
Functional significance of the intermediate conductance Ca 2+ -activated K + channel for the...
16
ION CHANNELS, RECEPTORS AND TRANSPORTERS Functional significance of the intermediate conductance Ca 2+ -activated K + channel for the short-term survival of injured erythrocytes Michael Föller & Diwakar Bobbala & Saisudha Koka & Krishna M. Boini & Hasan Mahmud & Ravi S. Kasinathan & Ekaterina Shumilina & Kerstin Amann & Golo Beranek & Ulrike Sausbier & Peter Ruth & Matthias Sausbier & Florian Lang & Stephan M. Huber Received: 9 April 2010 / Revised: 19 August 2010 / Accepted: 24 August 2010 / Published online: 21 September 2010 # Springer-Verlag 2010 Abstract Increased cytosolic Ca 2+ concentrations activate Gardos K + channels in human erythrocytes with membrane hyperpolarization, efflux of K + , Cl − , and osmotically obliged H 2 O resulting in cell shrinkage, a phenomenon referred to as Gardos effect. We tested whether the Gardos effect delays colloid osmotic hemolysis of injured eryth- rocytes from mice lacking the Ca 2+ -activated K + channel K Ca 3.1. To this end, we applied patch clamp and flow cytometry and determined in vitro as well as in vivo hemolysis. As a result, erythrocytes from K Ca 3.1-deficient (K Ca 3.1 −/− ) mice lacked Gardos channel activity and the Gardos effect. Blood parameters, reticulocyte count, or osmotic erythrocyte resistance, however, did not differ between K Ca 3.1 −/− mice and their wild-type littermates, suggesting low or absent Gardos channel activity in unstressed erythrocytes. Oxidative stress-induced Ca 2+ entry and phospholipid scrambling were significantly less pronounced in K Ca 3.1 −/− than in wild-type erythrocytes. Moreover, in vitro treatment with α-toxin from Staphylo- coccus aureus, which forms pores in the cellular membrane, resulted in significantly stronger hemolysis of K Ca 3.1 −/− than of wild-type erythrocytes. Intravenous injection of α- toxin induced more profound hemolysis in K Ca 3.1 −/− than in wild-type mice. Similarly, intra-peritoneal application of the redox-active substance phenylhydrazine, an agent for the induction of hemolytic anemia, was followed by a significantly stronger decrease of hematocrit in K Ca 3.1 −/− than in wild-type mice. Finally, malaria infection triggered the activation of K Ca 3.1 and transient shrinkage of the infected erythrocytes. In conclusion, K Ca 3.1 channel activ- ity and Gardos effect counteract hemolysis of injured erythrocytes, thus decreasing hemoglobin release into circulating blood. Keywords Intermediate conductance Ca 2+ -activated K + channel K Ca 3.1 . Red blood cell . Suicidal erythrocyte death . Calcium signaling . Scramblase Introduction Human erythrocytes express Ca 2+ -activated Gardos K + channels [1], which are believed to be generated by the intermediate conductance Ca 2+ -activated K + channel K Ca 3.1 (synonyms: SK4, IK, and KCNN4) [2, 3]. Gardos K + channels have been characterized in human erythrocytes by single channel recording [4–6]. Channel activity is voltage independent but dependent on the free intracellular cytosolic Ca 2+ concentration [Ca 2+ ] i . Ca 2+ acts via binding M. Föller : D. Bobbala : S. Koka : K. M. Boini : H. Mahmud : R. S. Kasinathan : E. Shumilina : F. Lang Department of Physiology, University of Tübingen, Tübingen, Germany K. Amann Department of Pathology, University of Erlangen-Nürnberg, Erlangen, Germany G. Beranek : U. Sausbier : P. Ruth : M. Sausbier Department of Pharmacology & Toxicology of the Institute of Pharmacy, University of Tübingen, Tübingen, Germany S. M. Huber (*) Department of Radiation Oncology, University of Tübingen, Hoppe-Seyler-Str. 3, 72076 Tübingen, Germany e-mail: [email protected] Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 DOI 10.1007/s00424-010-0878-1
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Functional significance of the intermediate conductance Ca 2+ -activated K + channel for the...

ION CHANNELS, RECEPTORS AND TRANSPORTERS
Functional significance of the intermediate conductanceCa2+-activated K+ channel for the short-termsurvival of injured erythrocytes
Michael Föller & Diwakar Bobbala & Saisudha Koka & Krishna M. Boini &Hasan Mahmud & Ravi S. Kasinathan & Ekaterina Shumilina & Kerstin Amann &
Golo Beranek & Ulrike Sausbier & Peter Ruth & Matthias Sausbier & Florian Lang &
Stephan M. Huber
Received: 9 April 2010 /Revised: 19 August 2010 /Accepted: 24 August 2010 /Published online: 21 September 2010# Springer-Verlag 2010
Abstract Increased cytosolic Ca2+ concentrations activateGardos K+ channels in human erythrocytes with membranehyperpolarization, efflux of K+, Cl−, and osmoticallyobliged H2O resulting in cell shrinkage, a phenomenonreferred to as Gardos effect. We tested whether the Gardoseffect delays colloid osmotic hemolysis of injured eryth-rocytes from mice lacking the Ca2+-activated K+ channelKCa3.1. To this end, we applied patch clamp and flowcytometry and determined in vitro as well as in vivohemolysis. As a result, erythrocytes from KCa3.1-deficient(KCa3.1
−/−) mice lacked Gardos channel activity and theGardos effect. Blood parameters, reticulocyte count, orosmotic erythrocyte resistance, however, did not differbetween KCa3.1
−/− mice and their wild-type littermates,suggesting low or absent Gardos channel activity inunstressed erythrocytes. Oxidative stress-induced Ca2+
entry and phospholipid scrambling were significantly lesspronounced in KCa3.1
−/− than in wild-type erythrocytes.Moreover, in vitro treatment with α-toxin from Staphylo-coccus aureus, which forms pores in the cellular membrane,resulted in significantly stronger hemolysis of KCa3.1
−/−
than of wild-type erythrocytes. Intravenous injection of α-toxin induced more profound hemolysis in KCa3.1
−/− thanin wild-type mice. Similarly, intra-peritoneal application ofthe redox-active substance phenylhydrazine, an agent forthe induction of hemolytic anemia, was followed by asignificantly stronger decrease of hematocrit in KCa3.1
−/−
than in wild-type mice. Finally, malaria infection triggeredthe activation of KCa3.1 and transient shrinkage of theinfected erythrocytes. In conclusion, KCa3.1 channel activ-ity and Gardos effect counteract hemolysis of injurederythrocytes, thus decreasing hemoglobin release intocirculating blood.
Keywords Intermediate conductance Ca2+-activated K+
channel KCa3.1 . Red blood cell . Suicidal erythrocytedeath . Calcium signaling . Scramblase
Introduction
Human erythrocytes express Ca2+-activated Gardos K+
channels [1], which are believed to be generated by theintermediate conductance Ca2+-activated K+ channelKCa3.1 (synonyms: SK4, IK, and KCNN4) [2, 3]. GardosK+ channels have been characterized in human erythrocytesby single channel recording [4–6]. Channel activity isvoltage independent but dependent on the free intracellularcytosolic Ca2+ concentration [Ca2+]i. Ca
2+ acts via binding
M. Föller :D. Bobbala : S. Koka :K. M. Boini :H. Mahmud :R. S. Kasinathan : E. Shumilina : F. LangDepartment of Physiology, University of Tübingen,Tübingen, Germany
K. AmannDepartment of Pathology, University of Erlangen-Nürnberg,Erlangen, Germany
G. Beranek :U. Sausbier : P. Ruth :M. SausbierDepartment of Pharmacology & Toxicology of the Institute ofPharmacy, University of Tübingen,Tübingen, Germany
S. M. Huber (*)Department of Radiation Oncology, University of Tübingen,Hoppe-Seyler-Str. 3,72076 Tübingen, Germanye-mail: [email protected]
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044DOI 10.1007/s00424-010-0878-1

to calmodulin, which is constitutively associated withGardos channels [7]. Gardos channels are highly K+ overNa+-selective (PK/PNa>100) and exhibit an inwardlyrectifying current–voltage relationship (when recorded withsymmetrical K+ solutions). Extracellularly applied TRAM-34 [8], charybdotoxin [9–11], and clotrimazole [12] inhibitKCa3.1 channels with IC50 values of 5–50 nM. However,these inhibitors are not completely specific for KCa3.1,since clotrimazole and TRAM-34, in the micromolar range,also inhibit nonselective cation channels [13, 14] andcharybdotoxin is a potent blocker of BK channels [15].
Total intracellular Ca2+ is low in human erythrocytes ascompared with nucleated cells [16–18]. This results from alow cytoplasmic Ca2+-buffering capacity [19, 20], the absenceof Ca2+-sequestering endoplasmic reticulum and mitochondria[21], and minimal endocytotic Ca2+-sequestering vesicles[22]. Due to the powerful Ca2+ extrusion by a high Ca2+
ATPase pump turnover [23, 24] and the low Ca2+ permeabil-ity of non-stressed erythrocytes, the cytosolic Ca2+ concen-tration ([Ca2+]i) usually remains below 100 nM [18, 23, 25],keeping the Gardos K+ channel activity low. As a conse-quence, human erythrocytes have a low membrane potentialand, thus, a high intracellular Cl− concentration [26].Activation of Gardos channels leads to K+ efflux withhyperpolarization of the erythrocyte membrane potential,which imposes an outwardly directed electrochemical Cl−
gradient across the erythrocyte membrane driving the efflux ofCl−. The cellular loss of KCl and osmotically obliged waterleads to erythrocyte shrinkage.
Stress stimuli such as low ionic strength (i.e., low Cl−
concentration), oxidative stress, hyperosmotic shrinkage[27], or glucose depletion [28] trigger the suicidal deathprogram of human erythrocytes by activating a nonselectivecation permeability, which could be identified by tracer fluxstudies [29–31], fluorescence optics, and patch-clamprecording [32]. At least in part, channel activation is dueto the formation of prostaglandin E2 [33]. Stimulation ofhuman erythrocytes with PGE2 increases [Ca2+]i [33, 34],leading to Gardos K+ channel-mediated erythrocyte shrink-age [33]. The shrinkage results in altered erythrocytedeformability and filterability [33–36] and participates inthe stimulation of cell membrane scrambling [3, 37].
Beyond Gardos channel activation, elevated [Ca2+]istimulates the neutral endopeptidase calpain [38, 39] andthe phospholipid scramblase [40, 41] and inhibits theaminophospholipid translocase [42] in erythrocytes. Thelatter builds up the phospholipid asymmetry of theerythrocyte membrane. Futhermore, Gardos-mediatederythrocyte shrinkage induces sphingomyelinase activityresulting in ceramide formation, which further potentiatesscramblase activity [43, 44]. Scramblase activation andtranslocase inactivation result in the breakdown of thephospholipid asymmetry. As a result, phosphatidylserine,
which usually resides in the inner leaflet, appears in theouter membrane leaflet [45]. Phosphatidylserine exposureat the cell surface is an “eat-me” signal. Thus,phosphatidylserine-exposing erythrocytes are rapidlyengulfed by macrophages and cleared from circulatingblood [46, 47].
Hemolysis of erythrocytes by α-hemolysin from Escher-ichia coli has been demonstrated to trigger activation ofP2X purinergic receptor in equine, murine, and humanerythrocytes and possibly of pannexins augmenting hemo-lysis [48]. α-Hemolysin-stimulated hemolysis is reportedlypreceeded by an increase in [Ca2+]i, Ca2+-stimulatedactivation of Gardos K+- and TMEM16A Cl− channels,and erythrocyte shrinkage, which delays hemolysis [49]. Bydelaying colloid osmotic hemolysis and fostering phospha-tidylserine exposure, the Gardos effect is thought to play apivotal role for the appropriate clearance of injurederythrocytes [50]. This might be particularly importantduring infection with hemolytic bacteria [51–53].
Analyzing KCa3.1-deficient mice, the present studyaimed to define the functional significance of KCa3.1Gardos channels for suicidal erythrocyte death and eryth-rocyte clearance. Known phenotypes of KCa3.1-deficientmice include an attenuated IgE-mediated activation of mastcells [54], enhanced resting tone of arterioles [55],improved glucose tolerance [56], attenuated unilateralureteral obstruction-induced renal fibrosis [57], attenuatedor even abolished Ca2+-mediated intestinal and colonicanion secretion [58], increased stool dehydration [59], andameliorated T cell-mediated colitis [60].
Materials and methods
Mice
KCa3.1-deficient mice (KCa3.1−/−) were generated as de-
scribed [61] and maintained at the animal facility of theDepartment of Pharmacology & Toxicology, Institute ofPharmacy, University of Tübingen. Either litter- or age-matched (6–12 weeks) wild-type and KCa3.1
−/− mice ofboth genders with hybrid SV129/C57BL6 background(always F2 generation) were randomly assigned to theexperimental procedures with respect to the Germanlegislation on animal protection. For Fig. 4c, erythrocytesfrom old mice (1–1.5 years) were compared with those ofyoung mice (6–12 weeks).
In vitro experiments
Erythrocytes and solutions Blood from KCa3.1−/− mice and
wild-type mice was drawn by retroorbital puncture andcollected in heparin-coated tubes. After three times of
1030 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044
washing in NaCl solution (containing in mM: 125 NaCl,5 KCl, 1 MgSO4, 32 N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid, HEPES/NaOH, 5 glucose, 1 CaCl2;pH 7.4) cells were suspended in NaCl solution. Ca2+
permeabilization of the erythrocyte membrane (patch-clampand flow cytometry experiments of Figs. 1 and 4) wasaccomplished by addition of 10 μM and 1 μM Ca2+
ionophore ionomycin to the NaCl solution, respectively.The effect of oxidative stress on the cytosolic free Ca2+
concentration ([Ca2+]i) and phospholipid scrambling (Fig. 5)was assessed by incubating the cells (0.4% hematocrit) for45 min at 37°C in the absence or presence of tert-butylhydroperoxide (0.1 mM; t-BHP) in NaCl solution orKCl solution (containing in mM: 130 KCl, 32 HEPES/KOH,5 glucose, 1 MgSO4, 1 CaCl2; pH 7.4). To test for sensitivityagainst Staphylococcus aureus α-toxin (Sigma, Schnelldorf,Germany, Fig. 6a, b), cells were incubated for 90 min at37°C in NaCl solution (0.4% hematocrit) in the absence or
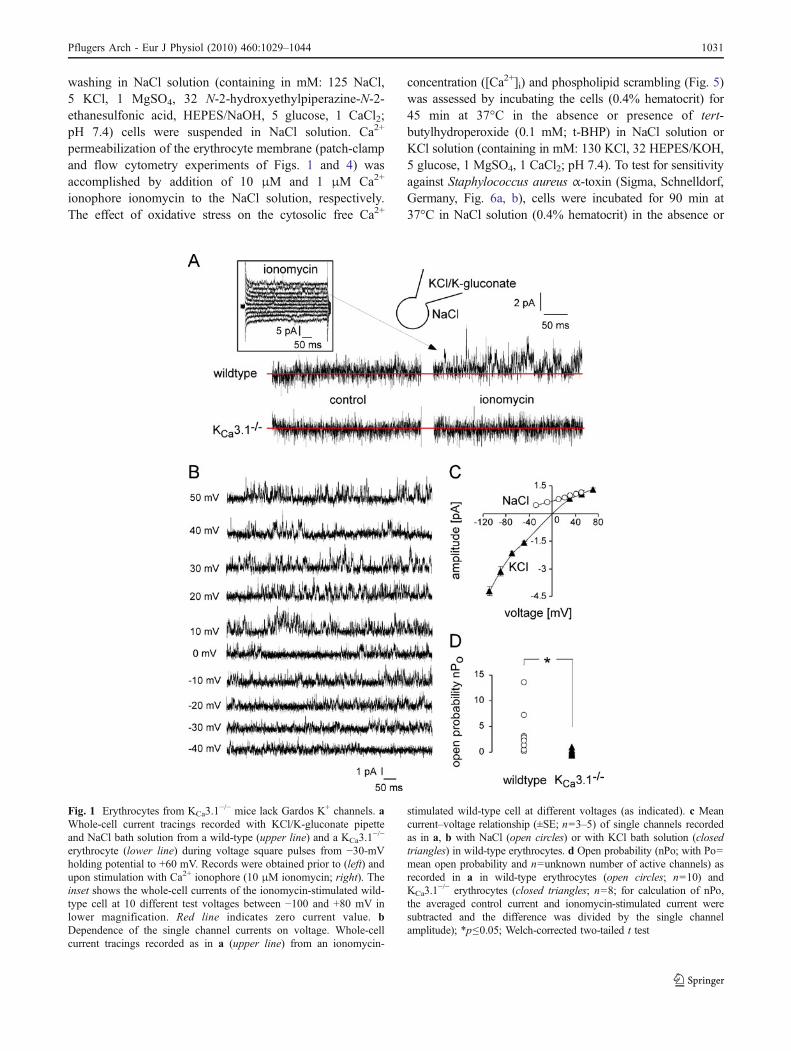
Fig. 1 Erythrocytes from KCa3.1−/− mice lack Gardos K+ channels. a
Whole-cell current tracings recorded with KCl/K-gluconate pipetteand NaCl bath solution from a wild-type (upper line) and a KCa3.1
−/−
erythrocyte (lower line) during voltage square pulses from −30-mVholding potential to +60 mV. Records were obtained prior to (left) andupon stimulation with Ca2+ ionophore (10 μM ionomycin; right). Theinset shows the whole-cell currents of the ionomycin-stimulated wild-type cell at 10 different test voltages between −100 and +80 mV inlower magnification. Red line indicates zero current value. bDependence of the single channel currents on voltage. Whole-cellcurrent tracings recorded as in a (upper line) from an ionomycin-
stimulated wild-type cell at different voltages (as indicated). c Meancurrent–voltage relationship (±SE; n=3–5) of single channels recordedas in a, b with NaCl (open circles) or with KCl bath solution (closedtriangles) in wild-type erythrocytes. d Open probability (nPo; with Po=mean open probability and n=unknown number of active channels) asrecorded in a in wild-type erythrocytes (open circles; n=10) andKCa3.1
−/− erythrocytes (closed triangles; n=8; for calculation of nPo,the averaged control current and ionomycin-stimulated current weresubtracted and the difference was divided by the single channelamplitude); *p≤0.05; Welch-corrected two-tailed t test
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1031
presence of α-toxin (10 hemolytic units/ml, HU/ml). Forwater-insoluble compounds, the solvent was applied asloading control.
Patch-clamp experiments The bath was grounded via abridge filled with NaCl bath solution (see above). Borosilicateglass pipettes (8–14 MΩ pipette resistance; GC150 TF-10,Clark Medical Instruments, Pangbourne, UK) manufacturedby a microprocessor-driven DMZ puller (Zeitz, Augsburg,Germany) were used in combination with a STM electricalmicromanipulator (Lang GmbH and Co KG, Germany).Currents were recorded at 35°C in fast whole-cell, voltage-clamp mode, and 3-kHz low-pass-filtered by an EPC-9amplifier (Heka, Lambrecht, Germany) using Pulse software(Heka) and an ITC-16 Interface (Instrutech, Port Washington,NY, USA). After a giga-ohm seal formation, the membranewas ruptured by additional suction.
The liquid junction potentials ΔE between the pipetteand the bath solutions, and between the salt bridge and thebath solutions were estimated as described earlier [62].Data were corrected for the estimated ΔE values. Whole-cell currents were evoked by 10 voltage pulses (400 or700 ms each) from −30 mV holding potential to voltagesbetween −100 and +80 mV. Applied voltages refer to thecytoplasmic face of the membrane with respect to theextracellular space. Inward currents, defined as flow ofpositive charge from the extracellular to the cytoplasmicmembrane face, are negative currents and depicted asdownward deflections of the original current traces.
Cells in Fig. 1a–d (open circles) were recorded with apipette solution containing (in mM) 80 KCl, 60 K-D-gluconate, 10 HEPES/KOH, 1 ethylene glycol-bis(β-
aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA),1 Mg-ATP, 1 MgCl2 (pH 7.2) and NaCl bath solution.Cells depicted in Fig. 1c (closed triangles) and Fig. 2 wererecorded with a further pipette solution (containing inmM: 140 K-D-gluconate, 10 HEPES/KOH, 1 EGTA,1 Mg-ATP, 5 MgCl2; pH 7.2) combined with NaCl(Fig. 2b, open circles) or KCl solution (see above) in thebath (Fig. 2a and closed triangles in Figs. 1c and 2b). TheKCa3.1 channel activity was either stimulated withionomycin (10 μM) or with the KCa channel opener 1-ethyl-2-benzimidazolinone (1-EBIO; 100 μM), both addedto the bath solution. For Fig. 1d, the open probability(nPo) of K+ channels was determined at 0 mV voltage bysubtracting the averaged current value under control conditionfrom that recorded thereafter in the presence of ionomycin(10 μM). The ionomycin-stimulated current fraction was thendivided by the single channel current amplitude (for KCa3.1
−/−
erythrocytes, the ionomycin-stimulated current fraction wasdivided by the mean single channel current amplitudedetermined in wild-type erythrocytes).
Forward scatter To assess Gardos K+ channel-mediatedchanges in erythrocyte size/volume, KCa3.1
−/− and wild-type erythrocytes were suspended in NaCl solution, and theforward scatter was recorded and analyzed by flowcytometry using a FACS-Calibur (Becton and Dickinson,Heidelberg, Germany).
Cytosolic free Ca2+ concentration For measurement of[Ca2+]i, control and oxidized KCa3.1
−/− and wild-typeerythrocytes were washed in NaCl solution and then loadedwith fluo3/AM (2 μM; Calbiochem; Bad Soden, Germany)
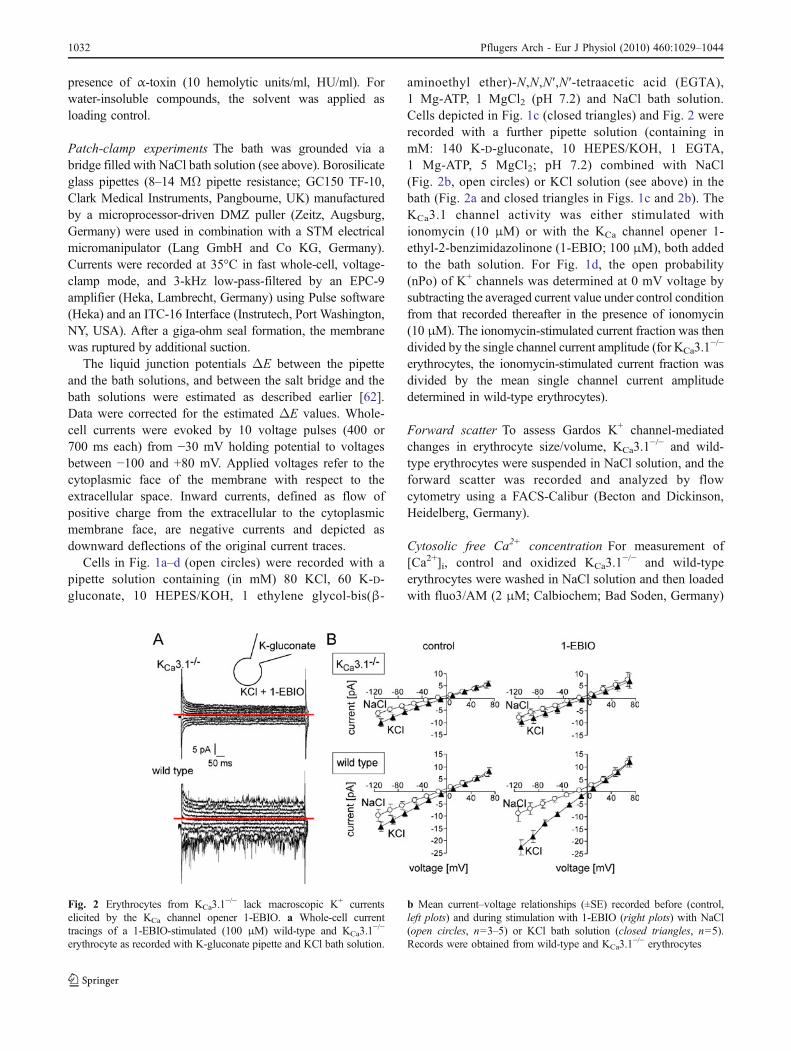
Fig. 2 Erythrocytes from KCa3.1−/− lack macroscopic K+ currents
elicited by the KCa channel opener 1-EBIO. a Whole-cell currenttracings of a 1-EBIO-stimulated (100 μM) wild-type and KCa3.1
−/−
erythrocyte as recorded with K-gluconate pipette and KCl bath solution.
b Mean current–voltage relationships (±SE) recorded before (control,left plots) and during stimulation with 1-EBIO (right plots) with NaCl(open circles, n=3–5) or KCl bath solution (closed triangles, n=5).Records were obtained from wild-type and KCa3.1
−/− erythrocytes
1032 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044

in NaCl solution for 15 min at 37°C. Thereafter, cells werewashed twice and resuspended in NaCl solution, and theCa2+-dependent fluo3 fluorescence intensity was measuredin fluorescence channel FL-1 (excitation at 488 nm,emission at 530 nm) and analyzed by geometric mean.For controlling fluo3 loading and quantification of theintracellular Ca2+ concentration, erythrocytes were stainedwith fluo-3/AM (Calbiochem, Bad Soden, Germany) inCa2+-free Ringer solution containing 4 mM glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 μMfluo-3/AM, and 1 mM of the Ca2+ ATPase inhibitor sodiumorthovanadate (Sigma, Schnelldorf, Germany) at a hemat-ocrit of 0.2% for 20 min at 37°C. After incubation, 100 μlof the suspensions was added to 2 ml of EGTA-buffered(4 mM) and 1 mM sodium orthovanadate-containingRinger solutions of defined free Ca2+ concentrations(ranging from 1 nM to 300 μM) adjusted according to thecalculations by WEBMAXC Standard program. Then,fluo3 fluorescence was measured before and after a 10-min incubation with 10 μM ionomycin in FL-1. For furthercontrol of dye loading, KCa3.1
−/− and wild-type erythro-cytes were loaded with BCECF (10 μM, Invitrogen,Karlsruhe, Germany) or fluo4/AM (2 μM; Invitrogen) inNaCl solution in the presence and absence of probenecid(5 mM, Sigma) for 30 min at 37°C and fluorescence wasmeasured by flow cytometry in FL-1 (excitation at 488 nm,emission at 530 nm) and analyzed by geometric mean.
Breakdown of the phospholipid asymmetry of the erythro-cyte membrane Phosphatidylserine appearance in the outermembrane leaflet was determined by annexin V-binding inflow cytometry. Control and oxidized KCa3.1
−/− and wild-type erythrocytes were washed and loaded for 20 min at37°C with annexin V-fluos (1:500 dilution; Roche Diag-nostics, Mannheim, Germany) in CaCl2-containing (5 mM)NaCl solution. After washing, annexin V fluorescence wasdetermined by flow cytometry in FL-1 and the percentageof annexin V-binding cells determined.
Hemolysis After incubation with α-toxin (10 HU/ml, for90 min at 37°C), cells were centrifuged at 400×g for 3 minand the supernatants were harvested. The hemoglobin (Hb)concentration of the supernatant was determined photomet-rically at 405 nm. Erythrocyte lysis in distilled water wasdefined as 100% hemolysis.
Blood parameters Erythrocyte number, packed cell volume,mean corpuscular volume, and blood hemoglobin concen-tration were determined using an electronic hematologyparticle counter (type MDM 905 from Medical DiagnosticsMarx; Butzbach, Germany) equipped with a photometricunit for hemoglobin determination. The gating was adjustedfor the application on murine erythrocytes.
Reticulocyte count EDTA–whole blood (5 μl) was added to1 ml Retic-COUNT (Thiazole orange) reagent from BectonDickinson. Samples were stained for 30 min at roomtemperature, and flow cytometry was performed accordingto the manufacturer’s instructions. Forward scatter (FSC),side scatter (SSC), and thiazole orange-fluorescence intensity(in FL-1) of the blood cells were determined. The number ofRetic-COUNT positive reticulocytes was expressed as thepercentage of the total gated erythrocyte populations. Gatingof erythrocytes was achieved by analysis of FSC vs. SSC dotplots using CellQuest software.
Measurement of osmotic resistance In a 96-well plate,2 μl erythrocyte pellets was exposed (2 min) tophosphate-buffered saline (PBS) solutions of decreasingosmolarity as prepared by mixing a PBS solution with adefined volume of distilled water. After centrifugation(500×g for 5 min), the Hb concentration of the super-natants was determined photometrically (at 405 nm) in anELISA reader.
In vivo experiments
α-Toxin-induced hemolysis The hemoglobin content ofthe plasma at 0 and 30 min after an i.v. administrationof α-toxin (1,800 U/kg body weight in PBS) wasdetermined using a commercial immunoperoxidase assay(mouse hemoglobin ELISA, Dunn Labortechnik,Asbach, Germany) according to the manufacturer’sinstructions.
Phyenylhydrazine-dependent hemolysis Hemolysis wasevoked by a single i.v. injection of 5 mg/kg body weightphenylhydrazine (Sigma) into another group of wild-typeand knockout mice. Before and 10 min or 12 weeks afterinduction of hemolysis, a blood specimen was collected,and the hematocrit was determined by centrifugation incapillaries.
In vivo clearance Erythrocytes from KCa3.1−/− and wild-
type mice were washed (NaCl solution), oxidized (seeabove), washed, and centrifuged, and the pellet was labeledwith 5-carboxyfluoresceine diacetate succinimidylester(5 μM; CFSE; Molecular Probes, Eugene, OR, USA).After washing twice and resuspending in NaCl solution(50% hematocrit), cell suspensions were re-injected intra-venously into the same mice, and clearance of CFSE-positive cells from peripheral blood was assessed by flowcytometry in FL-1.
Malaria infection For mouse infection, Plasmodium bergheiAnka-parasitized murine RBCs (2×106) as counted by flow
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1033

cytometry upon staining with the DNA/RNA-specific dyesyto16 (see below) were injected intraperitoneally intowild-type and KCa3.1
−/− mice. Parasitemia was determineddaily by syto16 staining (30 nM; Molecular Probes,Göttingen, Germany) and flow cytometry (FL-1) uponresuspending the erythrocytes in NaCl solution. In dotblots, non-infected cells, erythrocytes infected with earlystages (ring stages) and cells infected with late stages(trophozoites, schizonts) were differentiated by their low(background), intermediate, and high syto16 fluorescence,respectively (see Fig. 7a). On days 10–15 after infection,the parasite stage-dependent erythrocyte size was deter-mined by forward scatter. On infection day 18, the plasmaconcentrations of free hemoglobin, haptoglobin, and biliru-bin were determined by ELISA (mouse haptoglobinELISA, Kamiya Biomedical Company, Seattle, USA;bilirubin: photometric method (FUJI FDC 3500i, Sysmex,Norsted, Germany)) according to the manufacturer’sinstructions.
Results
Erythrocyte Gardos channels are thought to be encoded bythe KCNN4 gene. To directly test this assumption, whole-cell currents were recorded in erythrocytes from KCa3.1
−/−
and wild-type mice with KCl/K-gluconate solution in thepipette and NaCl Ringer solution in the bath. Records wereobtained before and after Ca2+-permeabilizing the erythro-cyte membrane by the addition of ionomycin (10 μM) tothe bath solution. In wild-type but not in KCa3.1
−/−
erythrocytes, ionomycin stimulated channel-mediated cur-rent transitions (Fig. 1a) that superimposed the very lowresting erythrocyte whole-cell currents (Fig. 1a, inset). Theionomycin-stimulated channels generated outward currenttransitions, which were apparent at voltages of greater thanor equal to −40 mV (Fig. 1b). Extrapolation of the meancurrent–voltage (I–V) relationship of the unitary currenttransitions (Fig. 1c) indicated a reversal potential at K+
electrochemical equilibrium (EK) and, thus, a K+ selectivity
for the channels. The slope of the I–V curve (as calculatedby linear regression between −20 and +50 mV) suggested aunitary conductance of 10±3 pS (n=4; Fig. 1c, opencircles) for the outward current.
When recorded with KCl bath and K-gluconate pipettesolutions (data from independent experiments shown inFig. 2), the mean I–V relationship of the channel amplitudeagain reversed at EK (around 0 mV) and exhibited inwardrectification with an inward and outward conductance (ascalculated by linear regression between −110 and −50 mVand between +30 and +70 mV) of 43±3 and 16±3 pS,respectively (n=5; Fig. 1c, closed triangles). Since for
human KCa3.1 similar inward rectification and conductan-ces have been reported [63], the present data stronglysuggest that the Ca2+-dependent K+-selective channel ofwild-type erythrocytes is encoded by KCNN4.
In the experiments on wild-type erythrocytes of Fig. 1a,the ionomycin-stimulated channel activity was highlyvariable between the individual cells but significantlydifferent from 0 (p=0.03; two-tailed one-sample t test;Fig. 1d, open circles). The observed low mean openprobability (nPo; n=number of active channels and Po=mean open probability of the individual channels) in therange of 3 (n=10; Fig 1d) of the Ca2+-permeabilized wild-type erythrocytes hinted to a very low number of functionalchannels per cell. In KCa3.1
−/− erythrocytes (n=8), in sharpcontrast, ionomycin did not increase outward currentswhich were significantly different from zero (p=0.8; two-tailed one-sample t test; Fig. 1d, closed triangles), indica-tive of absent K+ channel activity suggesting that KCa3.1 isthe only Ca2+-activated K+ channel subtype expressed bymurine erythrocytes.
To further assess the number of functional Gardoschannels, wild-type and KCa3.1
−/− erythrocytes werewhole-cell-recorded before and 5 min after bath additionof the KCa channel opener 1-EBIO (100 μM). Erythrocytesreportedly express nonselective cation channels with anabout twofold higher permeability for K+ over Na+ [64].Accordingly, the I–V curve recorded with NaCl bath and K-gluconate pipette solution reversed in non-stimulated wild-type and KCa3.1
−/− erythrocytes at slightly negativevoltages (−13±2 and −17±2 mV, n=4–5, Fig. 2b, leftplots, open circles). Stimulation with 1-EBIO did not affectthe whole-cell currents of both genotypes (Fig. 2b, rightplots, open circles) except for a small, not quite significantshift of the reversal potential towards EK in the wild-typeerythrocytes (to −23±4 mV). This small change in reversalpotential together with an apparently absent increase inoutward current suggests—at least under the chosenexperimental conditions—an only minor fractional KCa
current in wild-type erythrocytes.In KCl bath solution, inward whole-cell conductances
(between −110 and −50 mV) were similar in non-stimulatedwild-type and KCa3.1
−/− erythrocytes (135±19 vs 102±16 pS, n=5, Fig. 2b, left plots, closed triangles). Theinward conductance of 1-EBIO-stimulated wild-type eryth-rocytes, in contrast, exceeded that of stimulated KCa3.1
−/−
erythrocytes (225±44 pS, n=5 vs. 83±20 pS, n=5; p≤0.05,Welch-corrected two-tailed t test; Fig. 2a, b, right plots,closed triangles). Given the 45 pS inward conductance ofindividual channels (see above), the 1-EBIO-stimulatedconductance fraction of wild-type cells (about 100 pS) isequivalent to an overall channel activity of nPo=2, asimilarly low value as determined by the ionomycinexperiments of Fig. 1. Taken together, the electrophysio-
1034 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044
logical data confirm that KCa3.1−/− is functionally
expressed in mouse erythrocytes albeit in apparently verylow copy number and, secondly, that KCa3.1
−/− is the onlyKCa channel type in these cells.
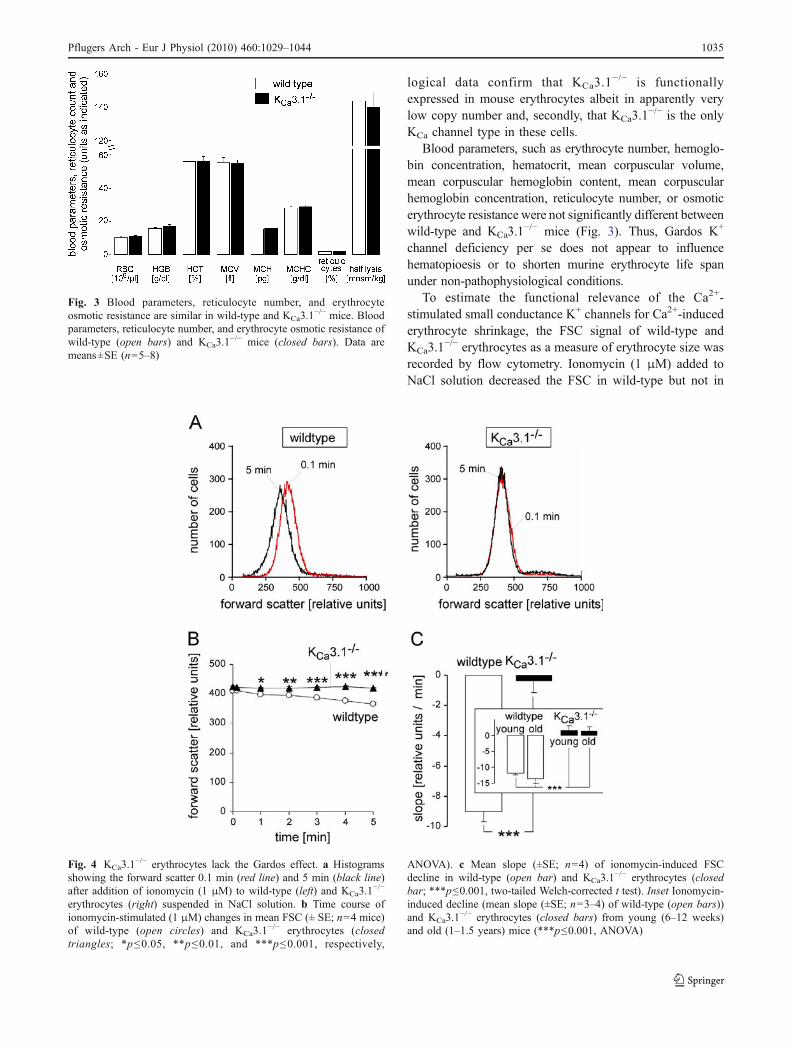
Blood parameters, such as erythrocyte number, hemoglo-bin concentration, hematocrit, mean corpuscular volume,mean corpuscular hemoglobin content, mean corpuscularhemoglobin concentration, reticulocyte number, or osmoticerythrocyte resistance were not significantly different betweenwild-type and KCa3.1
−/− mice (Fig. 3). Thus, Gardos K+
channel deficiency per se does not appear to influencehematopioesis or to shorten murine erythrocyte life spanunder non-pathophysiological conditions.
To estimate the functional relevance of the Ca2+-stimulated small conductance K+ channels for Ca2+-inducederythrocyte shrinkage, the FSC signal of wild-type andKCa3.1
−/− erythrocytes as a measure of erythrocyte size wasrecorded by flow cytometry. Ionomycin (1 μM) added toNaCl solution decreased the FSC in wild-type but not in
Fig. 3 Blood parameters, reticulocyte number, and erythrocyteosmotic resistance are similar in wild-type and KCa3.1
−/− mice. Bloodparameters, reticulocyte number, and erythrocyte osmotic resistance ofwild-type (open bars) and KCa3.1
−/− mice (closed bars). Data aremeans±SE (n=5–8)
Fig. 4 KCa3.1−/− erythrocytes lack the Gardos effect. a Histograms
showing the forward scatter 0.1 min (red line) and 5 min (black line)after addition of ionomycin (1 μM) to wild-type (left) and KCa3.1
−/−
erythrocytes (right) suspended in NaCl solution. b Time course ofionomycin-stimulated (1 μM) changes in mean FSC (± SE; n=4 mice)of wild-type (open circles) and KCa3.1
−/− erythrocytes (closedtriangles; *p≤0.05, **p≤0.01, and ***p≤0.001, respectively,
ANOVA). c Mean slope (±SE; n=4) of ionomycin-induced FSCdecline in wild-type (open bar) and KCa3.1
−/− erythrocytes (closedbar; ***p≤0.001, two-tailed Welch-corrected t test). Inset Ionomycin-induced decline (mean slope (±SE; n=3–4) of wild-type (open bars))and KCa3.1
−/− erythrocytes (closed bars) from young (6–12 weeks)and old (1–1.5 years) mice (***p≤0.001, ANOVA)
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1035
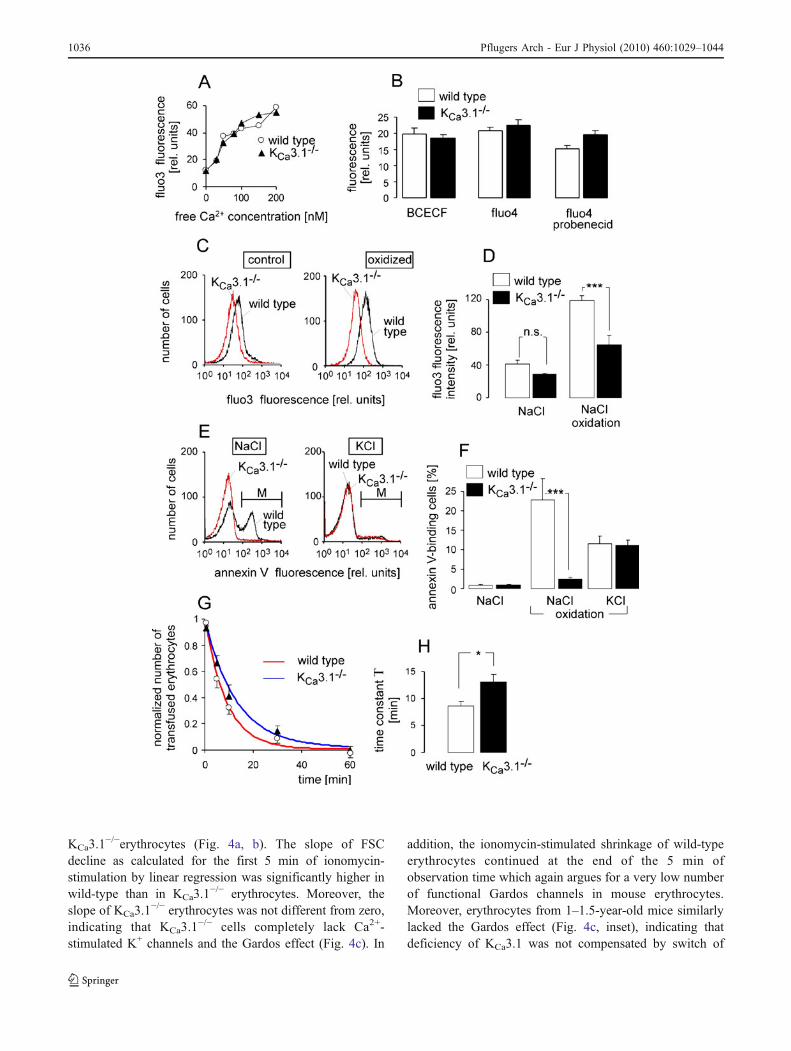
KCa3.1−/−erythrocytes (Fig. 4a, b). The slope of FSC
decline as calculated for the first 5 min of ionomycin-stimulation by linear regression was significantly higher inwild-type than in KCa3.1
−/− erythrocytes. Moreover, theslope of KCa3.1
−/− erythrocytes was not different from zero,indicating that KCa3.1
−/− cells completely lack Ca2+-stimulated K+ channels and the Gardos effect (Fig. 4c). In
addition, the ionomycin-stimulated shrinkage of wild-typeerythrocytes continued at the end of the 5 min ofobservation time which again argues for a very low numberof functional Gardos channels in mouse erythrocytes.Moreover, erythrocytes from 1–1.5-year-old mice similarlylacked the Gardos effect (Fig. 4c, inset), indicating thatdeficiency of KCa3.1 was not compensated by switch of
1036 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044

KCa channel expression during erythropoiesis with increas-ing mouse age.
The suicidal death program is thought to prevent colloidosmotic hemolysis of injured erythrocytes [65]. Erythrocyteshrinkage extends the survival of the dying erythrocyte, andbreakdown of the phospholipid asymmetry fosters itsrecognition and clearance by phagocytes [27]. To definethe KCa3.1 channel function for Ca2+-induced suicidaldeath program, wild-type and KCa3.1
−/− erythrocytes weresubjected to oxidative stress which reportedly activates Ca2+-permeable cation channels in erythrocytes [66]. To assesssuicidal erythrocyte death, cytosolic free Ca2+ ([Ca2+]i)concentration (Fig. 5a–c), and breakdown of the phospho-lipid asymmetry (Fig 5d, e) was measured in flow cytometryby the use of the Ca2+-sensitive dye fluo3, andphosphatidylserine-binding by use of a fluorescent annexinV protein, respectively. To test for fluo3 dye loading duringthe Ca2+ measurements, wild-type and KCa3.1
−/− erythro-cytes were suspended in media with various EGTA-bufferedfree Ca2+ concentrations, and the erythrocyte membrane waspermeabilized for Ca2+ with ionomycin (1 μM). As shownin Fig. 5a, the extracellular Ca2+-dependent fluo3 fluores-
cence intensity did not differ between wild-type andKCa3.1
−/− erythrocytes, suggesting equal dye loading andCa2+-dependent fluorescence in erythrocytes from bothgenotypes. Dye loading depends on esterase activity andon dye export capability e.g., via multidrug resistance(mdr) transport. To test for differences in these parameters,wild-type and KCa3.1
−/− erythrocytes were incubated withthe acetoxymethyl esters of the pH- and Ca2+-sensitivedye BCECF and fluo4, respectively (the latter dye wasincubated in the absence and presence of the mdr inhibitorprobenecid, 5 mM). As shown in Fig. 5b (left and middle),the BCECF and fluo4 fluorescence did not differ betweenwild-type and KCa3.1
−/− erythrocytes. In addition, prop-benecid did not change the fluo4 fluorescence (Fig. 5b,right), which argues against a significant export offluorescence dyes by mouse erythrocytes. Taken together,these control experiments strongly suggest similar loadingof wild-type and KCa3.1
−/− erythrocytes with fluo3.In non-oxidized wild-type erythrocytes, fluo3 fluores-
cence intensity was not significantly higher than in non-oxidized KCa3.1
−/− cells (Fig. 5c, left and Fig. 5d, first andsecond bars), suggesting a similar steady state [Ca2+]i inboth genotypes. Oxidative stress was followed by an almosttripling of the fluo3 fluorescence in wild-type erythrocytes(Fig. 5c, right and Fig. 5d), an effect significantly bluntedin KCa3.1
−/− cells (Fig. 5d).Annexin V-binding in flow cytometry demonstrated
breakdown of the membrane phospholipid asymmetry inonly about 1% of non-oxidized wild-type and KCa3.1
−/−
erythrocytes incubated in NaCl solution (Fig. 5f, first andsecond bar). Oxidation in NaCl solution dramaticallyincreased the percentage of annexin V-binding cells inwild-type erythrocytes (Fig. 5e, left and Fig. 5f, first vs.third bar) whereas having far less effect in KCa3.1
−/− cells(Fig. 5e, left and Fig. 5f, second vs. fourth bar). To testwhether this difference was due to Gardos-mediatedmembrane hyperpolarization and subsequent erythrocyteshrinkage, cells were oxidized in KCl solution. Ascompared with NaCl solution, oxidation in KCl solutiondecreased the percentage of annexin V-binding wild-type andincreased the percentage of annexin V-binding KCa3.1
−/−
erythrocytes, respectively, and, most importantly, abolishedthe difference in oxidation-stimulated annexin V-bindingbetween the genotypes (Fig. 5e, right and Fig. 5f, fifth vs.sixth bar). Taken together, the data suggest that Gardos K+
channel-mediated membrane hyperpolarization and/or eryth-rocyte shrinkage facilitate oxidative stress-induced Ca2+
entry and subsequent breakdown of the phospholipidasymmetry and, thus, contributes to the oxidative stress-triggered suicidal death program. As a consequence, suicidalerythrocyte death was impaired in KCa3.1
−/− erythrocytes.Exposure of phosphatidylserine at the erythrocyte sur-
face promotes the clearance of the dying erythrocyte [67].
R Fig. 5 Oxidized KCa3.1−/− erythrocytes execute an incomplete
suicidal death program and are less rapidly cleared from peripheralblood. a Dependence of the fluo3 fluorescence on [Ca2+]i. Thecalibration curve was recorded in wild-type (open circles) and withKCa3.1
−/− erythrocytes (closed triangles) with various EGTA-bufferedextracellular Ca2+-concentrations in Ca2+-permeabilized (10 μM ion-omycin) erythrocytes as described in the “Materials and methods”section. b Mean (±SE; n=12) BCECF (left) and fluo4 (middle, right)fluorescence recorded in the absence (left, middle) and presence(right) of the mdr inhibitor probenecid (5 mM). c Histograms showingCa2+-specific fluo3 fluorescence intensity of wild-type (black line) andKCa3.1
−/− erythrocytes (red line) as recorded by flow cytometry in theabsence (left) and presence of oxidative stress (0.1 mM tert-butylhydroperoxide, t-BHP, for 45 min; right). d Mean fluo3fluorescence intensity (± SE; n=8) of wild-type (open bars) andKCa3.1
−/− erythrocytes (closed bars) incubated as in a under controlconditions (first and second bars) or after oxidative stress (third andfourth bars). e Histogram of annexin V-binding to oxidized (0.1 mMt-BHP for 45 min) wild-type (black line) and KCa3.1
−/− erythrocytes(red line) incubated in NaCl (left) or in KCl solution (right). M is themarker indicating the population of cells considered as annexin V-binding. f Mean percentage (±SE; n=5–9) of annexin V-binding wild-type (open bars) and KCa3.1
−/− erythrocytes (closed bars) incubated(45 min at 37°C) in the absence (first and second bars) or presence ofoxidative stress (third to sixth bars) in NaCl (first to fourth bars) orKCl solution (fifth and sixth bars; ***p≤0.001, n.s. not significantlydifferent, ANOVA). g Disappearance of oxidized erythrocytes fromperipheral blood. Wild-type (open circles) and KCa3.1
−/− erythrocytes(closed triangles) were in vitro oxidized, labeled with the fluorescencedye CFSE, and intravenously injected into the same mice. The meannumber (± SE, n=8) of erythrocytes is normalized to the initiallytransfused number and plotted against the time. The time course ofdisappearance of wild-type and KCa3.1
−/− erythrocytes is fitted by amonoexponential decay (the fitted curves shown by red and bluelines), the calculated time constants Tau (±SE, n=5) of the averagedcurves are given for wild-type and KCa3.1
−/− mice by the open andclosed bars (h), respectively (*p≤0.05, two-tailed t test)
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1037
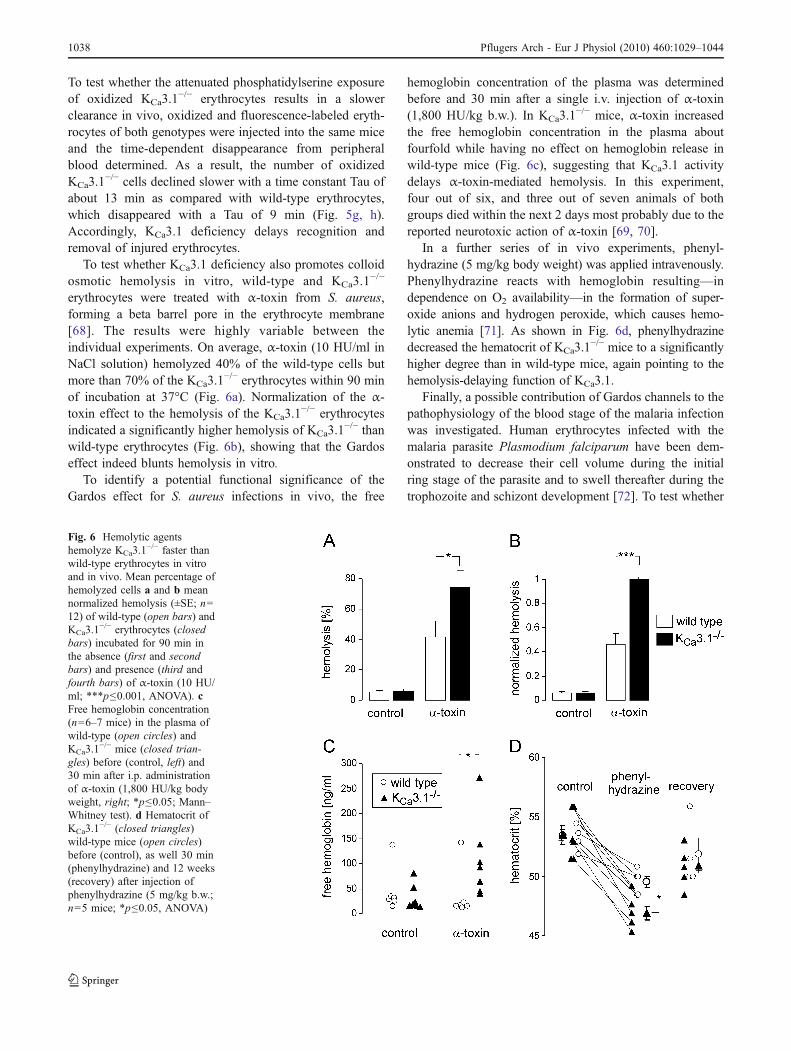
To test whether the attenuated phosphatidylserine exposureof oxidized KCa3.1
−/− erythrocytes results in a slowerclearance in vivo, oxidized and fluorescence-labeled eryth-rocytes of both genotypes were injected into the same miceand the time-dependent disappearance from peripheralblood determined. As a result, the number of oxidizedKCa3.1
−/− cells declined slower with a time constant Tau ofabout 13 min as compared with wild-type erythrocytes,which disappeared with a Tau of 9 min (Fig. 5g, h).Accordingly, KCa3.1 deficiency delays recognition andremoval of injured erythrocytes.
To test whether KCa3.1 deficiency also promotes colloidosmotic hemolysis in vitro, wild-type and KCa3.1
−/−
erythrocytes were treated with α-toxin from S. aureus,forming a beta barrel pore in the erythrocyte membrane[68]. The results were highly variable between theindividual experiments. On average, α-toxin (10 HU/ml inNaCl solution) hemolyzed 40% of the wild-type cells butmore than 70% of the KCa3.1
−/− erythrocytes within 90 minof incubation at 37°C (Fig. 6a). Normalization of the α-toxin effect to the hemolysis of the KCa3.1
−/− erythrocytesindicated a significantly higher hemolysis of KCa3.1
−/− thanwild-type erythrocytes (Fig. 6b), showing that the Gardoseffect indeed blunts hemolysis in vitro.
To identify a potential functional significance of theGardos effect for S. aureus infections in vivo, the free
hemoglobin concentration of the plasma was determinedbefore and 30 min after a single i.v. injection of α-toxin(1,800 HU/kg b.w.). In KCa3.1
−/− mice, α-toxin increasedthe free hemoglobin concentration in the plasma aboutfourfold while having no effect on hemoglobin release inwild-type mice (Fig. 6c), suggesting that KCa3.1 activitydelays α-toxin-mediated hemolysis. In this experiment,four out of six, and three out of seven animals of bothgroups died within the next 2 days most probably due to thereported neurotoxic action of α-toxin [69, 70].
In a further series of in vivo experiments, phenyl-hydrazine (5 mg/kg body weight) was applied intravenously.Phenylhydrazine reacts with hemoglobin resulting—independence on O2 availability—in the formation of super-oxide anions and hydrogen peroxide, which causes hemo-lytic anemia [71]. As shown in Fig. 6d, phenylhydrazinedecreased the hematocrit of KCa3.1
−/− mice to a significantlyhigher degree than in wild-type mice, again pointing to thehemolysis-delaying function of KCa3.1.
Finally, a possible contribution of Gardos channels to thepathophysiology of the blood stage of the malaria infectionwas investigated. Human erythrocytes infected with themalaria parasite Plasmodium falciparum have been dem-onstrated to decrease their cell volume during the initialring stage of the parasite and to swell thereafter during thetrophozoite and schizont development [72]. To test whether
Fig. 6 Hemolytic agentshemolyze KCa3.1
−/− faster thanwild-type erythrocytes in vitroand in vivo. Mean percentage ofhemolyzed cells a and b meannormalized hemolysis (±SE; n=12) of wild-type (open bars) andKCa3.1
−/− erythrocytes (closedbars) incubated for 90 min inthe absence (first and secondbars) and presence (third andfourth bars) of α-toxin (10 HU/ml; ***p≤0.001, ANOVA). cFree hemoglobin concentration(n=6–7 mice) in the plasma ofwild-type (open circles) andKCa3.1
−/− mice (closed trian-gles) before (control, left) and30 min after i.p. administrationof α-toxin (1,800 HU/kg bodyweight, right; *p≤0.05; Mann–Whitney test). d Hematocrit ofKCa3.1
−/− (closed triangles)wild-type mice (open circles)before (control), as well 30 min(phenylhydrazine) and 12 weeks(recovery) after injection ofphenylhydrazine (5 mg/kg b.w.;n=5 mice; *p≤0.05, ANOVA)
1038 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044
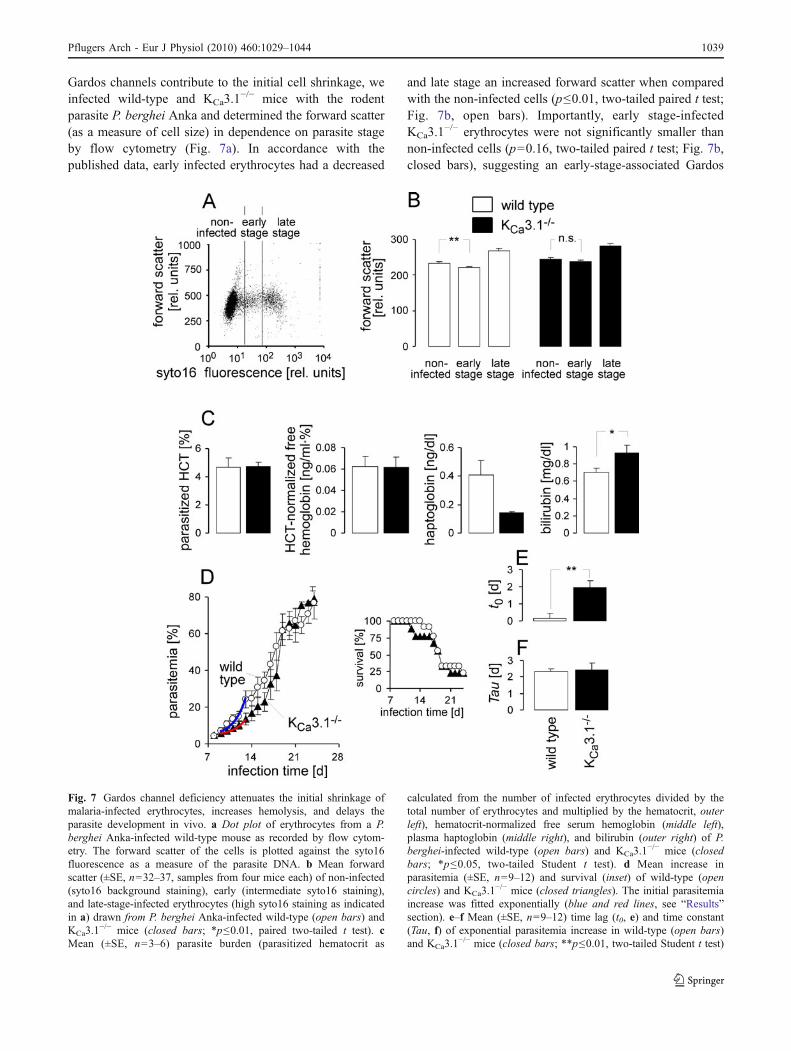
Gardos channels contribute to the initial cell shrinkage, weinfected wild-type and KCa3.1
−/− mice with the rodentparasite P. berghei Anka and determined the forward scatter(as a measure of cell size) in dependence on parasite stageby flow cytometry (Fig. 7a). In accordance with thepublished data, early infected erythrocytes had a decreased
and late stage an increased forward scatter when comparedwith the non-infected cells (p≤0.01, two-tailed paired t test;Fig. 7b, open bars). Importantly, early stage-infectedKCa3.1
−/− erythrocytes were not significantly smaller thannon-infected cells (p=0.16, two-tailed paired t test; Fig. 7b,closed bars), suggesting an early-stage-associated Gardos
Fig. 7 Gardos channel deficiency attenuates the initial shrinkage ofmalaria-infected erythrocytes, increases hemolysis, and delays theparasite development in vivo. a Dot plot of erythrocytes from a P.berghei Anka-infected wild-type mouse as recorded by flow cytom-etry. The forward scatter of the cells is plotted against the syto16fluorescence as a measure of the parasite DNA. b Mean forwardscatter (±SE, n=32–37, samples from four mice each) of non-infected(syto16 background staining), early (intermediate syto16 staining),and late-stage-infected erythrocytes (high syto16 staining as indicatedin a) drawn from P. berghei Anka-infected wild-type (open bars) andKCa3.1
−/− mice (closed bars; *p≤0.01, paired two-tailed t test). cMean (±SE, n=3–6) parasite burden (parasitized hematocrit as
calculated from the number of infected erythrocytes divided by thetotal number of erythrocytes and multiplied by the hematocrit, outerleft), hematocrit-normalized free serum hemoglobin (middle left),plasma haptoglobin (middle right), and bilirubin (outer right) of P.berghei-infected wild-type (open bars) and KCa3.1
−/− mice (closedbars; *p≤0.05, two-tailed Student t test). d Mean increase inparasitemia (±SE, n=9–12) and survival (inset) of wild-type (opencircles) and KCa3.1
−/− mice (closed triangles). The initial parasitemiaincrease was fitted exponentially (blue and red lines, see “Results”section). e–f Mean (±SE, n=9–12) time lag (t0, e) and time constant(Tau, f) of exponential parasitemia increase in wild-type (open bars)and KCa3.1
−/− mice (closed bars; **p≤0.01, two-tailed Student t test)
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1039

channel activation with subsequent loss of KCl and water.To test for an influence of the Gardos channel on themalaria-associated hemolysis of infected and/or non-infected bystander erythrocytes, the plasma concentrationsof free hemoglobin, haptoglobin, and bilirubin in groups ofn=6 and n=3 P. berghei-infected wild-type and KCa3.1
−/−
mice were analyzed, respectively. Groups with identicalmean parasite burden (Fig. 7c, outer left) were compared asdefined by the parasitized hematocrit (i.e., number ofinfected erythrocytes divided by number of total erythro-cytes multiplied by hematocrit). The free plasma hemoglo-bin did not differ between these groups when normalized tothe hematocrit (Fig. 7c, middle left). The concentration ofplasma haptoglobin tended to be lower, a difference almostreaching statistical significance (p=0.053, two-tailed,Welch-corrected t test) and that of bilirubin was signifi-cantly (p≤0.05; two-tailed, Student t test) higher inKCa3.1
−/− mice compared with wild-type mice (Fig. 7c,middle and outer right) pointing to a slightly pronouncedhemolysis in KCa3.1
−/− mice.To further study the functional significance of Gardos
channel activity for malaria infection, the increase inparasitemia of P. berghei Anka-infected wild-type andKCa3.1
−/− mice was monitored. As shown in Fig. 7d, theinitial phase of the parasitemia increase was fitted mono-exponentially (red and blue line in Fig. 7d) using thefollowing equation
y ¼ y0þ A � e t�t0ð Þ=Tau
(with y, y0, A, t, t0, and Tau being the percentage of syto16-positive cells, the percentage of the initially syto16-positivecells at the time of infection, the initial number of P.berghei Anka-infected cells shortly after infection anderythrocyte invasion, the infection time in days, the timedelay until onset of exponential growth in days, and thetime constant of monoexponential growth, respectively). Asshown in Fig. 7e, in KCa3.1
−/− but not in wild-type mice,the onset of exponential growth was delayed by about2 days. Thereafter, the parasites amplified with identicalkinetics (Fig. 7f) and survival did not differ between bothgenotypes (Fig. 7d, inset).
Discussion
A recent study on a different KCa3.1-deficient mouse model(also in a hybrid SV129/C57Bl6 background) showed areduced filterability, osmotic tolerance, and volume ofuntreated and Ca2+-permeabilized KCa3.1-deficient eryth-rocytes, respectively, when compared with the wild-typeerythrocytes [73]. The present study extends the recentwork by (1) directly showing KCa3.1 K+ channel activity
and characteristics in murine erythrocytes, (2) demonstrat-ing time-dependent KCa3.1-mediated shrinkage of Ca2+-permeabilized erythrocytes, (3) and defining KCa3.1-dependent Ca2+ entry into oxidized erythrocytes leadingto KCa3.1-dependent suicidal erythrocyte death and invivo clearance of the oxidatively injured erythrocytes. Inaddition, the present study demonstrates that KCa3.1delays the hemolysis of murine erythrocytes challengedby bacterial α-toxin or the oxidant phenylhydrazine invitro and/or in vivo. Moreover, our study reveals KCa3.1channel activation in the ring stage of infection by themurine malaria parasite P. berghei Anka, albeit withoutany significance for the course of infection.
Our data directly show that KCa3.1 is the only Ca2+-activated K+ channel type in mouse erythrocytes sinceKCa3.1-deficient erythrocytes lacked Ca2+-activated K+
channel activity and erythrocyte shrinkage. This is consistentwith a previous study demonstrating almost absent Ca2+-stimulated clotrimazole-sensitive 86Rb uptake by erythro-cytes from KCa3.1-deficient mice [74]. Similarly, humanreticulocytes as well as those differentiated in vitro fromerythroid progenitor cells contain KCa3.1 mRNA, but not themRNA of the small conductance Ca2+-activated K+ channel[2]. Ca2+-permeabilized human erythrocytes reportedlyexhibit a mean open probability of the Gardos channel ofnPo=3 [75]. In the present study, nPo of the Gardos channelin Ca2+-permeabilized mouse erythrocytes was highlyvariable between individual cells. On average, however,nPo was very similar to that reported in human erythrocytes,suggesting a similar Gardos channel expression in humanand murine erythrocytes.
The present observations reveal that blood parameters donot differ between wild-type and KCa3.1
−/− mice. This is incontrast with the blood parameters described for the otherKCa3.1-deficient mouse model [73] in which KCa3.1
−/−
erythrocytes displayed a larger mean corpuscular volumeand a mild macrocytosis. Albeit of identical background,this discrepancy might result from genetic differencesbetween the two mouse models. The other [73] but notour knockout mouse model carries the neomycin resistantgene (in place of exon 4 of the KCa3.1 gene [76]), whichmight impair erythrocyte viability.
Since in our model, KCa3.1-deficient erythrocytesapparently do not up-regulate other Ca2+-activated K+
channel types, mature mouse erythrocytes do not requireGardos K+ channel activity under unstressed conditions.Mature erythrocytes of other species, such as sheep ordonkey, even completely lack Ca2+-activated K+ channels[77], indicating that the function of mature mammalianerythrocytes does not critically depend on Gardos channelactivity.
The present study suggests a protective role of theKCa3.1 channel against hemolysis by phenylhydrazine.
1040 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044

Phenylhydrazine causes membrane damage by autooxida-tion followed by oxidation of membrane proteins andhemoglobin [78], possibly also by generation of superoxideradicals and hydrogen peroxide [71, 79]. Our results showthat phenylhydrazine-induced hemolysis is, to some extent,blunted in wild-type as compared with KCa3.1
−/− mice(Fig. 6d). In addition, a protective role of the KCa3.1,channel against α-toxin from the hemolytic bacteria S.aureus is suggested by the present data. α-Toxin belongs tothe cytolysins, which, similar to hemolysins of otherbacteria, perforate the plasma membrane of erythrocytesand other cells. It is known to be a major pathogeneticfactor during S. aureus infections [80]. In the present study,KCa3.1-mediated shrinkage transiently delayed hemolysisof α-toxin-perforated erythrocytes in vitro (Fig. 6a, b) andin vivo (Fig. 6c). Moreover, the Gardos K+ channel-mediated hyperpolarization of the erythrocyte membraneincreases the intracellularly directed driving force for Ca2+.Accordingly, [Ca2+]i was significantly reduced in KCa3.1
−/−
erythrocytes compared with KCa3.1+/+ upon oxidative
stress, a condition known to activate the Gardos K+ channel[65]. Elevated [Ca2+]i together with erythrocyte shrinkagestimulate breakdown of the phospholipid asymmetry andexposure of phosphatidylserine at the erythrocyte surface[81]. The functional significance of the Gardos effect forthis process is clearly shown by almost absent phosphatidyl-serine exposure in oxidized KCa3.1
−/− erythrocytes (Fig. 5).Phosphatidylserine exposure has been demonstrated to be aneat-me signal and to foster recognition and clearance ofinjured erythrocytes by macrophages [67]. Accordingly,oxidized wild-type erythrocytes were cleared faster thanKCa3.1
−/− erythrocytes (Fig. 5g). Since KCa3.1−/− erythro-
cytes oxidized in KCl solution displayed more phosphati-dylserine exposure than those oxidized in NaCl solution(Fig. 5f), it must be considered that other mechanisms maycontribute to annexin V-binding at unphysiologically highK+ concentration.
In summary, the present results suggest that KCa3.1-mediated short extension of erythrocyte survival in concertwith the observed KCa3.1-dependent exposure of phospha-tiylserine facilitated the in vivo clearance of injurederythrocytes prior to their hemolysis.
Activation of the Gardos channel is detrimental in sicklecell disease [82] or beta thalassemia [83], where it promoteserythrocyte deformation and limits erythrocyte survival,respectively. However, activation of the Gardos channel maybe advantageous in several conditions affecting the integrityof erythrocytes, such as septicemias. Most importantly, inview of the high incidence and mortality of septicemias [84],advantages appear to prevail the disadvantages of Gardoschannel expression in human erythrocytes.
Ca2+ ATPase pump activity of P. falciparum-infectednormal human erythrocytes reportedly remains functional
[85], and Ca2+ entry pathways reportedly are not stimulated[86]. Therefore, the cytosolic free Ca2+ concentrationremains below the threshold of Gardos channel activation[86]. In contrast, malaria-infected sickle cell trait (HbA/S)erythrocytes exhibit an elevated Ca2+ permeability in thering stage of infection, which triggers the suicidal deathprogram including cell shrinkage and phosphatidylserineexposure [67]. Premature suicidal death of infected HbA/Serythrocytes (that most probably involves Gardos channel-mediated erythrocyte shrinkage) fosters the clearance ofinfected cells and thus contributes to the mechanismsconferring the partial malaria resistance of individuals withsickle cell trait [67]. Malaria-infected normal mouseerythrocytes exhibit elevated Ca2+ permeabilities in thering stage of infection [67]. Consistently, the present studydemonstrated Gardos channel activation in those erythro-cytes (Fig. 7) which, however, did—except a slightlydelayed onset of exponential parasitemia increase—notalter the course of the disease. The plasma concentrationof haptoglobin and bilirubin suggest a slightly higherhemolysis in malaria-infected KCa3.1
−/− mice than ininfected wild-type mice. The free hemoglobin concentrationdid not differ between both genotypes, suggesting that thehemolysis rates did not exceed the hemoglobin bindingcapacities of haptoglobin. It must be kept in mind thatKCa3.1 deficiency is associated with blunted IgE-mediatedactivation of mast cells and may therefore affect theimmune system [54]. Moreover, KCa3.1
−/− mice suffer fromendothelial dysfunction [87]. Hence, the observed slightlyincreased hemolysis of malaria-infected KCa3.1
−/− micemay not only be the consequence of erythrocytic KCa3.1deficiency.
In conclusion, KCa3.1 is expressed in murine erythrocytesand mediates Ca2+-stimulated shrinkage and phosphatidyl-serine exposure. During normal unstressed erythrocyte life(i.e., in the absence of pathological hemolytic or oxidatingagents), KCa3.1 remains inactive. Under pathophysiologicalconditions such as murine malaria or S. aureus infection,however, KCa3.1 activity delays hemolysis and promotesrecognition and clearance of the dying erythrocyte.
Acknowledgements We thank Clement Kabagema for mousegenotyping and Syed Qadri for technical support. This study wassupported by the Deutsche Forschungsgemeinschaft Hu781/4-3 andLa 315/13-3, and the Carl-Zeiss-Stiftung.
References
1. Gardos G (1958) The function of calcium in the potassiumpermeability of human erythrocytes. Biochim Biophys Acta30:653
2. Hoffman JF, Joiner W, Nehrke K, Potapova O, Foye K, WickremaA (2003) The hSK4 (KCNN4) isoform is the Ca2+-activated K+
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1041

channel (Gardos channel) in human red blood cells. Proc NatlAcad Sci USA 100:7366–7371
3. Lang PA, Kaiser S, Myssina S, Wieder T, Lang F, Huber SM(2003) Role of Ca2+-activated K+ channels in human erythrocyteapoptosis. Am J Physiol Cell Physiol 285:C1553–C1560
4. Grygorczyk R, Schwarz W (1983) Properties of the CA2+-activated K+ conductance of human red cells as revealed by thepatch-clamp technique. Cell Calcium 4:499–510
5. Christophersen P (1991) Ca2(+)-activated K+ channel fromhuman erythrocyte membranes: single channel rectification andselectivity. J Membr Biol 119:75–83
6. Leinders T, van Kleef RG, Vijverberg HP (1992) Single Ca(2+)-activated K+ channels in human erythrocytes: Ca2+ dependenceof opening frequency but not of open lifetimes. Biochim BiophysActa 1112:67–74
7. Del Carlo B, Pellegrini M, Pellegrino M (2002) Calmodulinantagonists do not inhibit IK(Ca) channels of human erythrocytes.Biochim Biophys Acta 1558:133–141
8. Kaushal V, Koeberle PD, Wang Y, Schlichter LC (2007) The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microgliaactivation and nitric oxide-dependent neurodegeneration. J Neurosci27:234–244
9. Brugnara C, Armsby CC, De Franceschi L, Crest M, Euclaire MF,Alper SL (1995) Ca(2+)-activated K+ channels of human andrabbit erythrocytes display distinctive patterns of inhibition byvenom peptide toxins. J Membr Biol 147:71–82
10. Brugnara C, De Franceschi L, Alper SL (1993) Ca(2+)-activatedK+ transport in erythrocytes. Comparison of binding andtransport inhibition by scorpion toxins. J Biol Chem 268:8760–8768
11. Sugg EE, Garcia ML, Reuben JP, Patchett AA, Kaczorowski GJ(1990) Synthesis and structural characterization of charybdotoxin,a potent peptidyl inhibitor of the high conductance Ca2(+)-activated K+ channel. J Biol Chem 265:18745–18748
12. Brugnara C, de Franceschi L, Alper SL (1993) Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickleerythrocytes by clotrimazole and other imidazole derivatives. JClin Invest 92:520–526
13. Hill K, McNulty S, Randall AD (2004) Inhibition of TRPM2channels by the antifungal agents clotrimazole and econazole.Naunyn Schmiedebergs Arch Pharmacol 370:227–237
14. Schilling T, Eder C (2007) TRAM-34 inhibits nonselective cationchannels. Pflugers Arch 454:559–563
15. Tian L, McClafferty H, Chen L, Shipston MJ (2008) Reversibletyrosine protein phosphorylation regulates large conductancevoltage- and calcium-activated potassium channels via cortactin.J Biol Chem 283:3067–3076
16. Harrison DG, Long C (1968) The calcium content of humanerythrocytes. J Physiol 199:367–381
17. Bookchin RM, Lew VL (1980) Progressive inhibition of the Capump and Ca:Ca exchange in sickle red cells. Nature 284:561–563
18. Engelmann B, Duhm J (1987) Intracellular calcium content ofhuman erythrocytes: relation to sodium transport systems. JMembr Biol 98:79–87
19. Ferreira HG, Lew VL (1976) Use of ionophore A23187 tomeasure cytoplasmic Ca buffering and activation of the Ca pumpby internal Ca. Nature 259:47–49
20. Tiffert T, Lew VL (1997) Cytoplasmic calcium buffers in intacthuman red cells. J Physiol 500(Pt 1):139–154
21. Williamson P, Puchulu E, Penniston JT, Westerman MP, SchlegelRA (1992) Ca2+ accumulation and loss by aberrant endocyticvesicles in sickle erythrocytes. J Cell Physiol 152:1–9
22. Lew VL, Hockaday A, Sepulveda MI, Somlyo AP, Somlyo AV,Ortiz OE, Bookchin RM (1985) Compartmentalization of sickle-cell calcium in endocytic inside-out vesicles. Nature 315:586–589
23. Lew VL, Tsien RY, Miner C, Bookchin RM (1982) Physiological[Ca2+]i level and pump-leak turnover in intact red cells measuredusing an incorporated Ca chelator. Nature 298:478–481
24. Schatzmann HJ (1983) The red cell calcium pump. Annu RevPhysiol 45:303–312
25. David-Dufilho M, Montenay-Garestier T, Devynck MA (1988)Fluorescence measurements of free Ca2+ concentration in humanerythrocytes using the Ca2+-indicator fura-2. Cell Calcium 9:167–179
26. Hoffman JF, Laris PC (1974) Determination of membranepotentials in human and Amphiuma red blood cells by means offluorescent probe. J Physiol 239:519–552
27. Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM,Lang F (2005) Mechanisms of suicidal erythrocyte death. CellPhysiol Biochem 15:195–202
28. Foller M, Sopjani M, Koka S, Gu S, Mahmud H, Wang K, FlorideE, Schleicher E, Schulz E, Munzel T, Lang F (2009) Regulation oferythrocyte survival by AMP-activated protein kinase. Faseb JResource 23:1072–1080
29. LaCelle PL, Rothsteto A (1966) The passive permeability of thered blood cell in cations. J Gen Physiol 50:171–188
30. Jones GS, Knauf PA (1985) Mechanism of the increase in cationpermeability of human erythrocytes in low-chloride media.Involvement of the anion transport protein capnophorin. J GenPhysiol 86:721–738
31. Bernhardt I, Hall AC, Ellory JC (1991) Effects of low ionicstrength media on passive human red cell monovalent cationtransport. J Physiol 434:489–506
32. Lang KS, Duranton C, Poehlmann H, Myssina S, Bauer C, LangF, Wieder T, Huber SM (2003) Cation channels trigger apoptoticdeath of erythrocytes. Cell Death Differ 10:249–256
33. Lang PA, Kempe DS, Myssina S, Tanneur V, Birka C, Laufer S,Lang F, Wieder T, Huber SM (2005) PGE2 in the regulation ofprogrammed erythrocyte death. Cell Death Differ 12:415–428
34. Kaestner L, Tabellion W, Lipp P, Bernhardt I (2004) ProstaglandinE2 activates channel-mediated calcium entry in human erythro-cytes: an indication for a blood clot formation supporting process.Thromb Haemost 92:1269–1272
35. Li Q, Jungmann V, Kiyatkin A, Low PS (1996) Prostaglandin E2stimulates a Ca2+-dependent K+ channel in human erythrocytesand alters cell volume and filterability. J Biol Chem 271:18651–18656
36. Allen JE, Rasmussen H (1971) Human red blood cells: prosta-glandin E2, epinephrine, and isoproterenol alter deformability.Science 174:512–514
37. Schneider J, Nicolay JP, Foller M, Wieder T, Lang F (2007)Suicidal erythrocyte death following cellular K+ loss. Cell PhysiolBiochem 20:35–44
38. Siegel DL, Goodman SR, Branton D (1980) The effect ofendogenous proteases on the spectrin binding proteins of humanerythrocytes. Biochim Biophys Acta 598:517–527
39. Hall TG, Bennett V (1987) Regulatory domains of erythrocyteankyrin. J Biol Chem 262:10537–10545
40. Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ (1997)Molecular cloning of human plasma membrane phospholipidscramblase. A protein mediating transbilayer movement of plasmamembrane phospholipids. J Biol Chem 272:18240–18244
41. Woon LA, Holland JW, Kable EP, Roufogalis BD (1999) Ca2+sensitivity of phospholipid scrambling in human red cell ghosts.Cell Calcium 25:313–320
42. Zachowski A, Favre E, Cribier S, Herve P, Devaux PF (1986)Outside-inside translocation of aminophospholipids in the humanerythrocyte membrane is mediated by a specific enzyme.Biochemistry 25:2585–2590
43. Lang KS, Myssina S, Brand V, Sandu C, Lang PA, Berchtold S,Huber SM, Lang F, Wieder T (2004) Involvement of ceramide in
1042 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044

hyperosmotic shock-induced death of erythrocytes. Cell DeathDiffer 11:231–243
44. Lang KS, Myssina S, Lang PA, Tanneur V, Kempe DS, Mack AF,Huber SM, Wieder T, Lang F, Duranton C (2004) Inhibition oferythrocyte phosphatidylserine exposure by urea and Cl. Am JPhysiol Ren Physiol 286:F1046–F1053
45. Bratosin D, Estaquier J, Petit F, Arnoult D, Quatannens B, TissierJP, Slomianny C, Sartiaux C, Alonso C, Huart JJ, Montreuil J,Ameisen JC (2001) Programmed cell death in mature erythro-cytes: a model for investigating death effector pathways operatingin the absence of mitochondria. Cell Death Differ 8:1143–1156
46. Tanaka Y, Schroit AJ (1983) Insertion of fluorescent phosphatidyl-serine into the plasma membrane of red blood cells. Recognition byautologous macrophages. J Biol Chem 258:11335–11343
47. Schroit AJ, Madsen JW, Tanaka Y (1985) In vivo recognition andclearance of red blood cells containing phosphatidylserine in theirplasma membranes. J Biol Chem 260:5131–5138
48. Skals M, Jorgensen NR, Leipziger J, Praetorius HA (2009) Alpha-hemolysin from Escherichia coli uses endogenous amplificationthrough P2X receptor activation to induce hemolysis. Proc NatlAcad Sci USA 106:4030–4035
49. Skals M, Jensen UB, Ousingsawat J, Kunzelmann K, Leipziger J,Praetorius HA (2010) Escherichia coli alpha-hemolysin triggersshrinkage of erythrocytes via K(Ca)3.1 and TMEM16A channelswith subsequent phosphatidylserine exposure. J Biol Chem285:15557–15565
50. Lang F, Huber SM, Szabo I, Gulbins E (2007) Plasma membraneion channels in suicidal cell death. Arch Biochem Biophys462:189–194
51. Lang PA, Kaiser S, Myssina S, Birka C, Weinstock C, Northoff H,Wieder T, Lang F, Huber SM (2004) Effect of Vibrio para-haemolyticus haemolysin on human erythrocytes. Cell Microbiol6:391–400
52. Foller M, Shumilina E, Lam R, Mohamed W, Kasinathan R,Huber S, Chakraborty T, Lang F (2007) Induction of suicidalerythrocyte death by listeriolysin from Listeria monocytogenes.Cell Physiol Biochem 20:1051–1060
53. Kempe DS, Akel A, Lang PA, Hermle T, Biswas R, Muresanu J,Friedrich B, Dreischer P, Wolz C, Schumacher U, Peschel A, GotzF, Doring G, Wieder T, Gulbins E, Lang F (2007) Suicidalerythrocyte death in sepsis. J Mol Med 85:273–281
54. Shumilina E, Lam RS, Wolbing F, Matzner N, Zemtsova IM,Sobiesiak M, Mahmud H, Sausbier U, Biedermann T, Ruth P,Sausbier M, Lang F (2008) Blunted IgE-mediated activation ofmast cells in mice lacking the Ca2+-activated K+ channel KCa3.1.J Immunol 180:8040–8047
55. Wolfle SE, Schmidt VJ, Hoyer J, Kohler R, de Wit C (2009)Prominent role of KCa3.1 in endothelium-derived hyperpolarizingfactor-type dilations and conducted responses in the microcircu-lation in vivo. Cardiovasc Res 82:476–483
56. Dufer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G(2009) Enhanced glucose tolerance by SK4 channel inhibition inpancreatic beta-cells. Diabetes 58:1835–1843
57. Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J, MullerA, Kaistha A, Schmidt C, Raman G, Wulff H, Strutz F, Grone HJ,Kohler R, Hoyer J (2009) Renal fibrosis is attenuated by targeteddisruption of KCa3.1 potassium channels. Proc Natl Acad SciUSA 106:14518–14523
58. Matos JE, Sausbier M, Beranek G, Sausbier U, Ruth P, Leipziger J(2007) Role of cholinergic-activated KCa1.1 (BK), KCa3.1 (SK4)and KV7.1 (KCNQ1) channels in mouse colonic Cl− secretion.Acta Physiol (Oxf) 189:251–258
59. Flores CA, Melvin JE, Figueroa CD, Sepulveda FV (2007) Abolitionof Ca2+-mediated intestinal anion secretion and increased stooldehydration in mice lacking the intermediate conductance Ca2+-dependent K+ channel Kcnn4. J Physiol 583:705–717
60. Di L, Srivastava S, Zhdanova O, Ding Y, Li Z, Wulff H, LafailleM, Skolnik EY (2010) Inhibition of the K+ channel KCa3.1ameliorates T cell-mediated colitis. Proc Natl Acad Sci U S A107:1541–1546
61. Sausbier M, Matos JE, Sausbier U, Beranek G, Arntz C,Neuhuber W, Ruth P, Leipziger J (2006) Distal colonic K(+)secretion occurs via BK channels. J Am Soc Nephrol 17:1275–1282
62. Barry PH, Lynch JW (1991) Liquid junction potentials and smallcell effects in patch-clamp analysis. J Membr Biol 121:101–117
63. Huber SM, Tschop J, Braun GS, Nagel W, Horster MF (1999)Bradykinin-stimulated Cl− secretion in T84 cells. Role of Ca2+-activated hSK4-like K+ channels. Pflugers Arch 438:53–60
64. Huber SM, Duranton C, Lang F (2005) Patch-clamp analysis ofthe “new permeability pathways” in malaria-infected erythrocytes.Int Rev Cytol 246:59–134
65. Lang F, Gulbins E, Lerche H, Huber SM, Kempe DS, Foller M(2008) Eryptosis, a window to systemic disease. Cell PhysiolBiochem 22:373–380
66. Duranton C, Huber SM, Lang F (2002) Oxidation induces a Cl(−)-dependent cation conductance in human red blood cells. JPhysiol 539:847–855
67. Lang PA, Kasinathan RS, Brand VB, Duranton C, Lang C, KokaS, Shumilina E, Kempe DS, Tanneur V, Akel A, Lang KS, FollerM, Kun JF, Kremsner PG, Wesselborg S, Laufer S, Clemen CS,Herr C, Noegel AA, Wieder T, Gulbins E, Lang F, Huber SM(2009) Accelerated clearance of Plasmodium-infected erythro-cytes in sickle cell trait and annexin-A7 deficiency. Cell PhysiolBiochem 24:415–428
68. Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, GouauxJE (1996) Structure of staphylococcal alpha-hemolysin, a hepta-meric transmembrane pore. Science 274:1859–1866
69. Harshman S, Burt AM, Robinson JP, Blankenship M, HarshmanDL (1985) Disruption of myelin sheaths in mouse brain in vitroand in vivo by staphylococcal alpha-toxin. Toxicon 23:801–806
70. Chan KF, Lazarovici P (1987) Staphylococcus aureus alpha-toxin.1. Effect on protein phosphorylation in myelin. Toxicon 25:631–636
71. Shetlar MD, Hill HA (1985) Reactions of hemoglobin withphenylhydrazine: a review of selected aspects. Environ HealthPerspect 64:265–281
72. Lew VL, Macdonald L, Ginsburg H, Krugliak M, Tiffert T (2004)Excess haemoglobin digestion by malaria parasites: a strategy toprevent premature host cell lysis. Blood Cells Mol Dis 32:353–359
73. Grgic I, Kaistha BP, Paschen S, Kaistha A, Busch C, Si H, KohlerK, Elsasser HP, Hoyer J, Kohler R (2009) Disruption of theGardos channel (KCa3.1) in mice causes subtle erythrocytemacrocytosis and progressive splenomegaly. Pflugers Arch458:291–302
74. Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C,Alper SL, Melvin JE (2004) Physiological roles of the interme-diate conductance, Ca2+-activated potassium channel Kcnn4. JBiol Chem 279:47681–47687
75. Lew VL, Tiffert T, Etzion Z, Perdomo D, Daw N, Macdonald L,Bookchin RM (2005) Distribution of dehydration rates generatedby maximal Gardos-channel activation in normal and sickle redblood cells. Blood 105:361–367
76. Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I,Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C,Hoyer J, Kohler R (2006) Impaired endothelium-derived hyper-polarizing factor-mediated dilations and increased blood pressurein mice deficient of the intermediate-conductance Ca2+-activatedK+ channel. Circ Res 99:537–544
77. Brown AM, Ellory JC, Young JD, Lew VL (1978) A calcium-activated potassium channel present in foetal red cells of the sheep
Pflugers Arch - Eur J Physiol (2010) 460:1029–1044 1043

but absent from reticulocytes and mature red cells. BiochimBiophys Acta 511:163–175
78. Jain SK, Hochstein P (1980) Membrane alterations inphenylhydrazine-induced reticulocytes. Arch Biochem Biophys201:683–687
79. Cohen G, Hochstein P (1964) Generation of hydrogen peroxide inerythrocytes by hemolytic agents. Biochemistry 3:895–900
80. Bhakdi S, Tranum-Jensen J (1991) Alpha-toxin of Staphylococcusaureus. Microbiol Rev 55:733–751
81. Lang PA, Kempe DS, Tanneur V, Eisele K, Klarl BA, MyssinaS, Jendrossek V, Ishii S, Shimizu T, Waidmann M, Hessler G,Huber SM, Lang F, Wieder T (2005) Stimulation of erythrocyteceramide formation by platelet-activating factor. J Cell Sci118:1233–1243
82. Lew VL, Bookchin RM (2005) Ion transport pathology in themechanism of sickle cell dehydration. Physiol Rev 85:179–200
83. de Franceschi L, Rouyer-Fessard P, Alper SL, Jouault H, BrugnaraC, Beuzard Y (1996) Combination therapy of erythropoietin,hydroxyurea, and clotrimazole in a beta thalassemic mouse: amodel for human therapy. Blood 87:1188–1195
84. Sessler CN, Perry JC, Varney KL (2004) Management of severesepsis and septic shock. Curr Opin Crit Care 10:354–363
85. Tiffert T, Staines HM, Ellory JC, Lew VL (2000) Functional stateof the plasma membrane Ca2+ pump in Plasmodium falciparum-infected human red blood cells. J Physiol 525(Pt 1):125–134
86. Staines HM, Chang W, Ellory JC, Tiffert T, Kirk K, Lew VL(1999) Passive Ca(2+) transport and Ca(2+)-dependent K(+)transport in Plasmodium falciparum-infected red cells. J MembrBiol 172:13–24
87. Kohler R, Ruth P (2010) Endothelial dysfunction and bloodpressure alterations in K+-channel transgenic mice. Pflugers Arch459:969–976
1044 Pflugers Arch - Eur J Physiol (2010) 460:1029–1044




















