From Anti-allergic to Anti-Alzheimer's: Molecular Pharmacology of Dimebon
16
Current Alzheimer Research, 2010, 7, 97-112 97 1567-2050/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd. From Anti-allergic to Anti-Alzheimer’s: Molecular Pharmacology of Dimebon TM I. Okun 1, *, S.E. Tkachenko 1 , A. Khvat 2 , O. Mitkin 3 , V. Kazey 1 and A.V. Ivachtchenko 1 1 Avineuro Pharmaceuticals, Inc. 6605 Nancy Ridge Drive, Suite 126. San Diego, CA 92121, USA; 2 ChemDiv, Inc. 6605 Nancy Ridge Drive, San Diego, CA, USA; 3 Chemical Diversity Research Institute, Rabochaya St. 2a, 114401 Khimki, Moscow Reg, Russia Abstract: Dimebon, originally developed as an anti-histamine drug, is being re-purposed for new indications as an effec- tive treatment for patients suffering from Alzheimer’s and Huntington’s diseases, albeit with an as-yet unknown mecha- nism of action. We have performed molecular pharmacology profiling of this drug on a panel of 70 targets to characterize the spectrum of its activity, with the goal to possibly elucidate a potential molecular mechanism for the re-purposing of this drug candidate. We show that in addition to histaminergic receptors, Dimebon exhibits high affinity to a constellation of other receptors; specifically serotonergic, alpha-adrenergic and dopaminergic receptors. Good correlations with pub- lished literature were obtained for the affinity of Dimebon to inhibit butyrylcholinesterase, interact with H1and H2 recep- tors (Ki = 2 nM and 232 nM), and to block histamine-induced calcium fluxes in cells. Within serotonergic receptor sub- types, Dimebon shows highest affinity for 5-HT7 (Ki=8 nM) and 5-HT6 (Ki=34 nM) receptors, with the relative affinity rank-order of 5-HT7 > 5-HT6 5-HT2A = 5-HT2C > 5-HT1A = 5-HT1B > 5-HT2B=5-HT3. Dimebon also interacts with adrenergic receptor subtypes (rank-order: 1A (Ki = 55 nM)= 1B 2A (Ki = 120 nM) = 1D), and dopaminergic re- ceptor subtypes (rank-order: D1=D2S=D2L (Ki ~ 600 nM) >D3 D4.2>D4.4 D4.7). These results demonstrate a molecu- lar pharmacological basis for re-purposing of this drug to new therapeutic areas. The informed targeting of the combined molecular target activities may provide additional advantages for patients suffering from similar diseases syndromes. Un- derstanding the role that different pathways play in diseases with complex etiologies may allow for the rational design of multi-target drugs. Keywords and Phrases: Dimebon, Alzheimer’s disease, adrenergic receptors, dopaminergic receptors, histaminergic recep- tors, serotonergic receptors, calcium fluxes. 1. INTRODUCTION Dimebon TM (dimebolin hydrochloride) is an orally bioavailable antihistamine drug that was discovered in Rus- sia and has been used clinically there since 1983 [1]. Clinical observations of the elder patients treated with Dimebon TM led to realization that this drug can also be advantageous for persons suffering from Alzheimer’s and Huntington’s dis- eases. Alzheimer's disease (AD) is a devastating neurological condition characterized by a progressive decline in cognitive performance accompanied by behavioral and psychological syndromes, such as depression and psychosis. In animal models of Alzheimer's disease, Dimebon TM was confirmed to provide beneficial effects [2]. Preliminary results from human trials performed by Medivation have been promising [3]. As reported by Medivation [4], in six- month and twelve-month phase II trials “…Dimebon TM demonstrated significant improvement over placebo on all five efficacy endpoints” - memory, thinking, activities of daily living, behavior, and overall function. Currently, Di- mebon TM is in Phase III clinical trials. Besides the positive effect on AD patients, it was observed that Dimebon TM pos- sess pronounced antiarrhythmic properties [5] and its use could also be promising as cognition-enhancing drug in oth- erwise healthy individuals [6]. *Address correspondence to this author at the Avineuro Pharmaceuticals, Inc. 6605 Nancy Ridge Drive, Suite 126. San Diego, CA 92121, USA; E-mail: [email protected] Dimebon TM , 2,8-Dimethyl-5-[2-(6-methylpyridin-3-yl) ethyl]-2,3,4,5-tetrahydro-1 -pyrido[4,3-b]indole dihydro- chloride is a small molecule with physiochemical properties that have no violations of rule of 5 [7, 8]: MW < 500 (319.4), number of hydrogen bond acceptors < 10 (3) and hydrogen bond donors < 5 (3), and Log P < 5 (3.4). Dimebon TM also has a polar surface area (PSA) less than 60 (21.06 Å), which is considered to be favorable for penetrating the blood brain barrier (BBB). Unfortunately, there are only scarce data available that relate molecular pharmacology of Dimebon TM to its multi- functional effects on different aspects of CNS activity. It was shown that in addition to its interaction with H 1 receptors, Dimebon TM is able to i) inhibit butyryl cholinesterase (IC 50 =7.9 M) and, with a lesser affinity, acetyl cholinester- ase (IC 50 =42 M) [6], ii) interact with different therapeutic targets including blockage of neurotoxic effects of beta- amyloid (EC 50 =25 M) [6] and huntingtin proteins, iii) in- hibit L-type calcium channels (IC 50 =57 M) [9], iv) modu- late activity of AMPA and NMDA (IC 50 – 10-70 M) gluta- mate receptors [10], and v) block mitochondrial permeability transition pore (MPTP) [11]. The ability of Dimebon TM to block NMDA receptors and calcium channels was suggested as its primary protection mechanism of neuronal cells from glutamate exitotoxicity (particularly in Huntington’s pa- tients) and its ability to inhibit cholinesterase was suggested as being responsible for maintaining an elevated level of
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of From Anti-allergic to Anti-Alzheimer's: Molecular Pharmacology of Dimebon

Current Alzheimer Research, 2010, 7, 97-112 97
1567-2050/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd.
From Anti-allergic to Anti-Alzheimer’s: Molecular Pharmacology of Dimebon
TM
I. Okun1,*, S.E. Tkachenko1, A. Khvat2, O. Mitkin3, V. Kazey1 and A.V. Ivachtchenko1
1Avineuro Pharmaceuticals, Inc. 6605 Nancy Ridge Drive, Suite 126. San Diego, CA 92121, USA;
2ChemDiv, Inc. 6605
Nancy Ridge Drive, San Diego, CA, USA; 3Chemical Diversity Research Institute, Rabochaya St. 2a, 114401 Khimki,
Moscow Reg, Russia
Abstract: Dimebon, originally developed as an anti-histamine drug, is being re-purposed for new indications as an effec-tive treatment for patients suffering from Alzheimer’s and Huntington’s diseases, albeit with an as-yet unknown mecha-nism of action. We have performed molecular pharmacology profiling of this drug on a panel of 70 targets to characterize the spectrum of its activity, with the goal to possibly elucidate a potential molecular mechanism for the re-purposing of this drug candidate. We show that in addition to histaminergic receptors, Dimebon exhibits high affinity to a constellation of other receptors; specifically serotonergic, alpha-adrenergic and dopaminergic receptors. Good correlations with pub-lished literature were obtained for the affinity of Dimebon to inhibit butyrylcholinesterase, interact with H1and H2 recep-tors (Ki = 2 nM and 232 nM), and to block histamine-induced calcium fluxes in cells. Within serotonergic receptor sub-types, Dimebon shows highest affinity for 5-HT7 (Ki=8 nM) and 5-HT6 (Ki=34 nM) receptors, with the relative affinity rank-order of 5-HT7 > 5-HT6 5-HT2A = 5-HT2C > 5-HT1A = 5-HT1B > 5-HT2B=5-HT3. Dimebon also interacts with adrenergic receptor subtypes (rank-order: 1A (Ki = 55 nM)= 1B 2A (Ki = 120 nM) = 1D), and dopaminergic re-ceptor subtypes (rank-order: D1=D2S=D2L (Ki ~ 600 nM) >D3 D4.2>D4.4 D4.7). These results demonstrate a molecu-lar pharmacological basis for re-purposing of this drug to new therapeutic areas. The informed targeting of the combined molecular target activities may provide additional advantages for patients suffering from similar diseases syndromes. Un-derstanding the role that different pathways play in diseases with complex etiologies may allow for the rational design of multi-target drugs.
Keywords and Phrases: Dimebon, Alzheimer’s disease, adrenergic receptors, dopaminergic receptors, histaminergic recep-tors, serotonergic receptors, calcium fluxes.
1. INTRODUCTION
DimebonTM (dimebolin hydrochloride) is an orally bioavailable antihistamine drug that was discovered in Rus-sia and has been used clinically there since 1983 [1]. Clinical observations of the elder patients treated with DimebonTM led to realization that this drug can also be advantageous for persons suffering from Alzheimer’s and Huntington’s dis-eases. Alzheimer's disease (AD) is a devastating neurological condition characterized by a progressive decline in cognitive performance accompanied by behavioral and psychological syndromes, such as depression and psychosis.
In animal models of Alzheimer's disease, DimebonTM was confirmed to provide beneficial effects [2]. Preliminary results from human trials performed by Medivation have been promising [3]. As reported by Medivation [4], in six-month and twelve-month phase II trials “…DimebonTM demonstrated significant improvement over placebo on all five efficacy endpoints” - memory, thinking, activities of daily living, behavior, and overall function. Currently, Di-mebonTM is in Phase III clinical trials. Besides the positive effect on AD patients, it was observed that DimebonTM pos-sess pronounced antiarrhythmic properties [5] and its use could also be promising as cognition-enhancing drug in oth-erwise healthy individuals [6].
*Address correspondence to this author at the Avineuro Pharmaceuticals, Inc. 6605 Nancy Ridge Drive, Suite 126. San Diego, CA 92121, USA; E-mail: [email protected]
DimebonTM, 2,8-Dimethyl-5-[2-(6-methylpyridin-3-yl) ethyl]-2,3,4,5-tetrahydro-1 -pyrido[4,3-b]indole dihydro-chloride is a small molecule with physiochemical properties that have no violations of rule of 5 [7, 8]: MW < 500 (319.4), number of hydrogen bond acceptors < 10 (3) and hydrogen bond donors < 5 (3), and Log P < 5 (3.4). DimebonTM also has a polar surface area (PSA) less than 60 (21.06 Å), which is considered to be favorable for penetrating the blood brain barrier (BBB).
Unfortunately, there are only scarce data available that relate molecular pharmacology of DimebonTM to its multi-functional effects on different aspects of CNS activity. It was shown that in addition to its interaction with H1 receptors, DimebonTM is able to i) inhibit butyryl cholinesterase (IC50=7.9 M) and, with a lesser affinity, acetyl cholinester-ase (IC50=42 M) [6], ii) interact with different therapeutic targets including blockage of neurotoxic effects of beta-amyloid (EC50=25 M) [6] and huntingtin proteins, iii) in-hibit L-type calcium channels (IC50=57 M) [9], iv) modu-late activity of AMPA and NMDA (IC50 – 10-70 M) gluta-mate receptors [10], and v) block mitochondrial permeability transition pore (MPTP) [11]. The ability of DimebonTM to block NMDA receptors and calcium channels was suggested as its primary protection mechanism of neuronal cells from glutamate exitotoxicity (particularly in Huntington’s pa-tients) and its ability to inhibit cholinesterase was suggested as being responsible for maintaining an elevated level of

98 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
acetylcholine in Alzheimer’s patients (Huntington’s Out-reach Project for Education, at Stanford (HOPES, http://www.stanford.edu/group/hopes/treatmts/ antiglut/l7. html). In primary striatal neuronal cultures, DimebonTM was shown to inhibit NMDA receptors (IC50=10 μM), block volt-age-gated calcium channels (IC50=50 μM), and at 50 μM protect the cells from glutamate-induced apoptosis [12]. Though the described above targets are believed to be asso-ciated with a multiplicity of CNS disorders (such as stress, anxiety, mood, neurodegenerative diseases, and brain dam-age), the rather low affinity of DimebonTM to these targets (micromolar range as opposed to nanomolar affinities to H1 histamine receptor and many other receptors) makes it ques-tionable if DimebonTM’s CNS action can fully be explained by its effects on these targets alone. [12]
Therefore, a more detailed analysis of DimebonTM action on a broader panel of potential therapeutic targets could pro-duce valuable insight into the mechanisms of its CNS activ-ity as well as to shed light on molecular pharmacology re-ceptor activity fingerprint patterns that make the drug repur-posing possible.
2. MATERIALS AND METHODS
2.1. Materials
The reagents were from Sigma Chemical Company (St. Louis, MO) unless otherwise stated. All radiochemicals were from Amersham Radiochemicals (GE Healthcare Life Sci-ences, Pittsburgh, PA). Membrane preparations from cells either transfected with or endogenously expressed receptors and enzymes were from MDS Pharma Services (Taipei, Taiwan). SK-N-SH cells were from American Type Cul-tureCollection (Manassas, VA). Human recombinant tetracy-cline-inducible HEK-293 5-HT6R-T-REx cells were from Etogen Scientific (San Diego, CA).
2.2. Enzymatic Assays
Acetylcholinesterase [13, 14]. Source: human recombi-nant HEK-293 cells. Substrate: 700 μM acetylthiocholine. Pre-incubation Time/Temp: 15 minutes @ 25°C; reaction time/temp: 20 minutes @ 25°C; incubation buffer: 0.1 M Na3PO4, pH 7.4. Thiocholine formation was determined spectrophotometrically by reaction with 5,5'-dithiobis(2-nitrobenzoate).
Butyrylcholinesterase [15]. Source: human serum. Sub-strate: 560 μM S-butyrylthiocholine. Pre-incubation time/ temp: 15 minutes @ 25°C; reaction time/temp: 120 minutes @ 25°C; incubation buffer: 0.1 M Na3PO4, pH 7.4, 0.5% Tween 20. Thiocholine formation was determined spectro-photometrically by reaction with 5,5'-dithiobis(2-nitro-benzoate).
Monoamine Oxidase A (MAO-A) [16]. Source: human recombinant insect Hi5 cells. Substrate: 50 μM kynuramine. Pre-incubation time/temp: 15 minutes @ 37°C; reaction time/temp: 60 minutes @ 37°C; incubation buffer: 0.1 M K3PO4, pH 7.4. 4-hydroxyquinoline formation was quanti-fied spectrophotometrically.
Monoamine Oxidase B (MAO-B) [16]. Source: human recombinant insect Hi5 cells. Substrate: 50 μM kynuramine. Pre-incubation time/temp: 15 minutes @ 37°C; reaction
time/temp: 60 minutes @ 37°C; incubation buffer: 0.1 M K3PO4, pH 7.4. Formation of 4-hydroxyquinoline was quan-tified spectrophotometrically.
2.3. Radioligand Binding Assays
Adenosine A1 receptors [17]. Source: human recombi-nant CHO cells. Ligand: 1 nM [3H] 8-cyclopentyl-1,3-dipropylxanthine (DPCPX). Incubation time/temp: 90 min-utes @ 25°C; incubation buffer: 20 mM HEPES, pH 7.4, 10 mM MgCl2, 100 mM NaCl; KD: 1.4 nM, Bmax: 2.7 pmole/mg protein. Specific binding: 85%; non-specific ligand: 100 μM R-phenylisopropyl-adenosine (R(-)-PIA).
Adenosine A2A receptors [18]. Source: human recombi-nant HEK-293 cells. Ligand: 50 nM [3H]2-p-(2-carboxyethyl)phenethyl-amino-5'-N- ethylcarboxamidoade-nosine (CGX-21680). Incubation time/temp: 90 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM EDTA, 2 U/ml adenosine deaminase; KD: 64 nM, Bmax: 7 pmole/mg protein, specific binding: 85%; non-specific ligand: 50 μM adenosine-5 -N-ethylcarboxamide (NECA).
Adrenergic 1A receptors [19]. Source: Wistar rat sub-maxillary gland. Ligand: 0.25 nM [3H] prazosin. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.5 mM EDTA; KD: 0.17 nM, Bmax: 0.18 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM phentolamine.
Adrenergic 1B receptors [20]. Source: Wistar rat liver. Ligand: 0.25 nM [3H]prazosin. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.5 mM EDTA; KD: 0.31 nM, Bmax: 0.18 pmole/mg pro-tein, specific binding: 90%; non-specific ligand: 10 μM phentolamine.
Adrenergic 1D receptors [21]. Source: human recombi-nant HEK-293 cells (MDS Pharma Services, Taipei, Tai-wan). Ligand: 0.6 nM [3H]prazosin. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 0.58 nM, Bmax: 0.17 pmole/mg protein, specific bin-ding: 80%; non-specific ligand: 10 μM phentolamine.
Adrenergic 2A receptors [22]. Source: human recombi-nant sf9 cells. Ligand: 1.0 nM [3H] MK-912. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 12.5 mM MgCl2, 2 mM EDTA; KD: 0.6 nM, Bmax: 4.6 pmole/mg protein, specific binding: 95%; non-specific ligand: 10 μM 2-(2,6- dimethoxyphenoxyethyl) ami-nomethyl-1,4-benzodioxane (WB-4101).
Adrenergic ß1 receptors [23]. Source: human recombi-nant Rex 16 cells. Ligand: 0.03 nM [125I]cyanopindolol. In-cubation time/temp: 120 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, ).001% BSA, 1.5 mM CaCl2, 5 mM EDTA, 120 nM NaCl; KD: 0.041 nM, Bmax: 0.072 pmole/mg protein, specific binding: 95%; non-specific ligand: 100 μM S(-)-propranolol.
Adrenergic ß2 receptors [24, 25]. Source: human recom-binant CHO cells. Ligand: 0.2 nM [3H]4-(3-tertiary-butylamino-2-hydroxypropoxy)-benzimidazole-2-one hydro-chloride (CGP-12177l). Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 5 mM

From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 99
MgCl2, 0.5 mM EDTA, 120 nM NaCl; KD: 0.44 nM, Bmax: 0.437 pmole/mg protein, specific binding: 95%; non-specific ligand: 10 μM erythro-dl-1-(7- methylindan-4-yloxy)-3-isopropylaminobutan-2-ol (ICI-118551).
Bradykinin B1 receptors [26, 27]. Source: human IMR-90 cells. Ligand: 0.5 nM [3H] (Des-Arg10)-kallidin. Incubation time/temp: 60 min @ 25°C; incubation buffer: 20 mM HEPES, pH 7.4, 125 mM N-methyl-D-glucamine, 5 mM KCl, 1 mM 1,10-phenanthroline, 140 μg/ml Bacitracin; KD: 0.17 nM, Bmax: 0.55 pmole/mg protein, specific binding: 80%; non-specific ligand: 10 μM (Des-Arg9,Leu8)-Bradykinin.
Bradykinin B2 receptors [28, 29]. Source: human recom-binant CHO-K1 cells. Ligand: 0.2 nM [3H]Bradykinin. Incu-bation time/temp: 90 min @ 25°C; incubation buffer: 24 mM TES, pH 6.8, 1 mM 1,10-phenanthroline, 0.3% BSA; KD: 0.29 nM, Bmax: 2.0 pmole/mg protein, specific binding: 90%; non-specific ligand: 5 μM Bradykinin.
Calcium channel L-type, Benzothiazepine site [30]. Source: Wistar rat brain. Ligand: 2 nM [3H]diltiazem. Incu-bation time/temp: 3 hours @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.1% BSA; KD: 16 nM, Bmax: 0.21 pmole/mg protein, specific binding: 73%; non-specific ligand: 10 μM diltiazem.
Calcium channel L-type, Dihydropyridine site [31, 32]. Source: Wistar rat cerebral cortex. Ligand: 0.1 nM [3H]nitrendipin. Incubation time/temp: 90 min @ 25°C; in-cubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 0.18 nM, Bmax: 0.23 pmole/mg protein, specific binding: 91%; non-specific ligand: 1 μM nifedipine.
Calcium channel N-type [33]. Source: Wistar rat brain frontal lobe. Ligand: 10 pM [125I] -Conotoxin GVIA. Incu-bation time/temp: 30 min @ 4°C; incubation buffer: 20 mM Tris-HCl, pH 7.4, 0.5% BSA; KD: 0.05 nM, Bmax: 0.88 pmole/mg protein, specific binding: 96%; non-specific ligand: 0.1 μM -Conotoxin GVIA.
Dopamine D1 receptors [34, 35]. Source: human recom-binant CHO cells. Ligand: 1.4 nM [3H]7-chloro-3-methyl-1-phenyl-1,2,4,5-tetrahydro-3-benzazepin-8-ol (SCH-23390). Incubation time/temp: 2 hours @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 1.4 nM, Bmax: 0.63 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM (+)-Butaclamol.
Dopamine D2L receptors [36, 37]. Source: human recom-binant CHO cells. Ligand: 0.16 nM [3H]spiperone. Incuba-tion time/temp: 2 hours @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 0.09 nM, Bmax: 0.48 pmole/mg protein, specific binding: 85%; non-specific ligand: 10 μM haloperi-dol.
Dopamine D2S receptors [36, 37]. Source: human recom-binant CHO cells. Ligand: 0.16 nM [3H]spiperone. Incuba-tion time/temp: 2 hours @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 0.09 nM, Bmax: 1.6 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM haloperi-dol.
Dopamine D3 receptors [38]. Source: human recombinant CHO cells. Ligand: 0.7 nM [3H]spiperone. Incubation time/temp: 2 hours @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 0.36 nM, Bmax: 1.1 pmole/mg protein, specific binding: 85%; non-specific ligand: 25 μM (-)-sulpiride.
Dopamine D4.2 receptors [39, 40]. Source: human recom-binant CHO-K1 cells. Ligand: 0.5 nM [3H]spiperone. Incu-bation time/temp: 2 hours @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 0.32 nM, Bmax: 0.55 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM haloperi-dol.
Dopamine D4.4 receptors [39, 40]. Source: human recom-binant CHO-K1 cells. Ligand: 1.2 nM [3H]spiperone. Incu-bation time/temp: 2 hours @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 0.46 nM, Bmax: 0.63 pmole/mg protein, specific binding: 85%; non-specific ligand: 10 μM haloperi-dol.
Dopamine D4.7 receptors [39, 40]. Source: human recom-binant CHO-K1 cells. Ligand: 1.5 nM [3H]spiperone. Incu-bation time/temp: 2 hours @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 150 mM NaCl, 0.001% BSA; KD: 0.48 nM, Bmax: 0.77 pmole/mg protein, specific binding: 85%; non-specific ligand: 10 μM haloperi-dol.
Endothelin ETA receptors [41]. Source: human recombi-nant CHO cells. Ligand: 0.03 nM [125I]Endothelin-1. Incuba-tion time/temp: 2 hours @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.1% BSA, 0.5 mM CaCl2, 0.05% Tween-20; KD: 0.05 nM, Bmax: 0.35 pmole/mg protein, specific bind-ing: 90%; non-specific ligand: 0.1 μM Endothelin-1.
Endothelin ETB receptors [42, 43]. Source: human re-combinant CHO-K1 cells. Ligand: 0.1 nM [125I]Endothelin-1. Incubation time/temp: 2 hours @ 25°C; incubation buffer: 50 mM HEPES, pH 7.4, 1 mM CaCl2, 5 mM MgCl2, 0.5% BSA; KD: 0.085 nM, Bmax: 4.3 pmole/mg protein, specific binding: 75%; non-specific ligand: 0.1 μM Endothelin-1.
Epidermal Growth Factor receptors [44, 45]. Source: human A431 cells. Ligand: 0.08 nM [125I]EGF (human). In-cubation time/temp: 60 min @ 25°C; incubation buffer: 50 mM HEPES, pH 7.7, 1.2 mM CaCl2, 1.2 mM MgSO4, 5 mM KCl, 138 mM NaCl, 0.1% BSA; KD: 0.17 nM, Bmax: 5.5 pmole/mg protein, specific binding: 90%; non-specific ligand: 0.1 μM EGF (human).
Estrogen ER receptors [46]. Source: human insect Sf9 cells. Ligand: 0.5 nM [3H]estradiol. Incubation time/temp: 120 min @ 25°C; incubation buffer: 10 mM Tris-HCl, pH 7.4, 0.1% BSA, 10% glycerol, 1 mM DTT; KD: 0.2 nM, Bmax: 1400 pmole/mg protein, specific binding: 85%; non-specific ligand: 1 μM diethylstilbestrol.
GABAA receptors, Agonist site [47, 48]. Source: Wistar rat brain without cerebellum. Ligand: 1 nM [3H]muscimol. Incubation time/temp: 10 min @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 3.8 nM, Bmax: 1.8 pmole/mg pro-tein, specific binding: 90%; non-specific ligand: 0.1 μM muscimol.

100 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
GABAA receptors, Benzodiazepine site [49, 50]. Source: Wistar rat brain without cerebellum. Ligand: 1 nM [3H]flunitrazepam. Incubation time/temp: 60 min @ 25°C; incubation buffer: 50 mM phosphate buffer, pH 7.4; KD: 4.4 nM, Bmax: 1.2 pmole/mg protein, specific binding: 91%; non-specific ligand: 10 μM diazepam.
GABAB receptors, non-selective [51, 52]. Source: Wistar rat brain. Ligand: 0.6 nM [3H] [S-(R*,R*)]-[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-h ydroxypropyl](cyclohexyl-methyl) phosphinic acid (CGP-54626). Incubation time/temp: 20 min @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 2.5 mM CaCl2; KD: 2.3 nM, Bmax: 1.1 pmole/mg protein, specific binding: 80%; non-specific ligand: 100 μM CGP-54626.
Glucocorticoid receptors [53]. Source: human HeLa S3 cells. Ligand: 6 nM [3H]dexamethasone. Incubation time/temp: 120 min @ 25°C; incubation buffer: RPMI-1640, 10 mM HEPES, pH 7.2; KD: 5 nM, Bmax: 61,000 R/cell, spe-cific binding: 75%; non-specific ligand: 20 μM dexametha-sone.
Glutamate, Kainate receptors [54]. Source: Wistar rat brain (minus cerebellum). Ligand: 5 nM [3H]kainic acid. Incubation time/temp: 60 min @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 12 nM, Bmax: 0.35 pmole/mg pro-tein, specific binding: 80%; non-specific ligand: 1,000 μM L-glutamic acid.
Glutamate, NMDA receptors, Agonist site [55]. Source: Wistar rat cerebral cortex. Ligand: 2 nM [3H]D,L-(E)-2-amino-4-propyl-5-phosphono-3-pentenoic acid (CGP-39653). Incubation time/temp: 20 min @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 19 nM, Bmax: 2.3 pmole/mg protein, specific binding: 70%; non-specific ligand: 1,000 μM L-glutamic acid.
Glutamate, NMDA receptors, Glycine site [56]. Source: Wistar rat cerebral cortex. Ligand: 0.33 nM [3H] (E)-3-(2-phenyl-2-carboxyethenyl)-4,6-dichloro-1H-indole-2-carboxylic acid (MDL105519). Incubation time/temp: 30 min @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.7; KD: 6 nM, Bmax: 3.7 pmole/mg protein, specific binding: 85%; non-specific ligand: 10 μM MDL105519.
Glutamate, NMDA receptors, Phencyclidine site [57]. Source: Wistar rat cerebral cortex. Ligand: 4 nM [3H]1-(1-(2-thienyl)cyclohexyl)piperidine (TCP). Incubation time/temp: 45 min @ 25°C; incubation buffer: 10 mM Tris-HCl, pH 7.4; KD: 8.4 nM, Bmax: 0.78 pmole/mg protein, spe-cific binding: 94%; non-specific ligand: 1 μM disolcipine ((+)-MK-801).
Histaminergic H1 receptors [58]. Source: human recom-binant CHO cells. Ligand: 1.2 nM [3H]pyrilamine. Incuba-tion time/temp: 3 hours @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 2.5 mM MgCl2, 100 mM NaCl, 250 mM sucrose; KD: 1.1 nM, Bmax: 6.7 pmole/mg protein, specific binding: 94%; non-specific ligand: 1 μM pyrilamine.
Histaminergic H2 receptors [59]. Source: human recom-binant CHO-K1 cells. Ligand: 0.1 nM [125I]amino-potentidine. Incubation time/temp: 2 hours @ 25°C; incuba-tion buffer: 50 mM phosphate buffer, pH 7.4; KD: 0.45 nM, Bmax: 6.9 pmole/mg protein, specific binding: 90%; non-specific ligand: 3 μM tiotidine.
Histaminergic H3 receptors [60, 61]. Source: human re-combinant CHO-K1 cells. Ligand: 3 nM [3H]R(-)- -methylhistamine. Incubation time/temp: 120 min @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 0.04% BSA; KD: 2.4 nM, Bmax: 4.2 pmole/mg protein, spe-cific binding: 95%; non-specific ligand: 1 μM R(-)- -methylhistamine.
Imidazoline I2 receptors, Central [62]. Source: Wistar rat cerebral cortex. Ligand: 2 nM [3H]idazoxan. Incubation time/temp: 30 min @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.5 mM EDTA; KD: 4 nM, Bmax: 0.14 pmole/mg protein, specific binding: 85%; non-specific ligand: 1 μM idazoxan.
Interleukin (IL)-1 receptors [63]. Source: Mouse 3T3 cells. Ligand: 10 pM [125I] IL-1 . Incubation time/tTemp: 120 min @ 37°C; incubation buffer: RPMI 1640, 20 mM HEPES, pH 7.2, 0.1% sodium azide, 1% BSA; KD: 6 pM, Bmax: 8.2 fmole/mg protein, specific binding: 70%; non-specific ligand: 0.03 μM IL-1 .
Leukotriene, Cysteinyl CysLT1 receptors [64]. Source: human recombinant CHO-K1 cells. Ligand: 0.3 nM [3H]leukotriene D4 (LTD4). Incubation time/temp: 30 min @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 5 mM CaCl2, 5 mM MgCl2, 100 μg/ml Bacitracin, 1 mM benzami-dine, 0.1 mM PMSF; KD: 0.21 nM, Bmax: 3 pmole/mg pro-tein, specific binding: 93%; non-specific ligand: 0.3 μM LTD4.
Muscarinic M1 receptors [65, 66]. Source: human recom-binant CHO cells. Ligand: 0.8 nM [3H]N-methylsco-polamine. Incubation time/temp: 120 min @ 25°C; incuba-tion buffer: 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM EDTA; KD: 0.26 nM, Bmax: 2 pmole/mg protein, specific binding: 95%; non-specific ligand: 1 μM atropine.
Muscarinic M2 receptors [65, 66]. Source: human recom-binant CHO cells. Ligand: 0.8 nM [3H]N-methylsco-polamine. Incubation time/temp: 120 min @ 25°C; incuba-tion buffer: 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM EDTA; KD: 0.58 nM, Bmax: 5.1 pmole/mg protein, specific binding: 95%; non-specific ligand: 1 μM atropine.
Muscarinic M3 receptors [65, 66]. Source: human recom-binant CHO cells. Ligand: 0.8 nM [3H]N-methylsco-polamine. Incubation time/temp: 120 min @ 25°C; incuba-tion buffer: 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM EDTA; KD: 0.75 nM, Bmax: 5.4 pmole/mg protein, specific binding: 95%; non-specific ligand: 1 μM atropine.
Neuropeptide YY1 receptors [67, 68]. Source: human SK-N-MC cells. Ligand: 15 pM [125I]peptide YY. Incubation time/temp: 60 min @ 37°C; incubation buffer: 25 mM HEPES, pH 7.4, 1 mM MgCl2, 2.5 mM CaCl2, 0.1% BSA, 0.1 mg/ml Bacitracin; KD: 0.24 nM, Bmax: 0.58 pmole/mg protein, specific binding: 80%; non-specific ligand: 1 μM neuropeptide Y (human).
Neuropeptide YY2 receptors [69]. Source: human KAN-TS cells. Ligand: 10 pM [125I]peptide YY. Incubation time/temp: 120 min @ 37°C; incubation buffer: 25 mM HEPES, pH 7.4, 1 mM MgCl2, 2.5 mM CaCl2, 1 mg/ml Ba-citracin; KD: 0.012 nM, Bmax: 0.5 pmole/mg protein, specific binding: 90%; non-specific ligand: 1 μM porcine neuropep-tide Y (13-36).

From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 101
Nicotinic Acetylcholine receptors [70, 71]. Source: hu-man IMR-32 cells. Ligand: 100 pM [125I]epibatidine. Incuba-tion time/temp: 60 min @ 25°C; incubation buffer: 20 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM KCl, 1 mM MgSO4, 2.0 mM CaCl2; KD: 0.22 nM, Bmax: 0.46 pmole/mg protein, specific binding: 97%; non-specific ligand: 300 μM (-)-Nicotine.
Opiate (OP1) receptors [72]. Source: human recombi-nant CHO cells. Ligand: 0.9 nM [3H]naltrindole. Incubation time/temp: 120 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2; KD: 0.49 nM, Bmax: 8.6 pmole/mg protein, specific binding: 80%; non-specific ligand: 10 μM naloxone.
Opiate (OP2) receptors [73, 74]. Source: human re-combinant HEK-293 cells. Ligand: 0.6 nM [3H]diprenor-phine. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 0.4 nM, Bmax: 1.1 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM naloxone.
Opiate μ (OP3) receptors [75]. Source: human recombi-nant CHO-K1 cells. Ligand: 0.6 nM [3H]diprenorphine. In-cubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 0.41 nM, Bmax: 3.8 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM naloxone.
Phorbol Ester receptor [76]. Source: ICR mouse brain. Ligand: 3 nM [3H]4ß-phorbol 12,13-dibutyrate (PDBu). In-cubation time/temp: 60 minutes @ 25°C; incubation buffer: 20 mM Tris-HCl, pH 7.4, 5 mM CaCl2; KD: 8.7 nM, Bmax: 26 pmole/mg protein, specific binding: 80%; non-specific ligand: 1 μM PDBu.
Platelet Activating Factor (PAF) receptor [77]. Source: human platelets. Ligand: 0.12 nM [3H]PAF. Incubation time/temp: 180 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 100 mM KCl, 5 mM EDTA, 5 mM MgCl2, 0.25% BSA; KD: 0.13 nM, Bmax: 120 receptors/cell, specific binding: 90%; non-specific ligand: 1 μM PAF.
Potassium Channel [KATP] [78]. Source: hamster pancre-atic HIT-T15 beta cells. Ligand: 5 nM [3H]glyburide. Incu-bation time/temp: 120 minutes @ 25°C; incubation buffer: 50 mM MOPS, pH 7.4, 0.1 mM CaCl2; KD: 0.64 nM, Bmax: 1.0 pmole/mg protein, specific binding: 90%; non-specific ligand: 1 μM glyburide.
Potassium Channel HERG [79, 80]. Source: human re-combinant HEK-293 cells. Ligand: 1.5 nM [3H]astemizole. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 10 mM HEPES, pH 7.4, 0.8 mM MgCl2, 5 mM KCl, 130 mM NaCl, 1 mM Na-EGTA, 10 mM glucose, 0.1% BSA; KD: 6.8 nM, Bmax: 6.3 pmole/mg protein, specific bind-ing: 90%; non-specific ligand: 10 μM astemizole.
Purinergic P2X receptors [81, 82]. Source: New Zealand derived albino rabbit urinary bladder. Ligand: 8 nM [3H] ,ß-methylene ATP. Incubation time/temp: 30 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD1: 2.2 nM; KD2: 2.2 μM; Bmax1: 2 pmole/mg protein; Bmax2: 790 pmole/mg protein; specific binding: 80%; non-specific ligand: 100 μM ß, -methylene ATP.
Purinergic P2Y receptors [83]. Source: Wistar rat brain. Ligand: 0.1 nM [35S] ATP- S. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 15 nM; Bmax: 16 pmole/mg protein; specific bind-ing: 87%; non-specific ligand: 10 μM ADP-ßS.
5-Hydroxytryptamine (5-HT)1A receptors [48, 84]. Source: human recombinant CHO cells. Ligand: 1.5 nM [3H] 8-hydroxy-2-(di-n-propylamino)tertraline (8-OH-DPAT). Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.1% ascorbic acid, 0.5 mM EDTA, 10 mM MgSO4; KD: 2 nM, Bmax: 1.3 pmole/mg protein, specific binding: 75%; non-specific ligand: 10 μM metergoline.
5-HT1B receptors [85, 86]. Source: Wistar rat cerebral cortex. Ligand: 10 pM [125I]cyanopindolol. Incubation time/temp: 90 minutes @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 154 mM NaCl, 10 μM pargyline, 30 μM isoprenaline; KD: 0.19 nM, Bmax: 0.14 pmole/mg protein, specific binding: 70%; non-specific ligand: 10 μM serotonin.
5-HT2A receptors [87, 88]. Source: human recombinant CHO-K1 cells. Ligand: 0.5 nM [3H]ketanserin. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 0.2 nM, Bmax: 0.51 pmole/mg protein, specific binding: 90%; non-specific ligand: 1 μM mianserin.
5-HT2B receptors [87]. Source: human recombinant CHO-K1 cells. Ligand: 1.2 nM [3H]lysergic acid diethyla-mide (LSD). Incubation time/temp: 60 minutes @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 4 mM CaCl2, 0.1% ascorbic acid; KD: 2.1 nM, Bmax: 1.1 pmole/mg protein, specific binding: 80%; non-specific ligand: 10 μM serotonin (5-HT).
5-HT2C receptors [89]. Source: human recombinant CHO-K1 cells. Ligand: 1 nM [3H]mesulergine. Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 0.1% ascorbic acid, 10 μM pargyline; KD: 1.1 nM, Bmax: 4.9 pmole/mg protein, specific binding: 90%; non-specific ligand: 1 μM mianserin.
5-HT3 receptors [90, 91]. Source: human recombinant HEK-293 cells. Ligand: 0.69 nM [3H]3-(5-methyl-1H-imidazol-4-yl)-1-(1-methyl-1H-indol-3-yl)-1-propanone (GR-65630). Incubation time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 5 mM MgCl2; KD: 0.2 nM, Bmax: 11 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM tropanyl-3,5-dichlorobenzoate (MDL-72222).
5-HT4 receptors [92]. Source: Duncan Hartley derived Guinea pig striatum. Ligand: 0.7 nM [3H] -methyl-1H-indole-3-carboxylic acid, [1-[2-[(methylsulfonyl) amino] ethyl]-4-piperidinyl]methy ester (GR-113808). Incubation time/temp: 30 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 0.14 nM, Bmax: 0.13 pmole/mg pro-tein, specific binding: 80%; non-specific ligand: 30 μM 5-HT.
5-HT6 receptors [93]. Source: human recombinant HeLa cells. Ligand: 1.5 nM [3H]lysergic acid diethylamide (LSD). Incubation time/temp: 120 minutes @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM ascorbic acid, 0.001% BSA; KD: 1.3 nM, Bmax: 1.7 pmole/mg

102 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
protein, specific binding: 90%; non-specific ligand: 5 μM 5-HT.
5-HT7 receptors [94, 95]. Source: human recombinant CHO cells. Ligand: 5.5 nM [3H]LSD. Incubation time/temp: 120 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 0.5 mM EDTA; KD: 7.4 nM, Bmax: 0.95 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM 5-HT.
Sigma 1 receptors [96]. Source: human Jurkat cells. Ligand: 8 nM [3H]haloperidol. Incubation time/temp: 4 hours @ 25°C; incubation buffer: 5 mM potassium phos-phate; KD: 5.8 nM, Bmax: 0.71 pmole/mg protein, specific binding: 80%; non-specific ligand: 10 μM haloperidol.
Sigma 2 receptors [97]. Source: Wistar rat brain. Ligand: 3 nM [3H]ifenprodil. Incubation time/temp: 60 minutes @ 37°C; incubation buffer: 50 mM Tris-HCl, pH 7.4; KD: 4.8 nM, Bmax: 1.3 pmole/mg protein, specific binding: 85%; non-specific ligand: 10 μM ifenprodil.
Sodium Channel, Site 2 [98]. Source: Wistar rat brain. Ligand: 5 nM [3H]batrachotoxin. Incubation time/temp: 60 minutes @ 37°C; incubation buffer: 50 mM HEPES, 50 mM Tris-HCl, pH 7.4, 130 mM choline chloride, 5.4 mM KCl, 0.8 mM MgCl2, 5.5 mM glucose, 40 μg/ml LqTx; KD: 52 nM, Bmax: 0.7 pmole/mg protein, specific binding: 77%; non-specific ligand: 100 μM veratridine.
Tachykinin NK1 receptors [99]. Source: human recombi-nant CHO cells. Ligand: 0.25 nM [3H]1-(2-(3-(3,4-dichlorophenyl)-1-(3-isopropoxyphenylacetyl)piperidin-3-yl)ethyl)-4-phenyl-1-azabicyclo(2.2.2)octane chloride (SR-140333). Incubation time/temp: 90 minutes @ 25°C; incuba-tion buffer: 50 mM HEPES, pH 7.4, 1 mM MnCl2, 0.1% BSA; KD: 0.3 nM, Bmax: 10 pmole/mg protein, specific bind-ing: 85%; non-specific ligand: 2 μM 2-(diphenylmethyl)-N-((2-iodophenyl)methyl)-3-quinuclidinamine (L-703,606).
Transporter, Dopamine [100, 101]. Source: human re-combinant CHO-K1 cells. Ligand: 0.15 nM [125I](-)-2 -carbomethoxy-3 -(4-iodophenyl)tropane (RTI-55). Incuba-tion time/temp: 180 minutes @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 μM leupeptin, 10 μM PMSF; KD: 0.58 nM, Bmax: 0.047 pmole/mg protein, specific binding: 90%; non-specific ligand: 10 μM nomifens-ine.
Transporter, GABA [102]. Source: Wistar rat cerebral cortex. Ligand: 6 nM [3H] -aminobutyric acid (GABA). In-cubation time/temp: 20 minutes @ 25°C; incubation buffer: 50 mM HEPES, pH 7.4, 120 mM NaCl, 4 mM Ca(CH3COO)2, 10 μM isoguvacine, 10 μM S(-)-Baclofen; KD: 300 nM, Bmax: 60 pmole/mg protein, specific binding: 80%; non-specific ligand: 10 μM 1-(2-(((diphenylmethylene)amino)oxy)ethyl)-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid (NO-711).
Transporter, Norepinephrine [103]. Source: human re-combinant MDCK cells. Ligand: 0.2 nM [125I]RTI-55. Incu-bation time/temp: 180 minutes @ 4°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 μM leupeptin, 10 μM phenylmethanesulfonyl fluoride (PMSF); KD: 24 nM, Bmax: 2.5 pmole/mg protein, specific binding: 75%; non-specific ligand: 10 μM desipramine.
Transporter, 5-HT [104, 105]. Source: human recombi-nant HEK-293 cells. Ligand: 0.4 nM [3H]paroxetine. Incuba-tion time/temp: 60 minutes @ 25°C; incubation buffer: 50 mM Tris-HCl, pH 7.4, 120 mM NaCl, 5 mM KCl; KD: 0.078 nM, Bmax: 4.4 pmole/mg protein, specific binding: 95%; non-specific ligand: 10 μM imipramine.
2.4. Functional Assays
Histamine H1 receptors [106, 107]. Source: human neu-roblastoma cell line, SK-N-SH grown in Hybridoma Serum Free Medium. Cells were loaded at room temperature with calcium-sensitive dye, 4 μM Fura-2AM (Invitrogen, USA). DimebonTM was added either before or 30 sec after addition of 10 μM histamine, to measure its effect either on hista-mine-induced spike of calcium mobilization (Phase 1) or on the rate of calcium removal (Phase 2), respectively. Incuba-tion media: 145 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1.8 mM CaCl2, 30 mM HEPES, 11.2 mM D-glucose. Intra-cellular [Ca2+]i was assessed in ratiometric mode by Fura-2 fluorescence emission measured at 510 nm upon alternate excitation at 340 nm and 380 nm.
5-HT6 receptors [108]. Source: HEK-293 cells co-transfected with tetracycline-inducible 5-HT6 receptor T-Rex System (Invitrogen, Carlsbad, CA) were obtained from Eto-gen Scientific (San Diego, CA). The cells were grown in DMEM/10%FBS/1%AAS medium with Blasticidine S and Zeocin (both from Invitrogen, Carlsbad, CA). 5-HT6 receptor expression was activated by addition of tetracycline, as rec-ommended by the manufacturer, a day before the experi-ments. Incubation time/temp: 15 minutes @ RT°C with Di-mebonTM and additional 30 min @ RT°C with serotonin; incubation buffer: HBSS, 1 mM MgCl2, 1 mM CaCl2 100 μM IBMX, 5 mM HEPES, pH 7.4. Method: cAMP accumu-lation was assessed using cAMP LANCE assay kit (Perki-nElmer, USA) as recommended by the manufacturer.
hERG patch clamp [79]. Source: human recombinant hERG stably expressed in HEK-297 cells. Intracellular solu-tion: 130 mM KCl, 5 mM EGTA, 5 mM Mg Cl2, 10 mM HEPES, 5 mM Na-ATP, pH 7.2. Extracellular solution: 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 11 mM dextrose, 10 mM HEPES, pH 7.4. Voltage clamp meas-urements are performed at 25°C using PatchXpress 7000A (Molecular Devices, Sunnyvale, CA). hERG channels were activated by 2 second pulses to +20 mV from a holding po-tential of –80 mV, and peak tail currents were recorded upon re-polarization to –50 mV. This voltage-clamp pulse proto-col was performed continuously during the experiment (ve-hicle control, test compound, washout, and positive control additions). An inter pulse interval of 15 seconds allowed recovery from any residual inactivation. Test compounds were incubated with cells until the current reached a steady state level (3-8 minutes). After the final test compound con-centration was tested, test compound was washed out with continuous perfusion of extracellular solution for 3 minutes, followed by application of positive control (10 μM cis-apride). Data were analyzed using DataXpress software. Per-cent of control values were calculated based on current peak at each DimebonTM concentration relative to maximal tail current in the presence of vehicle control.

From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 103
2.5. Curve Fitting
Concentration-dependent curves of DimebonTM action were fitted with Prism 4 (GraphPad, San Diego, CA) using built-in sigmoidal dose-response (variable slope) function. Ki values for DimebonTM competitive binding were calculated based on the following Cheng-Prusoff equation:
50
1 /i
D
ICK
L K=
+
, where
IC50 is a half maximal concentration of DimebonTM calcu-lated from fitting the data, L is the concentration of the Radi-oligand, and KD is equilibrium constant for the Radioligand binding to the receptor under the same experimental condi-tions used in the competition experiments.
3. RESULTS.
3.1. Interaction of DimebonTM
with Histamine Receptors
A concentration-dependent displacement of radio-labeled ligands from their complexes with H1 and H2 receptors by DimebonTM is shown in Fig. (1). These data confirm the original report [1] showing that DimebonTM interacts with both H1 and H2 histaminergic receptors. Binding affinity of DimebonTM to H1 receptor (Fig. 1) determined in our ex-periments (IC50 = 3.8 nM) corroborates well with the data published by Medivation (IC50 = 3.4 nM) [109]. Though still in a nanomolar range, the affinity of DimebonTM to H2 recep-tor (IC50 = 287 nM) was almost two orders of magnitude lower than that to H1 receptor (Fig. (1)). Our data also show that at concentrations as high as 10 μM, DimebonTM does not interact with either H3 or H4 receptors Fig. (3). The ability of DimebonTM to interact with H2 receptors could provide an additional advantage to patients with gastrointestinal condi-tions such as peptic ulcers and gastro esophageal reflux dis-ease caused by histamine-induced gastric acid production through activation of H2 histamine receptors [110]. Next, we have studied the effect of DimebonTM on H1 receptor-induced calcium fluxes in a functional cell-based setting. We used SK-N-SH neuroblastoma cells, which respond to hista-mine with a robust calcium spike (Fig. (2)). Selective H1 receptor antagonist, d-chlorpheniramine, potently blocked the histamine-induced calcium mobilization in the cells with IC50 of 29 nM, which was in good agreement with data on its ability to displace mepyramine from mammalian brain mem-branes [111]. Though we failed to find in the literature any data indicating the histamine receptor subtype that is ex-pressed in SK-N-SH cells, it is well established that H1 re-ceptors are coupled through G q proteins with the calcium signaling machinery. Other histaminergic receptors, H2 and H3, are shown to act through, respectively, G s and G i/o [112, 113] modulating cAMP level in the cells. It was shown, though, that in mast cells endogenously expressing H4 recep-tor, histamine was able to also induce calcium mobilization [114]. In our experiments, neither H3/H4 antagonist, thioperamide maleate [115], nor H4 specific antagonist, 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methylpiperazine (JNJ 7777120) [116, 117] were able to block the histamine-induced calcium responses at concentrations as high as 10 μM (data not shown). Thus, based on the pharmacological subtype-specific inhibition profile of the histamine-induced responses in the SK-N-SH cells, we conclude that in this cell
line the endogenous histamine receptors that signal through calcium mobilization belong to the H1 type.
Fig. (1). Concentration-dependent displacement by DimebonTM of radio-labeled ligands, [3H]pyrilamine and [125I]aminopotentidine, from histamine H1 and H2 receptors, correspondingly. For the assay details, see Materials and Methods. Means±SEM (n=2) are shown.
In the cell-based functional assay, we have confirmed that DimebonTM antagonized histamine-induced calcium fluxes initiated through activation of H1 receptors (Fig. (2)). Added shortly before histamine, DimebonTM effectively blocked the histamine-induced mobilization of intracellular [Ca2+]i (Fig. (2A)). Addition of DimebonTM after the hista-mine-induced onset of the calcium spike, led to an apparent acceleration of intracellular calcium removal (Fig. (2B)), probably through blockage of capacitative calcium entry. Khankoeva et al. have previously described a similar finding that DimebonTM as well as other H1 receptor specific antago-nists blocked both the first and second phase of histamine-induced calcium spikes in cardiomyocytes with inhibition of the second phase being less sensitive to the blockers [118]. Our data corroborate well with this observation. In the SK-N-SH neuroblastoma cells, the potency of DimebonTM to block the second phase of calcium entry into the cells was an order of magnitude lower than that to block calcium mobili-zation from internal stores (Fig. (2C)). Faster removal of the calcium ions from cytoplasm (presumably, due to prevention of calcium ion influx) in the presence of the H1 receptor an-tagonist would accelerate the cell recovery after excitation and prevent the cells from the potentially cytotoxic effect of calcium overload. Earlier, one of us [119] showed a similar effect of 5-HT2A receptor antagonist, spiperone, to abolish second phase of calcium ion entry into (and accelerating calcium removal from) HEK-293 cells heterologously ex-pressing 5-HT2A receptor. Contrary to DimebonTM that is more potent to block first phase of the calcium ion spike induced by activation of the H1 receptor, spiperone exhibited higher potency to block the second phase of the the 5-HT2A receptor-induced calcium metabolism.
When comparing affinity of DimebonTM measured in the binding assay (Fig. (1), IC50=3.8 nM) with its potency meas-ured in the cell-based functional setting (Fig. (2C), IC50=158 nM), substantial difference can be seen. Such a discrepancy

104 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
between the affinity measured in an equilibrium binding as-say and potency measured in the non-equilibrium functional assay can result either from binding kinetics or from differ-ences in counter ligand concentrations (1.2 nM [3H]pyrilamine in binding experiments versus 10 μM hista-mine in the functional assay). Further work is needed to clar-ify this difference.
3.2. DimebonTM
Interacts with a Broad Panel of Thera-
peutic Targets
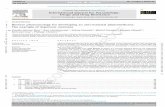
A specificity profile of DimebonTM was determined using a broad panel of 70 therapeutic targets that includes en-zymes, ion channels, neurotransmitter transporters, and G protein-coupled receptors (GPCR). The data show (Fig. (3)) that DimebonTM interacts with a broad spectrum of receptor classes. Besides with its earlier reported activity on butyryl-cholinesterase and histaminergic H1 and H2 receptors [1], DimebonTM interacts - although with relatively low affinities (~ 50% radioligand displacement at 10 M) - with some ion channels (benzothiazepine site of L-type Ca2+ channel, site 2 of sodium channel, and hERG potassium channel) and trans-porter for norepinephrine but not with other transporters. While showing no interaction with adenosine A1 and A2A receptors, DimebonTM binds to adrenergic (including imi-
dazoline I2 receptor) but not to ß adrenergic receptors. Di-mebonTM also effectively competes with specific radio-labeled ligands of dopaminergic and serotonergic receptors (Fig. (3)). We studied selectivity profiles of DimebonTM amongst the members of the respective therapeutic target constellations. Binding affinities of DimebonTM to the recep-tors were assessed by concentration-dependent competitive displacement of corresponding radio-labeled ligands in ac-cordance with published procedures briefly described in Ma-terials and Methods.
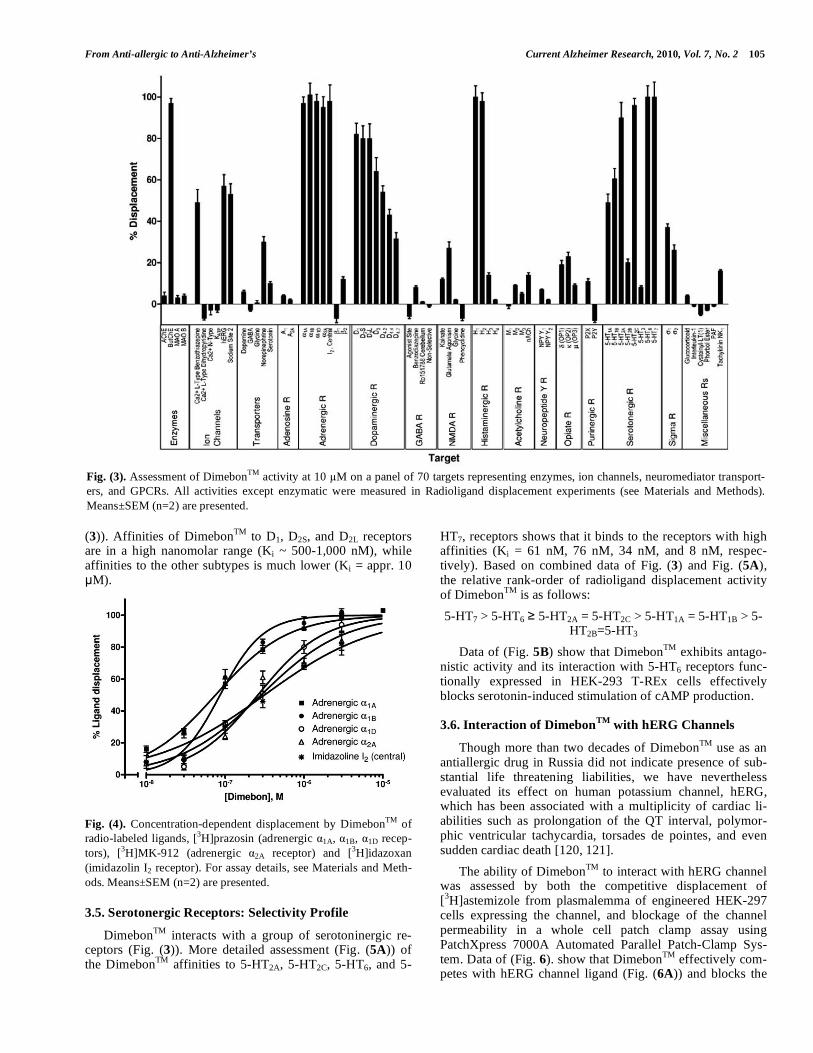
3.3. Alpha Adrenergic and Imidazoline Receptors: Selec-tivity Profile
DimebonTM potently competes for binding (Fig. (4)) with representatives of adrenergic receptors in the potency rank-order of 1A = 1B > 1D = 2A = I2 with approximately 3-fold preference for 1A, 1B receptors (Ki ~ 74-86 nM) over 1D,
2A and I2 receptors (Ki ~ 261-313 nM). No binding of Di-mebonTM to beta adrenoceptors was noticeable (see Fig. (3)).
3.4. Dopaminergic Receptors: Selectivity Profile
At 10 μM, DimebonTM moderately displaces correspond-ing radiolabeled ligands from dopaminergic receptors in the efficacy rank-order of D1=D2S=D2L>D3 D4.2>D4.4 D4.7 (Fig.
Fig. (2). Modulation by DimebonTM of histamine-induced [Ca2+]i signaling in SK-N-SH cells loaded with calcium sensitive ratiometric dye, Fura-2. Arrows indicate time points, at which the compounds are added to the cells. A. DimebonTM was added to the cells a few seconds before histamine and the peak values were assessed 30 s later to assess the inhibition effect on Phase 1 (dotted line). B. DimebonTM was added to the cells 30-35 s after histamine addition and the [Ca2+]i dissipation in the Phase 2 were assessed. C. Concentration-dependent inhi-bition by DimebonTM of histamine-induced calcium spikes (Phase 1) and acceleration of [Ca2+]i dissipation in the sustained phase (Phase 2). The rates of [Ca2+]i dissipation were calculated by fitting the data in (B) with one-phase decay equation built-in into Prism 4. Shown are Means±SEM (n=2) from one representative experiment.
From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 105
(3)). Affinities of DimebonTM to D1, D2S, and D2L receptors are in a high nanomolar range (Ki ~ 500-1,000 nM), while affinities to the other subtypes is much lower (Ki = appr. 10 μM).
Fig. (4). Concentration-dependent displacement by DimebonTM of radio-labeled ligands, [3H]prazosin (adrenergic 1A, 1B, 1D recep-tors), [3H]MK-912 (adrenergic 2A receptor) and [3H]idazoxan (imidazolin I2 receptor). For assay details, see Materials and Meth-ods. Means±SEM (n=2) are presented.
3.5. Serotonergic Receptors: Selectivity Profile
DimebonTM interacts with a group of serotoninergic re-ceptors (Fig. (3)). More detailed assessment (Fig. (5A)) of the DimebonTM affinities to 5-HT2A, 5-HT2C, 5-HT6, and 5-
HT7, receptors shows that it binds to the receptors with high affinities (Ki = 61 nM, 76 nM, 34 nM, and 8 nM, respec-tively). Based on combined data of Fig. (3) and Fig. (5A), the relative rank-order of radioligand displacement activity of DimebonTM is as follows:
5-HT7 > 5-HT6 5-HT2A = 5-HT2C > 5-HT1A = 5-HT1B > 5-HT2B=5-HT3
Data of (Fig. 5B) show that DimebonTM exhibits antago-nistic activity and its interaction with 5-HT6 receptors func-tionally expressed in HEK-293 T-REx cells effectively blocks serotonin-induced stimulation of cAMP production.
3.6. Interaction of DimebonTM
with hERG Channels
Though more than two decades of DimebonTM use as an antiallergic drug in Russia did not indicate presence of sub-stantial life threatening liabilities, we have nevertheless evaluated its effect on human potassium channel, hERG, which has been associated with a multiplicity of cardiac li-abilities such as prolongation of the QT interval, polymor-phic ventricular tachycardia, torsades de pointes, and even sudden cardiac death [120, 121].
The ability of DimebonTM to interact with hERG channel was assessed by both the competitive displacement of [3H]astemizole from plasmalemma of engineered HEK-297 cells expressing the channel, and blockage of the channel permeability in a whole cell patch clamp assay using PatchXpress 7000A Automated Parallel Patch-Clamp Sys-tem. Data of (Fig. 6). show that DimebonTM effectively com-petes with hERG channel ligand (Fig. (6A)) and blocks the
Fig. (3). Assessment of DimebonTM activity at 10 M on a panel of 70 targets representing enzymes, ion channels, neuromediator transport-ers, and GPCRs. All activities except enzymatic were measured in Radioligand displacement experiments (see Materials and Methods). Means±SEM (n=2) are presented.

106 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
channel conductance (Fig. (6B)) in a low micromole range of concentrations.
Fig. (5). A. Concentration-dependent displacement of radio-labeled ligands by DimebonTM from heterologously expressed human sero-tonin receptors, 5-HT7 ([3H]LSD), 5-HT6 ([3H]LSD), 5-HT2C ([3H]mesulergine), and 5-HT2A ([3H]ketanserin). B. Inhibition of serotonin-induced cAMP increase in HEK-293 T-REx cells trans-fected with functional human 5-HT6 receptor. Means±SEM, n=2, are presented.
4. DISCUSSION
Repurposing of approved drugs for new therapeutic areas became very popular within the pharmaceutical industry due to potential drastic reductions in the costs relative to de novo development of new drugs. The foundation for such an ap-proach is based on the fact that many drugs are rarely highly selective and often display activity to more than one thera-peutic target. At the same time, the established drugs have already been on the market for a long time and their adverse effects are very well established and, hence, redirecting of the established drug to a new therapeutic area does not re-quire additional expensive testing for toxicity, bioavailabil-ity, ADME etc.
As mentioned, DimebonTM, a tricyclic molecule (Fig. (7)), was originally developed in Russia as the antiallergic antihistamine drug with the typical side effect of causing drowsiness through blockage of neuronal H1 receptors. Aside from their role in pathogenesis of allergic reactions, these
receptors were shown to also participate in other pathologies such as gastroenterological, muscle, and CNS disorders [122].
Fig. (6). Interaction of DimebonTM with hERG channel. A. Com-petitive displacement of [3H]astemizole from hERG-HEK293 cell membranes. Means±SEM (n=2) are presented B. Blockage of hERG potassium channel conductance. For corresponding assay details, see Materials and Methods. Means±SEM (n=3) are pre-sented.
N
N
N
CH3H3C
CH3
2HCl
Fig. (7). Chemical structure of DimebonTM.

From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 107
It is well documented that classical antihistamines - espe-cially ones of the first- and second-generation - interact with and block many other receptors, for example, muscarinic acetylcholine receptors, -adrenergic receptors, and 5-HT receptors [123-125].
On the other hand, some tricyclic antidepressant drugs inhibiting reuptake of serotonin, norepinephrine and, to a lesser extent, dopamine, also bind to and inhibit many other receptors including, in particular, H1 receptors [126]. It has also been shown [127] that a large number of drugs structur-ally unrelated to tricyclics, which possess clinically proven antidepressant properties, share the ability to act as potent inhibitors of histamine receptors.
DimebonTM also exhibits moderate activity (in micromo-lar range) towards a few additional targets [6, 9-12]. Our data (Fig. (3)) as well as those by others [12] show that Di-mebonTM demonstrates binding propensity toward quite a broad spectrum of therapeutic targets.
The adrenergic receptors, play an important role in a variety of psychiatric diseases, migraine, schizophrenia, sleep disorders etc. [122], as well as in controlling benign prostatic hyperplasia [128], coronary stenosis [129, 130] and cerebellum-dependent neurological diseases [131]. It was also shown that stimulation of -2 adrenergic receptors im-proved memory and cognition in aged monkeys [132, 133]. Therefore, it is plausible to suggest that DimebonTM can be adventageous for treatment of these diseases as it is effective in blocking the receptors (Fig. (3)).
The imidazoline I2 receptors have been associated with neuroprotection during cerebral ischemia [134]. Indeed, Di-mebonTM was shown to protect neuronal cells from death caused by neurotoxic action of beta-amyloid [9]. Taking into consideration its relatively high affinity to these receptors (Fig. (4)), one could reasonably expect that DimebonTM might have additional positive effects in Alzheimer’s pa-tients who also suffer from associated mental disorders. On the other hand, potent antagonism of 1-adrenergic receptors with tricyclic and tetracyclic antidepressants [135] is associ-ated with orthostatic hypotension, which could be a negative indication for DimebonTM. Unlike some other antihistamines [125], DimebonTM does not interact with the muscarinic re-ceptors tested (Fig. (3)). In this respect, one of its few side effects noted in the literature - dry mouth or xerostomia, seems to be caused by reasons other than muscarinic receptor inhibition of salivary secretion [136].
Dopaminergic receptors that are also sensitive to Dime-bonTM (Fig. (3)) are recognized targets for psychotropic drugs such as antipsychotics and psycho stimulants [137]. These receptors are widely distributed in the brain and par-ticipate in controlling neural signaling. Dysfunction of do-paminergic neurotransmission in the CNS has been impli-cated in a variety of neuropsychiatric disorders, including social phobia [138], Tourette's syndrome [139], Parkinson's disease [140], schizophrenia [139], neuroleptic malignant syndrome [141], attention-deficit hyperactivity disorder (ADHD) [142], and drug and alcohol dependence [139, 143]. The receptors are also present in non-CNS tissues such as the cardio-pulmonary [144] and renal systems [145]. Agonists of D1-like and D2-like receptors could be beneficial for patients
with high blood pressure [145], whereas chronic use of the receptor antagonists could potentially lead to hypertension due to reduced dopamine activity in cardiovascular and renal systems. Therefore, further work will need to be carried out to determine if DimebonTM is an agonist or antagonist of the receptors.
As our data indicate (Fig. (3)), DimebonTM interacts with a broad class of serotonergic receptors. Polymorphisms of 5-HT2A and 5-HT2C receptors (T102C and C23S, respectively) are thought to be associated with different neuropsychiatric symptoms, including hallucinations and depression in pa-tients with Alzheimer’s disease [146-148] and may be re-sponsible for antipsychotic treatment responses in such pa-tients [149]. In in vivo animal models, it was shown that in-verse agonists of 5-HT2A/2C receptors exhibited antipsychotic and anti-dyskinetic activity [150]. The selective 5-HT2A re-ceptor antagonist, EMD281014, was able to dose-dependently improve working memory functions, which could indicate substantial therapeutic benefits to psychiatric patients. Thus, high affinity of DimebonTM to both 5-HT2A and 5-HT2C receptors (Fig. (5A)) manifests additional bene-fits of the drug when administered to patients with Alz-heimer’s disease.
5-HT6 and 5-HT7 receptors have recently been discov-ered, pharmacologically characterized [93, 151, 152], and their role in cognition, mood regulation, novelty seeking, and substance abuse has been proposed [153]. The receptors are almost exclusively expressed in the central nervous system [154]. Many typical and atypical antipsychotic agents showed high affinities to both types of the receptor [93, 94, 152] indicating a possibility that DimebonTM can also exhibit antipsychotic effects in AD patients. This makes the 5-HT6 and 5-HT7 receptors very attractive targets for antipsychotic intervention [155]. The receptor antagonists were shown to play a significant role in memory formation and in improv-ing learning consolidation and memory retention [156]. The blockade of 5-HT6 receptors is thought to lead to an im-provement of cognitive performance in a wide variety of learning and memory paradigms and also in anxiolytic and antidepressant-like activity. Recent reports show that 5-HT6 receptor antagonists demonstrate cognition-enhancing prop-erties [157], improvement of spatial recognition memory and reversal of age-related consolidation deficits of episodic-like memory [158]. This further highlights the therapeutic poten-tial of DimebonTM for treatment of AD patients.
Currently, Medivation and Pfizer are repurposing Dime-bonTM to treat Alzheimer’s and Huntington’s diseases. Pre-liminary clinical trial data (Phase II and Phase III) show that DimebonTM not only slows down but even reverses deterio-ration processes in some AD patients. As mentioned in the introduction, positive effects of DimebonTM on AD patients was initially assigned to its ability to interact with several targets that are thought to be involved in the disease and, in particular, with its ability to inhibit butyryl cholinesterase. Indeed, treatment of AD patients with cholinesterase inhibi-tors was shown to exhibit a strong negative correlation be-tween butyryl cholinesterase (but not acetyl cholinesterase) activity in their cerebrospinal fluid and the level of recogni-tion in the patients [159]. However, unlike other highly po-tent cholinesterase inhibitors under development for treat-

108 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
ment of AD [160], relatively high concentrations of Dime-bonTM required to inhibit the enzyme (IC50 in the range be-tween 1.3 μM in accordance with our data and 7.9 μM documented elsewhere [6]) makes it highly unlikely that its positive effect on the AD patients is mediated through bu-tyryl cholinesterase inhibition. We did not find data in avail-able literature indicating inhibition of butyryl cholinesterase in cerebrospinal fluid of the AD patients treated with Dime-bonTM, so this issue is to be clarified. For the same reason of low affinity, the other earlier suggested targets - blockage of neurotoxic effects of beta-amyloid [6], inhibition of L-type calcium channels [9], modulation of activity of glutamate receptors [10], and blockage of MPTP [11] - could be mar-ginally, if at all, responsible for the positive effects of Dime-bonTM on AD patients. Using a mice model [161] showed that DimebonTM can block TNF -induced changes in lipid metabolism and subsequent production of eicosanoids, ceramide, and ROS that may play a role in potentiating brain injury in AD patients.
Thus, in accordance with literature and our data, DimebonTM exhibits activity against a broad spectrum of therapeutically valid targets with micromolar to nanomolar potencies. Which of these targets are responsible for the beneficial effects of Dimebon in Alzheimer’s patients remains to be elucidated. The answer to this question could be gained from exposure levels of Dimebon in plasma and/or cerebrospinal fluid of patients with Alzheimer’s and Huntington’s diseases during clinical trials performed by Medivation. If effective DimebonTM concentrations were substantially below a threshold sensitivity of the low potency targets, such as cholinesterase, NMDA receptor, L-type channels etc., then that information would eliminate such targets from consideration. Unfortunately, we could not find such exposure levels of DimebonTM in available literature. An indirect indication that the DimebonTM exposure level in plasma should be below micromolar concentration range could be deduced from hERG channel blocking activity (Fig. (6)). Our data show that DimebonTM interacts with this channel in a micromolar range and hence the high micromolar levels of exposure would lead to cardiotoxicity, which has not been observed in the patients.
Successful repurposing of the antihistamine drug Dime-bonTM for treatment of Alzheimer’s and Huntington’s dis-eases raises some very interesting considerations regarding specificity and selectivity of drugs in general. Though tradi-tionally highly desirable, absolute specificity/selectivity of a drug might rarely be attainable. While “contamination” of the drug’s molecular pharmacology space with unintended targets is considered to be a negative factor in classical me-dicinal chemistry, it might also present an advantage in treat-ing patients with several associated impairments. It was shown, for example, that concomitant cholinergic and adren-ergic stimulation has positive effect on learning and memory performance by young and aged monkeys [162]. Instead of using a cocktail of several drugs, one could consider devel-opment of new drugs based on the principle of “all-in-one molecule.” Besides, for such a disease as Alzheimer’s, which has a very complex etiology with many neurotransmitter signaling pathways involved, such a poly targeted activity might be of special significance. J.J. Buccafusco and his col-leagues [163] suggested an idea of the development of multi-
functional compounds, which “will obviate the challenge of administering multiple single drug entities with potentially different degrees of bioavailability, pharmacokinetics, and metabolism.” DimebonTM seems to be an intriguing drug in such respects. Its chemical structure allows for elaborate combinatorial modifications and it would be of interest to study the possibility of directed systematic adjustments of its specificity/selectivity profile by structurally modifying the molecule with corresponding assessment of the AD treat-ment efficacy. Currently, we are working on a series of such modifications and results will be published elsewhere.
ACKNOWLEDGEMENTS
The authors wish to thank MDS Pharma Services for their analyses of DimebonTM in radio-binding assays.
REFERENCES
[1] Matveeva IA. Action of dimebon on histamine receptors. Farmakol Toksikol (in Russian) 46(4): 27-9 (1983).
[2] Lermontova NN, Lukoyanov NV, Serkova TP, Lukoyanova EA, Bachurin SO. Dimebon improves learning in animals with experi-mental Alzheimer's disease. Bull Exp Biol Med 129(6): 544-6 (2000).
[3] Doody R, Gavrilova S, Sano M, Thomas R, Aisen P, Bachurin S, et al. Effect of dimebon on cognition, activities of daily living, be-haviour, and global function in patients with mild-to-moderate Alz-heimer's disease: a randomised, double-blind, placebo-controlled study. Lancet 372(9634): 207-15 (2008).
[4] http://www.medivation.com/pipeline_alzheimer.html [accessed 03/ 04/2009]
[5] Galenko-Yaroshevskii PA, Chekanova OA, Skibitskii VV, Bar-tashevich VV, Khankoeva AI, Polyashova TI. Antiarrhythmic properties of Dimebox. Bull Exp Biol Med 119(4): 362-4 (1995).
[6] Bachurin S, Bukatina E, Lermontova N, Tkachenko S, Afanasiev A, Grigoriev V, et al. Antihistamine agent Dimebon as a novel neu-roprotector and a cognition enhancer. Ann NY Acad Sci 939: 425-35 (2001).
[7] Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeabil-ity in drug discovery and development settings. Adv Drug Del Rev 23: 3-25 (1997).
[8] Oprea TI, Davis AM, Teague SJ, Leeson PD. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci 41: 1308-15 (2001).
[9] Lermontova NN, Redkozubov AE, Shevtsova EF, Serkova TP, Kireeva EG, Bachurin SO. Dimebon and tacrine inhibit neurotoxic action of beta-amyloid in culture and block L-type Ca(2+) chan-nels. Bull Exp Biol Med 132(5): 1079-83 (2001).
[10] Grigorev VV, Dranyi OA, Bachurin SO. Comparative study of action mechanisms of dimebon and memantine on AMPA- and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull Exp Biol Med 136(5): 474-7 (2003).
[11] Bachurin SO, Shevtsova EP, Kireeva EG, Oxenkrug GF, Sablin SO. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann NY Acad Sci 993: 334-44 (2003).
[12] Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington’s disease. Mol Neurodegener 3(15): http://www.molecularneurodegeneration.com/content/3/1/15 (1995).
[13] Ellman GL, Courtney KD, Andres Jr, V, Featherstone RN. A new and rapid colorimetric determination of acetylcholinesterase activ-ity. Biochem Pharmacol 7: 88-95 (1961).
[14] Nadarajah B. The effect of pralidoximine chloride in the assay of acetylcholinesterase using 5,5’-dithio-bis(2-nitrobenzoic acid) (Ellman’s reagent). J Anal Toxicol 16: 192-3 (1992).
[15] Alcala MM, Vivas NM, Hospital S, Camps P, Munoz-Torrero D, Badia A. Characterisation of the anticholinesterase activity of two new tacrine-huperzine A hybrids. Neuropharmacology 44(6): 749-55 (2003).

From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 109
[16] Dorris RL. A simple method for screening monoamine oxidase (MAO) inhibitory drugs for type preference. J Pharmacol Methods 7(2): 133-7 (1982).
[17] Libert F, Sande JV, Lefort A, Czernilofsky A, Dumont JE, Vassart G, et al. Cloning and functional characterization of a human A1 adenosine receptor. Biochem Biophys Res Commun 187(2): 919-26 (1992).
[18] Varani K, Gessi S, Dalpiaz A, Borea PA. Pharmacological and biochemical characterization of purified A2A adenosine receptors in human platelet membranes by [3H]CGS21680 binding. Br J Pharmacol 117: 1693-701 (1996).
[19] Michel AD, Loury DN, Whiting RL. Identification of a single 1A-adrenoceptor corresponding to the 1A-subtype in rat submaxillary gland. Br J Pharmacol 98: 883-9 (1989).
[20] Garcia-Sainz JA, Romero-Avila MT, Hernandez RA, Macias-Silva M, Olivares-Reyes A, Gonzalez-Espinosa C. Species heterogeneity of hepatic 1A -, 1B -, and 1C -subtypes. Biochem Biophys Res Commun 186: 760-7 (1992).
[21] Kenny BA, Chalmers DH, Philpott PC, Naylor AM. Characteriza-tion of an 1D -adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br J Pharmacol 115: 981-6 (1995).
[22] Uhlen S, Porter AC, Neubig RR. The novel -2 adrenergic radio-ligand [3H]MK912 is 2C selective among human 2A, 2B. and
2C.¸ adrenoceptors. J Pharmacol Exp Ther 271: 1558-65 (1994). [23] Trimmer PA, Evans T, Smith MM, Kendall T, Harden TK,
Mccarthy KD. Combination of immunocytochemistry and radio-ligand receptor assay to identify -adrenergic receptor subtypes on astroglia in vitro. J Neurosci 4(6): 1598-606 (1984).
[24] McCrea KE, Hill SJ. Salmeterol, a long-acting Ñ.-adrenoceptor agonist mediating cyclic AMP accumulation in a neuronal cell line. Br J Pharmacol 110: 619-26 (1993).
[25] Fève B, Elhadri K, Quignard-Boulange A, Pairault J. Transcrip-tional down-regulation by insulin of the 3-adrenergic receptor expression in 3T3-F442A adipocytes: a mechanism for repressing the cAMP signalling pathway. Proc Natl Acad Sci USA 91: 5677-81 (1994).
[26] Horlick RA, Ohlmeyer MH, Stroke IL, Strohl B, Pan G, Schilling AE, et al. Small molecule antagonists of the bradykinin B1 recep-tor. Immunopharmacology 43(2-3): 169-77 (1999).
[27] Phagoo SB, Reddi K, Anderson KD, Leeb-Lundberg LM, Warbur-ton D. Bradykinin B1 receptor up-regulation by interleukin-1 and B1 agonist occurs through independent and synergistic intracellular signaling mechanisms in human lung fibroblasts. J Pharmacol Exp Ther 298(1): 77-85 (2001).
[28] Eggerickx D, Raspe E, Bertrand D, Vassart G, Parmentier M. Mo-lecular cloning, functional expression and pharmacological charac-terization of a human bradykinin B2 receptor gene. Biochem Bio-phys Res Commun 187(3): 1306-13 (1992).
[29] Hess JF, Borkowski JA, Young GS, Strader CD, Ransom RW. Cloning and pharmacological characterization of a human bradyki-nin (BK-2) receptor. Biochem Biophys Res Commun 184(1): 260-8 (1992).
[30] Schoemaker H, Langer SZ. [3H]Diltiazem binding to calcium channel antagonist recognition sites in rat cerebral cortex. Eur J Pharmacol 111: 273-7 (1985).
[31] Ehlert FJ, Roeske WR, Itoga E, Yamamura HI. The binding of [3H]nitrendipine to receptors for calcium channel antagonists in the heart, cerebral cortex and ileum of rats. Life Sci 30: 2191-202 (1982).
[32] Gould RJ, Murphy KMM, Snyder SH. [3H]nitrendipine-labeled calcium channels discriminate inorganic calcium agonists and an-tagonists. Proc Natl Acad Sci USA 79: 3656-60 (1982).
[33] Moresco RM, Govoni S, Battani F, Trivulzio S, Trabucchi M. Omegaconotoxin binding decreases in aged rat brain. Neurobiol Aging 11(4): 433-6 (1990).
[34] Dearry A, Gingrich JA, Falardeau P, Fremeau RT, Jr, Bates MD, Caron MG. Molecular cloning and expression of the gene for a human D1 dopamine receptor. Nature 347: 72-6 (1990).
[35] Zhou Q-Y, Grandy DK, Thambi L, Kushner JA, Van Tol HHM, Cone R, et al. Cloning and expression of human and rat D1 dopa-mine receptors. Nature 347: 76-80 (1990).
[36] Grandy DK, Marchionni MA, Makam H, Stofko RE, Alfano M, Frothingham L, et al. Cloning of the cDNA and gene for a human D2 dopamine receptor. Proc Natl Acad Sci USA 86: 9762-6 (1989).
[37] Hayes G, Biden TJ, Selbie LA, Shine J. Structural subtypes of the dopamine D2 receptor are functionally distinct: expression of the
cloned D2A and D2B subtypes in a heterologous cell line. Mol Endocrinol 6: 920-6 (1992).
[38] Sokoloff P, Giros B, Martres M-P, Bouthenet M-L, and Schwartz J-C. Molecular cloning and characterization of a novel dopamine re-ceptor (D3) as a target for neuroleptics. Nature 347: 146-151 (1990).
[39] Van Tol HHM, Bunzow JR, Guan H-C, Sunahara RK, Seeman P, Niznik HB, et al. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 350: 610-4 (1991).
[40] Van Tol HHM, Wu CM, Guan H-C, Ohara K, Bunzow JR, Civelli O, et al. Multiple dopamine D4 receptor variants in the human population. Nature 358: 149-52 (1992).
[41] Mihara K, Kondo T, Suzuki A, Yasui-Furukori N, Ono S, Sano A, et al. Relationship between functional dopamine D2 and D3 recep-tors gene polymorphisms and neuroleptic malignant syndrome. Am J Med Genet Part B: Neuropsychiatr Genet 117(1): 57-60 (2003).
[42] Cain MJ, Garlick RK, Sweetman PM. Endothelin-1 receptor bind-ing assay for high throughput chemical screening. J Cardiovasc Pharmacol 17 (Suppl. 7): S150-1 (1991).
[43] Chiou WJ, Magnuson SR, Dixon D, Sundy S, Opgenorth TJ, Wu-Wong JR. Dissociation characteristics of endothelin receptor ago-nists and antagonists in cloned human type-B endothelin receptor. Endothelium 5(3): 179-89 (1997).
[44] Massague J. Epidermal growth factor-like transforming growth factodr: II. Interaction with epidermal growth factor receptors in human placenta membranes and A431 cells. J Biol Chem 258: 13614-20 (1983).
[45] Dittadi R, Gion M, Brazzale A, Bruscagnin G. Radioligand binding assay of epidermal growth factor receptor: causes of variability and standardization of the assay. Clin Chem 36: 849-854 (1990).
[46] Obourn JD, Koszewski NJ, Notides AC. Hormone-and DNA-binding mechanism of the recombinant human estrogen receptor. Biochemistry 32: 6229-36 (1993).
[47] Enna SJ, Snyder SH. Influences of ions, enzymes and detergents on gamma-aminobutyric acid-receptor binding in synaptic membranes of rat brain. Mol Pharmacol 13: 442-53 (1976).
[48] Martinin C, Rigacci T, Lucacchini A. [3H]muscimol binding site on purified benzodiazepine receptor. J Neurochem 41 1183-5 (1983).
[49] Damm HW, Muller WE, Schlafer U, Wollert U. [3H]flunitrazepam: Its advantages as a ligand, for the identification of benzodiazepine receptors in rat brain membranes. Res Comm Chem Pathol Phar-macol 22: 597-600 (1978).
[50] Speth RC, Wastek GJ, Yamamura HI. Benzodiacepam receptors: temperature dependence of [3H]flunitrazepam binding. Life Sci 24: 351-7 (1979).
[51] Facklam M, Bowery NG. Solubiliaztion and characterization of GABAB receptor binding sites from porcine brain synaptic mem-branes. Br J Pharmacol 110: 1291-6 (1993).
[52] Mathivet P, Bernasconi R, Barry JD, Marescaux C, Bittiger H. Binding characteristics of Õ-hydroxybutyric acid as a weak but se-lective GABAB receptor agonist. Eur J Pharmacol 321: 67-75 (1997).
[53] Cidlowski JA, Cidlowski NB. Regulation of glucocorticoid recep-tors by glucocorticoids in cultured HeLa S3 Cells. Endocrinology 109: 1975-82 (1981).
[54] London ED, Coyle JT. Specific binding of [3H]kainic acid to recep-tor sites in rat brain. Mol Pharmacol 15 492-505 (1979).
[55] Sills MA, Fagg G, Pozza M, Angst C, Brundish DE, Hurt SD, et al.
[3H]CGP 39653: a new N-methyl-D-aspartate antagonist radio-ligand with low nanomolar affinity in rat brain. Eur J Pharmacol 192: 19-24 (1991).
[56] Siegel BW, Sreekrishna K, Baron BM. Binding of the radiolabeled glycine site antagonist [3H]MDS105,519 to homomeric NMDA-NR1a receptors. Eur J Pharmacol 312(3): 357-65 (1996).
[57] Goldman ME, Jacobson AE, Rice KC, Paul SM. Differentiation of [3H] phencyclidine and (+)-[3H]SKF-10047 binding sites in rat cerebral cortex. FEBS Lett 190: 333-6 (1985).
[58] De Backer MD, Gommeren W, Moereels H, Nobels G, Van Gom-pel P, Leysen JE, et al. Genomic cloning, heterologous expression and pharmacological characterization of a human histamine H1 re-ceptor. Biochem Biophys Res Comm 197(3): 1601-8 (1993).
[59] Ruat M, Traiffort E, Bouthenet ML, Schwartz JC, Hirschfeld J, Buschauer A, et al. Reversible and irreversible labeling and autora-diographic localization of the cerebral histamine H2 receptor using

110 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
[125I]iodinated probes. Proc Natl Acad Sci USA 87(5): 1658-62 (1990).
[60] Yanai K, Ryu JH, Sakai N, Takahashi T, Iwata R, Ido T, et al. Binding characteristics of a histamine H3-receptor antagonist, [3H]S-methylthioperamide: comparison with [3H](R) -methyl-histamine binding to rat tissues. Jpn J Pharmacol 65(2): 107-12 (1994).
[61] Zhu Y, Michalovich D, Wu H, Tan KB, Dytko GM, Mannan IJ, et al. Cloning, expression, and pharmacological characterization of a novel human histamine receptor. Mol Pharmacol 59(3): 434-41 (2001).
[62] Brown CM, MacKinnon AC, McGrath JC, Spedding M, Kilpatrick AT. 2-Adrenoceptor subtypes and imidazoline-like binding in the rat brain. Br J Pharmacol 99: 803-9 (1990).
[63] Chin J, Cameron PM, Rupp E, Schmidt JA. Identification of a high affinity receptor for native interleukin-1 and interleukin-1ß on normal human lung fibroblasts. J Exp Med 165: 70-86 (1987).
[64] Martin V, Sawyer N, Stocco R, Unett D, Lerner MR, Abramovitz M, et al. Molecular cloning and functional characterization of mur-ine cysteinyl-leukotriene 1 (CysLT(1)) receptors. Biochem Phar-macol 62(9): 1193-200 (2001).
[65] Luthin GR, Wolfe BB. Comparison of [3H]pirenzepine and [.H]quinuclidinylbenzilate binding to muscarine cholinergic recep-tors in rat brain. J. Pharmacol Exp Ther 228(3): 648-55 (1984).
[66] Buckley NJ, Bonner TI, Buckley CM, Brann MR. Antagonist bind-ing properties of five clonal muscarinic receptors expressed in CHO-K1 cell. Mol Pharmacol 35(4): 469-76 (1989).
[67] Sheikh SP, O'Hare MMT, Tortora O, Schwartz TW. Binding of monoiodinated neuropeptide Y to hippocampal membranes and human neuroblastoma cell line. J Biol Chem 264: 6648-54 (1989).
[68] Fuhlendorff J, Gether U, Aakerlund L, Langeland-Hohansen N, Thogersen H, Melberg SG, et al.[Leu31,Pro34]neuropeptide Y: a specific Y1 receptor agonist. Proc Natl Acad Sci USA 87: 182-6 (1990).
[69] Rose PM, Fernandes P, Lynch JS, Frazier ST, Fisher SM, Kodu-kula K, et al. Cloning and functional expression of a cDNA encod-ing a human type 2 neuropeptide Y receptor. J Biol Chem 270(39): 22661-4 (1995).
[70] Davila-Garcia MI, Musachio JL, Perry DC, Xiao Y, Horti A, London ED, et al. [125I]IPH, an epibatidine analog, binds with high affinity to neuronal nicotinic cholinergic receptors. J Pharmacol Exp Ther 282(1): 445-51 (1997).
[71] Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol 131(4): 729-39 (2000).
[72] Simonin F, Befort K, Gaveriaux-Ruff C, Matthes H, Nappey V, Lannes B, et al. The human Ó-opioid receptor: genomic organiza-tion, cDNA cloning, functional expression, and distribution in hu-man brain. Mol Pharmacol 46: 1015-21 (1994).
[73] Simonin F, Gaveriaux-Ruff C, Befort K, Matthes H, Lannes B, Micheletti G, et al. Kappa-opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology and expression pattern in the central nervous system. Proc Natl Acad Sci USA 92 (15): 7006-10 (1995).
[74] Maguire P, Tsai N, Damal J, Cometta-Morini C, Upton C, Loew G. Pharmacological profiles of fentanyl analogs at μ, , and opiate receptors. Eur J Pharmacol 213: 219-25 (2002).
[75] Wang JB, Johnson PS, Persico AM, Hawkins AL, Griffin CA, Uhl GR. Human mu opiate receptor: cDNA and genomic clones, phar-macologic characterization and chromosomal assignment. FEBS Lett 338: 217-22 (1994).
[76] Ashendel CL.The phorbol ester receptor: a phospholipid-regulated protein kinase. Biochim Biophys Acta 822: 219-42 (1985).
[77] Herbert J-M, Castro-Faria-Neto HC, Barbosa-Filho JM, Cordeiro RSB, Tibirica E. Pharmacological evidence for the putative exis-tence of two different subtypes of PAF receptors on platelets and leukocytes; studies with yangambin. J Lipid Mediat Cell Signal 17: 1-14 (1997).
[78] Gaines KL, Hamilton S, Boyd III AE. Characterizatrion of the sulfonylurea receptor on beta cell membranes. J Biol Chem 263: 2589-92 (1988).
[79] Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, et al. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74(1): 230-41 (1998).
[80] Finlayson K, Turnbull L, January CT, Sharkey J, Kelly JS. [3H]dofetilide binding to HERG transfected membranes: a potential high throughput preclinical screen. Eur J Pharmacol 430(1): 147-8 (2001).
[81] Bo X, Burnstock G. High- and low-affinity binding sites for [3H]-a,b-methylene ATP in rat urinary bladder membranes. Br J Pharmacol 101: 291-6 (1990).
[82] Ziganshin AU, Hoyle CHV, Bo X, Lambrecht G, Mutschler E, Baumert HG, et al. PPADS selectively antagonized P2X-purinoceptor-mediated responses in the rabbit urinary bladder. Br J Pharmacol 110: 1491-5 (1993).
[83] Boyer JL, Cooper CL, Harden TK. [32P]3’-O-(4-Benzoyl)benzoyl ATP as a photoaffinity label for a phospholipase C-coupled P2Y-Purinergic receptor. J Biol Chem 265(23): 13515-20 (1990).
[84] May JA, McLaughlin MA, Sharif NA, Hellberg MR, Dean TR. Evaluation of the ocular hypotensive response of serotonin 5-HT1A and 5-HT2 receptor ligands in conscious ocular hypertensive cy-nomolgus monkeys. J Pharmacol Exp Ther 306(1): 301-9 (2003).
[85] Hoyer D, Engel G, Kalkman HO. Characterization of the 5-HT1B recognition site in rat brain: binding studies with (-)[125I] iodocya-nopindolol. Eur J Pharmacol 118: 1-12 (1985).
[86] Pazos A, Hoyer D, Palacios JM. Mesulergine, a selective serotonin-2 ligand in the rat cortex, does not label the receptors in porcine and human cortex: evidence for species differences in brain sero-tonin-2 receptors. Eur J Pharmacol 106: 531-8 (1985).
[87] Bonhaus DW, Bach C, DeSouza A, Salazar FHR, Matsuoka BD, Zuppan P, et al. The pharmacology and distribution of human 5-hydroxytryptamine2B (5-HT2B) receptor gene products: compari-son with 5-HT2A and 5-HT2C receptors. Br J Pharmacol 115: 622-8 (1995).
[88] Saucier C, Albert PR. Identification of an endogenous 5-hydroxytryptamine2A receptor in NIH-3T3 cells: agonist-induced down-regulation involves decreases in receptor RNA and number. J Neurochem 68: 1998-2011 (1997).
[89] Wolf WA, Schutz JS. The serotonin 5-HT2C receptor is a promi-nent serotonin receptor in basal ganglia: evidence from functional studies on serotonin-mediated phosphoinositide hydrolysis. J Neu-rochem 69: 1449-1458 (1997).
[90] Boess FG, Steward LJ, Steele JA, Liu D, Reid J, Glencorse TA, et al. Analysis of the ligand binding site of the 5-HT3 receptor us-ing site- directed mutagenesis: importance of glutamate 106. Neu-ropharmacology 36: 637-47 (1997).
[91] Millerk WE, Fletcher PW, Teitler M. Membrane-bound and solubi-lized brain 5-HT3 receptor: improved radioligand binding assay us-ing bovine area postrema or rat cortex and the radioligand [3H]GR65630, [3H]BRL43694, and [3H]LY278584. Synapse 11: 58-66 (1992).
[92] Grossman CJ, Kilpatrick GJ, Bunce KT. Development of a radio-ligand binding assay for 5-HT4 receptors in guinea-pig and rat brain. Br J Pharmacol 109: 618-24 (1993).
[93] Monsma FJ, Jr, Shen Y, Ward RP, Hamblin MW, Sibley DR. Clon-ing and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol Pharmacol 43: 320-7 (1993).
[94] Roth BL, Craigo SC, Choudhary MS, Uluer A, Monsma FJ, Shen Y, et al. Binding of typical and atypical antipsychotic agents to 5- hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. Pharm Exp Ther 268(3): 1403-10 (1994).
[95] Shen Y, Monsma FJ, Jr, Metcalf MA, Jose PA, Hamblin MW, Sibley DR. Molecular cloning and expression of a 5-hydroxytryptamine 7 serotonin receptor subtype. J Biol Chem 268: 18200-4 (1993).
[96] Ganapathy ME, Prasad PD, Huang W, Seth P, Leibach FH, Gana-pathy V. Molecular and ligand-binding characterization of the s-receptor in the Jurkat human T lymphocyte cell line. J Pharmacol Exp Ther 289: 251-60 (1999).
[97] Hashimoto K, London ED. Further characterization of [3H]I fen-prodil binding to sigma receptors in rat brain. Eur J Pharmacol 236: 159-63 (1993).
[98] Catterall WA, Morrow CS, Daly JW, Brown CB. Binding of batra-chotoxin A 20- -benzoate to a receptor site associated with sodium channels in synaptic nerve ending particles. J Biol Chem 256: 8922-7 (1981).
[99] Patacchini R, Maggi CA. Tachykinin receptors and receptor sub-types. Arch Int Pharmacodyn 329: 161-84 (1995).
[100] Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci 14: 43-9 (1993).

From Anti-allergic to Anti-Alzheimer’s Current Alzheimer Research, 2010, Vol. 7, No. 2 111
[101] Gu H, Wall S, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem 269(10): 7124-30 (1994).
[102] Shank RP, Baldy WJ, Mattucci LC, Villani FJ, Jr. Ion and tempera-ture effects on the binding of gamma-aminobutyrate to its receptors and the high-affinity transport system. J Neurochem 54: 2007-15 (1990).
[103] Galli A, De Felice L, Duke B-J, Moore K, Blakely R. Sodium dependent norepinephrine induced currents in norepinephrine transporter transfected HEK293 cells blocked by cocaine and anti-depressants. J Exp Biol 198: 2197-212 (1995).
[104] Shearman LP, McReynolds AM, Zhou FC, Meyer JS. Relationship between [125I]RTI-55-labeled cocaine binding sites and the sero-tonin transporter in rat placenta. Am J Physiol 275(6 Pt 1): C1621-9 (1998).
[105] Wolf WA, Kuhn DM. Role of essential sulfhydryl groups in drug interactions at the neuronal 5-HT transporter. Differences between amphetamines and 5-HT uptake inhibitors. J Biol Chem 267(29): 20820-5 (1992).
[106] Malgaroli A, Milani D, Meldolesi J, Pozzan T. Fura-2 measure-ment of cytosolic free Ca2+ in monolayers and suspensions of various types of animal cells. J Cell Biol 105(5): 2145-55 (1987).
[107] Tsien RY. New tetracarboxylate chelators for fluorescence meas-urement and photochemical manipulation of cytosolic free Ca2+ concentrations. (In: DeWeer P and Salzberg B, Eds). Optical Meth-ods in Cell Physiology, New York: John Wiley & Sons, Inc. pp. 327-345 (1986).
[108] Routledge C, Bromidge SM, Moss SF, Price GW, Hirst W, New-man H, et al. Characterization of SB-271046: a potent, selective and orally active 5-HT6 receptor antagonist. Br J Pharmacol 130: 1606-12 (2000).
[109] http://files.shareholder.com/downloads/MDV/0x0xS1193125-08-101171/1011835/filing.pdf. (Accessed 03/04/2009).
[110] Mulholland MW, Simeone DM. Benign Gastric Disorders. In: Maingot R, Zinner MJ and Ashley SW, Eds. Maingot’s Abdominal Operations 11th ed USA: McGraw-Hill Professional, pp. 333-478 (2007).
[111] Tran VT, Chang RSL, Snyder SH. Histamine H1 receptors identi-fied in mammalian brain membranes with [3H]mepyramine. Proc Nati Acad Sci USA 75(12): 6290-4 (1978).
[112] Leurs R, Smit MJ, Timmerman H. Molecular pharmacological aspects of histamine receptors. Pharmacol Ther 66: 413-63 (1995).
[113] Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol 55: 1101-7 (1999).
[114] Hofstra CL, Desai PJ, Thurmond RL, Fung-Leung W-P. Histamine H4 Receptor mediates chemotaxis and calcium mobilization of mast cells. J Farmacol Exp Ther 305(3): 1212-21 (2003).
[115] Gbahou F, Vincent L, Humbert-Claude M, Tardivel-Lacombe J, Chabret C, Arrang J-M. Compared pharmacology of human hista-mine H3 and H4receptors: structure-activity relationships of hista-mine derivatives. Br J Pharmacol 147(7): 744-75 (2006).
[116] Shah C, McAtee L, Breitenbucher JG, Rudolph D, Li X, Lovenberg TW, et al. Novel human histamine H3 receptor antagonists. Bioorg Med Chem 12: 3309-12 (2002).
[117] Jablonowski JA, Grice CA, Chai W, Dvorak CA, Venable JD, Kwok AK, et al. The first potent and selective nonimidazole human histamine H4 receptor antagonists. J Med Chem 46: 3957-60 (2003).
[118] Khankoeva AI, Dukhanin AS, Galenko-Yaroshevskii PA. Mecha-nisms of histamine-induced increase of calcium level in cardio-myocytes. a relative efficacy of histamine receptor blockers. Bull Exp Biol Med 123(4): 357-9 (1997).
[119] Kaler G, Okun A, Okun I. Two temporal phases of [Ca2+]i modu-lation by 5-HT2A receptor differ in their affinities for both sero-tonin (agonist) and spiperone (antagonist). Abstract book of a sero-tonin club/brain research bulletin conference. Serotonin: From the Molecule to the Clinic, New Orleans, November 2-3, p.162 (2000).
[120] Brown AM. hERG Assay, QT liability, and sudden cardiac death. (In: Morganroth J, Gussak I, Eds.). Cardiac safety of noncardiac drugs: practical guidelines for clinical research and drug develop-ment. Totowa, NJ: Humana Press, pp. 67-81 (2005).
[121] Witchel HJ. The hERG potassium channel as a therapeutic target. Expert Opin Ther Targets 11(3): 321-36 (2007).
[122] Zheng CJ, Han LY, Yap CW, Ji ZL, Cao ZW, Chen YZ. Therapeu-tic targets: progress of their exploration and investigation of their characteristics. Pharmacol Rev 58: 259-79 (2006).
[123] Simons FER. H1-Antihistamines and the Central Nervous System. (In: Estelle F, Ed.). Histamine and H1-antihistamines in Allergic Disease, 2nd ed. USA: Informa Healthcare, pp. 337-388 (2002).
[124] Liu H, Farley JM. Effects of first and second generation antihista-mines on muscarinic induced mucus gland cell ion transport. BMC Pharmacol 5(8): (2005).
[125] Liu H, Qi Zh, Farley JM. Antimuscarinic actions of antihistamines on the heart. J Biomed Sci 13(3): 395-401 (2006).
[126] Richelson E. Tricyclic antidepressants block histamine H1 recep-tors of mouse neuroblastoma cells. Nature 274: 176-7 (1978).
[127] Kanof PD, Greengard P. Brain histamine receptors as targets for antidepressant drugs. Nature 272: 329-33 (1978).
[128] Price DT, Schwinn DA, Lomasney JW, Allen LF, Caron MG, Lefkowitz RJ. Identification, quantification, and localization of mRNA for three distinct alpha, adrenergic receptor subtypes in human prostate. J Urol 150(1): 546-51 (1993).
[129] Kern MJ, Horowitz JD, Ganz P, Gaspar J, Colucci WS, Lorell BH, et al. Attenuation of coronary vascular resistance by selective 1-adrenergic blockade in patients with coronary artery disease. J Am Coll Cardiol 5(4): 840-6 (1985).
[130] Julius BK, Vassalli G, Mandinov L, Hess OM. -adrenoceptor blockade prevents exercise-induced vasoconstriction of stenotic coronary arteries. J Am Coll Cardiol 33: 1499-05 (1999).
[131] Schambra UB, Mackensen GB, Stafford-Smith M, Haines DE, Schwinn DA. Neuron specific -adrenergic receptor expression in human cerebellum: implications for emerging cerebellar roles in neurologic disease. Neuroscience 135(2): 507-23 (2005).
[132] Arnsten AF, Cai JX. Postsynaptic -2 receptor stimulation im-proves memory in aged monkeys: indirect effects of yohimbine versus direct effects of clonidine. Neurobiol Aging 14(6): 597-03 (1993).
[133] Coull JT. Pharmacological manipulations of the 2-noradrenergic system. Effects on cognition. Drugs Aging 5(2): 116-26 (1994)
[134] Yu A, Frishman WH. Imidazoline receptor agonist drugs: a new approach to the treatment of systemic hypertension. J Clin Pharma-col 36: 98-111 (1996).
[135] Potter WZ, Padich RA, Rudorfer MV, Krishnan KRR. Tricyclics, tetracyclics, and monoamine oxidase inhibitors. (In: Stein DJ, Kupfer DJ, Schatzberg AF, Eds.). The American psychiatric pub-lishing textbook of mood disorders, USA: American Psychiatric Publishing; pp. 251-61 (2005).
[136] Eglen RM, Choppin A, Dillon MP, Hegde S. Muscarinic receptor ligands and their therapeutic potential. Curr Opin Chem Biol 3: 426-32 (1999)
[137] Girault J, Greengard P. The neurobiology of dopamine signaling. Arch Neurol 61(5): 641-4 (2004).
[138] Schneier FR, Liebowitz MR, Abi-Dargham A, Zea-Ponce Y, Lin SH, Laruelle M. Low dopamine D(2) receptor binding potential in social phobia. Am J Psychiatry157(3): 457-9 (2000).
[139] Kienast T, Heinz A. Dopamine and the diseased brain. CNS Neurol Disord Drug Targets 5(1): 109-31 (2006).
[140] Fuxe K, Manger P, Genedani S, Agnati L. The nigrostriatal DA pathway and Parkinson's disease. J Neural Transm Suppl 70: 71-83 (2006).
[141] Mihara K, Kondo T, Suzuki A, Yasui-Furukori N, Ono S, Sano A, et al. Relationship between functional dopamine D2 and D3 recep-tors gene polymorphisms and neuroleptic malignant syndrome. Am J Med Genet Part B: Neuropsychiat Gene 117(1): 57-60 (2003).
[142] Faraone S, Khan S. Candidate gene studies of attention-deficit/hyperactivity disorder. J Clin Psychiatry 67, (Suppl 8): 13-20 (2006).
[143] Hummel M, Unterwald E. D1 dopamine receptor: a putative neuro-chemical and behavioral link to cocaine action. J Cell Physiol 191(1): 17-27 (2002).
[144] Ricci A, Mignini F, Tomassoni D, Amenta F. Dopamine receptor subtypes in the human pulmonary arterial tree. Auton Autacoid Pharmacol 26(4): 361-9 (2006).
[145] Hussain T, Lokhandwala M. Renal dopamine receptors and hyper-tension. Exp Biol Med 228(2): 134-42 (2003).
[146] Holmes C, Arranz MJ, Powell JF, Collier DA, Lovestone S. 5-HT2A and 5-HT2C receptor polymorphisms and psychopathology in late onset Alzheimer's disease. Hum Mol Gene 7: 1507-9 (1998).

112 Current Alzheimer Research, 2010, Vol. 7, No. 2 Okun et al.
[147] Nacmiasa B, Teddea A, Forleoa P, Piacentina S, Guarnierib BM, Bartolib A, et al. Association between 5-HT2A receptor polymor-phism and psychotic symptoms in Alzheimer’s disease. Biol Psy-chiat 50(6): 472-5 (2001).
[148] Holmes C, McCulley M, Nicoll JA, Alder JT, Chen CPH, Francis PT. Genetic variation in the 5-HT2A receptor and altered neocorti-cal [3H] ketanserin binding in Alzheimer's disease. Neurosci Lett 420(1): 58-60 (2007).
[149] Engelborghs S, Holmes C, McCulley M, De Deyn PP. 5-HT2A receptor polymorphism may modulate antipsychotic treatment re-sponse in Alzheimer's disease. Int J Geriat Psychiatry 19(11): 1108-09 (2004).
[150] Weiner DM, Davis RE, Brann MR. Selective serotonin 2A/2C receptor inverse agonists as therapeutics for neurodegenerative dis-eases. US Patent 20060199842 (2006).
[151] Unsworth CD, Molinoff PB. Characterization of a 5-hydroxytryptamine receptor in mouse neuroblastoma N18TG2 cells. J Pharmacol Exp Ther 269: 246-55 (1994).
[152] Kohen R, Metcalf MA, Khan N, Druck T, Huebner K, Lachowicz JE, et al. Cloning, characterization, and chromosomal localization of a human 5-HT6 serotonin receptor. J Neurochem 66: 47-56 (1996).
[153] Ballaz SJ, Akil H, Watson SJ. Analysis of 5-HT6 and 5-HT7 recep-tor gene expression in rats showing differences in novelty-seeking behavior. Neuroscience 147(2): 428-38 (2007).
[154] Sleight AJ, Boess FG, Bos M, Bourson A. The putative 5-HT6 receptor: localization and function. Ann NY Acad Sci 861: 91-96 (1998).
[155] Woolley ML, Marsden CA, Fone KC. 5-HT6 receptors. Curr Drug Targets: CNS Neurol Disord 3: 59-79 (2004).
[156] Meneses A. Role of 5-HT6 receptors in memory formation. Drug News Perspect 14: 396-400 (2001).
[157] Upton N, Chuang TT, Hunter AJ, Virley DJ. 5-HT6 receptor an-tagonists as novel cognitive enhancing agents for Alzheimer's dis-ease. Neurotherapeutics 5(3): 458-69 (2008).
[158] Costa VDS, Duchatelle P, Boulouard M, Dauphin F. Elective 5-HT6 receptor blockade improves spatial recognition memory and reverses age-related deficits in spatial recognition memory in the mouse. Neuropsychopharmacology 34(2): 488-500 (2002).
[159] Giacobini E, Spiegel R, Enz A, Veroff AE, Cutler NR. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of pa-tients with Alzheimer's disease by rivastigmine: correlation with cognitive benefit. J Neural Trans 109: 1053-65 (2002).
[160] Ogura H, Kosasa T, Kuriya Y, Yamanishi Y. Comparison of inhibi-tory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butylcholinesterase in vitro. Methods Find Exp Clin Pharmacol 22(8): 609 (2000).
[161] Alessenko AV, Karatasso YO, Shevtsova EF, Shingarova LN, Bachurin SO. Protective effect of dimebon on the TNF- -induced lipid disorders in mice brain. Abstract Book, EHRLICH II –2nd World Conference on Magic Bullets, p. A-8 (2008).
[162] Terry AV, Jr, Jackson WJ, Buccafusco JJ. Effects of concomitant cholinergic and adrenergic stimulation on learning and memory performance by young and aged monkeys. Cereb Cort 3(4): 304-12 (1993).
[163] Youdim MBH, Buccafusco JJ. CNS Targets for multi-functional drugs in the treatment of Alzheimer’s and Parkinson’s diseases. J Neural Trans 112(4): 519-37 (2005).
Received: March 05, 2009 Revised: August 22, 2009 Accepted: August 23, 2009