectively.
Methylation analysis by DGGEPCR products of bisulfite-reacted p15, CDH and HIC1 promoterregions were analyzed using denaturing gradient gel electro-phoresis (DGGE) according to previously published proto-cols.14,15 In brief, the PCR products were loaded on a 10–70%denaturant gradient gel together with a fully methylated control(In vitro methylated DNA) and an unmethylated control(peripheral blood lymphocytes). After electrophoresis, gels werestained in Tris/EDTA buffer containing ethidium bromide andphotographed under ultraviolet transillumination. Samples werescored as methylated when bands or smears were present on thegels in the area below the band corresponding to unmethylatedDNA as reported in previous publications.14,15
Bisulfite pyrosequencingPyrosequencing of the p15 promoter region was performedusing the PSQHS96 system (Biotage AB, Uppsala, Sweden)including PyroGold reagents as previously described.16
Commercially available, validated primers were purchasedfrom Biotage AB. Sequences are available at the PyroMarkAssay Database (http://techsupport.pyrosequencing.com). Hot-Star Taq polymerase and HotStar Taqs Master Mix Kit (QiagenLtd, Hilden, Germany) were used to amplify 1.5ml of bisulfate-treated DNA. Cycling conditions were: (95 1C for 15 min,45� (95 1C for 20 s, 53 1C for 20 s and 72 1C for 20 s), 72 1Cfor 10 min, 4 1C). Samples were considered methylated at meanlevels of 415% methylation of the pyrosequenced CpG sites.
Methylation-specific melting curve analysisMethylation-specific melting curve analysis was performed aspreviously described for p15, CDH and HIC1 promoters, usingthe same primer pairs and PCR conditions as for DGGE.17
Global DNA methylation assay by LUMAGlobal DNA methylation was quantified using LUMA aspreviously described.18,19 A total of 500 ng genomic DNA wascleaved with HpaIIþ EcoRI or MspIþ EcoRI in separate reac-tions. After the digestion step, the extent of cleavage wasquantified by pyrosequencing. HpaII and MspI are methylation-sensitive isoschizomers. DNA methylation was defined fromthe HpaII/MspI ratio: fully methylated DNA gives a ratio thatapproaches 0 whereas if methylation is completely absent theratio approaches 1. The assay was carried out in duplicate.
Illumina HumanMethylation27 Beadchip arrayA total of 20 samples were also evaluated for genome-widepromoter methylation using the Illumina Infinium HumanMethy-lation27 Bead array. This analysis was made at the Bioinfor-matics and Expression Analysis core facility at the KarolinskaInstitute. The EZ DNA methylation kit (Zymo Research, Orange,CA, USA) was used for bisulfite conversion of 500 ng of DNA,and the remaining assay steps were performed as previouslypublished using Illumina-supplied reagents and conditions.20,21
The Illumina Infinium II Bead Array uses allele-specific annealingto either methylation-specific probes or non-methylation probesto detect the methylation grade of 27 578 individual CpG sitesspread across the promoter regions of 14 495 genes.
Statistical analysisCategorical clinical parameters and gene-specific variables wereanalyzed in relation to CR by chi-square analyses or Fisher’sexact test when appropriate. LUMA global methylation valueswere dichotomized at median. The contribution of genepromoter methylation and global methylation to DFS and OSwere assessed using the Kaplan–Meier method and comparedwith the log-rank test. Binary logistic regression was used formultivariate analysis of CR. Cox regression analysis was usedfor multivariate analysis of DFS and OS. Correlations werecalculated with Pearson’s correlation for continuous variablesand Spearman’s r for ordinal data. Multiple-group comparisonswere made using one-way analysis of variance. Statisticalcalculations were performed using the SPSS 16.0 software (SPSSInc., Chicago, IL, USA) except for array analysis. The Beadstudio2.0 software (Illumina Inc, San Diego, CA, USA) was used tonormalize the results of the Illumina Infinium assay. Normal-ization was made by subtracting the average signal of the 700negative controls from the probe signals. Differential methyla-tion analysis and construction of a volcano plot was performedusing the R statistical software package (www.r-project.org/) byapplying a t-test error model. CpG sites with a detection P-valueof 40.0001 were removed from analysis (n¼ 244). All P-valuesare two sided.
Results
Patient characteristicsThe study included 107 AML patients: 66 males and 41 females.Median age was 65.4 years (23–85 years). Nine patients hadtherapy-related AML and 98 were de novo AML. Baseline
Prognostic implications of DNA methylation in AMLS Deneberg et al
933
Leukemia

characteristics including molecular and cytogenetic character-istics are shown in Table 1.
Results of DGGE and LUMA methylation analysisDGGE showed methylation of the p15 gene promoter in 69(66%) of the evaluable AML cases (Table 2). In all, 69 (66%) ofthe patients were methylated in the CDH promoter and 57(51%) in the HIC1 promoter. There was a correlation betweenmethylation of p15 and methylation of HIC1 (R¼ 0.33,P¼ 0.001) but not between p15 and CDH or CDH and HIC1(P¼ 0.38, P¼ 0.94). The global DNA methylation levels asassessed by LUMA had a slightly left-skewed normal distributionpattern among the cases (Supplementary Figure 1). Age or sex
had no statistically significant effect on global LUMA methyla-tion levels. There was no difference in gene-specific or globalLUMA methylation levels between samples from peripheralblood or bone marrow (data not shown). Groups with differentinduction treatment protocols did not differ significantlyregarding the primary outcome variables, LUMA (P¼ 0.5), p15(P¼ 0.5), HIC (P¼ 0.2) or CDH methylation (P¼ 0.6). Neitherdid consolidation regimens, including whether the patientsreceived allogeneic stem cell transplantation or reducedchemotherapy consolidation, differ in LUMA (P¼ 0.5), p15(P¼ 0.8), HIC (P¼ 0.9) or CDH methylation rates (P¼ 0.6).Individual results for all experimental analyses and simpleoutcome variables are shown in Supplementary Table 1 andrepresentative examples of DGGE and LUMA in Figure 1.
Validation of DGGE by bisulfite pyrosequencing andmethylation-specific melting curve analysisTo validate the DGGE results, bisulfite pyrosequencing of thep15 promoter was performed in 29 of the samples. Pyrosequen-cing was concordant with DGGE in 23 out of 29 samples (79%,P¼ 0.003). The discordant samples were four cases methylatedby DGGE but considered negative by pyrosequencing, and twocases unmethylated by DGGE but positive by pyrosequencing.The eight samples evaluated with methylation-specific meltingcurve analysis for p15, CDH and HIC1 results were 100%concordant with DGGE. Examples of pyrosequencing results areshown in Figure 1.
Comparison of LUMA and Illumina whole-genomemethylation assaysIn 20 of the AML samples, methylation status of 27 578 mainlypromoter associated CpG sites, corresponding to 14 495 genes,was analyzed using the Illumina HumanMethylation27 Bead-Chip whole-genome assay. The methylation profile in the wholecohort was bimodal, as shown in Figure 2a. To minimizeoverlap between the two distributions, a cutoff at b40.5 was setto consider a CpG site hypermethylated. Global methylation, asassessed by LUMA, showed an inverse correlation with thenumber of hypermethylated promoter CpG residues, as assessedby the Illumina array, Pearson’s correlation 0.627, P¼ 0.003(Figure 2b). A volcano plot shows the imbalance with morepromoter-associated methylation in samples hypomethylatedglobally according to LUMA (Figure 2c), verifying an inversecorrelation between global hypomethylation and promoter CpGisland hypermethylation.
Table 1 Patient characteristics
Patient characteristics Non-MDS AML
Age, median (range), years 65 (23–85)Blast percentage, median (range) 68 (20–98)WBC count, median (range), � 10(9)/l 22 (0–368)Female/male 66/41Cytogenetic risk group (low/intermediate/high) 5/77/25CR rate 80 (75%)
FAB typeM0 8 (7%)M1 30 (28%)M2 30 (28%)M4 21 (20%)M5 8 (7%)M6 2 (2%)Not defined 8 (7%)
Molecular analysis (number positive/analyzed samples)FLT3-ITDa 29/95FLT3-TKD mutationa 10/94NPM1 mutation 34/93FLT3-ITD neg and NPM1 mut 18/93CEBPA mutation 7/95NRAS mutation 4/86
Abbreviations: AML, acute myeloid leukemia; CR, complete remission;CEBPA, CCAAT enhancer binding protein-a; FAB, French–American–British; FLT3, Fms-like tyrosine kinase 3; ITD, internal tandemduplication; MDS, myelodysplastic syndrome; NPM1, nucleophosmin;NRAS, neuroblastoma RAS viral oncogene homolog; TKD, tyrosinekinase domain; WBC, white blood cell.aTwo cases had simultaneous FLT3-ITD and FLT3-TKD mutation.
Table 2 Multivariate analysis of prognostic factors
CR HR (95% CI) DFS HR (95% CI) OS HR (95% CI) P-value CR/DFS/OS
Low LUMA methylation 5.9 (1.7–20.5) NR NR 0.005/NS/NSp15 methylation NR 0.44 (0.23–0.86) 0.41 (0.24–0.52) NS/0.02/0.001Age o55 years 7.3 (1.7–30.8) 0.31 (0.13–0.77) 0.24 (0.12–0.49) 0.007/0.01/o0.001Age 55–75 years 3.8 (0.99–14.4) 0.54 (0.24–1.22) 0.44 (0.23–0.84) 0.05/0.1/0.01Age 475 years 1 (reference) 1 (reference) 1 (reference)FLT3-ITD neg/NPM1 mutated 5.3 (0.9–30.6) 0.23 (0.09–0.60) 0.16 (0.07–0.39) 0.06/0.003/o0.001
Abbreviations: CI, confidence interval; CR, complete remission; DFS, disease-free survival; FLT3, Fms-like tyrosine kinase 3; HR, hazard ratio; ITD,internal tandem duplication; LUMA, luminometric methylation assay; NPM1, nucleophosmin; NR, not retained in multivariate analysis; NS,nonsignificant; OS, overall survival. Prognostic parameters retained at the last step in logistic regression (CR) and Cox regression (DFS and OS)analyses. Parameters included at step 1 in the backward stepwise analysis were: age, cytogenetic risk group, p15, CDH and HIC1 methylation,global LUMA methylation, deletions in chromosome 5 and/or 7 and FLT3-ITD alone and NPM1 mutation in the absence of FLT3-ITD (only forpatients with normal karyotype).
Prognostic implications of DNA methylation in AMLS Deneberg et al
934
Leukemia

Treatment outcomeCR was achieved in 80 (74%) of the patients: 43 (40%) of thepatients achieved a CR after one course of chemotherapy, 25(23%) after two and 12 (11%) after three courses or more. Themedian OS was 14.5 months (95% CI 8.4–20.6) and of patientsachieving CR, the median DFS was 22.8 months (95%confidence interval (CI) 10.9–34.7). The high-risk cytogeneticrisk group had a lower CR rate (56 vs 81%, P¼ 0.01) and shorterOS (10.4 vs 20.1 months, P¼ 0.01). FLT3, NPM1, CEBPA orNRAS mutations did not influence CR rate or OS except for thecombination of NPM1 mutation and lack of FLT3-internaltandem duplication (n¼ 18), who had a median OS that was notreached during the study period compared with 13.5 months(95% CI 10.5–16.5) in the remaining patients (P¼ 0.01).
Low levels of global DNA methylation by LUMA areassociated with a better CR ratePatients with low global LUMA methylation had a higher CRrate than those with high global LUMA methylation (82 vs 64%,P¼ 0.05). Traditional risk factors (age, cytogenetic risk group,FLT3-internal tandem duplication, mutant NPM1 in the absenceof FLT3-internal tandem duplication and occurrence of adeletion of chromosome 5q or 7) were entered into a binarylogistic regression model together with the experimentalparameters p15, CDH and HIC1 methylation and global LUMAmethylation. Low global LUMA methylation remained anindependent prognostic factor for achieving CR (hazard ratio(HR) 5.9, 95% CI 1.7–20.5, P¼ 0.005) together with age o55
years (HR 7.3, 95% CI 1.7–30.8, P¼ 0.007; Table 2). We alsoperformed a subgroup analysis of different age groups showingthe relationship between LUMA and CR to be age dependent(Figures 3b and c). Among patients younger than median age(65 years), 26 out of 27 (96%) with low global LUMAmethylation levels achieved CR when compared with 17 outof 24 (61%) with high global LUMA methylation (P¼ 0.02).In patients older than 65 years of age, there was no difference(66 vs 61%, P¼ 0.7). The better CR rates among patients withlow global LUMA methylation did not translate into better DFSor OS in the whole cohort (P¼ 0.6 and 0.9, respectively,Figure 3d). However, for patients younger than 65 years, wefound that OS and DFS were significantly better (P¼ 0.005 and0.04, respectively) among patients with low global LUMAmethylation (Figure 3e).
Methylation of p15 is associated with better DFS and OSThere were no differences in CR rates between patients withmethylated and unmethylated p15 (55/69 vs 24/35, P¼ 0.2). TheDFS was significantly longer in patients with methylated p15,with a median of 30 months when compared with 12 months forcases with unmethylated p15 (P¼ 0.03; Figure 4a). The medianOS was 23 compared with 11 months (P¼ 0.01; Figure 4b). In aCox regression analysis using the same risk factors as in thelogistic regression analysis above, p15 remained an independentprognostic factor for DFS (HR 0.44, 95% CI 0.23–0.86, P¼ 0.02)and OS (HR 0.41, 95%CI 0.24–0.71, P¼ 0.001; Table 2).Analysis of p15 by bisulfite pyrosequencing in 29 samples
PBL SSSI 1 2 3
Hpa ll digest Msp l digest
Hpa ll (C+G) Msp l (C+G)
Arb
itrar
y lig
ht u
nits
Arb
itrar
y lig
ht u
nits
116114112110108106104102100
116114112110108106104102100
4%
150
125
100
75
50
25
0
G G GTT GTATTTTG GGTTAGAG GGTTTTGAGTTC/T C/T C/T C/T C/T C/T C/T C/T GGT
5Sequence to analyze:
10 15 20 25 30 35 40
GA
E S G G G G G G G G G G GT T T T T T T T T T T T T TTC C C C C C CCA A A A A A A A
150
100
50
0
200
5 10 15 20 25 30 35 40
E S G G G G G G G G G G GT T T T T T T T T T T T TTC C C C C C CCA A A A A A A A
4% 4%3% 3% 3% 3%78% 76% 77% 67% 78% 70% 76%
S A C + G C + GT S A C + G C + GT
Step # 1 2 3 4 Step # 1 2 3 4
EcoRI (A) EcoRI (A)=0.916 =2.136
Figure 1 Representative examples of (a) denaturing gradient gel electrophoresis. PBL is DNA from peripheral blood monocytes, the negativecontrol. SSSI is SSSI-treated DNA, the positive control. Patient 1 is considered positive, 2 is negative and 3 is positive for DNA methylation. (b)Luminometric methylation assay (LUMA). Pyrograms show luminescence for MspI and HpaII, respectively. After normalization to EcoI, this samplehas a high ratio of HpaII/MspI (0.43), representing global hypomethylation. (c) Bisulfite pyrosequencing of the p15 promoter region for a negative(left) and a positive sample (right). Percentages indicate the degree of methylation at that CpG residue.
Prognostic implications of DNA methylation in AMLS Deneberg et al
935
Leukemia

confirmed the association between p15 methylation andsurvival (data not shown). HIC1 methylation showed no statis-tically significant association with CR. OS was better in HIC1methylated cases in univariate analysis (median 23 vs 11months), but there were no statistically significant associationsbetween HIC1 methylation and OS or DFS in multivariateanalysis (data not shown). There were no differences in clinicaloutcomes between patients with methylated and unmethylatedCDH (data not shown).
Increased genome-wide promoter hypermethylation isassociated with better DFS and OSAs our results suggested that increased gene-specific methyla-tion of the p15 promoter is associated with better outcome, wealso analyzed whether genome-wide promoter hypermethyla-tion was associated with CR rate, DFS or survival. Cases weredichotomized at median into two groups by the number ofhypermethylated (b40.5) CpG sites according to the Illuminaarray. There were no significant differences in age, cytogenetic
or molecular risk factors between the groups. CR rates did notdiffer between the groups, but the DFS was significantly longerin patients with more hypermethylation, with median notreached compared with 4 months for cases with less hyper-methylation (P¼ 0.02; Figure 4c). The median OS was notreached compared with 6 months (P¼ 0.005; Figure 4d). Theseresults suggest that not only increased p15 promoter methyla-tion, but also genome-wide promoter-associated hypermethyla-tion in general is associated with improved survival.
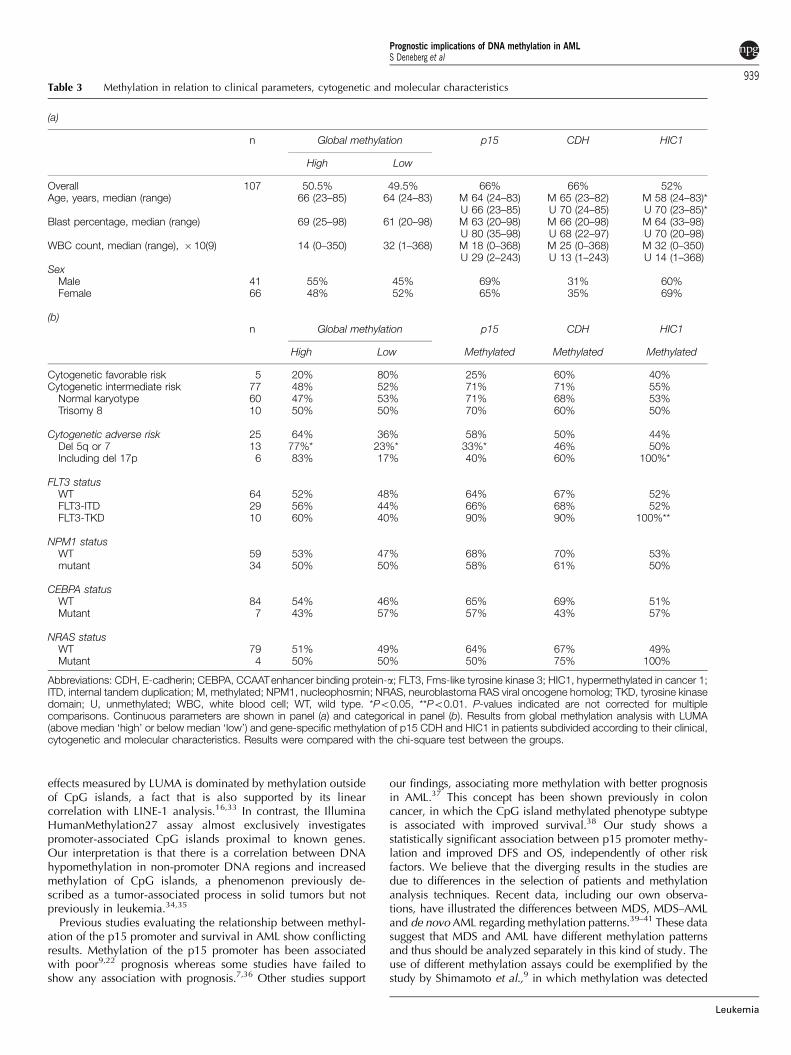
Associations between methylation status and geneticabnormalitiesWe also studied the relation between gene-specific methylationas well as global methylation by LUMA in relation to molecularand cytogenetic status of the AML cells. Table 3 shows theresults in patients subdivided according to cytogenetic andmolecular status. Although some associations could be shown,after Bonferroni correction, all associations were found to benonsignificant.
18001600
400200000800600400200
Average Beta
LUMA HPAII / MSPI
0.10 0.20 0.30 0.40 0.50 0.60 0.70 0.80 0.90p
-val
ue
(Lo
g 1
0)
4
3
2
1
0
-10 -5 0 5
Promoter CpG residue hypermethylation ratio (Log 2)
6000
7000
8000
9000
10000
11000
0.15 0.20 0.25 0.30 0.35 0.40 0.45
# C
pG
sit
es
# C
pG
sit
es a
vg B
eta
>0.5
Figure 2 Illumina HumanMethylation27 array results and comparison with global LUMA methylation. (a) The average number of CpG sitesmethylated at different b-values in the whole cohort shows a bimodal distribution. The b-value is a measure of the degree of methylation, andhigher means more methylation. (b) Correlation between the number of Illumina hypermethylated (b40.5) CpG sites and LUMA globalmethylation level in each patient analyzed by both. Pearson’s correlation 0.627, P¼0.003. Increasing LUMA values indicate less globalmethylation. (c) Volcano plot showing more Illumina global CpG site methylation in the group with low global LUMA methylation (dots positiveon the x axis) and less in the high global LUMA methylation group (dots negative on the x axis). The dotted lines indicate 0 and ±2 times differencebetween groups on a log2 scale, P-value of t-test statistics is indicated on a log10 scale on the y axis.
Prognostic implications of DNA methylation in AMLS Deneberg et al
936
Leukemia

Discussion
The role of DNA methylation in AML remains unclear. Studieson how methylation of gene promoters correlates with clinicaloutcome as well as with genetic and other clinical character-istics are few and data are somewhat conflicting.9,22 Exploration
of the epigenetic patterns in AML has the potential to define newbiological subgroups of AML or to provide new prognosticbiomarkers. In the light of the emerging therapeutical field ofepigenetically acting drugs, basic knowledge about methylationin leukemia is necessary to use these drugs efficiently and forrational study design. In this study we have analyzed the role of
Global DNAhypermethylation
Global DNAhypermethylation
Global DNAhypomethylation
918%
1835%
3465%
4282%
14%
729%
1771%
2696%
Global DNAhypomethylation
No CR
1.0
0.8
0.6
0.4
0.2
1.0
0.8
0.6
0.4
0.2
0.0
1.0
0.8
0.6
0.4
0.2
0.0
1.0
0.8
0.6
0.4
0 20 40 60 80 100 120
Disease free survival (months)
Disease free survival (months)
Overall survival (months)
Overall survival (months)
Global DNA hypomethylation
Global DNA hypomethylation
Global DNA hypermethylation
Global DNA hypomethylation n=27Global DNA hypermethylation n=24
Global DNA hypomethylation n=26Global DNA hypermethylation n=17
Global DNA hypermethylation
Su
rviv
alS
urv
ival
0 20 40 60 80 100 120
0 20 40 60 80 100
0 20 40 60 80 100
CR
No CRCR
All samples
Age 65 or below
Global DNAhypermethylation
833%
1667%
1139%
1761%
Global DNAhypomethylation
No CRCR
Age above 65
Figure 3 CR rates are higher in patients whose samples show global hypomethylation. The pie charts show CR rates in samples with globalmethylation levels higher than median (‘global DNA hypermethylation’) and global methylation levels lower than median (‘global DNAhypomethylation’). (a) The whole AML cohort (P¼ 0.05), (b) the same data for AML patients younger than 65 years (P¼ 0.02) and (c) for AMLpatients older than 65 years (P¼0.7). (d) Overall survival (P¼ 0.9) and disease-free survival (P¼ 0.6) in the whole AML cohort according to high orlow global DNA methylation by LUMA. (e) In patients younger than 65 years, OS (P¼ 0.005) and DFS (P¼0.04) were significantly better insamples with low global LUMA methylation levels. Patients were censored at the time of allogeneic stem cell transplantation.
Prognostic implications of DNA methylation in AMLS Deneberg et al
937
Leukemia

p15, HIC1 and CDH methylation, as well as that of global DNAmethylation, in a large cohort of genetically and clinically well-defined AML patients undergoing induction chemotherapy. Theanalysis was performed to study the effect of methylation onclinical outcome and response to therapy as well as its relationto genetic subtypes of AML. We also performed an analysis ofgenome-wide promoter-associated methylation using IlluminaHumanMethylation27 BeadChip array technology in a subset of20 patients.
Important novel observations in this study are the associationsbetween global DNA methylation as well as p15 promotermethylation and clinical outcome. Other novel findings werethe prognostic significance of the number of hypermethylatedCpG sites as measured by Illumina Infinium for DFS and OS aswell as the inverse association between global DNA methylationand genome-wide promoter methylation.
Our data suggest the existence of a subset of AML cases withgood prognosis, characterized by low global DNA methylationlevels paired with increased p15 methylation as well as genome-wide promoter-associated methylation levels, mainly discern-able in younger patients.
Global DNA hypomethylation is a common phenomenonin cancer4,23 and an indicator of good prognosis in acutelymphoblastic leukemia,24 whereas it is a poor prognosticmarker in breast cancer.25 In AML, a decrease in global DNAmethylation at relapse compared with diagnosis was observed inone study; however, these patients were selected on the groundsof having relapsed, which makes comparisons with our studydifficult.26 Mechanistically, global DNA hypomethylation mayhave a role in cancerogenesis through increased genomicinstability, activation of oncogenes or retrotransposons.27 Thereason for the increased CR rates in patients with global
hypomethylation observed in our study is unclear. There weresomewhat fewer patients with adverse and more with favorablekaryotype in the hypomethylated group, but these differenceswere not significant and were not retained in multivariateanalysis. Recent data show increased global LUMA methylationin cell lines resistant to daunorubicin in comparison to theirsensitive counterparts (own unpublished data), suggestive of apossible association with drug resistance. Further studies on thismatter are warranted, especially to determine whether demethyl-ating agents could sensitize resistant cells to conventionalcytostatic drugs in AML, which has already been shown in vitroin other cancers.28,29 Furthermore, our analysis showed thatglobal LUMA methylation data were prognostically importantmainly in patients younger than 65 years. One explanation forthis could be that intra-individual methylation variation accel-erates with increasing age, which could affect both global andgene-specific methylation and hence make cross-sectionalanalyses such as ours less predictive in an older population.30,31
Differences in correlations with CR versus survival for prog-nostic factors are not uncommon in AML. Although obviouslycorrelated, CR and survival represent somewhat different effectson the disease, which may explain the differences in outcomeon these variables. We also found an inverse relationshipbetween global methylation levels as measured by LUMAand CpG island-related genome-wide methylation levels asmeasured by Illumina HumanMethylation 27. LUMA analysis isbased on the HpaII/MspI recognition sequence, 50-CCGG-30,which are fairly well interspersed through the genome. Approxi-mately 50% of the CCGG sites are located in repetitiveDNA sequences and 50% in unique sequence,32 and eventhough they are accumulated at CpG islands, CpG islands onlycomprise about 1% of the genome.31 Therefore, methylation
1.0
0.8
0.6
0.4
0.2
0 20 40 60 80 600 20 40 80
Disease free survival (months)
Disease free survival (months)
Su
rviv
al
1.0
0.8
0.6
0.4
0.2
0.0
Su
rviv
al
1.0
0.8
0.6
0.4
0.2
0.0S
urv
ival
1.0
0.8
0.6
0.4
0.2
0.0
Su
rviv
al
0 50 100 150 200
Overall survival (months)
Overall survival (months)
0 50 100 150 200
p=0.029 p=0.011
p=0.005p=0.015
More CpG island methylationthan median n=9
More CpG island methylation thanmedian n=10
Less CpG island methylationthan median n=6
Less CpG island methylation thanmedian n=10
P15 P15Methylated n=55 Methylated n=69Unmethylated n=23 Unmethylated n=35
Figure 4 Prognostic role of gene-specific and global promoter methylation in AML patients. Kaplan–Meier diagrams showing the prognostic effectof p15 promoter methylation by DGGE on (a) disease-free survival (n¼104) and (b) overall survival (n¼ 78) as well as the number of CpG siteshypermethylated according to the Illumina Methylation array for (c) disease-free survival (n¼ 15) and (d) overall survival (n¼ 20).
Prognostic implications of DNA methylation in AMLS Deneberg et al
938
Leukemia
effects measured by LUMA is dominated by methylation outsideof CpG islands, a fact that is also supported by its linearcorrelation with LINE-1 analysis.16,33 In contrast, the IlluminaHumanMethylation27 assay almost exclusively investigatespromoter-associated CpG islands proximal to known genes.Our interpretation is that there is a correlation between DNAhypomethylation in non-promoter DNA regions and increasedmethylation of CpG islands, a phenomenon previously de-scribed as a tumor-associated process in solid tumors but notpreviously in leukemia.34,35
Previous studies evaluating the relationship between methyl-ation of the p15 promoter and survival in AML show conflictingresults. Methylation of the p15 promoter has been associatedwith poor9,22 prognosis whereas some studies have failed toshow any association with prognosis.7,36 Other studies support
our findings, associating more methylation with better prognosisin AML.37 This concept has been shown previously in coloncancer, in which the CpG island methylated phenotype subtypeis associated with improved survival.38 Our study shows astatistically significant association between p15 promoter methy-lation and improved DFS and OS, independently of other riskfactors. We believe that the diverging results in the studies aredue to differences in the selection of patients and methylationanalysis techniques. Recent data, including our own observa-tions, have illustrated the differences between MDS, MDS–AMLand de novo AML regarding methylation patterns.39–41 These datasuggest that MDS and AML have different methylation patternsand thus should be analyzed separately in this kind of study. Theuse of different methylation assays could be exemplified by thestudy by Shimamoto et al.,9 in which methylation was detected
Table 3 Methylation in relation to clinical parameters, cytogenetic and molecular characteristics
(a)
n Global methylation p15 CDH HIC1
High Low
Overall 107 50.5% 49.5% 66% 66% 52%Age, years, median (range) 66 (23–85) 64 (24–83) M 64 (24–83) M 65 (23–82) M 58 (24–83)*
U 66 (23–85) U 70 (24–85) U 70 (23–85)*Blast percentage, median (range) 69 (25–98) 61 (20–98) M 63 (20–98) M 66 (20–98) M 64 (33–98)
U 80 (35–98) U 68 (22–97) U 70 (20–98)WBC count, median (range), �10(9) 14 (0–350) 32 (1–368) M 18 (0–368) M 25 (0–368) M 32 (0–350)
U 29 (2–243) U 13 (1–243) U 14 (1–368)Sex
Male 41 55% 45% 69% 31% 60%Female 66 48% 52% 65% 35% 69%
(b)n Global methylation p15 CDH HIC1
High Low Methylated Methylated Methylated
Cytogenetic favorable risk 5 20% 80% 25% 60% 40%Cytogenetic intermediate risk 77 48% 52% 71% 71% 55%
Normal karyotype 60 47% 53% 71% 68% 53%Trisomy 8 10 50% 50% 70% 60% 50%
Cytogenetic adverse risk 25 64% 36% 58% 50% 44%Del 5q or 7 13 77%* 23%* 33%* 46% 50%Including del 17p 6 83% 17% 40% 60% 100%*
FLT3 statusWT 64 52% 48% 64% 67% 52%FLT3-ITD 29 56% 44% 66% 68% 52%FLT3-TKD 10 60% 40% 90% 90% 100%**
NPM1 statusWT 59 53% 47% 68% 70% 53%mutant 34 50% 50% 58% 61% 50%
CEBPA statusWT 84 54% 46% 65% 69% 51%Mutant 7 43% 57% 57% 43% 57%
NRAS statusWT 79 51% 49% 64% 67% 49%Mutant 4 50% 50% 50% 75% 100%
Abbreviations: CDH, E-cadherin; CEBPA, CCAATenhancer binding protein-a; FLT3, Fms-like tyrosine kinase 3; HIC1, hypermethylated in cancer 1;ITD, internal tandem duplication; M, methylated; NPM1, nucleophosmin; NRAS, neuroblastoma RAS viral oncogene homolog; TKD, tyrosine kinasedomain; U, unmethylated; WBC, white blood cell; WT, wild type. *Po0.05, **Po0.01. P-values indicated are not corrected for multiplecomparisons. Continuous parameters are shown in panel (a) and categorical in panel (b). Results from global methylation analysis with LUMA(above median ‘high’ or below median ‘low’) and gene-specific methylation of p15 CDH and HIC1 in patients subdivided according to their clinical,cytogenetic and molecular characteristics. Results were compared with the chi-square test between the groups.
Prognostic implications of DNA methylation in AMLS Deneberg et al
939
Leukemia

by methylation-specific PCR. DGGE is less sensitive thanmethylation-specific PCR but most likely better reflects thegeneral methylation levels in the highly variable promoter region.In addition, methylation-specific PCR may pick up very lowlevels of methylation that are not physiologically relevant.
Our AML cohort was defined with respect to the cytogeneticprofile as well as to the mutational status of FLT3, NPM1,CEBPA and NRAS. There were surprisingly few correlationsbetween the genetic characteristics of the patients and themethylation pattern, as analyzed by gene-specific methylationand global methylation by LUMA, suggesting that DNAmethylation is a phenomenon that occurs independently ofgenetic subgroup.
In conclusion, in this study, we evaluated a genetically andclinically well-defined, large AML cohort with respect to gene-specific and global methylation. Our results show that lowglobal DNA methylation levels correlate with an improved CRrate and that methylation of p15 correlates with better DFS andOS independently of other molecular and cytogenetic prog-nostic factors. We also show an inverse correlation betweenglobal DNA methylation and genome-wide promoter methyla-tion and an association between the number of hypermethylatedCpG sites measured genome-wide by the Illumina Human-Methylation27 array and prognosis. We suggest that methylationpatterns may be used as new tools to predict outcome in AMLpatients and that there are potentially new subgroups of AMLthat could be defined by methylation patterns. Still, furtherstudies on the prognostic and biological role of DNA methyla-tion in AML are warranted.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This study was supported by the Swedish Cancer Foundation,Swedish Research Council and Stockholm County Council.MK was supported by a KI post doc grant.
A special thanks to Professor Per Guldberg in Copenhagen forhis hospitality and help regarding methylation assay methodology.
AuthorshipStefan Deneberg collected clinical data, analyzed data andwrote the article. Michael Grovdal performed the DGGEanalyses and co-wrote the article. Mohsen Karimi performedthe LUMA, MS-MCA and pyrosequencing. Monika Janssonperformed DGGE analyses. Hareth Nahi helped collect clinicalinformation. Andrea Corbacioglu, Verena Gaidzik and Kon-stanze Dohner performed the molecular analyses. Christer Paulco-wrote the article and supervised sample collection andstorage. Tomas J Ekstrom performed the LUMA analysis andco-wrote the article. Eva Hellstrom-Lindberg helped design thestudy, assisted in data analysis and co-wrote the article. SorenLehmann designed the study and co-wrote the article.
References
1 Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloidmalignancies: pathogenetic and clinical implications. J Clin Oncol2005; 23: 6285–6295.
2 Rowley JD. The critical role of chromosome translocations inhuman leukemias. Annu Rev Genet 1998; 32: 495–519.
3 Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitororigin of human cancer. Nat Rev Genet 2006; 7: 21–33.
4 Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes ofsome human cancers from their normal counterparts. Nature 1983;301: 89–92.
5 Melki JR, Vincent PC, Clark SJ. Concurrent DNA hypermethylationof multiple genes in acute myeloid leukemia. Cancer Res 1999; 59:3730–3740.
6 Li Q, Kopecky KJ, Mohan A, Willman CL, Appelbaum FR, WeickJK et al. Estrogen receptor methylation is associated with improvedsurvival in adult acute myeloid leukemia. Clin Cancer Res 1999; 5:1077–1084.
7 Chim CS, Tam CY, Liang R, Kwong YL. Methylation of p15 andp16 genes in adult acute leukemia: lack of prognostic significance.Cancer 2001; 91: 2222–2229.
8 Galm O, Wilop S, Luders C, Jost E, Gehbauer G, Herman JG et al.Clinical implications of aberrant DNA methylation patterns inacute myelogenous leukemia. Ann Hematol 2005; 84 (Suppl 1):39–46.
9 Shimamoto T, Ohyashiki JH, Ohyashiki K. Methylation ofp15(INK4b) and E-cadherin genes is independently correlatedwith poor prognosis in acute myeloid leukemia. Leuk Res 2005;29: 653–659.
10 Grovdal M, Khan R, Aggerholm A, Antunovic P, Astermark J,Bernell P et al. Negative effect of DNA hypermethylation on theoutcome of intensive chemotherapy in older patients with high-riskmyelodysplastic syndromes and acute myeloid leukemia followingmyelodysplastic syndrome. Clin Cancer Res 2007; 13: 7107–7112.
11 Vardiman JW, Harris NL, Brunning RD. The World HealthOrganization (WHO) classification of the myeloid neoplasms.Blood 2002; 100: 2292–2302.
12 Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A,Bullinger L et al. Mutations and treatment outcome in cytogeneti-cally normal acute myeloid leukemia. N Engl J Med 2008; 358:1909–1918.
13 Zeschnigk M, Lich C, Buiting K, Doerfler W, Horsthemke B. Asingle-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at theSNRPN locus. Eur J Hum Genet 1997; 5: 94–98.
14 Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P.Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER isfrequent in myelodysplastic syndrome and predicts poor prognosisin early-stage patients. Eur J Haematol 2006; 76: 23–32.
15 Cremonesi L, Firpo S, Ferrari M, Righetti PG, Gelfi C. Double-gradient DGGE for optimized detection of DNA point mutations.Biotechniques 1997; 22: 326–330.
16 Geli J, Kiss N, Karimi M, Lee JJ, Backdahl M, Ekstrom TJ et al.Global and regional CpG methylation in pheochromocytomas andabdominal paragangliomas: association to malignant behavior.Clin Cancer Res 2008; 14: 2551–2559.
17 Guldberg P, Worm J, Gronbaek K. Profiling DNA methylation bymelting analysis. Methods 2002; 27: 121–127.
18 Karimi M, Johansson S, Stach D, Corcoran M, Grander D,Schalling M et al. LUMA (LUminometric Methylation Assay)–ahigh throughput method to the analysis of genomic DNAmethylation. Exp Cell Res 2006; 312: 1989–1995.
19 Karimi M, Johansson S, Ekstrom TJ. Using LUMA: a Luminometric-based assay for global DNA-methylation. Epigenetics 2006; 1: 45–48.
20 Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KLet al. Highly parallel SNP genotyping. Cold Spring Harb SympQuant Biol 2003; 68: 69–78.
21 Fan JB, Gunderson KL, Bibikova M, Yeakley JM, Chen J, WickhamGarcia E et al. Illumina universal bead arrays. Methods Enzymol2006; 410: 57–73.
22 Wong IH, Ng MH, Huang DP, Lee JC. Aberrant p15 promotermethylation in adult and childhood acute leukemias of nearly allmorphologic subtypes: potential prognostic implications. Blood2000; 95: 1942–1949.
23 Issa JP. CpG island methylator phenotype in cancer. Nat RevCancer 2004; 4: 988–993.
24 Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA,Navarro G, Garate L et al. Promoter hypermethylation and globalhypomethylation are independent epigenetic events in lymphoidleukemogenesis with opposing effects on clinical outcome.Leukemia 2006; 20: 1445–1448.
Prognostic implications of DNA methylation in AMLS Deneberg et al
940
Leukemia

25 Soares J, Pinto AE, Cunha CV, Andre S, Barao I, Sousa JM et al.Global DNA hypomethylation in breast carcinoma: correlationwith prognostic factors and tumor progression. Cancer 1999; 85:112–118.
26 Kroeger H, Jelinek J, Estecio MR, He R, Kondo K, Chung W et al.Aberrant CpG island methylation in acute myeloid leukemia isaccentuated at relapse. Blood 2008; 112: 1366–1373.
27 Costello JF, Plass C. Methylation matters. J Med Genet 2001; 38:285–303.
28 Shang D, Liu Y, Matsui Y, Ito N, Nishiyama H, Kamoto T et al.Demethylating agent 5-aza-20-deoxycytidine enhances suscept-ibility of bladder transitional cell carcinoma to Cisplatin. Urology2008; 71: 1220–1225.
29 Festuccia C, Gravina GL, D’Alessandro AM, Muzi P, Millimaggi D,Dolo V et al. Azacitidine improves antitumor effects of docetaxeland cisplatin in aggressive prostate cancer models. Endocr RelatCancer 2009; 16: 401–413.
30 Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar MLet al. Epigenetic differences arise during the lifetime of mono-zygotic twins. Proc Natl Acad Sci USA 2005; 102: 10604–10609.
31 Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T,Cui H et al. Intra-individual change over time in DNA methylationwith familial clustering. JAMA 2008; 299: 2877–2883.
32 Fazzari MJ, Greally JM. Epigenomics: beyond CpG islands. NatRev Genet 2004; 5: 446–455.
33 Romermann D, Hasemeier B, Metzig K, Gohring G, Schlegelberger B,Langer F et al. Global increase in DNA methylation in patients withmyelodysplastic syndrome. Leukemia 2008; 22: 1954–1956.
34 Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358: 1148–1159.35 Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev
Cancer 2004; 4: 143–153.36 Olesen LH, Aggerholm A, Andersen BL, Nyvold CG, Guldberg P,
Norgaard JM et al. Molecular typing of adult acute myeloidleukaemia: significance of translocations, tandem duplications,methylation, and selective gene expression profiling. Br JHaematol 2005; 131: 457–467.
37 Kroeger H, Jelinek J, Komblau SM, Bueso-Ramos CE, Issa JP.Increased DNA methylation is associated with good prognosisin AML. 49th Annual Meeting of the American-Society-of-Hematology, 2007, Atlanta, GA, 2007, p 595.
38 Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda Met al. CpG island methylator phenotype, microsatellite instability,BRAF mutation and clinical outcome in colon cancer. Gut 2009;58: 90–96.
39 Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E et al.MDS and secondary AML display unique patterns and abundanceof aberrant DNA methylation. Blood 2009; 114: 3363–3364.
40 Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe Cet al. Aberrant DNA methylation is a dominant mechanism inMDS progression to AML. Blood 2009; 113: 1315–1325.
41 Deneberg S, Grovdall M, Jansson M, Gaidzik VI, Corbacioglu A,Nahi H et al. Different incidence and implications of DNAhypermethylation in de novo AML compared to high-risk MDSand AML following MDS. 50th Annual Meeting of the American-Society-of-Hematology, 2008, 06–09 December, San Francisco,CA, 2008, p 1146–1147.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
Prognostic implications of DNA methylation in AMLS Deneberg et al
941
Leukemia




















