
Gap junction endocytosis and lysosomal degradation of connexin43-P2 in WB-F344 rat liver epithelial...
8
Carcinogenesis vol.17 no.9 pp.1791-1798, 1996 ORIGINAL ARTICLES Gap junction endocytosis and lysosomal degradation of connexin43-P2 in WB-F344 rat liver epithelial cells treated with DDT and lindane Xiaojun Guan and Randall J.Ruch 1 Department of Pathology, Health Education Building, Room 202, Medical College of Ohio, 3000 Arlington Avenue, Toledo, OH 43699, USA 'To whom correspondence should be addressed Treatment of WB-F344 rat liver epithelial cells with DDT (l,l-bis(p-chlorophenyl)-2,2,2-trichloroethane) or lindane induces a loss of gap junction plaques and a decrease in the phosphorylated gap junction protein connexin43-P2 (Cx43-P2), which is associated with the plaques. In this study we have considered several mechanisms. The loss of junctional plaques could be due to disaggregation of junctional particles or to endocytosis of the plaques, while the loss of Cx43-P2 could be due to dephosphorylation or degradation. lmmunohistochemical analyses of DDT- or lindane-treated cells revealed a reduction in plasma mem- branous Cx43-positive gap junction plaques coincident with the appearance of Cx43-positive punctate cytoplasmic structures. The cytoplasmic Cx43-positive structures even- tually disappeared after 4 h treatment Diffuse Cx43- positive plasma membranous staining was not seen follow- ing DDT or lindane treatment Western blot analyses of these cells indicated that Cx43-P2 decreased in a time- dependent manner that paralleled the disappearance of gap junction plaques from the plasma membrane. The loss of Cx43-P2 was not due to dephosphorylation, since no increase in non-phosphorylated (Cx43-NP) or other phos- phorylated (Cx43-Pl) forms of the protein were evident The decrease in Cx43-P2 and the disappearance of cytoplasmic Cx43-positive structures were prevented by colchicine and chloroquine, which suggests that Cx43-P2- containing plaques were internalized and degraded in lysosomes. In addition, two small (-18 and -22 kDa) bands appeared in Western blots coincident with the loss of Cx43- P2 and may be degradation products of the protein. These immunohistochemical and biochemical data strongly suggest that the loss of gap junction plaques and of Cx43- P2 in WB-F344 cells treated with DDT and lindane were due to endocytosis of the plaques and degradation of Cx43- P2 in lysosomes. Introduction Gap junctions are clusters of plasma membrane channels that connect the interiors of adjacent cells and permit the diffusion of small (< 1 kDa) ions and molecules between coupled cells (1). This intercellular exchange, or gap junctional intercellular communication (GJIC*), is involved in the regulation of •Abbreviations: GJIC, gap junctional intercellular communication; Cx43, connexin43; DDT, 1,1 -bis(/>-chlorophenyl)-2,2,2-trichloroetriane; LND, lindane; DMSO, dimethylsulfoxide; CHX, cycloheximide; TPA, 12-0-telrade- canoy Iphorbol-13-acetate. © Oxford University Press cellular growth and neoplastic transformation in addition to many other physiological roles (2-5). Cellular proliferative states (e.g. two-thirds partial hepatectomy) and agents that enhance cellular proliferation (e.g. certain growth factors and oncogenes) reduce GJIC (3-5). Most neoplastically trans- formed cells have reduced levels of GJIC compared with non- transformed cells (2). This latter defect is often the result of reduced expression of connexins (6), which are the protein- aceous subunits of gap junctional channels (6). Recent studies have shown that the enhancement of connexin expression and GJIC by transfection with connexin cDNA expression vectors restored a more normal phenotype to neoplastic cells. This was evidenced by reduced growth in vitro and/or decreased tumorigenicity (7-13). In contrast, inhibition of GJIC by stable transfection with a connexin antisense cDNA or by treatment of cells with connexin antisense oligonucleotides has resulted in abnormal growth regulation in vitro (14,15). Thus, direct and indirect evidence indicates that GJIC is involved in growth regulation and neoplastic transformation. Many agents have been reported to inhibit GJIC both in vivo and in vitro. These agents include numerous non-genotoxic rodent carcinogens, such as phthalate esters, pesticides, barbit- urates, peroxides, polychlorinated and polybrominated biphenyls and phorbol esters (reviewed in 16-18). Because of the involvement of GJIC in growth regulation and neoplastic transformation, inhibition of GJIC by these agents may be important in their ability to enhance neoplastic transformation. Since several of these compounds may play a role in human carcinogenesis (19,20), it is important to understand how they inhibit GJIC. This will facilitate extrapolations between human and animal results and may lead to strategies designed to prevent the inhibitory effect. To date, however, the mechanisms of action of these agents on gap junctions are poorly understood. We have been examining the mechanisms by which pesti- cides and other agents inhibit GJIC in WB-F344 rat liver epithelial cells (21-24). These cells are good models to study chemical effects on gap junctions because they have large gap junctions and are highly coupled. The junctions are comprised of connexin43 (Cx43), although a small amount of connexin26 can also be detected under certain culture conditions (21,22). In previous studies, we have reported that pesticides such as l,l-bisO-chlorophenyl)-2,2,2-trichloroethane (DDT), lindane (LND; y-hexachlorocyclohexane), heptachlor epoxide and dieldrin induced the loss of Cx43-containing gap junctions from these cells in a dose- and time-dependent manner (21- 24). These changes occurred 1-4 h after treatment and were detected immunohistochemically using an anti-Cx43 mono- clonal antibody. The loss of gap junctions was not due to the inhibition of Cx43 gene transcription, but was correlated with a decrease in Cx43-P2, the phosphorylated form of the protein that is thought to be the major component of large junctional plaques (25). 1791 by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 29, 2013 http://carcin.oxfordjournals.org/ Downloaded from
Transcript of Gap junction endocytosis and lysosomal degradation of connexin43-P2 in WB-F344 rat liver epithelial...

Carcinogenesis vol.17 no.9 pp.1791-1798, 1996
ORIGINAL ARTICLES
Gap junction endocytosis and lysosomal degradation ofconnexin43-P2 in WB-F344 rat liver epithelial cells treated withDDT and lindane
Xiaojun Guan and Randall J.Ruch1
Department of Pathology, Health Education Building, Room 202, MedicalCollege of Ohio, 3000 Arlington Avenue, Toledo, OH 43699, USA
'To whom correspondence should be addressed
Treatment of WB-F344 rat liver epithelial cells with DDT(l,l-bis(p-chlorophenyl)-2,2,2-trichloroethane) or lindaneinduces a loss of gap junction plaques and a decrease inthe phosphorylated gap junction protein connexin43-P2(Cx43-P2), which is associated with the plaques. In thisstudy we have considered several mechanisms. The lossof junctional plaques could be due to disaggregation ofjunctional particles or to endocytosis of the plaques, whilethe loss of Cx43-P2 could be due to dephosphorylation ordegradation. lmmunohistochemical analyses of DDT- orlindane-treated cells revealed a reduction in plasma mem-branous Cx43-positive gap junction plaques coincidentwith the appearance of Cx43-positive punctate cytoplasmicstructures. The cytoplasmic Cx43-positive structures even-tually disappeared after 4 h treatment Diffuse Cx43-positive plasma membranous staining was not seen follow-ing DDT or lindane treatment Western blot analyses ofthese cells indicated that Cx43-P2 decreased in a time-dependent manner that paralleled the disappearance ofgap junction plaques from the plasma membrane. The lossof Cx43-P2 was not due to dephosphorylation, since noincrease in non-phosphorylated (Cx43-NP) or other phos-phorylated (Cx43-Pl) forms of the protein were evidentThe decrease in Cx43-P2 and the disappearance ofcytoplasmic Cx43-positive structures were prevented bycolchicine and chloroquine, which suggests that Cx43-P2-containing plaques were internalized and degraded inlysosomes. In addition, two small (-18 and -22 kDa) bandsappeared in Western blots coincident with the loss of Cx43-P2 and may be degradation products of the protein.These immunohistochemical and biochemical data stronglysuggest that the loss of gap junction plaques and of Cx43-P2 in WB-F344 cells treated with DDT and lindane weredue to endocytosis of the plaques and degradation of Cx43-P2 in lysosomes.
Introduction
Gap junctions are clusters of plasma membrane channels thatconnect the interiors of adjacent cells and permit the diffusionof small (< 1 kDa) ions and molecules between coupled cells(1). This intercellular exchange, or gap junctional intercellularcommunication (GJIC*), is involved in the regulation of
•Abbreviations: GJIC, gap junctional intercellular communication; Cx43,connexin43; DDT, 1,1 -bis(/>-chlorophenyl)-2,2,2-trichloroetriane; LND,lindane; DMSO, dimethylsulfoxide; CHX, cycloheximide; TPA, 12-0-telrade-canoy Iphorbol-13-acetate.
© Oxford University Press
cellular growth and neoplastic transformation in addition tomany other physiological roles (2-5). Cellular proliferativestates (e.g. two-thirds partial hepatectomy) and agents thatenhance cellular proliferation (e.g. certain growth factors andoncogenes) reduce GJIC (3-5). Most neoplastically trans-formed cells have reduced levels of GJIC compared with non-transformed cells (2). This latter defect is often the result ofreduced expression of connexins (6), which are the protein-aceous subunits of gap junctional channels (6). Recent studieshave shown that the enhancement of connexin expression andGJIC by transfection with connexin cDNA expression vectorsrestored a more normal phenotype to neoplastic cells. Thiswas evidenced by reduced growth in vitro and/or decreasedtumorigenicity (7-13). In contrast, inhibition of GJIC by stabletransfection with a connexin antisense cDNA or by treatmentof cells with connexin antisense oligonucleotides has resultedin abnormal growth regulation in vitro (14,15). Thus, directand indirect evidence indicates that GJIC is involved in growthregulation and neoplastic transformation.
Many agents have been reported to inhibit GJIC both in vivoand in vitro. These agents include numerous non-genotoxicrodent carcinogens, such as phthalate esters, pesticides, barbit-urates, peroxides, polychlorinated and polybrominatedbiphenyls and phorbol esters (reviewed in 16-18). Because ofthe involvement of GJIC in growth regulation and neoplastictransformation, inhibition of GJIC by these agents may beimportant in their ability to enhance neoplastic transformation.Since several of these compounds may play a role in humancarcinogenesis (19,20), it is important to understand how theyinhibit GJIC. This will facilitate extrapolations between humanand animal results and may lead to strategies designed toprevent the inhibitory effect. To date, however, the mechanismsof action of these agents on gap junctions are poorly understood.
We have been examining the mechanisms by which pesti-cides and other agents inhibit GJIC in WB-F344 rat liverepithelial cells (21-24). These cells are good models to studychemical effects on gap junctions because they have large gapjunctions and are highly coupled. The junctions are comprisedof connexin43 (Cx43), although a small amount of connexin26can also be detected under certain culture conditions (21,22).In previous studies, we have reported that pesticides such asl,l-bisO-chlorophenyl)-2,2,2-trichloroethane (DDT), lindane(LND; y-hexachlorocyclohexane), heptachlor epoxide anddieldrin induced the loss of Cx43-containing gap junctionsfrom these cells in a dose- and time-dependent manner (21-24). These changes occurred 1-4 h after treatment and weredetected immunohistochemically using an anti-Cx43 mono-clonal antibody. The loss of gap junctions was not due to theinhibition of Cx43 gene transcription, but was correlated witha decrease in Cx43-P2, the phosphorylated form of the proteinthat is thought to be the major component of large junctionalplaques (25).
1791
by guest on October 29, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 29, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 29, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 29, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 29, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 29, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 29, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 29, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
X.Guan and KJ.Ruch
The loss of gap junctions in these cells could be due tothe disaggregation of particles in junctional plaques or toendocytosis of the plaques, whereas the decrease in Cx43-P2could result from Cx43-P2 dephosphorylation or degradation.In the present study, we have further examined how DDT andLND induce the loss of gap junctions and Cx43-P2 in WB-F344 cells with these possibilities in mind. Our data indicatethat these agents induce gap junction endocytosis and Cx43-P2 degradation in lysosomes.
Materials and methodsReagents
All reagents and tissue culture medium components were purchased fromSigma Chemical Co. (St Louis, MO) unless noted otherwise.
Cell culture
WB-F344 rat liver epithelial cells were cultured in Richter's improved minimalessential medium (Irvine Scientific, Santa Ana, CA) supplemented with 5%fetal bovine serum and 50 |ig/ml gentamicin sulfate. The cells were maintainedin standard tissue culture dishes (Falcon/Becton-Dickinson) and passaged bytrypsinization as described (21).
Treatment of cells with test agents
WB-F344 cells were cultured in 35 mm dishes for the dye coupling assaysand immunohistochemistry or in 100 mm dishes for the biochemical assays.At -90% confluence, the cells were treated with the test agents. DDT andLND were first dissolved in dimethylsulfoxide (DMSO) and then mixed intothe culture medium (1 |il test agent/ml medium). Control cultures were treatedwith DMSO (1 nl/ml medium). Cycloheximide (CHX), colchicine andchloroquine were first dissolved in sterile deionized water and then appliedto the cells (1 (il/ml medium).
Dye coupling assay of GJIC
Fluorescent dye coupling levels in WB-F344 cells were determined bymicroinjection of Lucifer Yellow CH dye exactly as described (21).
Immunohistochemical staining of gap junctions
Cx43 was detected in WB-F344 cells by indirect immunofluorescent stainingusing a mouse anti-Cx43 monoclonal primary antibody (Zymed Corp., SouthSan Francisco, CA) and a fluorescein isothiocyanate-conjugated rabbit anti-mouse IgG secondary antibody (Jackson Immunoresearch, West Grove, PA)as described (21). The cells were viewed and photographed under epifluorescentillumination through a 100X oil immersion objective lens mounted on aNikon Diaphot microscope.
Western blot analyses of Cx43
Western blot analyses of Cx43 in WB-F344 cells were performed usingplasma membrane-enriched fractions and whole cell extracts. Membrane-enriched fractions were prepared using alkaline hypotonic buffer exactly asdescribed (21). Whole cell preparations were generated by harvesting the cellsby scraping in phosphate-buffered saline, collecting the cells by centrifugation(1500 r.p.m), sonicating the cell pellet in 0.5 ml alkaline hypotonic bufferand adding 55 |il 20% SDS to the sonicate (final SDS concentration 2%).
Membrane-enriched and whole cell samples (10 |ig protein/sample) werecombined with an equal volume of 2X Laemmli sample buffer and separatedby SDS-PAGE on 12% polyacrylamide gels under reducing conditions.Proteins were transferred to Immobilon-P membranes (Millipore Corp.) byelectrophoresis and Cx43 was detected using a mouse monoclonal anti-Cx43antibody (Zymed) and a Western blot detection kit (Amersham Corp., ArlingtonHeights, IL) as described (21)
Results
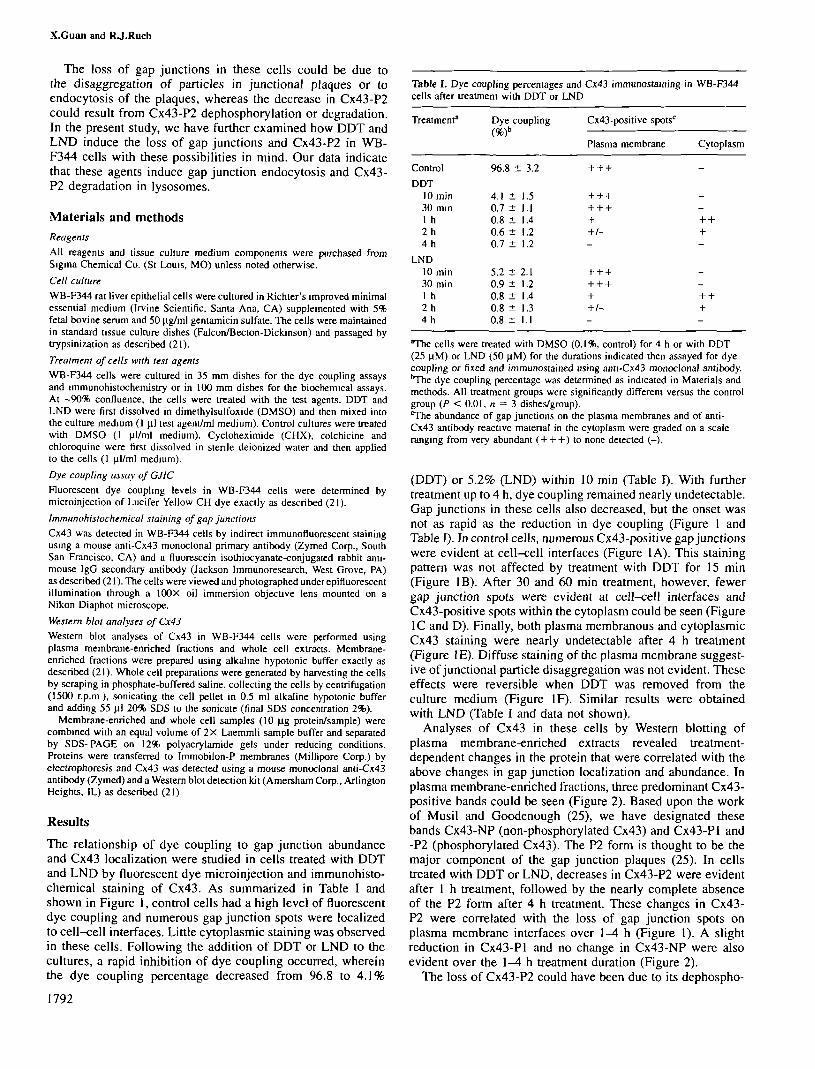
The relationship of dye coupling to gap junction abundanceand Cx43 localization were studied in cells treated with DDTand LND by fluorescent dye microinjection and immunohisto-chemical staining of Cx43. As summarized in Table I andshown in Figure 1, control cells had a high level of fluorescentdye coupling and numerous gap junction spots were localizedto cell-cell interfaces. Little cytoplasmic staining was observedin these cells. Following the addition of DDT or LND to thecultures, a rapid inhibition of dye coupling occurred, whereinthe dye coupling percentage decreased from 96.8 to 4.1%
1792
Table I. Dye coupling percentages and Cx43 immunostaming in WB-F344cells after treatment with DDT or LND
Treatment"
Control
DDT10 min30 min1 h2 h4 h
LND10 min30 min1 h2h4 h
Dye
(%)
96.8
4.10.70.80.60.7
5.20.90.80.80.8 :
coupling Cx43-positive spots0
Plasma membrane Cytoplasm
±3.2 + + +
t 1.5 + + +t 1.1 + + +t 1.4 + + +t 1.2 +/- +t 1.2
t 2.1 + + +t 1.2 + + +t 1.4 + + +t 1.3 +/- +t 1.1
The cells were treated with DMSO (0.1%, control) for 4 h or with DDT(25 |iM) or LND (50 \iM) for the durations indicated then assayed for dyecoupling or fixed and immunostained using anu-Cx43 monoclonal antibody.'The dye coupling percentage was determined as indicated in Materials andmethods. All treatment groups were significantly different versus the controlgroup (P < 0.01, n = 3 dishes/group).The abundance of gap junctions on the plasma membranes and of anti-Cx43 antibody reactive material in the cytoplasm were graded on a scaleranging from very abundant (+ + +) to none detected (-).
(DDT) or 5.2% (LND) within 10 min (Table I). With furthertreatment up to 4 h, dye coupling remained nearly undetectable.Gap junctions in these cells also decreased, but the onset wasnot as rapid as the reduction in dye coupling (Figure 1 andTable I). In control cells, numerous Cx43-positive gap junctionswere evident at cell-cell interfaces (Figure 1A). This stainingpattern was not affected by treatment with DDT for 15 min(Figure IB). After 30 and 60 min treatment, however, fewergap junction spots were evident at cell-cell interfaces andCx43-positive spots within the cytoplasm could be seen (Figure1C and D). Finally, both plasma membranous and cytoplasmicCx43 staining were nearly undetectable after 4 h treatment(Figure IE). Diffuse staining of the plasma membrane suggest-ive of junctional particle disaggregation was not evident. Theseeffects were reversible when DDT was removed from theculture medium (Figure IF). Similar results were obtainedwith LND (Table I and data not shown).
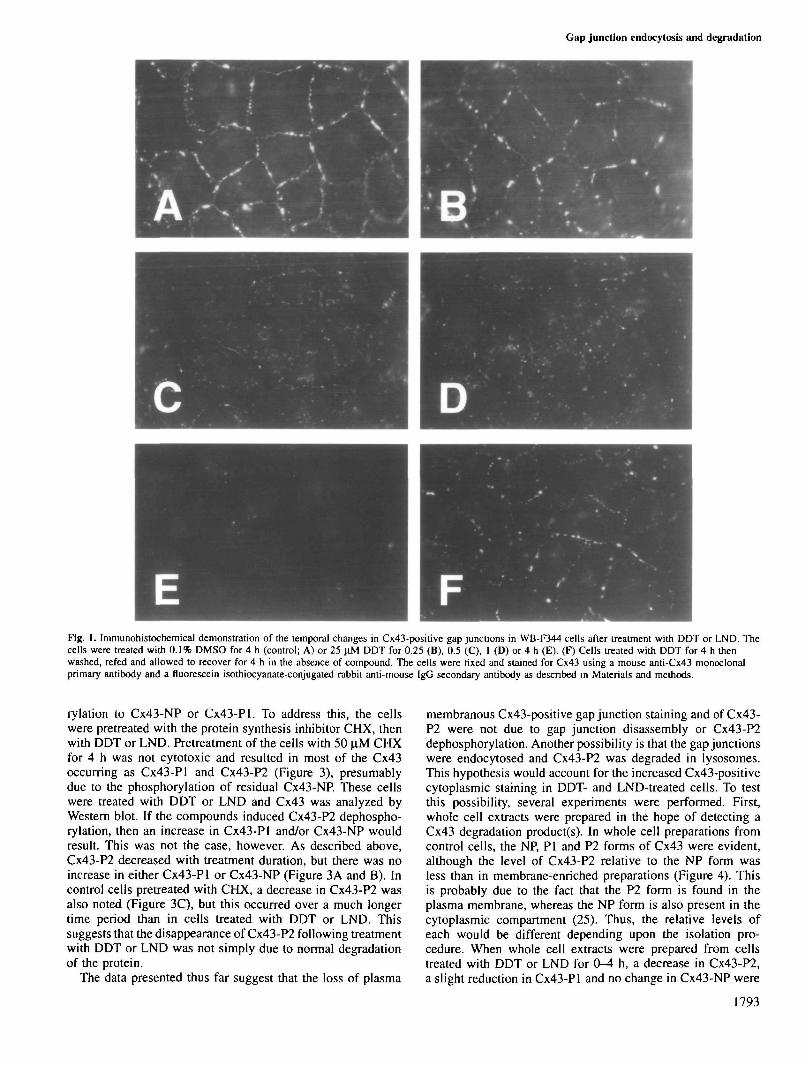
Analyses of Cx43 in these cells by Western blotting ofplasma membrane-enriched extracts revealed treatment-dependent changes in the protein that were correlated with theabove changes in gap junction localization and abundance. Inplasma membrane-enriched fractions, three predominant Cx43-positive bands could be seen (Figure 2). Based upon the workof Musil and Goodenough (25), we have designated thesebands Cx43-NP (non-phosphorylated Cx43) and Cx43-Pl and-P2 (phosphorylated Cx43). The P2 form is thought to be themajor component of the gap junction plaques (25). In cellstreated with DDT or LND, decreases in Cx43-P2 were evidentafter 1 h treatment, followed by the nearly complete absenceof the P2 form after 4 h treatment. These changes in Cx43-P2 were correlated with the loss of gap junction spots onplasma membrane interfaces over 1-4 h (Figure 1). A slightreduction in Cx43-Pl and no change in Cx43-NP were alsoevident over the 1-4 h treatment duration (Figure 2).
The loss of Cx43-P2 could have been due to its dephospho-
Gap junction endocytosis and degradation
Fig. 1. Immunohistochemical demonstration of the temporal changes in Cx43-positive gap junctions in WB-F344 cells after treatment with DDT or LND. Thecells were treated with 0.1% DMSO for 4 h (control; A) or 25 uM DDT for 0.25 (B), 0.5 (C), 1 (D) or 4 h (E). (F) Cells treated with DDT for 4 h thenwashed, refed and allowed to recover for 4 h in the absence of compound. The cells were fixed and stained for Cx43 using a mouse anti-Cx43 monoclonalprimary antibody and a fluorescein isothiocyanate-conjugated rabbit anti-mouse IgG secondary antibody as described in Materials and methods.
rylation to Cx43-NP or Cx43-Pl. To address this, the cellswere pretreated with the protein synthesis inhibitor CHX, thenwith DDT or LND. Pretreatment of the cells with 50 .̂M CHXfor 4 h was not cytotoxic and resulted in most of the Cx43occurring as Cx43-Pl and Cx43-P2 (Figure 3), presumablydue to the phosphorylation of residual Cx43-NP. These cellswere treated with DDT or LND and Cx43 was analyzed byWestern blot. If the compounds induced Cx43-P2 dephospho-rylation, then an increase in Cx43-Pl and/or Cx43-NP wouldresult. This was not the case, however. As described above,Cx43-P2 decreased with treatment duration, but there was noincrease in either Cx43-Pl or Cx43-NP (Figure 3A and B). Incontrol cells pretreated with CHX, a decrease in Cx43-P2 wasalso noted (Figure 3C), but this occurred over a much longertime period than in cells treated with DDT or LND. Thissuggests that the disappearance of Cx43-P2 following treatmentwith DDT or LND was not simply due to normal degradationof the protein.
The data presented thus far suggest that the loss of plasma
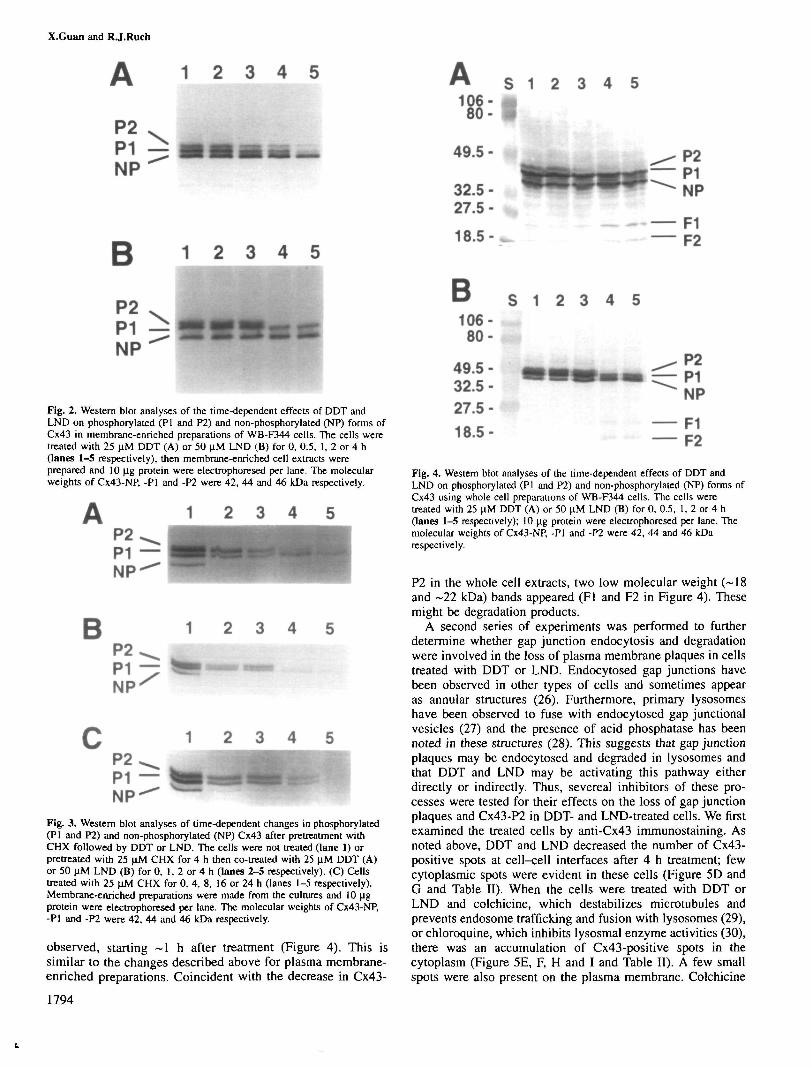
membranous Cx43-positive gap junction staining and of Cx43-P2 were not due to gap junction disassembly or Cx43-P2dephosphorylation. Another possibility is that the gap junctionswere endocytosed and Cx43-P2 was degraded in lysosomes.This hypothesis would account for the increased Cx43-positivecytoplasmic staining in DDT- and LND-treated cells. To testthis possibility, several experiments were performed. First,whole cell extracts were prepared in the hope of detecting aCx43 degradation product(s). In whole cell preparations fromcontrol cells, the NP, PI and P2 forms of Cx43 were evident,although the level of Cx43-P2 relative to the NP form wasless than in membrane-enriched preparations (Figure 4). Thisis probably due to the fact that the P2 form is found in theplasma membrane, whereas the NP form is also present in thecytoplasmic compartment (25). Thus, the relative levels ofeach would be different depending upon the isolation pro-cedure. When whole cell extracts were prepared from cellstreated with DDT or LND for 0-4 h, a decrease in Cx43-P2,a slight reduction in Cx43-Pl and no change in Cx43-NP were
1793
X.Guan and RJ.Ruch
1 2 3 4 5
P2P1 —NP
B 1 2 3 4 5
Fig. 2. Western blot analyses of the time-dependent effects of DDT andLND on phosphorylated (PI and P2) and non-phosphorylated (NP) forms ofCx43 in membrane-enriched preparations of WB-F344 cells. The cells weretreated with 25 nM DDT (A) or 50 |iM LND (B) for 0, 0.5, 1, 2 or 4 h(lanes 1—5 respectively), then membrane-enriched cell extracts wereprepared and 10 |ig protein were electrophoresed per lane. The molecularweights of Cx43-NP, -PI and -P2 were 42, 44 and 46 kDa respectively.
B 1 2 3 4P2P1 —NP
Fig. 3. Western blot analyses of time-dependent changes in phosphorylated(PI and P2) and non-phosphorylated (NP) Cx43 after pretreatment withCHX followed by DDT or LND. The cells were not treated (lane 1) orpretreated with 25 |lM CHX for 4 h then co-treated with 25 |iM DDT (A)or 50 nM LND (B) for 0, 1, 2 or 4 h (lanes 2-5 respectively). (C) Cellstreated with 25 uM CHX for 0, 4, 8, 16 or 24 h (lanes 1-5 respectively).Membrane-enriched preparations were made from the cultures and 10 Jigprotein were electrophoresed per lane. The molecular weights of Cx43-NP,-PI and -P2 were 42, 44 and 46 kDa respectively.
observed, starting ~1 h after treatment (Figure 4). This issimilar to the changes described above for plasma membrane-enriched preparations. Coincident with the decrease in Cx43-
1794
S 1 2 3 4 5
49.5- m
32.5-27.5-
1 8 . 5 - ^
P2P1NP
F1F2
B S 1106- m$80-
49.5-32.5-27.5-18.5-
2 3 4 5
P2P1NP
F1F2
Fig. 4. Western blot analyses of the time-dependent effects of DDT andLND on phosphorylated (PI and P2) and non-phosphorylated (NP) forms ofCx43 using whole cell preparations of WB-F344 cells. The cells weretreated with 25 ujvl DDT (A) or 50 \M LND (B) for 0, 0.5, 1, 2 or 4 h(lanes 1-5 respectively); 10 u.g protein were electrophoresed per lane. Themolecular weights of Cx43-NP, -PI and -P2 were 42, 44 and 46 kDarespectively.
P2 in the whole cell extracts, two low molecular weight (~18and ~22 kDa) bands appeared (Fl and F2 in Figure 4). Thesemight be degradation products.
A second series of experiments was performed to furtherdetermine whether gap junction endocytosis and degradationwere involved in the loss of plasma membrane plaques in cellstreated with DDT or LND. Endocytosed gap junctions havebeen observed in other types of cells and sometimes appearas annular structures (26). Furthermore, primary lysosomeshave been observed to fuse with endocytosed gap junctionalvesicles (27) and the presence of acid phosphatase has beennoted in these structures (28). This suggests that gap junctionplaques may be endocytosed and degraded in lysosomes andthat DDT and LND may be activating this pathway eitherdirectly or indirectly. Thus, severeal inhibitors of these pro-cesses were tested for their effects on the loss of gap junctionplaques and Cx43-P2 in DDT- and LND-treated cells. We firstexamined the treated cells by anti-Cx43 immunostaining. Asnoted above, DDT and LND decreased the number of Cx43-positive spots at cell-cell interfaces after 4 h treatment; fewcytoplasmic spots were evident in these cells (Figure 5D andG and Table II). When the cells were treated with DDT orLND and colchicine, which destabilizes microtubules andprevents endosome trafficking and fusion with lysosomes (29),or chloroquine, which inhibits lysosmal enzyme activities (30),there was an accumulation of Cx43-positive spots in thecytoplasm (Figure 5E, F, H and I and Table II). A few smallspots were also present on the plasma membrane. Colchicine

Gap junction endocytosis and degradation
Fig. 5. Immunohistochemica] demonstration of the effects of colchicine and chloroquine on the loss of gap junctions in WB-F344 cells treated with DDT. Thecells were treated with 0.1% DMSO (control; A), DMSO plus 30 uM colchicine (B), DMSO plus 50 ^M chloroquine (C), 25 nM DDT (D), DDT pluscolchicine (E), DDT plus chloroquine (F), 50 |iM LND (G), LND plus colchicine (H) or LND plus chloroquine (I) for 4 h. After treatment, the cells werefixed and immunostained for Cx43 as described in Materials and methods.
Table II. Effects of colchicine and chloroquine on the inhibition of dyecoupling and loss of gap junctions induced by DDT and LND in WB-F344cells
Treatment1 Dye coupling Cx43-positive spotsc
Plasma membrane Cytoplasm
ControlColchicineChloroquineDDTDDT +colchicineDDT +chloroquineLNDLND +colchicineLND +chloroquine
98.8 ± 4.498.5 ± 1.798.4 ± 0.6
1.9 ± 1.80.8 ± 1.4
1.2 ± 2.1
1.2 ± 2.10.7 ± 1.3
1.2 ± 2.0
The cells were treated with DMSO (0.1%, control), colchicine (30 uM),chloroquine (50 uM), DDT (25 uM) and/or LND (50 uM) for 4 h thenassayed for dye coupling or fixed and immunostained using anti-Cx43monoclonal antibody.bThe dye coupling percentage was determined as indicated in Materials andmethods. All groups treated with DDT or LND were significantly differentversus the control group (P < 0.01, n = 3 dishes/group).The abundance of gap junctions on the plasma membranes and of anu-Cx43 antibody reactive material in the cytoplasm were graded on a scaleranging from very abundant (+ + +) to none detected (-).
and chloroquine treatment alone had no effect on Cx43immunostaining (Figure 5A-C and Table II)- Coincident withthe abilities of colchicine and chloroquine to cause the accumu-lation of Cx43-positive spots in DDT- and LND-treated cells,Western blot analyses of the cells revealed that both drugsalso prevented the loss of Cx43-P2 induced by DDT and LND(Figure 6). These results suggest that the loss of gap junctionplaques and of Cx43-P2 in DDT- and LND-treated cells wasdue to endocytosis and degradation of plaques in lysosomes.
Discussion
In this study, we have shown that the inhibition of GJ1C (dyecoupling) induced by DDT or LND occurred rapidly and wasinitially not due to gross structural changes in the size,location or number of gap junction plaques or the quantity orphosphorylation status of Cx43. Subsequent treatment, how-ever, led to the disappearance of plasma membrane junctionalplaques, the appearance of Cx43-positive spots in the cyto-plasm, which eventually disappeared, and the disappearanceof Cx43-P2. These time-dependent changes in dye coupling,Cx43 immunostaining and Cx43-P2 status have been describedin WB-F344 and other types of cells treated with severalchemical inhibitors of GJIC (21-24,31-34), but the mechan-ism^) was not determined. In this study, we have consideredwhether the disappearance of plasma membrane plaques wasdue to their disassembly into individual particles or to endo-cytosis and whether Cx43-P2 was dephosphorylated ordegraded.
Based upon the immunohistochemical data, we believe the
1795

X.Guan and RJ.Ruch
1 2 3 4 5 6
P2P I -MP^
B
P2P I -MP^
1 2 3 4 5 6
•««ii
Fig. 6. Western blot analyses of the effects of colchicine and chloroquine onthe loss of Cx43-P2 in WB-F344 cells treated with DDT or LND. (A) Cellstreated with 0.1% DMSO (control, lane 1), 30 nM colchicine (lane 2),25 nM DDT (lane 3), 50 |iM LND (lane 4), DDT plus colchicine (lane 5)or LND plus colchicine (lane 6) for 4 h. (B) Cells treated with 0.1%DMSO (control, lane 1), 50 U.M chloroquine (lane 2), 25 nM DDT (lane 3),50 U.M LND (lane 4), DDT plus chloroquine (lane 5) or LND pluschloroquine (lane 6) for 4 h. After treatment, membrane-ennchedpreparations were made from theses cells and 10 |ig protein wereelectrophoresed per lane. Non-phosphorylated (NP) and phosphorylated (PIand P2) forms of Cx43 are marked; their molecular weights are 42, 44 and46 kDa respectively.
junctional plaques were endocytosed rather than disassembled.Disaggregation and dispersal of junctional particles in theplasma membrane would result in diffuse Cx43-positiveimmunostaining of the plasma membrane (35,36) and we sawno evidence for such staining in DDT- and LND-treated cells(Figures 1 and 5). Also, Cx43-positive structures appeared inthe cytoplasm coincident with the loss of Cx43-positive spotsfrom the plasma membranes (Figures 1 and 5). These structuresaccumulated in cells that were also treated with colchicine andchloroquine, suggesting they were endocytosed gap junctionvesicles on the pathway to degradation in lysosomes. Theendocytosis of gap junction plaques and fusion of the vesicleswith lysosomes has been observed in other systems (26-28),including cells treated with other tumor promoters, such asphorbol esters (22,31) and butylated hydroxytoluene (24). Thepatterns of immunostaining we observed are thus consistentwith the literature and the interpretation that gap junctionplaques were endocytosed and not disassembled into individualjunctional particles in cells treated with DDT or LND. However,this does not preclude the possibility that other agents mighthave different effects on gap junctions (discussed below).
We also conclude that the loss of Cx43-P2 was due to itsdegradation rather than dephosphorylation. First, two putative
1796
degradation products of Cx43-P2 were detected in cells treatedwith DDT and LND. These ~ 18 and -22 kDa peptides appearedcoincident with the loss of Cx43-P2 in whole cell extracts,but were not evident in plasma membrane-enriched prepara-tions. This suggests that the two peptides were derivatives ofC43-P2 and were localized in the cytoplasmic and/or nuclearcompartment. Since Cx43 immunoreactivity was not detectedin nuclei, the small peptides were most likely cytoplasmic,consistent with the degradation of Cx43-P2 in lysosomes.Secondly, no increase in Cx43-NP or Cx43-Pl was apparentin cells treated with CHX and then DDT or LND, indicating thatthe loss of Cx43-P2 was not due simply to dephosphorylation.However, it is possible that dephosphorylation occurred andtriggered rapid Cx43-P2 degradation; such a short-liveddephosphorylated species might not have been detected byWestern blot. We do not believe this is the case, however,since the protein phosphatase inhibitor okadaic acid had noeffect on the loss of Cx43-P2 in cells treated with DDT orLND (data not shown). Thirdly, colchicine and chloroquineprevented the loss of Cx43-P2, which indicates that micro-tubules and lysosomes were involved in the disappearance ofCx43-P2. These three lines of evidence strongly suggest thatDDT and LND induced the loss of Cx43-P2 by gap junctionendocytosis and degradation in lysosomes, rather than bydephosphorylation.
This may not be the case with other inhibitors of GJIC,however. 18P-Glycyrrhetinic acid, which also inhibits dyecoupling and decreases gap junctions and Cx43-P2 in WB-F344 cells, induces gap junction disassembly and Cx43-P2dephosphorylation (37). The disappearance of gap junctionplaques in these cells is accompanied by the appearance ofdiffuse Cx43-positive staining of the plasma membrane atcell-cell borders. In addition, treatment of WB-F344 cells withthe phorbol ester 12-0-tetradecanoylphorbol-13-acetate (TPA)is accompanied by the rapid (within 15 min) endocytosisof gap junction plaques and the appearance of a novelphosphorylated form of Cx43, Cx43-P3 (22,24). There was noevidence, however, that TPA reduced Cx43-P2 content orinduced its degradation.
Little is known regarding connexin degradation. A recentstudy by Laing and Beyer (38) suggests that in normalrat kidney cells, degradation of Cx43 principally occurs byubiquitination of the peptide and degradation in proteosomes.Their data also indicate that degradation of Cx43 can occur inlysosomes, but that this is a minor route. In WB-F344 cellstreated with DDT or LND, however, our data suggest thatCx43 degradation occurs principally in lysosmes. This isbecause chloroquine nearly completely prevented the disap-pearance of Cx43-P2 (Figure 6). The differences between ourdata and that of Laing and Beyer (38) may be due to the factthat DDT and LND shift Cx43 degradation from the ubiquitin/proteosome pathway to the endosome/lysosome pathway orthat the former pathway may be a minor or non-existentpathway in WB-F344 cells. It would be interesting to testwhether DDT or LND induce Cx43-P2 degradation in normalrat kidney cells and if this could be inhibited by chloroquine.
Connexins may be degraded by other pathways. Connexin32has been reported to be degraded by calpains (39). However,this does not appear to be the case for Cx43 in normal ratkidney (38) or WB-F344 cells. Calpain inhibitor I did notprevent the loss of Cx43-P2 in WB-F344 cells treated withDDT or LND (Guan and Ruch, unpublished results).
The appearance of two putative Cx43-P2 fragments in DDT-

Gap junction endocytosis and degradation
and LND-treated cells (Figure 4) suggests that Cx43 may bedegraded by at least two steps, the first step generating the~22 kDa fragment and the second resulting in the ~18 kDapiece. It is important to note that the epitope of the Cx43monoclonal antibody is located within amino acid residues252-271 and, thus, must be present within both fragments. Inagreement with our data, Goldberg and Lau (40) have describeda phosphorylated 17 kDa degradation product of Cx43 innormal and v-s/r-transformed Rat-1 fibroblasts. We do not yetknow, however, if the 18 and 22 kDa fragments we observedare phosphorylated or related to the 17 kDa fragment. Furtherstudies using other site-specific antibodies are necessary toelucidate the various steps of Cx43 degradation in lysosomes.
It will also be interesting to determine the 'trigger' mechan-ism for gap junction endocytosis and whether this is relatedto the initial inhibitory effects of DDT and LND on dyecoupling, which do not involve gross structural changes in thejunctions or Cx43 status. It will also be of interest to determinethe role that gap junction endocytosis and degradation play inthe in vivo effects of DDT or LND on gap junctions (41-43)and the carcinogenic and toxicological properties of thesecompounds.
AcknowledgementsThe authors gratefully acknowledge the technical assistance of MarthaFernstrom. This study was supported by the National Institutes of Health(grant CA57612).
Referencesl.Loewenstein.W.R. (1987) The cell-cell channel of gap junctions. Cell, 48,
725-726.2. Loewenstein,W.R. (1979) Junctional intercellular communication and the
control of growth. Biochim. Biophys. Ada, 560, 1-65.3,TroskoJ.E. and Chang.C.C. (1984) Adaptive and nonadapuve consequences
of chemical inhibition of intercellular communication. Pharmacol. Rev.,36, 137S-144S.
4. Ruch,R.J. (1994) The role of gap junctional intercellular communicationin neoplasia. Annh Clin. Lab. Set., 24, 216-231.
5. Yamasaki.H. (1990) Gap junctional intercellular communication andcarcinogencsis. Carcinogenesis, 11, 1051-1058.
6.White,T.W., Bruzzone.R. and Paul.D.L (1995) The connexin family ofintracellular channel forming proteins. Kidney Int., 48, 1148-1157.
7.Zhu,D., Caveney.S., Kidder.G.M. and Naus.C.C.G. (1991) Transfection ofC6 glioma cells with connexin 43 cDNA: analysis of expression,intercellular coupling, and cell proliferation. Proc. Nail Acad. Sci. USA,88, 1883-1887.
8. Mehta,P.P., Hotz-Wagenblatt,A., Rose.B., Shalloway.D. andLoewenstein.W.R. (1991) Incorporation of the gene for a cell-cell channelprotein into transformed cells leads to normalization of growth. J. Membr.Biol., 124, 207-225.
9. Eghbali,B., KesslerJ.A., Reid.L.M., Roy.C. and Spray,D.C. (1991)Involvement of gap junctions in tumorigenesis: transfection of tumor cellswith connexin 32 cDNA retards growth in vivo. Proc. Natl Acad. Sci.OS4, 88, 10701-10705.
10. Naus.C.C.G., Elisevich.K., Zhu.D., Belliveau,DJ. and Del Maestro.R.F.(1992) In vivo growth of C6 glioma cells transfected with connexin43cDNA. Cancer Res., 52, 4208-4213.
ll.Sugiura,H. and Joyner,R.W. (1992) Action potential conduction betweenguinea pig ventricular cells can be modulated by calcium current. Am. J.Physiol. Heart Circ. Physiol., 263, H1591-H1604.
12.Mesnil,M., Kmtovskikh.V., Piccoli.C, Elfgang.C, Traub.O, Willecke,K.and Yamasaki.H. (1995) Negative growth control of HeLa cells byconnexin genes: connexin species specificity. Cancer Res., 55, 629-639.
13.Chen,S.C, Pelletier.D.B., Ao,P. and Boynton.A.L. (1995) Connexin43reverses the phenotype of transformed cells and alters their expression ofcyclin/cyclin-dependent kinases. Cell Growth Differential., 6, 681-690.
14.Goldberg,G.S., Martyn.lCD. and Lau,A.F. (1994) A connexin 43 antisensevector reduces the ability of normal cells to inhibit the foci formation oftransformed cells. Mol. Carcinogen., 11, 106-114.
15.Ruch,RJ., Guan.X. and Sigler,K. (1995) Inhibition of gap junctional
intercellular communication and altered growth in Balb/c 3T3 cells treatedwith connexin43 antisense oligonucleotides. Mol. Carcinogen., 14, in press.
16.TroskoJ.E., Chang.C.C., Madhukar.B.V. and KlaunigJ.E. (1990) Chemical,oncogene and growth factor inhibition of gap junctional intercellularcommunication: an integrative hypothesis of carcinogenesis. Pathobiology,58, 265-278.
17.KIaunigJ.E. and Ruch.RJ. (1990) Role of intercellular communication innongenotoxic carcinogenesis. Lab. Invest., 62, 135—146.
18.Budunova,I.V. and Williams.G.M. (1994) Cell culture assays for chemicalswith tumor promoting or inhibiting activity based on the modulation ofintercellular communication. Cell Biol. Toxicol., 10, 71-116.
19.Wolff,M.S., Toniolo.P.G., LeeJE.W., Rivera,M. and Dubin.N. (1993) Bloodlevels of organochlorine residues and risk of breast cancer. J. Natl CancerInst., 85, 648-652.
20.Garabrandt, D.H., HeldJ., Langholz.B., PetersJ.M. and Mack,T.M. (1992)DDT and related compounds and risk of pancreatic cancer. J. Nail CancerInst., 84, 764-771.
21.Ruch,RJ., Bonney.W.J., Sigler.K., Guan,X., MatesicJ)., Schafer.L.D.,DuponuE. and TroskoJ.E. (1994) Loss of gap junctions from DDT-treatedrat liver epithelial cells. Carcinogenesis, 15, 301-306.
22.Matesic,D.F., Rupp.H.L., Bonney.W.J., Ruch.RJ. and TroskoJ.E. (1994)Changes in gap junction permeability, phosphorylation, and numbermediated by phorbol ester and non-phorbol ester tumor promoters Mol.Carcinogen., 10, 226-236.
23.Guan,XJ., Bonney.WJ. and Ruch,RJ. (1995) Changes in gap junctionpermeability, gap junction number, and connexin43 expression inlindane-treated rat liver epithelial cells. Toxicol. Appl. Pharmacol., 130,79-86.
24.Guan,X., HardenbrookJ., Fernstrom,MJ., Chaudhuri.R., Malkinson^A.M.and Ruch.RJ. (1995) Downregulation by butylated hydroxytoluene of thenumber and permeability of gap junctions in cell lines derived from mouselung and rat liver. Carcinogenesis, 16, 2575-2582.
25.Musil,L.S. and Goodenough.D.A. (1991) Biochemical analysis ofconnexin43 intracellular transport, phosphorylation, and assembly into gapjunctional plaques. J Cell Biol., 115, 1357-1374.
26. Larsen.W.J and Risinger,M.A. (1985) The dynamic life histories ofintercellular membrane junctions. Mod. Cell Biol., 4, 151-216.
27. Vaughan.D.K. and Lasater.E.M. (1990) Renewal of electrotonic synapsesin teleost retinal horizontal cells. J. Comp. Neural., 299, 364—374.
28. Larsen,WJ. and Hai-Nan (1978) Origin and fate of cytoplasmic gapjunctional vesicles in rabbit granulosa cells. Tissue Cell, 10, 585—598.
29.Elkjaer,M.L., Birn,H., AgreJ., Christensen,E.I. and Nielsen.S. (1995)Effects of microtubule disruption on endocytosis, membrane recycling andpolarized distribution of Aquaporin-1 and gp330 in proximal tubule cells.Eur. J. Cell Biol., 67, 57-72.
30. Nishimura.Y., Kato.K., Furuno.K. and Himeno.M. (1995) Biosynthesis andprocessing of cathepsin D in primary cultures of rat hepatocytes. Biol.Pharm. Bull., 18, 825-828.
31.Berthoud,V.M., Ledbetter.M.L S., Hertzberg,E.L. and SaezJ.C. (1992)Connexin43 in MDCK cells: regulation by a tumor-promoting phorbolester and Ca2+. Eur. J. Cell Biol., 57, 40-50.
32. Wamgard.L., Hemming.H., Flodstrom.S., Duddy.S. and Kass.G.E.N. (1989)Mechanistic studies on the DDT-induced inhibition of intercellularcommunication. Carcinogenesis, 10, 471-476.
33.Budunova,l.V., Williams.G.M. and Spray.D.C. (1993) Effect of tumorpromoting stimuli on gap junction permeability and connexin43 expressionin ARL 18 rat liver cell line. Arch. Toxicol., 67, 565-572.
34.Rivedal,E., Yamasaki.H. and Sanner.T. (1994) Inhibition of gap junctionalintercellular communication in Syrian hamster embryo cells by TPA,retinoic acid and DDT. Carcinogenesis, 15, 689-694.
35.Yancey,S.B., Easter.D. and RevelJ.-P. (1979) Cytological changes ingap junctions during rat liver regeneration. / . Ultraslrucl. Res., 67,229-242.
36. Lane,N.J. and SwalesJ-.S. (1980) Dispersal of junctional particles, notintemalization, during the in vivo disappearance of gap junctions. Celt,19, 579-586.
37.Guan,X., Wilson.S., Schlender.K.K. and Ruch,RJ. (1996) Gap junctiondisassembly and connexin43 dephosphorylation induced by 18p"-glycyrrhetinic acid. Mol. Carcinogen., in press.
38. LaingJ.G. and Beyer,E.C. (1995) The gap junction protein connexin43 isdegraded via the ubiguitin proteasome pathway. J. Biol. Chem., 270,26399-26403.
39.Elvira,M., DiezJ.A., Wang.K.K.W. and VillaloboA (1993) Phos-phorylation of connexin-32 by protein kinase C prevents its proteolysisby u-calpain and m-calpain. /. Biol. Chem., 268, 14294-14300.
40.Goldberg,G.S. and Lau.A.F. (1993) Dynamics of connexir»43 phos-phorylation in ppcXT'^-transformed cells. Biochem. J., 295, 735-742.
1797

X.Guan and RJ.Ruch
41.Sugie,S., Mori,H. and Takahashi,M. (1987) Effect of in vivo exposure tothe liver tumor promoters phenobarbital or DDT on the gap junctions ofrat hepatocytes: a quantitative freeze fracture analysis. Carcinogenesis, 8,45-51.
42.Tateno,C, Ito.S., Tanaka.M., Oyamada,M. and Yoshitake,A. (1994) Effectsof DDT on hepatic gap junctional intercellular communication in rats.Carcinogenesis, 15, 517-521.
43.Krutovskikh,V.A., Mesnil.M., Mazzoleni.G. and Yamasaki.H. (1995)Inhibition of rat liver gap junction intercellular communication by tumor-promoting agents in vivo: association with aberrant localization of connexinproteins. Lab. Invest., 72, 571-577.
Received on January 3, 1996; revised on April 16, 1996; accepted on June10, 1996
1798
















