
François GOUN.AND - International Nuclear Information ...
236
ORSAY n° d'ordre : UNIVERSITE DE PARIS-SUD CENTRE D'ORSAY THESE présentée Pour obtenir LE GRADE DE DOCTEUR ES SCIENCES PHYSIQUES par François GOUN.AND ETUDE DE QUELQOES PROCESSUS COLLISIONNELS, A ENERGIE THERMIQUE, METTANT EN JEU DES ATOMES .ALCALINS EXCITES OU TRES EXCITES Soutenue le 29 mai 1980, devant la Commission d'Examen: MM. S.. FENEUn.LE Président J. BAUDON Rapporteur A. OMONT A. TRAMER Examinateurs C. MANUS J . BEBL.Ai."IDE T.F.GALLAGHER Invité
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of François GOUN.AND - International Nuclear Information ...

ORSAY
n° d'ordre :
UNIVERSITE DE PARIS-SUD
CENTRE D'ORSAY
THESE
présentée
Pour obtenir
LE GRADE DE DOCTEUR ES SCIENCES PHYSIQUES
par
François GOUN.AND
ETUDE DE QUELQOES PROCESSUS COLLISIONNELS, A ENERGIE THERMIQUE,
METTANT EN JEU DES ATOMES .ALCALINS EXCITES OU TRES EXCITES
Soutenue le 29 mai 1980, devant la Commission d'Examen:
MM. S.. FENEUn.LE Président
J. BAUDON Rapporteur
A. OMONT
A. TRAMER Examinateurs C. MANUS
J . BEBL.Ai."IDE
T.F.GALLAGHER Invité

REMERCIEMENTS
Je voudrais tout d'abord remercier Monsieur 1e Pofesseur
S. FENEUILLE, Directeur du laboratoire Aimé Cotton qui m'a fait
1'honneur d'accepter ie patronnage scientifique de ce travail.
Je remercie très vivement Messieurs 1es Professeurs
A. OMONT et J. BAUDON et Monsieur A. TRAMER, Ma.!tre de Recherches
au CNRS, qui ont bien vouJ.u accepter de faire partie du jury.
C-e travail a été réaiisé dans 1es 1aboratoires du Service
de Physique Atomique du Centre d'Etudes Nuc1~aires de Sac1ay, sous
1a.direction de Monsieur C. MANUS. Je tiens à 1ui exprimer toute ma
reconnaissance pour 1'accuei1 qu'i1 a bien vouJ.u m'y réserver et
pour 1'int~r3t constant avec 1eque1 i1 a suivi l'évolution de ces
études.
J'exprime toute ma gratitude à Monsieur J. BERLANDE pour
1es conseils qu'i1 m'a prodigués et pour son soutien constant et
averti pendant toute 1a préparation de cette thèse.
Que Messieurs J. CUVELL.IER et M. HUGON qui ont participé
à cette étude à différents stades de son dérouJ.ement soient remer
ciés pour leur précieuse co11aboration.
Je tiens à remercier Messieurs J. PASCALE et E. De PRUNELE
pour 1eur aide et 1eurs conseils dans 1'interprétation théorique des
résultats expérimentaux.

Je réserve ici une place privilégiée à Monsieur
P.R. FOU:RNIER pour son assistance technique et pour la part impor
tante qu'il a prise dans l'obtention et le traitement des données.
Que Messieurs A. HOURDIN, J.P. FELIX et M. AHR'WEn..LER qui ont éla
boré les dispositifs électroniques nécessaires à l'obtention et au
traitement des données soient particulièrement remerciés pour leur
précieuse collaboration. Je rends hommage à notre souffleur de
verre, Monsieur J. DELCEER, dont les qualités exceptionnelles nous
ont permis de construire et de modifier dans les meilleurs condi
tions nos dispositifs expérimentaux.
C'est enfin avec plaisir que je remercie Madame D. BONGUET
qui a assuré le travail ingrat de la frappe du manuscrit avec beau
coup de compétence et de gentillesse.

Chapitre I - INTRODUCTION
Références du chapitre I
SOMMillŒ
Chapitre II - GENERALITES ; NOTATIONS ET DEFL~TIONS.
II.1 - Spectroscopie des atomes aJ.caJ.ins.
II.2 - Processus collisionnels étudiés et notations.
Chapitre III - LES MODELES THEORIQUES.
III.1 - Introduction.
III.2 - GénéraJ.ités.
III.3 - Les potentiels adiabatiques.
III.3.1 - Une approche simple.
III.3.2 - CaJ.culs élaborés des termes moléculaires.
a) Méthode "ab initio".
b) Méthodes semi-empiriques.
III.4 - Traitement du problème collisionnel.
III.4.1 - Approche semi-classique.
a) Formulation.
b) Solution anaJ.ytique.
c) Approche générale.
d) Intér~t et vaJ.idité.
III.4.2 - Approche quantique.
Pages
1
10
13
13
17
19
19
20
22
23
25
25 . 26
29
29
30
32
37
37
38

III • .5 - Le cas'pa.rticuJ.ier des états fortement excités.
Ill.,5.1 - Potentiel. d'interaction et états de Rydberg.
a) Le pseudo-potentiel. de Fermi.
b) Potentiel. d'échange atome excité-atome.
c) Inf"J.uence du coeur ionique.
d) Remarques final.es.
III.,5.2 - Général.isation du traitement de J.a col.J.ision au cas des états de Rydberg.
a) Approche semi-cl.assique.
b) Approche quantique.
c) Cas des col.J.isions al.cal.in excité-al.cal.in~
d) Remarques final.es.
III • .5.3 - ModèJ.es spécifiques aux états de Rydberg.
a) ModèJ.e de Flannery.
b) ModèJ.e de De PruneJ.é-PascaJ.e.
c) ConcJ.usion rel.ative aux modèl.es.
Références du chapitre III.
Chapitre IV - TECHNIQUES GENERALES D'EXPERDŒNTATION.
IV.1 - Introduction.
IV.2 - Création séJ.ective des états excités.
IV.3 - Le b&ti de mesures.
IV.3.1 - CeJ.J.uJ.e.
IV.3.2 - B&ti de pompage.
IV.3.3 - Dispositifs de contr8J.es et de mesures.
IV.4 - Le dispositif' de détection.
IV.4.1 - Optique de col.J.ection.
IV.4.2 - Système d'anal.yse des longueurs d'onde.
IV.4.3 - Les photomuJ.tipJ.icateurs.
IV.4.4 - AJ.ignement de J.'ensembl.e du système optique.
IV.4.5 - EJ.ectronique de détection.
a) Fluorescence total.e.
b) Fluorescence résol.ue dans J.e temps.
Pages
39
40
40
42
43
4:3
44
44
47
.52
53
.53
.53
.58
60
61
6.5
65
65
69
71
72
72
73
73
74
76
77
78
81
81

IV.4.6 - Rég1agè et éta1onnage d'une cha!ne de comptage.
Références du chapitre IV
Chapitre V - LA MESURE DES SECTIONS EFFICACES.
V.1 - Introduction.
V.2 - Les équations généra1es.
v.3 - F1uorescence réso1ue dans 1e temps.
Pages
82
87
89
89
89
94
v.3.1 - Généra1ités. 94
v.3.2 - Les méthodes de dépoui11ement. 96
v.3.3,- Transfert d'excitation et f1uorescence réso1ue dans 1e temps. 101
V.3.4 - Dépopu1ation co11isionne11e tota1e (quenching) et f1uorescence réso1ue dans 1e temps. 104
V.4 - F1uorescence non réso1ue dans 1e temps.
V.4.1 - Généra1ités.
V.4.2 - Transfert d'excitation et f1uorescence non réso1ue
108
108
dans 1e temps. 109
V.4.3 - Quenching et f1uorescence non réso1ue dans 1e temps~ 112
v.5 - Sources d'erreur et précision des mesures. 113
v.5.1 - Phénomènes 1iés à 1a source d'excitation. 114
v.5.2 - Mesures des températures et des pressions. 119
v.5.3 - Causes d'erreur dues à 1'optique de détection. 123
V.5.4 - Erreurs dues à 1'é1ectronique de comptage. 128
v.5.5 - Erreurs dues au dépoui11ement des données. 130
V.5.6 - Erreurs systématiques. 131
Références du chapitre V 133
Chapitre VI - RESULTATS ET DISCUSSION. 1 35
VI.1 - Introduction. 135
VI.2 Etude d'un groupe de niveaux faiblement excités
( 7P-6D) du , . ce sium.
VI.2.1 - Présentation du travai1.
VI.2.2 - Article 1
VI.2.3 - Comp1éments.
136
136
137
149

VI.2.4 - Comparaisons complémentaires.
a) RésuJ.tats expérimentaux.
b) RésuJ.tats théoriques.
VI.2.5 - Conclusion.
VI.3 - Etude de la dépopu1ation co1lisionnel1e totaJ.e des
niveaux de Rydberg.
VI.3.1 - Présentation du travai1.
VI.3.2 - Travai1 expérimentai.
a) Que1ques aspects radiatifs du comportement des états de Rydberg.
b) Etude collisionnelle des états de Rydberg P Articles 2 et 3.
c) Etude co1lisionne1le des états de Rydberg S du rubidium.
Pages
153
153
155
156
157
157
160
160
168
183
VI.3.3 - Discussion de l'ensemble des résuJ.tats collisionnels. 185
a) Nature des canaux de réaction.
b) Quelques remarques qualitatives.
c) Col1isions alcalin-alcalin.
d) Co11isions alcalin-gaz rare.
e) Modè1e de Gal1agher-Cooke.
f) Résumé.
Références du chapitre VI
Chapitre VII - CONCLUSION.
Etalonnage de la transmission en longueur d'onde d'une voie de mesure.
Correction d'effusion thermique.
186
187
188
192
206
208
209
213
215
217
AEJ2.endice I
AEJ2.endice II
AEI?,endice III
AEI?,endice IV
Calcul. de l'absorption des raies de fluorescence. 221
Appendice V
Mesure de la densité d'atomes alcalins par absorption de la raie de résonance. (Méthode de la largeur équivalente).
Durée de vie radiative des atomes alcalins Article 4.
225
229

Chapitre I
INTRODUCTION
L'étude des collisions atomiques à énergie thermique revGt
une grande importance tant du point de vue pratique que théorique.
Les processus collisionnels peuvent en effet jouer un rSle important
dans les vapeurs neutres aussi bien que dans les gaz ionisés parce
qu'ils influent directement sur les propriétés macroscopiques et donc
sur le comportement du milieu. Des domaines aussi variés que l'astro
physique (physique du milieu interstellaire, physique des atmosphères
planétaires •.• ), la physique atomique, la physique des gaz ionisés
(élaboration de modèles pour l'évolution des plasmas) et la physico
chimie (collisions réactives, séparation isotopique) sont ainsi con
cernés. Ces études présentent aussi un intér3t plus fondamental. Com
plémentaires des expériences de spectroscopie (qui concernent les
propriétés d'un atome isolé : niveaux d'énergie, écarts d~ structure
fine ou hyperfine, polarisabilité ••• ), elles permettent en liaison
avec des études théoriques~ de mieux conna.!tre les forces d'interac
tion entre atomes mises en jeu.
Nous nous sommes, dans cette étude, limités au domaine, déjà
très vaste, des transferts collisionnels que l'on peut schématiser par
la réaction
A*(i) + B --,..A*(j) + B + t.E (Al)
où A*(i) et A*(j) représentent un atome A dans deux niveaux d'excita
tion électronique i et j différents et Bun atome neutre dans son état
fondamental. En particulier nous ne parlerons pas ici des réactions de
transfert du type :

- 2 -
A* + B ~ A + B* + D.E (A2)
où A retombe à 1 1 état fondamenta1, B étant excité dans 1'état B* après
co1lision. Notre étude a aussi porté sur la dépopu1ation co11isionne11e
tota1e (quenching) d'états excités. En ce cas 1es états finaux (neutres
ou ioniques) ne sont pas étudiés et 1e processus prend a1ors uniquement
en compte 1a disparition des espèces A*, due à 1a co1lision.
Nous a1lons maintenant donner un bref aperçu de 1a situation
du domaine couvert par notre étude en 1972, tant du point de vue expé
rimentai que .théorique. Le transfert d'excitation et le quenching des
raies de réson_ance· ont fait l'objet de nombreux travaux durant les tren
te premières années de ce sièc1e1 • En particu1ier 1a première preuve
qua1itative de 1'existence d'un transfert d'excitation (de type (A2))
a été apportée par Caria et Franck2. Après une 1ongue période de som
meil, due à 1a fois à 1'absence de techniques expérimenta1es appropriées
autant qu'à 1 1 état embryonnaire de la théorie 1es travaux de R. Seiwert3
(1956) remettent 1e sujet à l'ordre du jour. Mais il revient à
L. Krause et à son équipe d'avoir débuté les études expérimenta1es sys
tématiques dans ce domaine4 • Conjointement E.E. Nikitin et ses colla
borateurs développaient dès 1965 1es premiers modè1es théoriques5 •
Ces travaux ont essentiellement porté sur 1es transitions de structure
fine des atomes a1ca1ins dans leur premier doub1et résonnant, que l'on
peut schématiser par :
A(nP1 / 2 ) + B '½ /2 ~ 3/2 ( ) + B + ~E
====== A nP 3/2 Q3/2 -1/2
(A3)
où D.E représente l'écart d'énergie asymptotique entre 1es niveaux
nP1; 2 et nP3
; 2 . Les expériences ont été effectuées sur des vapeurs
neutres, en ce1lu1e, 1es sections efficaces obtenues (Q1 ;2
_3
; 2 et
Q312 _112
respectivement) étant donc moyennées sur 1a distribution
de vitesse (maxwellienne) des particules en co1lision, pe~dant ainsi
une partie de 1'inf'ormation contenue dans 1e processus microscopique.
La technique expérimenta1e emp1oyée fut celle, très simple de princi
pe (voir Chapitres IV et V) de la fluorescence sensibilisée. L'atome
B choisi fut, soit l'atome a1ca1in dans son état fondamenta1 (n0s112
),
soit un atome de gaz rare. Le coup1e a1ca1in-gaz rare présente un
intér3t théorique particulier du fait de sa relative simplicité ; en

- 3 -
effet la détermination du potentiel d'interaction,c.'est-à-d.ire, comme
nous le verrons, la résolution de la partie statique du problème,peut
se faire en considérant un système à trois corps (coeur de l'a1ca1in,
é1ectron périphérique et atome de gaz rare). Ainsi E.E. Nikitin et
ses collaborateurs purent proposer des modèles ana1ytiques relative
ment simples de potentie1. L'importance relative des différentes in
teractions susceptibles de donner lieu au transfert {interaction dipo
laire à longue portée, forces d'échange) fut estimée et les sections
efficaces de transfert ca1cu1ées par des méthodes semi-classiques6 ' 7 .
Ces approches théoriques furent étendues aux col1isions a1ca1in-a1ca
lin8, pour 1esque11es des résu1tats étaient disponib1es, ma1gré 1a
p1us grande complexité du système (problème à quatre corps). Du point
de vue expérimentai 1es travaux du groupe de Windscr9- 12 (1966-1967)
furent complétés par une remarquable étude, en cellu1e, de la varia
tion des sections efficaces de transfert avec la température pour les
coup1es Rb-Gaz rare et Cs-Gaz rare effectuée par A. Ga11agher13 (1968).
I1 revient à M. Pimbert14
(1972) d'avoir étendu ce type d'é
tude à des niveaux plus excités que 1es premiers doub1ets résonnants,
par des mesures relatives au transfert intramu1tip1et du troisième dou
blet résonnant (8P) du césium. On lui doit aussi les premières mesures
de transfert extramu1tiplet (de type 8P ..... 7D). Là aussi, Nikitin et
ses collaborateurs proposèrent conjointement des modèles théoriques15 , 16 •
Des études de transfert col1isionnel furent aussi effectuées
dans des gaz ionisés (post-décharge d'hélium en particu1ier). L'inter
prétation en est toujours délicate car, vue la complexité du milieu
(présence d'ions et d'électrons), de nombreux mécanismes doivent 3tre
pris en compte, ce qui rend difficile l'obtention d'informations pré
cises sur les transferts de types (A1) et (A2). Citons ici le remar
quable travail de Wel1enstein et Robertson17 • Notons enfin ~es études
expérimenta1es, concernant les atomes a1ca1ins, effectuées dans les
flammes18 . Ces expériences posent de nombreux problèmes d'interpréta
tion19.
Nous voudrions maintenant effectuer une rapide comparaison
entre les résu1tats théoriques et expérimentaux que nous avons mention
nés, concernant les transferts collisionnels mettant en jeu des atomes
a1ca1ins excités, car celle-ci nous permettra d'introduire les motiva
tions essentielles du début de notre travail. Considérons tout d'abord

- 4 -
le tableau I où sont reportés~en particulier,les résultats relatifs
aux premiers doublets résonnants des aJ.caJ.ins (pour lesquels l'écart
énergétique AE entre les deux composantes de structure fine varie de
17 cm-1 pour le sodium à 554 cm-1 pour le césium, encadrant ainsi
l'énergie thermique moyenne disponible lors de la collision, qui vaut
environ 400 cm-1 ). On peut dire que l'écart énergétique AE appara!t
comme un paramètre important: plus ~E est faible, plus la section ef
ficace est grande. Cette variation appara!t m3me très régulière dans
le cas des collisions aJ.caJ.in-aJ.ca1in, en accord avec la règle empi
rique de Franck20 • Ces remarques, ainsi que la variation des sections
efficaces en fonction de la température13 peuvent recevoir des inter
prétations qua1itatives simples14 , liées aux différentes vaJ.eurs du
paramètre de Massey21 • De m3me la variation des sections efficaces en
fonction de la nature du gaz rare reçut des explications très qua1ita
tives9110. Le tableau II montre, pour quelques couples al.cal.in-gaz rare,
la comparaison des résultats expérimentaux et des cal.culs théoriques
effectués à l'aide de potentiels semi-empiriques. Eu égard à la simpli
cité de l'approche théorique utilisée, l'accord obtenu appara!t assez
satisfaisant. Il faut bien noter que cette comparaison concerne des
sections efficaces moyennées (aussi bien sur les sous-niveaux initiaux
et finaux que sur la distribution de vitesse des particules en colli
sion, les expériences étant effectuées en cellule), ce qui en restreint
la portée. Compte tenu de cette importante restriction, l'ensemble des
résultats théoriques obtenus par le groupe de Nikitin6 ' 7 permettait
cependant de penser qu'en ce qui concerne les niveaux de résonance des
aJ.ca1ins les mécanismes d'interaction fondamentaux étaient assez bien
compris, du moins dans le cas des collisions aJ.ca1in-gaz rare.
Les résultats obtenus par M. Pimbert14 , relatifs à des ni
veaux nettement plus excités montrent une situation très différente de
celle observée pour les niveaux de résonance. Ils sont aussi reportés
dans le tableau I. Si les sections efficaces relatives au couple Cs*-Cs
semblent suivre la variation précédemment observée en fonction de AE,
les sections efficaces Cs*-GR présentent de notables anomaJ.ies; en
particulier les transitions 8P ~7D (AE ~ 350 cm-1
) montrent des sec
tions efficaces anormaJ.ement grandes. De plus la comparaison entre va
leurs théoriques et expérimental.es, présentée dans le tableau III lais
se subsister un désaccord profond.

Al.cal.in
*Na
*K
Cs
*Rb
Cs
*Cs
- .5 -
Tab1eau I
Sections efficaces (en A2 ) de transfert d'excitation . 9-12 14 pour 1es atomes a1ca1ins ' •
Perturbateur
Transition ~(cm-1 ) T( °K) Al.cal.in He Ne Ar Kr
JP1/2 - JPJ/2 17 '.398 5J2 86 67 110 85
4P1 / 2 - 4PJ/2 57 J68 J70 59 • .5 14. J J6.7 61.4
8P, / 2 - 8PJ/Z 8J 420 190 J4 4.4 .5 • .5 4.,5
5P1; 2 - 5PJ/2 2J8 J40 .5J 7.6 X 10-2 1.7x10-J 1.ox10-J 6.4 X 10-4
SPl /2 - 70J/2 JJ9 420 21 1.4 8 X 10:_2 0.29 O.J7
6P1/ 2 - 6PJ/2 .554 J1, 6.4 .5,7x10-.5 1.9x10-5 1,6 x,o-5 8-.J X 10-.5
Les transitions concernant les premiers doublets résonnants sont indiquées par le signe*
Xe
90
104
15
7.9x10-4
1 .8
7.2x 10 -5

- 6 -
A1ca1in Perturbateur T( °K) Qexp Qth
Na Ar. 398 110 120
K Ne 368 14. 3 7.0
Rb He 340 7.6 X 10-2 -1 1.0 X 10 .
Rb Ne 340 1 . 7 X 10-J 0.8 X 10-3
Tableau II
Comparaison entre sections efficaces théoriques Qth6 et
expérimenta1es Q 10- 12 pour la transition de structure exp
fine P1; 2 - P3; 2 du premi.er doublet résonnant ~: quelques
a~omes a1ca1ins .: Qth et Q sont en A. exp
Perturbateur Qth Qexp -
He 2. 1 34
Ne 4.4
Ar 0.15 5.5
Kr 4.5
Xe 0 .11 15
Tableau III
Comparaison entre sections efficaces (en Â2) théoriques15 , 16 Qth
et expérimenta1es14
Qexp pour la transition (8P1 ; 2 --sP3
; 2 ) du
césium induite par collision avec les gaz rares (T = 4.20 K).

- 7 -
En résumé, les col1isions mettant en jeu les niveaux réson
nants des alcalins, qui sont très bien isolés dans les diagrammes
d'énergie, semblaient assez bien comprises, eu égard à la relative
méconnaissance du potentiel d'interaction, à la relative simplicité du
traitement de la partie dynamique du problème de la collision (méthode
de type Landau-Zener-Stttcke1berg) et au caractère moyenné des résu1tats
expérimentaux (de fait l'apparition de courbes de potentiel adiabati
ques alcalin-gaz rare réalistes pour les premiers doublets résonnants22
et l'utilisation de traitements dynamiques semi-classique23 ou quanti
que24 plus élaborés permirent d'améliorer encore la comparaison entre
théorie et expérience ainsi que de préciser l'importance des différen
tes interactions responsables du transfert). Les résu1tats obtenus
pour les niveaux excités, donc moins bien isolés de leurs voisins dans
le diagramme énergétique,montraient clairement la nécessité d'une bon
ne connaissance du potentiel d'interaction pour les états excités,
pour lesquels on pouvait supposer des couplages statiques entre niveaux
beaucoup plus importants que dans le cas des niveaux l.es plus bas. En
effet les calcu1s menés par E.E. Nikitin15 à partir de potentiels très
empiriques prenant en compte certains couplages statiques supposés im
portants conduisaient à des sections efficaces en net désaccord avec
l'expérience (Tableau III). La nécessité de poursuivre conjointement
un effort théorique et expérimental dans ce domaine apparaissait donc
très clairement.
Nous sommes maintenant à m3me d'exposer les motivations et
les buts poursuivis dans ce travail, ainsi que d'en exposer succincte
ment le dérou1ement. Le but essentiel est l'obtention de données pré
cises sur les propriétés collisionnelles des états excités, ce en liai
son constante avec les approches théoriques développées conjointement
dans notre laboratoire par J. Pascale et ses collaborateurs. Le déve
loppement de sources lumineuses accordables à haute intensité (laser
à colorant (dye)) permettait tout d'abord d'envisager le peuplement de
nombreux niveaux excités ou très excités des atomes alcalins, autori
sant ainsi un développement très souple du programme de travail, en
fonction de l'évolution des données théoriques. De plus la détection
optique des états atomiques excités ne pose pas de ·problèmes particu
liers, du moins pour des valeurs du nombre quantique principal n pas
trop élevées (n ~ 25). La technique de la fluorescence sensibilisée,
en cellu1e, fut choisie car notre laboratoire en possédait bien les

- 8 -
techniques de base après 1e travai1 effectué par M. Pimbert. Cette
méthode dut cependant être adaptée aux prob1èmes particu1iers posés
par 1es sources d'excitation (1aser dye pu1sé). I1 faut noter ici que
cette approche n'était pas 1a seu1e envisa.geab1e. Ainsi des études de
profi1s de raie25 peuvent permettre d'obtenir des renseignements sur
1es potentie1s d'interaction. Les mesures co11isionne11es par techni
que de faisceaux constituaient l.llle autre approche possible ; les infor
mations obtenues sont beaucoup plus précises que ce11es fournies par
1es études en ce11u1e, car non moyennées sur 1es vitesses (on peut m3me,
en jouant sur 1a po1arisation de 1a source d'excitation et de 1a 1umière
observée, étudier des processus mettant en jeu des sous-niveaux bien
définis). La comparaison entre va1eurs théorique et expérimenta1e est
beaucoup p1us fine. De te11es expériences, en fait comp1émentaires de
ce11es effectuées en ce11u1e mais beaucoup p1us diffici1es à mettre en
oeuvre et à exp1oiter, sont d'ai11eurs actue11ement en cours dans notre
laboratoire. La raison théorique essentie1.le du choix du couple a1ca1in
gaz rare fut, outre sa re1ative simp.licité déjà mentionnée, 1e travai1
amorcé par J. Vandep1anque et J. Pasca1e en vue d'amé1iorer et d'étendre
à des niveaux p1us excités que 1es premiers doub.lets résonnants .lamé
thode proposée par W .E. Bayl.is22 pour 1' obtention des po.tentie.ls adia
batiques des coup1es a1ca1in-gaz rare. En effet l.'obtention des courbes
de potentie.l constitue 1a première étape (.la seconde étant 1e traite
ment de 1a dynamique du prob1ème) de tout ca1cu1 précis des sections
efficaces de transfert d'excitation. Ce travai126 , pub1ié en 1974,
permit de comprendre et de guider 1a première expérience que nous avons
entreprise sur le groupe de niveaux (7P-6D) du césium27 . Ce11e-ci four
nit de très intéressantes informations sur l'influence des couplages
statiques entre niveaux excités (qui déterminent l.a forme des courbes
de potentie1) sur la va1eur des sections efficaces. A la suite de ce
travai1 i1 nous sembla indispensable, comme nous le verrons,d'étendre
ce type de mesure à des niveaux hautement excités (niveaux de Rydberg)
pour lesquels très peu de résu1tats expérimentaux étaient disponibles.
L'intér3t de ces états tant du point de vue expérimentai que
théorique appara!t très grand. Leur importance en astrophysique et en
physique des plasmas avait été maintes fois sou1ignée. Leur "dimension 11
0
m3me (N 100-1000 A), inhabituelle en physique atomique permettait les
hypothèses .les plus diverses quant à leurs propriétés collisionnel1es.
Après une étude préliminaire (1976) sur l.'état 10P du potassium28 , une

- 9 -
étude systématique des états de Rydberg du rubidium fut entreprise.
Ce11e-ci a été rendue possib1e par 1a mise en oeuvre de techniques
é1aborées de détection de signaux faibles réso1us dans 1e temps.
E11e se poursuit encore actue11ement dans notre laboratoire. Nous
présentons ici 1es résultats relatifs aux états n.P (12 ~ n 4 22) et
nS (12 ~ n ~ 18) du rubidium (1976-1979), que nous comparons à diver
ses approches théoriques. Nous avons aussi étudié, avec M. Hugon,
outre les états nS, les états nD et nF du rubidium. Ces derniers ré
sultats sont présentés dans la thèse de M. Hugon. Le travail que nous
présentons forme cependant, comme nous le verrons, un ensemb1e cohé
rent qui a permis de dégager quelques aspects originaux des processus
co1lisionnels mettant en jeu des états de Rydberg.
Le vaste domaine des collisions atomiques à énergie thermi
que continue de susciter de nombreuses études tant expérimentales que
théoriques. Nous n'avons pas abordé certains aspects de celui-ci·:
collisions dépolarisantes, collisions en présence de champ magnétique,
transfert entre sous-niveaux •.• Cependant 1e présent travai1, qui
couvre des situations très diverses (niveaux faib1ement ou fortement
excités, perturbateurs variés) a permis, en liaison avec 1e déve1oppe
ment des travaux théoriques, de mieux dégager 1es principales interac
tions physiques mises en jeu, améliorant ainsi notre connaissance de
çes processus collisionne1s.
Nous avons tenu compte, lors de 1a rédaction du présent
manuscrit, de l'évolution très rapide du domaine traité, tant en ce qui
concerne 1es techniques expérimentales que la compréhension des phéno
mènes physiques. Ainsi nous avons vo1ontairement abrégé 1a description
de certains aspects expérimentaux et que1ques discussions physiques
qui, si elles ont joué un rSle important dans le déroulement histori
que de notre travai1, ne sont plus, en 1980, de que1que actualité.
Dans le m3me esprit et afin de fournir au lecteur une vue d'ensemble du
sujet traité p1ut$t qu'un aperçu du déve1oppement chrono1ogique de notre
recherche nous avons adopté le p1an suivant. Après un bref chapitre
consacré à des généralités et des notations qui nous seront uti1es tout
au long de l'exposé (chapitre II), nous analyserons au chapitre III 1es
problèmes théoriques que posent 1es collisions atomiques à énergie ther
mique. Les deux chapitres suivants (IV et V) seront consacrés à la des
cription et à la discussion des techniques expérimentales. Enfin l'en
semble de nos résultats expérimentaux sera présenté et analysé de ma-
nière détail1ée (chapitre VI) avant de conc1ure.

- 10 -
REFERENCES DU CHAPITRE I
1 - A.C.G. MITCHELL et M. ZEMANSKY, Resonance radiation and excited atoms. Cambridge University Press, London (19j4)~
2 - G. C.ARIO et J. FRANCK, Z. Phys. 2.J., 747 (1929).
3 - R. SEIWERT, Ann. Phys. 1.§., 54 (1956).
4 - L. KRAUSE, App1. Opt. 2, 1375 (1966).
5 - E.E. NIKITIN, J. Chem. Phys. !!:,l, 744 (1965).
6 - E.I. DASHEVSKAYA et E.E. NIKITIN, Opt. Spektrosk 22, 866 (1967). (Opt. Spectrosc. 22, 473 (1967)).
7 - E.I. DASEEVSKAYA, E.E. NIKITIN et A.I. REZNIKOV,
J. Chem. Phys. 53, 1175 (1970).
8 - E.I. DASEEVSKAYA, A.I. VORONIN et E.E. NIKITIN,
Can. J. Phys. 47, 1237 (1969).
9 - M. CZAJKOWSKI, D.A. McGILLIS et L. KRAUSE,
Can. J. -Phys. :±:±., 91 ( 19€6) .
10 - B. PITRE, A.G.A. RAE et L. KRAUSE, Can. J. Phys. 44, 731 (1966).
11 - G.D. CHAPMAN et L. KRAUSE, Can. J. Phys. 44, 753 (1966).
12 - J. PITRE et L. KRAUSE, Can. J. Phys. 45, 2671 (1967).
13 - A. GALLAGEER, Phys. Rev. 172, 88 (1968).
14 - M. PIMBERT, J. Phys. (Paris) ,ll, 331 ( 1972).
15 - E.E. NIKITIN et A.I. REZNIKOV, Chem. Phys. Lett. ~, 161 (1971).
16 - A.A. ZEMBEKOV et E.E. NIKITIN, Chem. Phys. Lett. ~' 213 (1971).
17 - H.F. WLL.ENSTEIN et W.W. ROBERTSON, J. Chem. Phys. 56, 1072 (1972).
18 - H.P. HOOYMA.YERS et C.T. ALIŒMADE, J. Quant. Spectrosc. Rad.
Transf. 6, 847 (1966).

- 11 -
19 - D.R. J'ENKINS, Proc. Roy. Soc. A306, 413 (1968).
20 - J. FRANCK, Naturwiss. 14, 211 (1929).
21 - M.S.W. MASSEY, E1ectronic and Ionie Impact Phenomena T. Ill,
p. 1925, Oxford, C1arendon University Press (1969).
22 - W.E. BAYLIS, J. Chem. Phys. 51, 2665 (1969).
23 - F. MASNOU-SEEUWS, J. Phys. B3, 1437 (1970).
24 - R.H.G. REID et A. DALGA:RNO, Chem. Phys. Lett. 6, 85 (1970).
25 - R.E.M. EEDGES, D.L. DRUMMOND et A. GALLAGEER,
Phys. Rev. A6, 1519 (1972).
26 - J. PASCALE et J. VANDEPLANQOE, J. Chem. Phys. 60, 2278 (1974).
27 - J. C'UVELLIER, P.R. FOURNIER, F. GOUNAND, J. PASCALE et J. BEBLANDE,
Phys. Rev. fil, 846 (1975).
28 - F. GOUNAND, J. CUVELL:IER, P.R. FOURNIER et J. BERL.ANDE,
J. Phys. (Paris) 2Z, L169 (1976).

Chapitre II
GENERALITES NOTATIONS ET DEFINITIONS.
Il nous a paru utile d'indiquer dans un chapitre séparé les
caractéristiques spectroscopiques essentielles, pour notre propos, des
atomes alcalins ainsi que de donner un bref aperçu des phénomènes co1-
lisionnels que nous avons étudiés. Ainsi seront introduits les paramè
tres et les notations nécessaires à la lecture de notre travail, ce
qui nous évitera par la suite d'alourdir l'exposé.
II.1. SPECTROSCOPIE DES ATOMES ALCALINS.
Un état atomique sera noté, par exemple, 72P1; 2 indiquant
les nombres quantiques n = 7, 1 = 1, j = 1/2. L'indice 2 qui indique
la valeur du spin (2s + 1) sera le plus souvent omis pour des raisons
de commodité. Quand nous ne nous intéresserons pas explicitement à la
structure fine, la valeur de j n'appardtra pas : on parlera ainsi du
doublet 7P (au lieu de 72P1; 2 et 72l,3; 2 ). Sauf exception nous n'aurons
pas à nous préoccuper de l'influence du spin nucléaire (structure hyper
fine).
Chaque niveau est caractérisé par la valeur de son énergie , , -1
E 1 . que 1 1 on notera en general en cm , le niveau fondamental n, 'J
n0
2s1 ; 2 (n0
= 3, 4, 5, 6 respectivement pour Na, K, Rb et Cs) étant
pris comme référence : Eno,o, 1 ; 2 = O. Chaque alcalin est donc carac
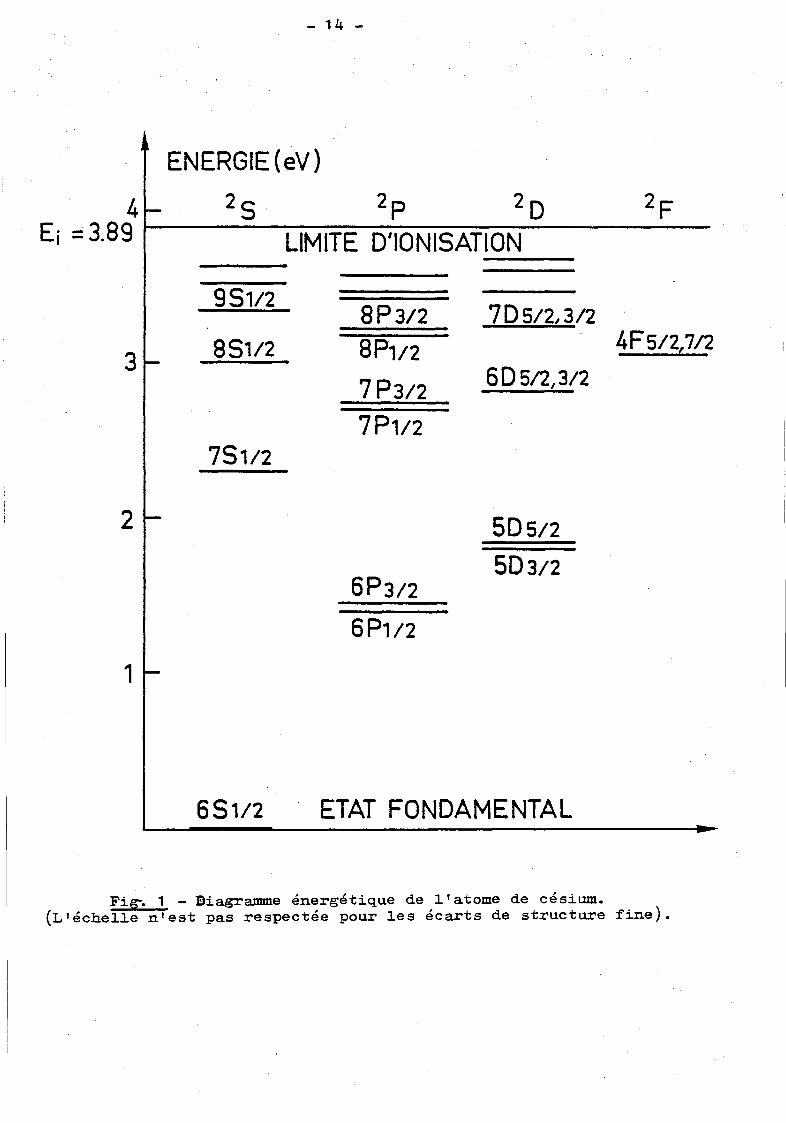
térisé par son diagramme énergétique, dont un exemple est donné par
la figure 1, dans le cas du césium. On voit que le premier doublet
résonnant 6P est très bien isolé dans le spectre, les niveaux les plus
proches étant distants de plusieurs milliers de cm-1• Le deuxième dou
blet (7P) n'est séparé, lui, que d'environ 700 cm-1
du doublet 6D. Plus
on monte en valeur den, plus les niveaux. d'énergie sont resserrés.
Ei :::3.894
~
3 J-
2
1
- 14 -
ENERGIE (eV)
25
95112
8S112
7S112
6S112
2p 2o
LIMITE D'IONISATION
8P312 8P112
7P3/2 7P112
6P3;2
6P112
705/2.,3/2
~Ds~1 ~12
5Ds12
503;2
ETAT FONDAMENTAL
2F
4Fs12,112
Fig-. 1 - Diagramme énergétique de i 1 atome de césium. (L 1 échei1e n'est pas respectée pour ies écarts de structure fine).

- 15 -
Pour une va1eur del déterminée(~ 1) l'écart de structure fine décroît
aussi très sensiblement avec la va1eur den. Ainsi l'écart
fine dans le cas du césium est de 554 cm-1 pour le doublet
pour le 8P et de 2 cm-1 pour le 19P. Notqns pour finir que
de structure
6 -1 P, 83 cm
ces écarts
de structure fine diminuent avec la masse de l'atome a1ca1in: ainsi,
pour le premier doublet résonnant du sodium (3P) il est de 17 cm-1 au
lieu de 554 dans le cas du césium {6P).
ll est intéressant d'introduire dès maintenant les notions
de nombre quantique effectif n* et de défaut quantique o1 • Nous aurons
souvent à y faire appel dans le cas des niveaux fortement excités
(va1eur élevée den), dits niveaux de Rydberg. Ces notions permettent
de reconstruire le diagramme d'énergie de l'atome a1ca1in à l'aide
d'un nombre très réduit de paramètres. On peut définir le nombre quan
tique n* par:
1 -n*2
= Ei - En,1
RA. (B1)
où Ei et RA. sont, respectivement, les énergies d'ionisations et la cons
tante de Rydberg de l'a1ca1in considéré, En,l·étant l'énergie du niveau
considéré (nous négligeons ici la structure fine).
Le défaut quantique o1 , caractéristique d'une série del
donné (états D par exemple) s'exprime par:
ô1 = n - n* = a0
+ a, x +a2 x2 + • • • (formul.e de Ri tz étendue)
où x = (1/n*) 2 . Pour n (ou n*) élevé on a donc
ô = l n - n* ,J a
0
(B2)
(B3)
c'est-à-dire que le défaut quantique peut-~tre considéré comme cons
tant. Le tableau IV fournit les valeurs de a pour les différents nio veaux des atomes alcalins.

- 16 -
s p D F G
1 .35 0.855 -2 1 .45 X 10-3 4.25 X 10-4 Na 1 .52 X 10 ·
K 2.18 1. 71 0.277 9.7 X 10-3 3.0 X 10-3
Rb 3 .13 2.65 1 .35 1 • 6 .:x: 10-2 7.5 X 10-2
Cs 4.05 3.57 2.47 4 -2 3. X 10 2.7 X 10 -2
Tableau IV
Valeurs de a {éq. {BJ)) pour les différentes séries spectrales 0
des atomes alcalins.
On remarque que pour 1;,. 2 dans le cas du sodium et pour 1;. 3 pour les
autres alcalins les défauts quantiques sont très petits indiquant alors
un caractère 1:l.ydrogénoïde. L'éauation (B1) indique aussi g_ue, pour
une série donnée l'écart d'énergie entre les plus proches voisins varie
en 1/n*3 , c'est-à-dire décro!t très rapidement. En ce qui concerne les
écarts d'énergie entre niveaux voisins (de 1 ~) on peut donner, pour
fixer les idées l'ordre de grandeur suivant, correspondant à n* ~ 20
dans le rubidium: les niveaux non hydrogénoïdes (S, P, D) sont dis
tants d'environ 10 cm-1 alors que le niveau F {hydrogénoïde) est situé
à~ 0,2 cm-1 du niveau G voisin. Notons, pour terminer, que la structu
re fine est extr~mement faible dans le cas des états excités {ainsi le
niveau 20P du rubidium présente un écart de structure fine d'environ
0.6 cm-1 , à comparer avec l'écart d'énergie de l'ordre de 10 cm-1 avec
les autres niveaux les plus proches) ; le tableau V donne la correction
à apporter à a ~ ô1 pour prendre en compte la structure fine des états 0 -
P et D des alcalins.

- 17 -
p D
Na 10-3 1 o-5
K 3 X 10-3 10-4
Rb 1.3 X 10-2 ,o-3
Cs 3 X 10-2 10-2
Tab1eau V
Va1eurs approchées de 1'écart Aô entre 1es niveaux de structure fine des doublets Pet D des a1ca1ins.
II.2. PROCESSUS COLLISIONNELS ETUDIES ET NOTATIONS.
-- -
Nous avons effectué l'étude expérimenta1e de deux types de
processus. Le premier est 1e transfert co11isionne1 entre niveaux
é1ectroni9._ues, défini par :
A*(i) + B
Qi ..... j
Q. ~i J
A.*(j) + B + AE (B4)
où A*(i) représente un atome a1ca1in dans l'état i, A*(j) un atome a1-
ca1in dans l'état jet B l'atome perturbateur (gaz rare ou a1ca1in dans
l'état fondamenta1 n s1 ; 2 ). Q. . et Q .. sont les sections efficaces 0 J.-J J+J.
associées au processus et D.E l'écart d'énergie entre les états atomi-
ques i et j. Si les états i et j sont les deux niveaux d'un m3me dou
blet on a affaire à une transition de structure fine intramuJ.tiplet;
par exemple :
Cs(7P1 / 2 ) +Ar~ Cs(7P3; 2 ) + Ar - 180 -1 cm
sinon nous parlerons d'une transition extramuJ.tiplet
Cs(7P1 ; 2 ) + Ar~ Cs(6n312 ) + Ar - 820 -1 cm
soit, par exemple :

- 18 -
Les sections efficaces Q. J -i
et Q. . sont reliées par le principe 1 ~J
du bilan détaillé :
-Qj_i
g. .:.J. exp gi
-6.E .. ll
kT (B5)
où g. et g. sont les poids statistiques des niveaux i et j (c'est-à-1 J
dire 2j. + 1 et 2j. + 1), 6.E .. l.' écart entre l.es niveaux, k la cons-1 J J1 l
tante de Boltzmann (k = 0.695 cm- /°K) et Tl.a température de la va-
peur dans la cel.lule (en °K).
Le deuxième processus étudié est la dépopulation collision
nelle totale (ou quenching) d'llll niveau donné, soit :
A* (i) + B Qq (i),.. ( F ) (B6)
où A*(i) représente l'alcalin dans le niveau i et (F) indique les pro
duits finaux de la réaction, sans référence explicite à leur nature,
neutre (par exemple : états atomiques voisins) ou ionique. Q (i) repré-q sente l.a section de quenching du niveau i, somme de toutes les sections
efficaces des processus inélastiques possibles ..
Mentionnons pour terminer le processus de mélange collision
nel de moment angulaire, que nous aurons à discuter en comparaison avec
nos résultats, et que l'on peut schématiser par
A*(n,l) + B ~ A*(n,J. 1 > l.) +. B .:t. 6.E (B7)
où A*(n,J.) représente l'atome alcalin dans J. 1 état (n,l.), pour lequel le
défaut quantique est f'aibl.e (état 11hydrogénoïde 11 mentionné pl.us haut)
et A*(n,l.' > 1) l.'atome alcalin dans un des états (non spécifiés) voi
sins, de m~me valeur den, mais de moment angulaire supérieur à l..

Chapitre III
LES MODELES THEORIQUES.
III.1. INTRODUCTION.
Nous ailons présenter un exposé succinct des différents pro
blèmes théoriques posés par l'étude des co11isions entre particu1es
lourdes aux énergies thermiques et des diverses méthodes proposées pour
les résoudre. Un bref exposé générai permettra de scinder en deux gran
des catégories 1es prob1èmes posés : nous traiterons d'abord 1'aspect
statique (connaissance du potentiel d'interaction) puis 1'aspect dyna
mique (réso1ution de l'équation de Schrodinger) de la co11ision. Dans
chacune de ces deux parties l'exposé suivra, en générai, un ordre de
complexité croissante. Nous indiquerons, à chaque étape, les limita
tions et la vaiidité des approches considérées, ainsi que les app1ica
tions possibles. Une dernière partie sera dévolue au cas particu1ier
des collisions mettant en jeu des états excités pour lesquels l'exten
sion des méthodes exposées en début de chapitre ne va pas sans problè
mes. Nous anaiyserons aussi dans cette partie des modèles particu1iers
développés spéciaiement pour le cas des états de Rydberg. Dans tout
ce chapitre nous ne détaillerons que les méthodes et les approches
que nous avons effectivement utilisées, ainsi que les points encore peu
ou ma1 connus (collisions mettant en jeu des états de Rydberg). Pour le
reste nous renverrons le lecteur aux références qui nous ont paru im
portantes. Sauf mention spéciaie, nous considérerons ici le cas des
collisions aicaiin excité-gaz rare conduisant à des transferts de po
pu1ation entre niveaux, laissant en particulier de c$té le vaste domai
ne .des col1isions entre sous-niveaux de type (J,m.) - (J',m.,) que J J
nous n'avons pas étudié expérimentaiement.

- 20 -
III • 2 • GENERALITES.
Dans le cas des collisions thermiques la vitesse relative v
des particules en collis~on est très inférieure à la vitesse v d'un e électron quelconque du système (en fait, dans le cas des collisions
alcalin-gaz rare, seule intervient la vitesse ve de l'électron externe
de l'alcalin, le coeur Â+ et le gaz rare G ne comportant que des cou
ches complètes). Cette hypothèse (ve ~ v) a un caractère très général
et vaut aussi dans le cas des états de Rydberg (par exemple à
T = 4oo°K, pour les collisions Na-He on a v/ve ~ 0.1 pour n ~ 100).
On a alors intérêt à considérer les états de la quasi-molécule formée
par les deux atomes interagissant, les noyaux étant supposés immobi
les à la distance R. Le mouvement relatif des noyaux sera ensuite trai
té comme une perturbation. L'approximation qui néglige le mouvement
des noyaux est l'approximation de Born-Oppenheimer1
• Nous allons rap
peler brièvement sa formulation, ce qui nous permettra d'introduire
la notion très importante d'états adiabatiques.
L'équation de Schrëdinger (dans le référentiel du centre de
masse) s'écrit:
( + ) . ~~ c- ) H'l' r,R; t = in IT r,R; t ( C1)
où H représente le hamiltonien total du système et ~(r,R;t) la fonction
d'onde totale, r représentant les coordonnées électroniques. Il est in
téressant de mettre le hamiltonien H sous la forme :
H(r,R) = T(R) + He1 (r,R) (c2)
où T(R) est l'opérateur d'énergie cinétique des noyaux; He1 (r,R) tient
compte de toutes les autres interactions (en particulier il contient
l'interaction électrostatique entre les noyaux). On définit alors les
valeurs propres et les fonctions propres de He1 ,par
H 1("1°,R) ~ (r,R) = V (R) ~ (r,R) e n n n (c3)
où~ (r,R) et V (R) sont les fonctions d'onde et les énergies adiaba-n n tiques du système pour une valeur donnée de R, qui intervient ici com-
me paramètre. Les~ (l,R) forment une base complète, soit : n

- 21 -
< ~ni ~m> = Ônm (c4)
où les bra-kets correspondent à l'intégration sur les coordonnées élec
troniques r. La résolution de l'équation (c3) constitue la première
étape du problème, étape que nous pouvons appeler statique, puisque
s'effectuant à R fixe. Il est fondamenta1 de voir que les états adia
batiques sont des combinaisons linéaires des états propres de HA(1').
Les coefficients de ces combinaisons linéaires étant fonction de R, il
en résu1te des couplages entre états adiabatiques, que nous appellerons
couplages statiques. En effet He1 ('r,R) peut s'écrire
He1 (1°,R) = HA(r) + VI(r,R) (C5)
ici HA(r) représente le hamiltonien électronique de l'atome a1ca1in
isolé et VI(~,R) rend compte de l'interaction a1ca1in-gaz rare. Les
fonctions propres et les va1eurs propres de HA(r) sont les fonctions
d'onde et les énergies des niveaux de l'atome a1ca1in et ne sont fonc
tions propres et va1eurs propres de He1 (1,R) qu'asymptotiquement,
pour R -~. Ainsi lors de la résolution de l'équation. (C3), qui revient
à diagona1iser H 1 (r,R),,les fonctions adiabatiques~ (r,R) appardtront e n comme des combinaisons linéaires des fonctions propres de HA(r), avec
des coefficients dépendant de la forme explicite de VI(~,R), c'est-à
dire, en particu1ier, de R.
Nous pouvons maintenant développer la fonction d'onde tota1e
~(r,R;t) sur la base adiabatique~ (~,R) soit: n
p(~,R;t) = L ~m(1,R)'f-'m(R;t) m
Reportant (c6) dans (C1) et tenant compte de (C2), (C3) et (c4) on
obtient:
{T(R) + V (R)} 141 (R;t) + I C 'lt' 1 (R;t) n n n' nn n
. cl'jJ n = 11i ~ (R; t)
avec
Cnn' =.::: ~ni T(R)I ~n' >
(c6)
(c7)

- 22 -
L'équation (C7) rend compte de la dynamique de la collision. Sa résolu
tion constitue la deuxième étape du problème. L'approximation de Born
Oppenheimer consiste à négliger le mouvement des noyaux, c'est-à-dire
à considérer C , = O. L'équation (c7) montre qu'alors l'état électro-nn nique du système restera inchangé au cours de la collision, c'est-à-
dire que, par exemple :
ï(r,R;t) = ~ (r,R)~ (R;t) n n
Dans le cas contraire (C , ~ 0) on peut dire que le mouvement des nn
(es)
noyaux induit, par l'intermédiaire du terme de couplage dynamique C , , nn
des transitions électroniques non adiabatiques* au cours de la colli-
sion. La fonction d'onde totale devient alors lllle combinaison des fonc~
tions adiabatiques, à partir de laquelle on calculera les probabilités
de transition.
Il est clair que le problème des transitions non adiabatiques
peut 3tre scindé en deux parties distinctes :
- lllle partie statique, qui correspond à l'obtention, par résolu
tion de l'équation (C3), des états adiabatiques et de leurs énergies,
R étant fixé.
- lllle partie dynamique, qui correspond à la résolution du système
d'équations couplées (c7).
Nous allons maintenant développer plus en détail ces deux
aspects.
llI.J. LES POTENTIELS ADIABATIQUES.
La résolution de (CJ) dans le cas du couple alcalin-gaz rare
demande en général la mise en oeuvre de techniques numériques élabo
rées. Nous allons poser le problème puis indiquer dans les paragraphes
suivants les différentes approches utilisées, en commençant par la plus
simple, ainsi que quelques caractéristiques générales des résultats
obtenus, qui nous seront utiles par la suite.
* L'approximation de Born-Oppenheimer est équivalente, au..~ énergies thermiques, à l'approximation adiabatique qui consiste à négliger seulement Cnn, avec n ~ n', c'est-à-dire à inclure Cnn dans le premier membre de (c7). On peut montrer en effet que ce terme est toujours négligeable conduisant à l'équivalence des deux approximations.

- 23 -
Nous a11ons d'abord considérer les collisions de structure
fine dans le premier doublet des atomes a1ca1ins, soit :
A(n2
P1 / 2 ) + G A(n2PJ/2 ) + G - t:.E.
En effet celles-ci ont fait les premières l'objet d'études
expérimentales et théoriques. Cela ne restreint pas la généralité de
ce qui va suivre, tout en permettant un exposé plus précis. Les solu
tions asymptotiques (R -~) de l'équation (C3) sont évidemment les
états atomiques du système (le terme VI(r,R) étant nul dans (C5)). Le
problème consiste à trouver les termes moléculaires V (R) qui sont n associés asymptotiquement aux états atomiques 2P.(A) + 1 s (G), le gaz
J 0
rare restant toujours à l'état fondamental. On peut montrer facilement
que le hamiltonien total devant commuter avec .r2 et J (valeurs corresz pondant à l'atome alcalin, le gaz rare ne comportant que des couches
complètes), Jet m. restent les bons nombres quantiques. Les états de J
même\ mjl ayant même énergie il y a autant de termes moléculaires asso-
ciés asymptotiquement aux états atomiques 2P.(A) + 1 s (G) que de va-J 0
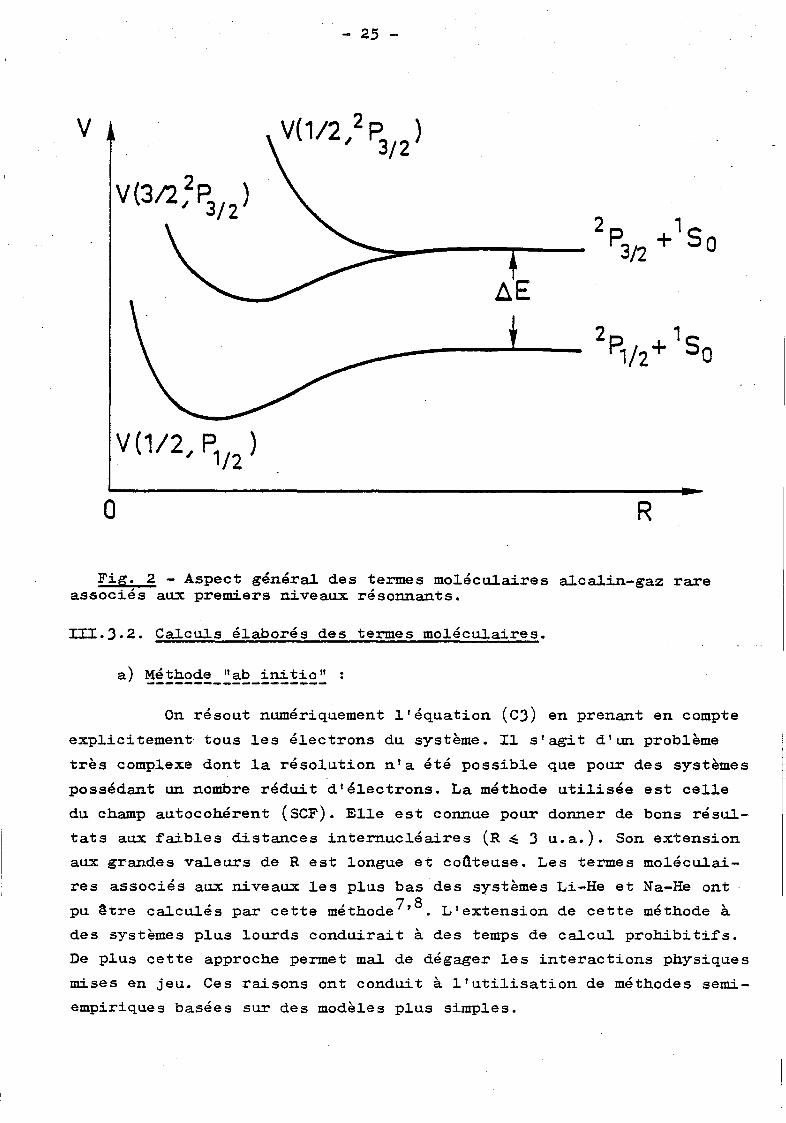
leurs de I mjl• Ainsi il y a deux termes associés à l'état 2
P3; 2 (A)
(nous omettons désormais 1 s
0(G)) : V(l/2, P3; 2) et V(J/2, P3; 2 ), la
va1eur de l~l correspondant au premier indice*.
III.J.1. Une approche simple.
E.E. Nikitin et ses collaborateurs3 ' 4 ont développé une ap
proche particulièrement simple ·qui s'est avérée très fructueuse avant
l'apparition de calculs plus élaborés pour les termes moléculaires
alcalin-gaz rare. Elle consiste à mettre en évidence explicitement l'in
teraction spin-orbite puis à prendre des approximations analytiques
simples pour l'interaction restante.
*ces états moléculaires sont souvent notés 2r1 ;2
et 2ir312
en notation
Herzberg ( 2S+lAQ, où S représente le spin total, A rendant compte de
la_valeur de m1 , composante du moment orbital sur l'axe internucléaire,
ici O et 1 respectivement, et Q la valeur de jm.f). Cependant, comme J
l'ont montré Pascale et Vandeplanque 2 cette notation est ambigüe car
m1 n'est pas toujours (VR) un bon nombre quantique. Nous indiquerons,
quand cela sera nécessaire,la correspondance entre les différentes
notations.

- 24 -
Le hamiltonien électronique total peut s'écrire
Hl= H + W (c9) e o
où H représente la partie électrostatique et W l'interaction spino
orbite. Les états propres et les fonctions propres de H peuvent 0
s'écrire :
V! = <. p O j HO j p O >
V 'Il = <. p 1 1 HO I p 1 > = < p -1 1 Ho I p - 1 > ( C10)
où I P0>, 1 P
1> représentent les fonctions d'onde électroniques lfJ,
.:t. .,m1 (avec l = 1) (sans tenir compte du spin, qui n'intervient que dans w). La diagonalisation de H0 + W sur la base (P3; 2 , ± 1 ; 2 ; P3; 2 , .:t. J/2 P1 ;
2, + 1 ; 2 ) permet d'obtenir les termes moléculaires sous J.a forme
( 2 ) 1[ AE ] ,2 V 1/2, P1 ; 2 = 2 V11
+ VI - 3 - AU souvent note 11112
v(1/2, 2
P 3; 2 ) = ! [v11 + VI - ~ - Au] souvent noté 2
I 1 ; 2 ( C11)
2 AE V(J/2, PJ/2 ) = V11 + "3'" , 2
souvent note 11312
où AE représente l'écart de structure f'ine ; AU est déterminée par:
(AU) 2 = ( V 11
- VI ) 2
- ! .ô.E ( V 11
- VI ) + ( AE ) 2 ( C12)
Cette décomposition ne J.aisse subsister que deux inconnues VI
et V11
• A moyenne (R>5 u.a.) et grande distance interatomique (seules
régions intéressantes dans le cas des collisions thermiques) les seules . .
interactions à prendre en compte sont l'interaction électrostatique
(Van der Waals à grande distance) et l'interaction d'échange, due à
l'indiscernabilité des électrons de l'alcalin et du gaz rare (qui cor
respond, en toute rigueur, à la procédure d'antisymétrisation de la
fonction d'onde totale). Des formes analytiques simples ("potentiel
exponentiel" à moyenne distance interatomique) ont été utilisées5 ' 6 •
Leur validité, ainsi que J.a sensibiJ.ité des calculs vis-à-vis des pa
ramètres retenus a déjà été largement discutée6 , 3 , 4 • La figure 2 pré
sente l'aspect général des termes moléqulaires ainsi obtenus.
- 25 -
V V(1/2} p3/2)
V(3/2:~/2)
V (1/2., P,12
)
0
iiE
2 p +1So 3/2
2 1 Pi12+ Sa
R
Fig. 2 - Aspect générai des termes mo1écu1aires a1ca1in-gaz rare associés aux premiers niveaux résonnants.
III.J.2. Ca1cu1s élaborés des termes molécu1aires.
a)~~!~~~=-:~~-!~!!~::
On résout numériquement l'équation (C3) en prenant en compte
explicitement tous les électrons du système. Il s'agit d'un problème
très complexe dont la réso1ution n'a été possible que pour des systèmes
possédant un nombre réduit d'électrons. La méthode uti1isée est cel1e
du champ autocohérent (SCF). Elle est connue pour donner de bons résu1-
tats aux faibles distances internucléaires (R ~ 3 u.a.). Son extension
aux grandes va1eurs de Rest longue et codteuse. Les termes mo1écu1ai
res associés aux niveaux les plus bas des systèmes Li-He et Na-He ont
pu 3tre ca1cu1és par cette méthode 7 ' 8 . L'extension de cette méthode à
des systèmes plus lourds conduirait à des temps de ca1cu1 prohibitifs.
De plus cette approche permet ma1 de dégager les interactions physiques
mises en jeu. Ces raisons ont conduit à l'utilisation de méthodes semi
empiriques basées sur des modèles plus simples.

- 26 -
b) ~!~~~~=~-~=~==~E!!!S~=~· L'idée essenti.elle est de considérer que seuJ. l'électron de
vaJ.ence de l'al.cal.in joue un rS1e et de traiter ainsi un prob1ème à
trois corps (l'électron de vaJ.ence de l'aJ.caJ.in, le coeur ionique A+
et le gaz rare G considéré comme une sphère polarisable) comme il est
indiqué sur la figure 3.
e-_. r
R
ro
Figure 3 Modèle à trois corps
de ·représentation dÙ couple_. al.cal.in-gaz rare
Le problème revient à diagonal.iser He1 (r,R) (éq. (C5)) sur
1a base des fonctions propres (atomiques) de HA(r). Comme noté précé
demment le potentiel d'interaction VI(r,R) introduit al.ors des coupla
ges statiques entre les fonctions d 1 ondes molécuJ.aires adiabatiques.
La résolution numérique requiert la solution préaJ.able des problèmes
suivants :
- Choix des fonctions d'onde atomiques ;
- Limitation de la base des états atomiques (théoriquement infinie)
à prendre en compte ;
- Détermination de 1a forme explicite de VI(r,R).
Ce modèle a tout d 1 abord été utilisé par Baylis9 pour le cal.cul des
termes moléculaires associés à l'état fondamental. 2s112 et au premier
doublet excité 2P1;2
, 312 des atomes al.cal.ins perturbés par un gaz rare.
Baylis a utilisé une base réduite aux trois états atomiques précités,
les fonctions d'onde de type Bates-Damgaard et un potentiel d 1 interac
tion comprenant essentiellement deux termes. Le premier rend compte de

- 27 -
l'interaction électrostatique, de type Van der Waals pour les grandes
valeurs de R, le second, important aux faibles valeurs de R, est un
pseudo-potentiel répuJ.sif10 qui remplace la procédure d'antisymétrisa
tion de la fonction d'onde totale (automatiquement prise en compte dans
les calcuJ.s ab-initio) et rend compte des forces d'échange. Le seuJ.
paramètre ajustable, r, est déterminé pour reproduire au mieux la 0
profondeur (déterminée expérimentalement) du puits de potentiel du
terme molécuJ.aire associé à l'état fondamental n0
2 s112
• Les potentiels
de Baylis ont été utilisés lors du calcuJ. des sections efficaces de
transfert de structure fine des premiers doublets alcalins.
Cette méthode a été généralisée par J. Pascale et
J. Vandeplanque 2 qui ont obtenu les termes molécuJ.aires associés à de
nombreux états excités pour tous les couples alcalin-gaz rare, ce
gr!ce à l'utilisation d'une grande base d'états atomiques. Le point
le plus intéressant de ce calcuJ. est la mise en évidence de l'influen
ce des couplages statiques qui donnent lieu à l'apparition de structu
res plus ou moins prononcées dans les termes molécuJ.aires. Un exemple
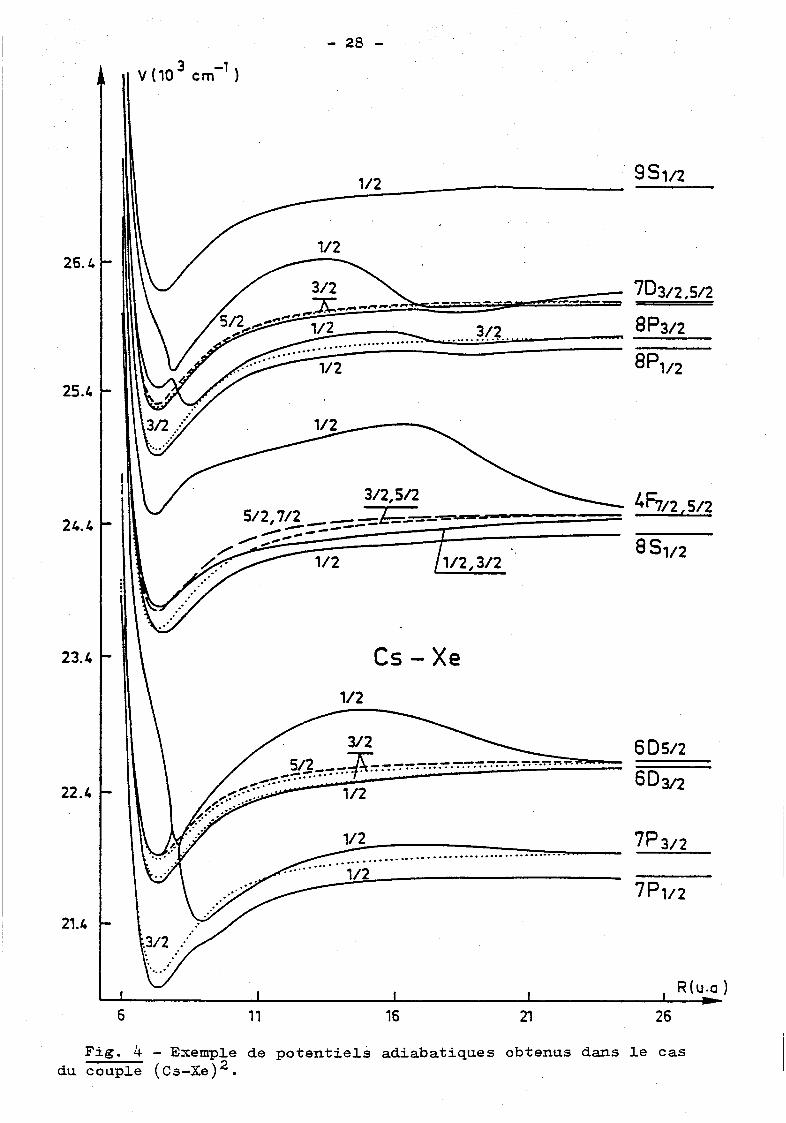
en est donné figure 4 pour le couple (Cs-Xe). Ainsi un pseudo-croise
ment marqué, pour R ~.8 u.a., appara!t entre les termes V(1/2,7P3; 2 )
et V(1/2, 6n312
). Nous reviendrons sur ce point lors de l'analyse de
nos résuJ.tats expérimentaux concernant les transitions 7P +6D. Il est
à noter que,m3me pour le premier doublet résonnant, les calcuJ.s de
Pascale et Vandeplanque diffèrent notablement de ceux de Baylis indi
quant clairement l'influence du choix de la base sur les résuJ.tats
obtenus. Ces calcuJ.s ont été très largement utilisés ces dernières
années. Ils ont permis d'expliquer de manière satisfaisante de nom
breux résuJ.tats expérimentaux, ce particuJ.ièrement dans le cas des gaz
rares lourds. Les résul.tats maintenant disponibles concernant aussi
bien des mesures de sections efficaces obtenues en celluJ.e11 ' 12 ou par
méthode de faisceaux13 , que des mesures d'élargissement des ailes
lointaines des raies de résonance14
, ou l'observation des bandes asso
ciées aux excimères alcalin-gaz rare15 ou encore la spectroscopie
laser des quasi-molécuJ.es alcalin-gaz rare16 permettent des tests très
divers de ces potentiels (ce aussi bien en fonction de R que de la na
ture du gaz rare). Il est vraisemblable que, particuJ.ièrement pour les
gaz rares légers et aux faibles valeurs de R, des améliorations sont
nécessaires pour arriver à un meilleur accord entre résuJ.tats expéri
mentaux et théoriques. De fait un calcul très récent, reprenant la
- 28 -
V(10 3 cm-1 )
1/2
1/2 26.4
3/2 ~········-··~...!.
25.4
24.4 5/2, 7/2 3/2,Sn -----'=== -------~-=--
___ ww--' ,,_...,
,,.,... :..----~ 1/2 /1/2,3/2
23.4 Cs -Xe
22.4
...........................
21.4
6 ,, 16 21
~-~~--
9S112
703/2,5/2
8P312
4F112,s12
8S112
605/2
60312
7P312
7P112
R { u.a )
26
Fig. 4 - Exemple de potentiels adiabatiques obtenus dans le cas du couple (cs-Xe) 2 •

- 29 -
m~me méthode mais uti1isant des fonctions d'onde p1us élaborées et un
terme d'échange légèrement différent a été effectué17 , mais les tests
théorie-expérience ne sont pas encore assez nombreux pour permettre
de conc1ure à une amélioratiqn notable des potentiels ainsi obtenus.
D'autres ca1cul.s sont disponibles pour certains couples et
pour des niveaux peu excités18
(niveaux résonnants). I1s diffèrent
essentiel1ement des précédents par 1a forme des termes d'interaction
à courte portée qui sont les plus diffici1es à évaiuer. Ainsi des po
tentie1s modè1es (interaction e--G entièrement attractive) ont été
uti1isés par C. Bottcher19 (cas Na-He) et très récemment par F. Masnou
et ai. 20 (cas Na-Ne). Dans ce dernier cas l'accord obtenu avec les ré
su1tats expérimentaux16 constitue un bon test de la méthode, du moins
pour des va1eurs de R de l'ordre de 10 u.a.
Le modè1e à trois corps s'est avéré très fructueux car i1
permet l'obtention des courbes de potentiel adiabatique associées à
de nombreux états atomiques. Les résu1tats obtenus par les diverses
méthodes citées peuvent cependant différer notablement et seu1e une
1arge confrontation avec 1es nombreux résu1tats expérimentaux mainte
nant disponib1es peut permettre de conc1ure vcµab1ement quant à leur
efficacité.
III.4. TRAITEMENT DU PROBLEME COLLISIONNEL.
L'étude des transitions non adiabatiques induites entre ter
mes électroniques par 1e mouvement des noyaux (rupture de 1'approxima
tion de Born-Oppenheimer) peut se traiter essentie11ement de deu.~ ma
nières
- soit on uti1isera 1'approche semi-classique
- soit on uti1isera 1'approche quantique.
III.4.1. Approche semi-c1assigue.
E11e consiste à séparer le système lent (mouvement des noyaux),
qui sera traité de manière classique, c'est-à-dire par l'introduction
d'une trajectoire R(t) et le système rapide (mouvement électronique)
qui sera traité quantiquement. Nous al1ons tout d'abord rappe1er briè
vement la formu1ation générale de cette app~oche.

- JO -
a) !~~~~!:!:2!!-.
Le hamiltonien électronique Hel (éq. (C3)) devient fonction
du temps par l'intermédiaire de R(t) ; il obéit à l'équation de
Schrëdinger :
- - • ë) --H 91
( r , R ( t ) ) $6 ( r , t ) = :l.'.fi ît ~{ r , t )
Les solutions de l'équation (C3) (fonctions d'ondes adiabatiques
~ (r,R)) formant une base complète on peut écrire n
~(r, t) = ~ an ( t) ~n (r",R) exp ( - ½ f V0
(R)dt)
( C13)
( C14)
Le problème consiste alors à trouver les coefficients a (t) (1 a (t)l 2 n n
représentant la probabilité de trouver le système dans l'état adiaba-
tique n à l'instant t). Une manipulation analogue à celle conduisant
à l'équation (C7) donne :
da ( i in ___,E; : L C I a I exp - ..,: dt , nn n a n J\v
0,-vn)dt)
-oo
avec C nn ' = <. ji1 n / - iii .]_ 1 $6 > ( C1 5 )
3t n'
Le système (C15) est l'analogue semi-classique des équations
(c7). Le problème se ramène ainsi à la résolution d'un système (d'ordre
théoriquement infini) d'équations différentielles couplées, les ji1 et n V étant supposés connus (paragraphe III.3). Notons que les couplages n statiques apparaissent dans le terme exponentiel, les C , rendant nn compte des couplages dynamiques. On peut les expliciter aisément en
utilisant les notations de la figure 5.
L'opérateur -ih 'ft peut s'écrire
c) - -- ih î"-ë -. o . . 1
i1i. R ) R - 11i ji1 ~ ( C16)
le premier terme correspond au couplage radial, le second au couplage
rotationnel, qui vaut :

- 31 -
• cl _ bv J - ifl ~ ~ - R2 Y ( C17)
où v est la vitesse relative des particu.1.es en collision. On peut mon
trer facilement que {R (couplage radial) ne couple que les états molé
cu.1.aires de m3me symétrie (~jmjl = 0) alors que~ (couplage rotation
nel) ne couple que les états de symétrie différente (~\mjl = ~ 1), sauf
si I m.\ = 1/2, cas pour lequel il existe mame si ~jm.l = o. J J
y
A >>' • X
z
Fig. 5 - Paramètres de la collision dans l'approche semi-classique. La trajectoire est supposée rectiligne, de paramètre d'impact b.
Le système (C15) doit, en général, 3tre résolu numériquement.
Il existe cependant des cas où une solution analytique est possible.
C'est cette première situation que nous allons à'abora envisager.

- J2 -
b) ê~!~~!~~-~~r~!S~=:
Cel1e-ci n'est possible que si deux conditions supplémentai
res son~ réa1isées
- i1 n'y a que deux termes électroniques fortement coup1és ; le
système (c15) se réduit à deux équations coup1ées
- le couplage adiabatique est loca1isé à une zone de faible éten
due autour d'un point R0
•
Ces conditions sont réa1isées si l'on a croisement ou pseudo-croise
ment de deux termes électroniques v1 et v2 • On peut a1ors appliquer
1e modèle Lzs22 , 23 , 24 , que l'on peut formuler ainsi suivant la symétrie
des termes considérés
i) v1
et v2
sont de symétrie différente
On peut avoir croisement (Fig. 6) et le terme de couplage dy
namique c12 (rotatioIUlel) est doIUlé par
V
V
Re
c12 = :; <~1IJyl~2>
v,
Vz R
Figure 6
Croisement de deux termes moléculaires.
( C18)

- 3J -
1e système (C15) se réduisant a1ors à
. da_, bv !î1 !î1 ( i Jt (V2-v1 )dt) ifi. dt = a2 R2 < 1 I J Y 1 2 > exp - 1i C -eo
da2 b ( . f t (v1-V 2 )dt) in~= a, R; < ~2 1Jyl~1> exp - ½ C -0,
(C19)
Supposons, de p1us (modè1e dit 1inéaire)
V 1 = -F 1 b.R + V
(c20)
v2 = -F2
AR + V
où F1 et F2 représentent 1es pentes au point de croisement et~ vaut :
AR = R -R0
= vR t ( C21)
où vR' vitesse radia1e, en R = R0
, supposée constante est donnée par:
VR = vJl (~Y V E (C22)
E représente 1 1 énergie cinétique 1/2 M 2 V ' M étant 1a masse réduite du
système (A- G). Le système (C19) se résout a1ors aisément (avec 1es
conditions initia1es a,(-M) = 1 et a 2 (-~) = 0) donnant pour 1a proba
bi1ité de transition 1 ..... 2 en R C
2 ( 2n(C12)2 ) p12 = I a2(+~)I = exp - 1i v~IF
1-F~I = exp(- ~/vR) (C2J)
La probabi1ité tota1e s'obtient par double passage en R ; si 1es termes C
d'interférence sont nég1igés25 (c'est-à-dire si R0
est situé 1oin du
"turning point") on a
q, = 2P 1 2 ( 1 -P 1 2) (C24)
La section efficace ~12 (v) est obtenue par intégration sur le paramètre
d'impact, soit :

- :)4 -
lb
max
o-12 (v) = 411
0
-o:/vR -o:/vR e (1-e )b db (c25)
où b correspond à la racine de l'équation (C22). La section effica-max
ce moyennée sur une distribution de Maxwell (cas des mesures en cellule)
est donnée par:
cr12(T) = J:,2(E) Eseuil
e-E/kl' E --dE k2T2
(c26)
Nous verrons au chapitre VI un exemple d'application de ce modèle ainsi
que la procédure de calcul de l'expression (C25).
ii) v1 et v2
sont de m~me symétrie :
On peut avoir pseudo-croisement (Fig. 7) et le terme de cou
plage dynamique c12 est alors de type radial.
V 1
V
20
2 2
R Re
'Fig. 7 - Pseudo-croisement de deux termes moléculaires.

- 35 -
Sur la figure 7 les termes adiabatiques sont notés 1 et 2 et les ter
mes que nous appellerons diabatiques (c'est-à-dire les termes qui peu
vent se croiser .à l'ordre 0) 1° et 2°. L'hamiltonien, développé sur
la base diabatique peut s'écrire sous la forme matricielle :
(
H,1 jHf ~o =
H21
1¾2) H22
(c27)
avec :
H .. = <~';>IHI~~ > 1J 1 J
La diagonalisation de ~Hl~ permet facilement de montrer que
H, 2 = 6. V = H21 (C28)
On peut développer la fonction d'onde sur la base diabatique, soit :
'I' = ", !ilr exp ( - ! J \,, 1 dt) + a2 !il~ exp ( - i -OIi
JtH22 dt) '-<JO
Le système (C15) se réduit alors à
iii :; = a 2 H21 exp ( - ! j \a22-a, 1 )dt)
da2
i-:h dt
-0:I
= ", JI, 2 exp ( --½ J\a, 1 -H2)dt) -cc,
(c29)
(c30)
Si on suppose de plus (modèle linéaire) que 1es termes diabatiques sont
linéaires au voisinage de R, et que 6.V peut @tre considéré comme l.llle C
constante, c'est-à-dire que :
lHt~o =(V- F1AR
6.V
6.V )
V- F2
AR
(c31)

- 36 -
1e système (CJO) est forme11ement identique à (C19) et sa réso1ution
donnera:
p12 = exp (- 2n(6.V)2 ) = exp ( -j3/vR) 'fi vRIF1 -F2 1
(CJ2)
On est ainsi ramené au cas précédent avec j3 en lieu et p1ace de~
(cf. éq. ( C2J) ) •
La validité du modèle LZS linéaire a été largement discu
tée25,26; e11e se ramène toujours aux points suivants:
- variation 1inéaire des termes é1ectroniques au voisinage de R C
- vitesse vR supposée constante au voisinage de R0
; on est donc
1oin du "turning point";
- terme de couplage dynamique supposé constant au voisinage de R. C
Ces hypothèses sont restrictives et la méthode ne s'applique qu'à des
situations particu1ières qui présentent des croisements ou des pseudo
croisements bien localisés (cf. Fig. 4).
Notons enfin qu'une solution analytique du système (C15) est
possib1e dans 1e cas de deux états dont les termes molécu1aires présen
tent des croisements (ou pseudo-croisements) p1us étendus (c'est-à-dire
que la zone de non-adiabaticité s'étend de manière notab1e autour de
R0
) : il s'agit du modèle LZS exponentiel déve1oppé par Nikitin et ses
co11aborateurs25 • Il a été appliqué avec succès au cas des transitions
de structure fine des premiers doub1ets alcalins, la partie statique du
problème étant traitée de manière approchée (paragraphe III.J.1). Ne
l'ayant pas utilisé nous renvoyons le lecteur intéressé à la référen
ce 25.
Les deux versions du modèle LZS ne s'appliquent cependant
qu'à des cas bien spécifiques (deux niveaux présentant des couplages
statiques bien localisés). Seu1 un calcul semi-classique comp1et per
met de s'affranchir des restrictions que nous avons mentionnées.

- 37 -
) , , c ~EEE2S~~-~~~~E~~
E11e consiste à résoudre numériquement 1es équations coup1ées
du système (c15). Les prob1èmes sou1evés par cette méthode, outre 1es
prob1èmes numériques c1assique, sont 1es suivants :
- choix des potentie1s (voir paragraphe III.:J)
- choix de 1a base pour 1'expression exp1icite de (c15). On peut
choisir 1a base adiabatique mo1écu1aire27 , 1a base atomique29 , ou m3me
une base qui diagona1ise une partie de Hei et qui peut rendre p1us
simp1e 1e ca1cu1 des C ,, du moins pour certaines zones de distance IUl
. t 1' . 28 in ernuc eaire ;
- 1imitation de 1a base pour réduire 1e nombre d'équations coup1ées
à résoudre. On peut ainsi prendre une base comportant un nombre d'états
p1us faib1e que ce1le utilisée 1ors de 1a réso1ution de 1a partie sta
tique du prob1ème. Certains couplages statiques avec des états voisins
seront ainsi pris en compte, m3me si ceux-ci ne sont pas inclus dans
1e traitement dynamique du prob1ème 29;
- choix de la trajectoire. On peut choisir une trajectoire recti
ligne ou une trajectoire définie par un des tèrmes adiabatiques28 , 29 •
Les effets de trajectoire sont surtout importants pour 1es états peu
excités.
Une te1le approche a été utilisée aussi bien pour 1es doublets réson
nants28,29 que pour 1es états p1us excités29 .
d) ~!~~!!_~!_!~!~!~~-
Ce1le-ci est particulièrement bien adaptée à l'étude des co1-
lisions thermiques alcalin-gaz rare, son hypothèse de base (séparation
des mouvements électronique et nuc1éaire) restant valide m3me pour des
états très excités. Elle permet de bien mettre en évidence l'importance
re1ative des différents couplages dynamiques en fonction de R, apportant
ainsi a postériori une certaine justification des approches simplifiées
(de type LZS). Il faut cependant noter ses principaux inconvénients:
- son extension à des états très excités est rendue difficile par
la nécessité de résoudre alors un système contenant un grand nombre
d'équations couplées ;

- :38 -
- 1e choix de 1a trajectoire peut introduire une erreur non nég1i
geable, particu1ièrement dans 1e cas des états peu excités ;
- e11e n'est va1ab1e que si l'écart énergétique asymptotique AE
entre les niveaux considérés est petit devant l'énergie cinétique re
lative E. Ce11e-ci est, pour 1es expériences en cellu1e d'environ
4 -1 . . . J\"C' 1 -1* 00 cm • On ne peut donc ut111ser cette approche que si ~ .$, 00 cm •
Pour pa11ier aux deux derniers inconvénients mentionnés une
approche entièrement quantique peut 3tre envisagée.
III.4.2. Approche quantique.
On doit a1ors résoudre numériquement le système (c7). La for
mu1ation du prob1ème a été faite par R.H.G. Reid30 , suivant les travaux
de A.M. ArthJ..Œs et A. Da1garno31 • L'idée est de résoudre une équation
de Schrëdinger du type "ondes partie1les" en uti1isant un développement
du potentie1 d'interaction en po1ynSmes de Legendre, ce qui ne laisse
subsister que certains couplages entre ondes partiel1es. La formu1ation
de Reid est développée sur une base atomique mais une base molécu1aire
peut aussi 3tre utilisée. Dans le cas des transitions intramu1tiplets
entre niveaux supposés bien iso1és le système se ramène alors à la ré
solution de deux systèmes de trois équations différentiel1es couplées.
Il n'y a alors plus de problème de trajectoire et les valeurs calcul.ées
pour des énergies voisines du seui1 prennent bien en compte tous les
effets d'interférence, ce qui n'est pas le cas de la méthode semi
classique. Par contre la résolution numérique pose des problèmes quand
l'énergie croît, car, comme il est bien connu, le nombre d'ondes par
tiel1es qui contribuent à la section efficace augmente notablement. Il
faut noter que les méthodes semi-classique et quantique obtiennent des
résu1tats similaires pour une énergie notablement supérieure à l'éner
gie du seuil. La méthode quantique a été utilisée uniquement pour les
premiers doublets résonnants des alcalins; des potentiels adiabatiques
de Baylis et ceux de Pascale et Vandeplanque ou des potentiels semi
empiriques ont été pris en compte, suivant les auteurs. Les résu1tats
* L'approche simplifiée de Nikitin a pu cependant ~tre étendue à des cas où AE/EN 1, en considérant la queue de la distribution Ma..""CWellienne de la vitesse relative des particules en collision.

- 39 -
sont en accord, en général., satisfaisant avec les résu1tats expérimen
taux. L'extension d'une telle méthode aux états fortement excités se
heurte, essentiellement, au m~me problème que la méthode semi-classique,
à savoir la nécessité d'introduire un grand nombre d'états dans le cal.
cu1 de la section efficace conduisant ainsi à la résolution d'un sys
tème contenant un grand nombre d'équations différentielles couplées.
III.5. LE CAS PARTICULIER DES ETATS FORTEMENT EXCITES (états de Rydberg).
Tous les problèmes posés par les collisions à énergie thermi
que mettant en jeu des états de Rydberg sont liés plus ou moins direc
tement au fait que, dans ce cas, le nombre d'états accessibles après
la collision, si on considère simplement les énergies asymptotiques
des niveaux de l'al.cal.in et l'énergie moyenne des particu1es en colli
sion (N 400 cm-1 ), devient grand. Ceci se répercute aussi bien sur
l'aspect statique (potentiel d'interaction) que sur l'aspect dynamique
(traitement de la collision) de la question. Nous traiterons tout
d'abord ces deux aspects dans l'esprit des chapitres précédents en
mentionnant les méthodes qui dérivent de celles que nous venons d'ana
lyser. Nous terminerons en indiquant des modèles qui ont été développés
plus spécifiquement dans le cas des états de Rydberg. Dans tout ce cha
pitre nous n'indiquerons pas, ou que très brièvement, les résultats ex
périmentaux correspondant, quand ils sont disponib1es. Les comparaisons
théorie-expérience seront développées dans la discussion des résu1tats
expérimentaux. Il est cependant à noter que, dans la plupart des cas,
les cal.cu1s théoriques ont été développés en liaison étroite avec les
travaux expérimentaux, comme nous le mentionnerons dans le courant de
notre exposé. Nous donnerons quelques indications relatives au cas où
le perturbateur est l'atome al.cal.in à l'état fondamental., notre travail
expérimental. ayant aussi porté sur ce point.
Avant de poursuivre l'exposé nous voudrions tout d'abord
mentionner l'idée très simple que l'on se faisait général.ement de ce
problème avant le début des travaux expérimentaux. La simple considé
ration de la taille géométrique de l'atome dans un état de Rydberg
laissait à penser que sa stabilité vis-à-vis des collisions devait dé
croître très rapidement avec l'état d'excitation, le rayon de l'atome
croissant approximativement comme n 2 • Plus exactement la section effi-

1i; '1 !I:
'!I i
111
li 11
Ili,
1'
l!i
I
l' 1: 11
1
1
li.
1il ,1,I
Il
!' Il
l'i ,,
i: i. i,,
i
11
1
1
! 'il
1.
,, 1 1:
'
- 40 -
cace associée à un processus collisionnel devait refléter plus ou moins
directement la section géométrique de l'atome erg
2. n*2 [ 2 cr = n < r > = 'Il - 5n* + 1 - 31(1 + 1 )]
g 2 (c33)
où~ est en wlités atomiques (c'est-à-dire exprimée en n a 2 - 0.88 12 ). g O - .
Ainsi pour un nombre quantique de 1 'ordre de 20 on obtient erg ~ 106 .
Notons que a-g croît proportionnellement à n*4 • Les travaux tant expé
rimentaux que théoriques ont modifié considérablement cette vision sim
pliste du problème.
III.,5.1. Potentiel d'interaction et états de Rydberg.
Dans le cas des états de Rydberg le modèle à trois corps
(e- de valence, coeur de l'alcalin et gaz rare) semble particulièrement
adapté pour l'obtention du potentiel d'interaction, l'électron de va
lence se trouvant, en moyenne·, à grande distance du coeur ionique A+.
L'utilisation du modèle de Baylis, généralisé par Pascale et Vand.eplan
que est cependant d'application difficile, vue la nécessité d'inclure
dans la base atomique un grand nombre d'états, ce qui conduit à des
difficu.ltés de calcul prohibitives. Notons cependant que des potentiels
adiabatiques ont pu 3tre obtenus pour des valeurs den de l'ordre de 12
en utilisant une base atomique réduite aux états les plus proches de
celui que l'on désire calculer32 . La précision d'une telle procédure
semble difficile à évaluer et sa généralisation à des valeurs den plus
élevées apparaît problématique. On ~st donc amené à faire d'autres
hypothèses simplificatrices en vue d'obtenir des potentiels approchés.
a) ~=-E~=~~~:E~!=~!!=!-~=-!=~:
Il consiste à approcher l'interaction électron-gaz rare par
un pseudo-potentiel de la forme 33 ,34 :
( .... -V = 2nLcS r - R) (C34)
-où r représente la position de l'électron, R celle du gaz rare et L la
longueur de diffusion, à énergie nulle, pour le gaz rare considéré.
Cette expression particulièrement simple permet, comme nous le verrons,
de notables simplifications de calcul lors du traitement de la partie
dynamique du problème. En particulier les éléments de matrice de ce
pseudo-potentiel s'expriment très simplement :

- 41
* V .. =<ilVlj) = 2nL 4].o/. 1J 1 J
(C:34)
Ce potentiel. a été tout d'abord introduit par Fermi35 , puis
amél.ioré quel.que peu par l.a suite36- 37 , pour traiter l.e probl.ème de
l. 1 é1argissement et du dépl.acement des raies spectral.es de 1a série
principal.a (n S - nP) des atomes aJ.caJ.ins, à forte pression de gaz 0
perturbateur. Pour des vaJ.eurs él.evées de n (n ~30) un bon accord a été
en général. obtenu avec 1es vaJ.eurs e:xpérimentaJ.es38 •
Ce type de potentiel. permet de retrouver, au premier ordre
de perturbation l.a section efficace de diffusion él.astique à énergie
nu11e él.ectron-gaz rare, soit :
Cïo = 4nL2 (c36)
Ceci ne signifie nu11ement que l.e calcul des perturbations s'applique à
un tel. type de potentiel.. De fait 1es termes suivants du dével.oppement
perturbatif ne sont pas définis39 • Ce potentiel. appara!t donc comme un
potentiel. modèl.e mathématique permettant de rendre compte de l.'interac
tion supposée très l.ocalisée (présence de 1a fonction 8)entre 1'él.ec
tron et l.e gaz rare.
L'empl.oi de ce pseudo-potentiel. pour l.e traitement des col.li
sions mettant en jeu des états de Rydberg appelle quel.ques remarques . t t 40 impor an es :
- Le pseudo-potentiel. de Fermi impl.ique que, 1ors de l'interaction
électron externe-gaz rare, seu.J.e l.a diffusion de l.'onde S. intervient
(puisqu'il correspond à la diffusion élastique à énergie nul.le, cf.
éq. (c36». Ceci suppose que la l.ongueur d'onde de De Broglie de l'élec
tron soit grande devant l.a portée du potentiel électron-gaz rare41 •
Cette approximation, dite de 1a l.ongueur de diffusion, n'est vaJ.abl.e
que dans l.e cas de l.'hél.ium, pour l.equel la section efficace de diffu
sion élastique à basse énergie est sensiblement constante42 • Pour J.es
autres gaz rares (qui présentent tous un effet Ramsauer42 pl.us ou moins
prononcé) il est possible de construire un potentiel. prenant en compte
1a diffusion des ondes partielles d'ordre supérieur34 , 43 mais cel.a com
plique notablement la résol.ution numérique du traitement dynamique de
la collision, comme nous le noterons lors de la discussion de celui-ci.

1
1
1 1
'
1
1 1 1 J.
1
1
Il' Il 1
! I;
ill I' 1.
- 42 -
- Le pseudo-potentiel. de Fermi empJ.oyé dans J.e traitement de J.a
col.l.ision négl.ige J.'effet du coeur ionique durant J.a diffusion de
J.'éJ.ectron externe par J.e perturbateur puisqu'il. J.a décrit en terme de
diffusion éJ.ectron J.ibre-perturbateur. Derouard40
a montré que ceci
équival.ait à "J.'impul.se approximation1144 et n'était val.abl.e que pour
des distances internucl.éaires R suffisamment grandes (~ 100 u. a.).
Ainsi l'empl.oi du pseudo-potentiel. de Fermi n'est justifié que pour
des col.J.isions mettant en jeu des états suffisamment excités (n~10).
Enfin, m3me dans ce cas, seul.es J.es col.l.isions à grand paramètre d'im
pact_ (J.ors d'un traitement semi-cl.assique) seront prises en compte cor
rectement. Il. importe donc de vérifier qu'el.J.es seul.es contribuent no
tabl.ement à l.a vaJ.eur del.a section efficace.
Le "potentiel." de Fermi apparatt donc comme un potentiel. J.o
cal.isé permettant de rendre compte, à J.'aide des données del.a diffu
sion él.astique, de J.'interaction d'échange atome-atome à grande dis
tance pour J.aque11e des formul.ations pl.us général.es ont été proposées.
b) ~~!!~!!!!_~~~~~~~=-~!~~!-=~~!!~=~!~~=:
Général.isant J.es résuJ.tats de Ovchinikova44 et Smirnov45 ,
Janev43 a obtenu J.'expression général.a de l.'interaction d'échange
atome excité-atome, vaJ.abl.e pour R suffisamment éJ.evé, ce en fonction
de l.'état éJ.ectronique de l.'atome excité, del.a longueur de diffusion L
du perturbateur ainsi que de sa pol.arisabil.ité ~. Les expressions obte
nues sont valabl.es si le potentiel. d'ionisation de l.'aJ.caJ.in est faibl.e
devant cel.ui de J.'atome perturbateur {ce qui est vérifié dans le cas
des gaz rares). Il. est intéressant de noter que dans le cas où J. = m = o
( étatet en considérant que E. <.< 1 J. 6 eV (E. étant l. 'énergie d' ioni-1 1
satio· J.'aJ.cal.in) J.e cal.cul. de Janèv donne :
V(R) ~ 2nLIV2 (R)
qui coïncide avec l.'expression (C35) montrant ainsi que J.e pseudo
potentiel. de Fermi peut 3tre considéré, au moins dans un cas particu
lier, comme une expression approchée du potentiel. d'échange.

_ 43 -
c) !~!~!~~=-~~-~~=~!_!~~S~=:
L'interaction coeur ionique-perturbateur est constituée
principal.ement d'un terme é1ectrostatique qui peut s'écrire34 (pour
de grandes val.eurs de R et dans 1e cas où 1e perturbateur ne présente
pas de dipo1e permanent) :
U(R) = - ~4 (en u.a.) 2R
( C37)
où~ représente 1a po1arisabilité du perturbateur. On peut supposer
que, pour n suffisamment grand,ce terme vient simplement s'ajouter au
pseudo-potentiel de Fermi, ce qui revient à sommer séparément 1es in
teractions e--perturbateur et A+-perturbateur. Ceci n'est évidemment
p1us justifié pour 1es faib1es val.eurs den, pour 1esque1les i1 con
vient d'utiliser un potentie1 plus réal.iste obtenu à partir de 1'in
teraction à trois corps (type potentiel de Pascal.e-Vandep1anque par
exemple). Le terme U(R) est en général. faib1e devant 1e potentiel de
Fermi (terme d'échange).
Notons que 1e cal.cu1 de Janev inc1ut, d'une certaine manière,
1'inf1uence de A+ par 1'intermédiaire du paramètre ~43 • Le terme
d'échange, comme 1'a montré Smirnov45 , reste supérieur à l'interaction
Van der Waal.s m3me pour 1es très grandes val.eurs de R, ce qui fait que
1'on peut général.ement nég1iger celle-ci.
d) ~=~~S~!~_f!~~=~:
Les méthodes classiques d'éval.uation de potentie1 semblent
difficiles à général.iser au cas des états de Rydberg du fait du grand
nombre d'états atomiques à prendre en compte. Le fait que l'électron
externe soit, en moyenne, très éloigné du coeur conduit à sommer sim
plement 1es deux interactions e--G et A+--0·, la première étànt considé
rée comme prépondérante. I1 n'est cependant pas évident, a priori, que
pour des val.eurs très élevées den celle-ci soit seule à considérer,
comme nous le verrons par la suite. Pour l'interaction e--G le pseudo
potentiel de Fermi (ou ses variantes) semble particulièrement adapté,
de par sa forme simple, au traitement des collisions mettant en jeu
des états de Rydberg. Notons pour terminer que, dans le cas où le pertur
bateur est l'atome al.cal.in dans son état fondamental., le problème est
plus complexe du fait, d'une part de 1a forte val.eur de la polarisabili-

1
·11:.!
il
il \;: 11' 1
H
,1
Ili li jjl
1
::,: ,1
1:
11.:.: i' il!i il' 1 1
li 1 1
1
il
11:
ii' j
1 1 i
l 1
1
11 ~. l
i
i Il
Il !1. :1 ,,, :, !!
1
1 !
- 44 -
té~ (inf'1uence du coeur ionique) et d'autre part du manque de données
précises re1atives à la diffusion e--a1ca1in.
III.5.2. Généra1isation du traitement de la.co1lision au cas des états
de Rydberg.
Nous traitons dans ce paragraphe de l'extension des méthodes
que nous avons précédemment décrites (paragraphe III.4) au cas des
états de Rydberg. La méthode semi-c1assique et 1a méthode quantique
ont été utilisées. Nous les ana1yserons successivement en soulignant
à chaque fois 1es difficultés rencontrées, propres aux états de
Rydberg.
a) ~EE~~=~=-~=~==!~~~!S~! :
Cette méthode a été tout d'abord proposée par Gersten46 pour
rendre compte du mélange collisionnel de moment angulaire du sodium en
collision avec des gaz rares, processus que l'on peut représenter par :
Na(n,1 = 2) + G --- Na(n,l > 2) + G (C:38)
Les états nD du sodium sont, comme nous l'avons mentionné, hydrogénoï
des, c'est-à-dire très proches des niveaux (n,l> 2) en énergie, ce qui
fait que 1'on peut considérer, en première approximation, ces états
comme seuls intervenant dans 1e ca1cul. On utilise 1e potentiel de
Fermi (éq. (C:34)) et la trajectoire est définie comme rectiligne par
- ..,.. -R = c + vt (c:39)
ce qui est bien justifiée, le potentie1 A+-G (éq. (C37)) étant négligé.
L'équation de Schrëdinger s'écrit :
i1i a°t l'i'> = (Ho+ V) I '+' .> (c4o)
On peut développer I~> sur 1es états propres f n,l,m> de H. On obtieno dra ainsi l.lll système de n 2 -4 équations couplées. Gersten suppose deux
cas : pour les grandes va1eurs de b (b ~ b ) il utiJ.ise l.lll traitement 0
perturbatif au premier ordre qui donne pour l'amplitude fina1e de pro-
babilité de transition de l'état I n,2,m'> à l'état I n,l,m:>47 :

_ 45 -
m' C1m(+oo) - -
+oit
½ i~ < n1ml VI n2m'> exp ( - ½(enJ.m-tn.2m' )dt) ( C41)
où tnJ.m et tn2m, représentent 1es énergies des états atomiques (états
propres de H) ; ceux-ci étant tous hydrogénoides, Gersten nég1ige ce 0 .
terme (on peut montrer que cette approximation est va1ab1e m@me pour
des va1eurs faibl.es de n, comme ce11es qu'il. considère : 4 ~ n ~ 7), ce
qui conduit ' a :
m' c1m(+411) =
i - 1i
+• 1. < n1m1Vln2m'>dt (C42)
Les fonctions d'ondes hydrogénoides sont uti1isées dans 1 1 éva1uatîon
des é1éments de matrice à 1'aide de 1 1 équation (c35). La section effi
cace correspondante s'écrira, après moyenne sur 1es états initiaux:
2n a-2 = 5 L
1,m,m' [
+"'
b 0
le~~( +o-) 12 b db (C43)
Dans 1e cas des faib1es paramètres d'impact on suppose que 1a probabi-
1i té de transition est constante tant que ceJ.ui-ci reste iIJŒ'érieur à
tme certaine va1eur b 1 , que nous définissons p1us 1oin, et s'obtient
simp1ement en considérant la mu1tip1icité des états initiaux et finaux,
soit:
2 n - 9
o-: = 2n 2 4 1 n -
f, 0
b db 2 = 11 b1
La section efficace tota1e pour J.e processus (C38) s'écrit a1ors
cr = 0-1 + 0-2
(c44)
(C45)
Le paramètre b1 s'obtient par continuité en écrivant que, en b0
, les
deux ampJ.itudes de probabi1ité sont éga1es, soit ((C43) et (c44)) :
1,,m' 12 2 5 L clm(+a.) = n - 9 n 2
- 4 (c46)

il' 1:, l:1 1: 1
- 46 -
Il. reste al.ors à passer du repère mobile au repère fixe, ce qui, dans
1e cas d'un ca.l.cu1 perturbatif au premier ordre, se fait sans intégra
tion supp1émentaire. Les résu1tats obtenus sont en accord re1ativement
satisfaisant avec J. 'expérience 48
mais 1.es ca.l.cu1s sont 1.imi tés à n ~ 7.
Cette méthode a J.'avantage d'une relative simplicité et son extension
à d'autres types de processus, sous réserve de sa va.l.idité d'applica
tion, est possible.
Omont34 , après avoir longuement discuté l'emploi du pseudo
potentie1 de Fermi (cf. paragraphe III.5.1) et 1es différentes appro
ches dynamiques possibles pour le traitement des collisions mettant en
jeu des états de Rydberg, a uti1isé la méthode semi-c1assique pour
éva.l.uer 1es sections de quenching. Dans le but d'obtenir des expres
sions ana.l.ytiques simp1es i1 a utilisé les fonctions d'onde J.W.K.B.
et remp1acé certaines sommes discrètes par des intégral.es. Les hypo
thèses nécessaires à la résolution analytique du problème limitent le
champ d'app1icabilité des résu1tats obtenus à des valeurs den élevées
(n*125). Cependant, outre 1eur simplicité d'emploi, ces formules met
tent en évidence le comportement asymptotique des sections efficaces,
aussi bien en fonction de la vitesse re1ative des particu1es en colli
sion que de J.a va.l.eur den. Ces comportements seront, dans une étape
expérimentale ultérieure, très intéressants à vérifier. Notons enfin
que, dans le cas du mélange collisionnel de moment angul.aire49 , la dé
pendance prévue en ~-3 des sections efficaces est bien.observée expéri
mentalement (dans le cas de l.'hélium), aussi bien qu'obtenue par d'au
tres approches théoriques (méthode quantique d'Hickman), comme nous
le verrons au chapitre VI.
Derouard et Lombardi40 ont étendu le cal.cul. de Gersten aux
niveaux plus excités (8~n~14) et ont comparé leurs résu1tats aux ex
pressions analytiques obtenues par Omont, et aux résultats expérimen
taux de Gal.1.agher48 , 49 • Leur travail. montre, en particu1ier, que J.es
collisions à faible paramètre d'impact contribuent très peu au résultat
final., ce qui justifie l'emploi du pseudo-potentiel de Fermi et indi
que que l.'hypothèse de collisions fortes utilisée par Gersten à faible
paramètre d'impact n'est pas, dans ce cas, justifiée. Les expressions
obtenues pour les sections efficaces donnent une formulation générale
de la méthode semi-classique, applicable au cal.cul. des différents ca
naux de désexcitation collisionnelle d'un état de Rydberg. Il. est mon-

- 47 -
tré de plus que l'approximation de la longueur de diffusion n'est va
lable que dans le cas de l'hélium et qu'une telle méthode ne peut s'ap
pliquer que pour des transitions inélastiques dont l'écart d'énergie
ne dépasse pas quelques dizaines de cm-1• Notons enfin que les résul
tats obtenus sont en bon accord avec l'expression asymptotique proposée
par Omont à condition d'effectuer une légère correction due aux va1eurs
relativement peu élevées du nombre quantique des niveaux étudiés
(8~n,15).
b) ~EE!~~~!-S~~~!S~!:
Le premier ca1cul quantique a été effectué par 01son50 • Il
concerne le processus collisionnel décrit par l'équation (C:38). Pour
donner au problème une formu1ation simple Olson a posé les hypothèses
suivantes
- seule est prise en compte la réaction
Na(nD5
; 2 ) + G --... Na(nF 512 ) + G + AE (C47)
- le forma1isme molécu1aire est utilisé, mais de manière simplifiée,
le couplage rotationne1 étant négligé et 1es trois couplages radiaux
(qui prennent en compte les états molécu1aires de m3me 1 mjl, soit
(1/2, 1/2), (3/2, 3/2) et (5/2, 5/2), asymptotiquement associés-aux
états atomiques nD5
; 2 et nF5
; 2 ) étant supposés identiques.
- l'éva1uation de 1'interaction d'échange se fait à l'aide du po
tentiel asymptotique de-Janev.
Les résu1tats obtenus sont en accord très satisfaisant avec 1'expérien
ce49, rendant bien compte, en particu1ier du maximum de la section ef
ficace obtenue, en fonction den, pour 1es trois gaz rares légers
(He, Ne, Ar), seu1s cas traités. La méthode nous semb1e cependant très
critiquable pour ies raisons suivantes ~
elle restreint arbitrairement le nombre des états pris en
compte. En particu1ier la transition présentant le plus fa:Lble écart
d'énergie serait la transition nD3
; 2 ~nF5
;2
;
. elle néglige le couplage rotationnel, qui peut--~tre impor
tant m3me pour des grandes va1eurs de R, comme l'a montré récemment
Pasca1e, dans le cas de deux niveaux très voisins du sodium (transi
tion 4n312 _,. 4n512 )

i,
;1
:1
jj I' 1
1
I! 1:
- 48 -
. el.J.e util.ise une approximation des potentiel.s de Janev
qui correspond, en fai~ à des états S, et non à des états D ou F, ce
qui revient à considérer, comme J.e note d'ail.l.eurs Ol.son, un simpl.e
potentiel. de Fermi.
La compl.exité del.a formul.ation quantique a conduit 01son à
poser des hypothèses très restrictives, pour J.e moins discutabl.es,
mais. 1'accord théorie-expérience obtenu appar~t très satisfaisant.
Conscient de ces difficuJ.tés A.P. Hickman a dével.oppé une formu.J.ation
quantique51 rigoureuse du prob1ème, en uti1isant J.e dével.oppement du
pseudopotentiel. de Fermi en po1ynSmes de Legendre. La réso1ution numé
rique du système d'équations coupl.ées n'a cependant été possib1e que
pour n ~ 8. Les val.eurs de sections efficaces obtenues diffèrent assez
peu de ce11es obtenues par 01son mais amé1iorent encore 1'accord théo
rie expérience. Pour des val.eurs den supérieures à 8, Hickman a propo
sé l'util.isation de la première approximation de Born, en util.isant
toujours le pseudopotentiel de Fermi. Les résuJ.tats obtenus51 pour
10~ n 615 dans J.e cas de l'hél.ium sont en très bon accord avec 1es ré
su.J.tats expérimentaux49 . Cette approche nous a sembl.é très intéressante
et nous l'avons util.isée dans le cas des états P. et S du rubidium.
Avant de discuter sa val.idité et ses avantages nous al.l.ons en présen
ter 1a formul.ation 1a pl.us général.a.
Soient i et f J.es états initial. et final. del.a col.lision. La
section différentiel.J.e de diffusion inél.astique, à 1a première appro
ximation de Born, s 1 écrit52 :
do". k 2 k 1 / . (+ - i- ,2 ~ _ _f l 12 ( m ) f 1 k. -kf r'
dQ· - k. Aif = --2 k vf. (r') e i . d'1' 1 2nn i 1
(C48)
avec :
ufi (r') = ! '+';(t:) u(~' ,;) Y\ ('l)dr (C49)
- -où m représente la masse réduite des particul.es en col.l.ision, ki et kf
J.es vecteurs d'ondes avant et après col.l.ision (de modul.es ki et kf),
1 1 l.a variabl.e dynamique* (distance internucl.éaire) et l' (i.e. r, G, lf)
* Nous uti1isons ici, EOur l.a distance internucl.éaire, ~', au J.ieu del.a notation habituel.l.e R, pour facil.iter une comparaison directe avec J.es résuJ.tats de 1a référence 51.

- 49 -
1e rayon vecteur de 1'é1ectron externe. Ufi représente l'élément de
matrice du potentie1, dont 1a forme n'est pas encore exp1icitement
spécifiée et Aif 1'amp1itude de diffusion. Les notations sont données
sur 1a figure 8.
.... r'
e-
Fig. 8 - Modè1e à trois corps pour l'interaction atome de Rydberg-perturbateur.
Si on peut obtenir le déve1oppement de U(r,r') en polyn8mes
de Legendre, soit :
u(~,r') = t ui\ (;) P) (r,t 1) (C50)
où r représente le module de~, alors il est facile de montrer (on uti-
1ise le développement de PÀ et des ondes planes en harmoniques sphéri
ques, ainsi. que la formule de normalisation des harmoniques sphéri
ques52) que l'amp1itude de diffusion peut s'écrire :
Aif = - ; ~i'° f j).(Kr') V/i"•)r•2dr• r 1\1;(:i')P;,.(i;°) 'f\(i!'•)a.1'• ( C51)
'~( ) -~ , ou K de module K vaut ki-kf; jÀ represente la fonction de Bessel
d'ordre À • Une formule similaire a été obtenue par Crawford.5:3 dans le
cas é1astique. On déve1oppe ensuite le potentie1 de Fermi en polyn8mes
de Legendre soit :

'•1
1
"!
1111
1
!Il
:1,
1,
,tli 1
1
11
u( :\) = (2).+1) 2
- 50 -
[" P~ (1-1•) U sinQdQ
où U est dormé par l'équation (C34). En utilisant le développement de
la fonction Sen pol.yn8mes de Legendre, soit54 :
s (r-r' > _ scr-r'} I {21.+1) p (r-~'> - rr' 4n l. l.
on obtient
u(,.) _ (2).+1) L ô(r-r 1 )
- 2 rr'
soit finalement :
u(~,r') =L (2.\+1) ). 2
L2 5(r-r') P";,.(l,r1 )
r' (C52)
qui est l'analogue de (C50) pour le potentiel de Fermi. L'équation
(C52) est équivalente au développement dérivé par Hickman d'une manière
pl.us complexe. Si on caractérise les états i et f par les nombres quan
tiques (n, 1., m) et (n', l', m') respectivement, on obtient à partir
de (c51) et (c52) :
A(nJ.m-n' 1 'm'l 9) = - ~ /(21+1 )(21 1 +1) L (i) ).(-1 )m' ( 1
~ ~ 0
l. ' À)(1 1 ' ;.. ) IÀ o o m -m- o
avec
IÂ = f~ Rn, 1 ,(r)j,(Kr)Rnl.(r) r2dr
0
où 9représente l'angle de diffusion.
(C53)
Nous avons utilisé pour le calcul. les fonctions d'ondes hydrogénoïdes,
de partie radiale Rn'l.' et Rnl. respectivement. En moyennant sur met m'
on obtient :
der ( dQ nl.~n'l.'19) k'
= k(21.+1) I m,m'
2 IA(nl.m .. n'l'm' 1 e) 1

- .51 -
où nous avons remplacé k. et kf par k et k' respectivement. Les règles
de sommation sur les coe~ficients 3j54 permettent d'obtenir fina1ement
mL 2 1 (l l' ~ )
2
~(ni .... n'l' 1 e) = (1:l.2) ~ (21'+1) t (2~+1) 0 0 0 II).1 2 (C.54)
La section tota1e s'obtient a1ors par la moyenne angu1aire classique
'l"t
a-(nl. ... n'l.') = 2n l ;/g sin8 d8
0
(C.5.5)
Nous développerons dans le chapitre VI les procédures de caJ.cu1 que
nous avons utilisées pour l'évaJ.uation numérique des équations (C.54) et (è.55).
L'avantage principa1 de cette méthode, outre sa simplicité
d'emploi, réside dans le fait de pouvoir obtenir les sections efficaces
de collision relatives à des processus bien définis. On peut donc ana
lyser ainsi, par comparaison, l'importance relative des différents ca
naux de réaction intervenant dans la désexcitation collisionnelle to
taie. Par contre si de nombreux canaux sont à prendre en compte les
temps de caJ.cu1 s'en trouveront notablement accrus. Le domaine de va
lidité de cette approche est difficile à définir avec précision. Le
premier problème vient de l'emploi du pseudopotentiel de Fermi. Nos
discussions précédentes relatives à sa va1idité n'appellent pas de re
marques supplémentaires. Notons simplement que l'extension de cette
approche aux gaz rares autres que l'hélium ne pose pas de difficultés
de principe. On peut en effet inclure simplement, comme l'a fait ré
cemment Hickman55 , les effets dus à la polarisation du gaz rare en uti
lisant un développement de l'amplitude de diffusion du gaz rare en
fonction de l'énergie56 • L'accord obtenu entre résultats théoriques
et expérimentaux dans le cas du néon et de l'argon est des plus satis
faisant55. Plus difficile nous semble ~tre de justifier l'emploi de
la première approximation de Born (bien connue pour donner de bons ré
su1tats pour les collisions à haute énergie) dans le traitement de col
lisions à énergie thermique. Il est certain que le bon accord théorie
expérience obtenu, comme nous le verrons, dans de nombreux cas ne peut
servir à justifier son emploi. Mentionnons seu1ement le fait que les
transferts d'énergie interne mis en jeu sont au plus de quelques cm-1

"I '
1
1
I" 1
il
il
- 52 -
a1ors que 1 1 énergie cinétique disponib1e est voisine de 400 cm-1 , ce
qui correspond, d'une certaine manière,à une situation haute énergie.
I1 convient cependant de noter que, au contraire du cas haute énergie,
ce sont 1es co11isions à grand paramètre d'impact qui contribuent 1e
p1us à 1a section efficace (de p1us, comme nous 1'avons mentionné, 1e
potentie1 de Fermi ne peut rendre compte que des interactions à grande
distance). De Prune1é39 et Hickman55 ont essayé de discuter 1a va1idi
té de cette approche. I1 nous semb1e cependant que ce prob1ème reste
ouvert.
Notons enfin que Matsu.zawa57 a déve1oppé une approche basée
sur 1 1 approximation soudaine ( "impuJ.se approximation"). Ainsi que 1 1 a
noté de Prune1é39 i1 nous semb1e que son traitement est équiva1ent à
1a méthode que nous venons de décrire. De fait 1es résuJ.tats numériques
obtenus pour divers processus co11isionneis58 (mé1ange des moments an
guJ.aires dans 1es cas du sodium et du rubidium.) sont 1es mêmes que
ceux que nous avons pu ca1cuJ.er à 1'aide de 1 1 approche d'Hickman.
c) Cas des co11isions a1ca1in excité-a1ca1in
Tout ce que nous avons dit précédemment concerne 1e cas où
1e perturbateur est un gaz rare. Une première difficu1té vient du fait
de 1'identité du perturbateur et de 1a cib1e qui peut donner 1ieu à un
transfert résonnant. Les sections efficaces correspondantes(~ 1o-14cm2 )
sont cependant notab1ement inférieures aux sections efficaces de dépopu-
1ation co11isionne11e observées(> qq 10-13 cm2 ). La deuxiè~e difficuJ.té
est 1iée à 1a grande portée de 1'interaction e--a1ca1in (ainsi L dans
1e cas du rubidium. est de 1'ordre de 30 u.a. à comparer à 1-5 u.a. dans
1e cas des gaz rares) qui rend très discutab1e 1 1 introduction d'un po
tentie1 fortement 1oca1isé, de type Fermi. De p1us 1es données précises
sur 1a diffusion é1astique e- 1ibre-a1ca1in à basse énergie font dé
faut. On sait seu1ement42 , 59 que 1es sections efficaces correspondantes
(N 10-13 cm2 ) sont très supérieures à ce11es re1atives aux gaz rares
et présentent de très fortes variations en fonction de 1'énergie dans
1a zone (0-0.2 eV). Notons pour terminer que Omont34 a mentionné 1'exis
tence probab1e de pseudo-croisements à grande distance entre 1es cour
bes de potentiel adiabatiques a1ca1in excité-a1ca1in qui pourraient
expliquer 1'importance des sections efficaces obtenues.

- 5:l -
d) ~!~~S~!~_f!~~!~:
I1 appardt c1airement que 1'extension des méthodes usuelles
de traitement des collisions thermiques au cas des états fortement
excités se heurte à de multiples difficultés aussi bien dans la partie
statique que dans la partie dynamique du problème. Les hypothèses
simplificatrices nécessaires (emploi d'une forme relative simple de
potentiel et de méthodes perturbatives) sont souvent d.iffici1es à jus
tifier a priori. Les modèles déve1oppés spécialement pour le cas des
états de Rydberg, que nous al1ons maintenant décrire brièvement, per
·mettent, dans une certaine mesure, de s'affranchir des difficultés
soulevées par l'emploi des méthodes que nous venons de décrire. Ce
pendant leur emploi donne accès, en général, à des informations plus
1imitées.
III.5.3. -Modèles spécifiques aux états de Rydberg.
) , 60-61 ( . ) a ~~~~!!-~!-!!~!!]: Binary Encounter Theory
Ce modèle de choc binaire instantané requiert les hypothèses
suivantes
- l'interaction A+-G est négligée ;
- la portée de l'interaction e- externe-Gest supposée petite
devant la distance moyenne e--A+ ce qui correspond à:
L « <r>
où Lest la longueur de diffusion e--G
- le temps de la col1ision e--G est supposé petit devant la pério
de de révolution (orbiting time) de l'électron autour de A+.
ilors le processus collisionnel peut 3tre décrit comme suit : juste
avant la collision la vitesse de l'électron est déterminée par l'in
teraction coulombienne e--A+. Durant le choc très bref, cette interac
tion est négligée et on considère la collision élastique e--G. Juste
après le choc on considère que l'énergie potentielle (e--A+) n'a pas
varié, ce qui est bien conforme aux hypothèses faites. Ainsi le gain
(ou la perte) d'énergie cinétique du mouvement relatif (e--A+) repré
sente l'énergie disponible pour effectuer une transition quantique.
Cette variation d'énergie cinétique et donc d'énergie interne de la

il!
- 54 -
paire (e--A+) peut se faire de manière continue. On considérera (comme
i1 est habitue1 de 1e faire dans ce type de modè1e) que 1e caractère
discontinu de la transition entre niveaux quantiques s'introduit de
1a manière suivante. Soit E. 1 1 énergie du niveau initia1 et E 1 1 éner-1 n
gie du niveau fina1: on aura transition de l'état i à 1 1 état n si
1 1 énergie interne de 1a paire é1ectron-A+ a varié d'une quantité AE
te11e que
E - E., ~E, E l - E. n i n+ i
où E 1 représente l'énergie du niveau immédiatement supérieur au nin+ veau n (voir Fig. 9).
E n+1
En ~E
L Ei
Fig. 9 - Passage continu-discontinu dans l'approche de F1annery.
La section efficace o-( i -+n) s'obtiendra ainsi en intégrant sur la par
tie hachurée ~(AE) soit :
<r(i - n) 1E 1 -E. n+ J.
= a-(AE)
E -E n i
d(AE)
où ~(AE) est 1a section efficace correspondant à une variation d'éner
gie interne de la paire (e--A+) d'i.me quantité comprise entre
AE et AE + d(AE). La mécanique classique permet d'obtenir pouro-(AE)
l'expression suivante :

cr(AE) = AE
uv2
+ ·1g 2dg g 1/2
g- («2+/32)
- 55 -
~+.
f o-(g,IJI) d(cos'Y) . 1/2
'f- [< cos\V+ -cos ) ( cos -cosljl-)J
(c56)
où g représente la vitesse relative (e--G), v la vitesse du gaz rare,
u celle de l'électron périphérique, ~(g,~) la section efficace diffé
rentielle élastique (e--G), ~ et /3 des paramètres déterminés par les
conditions initiales
1 ( 2 2 1 - a 2) ~ = 2 Me--G u -v + î+a g
~MA+ a= me_(Ma, +MA++ me_) (c57)
1 /3 = 2 Me--G
g2(2u2+2v2-g2) - (u2-v2)2 1/2
où~, MA+ et me_ sont les masses respectives du gaz rare, du coeur
ionique et de l'électron périphérique et Me--G la masse réduite du
système (e--G). Les bornes d'intégration de l'équation (c56) dépendent
des conditions initiales et correspondent à la conservation de l'éner
gie pour le système global39, 60, 61 •
La formulation précédente appelle quelques remarque~:
- La mécanique classique est seule utilisée. Le caractère quanti
que de la collision n'appara.!t que sous deux formes : l'interaction
coulombienne (e--A+) détermine la distribution de vitesse f(u) de
l'électron périphérique et la connaissance de la section différentiel
le élastique (e--G) est requise.
- Si u et v sont fixés, AE est imposé par la conservation de
l'énergie et seuls certains niveaux seront classiquement accessibles.
En fait la distribution de vitesse quantique f'(u) s'étendant à l'infi
ni tous les niveaux, y compris ceux du continuum sont accessibles avec
une certaine probabilité.
- La théorie de Flannery ne considère que des niveaux hydrogénoï
des dégénérés caractérisés par un seul nombre quantique n. Alors on
peut montrer que le principe du bilan détaillé est bien vérifié pour
les sections efficaces ~(n -n+1) et ~(n+l ~n). L'extension de lamé
thode aux niveaux (n,1) des alcalins est difficile et le principe du

!111
1
.11
- 56 -
bi1an détai11é ne sera pas a1ors automatiquement vérifié. Cette remarque
est importante car elle limite quelque peu l'applicabilité de la métho
de. Celle-ci permet cependant d'obtenir la section efficace tota1e de
collision relative à un niveau quelconqÙe (n,1) comme le mentionne la
remarque suivante.
- Comme l'a montré de Pruneié39 la description m3me du processus
collisionnel revient à considérer comme implicite l'hypothèse suivante
le taux de réaction du système (e--A+) avec le gaz rare est éga1 à ce
lui qe l'électron externe avec le gaz rare. Plus explicitement cela re
vient à considérer, après intégration sur les variables de vitesse et
angulaires que le taux tota1 de collision (c'est-à-dire collisions
élastiques+ co11isions inélastiques) a1ca1in très excité-gaz rare
vaut celui du système électron périphérique gaz rare soit*:
V ~(V) =<v e f +oi
a-> = e nl o cre
V e
f(v )dv e e (C58)
où V représente la vitesse relative a1ca1in-gaz rare, Qnl(v) la section
e:f:ficace tota1e (élastique+ inélastique) de l'état (n,1), ~ la section e
de dif:fusi.on libre électron gaz rare ( qui dépend, en généra1J de v ) e
et ve et :f(ve) la vitesse de l'électron et sa distribution quantique
pour l'état (n,1) considéré. Ceci suppose que v >> V (c'est-à-dire que e
la vitesse relative e--gaz rare est celle de l'électron) ce qui est
vrai m3me pour des va1eurs très élevées de n (n~ 100 dans le cas Na-He
par exemple, pour des expériences en cellule ou T ~ 450 K). Si les
processus élastiques peuvent 3tre considérés comme négligeables, la
section de quenching sera donnée simplement par ~(v), qu'il suf:fit
a1ors d'intégrer sur une distribution de Maxwel1 correspondant à la
température de l'expérience. Si les processus élastiques peuvent 3tre
éva1ués, ou mesurés par ailleurs on obtient :
~(v) = ~(v) + Çast.(v) (c59)
où~ et ~ast
• représentent les sections efficaces de quenching et
élastique de l'état (n,1) considéré.
* . , , 62 Cette expression est ana1ogue a celle trouvee par M. Matsuzawa dans le cas de l'ionisation des états de Rydberg par des molécules polaires, en utilisant une méthode· simplifiée, dérivée de l'approximation d'impact, que nous ne développerons pas ici.

- 57 -
Le modèle de F1annery est très intéressant car, si on se li
mite au cas des niveaux caractérisés par le seul. nombre quantique prin
cipal n, il permet le calcul. des sections efficaces de transfert entre
niveaux, de la section efficace d'ionisation et de la section efficace
de dépopulation totale. Les hypothèses de base sont assez facilement
satisfaites, m3me pour des valeurs relativement faibles den. Ainsi.
pour n = 10, <r> vaut de l'ordre de 100 u.a. et le modèle peut donc
décrire les collisions avec les gaz rares pour lesquels la portée de
1'interaction est inférieure ou égale à environ 7 u.a. (cas du xénon).
Des formul.es approchées valab1es pour 1es grandes valeurs den (n ~ 50)
peuvent 3tre obtenues60 . Mentionnons que dans le cas oua-(g,~) dépend
peu de~ l'expression générale se simplifie notablement, conduisant à
cr(AE) =~ uv2 1
g+ a-(g) g2dg
g - ( ~2 +13 2 ) 1 / 2 (C60)
Si de plus o-(g) dépend peu de l'énergie dans l'intervalle g+-g- (ce qui
est le cas de l'hélium pour 1equel la section efficace élastique de
diffusion électronique varie peu à basse énergie), on peut alors sortir
~(g) de 1'intégrale. Dans 1e cas contraire (présence d'un minimum de
Ramsauer à basse énergie) 1'intégration doit 3tre effectuée numérique
ment à partir d'une des deux équations. Ce·modèle a été appliqué par
Fla.nnery au cas des collisions entre atome d'hydrogène excité (n~10)
et atome d'hydrogène ou d'hélium à l'état fondamental. Notons que
certaines expressions obtenues à partir de ce modèle peuvent,con:nne l'a
montré Omont, se déduire d'un traitement perturbatif, au premier ordre,
associé à la formulation semi-classique. Ainsi l'expression 68 de la
référence 60 (valable pour n~50) est formellement identique à l'équa
tion 3.34 de 1'artic1e de revue d'Omont34 . Nous pouvons dès maintenant
indiquer (nous reviendrons sur ce point lors de la discussion de nos
résultats expérimentaux) que le modèle de F1a.nnery ne semble pas capa
ble de rendre compte de manière satisfaisante des résultats expérimen
taux disponibles.

1 ,111
1
- 58 -
b) ~2~~!=-~=-~=-E~~=!~:~!~~~=~=: Le désaccord observé entre le modèle de Flannery et les va
leurs expérimental.es et la difficulté d'obtenir les sections efficaces
de dépopulation total.e des états de Rydberg par les méthodes habituel
les de la théorie des-collisions ont amené E. de Prllllelé et J. Pascal.a
à formuler llll modèle différent de celui de Flannery, bien que faisant
appel aux m3mes hypothèses de base.
On suppose que la portée de l'interaction (e--G) est faible
devant la distance moyenne de l'électron ou noyau A+ et que l'interac
tion A+-G peut 3tre négligée. L'interaction (e--G) est ainsi supposée
suffisamment local.isée pour que la collision (e--G), qui rend compte
de l'interaction entre l'atome très excité et le perturbateur, puisse
3tre décrite par un modèle de contact. Ainsi si l'électron, à l'ins-
tant
r 0
~ rentre en contact avec la sphère, centrée en G, de rayon
= (<a-e>n1/n)1
/2
, où <o-e'n1 représente la section efficace de diffu
libre (e--G), moyennée sur la distribution de vitesse relative de sion
l'électron par rapport au perturbateur, on considérera qu'il y a eu
collision (élastique ou inélastique). Plus exactement la probabilité
qu'une collision (élastique ou inélastique) ait lieu à l'instant t
vaut :
P(t) = 14'n1m(R)l 2 AV ( C61)
où AV représente l'élément de volume compris entre les deux sphères
{centrées en G) de rayon r et r + dr. L'élément dr est pris égal. à 0 0 0 0
dr : < V ) dt o e n1
(c62)
où <ve>n1 représente la vitesse moyenne de l'électron pour l'état {n,1)
considéré. La justification de l'équation précédente ne peut 3tre faite,
a priori, puisque l'on décrit l'électron par sa fonction d'onde~ 1
, n, ,m
et que d'autre part le modèle de contact conduit à lui assigner une
trajectoire. Etant donné la val.eur der, on est conduit à: 0
P ( t) = 4< O-e > n1 < v 8 > n1 j ~ n1m ( R) I 2 dt
Si, de plus on suppose < ve>n1 >> V (vites se relative A+ -G), ce qui est
vérifié m3me pour des val.eurs den très élevées (n ~ 100), on peut,

- 59 -
assignant à l'atome une trajectoire rectiligne, de paramètre d'impact b,
obtenir la section efficace totale de collision par intégration le long
de la trajectoire rectiligne, en effectuant les moyennes angulaires
classiques (passage du repère mobile au repère fixe). Nous ne donnons
ici que le résuJ.tat final
Q ~ OT~t (V) ~ Q. sup ~ ""nJ. 9' inf
avec
100 +~
Qsup =2n o b db [1-exp(- ;<ve>n1<a-e>nl. 4"n f.~9.!i_(/b2+s2)ds)]
et (C6J)
l QO [ b db 4 Q.inf = 2n 21+1 1-exp( - v<v e> ni <a-e > ni
0
+oit
21+1 f 2 / 2 2 ] îm""" _'1.ni (
0
b +s )ds)
où inl représente la partie radiale de la fonction d'onde de l'élec
tron périphérique (des fonctions d'onde hyd.rogénoides sont utilisées
pour des raisons de commodité) et s 2 = b2 + R~, R étant la distance
(A+-B).
Quelques remarques relatives à ces formules s'imposent:
- seuJ.es les bornes·inférieure et supérieure ont été calcuJ.ées,
car l'obtention de ~t(v) nécessiterait des intégrations angulaires
longues et conteuses ;
- sil= o, c'est-à-dire pour les états S, les intégrations angu
laires sont inutiles car on obtient~ = Q.2• (Ceci correspond à une sy
métrie de l'interaction, mais n'est pas, en règle générale, valable.
Ce type de symétrie a parfois été supposé, par exemple par Olson,
pour simplifier les calcuJ.s des moyennes angulaires) ;
- sil~ o, il est facile de montrer que Q. .... Q.._..,. en dévelop-sup 1.1..u.
pant l'exponentielle des :f'ormul.es (C6J) ce d'autant plus vite que n
est grand ( car < Ve >ni"' !) et que < cre >ni est petite (c'est-à-dire que
l'interaction e--G est à plus courte portée ; ainsi on aura Q ~Q._..,., sup J.u.J. -
pour une valeur den plus petite dans le cas de l'hélium, que du xénon,
par exemple). Le calcuJ. numérique montre que Q ~Q._..,. assez rapide-sup J..1..u.
ment : dans le cas des couples Na(nD) - He et Na(nD) - Xe on obtient
Q.sup ~Qinf (à 10% près) pour n ~ 10 et 25 respectivement.

- 60 -
- On peut obtenir une borne supérieure de ~t (v)' par J. 'expression :
v ~t(v) ~ 4 <ve>n.1. <.0-e>n.1. (C64)
et le calcul. montre que J.'on tend ~ers cette limite pour des valeurs
suffisamment grandes den. Cette expression est très intéressante, car
elle se compare directement à l'expression (C58) déduite du modèle de
Flannery. On constate, en particulier, que la dépendance de Q:t(v) en
fonction de V, prédite par les deux modèles, est la m~me, la valeur
absolue différant d'un facteur 4.
- Comme dans le cas du modèle de Flannery la section efficace to
tale calculée prend en compte aussi bien les processus élastiques que
les processus inélastiques et les m@mes remarques peuvent 3tre faites
concernant l'obtention de la section efficace de quenching (processus
inélastiques uniquement).
c) Conclusion relative aux modèles
Les deux modèles que nous avons décrits font appel aux mêmes
hypothèses de base mais diffèrent nettement dans. leur formuJ.ation. Le
modèle de Flannery utilise essentiellement la mécanique classique alors
que celui de de Prunelé-Pascale, bien qu'il décrive la collision comme
collision de contact, formuJ.e le problème de manière analogue au_ traite
ment semi-classique. L'approche de Flannery autorise l'obtention des
sections efficaces de transfert entre niveaux individuels, mais ce,
seulement dans le cas où ils sont caractérisés par un seul nombre quan
tique. Les deux modèles qui incluent facilement les données de la dif
fusion élastique e--perturbateur à basse énergie conduisent à des ex
pressions approchées très simples pour la section efficace totale. La
précision de tels modèles est difficile à établir a priori et seuJ.e la
confrontation avec des résultats expérimentaux permettra un test vala
ble. Notons enfin que l'extension de ces modèles au cas des collisions
alcalin excité-alcalin ne pose pas de problèmes de principe, à condi
tion de satisfaire leurs hypothèses de base, ce qui conduit à ne les
appliquer que pour des nombres quantiques notablement plus élevés que
pour le cas alcalin-gaz rare (la portée de l'interaction e--alcalin
étant notablement plus grande que celle de l'interaction e--gaz rare).

- 61
REFERENCES DU CHAPITRE III
1 - M. BORN et J.R. OPPENHEIMER, Ann. Phys. 84, 457 (1927) .
2 - J. PASCALE et J. VANDEPL.ANQDE, J. Chem. Phys. 60, 2278 (1974).
.'.3 - E.E. NIXITIN, J. Chem. Phys. ~' 744 (1965).
4 - E.E. NIKITIN, The Excited State in chemical. Physics, Chapitre V,
John Wi1ey and Sons (1975).
5 - J. C.ALLAWAY et E. BAUER, Phys. Rev. A140, 1072 (1965).
6 - B.M. SMIRNOV, Sov. Phys. J.E.T.P. 24, .'.314 (1967).
7 - M. KRAUSS, P. MALDONADO et A.C. WAHL, J. Chem. Phys. 54, 4944 (1971).
8 - s.B. SCHNEIDERMAN et H.H. MICHELS, J. Chem. Phys. 42, .'.3706 (1965).
9 - W.E. BA'YLIS, J. Chem. Phys. 2..1., 2665 (1969).
10 - P. GOMBAS, Pseudopotential.e , Springer, New York ( 1967).
11 - F. BIRABEN, K. BEROFF, E. GliCOBINO et G. GRYNBERG,
J. Phys. (Paris) .'.39, L108 (1978).
1 2 - J • CUVELLIER, P. R. FOURNIER, F. GOUNAND, J . PASCALE et: J • BERLANDE ,
Phys. Rev. fil, 846 (1975).
1.'.3 - J. CUVELLIER, J. BERL.Al.'IDE, C. BENOIT, M.Y. PERRIN, J.M. MESTD.AGH et J. de MESMAY, J. Phys. ~' L461 (1979).
14 - D.L. DRUMMOND et A. G.ALLAGHER, J. Chem. Phys. 60, .'.3426 (1974).
15 - B. SAY.ER, M. FERRAY et J. LOZINGOT, J. Phys. fil, 227 (1979).
16 - R. AEMAD-BITAR, W.P. LAPATOVICH, D.E. PRITCHARD and I. RENHORN,
Phys. Rev. Lett . .'.39, 1657 (1977).
17 - E. CZUCHAJ et J. SIENKIEWICZ, z. Naturf'orsch. J4a, 694 (1979).
18 - A.R. MALVERN and G. PEACH, Proceedings of the tenth ICPEAC Paris
(1977).

li
1
l: il iil :11 ;I :1 il :11
i' 1 1,
11
11 1' 11 il j,i
- 62 - ·
19 - C. BOTTCHER, Chem. Phys. Lett. !§., 457 (1973).
20 - F. MASNOU-SEEUWS, M. Pim..IPPE and P. V ALIRON,
Phys. Rev. Lett. 41, 395 (1978).
21 - J. PASCALE et P.M. STONE, J. Chem. Phys. 65, 5122 (1976).
22 - L.D. LANDAU, Phys. z. Sowjet. 2, 46 (1932).
23 - C. ZENER, Proc. Roy. Soc. A137, 696 (1932).
24 - E.C.G. STÜCKELBERG, He1v. Phys. Acta 2, 369 (1932).
25 - E.E. NIKITIN, Theory of e1ementary atomic and mo1ecuJ.ar processes
in gases, C1arendon Press, O:x::f'ord (1974)~
26 - M.S. Cim..D, Mo1. Phys. 24, 18 (1972).
27 - F.H. MIES, Phys. Rev. Kl_, 942 (1973).
28 - F. MASNOU-SEEUWS et R. McCARROLL, J. Phys. fil, 2230 (1974).
29 - J. PASCALE et P.M. STONE, J. Chem. Phys. 62, 4809 (1975).
30 - R.H.G. REID, J. Phys. B6, 2018 (1973).
31 - A.M. ARTHURS et A. DALGARNO, Proc. Roy. Soc. A256, 540 (1960).
32 - J. PASCALE, Communication privée.
33 - O.B. FIRSOV, Zh. Eksp. Teor. Fiz. 21, 627 (1951).
34 - A. OMONT, J. Phys. (Paris) 38, 1343 (1977).
35 - E. FERMI, Nuovo Cimento 1!., 157 (1934).
36 - V.A. ALEKSEEV et I.I. SOBEL'MAN, Sov. Phys. JETP 22,882 (1966).
37 - G.K. IVANOV, Opt. Spektrosk. 40, 965 (1976)
(Opt. Spectrosc. 40, 554 (1976)).
38 - M.A. MAZING et F.D. SERAPINAS, Sov. Phys. JETP JJ, 294 (1971).
39 - E. de PRUNELE, These de 3ème cyc1e, Paris (1979).

- 63 -
40 - J. DEROUARD et M. LOMBARDI, J. Phys. fil.!, 3875 (1978).
41 - O. HINCIŒLMANN et L. SPRUCH, Phys. Rev. ~' 642 (1971).
42 - H.S.W. MASSEY, E1ectronic and Ionie Impact Phenomena, Vo1. 1,
Chap. 6, Oxford, New-York (1969) ..
43 - R.K. JAN.EV, J. Phys. B4, 215 (1971).
44 - M.Y. OVCEINIKOVA, Sov. Phys. JETP g, 181 (1966).
45 - B.M. SMIRNOV, Sov. Phys. JETP !2,, 692 (1964).
46 - J.I. GERSTEN, Phys. Rev. A14, 1354 (1976).
47 - L. LANDAU et E. LIFSCHITZ, Mécanique Quantique, Ed. Mir,
Moscou (1967).
48 - T.F. GALLAGHER, S.A. EDELSTEIN et R.M. HILL, Phys. Rev. Lett.
35, 644 (1975).
49 - T.F. GALLAGHER, S.A. EDELSTEIN et R.M. HILL, Phys. Rev. A15,
1945 (1977).
50 - R.E. OLSON, Phys. Rev. A15, 631 (1977).
51 - A.P. HICKMAN, Phys. Rev. A18, 1339 (1978).
52 - C.J. JOACHAIN, Quantum Co11ision Theory
North Ho11and/Amer;can E1sevier 1975.
53 - O.H. CRAWFORD, A. D.ALGARNO et P.B. HAYES, Mo1. Phys. il, 181 (1967).
54 - A.R. EDMONDS, Angu1ar Momentum in Quantum Mechanics, Princeton University Press (1957).
55 - A.P. HICKMAN, Phys. Rev. A19, 994 (1979).
56 - T.F. O'MALLEY, Phys. Rev. 130, 1020 (1963),
57 - M. MATSUZAWA, J. Phys. B12, 3743 (1979).
58 - M. MATSUZAWA, Communication privée.
59 - I.I. FABRIKANT, Phys. Lett. 58A, 21 (1976).

1 '
- 64 -
60 - M.R. FLANNERY, .Ann. Phys. (New-York) .fil., 465 (1970).
61 - M.R. FLANNERY, J. Phys. B4, 892 (1971) •
62 - M. MATSUZAWA, J. Phys. Soc. Japan, 32, 1088.(1972). •
63 - E. de PRtJNELE et J. PASCALE, J. Phys. B12, 2511 (1979).

Chapitre IV
ŒECHNIQlJES GENERALES D'EXPERIMENTATION.
IV.~. INTRODUCTION.
L'étude expérimental.e d'llll.e réaction de type
A*(i) + B -- A*{j_) + B ±. ~
fait appe1 à des techniques très général.es dont nous al.1ons exposer 1es
bases principa1es dans 1e présent chapitre. On peut, forme11ement, 1es
diviser en trois groupes:
- création sé1ective des espèces A*(i}
- mise en oeuvre du processus co11isionne1 et contrSie des
différents paramètres
- détection sélective des densités de population des espèces A*(j). et A*(i) en vue de l'obtention des sections de transfert ou de quenching
(Chapitre v).
Pour chacllll. des trois groupes nous indiquerons, brièvement, les diffé
rentes techniques possib1es puis nous décrirons, p1us précisément, ce1-
1es que nous avons utilisées au cours de cette étude.
IV. 2. CREATION SELECTIVE DES ETATS EXCITES.
Celle-ci peut s'effectuer de deux manières. La première con
siste à utiliser une lampe à résonance contenant le m3me atome A que
celui que l'on désire exciter. La raie optique désirée est ensuite
isolée et utilisée pour la photo-excitation sélective de type :
A+ hV ~A*(i)

- 66 -
Cette méthode a été utilisée de manière très générale pour l'étude de
tous les premiers doublets alcalins par le groupe de L. Krause. Elle
présente cependant de notables inconvénients. Le taux de photo-excita
tion demeure très faible et l.es exp-ériences requièrent donc des- tech
niques de détection élaborée. Leurs caractéristiques spectrales doi
vent 3tre bien contr81ées et connues (problème d'auto-absorption, lar
geur spectrale) sous peine d'entrdner des erreurs notables dans les
mesures, comme l'a montré le travail de A. Gallagher1 • Mais le princi
pal inconvénient de cette technique réside dans son peu de souplesse.
On ne peut en effet obtenir une densité suffisante d'atomes A*(i) que
pour les transi tians de résonance fto_rtes forces d'oscillateur) ce qui_
limite très notablement les possibilités d'études. L'équipe de Windsor
a pu étudier ainsi toutes les transitions résonnantes des atomes alca
lins et, plus récemment, des travaux sur les seconds doublets (6P du
rubidium et 7P du césium) ont été publiés par cette m3me équipe2·' 3 •
Notons, pour en terminer, que cette technique est aussi utilisable dans
le cas d'une coïncidence quasi-exacte entre deux raies optiques d'ato
mes différents. C'est cett~ méthode qui a été·utilisée par M. Pimbert4
pour étudier le niveau 8P du césium (coïncidence entre la transition
6S-8P du césium et J3P-a3s de l'hélium à 3889 A). De telles coïnciden
ces sont rares et les possibilités donc très limitées, les inconvé
nients étant l.es m3mes que ceux notés précédemment.
La deuxième méthode consiste à utiliser des l.asers à col.o
rant accordabl.es (l.aser 11 dye 11)5 ' 6 • De fait ce type de source lumineuse
intense commençait à se dével.opper au début de notre travail et l'évo
lution très rapide de ce domaine 7 ' 8 permettait d'envisager, dans un
avenir proche, leur utilisation pour le pompage efficace de nombreuses
transitions dans l.es atomes alcalins. Désireux d'avoir la possibilité
~'étudier des situations expérimentales diverses, nous avons donc choi
si ce type de source, dont les caractéristiques (puissance, largeur
spectrale) permettent d'obtenir des populations notables d'états excités,
ou très excités, pour l.esquel.s l.es raies d'excitation ont des forces
d'oscillateur très faibles.
Nous n'avons utilisé que des lasers 11 dye" travailJ.ant en im
puJ.sions, le développement des lasers "dye" continus ayant été plus
tardif que celui des lasers puJ.sés, leurs caractéristiques étant de
plus moins bien adaptées, en général, aux types d'expériences que nous
avons effectuées. Nous ne rappellerons ici ni le principe, ni l'histo-

- 67 -
rique des lasers à col.orants. Le lecteur intéressé pourra consuJ.ter,
entre autres, la référence 9. Le domaine des lasers à col.orants ayant
évolué très rapidement une description détaillée des trois sources
lumineuses que nous avons utilisées ne nous para!t pas présenter un
grand intér3t et nous nous contenterons d'en rappeler très brièvement
les caractéristiques essentielles.
1) Laser 1 : Il s'agit d'un laser pulsé pompé par lampe flash9 ,
qui fut développé par le groupe de physique moléculaire de l'école
Polytechnique, aucun laser commercial n'étant alors disponible pour
l'étude du deuxième doublet résonnant du césium (7P1 ; 2 , 3; 2 ). Les lon
gueurs d'onde des transitions d'excitation (6s1 ; 2 -0
7P1 ;2
) et
(6s112 + 7P3
; 2 ) sont, respectivement, 4593 et 4555 A. Les principales
caractéristiques de ce laser sont rappelées dans le tableau VI.
Excitation: lampe flash
Colorant: solution 10-3 M de 4-6 diméthyl 7 méthyla.minocoumarine
dans un mélange (2:1) eau-éthanol. 0
Gamme spectrale couverte 0
4510 - 4630 A
Largeur spectrale (A) 1
Durée d'impulsion à mi hauteur (µs) : 1 • 5
Energie par impulsion (µJ) : 50
Puissance cr@te (kW) : IV J•5 1Q
Taux de répétition (Hz) : 3
Tabl.eau VI
-2
Principales caractéristiques du Laser 1
2) Laser 2 : Ce laser puJ.sé pompé par l.ampes flash fut construit
dans notre laboratoire par V. Neumann.10 , en vue de permettre l'étude
des états très fortement excités des atomes alcalins. Nous l'avons
utilisé pour photoexciter le niveau 10P du potassium (transition d'exci-o
tation 4s1 ; 2 ~ 10P1; 2 , 312 ; À~ JOOO A). Le doublement de fréquence
s'effectue à l'extérieur de la cavité à l'aide d'un cristal non linéai~

- 68 -
re (ADA) dont 1e "phase-matching" est assuré par 1a variation de 1a
température du crista1. Le tab1eau VII regroupe 1es principa1es carac
téristiques de ce 1aser.
Excitation: 1ampe f1ash
Co1orant : soiution aqueuse 10-4 M de rhodamine 6G
(+ 4% d'a.mmonyx LO et 1% d'a1coo1 éthy1ique)
Dispositif doub1eur de fréquence extra-cavité, par cristai ADA
(phase-matching en température)
Caractéristigues Laser_ non doub1é _.{_;>.., 6000 Al
0.8
• Laser_ doub1é _1,i\ ,., JOOO Al
Largeur spectra1e {Â)
Durée d 1 impu1sion à mi hauteur (µs)
Energie par impu1sion (mJ)
Puissance crête {kW)
Taux de répétition {Hz)
J.7
110
25
0.15
Tab1eau VII
Principa1es caractéristiques du Lase.r 2
o.4
J. 1
8
J. 1
0.15
J) Laser 3: Les difficul.tés d 1 uti1isation du 1aser 2 (en particu-
1ier son faible taux de répétition et 1a durée de vie re1ativement
courte des 1ampes f1ash) ainsi que 1es progrès techno1ogiques réa1isés
dans 1e domaine des lasers 11dye 11 nous ont conduits à utiliser, pour
poursuivre nos études sur les états de Rydberg, un matérie1 commercia1.
Il s'agit d'un 1aser "dye" Mo.lectron DL.200 pompé par un laser azote de
forte puissance (Mo1ectron UV1000). Ce système très fiab1e et très sou
p1e d'uti1isation nous a donné toute satisfaction. Nous avons ainsi pho
toe:x:cité, dans le cas du rubidium, 1es états nP (12~n~22) par e.xcita-=-0
tation directe à partir du niveau fondamenta1 5S (Â ~ 3000 A) et 1es
états nS ( 12 ~ n ~ 18) ; dan-: ce dernier cas 1e peup1ement s I effectue en
deux éche1ons : on peup1e tout d 1 abord0
1e niveau de résonance 5P312 ,
à partir du fondamenta1 5S, (À= 7800 A), puis, de là, les états nS 0
(À N 5000 A). Les deux lasers DL200 uti1isés sont pompés par le m~me
laser azote, dont 1e faisceau est divisé à l'aide d'une lame sépara
trice. Le tab1eau VIII fournit les principa1es caractéristiques du
1aser 3.

- 69 -
Excitation: laser azote (durée d'impulsion 10 ns)
Di.spositi.f doubleur de fréquence : extra-cavité, par cristal. KDP (ph ... .,;c-matchil'l(; an{:ulaire)
Caractéristiqucs_typiqucs
d'utilisation -------------Colorant
Gonccntr~tion {M/1) Solvant
Puissance de pompe {kW)
Largeur spectral.a (Â)
Durée d'impul.sion à mi-hauteur (ns)
Energie par impul.sion (µJ)
Puissance crête (kW)
Taux de répétition (Hz)
À,. JOOO A (laser doublé)
Rhodamine B
5.10-J
Ethanol
900
O.J
J
10
J
JO
TabJ.eau VIII
• À"' 5000 A
(la.,;cr non doublP)
Coumarine fluorée (500) 10-2
Ethanol
250
0.5
6
60
10
JO
• l-~OOA
(la~cr non doublé)
M. DOTC (iodure)
2.4 ,o-J DMSO
500
0.5
6
15
2.5
JO
Principal.es caractéristiques du Laser 3, dans des conditions typiques 0
d'utiJ.isation: excitation des niveaux P du rubidium ( X N 3000 A)
et des niveaux S du rubidium 0
(premier écheJ.on : )1 ,v 7800 A ; deuxième écheJ.on 0
À N 5000 A).
Quel que soit le J.aser utilisé i1 a été pJ.acé dans t.m.e cage
de Faraday (type cel1uJ.aire en cuivre, double écran, de réjection mi
nimaJ.e 100 db, :fabriquée par J.a SIDT) a:fin de ramener 1es bruits para
sites é1ectriques à un niveau compatib1e avec l'empJ.oi d'une cha!ne
de comptage de photons.
IV.J. LE BATI DE MESURES.
I1 comprend, pour l'essentieJ., une ceJ.1uJ.e de mesure en verre,
un groupe de pompage qui permet d'y maintenir en vide poussé, un sys
tème d'introduction de gaz et des dispositifs de mesure de pression et
de température. La ceJ.J.uJ.e se trouve dans une étuve réguJ.ée en tempé
rature. La :figure 10 donne le schéma général du dispositif qui est
réalisé en quartz {celJ.uJ.e)et pyrex.

0 bservation
Fuite reglable
Q,I _.
- 70 -
, ,.-/,-/
Etuve (300 °C )
tube d'absorption
P?2'"2 ¾''' 'W ,, "• .1
Jauge
l 11~11v~~~~c:?'c::00il lfa'.;1/«P«~#./~//.-:0âl 1~ .o
Q,I '..... Q,I
E 0 C 0 ~
0
alcalin (T~140°C) 0
gaz rares
C[) 1 Pompe o palettes
Fig. LO - Schéma général du dispositif expérimental.
Jauge
Pompe à diffusion d'huile

- 71 -
:c..v.3.1. Cellu1e (Fig. 11).
@
Vers pompage
G)
1cm
Figure 11 Cellule de mesure.
Le corps de la cellule est en suprasyl 1 (silice de très fai
ble taux de fluorescence, ce qui permet de diminuer l'influence de la
lumière parasite). Les faces ont été soudées par adhérence moléculaire.
La forme, en "toit", permet, dans le cas d'une excitation laser par la
face 1 de piéger de manière assez notable le faisceau lumineux après
passage dans 1a zone d'observation. Nous avons aussi utilisé la face 2
comme face d'entrée pour le faisceau excitateur (dans le cas où la dé
tection s'effectue sur une transition optique de longueur d'onde diffé
rente de celle d'excitation, ce qui rend moins critique les problèmes
dus à la lumière parasite). L'observation s'effectue par la face 3.
Cette cellule est placée à l'intérieur d'une étuve dont la température
peut-~tre maintenue constante à z2°C. Elle est en communication avec un
appendice qui contient le métal alcalin liquide et dont on peut régu
ler à +o.5°c la température de manière indépendante de celle de l'étu-

- 72 -
ve, ce qui permet de fixer 1a pression et donc 1a densité. La tempéra
ture de l'étuve est toujours supérieure à celle de l'appendice pour
éviter 1a condensation de 1'a1ca1in sur les parois de la celluJ.e.
IV.J.2. Bati de pompage.
Il comprend une pompe à pa1ettes double-étage (ilcatel 2012)
suivie d'une pompe à diffusion d'huile (SOGEV D80) surmontée d'un baf
fle à fréon. Une rampe sur laquelle sont soudées les bouteilles de gaz
perturbateur (gaz rare) peut-3tre mise en communication avec la cellu
le par l'intermédiaire d'une fuite réglable. Les gaz utilisés (gaz
industriel de la Courneuve; bouteille de 1 1 sous pression atmosphé
rique) ont un taux d'impureté inférieur à 3 ppm pour l'hélium et 7 ppm
pour les autres gaz rares. Avant introduction du gaz rare dans la ram
pe, celle-ci ainsi que la celluJ.e de mesure sont étuvées à J00°C. Un
vide résiduel de 5.10-7 Test obtenu.
IV.J.J. Dispositifs de contr81es et de mesures.
Différents thermocouples permettent de contrSler et de mesu
rer la température de l'étuve principa1e et de l'appendice contenant
l'a1ca1in. La pression de vapeur saturante de celui-ci est ainsi con
nue d'après la température de l'appendice. Les formuJ.es utilisées sont
les suivantes :
P 1 , . 11
- our e cesium
p = 2.45 108
e-8910/T Cs VT
- Pour le potassium12
log PK= 7.40 -444s.z
T
- Pour le rubidium 12 . .
4c;?o -4 log PRb = 15.88 - ~ + 5.866 10 T - 2.991 log T
Dans ces formules les pressions sont exprimées en mm de Hg (Torr) et
T en °K. En admettant que la vapeur a1ca1ine se comporte comme un gaz
parfait la densité N d'atomes en phase vapeur dans la celluJ.e est
donnée par

- 73 -
N = 9.66 x 1018 ~
Tet
où p est exprimé en Torr et Tet (température de l'étuve) en °K.
- Une deuxième mesure de la densité d'atomes a1ca1ins peut-
3tre effectuée en mesurant l'absorption de la raie de résonance dans
une ce11u1e contenue dans l'étuve. Le détail de la mesure est donnée
dans l'appendice IV. Nous ne nous sommes servis de ce diagnostic que
dans la dernière partie de cette étude (états de Rydberg du rubidium).
- Diverses jauges à ionisation permettent les mesures de vi
de dans différentes parties du b!ti.
- La pression de gaz rare est mesurée à l'extérieur de l'étu
de à l'aide d'un manomètre à huile de silicone (15.54 mm= 1 mm de Hg),
à très faible tension de vapeur (410-ST à 20°C). Celui-ci permet, à
l'aide d'un cathétomètre, la mesure de pression dans la gamme (2.10-3
à 30 T).
- Les différentes parties du b!ti peuvent 3tre isolées à
l'aide de vannes (Fig. 10).
IV.4. LE DISPOSITIF DE DETECTION.
Celui-ci a pour but la collection et 1'ana1yse des signaux
lumineux émis par les atomes excités dans 1es états A*(i) et A*(j). I1 comprend, pour 1'essentie1, trois parties:
- une optique de collection
un système séparateur qui permet d'isoler 1es raies optiques,
émises par les atomes excités, que l'on désire ana1yser;
- le détecteur proprement dit (photomu1tip1icateur) et son élec
tronique associée.
IV.4.1. Optique de collection.
Nous utilisons une lentille de 50 mm de foca1e et de diamètre
50 mm, en quartz. Celle-ci est positionnée, à l'intérieur de l'étuve
principa1e, à l'aide d'une pièce de centrage, el1e-m~me solidaire d'un
panneau démontable de l'étuve principa1e. La lentille est réglable de
l'extérieur (m~me si l'étuve est chauffée). La zone d'observation,de

- 74 -
faib1e étendue, est située au foyer de cette 1enti11e, et on obtient
donc à 1a sortie de 1 1 étuve un faisceau para11è1e. Si 1e dispositif
séparateur est constitué d'i.m fi1tre interférentie1, qui travai11e en
1umière para11èle, on forme, après passage du fi1tre interférentiel,
1'image de la zone d'observation sur la photocathode du photomu1tipli
cateur (P.M.) à 1'aide d'une seconde lenti11e. Si 1e P~M. uti1isé pos
sède une photocathode large on n'uti1ise pas de dispositif de refoca-
1isation, mais seu1ement un système de diaphragmes pour éviter 1'i11u
mination des bords de la photocathode, ce qui peut donner lieu à des
émissions parasites g3nantes. Si le dispositif séparateur est consti
tué d'un monochromateur, on forme 1'image de la zone d'observation sur
la fente d'entrée du monochromateur.
Le dispositif que nous avons uti1isé comprend deux voies de
mesure (observation simu1tanée possible des signaux émis par les espè
ces A*(i) et A*(j»; ce11es-ci sont réa1isées à 1'aide d'une lame sépa
ratrice (absorption de l'ordre de 20~; transmission 50-50 ou 70-JO en
intensités relatives suivant la lame utilisée), p1acée sur un support
orientable, dans une botte noircie intérieurement. La 1ame séparatrice
travaille en faisceau para11èle. De plus des de~sités neutres peuvent
Atre insérées sur le parcours des deux faisceaux pour atténuer ceux-ci,
si nécessaire. L'ensemble du dispositif- optique (schématisé sur la
figure 12) est rendu étanche à la lumière extérieure à l'aide de tubes
noircis intérieurement, ce qui permet d'obtenir un niveau de lumière
parasite particulièrement bas.
IV.4.2. Système d'analyse des longueurs d'onde.
Celui-ci peut comprendre, sur chacune des voies, soit un
filtre interférentiel, soit un monochromateur, soit les deux, suivant
les nécessités de l'expérience. En effet, si le signal que l'on désire
observer est fort (cas de la fluorescence directe émise par le niveau
A*(i) que l'on excite), un filtre interférentiel peut s'avérer avoir
une réjection suffisante, les intensités émises par les autres espèces
excitées A*(j) étant, en généra1, très faibles vis-à-vis du signal étu
dié. Dans le cas contraire un monochromateur est nécessaire pour iso
ler les raies observées. Dans le cas de signaux très faibles, de lon
gueur d'onde proches de raies intenses, un filtrage préa1able effectué
à l'aide d'un filtre interférentiel peut s'avérer utile pour éviter
les problèmes dus à la diffusion à l'intérieur du monochromateur, m~me

Etuve
Cellule
Densité
Filtre
- 75 -
Separotrice Monochro moteur
Densité
Filtre
1 .. Cryostots ---~
• 1 1 Photomultiplicoteurs 1 1 •
.Fig. 12 - Ensemb1e du_dispositif de détection optique.

- 76
si la résolution de celui-ci est théoriquement suffisante. Il convient,
dans to.us les cas, de s'assurer expérimenta1ement que la configuration
adoptée permet une ana1yse correcte des signaux.
Nous avons utilisé des filtres interférentiels Wliquement
dans le cas de l'observation de la fluorescence directe issue des ni
veaux 7P112 ou 7P .'.3/2 du césium. ceu:;-ci sont centrés à ·~ 0
=
(raie 7P112 ~ 6s1 ; 2 ) ou )0
= 4555 0 A (raie 7P.3/z --i- 6s1 ; 2 ),
La largeur à mi."":-hauteur est de 20 A environ,·ia réjection à
~ étant de l'ordre de 2%. I1s sont munis de plus de verres 0
0
459.'.3 A 0
ce à +2. A. 0
20 A de
colorés
pour assurer le blocage de tout 1e spectre visible. Leurs caractéristi-
ques exactes ont été vérifiées, dans les conditions de fonctionnement
expérimenta1es. I1 est, en particulier, très important de les utiliser
en lumière para1lè1e, le centrage étant sensible à l'angle d'incidence.
Les monochromateurs à réseau utilisés nous ont été fournis
par la maison Stehle. Leur foca1e est de 500 mm, leur ouverture de
F/7.1. Les fentes, à lèvre (h = 20 mm), sont de largeur fixe. Nous dis
posons d'un jeu de fentes de largeur, 20, 50, 100, 200, 500, 1000 et 0
2000 µ. La dispersion est d'environ 10 A/mm. Les réseaux (Jobin-Yvon
M27 ou M29) ont 1200 traits par mm et la partie.gravée est de
70 x 70 mm. Les longueurs d'onde de blaze sont respectivement 3300 et 0 0
6,'.300 A. On peut ainsi explorer tout le spectre visible de 2500 à 9000 A
environ. Les fonctions d'appareil ont été mesurées dans les conditions
de travail et sont très proches de celles données par le fournisseur.
Le taux de réjection (qui dépend de la largeur des fentes utilisées)
est meilleur que 10-4 dans les conditions de l'expérience.
IV.4 . .'.). Les photomultiplicateurs.
Le choix du photomultiplicateur dépend essentiellement du
domaine spectral étudié et du mode de détection envisagé (analogique
ou digital). Deux types de photomultiplicateur ont été montés sur
l'expérience suivant les longueurs d'onde à étudier. Nous indiquons
tout d'abord leurs principa1es caractéristiques.
- 56DVP (Radiotechnique). Il s'agit d'un tube à 14 étages possé
dant une photocathode bia1ca1ine (réponse type D; domaine spectra1 0
utile : .'.)000 - 5000 A) particuJ.ièrement adaptée pour travailler dans
le bleu. La photocathode, fronta1e, a un diamètre de 42 mm. Le gain
est typiquement de l'ordre de 108 , et l'efficacité quantique d'environ

- 77 -
a 25% (à 4000 A). La largeur d'impulsion se situe à environ 6 ns à mi-
hauteur, le temps de montée demeurant inférieur à 2 ns. Refroidi à
-25°C le bruit est d'environ 3 kcs (mesuré à l'aide de la cha.1ne de
comptage préalablement réglée). La tension d'utilisation vaut environ
2000 V.
· - C31034(RCA). Il. s'agit d'un tube à 12 étages (dynodes cuivre
béryll.ium à structure en ligne, muni d'une photocathode dopée à l'ar
séniure de gall.ium).Cel.1.e-ci est de dimensions réduites (4 x 10 mm) , . a
mais présente une réponse spectrale très étendue (2000 à 9000 A envi-a
ron). L'efficacité quantique est de 15~ environ (à 5000 A) et le gain
(modèl.e trié) de 106 à 1500 V. Le refroidissement à -20°C permet de
réduire le bruit de fond à environ 20 es. La largeur d'impulsion est
d'environ 5 ns à mi-hauteur, pour un temps de montée inférieur à 1.8 ns. Si on le compare au 56DVP 1.es inconvénients sont 1.es suivants:
• focalisation pl.us critique due à 1.a dimension réduite de
la photocathode;
• gain pl.us faible qui nécessite une amplification pl.us gran
de pour l'utilisation en camp~age de photon, avec les inconvénients
qui en découlent (risques d'accrochage en particulier) ;
. fragil.ité beaucoup pl.us grande (courant maximum admissible
environ 100 fois pl.us faible) et usure non négl.igeable (due au dopage
de la photocathode) qui nécessite de fréquents contrSl.es.
Tous ces inconvénients sont en fait largement compensés par la soupl.es
se d'emploi due à l'étendue spectrale de la photocathode. Quel que soit
le type de photomultipl.icateur utilisé celui-ci se trouve placé dans
une enceinte de refroidissement à effet Peltier, munie d'un dispositif 1 optique approprié permettant, dans le cas de l'utilisation d'un mono
chromateur, la conjugaison de la fente de sortie de celui-ci sur la
photocathode du photomultipl.icateur. Les c!bl.ages util.isés pour la ré
partition de la haute tension sur les différentes dynodes ont été étu
diés en fonction des conditions de travail. (les sources lumineuses
travail.1.ant en impulsion) pour obtenir, dans chaque cas particulier,
une réponse présentant le minimum de distorsion (rebondissements •.. ).
IV.4.4. Al.ignement de l'ensemble du système optique.
Celui-ci est effectué à l'aide d'une expérience préliminai
re permettant de simuler les conditions réelles d'observation. Pour ce
faire on utilise un laser di alignement He-Ne muni d'un afocal permet-

- 78 -
tant d'obtenir un faisceau de diamètre~ 20 mm que l'on focaJ.ise à
l'endroit, préaJ.ab1ement repéré, de la photocathode du photomu1tipli
cateur et l'on suit ensuite 1e faisceau, ce qui permet 1e rég1age suc
cessif de tous les éléments optiques précédemment anaJ.ysés et la con
jugaison fina1e avec la zone d'observation. Ce rég1age long et minu
tie~ doit 3tre repris à chaque fois qu'une modification importante
est faite sur le système de détection (changement de photomu1tiplica
teur en particu1ier, ou ouverture de l'étuve principaJ.e.
IV.4.5. Electronique de détection.
Le choix de ce11e-èi-dêpend essentiellement de deux paramètres
- le type de détection envisagé : anaJ.ogique ou digitaJ.;
- le type d'information à mesurer: fluorescence totaJ.e, ou réso-
lue dans le temps.
En ce qui concerne le premier paramètre, le choiX du mode anaJ.ogique
ou digitaJ. est déterminé suivant le type d'expérience envisagée (si
gnaux faibles ou forts) et le type de source uti1isée (continue ou pu1-
sée). Différentes méthodes sont possibles et ont été, de fai~ utilisées
• utilisation d'un picoampéremètre enregistreur (cas d'une source
continue ;
• technique d'échanti11onnage (cas d'une source pu1sée) ;
. technique de comptage de photons (source continue ou pu1sée).
Cette dernière est la plus avantageuse dans le cas de la détection de
signaux très faib1es. El1e se pr3te, de plus, particu1ièrement bien
aux expériences conduites avec une source lumineuse pu1sée. Toutes nos
expériences ont été effectuées avec ce mode de détection qui est par
ticulièrement souple et bien adaptée au..~ mesures que nous avons effec
tuées ; en effet cette méthode digitaJ.e de détection permet, dans le
cas de l'utilisation d'une source pu1sée, 1a suppression quasi-tota1e
de tous les signaux parasites. Cette tecbnique13 , 14 est bien connue
maintenant et nous n'en décrivons que brièvement les caractéristiques
en insistant plus particulièrement sur les performances des éléments
que nous avons uti1isés.

- 79 -
Quel que soit le type d'informations à mesurer (fluorescence
tota1e ou résolue dans le temps) la chatne de comptage comprend tme
horloge, llll amplificateur et llll discriminateur à seuil réglable. Le
dispositif est schématisé sur la figure 13. ·
- L'horloge remplit les fonctions suivantes
. elle délivre tme impulsion électrique de commande du laser
à t 0
• elle ouvre à t0
+ t une porte de largeur C:i.t durant laquelle
la chatne peut compter (ouverture des discriminateurs). Ce dispositif
est rendu nécessaire par le fait que l'impulsion lumineuse débute avec
un certain retard par rapport à t {ce retard est de l'ordre de 20 µs 0
dans le cas des lasers 1 et 2 et de 1 µs dans le cas du laser 3), ce
retard pouvant de plus varier (jitter) d'tm tir à l'autre (le jitter
est d'environ 10 µs pour les lasers 1 et 2 et de l'ordre de 5 ns dans
le cas du laser 3). Le temps d'ouverture de la porte-C:i.t d~end--du r;;:-------·· -
tard et de son jitter d'une part, et de la durée du phénomène à analy-
ser d'autre part
elle permet le réglage de t et L:i.t ainsi que la programma
tion préa1able du nombre total de tirs laser à analyser (précompte).
Cette horloge a été conçue et construite par notre groupe d'assistance
électronique.
- Un amplificateur rapide {en effet les durées de vie typique des
niveaux que nous avons observés varient de 50 ns à 20 µs, nécessitant
l'emploi d'un dispositif rapide de comptage) 100 MHz. Nous avons utili
sé soit llll amplificateur SAIP 7016, à gain continllment variable de
1 à 100, soit llll amplificateur LETI à gain fixe de 10.
Un discriminateur à seuil variable qui effectue la mise en for
me des impulsions et permet la discrimination de celles-ci, à l'aide
d'tm seuil continOment variable de 50 à 500 mV. SeuJ.es les impulsions
de hauteurs supérieures au seuil sont disponibles à la sortie, pour le
comptage. Nous avons utilisé soit un discriminateur SAIP 7013 soit un
discriminateur fourni par le L.ETI.

CELLULE
1
l . 1
1
1
l
PM
-·~
IMPRIMANTE
LASE R
to ---
V 1'
to
t1
t2 h àt=20Tc
to+t --u,
t' C
- 80 -
DISCRIMINATEUR
AMPLIFICATEUR
ECHELLE SEPARATEUR DE DE
COMPTAGE VOIES
1 t2 r_J
ANALYSEUR 1 r-20 CANAUX
1 t COMMANDE DU
1 CONTROL COMPTEUR 1
' 1 1 1
HORLOGE 1 CALCULATEUR
1 1 1 1 1 1 1
t1 1
GENERATEUR - _J TELETYPE
t
Fig. 13 - Schéma général de l'électronique de détection. Les deux possibilités de mesure (fluorescence résolue ou non en temps) sont représentées sur la figure.
---C
E

- 81 -
L'élément final. de la chdne de comptage dépend, lui, du type
d'informations à mesurer.
a)!!~~~!~==~==-!~!~=
Il s'agit al.ors de mesurer le nombre total. de coups reçus
durant l'ouverture de N portes de largeur ~t (N étant le nombre de tirs
lasers pris en compte. Cette val.eur de précompte est affichable sur
l'horloge). Nous envoyons le signal. de sortie du discriminateur sur
lllle échelle rapide 100 MHz de type ECHO 100 (CRC). L'information final.a
est visual.isée par affichage digital. et transférée sur lllle imprimante
papier (ADDO).
b) !!~~~=!==~==-~~!~!~=-~~~-!=-!=~E~: Deux méthodes peuvent 3tre envisagées :
emploi d'un convertisseur temps-amplitude : le premier coup
reçu arr~te la charge linéaire d'une capacité dont la tension est al.ors
relue, puis digital.isée, ce à chaque tir laser. Des appareils commer
ciaux de ce type sont disponibles. Le principal. inconvénient réside
dans la faible sensibilité du.dispositif (un coup par tir laser, au
maximum) qui nécessite des temps d'accumulation assez longs;
- emploi d'un multicanaux rapides : on prend al.ors en c.ompte
tous les coups reçus durant llll tir laser (abstraction faite du temps
mort global. de l'électronique) que l'on classe suivant leur temps
d'arrivée par rapport à une référence fixe.
Nous avons utilisé cette technique qui permet de réduire notablement
le temps d'accumulation total., dans le cas où l'on reçoit plus d'un
coup par tir laser. Aucun appareil commercial. de ce type n'ayant les
performances requises (largeur de canaux N 100 ns), le groupe d'assis
tance électronique nous a fourni, après étude et réal.isation,un ana
lyseur 20 canaux rapide dont la largeur des canaux peut varier de 50 ns
à 2 µs, ce à l'aide d'une ligne à retard étal.année. Ce matériel permet
donc l'anal.yse de signaux de constante de temps égal.e ou supérieure à
250 ns environ. Le temps de recouvrement entre les canaux est négligea
ble(~ 0.1 ns). Les informations reçues sont transférées à un ordina
teur PDP15 (Digital. Corporation) qui effectue le moyennage sur le pré
compte ainsi que différents cal.culs aIU1exes (écart-type, .. ). La sortie

- 82 -
fina1e s'effectue sur une imprimante (Télétype). Les résu1tats sont
aussi stockés sur bande magnétique pour traitement u1térieur. La lar
geur exacte des canaux est déterminée (à mieux que 0.5~) à l'aide du
tiroir de comptage 525 MHz d 1 1.m oscilloscope Tektronix 7904. Le top
de synchronisation de l'ana1yseur est fourni par l'horloge. Pour s'as
surer, en permanence, du bon fonctionnement de l'ensemble, on effectue
simu1tanément la mesure de la f1uorescence tota1e à l'aide du disposi
tif précédemment décrit (en ouvrant 1.me porte de comptage de durée
4lt = 20 ?: A où ?:' A représente la largeur des ~anaux de 1 1 ana1yseur, ce
en synchronisme avec l'ouverture de l'ana1yseur).
IV.4.6. Réglage et éta1onnage d 1 1.me cha!ne de comptage.
Le réglage s'effectue suivant la procédure classique qui con
siste à trouver les va1eurs de gain et de discrimination conduisant à
1'apparition d'un pa1ier de comptage pour la courbe n = f(V), où n
représente 1a fréquence de COIJ1î)Jage (pour 1.m signa1 continu, obtenu
par exemple à 1'aide d'une lampe à ruban de tungstène) et V la tension
tota1e appliquée au photomultiplicateur. On se place a1ors à 1.me haute
tension correspondant à une va1eur moyenne du pa:1ier obtenu (Fig. 14). Le réglage obtenu est ensuite vérifié à l'aide d'un oscilloscope 500 MHz qui permet de s'assurer que le seuil de discrimination est correct en
visua1isant les éventuels échos dus aux rebondissements du photomu1ti
p1icateur, ce à la sortie du discriminateur. On vérifie fina1ement la
bonne linéarité de l'ensemble à l'aide de densités neutres éta1onnées.
Un ensemble de va1eurs typiques est le suivant :
Photomu1tip1icateur
56DVP
C:31034
THT
2:)00 V
1800 V
Gain
10
60
Seuil
50 mV
150 mV
Le para.mètre essentiel de la cha!ne de comptage qui reste
a1ors à mesurer est son temps mort ~M. La connaissance de celui-ci est
en effet requise_pour le ca1cu1 des éventuelles corrections 15 à apporter
aux comptages·obtenus. Le para.mètre ~M est essentiellement déterminé
par les caractéristiques du discriminateur. En effet, si deux impul
sions sont trop rapprochées le discriminateur n'a pas le temps de se
l 10
7~
10 6
,os
4 10
N (coups)
2
- 8:3 -
_____ ---4._..____.--t-_..,. __ d=O
PM 56 DVP Seuil= 50 mV Gain de l'ampli =10
2,35
d=1
d=2
V( kV)
2,5
Fig. 14 - Pal.iers de comptage obtenus avec un photomu.ltip1icateur 56DVP pour différents éc1airements. Les conditions de travai1 retenues sont indiquées par 1e pointi11é vertical.,

- 84 -
réarmer et une seuJ.e impuJ.sion est al.ors prise en compte par l'échelle
(ou par le système muJ.ticanaux). La fréquence de comptage vraie n sera 0
al.ors différente de la fréquence de comptage mesurée n (pour une lumiè-
re continue).
La relation entre net n dépend du mode de réarmement du dis-e criminateur. Celui que nous utilisons fonctionne en mode paral.ysable
(temps mort de type autoprolongé ; pour les différents types de temps
mort et les corrections, parfois complexes, à apporter aux comptages
mesurés, nous renvoyons le lecteur intéressé à la référence 15) : si
à t le signal. d'entrée du discriminateur passe au-dessus du seuil, 0 .
le discriminateur enverra dans l'échelle une impulsion et ne sera capa-
ble de prendre en compte le dépassement de seuil suivant qu'un temps
mort ~Maprès que le signal. soit repassé au-dessous du seuil. On conçoit
bien que si plusieurs photons arrivent quasi-simultanément l'impulsion
à l'entrée du discriminateur sera élargie par rapport à celle due à
l'arrivée d'un seul photon et le discriminateur sera aveugle plus long
temps, perdant ainsi un certain nombre de photons. Ce mode de fonction
nement conduit à la relation15 :
n = n 0
-n?: e o M
si l'on suppose que l'arrivée des photons est un processus poissonnien.
La mesure de eM s'effectue à l'aide d'une lampe continue bien
stabilisée. On mesure simuJ.tanément le courant I débité par le photo
muJ.tiplicateur (I"' n ) et la fréquence de comptage n, ce pour diffé-o
rents éclairements. Théoriquement la courbe n = f(n) présente un mac
ximum n tel que n = 1 /e t:M permettant d'obtenir CM. En fait ce max max maximum correspond en général à un éclairement supérieur à l'éclaire-
ment maximum admissible. Pour s'affranchir de cette limitation on utili
se la méthode suivante: on trace 1/n en fonction de 1/n (ou de 1/I). 0
La relation entre n et n montre que si n t: M « 1 on a : 0 0 ~
1 1 = n n
0
+ ~M
Ainsi l'ordonnée à l'origine de la courbe fournit ~M' si on
se limite à la partie linéaire (voir Fig. 15). On s'assure, a postério
ri, que les points pris en compte vérifient bien la condition n ~ «1. o M
La différence (1/n - 1/n) définit, en fait, un temps caractéristique 0
de la résolution de la cha!ne, pour l'éclairement n0
• Ce temps e vaut

50 f- ..1.. (10-8 S) n
40
30
20
10
0 - 1 't'M--=25 ,a-as
~0 I
5
- 85 -
10
PM 56 DVP
THT = 2350 V
Seuil= 50 mV Gain de L'amplificateur= 10
15
1/I ( l-L A)
20
Fig. 15 - Détermination du temps mort ~M d'une cha.tne de comptage.

- 86 -
1 ..1.. = 9 = Ïi - no
n ~M 1 0 -e
no
Ce temps n'est indépendant den que·pour l.es faibl.es taux de comptage 0
(n0 ~ <<. 1) pour l.esquel.s il. s'identifie à ~-
Nous avons mesuré des val.eurs de ~M comprises, suivant l.es
photomul.tipl.icateurs et l.es éJ.éments él.ectroniques util.isés (ampl.ifi
cateur et discriminateur) entre 12 et 30 ns pour l.'ensembl.e del.a chd
ne, ce qui est bien compatibl.e avec 1.es caractéristiques du matériel.
100 MHz util.isé. En effet 1.a rel.ation entre net n ne tient pas compte 0
del.a l.argeur effective des impul.sions qui entrent dans l.e discrimina-
teur. Cel.l.e-ci (déterminée par l.e photomul.tipl.icateur et l.'ampl.ifica
teur) est comprise entre environ 6 et 15 ns et vient s'ajouter au temps
mort du seul. discriminateur (de l.'ordre de 10-12 ns). La procédure ef
fective de correction de comptage, dans l.e cas d'l.ll'le l.umière non con
tinue, est discutée au chapitre V.

- 87 -
REFERENCES DU CHAPITRE IV
1 - A. GALLA.GEER, Phys. Rev. ill, 88 (1968). ;,-:.,
2 - I. SIAR.A, E.S. ERYCYSHYN et L. KRAUSE, Can. J. Phys. 2Q., 1826 (1972).
3 - P.W. PACE et J.B. ATKINSON, Can. J. Phys. 52, 1641 (1974).
4 - M. PIMBERT, J. Phys. (Paris) 33, 331 (1972).
~
5 - F.P. SCHAFER, W. SCHMIDT et J. VOLZE,
App1. Phys. Lett. 9, 306 (1966).
6 - J.F. HOLZRICHTER et A.L. SCHAWLOW,
Ann. N.Y. Acad. Sei. 168, 703 (1970).
" 7 - D. BASTING, F.P. SCHAFER et B. STE:ŒR, App1. Phys. J, 81 (1974).
8 - O. K!LDEBRAND, Opt. Comm. 10, 310 (1974).
9 - Dye Lasers, 2ème édition. Topics in App1ied Physics,
Edité par F.P. Scha:f'er, Springer-Ver1ag (1977).
10 - V. NEUMANN, Thèse de 3ème cyc1e, Paris (1975).
11 - W.B. NOTTINGHAM, Thermo-E1ectron Engineering Corporation,
Techn. Rep. TEE 7002-5 (1960).
12 - A.N. NESMEYAL~OV, Vapor Pressure of the E1ements, Academic Press
Inc., New-York (1963).
13 - J.F. JAMES, Month. Not. Roy. Astron. Soc. 1.lZ, 15 (1963).
14 - L. BIRENBAUM et D.B. SCARL, App1. Opt. g, 519 (1973).
15 - V.I. GOLDANSICTI, A.V. KUTSENKO et M.I. PODGORETSICTI,
Counting Statistics of Nuc1ear Particles, Hindustan Pub1ishing
Corporation, Delhi (1962).

Chapitre V
LA MESURE DES SECTIONS EFFICACES.
V.1. INTRODUCTION.
Ce chapitre a pour but d'examiner les différentes méthodes
possibles d'obtention des sections efficaces de transfert collision
nel ou de quenching à partir de mesures de fluorescence. Au cours de
l'exposé nous nous attacherons tout particulièrement à souligner les
avantages et les inconvénients liés à chaque méthode, ce en fonction
du problème considéré (transfert ou quenching, niveaux très excités
ou non). Après avoir rappelé les équations générales qui régissent
l'évolution des populations des états excités nous classerons les mé
thodes possibles en deux grandes catégories suivant que la fluores
cence est ou non résolue dans le temps. Nous examinerons ensuite cha
cune des deux catégories en détaillant plus complètement les méthodes
que nous avons effectivement utilisées, ce qui nous évitera d'alour
dir l'exposé du chapitre VI qui ne donnera que les détails spécifi
ques aux mesures que nous avons effectuées. Nous terminerons ce cha
pitre par un exposé relatif à la précision des mesures, où nous pas
serons en revue les causes d'erreur possibles.
V. 2. LES EQUATIONS GENERALES.
Les expériences concernant les transferts collisionnels en
tre niveaux de structure fine des premiers doublets alcalins s'inter
prètent aisément à l'aide d'llll modèle à deux niveaux. Un tel modèle
se justifie parfaitement, le premier doublet excité des atomes alca
lins étant très bien isolé de tout autre niveau dans le diagramme éner
gétique (les niveaux les plus proches se situent à plusieurs milliers

- 90 -
de cm-1 , l'énergie thermique moyenne disponible lors de la collision
n'étant que d'environ 400 cm-1
). Un tel modèle n'est pas justifié
lorsque l'on considère des niveaux atomiques très excités pour les
quels il existe toujours plusieurs niveaux accessibles lors de la col
lision. Nous a11ons donc considérer tout d'abord un modèle très géné
ral prenant en compte un nombre arbitraire de niveaux. Une te11e for
mulation permettra de dégager le principe générai des différentes
méthodes possibles. Des hypothèses simplificatrices, propres à des
situations expérimentales déterminées, qu'il conviendra de justifier
dans chaque cas particulier, permettront une interprétation simple
des diverses données expérimentales.
Considérons un niveau i, soit n. (t) sa population à l'ins-1.
tant t. Les processus suivants le couplent aux autres niveaux atomi-
ques
- ~~E~E~~!!~~-!~~~!!!! : elle est caractérisée par les coeffi
cients d'Einstein A .. (homogènes à une fréquence), ceux-ci n'étant l.J
différents de zéro que si E.~ E., les règles de sélection usuelles J l.
étant de plus satisfaites (Al=+ 1, Aj = O, + 1). - -- processus de photoexcitation: nous les noterons~-. (homogènes
---------------------------- l.J à une fréquence) ; ils représentent soit le processus d'excitation
lui-même, soit les transferts par rayonnement IR (de type corps noir)
dus à la cellule elle-même1
(les mêmes règles de sélection que dans
le cas de la dépopulation radiative sont donc valables). Nous néglige
rons ici 1 1 influence de 1 1 émission st.imulée induite par le rayonnement
excitateur. Nous ne prendrons pas en compte la photoionisation de
l'état excité (ionisation multiphotonique de l'atome a1ca1in) : en
effet les lasers utilisés étant de faible puissance on peut montrer
aisément que les fréquences de destruction des états excités par ce
processus demeurent toujours très faibles (même dans le cas des états
de Rydberg) devant les fréquences de dépopulation radiative. Pour ces
calculs nous avons utilisé les sections efficaces de photoionisation
calculées par la méthode du défaut quantique2 .
- !E~~f~E!~-~2±±!~!2~~!~ : ils sont caractérisés par une fréquen
ce de collisions K .. que l'on peut écrire : l.J
Kij = NB fa, q .. (v). v. f'(v)dv l.J
0
(E1)

- 91 -
où NB représente la densité de particules perturbatrices induisant la
collision (i + j), f(v) la fonction de distribution de la vitesse re
lative v des particules en collision et q .. (v) la section efficace de 1J
transfert pour la réaction
A*(i) + B -A*(j) + B - ~-. 1J
(E2)
où A*(i) et A*(j) représentent l'atome aJ.caJ.in dans les états i et j,
B la particule perturbatrice et~-. l'écart énergétique (asymptotique) 1J
entre les niveaux i et j considérés. La particule B peut 3tre :
• l'atome aJ.caJ.in dans son état fondamentaJ. (collision aJ.caJ.in excité
aJ.c aJ.in) ;
• wi atome de gaz rare (collision aJ.caJ.in excité-gaz rare) ;
. wie impureté résiduelle (atomique ou moléculaire) : ce processus
est très peu probable vue la pureté des gaz introduits et le bon vide
résiduel (5.10-7 T) obtenu. Nous le discuterons cependant dans les
causes d'erreur possibles ;
• wie molécule d'aJ.caJ.in: aux pressions de vapeur ou nous avons
travaillé la dimérisation de l'aJ.caJ.in est wi processus très peu pro
bable et la densité de molécules présentes est trop faible pour en
.traîner wi transfert mesurable. Nous négligerons donc ce processus;
wi atome aJ.caJ.in excité (collision aJ.caJ.in excité-aicaJ.in excité)
les taux de photoexcitation étant faibles dans nos expériences on peut
penser que ce type de collision n'influe pas (ce bien que les sections
efficaces correspondantes soient très maJ. connues) sur nos mesures.
Il faut cependant noter que ce processus conduirait à une variation
quadratique des signaux de fluorescence observés en fonction de la
densité d'atomes aJ.caJ.ins (au lieu de la variation linéaire que nous
discuterons plus loin). Nous n'avons jamais observé une telle varia
tion, ce qui justifie la non prise en compte de ce processus;
. une particule chargée :: ion aJ.caJ.in ou électron, créé par photo
ionisation du niveau excité étudié. Bien que ce processus soit peu
probable, comme nous 1 r avons mentionné~· il. convient d~ noter que les
collisions alcaJ.in excité-particule chargée (e- en particulier) sont
très efficaces. Nous avons estimé la contribution de ces collisions,
d'wie part à l'aide des densités de particules chargées caJ.cul.ées
d'après les sections de photoionisation2 , d'autre part à l'aide de
vaJ.eurs estimées de leurs sections efficaces3 ' 4 . Cette contribution

- 92 -
peut toujours 3tre nég1igée devant 1e terme de dépopu1ation radiative.
Notons que, là aussi, une con:f'irmation expérimentale est possible. En
effet la recombinaison des ions créés par photoionisation doit donner
1ieu à l'apparition de raies de fluorescence issues de niveaux d'éner
gies notablement supérieures à cel1e du niveau i considéré. Nous n'avons
pas observé ce phénomène, à 1'aide de notre dispositif expérimental..
En résumé nous ne retiendrons dans notre formu1ation généra
le que les deux premiers types de transfert col1isionnel que nous
avons mentionnés. Dans ces cas, les expériences étant effectuées en
ce11u1e 1a fonction de distribution f( v) (.éq. (E1)) peut 3tre consi
dérée comme maxwe11ienne et l'équation (E1) peut se simp1ifier en dé
finissant pour 1a réaction (E2) une section efficace moyenne Q .. par: l.J
avec :
K .. = l.J NB Qij VA-B-
VA-B = 8kT
rt µA-B
(E:3)
(E4)
vA-B· représentant 1a vitesse moyenne des partict.ù.es en co11ision, µA-B 1eur masse réduite, k 1a constante de Bo1tzmann et T(en °K) 1a tempé
rature du gaz.
Avec les hypothèses que nous venons de discuter, l'évo1ution
de la densité de popu1ation d'un état i est alors donnée par 1'équa
tion différentie11e linéaire du premier ordre :
dn. ( t) 1
dt = L n.(t) (K .. +A .. +~ .. ) - n.(t) L (K .. +A . . +yj . . ) ·..1.· J Jl. Jl. Jl. 1 . ..1,. l.J l.J l.J Jr1 Jr1
(E5)
Le premier terme du membre de droite représente 1es diverses
contributions à la popu1ation de 1 1 état i, 1e second les contributions
à sa dépopu1ation. La base du déve1oppement n'est pas spécifiée et
eng1obe tous les états j possibles. On obtient donc un système coup1é
d'équations 'linéaires du premier ordre.
En régime stationnaire (cas d'une excitation continue par
exemp1e) le système (E5) peut s'écrire, à l'équilibre (c'est-à-dire dn·
quand dt1
= o,pour tout i)

- 9J -
N. I ( K .. + A . . + !i1 .. ) = l. . .l · l.J l.J l.J
Jrl. I j-,ii
N. (K .. +A .. +!ii .. ) J Jl. Jl. Jl.
(E6)
où N. et N. représentent les popul.ations des états i et j, à l'équi-l. J
libre. On.obtient alors un système couplé d'équations linéaires.
Le cas du régime stationnaire peut s'étendre sans difficul
tés à une expérience effectuée à l'aide d'une source pul.sée, moyen
nant certaines hypothèses. Si le système de détection permet d'obte
nir les popul.ations intégrées N. définies par: l.
Ni = Jt2 t1
n.(t)dt l.
(E7)
où t1
et t2
représentent deux instants encadrant l'impul.sion excitatri
ce et tels que la quantité (t2-t1 ) soit très supérieure au..~ constantes
de relaxation de tous les états pris en compte dans le modèle, l'inté
gration du système (E~) conduit alors à un système formellement iden-~
tique à (E6) .
Les équations (E5) et (E6) appellent quelques remarques très
générales
- la fréquence de collisions K .. comprend, dans le cas le plus l.J
générai, deux termes, l'un dd aux collisions alcalin-alcalin toujours
présentes, l'autre dd aux collisions alcalin-gaz rare, soit :
K .. l.J
G = NG Qij VA-G + NA Qij VA-A (ES)
où NG(NA) représente la densité d'atomes de gaz rare (d'alcalin à
l'état fondamental; NA est supposé constant ce qui implique un faible
taux d'excitation), Q~.(~.) les sections efficaces de transfert. i-j l.J J . . - -
dues aux collisions alcalin-gaz rare (alcalin-alcalin) et vA-G(vA-A)
la vitesse moyenne alcalin-gaz rare (alcalin-alcalin). La détermina
tion des QI:. se fait en opérant à pression de gaz rare nul.le (NG = 0) l.J
ce qui limite K .. à un seul terme. La détermination des Q~. s' effec-l.J l.J
tue à pression d'alcalin constante (NA= cte), généralement très fai-
ble, ce qui revient, formellement, à ajouter un terme constant au..~
A . . ou ~ .. dans les équations (E.5) ou (E6). l.J l.J

- 94 ~
- Les A .. sont des constantes supposées connues, qui dépendent 1J
des niveaux considérés.
- La détermination des~-. est difficile, mais. leur influence sur 1J
la mesure des sections efficaces est en général très faible et on peut
souvent les considérer comme de simples corrections à apporter aux
A .. correspondants. 1J
- Les valeurs des K .. ne sont pas toutes indépendantes car les 1J
sections Q .. et Q . . , pour llll m3me gaz perturbateur, sont reliées par J1 1J
l'expression du bilan détaillé, qui exprime le principe de microréver-
sibilité, soit:
Q.. g, ( t.E. ·) -,1 1 11 ~ = - exp - ~ Q.. g, kT 1J J
(E9)
où g. et g. représentent les poids statistiques respectifs des niveaux 1 J
i et j.
Les méthodes de mesure des sections efficaces par fluores
cence se déduisent très simplement des équations générales que nous
venons d'établir. Si on se limite au cas d'une excitation lumineuse
pulsée on peut les classer en deux grandes catégories suivant que l'on
mesure l'évolution temporelle de la densité d'atomes (d'llll ou plusieurs
états excités) où la population intégrée. Dans le premier cas l'obten
tion des sections efficaces s'effectue à partir des équations (E5),
dans le second à partir des équations (E6). Nous allons maintenant
analyser en détail les deux possibilités.
V. 3. FLUORESCENCE RESOLUE DANS LE TEMPS.
v.3.1. Généralités.
On a pour donnée expérimentale llll signal S(t), obtenu à par
tir d'une des techniques générales discutées au chapitre précédent
(voir aussi référence 5), qui est de la forme :
S(t)=a:n.(t) 1
(E10)
où n.(t) représente la population d'un état i donné et a: est lllle consi
tante dépendant des caractéristiques de l'appareillage : angle solide
de détection, transmission du système optique, efficacité quantique du

- 95 -
photomultiplicateur ••. En fait~ ne joue en général. aucl.ll'l r8le dans
la mesure des sections efficaces, ce qui veut dire que la méthode
n'implique aucun étalonnage particulier du système d'observation. Le
problème est de remonter de la connaissance de n.(t) aux valeurs des 1
K .. (donc des Q .. ). Faisons d'abord quelques remarques général.es avant 1J 1J
de passer aux applications possibles de la méthode. La résolution du
système (ES) pose deux problèmes très généraux:
a) connaissance des conditions initial.es
b) limitation de la base d'états à prendre en compte.
a) Les équations (ES) sont, en général., résolues avec les condi
tions initial.es suivantes :
n.(O)=N l. 0
n . ( O) = 0, 'v j ;:{ i J
(E11)
Ces conditions impliquent la séparation complète des proces
sus d'excitation et d'observation, c'est-à-dire que l'on peut ignorer
la forme exacte de l'impulsion d'excitation et que l'observation s'ef
fectue après l'impulsion d'excitation. Elles sont toujours facilement
satisfaites. En effet, si l'on excite un niveau de durée de vie assez
courte (quelques dizaines de nanosecondes) on peut effectuer l'excita
tion à l'aide d'l.lil laser pulsé en interposant de plus un dispositif
optique de coupure (cellule de Pockels par exemple) dont le front de
descente est très raide (N qq ns). Dans le cas d'un niveau très excité
(dont la durée de vie typique est de l'ordre de la microseconde ou
plus) l'utilisation d'un laser à colorant (pompé par laser à azote)
dont la durée d'impulsion est de l'ordre de S ns permet de satisfaire
aisément aux conditions (E11). Il convient cependant de vérifier que
les niveaux j n'ont pas eu le temps de se peupler notablement par col
lisions durant la durée de l'impulsion laser, ce pour remplir la deu
xième condition (E11). Lors des mesures que nous avons effectuées par
fluorescence résolue dans le temps les conditions initiales (E11) sont
toujours satisfaites.

- 96 -
b) La 1imitation de 1a base d'états est un prob1ème spécifique à
chaque situation expérimenta1e et qui doit 3tre examinée avec soin
car i1 conditionne 1a va1idité du dépoui11ement effectué. Nous discu
tons p1us 1oin que1ques cas particuliers.
I1 est faci1e de voir que 1a so1ution 1a plus généra1e du
système (E5) est de 1a forme :
n. ( t) = ~ -t /c 1
.L.. ~ e k k
(E12)
c'est-à-dire que 1e signa1 observé, se présente comme une combinaison
1inéaire d'exponentielles de constantes de temps différentes. Le pro
blème générai posé par 1es méthodes de fluorescence résolue dans le
temps est al.ors 1e suivant : comment extraire les constantes de temps
~ de 1a courbe S(t)? Nous a1lons maintenant examiner la question en
détail en donnant 1es principa1es méthodes et en indiquant 1es solu
tions que nous avons effectivement retenues.
v.3.2. Les méthodes de dépoui11ement.
Les signaux S(t) présentent quatre caractéristiques princi
pa1es qui sont très importantes pour le dépouillement :
- 1e nombre d'informations est limitée aux nombres de canaux.
Nous avons uti1isé pour nos mesures un ana1yseur 20 canaux (chapitre IV),
ce qui1 nous 1e verrons,,à influé sur les méthodes retenues ;
- 1'interva11e de temps ana1ysé est fini ;
- 1 1 information contenue dans un cana1 est affectée d'une erreur,
en générai statistique ;
- 1e signa1 contient de p1us un bruit constant dd au photomulti
plicateur. Le signa1 est donc en fait de 1a forme :
s(t) = C 0
+ I k
-t/?:' ~ e k (E13)
Le problème de l'extraction des¾: et ~k est comp1exe. Ceci
est essentiellement dO au fait que 1es paramètres à déterminer présen
tent un fort degré de corrélation. Autrement dit un changement notable
de l'un peut être compensé par une variation d'un autre paramètre sans
changement appréciable de la forme de la courbe obtenue6 • Ainsi les

- 97 -
deux courbes (0.75 e-t/5 •5 +0.25 e-t/S) et (0.75 e-t/6 •7 + 0.25 e-t/4 .5)
ne diffèrent pas de plus de 2.10~4 en valeur relative, ce qui est évi
demment très inférieure au niveau de bruit statistique des courbes ex
périmentales. Ainsi la confiance dans les résultats obtenus à partir
de courbes expérimentales est-elle assez limitée si l'on ne dispose
pas, au départ, d'estimations assez précises (obtenues par exemp1e de
manière graphique). Toutes les méthodes d'ajustement numérique sont
plus ou moins sensibles aux quatre caractéristiques des courbes expé
rimentales que nous avons mentionnées :
- le nombre limité de canaux limite le nombre d'exponentielles
que l'on peut extraire de la courbe S(t). Ainsi, on peut montrer que,
quelle que soit la méthode, il est nécessaire d'avoir au moins une
centaine de canaux pour pouvoir prendre en compte deux exponentielles,
si le niveau de bruit statistique moyen est compris entre 1 à 10~
(ce qui est le cas le plus fréquent). Ne disposant que d'un analyseur
20 canaux nous nous sommes donc limités à des situations expérimenta
les telles que S(t) soit de la forme :
S(t) = c + ~ e-t/~ 0
(E14)
ce qui correspond à la seule mesure de la dépopulation totale d'un
niveau (voir paragraphe V.3.4).
- la limitation de l'intervalle d'analyse élimine en général tou
tes les méthodes (transformation de Fourier) qui utilisent un passage
de l'espace des temps à l'espace des fréquences, qui nécessitent, pour
limiter une forte _erreur de troncature, un temps d'observation très
supérieur (de l'ordre de 10 fois) à la constante de temps la plus lon
gue.
- les erreurs statistiques de comptage (comprises en général en
tre 1 et 10%) augmentent en fait le nombre de solutions possibles, ce
qui rend nécessaire des estimations probables assez fines pour obte
nir des valeurs correctes, ce quelle que soit la méthode choisie.
- on a, enfin, intérêt à mesurer C , pour effectuer l'ajustement 0
numérique sur une équation de type (E12) plutôt que (E1J). Ceci est en
général possible gr~ce à des mesures d'étalonnage _préalables. Sinon,
toutes les méthodes s'avèrent sensibles à C m@me pour de faibles va-o leurs relatives de cette constante vis-à-vis des taux de comptage.

- 98 -
En résumé, avant de passer à une brève énumération des dif
férentes méthodes possibles, nous voulons souligner la difficulté réel
le du problème. Le choix de la méthode est, en fait, un cas d'espèce,
leur efficacité étant souvent fonction des caractéristiques propres à
chaque problème: niveau de bruit, valeurs relatives des différentes
constantes.de temps (certaines méthodes sont plus appropriées quand
celles-ci sont voisines) ~·· Seuls des essais de simulation nombreux,
aussi proches que possible des situations expérimentaJ.es à traiter,
permettent de justifier le choix d'une méthode.
Nous allons donner maintenant une liste rapide des méthodes
possibles en renvoyant le lecteur aux références de base. Nous indi
querons pour terminer le choix que nous avons retenu, en fonction des
contraintes propres à notre expérience.
- ~~!~~~=-~~~E~S~! Elle n'est efficace que dans des cas très sim-
ples :
Elle consiste à extraire directement la (ou les) constante(s)
de temps à partir du graphe Log S = f(t) •
• Le signal ne contient qu'une exponentielle
donne alors directement la constante de temps •
la pente de la droite
• Le signal contient deux exponentielles de constante de temps nota-
blement différentes et l'intervalle d'analyse est suffisamment grand
pour permettre l'évaluation de la constante de temps la plus large.
Alors une simple soustraction suivie d'un ajustement linéaire permet
l'obtention de la constante la plus courte. Dans ce dernier cas un
lissage par intégration peut permettre d'améliorer la contraste entre
les deux contributions5 .
, 1 7 Methode des moments • -------------------Méthode des fonctions modulatrices8 • ----------------------------------
- ~~!~2~=-E~-~E~~f2~~~!2~-~=-!2~!=E:_~~-~=-~~E!~s=:~-
- ~~~~~~=-E~-~~!~~=~-~~~~~L-~~~-!!~~~~=i-E~~~~~~=6,11,12 Cette méthode très générale s'est avérée la plus efficace lors des
tests que nous avons effectués. Elle consiste11 à minimiser la valeur
de :

- 99 -
*2 = I ) _1_ ( s. - s < t. ))
2 I . ,_2 1. 1. ' 1. V•
(E15) 1.
par rapport aux différents paramètres de S(t) (éq. (E13). ~ 2 représen-1.
te ici la variance de 1 'observation S. du canal. i. La pondération 1 /cr. 2 J. J.
est très importante car elle permet de stabiliser très notablement la
méthode en fonction du taux de bruit statistique. Différents algorithmes
de minimisation sont possibles11 , qui nécessitent tous de fournir des
estimations préalables (en général. obtenues graphiquement) des constan
tes de temps pour faciliter la convergence. Des programmes très per- ·
b , , , , d' , 12 formants ases sur cette methode ont ete eveloppes
L'utilisation que nous avons faite de la méthode de fluores
cence résolue dans le temps a été essentiellement déterminée par l'ap
pareillage dont nous disposons (analyseur 20 canaux décrit au chapitre
précédent, paragraphe IV.4.5). Il est apparu lors des simulations que
nous avons effectuées sur des signaux du type de l'équation (E13) qu'il
serait illusoire de chercher à obtenir l'extraction de plus d'une cons
tante de temps des 20 points de mesure. Nous nous sommes donc toujours
limités lors du dépouillement quantitatif à des courbes de type (après
soustraction du bruit de fond C) 0
s(t) = a: e-t/"C. (E16)
Nous verrons dans les paragraphes suivants à quel type d'information
un tel signal. est directement relié. La procédure de dépouillement
adoptée est al.ors la suivante (c'est en fait une variante particuliè
rement simple de la méthode des moindres carrés)
N f f t , . 1
. , . d , , 11 , 1 3 , 1 4 - ous e ec uons une regression 1.nea.ire pan eree
sur la droite :
t. 1.
Log Si= Log a: - ~
où Si représente le comptage du canal. i et ti = i x ~A où ~A représente
la largeur des canaux. L'incertitude est supposée négligeable sur t. 1.
(la calibration de la largeur des canaux est effectuée à mieu.~ que
0.5%). Le poids w. de l'ordonnée Log S. est pris égal à: J. 1.
w -i -1
(Log(S.) - Log(S.-VS.)) 1. J. 1.
(E1 7)

- 100 -
qui suppose lllle variance poissonnienne pour le taux de comptage S. 1
(ce qui est bien vérifié). Le coefficient ~(et éventuellement Log~)
est ensuite obtenue à partir des équations classiques: (en posant
- Log S · ) : , • ' Y. - i L.. t Y.
1 W. i 1 i 1
! = ' '2 t: L.. w t.
i i 1
I wi Yi i avec y'. = y. _ ,
1 1
L._ W. 1
I wi ti i
1 t -t. = i '
1 L._ W. 1
(E18)
(E19)
L'évaJ.uation de l'erreur standard sur~ peut se faire à partir des
équations données par ~i11iamson14 . Pour s'assurer que la courbe ex
périmentaJ.e est représentée de manière correcte par lllle équation de
type (E11) nous avons toujours caJ.culé le coefficient de corrélation11
donné par:
1
~ wi Yi ti I ~1 (E20)
r = 1 v2. '2 y2. t'2x . w. Y. . W. 1. 1 1 1 1 1
et n'avons retenu que les observations pour lesquelles r ~ 0.9. Nous
avons enfin vérifié que la vaJ.eur de~ ainsi caJ.culée était proche de
celle obtenue par lllle évaJ.uation graphique. SignaJ.ons enfin que lamé
thode adoptée donne des résultats en généraJ. très voisins (écart rela
tif~ 5~) de ceux obtenus par la méthode des moments ou par celle des
fonctions modulatrices, du moins si le bruit statistique est faible.
En effet ces deux dernières méthodes y sont beaucoup plus sensibles
que celle des moindres carrés.

- 101 -
v.3.3. Transfert d'excitation et fluorescence résolue dans le temps.
L'obtention d'une section efficace de transfert d'excitation
par la méthode de fluorescence résolue dans le temps présente I.Ul;Cas
particulier intéressant d'application de cette méthode. Nous allons
montrer rapidement dans quelles conditions cette obtention est possible
et pourquoi elle est, en général, d'utilisation limitée et délicate.
Les notations sont données sur la figure 16. Elles appellent
les remarques suivantes
(rA)2 = A2 K12 K21 K2
(rA) 1=A1
K1
Fig. 16 - Modèle à deux niveaux pour l'étude du transfert d'excitation.
- on supposera que le niveau 1 est peuplé très rapidement ce qui
permet de négliger le détail du processus excitateur et conduit aux
conditions initiales suivantes (cf. équation (E11)
n.,(o) = N 0
n2
(o) = o (E21)
- les fréquences collisionnelles (Eq. (EJ) K, 2
et K21
correspon
dent au transfert 1 + 2 et 2 ~ 1 • K, ( ou K2
) représente la dépopula
tion collisionnelle de l'état 1 (ou 2) vers les niveaux autres que 2
(ou 1). A1 et A2
représentent les durées de vie radiatives des états
1 et 2.

- 102 -
- 1e modè1e choisi imp1ique en outre qu'il n'y ait pas de repopu
lation co1lisionne1le des niveaux 1 et 2 par d'autres niveaux et que
1 et 2 ne soient pas coup1és eritre eux radiativement (cas par exemp1e
d'';llle_ transition de structure fine P1 ; 2 ~P3; 2 ), 1e système (E5) s'ecrxt aiors:
dn1 dt = K21 n2 - ( ~ 2 + K1 + A, ) ~
(E22)
~ ërt° = K,2 ~ - (K21 + K2 +A2)n2
(~2
et K21
sont reliés par le principe du bi1an détaillé donné par
(EJ) et (E9)); 1a so1ution de (E22), en tenant compte des conditions
initiaies (E21) s'écrit :
avec
2R1 ,2 = -
R t n1 ( t) = c, e 1 + c2
R2
t e
n2(t) = c
3 e ~t R
2t
- e
(E2J)
( K, ~ +K1 +~ +K21 +K2 +A2) + V ( K21 +K2 +A2 -~ 2-K1 -~ ) 2
+ 4Ks 2K21
On obtient donc pour n 1 (t) et n 2 (t) des combinaisons linéaires de deux
e.xponentiel1es (plus généraiement on obtient une combinaison linéaire
comprenant un nombre d'exponentielles égai au nombre de niveaux expli
citement pris en compte dans le système (E5)). Les inconnues sont au
nombre de J : ~2
, K, et K2 . La méthode consiste à mesurer R1 et R2
en fonction de la pression de gaz perturbateur à partir des courbes
expérimentaies et à en extraire 1es vaieurs des sections efficaces
~ 2 , ~ et Q2 . ~ et R2 sont en générai des fonctions quadratiques de
la pression. Cette méthode est largement discutée dans les références
2 et 15 (interprétation matricielle) et n'appelle de notre part que
quelques remarques plus spécifiques :
. Le modèle le plus générai nécessite un appareillage pos
sédant un nombre suffisant de canaux pour une extraction correcte des
constantes de temps (voir paragraphe V.J.2) ; celles-ci sont en géné
rai du m~me ordre de grandeur ce qui peut faciliter le choix de lamé
thode de dépouillement des courbes.

- 103 -
. L'observation,à très basse pression,de l'évolution des po
pu1ations n1 (t) et n 2 (t) permet d'obtenir les valeurs de A, et A2 ,
c'est-à-dire que la méthode ne nécessite pas leur connaissance a priori,
ce qui peut ~tre un avantage notable.
. Si les deux niveaux sont très bien isolés dans le spectre
éne~gétique (cas des premiers niveaux résonnants des alcalins) on peut,
de plus, considérer K, et K2 comme négligeables devant K12
et K21 , ce
qui simplifiera notablement la procédure d'ajustement. De telles situa
tions sont cependant beaucoup plus rares pour des niveaux très exci-
t , 15,16 es •
• Il est facile de voir (en considéra.nt la première équa
tion de (E22)) qu'à suffisamment basse pression on a
I½(t)tv e -(K, 2+K, +A,) t
(E24)
c'est-à-dire que ~(t) ne comporte qu'une exponentielle de constante
de temps :
1 ë = ~2 + ~ + ~ (E25)
La variation de 1/C en fonction de la densité de gaz perturbateur est
donc linéaire, d'ordonnée à l'origine~ (extrapolation à pression
_nu1le) et de pente K12 + ~ : on obtient donc (cf. éq. (E3)) la section
de dépopu1ation totale du niveau 1, qui n'est égale à la section de
transfert que si~ est négligeable (cas de la remarque précédente).
. La sensibilité de la méthode est directement reliée aux
valeurs relatives des constantes collisionnelles K et radiatives A.
Autrement dit il faut pouvoir élever suffisamment la pression pour
que les durées de vie des niveaux soient notablement modifiées par
les collisions.
En conclusion nous pouvons dire que la mesure des sections
efficaces de transfert d'excitation par fluorescence résolue dans le
temps est une mesure délicate nécessita.nt d'une part un appareillage
performant, d'autre part une situation expérimentale particulière
(système à deux niveaux). Dans le cas le plus général (plus de deux
niveaux notablement couplés) elle nécessiterait le dépouillement de
courbes comportant trois exponentielles (ou plus), problème dont nous
avons mentionné l'extr~me complexité. Notons en:f'in que m~me dans le

- 104 -
cas des co11isions de structure fine des premiers doub1ets a1ca1ins
son emp1oi est rendu diffici1e par 1a faib1e durée de vie de ceux-ci
(N 20 ns) qui exige un système possédant une exce11ente réso1ution
tempore11e.
v.3.4. Dépopu1ation co11isionne11e tota1e (quenching) et f1uorescence
résolue dans 1e temps.
Le système d'équations (E5) peut, dans un cas, prendre une
forme particu1ièrement simple. Soit i 1e niveau initia1ement peuplé.
Si on peut nég1iger tous 1es termes de repopu1ation nous pouvons
écrire :
dn i ( t) = -ni ( t) ( )~i dt
A .. J.J
+ L K.·) j~i J.J
nous avons ici omis les termes de type~-., que J.J
vent, soit négliger, soit inclure dans 1e terme
l'on peut le p1us sou
radiatif L A . .. Notons j~i J.J
A. et K. 1a somme des contributions radiative J. J.
et co11isionne11e ; on
obtient a1ors:
dni ( t) = -ni ( t) ( Ai + Ki) (E26)
dont la solution, en supposant les conditions initia1es (E11) remplies,
s'écrit
n. ( t) = N J. 0
-(A.+K.)t e i i (E27)
On observera donc une décroissance monoexponentie11e du signa1, de
constante de temps :
1 ;:- = A. + K. .... J. J.
Ici seu1 K. dépend de la pression de gaz perturbateur et l'équation J.
précédente peut s'écrire
! - 1 ~ - ~+K. 0 1
(E28)
où 1/~ représente la durée de vie radiative du niveau i. Suivant les 0
remarques du paragraphe V.3.1 l'expression la plus généra1e de K. 1
s'écrit :

-_10.5 -
G- A Ki= NG Qi vA-G + NA Qi vA-A (E29)
La considération de (E28) et (E29) conduit à J.a procédure expérimenta
le, particuJ.ièrement simple, suivante : on mesurera tout d'abord, sans
introduire de gaz rare, J.a variation de J.a durée de vie effective du
niveau 1/~ en fonction de J.a densité d'alcalin NA. La pente de la
droite ainsi obtenue permet J.a détermination de Q~ et l'ordonnée à l.
l'origine (extrapolation à densité nuJ.J.e) J.a mesure de J.a durée de
vie radiative 1/C du niveau i considérée. On opérera ensuite à den-o
sité d'alcalin constante (choisie aussi faible que possible, pour
assurer la validité de l'équation (E26) ; voir ci-dessous) en faisant
varier la densité de gaz rare NG. Le second terme de l'équation (E29)
reste alors constant et s'ajoutë à 1/~ pour fournir une nouvelle 0
ordonnée à l'origine, la pente de la droite permettant d'obtenir Q~. l.
Le principal avantage de cette méthode réside dans sa grande
simplicité qui permet l'utilisation d'un nombre limité de canaux. C'est
pourquoi nous l'avons choisie pour J.a mesure de 1a dépopuJ.ation coJ.1i
sionnel1e totale des états de Rydberg du rubidium. La mesure de~ à
partir de courbes expérimentales s I effectue à l'aide d I une méthode --de
régression linéaire pondérée qui a été décrite précédemment.
Les inconvénients sont tous liés aux conditions de validité
de l'équation (E26) ; on peut cependant à partir des courbes expérimen
tales définir la zone de pression dans J.aque11e ces conditions sont
satisfaites. Nous allons maintenant discuter de manière plus détai11ée
ces quelques points.
Les conditions initiales (E11) sont facilement satisfaites,
particulièrement pour les états de Rydberg dont les durées de vie C
sont de l'ordre de la µs, la durée de l'impuJ.sion laser utilisée étant,
dans notre cas de l'ordre de 5 ns. L'équation (E26) suppose essentiel
lement que les niveaux j ne contribuent pas notablement à J.a repopuJ.a
tion de l'état i considéré. En toute rigueur elle est valable à t = o
(les conditions (E11) étant satisfaites), c'est-à-dire que J.a mesure
d J. d , . , l . thmi ( 1 dni) e a erivee ogari que ii'-:- dt fournit bien la valeur de 1. t=o
(A.+ K.). Cependant la détermination de cette dérivée à partir de don-1. J..
nées expérimentales est délicate et nous avons préféré utiliser l'équa-
tion (E26) après avoir au préalable déterminé expérimentalement son
domaine de validité pour chaque gaz perturbateur étudié. Nous avons

- 106 -
opéré comme suit. Nous travai11ons d'abord à très.faible pression,
zone dans laquel1e 1/~ est très voisin de 1/c0
• Nous augmentons en
suite la pression, la durée d'observation restant la m@me. On observe
la diminution de la durée de vie effective (Eq(E28)), la courbe
Log S = f(t) restant linéaire (taux de corrélation ~0.9). A partir
d'une certaine pression l'effet de la repopulation se fait sentir par
une disxorsion de linéarité qui correspond à l'apparition d'une cons
tante de temps apparente plus longue (Fig. 17, extraite de l'article 3 voir chapitre VI). On obtient ainsi la zone de validité de l'équation
(E27) pour la durée d'observation considérée (on peut d'ailleurs aug
menter cette zone de validité en se limitant à une durée d'observation
plus courte, méthode qui conduit, à la limite, à la mesure de la déri
vée logarithmique). Nous nous sommes toujours limités à des mesures
de~ comprises entre~ et 2~ en nous étant toujours assurés de la 0 0
bonne linéarité des courbes Log S = f(t).
Pour les états de Rydberg, pour lesquels la repopulation
s'effectue, en général, à partir de plusieurs niveaux j, il est très
difficile de tirer des informations de la constante de temps apparente
car elle représente en fait la somme de plusieurs contributions diffé
rentes. La seule exception à cette remarque est fournie par le mélange
collisionnel des moments angulaires13 , pour lequel la repopulation
peut-3tre attribuée de manière précise à un niveau j*. On est alors
ramené à un problème de type deux niveaux, c'est-à-dire au paragraphe
précédent.
Signalons que la repopulation du niveau i implique de com
mencer l'observation le plus t8t possible après l'impulsion laser, ou
du moins après un temps très faible devant la durée de vie effective
1/~ afin d'avoir la meilleure dynamique possible. Ceci est assez aisé 0
pour les niveaux très excités pour lesquels 1/~0 est de l'ordre de la
micro-seconde, l'observation pouvant commencer quelques dizaines de
nanosecondes après le tir laser (pour éviter l'influence des bruits
électriques parasites dus à la décharge du laser de pompe, ou la lu
mière parasite intense créée par le laser d'excitation).
* Ce niveau j est, en fait, un groupe de niveaux que l'on peut traiter de manière globale.
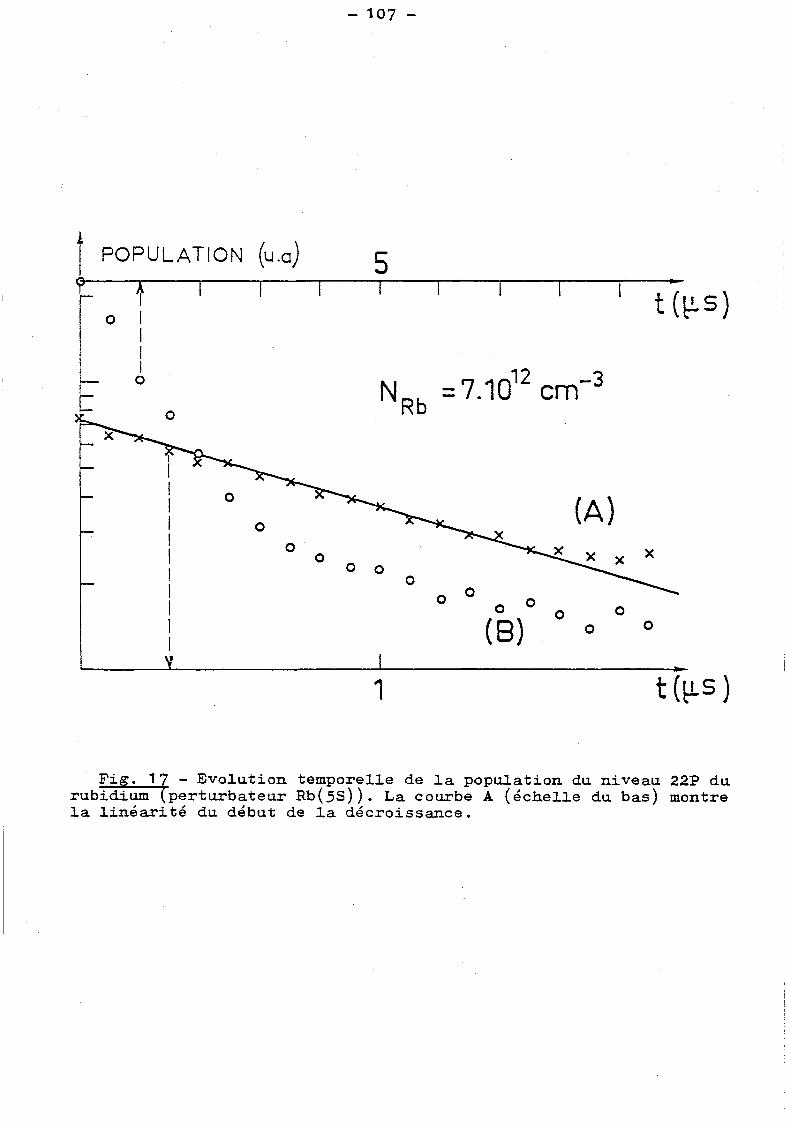
- 107 -
t POPULATION (u.a) 5 .. 1 1 1 t(µ_s)
1 ! ? A 1 1 1 1
0 1
1
1
=7.10,2cm -3 NRb
1 0 c
0
0 ""~-- (A) "~ 0
0 ~ ~ ~~ X 0 0 0
0 0
0 0 0 0 0 (8) 0 0
1
'
1 t (lJ-5)
Fig. 17 - Evolution temporelle de la population du niveau 22P du rubidium (perturbateur Rb(5S)). La courbe A (échelle du bas) montre la linéarité du début de la décroissance.

- 108 -
La mesure des sections de quenching par fluorescence résolue
dans 1e temps a 1'avantage de 1a simp1icité mais nécessite que1ques
précautions, quant à son emp1oi. E11e fournit de p1us une information
moins détai11ée puisque le quenching représente la somme· des divers
processus iné1astiques pouvant donner 1ieu à 1a dépopu1ation du niveau
étudié.
V.4. FLUORESCENCE NON RESOLUE DANS LE TEMPS.
V.4.1. Généra1ités.
La donnée expérimenta1e S. est proportionnel1e à la popu1a-1
tian intégrée N. du niveau i soit 1
S. = a-. N. 1 ~ 1
(E:30)
Les N. sont donnés par 1e système (E6) et~- est une constante dépen-1 1
dant d'une part de l'intensité de la transition d'observation choisie,
d'autre part de l'efficacité globa1e du système de détection (ang1e
so1ide d'observation, transmission du système optique à 1a longueur
d'onde d'observation Âib, efficacité quantique du photomu1tip1icateur . 0 S
(pour  = ?/·b ) , etc ••• ) . On peut écrire 0 S
~. = A?bs T(Ài ) 1 1 obs (E:31 )
où A?bs représente 1e coefficient d'Einstein de 1a transition d'obser-1
vation et T(Â b) 1'efficacité g1oba1e du système d'observation à . 0 S
 = Â1 b. La va1eur de~- étant impossib1e à ca1cu1er a priori, on 0 S 1
opérera en effectuant 1e rapport de deux signau..~ S. et S. issus des 1 J
niveaux i et j respectivement soit, avec un dispositif de comptage :
S. .2:. = R .. s. 1J
J
A?bs - J. --Aobs
j
T(:\~bs)
T(À!bs)
N. 1
w:J
(E:32)
La méthode consiste à mesurer la variation de R .. en fonction de 1a 1J
densité du gaz perturbateur, en ayant, au préa1ab1e, éta1onné 1e sys-
tème pour obtenir 1a transmission relative du système aux deux longueurs
d'onde d'observation c'est-à-dire le rapport (T(Âib ))/(T(Àjb) ). 0 S · 0 S
L'obtention des sections efficaces s'obtient donc en deux étapes. La
première consiste à ca1cu1er N./N. à partir de la donnée expérimentai J

- 109 -
le R .. , la seconde comportant la résolution du système (E6), à l'aide 1J .
d'hypothèses appropriées, pour remonter des valeurs de N./N. aux taux 1 J
de transfert. La première différence importante entre les méthodes de
fluorescence résolue et non résolue réside dans le fait que cette der-·
nière nécessite la connaissance d'une part des coefficients d'Einstein
des transitions d'observation (ou du moins de leur rapport) et d'autre
part de la transmission relative du système de détection pour les deu.~
longueurs d'onde d'observation. Nous verrons la mesure de celle-ci
dans le cas particulier de notre dispositif expérimental au chapitre
suivant. Par contre l'obtention de N./N., donnée de base, ne nécessite 1 J
qu'une mesure, celle du rapport des taux de comptage R .. , alors que 1J
l'obtention d'une durée de vie effective t nécessitait le dépouille-
ment d'une courbe S(t), avec des hypothèses ~ppropriées. Ces remarques
sont directement liées à la structure des équations (E5) et (E6) :
le système (E5) est un système d'équations différentielles linéaires
homogènes que l'on résout pour des conditions initiales données alors
que les équations (E6) forment un système d'équations linéaires, l'in
tégration ayant déjà été effectuée (pour des conditions finales déter
minées).
V.4.2. Transfert d'excitation et fluorescence non résolue dans le temps.
Nous reprenons ici l'exemple du paragraphe v.3.3 et utilise
rons les m~mes notations (le niveau 1 étant toujours supposé le niveau
pompé). L'équation (E6) donne
N2- (K21 + K2 + A2) = N1 ~2
soit N2
-N1
K12
A2 + K2 + K21 (E33)
Cette équation a une structure particuJ.ièrement simple. Les taux de
collision K étant fonctions linéaires de la densité N d'atomes pertur
bateurs, on obtient pour N2/N1 une variation de la forme b !'1~cN par
ticulièrement facile à ajuster par les méthodes de minimisation clas
sique (moindres carrés avec pondération). Faisons quelques remarques
importantes :

- 110 -
- à basse densité on peut négliger (K2 + K21 ) devant A2
et une
variation linéaire est alors obtenue, soit :
N2
N1 =
Ki2 A2
où K,2
est donnée par l'équation (EJ). La pente de la droite
(EJ4)
N2
/N1 = f(N) donne alors ~ 2 à condition que la durée de vie du niveau
2 (~2
= 1/A2 ) soit connue, ce qui n'était pas nécessaire dans le cas
de la méthode par fluorescence résolue. Un avantage très important de
la méthode appara!t : son extr3me sensibilité. En effet il est possi
ble, à l'aide des dispositifs de comptage, de mesurer des valeurs très
faibles de N2 /N1 (N 10-4 ), c'est-à-dire que la zone linéaire est très
facilement accessible et que l'on mesure ainsi K, 2 dans une zone où
la variation de la durée de vie du ni veau 2, due au terme de repopul.a
tion K2 + K21 , serait indétectable, c'est-à-dire que la méthode de
fluorescence résolue dans le temps serait totalement inapplicable.
- la détermination de K2 + K21 à partir de la courbure du graphe
N2/N1 = f(N), si elle est théoriquement possible,s'avère de fait assez
délicate car il faut se limiter, sauf cas particulier, au début de la
courbure, le modèle à deux niveaux n'étant, en général, plus justifié
à très forte densité de gaz perturbateur. On obtient donc généralement
une estimation de (K2
+ K21 ) (c'est-à-dire de K2 puisque K21 est relié
à K,2
par le principe de microreversibilité).
- nous avons effectivement utilisé la méthode qui découle de
l'équation (EJ4) pour déterminer les sections efficaces de structure
fine entre les deux niveaux 7P1 ; 2 et 7P312 du césium. Le dépouillement
s'effectue alors par une régression linéaire classique (déjà décrite
au paragraphe V.J.2), après s'~tre assuré de la validité de l'équation
(EJ4) à l'aide du coefficient de corrélation r).
- la méthode peut se généraliser aisément à un modèle à plus de
deux niveaux (nous en verrons un exemple au chapitre suivant), en me
surant successivement les valeurs des différents R .. en fonction de N. J..J
Ceci découle très simplement de la structure linéaire du système (E6).
Il faut cependant dans ce cas prendre garde à la possible propagation
des erreurs (voir .chapitre VI, article 1).

- 111 -
- notons enf'in que Krause et ses collaborateurs ont utilisé, dans
toutes leurs études, une procédure légèrement différente pour obtenir
les sections efficaces. Elle consiste à mesurer successivement, en
fonction de la densité N de gaz perturbateur, N2
/N1 (le niveau 1 étant
le niveau pompé) puis N1 /N2 (le niveau 2 étant le niveau pompé). En
supposant, de plus, que K2 et K., sont négligeables devant, respective
ment, K21 et K12 , on extrait alors Iti 2 et K21 par la résolution des
deux équations de type (E33), à N donné. Enfin la pente des graphes
Iti 2 (N) et K21 (N) fournit Q., 2 et Q21 . Cette méthode a un avantage :
elle permet d'extraire K12 et K21 de mesures effectuées dans les zones
où N2
/N1 et N1 /N2 ne sont pas des fonctions linéaires de N (mais des
fonctions de la forme aN/b + cN). Ceci peut permettre ainsi d'opérer
dans une zone où l'erreur sur la mesure de N sera très faible. Elle
présente cependant deux inconvénients. Tout d'abord il faut pouvoir
pomper les niveaux 1 et 2 (alors que, par exemple, la mesure de Q., 2 à
très basse pression ne nécessite que le peuplement du seul niveau 1). Mais surtout il faut supposer que K2 et~ sont négligeables devant
K21 et K12
, hypothèse qui n'est vérifiée, a priori, que si les niveaux
1 et 2 sont très bien isolés énergétiquement de leurs voisins. Ainsi,
si cette procédure s'avère intéressante pour les premiers niveaux ré
sonnants des alcalins, son emploi n'est pas justifié pour l'étude des
niveaux plus excités, pour lesquels elle peut conduire à des valeurs
erronnées (voir chapitre VI). Dans ce dernier cas la seule méthode
sure est la mesure de N1/N2 à basse pression (EJ4) ; on s'assurera de
sa validité, a postériori, par le calcul du coefficient de corrélation
comme il a été dit plus haut.
En résumé cette méthode est d'un emploi simple et son extr~
me sensibilité permet d'opérer facilement dans des zones linéaires où
~ 2 est aJ.ors facile à extraire (la fluorescence résolue dans le temps
ne donnait alors que le quenching et non le transfert, dans le cas le
plus généraJ.). Elle nécessite cependant un étalonnage préalable du
système ainsi que la connaissance de certaines données spectroscopi
ques.

112. -
V.4.J. Quenching et f1uorescence non réso1ue dans le temps.
Différentes méthodes sont possibles pour 1a mesure de la
dépopulation collisionnelle tota1e. Elles dérivent toutes, quant à
leur principe, de la méthode de Stern-Vo1mer17 . Celle-ci consiste à
mesurer le rapport des signaux de fluorescence émis par 1e niveau à
étudier en l'absence et en présence de collisions. Dans le premier
cas le signa1 S est de la forme 0
so a:: = !'A
où a:: est une constante dépendant du taux d'excitation et du dispositif
expérimentai. Si on mesure maintenant la fluorescence en présence de
gaz perturbateur, dans les m3mes conditions expérimenta1es, on obtien
dra
a:: S = 'tA + K
où K représente l'effet de la dépopulation tota1e sur le niveau consi
déré. L'équation de base de la méthode s'écrit a1ors très simplement :
K §.. = 1 + !'A so (EJ5)
La mesure de S/S en fonction de N, densité de gaz perturbateur, doit 0
donc donner une droite, d'ordonnée à l'origine 1 et de pente Qv/rA
(puisque K = NQv). Si la durée de vie 1/~A est connue, on obtient a1ors
Q section de dépopulation collisionnelle tota1e du niveau considéré.
L'équation (EJ5) suppose que seuls sont pris en compte les termes de
dépopulation, les termes de repopulation étant tous supposés négligea
bles. On peut montrer1 que l'expression la plus généra1e de s/s est 0
en fait une série en puissances de N dont l'expression (EJ5) repré-
sente l'expression limitée au premier ordre. La forme la plus généra1e
de la série est de peu d'utilité pratique et la méthode n'a guère été
utilisée qu'en se limitant à l'approximation linéaire.
Nous avons effectivement utilisé une méthode de type Stern
Volmer pour étudier la dépopulation collisionnelle tota1e du niveau
10P du potassium (voir chapitre VI). Les principaux inconvénients de
la méthode sont les suivants :
·"'

- 11 J -
- la validité de l'équation (EJS) suppose des conditions assez
restrictives et parfois difficiles à remplir. Il faut ~tre sô.r que le
taux d'excitation reste le m~me lors des mesures de Set de S, ce quel 0
que soit N ce qui suppose une réabsorption négligeable de la raie
d'observation en fonction de N (condition difficile à remplir dans
le cas des raies de résonance, par exemple) ainsi qu'une absorption
constante du faisceau excitateur. Cette dernière restriction implique
que la source d'excitation ait une intensité constante et que la raie
d'absorption ne soit pas élargie par collision, d'où la nécessité de
conna.1tre les caractéristiques d'élargissement de pression de la raie
d'excitation.
- il faut conna.1tre la durée de vie 1 /A pour obtenir Q, ce qui
n'est pas le cas quand on utilise la méthode de fluorescence résolue
dans le temps (qui fournit la durée de vie par extrapolation a pres
sion nulle comme nous l'avons vu).
- on doit se limiter à la zone linéaire en pression de S/S. 0
Cette dernière condition n'est pas trop restrictive puisque l'on peut
aisément la vérifier expérimentalement.
En résumé, la méthode de Stern-Valmer de mise en oeuvre et
de dépouillement très simple (régression linéaire) présente l'inconvé
nient de nécessiter la connaissance préalable -de certaines données
(durée de vie, élargissement collisionnel) pour justifier son emploi
aussi bien que pour extraire la section de quenching. De ce point de
vue la méthode de fluorescence résolue dans le temps nous semble, en
général, préférable.
V.3. SOURCES D'ERREUR ET PRECISION DES MESURES.
Dans ce paragraphe nous examinons successivement les diffé
rents phénomènes susceptibles de perturber les mesures et les causes
d'erreurs expérimentales. Nous ne donnerons, sauf cas particulier, une
évaluation précise de ces dernières que pour celles qui sont communes
à toutes les mesures que nous avons effectuées. Les évaluations quan
titatives des erreurs plus spécifiques aux situations expérimentales
envisagées ainsi que l'évaluation de l'erreur globale sur le résultat
final de la mesure, seront données au chapitre VI.

- 114 -
v.5.1. Phénomènes liés à la source d'excitation.
-~~~=~-~E=~!~~=: 0
La largeur de celle-ci est de l'ordre de 1 A pour le laser 1
(excitation des raies 6s112 ~ 7P1 ; 2 et 651 ; 2 - 7P3; 2 du césium), de 0
o.4 A pour le laser 2 (excitation de la raie 451 ; 2 ~10P du potassium), 0
de 0.3 A pour le laser 3 double en fréquence (excitation des niveaux . . 0
de Rydberg nP du rubidium) et de 0.5 A sans doublement de fréquence
(excitation à deux échelons des niveaux de Rydberg 5 du rubidium). La
largeur des raies d'absorption atomique de structure fine, principale
ment due à l'effet Doppler, est de l'ordre de 5.10-3 A (elle est par
exemple de 6.5 10-3 pour les raies de structure fine du niveau 7P du
césium, et 460 K et de 5.6 10-3 A pour le niveau 22P du rubidium).
On peut donc considérer, à une très bonne approximation,que l'ensemble
de la raie est excitée de manière uniforme et qu'aucune sélection en
vitesse n'est donc effectuée par le processus d'excitation. Le seul
problème lié à la largeur de la source est rencontré pour les expé
riences concernant les niveaux de Rydberg P. En effet l'écart de struc
ture fine entre les raies correspondant au..~ niveaux P1 ;2
et P3
; 2 est
de l'ordre de grandeur de la largeur de la raie d'excitation pour les
plus faibles valeurs den (nN10). Nous avons montré expérimentalement
qu'aucune variation systématique n'était décelable dans les résultats
obtenus en balayant la raie d'excitation sur l'ensemble du doublet.
Il est donc légitime de considérer alors les résultats obtenus comme
concernant l'ensemble du niveau P, sans références à la structure
fine.
- ~~E~!!!!~~-=E!=!~~!:
La réparti tian de la puissance lumineuse dans la largeur de 0
la raie d'excitation (N 0.5 A) est loin d 1 @tre homogène dans le cas
d'un laser "dye" pulsé ; cette réparti tian présente en fait une structu
re complexe de modes. Celle-ci pourrait entra.!ner une excitation pré
férentielle de certaines classes de vitesse. Nous ne pensons pas que
ce phénomène soit important pour le type d'expériences que nous avons
effectuées. Expérimentalement nous avons bien observé que les sections
efficaces de transfert d'excitation vérifiaient le principe du bilan
détaillé. De plus le temps de corrélation du champ électrique (c'est
à-dire l'inverse de la largeur spectrale de l'impulsion excitatrice)

- 115 -
étant très petite devant la durée de l'impulsion on peut montrer18 que
le champ électrique peut ~tre décrit par une fonction aléatoire, ce
qui revient à supposer une excitation de type "broad-line".
Stabilité_en_longueur_d'onde_et_en_puissance.
Celle-ci est importante à considérer dans toutes les méthodes
utilisant des rapports de fluorescence (transfert par fluorescence non
résolue en temps, méthode de Stem-Valmer). La stabilité en longueur
d'onde des lasers que nous avons utilisés s'avère en général suffisan
te pour que l'on reste centré sur la raie d'excitation durant le temps
d'obtention des données (de 1 à 90 mn suivant les expériences envisa
gées). Par contre la stabilité en puissance des lasers pulsés n'est
pas suffisante pour opérer les mesures en séquence quand la connaissan
ce de deux signaux de fluorescence est requise. En effet on a d'une
part une dérive lente de la puissance due à la dégradation du colorant,
particulièrement dans le cas des lasers 1 et 2 pompés par flashs,
d'autre part une instabilité d'un coup sur l'autre qui peut atteindre
20%, toujours dans le cas des lasers 1 et 2. Nous avons donc toujours
effectué la mesure simultanée des deux signaux dont nous désirons le
rapport, ce en utilisant deux voies de mesure, nous affranchissant
ainsi de tous les problèmes liés aux instabilités de la source. Notons
pour terminer que la stabilité en puissance est sans importance dans
le cas de mesures par fluorescence résolue dans le temps.
- Puissance de la source excitatrice.
Celle-ci peut donner lieu à deux types de difficulté. L'une
est directement reliée au taux d'inversion obtenu, c'est-à-dire à la
valeur de T. = N* /N , N* représentant la densité d'atomes alcalins inv o .
excités dans l'état considéré et N la densité d'atomes alcaJ.ins 0
telle que l'on peut la déterminer à partir de la température de l'ap-
pendice qui le contient. Si la puissance de la source excitatrice de
vient trop forte le taux d'inversion peut atteindre une valeur telle
que l'on ne peut plus considérer la densité d'atomes alcaJ.ins au ni
veau fondamental comme constante (et égale à N ). Il convient alors 0
de modifier les équations développées au chapitre précédent et, en
particulier, la mesure des sections efficaces alcalin-alcalin se
trouve notablement modifiée, la densité d'atomes au niveau fondamen
tal variant alors avec le temps (elle vaut N - N*(t)). Toutes nos 0

- 116 -
expériences ont été conduites en utilisant des puissances lasers tel
les que les taux d'inversion obtenus n'ont jamais été supérieurs à
5.10-J ce qui nous permet de négliger complètement cet effet.
La deuxième difficuJ.té vient du fait que m~me pour des va
leurs très faibles de T. la va1eur absoiue de N* peut ~tre suf'fisan-inv te pour donner lieu à ded effets coopératifs de superradiance, parti-
cu1ièrement importants dans le cas des états de Rydberg19 . Nous re
viendrons sur la superradiance au chapitre VI. Contentons-nous ici
de mentionner que celle-ci modifie profondément le comportement radia
tif de la vapeur et donc les conditions d'applicabilité des équations
de base que nous avons précédemment établies. En particuJ.ier la super
radiance donne lieu à une dépopulation des niveaux excités, qui s'ef
fectue alors avec des constantes de temps beaucoup plus rapides
(~10 ns) que celles relatives à la relaxation radiative classique
(N 100 ns pour les états les plus bas et N quelques µs pour les états
de Rydberg). Pour ce qui concerne notre travail expérimental nous pen
sons que les résuJ.tats obtenus ne sont pas affectés par ce phénomène,
ce pour les raisons suivantes.
Les mesures relatives aux niveaux bas (7P -6D du césium) ont
été effectuées par la méthode de la fluorescence non résolue en temps.
Celle-ci a pour point de départ la mesure du rapport des intensités de
deux raies de fluorescence. Nous avons montré que ce rapport ne présen
te aucune variation systématique, aux erreurs statistiques près, lors
que l'on fait varier la puissance du laser excitateur (et donc la den
sité N* d'atomes excités) dans un rapport de 1 à 50. '
Le problème s'avère plus délicat pour les états très excités,
pour lesquels les phénomènes de superradiance sont notablement plus
importants19 . Les mesures ont alors été conduites par la méthode de
fluorescence résolue en temps qui permet de s'affranchir, dans une
certaine mesure, de l'influence éventuelle de la superradiance. En
effet, celle-ci ayant des constantes de temps très courtes (,v 10 ns),
il suffit de commencer l'observation environ 100 ns après l'excitation
laser pour pouvoir négliger cet effet19 . De plus nous avons montré
(Appendice V) que les durées de vie radiatives mesurées dans ces con
ditions sont en bon accord avec les prévisions théoriques. Nous nous
sommes cependant assurés que les durées de vie mesurées dans ces con
ditions ne présentaient aucune variation systématique décelable en fonc-

- 117 -
tion de la puissance de la source d'excitation. Notons, enfin, que
dans le cas des états P très excités les conditions d'excitation
(laser UV de faible puissance, forces d'oscillateur de la série
nS - nP très faibles) sont telles que la densité d'atomes excités N*
est faible et donc la superradiance peu importante. De fait, nous
avons bien observé que la variation N = f(t) de la densité des atomes
excités en fonction du temps ne présentait pour ainsi dire pas de
comportement anormal m~me juste après le tir laser, au contraire des
états S pour lesquels une ouverture retardée du dispositif. de mesure
est nécessaire pour s'affranchir de la dépopulation rapide due à la
superradiance.
- Polarisation de la source d'excitation
Les équations générales que nous avons établies au paragra
phe V.2 ne prenaient en compte que la relaxation de la population des
états excités. En fait la description la plus complète du système ne
peut se faire qu'à partir de la matrice densité du système20 , 21
Celle-ci contient, en général, non seulement des termes diagonaux re
présentant la population des sous-niveaux, mais aussi des termes non
diagonaux (cohérence) qui peuvent contribuer au signal de fluorescen
ce obtenue. Le calcul du signal de fluorescence peut se faire à partir
d , . . ' ' d ' d h' 21 d , es equations generales onnees ans lat ese de M. Dumont , ans ~e
cas des expériences de fluorescence non résolue en temps, ou dans les
références22 , 23 dans le cas des expériences de fluorescence résolue
en temps. Dans les deux cas le formalisme utilisé est celui des opéra
teurs tensoriels irréductibles qui permet de s'adapter à la symétrie
sphérique du problème. Rappelons que l'ordre tensoriel k ne peut pren
dre que les valeurs O, 1 ou 2, correspondant respectivement à la popu
lation, l'orientation et l'alignement du niveau considéré. Nous ne dé
taillerons pas les calculs assez fastidieux qui nous ont permis l'éva
luation quantitative des effets liés à la polarisation de l'excitation
et de la détection dans nos expériences, mais nous préciserons bien
le type de perturbations que peuvent donner de tels effets ainsi que
les raisons expérimentales qui nous ont conduits à les négliger,
raisons que confirment bien les évaluations théoriques.
Dans le cas des expériences de fluorescence non résolues en
temps, les sections efficaces de transfert d'excitation telles que
nous les avons définies au paragraphe V.2 représentent en fait la

- 118 -
moyenne- des sections efficaces de transition entre ~-sous-ni veaux Zeeman 1
de type (J,mJ ~J ,mj 1 ). Ceci ne sera légitime que si l'anisotropie
introduite par le pompage demeure faible. Dans le cas précis de nos
expériences sur les niveaux 7P1; 2 et 7P3
; 2 du césium, effectuées en
lumière non polarisée, ce tant à la détection qu'à l'excitation on
peut montrer.que ces effets de polarisation seront très faibles. Si
on peuple le niveau 7P1; 2 à partir du niveau fondamental 6s112
, on
ne crée que des termes diagonaux dans la matrice densité de l'état
excité (ceci est caractéristique des transitions J = 1/2 --+-J 1 = 1/2, dans le cas d'wie excitation linéaire, et donc aussi dans le cas d!une
excitation non polarisée2 j) et la description en terme de population
est donc correcte. Si l'on excite le niveau 7P3; 2 en lumière non pola
risée une partie du signal de fluorescence directe observé est da à
la composante d'ordre 2 (alignement) de la.matric~·densité. Utilisant
les équations de la référence 20, en tenant compte du spin nucléaire
I = 7/2 du césium, nous avons calculé que la contribution de l'aligne
ment au signal de fluorescence directe est au maximum de 5,5~ du si
gnal total, ce en négligeant toute relaxation collisionnelle. Les sec
tions efficaces de destruction de l'alignement24 par collision étant
au moins un ordre de grandeur plus grandes que les sections efficaces
de transfert, cet alignement résiduel sera en fait rapidement détruit
par collisions et le transfert d'excitation peut 3tre décrit à partir
des équations du paragraphe V.2 avec une très bonne approximation. No
tons pour terminer que les sections efficaces de transfert
(7P1 ; 2 ~7P3; 2 ) obtenues en peuplant les niveaux 7P1 ; 2 et 7P3; 2 sont
en bon accord avec le principe du bilan détaillé, ce qui constitue wie
vérification expérimentale du fait que les effets éventuels de la po
larisation sont bien négligeables.
Le cas des expériences de fluorescence résolue en temps ef
fectuées sur le rubidium est un peu plus complexe à analyser. Le signal
de fluorescence observé I(t) peut s'obtenir aussi en termes d'opérateurs
tensoriels irréductibles22123 . L'excitation est polarisée linéairement,
la détection s'effectue à angle droit en lumière non polarisée. Là aus
si seuls les ordres k = o (population) et k = 2 (alignement) peuvent
contribuer au signal observé. Le terme dn à l'alignement a deux consé
quences ; la première est l'apparition d'une constante de rela..~ation
r( 2 ) différente de r(o), ce qui donnerait alors un signal de fluores
cence comportant deux exponentielles, ce m~me en l'absence de tout effet

- 119 -
de repopu1.ation25 • En fait rC 2 ) ne sera notablement différent de r(o)
que si la raie d'observation est fortement réabsorbée ce qui n'est
pas le cas dans nos expériences. Ceci est bien confirmé expérimenta
lement : les courbes obtenues en l'absence d'effets de repopû.l.ation
montrent bien une décroissance monoexponentielle du signal. La deu
xième conséquence est l'apparition d'une structure modulée22 , due à
des battements quantiques, qui se superpose à la décroissance obser-
vée. Cette structure sera complexe dans le cas du rubidium car celui-
ci possède deux isotopes ( 85Rb et 87Rb) de spins nucléaires différents
(I = 5/2 et I = 3/2, respectivement) ; notre dispositif d'observation
ne possèdant que vingt canaux, cette structure sera en fait très
moyennée à l'observation. Nous avons cependant calculé la profondeur
maximale de modulation attendue dans le cas du peuplement des états P
du rubidium en bande large (sans résolution de la structure fine)
celle-ci sera, au maximum, de 12~ sans tenir compte de la résolution
expérimentale qui abaissera encore notablement cette valeur. De fait
les courbes expérimentales obtenues ne présentent aucune structure
systématique, à la précision des mesures prés. Notons pour terminer
qu'aucune perturbation n'est attendue dans le cas des états S du rubi
dium. En effet le peuplement du niveau intermédiaire 5P3
; 2 s'effec~ue
de manière isotrope, à cause de la réabsorption de la raie de résonan
ce26, et la transition 5P3
; 2 ~ns112
n'introduit aucun effet de pola
risation, le terme d'alignement étant nuJ. dans le cas (J= 3/2 ~J' = 1/2).
V.5.2. Mesures des températures et des pressions.
TemEérature_~~-!~_vapeur.
Celle-ci est connue à= 2°K à l'aide des thermocouples pla
cés sur la cellule de mesure. L'erreur relative, qui en découle, sur
la connaissance de la vitesse moyenne des particules en collision ain
si que sur les densités d'atomes alcalins est donc de l'ordre de 0.3%, ce qui est pratiquement négligeable.
Température_de_l'appendice_contenant_l'alcalin.
La régulation de celle-ci est effectuée à= 0.1°C, mais sa
valeur absolue n'est pas connue avec une grande précision. En effet les
thermocouples sont placés à l'extérieur de la paroi de verre et un gra
dient de température de l'ordre de 1 à 2°C peut exister entre la tempé-

- 120 -
rature de régulation affichée et la température réelle de la surface
de l'alcalin qui détermine sa tension de vapeur et donc sa densité.
Tension_de_vapeur_de_l'alcalin.
La détermination précise de la pression d'atome alcalin est
un problème difficile. Comme nous venons de le voir la température de
celui-ci n'est pas connue à mieux que 1 à 2°C près, ce qui entraîne
une incertitude de l'ordre de 15% si la pression est déterminée à par
tir de la formule donnant la tension de vapeur en fonction de la tem
pérature. Mais,de plus,cette formule est déterminée par ajustement
numérique à des résultats expérimentaux d'origine et de précision di
verses. Enfin les interactions entre l'atome alcalin et le matériau
de la cellule peuvent donner lieu à des réactions diverses dont l'in
fluence s.ur la tension de vapeur demeurent difficiles à évaluer*.
Dans le cas où nous désirions connaitre avec précision la pression
d'atomes alcalins (mesures des sections efficaces alcalin-alcalin pour
les niveaux de Rydberg du rubidium), nous avons estimé nécessaire de
vérifier par mesure d'absorption les valeurs déduites de la courbe
de tension de vapeur. Une telle vérification a déjà été effectuée27 ,
dans le cas du rubidium et dans la gamme de pression où nous avons
travaillé. Gallagher et Lewis, utilisant une méthode sophistiquée
d'absorption, ont conclu à la validité de l'équation, donnant la ten
sion de vapeur, proposée par Nesmeyanov, ce avec une précision de
l'ordre de 10~. Notre but étant non de déterminer la densité avec une
très bonne précision mais seulement d 1 $tre sô.r que la densité déduite
de la température de l'appendice n'était pas très différente de celle
obtenue par une autre méthode, ce qui aurait indiqué des interactions
importantes entre l'alcalin et le verre de la cellule, nous avons uti
lisé la méthode, relativement simple, de la largeur équivalente. Sa
mise en oeuvre est exposée en détail dans l'appendice IV. Elle permet
de s'affranchir de la fonction d'appareil et autorise, avec une réso-o
lution de l'ordre de O.J A, la mesure de pressions supérieures à
* Pour nous affranchir de ces effets nous avons toujours saturé les pa-rois de la cellule en alcalin en plaçant tout d'abord la cellule pendant quelques heures à forte pression d'alcalin2 7 (N 5.10-4 T). Celle-ci doit cependant ~tre suffisamment faible pour éviter toute attaque chimique (jaunissement de la cellule) du verre par l'alcalin (pressiontv5.10-J T).

- 121 -
5.10-5 T. Nous avons observé, pour des pressions comprises entre
6.10-5 et 10-3 T, un bon accord entre les valeurs déduites de cette
méthode et celles fournies par la courbe de tension de vapeur. L'écart
maximal observé a été de 15%. Nous avons· considé~é que la densité de
rubidium était connue.avec une précision meilleure que 25~, ce dans
( -6 -4 ) toute notre zone de travail 8.10 - 5.10 T, la courbe de pression
de vapeur ayant une précision de l'ordre de 10%27 . Notons,pour termi
ner que, si la mesure par absorption n'a pu gtre conduite, faute de
sensibilité, à des pressions inférieures à 6.10-5 T, les valeurs des
durées de vie radiative déduites par extrapolation à pression d'alca
lin nulle sont en bon accord avec les estimations théoriques, ce qui
indique que,m3me aux pressions de travail les plus basses (8.10-6 T),
les pressions déduites de la température de l'appendice sont correctes.
Pression_de_gaz_rare.
La hauteur de la colonne d'huile est estimée avec une erreur
absolue maximale de 5.10-2 mm, ce qui correspond à une pression d'en
viron 3.10-3 T. L'erreur relative dépend donc de la pression de travail
mais n'excède pas 10~ pour des pressions supé~ieures à 3.10-2 T. Nous
n'avons, en général., utilisé ce dispositif que pour des pressions su
périeures à 10-1 T. L'erreur relative est alors négligeable. Pour des
pressions inférieures à 10-1 T nous avons utilisé un manomètre à ca
pacitance (M.K.S. Baratron) qui permet de mesurer les pressions dans
la gamme 5.10-4 - 5.10-1 T avec une précision meilleure que 5% (l'er
reur relative est en fait de l'ordre de 2~, dès que la pression dépas
se 2.10-J T, valeur pour laquelle la dérive électronique du zéro de
l'appareil devient tout à fait négligeable). Nous avons vérifié, dans
la gamme de pressions commune au.~ deux dispositifs que ceux-ci con
duisaient à des valeurs identiques, aux erreurs de mesure près.
Il est nécessaire lors de la mesure de la pression d'atten
dre un temps suffisamment long avant d'effectuer celle-ci, pour per
mettre la stabilisation de la pression dans l'enceinte de mesure. En
effet lors de l'introduction d'une forte pression de gaz rare l'alca
lin est repoussé par effet de piston et il faut attendre qu'il ait
rediffusé dans l'enceinte avant d'effectuer la mesure. Le phénomène
est facilement observable expérimentalement et peut se calculer par
un modèle de diffusion simple. Le temps d'établissement de l'équilibre
est de l'ordre de la minute pour les gaz rares légers mais peut attein-

- 122 -
dre 1 h JO aux plus fortes pressions de xénon auxquelles nous ayons
travaillé (aj 20 T, lors des expériences de transfert 7P - 6D).
Une dernière source d 1 erreur dans la mesure de la pression
de gaz rare provient de l'effusion thermique : la mesure s 1 effectue
à la température ambiante T alors que le gaz dans l'enceinte est à 0
la température T. Les deux pressions ne sont égales que pour un écou-
lement visqueux du gaz. En régime moléculaire la pression p à l'inté
rieur de l'enceinte est reliée à la pression lue p par : 0
P = P 0 Fe Ainsi l 1 erreur relative peut atteindre 40% pour T = 600 K. Une correc
tion s'évère donc nécessaire, d'autant que les gammes de pressions de
travail recouvrent les deux types de régime (ainsi pour l'hélium à
600 K la pression de transition entre les deux régimes est de l'ordre
de 0.22 T; elle est de 4.4 10-2 T pour le xénon). Nous avons effectué
cette correction à l'aide d 1 un modèle simple. La méthode est exposée
dans l'appendice II. La précision de la correction est difficile à
évaluer. On peut cependant penser qu'elle n 1 affecte pas de plus de J% la précision de J.a mesure de la pression de gaz rare, telle que nous
l'avons évaluée au début de ce paragraphe.
L'erreur finale sur la pression de gaz rare dépend de la va
leur absolue de celle-ci mais reste comprise entre Jet 10~, pour l'en
semble des mesures que nous avons effectuées.
Influence_éventuelle_du_vide_résiduel_et_de_la_pureté_des_gaz
utilisés.
La présence d 1 impuretés moléculaires peut modifier les résul
tats obtenus, leurs sections efficaces collisionnelles pouvant gtre de
plusieurs ordre de grandeur supérieures à celles des gaz rares (en
particulier dans le cas des états de Rydberg). Il convient donc d 1 ob
tenir un vide résiduel aussi faible que possible. Celui-ci était de
5.10- 7 T pour toutes les expériences, sauf celles concernant les états
S du rubidium pour lesquelles u...~e amélioration du bâti de pompage a
permis de le porter à 5.10-8 T. :fous avons mon,:;ré expérimentalement
que mgme dans le cas le plus cri tique (ni veau 22P du rubidium) l 1 erreur
résultante ne dépassait pas ') al. ..) ,:a. Elle est parfaitement négligeable pour
tous les autres niveaux que nous avons étudiés.

- 123 -
Les gaz rares utilisés sont stockés à une pression d'environ
500 T dans une enceinte dans laquelle un vide meilleur que 5.10-7 Ta
été au préalable obtenu. Les impuretés moléculaires présentes dans
les gaz utilisés était de 7 ppm pour Ne, Ar, Kr et Xe et de 3 ppm
pour He, l'effet de celles-ci demeure complètement négligeable.
v.5.3. Causes d'erreur dues à l'optique de détection.
Diffusion_ thermique ( "Thermal escape n).
Les durées de vie des niveaux que nous avons étudiés sont
comprises entre une centaine de nanosecondes (niveaux 7P-6D du césium)
et plusieurs microsecondes (niveaux de Rydberg P du rubidium). Ainsi
il peut arriver que les atomes excités puissent s'échapper du volume
d'observation (dont les dimensions linéaires typiques sont de l'ordre
de 2 mm) avant d'avoir pu ~tre observés (pour une vitesse typique de
105 cm/s l'atome parcourt 1 mm en 1 µs), ce qui peut donner lieu d'une
part à une diminution du signal total observé, d'autre part à une dis
torsion notable de la courbe de variation de la fluorescence en fonc
tion du temps. Dans le cas. des niveaux faiblement excités ce phénomène
est négligeable. Il n'en est plus de m~me pour les niveaux de Rydberg.
On peut s'affranchir en partie de cet effet en utilisant un faisceau
excitateur de grandes dimensions par rapport au temps de parcours
moyen d'un atome excité. Nous avons effectué une correction numérique
qui tient compte du volume d 1 observation, de la durée de vie observée
et des caractéristiques géométriques de l'excitation. La méthode uti
lisée est décrite dans la référence 28. Notons cependant que la correc
tion maximale effectuée (niveaux 22P et 18S du rubidium) ne dépasse pas
12%. La précision de la correction étant de l'ordre de 10% (due à la
définition géométrique des faisceaux d'excitation et de détection)
l'erreur finale sur la mesure de la durée de vie n'excède pas 1%.
Absorption_des_raies_d'observation.
Celle-ci peut se manifester de deux manières : atténuation
de l'intensité du signal observé et modification de la constante de
temps de celui-ci. Le problème n'est critique que pour les transitions
relatives à des niveaux faiblement excités, qui possèdent des forces
d'oscillateur assez fortes. C'est le cas de nos mesures concernant les
niveaux 7P-6D du césium. Les mesures ayant été effectuées par fluores-

- 124 -
cence non résolue dans le temps, la prise en compte du phénomène né
cessite le calcul du rapport des absorptions relatives des deux raies
d'observation choisies qui constitue un des termes du rapport
(T(~ib ))/(T(~jb )) de l 1 équation (EJ2) (le deuxième terme qui vient 0 S O S
de la transmission optique du système de détection à ces deux longueurs
d'onde sera discuté au paragraphe suivant). L 1 absorption relative d'une
des raies d'observation peut se calculer par la formule :
I I
0
= 1
rn J+=
-<>o
e-q2 e -k(v)l dq (EJ6)
où l représente la longueur moyenne d'absorption (J mm dans le cas de
nos expériences) et k(v) le coefficient d'absorption. L'expression de
ce dernier doit tenir compte à la fois de l'élargissement Doppler et
de l'élargissement collisionnel de la raie (celui-ci étant fonction
à la fois de la pression d'alcalin et de la pression de gaz rare). On
obtient pour k(v) le profil de Voigt classique (convolution de la
gaussienne due à l'effet Doppler et de la Lorentzien,.~e due à l'effet
des collisions) qui permet d'écrire (EJ6) sous la forme :
i+ oo ( + QG 2 \ - 1 2 . k al -y ~ = , e-q exp - on r 2 e 2 dyj dq (EJ7) o Yn
00 -J_
00 a + (q-y)
où k, représentant le coefficient d'absorption au centre de la raie, 0
est donné par
avec
k 0
= _2_ V Lo~2 g2 À; NA ~\) - ---D n g 1 Sn
~\) = D
2V _o C
v 2RT Log2
(EJS)
(EJ9)
(g1 et g 2 représentent les poids statistiques des niveaLLX supérieur
et inférieur de la transition, ~ la densi~é d'atomes alcalin dans le
niveau inférieur, A le coefficient d'Einstein de la transition,
et  respectivement la fréquence et la longueur d'onde) 0
a, dans l' équa~ion (EJ7), est donné par
b. \) C
a = D.V V Log2 D
v 0
(E40)

- 125 -
où ~~cet ~~D représentent respectivement les élargissements collision
nel et Doppler.
La procédure détaillée de calcul de l'équation (EJ7) est
donnée en appendice III. Cette correction d'absorption appelle quel
ques remarques générales
- Dans toutes nos expériences le rapport I/I n'est jamais infé-o
rieur à 0,7 et l'erreur sur le calcul de ce rapport n'excède pas 20~.
Cette erreur provient essentiellement de l'incertitude sur la valeur
de la densité N d'atomes de césium.
- Ce calcul ne concerne que les raies 7P1 ; 2 ~6s112 et
7P3
; 2 ~6s112 ; les raies d'observation du niveau 6D (6D - 6P) ne
sont pas réabsorbées le niveau 6P n'étant pas peuplé.
- Le calcul nécessite la connaissance des constantes d'élargisse
ment collisionnel des niveaux considérés (7P1 ; 2 et 7P3
~2 dans notre
cas). Celles-ci sont assez bien connues dans notre cas 9,J0,31 . Notons
que l'élargissement d~ aux collisions césium-césium est faible devant
l'effet Doppler à notre pression de travail et qu'ainsi seul l'élar
gissement césium-gaz rare est à considérer, du moins aux plus fortes
pressions de gaz rare (N qq. Torrs).
- La valeur de I/I dépend aussi du coefficient d'Einstein de la 0
transition considérée. Celui-ci n'est pas toujours bien connu, ce qui
peut entra!ner une erreur assez importante sur le calcul de l'absor
ption. Le problème du choix des coefficients d'Einstein sera discuté
en détail au paragraphe V.5.6.
Dans le cas des expériences concernant les états fortement
excités on peut montrer facilement que l'absorption joue un r$le par
faitement négligeable. Ainsi pour le niveau 12P (cas le plus défavo
rable), la seule transition réabsorbée (12P ~ 5s) possède une longueur
d'absorption de l'ordre de 25 cm, à la plus forte densité de rubidium
à laquelle nous ayons travaillé. Le chemin optique dans la cellule
était de l'ordre de 0,5 cm l'absorption résultante est bien négligea
ble. Comme, de plus, cette transition ne représente que 15~ de l'émis
sion spontanée totale du niveau 12P, l'erreur ma.timale sur la durée
de vie mesurée (par fluorescence résolue dans le temps) n'excèdera
pas 0.3%. Un calcul similaire montre que cet effet est aussi parfaite
ment négligeable dans le cas des niveaux nS du rubidium (la transition

- 126 -
pouvant gtre réabsorbée étant alors la transition ns112 -3PJ/2 , le
niveau 5PJ/2
pouvant être, à forte densité de rubidium, notablement
peuplé quelques microsecondes après l'excitation).
Transmission_du_système_optique.
Comme nous l'avons mentionné précédemment, seule la mesure
de section de tr.ansfert par fluorescence non résolue dans le temps
nécessite la connaissance de la transmission du système optique. En
fait, seul est requis le rapport des transmissions du système aux
deux longueurs d'onde d'observation (cf. (EJ1). Si l'on excepte
l'absorption possible dans la cellule de mesure (paragraphe précédent),
ce rapport est entièrement déterminé par le dispositif de détection
(éléments optiques des deux voies de mesure, efficacité relative du
système monochromateur, photomultiplicateur, cha!ne de comptage aux
deux longueurs d'onde considérées). La mesure de ce rapport s'effec
tue de la manière suivante : on observe tout d'abord le m~me signal
de fluorescence à une des longueurs d'onde de travail sur les deux
voies, ce qui permet de déterminer le rapport géométrique de collec
tion de celles-ci, ce dans les conditions de travail. L'erreur qui
en résulte correspond à celle obtenue sur un rapport de comptage.
Pour un temps d'accumulation assez long elle n'excède jamais ï%. On
mesure ensuite la variation de transmission d'une des voies entre les
deux longueurs d'onde de travail, ceci, en continu, à l'aide d'une
lampe tungstène stabilisée. La précision de la mesure, décrite en
appendice I, est de l'ordre de 3%. Un dernier point reste à noter
dans certaines expériences il s'est avéré nécessaire d'insérer dans
une des voies de mesure, soit des densités neutres, pour éviter la
saturation des échelles de comptage, soit des filtres interférentiels
pour limiter la lumière parasite, ce qui modifiait la transmission de
la voie correspondante par rapport à la configuration de Téférence
servant à la mesure du rapport géométrique. La transmission des den
sités neutres a été mesurée, dans une expérience préalable, ce aux
longueurs d'onde d'utilisation. De même pour la transmission des fil
tres interférentiels. Il est à noter qu'il faut faire travailler ceux
ci en lumière parallèle,et non convergente, si l'on ne veut pas dépla
cer leur courbe de transmission par rapport à celle mesurée lors de
leur étalonnage. L'erreur résultante due à l'insertion de ces élémen~s
optiques n'excède pas J~. On peut finalement considérer que le rapport
des ~ransmissions des deux voies de mesure est connu à mieux que 13~.

- 127 -
~~~!~~~-E~~~~~~-
Bien que la conception (forme et matière) des cellules de
mesure soit prévue pour limiter au maximum la lumière parasite, il
n'a pas été possible de supprimer complètement cette dernière essen
tiellement lors des mesures concernant les états peu excités du césium.
En effet les études concernant les états de Rydberg ont été effectuées
à l'aide de lasers à colorants de faible largeur d'impulsion (de l'or
dre de 8 ns alors que les décroissances observées ont des durées typi
ques de l'ordre de la microseconde) ; dans ce cas il suf'fit d'ouvrir le
dispositif de mesure quelques dizaines de nanosecondes après le (ou les)
pulse(s) d'excitation (ce délai est nécessaire pour s'affranchir d'éven
tuels "after pulses" dus à un éclairement intense du photomultiplica
teur) pour éliminer complètement cette dernière. Notons que dans ce
cas seuls les états S sont concernés car la transition d'observation
(ns1 ; 2 -5P1 ; 2 ) es~ ~ssez proche de la transition d'excitation
(nP3/2 -5s1;2) pour que le taux de réjection du monochromateur soit
insuffisant pour éliminer toute la lumière diffusée due à l'impulsion
d'excitation. Dans le cas des états de Rydberg nP, la transition
d'observation (nP - 4D) se situe à plusieurs .milliers d'Angstrëms
de la transition d'excitation (5s112 -nP), ce qui permet d'éliminer
complètement l'effet de la lumière parasite.
Dans le cas des états (7P-6D) on a toujours à observer la
transition d'excitation (fluorescence directe), car les autres raies
possibles d'observation se situent dans le proche infrarouge, au-delà
de la zone de sensibilité des photocathodes disponibles. Dans ce
dernier cas la procédure de correction utilisée est la suivante : on 0
décale la longueur d'onde du laser d'excitation de J A par rapport à
la raie atomique (ceci se fait à l'aide d'un réticule gradué placé sur
le spectrographe de contr8le de la longueur d'onde) et on enregistre
le signal, puis on revient sur la raie, et la procédure est itérée.
Ceci permet de moyenner les fluctuations éventuelles de puissance du
laser durant la mesure. La lumière parasite est ensui te soustraite du
signal, en tenant compte de la variation de transmission du filtre 0
interférentiel (entre À et  + J A) préalablement déterminée. Cette 0 0
procédure nous a donné toute satisfaction, compte tenu du fait que la
lumière parasite n'excède jamais 20% du signal observé. La soustraction
de cette lumière parasite entra!ne une erreur supplémentaire sur le
comptage final qui n'excède pas 2~.

- 128 -
v.5.4. Erreurs dues à l'électronique de comptage*.
Tout comptage N est affecté d'une erreur statistique v'N qui
exprime simplement la variance de la loi de Poisson (l'arrivée des
photons constituant des événements statistiquement indépendants). Il
faut cependant effectuer, aux forts taux de comptage, une correction
qui tient compte du temps mort '1-1:de la chaîne électronique. Physique
ment cela implique que la cha!ne reste aveugle pendant un temps_~après
le comptage d'une impulsion. Ainsi si une impulsion est comptée à t= t , 0
tous les photons arrivant dans l'intervalle (t , t +~M) ne seront pas 0 0
pris en compte. Ce temps mort ~Mest essentiellement déterminé par le
temps de réarmement du discriminateur. Les discriminateurs utilisés
sont capables de compter des impulsions régulièrement espacées jusqu'à
des cadences d'environ 100 MHz, ce qui indique un temps mort d'environ
10 ns. En fait celui-ci sera légèrement supérieur du fait de la résolu
tion temporelle du photomultiplicateur et de la vitesse de réponse
réelle de l'amplificateur. Il convient donc de mesurer le temps mort
réel ~M de la cha.1ne utilisée. Les méthodes utilisées, ainsi que les
corrections de comptage correspondantes sont bien connues et nous ne
les décrirons que succinctement.
Dans le cas d'une excitation continue (lampe spectrale par
exemple) si on appelle n la fréquence réelle de comptage et n la fréo quence observée on a la relation de base 31
-n -z: n = n e o M
0 (E41)
net ~M étant connus, on peut déterminer n0
(par exemple à l'aide d'une
table numérique). La rela tian (E41) sert aussi à déterminer l:'M. On
éclaire le photomultiplicateur à l'aide d'une lampe continue bien sta
bilisée et on observe, pour différents éclairements, d'une part la va-
leur du courant I (qui est proportionnelle à n ), d'autre 0
quence de comptage n mesurée par la cha!ne. Le graphe n =
part la fré
f'(n) possède 0
un ma..""Cimum pour nmax = 1 / e "'M' 0 J:
ce
ne
qui permet de déterminer ~MJà condition
que la fréquence maximale n 0
*
corresponde pas à un courant supérieur
Nous reprenons ici, pour plus de clarté, quelques points que nous avons mentionnés au paragraphe IV.4.6.

- 129 -
à celui que peut débiter le photomultiplicateur ce qui est souvent le
cas. On peut alors utiliser la relation approchée :
1 1 = n n
0
+ ~ avec no 1-i<~ 1 (E42)
On tracera la courbe 1/n = f(1/I) (Fig. 15) dont l'ordonnée à l'origi
ne (obtenue à partir de la zone linéaire, qui correspond à des tau.~
de comptage relativement peu élevés) fournira la valeur de ~M. C'est
cette dernière méthode que nous avons généralement utilisée. Elle
nous a fourni des valeurs de ?:M allant de 12 à 30 ns suivant les dis
criminateurs et les photomultiplicateurs utilisés. Une vérification
simple consiste à reporter alors la valeur den (recalculée d'après 0
(E49), avec la valeur de~ trouvés) en fonction du courant correspon-
dant. On se rend alors bien compte que m~me à l'aide d'une cha!ne
"100 MHz" la correction de comptage est importante ( IV 20;,) pour des
fréquences n de l'ordre de 10 MHz. Il convient maintenant de voir 0
quel.le correction doit ~tre appliquée dans le cas de nos expériences,
c'est-à-dire pour une excitation .lumineuse pulsée. Nous étudierons
successivement le cas de la fluorescence non résolue, puis résolue
en temps.
Dans le premier cas nous avons utilisé directement la for
mule (E41) en définissant la fréquence de comptage n de la manière
suivante. Soit x le nombre de tirs lasers, N le nombre total de coups
comptés. La durée de l'impulsion laser ( N 1. 5 µs à mi-hauteur) étant
très supérieure à la durée de vie des niveaux. 7P ou 6D (~ 0.1 µs) et
la largeur des portes de comptage étant de 100 µs, nous avons considé
ré que la fréquence moyenne de comptage était donnée approximativement
par :
n - N - -6 x;icl.510
(E43)
L'équation (E41) nous fournit alors n, c'est-à-dire N (d'après (E43)), 0 0
le comptage réel. Ceci ne constitue évidemment qu'une approximation,
mais celle-ci nous a paru suffisante, dans la mesure où nous n'avons
jamais travaillé à des tau.~ de comptage tels que la correction soit
supérieure à 20%. Nous avons vérifié que cette approximation de
"largeur équivalente" était suffisante de la manière suivante. Les
deu.~ chaînes de comptage préalablement réglées et leurs temps morts

- 1 JO -
~1 et ~ 2 ayant été déterminés, nous avons mesuré le rapport géométrique M M
des deux voies en observant la m~me raie de fluorescence, ce à faible
taux de comptage (corrections de temps mort négligeables). Pour ce fai
re nous avons atténué le laser d'excitation à l'aide de densité neutre.
La valeur du rapport des deux voies a été déterminée à environ 5~ près,
en accumulant un temps suffisant. Puis nous avons remesuré ce rapport,
pour des valeurs croissantes de la puissance du laser d'excitation et
avons vérifié, qu'aux erreurs statistiques près (N 5%) ce rapport res
tait bien constant, ce m~me pour des taux de comptage N1
et N2
élevés,
pour lesquels les corrections de comptage atteignaient JO~. Notons
qu'une correction précise tenant compte de la forme exacte de l'impul
sion d'excitation ne pose pas de problèmes particuliers et peut ~tre
effectuée par intégration numérique32 . La correction que nous avons
retenue s'avère cependant très suffisante.
Le cas des mesures à l'aide de l'analyseur multicanaux
(fluorescence rasolue en temps) peut se traiter de manière similaire.
La largeur des canaux est connue avec tme très bonne précision
(N 5.10-3 , après le temps nécessaire à la stabilisation de la ligne
à retard qui est de l'ordre de trente minutes). On peut donc remonter
à la fréquence instantanée n, qui peut gtre ·considérée comme constante
à une très bonne approximation, la largeur d'un canal étant nettement
inférieure à la constante de temps de décroissance. La correction dans
le cas des états de Rydberg n'a jamais dépassé 10~ (ce pour les pre
miers canaux).
V.5.5. Erreurs dues au dépouillement des données.
Le dépouillement de nos données (points expérimentaux affec
tés de barres d'erreurs verticales et, éventuellement, horizontales,
calculées d'après les considérations données ci-dessus) a cons±sté
essentiellement à effectuer des ajustements par moindre carré à des
formes analytiques très simples (y= a.x + b, y= b a.x par exemple), x+c formes que nous avons données lors de la discussion des méthodes de
mesure. Ces dépouillements sont très classiques et nous ne reviendrons
pas sur leur détail. Le seul point que nous voudrions discuter ici est
celui de la précision obtenue sur le, (ou les) paramètre(s) ainsi
déterminé(s).

- 131 -
Prenons le cas simple de la régression linéaire. Le calcul
de l'erreur sur la pente de la droite, effectuée par les méthodes
classiques de la statistique8 , conduit bien souvent à une erreur fai
ble. Ce résultat nous semble valable quand les erreurs sur les points
sont statistiques, ce qui est le cas par exemple pour la détermination
de la durée de vie d'un niveau à partir du tracé semi-logarithmique
Log(N) = f(t). Par contre quand les erreurs sur les points expérimen
taux incluent de nombreux phénomènes (transmission optique, absorption,
mesure de pression ... ) l'erreur résultante (sur une section efficace
par exemple) semble physiquement peu réaliste. Dans ce dernier cas
nous avons préféré à la méthode purement statistique une estimation
graphique de l'erreur finale qui, si elle para.!t plus subjective, nous
semble beaucoup plus réaliste du point de vue physique.
Nous avons adopté la mgme position dans les cas d'ajustements
à des formes plus complexes qu'une droite simple en faisant tracer·des
réseaux de courbes paramétrées pour visualiser l'influence des diffé
rents paramètres et en déduire l'erreur relative sur le résultat final
ce en fonction des barres d'erreurs individuelles préalablement déter-. ,
minees.
v.5.6. Erreurs systématiques.
Les deux causes principales d'erreur systématique que nous
discuterons concernent d'une part la validité des modèles utilisés et
le choix des coefficients d'Einstein, ce dernier ne pouvant affecter
que les mesures de fluorescence non résolue en temps.
Nous avons déjà discuté certaines conditions de validité des
modèles utilisés lors de leur description. Nous donnerons au chapitre
suivant des renseignements plus spécifiques aux situations expérimen
tales que nous avons rencontrées. Remarquons que seuls des modèles très
simples, à nombre très li.mité de paramètres ont été utilisés. L'effort
expérimental a donc porté comme nous le verrons sur la vérification
de l'absence de phénomènes parasites pouvant perturber de manière no
table ces modèles simples. Il nous semble donc peu probable qu'une
erreur systématique notable puisse provenir des modèles employés pour
le dépouillement des données.

- 132 -
Une cause importante d'erreur systématique dans le cas des
mesures de fluorescence non résolue dans le temps provient du choix
des coefficients d'Einstein. Dans le cas des mesures de transfert il
faut conna.1tre d'une part le rapport des coefficients d'Einstein cor
respondant au..~ deu..~ raies observées (évaluation du rapport des popu
lations; cf Eq. (E32)) et d'autre part la durée de vie du niveau
correspondant à la fluorescence sensibilisée (~ 2 = 1/A2
cf. Eq. (EJJ)).
En ce qui concerne la mesure du quenching, seule la connaissance de
la durée de vie du niveau est requise (Eq. (E35)) (Les coefficients
d'Einstein sont aussi nécessaires au calcul de la réabsorption éven
tuelle des raies observées, mais celle-ci ne représente, en général,
qu'une correction et le problème du choix des coefficients A ne pré
sente pas ici le caractère très fondamental que nous venons de men
tionner). En général les durées de vie des états excités peuvent $tre
calculés à l'aide de méthodes théoriques assez simples (méthode de
type Bates Damgaard par exemple33 ) qui fournissent de bonnes estima
tions. De plus des déterminations expérimentales précises sont sou
vent disponibles (tel était le cas des niveaux 7P du césium). Par con
tre la mesure de la détermination théorique précise de coefficients A
relatifs à des transitions données constitue un problème difficile.
Ainsi les valeurs théoriques présentent souvent une assez grande dis
persion (nous en verrons des exemples) et il peut en résulter des
erreurs systématiques impor~antes sur l'évaluation des rapports de
population à partir de (E32). Notons qu'il est parfois possible de
justifier expérimentalement le choix d'un système particulier de coef
ficients A, en opérant à pression suffisamment élevée de gaz pertur
bateur pour mettre à l'équilibre les deux niveaux i et, j. Le rapport
des populations est alors donné par le principe du bilan détaillé
(Eq. (E9)) et la mesure de (R .. ) fournira alors le rapport iJ eq
A.obs/A_obs (Eq. (EJ2)), Mais, dans tous les cas, il importe de bien i J
spécifier les coefficients choisis en justifiant, si possible, de ma-
nière expérimentale les raisons de ce choix.

- 1 JJ -
REFERENCES DU CHAPITRE V
1 - M •. PIMBERT, J. Phys. (Paris), JJ, 331 (1972).
2 - A. BURGESS et M.J. SEATON, Mon. Not. R. Astron. Soc., 120,
121 ( 1960) .
3 - I.C. PERCIVAL, Nucl. Fusion., f, 182 (1966).
4 - H.E. SARAPH, Proc. Phys. Soc., 83, 763 (1964).
5 - A. CORNEY, Adv. in Electronics and Electron Physics, 29, 115 (1970).
6 - A. GRINV.ALD et I.Z. S'l:EINBERG, Anal. Biochemistry, 22.,, 583 (1974).
7 - R.D. DYSON et I. ISENBERG, Biochemistry, 10, 3233 ( 19_71).
8 - B. VALEUR et J. MOIREZ, J. de Chimie Physique, 70, 500 (1973).
9 - D. G. GARDNER, J . C . GARDNER, G. LAUSH et W. W. MEINKE ,
J. Chem. Phys.,2!_, 978 (1959).
10 - A. GAFNI, R.L. MODLIN et L. BRAi.'ID, Biophys. J., !2., 263 (1975).
11 - P.R. BEVINGTON, Data Reduction and Errer Analysis for the Physical
Sciences, Mc Graw-Hill (1969).
12 - D.J.G. IRWIN et A.E. LIVINGSTON, Camp. Phys. Comm., 1, 95 (1974).
13 - D. YORK, Can. J. Phys., 44, 1079 (1966).
14 - J.H. Wil,LIAMSON, Can. J. Phys., 46, 1845 (1968).
15 - L.R. PENDRILL, J. Phys. E10, L469 (1977).
16 - T.F. GALLAGHER, S.A. EDELS'l:EL.~ et R.M. HILL, Phys. Rev., A15,
1945 (1977).
17 - D. S'l:ERN et M. VOLMER, Z. Physik, 20, 183 (1919),
18 - M. GROSS, Thèse Jème cycle, Paris (1975).

- 134 -
19 - F. GOUNAND, M. HUGON, P.R. FOURNIER et J. BERLAl.'IDE,
J. Phys. B12, 547 (1979).
20 - F. GOUNA.i.'ID, Polarisation de la lumière de fluorescence,
Rapport Interne CEA (1972).
21 - M. DUMONT, Thèse, Paris (1971).
22 - L.R. PENDRILL et G.W. SERIES, J. Phys. ~' 4049 (1978).
23 - R. LUYP.AERT et J. VAN CRAEN, J. Phys. B1 O , __ J627 (1977).
24 - E.L. ALTM:.Ai.~ et Y.V. EVDOKIMOV, Opt. Spectrosc. 28, 431 (1970).
25 - J.S. DEECH et W.E. BAYLIS, Can. J. Phys., 49, 90 (1971).
26 - J.S. DEECH, R. LUYP.AERT, et G.W. SERIES, J. Phys. B8, 1406 (1975).
27 - A. GALLAGHER et E.L. LEWIS, J.,Opt. Soc. Am. 63, 864 (1973).
28 - J.L. CURTIS et P. ERMAN, J. Opt. Soc. Am. 67, 1218 (1977).
29 - J.L. LEMAIRE et F. ROSTAS, J. Phys. B 4, 555 (1971).
JO - Y.V. EVDOKIMOV, Opt. Spektrosk. 24, 832 (1968).
(Opt. Spectrosc. 24, 448 (1968).
J1 - V.I. GOLDANSKII, A.V. KUTSENKO et M.I. PODGORETSKII,
Counting statistics of nuclear particles, International mono
graphs on advanced mathematics and physics, Hindustan publis
hing corporation, Delhi (1962).
J2 - M.Y. PERRIN, Thèse de Jème cycle, Paris (1979).
JJ - D.R. BATES et A. DAMG.AARD, Phil. Trans. R. Soc., .A.242, 101 (1949).

Chapitre VI
RES1JLTATS ET DISCUSSION.
VI.1. INTRODUCTION.
Nous venons d'exposer de manière détaillée différentes appro
ches théoriques et expérimentales au problème très général des colli
sions inélastiques aux énergies thermiques. Le but du présent chapitre
est de présenter de manière aussi cohérente que possible un ensemble
de résultats concernant ce type de collisions en faisant ressortir les
principales idées physiques que seule une confrontation théorie-expé
rience permet de dégager. Dans cette optique nous nous attacherons
moins à une description phénoménologique des résultats obtenus qu'à
une étude comparative de ceux-ci avec les données expérimentales obte
nues par d'autres équipes et surtout à une confrontation avec les ré
sultats fournis par les approches théoriques disponibles.
Le chapitre se subdivise ainsi : une première partie sera
consacrée aux niveaux faiblement excités tandis que la seconde traite
ra des états de Rydberg (un bref exposé des résultats relatifs aux
phénomènes de superradiance précédera la description et la discussion
des résultats proprement collisionnels) ; enfin une conclusion synthé
tique terminera le chapitre. Pour chaque partie nous indiquerons
d'abord les motivations principales de notre travail avant d'insérer
les publications correspondantes (dans le cas des états de Rydberg
nous donnerons les résultats des mesures concernant les états S du ru
bidium, ceux-ci n'ayant pas encore été publiés). Nous inclurons quel
ques compléments tant expérimentaux que théoriques, que nous n'avons
pu insérer, faute de place, dans les publications et qui nous parais
sent nécessaires à la bonne compréhension de l'exposé, ainsi que la
comparaison avec des résultats expérimentaux ou théoriques parus après

- 136 -
ces publications. Une conclusion donnera enfin les idées physiques es
sentielles que nous avons pu dégager soit sous forme de rappel, soit
à l'aide de résultats, ou d'idées, postérieurs aux publications.
VI.2. ETUDE D'UN GROUPE DE NIVEAUX FAIBLEMENT EXCITES (7P-6D) DU
CESIUM1
' 2 .
VI.2.1. Présentation du travail.
Les principales motivations du présent travail ont été men
tionnées dans l'introduction. Nous nous contenterons de les rappeler
brièvement tout en complétant certains points.
Le choix du groupe de niveaux étudiés (7P1; 2 , 3; 2 - 6D3; 2 , 5; 2 du césium) a été dicté par des considérations aussi bien expérimenta
les que théoriques. Il semblait tout d'abord possible de peupler sélec-o
tivement les deux compos~tes (6s112 ~7P112 , À= 4593 A et
6s112 __.7p312
, À= 4555 A) du doublet 7P à l'ai_de d'un laser à colorant
pulsé utilisant Wle coumarine appropriée, bien qu'aucun laser cominer
cial ne fut disponible à ces longueurs d'onde. Une étude menée conjoin
tement avec le laboratoire de Physique Moléculaire de l'école Polytech
nique (M. Meyer) aboutit à la construction du laser 1 (décrit au cha
pitre III) dont nous avons déjà donné les principales caractéristiques.
Le groupe de niveaux retenu présentait l'avantage d 1$tre bien isolé de
tous les autres niveaux dans le spectre énergétique du césium (l'écart -1 avec le niveau le plus proche, 8s112 , est en effet de 1700 cm , ce
qui est très supérieur à l'énergie disponible lors de la collision
(N 400 cm-1 )). La possibilité de n'avoir à prendre en compte que quatre
niveaux dans le dépouillement des données présentait un intér~t sup
plémentaire. Enfin et surtou~ les premiers calculs de J. Pascale et
J. Vandeplanque montraient nettement l'existence de couplages variés
entre ces niveaux, particulièrement importants dans le cas des gaz ra
res lourds. En résumé, ce groupe de niveaux sembla~t particulièrement
adapté à une première confronta,tion détaillée entre théorie et expé-
rience une meilleure connaissance des interactions mettant en jeu
des états fortement excités en était attendue. Notons enfin, du point
de vue expérimental, qu'une telle expérience représentait, à notre con
naissance, la première étude de collisions à énergie thermique utili
sant un laser à colorant accordable conune source d'excitation.

- 137 -
Les résultats préliminaires, relatifs au transfert intramul
tiplet (7P1; 2 ~ 7P3
; 2 ) furent publiés en 19731
et l'étude complète
du groupe (7P-6D) en 19752 . Cette deu..tième publication est incluse
dans le présent travail.
VI.2.2. Article 1.

- 138
PHYSICAL REVIE\\" A \' 0 L l" '.\f E 11 . ~ l' '.\1 B E R 3 l\lARCH 1975
Inelastic collisions involving excited cesium atoms at thermal energies
J. Cuvellier, P. R. Fournier, F. Gounand, J. Pascale, and J. Berlande C.intre d'Etudes Nucléaires de Saclay, Service de Physique Atomique-BP No.2, 91190-Gif-sur-Yvelle, Fra.,rce
(Received 29 July 1974)
Cross sections for the 7 P 112 - 6D lll and 7 Pm - 6D 312 transitions, induced at thermal energies by cesium-rare-gas collisions, have been measured over the temperature range from 420 to 615 K. The values of the cross sections are found to be surprisingly high when considering only the energy defect of the levels involved. Moreover, the cross sections for Cs-He collisions exhibit very different behavior than those corresponding to heavier rare gases. A discussion with the aid of theoretical adiabatic potenrial curves explains the main features of the experimental results. Theoretical calculations of the cross sections have been carried out for some cases, and give results in satisfactory agreement with the cxperimental ,·alues.
1. INTRODUCTION
Recent experimental and theoretical work on inelastic atomic collisions at thermal energies involving highly excited atoms is of great interest in efforts to obtain a better knowledge of interatomic forces. l-3
Numerous experiments4 have been performed on the transfer of excitation between the finestructure levels of the first 2P doublet of the alkali metals when they undergo a collision with a raregas atom, i.e.,
A(2Pl 12 )+X ! A(2P 312)+X-AE, Q'
where A is an alkali-metal atom, AE the finestructure splitting, Xis a rare-gas atom in its ground state, and Q and Q' are the cross sections associated with these processes. The general feature clearly shown by the experimental results is that Q and Q' are directly related to the magnitude of AE. However, for a better understanding of these inelastic processes a study of higher excited levels was necessary. For this reason finestructure transitions in the second and third 2P doublets of cesium, induced by collisions with rare-gas atoms, have been studied in our laboratory .1
•5 The results have shown that the absolute values of the cross sections depend not only on the energy defect AE. but also on the position of the doublet with respect to the other levels in the cesium energy diagram (Fig. 1 ). It has been suggested that coupling with neighboring levels might occur.3
A reliable theoretical computation of these collisional cross sections has been done only for the first resonant doublet of the lightest alkali metals. In that case the simplified potential-energy curves used in the calculations seem to be correct in the range of internuclear distances where the transitions are the most probable. s-e
11
No agreement was obtained with similar calculations for collisions involving higher excited states. 3
In view of these results it is obvious that the theoretical calculation of the cross sections requires a fairly good knowledge of the adiabatic potential curves of the alkali-rare-gas atom pairs. In arecent calculation of the alkali-rare-gas adiabatic potentials the importance of coupling between neighboring levels, giving rise to structure in the potential-energy curves, was clearly pointed out. 9 •
10 An examination of the curves suggests, in particular, that an experimental study of extramultiplet collisional transitions between the 72P and 62D levels of cesium is of interest. The potential curves asymptotically correlated to these l~vels show a pronounced structure due to coupling between these levels.
The possibility, for the first time, of comparing theoretical predictions with experimental results has led us to complete our previous investigation of intramultiplet transitions concerning the second resonant doublet of cesium with the experimental study of the (72P - 62 D) transitions. In this article we report on the experiment and discuss the results with the help of the potential-energy curves.
Il. DESCRIPTION OF EXPERIMENT
A. Method
We are concerned with the collisional process Q-•
Cs(j)+X ,d; Cs(i)+X-~E, (1) QiJ
where Xis a rare-gas atom in its ground state and ~E is the energy defect between the j and i excited levels. The cross section for the transfer of excitation !rom j to i is denoted by Q ;i ; the reverse by Qi;. Figure 2 shows the levels involved in the process: In our case the in-going channel is either the 72P 112 or 72P 312 lev el. The population of these
846
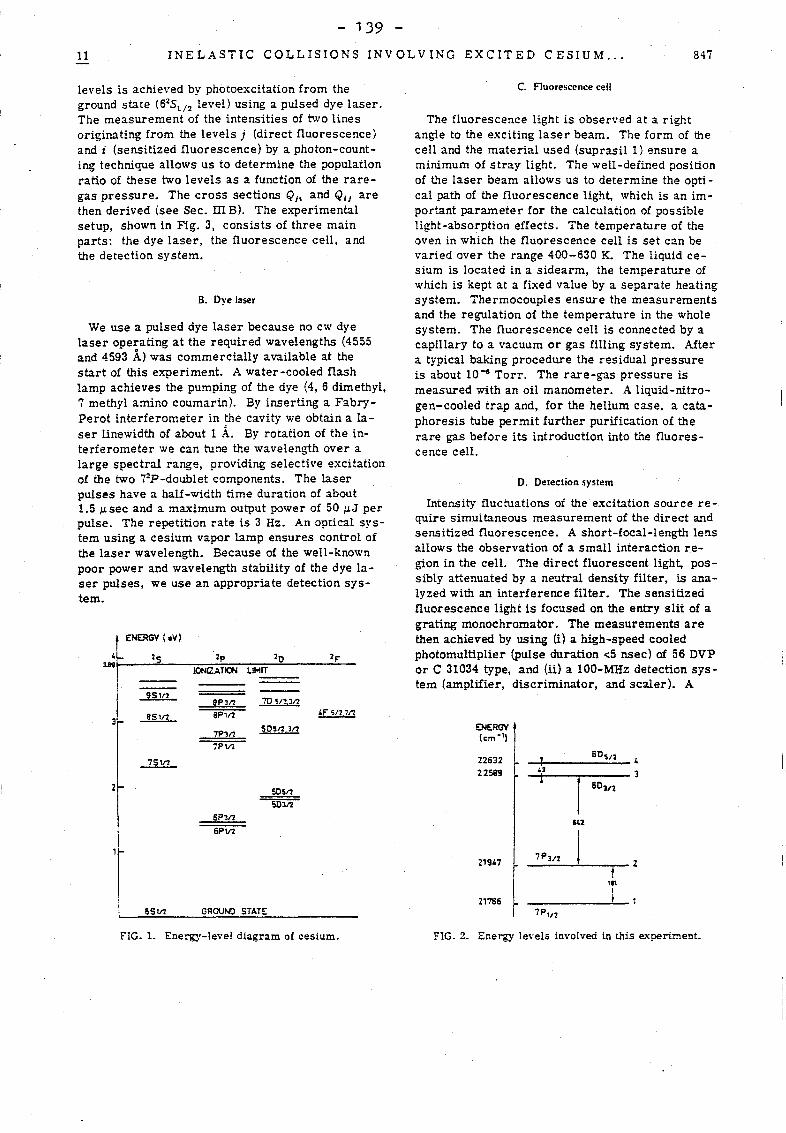
- 139 -
11 INELASTIC COLLISIONS INVOLVING EXCITED CESIUM ... 847
levels is achieved by photoexcitation from the ground state (625112 level) using a pulsed dye laser. The measurement of the intensities of two lines originating from the levels j (direct fluorescence) and i (sensitized fluorescence) by a photon-counting technique allows us to determine the population ratio of these two levels as a function of the raregas pressure. The cross sections Q,; and Q;J are then derived (see Sec. m B). The experimental setup, shown in Fig. 3, consists of three main parts: the dye laser, the fluorescence cell, and the detection system.
B. Dye laser
We use a pulsed dye laser because no cw dye laser operating at the required wavelengths (4555 and 4593 Â) was commercially available at the start of this experiment. A water-cooled flash lamp achieves the pumping of the dye (4, 6 dimethyl, 7 methyl amino coumarin). By inserting a FabryPerot interferometer in the cavity we obtain a laser linewidth of about 1 Â. By rotation of the interferometer we can tune the wavelength over a large spectral range, providing selective excitation of the two 72P-doublet components. The laser pulses have a half-width time duration of about 1.5 µ sec and a maximum output power of 50 µJ per pulse. The repetition rate is 3 Hz. An optical system using a cesium vapor lamp ensures control of the laser wavelength. Because of the well-known poor power and wavelength stability of the dye laser pulses, we use an appropriate detection system.
ENERGV (eV)
u:b 25 2p 2o 2F
IONCZATION LJMIT -- --9S111 --
8P1n 70 Sil 3/?
I -1fila_ aP111 4F Sil 7rz
_m,a. 6Dsn.1n
7PVÎ
~
~ ~
SPl/'?
6PV2
ss,n GROUNO STATE
FIG. 1. Energy-level diagram of cesium.
C. Fluorescence cell
The fluorescence light is observed at a right angle to the exciting laser beam. The !orm of the cell and the mate rial used (suprasil 1) ensure a minimum of stray light. The well-defined position of the laser beam allows us to determine the optical path of the fluorescence light, which is an important parameter for the calculation of possible light-absorption effects. The temperature of the oven in which the fluorescence cell is set can be varied over the range 400-630 K. The liquid cesium is located in a sidearm, the temperature of which is kept at a fixed value by a separate heating system. Thermocouples ensure the measurernents and the regulation of the temperature in the whole system. The fluorescence cell is connected by a capillary to a vacuum or gas filling system. After a typical baking procedure the residual pressure is about 10~ Torr. The rare-gas pressure is measured with an oil manometer. A liquid-nitrogen-cooled trap and, for the helium case, a cataphoresis tube permit further purification of the rare gas before its introduction into the fluorescence cell.
D. Detection system
Intensity fluctuations of the excitation source require simultaneous measurement of the direct and sensitized fluorescence. A short-focal-length lens allows the observation of a small interaction region in the cell. The direct fluorescent light. possibly attenuated by a neutral density filter, is analyzed with an interference filter. The sensitized fluorescence light is focused on the entry slit of a grating monochromator. The measurements are then achieved by using (i) a high-speed cooled photomultiplier (pulse duration <5 nsec) of 56 DVP or C 31034 type, and (ii) a 100-MHz detection system (amplifier, discriminator, and scaler). A
ENERGV (cm-1)
22632 ~ sns,2 4
22589 43 3 1
1 6D1n
5'2
1
21947 L 7P1n l 2
1'1 1
21756 ~ + 7?112
FIG. 2. Energy levels involved in this experiment.

- 140 -
848 J. CUVELLIER et al. 11
trigger system opens the counter during a time greater than the light-pulse duration, but sufficiently short to ensure a \1 ery low dark-noise level. Preliminary experiments permit the calibration of the whole system.
Ill. DATA ANALYSIS
The cross sections for excitation transfer are obtained after a two-step procedure. First, from the experimental results of the intensity ratio of sensitized to direct fluorescence light the ratios of the i- to j-Jevel populations are deduced as a function of the rare -gas pressure. Then the cross sections are àetermined from the population ratios by using a four-level coupling model.
A. Determination of population ratios ·
The counting measurements are made in pulsed mode, with a pulse-time àuration greater than the lifetimes of the studied excited levels. The counting apparatus operates during a time -;- which covers completely the excitation light pulse. If 'Jl(j) is the instantaneous population of atoms lying in the j level. N(j) = J/ 'Jf.(j)dl represents the total number of atoms lying in the j level àuring the laser pulse and Ai_.N(j) is the total number of photons spontaneously emitted in the transition from the level j to the level k (where Ai-• is the Einstein coefficient). The detection system, tuned to the j- k transition, gives a counting signal C(j) proportional to A
1_,,N(j). In the same way, we ob
tain for the i - l transition a counting signal C(i) proportional to A 1_ 1N(i).
We can write
N(i) A;-• aC(i) N(j) = A
1_
1 Xi yii i3C(j)' (2)
Oven
where Y ii represents the relative efficiency of the whole detection apparatus, taking into account the attenuation of the beams, the geometical arrangement of the two detection channels, and their relative sensitivity. X1 takes into account the influence of light absorption. The quantities a and /3 are corrective factors for the evaluation of the real counting rate when the dead time of the electronic apparatus is taken into account (the dead time has been previous ly found to be 10 nsec ).
The determination of Yu _is achieved by appropria te experiments. Measurements are performed with the sidearm containing liquid cesium at a temperature of 90 °C. This corresponds to a cesium pr~ssure of about 3 x 10-4 Torr [we use Nottingham' s formula11 j. Because the optical path of the fluorescence light is about 3 mm, the absorption of the direct fluorescence (72P 112 - 62S112 or 72P 312 - 62S112 transition) is not negligible as it is in the case for the sensitized fluorescence (62 D312
- 62P 112 transition). We calculate the X, coefficient using the results given by Refs. 12-14, including, when necessary. in our calculation the pressure broadening of the cesium lines b~· the rare gas. The calculated value of Xi does not differ from unity by more than 20%.
We used in our calculations the A values given by Warner. 15 The choice of A values will be discussed later (see Sec. m C 4 ). The measurements of the rare-gas pressure, using an oil manometer, need correction because of thermal transpiration, the manometer not being at the same temperature as the fluorescence cell. Knowledge of the flow regime (molecular, viscous, or intermediate) allows us, by an interpolation method, to determine the corrective factor under the experimental conditions.16'17
ûJ _ Monochromator
· Splitter ___ ~ ----7------~ 1 /1 / I ·
--1
\ 1 • -L Oens1ty 1 r,tter -,-1 o.,,,1y J...rn.., , / \ 1 f&t:;Cryostat
~ 1 / 1 / ?hotomultiplier Cryos ..:i__1,
F"aroday cage
H_f'. Fïlter
1 ' l" • 1 ?hotomult1p 1e,. 1 1 1 1
---}'
Amplifier
Sccler
Printer
s· ,gnol • generator
FIG. 3. Experimental setup.

- 141
11 INELASTIC COLLISIONS INVOLVING EXCITED CESIUM ... 849
B. Derivation of cross sections
The results of the previous experiments concerning intramultiplet transitions induced in alkali-rare-gas collisions are, in general, interpreted using a two-level scheme (i.e., j and i levels of Eq. (1 )]. This scheme is well justified in the case of the first resonant doublet but must be modified in the case of higher excited levels because of the influence of other neighboring levels on the population ratios. We will describe here a more general mode! which allows us to analyze the experimental data when one has to consider the influence of a great number of levels. We will then apply the results obtained to the case of the 72P 112 • 3 ; 2
and 62 D312 , 512 levels invol ved in this study (the se four levels are well separated from the others in the energy diagram of cesium).
Let us consider a large number of atomic levels i, j, . . . . The following processes 1re supposed to couple these levels: .
(a) Collisional processes. These are characterized by a frequency K;; with K;; =N,ïJQ;;, where N
1 is the rare-gas pressure, v the mean velocity
of the relative motion of the colliding particles, and Q;; is the Maxwellian averaged cross section for the j- i collisional transition considered.
(b) Radiative processes. These are characterized by the Einstein coefficients A;; (some of them equal to zero).
(c) Photoexcitation processes. These are denoted by <fi;;· These terms represent radiative pumping due to the cell considered as a black body.
We can write the population balance of the level i in the form
L N(p )(Kp; +Api+ 'Pp;)= N(i) L (K;, +A;,.+ 'P;,) , - i , - i
= N(i)(K; +A;+ 'P;),
with
LK;p=K;, LA;,=A;, L<l>;,='P;, p•i p•, paj
N(i) and N(p) are, as defined previously, the integrated populations of levels (i) and (p) during the measurement time r.
Denote the photoexcited level by j, the level corresponding to the sensitized fluorescence by i, and the ratio N(k)/N(l) by R.,. We can write, using the preceding formula,
R;; = L Rp;ap;, , ... i
at>, = (Kp; +A.,;+ 'P.o; )/(K; +A; + </>;).
(3)
(4)
Note, finally, that the principle of detailed balancing can be written
(R;;)<q=(Kji) =Q;; =gie<-U;;l•Ti, (5) K;; ,4 Q;; g;
where g; and g; are the statistical weights of the i and j levels, 6.E; 1 is the energy defect between these two levels, and the eq subscript indicates that the corresponding quantities are defined at Boltzmann equilibrium at the gas temperature T.
Applying this model to the four levels 72P 112 , 312
and 62D3 ; 2 , 5; 2 and using the notation of Fig. 2, Eq. (3) can be written
R31 = 0:13 + R21 Ci23 + RH Ct.43 •
Since R 41 = R43 R31 , one obtains
R31 = (et.13 + ~1 Ct.23 )/(1 - R130!43)
and, finally, using Eq. (4),
R _ (Kp + 'Pp) +;&1 (K.z;i + ,P,3) J1 - K3 -R13Ku+A3 .
A similar expression can be obtained for R32' The experiment determines R31 (and R 32 ) as a
function of the rare-gas pressure N1 (note, in particular, that q:> 13 = R31 A 3 when N, = 0). Previous measurements (5) determine ~ 1 (and R12 ) as a function of Nr- The above formula can now be written
_(N,I'Q13+</)13)+~1(N:ïiQ:i3+<.tl23) () R31- N,v(Q3-R13Q43)+~ ' 6
where Q 3 is the total collisional depopulation cross section of level 3.
The preceding analysis omits the influence of (Cs-Cs) collisions. Strictly speaking, the collisional frequency K;; must b.e written
K1; =N1ûQ;; +N,,vr,QJ; r, Nr, is the cesium density, ïJ,, is the mean velocity of the relative motion of two colliding cesium atoms, and Q;;,, is the Ma.'<".vellian averaged cross section for the j - i transition induced by (Cs-Cs) collisions. In our experiment the cesium density N,,is kept at a fixed value. The effects of (Cs-Cs) collisions in Eq. (6) are (i) introduction into the numerator of two constant terms which can be regarded as included in the experimental values of cp 13 and rt, 23 , and (ii) introduction of an additive term to A3 in the denominator. One can see easily that this term is quite negligible if one refers to the value of N(/"'1013 cm -3
).
We now cons1der the analysis of the experimental data using Eq. (6). Because of experimental di.fficulties in determining the population of level 4 we do not measure R13 • However, since the energy defect t:,.E34 is small (43 cm -1
), one can consider that Boltzmann equilibrium between these two levels is rapidly obtained. So, using Eq. (5) we can
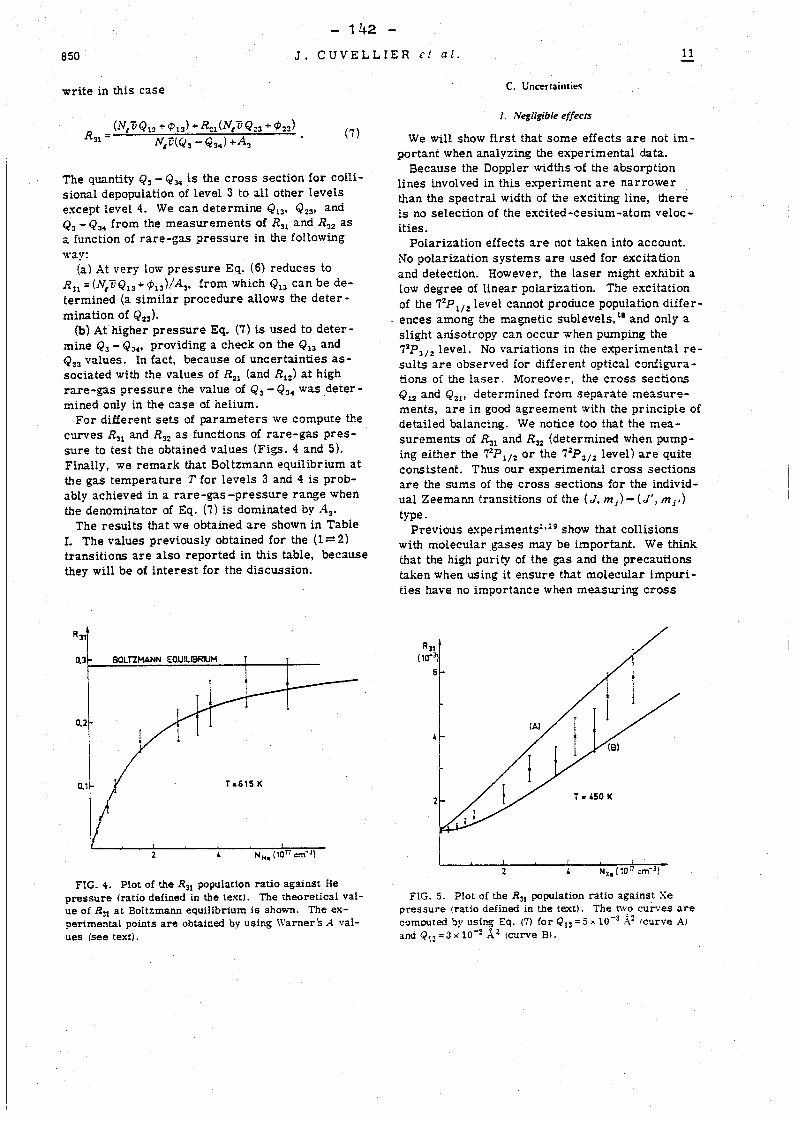
- 142 -
850 J. CUVELLIER et al. 11
write in this case
(N,TIQ13 +4>13) +R21(N/jQ23 +4>23) R31= N,ï'(Q3-Q34)+A3
(7)
The quantity Q3 - Q34 is the cross section for collisional depopulation of level 3 to all other levels except level 4. We can determine Q13, Qw and Q
3 - Q34 from the measurements of R31 and R32 as
a function of rare-gas pressure in the following way:
(a) At very low pressure Eq. (6) reduces to R
31 =(N,vQ13 +<P13 )/A3• from which Q13 can be de
termined (a similar procedure allows the de ter -mination of Q23 ).
(b) Athigher pressure Eq. (7) is used to determine Q3 - Q34, providing a check on the Q13 and Q13 values. In fact, because of uncertainties associated with the values of R21 (and R 12 ) at high rare-gas pressure the value of Q3 - Q34 was . de ter -mined only in the case of helium.
For different sets of parameters we compute the curves R31 and R 32 as functions of rare-gas pressure to test the obtained values (Figs. 4 and 5). Finally, we remark that Boltzmann equilibrium at the gas temperature T for levels 3 and 4 is probably achieved in a rare-gas-pressure range when the denominator of Eq. (7) is dominated by A 3 •
The results that we obtained are shown in Table I. The values previously obtained for the (1=2) transitions are also reported in this table, because they will be of interest for the discussion.
R31
0.3~ BOLTZMANN EQUILIBRl.JM î l
0.2
"'[ I Ta61S K
V 1 1 1 2 4 N "• ( 1011 cm·lJ
FIG. 4. Plot of the R31 population ratio against He pressure (ratio defined in the textl. The theoretical value of R31 at Boltzmann equilibrium is shown. The experimental points are obtained by using \Varner's A values (see text).
C. Uncertainties
/. Negligible effects
We will show first that some effects are not important when analyzing the experimental data.
Because the Doppler widths -of the absorption Unes involved in this experiment are narrower . than the spectral width of the exciting Une, there is no selection of the excited-cesium-atom velocities.
Polarization effects are not taken into account. No polarization systems are used for excitation and detection. However, the laser might exhibit a low degree of linear polarization. The excitation of the 72P
112 level cannot produce population differ
ences among the magnetic sublevels, 18 and only a slight anisotropy can occur when pumping the 72P 312 level. No variations in the experimental results are observed for different optical configurations of the laser. Moreover, the cross sections Q12 and Q217 determined from separate measurements, are in good agreement with the principle of detailed balancing. We notice too that the measurements of R31 and R 32 (determined when pumping either the 72P 1 n or the 72P 312 lev el) are quite consistent. Thus our experimental cross sections are the sums of the cross sections for the individual Zeemann transitions of the ( J, mi)- ( J', mi,) type.
Previous experiments1•19 show that collisions
with molecular gases may be important. We think that the high purity of the gas and the precautions taken when using it ensure that molecular impurities have no importance when measuring cross
R31
(10-3)1
6
2
4 Nx. ( ,on-~m"l)
F1G. 5. Plot of the R31 population ratio against Xe pressure (ratio defined in the text). The two curves are computed by using Eq. (7) for Q13 = 5 x 10-3 A2 tcurve A) and Q13 =3xl0-2 Â 2 (curve B).

11
.:; > ., .. r? "' ., :S -0
~ ~ i5.. 8. CJ
-,::,
~ ~ "' 0 CJ
c::
3 ., -5 "' ë: ., u: ., ... C. ., ... ...
r::::.-;:; <:>
w «
,:
"' ,:
~ CJ ., "' "' "' 0 ...
C,.)
..; r.,J ,..J
~ < ,...
., X
... :t!
:. <
., z
.Sl
,:
:§ .; C: d :. ,...
- 143 -
INELASTIC COLLISIONS INVOLVING EXCITED CESIUM ... 851
1
N 1 o_
1 1 ,-1 1 0 0 X 0 ...-f..,... M..-X X VI X ô ô . â;
. . r, . .... .... f Q
+l+l~+I tôt--~ •• li) •
E !!; M ~
N N N 1 1 1 00 0 ....... -. -X X I X -- 0 -0 t'- .... t.Q
N ...; ~ 0 +1 +1 VI +1 .... r- .... . . . St~
.. 1 1 1 0 o .. 0 ......... .... ~.! ~ !. ~ e',l X N 0 Q N...; +I ~ 'Il +I "'0
==
1 1 00 ........
r-
~
~
1 0 X X
~~1~ C O _. N
+I +I -~ +I (X)~ e cc
1 1 00 ........ X X 00 00 i'.ô
~NO O,...; +I +I +I +I +I N .._ a.r.l C- 0 ........... --~~~
N
r::::.-;;
~ !:.: N -:..:;,
e,-;:,;:,t
;:~~~ i f t t t f3 N N""' N-
::::-;;::::-;; 1 Q., Q., o.. 0.., ~ e- c- t- r:- ~
~ 0 .,., ..,.
1 f.,
Q <:>
w
1 1 0 0 .... .... X X ~~ 00 +I +I .... r-
t!. .::
... 1 -o, .... 0 X,... r- X VI ii: .....
'i' +I 0 L'O ..... MC
- 1 1 0 ~ .... X X
Nê7i'N~N +1+1-tt+l+I C"': """ c:o a, t:) _ _. __ _
N .... ~
- N N NI(',:;) ;:, :::: ;:, -;; t ~;:Sfil N
f t t t 9° N N - N(.; ::::;:;::~-
:...o..°"' o.. '-C-- t'"- ~ t'- ~
~ .,., ;;
f.,
~ w
sections greater than 10-19 cm2• Thus only the
value obtained for Q 23 in the case of neon could be significantly affected by residual molecular impurities.
The model used to determine the cross sections (see Sec. III B) assumes that no radiative cascade effect !rom upper levels exists. Ions, possibly created by the excitation process, could produce after recombination atomic excited levels. We verified by looking at transitions coming from upper levels that no cascade effect appeared under our experimental conditions.
2. Causes of experimenta/ e"ors
The experimental errors corne essentially from the determination of the ratios N(i)/N(j) as given by Eq. (2) and the densities N: and Ne,.
(i) The uncertainty associated with the real counting rate is between 3 and 15%, taking into account a dead-time correction and statistical error. The calculation of the effect of light absorption has an uncertainty of 20%, mainly due to the uncertainty in the Cs density value. This absorption effect contributes only 20% in the determination of the X; value. So we evaluate the error in X; to be less than 5%. The coefficient Yi; is more difficult to determine with accuracy. Its determination requires three steps: (a) The measurement of how light intensity is split between the sensitized-fluorescence and the direct-fluorescence channels; this ratio depends upon the geometrical arrangement of the two channels. The resulting uncertainty is the same as for the counting rate (about 1Cl%). (b) The determination of the sensitivity as a function of wavelength of the channels, using a tungsten lamp. The resulting uncertainty does not exceed 5%. (c) The calibration of the neutral density filter used in the path of the direct fluorescence light. The uncertl.inty that results is less than 3%. Thus the determination of Yu is achieved with about 15% uncertainty. One can consider that the population ratio corresponding to an experimental point is known with 20-30% accuracy for a given set of A coeificients.
(ii) The pressure measurements are more accurate. The determination of the rare-gas pressure N1 , using a cathetometer, is made with great accuracy, and the thermal-transpiration correction is sufficiently elaborate to allow the determination of N1 with less than 5% uncertainty. A diffusion time sufficient to ensure pressure homogeneity in the measurement cell is necessary. An accurate determination of the cesium density is not required in our experiments as the rare-gas pressure determines the population ratios. The temperature regulation of the sidearm containing liquid cesium

- 144 -852 J. CUVELLIER et al. 11
ensures sufficient stability of the cesium density. However. the absolute value of Ne, is not precisely known, leading, as noted previously, to an uncertainty in the determination of the X 1 coefficient.
3. Estimation of errors concerning Q 13 , Q 23,' and
Q3-Q34
We use the following procedure. A set of curves of R13 (and Rz3 ) is plotted as a function of rare-gas pressure, for different values of the cross sections (Eqs. (6) and (7)]. By comparing these curves with the experimental points and their cor -responding error bars, we determine the maximum error compatible with our measurements. This implies the validity of the mode! we have used to derive Eqs. (6) and (7). This point has been discussed in a previous section.
4. Cho/ce of Einstein coefficients
We now discuss the choice of Einstein coefficients. Knowledge of the se coefficients is· required in each step of the interpretation of the experimental results: for the determination of the population ratios, the calculation of the light absorption correction, and the derivation of the cross sections. There are large discrepancies between the A values given by different authors (Table II). Consequently, systematic errors can result from the choice of a particular set of A values. It is therefore necessary at this point to state the set of A values we use and the reasons for our choice.
We discarded first the A coefficients given by Heavens20 because they do not give Q12/Q 21 ratios in agreement with the principle of detailed balancing (Eq. (5)]. An accurate determination of this ratio requires knowledge of the ratio of the A coefficients for the (72P 3 ; 2 - 62S112 ) and (72P 112
- 625112 ) transitions with a sufficient accuracy. The values of this ratio, obtained when using separately the results of Stone, 21 Warner, 15 Norcross, 22
and Weisheit, 23 are close, although the values obtained by these authors for each of the A coefficients düfer significantly. Sorne experimental measurements of this ratio are also available. 22
•24
• 25
The average experimental value is about 2.45, which is close to the theoretical values given by the four authors previously quoted. Note that the
value obtained by Heavens is 1.40. The problem of the choice of an A set is posed
in a different way when considering the determination of Q13 and Q23• The Einstein coefficients associated with the (72P 112 -62S112 ) and (72P 3; 2
- 625112 ) transitions have been calculated by the previously quoted authors and the coefficients as -sociated with the (62D312 - 6 2P 112 ) transition only by Warner and Stone. To interpret the data we must know the ratios of (Ai;20312 - 62P 112 ) to (A-r2P3/2_5251;2l and (At;203;2-62P112> to (Ai2P1;2
- 62S112 ). We calculated them from Warner and Stone·s results; in the cases of Weisheit and Norcross we used A values for the (62D312 - 62P 112 )
transition given by either Warner or Stone, which are close. The choice of the A set is made by comparison of the equilibrium ratios given by Eq. (5) and the experimental ratios R31 and R32 determined with the various sets of A values. The ratios should be very close to equilibrium values at high pressure of helium. The values of R31 and R32 can be seen as constant for helium pressure greater than 30 Torr (the higher-pressure mea-. surements are performed at about 45 Torr). In these experimental conditions R31 and R32 are derived only by using Eq. (2), without reference to any model, providing a direct measurement of the ratio of the two corresponding A values. The Warner coefficients provide the best agreement for the R31 and R32 values (Fig 4 ). It should be mentioned that in the case of the measurements of R12 and R21 at high p·ressure of helium all the systems. except Heavens' s, gave reasonable agreement with the experimental measurements.
Note that the theoretical lifetimes of both the 72P 312 and 72P 112 levels. calculated using Warner's values, are also in good agreement with the experimental determination.20
-28 As far as we know,
no measurements of the 62 D312 lüetime have been reported.
For all these reasons we use Warner' s A values for the analysis of our experimental data.
IV. DISCUSSION
Previous experimental results concerning the collisional transitions between 2P 112 and 2P 312 levels of the alkali metals, show clearly that the values for the cross sections, for a given rare-gas
TABLE II. Theoretical values of Einstein'sA coefficients (106 sec-1).
Transition Ref. 20 Ref. 21 Ref. 15 Ref. 23 Ref. 22
7Pv2- GSv2 2.12 0.90 1.58 0.65 0.76 7P312- 6Sv2 2.97 2.79 4.18 1.69 1.67
6D312- 6Pv2 12.i 12.9 9.86

- 145 -11 INELASTIC COLLISIONS INVOLVING EXCITED CESIUM ... 853
TABLE III. Experimental cross sections (Â2) for various fine-structure collisional transitions. =
AE (cm-1) T (K) He
Na 3P112-3P312 17 397 86 K 4P 112 - 4P 312 58 368 59.5 Rb 6Pu2- 6P312 77 430 29.3 Cs 8P 112 - 8P 312 83 420 34 Cs 7P 112 - 7P 312 181 450 12 Rb 5P 112 - 5P 312 238 450 lx 10-1
Cs 6P 112-6P312 554 450 l.5X 10-4
atom, decrease with increasing energy defect ~ (see Table Ill). However, the vari.ation of the cross-section values as a function of the energy defect is found to be different for each rare-gas atom. Therefore reference to the energy defect alone is insufficient for the discussion of the values of the experimental cross sections. The cross sections obviously depend on the forces of interac -tion between the rare-gas and alkali atoms. The nature of the rare-gas atom and the positions of the initial and final lei•els in the energy diagram of the alkali atom therefore have to be considered.
Theoretical calculations of the intramultiplettransition cross sections have previously been carried out for the first resonant doublets of the alkali metals. 6
-8 Good agreement was obtained in
the case of the lightest alkali metals, for which the energy defect involved in the transition is small. These calculations generally used adiabatic potential curves which have been obtained asymptotically and did not take into account possible coupling with neighboring levels. Thus these potential-energy curves did not show any structure. Simplüied potential-energy curves have also been used for the determination of intramultiplet transitions in the third resonant doublet of cesium, 3 but no agreement has been obtained between theory and experiment. 1 As far as we know, no calculatien of cross sections concerning both the intraand intermultiplet processes associated with the four levels considered here has been carried out. Recently, extensive molecular potentials for the alkali metal-rare-gas -a tom pairs have been calcula ted. 9 •
10 They are correlated at large internuclear distances with numerous exciteà levels of the alkali metals. The adiabatic potential-energy curves generally show a structure, more or less pronounced, according to the alkali-metal-raregas-atom pair which is considered (see, for example, Fig. 6). This structure is clearly àemonstrated to be due to coupling between neighboring levels. Because the first resonant doublets are more isolated from the other levels than the higher excited 2P doublets, the corresponding adiabatic potential-energy curves are seen to be more regu-
Ne Ar Kr Xe References
67 110 85 89.8 4 14.3 36.7 61.4 104 4 10.3 24 23.3 43.9 2
4.4 5.5 4.5 15 1 l.8X 10-I l.2X 10-I 9.1 X 10-z 6.5x 10-1 5
4X 10-l lx 10-3 5x 10-4 6x 10-• 29 4 X 10-~ 29
lar, with, however. some structure. A calculation of the intramultiplet cross sections. using these potential curves, is in progress.
Let us consider now the results obtained for the intermul tiplet
(72P1;2 ~ 52D3;2l
and
(72P3/2 ~ 52DJ;2l
POTENTIAL ENERGY (10 1 cm-')
~ 2 112
(a)
60512
22,5~~
22~ ~ 312 111 7Pi11
312 112 Ç,-He 50m
1/2 7P •12
9 12 15 18 21 21. INTERNUO.EAR SEPARATION( a.u.)
POTENTIAL ENERGY (1o'cm- 1) (b)
512 in 60s11
1n J/2 1n
1/2
~ 60111
7P111 e.,
9 1'2 15 18 21 21. INTERNUCLEAR SEPARATION (a.u.)
! POTENTIAL ENERGV (10 1cm- 1) (cl z3f11 \ 112.....--
22.5
-------1/2
7P111 7Py1
9 12 15 18 21 24 INTERNUCLEAR SE?ARATION ( a.u )
FIG. 6. Potencial-energy curves that correlate asymptotically with the 7 P 1/ 2, ï P 312 , 5D312, and 5D512 states of cesium. taken from Ref. 10. A îull discussion of these curves is found in Ref. 9. 1a) Cs-He; (bl Cs-Ar; 1c1 Cs-Xe.
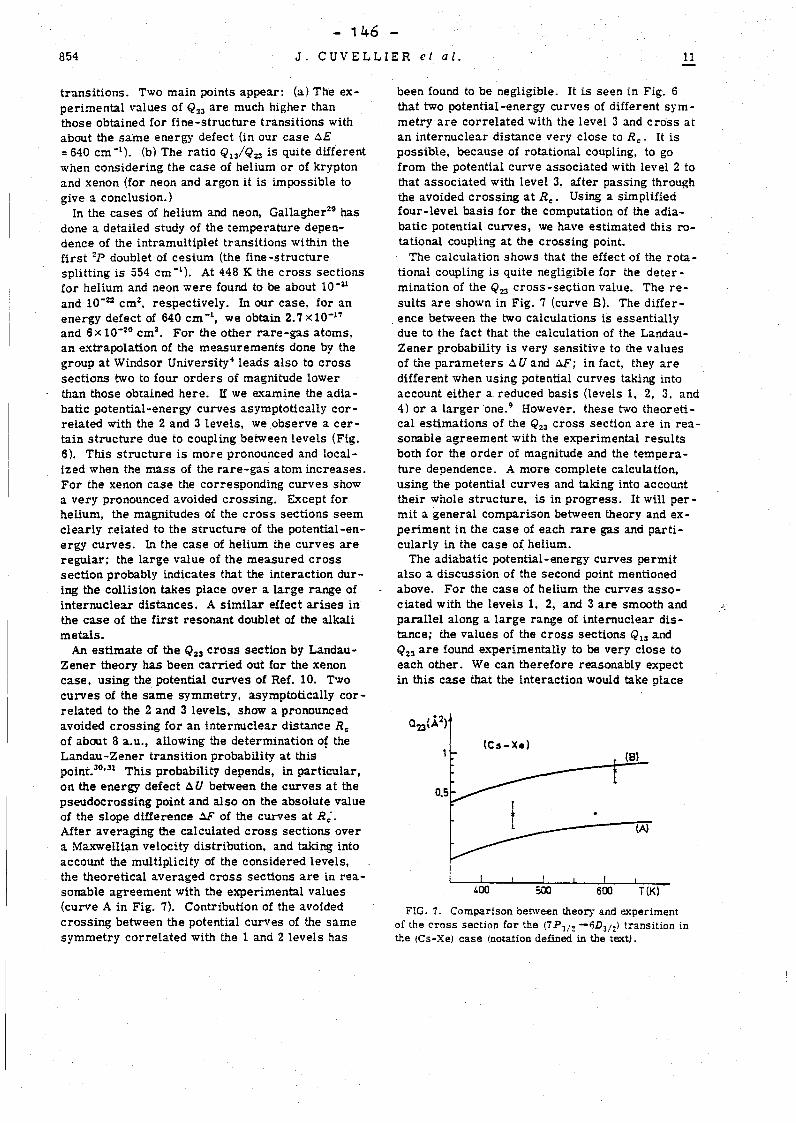
- 146 -854 J. CUVELLIER et al. 11
transitions. Two main points appear: (a) The experimental values of Q23 are much higher than those obtained for fine-structure transitions with about the same energy defect (in our case t::.E = 640 cm -1 ). (b) The ratio Q1/Q23 is quite different when considering the case of helium or of krypton and xenon (for neon and argon it is impossible to give a conclusion.)
In the cases of helium and neon, Gallagher29 has done a detailed study of the temperature dependence of the intramultiplet transitions within the first 2P doublet of cesium (the fine-structure splitting is 554 cm -1
). At 448 K the cross sections for helium and neon were found to be about 10-21
and 10-22 cm2, respectively. In our case, for an energy defect of 640 cm - 1
, we obtain 2. 7 x 10-17
and 6 x 10-20 cm2• For the other rare-gas atoms,
an extrapolation of the measurements done by the group at Windsor University 4 leads also to cross sections two to four orders of magnitude lower than those obtained here. If we examine the adiabatic potential-energy curves asymptotically correlated with the 2 and 3 levels, we observe a certain structure due to coupling between levels (Fig. 6). This structure is more pronounced and localized when the mass of the rare-gas a tom increases. For the xenon case the corresponding curves show a very pronounced avoided crossing. Except for helium, the magnitudes of the cross sections seem clearly related to the structure of the potential-energy curves. In the case of helium the curves are regular; the large value of the measured cross section probably indicates that the interaction during the collision takes place over a large range of internuclear distances. A similar effect arises in the case of the first resonant doublet of the alkali metals.
An estimate of the Q23 cross section by LandauZener theory has been carried out for the xenon case, using the potential curves of Ref. 10. Two curves of the same symmetry, asymptotically cor -related to the 2 and 3 levels, show a pronounced avoided crossing for an internuclear distance Re of about 8 a.u., allowing the determination o; the Landau-Zener transition probability at this point. 30
•31 This probability depends, in particular,
on the energy defect t::.U between the curves at the pseudocrossing point and also on the absolute value of the slope difference t::.F of the curves at Re .. After averaging the calculated cross sections over a Maxwellian velocity distribution, and taking into account the multiplicity of the considered levels, the theoretical averaged cross sections are in reasonable agreement with the experimental values (curve A in Fig. 7). Contribution of the avoided crossing between the potential curves of the same symmetry correlated with the 1 and 2 levels has
been found to be negligible. It is seen in Fig. 6 that two potential-energy curves of different symmetry are correlated with the level 3 and cross at an internuclear distance very close to Re . It is possible, because of rotational coupling, to go from the potential curve associated with level 2 to that associated with level 3, after passing through the avoided crossing at R0 • Using a simplified four-level basis for the computation of the adiabatic potential curves, we have estimated this rotational coupling at the crossing point.
The calculation shows that the effect of the rotational coupling is quite negligible for the deter -mination of the Q23 cross-section value. The results are shown in Fig. 7 (curve B). The differ-
. ence between the two calculations is essentially due to the fact that the calculation of the LandauZener probability is very sensitive to the values of the parameters t::.U and AF; in fact, they are different when using potential curves taking into account either a reduced basis (levels 1, 2, 3, and 4) or a larger one. 9 However. these two theoretical estimations of the Q23 cross section are in reasonable agreement with the experimental results both for the order of magnitude and the temperature dependence. A more complete calculation, using the potential curves and taking into account their whole structure, is in progress. It will permit a general comparison between theory and experiment in the case of each rare gas and particularly in the case of. helium.
The adiabatic potential-energy curves permit also a discussion of the second point mentioned above. For the case of helium the curves asso-ciated with the levels 1, 2, and 3 are smooth and ,: parallel along a large range of internuclear dis-tance; the values of the cross sections Q 13 and Q23 are found experimentally to be very close to each other. We can therefore reasonably expect in this case that the interaction would take place
C23{Â2)
(Cs-Xe) (8)
{A)
400 500 600 T(K)
FIG. 7. Comparison between theory and experiment of the cross section for the (7 P 3; 2 -6D3; 2) transition in the (Cs-Xe) case (notation defined in the text).

- 147 -11 INELASTIC COLLISIONS INVOLVING EXCITED CESIUM ... 855
over a large range of internuclear distance for both the 1 - 3 and 2 - 3 transitions. For xenon and krypton the curves associated with the levels 1, 2. and 3 show- structure. Moreover. the structure is different when considering the curves associated with the levels 1 and 2. In these cases the cross sections QL 3 and Q23 are found experirnentally to be very düferent. In the case of xenon, where an avoided crossing at about 9.25 a.u. is seen between the same syrnmetry curves associated with the levels 1 and 2. it has been possible to estimate theoretically the cross section Q13 • This has been done, as before, taking into account the avoided crossings shown by the potential-energy curves associated with levels 1 and 2 and 2 and 3, respectively. The theoretical results are in good agreement with the experimental estimates (Fig. 8).
It is clear that our analysis of the results concerning intermultiplet transitions is essentially based on the structure of the potential curves invol ved in the transition. The same analysis for any intramultiplet transition might be used if the corresponding curves show such strucb.1re.
The last point to be discussed is the value obtained for Q3 -Q 34 in the case of helium. We can write
Q3 -Q34 =Q31 +Q32-..Qq,
where Q • represents the collisional depopulation of level 3 to all levels other than those considered here (levels 1, 2, and 4). Using the principle of
detailed balancing we can cal culate Q 31 + Q n from our results and compare the value obtained with Q 3 - Q,w This permits an estimate of Q •. One can see that no collisional depopulation of level 3 to any level other than 1, 2, or 4 occurs. It is well known that large "quenching" cross sections are obtained in the case of a molecular-perturber gas (in particular, nitrogen). 1
•19 Moreover, a theoret
ical calculation using simplified potential curves has predicted large "quenching" cross sections for highly excited alkali-metal atoms colliding with rare-gas atoms. 3 The fact that the (Cs-He) potential-energy curves have no localized structure does not permit a conclusion before a more complete calculation is performed. For the other rare gases at thermal energies the potential curves show only coupling between the four levels considered here. Thus it appears that the corresponding cross sections Q. would probably be small.
a,JÂ2J
(Cs-Xe)
400 500 600 T(K)
FIG. 8. Comparison between theory and experiment of the cross section for the (7 P 1; 2 -6D312) transition in the (Cs-Xe) case (notation defined in the text).
V. COl'iCUJSIOl\
This ex-perimental study of inelastic collisional processes involving 72P and 62 D cesium atoms and ground-state rare-gas atoms at thermal energies leads to surprising results if one considers only the energy defect for these processes. However. if the experimental results are discussed using the theoretical adiabatic potential-energy curves obtained recently, a satis fac tory explanation of the main features of this work is obtained. The potential curves show various structures due to coupling betv:een neighboring levels. The structure
is of diff erent importance for different rare-gas a toms. This explains (a) the possibility of having a large cross section, even in the case of a large energy defect between the considered levels, and (b) the dependence of the Q13/Q 23 ratio on the nature of the rare gas.
In the case of the (Cs-Xe) pair the potential-energy curves show two pronounced avoided crossings. By using the Landau-Zener formula to evaluate the transition probabilities at these points, the Q13 and Q23 cross sections are found to be in reasonable agreement with the experimental values. This provides a first check of the reliability of the theoretical potential curves, the accuracy of which was difiicult to determine a priori.
ACKNOWLEDGME:S:T
We wish to thank Dr. P. M. Stone for a critical reading of the manuscript.

- 148 -856 J. CUVELLIER et al. 11
1~1. Pimbert, J. Phys. (Paris) 33, 331 (19721. 2I. Siara, E. S. Hrycyshyn, andL. Krause, Can. J. Phys.
50, 1828 (1972). 3~E. Nikitin and A. J. Reznikov, Chem. Phys. Lett . .§..
161 (1971). ~P. L. Lijnse, in Report No. 398, Fysisch Laboratorium,
Rijksuniversiteit Utrecht, 1972 (unpublished), and references therein.
5 J. Cuvellier, P. R. Fournier, F. Gounand, and J. Berlande, C. R. Acad. Sei. B 276, 855 (l9i3).
6E. I. Dashevskaya and E. E~ikitin, Opt. Spektrosk. 22, 866 (1967) (Opt. Spectrosc. 22, 473 (1967)1. -
~E. I. Dasbe,•skaya, E. E. Nikitin, and A. I. Reznikov, Opt. Spectrosk. 29, 1016 (1970) [Opt. Spectrosc. 29, 540 (1970)1. - -
8F. Masnou-Seeuws, J. Phys. B 3, 1437 (1970). 9J. Pascale and J. Vandeplanque;-" J. Chem. Phys. 60,
2278 (1974). 10J. Pascale and J. Vandeplanque, Commissariat à l'
Energie Atomique Report (unpublished) (available on request from the present authors).
11 w. B. Nottingham, Thermo-Electron Engineering Corporation Technical Report No. TEE 7002-5, 1960 (unpublished); see also Ref. 1.
12J. L. Lemaire and F. Rostas, J. Phys. B 4, 555 (1971). 13F. Lambert, C. R. Acad. Sei. B 274. 1406-(1972). 11Y. V. Evdokimov, Opt. SpektrosLJ4, 832 (1968) [Opt.
Spectrosc. 24, 448 (1968)1. -
15T. H. Warner, Mon. r-ot. R. Astron. Soc. ill, 115 (1968).
16T. Edmonds and J. P. Hobson. J. Vac. Sei. Technol. .t 182 (1965),
17 E. H. Kennard, Ki11ctic Theory oj Cases (McGraw-Hill, New York, 1938).
18G. W. Series, Rep. Prog. Phys. 22, 280 (1959). 19J. L. Rocchiccioli, C. R. Acad. Sci. (Paris) fil, 787
(1972). 200. S. Heavens, J. Opt. Soc. Am. 51, 1058 (1961). ?tp_ M. Stone, Phys. Rev. 127, 1151 (1962). 220. W. Norcross, Phys. R-;;:- A]., 606 (1973); and
t"eferences therein. 23J. C. Weisheit, Phys. Rev. A 5. 1621 (1972). 21 L. Agnew, Bull. Am. Phys. Soë. 11, 327 (1966). 25R. J. Exton, dissertation (West Virginia University,
1972) (unpublished). 26R. W. Schmieder, A. Lurio, and W. Happer, Phys. • Rev. A ~. 1216 (1970). • 7E. L. Altmann and S. A. KazantseY, Opt. Spektrosk. 28.
805 (1970) [Opt. Spectrosc. 28, 432 (1970) J. -28E. L. Altman and M. Chaika-:-Opt. Spektrosk. 19, 968
(1965) [Opt. Spectrosc. 19, 538 (1965)1. -29 A. Gallagher, Phys. Re;:- 172, 88 (1968). 30c. Zener, Proc. R. Soc. A 137, 696 (19321; L. O.
Landau, Phys. z. Sowjetunion 2. 46 (1932). 31 L. Landau and L. Liischitz, Qu~ntum Mechanics (Per
gamon, London, 1958).

- 149 -
VI.2.J. Compléments.
Il ne nous semble pas nécessaire de commenter la partie ex
périmentaJ.e de l'article, le chapitre V ayant fourni tous les rensei
gnements nécessaires. Seule la partie théorique appelle quelques
éclaircissements.
Nous aJ.1ons exposer de manière détai11ée le cal.cul de type
Landau-Zener que nous avons effectué pour 1a transition 7P3
; 2 -6n312
(Fig. 7 de l'article 1) dans 1e cas (Cs-Xe) pour 1eque1 les courbes de
potentiel adiabatique présentent une structure très marquée qui justi
fie l'emploi de cette approche théorique.
Le premier cal.cul effectué (courbe (A)) ne prend en compte
que 1e couplage radial. entre les courbes de même symétrie (1/2) asso
ciées asymptotiquement aux niveaux 7P3
; 2 et 6n312
qui présentent un
pseudo-ctoisement marqué en Re"" 8 u.a. La probabilité de transition
P12 est donnée par l'équation (C32)
P ( -2'1't ~v2 ) = exp 12 iivlF-FI
R 1 2
(F1)
avec
VR = V , ! ( b )
2 V y1 - a; - Ë (F2)
les notations étant celles du chapitre III.
Pour une énergie E donnée on obtient cr par intégration sur
le paramètre d'impact b, soit :
cr(E) = 4'1't lb max
p12(l - p12)
0
b db (FJ)
avec b 2
( ;~~) = 1 V - E (F4)
on effectue les changements de variable suivants
(t/ = u

- 1.50 -
P12 = exp ( v, -a ) - u - ;
avec
2'11 Av2 a= 1i vlF1 - F 2 1
L'équation (F3) s'écrit a1ors:
Cf"= 2'11 R2 C
-a -a i ·1-v/E 1 )i o exp ( V 1 - u- : ) 1 - exp ( V 1 - u - i du
Fina1ement les deux changements de variable successifs suivants
puis
permettent d'obtenir
a-(E)
avec
= 4'11 R.2 C
(1 - v)_, /2
u--E = X
X
( -)-1/2 = Y
1- V E
(1 - ;) ["' e -by (1 - e-by) y-J dy
1
b - ( -)-1/2 - a 1 V - Ë
- . ;11 AV: - (1-;)1 2 ~ vlF1- F2l
(F.5)
(F6)
L'équation (F.5) représente la forme la plus adaptée au ca1cul numérique,
l'intégra1e
! IX) -by ( -by) e 1 - e
1
y-3 dy
étant tabulée9 • Les quantités AV, V et [F1 - F2 l sont déduites des cour
bes de potentiel. On détermine enfin la section efficace o-(T) par inté
gration numérique sur la distribution de Maxwell (Eq.(C26)) soit :

- 151 -
a-(T) = f'CT(E) exp( - :)
seuil
E - dE k2T2
(F7)
Ce calcul correspond au pseudo-croisement (1/2 - 1/2). Le m~me résultat
sera obtenu pour le pseudo-croisement ((- 1/2) - (- 1/2)). Comme il y
a en tout quatre états initiaux (1/2, - 1/2, 3/2, - 3/2) associés
asymptotiquement au niveau 7P3; 2 on doit moyenner sur ces états et ie
résultat final devient
a-( 7P 312 ~ 6D3/ 2 ) = ! a-(T) (F8)
On obtient ainsi la courbe (A) de la figure (Article 1).
Le calcul précédent néglige les couplages rotationnels, pos
sibles entre .courbes moléculaires de symétrie différente. En utilisant
les courbes de potentiel obtenues à partir d'une large base d'états
atomiques8 , il était difficile de calculer la probabilité de transi
tion (7P312 --+-6D3; 2 ) étant donné la complexité de la zone présentant
les croisements (ou pseudo-croisements). Le calcul (courbe (B) de la
figure 7, Article 1) a donc été mené à partir des potentiels adiaba
tiques obtenues à partir d'une base à quatre ·états atomiques (7P112
,
7P3; 2 , 6n312 , 6D5; 2 ). Ceux-ci sont présentés figure 18.
V( u .a.)
/
/ /
_,,,,,..,,,,.. ,,,,, ,,,,,.,,,,,.,,,,,. 3/2
Jso512
312 -- --- J s D P2 _,- _,,,-· .---_;:.-.----- 312 ,,, /i/2
.___ __ 7P3;2
Fig ... 18 - Croisements et pseudo-croisements des courbes de potentiel (Cs-Xe) associées aux niveaux 6D3; 2 , 5; 2 et 7P3; 2 ·

- 152 -
Aux points P1 et P 4 1.a probabilité de transition (coupl.age
radiai) est fournie par !.'équation (C32), tandis qu'en P2 et P3 (cou
plage rotationnel.) el.le est donnée par !.'équation (c23). Si on appel.le
~1 , 12
, ; et ~4 1.es probabilités de transition en P1 , P 2 , P3
et P4 on obtient pour 1.es probabilités g1oba1es de passage du niveau 7P3;
2 au niveau 6D3; 2
e>(!C7P3;2) -½< 6D3/2)) =
S'1 < 1-11 > ) 1 + (<1 _,2>2 + !I'~ 1 < 1-\!':i>2 + !I'; C< 1-!1'4>2 + !PD O l (F9)
et
t:1t 1 3 ~(2(7P3/2) ....,-2( 6D3/2)) =
!1192<1-9'2> )1 + ~1-e>:i>2 + ~ i <1-!14>2 + !l':u l (F10)
La première équation donne 1.a probabilité de sortie sur la courbe mo-
1écu1aire 1/2 associée au niveau 6D312 , 1.a seconde celle de sortie sur
1.a courbe mol.écu1aire 3/2 associée à ce m~me niveau, 1.a voie d'entrée
étant, dans les deux cas, 1.a courbe mo1écu1aire 1/2 associée au ni
veau 7P312 . On a ensuite intégré (numériquement) sur 1.e paramètre
d'impact b, puis sur la distribution de Maxwell.. Après moyenne sur
1.es états initiaux et sommation sur 1.es états finaux on obtient 1.a
courbe (B) de 1a figure 7 (Artic1e 1). Le caicul. a montré que 1a con
tribution des coup1ages rotationne1s était nég1igeab1e, c'est-à-dire
que la probabilité donnée par (F10) ne contribuait pas au résultat
fina1 (ce qui correspond à ~2 = 0) et que l'équation (F9) pouvait se
réduire à 29'1 (1-P1 ), ce qui correspond aux condi tians du ca1cu1 pré
cédent (courbe (A)). La différence observée entre 1es deux courbes
ne provient, comme mentionné dans l'article, que du fait que les va-
1.eurs C::..V et jF1 -F2 1 ne sont pas 1.es m~mes -(au point 1) suivant que
l'on utilise des potentiels obtenus à partir d'une 1.arge base d'états
atomiques ou d'une base réduite. De fait il. est facile de se rendre
compte sur !.'équation· (F1) de 1a sensibilité de P à ces deux paramètres.
Notons enfin que, contrairement à ce qui est noté dans 1'ar
ticl.e 1, aucun ca1cu1 de sections efficaces relatives aux transitions
étudiées et prenant en compte 1.es courbes de potentiel. de Pascaie et
Vandepl.anque n'a été effectué. Nous reviendrons sur ce point au para
graphe suivant.

- 153 -
VI.2.4. Comparaisons complémentaires.
a) ~~~~~~!~-=~E~!!~=~~~~-
une étude des sections de transfert ~2
et Q21 intramu1ti
plet, ainsi que de leur variation avec la température a été publiée
par I.N. Siara et a1. 1 O en 1974 ( ce.tte étude util~se aussi la méthode
de fluorescence non résoJ.ue en temps, mais la source d'excitation est
wie lampe à décharge césium-argon, technique dont nous avons sou1igné
les inconvénients). La comparaison avec nos résu1tats expérimentaux,
précédemment publiés1
, montre Wl bon accord générai, la différence
la pl.us marquée ("' 30'1') étant observée dans le cas du xénon (Tableau
IX). Les différences (au pl.us éga1es à JO%) peuvent, à notre avis,
s'expJ.iquer par J.es deux remarques suivantes:
Perturbateur N , 1 os resu1tats Si ara et a1. 1 O
He 12 15
Ne 0 .18 O .15
AI 0.12 0 .11
Kr 0.091 0.080
Xe 0.65 0.50
Tableau IX
Comparaison de nos résu1tats , . t 1 experimen aux aux mesures
de Siara et ai. 10 pour le transfer-t intramu1tipJ.et (7P1; 2 -7P
3; 2 )
du césium induit par collision avec les gaz rares.
Les sections efficaces sont en 12 . T = 450 K.
- Les auteurs n'ont pas utiJ.isé le m3me système de coefficients
d'Einstein pour dépouilJ.er leurs mesures. Si la prise en compte des
durées de vie expérimenta1es peut se justifier aisément (elles sont en
effet plus précises, dans le cas des niveaux 7P, que les déterminations
théoriques. Le système que nous avons retenu (Warner) fournissait ce-

- 154 -
pendant des va1eurs de rA proches de cell~s déterminées expérimenta1e
ment11), il nous semble peu cohérent d'utiliser pour la détermination
des rapports de branchement (cf. éq. (E32)) la moyenne des va1eurs
obtenues par deux théories différentes (Heavens et Warner en l'occu
rence). Notons enfin que les raisons du rejet des va1eurs proposées
par Norcross (qui sont certainement les plus précises, mais qui sont
ma1heureusement incomplètes2 ) sont~à 1 1 évidence,erronées. Ces quelques
remarques sou1ignent bien, sur LUl cas précis, l'importance du choix
des coefficients d'Einstein sur la détermination des sections efficaces.
- P1us fondamenta1 nous semb1e 3tre 1e fait que 1es auteurs ont
comp1ètement nég1igé 1'inf1uence éventue11e des niveaux 6D. Or les
résu1tats que nous avons obtenus montrent clairement, en particu1ier
dans le cas des gaz rares lourds (pour lequel les sections intra et
extramu1tiplet sont du m~me ordre de grandeur), que l'influence des
niveaux 6D n'est pas nég1igeable dans la zone de pression (1 à 10 Torrs
dans le cas du xénon) où ces auteurs ont effectué leurs mesures. De
plus i1 ne semble pas qu'i1s aient tenu compte de l'élargissement de
pression césium-gaz rare qui n'est pas entièrement négligeab1e au vu
de leurs conditions expérimenta1es. Cette deuxième remarque met bien
en évidence l'importance du choix du modèle retenu pour le dépoui11e
ment.
Le deuxième travail expérimentai 1 2, publié en 1975, concer
ne l.Uliquement les sections de transfert ~ 2 et Q21 relatives aux colli
sions (Cs-Xe). Les sections reportées, respectivement ~ 2 = 0.150 1 2
et Q21 =0.16712 , sont à comparer aux va1eurs voisines de 0.60 1 2 que
nous avons mesurées (Tableau Ide l'article 1). M~me si l'on tient
compte du fait que nos mesures ont été effectuées à 450°K, au lieu de
325°K pour cel1es de Kielkopf, (la variation de Q12 ou Q21 en fonction
de la température demeure assez faible dans cet interva1le10) LUl dé
saccord très no~able subsiste (environ LUl facteur 3). Il nous semble
devoir ~tre attribué, là aussi, à la non prise en compte de l'influen
ce des niveaux 6D; ce fait est d'autant plus grave que les mesures
ont été effectuées entre 1 et 150 Torrs, c'est-à-dire à très forte
pression. Il est en effet facile de voir, d'après nos résu1tats, qui
indiquent des sections efficaces 7P -6D du m~me ordre de grandeur
que les sections de transfert intramu1tiplet, dans le cas du xénon,
que le modèle à deux niveaux utilisé par Kielkopf n'est pas va1able.

- 155 -
Les durées de vie mesurées, à haute pression, correspondent certaine
ment à un mélange de niveaux. Il est très intéressant de mentionner
que de telles pressions étaient bien requises pour l'emploi de lamé
thode de fluorescence résolue dans le temps, les niveaux bas possé
dant de très fortes constantes radiatives. Ceci sou1igne clairement
les inconvénients de ce~te méthode pour l'étude du transfert entre
niveaux faiblement excités, point déjà discuté au chapitre V.
b) ~~~~!~!~-È~~~~S~=~·
La comparaison satisfaisante obtenue, dans le cas du xénon,
entre résu1tats expérimentaux et résu1tats théoriques (fournis par la
méthode L.Z.S.) indique clairement l'importance des couplages stati
ques entre niveaux excités et donc la nécessité de disposer de bonnes
courbes de potentiel. Cependant la méthode L.Z.S. s'avère d'un emploi
restreint, puisqu'elle exige la présence de structures localisées
dans les potentiels adiabatiques. Une comparaison complète requiert
donc l'emploi d'approches théoriques plus élaborées. La méthode semi
classique ne peut 3tre utilisée car les seuils de réaction (180 cm-1
pour le transfert intramu1tiplet et 640 cm-1 pour le transfert
7P3; 2 ~6D 12 ) sont voisins de l'énergie thermique disponible
(N 400 cm-1), ce qui conduirait à une forte surestimation des sections
efficaces. Ainsi seu.J.e la méthode quantique prenant en compte des
courbes de potentiel réalistes, asymptotiquement associées aux niveaux
atomiques 7P et 6D permettrait une comparaison d'ensemble. Une telle
approche a été utilisée avec succès pour les premiers doublets réson
nants13. Sa formulation est maintenant bien connue. Son emploi deman
derait sans doute des temps de calcul importants, dus à la nécessité
d'inclure les courbes de potentiel associées à quatre états atomiques
(au lieu de deux dans le cas des premiers doublets) mais ne pose pas
de difficu1tés de principe. Malheureusement ces calcu1s n'ont pas été
effectués. En résumé seule la méthode L.z.s. a été utilisée; grâce à
l'emploi de potentiels élaborés elle a fourni, dans le seu1 cas où
elle est applicable, des estimations satisfaisantes.
Les seu1s autres calcu1s6114 concernant des états plus exci
tés que les premiers doublets résonnants sont relatifs au transfert
intramu1tiplet du deuxième doublet du rubidium (6P; AE = 77 cm-1 ) et
du troisième doublet du césium (8P ; AE = 83 cm-1 ). Le cas de ce der
nier est intéressant car sa situation dans le diagramme énergétique

- 1.56 -
est assez anal.ogue à celle du groupe de niveaux que nous avons étudié,
les niveaux 7D n'étant distants que de 330 cm-1 . Il nous semble inté
ressant de commenter les résu1tats théoriques obtenus car ils viennent
confirmer, dans une situation anal.ogue, les conclusions que nous venons
de tirer, à savoir l'importance des couplages statiques entre niveaux
excités sur les sections efficaces de transfert collisionnel. Notons
tout d'abord que l'approche semi-classique peut 3tre utilisée, l'écart
intramu1tiplet (83 cm-1
) étant petit devant l'énergie thermique dis
ponible (N 400 cm-1 ). Un premier cal.cu1~ effectué à partir de poten
tiels empiriques,conduit à un désaccord très notable avec les vaJ.eurs
expérimental.es5 : facteur 35 dans le cas de l'argon et 120 dans le cas
du xénon. Un second cal.cul.14
utilisant les potentiels de Pascal.e et
Vandeplanque8 {qui montrent à l'évidence des couplages statiques nota
bles entre les niveaux 8P et 7D) atténue très sensiblement le désac
cord: facteur 2 pour l'argon et 2,5 pour le xénon. L'écart restant
peut 3tre attribué, comme les auteurs l'ont noté, au fait que, pour
des raisons de temps de cal.cul., le traitement dynamique de la collision
n'a été effectué qu'en prenant en compte les potentiels adiabatiques
associés aux états atomiques 8P {ces potentiels étant obtenus, eux,
à partir d'une large base d'états atomiques) : a~nsi les états 7D sont
ignorés dans la partie dynamique du problème. Là aussi aucun cal.cul.
quantique n'a été effectué mais Pascal.a et Stone14
ont montré l'équi
vaJ.ence des résul.tats fournis par les deux approches (dans le cas du
niveau résonnant du sodium) dès que l'énergie de la collision est
notablement supérieure à celle du seuil.
VI.2.5. Conclusion.
Les résul.tats expérimentaux que nous avons présentés ont per
mis de mettre en évidence l'importance essentielle des couplages sta
tiques-entre niveaux excités sur les processus de transfert collision
nel. En effet on peut expliquer ainsi les différences très notables
de comportement {en particul.ier en fonction de l'écart d'énergie asymp
totique et de la nature du gaz rare) entre les sections efficaces re
latives aux premiers doublets résonnants et celles relatives aux ni
veaux plus excités. La possibilité d'obtenir des vaJ.eurs élevées pour
des sections efficaces de transfert entre niveaux énergétiquement éloi
gnés (par rapport à l'énergie thermique disponible) a été mise en évi
dence expérimental.ement (notons que dans le cas de l'hélium une situa-

- 157 -
tien comparable a été observée par Wallenstein et Robertson15 , en bon
accord avec des ca.lcu1s théoriques16 effectués à l'aide de courbes
de potentiel élaborées).
Les ·traitements dynamiques du problème collisionnel dévelop
pés pour les premiers niveaux résonnants peuvent 3tre étendus au cas
des niveaux plus excités, mais ils nécessitent la solution préalable
de la partie statique du problème, c'est-à-dire la détermination de
courbes de potentiel réalistes. Cel1e-ci représente un problème com
plexe du fait de la nécessité d'inclure une large base d'états atomi
ques pour prendre en compte les couplages statiques entre états voi
sins. Nous avons montré que, dans le cas où ces couplages sont très
localisés une approche théorique simple pouvait conduire à un accord
satisfaisant avec l'expérience. Cependant, pour les niveaux que nous
avons étudiés, seu1e une approche quantique utilisant des courbes
molécu1aires associées aux quatre niveaux atomiques 7P1 ; 2 , 3; 2 et
6D3
;2
,5
;2
permettrait une comparaison généra.le entre théorie et expé
rience. Un tel traitement semble possible mais n'a pas encore été ef
fectué.
VI.3. ETUDE DE LA DEPOPULATION COLLISIONN.ELLE TOTALE DES NIVEAUX DE R'YDBERG.
VI.3.1. Présentation du travail.
Les raisons de l 1 intér3t pour l'étude des états de Rydberg
sont mu1tiples. Sans prétendre 3tre exhaustif i1 nous para!t intéres
sant d'en mentionner certaines avant de présenter les motivations et
les buts essentiels de notre étude.
La définition m3me d'état de Rydberg varie d'un auteur à
l'autre. Nous considérerons qu'un atome est dans un état de Rydberg
quand l'électron excité se trouve, en moyenne, loin du noyau, c 1 est
à-dire que <r> a une va.leur élevée. La limite entre état excité et
état de Rydberg est parfaitement arbitraire. Dans la suite de l'exposé
nous considérerons que les états de Rydberg correspondent à des atomes
pour lesquels la va.leur du nombre quantique principal est supérieure
ou égale à 10, ce qui conduit à des va.leurs de :
1 2 < r > = 2 ( :Jn - 1 ( l + 1 ) )

- 158 -
aJ.lant de 105 à 150 u. a., suivant la vaJ.eur de 1. Un premier in tér@t
des états de Rydberg vient de l'image très simple que l'on peut s'en
faire : un électron externe très loin d'un coeur ionique. Cette image
montre tout de suite le parti que l'on pourra tirer du caractère hy
drogénoïde du système. De fait les propriétés spectroscopiques des
états de Rydberg sont en générai très bien prédites par la généraJ.i
sation des propriétés correspondantes de l'hydrogène1 ~ en tenant sim
plement compte de l'in:f'luence du coeur à l'aide du défaut quantique,
c'est-à-dire en remplaçant n par n* = n - o1 (voir chapitre II). De
ce point de vue un atome de gaz rare dans un état de Rydberg diffère
peu d'un atome aJ.caJ.in, dans un état den* voisin, ce qui est loin
d'3tre le cas pour les états peu excités. Nous avons en:f'in vu le par
ti que l'on peut tirer de la simplicité d'une telle image lors de
l'approche des problèmes co11isionnels.
Un deuxième aspect intéressant de ces états, qui découle di
rectement de la définition que nous en avons donnée, vient de ce que
la simple extrapolation du modèle hydrogénoïde aux fortes vaJ.eurs de
n conduit à leur attribuer des propriétés pour le moins inhabituelles
en Physique Atomique : très longue durée de vie,. polarisabili té géan
te, grande sensibilité aux champs électrique ou magnétique, ionisa
tion très facile ••• La mise en évidence expérimentaJ.e et la vérifica
tion de telles caractéristiques justifiaient à elle seule la légitime
curiosité du physicien. Les collisionnistes eux-aussi ne pouvaient
manquer d 1 3tre attirés par de tels états: leur stabilité collision
nelle est-elle très grande ou très faible?
Notre dernière remarque nous amène à mentionner l'intér@t
pratique lié à la connaissance des propriétés des états de Rydberg.
Ceux-ci en effet jouent un rSle important en astrophysique, où leur
mise en évidence expérimentaJ.e (raies radio de recombinaison corres
pondant à des transitions n .. n-1 pour des vaJ.eurs den de l'ordre de
100) dans certaines régions de l'espace (région H en particulier) re
monte à de nombreuses années. Un autre domaine où il est important de
connaitre les propriétés des états de Rydberg est celui de la phys.i.que
des plasmas (plasmas d'intér@t astrophysique, fusion contrSlée ••. )
en effet toute modélisation de plasma nécessite la prise en compte
des états proches du continuum qui ont une in:f'luence importante lors
de la recombinaison collisionnelle-radiative. Un dernier centre d'in
tér@t pratique, qui constitue une motivation importante pour de tels

- 1.59 -
travaux, particu1ièrement outre-atlantique, est celui de la séparation
isotopique par laser. Sans entrer dans le détail on peut dire que la
création de fortes densités d'états de Rydberg constitue une étape
importante du processus. Ainsi la connaissance de tous les termes de
perte (superradiance, co11isions) est requise pour estimer de manière
réaliste les possibilités pratiques et le rendement du processus de
séparation isotopique.
Si toutes les considérations que nous venons de faire suffi
sent à justifier l 1 intér3t de te11es études, il convient de sou1igner
que le démarrage du travail expérimental sur le sujet n'a été rendu
possible que par les progrès constants de la technologie des lasers
à colorant, seuies sources lumineuses capables de créer de manière
sélective des densités notables d'atomes très excités. Ainsi le début
des travaux expérimentaux concernant les états de Rydberg est très
récent (1973 environ). Avant cette période ces états avaient bien été
observés au laboratoire dans des plasmas ou des post-décharges, mais
le caractère non sélectif de telles expériences n'avaient pas permis
une étude systématique précise de leurs propriétés.
En ce qui concerne les motivations ~ropres à notre travail
i1 faut mentionner de plus que l'étude des propriétés collisionnelles
de ces états s'inscrivait parfaitement dans la ligne de nos activités
de recherche. Notre travail sur les états excités du césium nous
avait permis de nous familiariser avec les techniques d'excitation
laser et de détection de signaux de fluorescence et de réfléchir sur
les possibilités d'approches expérimentale et théorique du problème
des collisions mettant en jeu des états hautement excités. De plus
la mise en ~vidence d'un comportement collisionnel notablement diffé
rent entre le premier doublet résonnant et les doublets suivants nous
amenait très naturellement à étendre nos études vers des niveaux de
nombre quantique principal n plus élevé.
Quand nous avons entrepris ce travail le seul résultat con
cernant les collisions mettant en jeu des atomes alcalins très excités,
aux énergies thermiques,était celui de T.F. Gallagher18 et al. qui
indiquait pour les sections de mélange collisionnel des états nD
(5 6 n ~ 10) du sodium par les gaz rares une dépendance en n4
(c'est
à-dire reliée à la taille géométrique de l'atome nn4a 2 ). On pouvait 0
se demander si une telle dépendance était encore valable pour des va-

- 160 -
1eurs den supérieures à 10. En effet, un tel comportement conduisait
rapidement à des sections efficaces de co11ision très é1evées indi
quant a.1ors une très faib1e stabilité de ces états, avec toutes les
imp1ications qui en découlent (difficultés d'observation en particu-
1ier). Notons aussi qu'à 1a m~me époque West et ai. 19 avait mis en
évidence l'importance de 1'interaction e--perturbateur lors des colli
sions(atomes de Rydberg-molécules~ Les études spectroscopiques avaient,
quant à elles, permis de vérifier que la théorie hydrogénoide permet
tait bien en généra.1 de rendre compte des observations expérimenta.1es
(durées de vie20 , po1arisabi1ité21, interva.11e de structure fine22
).
VI.3.2. Travail expérimenta.1.
Avant de présenter notre travail expérimenta.1 relatif aux
collisions mettant en jeu des états de Rydberg, nous a.11ons exposer
un ensemb1e de résultats re1atifs aux propriétés radiatives des états
très excités. Ces résultats, s'ils ne constituent pas la partie essen
tielle de notre travail n'en sont pas moins importants car i1s concer
nent des phénomènes dont il convient de tenir compte et d'éva1uer l'im
portance lors de toute expérience mettant en jeu des états de Rydberg.
Ils apportent de plus des renseignements intéressants pour la bonne
compréhension du comportement des états de Rydberg, dans un environ
nement expérimenta.1 déterminé.
a) S~!!S~=~-~~E!~!~-!~~~!!f~-~~-~~~E~~!!~!~!-~=~-~!~!~-~= ~~~=~~·
1) Photoionisation.
La réabsorption par un atome très excité d'un photon corres
pondant à 1a (ou aux) longueur(s) d'onde d'excitation peut donner 1ieu
à la photo~onisation de celui-ci. On a a.1ors création d'un photoélec
tron. Si une densité notable d'électrons était ainsi produite les col
lisions (états de Rydberg-électron) pourraient affecter sensiblement
les résultats obtenus. Pour éva.1uer quantitativement un te1 effet nous
avons d'abord ca.1culé les sections efficaces de photoionisation des
états de Rydberg que nous avons étudiés à l'aide de la méthode de 23 .
Burgess et Seaton • Dans le cas des états P llne seule longueur d'onde
est à considérer, a.1ors que pour 1es états S nous avons ca.1culé les
sections efficaces relatives aux deux longueurs d'onde Â1 et i 2 , l'exci-

- 161 -
tation optique comprenant aJ.ors deux échelons. Celles-ci varient de
2.5 10-19 cm2
(niveau 12S photoionisé par la première impuJ.sion laser 0
à ~1 = 7800 A; la section de photoionisation à Â = ~2 est toujours
beaucoup plus faible que celle obtenue à~= Â1 , car elle correspond
à une énergie très supérieure à l'énergie du seuil de la réaction) à
1 -21 2 ( . ) , quelques O cm ru.veau 12P. Dans le cas le plus defavorable
(niveau 12S) le rapport de la densité de photoélectrons créés à celle
des atomes très excités n'excède pas 3 x 10-5 . En utilisant les va
leurs de sections efficaces (e--atome) caJ.cuJ.ées par Saraph2 ~ recom
mandées par Perciv~2 ~ on peut évaJ.uer le taux de collision corres
pondant et le comparer au taux mesuré. L'erreur résuJ.tante n'excède
pas 3% dans le cas du niveau 12S et est parfaitement négligeable pour
tous les autres niveaux excités. Notons que l'effet des collisions
(e--atome) revient en fait à ajouter un terme de dépopuJ.ation au se
cond nombre de l'équation (E28), terme d'ailleurs indépendant de la
pression de gaz rare et toujours très inférieur à K .• l.
2) Rayonnement du corps noir.
26 t , , 1 "nf d T.F. Gallagher et al. ont mon re recennnent li luence u
rayonnement du corps noir sur la durée de vie·radiative de l'état con
sidéré, l'effet étant particuJ.ièrement important dans le cas des états
de Rydberg. La prise en compte de cet effet revient à remplacer 1/~ 0
(Eq.(E.28)) par 1/~ + 1/~D où~ représente la durée de vie rad~ative 0 0
et ~Dun terme correctif' calcuJ.able à partir des éléments de matrice
dipolaire reliant l'état considéré à tous ceux auxquels il se trouve
radiativement connecté. Cet effet n'affecte en rien la mesure des sec
tions efficaces puisqu'il revient à ajouter un terme constant au second
membre de l'équation (E28). SeuJ.es les durées de vie radiative dédui
tes de l'ordonnée à l'origine des courbes 1/e = f(NA) peuvent s'en
trouver affectées. Nous discuterons ce point dans l'appendice V.
3) Durée de vie radiative des états de Rydberg.
Comme nous l'avons mentionné au cours de l'exposé la métho
de de fluorescence résolue dans le temps permet d'obtenir, par extra
polation à densité nuJ.le d'alcalin, les valeurs des durées de vie ra
diative des niveaux étudiés. Les résuJ.tats obtenus sont rassemblés dans
le tableau X. Nous discuterons les résultats obtenus et les compare
rons à des valeurs théoriques dans l'appendice V, les mesures de durée
de vie radiative n'étant pas le but principaJ. de notre travail.

- 162 -
L'obtention de ce11es-ci n'est cependant pas sans intér3t, 1eur con
naissance étant requise dans de nombreux domaines de la physique :
astrophysique, physique des p1asmas, physique atomique (dépouillement
des expériences de transfert d'excitation par 1a méthode de fluorescen
ce non réso1ue dans 1e temps par, exemp1e).
Niveau "C 0
12P 1.55 .:!: 0.20
14P 2.60 + o.4o
17P 6.40 ± 1 .30
22P 14 .:!: 5
12S o. 77 ± O .15
14S 1.26 .:!: 0.25
16S 2 . 1 9 .:!: O . 50
18S 3.30 .:!: 0.70
Tab1eau X
Durée de vie radiative~ (en µs) d'états Set P très excités
0du rubidium.
4) Superradiance et états de Rydberg.
- - -- ,._ -
Bien que, chrono1ogiquement, cette étude se p1ace à 1a fin
de notre travai1 i1 nous para!t uti1e de 1'insérer à cette p1ace pour
deux raisons :
i) 1a superradiance est un phénomène radiatif et non co11isionne1,
mais i1 est important d'en bien conna!tre les effets pour aborder 1'é
tude expérimenta1e des phénomènes col1isionnels, particulièrement dans
le cas des expériences en ce1luJ.e (où 1es densités d'atomes excités
peuvent atteindre des va1eurs re1ativement é1evées). La prise en comp
te de cet effet conditior..ne dans de nombreux cas la procédure expéri
menta1e à uti1iser lors des études de co11isions. Notons, que fortui
tement, la superradiance, comme nous l'avons indiqué au chapitre V,
jouait peu de r8le dans le cas des états de Rydberg P par lesquels

- 163 -
nous avons commencé notre travail (en 1976, date à laquelle les étu
des expérimentales des phénomènes de superradiance n'en étaient qu'à
leur début), essentiellement à cause de la faible efficacité du pro
cessus de peuplement.
ii) l'exposé relatif aux résultats collisionnels pourra 3tre ain
si présenté sous la forme d'un ensemble cohéren~.
La superradiance est un phénomène d'émission spontanée coopé
ratif: dans certaines conditions un ensemble d'atomes présentant wie
inversion de popu1ation sur une transition donnée peut émettre de ma
nière directive un rayonnement beaucoup plus intense et de constante
de temps beaucoup plus rapide qu'il ne le ferait si les atomes pou
vaient 3tre considéré comme indépendants les uns des autres (émission
dipolaire classique). Cet effet, prévu dès 1954 par Dicke27 , fait
l'objet de très nombreuses publications théoriques. Les travaux expé
rimentaux sur ce sujet ne sont apparus que beaucoup plus récemment28 ' 2~
Il s'agit d'un problème à N corps très complexe pour lequel
des approches simplifiées aussi bien semi-classique30 que quantique31
ont été proposées. Nous renvoyons le lecteur intéressé par la discus
sion des approches théoriques au travail de P~llet32 • Nous utiliserons
ici wie formu1ation très simple, mais suffisante pour notre propos,
qui est essentiellement de montrer les principaux effets de la super
radiance sur l'étude expérimentale des états de Rydberg, sans nous
préoccuper des problèmes très complexes32 posés par une interprétation
quantitative précise de celle-ci (dégénérescence des niveaux, effets
de polarisation, régimes différents de superradiance ..• ). Une émission
superradiante se produit dans Wl échantillon de longueur l si le temps
caractéristique ~R de couplage des dipSles (qui interagissent sur la
longueur l avec le champ électromagnétique servant à la création des
atomes excités, de densité n) est beaucoup plus petit que leur temps
de déphasage T;. ~Rest donné par3°, 31
?:R Sn
= n.1À2 A
où~ et A sont, respectivement, la longueur d'onde et le coefficient
* d'Einstein de la transition superradiante. Pour nos expériences T2 correspond au déphasage Doppler29 (T; """À). Il faut donc que la condi
tion suivante soit remplie:

- 164 -
* T2 R=-»1
C:R avec R,,..., ~3
L'émission superradiante présente les caractéristiques suivantes
- elle est directive (colinéaire au faisceau excitateur) et non
isotrope,comme dans le cas de l'émission dipolaire classique ;
- son intensité est proportionnelle à n 2 (et non à n comme dans
le cas dipolaire) ;
- elle est retardée d'un temps ~D par rapport à l'impulsion d'exci
tation initiale, avec :
81"tK ~ = K't:R = nl).2A
où K est un facteur qui dépend de l'approche théorique choisie mais qui,
dans tous les cas, est à peu près indépendant den. Cette émission di
rective, intense et brève (la forme temporelle de l'émission superra
diante est complexe, mais sa durée n'excède pas quelques~) modifie
complètement le comportement radiatif de la vapeur.
La première mise en évidence de la superradiance dans les
atomes alcalins est da à Gross et al. 29 , qui ont étudié les niveaux
* peu excités du sodium. Pour ceux-ci les valeurs de T2 et de ~D sont
faibles(~ 10 ns). Les états de Rydberg présentent une situation parti
culièrement favorable à l'observation de transitions superradiantes,
car les écarts énergétiques entre niveaux voisins sont faibles, ce qui
conduit à l'existence de nombreuses transitions radiatives de grande
* longueur d'onde. Ainsi la condition T2/~R )> 1 est facile à remplir
car ce rapport varie comme i 3 • De plus les valeurs de Sl que l'on peut
attendre sont plus grandes que dans le cas des états faiblement exci
tés rendant ainsi l'expérimentation plus aisée. En contrepartie l'in
terprétation quantitative devient très délicate car de nombreu:s.es
transitions peuvent donner lieu à une émission superradiante et les
modèles théoriques, déjà complexes, sont développés pour des situa
tions expérimentales beaucoup plus simples (système à deux niveaux).
Notre expérimentation33 a porté sur les états de Rydberg du
rubidium. Sans restreindre la généralité de nos observations nous al
lons considérer un niveau particulier (12S)1 supposé peuplé en un temps
très bref (N 5 ns; le processus de photoexcitation s'effectue en deux

- 165 -
étapes à l'aide de lasers pul.sés, comme nous l'exposerons plus loin).
Le diagramme énergétique est représenté sur la figure 19. Nous avons
observé la variation temporelle de la densité de popul.ation aussi bien
du ni veau 12S que des ni veaux voisins radiati vement connectés, en dé
tectant les raies de fluorescence (dans le domaine visible) issues de
ceux-ci à l'aide d'un système similaire à celui utilisé pour les étu
des collisionnelles. La seul.a différence vient de l'utilisation d'un
ana1yseur rapide de transitoires (Tektronix R7912) à la place du sys
tème de comptage. Un résu.ltat typique est montré figure 20. Les obser
vations, effectuées pour des densités d'atomes excités comprises entre
108 et 1012 cm-3 conduisent aux conclusions suivantes:
- les niveaux radiativement connectés au niveau pompé sont peuplés
très rapidement par cascades superradiantes. La figure 20 montre les
effets de la cascade 12S - 11 P +- 9D -fi- 7F. En effet nous n'observons
pas directement les transitions superradiantes (leur longueur d'onde~
de l'ordre de 20 µ, nécessiterait l'emploi de détecteurs infrarouges
très rapides) mais leur effet sur la popul.ation des niveaux. Le carac
tère superradiant de ces émissions a été cependant clairement mis en
évidence en étudiant, dans une grande gamme, la variation du délai
après lequel la popul.ation des états excités e·st maximum, ce en fonc.
tion aussi bien den que del. Dans les deux cas la variation attendue
( -1 -1) , , , 33 CD N n l a bien ete observee .
- le niveau initia1ement peuplé aussi bien que les niveaux voisins
radiativement connectés ne sont pas totaiement dépeuplés par superra
diance. Ainsi l'observation d'une décroissance radiative classique (de
constante de temps de l'ordre de la µs pour les états étudiés) est pos
sible quelques dizaines de nanosecondes après l'impul.sion de pompage.
La figure 20 montre même que le niveau 7F n'est pas dépeuplé par super
radiance et présente une dépopul.ation radiative norma1e (la durée de
vie mesurée est en bon accord avec les estimations théoriques), ce pour
des conditions expérimenta1es appropriées ;
- les retards observés sont en bon accord avec les estimations
théoriques del.a référence 31 (va1ables en toute rigueur pour des sys
tèmes à deux niveaux) : cette comparaison est cependant ma1aisée car
il est très difficile d'estimer n à mieux qu'un facteur 3. Une mesure
t ' , t 3 4 .&'f , ' b . res recen e , e~ ectuee sur un systeme eaucoup plus simple, a con-
firmé de manière précise ce dernier point.

- 166 -
E ( cm-1) 1
125
10 D
320001- ~11P
~ 8F 115~ ........ -
90
10 P
7F 105
80
31000 gp
Fig. 19 - Diagramme énergétique de l'atome de rubidium. Les flèches indiquent les principales transitions superradiantes mises en évidence après peuplement du niveau 12S.
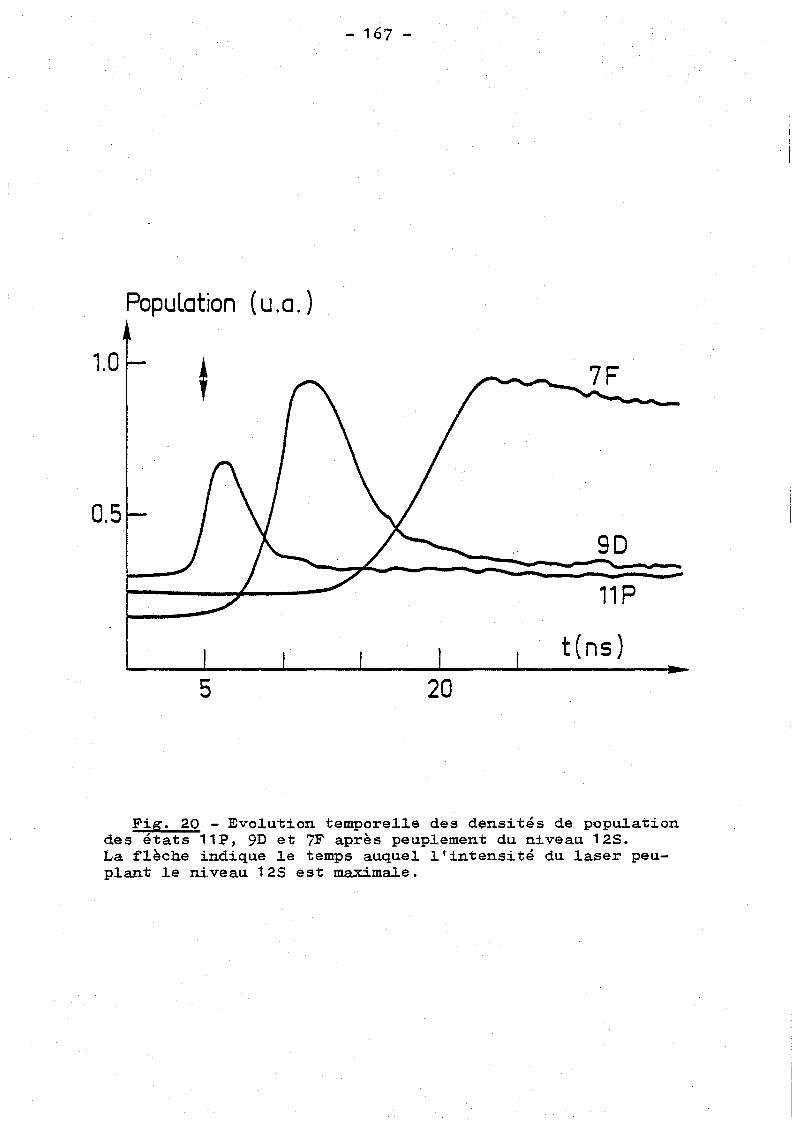
Population ( u .a.)
1.0 t
0.5
5
- 167 -
20
90
11 P
t ( ns)
Fig. 20 - Evo1ution temporelle des densités de population des états 11 P, 9D et 7F après peuplement du ni veau 12S. La f1èche indique 1e temps auque1 l'intensité du 1aser peuplant le niveau 12S est maxima1e.

- 168 -
En résumé, le bref aperçu des résultats relatifs à la super
radiance dans les états de Rydberg conduit à la conclusion importante
suivante : m~me à relativement faible densité d'atomes excités le com
portement radiatif de la vapeur est complètement modifié. Les niveaux
voisins radiativement connectés sont très efficacement peuplés dans
des temps (N 20 ns dans le cas présent) très inférieurs à ceux atten
dus("" 1 - 2 µs ici) d'un comportement radiatif classique. Il convient
de tenir compte de ces effets dans l'interprétation de toute expérien
ce de fluorescence résolue dans le temps. En particulier l'utilisation
des méthodes décrites au chapitre précédent requiert de débuter l'ob
servation un temps suffisamment long après le pulse excitateur (si on
a, par ailleurs, observé que la densité d'atomes excités était suffi
sante pour donner lieu à l'apparition de transitions superradiantes). --
Un dernier point à mentionner est que la superradiance peut 3tre uti-
lisée pour peupler efficacement des niveaux difficilement accessibles
par des méthodes classiques. Ainsi Hugon et al. ont pu étudier les
propriétés aussi bien radiatives35 que collisionnelles36 des états de
Rydberg F du rubidium.
b) Etude collisionnelle des états de Rydberg P; Articles 2 et 3. -------------------------------------------------------------Nous insérons les deux articles que nous avons publié sur
la dépopulation collisionnelle des états de Rydberg P. Le premier
(Article 2) concerne le niveau 10P du potassium, le second (Article 3)
présente une étude plus complète de quatre niveaux nP (12 4 n '= 22) du
rubidium. Le choix des niveaux étudiés répond à divers critères. Nous
désirions commencer l'étude des états de Rydberg par un niveau den
peu élevé pour effectuer la transition avec les niveaux faiblement
excités étudiés précédemment. Nous voulions cependant démontrer les
possibilités d'études des propriétés collisionnelies de niveaux pour
lesquels les forces d'oscillateur des transitions d'excitation étaient
faibles. Le niveau 10P du potassium répondait bien à ces conditions. 0
La l.ongueur d'onde d'excitation (À(4S ~ 10P) ,v 3000 A) permettait
l'utilisation d'un lasen à rhodamine doublé en fréquence (laser 2).
Les difficultés d'utilisation de ce laser, ainsi que les progrès tech
nologiques rapides en ce domaine, nous ont rapidement conduits à lui
préférer un matériel commercial (laser 3) plus facile d'emploi. Notre
choix s'est alors porté sur le rubidium, pour la poursuite de nos étu
des, car les niveaux de Rydberg S, Pet D étaient facilement accessi-

- 169 -
bles par excitation à un (états P) ou deux (états Sou D) éche1ons à
1'aide de ce matériel. Ce choix permettait ainsi un développement très
souple de notre programme de recherches.
L'importance relative de ces deux articles est très inégale.
L'étude du ni veau 1 OP présent.e le caractère d I une expérience prélimi
nai:e. La technique employée (f1uorescence non résolue dans le temps)
est d'un emploi dé1icat pour la mesure du quenching (voir chapitre V) ;
nous l'avons abandonnée dès que nous avons disposé de l'analyseur mul
ticanaux rapide. L'aspect expérimental de ce travail ne nous para.!t pas
mériter de compléments particuliers et nous commenterons 1es résultats
obtenus lors de la discussion générale. Notons cependant que ce travail
fournissait la première mesure de la section de quenching (alcalin
alcalin) d'un état de Rydberg ainsi que 1a première mise en évidence
de la stabilité de certains de ces états vis-à-vis des collisions
avec les gaz rares~ En effet les sections efficaces mesurées pour les
gaz rares (,v 10 12 ) apparaissaient de deux ordres de grandeur infé
rieures à celles mesurées par T.F. Gallagher et al.18 dans 1e cas des
états D du Na, seuls résultats relatifs aux états de Rydberg dispo
nibles à l'époque. Nous reviendrons plus loin sur ce point. Notons
pour terminer une erreur dans la figure 3 de l'article 2: la courbe
de potentiel relative à la paire (K+ + K-) doit évidemment présenter
un caractère coulombien attractif.
L'étude des états P du rubidium (article 3) présente un ca
ractère beaucoup plus complet et systématique que le travail précédent.
Nous avons utilisé une méthode de fluorescence résolue dans le temps,
particulièrement bien adaptée aux mesures des sections de quenching.
Les procédures sont détaillées aux chapitres IV et V et n'appellent
pas de nouveaux commentaires.

- 170 -LE JOl"R"•'- DE PH\S{Ql"E - Lf:·n·1u:s
Clasrnicati,,n Ph\'Sics .~r,cruus .s.~;:n - s.:~o
TO~IE 37. Jt.:lllET·AOL'T 1976. PAGE L-16~
COLLISIONAL DEPOPULATION OF A HIGH-LYING P STATE OF POTASSfu~I
F. GOUNAND. J. CUVELLIER. P. R. FOURNIER and J. SERLANDE
Centre d'Etudes Nucléaires de Saclay, Service de Physique Atomique. B.P. 2, 91190 Gif-sur-Yvette, France
(Reçu le 5 avril 1976, accepté le 3 mai 1976)
Résumé. - On a mesuré les sections efficaces de dépopulation collisionnelle totale du ni~·eau 10 P du Potassium. à 470 K. induite par collision avec des atomes de Potassium ou de gaz rare : Q (K) = 6000 ± 3000A2
: Q (He)= 18 ± 6A 2; Q (Ar)= 8 ± 3 A2
: Q (Xe)= .W ± 15A2 •
Lïmportance des transferts d'excitation électronique vers les niveaux voisins est indiquée. Il est montré que l'électron de valence a un comportement quusi-libre.
Abstract. - T ota! cross-sections for the collisional depopulation of the 10 P level of Potassium by Potassium and rare gases have been measured. with the following results : Q ( K) = 6 000 ± 3 000 A 2 :
Q (He)= 18 ± 6 A2; Q (Ar)= 8 ± 3 A2
; and Q (Xe)= .W ± l5 A2• The importance of the elec
tronic excitation transfer to neighbouring levels is clearly demonstrated as well as the influence of the quasi-free behaviour of the valence electron.
Because the outer electron is. in a very large, weakly bound orbit. atoms in high-lying or Rydberg states exhibit rather unique properties : long radiative lif etimes, giant polarizabilities and great sensitivity to various perturbations (electric field. collision processes ... ). Investigations of some of these properties have already begun (1, 2].
Experiments on excitation transfer and total collisional depopulation (quenching) of lower excited states of alkali atoms have been perfonned in our laboratory [3]. At the same time, the theoretical calculation of a!kali-rare gas potential curves [4] permits a satisfactory approach to the problem of ine!astic collisions at thermal energies. The extension of such experimental and theoretical studies to Rydberg states seems to be very interesting. We report here. for the first tune, an experimental study of the collisional depopulation of the 10 P level of Potassium due to collisions with Potassium or rare gas atoms in their respective ground States.
We have measured the total quenching cross-sections and have also determined the cross-section for a particular quenching process : the electron excitation transfer to a neighbouring, well-defined., atomic level. We are concemed with two reactions : one involving the total quenching
K(!O P) + X~ K('i6 10 P) + X
where K( 10 P) is a Potassium atom lying in the 10 P level. X either a ground-state Potassium ( K) or rare gas (G) atom. Qx the quenching cross-section. and
K( ::/= 10 P) a Potassium atom in its final (ionic or neutral) state, and the second reaction involving the particular specific deactivation
K(lO P) + X Qx(lOP-n, K(f) + X - 6.E
where K(f) is a Potassium atom lying in the final f atomic leve!, Qx(lO P - f) the cross-section associated with the considered process and M the energy splitting between the two levels. The K and G subscripts will indicate that the corresponding quantities refer to (Potassium-Potassium) and (Potassium-Rare gas) collisions respectively. Figure 1 shows the levels involved.
The experimt:ntal set-up shown in figure 2 is quite similar to the one described in ref. [3]. A reference cell, filled with Potassium and kept at a fixed temperature, allows us to monitor both the wavelength and the intensity of the light source. which consists of a flash-lamp pumped dye laser (Rhodamine 6 G). associated with an extra-cavity frequency doubling device that"' uses a non-linear crystal (ADA) [5]. The UV pulses (À = .2 992 A) have a half-width time duration of about 3. ï µs, an output power of 0.5 mJ per pulse and a linewidth of 0.4 A. The repetition rate is one pulse per 6 s. The power is enough to t:nsure a high population of the 10 P level in spite of the very low oscillator strength. which is a typical feature of the Rydberg states. To oµr knowledge it is the first time that the population of a highly ~xcited P state of an alkali atom is directly achieved by selective optical excitation.

- 1 71 L•I i(I JOCR;";AL DE PHYSIQUE ·· 1.1-:n·R[S N" i-~
f Energy (cm-1
)
Ionisation limit (35009.78)
34000
12 P 10 f
T
___ 12 s __ 10 d
_111
33500
11 P
_ 10p
-10s
33 000 __ gp
9d
Bd
9f
Bf
FIG. l. - Energy levels of the Potassium acom involved in this experiment.
Oven
p
p
laser 2992.A
p
F1G. 2. - Schemacic diagram of the apparatus : P : monochromator and high-speed photon counting system; T : trigger.
We observed the population of the JO P Ievel by looking at the direct fluorescence of the (10 P - 3 D) transition (/. = 8 418 A). At Potassium densities of about 1013 cm- 3, numerous other optical transitions. coming from upper levels (9 D, 11 S. 9 F. 11 P. I O D, 12 S, 12 P) collisionally populated from the I OP are found to be prescnt. We did not study the lower levels bccause of the cascading cfl'ects that occur (except for the 8 F level). The variation of the population of the 9 D. 11 S. 9 F and 11 P levels. due to the addition of rare gas dem.itit!S of ahouc 10 15 cm- 3
•
has also heen observed. Ali the experiments are performed at a u:mperature of 470 K.
The determin:.ition of the Qi.. and QG quenching
cross-section is achieved by looking at the variation in the intensity ratio of the fluorescence line (i.. = 8 418 A) from each of the two cells, as a fonction of Potassium or rare gas density in the experimental cell (in the later case the Potassium density is kept at a fixed value). We modify the classical Stem-Volmer method to our experimental conditions. The requirements for its valîdity [6] (low stimulated emission, no absorption or collisional broadening, low repopulation of the 10 P level, ... ) are fullfilled in a satisfactory way. The results we obtain are shown in table 1. For the derivation of the crosssection we use the radiative lifetime of the l O P Jevel as computed from the results of Anderson and Zilitis [7], since their computation also produce good agreement with the Sodium lifetime results (l].
TABLE 1
C omparison between the 10 P quenching cross-sections and the total e/aslic scattering cross-sections
for electrons of 0.15 eV energy
Process Q (A2) Process Q. (A2)
- - -K-K 6000±3000 e--K ::::: 1 000 K-He 18 ± 6 e--He ::::: 6 K-Ar 8 ± 3 e--Ar ::::: 1.5 K-Xe 40 ± 15 e--xe ::::: Il
We have also determined the excitation transfer cross-section from the l O P level to the group of levels (9 D, 11 S), when the collision partner is a groundstate Potassium atom. The experimental method ( sensitized fluorescence) and the derivation of the ~ross-section are quite similar to those described in ref. [3]. The obtained value is
QK(lO P - (9 D, 11 S)) = (800 ± 400) A2 •
ln the following discussion we will refer to neutral and ionic channels for reactions whose final product is, respectively, an atom lying in a well-de.fined excited state or an ion. The value of QK (6 x 10- 13 cm- 2
)
is very high (the geometrical cross-section value is about 9 x 10- 13 cm2). Two types of mechanisms (neutral or ionic channel) can achieve the collisional depopulation of the 10 P level. We can observe numerous neutral channels and we measure one of them to be about 10 ~,~ of the quenching. When considering the statistical weights and the respective position in the energy diagram of the energetically accessible levels, one can see that the neutral channels account for an important part of the 10 P level quenching.
Lee and Mahan [8] have sho\\ln that the two following reactions (ionic channels) are present (see Fig. 3) :
K( 10 P) ~ K(4 S) - Ki + e
K( 10 P) + K(4 S) - K ~ + K -
( l) (aSSüCÏalÎVe ioniz.alion)
(!)
- 172 -'.'!·· 7-l! COLUSIO'\i .. \L DEPOPCLA T!ON OF A HIGH-L Yl:--G P ST.-.\TE L-1-1
but the values vf the corresponJing cross-section are not kno .... n. A recent paper [9] indicates that the associative ionization cross-section for the 7 D level of Cesium is about 3 x w- ts cmz. In the case ofHelium the cross-sections are of the same order of magnitude. Even if the cross-sections for reactions (1) and (2) would reach lO- 14 cmz, the quenching of the 10 P would still remain mainly due to inelastic collisions giving rise to neutral channels. Thus. since the ionic channels do not contribute more than a few percem to the total 10 P depopulation value, collisional ionization does not represent the main quenching process for this level.
-> ..S!. >,
f:' a, C":
Lù
-\-\ \
t
\\\ \ ,,,
\ \\ \ \\ \ ,, \ ,, ///
\ , ..... .,,, // ,__.-,.,,, 2 l
4 1
5 l
8 !
0
lntemuclear distance (Al
10 1-.,.
FIG. 3. - Estimatcd potential energy curves for K 2 and K~--
We will now discuss the values obtained for QG. When considering the data concerning the potential curves of the (K + -rare gas) ions (1 OJ, the ionic channels seem to be energetically forbidden, since the 10 P level lies about l 600 cm - 1 below the ionization limit. Thus only neutral channels can contribute to the quenching. Sorne of these have been observed. The Q0 cross-sections (see Table I) are noticeably higher than the values reported in the case of lower excited levels of alkali atoms (11], for which the number of energetically accessible channels are less numerous. The Potassium-rare gas energy curves correlated to the levels under study have already been obtained using the method described in ref. (4]. We hope that a comparison between experimental and theoretical excitatiçm transfer cross-sections, calculated from the potential energy curves, will be possible in the near future. ·
The outer electron of an alkali atom lying in a Rydberg state is very weakly bound (the average value of the orbital radius is about 55 A in our case).
Thus it set:ms very interesting to compare the values of QK and QG to the values of the elastic scattering cross-se1:tions of electrons on the corresponding targcts at an incident energy of the ele1:tron e4ual to thosè of the valence electron in tht! 10 P level ( ~ 0.15 eV). These cross-sections [ 12] are also reporteJ in table I. Two main features appear. First, the quenching cross-sections are always higher than the com:sponding elastic scattering cross-sections Qe; second, the quenching cross-sections behave quite similarly to the corresponding Qe, in that there is a great difference between the Potassium-Potassium and Potassium-rare gas cross-section values and a similar variation as a function of the considered rare-gas. One cannot attribute the low values of QG to the absence of ionic cbannels, since they only make a smaU contribution to the high QK value. Instead, these low values seem related to the quasifree behaviour of the valence electron, and its weak interaction with. the noble gases in our experimental conditions. Studies conceming the collisional broadening and shifts of the Rydberg states of the alkali atoms (13] have clearly pointed out the importance of the quasi-free behaviour of the outer electron. The comparison mentioned here suggests future e.'<perimental as well as theoretical investigations. The determination of the quenching cross-sections as a function of the principal quantum number n (i.e. the energy of the valence electron) would permit one to check the reliability of the comparison with the elastic scattering of slow electrons. In particular the behaviour of QG would noticeably differ according to whether the elastic scattering by the considered gases exhibit or do not exhibit the Ramsauer-T ownsend eff ect. The proposed analogy however would not give rise to quenching cross-sections directly related to the geometrical cross-sections (i.e. - n~), since the elastic scattering cross-sections of very low energy e!ectrons exhibit various behaviours, according to the nature of the targets. A theoretical approach, taking into account the quasi-free behaviour of the valence electron pointed out in this study, seems to be possible. Although previous theoretical treatments have called this fact to mind, the experimental results reported here clearly point out the necessity of inc!uding this behaviour.
We wish to thank Dr. Gutcheck for a critical reading of the manuscript.

L-lï:
- 173 -JOL·R~AL DE PHYSIQL'E I.ETrRf:"i N° 7-X
Reforences
(1] GALLAGHER. T. F .. EDELSTF.l~. S. A. and H11.L. R. M .• Phys. · Re,·. À 11 (1975) 1504; Phys. Rev. Leu. 35 (1975) 644.
[2] FABRE. C. and HAROCHE. S .. Opt. Commun. 15 ()975) ::-4; and
DucAS. T. W., LITTMAN, M. G., FR.EEMAN, R. and KLEPPNER. D .. Ph,vs. Re,•. Lett. 35 (1975) 366.
[3] CL'YELLIER. J., FOURNIER, P. R .• GouNAND. F .• PASCALE. J. and BERLANDE, J., Phys. Rev. A Il (1975) 846.
(4] PASCALE. J. and VANOEPLANQUE., J., J. Cliem. Phys. 60 (1974) 2278.
(5] NEt:MANN. V .• Thesis. Paris (1975). Unpublished. [6] MASSEY. H. S. W .. Elecrronic and lo11ic Impact Plrenomena.
Vol. 3 (Oxford), p. 1497. 1969.
[7] A~DERSON, E. M. and ZILITIS. V. A .• Op1. Specirosc. 16 (1964) 99.
[8] LEE. Y. T. and MAHAN. B. H .• J. Chem. Phys. 42 /1965) 2893. (9) DOBROLEZH, B. V .• KLYUCHAROV, A. N. and SEPMAN, V. Y.,
Opt. Spectrosc. 6 (1975) 630. [10] MASON, E. A. and SHAMP. H. W .. Ann. Plzys. 4 (1958) 233. [11] LIJNSE. P. L., in Report No 398, Fysich Laboratorium Ryksu
niversiteit Utrecht (1972) (unpublished) and references therein.
[12] See ref. [6] Vol. 1. Chapter 6. (13) MAZING. M. A. and SEREEPINAS. P. D., S01·. Phys. J.E.T.P.
33 (1971) 294.

PHYSICAL REVIEW A
174 -VOLUME 15, NUMBER 6 JUNE 1977
Collisional depopulation of Rydberg P states of rubidium at thermal energies
F. Gounand, P. R. Fournier, and J. Berlande C.entre d'Etudes Nucléaires de Saclay. Service de Physique Atomique. B.P. 2-91190. Gif-sur-Yverte. France
(Received 12 November 1976)
We have measured total cross sections for the collisional depopulation of high-lying nP states (12::; n ::; 22) of rubidium induced by thermal collisions with ground-state rubidium or rare-gas atoms. lt is observed that for n 2: 14 the rubidium-rare-gas cross sections do not vary with the principal quantum number, contrary to previous hypotheses. Our results are discussed using the simple argument of quasifree behavior of the valence electron involving either an elastic-scattering or a binary-encounter approach. However, the disagreement observed between the experimental and the computed cross sections suggests that a three-body approach is generally needed for a good understanding of collisional processes involving highly excited states.
L INTRODUCTION
The interest in studying the collisional properties of Rydberg states has been clearly pointed out by recent experimental1
-3 and theoretical4
~ works. In a previous Letter3 concerning the total collisional depopulation of a high-lying P state of potassium due to a perturbing gas (potassium or rare-gas atom in its ground state), we have shown that the quenching cross-section values vary, as a function of the nature of the perturber, as the corresponding electron-atom elastic scattering cross-section values. To study this similarity in more detail and to obtain more information on the collisional processes involving Rydberg states, we began a study of the collisional quenching, at thermal energies of highly excited P states of rubidium atoms as a function of both the binding energy of the valence electron and the nature of the perturbing atom trubidium or rare-gas atom in its ground state). These states are created in a rubidium vapor by photoexcitation from the 55 ground state. We have chosen rubidium because the oscillator strengths of the 55-nP transitions are much larger than the corresponding ones in the case of potassium, thus making possible such measurements with a high sensitivity.
Various collisional mechanisms lead to the depopulation of a given highly excited Level: excitation transfer to a neighboring level, direct or associative ionization, etc. To our knowledge no data concerning the quenching of highly excited levels are available. SUch data are of great practical interest in numerous areas (astrophysics, ionized gases). Moreover, they permit the evaluation of the importance of the collisional effects in experiments dealing with other properties of Rydberg levels (lüetime or fine-structure measurements, for example). Finally, the quenching-rate coefficients are essential parameters in the analysis of experimental data concerning a particular calli-
15
sional process (electronic excitation transfer, for exa.mple). The quenching cross-section values, which are upper limits for the cross-section values associated with particular inelastic channels, and their behavior as a function of the main parameters of the collision (binding energy of the outer electron, nature of the perturbing gas) provide, as we will see, a most interesting check of the available theoretical _ approaches.
ll. EXPERIMENTALSET-UP
We have measured the total quenching cross section corresponding to the process:
Rb(nP)+X~Rb(=nP)+ X, (1)
where Rb(nP) is a rubidium. atom excited to the nP state; X, either a rubidium ground state (Rb) or rare-gas (G) atom; Qx(nP), the quenching cross section of the nP Level; and Rb ( .=nP), a rubidium atom excited to astate (neutral or ionic) düferent from the initial one. The quenching cross section Qx(nP) is defined, as usual in a gas-cell experiment, from the relation
K~(nP) = Qx(nP)v-ai.-z, (2)
where K~(nP) is the rate constant for reaction (1), and Ü-ai,.r is the Maxwellian-averaged relative velocity of the two colliding atoms. The values of the K~(nP) rate constants are deduced (see Sec. m A) from the measurement of the effective lifetime of the nP Level as a function of the density of the perturbing atom.
The experimental set-up is shown in Fig. 1. A nitrogen laser (Molectron UV 1000), and a Rhodamine B dye laser, followed by an extra-cavity frequency doubling device using a KDP crystal produce the uv pulses (,\ - 3000 À). These pulses have an average duration of about 3 nsec, with an energy of 10-5 J per pulse and a linewidth of 0.3 À. The repetition rate can be varied up to 30 Hz. The uv
2212

- 175 -
15 COLLISIONAL DEPOPULATION OF RYDBERG P STATES OF •.• 2213
power is high enough to insure a noticeable population of some highly excited P ~tates of rubidium. The uv beam passes through the experimental cell containing rubidium vapor. This cell is set in an oven maintained at a fixed temperature (T = 460"K). The liquid rubidium is located in a sidearm, the temperature of which can be kept at a fixed value (over the range 330-400 ° K) by a separate heating system. Thermocouples provide the measurement and the regulation of the temperature in the whole system. The measurement cell is connected by a capillary to a vacuum or gas filling system. After a typical baking procedure the residual pressure is about 5 x 10·7 Torr. The rare-gas pressure, ranging from 2 x 10·3 to a few Torr, is measured with an oil-manometer and the rubidium vapor pressure, ranging from 8 x 10-e to 5 x 10-« Torr, is determined by the temperature of the sidearm containing liquid rubidium. 7 The fluorescence light is analyzed with a grating monochromator which selects a Une originating from the pumped level (we observe the nP-4D transition in the 7000-7600-Â range). The lüetime measurements are then made by using a high-speed cooled photomultiplier (C 31034 type), a 100-MHz counting system (amplüier and discriminator) and a twenty channel scaler connected to a POP 15 computer. The channel width can be varied from 40 nsec up to several J,LSec according to the experimental requirements. A trigger system starts the analysis a short time (-15 nsec) after the laser pulse.
Ill DATA ANALYSIS
A. Decay frequency determinations
As mentioned previously, the quenching crosssections were obtained by measuring the effective lifetime of the nP excited level as a function of the density of the perturbing gas. We can write
1 1 1 1 -=-+--+-, T Trad TRII TG
(3)
where T is the measured lifetime and Trad the radiative lifetime (1/T rad =r;1A 1 where the A;' s are the Einstein coefficients of all the lines originating from the nP level). The last two terms on the right side of Eq. (3) represent the effect of collisions with rubidium and rare-gas atoms, respectively, on the lifetime. The se terms can be written as
1/T R11: NRIIQRll(nP}vRll-Rll '
1/T G =N eQdnP)vRII-C"1 (4)
where NR11 and Ne are the rubidium and rare-gas densities; ii'Rb•Rll and vR11-c are the Maxwellian averaged velocities of the relative motion of the two colliding atoms for the {Rb-Rb) and (Rb-G) colli-
Cell
Monochromator
Amplifier Discriminator
Nitrogen Laser
+ Oye Laser
+K.O.P. Crystal
Trigger System
POP15 Computer
Multichannel Analyser
FIG. 1. Schematic diagram of the apparatus.
sions, respectively. Equation (4) implies that other atomic levels do not contribute radiatively or collisionally to the population of the considered nP level, that is to say no "repopulation" of the nP level is present (when originating from more highly excited level this repopulation is also called cascading).
Equations (3) and (4) show that the variation of 1/T as a function of NR11 (with Ne =O) is linear with a slope proportional. to Qa11(nP). Similarly, the variation of 1/T as a function of NG (withNl\11 =Ct) allows the determina.tion of Qc(nP). Moreover, the extrapolation of the 1/T value to zero rubidium pressure yields the natural radiative lifetime of the col".sidered level. Results concerning the T..,.
values are published elsewhere. 8 They range from l. 55 µsec for n = 12 to 14 J,Lsec for n = 22. Repopulation effects can be seen in Fig. 2, which shows semi-log plots of the population decay of the 22P level for two different time scales. The linèar decay of log (I1) seen at early times (curve A) indicates that repopulation effects are almost negligible during that time. At late times {curve B) the effect of repopulation of the level becomes apparent. This is in accordance with the well-known fact that, in a first approximation, repopulation effects become noticeable after a time on the order of the effective lüetime of the repopulating level. For the rare gas the lowest pressure investigated was 2 x 10-3, Torr. At this pressure no departure from the decay obtained for zero rare-gas pressure was observed in the case of the three gases investigated here. Then the rare-gas pressure was increased by small amounts in order to follow
- 176 -:?.214 F. G O l' l'i A N IJ , P. R . F O U R N I E R , A N D J . B ER LA :"i D E 15
5 -Ill l O l -i: ::, .ci ... 2
t (p.s)
NRb :7.1012 cm-3
.= >, :t: Ill C: QI -.!: ~
0 0
0 • 0 •
0 0 ••
(8) 0 0 0 0
, t (p.S)
FIG. 2. Experimental decay curves observed for the 22 P state in rubidium (perturber: ground-state rubidium). Points (A) refer to the lower scale, points (B)
to the upper scale.
accurately the 'T variation and to detect any anomalous behavior. For the measurement of Qc(nP) we chose the lowest possible rubidium pressure and verüied that a signüicant change in the rubidium pressure did not affect the value of Qc(nP). Finally, the slope of the curves (i.e., see Fig. 3) determining QRb(nP) and Qc(nP) were obtained from a classical least-square fitting procedure.
B. Uncertainties
J. Neg/igib/e effects
We would like first to discuss the influence on the measured lüetime of impurity gases which may be present in the cell or in the rare gas.
The average background pressure in the cell is 5xl0-,Torr, as indicated previously. When only rubidium vapor is present, collisions between excited rubidium atoms and impurities would add a constant term (l/'T1m11 =N1m•v'Q1-{nP)] to the right si.de of Eq. (3) and therefore affect the measured 1/'T rad at very low rubidium pressures. Experimentally it is observed8 that the measured values of 1/r rad do not exhibit a constant devi.ation from the corresponding theoretical values.9 Moreover the variation of the Trad as a function of the effective quantum number n* is in good agreement with the theoretical prediction of the quantum defect theory (i.e., 'Tn4-n*3}. Finally, during one set of measurements, a small leak in the vacuum system gave ri.se to an increase in the background pressure to 10-s Torr. Such an increase reduced the observed T of the 17P level by 30% for a rubidium
-pressure of 8 x lQ-6 Torr. From this observation one can estimate the quenching cross sections for the residual impurities to be on the order of 4 x 10-t2 cm2 • This indicates that for a resi.dual pressure of 5 x 10-1 Torr the T measured for very low rubidium pressure would be affected by less
than 3% for the 22P level and much less for the other levels. But it does not affect the values of QRb(nP) which are deduced from the slopes of the 1/T = /(NRb) curves (since the background pressure would be only adding a constant value to the observed 1/'T decay).
The concentration of the molecular impurities in the rare gas used (Gaz Industriels de la Courneuve) is less than 7 ppm for neon and argon and less than 3 ppm for helium. In view of the quenching cross sections measured for the rare gases only residual impurities having quenching cross sections on the order of 10-10 cm2 could affect the results of this work. Such cross sections look most improbable and are well above the previously deduced value (4 x 10-12 cm2}. Before its introduc"tion into the measuring cell the rare gas is stored at a pressure of about 500 Torr in a tank where a residual vacuum of 5 x 10-1 Torr was obtained. Therefore a negligible pollution of the rare-gas occurs before it is introduced in the cell.
We are led to concludè that under our experimental conditions impurity gases do not affect signüicantly the measurements.
The nP-4D fluorescence emissions, used to monitor the population decay of the nP levels, are not trapped because their terminal state is not the ground state.
The determination of 1/T Rb from the difference between the measured 1/T and 1/T n,1 [see Eq. (3)] was done assuming that 1/T rad does not change when varying the rubidium pressure. It is easy to show that this introduces a negligible error in view of the magnitude of the trapping effect. Only the nP - 5S transitions are trapped. In the most unfavorable case (12P - 5S transition) the optical path in the cell {-0.5 cm) is much smaller than the absorption length (25 cm, assuming A12p.. 5s = 6.5 x 104 sec-1
, for the highestrubidium density investi-
1/-r (10 6 5-l)
0.5
NRb(1013 cm-3)
0.5
FIG. 3. Plot of the inverse of the experimental effective lifetime vs rubidium pressure for the 17 P level in rubidium.

- 177 -15 COLLJSJONAL DEPOPULATION OF RYDBERG P STATES OF .•. 2215
gated i.e., -1013 cm-s). Moreover this transition contributes only for 15% to the spontaneous emission rate 6 1 A 1 of the 12P lev~l.9 Therefore the maximum.error due to trapping when varying the rubidium density is less than 0.3% of the 1/Tra<S for the 12P level.
Orùy one maximum in the fluorescence intensity is observed when the laser wavelength is swept across the absorption line. We measured the lüetime as a function of the laser wavelength across the absorption line for the 12P level, whose finestructure interval between the 2P 112 and 2P 312 sublevels is on the order of the laser linewidth, and no systematic variation was observed, within experimental error. Thus we consider that the n 2 P 1 12 and n 2 P 3 12 sublevels are both in vol ved in this experiment.
The uv light from the laser exhibits a high degree of linear polarization. The experimental observation were made at a right angle to the uv beam with a detection system insensitive to polarization. By using the multipole expansion of the time dependence of the fluorescence light, one can show that the well-known effects of polarization10
are not important under the present experimental conditions. This was also deduced from the experimental curves obtained.
Although a recent work11 indicates the possible existence of cooperative effects when the laser power is high enough, such effects should not be expected for the present experimental conditions. This was confirmed by measuring the lüetime T
for düferent energies of the exciting beam (attenuating the beam by a factor up to 20) and observing no systematic variation.
During a typical lüetime T the excited atoms can travel over distances on the order of some millimeters. This may introduce a systematic error in the measurement of T, if the atoms escape from the observed volume before radiating. If this is the case a corrective term has to be added to the right side of Eq. (3). In order to reduce any such effect, we used an exciting beam whose dimensions (-15 x 7 mm) were much larger than those of the volume observed by the optical detection system. An almost homogenous population density of excited species is thus insured in the observed volume. We estimate that the resulting error for the largest measured lüetimes ( -7 µsec) would not exceed 10%.
2. E.xperimenral erron
The experimental errors associated with the measured values of Q;c(nP) corne mainly from the accuracy on the 1/T values (coming from counting errors) and from the determination of NRb and Ne. The uncertainty associated with the counting rate
is between ±3 and :tll %, taking into account the dead time of the counting apparatus and the statistical error. The calibration of the time scale of the 20 channel scaler is made with an accuracy of ±1%. Thus one can consider that the determination of a lüetime T value [deduced !rom the log(I1) =/(t) curve] is achieved with better than ±12% accuracy [the relative error on the log(I1) values being smaller than 11% in view of thê obtained counting rates]. The determination of the rare gas pressure is made with an accuracy of better than :t:5% for P c ~ 10--z Torr. The thermal transpiration correction (the oil-manometer not being at the same temperature as the measurement cell) is sufficiently accurate to allow finally the determination of N c in the measurement cell with better than :t:8% uncertainty. We can thus estimate the accuracy of the QG(n.P) values to be about ±20% in the worst cases.
The rubidium density is derived from the temperature of the sidearm containing liquid rubidium. Gallagher and Lewis12 have shown that with proper experimental care, such a determination can be trusted in the lowest range investigated here (-1095 Torr). Precautions similar to those reported in Ref. 12 were taken to insure that the results are independent of the temperature history of the measuring cell. Moreover we verüied that the density measurements agreed within ±15% with those determined from absorption measurements of the resonance line for vapor pressures greater than 6 x 10-s To:rr (the pressure range in the experiment was 8 x 10"'4 to 5 x 10-4 Torr). Our apparatus is not sensitive enough to allow measurements below this pressure. -However, the Tr14 values, as obtained by the extrapolation of 1/T values (measured for 8 x 10-4 -:i. PRb -:i. 5 x 10-• Torr) do not exhibit any anomalous behavior, as pointed out previously. In conclusion we estimate that the determination of NRb is achieved with better than ±25% accuracy (including both the deviation bet:ween the absorption measurements and the calculated vapor pressure (±15%), and an estimated confidence limit of 10% for the vapor pressure curve), giving rise to a total uncertainty of ±37% for the QRb(nP) values in the worst cases.
IV. RESULTS AND DISCUSSION
Table I shows our experimental results. We have chosen to consider exclusively the n = 12, 14, 17, and 22 levels whose ionization energies scale down smoothly. This permits the extension of our previous work3 towards more highly excited levels while keeping a reasonable data collection time (although this is no longer the case for n> 22). The following points can be noted from Table I.

- 178 -
.:?!!16 F. G O U N A N D , P. R . F O C R N I E R. A N U J . B E R LA N O E 15
TABLE I. Quenching cross sections Qx<nP) for nP states in rubidium. All the cross sections are in Â2• E 1 is the ionization energy of the considered level.
Perturber X · Level E1 (eV) n' Rb He Ne Ar
12P 0."19 2.1 X 104 (2.3 ± 0.8) X 103 38:1:7 4.9:1:0.9 15:1:3 14P 0.11 3.8 X
0
104 (J .1 i: 2.5) X 103 56±10 12 :!:2.4 25:!:5 17P 0.066 8.3 X 104 (1.00:i: 0.35) X 104 58±11 11 :1:2.1 28:!:5 22P 0.036 2.3 X 105 (1.6 :: 0.6) X 104 60:!:12 13 i: 2.5 30:!:6
(i) The Qab(11P) cross-section values are about two orders of magnitude larger than the corresponding Qc(nP) values.
(ii) The behavior of the Qab(nP) values as a function of n is different from the corresponding behavior of the Qc(nP) values. A signüicant increase is observed in Qa11(nP) with n that is not directly related to the geometrical size of the excited atom, i.e., -n4
• The Qc(nP) values seem to saturate or even to slightly decrease, within experimental errer.
(iii) For any given level the following inequalities hold:
QRb » QH•> Q,._r > QN• •
Before discussing these results, we would first like to point out that knowledge of the nature of the end products for the quenching process would be of great help for developing experiments concerning the collisional properties of Rydberg states and for the elaboration of theoretical treatments concerning these processes. We wish, therefore, to consider the various processes responsible for the quenching of a highly excited nP state and their relative efficiencies. Two different types of mechanism are involved: (a) The first mechanism leads to neutral end products [the neutral channels of reaction (1)]. There is a transfer of electronic excitation !rom the nP state to other rubidium bound states. (b) The second mechanism leads, when energetically possible, to ionic end products [the ionic channels of reaction (1)].
We shall show that the second mechanism can be considered as negligible with respect to the first one, under the present experimental conditions.
Consider first the direct ionization process:
Rb(nP)+X-Rb•+X+e·, (5)
where X is a ground-state rubidium or rare-gas atom. Reaction (5) is energetically possible for n;,, 19 if one considers a collision occurring with the relative velocity of the reactants equal to the Maxwellian averaged velocity. A recent theoretical work13 has shown that the cross section <11 for reaction (5) is always less than the cross section <1 for a free electron elastically scattered by atom
X. Equation (9) of Ref. 13 gives <1; = <1</J(x), where c;t>(x) = 2.5x3
• The parameter x (roughly the ratio of the atomic velocity of rubidium and X atoms ta the valence electron velocity in its nP orbit) is of the order of lQ"'l for the present experimental conditions. Considering the values of the elastic cross section a,14
'15 which are about one order of
magnitude smaller than the cross sections Qx(nP) reported here, the contribution of reaction (5) to the total quenching can therefore be regarded as completely negligible.
Lee and Mahan16 have shown that, for the levels considered in this study (12 ~ n ~ 22), the two following reactions, leading to ionic end products, are present in a pure rubidium vapor:
Rb(nP)+Rb(5S)- Rbi+e·
(associative ionization), (6)
Rb(nP)+ Rb(5S)- Rb•+Rb-. (7)
The values of the corresponding cross sections have not been measured, but the results indicate that the. two reactions have comparable efficiencies. Experimental studies17 recently have shown that the associative ionization cross sections are about 10·15 cm2 for n -10-12 in the case of the cesium nP levels. Assuming for the 12P level of rubidium an associative ionization cross section on the same order of magnitude, it can be concluded that processes (6) and (7) contribute negligibly to the collisional depopulation of this level, since the corresponding cross section for this process is 2.3 x 10-u cm2 • The same conclusion should also hold for the highest level (22P) investigated here in view of the behavior of the associative ionization cross sections as a function of n. Thus the quenching reactions leading to neutral products seem ta be more efficient than those leading to ionic products in the case of Rb-Rb collisions.
As far as we know, there is no information concerning the efficiency of the reaction:
Rb(nP)+G-(Rb• -G)+e·, (8)
where G is a ground-state rare-gas atom. Considering the theoretical potential curves for the

- 179
15 COLLISIONAL DEPOPULATION OF RYDBERG P STATES OF ... 2217
~ 0 2)
He 1 a (A
10D{-~ 5
E{eV) 500 t . 1 1
E{eV)
0.1 0.2 4a 0.1 02 4b
~( {l2, 0Ne
: E(eV)
05 f- __,/
E(eV) 1 1 1 -0.1 0.2
4c 0.1 02 4d
FIG. 4. Electron-atom elastic scattering cross sections vs electron energy E for Rb(4a), He(4b), Ne(4c) and Ar(4d).
(Rb• -G) ionic complex18 and the Maxwellian averaged relative velocity of the two colliding atoms, it seems that this ionic channel is energetically accessible when n ~ 19 for Rb-He and Rb-Ne collisions and when n ~ 15 for Rb-Ar collisions. Because the QH.,Ne,Ar(nP) quenching cross-section values show no apparent discontinuity when going from the 14P to the 22P level, one may conclude that process (8) gives a small or negligible contribution to the collisional quenching of the considered nP levels.
To summarize, we believe that the ionic channels represent only a minor contribution to the depopulation of the highly excited nP states and that the depopulation is therefore due mainly to excitation transfer to neutral atomic states. We arrived at the same conclusion in a previous work3 concêrning the total quenching cross section of the lOP level of potassium, in which we showed that several neighboring levels were noticeably populated under similar experimental conditions.
The outer electron of an alkali atom lying in a Rydberg state is very weakly bound (the average orbit radius is about 102 Â), and only its velocity distribution, which is non-Maxwellian, accounts for the discrete character of the energy spectrum. This quasüree behavior is often discussed in the literature and is the basis of most of the theoretical works on Rydberg states. Recently such a theoretical approach5 has led to an excellent agreement with experimental results2 concerning collisions of highly excited F states of xenon atoms and SF 6
molecules and the subsequent formation of SF; and xe• ions. However, no systematic evidence of this quasüree behavior of the outer electron has been demonstrated for atom-atom collisions. It is interesting to see now ü our results can be explained using this argument, i.e, how elastic rate constants for free-electron noble gas (or rubidium)
scattering compare with the quenching rate constants deduced from our measurements. For this comparison to be of some value, however, it is necessary that the change in potential energy resulting from the quenching process is small compared with the collision energy. For a collision energy of about 410 cm·1 , only the 22P level fulfills this requirement, since the potential energy change corresponding to a transition to the 21D level is + 9 cm-1 and to the 19F level is -10 cm-1. We thus restrict the comparison to this case.
Figure 4 shows the well established (e·-rare gas) elastic scattering cross sections,14 as a function of energy in the range of interest, and the (e·-rubidium) elastic scattering cross section for which few reliable quantitative results are available.15 In the latter case a rapid increase of the cross section is observed with decreasing energy. In Table Il we compare the Maxwellian averaged rate constants K~(22P) [deduced from our measurements; see Eq. (2)] and the corresponding electron scattering rate constants¾ (22P) which are derived from
~{22P)= 1o· vu:r(v}f(v)dv, (9)
where u:r(v) is the elastic scattering cross section and f(v) the relative velocity distribution19 of the outer electron for the 22P level considered. The comparison shows that the ~(22P) values e."'thibit the qualitative features (i) and {iii) mentioned at the beginning of this section, although the absolute values of ~(22P) and K~(22P) noticeably düfer (from a factor of 3 ta 20). Even for the highest level investigated here, the changes in potential energy resulting from the quenching process {+ 9 and -10 cm·1 ) are too large to allow a reliable treatment in terms of elastic scattering.
In Table m we report the values of the quenching rate constants K8H! (nP) obtained for the helium case from the binary-encounter theory for atomatom inelastic collisions developed by Flannery. 6
In this theoretical approach, (a) the elastic scattering of the outer electron by the perturbing atom is taken into account, {b) the e--alkali core Cou-
TABLE II. Quenching rate coDStants Kj 3.Ild valence electron-atom elastic rate constallts K! for the 22P sta.te of rubidium. All values are in cm3 sec·1•
Perturber X Kj Kg
Rb 7 .6 X 10-8 77 X 10•B
He 9.7 X 10•10 40 X lO•IO
Ne 1.01 X lO•IO 3Sx 10·10
Ar 1.8 X 10•10 36 X lO•IO

180 -
2218 F. G O U N A N D , P . R . F O U R N I E R , A N D J . B E R L A N D E 15
TABLE m. Quenching rate constants K~. and binaryencounter rate constants K{J.F. for Rb-He collisions. All values are in cm3 sec"1•
Level xj. KBF. He
12P 6.0 X lO•IO 27 X 10•\0
14P 8 .9 X lO•tO 41 X 10•IO
17P 9.2 X 10"\0 64 X lO"IO
22P 9.7 X lO•tO. 88 X 10•\0
lombic interaction is used to determine the velocity distribution of the valence electron, (c) the interaction between the alkali core and the perturbing atom is ignored, and (d) the inelastic collision ·is treated by classical mechanics.
Flannery has used this approach to study collisions between excited (n ~ 10) and ground-state hydrogen atoms at thermal energies. We have calculated, according to Ref. 6, the rate coefficient x:! by summing the rate coefficients associated with the various inelastic processes which give noticeable contributions to the x:! values. We have considered the helium case because the {e"-He) elastic cross section can effectively be taken as a constant [see Fig. 4(b)] thereby simplifying the calculations. The calculated rate constants X: and the ~ values vary the same way with n, but the Kf values are always larger than the experimental ones by a factor between 4.5 and 9.
It appears that a simple two-body theory, involvûig oruy the alkali valence electron and the perturbing atom is unable to correctly reproduce the present e.xperimental results. [The large cross sections obtained for the Rb(nP)-Rb{SS) collisions may be partly due to the fact that the reactants are of identical atomic species.] Therefore, the alkali core should also be considered in the theoretical treatment of the collision problem. This has also been recently pointed out by Smirn~ following his study of the quenching of highly excited hydrogen atoms by ground-state helium atoms. A theory based on a three-body interaction model, involving the alkali core, the valence electron, and the perturbing atom is expected to yield a better agreement with the present experimental results. In particular, a formulation of the collision problem in terms of adiabatic potentials of the a.lkali-perturbing atom system seems to be well suited to the study of these collisions at thermal energies.
The oruy previous e.xperimental works dealing with atom-atom collisions involving Rydberg states are those of Gallagher et al.,1• 21 who have studied the collisional angular momentum mL-cing of highly excited n D states (6 ~ n ~ 15) in sodium induced by collisions with rare gases, that is to say the trans-
fer of electronic excitation of the (n; l = 2) state towards the group of neighboring levels (n; l > 2). For this particular process (which represents, as pointed out by Gallagher et al., 1 the major part of the collisional quenching of the ,zD states) very high cross-section values (of the order of 10·13
cm2) have been measured. The cross sections are
found to rise rapidly with increasing n to a maximum at n = 10 and from there to decrease slightly for higher n values. A similar behavior of our quenching cross sections for increasing values of n was observed, although they are one to two orders of magnitude lower than those reported by Gallagher et al., and refer to a düferent collisional process. One has to be careful when comparing the two experiments because the investigated levels occupy qui te düferent positions in their respective energy diagram. The nD states of sodium investigated by Gallagher et al. are very close to the (n; l > 2) states (the energy defect is less than 1 cm·1 for n -12) but are well isolated from the other states. In contrast the nP states of rubidium are well isolated from all the other states (at least 9 cm"1 for n = 22). Thus one might expect the collisional quenching cross sections of the nP Rb states to be lower than those of the corresponding n D states for the Na atoms. Our results and those of Gallagher et al. are therefore not at all contradictory. In other words the order of magnitude difference between the results of the two experiments may be due in part to the fact that electronic transition in Na-rare-gas collisions take place at internuclear distances much larger than for the Rb-rare-gas collisions.
Table N shows the rate coefficients ~! deduced from the experimental results of Gallagher et al.21
in the case of helium and the rate coefficients ~. calculated according to Eq. (9)(the scattering of
TABLE IV. Experimental rate constants x0 a. for the collisional angular momentum mixing of highly excited D states of sodium in collision with helium (results of Gallagher et al., see Ref. 21). K• is deduced from Eq. (9) (see text), K~ from Ref. 23. All values are in cm3 sec"1•
Level KGa. He K~. xg!
6D 5.7 X 10"9 2.0 X 10"8 l.O x 10·8
1D 1.2 X 10"8 1.8 X 10-8 3.4 X 10"8
8D 1.6 X 10-8 1.5 X 10-8 2.9 X 10"8
9D 1.6 X 10"8 1.3 X 10-8 1.7 X 10"8
lOD 3.3x10"8 l.2 X 10-8 1.5 X 10-8 llD 2.8 X 10-8 l.l X 10-8 1.3 X 10"8
12D 2.5 X 10-8 1.0 X 10"8 1.2 X 10-8 13D 2.5 X 10-8 9.3 X 10"9 1.2 X 10"8
14D 2.0 X 10"8 8 .6 X 10"9 l.l X 10"8
15D 2.0 X 10•8 8.1 X 10"9 1.1 X 10-3

- 181
15 COLLISIO'.\AL UEPOPULATIO~ OF RYDBERG F STATES OF ... 2219
the quasüree electron on the helium atom). It is seen that for n:;;,. 8 these rate coefficients are in agreement within a factor B, 1 ~ B ~ 3, better than the similar comparison for the Rb-He collision (see Table II). This is not surprising in view of the potential energy changes involved in the angular momentum mixing process studied by Gallagher et al. A recent two-state quantum mechanical calculation by Olsonz2, 23 using asymptotic adiabatic potential curves for the Na-rare-gas systems is in reasonable agreement with all the experimental results of Gallagher et al. (see Table IV where only Na-He results are reported). It is interesting to note that the rate constants ~. for Na(nD)-He collisions agree within 30% with the corresponding rate constants ~~ deduced from the theoretical values obtained by Oison for n > 8. For heavier rare gases, elastic rate constants compare less satisfactory with experime;tal and Olson values. This reflects the growing importance of both the polarization effects (Van der Waals interaction) and the electron-rare-gas exchange interaction with increasing mass of the rare gas, the latter being signüicant even at large internuclear distances. 24
V. CONCLUSION
Collisional quenching cross sections of highly excited nP states of rubidium (12 ~ n ,;; 22) have been measured at thermal energies as a function of n and the nature of the perturbing atom. It is observed that the cross-section values do not increase with n as the geometrical size of the ex-
1T. F. Gallagher, S. A. Edelstein, and R. M. Hill, Pllys. Rev. Lett. 35, 644 (1975).
z -W. P. West, G. W. Foltz, F. B. Dunning. C. J. Latimer, and R. F. Stebbings, Phys. Rev. Lett. 36, 854 (1975).
3F. Gounand, J. Cuvellier, P. R. Fourn~. and J. Berlande, J. Phys. (Paris) 37, Ll69 (1976).
4L C. Percival and D. Richards, Adv. At. Mol. Phys. il, 1 (1975).
5M. Matsuzawa, J. Phys. Soc. Jpn. 32, 1088 (1972). 6M. R. Flannery, Ann. Phys. (N. Y.)61, 465 (1970). 7A. N. Nesmeyanov, Vapor pressure of the elements
(Academic, New York, 1963). 8F. Gounand, P. R. Fournier, J. Cuvellier, and J. Ber
lande, Phys. Lett. 59A, 23 (1976). 9E. M. Anderson and V. A. Zilitis, Opt. Spektrosk. 16,
382 (1964) ( Opt. Spectrosc. 1!, 211 (1964)]. -u • See for example, J. S. Deech, R. Luypaert, and G. W. Series, J. Phys. B 8, 1406 (1975).
11 :.\l. Gross, C. Fabrë, P. Pillet, and S. Haroche, Phys. Rev. Lett. 36, 1035 (1976).
12A. Gallagh;;- and E. L. Lewis, J. Opt. Soc. Am.~. 864 (1973).
13 B. M. Smirnov, Invited lectures, review papers, and progress reports of the Ninth International Conference
cited atom (i.e., as n4). In particular the cross
sections for Rb-rare-gas collisions seem to reach a constant value or even to slightly decrease.
The quenching of these nP states has been shown to be mainly due to electronic excitation transfer to neighboring atomic levels. In view of the fact that these levels are close in energy to the nP states we find the Rb-rare-gas cross-section values surprisingly small.
No simple model, i.e., a two-body model taking into account only the interaction of the quasüree valence electron with the perturbing atom, can explain in a satisfactory way our experimental results. The Rb-rare-gas quenching processes occur at internuclear distances for which a theoretical calculation must apparently make use of adiabatic potential curves derived from a three-body model (valence electron, core of Rb, perturbing atom). For some processes having large cross sections a simpler approach using asymptotic potentials may lead to a sat_isfactory agreement with experiment.
ACKNOWLEDGMENTS
We acknowledge the help of J. Cuvellier during the measurements and are indebted to M. Ahrweiller and J. P. Felix for developing and building the acquisition system. We would like to thank J. Pascale for helpful discussions and valuable comments, and T. F. Gallagher for providing his da.ta prior to publication. We wish to thank L. Pitchford and R. A. Gutcheck for carefully reading the manuscript.
on the Physics of Electronic and Atomic Collisions, edited by T. S. Risley and R. Geballe (University of Washington Press, 1975), p. 701.
uH. S. W. Massey, Electronic and Ionie Impact Phenomena (Oxford, New York, 1969), Vol. 1, Chap. 6. See also D. E. Golden and H. W. Bandel, Phys. Rev. 149, 58 (1966); and C. Sol, F. Devos, and J. C. Gauthier, Phys. Rev, A 12, 502 (1975).
15L. C. Balling, Phys. Rev.179, 78 (1968). A more recent calculation by I. I. Fabrikant [ Phys. Lett. 58A, 21 (1976)] indicates cross section about 50% higher than those calculated by Balling. This discrepancy is not important for the purpose of this work.
18Y. T. Lee and B. H. Mahan, J. Chem. Phys. g, 2893 (1965).
17E. E. Antonov, Y. P. Korchevoy, V. I. Lukashenko, and I. N. Hilko, I'Toceedings of the Twelfth International Conference on Ionization Phenomena in Gases, p. 33, Eindhoven, 1975, edited by J. G. A. Holschen and D. C. Schram, (North Holland, American Elsevier, Amsterdam, 1975).
18J. Pascale (private communication). 19B. Podolsky and L. Pauling, Phys. Rev. 34, 109 (19291. 20v. A. Smirnov, Opt. Spektrosk. !Z,, 407 \1974) ( Opt.

- 182 -
2220 F. G O U N A N D , P . R • F O U R N I E R , A ~ D J . R E R LA N D E
Spectrosc. 37, 231 (1974)). 21 T. F. Gallagher, S. A. Edelstein, and R. M. Hill, Phys.
Rev. A 15, 1945 (1977). 22S. A. Edelstein, T. F. Gallagher, R. E. Oison, and
R. M. mu: Abstracts of the Fifth International Conference on Atomic Physics, edited by R. Marrus, M. H. Prior, and H. A. Shugart (Berkeley, 1976), p. 240.
23R. E. Olson, Phys. Rev. A il, 631 (1977).
24 The adiabatic potential curves for the alkali-helium systems are rather nat. J. Pascale and J. Vandeplanque, J. Chem. Phys. ~. 2278 (1974). See also J. Pascale and J. Vandeplanque, CEA Report (unpublished), available on request from the authors. The curves for the other alkali-rare gas systems exhibit a structure which becomes more pronounced with increasing mass of the rare-gas atom.
15

- 183 -
c) ;!~~=-~~!!!~!~~=!!=-~=!-~!~E~-~=-~I~~=~~-§-~~-~~~!~!~·
Nous avons comp1été 1e travai1 effectué sur 1es états P du
rubidium par des mesures simi1aires sur 1es états S (12~ n~18). Le
but de ces nouvel1es expériences est de confirmer les conc1usions re
latives au précédent travail et de disposer d'un assez 1arge ensemble
de données en vue d'une confrontation p1us systématique avec l'appro
che théorique récemment déve1oppée par Hickman37 . Un point intéressant
à mentionner, qui justifie l'intér~t du choix des états S, est 1eur
re1ative proximité, dans le diagramme d'énergie, d'états hydrog~noï~es
de forte multiplicité (en effet le défaut quantique des états nS est
de 3.15 indiquant la proximité des états hydrogénoïdes fortement dé
générés (n-3) F, G, H .•• ). La situation présentée par les états S
apparaît donc, de ce point de vue, intermédiaire entre les états P
bien isolés (de défaut quantique 2.65) et 1es états hydrogénoïdes
toujours très proches énerg~tiquement des niveaux voisins (te1s les
niveaux D du sodium étudiés par Ga1lagher18 , et pour lesquels l'appro
che théorique d'Hickman avait été développée). Les états S sont créés
sélectivement è l'aide de deux lasers à colorants (de type 3) pompés
par le m~me laser à azote (Molectron UV1000), suivant le schéma clas
sique de la figure 21. La première étape consiste à peupler le niveau 0
5P3
;2 {) 1 = 7800 A; le colorant utilisé est une solution de méthyl
DOTC 2.4 10-3 M dans du DMSO). Le niveau ns112
est ensuite peup1é à
partir de ce niveau de résonance à l'aide de la seconde impulsion la-o
ser (12
N 5000 A; le colorant utilisé est une solution de coumarine
500, 10-2 M dans l'alcool éthylique). La seconde impulsion est retar
dée optiquement d'environ 4 ns par rapport à la première, ce afin
qu'une population notable d'atomes dans l'état 5P312
soit atteinte
avant l'arrivée de la seconde impulsion lumineuse. Les énergies res
pectives des deux impulsions sont d'environ 15 et 60 µJ, la largeur 0
spectrale étant de 0,5 A. La décroissance de la population de l'état
ns112
considéré se fait en observant la f1uorescence émise sur la
transition ns112 ~5P1 ; 2 , c'est-à-dire à une longueur d'onde diffé
rente de la transition d'excitation, ce qui permet de s'affranchir en
grande partie de la lumière parasite.
La procédure expérimentale et les causes d'erreur ont été
largement discutées au chapitre V. Les seules modifications que nous
avons apportées au b~ti de mesure pour cette étude sont les suivantes
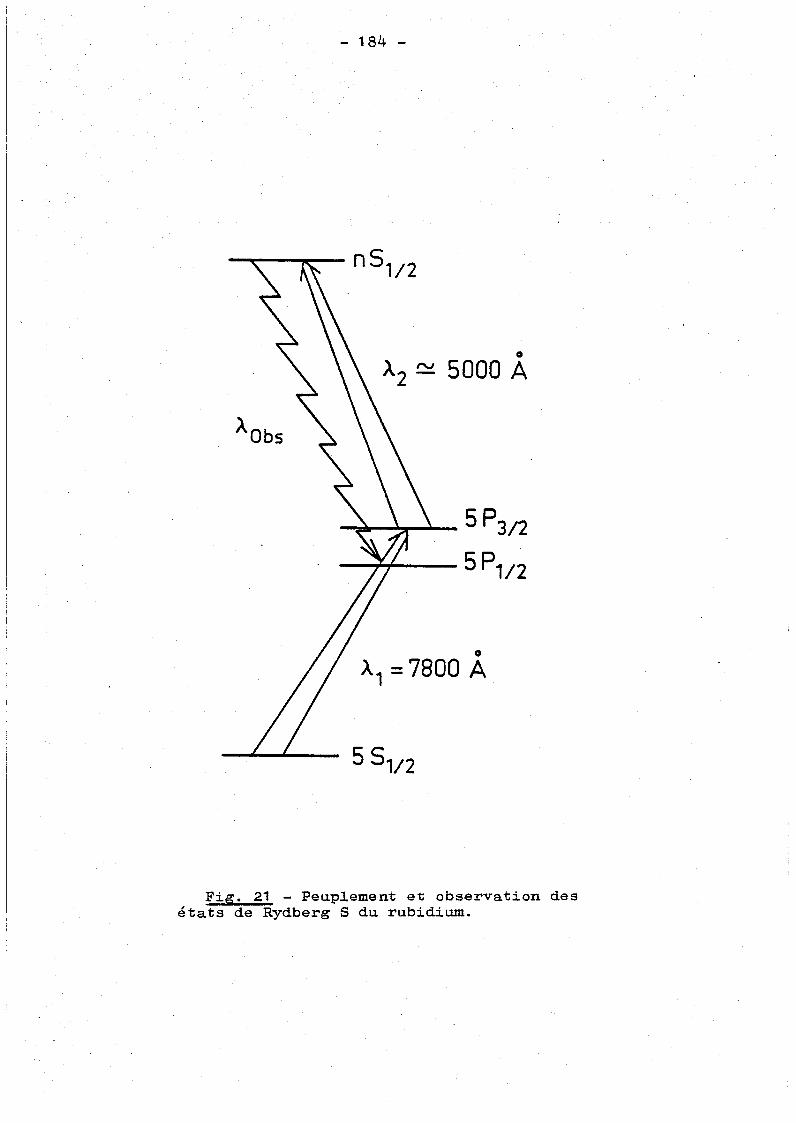
- 184 -
~-~ nS112
ÀObs
0
À 2 ~ 5000 A
, , 5P312 ... ½il .
5P1;2 -rr--
0
À 1 = 7800 A.
s s,12
Fig. 21 - Peup1ement et observation des états de Rydberg S du rubidium.

- 185 -
- un deuxième groupe de pompage a été mis en place permettant
d'obtenir un vide résiduel de 5 x 10-8 T, c'est-à-~ire d'un ordre de
grandeur inf'érieur à celui obtenu lors des expériences précédentes
- af'in de permettre des mesures à très basse pression de gaz
rare, nous avons remplacé le manomètre à huile par un manomètre à ca
pacitance MKS Baratron qui permet la mesure des pressions dans la
zone 10-4 à 1 T avec une excellente précision(~ 1-3%).
Le tableau XI donne les résultats expérimentaux
Perturbateur
Niveau Hélium Rubidium
12S ( 1 . 1 O + O. 20) 10-14 (1.40 + o.40)10-12
14S (2.05 ± 0.50)10-14 (2.10 + o.45)10-12
16S ( 1 . 4 5 !. 0 • 40 ) l O - 1 4 (3.90 + 1.00)10-12
18S (3.25 + 0.65)10-14 (7.00 ± 1.40)10-12
Tableau XI
Sections efficaces de quenching (en cm2 ) des niveaux S très excités du rubidium.
VI.3.3. Discussion de l'ensemble des résultats collisionnels.
Aucune autre mesure expérimentale de section efficace de dé
population collisionnelle totale, relative aux niveaux que nous avons
étudiés, n'étant, à notre connaissance,disponible nous allons mainte
nant discuter l'ensemble des résultats que nous avons obtenus. Ceux-ci
sont reportés dans le tableau XII.

- 186 -
Perturbateur
El,;ment Niveau Alcal.in à. He :-.e Ar Xe l'état fondamental
K 1 OP ( 6 j: J) x 1 OJ 18 .:, 6 8 .:. J 40.:. 15
Rb 12P (2.J !, 0.8) X 1QJ J8;;: 7 4.9.:: 0.9 1 5 .::: J
Rb 14P (i.1 !, 2.5) X 1QJ 56.:, 10 12 ;;: 2. 4 25.:. 5
Rb 1 ïP (1 .00 !, O.JS) X 10 4 58 ;;: 11 11 .:, 2. 1 28;;: 5
Rb 22P (1 .6 + 0.6) X 104 60 .:, 12 1J.:, 2.5 JO.:: 6 4
Rb 12S (1 .4 !, 0.4) X 10 110.:, 20
Rb 14S (2.10 + 0.45) X 104 205 !. 50
Rb 16S (J.9 !, 1.0) X 104 145 .:, 40
Rb 18S (7.Q !, 1,4) X 104 J25.:. 65
Tableau XII
VaJ.eurs expérimental.es (en A2 ) des sections efficaces de quenching de quelques niveaux de Rydberg des atomes aJ.caJ.ins.
a) ~~!~~-~~~-=~~~-~~-E~~=~!~~·
Nous avons mesuré essentiellement* des sections efficaces
de quenching. Celles-ci prennent en compte l'ensemble des processus
inélastiques qui donnent lieu à la dépopulation du niveau considéré.
Essentiellement deux types de mécanisme sont à considérer
- le premier conduit à des voies de sortie neutre (transfert
d'excitation)
- le second conduit à des voies de sortie ioniques. ,
Les différentes voies de sortie ioniques possibles sont discutées dans
l'article 3. Ne disposant pas de données nouvelles relatives à ce se
cond type de mécanisme, nous nous bornerons ici à rappeler nos deux
conclusions essentielles :
* ( Une seule mesure de section de transfert en collision aJ.caJ.in-aJ.caJ.in) est reportée dans l'article 2 (quelle que soit la technique expérimentaJ.e choisie, de telles mesures sont, sauf cas particulier, très délicates, coxmne il a été montré au chapitre v).

- 187 -
1) il semble que le second type de mécanisme contribue de
manière très faible à la dépopuJ.ation des. niveaux de Rydberg que nous
avons étudiés, ce aussi bien dans le cas des collisions al.cal.in-al.cal.in,
qu'al.cal.in-gaz rare. Ce point mériterait une con.f'irmation expérimenta
le précise. Une expérience destinée à mesurer les sections efficaces
relatives à ce second type de mécanisme a été entreprise dans notre
laboratoire
2) les transferts d'excitation, qui constituent la part pré
pondérante du quenching, s'effectuent vers les niveaux énergétiquement
les plus proches du niveau étudié. Ce fait, assez intuifif, du moins
en ce qui concerne les collisions atome-atome, a été clairement mis
en évidence en observant les signaux de fluorescence émis par les ni
veaux autres que le niveau initialement peuplé.
b) S~!!S~!!-!!~~S~=~-S~~!E~E!!!~·
Le tableau XII qui regroupe l'ensemble des résuJ.tats expéri
mentaux permet de tirer trois conclusions qual.itatives importantes :
- Les sections efficaces relatives aux collisions al.cal.in
al.cal.in sont toujours très supérieures (d'env±ron deux ordres de gran
deur) à celles relatives aux collisions al.cal.in-gaz-rare. Notre travail
apporte la première mise en évidence expérimental.a de ce fait. Tous les
travaux postérieurs, comme nous le verrons plus loin,ont con.f'irmé ce
point. Cette constatation met clairement en relief l'importance de
l'interaction e- externe (quasi-libre)-perturbateur dans le processus
de désexcitation collisionnelle des états de Rydberg (interaction dont
l'importance a déjà été mentionnée au chapitre III). En effet il est
bien connu que les sections de diffusion élastique e--al.cal.in sont
très supérieures aux sections e--gaz rare, ceci étant d~ au fait que
la portée de l'interaction croît avec la polarisabilité de l'atome
considéré.
- Les sections efficaces relatives aux collisions al.cal.in
gaz rare sont faibles (quelques 10-14 cm2 au plus). Cette première mise
en évidence de la relative stabilité de certains états de Rydberg vis
à-vis de ce type de collision contraste avec la situation observée par
T.F. Gal.lagher et ai. 38 dans le cas des états D du sodium (mixing col
lisionnel de moment angulaire) pour lesquels les sections efficaces , -13 2 mesurees sont de l'ordre de 10 cm.

- 188· -
- Le comportement, en fonction du nombre quantique principal
n (ou, plus précisément en fonction du nombre quantique effectif n*)
appara.!t notablement différent suivant que l'on considère les colli
sions alcalin-a1calin ou a1ca1in-gaz rare. Dans le premier cas on
observe une croissance importante de la section efficace en fonction
den*; dans le second les sections efficaces apparaissent sensible
ment constantes. Pour ce dernier cas un comportement notablement dif
férent est observé pour les états D du sodium38 : les sections effi
caces croissent très rapidement jusqu'à n* tV 10 pour décro!tre ensui te
sensiblement.
Au vu de ce qui précède on peut dire que les sections effi
caces, pour un niveau donné, leurs valeurs absolues aussi bien que
leurs variations en fonction den* peuvent différer très sensiblement
suivant le moment angulaire orbita1 1 (c'est-à-dire suivant la posi
tion des niveaux considérés dans le spectre énergétique de l'atome
alcalin). Il convient maintenant par une discussion plus quantitative
faisant appel aux résultats théoriques, quand cela est possible,
d'expliquer ces différentes observations.
c) Collisions alcalin-a1ca1in. --------------------------Nous commençons la partie centra1e de notre discussion par
le cas des collisions alcalin-alca1in avant de traiter le cas des col
lisions a1calin-gaz rare. Cette séparation bien qu'elle corresponde,
nous venons de le voir, à certains aspects de la réa1ité expérimentale
est quelque peu arbitraire. En effet, nous avons vu, au chapitre III,
que toutes les approches théoriques que nous avons mentionnées, qui
ont été développées pour le cas des collisions alcalin-gaz rare, peu
vent, en principe, 3tre étendues au cas des collisions alcalin-alcalin.
Cependant cette extension reste à faire et pose de nombreux problèmes
(cf. chapitre III, paragraphe III.5.2.c), bien que l'interaction prise
en compte (e- externe-perturbateur) soit de m~me nature dans les deux
cas, ne différant que par son intensité. Ne pouvant nous comparer,
pour ces collisions, qu'à des estimations particulièrement grossières,
nous avons pensé qu'il était plus logique de traiter à part cette ca
tégorie de collisions.

- 189 -
Les grandes valeurs des sections efficaces al.cal.in-al.cal.in
ainsi que leur augmentation rapide en fonction den* amènent naturel
lement à les comparer aux sections géométriques (~ n*4) des niveaux
correspondants. Physiquement ceci revient à dire que l'interaction e
quasi-libre-al.cal.in est suffisamment forte (les sections efficaces e
libre-alcalin sont, à très basse énergie, de l'ordre de 103 A2 ) pour
que la section efficace de collision inélastique reflète plus ou moins
la taille de l'atome de Rydberg. Cette image est évidemment très gros
sière, mais, en l'absence de tout résultat théorique, il semble raison
nable de regarder si elle peut rendre compte des résultats observés.
La figure 22 montre la comparaison dans le cas des états nS.
Nous avons pris pour section géométrique, comme il est habituel,
n<r2 >, où <r2> représente la moyenne du carré de la distance e de
Rydberg-coeur ionique et vaut
< r 2) = ½ n*
2 [5n*2
+ 1 - 31(1+1 )] ~ ~ n*4 en u.a.
Notons que, n'ayant étudié que des niveaux pour lesquels n* ~ 10, cette
section géométrique ne dépend à peu près pas de .l. L'accord entre sec-
tion de quenching et section géométrique es~
situation semblable a été observée par Deech
états S et n312
du césium* (8 ~ n ~ 14) et par
états n312 du césium ( 6 ~ n 4 10).
très satisfaisant. Une
et al. 39 dans le cas des 40 Tam et al. pour les
Dans le cas des états P du rubidium le tableau XIII indique,
par contre, une différence sensible entre section de quenching et sec
tion géométrique. Ceci n'a rien, a priori, de surprenant, la section
géométrique ne devant pas 3tre considérée comme une estimation théori
que mais comme un élément grossier de comparaison. En particulier elle
ne tient aucun compte de la valeur del, c'est-à-dire, pour une valeur
donnée den*, de l'environnement énergétique du niveau considéré. Celui
ci, à l'évidence, joue un rSle important dans le cas des collisions
avec les gaz rares (voir paragraphe précédent) et, en l'absence de tout
support théorique valable, nous n'avons pas de raison de penser, a
priori, qu'il n'en est pas de m~me, dans une certaine mesure au moins,
* La comparaison de résultats relatifs à des niveaux du rubidium et du césium se justifie du fait de l'importance de l'interaction e--alcalin (quel que soit l'alcalin) comme il a été noté au début du paragraphe.
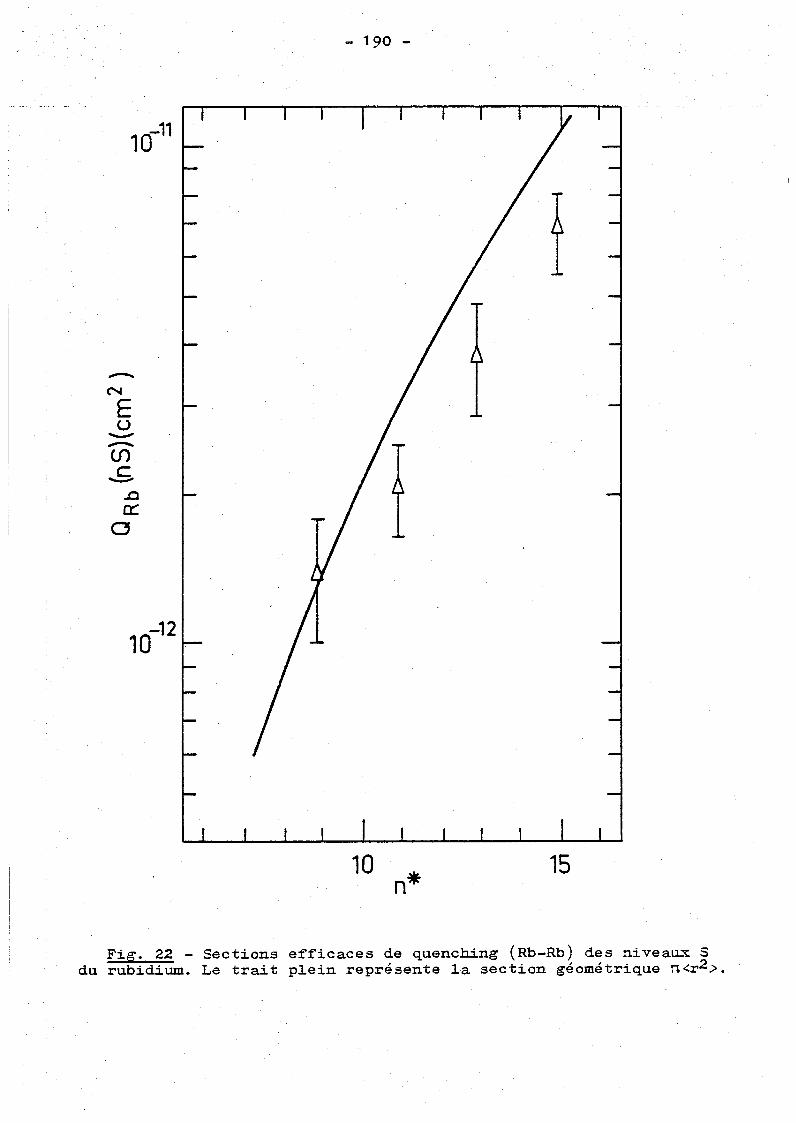
10-11
-N
E u --U')
...s ..c 0::::
0
10-12
- 190 -
10 n*
15
Fig. 22 - Sections efficaces de quenching (Rb-Rb) des niveau_~ S du rubidium. Le trait p1ein représente la section géométrique n<r2>.

- 191 -
pour les collisions alcalin-alcalin. Ce point est conforté par lare
marque suivante. Les niveaux S du rubidium et n312 du césium, pour les
quels les sections de quenching sont en bon accord avec les sections
géométriques, sont tous deux proches de niveaux voisins (F,G,H .. pour
les états nS (Rb) dont le défaut quantique 3.15 est proche d'wi entier,
et n512 pour les états nn312 (Cs) l'écart de structure fine étant très
faible}, ce qui n'est pas le cas des états nP (Rb) dont le défaut quan
tique vaut 2.65. Ceci est en accord avec les observations de Pendri1141
et de T·am et al. 40 qui ont montré que la dépopulation des niveaux
nn312 se faisait principalement vers les niveaux nn512 indiquant de
ce fait wie section de quenching des niveaux nD, sans référence à la
structure fine, notablement inférieure à la section géométrique. Or
ceux-ci présentent Wl défaut quantique de 2,47, indiquant Wl relatif
isolement énergétique vis-à-vis des niveaux voisins.
Niveau Qexp 2 n<r>=%, eo.
12P (2.3 ~ 0,8) X 10J 1 , 65 X 10 4
14P (7,1 ~ 2,5) X 10J J ,·60 X 104
17P ( 1 , 00 ,:t. 0, 35) X 1 o4 9.20 X 10 4
22P (1,6 + 0,6) X 10 4 J, 10 X 1 o5
Tableau XIII
Comparaison entre les valeurs expérimentales des sections de quenching rubidium-rubidium et la section géométrique de
l'atome, pour les états nP du rub¼dium. Les valeurs reportées sont en A2 •
Ne disposant d'aucune approche théorique, il semble diffici
le de pousser plus loin la ~iscussion. Notons simplement, pour termi
ner, que l'importance des sections efficaces alcalin-alcalin entraine
llll.e modification notable des durées de vie effective des états de
Rydberg, ce même à des densités relativement faibles (rJ10-5 T). Il
importe donc d'effectuer la mesure des sections efficaces alcalin-
gaz rare à très basse pression d'alcalin si l'on ne veut pas ~tre g~né
par l'effet des collisions alcalin-alcalin.

- 192 -
d) 2~!!!~!~~~-~~~!~=~!~-~~~!·
Nous partirons du point principa1 de la discussion de l'ar
ticle 3: les résu1tats obtenus par T.F. Ga1lagher et ai.38 pour les
états nD (Na) (Fig. 23) sont très différents de ceux que nous obtenons
pour les états nP (Rb) ce aussi bien en ce qui concerne la vaJ..eur abso
lue des sections efficaces (N103 Â2 pour les niveaux nD (Na) et
N101 Â2 pour les niveaux nP (Rb» que leur variation en fonction den*
(décroissance pour n*> 10 dans le cas des états nD (Na) en collision
avec les gaz rares légers, légère croissance ou saturation dans le cas
des états nP (Rb)). Ces différences doivent atre attribuées à la posi
tion des niveaux considérés dans leur diagramme énergétique respectif.
En effet 1 1 écart entre les niveaux nD (Na) ~ui sont quasi-hydrogénoï
des ; 5= 0.02) et leurs voisins nF,G,H ... est de l'ordre du cm-1
aJ.ors que les niveaux nP (Rb) sont distants d'une dizaine de cm-1
des
niveaux les plus proches ((n-1)D et (n-3)F,G,H ... ).
Cette importance de l'environnement énergétique a reçu des
con:f':irmations expérimenta1es très nettes.
- Nos mesures relatives aux états nS (Rb) ·(Tableaux XI et XII)
montrent (pour les collisions Rb-He) des vaJ..eurs de sections efficaces
(N102 Â2 ) intermédiaires entre celles relatives aux états nP (Rb) et
celles obtenues pour les niveaux nD (Na). De fait, du point de vue
transfert d'énergie, les niveaux nS (Rb) (pour lesquels o = 3.15) pré
sentent une situation intermédiaire entre les niveaux nP (Rb) (pour
lesquels S = 2.65 indiquant un relatif isolement énergétique des ni
veaux correspondants) et nD (Na) (6 = 0.02 indiquant la quasi-coïnci
dence énergétique avec les états nF,G,H ... ).
- M. Hugon et ai. 36 ont observé pour les niveaux hydrogénoides
nF (Rb) (6 = 0.02) un comportement analogue à celui des niveaux nD (Na),
tout en étendant les mesures au cas des perturbateurs lourds (xénon
et rubidium).
- T.F. Gallagher et ai.42 ont mesuré pour les niveaux non-hydro
génoïdes nS (Na) (o = 1 .JO) des va1eurs de l'ordre de 10 Â2 (pour
nN10) pour les collisions Na-He, confirmant ainsi nos mesures rela
tives aux états nP (Rb) (ô = 2.65), les défauts quantiques indiquant
pour les deux séries,un environnement énergétique similaire.

5000 r
2000~
~
~ 200~ I
r 2000.
1000
500 I 200
5
- 193 -
He l 2 ~ Î ~ i 1 2
Ne I I I I I I I I
T I I Ar - ,- I I
I
10 n
15
Fig. 23 - Sections efficaces (en A2 ) de mélange collisionnel de moment angulaire des états D du sodium en collision avec les gaz rares38.

- 194 -
Tout ceci montre clairement que les sections de dépopu1ation
pour un perturbateur donné (en particu1ier pour l'hélium, pour lequel
on dispose d'un ensemble très complet de mesures), et quel que soit
l'aJ.caJ.in considéré, varient de manière monotone avec la position du
niveau étudié dans le diagramme énergétique. Cette observation fonda
mentaJ.e nous a servi de base pour le choix d'une approche théorique
comme nous aJ.lons le discuter maintenant.
Nous aJ.lons tout d'abord restreindre la discussion au cas
des collisions aJ.caJ.in-hélium, ce pour deux raisons :
- nous disposons pour ce couple d'wi ensemble complet de
résultats expérimentaux;
- comme nous l'avons déjà largement discuté (chapitre III)
les approches théoriques ont, dans ce cas, une formulation plus simple
(approximation de la longueur de diffusion).
Les résultats expérimentaux obtenus pour le quenching des
états hydrogénoïdes nD (Na) 38 (processus quasi-élastique de mélange
collisionnel de moment angulaire
par diverses
et aJ.. 43 ) ou
avec les états nF, G, H, ... voisins)
approches théoriques de type semi
quantique (Olson44 , Hickman37, sont bien expliqués
classique (Derouard
Matsusawa45 ) et par le modèle de de Prunelé-PascaJ.e, toutes approches
ne prenant en compte que l'interaction entre l'électron quasi-libre et
l'atome d'hélium. Les résultats récents obtenus par Hugon et ai. 36
pour les états hydrogénoïdes nF (Rb) sont aussi bien reproduits par
le modèle de de Prunelé-PascaJ.e46 et par une approche de type Hickman.
Au vu de la nette correlation, précédemment mentionnée, entre
la vaJ.eur absolue des sections efficaces de quenching et l'écart éner
gétique du niveau considéré avec ses proches voisins, il nous semble.
raisonnable de penser que la nature de l'interaction responsable de
la dépopulation des états hydrogénoïdes nD (Na) ou nF (Rb) est la
m~me que celle responsable du quenching des états non hydrogénoïdes
nS (Rb) et nP (Rb). En effet i1 n'y a, à notre avis, aucune raison de
considérer que l'interaction e--He, qui joue à l'évidence le rSle fon
damentaJ. dans le quenching des états hydrogénoïdes, ne jouerait qu'un
rSle mineur dans le cas des états non hydrogénoïdes, l'extension moyen
ne des fonctions d'onde (c'est-à-dire< r>) étant la m~me (pour des n*
comparables) dans tous les cas. La seule différence vient de la posi-

- 195 -
tion du niveau étudié dans 1e diagramme énergétique de 1'a1ca1in consi
déré. Nous avons donc ca1cu1é 1es sections efficaces de quenching K*-He
et Rb*-He des niveaux que nous avons mesurés à l'aide de la méthode
qui nous a semblé 1a plus simple, c'est-à-dire de la méthode quantique
développée par Hiclanan37 (première approximation de Born+ potentiel
de Fermi). Il est cependant probable que les autres méthodes, décrites
au chapitre III, qui prennent en compte 1a seu1e interaction e--He
donneraient des résu1tats comparables (ceci est en tout cas vérifié
pour 1es niveaux hydrogénoïdes37 , 43 , 46 ). Notons enfin que l'utilisa
tion du modèle de de Prune1é-Pascale est délicate pour les états non
hydrogénoïdes (cf. chapitre III) ; quant au modèle de Flannery son
emp1oi pour des niveaux définis par deux nombres quantiques net l pose
de nombreux problèmes et sa version simplifiée (Eq. (C58)) conduit à
des va1eurs nettement sous-estimées (cf. article 3 et référence 46).
Nous avons développé la formuiation générale de l'approche
d'Hiclanan au chapitre III (paragraphe III.J.2.b). Outre sa re1ative
simplicité, dans le cas de l'hélium, elle a l'avantage de fournir des
renseignements sur 1'importance relative des différents canaux de dés
excitation. Nous allons maintenant donner les procédures pratiques de
calcu1 que nous avons adoptées.
Rappelons tout d'abord les équations de départ :
. 2 l 1 1 J.. )2 : (n,l ...-n',l'( e) =(~) ~1
(21'+1) ~(2À+1) (o O O II).1 2 (F11)
avec
Il-= f"'Rn'J.'(r) j,.(Kr) Rnl.(r) r 2 dr (F12)
0
La section totale étant obtenue par:
O"(nl -.. n' l') = 2'1"'t in
dCY' dQ sine d0
0
(F13)
Il faut tout d'abord évaJ.uer I~. Nous avons utilisé des fonctions d'on
de hydrogénoïdes, soit 47 :

Rnl(r) 21+1
= nJ.+2 (21+1) !
- 196 -
(n+J.)! rl e-r/n F (-n+J.+1, 21+2 11
(n-l.-1) !
J. -r/n ( 2r) = Anl r e F11 -n+J.+1, 21+2; n 48
On a de plus
j). (Kr) = J;,.+112(Kr) v 2~
L'équation (F12) s'écrit al.ors:
I = A A .(K'K-1/2 fa, l.+1.'+3/2 -(1/n+1/n 1 )r J (Kr) :,. nl. n, l., Y 2 r e .À +1 /2
0
F 11 ( -n+l.+1 , 21.+2 !~) F11 (-n'+l. 1 +1, 21 1 +2
- A A ,rr;;' -1/2 ( - nl. n, l., V 2 K J ~ ,K)
Les F11 étant des poJ.yn8mes finis J (~,K) peut s'écrire
d : n+n 1 -1.-l.' -2 max
J(Â ,K) = I d=o
Ad Jd(;.,K)
~) n
2r)dr n'
où Ad est un coefficient cal.culab1e à partir des paramètres des F11 et
Jd(À,K) est donné par
Jd(\,K) = f"' r1+1' +J/2+d
0
-(1/n+1/n')rJ / (Kr) dr e À +1 2
Cette intégral.e est anal.ytique49 et permet, en posant
1 1 1 - = - + -N n n'
d'obtenir final.ement
Vî Â+1 /2 7\ +l.+l 1 +J+d J (~ K) = K N À! (l K2N2)-l.-l'-d-2
d ' 7 ~ 1 ..... 1 1 1 + -
( i,-J.-l'-1-d Â-1-1'-d F21 2 '
À + 2. 2
-K2N2)

- 197 -
ce qui résout le cal.cul de I~. L'équation (F1.3) s'écrit al.ors
da(nJ. ""':._n'i'IK) =( ;:'.;)2 ~' ( 2i'+l) A!iA!,i,!('i+2i•+6
I ;>.
2 2 À~ Â ! i 2 (K~2)). ( 1 l' 1')2
( 2 À ! 2 Ï.+ 1 ) ! 0 0 0
n+n'-1-1'-2 "°' d 2.__2 -1-1' -d-2 L AdN (À +l+l '+2+d) ! ( 1 +K~ )
d=o
( ~-1-1 1 -1-d Â-1-1'-d  .3
F21 2 ' 2 ; + 2 -K2ii2)12 (F14)
pour éval.uer l'équation (F1J) on utilise la relation:
K2 = k 2 + k'
2 - 2kk' cose
qui permet d I obtenir, en posant . . K2N2 = x
N2
1 k+k' 12
cr(n1 ...,.n'l') = 'l't
kk'·N2 I . do-(n1 + n'l'I K) dQ dx
N2
\k-k' 1 2
En tenant compte de (F14) on obtient final.ement
2 l+l' 2 g{nJ. - n' 1) = (~) A!r'!,
1, ( 2i, +1 )N21+21' +4 L 22.\c,, ! )2 (l. 1
1
).)
}.=Il-l'i (2.,.)! (2,.+1)! o o o
N2 (k+k' )2
> .A.di.<i(l+l+l'+2-tti)! (1+x)-l-l'-2 F (:;>.-l-l'-1-d f ln+n•-1-1 1 2
d=o 21 ., • ?.-1-1 1 -d
2 ;À+.l. )\2 2. -x
N2 lk-k' 12
(F15)
(F16)
(F1 7)
(F18) x'- dx
On obtientc:r(n1 ~n'l') en u.a. (na2 = 0.88 A.2 ) en utilisant les parao mètres suivants

- 198 -
1i = 1
k = 1823 µv
V= 6.650812 10-5 n k 12 = k
2 + 2µ (E1 -E2 )
E = 1
E J. - E. n 1
219475
E - E. n'l' 1
; E2 = 219475
oùµ représente 1.a masse réduite (en g) des particuJ.es en coJ.J.ision, , ( -1 ) T J.a temperature de J.a ce1.J.uJ.e en °K; EnJ.' En'1.' et Ei en cm re-
présentent J.es énergies des niveaux de départ et d'arrivée, et 1.'éner
gie d'ionisation de 1'a1ca1in considéré.
Faisons queJ.ques remarques:
- La borne supérieure d'intégration peut-~tre étendue à J.'inf'ini ;
1.a vaJ.eur numérique de 1.'intégraJ.e n'étant sensibJ.e qu'à J.a borne infé
rieure (qui rend compte du défaut d'énergie entre 1.es deux niveaux con
sidérés).
- A 1.' approximation de 1a J.ongueur de diffusion, a- (:hl. ~ n 11.') varie
comme l.e carré de cel.J.e-ci (équation (F18)). Cependant l'extension du
caJ.cuJ. que nous avons présenté aux gaz rares autres que J.'hél.ium néces
site de prendre en compte l.es termes de pol.arisation51 et compl.ique
assez notab1.ement l.e caJ.cuJ. (voir chapitre III, paragraphe III.5.2.b).
- La fonction hypergéométrique F 21 se réduisant à un poJ.ynSme en
x on peut obtenir une expression entièrement anaJ.ytique pour l'équation
(F18), soit :
o-( nl. , n I l ' ) ( 2
- mL 2 2 - hk). AnJ.An'l'(2J.'+1)N21.+2J.'4I I'
f-1~" 22:>.(À!)2 (1. l.' À )2 2i\ ! (2).+1)! O O o
"" d+d' L AdAd,N ()+l+l 1 +2+d)! (À+l+l 1 +2+d')! d,d'
I • • 1
J.. 'J..
a:. ex:. 1 J. J.. I
j
/3 . j-d-d 1 -21.-21' -J
[xj-d-d' -21-21' -J JN: (k+k' ) 2 + 1
N (k-k' )2 +1
(F19)

- 199 -
où Ad, Ad'' OC. tt., et~- sont des coefficients dépendants des paramètres l. l. J
des fonctions d'ondes et des divers indices. Là aussi il suffit d'éva-
luer l'expression pour la borne inférieure (x = N2 (k-k 1 )2 + 1). Nous
avons pu, de cette manière1 calcuJ.er les sections de mélange collision
nel de moment anguJ.aire des états D du sodium, jusqu'à n = 10 ; ceci
nous a permis de vérifier la précision de notre programme numérique
(voir ci-après). Un tel calcuJ. n'est plus possible pour des valeurs de
n supérieures à 10, le nombre de termes à sommer devenant prohibititif
et.donnant lieu à des erreurs d'arrondi notables dans l'évaluation de
la série alternée ( éq. (F11)). En conséquence, nous avons adopté la
procédure suivante. Après évaluation de la fonction hypergéométrique
F21 par un algorithme classique, l'intégration de la formuJ.e (F18) est
effectuée numériquemen~ en double précision,à l'aide d'un algorithme à
pas variable utilisant une méthode de Newton-Coates à huit points50 .
Un temps de calcuJ. typique est de l'ordre de 2 s pour n, n'~15. Nous
avons tout d'abord vérifié que nous obtenions bien les m3mes valeurs
numériques que celles données par Hickman37 dans le cas du mélange
collisionnel de moment anguJ.aire des états D du sodium.
Le tableau XIV donne les résuJ.tats numériques que nous avons
obtenus dans le cas des niveaux 10D et 15D du sodium. Notons que les
sections partielles (que l'article d'Hickman37 ne fournit pas) décrois
sent lentement avec le moment orbital l du niveau d'arrivée, observa
tion en bon accord avec les résuJ.tats de l'approche semi-classique43 •
~iveau: départ 1 Moment angulaire l du niveau (n,l) d'arrivée Section 1 9exp 3 4 5 6 7 8 9 10 11 12 13 14 totaJ.e
1
10D 420 450 443 411 380 357 .339 2800 / 2200 ;:: 800
1 .5D 102 106 1 01 93 87 81 iï 74 71 69 67 6.5 992 1380 !. 200
Tableau XIV
Sections efficaces théoriques (en A2 ) des transitions de mélange de moment anguJ.aire de deu..~ niveaux D du sodium en collision avec
l'hélium. La section totale est à comparer aux valeurs expérimentales Q de la référence J9. exp

- 200 -
La décroissance observée pour 1a section tota1e s'exp1ique par le fait
que les sections partielles (nD -nH par exemple) diminuent rapidement
avec n (environ comme n-4) dès que l'écart énergétique est de l'ordre
du cm-1 . Cette diminution n'est qu'en partie compensée par le fait· que
le nombre de transitions partielles à sommer augmente comme n. On ob
tient ainsi (pour n ~ 10) pour le processus de mélange collisionne1 de
moment angu1aire, une section tota1e qui décro!t environ en n-3.
Pour le ca1cu1 des sections efficaces mettant en jeu des ni
veaux non hydrogénoides (notre cas) nous avons effectué une interpola
tion numérique entre les va1eurs e~tières encadrant les nombres quan
titifs effectifs n* des états considérés. Les résu1tats obtenus pour
les sections efficaces partielles (ni -n'l') vérifient, dans tous les
cas, d'une manière satisfaisante le principe du bilan détaillé (à mieux que 5~). Les tableaux X:V., XVI et XVII donnent le détail des ré
su1 tats numériques obtenus ainsi que les écarts d'énergie mis en jeu
et les figures 24, 25 et 26 les niveaux pris en compte.
Le tableau XVIII montre la comparaison entre résu1tats théo
riques et expérimentaux. Conscient des difficu1tés de principe posés
par l'extension de cette méthode à des transitions relativement iné
lastiques, nous avons indiqué sur le tableau XVIII le niveau qui con
tribue le p1us à 1a dépopu1ation co1lisionne1le tota1e ainsi que son
écart d'énergie en cm-1 avec 1e niveau étudié. Il appara!t très clai
rement que l'approche théorique utilisée permet d'obtenir une estima
tion raisonnable des sections efficaces (à mieux qu'un facteur 3,
dans le cas le plus défavorable). De plus elle rend très bien compte
de 1a faible variation observée des sections efficaces en fonction de
n. Les tableaux X:V à XVIII permettent aussi de tirer un enseignement
important sur la nature des voies de sortie. On constate en effet que
1e niveau qui contribue 1e plus au quenching est toujours le niveau
réservoir (F,G,H ••. ) le plus voisin, ce qui met en évidence l'impor
tance de 1a mu1tiplicité du niveau fina1 dans le processus de dépopu
lation. Afin de confirmer les bons résu1tats obtenus par cette appro
che nous avons calculé les sections de dépopulation tota1e des niveaux
10S(Na) et 11S(Na) : elles valent respectivement 16 et 26 12 , en très
bon accord avec les valeurs expérimenta1es42
(14.4 = J.O et 21 = 5 12,
respectivement). Le calcul montre aussi que, pour ces états, la majeu
re partie de la dépopulation s'effectue vers les niveaux de forte mu1-

Niveau de
départ
10P
- 201 -
Niveau..~ d'arrivée
8F ,G,H ••.
15.2
(120)
9D
0.1
(160)
Tab1eau XV
Section tota1e
15. 3
Sections efficaces théoriques (en Â2 ) partie11es et tota1e de dépopu1ation du niveau 10P du potassium en co11isian avec 1 1hé1ium.
Les nombres entre parenthèses indi~uent,. four chaque section partie11e 1.
1e modu1e de 1 1 écart énergétique len cm-) entre 1e niveau de départ et 1e niveau d'arrivée. Le ca1cu1 uti1ise 1a première approximation
de Born et 1e potentie1 de Fermi. La température du gaz est T = 470 K.
90
l 10P!
8 F:G.,H.,. ..
Fig. 24 - Niveaux pris en compte dans 1e ca1cu1 de 1a section efficace de quenching du niveau 10P du potassium (cf. Tab1eau XV).

- 202 -
Niveau de départ Niveaux d'arrivée Section totale (nP) (n-J)F,G,H •.• (n-1 )D
12P 14. 9 7.7 22.6 (103) (80)
14P J6.J 4.4 40.7
(57) (44)
17P 60.8 2.J 63.1
(32) (22)
22P 81. 3 1 . 0 82.J
( 11 ) (9)
Tableau XVI
Sections efficaces théoriques (en Â2 ) partielles et totales de dépopu1ation des niveaux nP du rubidium en collision avec
l'hélium. Les nombres entre parenthèses indiquent, pour chaÎue section partielle, le modu1e de l'écart énergétique (en cm-) entre le niveau de départ et le niveau d'arrivée. Le calcu1
utilise la première approximation de Born et le potentiel de Fermi. La température du gaz est T = 460 K.
----(n+1) D
~
( n - 3 ) G G, H, ...
Fig. 25 - Niveaux pris en compte dans le calcul de la section efficace de quenching des niveaux nP du rubidium (cf. Tableau XVI).

- 203 -
Niveau de départ Xiveau.x d'arrivée Section ns nP (n-1 )P (n-2)D (n-1 )D 1 (n-:3)F,G, ••• {n-4)F,G, ••. totaJ.e
!
12S 2.8 J.2 1 5. 4 o. 1 1 267 < ,0-2 289 (1 40) (1 79) (67) (220) 1 (J7) (J25)
14S 1 • 7 1. 7 6.6 2.1 :366 0.5 1 :378 ·( 77) (96) ( :37) (121) ( 20) (247) l
16S 1 .o 1.2 4.o 1.2 J80 J.2 :391 (47) (57) (2:3) (74) ( 12) (101)
18S 0.7 0.8 2.7 0.6 :3:38 6.o '.349 (J1) (:37) ( 1 5) (50) (8) (65)
Tab1eau XVII
Sections efficaces théoriques (en Â2 ) partie11es et tota1es de dépopu1ation des niveaux nS du rubidium en co11ision avec
1'hé1ium. Les nombres entre parenthèses indiquent, pour chaÎue section partie11e, 1e modu1e de l'écart énergétique (en cm-) entre 1e niveau de départ et 1e niveau d'arrivée. Le ca1cu1
uti1ise 1a première approximation de Born et 1e potentie1 de Fermi. La température du gaz est T ~ 520 K.
nP (n -1) D
[iifil (n-2) D (n-3)FGH··· I I '/
(n-1) P
(n-L.) F;G,H, ...
Fig. 26 - Niveaux pris en compte dans le ca1cu1 de la section efficace de quenching des niveaux nS du rubidium (cf. Tab1eau XVII).
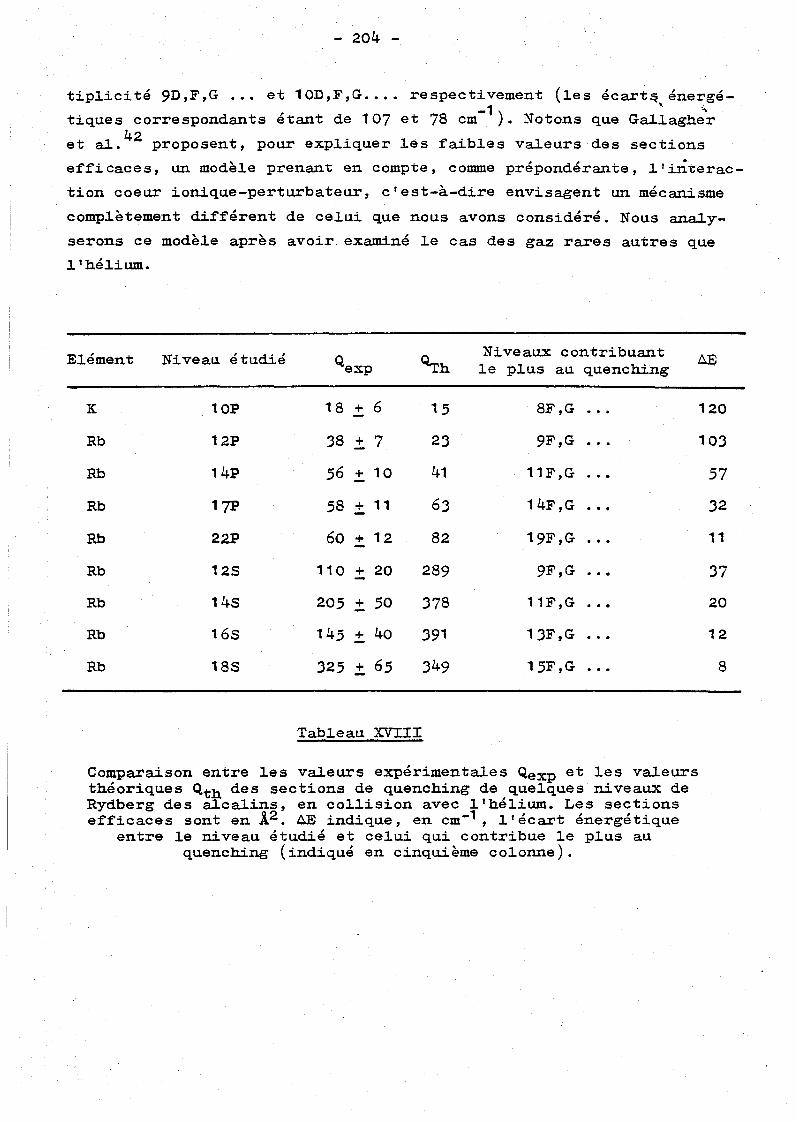
- 204 -
tipl.icité 9D,F,G •.• et 10D,F,G •••. respectivement (l.es écart~ énergé-1 '· '
tiques correspondants étant de 107 et 78 cm-). Notons que Gall.agh;r
et al. 42 proposent, pour expliquer l.es faibles valeurs des sections
efficaces, un modèl.e prenant en compte, c0Dm1e prépondérante, l'interac
tion coeur ionique-perturbateur, c'est-à-dire envisagent un mécanisme
complètement différent de cel.ui que nous avons considéré. Nous analy
serons ce modèle après avoir examiné le cas des gaz rares autres que
l. 'hél.ium.
Elément Niveau étudié Qexp 'trh Niveaux contribuant
~ le plus au quenching
K 10P 18 ± 6 15 8F,G •..
Rb 12P 38 + 7 23 9F ,G ...
Rb 14P 56 + 10 41 11F,G ..•
Rb 1 7P 58 ± 11 63 14F ,G •••
Rb 22P 60 + 12 82 19F ,G •••
Rb 12S 110 z. 20 289 9F,G •.•
Rb 14S 205 + 50 378 11F,G .••
Rb 16S 145 + 40 391 13F ,G •.•
Rb 18S 325 + 65 349 15F,G ..•
Tableau XVIII
Comparaison entre les valeurs expérimentales Qexp et les valeurs théoriques Qth des sections de quenching de quelques niveaux de Rydberg des alcalins, en collision avec l'hélium. Les sections efficaces sont en 12 . ~ indique, en cm-1 , l'écart énergétique
entre l.e niveau étudié et celui qui contribue le plus au quenching (indiqué en cinquième col.orme).
120
103
57
32
11
37
20
12
8

- 20.5 -
La non-va1idité de l'approximation de la longueur de diffu
sion entra!ne a1ors des complications notables de ca1cul, dues à la
nécessité d'inclure des termes correctifs de polarisation au pseudo
potentiel de Fermi. Une telle approche a cependant été formulée par
Hiclanan51 dans le cas des états hydrogénoïdes et a fourni des va1eurs
en bon accord avec les mesures expérimenta1es relatives à la dépopu
lation des états nD(Na) en collision avec le néon et l'argon. Nous
n'avons pas développé une approche correspondante dans le cas des ni
veaux non hyd.rogénoïdes nP(Rb). Cependant le tableau XII permet de
faire quelques remarques.
- En ce qui concerne le néon les sections de quenching sont net
tement in:f'érieures à celles obtenues dans le cas de l'hélium (d'un
facteur 5 environ). Une situation semblable est observée pour les
états nD(Na) 38 . De fait, à basse énergie, la section de diffusion e--Ne
est sensiblement in:f'érieure à la section e--He. Le ca1cul d'Hiclanan51
fournit pour les états nD(Na) des va1eurs théoriques en bon accord
avec l'expérience. Notons que la correction de polarisation doit 3tre
assez faible car, d'une part le néon est peu polarisable, d'autre part
un ca1cul semi-classique effectué à l'approximation de la longueur de
diffusion43 conduit, lui aussi à un bon accord avec l'expérience. On
peut donc raisonnablement penser que l'interaction prépondérante es~
bien l'interaction e--Ne et qu'un ca1cul semblable à celui d'Hiclanan
conduirait à un accord satisfaisant avec les résultats observés.
En ce qui concerne l'argon les sections de quenching des niveaux
nP(Rb) sont in:f'érieures à celles de l'hélium (d 1 1.m facteur 2 environ).
La situation inverse est observée pour les états nD(Na), en bon accord
avec les résultats d'Hiclanan51 • A basse énergie (0.0.5 eV) la section
de diffusion élastique e--Ar varie rapidement (effet Ramsauer pronon
cé) en encadrant les va1eurs correspondantes pour l'hélium. De plus
sa polarisabilité est notablement plus forte que celle du néon. Il est
donc probable que le terme de correction de polarisation est important.
Rien ne permet de penser, sans éva1uer ce terme de manière précise,
que là-aussi l'interaction prépondérante ne soit pas l'interaction
e--Ar.

- 206 -
e) ~~~~!=-~=-~~!~~=!=~~~~=~=· Avant de résumer notre discussion il nous para!t indispensa
ble de discuter le modèle proposé par Gal.lagher et ai.42 pour expliquer
les faibles sections efficaces obtenues (pour les collisions alcalin
gaz rare) dans le cas de niveaux non hydrogénoïdes (nS(Na)). Ce modèle
postule pour ces états un mécanisme totalement différent de celui cor
respondant au cas des niveaux non hydrogénoides: l'interaction prépon
dérante serait al.ors l'interaction coeur ionique-perturbateur et non
1. 1 interacti.on e--perturbateur. Nous allons tout d'abord analyser les
arguments avancés pour 1.e changement de la nature de l'interaction à
prendre en compte suivant que les niveaux sont hydrogénoïdes ou non.
Les auteurs notent tout d'abord,fort justement, que deux pos
sibilités sont à considérer pour expliquer les résultats obtenus soit
une faible probabilité de transition à grande distance internucléaire
(pour des paramètres d'impact comparables à< r>, pour parler en terme
de théorie semi-classique), soit une forte probabilité de transition à
faible distance internucléaire. Ce dernier cas conduit, ce qu'ils font,
à prendre en compte l'interaction coeur ionique-perturbateur, qui est
bien une interaction à courte portée52 • Les de~ raisons invoquées pour
le rejet a priori de la première hypothèse (interaction à grande dis
tance internucléaire) ne nous semblent pas val.ables. La première,
d'ordre théorique, vient d'une estimation effectuée par Hickman44 qui
conduirait à de très faibles valeurs pour les sections efficaces
nS -t nP dans le cas du sodium. Il faut noter que ce cal.cul est effec
tué à l'aide de fonctions d'ondes très approchées (Eq. 49 de la réfé
rence 44) qui ne sont réalistes que pour des valeurs der notablement
supérieures à .::r > ce qui entache certainement le cal.cul d'une erreur
notable. De plus rien ne dit, et nos cal.culs prouvent le contraire,
que ces transitions sont celles qui contribuent le pl.us au quenching
des états nS du sodium (en fait, comme nous l'avons indiqué précédem
ment ce sont les niveaux fortement dégénérés (n-1)F,G,H ••. qui appor
tent la contribution la plus notable). La deuxième raison invoquée se
rait que l'on devrait observer dans le cas d'une interaction à grande
distance internucléaire une variation notable des sections efficaces
en fonction den. Ceci n'a rien d'évident a priori et la première ap
proximation de Born utilisée avec le potentiel de Fermi montre, que
pour des écarts d'énergie compris entre environ 0.2 et 0.02 k:T, les

- 207 -
sections efficaces entre un niveau non hydrogénoïde et le niveau de
forte mu1tiplicité le plus voisin ne varient pas notablement avec n.
Une décroissance rapide avec n ne sera observée que si 1a borne infé
rieure de 1'intégrale intervenant dans 1e calcu1 de 1a section effi
cace (Eq.(F18)) peut 3tre prise égale à zéro, c'est-à-dire pour des
transitions quasi-élastiques. Ces remarques étant faites nous al1ons
analyser maintenant 1e modèle proposé.
Quand l'atome d'hélium s'approche du coeur ionique de l'al
calin i1 y a formation d'un dipole transitoire dont 1e champ électri
que a pour principal effet d'opérer une séparation des niveaux de
l'atome alcalin, phénomène assez analogue à l'effet Stark classique.
Ce dip81e transitoire peut alors induire une transition entre deux
états voisins, ce par un phénomène de croisement de niveaux analogue
à l'ionisation Stark des états de Rydberg par un champ électrique pu1-
sé. Une évaluation, très grossière, des sections efficaces à partir 0
de ce modèle conduit à une valeur, indépendante den, voisine de 7 A
dans le cas des col1isions (Na-He) (à comparer à la valeur expérimeno
tale 21 A pour le niveau 10S). J:1 faut noter que Gal1agher et al.
observent une variation non négligeab1e de 1a section efficace de 0
quenching en fonction den (celle-ci passe de·7.3 A pour le niveau 9S 0
à 21 A pour 1e 11S) ce qui rend 1a comparaison assez problématique.
Nous avons essayé d'estimer, à l'aide de ce modèle, les sections effi
caces dans le cas des niveaux Set P du rubidium. Uti1isant les valeurs
publiées par Waldman et al52 pour le potentiel Rb+-He nous estimons
que ce processus conduirait, dans les deux cas, à une section de dépo-o
pu1ation, constante avec n, au plus égale à 20 A, ce qui, du moins
pour les éta'ts nS(Rb), est d'environ un.ordre de grandeur inférieur
à nos valeurs expérimentales.
Concluons cette discussion par quelques remarques:
- pour les valeurs den les plus faibles étudiées par Gal1agher
et al. ( 6 !: n "9) il nous para!t plus que probable que l'approche la
p1us correcte est une approche trois corps (de type pseudopotentiel)
te11e que cel1es que nous avons décrites pour les niveaux peu excités
( 7P - 6D du césium par exemp1e) ;
- pour les valeurs den les plus élevées (n~10) i1 nous para!t
diffici1e de négliger complètement l'interaction e- externe-hélium
m~me quand les sections efficaces mesurées sont faibles (~ de la di-02
zaine d'A ) .

- 208 -
f') !!~~~~.
Nous avons clairemen.t montré l'importance de l I interaction
e- externe-perturbat~ur sur la dépopulation collisionnelle des états
de Rydberg. Une approche théorique simple a permis, dans le cas des
collisions alcalin-hélium, d'obtenir de bonnes estimations des sec
tions ef'f'icaces de dépopulation. Notre travail permet d'expliquer
les comportements dif'f'érents des sections ef'f'icaces en fonction de
la position du niveau étudié dans le diagramme énergétique de l'ato-
me alcalin. En particulier, l'importance des écarts énergétiques asymp
totiques entre les niveaux concernés et l'influence de la multiplicité
du niveau d'arrivée ont été clairement dégagées.

- 209 -
REFERENCES DU CHAPITRE VI
1 - J. CUVELLIER, P.R. FOURNIER, F. GOUN.AND et J. BERLANDE,
CR Acad. Sc. Paris 276, 855 (1973).
2 - J. CUVELLIER, P.R. FOURNIER, F. GOUNAND, J. PASCALE et J. BERL.ANDE,
Phys. Rev. A11, 846 (1975). -3 - L. KRAUSE, VII ICPEAC Invited Papers and Progress Reports,
édité par T.R. Govers et F.J. de Heer, North-Ho11and,
Amsterdam (1972).
4 - E.E. NIKITIN, The Excited State on Chemical. Physics,
édité par J.W. Mac Gowan, Wiley, New-York (1975}.
5 - M. PIMBERT, J. Phys. {Paris) 33, 331 (1972).
6 - E.E. NIKITIN et A.I. REZNIKOV, Chem. Phys. Lett. 8, 161 (1971).
7 - W.E. BA'YLIS, J. Chem. Phys. 51, 2665 (1969).
8 - J. PASC.ALE et J. VANDEPLANQDE, J. Chem. Phys. 60, 2278 (1974). -9 - R.E. OLSON, F.T. SMITH et E. BAUER, App1. Opt. !Q, 1848 (1971).
10 - I.N. SI.ARA, H.S. KWONG et L. KRAUSE, Can. J. Phys. 8, 945 (1974).
11 - P.W. PACE et J.B. ATKINSON, Can. J. Phys. 52, 1641 (1974).
12 - J.F. KIELKOPF, J. Chem. Phys. g_g, 4809 (1975).
13 - R.E. OLSON, Chem. Phys. Lett. 22., 250 (1975).
14 - J. PASCALE et P.M. STONE, J. Chem. Phys. 65, 5122 (1977).
15 - H.F. w.ELLENSTEIN et W.W. ROBERTSON, J. Chem. Phys., 56, 1072 (1972).
16 - W.J. STEETS et N.F. L.AJ.~, Phys. Rev. fil, 1994 (1975).
17 - H.A. BETHE et E.E. SAL.PETER, Quantum Mechanics of one and two
electron atoms, Springer Verlag (1957).

- 210 -
18 - T.F. G.ALLAGHER, S.A. EDELSTEIN et R.M. BILL,
Phys. Rev. Lett. 35, 644 (1975).
19 - W.P. w.EST, G.W. FOLTZ, F.B. DUNNING, C.J. LATIMER et R.F. STEBBINGS,
Phys. Rev. Lett . .d§., 854 (1976).
20 - T.F. G.ALLAGHER, S.A. EDELSTEIN et R.M. BILL,
Phys. Rev. ,fil, 1504 (1975).
21 - C. FA.BBE et S. HAROCHE, Opt. Comm. 15, 254 (1975).
·22 - S.M. CURRY, C.B. COLLINS, M. Y. MIRZA, D. POPESCU et I. POPESCU,
Opt. Comm. !..2., 251 (1976).
23 - A. BURGESS et M.J. SEATON, Mon. Not. R. Astron. Soc. f20, 121 (1960).
24 - H.E. SARAPH, Proc. Phys. Soc. 83, 763 (1964).
25 - I.C. PERCIVAL, Nucl. Fusion, 2, 182_(1966).
26 - T.F. G.ALLAGHER et W.E. COOKE, Phys. Rev. Lett. 42, 835 (1979).
27 - R.H. DICIŒ, Phys. Rev. 93, 99 (1954).
28 - N. SKRIBAl.~O'WITZ, I. P. HERMAN, J . C . MacGil..LIVRAY et M. S. FELD,
Phys. Rev. Lett. 30, 309 (1973).
29 - M. GROSS, C. FABRE, P. Pn..LET et S. HAROCHE,
Phys. Rev. Lett. 36, 1035 (1976).
JO - J.C. MacGil..LIVRAY et M.S. FELD, Phys. Rev. A14, 1169 (1976).
31 - R. BONIFACIO et L.A. LUGIATO, Phys. Rev. fil, 1507 (1975).
32 - P. Pn..LET, Thèse de 3ème cycle, Paris (1977).
33 - F. GOUN.AND, M. HUGON, P.R. FOURNIER et J. BERLANDE,
J. Phys. B12, 547 (1979).
34 - Q.H.F. VREREN et M.H.F. SCHtJlJRMAN, Phys. Rev. Lett. 42, 224 (1979).
35 - M. HUGON, F. GOUNAND et P.R. FOURNIER, J. Phys. B11, L605 (1978).
36 - M. HUGON, F. GOUN.AND, P. R. FOURNIER et J. BERL.A.i.'IDE,
J. Phys. B12, 2707 (1979).

- 211 -
37 - A.P. EJ:CXMAN, Phys. Rev. !!.§_, 1339 (1978).
38 - T.F. G.ALLAGHER, S.A. EDELSTEIN et R.M. HILL,
Phys. Rev. A15, 1945 (1977).
39 - J. S. DEECH, R. LUYPAERT, L.R. PENDRD.,L et G. W. SERIES,
J. Phys. B10, L137 (1977).
40 - A.C. TAM, T. YABUZAKI, S.M. CURRY, M. HOU et W. HAPPER,
Phys. Rev. A17, 1862 (1978).
41 - L.R. PENDRD.,L, J. Phys. B10, L469 (1977).
42 - T.F. GALLAGHER et W.E. COOIŒ, Phys. Rev. fil, 2161 (1979).
43 - J. DEROUA.RD et M. LOMB.ARDI, J. Phys. B11, 3875 (1978).
44 - R.E. OLSON, Phys. Rev. A15, 631 (1977).
45 - M. MATSUSAWA, J. Phys. fil, 3743 (1979).
46 - E •. de PRUNELE et J. PASCALE, J. Phys. fil, 2511 (1979).
47 - L. LANDAU et E. LIFCEJ:TZ, Mécanique Quantique p. 416, Ed. Mir,
Moscou (1967).
48 - C.J. JOACHAIN, Quantum Co11ision Theory, North Ho11and/American E1sevier (1975).
49 - I.S. GRADSHTEYN et I.M. RYZEIK, Tab1e of IntegraJ.s Series and Products, Academic Press, New-York (1965).
50 - G.E. FORSYTEE, M.A. MALCOL~ et C.B. MOLER, Computer Methods for
MathematicaJ. Computation, Prentice Ha11 (1977).
51 - A.P. EJ:CKMAN, Phys. Rev. A12, 994 (1979).
52 - M. WALDMAN et R.G. GORDON, J. Chem. Phys. 71, 1325 (1979).

Chapitre VII
CONCLUSION
Nous avons présenté un ensemble de mesures relatives aux pro
priétés collisionnelles des atomes aJ.caJ.ins excités ou très excités,
au.~ énergies thermiques, la cible étan~ soit un atome de gaz rare, soit
l'atome aJ.caJ.in dans son état fondamentaJ.. Notre travail couvre un en
semble très large de situations expérimentaJ.es ( 7 4 n <.. 22 ; 0 ~ l • 2),
ce qui nous a permis, en liaison étroite avec les développements théo
riques, de bien dégager les problèmes essentiels posés par ce type de
collision.
Nous avons été amenés dans la discussion des approches théo
riques à faire la distinction, à notre avis essentielle, entre la par
tie statique (détermination du potentiel d'interaction) et la partie
dynamique (caJ.cul des sections efficaces) du problème. Notre travail
expérimentai relatif aux niveaux (7P-6D) du césium a mis en évidence
la nécessité d '~une bonne connaissance du potentie.l d'interaction, qui,
seule,peut permettre de rendre compte des différences très notables
de comportement observées entre .les niveaux résonnants et les niveaux
excités. En effet, pour ces derniers, la présence de niveaux voisins
entra!ne l'apparition de couplages statiques importants. Nous avons
pu, pour une telle situation, obtenir, dans un cas particulier, un
bon accord entre vaJ.eurs théoriques et expérimentaJ.es.
Pour les états relativement peu excités (n~10) on dispose
maintenant de potentiels, certes perfectibles, mais déjà suffisamment
élaborés pour permettre d'envisager des comparaisons détai.l.lées entre
théorie et expérience, le traitement de .la partie dynamique ne pose
plus que des problèmes techniques .liées au nombre d'équations couplées
à prendre en compte. Dans cet esprit le groupe de niveaux étudiés
(7P -6D du césium) nous semble particulièrement intéressant à étudier

- 214 -
à l'aide de techniques plus fines (jets croisés) que celle que nous
avons utilisée (mesure en cellule).
Pour ce qui est des états de Rydberg des approximations sé
vères sont nécessaires aussi bien pour la résolution de la partie sta
tique que de la partie dynamique, le nombre de niveaux à prendre en
compte rendant quasiment impossible l'extension des modèles utilisés
pour les états peu excités. Notre travail a montré l'importance essen
tielle de l'interaction (e- externe-perturbateur) sur le processus de
désexcitation collisionnelle des états de Rydberg. Un traitement dyna
mique basé sur la première approximation de Born et ne prenant en
compte que cette interaction a permis de rendre compte de façon satis
faisante de l'ensemble des résultats disponibles pour le couple alcalin
hélium (mesures effectuées aussi bien dans notre laboratoire que par
des équipes étrangères), cas qui pose les problèmes numériques les
moins ardus. Une vue globa1e du problème de la désexcitation collision
nelle des états de Rydberg a ainsi été dégagée.
Il importe maintenant d'approfondir les procédures numériques
relatives aux approches théoriques afin de pouvoir étendre les compa
raisons théorie-expérience aux situations expérimenta1es les plus dif
verses (perturbateurs hautement polarisables). Notons enfin l'intér~t
présenté par l'extension des mesures à des valeurs élevées den
(N 30-40). En effet toutes les approches existantes prévoient l.llle dé
croissance de la section de dépopulation, pour des valeurs den suf
fisamment grandes. Dans ce cas la diminution constante de la contribu
tion de l'interaction e--perturbateur devrait permettre, outre la véri
fication du comportement asymptotique des théories existantes, de met
tre en évidence l'influence d'autres interactions (coeur-ionique per
turbateur), encore masquées dans les études menées jusqu'à présent.

Appendice I
ETALONNAGE DE LA TRANSMISSION EN LONGUEUR D'ONDE D'UNE VOIE DE MESURE.
Celui-ci s'effectue, en comptage, à l'aide d'une lampe à fi
lament de tungstène. On mesure les comptages c1 et c2
à deux longueurs
d'onde i 1 et Â2
• La transmission relative s'en déduit en tenant compte
de la variation de luminance de la source éta1on entre ~1 et À2
• La
lampe est a1imentée à l'aide d'une alimentation stabilisée. Quand
l'équilibre thermique est établie, on mesure la température TÂ du fila
ment à l'aide d'un pyromètre optique. La mesure fournit la température
Ti. de brillance (ou de luminance) qu'il convient de corriger pour obte
nir la température T du corps noir équiva1ent1
en tenant compte du
facteur d'absorption~~ du tungstène (loi de Kirchoff) :
T = b T~ log(e)
b log (e) - À T). log(~.,.) (A)
où À représente la longueur d'onde de travail du pyromètre (0.65 µ)enµ
TÀ la température lue (comprise entre 1600 et 1800°K)
~Â le facteur d'absorption du tungstène (qui varie linéairement
avec T de o.426 pour T = 2600°K à o.448 pour T = 1600°K)
b un coefficient numérique va1ant 1.4388 104 •
Une fois T connue, on peut ca1culer la luminance ·aux longueurs d'ondes
choisies par la formule de Planck:
L Â = ( 1 000 Â ) - 5
où L est exprimée en W/cm2 .A.Sd
T en °K i, en µ
a b7ÂT _ 1 e
(B)

avec a = 1.197 1015
b = 1.4388 104
- 216 -
Comme on opère en comptage i1 faut ca.lcu1er la va.leur de (À LÀ)
puisque, par exemple c1 = W~/hc où West proportionnel à LÀ1 • La
transmission relative aux longueurs d'onde ~1 et Â2 est donnée par
= c1 À 2L).2
C2 À1L;>,.1 (c)
Nous avons normalisé les transmissions à une 1ongueur d'onde 0
donnée (455.5 A, qui correspond à la transition 7P3
; 2 ~6s112 du cé-
sium), puisque nous n'avions besoin que du rapport de ce1les-ci. Nous
avons toujours effectué les mesures pour différentes températures du
filament. La dispersion maxima.le obtenue sur les transmissions rela
tives est de l'ordre de 3~.
Référence :
1 - P. FLEURY et J. P. MATHIEU,
Cha.leur, Thermodynamique, états de la matière, Eyrolles, Paris (19.59).

Appendice II
~ORRECTION D'EFFUSION THERMIQUE.
Rappelons que cette correction est rendue nécessaire par le
fait que la mesure de la pression dans la cellu1e de mesure s'effectue
à une température différente de celle de l'étuve qui contient la cel
lu1e. Si on appelle P1 la pression mesurée à la température T1 (tempé
rature ordinaire~ 300 K) et P2 la pression réelle dans l'enceinte, à
la température T2 (qui peut varier de 450 à 620 K) on a:
- soit P2 = P1 dans le cas où l'écou1ement entre la cellu1e
et l'enceinte de mesure s'effectue en régime
visqueux;
- soit PR P2 = T
2 si le régime d'écou1ement est molécu1aire.
1 1
(A)
(c)
Le problème est difficile à résoudre de manière rigoureuse et les vé
rifications expérimentales sont délicates. Nous renvoyons le lecteur
intéressé à la référence 1.
Nous avons adopté la procédure de correction suivante.
Appelons D le diamètre du tube dans la zone de transition entre les
températures T1 et T2
(sortie de l'étuve). Nous calculons d'abord le
para.mètre oc qui détermine le rapport des conductances (en cm3/s) pour
les deux types de régime, visqueux [v] ou molécu1aire [MJ, soit:
[v] ct = [MJ
Ensuite nous avons adopté l'interpolation suivante
(D)

- 218 -
p2 0: + VT2/T1 P1 ~ a: + 1 (D)
qui vérifie bien, dans les cas limites, les formu.les (A) et (B) et
revient à considérer, de plus, que, quand~= 1, la pression réelle
est la moyenne des pressions obtenues respectivement en régimes visqueux
et moléculaire. L'équation (D) n'a pas de justification thermodynamique.
Elle constitue seu.lement une hypothèse raisonnable1 • L'erreur finale
sur l'évaluation du rapport P2/P1 est, de ce fait, très difficile à
estimer. Il faut cependant noter que, d'une part, le rapport P2/P1 ne
dépasse pas 1 .4, d'autre part que, pour de nombreuses séries de mesures,
nous avons opéré en régime "pur", c'est-à-dire qu'aucune correction
n'était nécessaire. Nous pensons que, même dans la zone de transition
entre les deux régimes (où le rapport P2/P1 est de l'ordre de 1.2,
au plus), l'application de l'équation (D) n'entra.!ne pas une erreur
supérieure à 5% sur la COIUlaissance du rapport P2/P1 •
on a
Le calcul numérique de la correction s'effectue comme suit1
0: = 5.875 10-2 X (1 + 1 .24 x) (1 + x)
X = D"\P fi ;r-1/2
(E)
où D s'exprime en cm, P = (P1 + P2 )/2 en baryes, i(coefficient de vis
cosité) en poises, M (masse molaire) en grammes, T=(T1 + T2)/2 en °K.
Nous donnons dans le tableau I d'une part les valeurs de 1 /l) {M/R pour les différents gaz rares (à T = 273°K), ainsi que la pression P, pour laquelle 0: = 1 (ce pour T1 = JOO K, T
2 = 600 K et D = 1)
1/~ VM/R P (a:= 1) en Torr
-He 1 .1 7 0.22
Ne 1 . .58 O .16
Ar J.02 0.085
Kr 4.46 0.058
Xe 5.82 0.044
Tableau 1

- 219 - -
(Dans le tableau 1 on donne !1 pour T = 273 K. Dans le calcul il faut
prendre "'\(T) = "f\(273) V T/273).
Nous avons utilisé pour le calcu1 de~ une méthode itérative,
puisque P2
(donc P) n'est pas connu a priori; on a donc cal.cu1é ~ (P1 ,T)
et on en a déduit une première valeur de P2
, qui permet le calcul de
(P) dr 1 • On recalcule alors~ avec cette nouvel1e valeur et la pro-or e cédure est itérée jusqu'à convergence de P, d'où l'on déduit P
2• La
convergence est, en fait, très rapide et, m~me dans le cas d't.Ul régime
intermédiaire, cinq itérations sont suf'fisantes pour assurer la stabi
lité de P à mieux que 10-4 . Le dernier point à mentionner est la défi
nition assez arbitraire de T. Nous avons vérifié que~ est en fait peu
sensible à T; ainsi, pour des conditions typiques, oc(T = 380) = 0~81
et oc(T = 620) = 0.79. Ainsi la définition que nous avons adoptée pour
T n'entra.!ne qu'une erreur nég1igeable sur le résultat final.
Référence :
1 - C.M. VAN ATTA, Vacuum Science and Engineering, Mc Graw-Hi11,
New-York ( 1965).

Appendice III
,C.Al.,CUL DE L'ABSORPTION DES RAIES DE FLUORESCENCE.
Ce ca1cul. de correction n'est nécessaire que dans le cas des
états faiblement excités. Nous l'avons effectué dans le cas de la réab
sorption des raies (7P1 ; 2 ~6s112 ) et (7P3
; 2 -6s112) du césium. Les
expériences ont été conduites à une pression de césium su;f'fisamment
basse pour que seul.e la prise en compte de l'élargissement Doppler soit
nécessaire. Par contre les pressions de gaz rare utilisées sont telles
qu'il convient de tenir compte de l'élargissement collisionnel da au
gaz rare, dès que la pression dépasse, environ,1 Torr.
L'absorption I/I peut s 1 écrire1 0
I 1
¾= vr: f·~ e-q2
-oo
-k('J)l dq e (A)
où 1 représente la longueur moyenne d'absorption (3 mm) et k(Y) le coef
ficient d'absorption qui peut s'exprimer par le profil de Voigt classi
que (convolution de la Lorentzienne due à l'effet des collisions et de
la gaussienne due à l'effet Doppler, l'élargissement naturel de la raie
étant négligeable) :
k( \)) k a
0 =7 r<o -y2
a2: (q-y)2 -œ
dy (B)
où a et q représentent deux paramètres sans dimensions, reliés à la
fréquence centra1e de la raie v0
et aux élargissements Doppler A~D et
collisionnel A~c par:

- 222 -
AV~ a _ __9. Log2 - Av
D
2(V-'J0
)
yLog2 q = A\)D
(c)
et k le coefficient d'absorption au centre de la raie, soit 0
2 ~g2 )..2 N A
k 0
o = AvD g1 ~ (D)
Ici g2
et g1 sont les poids statistiques des niveaux supérieur et in
férieur de la transition considérée, de longueur d'onde À et de coef-o ficient d'Einstein A. N représente la densité d'atomes de césium, à
l'état fondamenta1. Fina1ement 1es élargissements Dopp1er et collision
nel sont donnés par
2~0 y 2RT Log2 A~D = C M
(E)
A\) = 1S NG C
où c représente 1a vitesse de 1a 1umière, K 1a constante des gaz par
faits, T 1a température de 1a vapeur (en °K), M 1a masse mo1aire du
césium, NG 1a densité de gaz rare et 1S la constante d'é1argissement
co11isionne1le correspondante. Les va1eurs de 1S sont données dans les
références 2 et 3.
De nombreuses méthodes sont possibles pour 1'éva1uation numé
rique du profil de Voigt4- 6 • Elles dépendent essentie11ement de la va
leur du paramètre a. Celle-ci ne dépassant pas 5 nous avons évaiué (B) à 1'aide de la série de Zemansky7 , soit
k(v)
avec
et
00
= ka I n=o
C ( a) n
e-q2
n!
2n 9.
2ea t2 2 2r C0
(a) = y:;; e- dt= ea erfc(a)
a
cn(a) = ~-1 (~ - 2a Cn_1 (a))
(F)

- 223 -
Nous avons pris 20 termes dans le ca1cu1 de la série, après avoir véri
fié que seu1e la précision du ca1culateur affectait a1ors le résu1tat
La quadrature fina1e (éq. (A)) est effectuée par la méthode de Gauss
Hermite à 25 points8 , particu1ièrement bien adaptée au ca1cul des in
tégra1es de la forme :
l+GI
-x2 -œ e :f'(x) dx •
Nous n'avons pas utilisé cette méthode pour le calcu1 du profil de
Voigt (éq.(D)) qui est aussi de cette forme, car pour a tendant vers 0
elle ne redonne pas le profil de Gauss9 .
Références
1 - A.C.G. MITCHELL et M.W. ZEMANSKY, Resonance radiation and excited
atoms, Cambridge University Press, 2ème édition (1961).
2 - J.L. LEMAIRE et F. ROSTAS, J. Phys. B4, 555 (1971).
3 - Y.V. EVDOKIMOV, Opt. Spektrosk 24, 832 (1968).
(Opt. Spectrosc. 24, 448 (1968)).
4 - W. LOCHTE-HOLTGREVEN, Editor, Plasma diagnostics, North-Ho11and
(1968).
5 - J.J. OLIVERO et R.L. LONGBOTHOM, J. Quant. Spectres. Radiat.
Transfer. !1, 233 (1977).
6 - J.H. PIERLUISSI et P.C. VANDERWOOD, J. Quant. Spectrosc. Radiat.
Transfer. 18, 555 (1977).
7 - M.W. ZEMANSKY, Phys. Rev. 36, 219 (1930).
8 - z. KOPAL, Numerica1 Ana1ysis, Chapman and Hall, Londres (1961).
9 - F. LAMBERT, Thès~ Université Paris VI, Paris (1975).

Appendice IV
MEStmE DE LA DENSITE D'ATOMES ALCALINS PAR ABSORPTION DE LA RAIE
DE RESONAl.'iCE (METHODE DE LA LARGEUR EQUIVALENTE).
Nous avons uti1isé cette méthode pour obtenir une deuxième
mesure de 1a densité d'atomes al.cal.ins, 1a première détermination étant
obtenue à partir de 1a courbe de tension de vapeur saturante de 1'al.
cal.in considéré. La méthode de 1a 1argeur équival.ente présente 1 1 avan
tage d'une mise en oeuvre re1ativement simpJ.e. De pJ.us eJ.1e permet de
s'affranchir de 1'inf'1uence del.a fonction d'apparei1, toujours dé1i
cate à déterminer avec précision. Nous exposerons d'abord brièvement
l.a méthode 1, qui est bien connue. Nous donnerons ensui te l.e ca1cu1 théo
rique qui permet, du résu1tat del.a mesure, de remonter à 1a densité
d'atomes al.cal.ins. Les équations général.es seront données, ainsi que
l.a procédure numérique retenue dans 1e cas particu1ier de 1a raie de
résonance ( 5P .'.3/2 - 5S112 ) du rubidium.
La méthode consiste, après enregistrement à 1'aide d'une 1am
pe à fi1ament de tungstène du profi1 d'absorption del.a raie, à ca1cu-
1er J.a 1argeur équival.ente du profi1 obtenu. Ceci nécessite un étal.on
nage précis en l.ongueur d'onde du défiJ.ement du monochromateur ainsi
quel.a détermination, par p1a.nimètrie de l.'aire du profiJ. d'absorption
(Fig. 1) : l.a J.argeur équival.ente L est te1J.e que J.es deux aires ha-eq churées soient égal.es.
I
Fig. 1
Ào À

- 226 -
La f'ormu1e permettant J.e caJ.cu1 théorique de L est la suivante eq
[
+<io
L = ( 1 ek ( \J ) l J eq L - dV
0
(A)
où J. représente J.a longueur d'absorption et k(V) J.e coefficient
d'absorption. Nous avons vu J.'expression de ceJ.ui-ci dans J. 1 appendice
précédent. En fait J.e caJ.cu1 précis de k(v) doit tenir compte de J.a
structure hyperfine de ia raie de résonance (cette remarque est sur
tout vraie à basse pression d'alcalin typiquement pour p <- 5.10-4 T).
Dans J.e cas du rubidium on doit aussi tenir compte de J.a présence de
deux isotopes naturels si bien que la forme la plus complète de k(v)
devient :·
k( \J) ""' ""' a .. = L.. L.. p .. k (i,j) ..2:.:1. i j iJ o n f
+• 2 -y
a~. + e (q .. -y)2 _a, 1J 1J
dy (B)
où la sommation suri co~respond aux différents isotopes et ceJ.J.e sur
j aux différentes composantes hyper:f'ines. p .. est le poids statistique 1J
de J.a composante j de l'isotope i. k (i, j), a .. et q .. sont reJ.atifs à 0 1J 1J
cette composante. Leurs expressions sont données par les équations
(c)· et (D) de l'appendice III. Pour simplifier J.'écriture nous note
rons H .. .le profil. de Voigt de la composante ( i, j). On obtient alors : 1J
L = l 00
) 1 - exp 1-J. L L p. . k ( i, j) H .. J l d V eq L. . . 1J o 1J ) 0 1 J
avec Hij = a~j [46 _2 __ e_-_Y_2_~2 dy (c)
a .. + (q .. -y) - 1J J.J
La densité N d'alcalin intervient dans a .. (eqs (c) et (E) de l'appen-1J
dice III) et on est donc conduit à calculer Leq pour différentes valeurs
de N pour construire J.a courbe L = f(N) eq tion de N à partir de la mesure de L eq
qui servira à la détermina-

- 227 -
Nous a11ons traiter maintenant 1e cas de 1a raie
(5P3
; 2 -5s1 ; 2 ) du rubidium. Si on nég1ige, ce qui est parfaitement
justifié, 1a structure hyperfine de 1 1 état excité, 1a raie présente
quatre composantes dont 1es caractéristiques2 sont données dans 1e
tab1eau 1.
0
Isotope Spin nuc1éaire Moment é1ectronique tota1 Longueur d'onde (A) (F = I + J)
85Rb 5/2 2 7800.232
3 7800.294
87Rb 3/2 1 7800.182
2 7800.321
Tab1eau 1
L'abondance re1ative des isotopes 85 et 87 est [ 85RJ;[87Rb] = 2.57.
Nous avons supposé enfin 1e même é1argissement co11isionne1 ~ pour 1es
quatre composantes (éq. (E) de 1 1 appendice III). Ce1ui-ci est connu
avec précision3 • Contrairement au cas de 1'appendice III, 1a procédure
choisie pour 1'éva1uation de H .. dépend à 1a fois de a .. et de q .. , l.J l.J l.J
car même 1es grandes va1eurs de q .. contribuent au résu1 tat fina1 l.J (1e terme e-42 de 1'équation (A) de 1'appendice III annu1ait 1 1 inf1uen-
ce de ces va1eurs). Or i1 est faci1e de se rendre compte que 1e ca1cu1
de 1a série de Zemansky est diffici1e dans ce cas. La procédure suivan
te a été retenue. Les va1eurs de a .. étant toujours inférieures à 10-1
l.J 1 3 (1e ca1cu1 est effectué pour 6 .1 O 1 O, N !: 7. 10 ) , H .. est ca1cu1é par
l.J 1a série de Zemansky (éq. (F), Ap. III) pour q ~ 4.2. Pour 1es grandes
va1eurs de q nous uti1isons 1a série suivante4 :
H .. l.J
2 q .. - l.J = e 2a. · i 1 + ---=.J.. -+ 2 fiï" 2qij
1 • :! (2q-?. )2
l.J + •• • l ( D )

- 228 -
que nous limitons au huitième terme. La précision obtenue sur
l'aide de cette procédure est meilleure que 10-7 . L'obtention
H .. à J.J
de L eq s'effectue par une intégration numérique à pas variable*, basée sur
une interpolation polynomiale du huitième degré5 . L'algorithme retenu
a le gros avantage de s'adapter à la forme de l'intégrant et de ne
prendre qu'un nombre de points minimum pour obtenir la précision€
choisie (~ = 10-4 ).
Références :
1 - R.M. :mLL, J. Quant. Spectrosc. Radiat. Transfer ,il, 19 (1979).
2 - H.M. GIBBS et R.J. HULL, Phys. Rev. 153, 132 (1967).
3 - K. NIEMAX et G. PICHLER, J. Phys. B8, 179 (1975).
4 - A.C.G. MITCHELL et M.W. ZEMAl.~SKY, Resonance radiation and excited
atoms, Cambridge University Press, 2ème édition (1961) p. 321.
5 - G.E. FORSYTEE, M.A. MALCOLM et C.B. MOLER, Computer Methods for
Mathematical Computation, Prentice Hal.l Inc. (1977) p. 97.
* La borne supérieure d'intégration de l'équation (c) a été prise égale
à 50 VD 1 , ce qui permet la détermination de L avec une préci-opp er eq sien relative meilleure que 10-5 .

Appendice V
DUREE DE VIE RADIATIVE DES ATOMES ALCALINS ARTICLE 4.
Le ca1cul. théorie de 1a durée de vie d'un état excité ire
quiert 1a connaissance de tous 1es é1éments de matrice dipo1aire de
type <ilRlj>, où j représente un état radiativement connecté à 1'état
i considéré. Dans 1e cas des états de Rydber~ i1 faut ca1cul.er de nom
breux é1éments de matrice pour obtenir 1a durée de vie~. Ainsi seul.es
des méthodes simp1es peuvent 3tre uti1isées ,- te11e ce11e proposée par
Bates et Damgaard1
qui nous a permis d'obtenir 1es é1éments de matrice
radiaux des atomes a1ca1ins pour n, 28. Nous insérons cet artic1e2
(Artic1e 4) à 1a fin de cet appendice. I1 n'appe11e pas de commentai
res particuJ.iers. A partir de ces é1éments de matrice on peut obtenir,
entre autres, 1a durée de vie radiative des états de Rydberg que nous
avons étudiés. La comparaison entre résuJ.tats théorique et expérimen
taux est donnée dans 1e tab1eau 1
Niveau ~xp (µs) ë:'th ( µs) ?;ca1 ( µs)
12P 1. 55 !, O. 20 2.40 1.69
14P 2.60 + o.4o 4.23 2.64
17P 6. 40 !. 1. 30 8.41 4.50
22P 14.o !. 5.0 21 .1 9.00
12S O. 77 !. O .15 0.887 0.732 14S 1. 26 !, 0. 25 1. 60 1. 23
16S 2.19 !, 0. 50 2.62 1. 89
18S 3. 30 ± O. 70 4.00 2.73
Tab1eau 1
Comparaison entre 1es durées de vie expérimenta1e Cexp et théorique ~th pour que1ques états de Rydberg du rubidium.
Voir 1e texte pour 1a signification de Cca1·
. 1
1

f'
- 230 -
Le tableau 1 montre une variation similaire en fonction de
n* entre~ et ~th' bien que les va1eurs absolues, en particu1ier exp dans le cas des niveaux P, diffèrent assez sensiblement. Nous avions
déjà sou1igné ce fait dans la référence J. Très récemment Ga1lagher
et Cooke4 ont montré l'in:fluence que pouvait avoir le rayonnement du
corps noir sur la mesure des durées de vie radiative. Celle-ci peut
s'exprimer par la relation
1 -= 'Cca1
1 1 -+-?:th ?:CN
où "C.ca1 représente la durée de vie du niveau à la température de l'ex
périence (460 K pour les états P, 520 K pour les états S), ~th la durée
de vie ca1cu1ée, et ~CN une correction dépendant de la température T
et dont le ca1cu1 requiert principa1ement la prise en compte des élé
ments de matrices radiaux entre le niveau considéré et ses plus pro
ches voisins. Nous avons pu ca1culer ce terme correctif' à partir des
élémeAts de matrice que nous avions ca1cu1és2 (Article 4). Le tableau 1
montre les va1eurs obtenues pour ccai· Bien que le ca1cu1 de cCN' à
l'aide de la méthode de la référence 2, soit certainement moins précis
que le ca1cu1 de C~h (la méthode utilisée est en ef'f'et peu adaptée au
ca1cu1 des éléments radiaux entre niveaux voisins, qui, d'autre part,
contribuent peu à la va1eur de eth), la comparaison entre ?:' a1 et ~ c exp montre un accord satisfaisant. Un tel accord montre l'importance du
rayonnement du corps noir dans le cas des états de Rydberg et con:firme
la possibilité d'obtention à l'aide d'une approche théorique simple
(de type Bates-Damgaard) de bonnes estimations pour la durée de vie
radiative des états de Rydberg des atomes a1ca1ins.
Réf'érences :
1 - D.R. BATES et A. DAMG.AARD, Phil. Trans. R. Soc. A242, 101 (1949) .
2 - F. GOUN.AND, J. Phys. (Paris) 40, 457 (1979).
3 - F. GOUN.AND, P.R. FOURNIER, J. CUVELLIER et J. BERLANDE,
Phys. Lett. 59A, 23 (1976).
4 - T.F. GA.LLAGHER et W.E. COOKE, Phys. Rev. Lett. 42, 835 (1979).
. 5 - F. GOUNAND, M. HUGON et P.R. FOURNIER, soumis pour publication au
J. Phys. (Paris) (1979).

t.E JOURNAL DE PHYSIQUE
Classification Physics Abstracts 32.70
- 231 TOME 40, MAI 1979, PAGE 457
Calculation of radial matrix elements and radiative lif etimes for highly excited states of alkali atoms using the Coulomb approximation
F. Gounand
Cencre d'Etudes Nucléaires de Saclay, Service de Physique Atomique, B.P. n" 2, 91190 Gif sur Yvette, France
(Reçu le 2S octobre /978, révisé le J janvier 1979, accepté le 22 janvier 1979)
Résumé. - Les éléments de matrice radiaux mettant en jeu des états fortement excités des atomes alcalins sont calculés à l'aide de l'approximation Coulombiennc. Le choix du critère de coupure a fait l'objet d'une étude détaillée. Ces éléments de matrice (disponibles sur demande adressée à l'auteur) peuvent être utilisés pour le calcul des forces d'oscillateur, durées de vie, rapports de branchement, polarisabilités des états de Rydberg des alcalins. Nous présentons le calcul des durées de vie radiative des états S, P, D et F très excités (10 ~ n ~ 28) des atomes alcalins. Les durées de vie calculées sont en bon accord avec les résultats expérimentaux disponibles.
Abstract. - Radial matrix clements have bcen computed involving highly cxcited states of alkali atoms. An entircly analytical Coulomb mcthod has bccn uscd. Particular attention has bccn paid in the choice of the eut criterion. The extensive data are available from the author upon request and can be used to compute, for example, oscillator strcngths, radiative lifetimes, branching ratios and polarizabilities of alkali Rydberg states. In order to test the results. radiative lifetimes have bccn derived for highly e."tcited (10 ~ n ~ 28), S, P, D and F States of alkali atoms. The computed Iifetimes are reported and arc observed to be in good agreement with the :i.vailable experimental data.
l. Introduction. - The interest in studying the properties of Rydberg atoms has been clearly pointed out by recent experimental and theoretical work [l-5]. A knowledge of oscillator strengths, radiative lif etimes and polarizabilities for highly excited states is often needed in these studies. All these quantities require the calculation of radial matrix elements involving highly excited levels. There exists in the literature some theoretical results, mainly concerning the oscillator strengths, but they are, in general. limited to the lowest states (6, 7] (n - 10) or to some particular spectroscopie series (8-1 OJ. Only Anderson and Zilitis [11, 121 have reported extensive oscillator strength calculations (up ta n - 18) made by using a parametric potential. But it seems difficult to extrapolate their results to more highly excited states for which experimental results begin to appear [I 3-15]. Thus extensive tables of radial matrix elements involving alkali Rydberg states would clearly be of some interest for the physicists working in this area of research. To our knowledge such results are not presently available for highly excited states.
The use of an eJaborate theoretical mode! taking into account accurate radial wave functions (i.e. including core polarization as well as spin-orbit etfects) would require prolong<!d computation time.
Thus, only approximate methods can be considered for extensive calculations of radial matrix elements. The well known Coulomb approximation [16] (here after referred as C.A.) seems to provide the best compromise between computation time and accuracy. We will see that a proper choice of the eut-off criterion for the asymptotic expansion (see § 2) and the use of high precision subroutines allow the obtainment of the radial matrix elements even for high principal quantum number n values. We have computed all the radial matrix elements involving S, P, D and F levels of alkali atoms up to n = 28. For obvious reasons all these elements are not listed here but are available upon request from the author (17]. We report here, as an example of application, the results of calculations concerning the radiative tifetimes of highly excited (10 ~ n ~ 28) S, P, D and F levels of alkali atoms. Only states with n ~ 10 have been considered (somewhat arbitrarily) as Rydberg states and are therefore concerned in the present work. However, the calculations have also been performed for low-lying states.
2. Method. - We use the very well known metho<l first proposed by Bates and Damgaard (16]. The basic idea was that asymptotic fonns of bound state Cou-

- 232 -
458 JOURNAL DE PHYSIQUE N° 5
lomb wave functions can be used because the contribution to the dipole integral from the region of space near the origin (i.e. for r -.. O). where this form is invalid. is smalt. We only summarize now the main features of the method (18].
We want to compute
S = ( n' l' 1 r I ni) = f '° PnAr) rPn1(r) dr. (2.1) 0 .
Using the asymptotic form of the bound state Coulomb wave functions P.Ar) and P.1(r) one finally obtains
( 1 1) _(,...+11•+2-p)
s = KK' L A - + - X p=o li n*' n•
x I'(n*' + n• + 2 - p)
= KK' L T(p) (2.2) ll""o
where K and K' are the nonnalization factors [19] corresponding respectively to the (n, l) and (n', l') levels and n• and n•' their effective quantum numbers (n• = n - ô, ô being the quantum defect). AP is given by
A = li I r.l'
forr +r' "'P
li
a, a,, = L a, "r-11 rsO
(2.3)
where a, (and a,,) are coefficients that are calculated from the following equations :
a0 = 1
a,= '1r-{;:) [!(/ + 1) -_(n• - t) (n• - t + l)].
(2.4)
It is well known that the numerical results are sensitive to the number of tenns included in the summation (2. 2) and that it is very difficult to give any precise justification for the choice of a ·given eut criterion. In their original paper (16] Bates and Damgaard used the condition (n*' + n* - p) ~ 1. However, one can easily see that the first neglected tenn is not always small compared to the sum of the others. The behaviour of the asymptotic expansion (2.2) has been investigated for ail the following series of Na, K. Rb and Cs : S - P, P - D, D - F and F - G. When considering the sign, the same beha viour was observed : first the sign of T(_p) alternates, then for some value of p the terms keep the same sign until p0, the value for which the sign changes. For Na. Rb and Cs we use the eut-off criterion proposed by Bebb [18], i.e. one terminates the series for p = Po - l and adds the average of the two following tenns (of opposite sign). Thus :
[
p0-t l J S = KK' ~ Tt._p) + 2 (T(p0) + T(p0 + l )) .
(2. 5)
In the case of potassium it is observed that T(p0 + l) is always much greater than T(p0), indicating that the expansion can be assumed to termina te exactly. In this later case we use :
110
s = KK' L T(p) . (2.6) 0
Our choice was confirmed by considering the very sensitive (n0 S - n P) oscillator strength series (see below for the calculation) for which elaborate calculations [9, lOJ as well as precise determinations [8, 20] are available. The procedure chosen appears to be the most efficient when using an entirely analytical C.A. method.
For a given allcali atom the basic parameter of the C.A. method is the effective quantum number n•, which is determined from the energy levels E •. , [19]. For the lower states accurate data are available [21-23] allowing the obtainment by a numerical fit, of an extended Ritz formula :
<5, = n - n* = ao + a1(n•r 2 + + az(n*r 4 + a3(n•)-6 (2. 7)
where ô1 is the quantum defect depending only on /. We solved (2. 7) for n• and verified that the corresponding E..,1 values were in good · agreement with those quoted in the tables of Moore (24] or, when available, with recent experimental results [25, 26]. This method provides a consistent set of energy values and eliminates some suspicious values appearing in the available tables.
3. Results and accuracy. - The calculations were perfonned on a CDC 7600 computer. Double precision was used, corresponding to about 27 decimal. digits. The r functions were computed using the results of refs. [27, 28]. Such a high precision is required for high n values. The use of single precision limited the calculations to n < 17. Ail the matrix elements werc obtained in a time less than 100 s.
It is obvious that the accuracy of the results depends upon the E,.J values used. Obtaining these from an extended Ritz formula eliminates local accidents. The accuracy of the C.A. method has been widely discussed (7, 29]. The main cause of uncertainty is due to the choice of the eut criterion, for which no precise theoretical justification can be given. T o try to avoid such a problem, numerical C.A. methods have been proposed (7, 29]. But they require computing times much greater than the present method. the accuracy of which is observed to be comparable even for the case of the very sensitive S - P series.
4. Application to the determination of radiative lifetimes. - The oscillator strengths and Einstein coefficients are given by the following equations [30] (one considers two levels a and b, with Eb > E.) :
r _ 303. 75 1 S l.z max (/ •• t.,,) ( ) Ja-b - --------- 4. 1
g3

- 23:3 -N° 5 RADIAL MATRIX ELEMENTS AND LIFETIMES OF RYDBERG LEVELS ~59
and 6.670 2 x 10 15 g. h-b Âi,-a = •2
A. gb (4.2)
where À. (in A) is the wavelength of the (b -+ a) transition. S (in a.u.) the dipole integral given by (2. 2) :i!ld g the statistical weight of the level (i.e. 2 / + 1). The lif etime of a given level is calculated by :
l (4.3) 't'i, =
IAi,-, f
Table I. - Lifetimes r (in 10-6 s)for rubidium excited states.
n s p D F
- - - - -10 0.427 · 1.19 1.07 0.686 11 0.628 1.71 1.41 0.904 12 0.887 2.40 1.83 1.17 13 1.21 3.23 2.33 1.47 14 1.60 4.23 2.91 l.83 15 2.07 5.41 3.61 2.24 16 2.62 6.80 4.40 2.71 17 3.26 8.41 5.30 3.25 18 4.00 10.2 6.32 3.85 19 4.85 12.3 7.47 4.52 20 5.81 14.7 8.75 5.26 21 6.88 18.0 10.2 6.08 22 8.08 21.1 11. 7 6.98 23 9.41 24.5 13.5 7.97 · 24 10.9 28.3 15.4 9.04 25 12.5 32.5 17.4 10.2 26 14.2 37.0 19.7 11.5 27 16.2 41.9 22.1 12.8 28 18.3 47.2 24.8 14.3
where the surnmation holds for all levels f (Er < Er,) radiatively connected to the level b. Table I gives the computed Iifetimes in the case of rubidium and table Il reports the r0 and ,:i: parameters obtained from a numerical fit of the r values (10 ~ n ~ 28) to the formula (which is currently used to fit the experimental data [3 lJ)
't' = •on*". (4.4)
AU the ci: values are close to 3 which is the value expected in the limit of n large from the consideration of the oscillator strength sum rules [8] ( 1
). Comparison between some recent experimental results and our data are reported in table III for sodium and rubidium.
Table Il. - a; and •o (in 10-9 s) parameters of the equation 't' = •o n- (see text) for the a/kali atoms.
s p D F - - -
Na IX 3.00 3.11 2.99 2.96
•o 1.38 8.35 0.96 1.13
K IX 3.00 2.78 2.82 2.95
•o 1.32 6.78 5.94 0.83
Rb a; 2.94 3.02 2.85 2.95
•o 1.43 2.76 2.09 0.76
Cs a; 2.96 2.94 2.93 2.94
•o 1.43 4.42 0.96 0.69
( 1) The consideration of the partial sum rule (eq. (3) of ref. [8D for the (n0 S-nP) series easily explains (the first tenn being close to unity, and the sum being unity) the extreme sensitivity of thesc series to canœllation effects in any method of calculation.
Table III. - Comparison between experimenta/ and theoretica/ lifetimes for various highly excited a/kali levels. Ail !ifetimes are given in 10-6 s.
Exp. Th. Th. Alkali Level This work [32, 14, 13, 15] [l 1. 12] [7]
- - - -Na IOS 0.90 1.02 ± 0.05 1.06 0.91 Na l 1S 1.25 1.28 ± 0.13 1.43 1.26 Na 13S 2.18 2.27 ± 0.17 2.52 Na 100 0.92 0.97 ± 0.035 0.95 0.92 Na 120 1.59 l.65 ± 0.15 1.58. 1.57 Na l3D 2.02 _2.12 ± 0.40 2.09 Na 13F 2.26 2.27 ± 0.40 Na 14F 2.81 2.64 ± 0.45 Na 15F 3.45 3.54 ± 0.50
Rb I2P 2.40 1.55 ± 0.20 2.22 Rb 14P 4.23 2.60 ± 0.40 3.95 Rb 17P 8.41 6.40 ± 1.30 7.93 Rb 22P 21.l 14.0 ± 5.0 Rb l IF 0.90 0.90 ± 0.14 Rb 13F l.47 1.62 ± 0.24 Rb 15F 2.24 l.96 ± 0.29 Rb l7F 3.25 2.96 ± 0.44 Rb l9F 4.52 4.00 ± 0.80 Rb 2IF 6.08 6.70 ± l. iO

460 - 2J4 -
JOURNAL DE PHYSIQUE N"'
Ali the computed values agree well with the experimental ones, except for the P levels of rubidium, for which better agreement is obtained by the parametric potential method. But more results are needed to draw any firm conclusion. The results concerning the F levels agreé quite well with the measured values. This is due to the fact that these levels are almost hydrogenic, even for the heaviest alkalis.
5. Conclusion. - Radial matrix elements have been computed for highly excited alkali atoms over a wide range of principal n and orbital / quantum number values. Such data should be of interest for any one working in the field of Rydberg states. The method uses an entirely analytical C.A. method. Particular
attention has been paid to the choice of the eut criterion. We have also derived the radiative !if etimes of highly excited alkali atoms. The corriputed values are observed to be, in general, in agreement with the experimental results. For higher n values (n > 30) the use of the C.A. method is possible but requires computation procedures different from the brute force one used in the present article [33-35]. We should finally Iike to emphasize that the usefulness of the present work lies mainly in the completeness and the self consistency of the results.
Acknowledgments. - The author would like to thank .Dr. J. Berlande for helpful discussions.
References
[1] GALLAGHEll. T. F., Eoa.smN, S. A. and HILL, R. M., Phys. ReY. Lett. 3S (1975) 644.
[2) GALLAGHEll, T. F., EoEt.STEIN, S. A. and HILL, R. M., Phys. ReY. A 15 (1977) 1945.
[3] FABRE. C., HAROCHE. S. and Gov, P., Phys. Rei•. A 18 (1978) 229.
[4] OMONT, A., J. Physique 38 (1977) 1343. [5] GoUNAND, F., HUGON, M., FOURNIER. P. R. and BERLANDE. J .•
J. Phys. B. (to be publishcd). [6] WARNER,B.,Mon. Not. R. Astron. Soc.139(1968) ll5. [7) 1.INDGARD, A. and NŒLSEN, S. E., Atom. Data Nucl. Data
Tables 19 (1977) 534. [8] MARR. G. V. and CREEK, D.M., Proc. R. Soc. A 304 (1968) 245. [9] WEISHEIT, J. c., Phys. ReY. AS (1972) 1621.
[10] NoRCllOSS, D., Phys. ReY. A 7 (1973) 606. [11] ANDERSON, E. M. and ZluTIS, V. A., Opt. Spektrosk. 16 (1964)
177; Opt. Spectrosc. 16 (1964) 99. [12) ANDERSON, E. M. and Zn.ms, V. A., Opr. Spekzrosk. 16
(1964) 382; Opt. Spectrosc. 16 (1964) 211. [13] GoUNAND, F., FoUllNIER, P. R., CUVELLIEll. J. and BElll.ANDE.
J., Phys. Lett. 59A (1976) 23. [141 GALLAGHEll, T. F., CootŒ, W. E. and Eoa.smN, S. A., Phys.
ReY. A 17 (1978) 904. [15] HUGON, M .. GouNAND, F. and FOUllNlEll, P. R •• J. Phys. B 11
(1978) L-605. [16] BATES, D. R. and DAMGAAilD, A., Phil. Trans. R. Soc. A 242
(1949) 101.
[17] GoUNAND, F., unpublished C.E.A. report. (18] BEBB,H. B.,Phys. ReY.149(1966)25. [19) BURGESS, A. and SEATON, M. J., Mon Not. R. Astron. Soc. 120
(1960) 121. [20] PtCHLER, G., J. Quant. Spectrosc. Radiat. Transfer 16 (1976)
147. [21] R!sBERG, P., Ark. Fys. 10 (1956) 583. [22] JOHANSSON, l., Ark. Fys. 20 (1961) 135. [23] Kl.aMAN, H.,J. Opt. Soc. Am.Sl (1962) 441. [24] MOORE, C.E., Atomic Energy Levels (NBS Washington) 1971. [25] FERGUSON, A. I. and DUNN, M. H., Opt. Commun. l3 (1977)
227. [26] MrRZA, M. Y. and OUUY, W. W., J. Phys. B. 11 (1978) 1917. [27] WRENCH, J. W., Math. Comp. 22 (1968) 620. [28] SPIRA, R., Math. Comp. 25(1971) 317. [29] FRIEDRICH, H., KATTERBACH, K. and TREl'FTZ. E., J. Quant.
Spectrosc. Radiat. Transfer 10 (1970) 11. [30) CoRNEY, A., Ath. Electron. Electron P:tys. 29(1970) 119. [311 DEECH, J. S., LUYPA.ERT, R., PENDR1LL, L. R. andSl!RJES, G. W.,
J. Ph_vs. B. 10 (1977) L-137. [32] GALLAGHEll. T. F., EoEl.STEIN, S. A. and Hiu., R. M., Phys.
Rev. A 11 (1975) 1504. [33] EoMONDS, A. R. and K.El.LY, B. A. 1978 (to be publishcd). [34] PICAllT, J., EoMONDS, A. R. and TRAN MINH, N., J. Phys. B.
11 (1978) L-ti51. [35] VAN REGEMORTER, H., HOANG Bura, D. and Plluo'HOMME, M.,
(1978) to be published.




















