
WASP regulates suppressor activity of human and murine CD4+CD25+FOXP3+ natural regulatory T cells
Upload
sacklerinstituteCategory
view
1download
0
Foxp3 exploits a preexistent enhancer landscape for regulatoryT cell lineage specification
Robert M. Samstein1,2,*, Aaron Arvey1,*, Steven Z. Josefowicz1,2,*, Xiao Peng1,2, AlexReynolds3, Richard Sandstrom3, Shane Neph3, Peter Sabo3, Jeong M. Kim4, Will Liao5,Ming O. Li2, Christina Leslie6, John A. Stamatoyannopoulos3, and Alexander Y.Rudensky1,2,**
1Howard Hughes Medical Institute, Memorial Sloan-Kettering Cancer Center, New York, NY10065, USA2Immunology Program, Memorial Sloan-Kettering Cancer Center, New York, NY 10065, USA3Department of Genome Sciences, University of Washington, 1705 NE Pacific Street, Seattle,WA 98195, USA4Genentech, San Francisco, CA 940805Department of Applied Mathematics & Statistics, Stony Brook University, Stony Brook, NY11794, USA6Computational Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY10065, USA
SummaryRegulatory T (Treg) cells, whose identity and function are defined by the transcription factorFoxp3, are indispensable for immune homeostasis. It is unclear whether Foxp3 exerts its Treglineage specification function through active modification of the chromatin landscape andestablishment of new enhancers or by exploiting a pre-existing enhancer landscape. Analysis ofthe chromatin accessibility of Foxp3-bound enhancers in Treg and Foxp3-negative T cells showedthat Foxp3 was bound overwhelmingly to pre-accessible enhancers occupied by its cofactors inprecursor cells or a structurally related predecessor. Furthermore, the bulk of Foxp3- bound Tregcell enhancers lacking in Foxp3− CD4+ cells became accessible upon T cell receptor activationprior to Foxp3 expression with only a small subset associated with several functionally importantgenes being exclusively Treg cell-specific. Thus, in a late cellular differentiation process Foxp3defines Treg cell functionality in an “opportunistic” manner by largely exploiting the preformedenhancer network instead of establishing a new enhancer landscape.
IntroductionLineage-specifying transcription factors (TFs) are defined by their sufficiency and necessityto establish cell identity, coordinate cellular differentiation, and maintain developmentally
© 2012 Elsevier Inc. All rights reserved.**To whom correspondence should be addressed.*These authors contributed equally
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to ourcustomers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review ofthe resulting proof before it is published in its final citable form. Please note that during the production process errors may bediscovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
NIH Public AccessAuthor ManuscriptCell. Author manuscript; available in PMC 2013 March 28.
Published in final edited form as:Cell. 2012 September 28; 151(1): 153–166. doi:10.1016/j.cell.2012.06.053.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

established transcriptional programs. Differential use of regulatory elements defines mostpreviously studied lineage specific gene expression programs (Odom et al., 2004;Heintzman et al., 2009; Heinz et al., 2010; Natoli, 2010; Thurman et al., 2012). Thus, itseems reasonable to suggest that lineage-specifying TFs establish distinct differentiated cellstates by setting up novel enhancer repertoires (Mercer et al., 2011). On the other hand,some activation induced transcription factors such as the glucocorticoid receptor largelyutilize pre-established enhancers to impart changes in gene expression (John et al., 2011).These considerations raise the question of whether a late-acting differentiation factor likeFoxp3 exerts cell lineage specification function by actively remodeling the chromatinlandscape and establishing a distinct new set of enhancers or by exploiting an enhancerlandscape prepared in precursor cells by their earlier developmental history.
Foxp3, an X-chromosome encoded member of the forkhead TF family, controlsdifferentiation and function of regulatory T (Treg) cells (Littman and Rudensky, 2010). Thisdistinct and stable lineage of suppressive CD4+ T cells is characterized by a unique geneexpression program and serves as a critical guardian of immune homeostasis (Josefowiczand Rudensky, 2009; Rubtsov et al., 2010). Treg cell depletion in normal adult mice resultsin a fatal lympho- and myeloproliferative disorder with widespread inflammatory lesions(Kim et al., 2007). Foxp3 is both necessary and sufficient to confer suppressor capacity tonaïve CD4+ T cells (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003; Gavin et al.,2007). Foxp3 is induced during thymic differentiation or upon activation of peripheral CD4+
T cells in response to T cell receptor (TCR) stimulation in combination with several othersignals including IL-2 and TGF-β. Furthermore, forced expression of Foxp3 conferssuppressor function to Treg precursor cells and Foxp3 ablation in mature Treg cells resultsin loss of lineage identity and immunosuppressive phenotype (Fontenot et al., 2003;Williams and Rudensky, 2007). However, an understanding of how Foxp3 coordinates thedifferentiation of Treg cells and their distinct suppression program is lacking.
We examined chromatin accessibility of Foxp3 bound enhancers in Treg cells and Foxp3−
CD4+ T cells, which serve as precursors during extra-thymic Treg cell generation. Genome-wide analysis of Foxp3 binding sites using chromatin immunoprecipitation followed byhigh-throughput sequencing (ChIP-seq) was combined with genome-wide analysis ofenhancers using DNase I hypersensitive site sequencing (DNase-seq). We found that Foxp3was bound overwhelmingly to enhancers already accessible in precursor CD4+Foxp3− Tcells prior to Foxp3 expression with only 2% of all Foxp3 bound enhancers observed inFoxp3+ Treg cells, but not in resting Foxp3-negative T cells. However, even theseseemingly Treg-specific sites were mostly established in a Foxp3-independent manner inresponse to TCR signaling except for a small subset of exclusively Treg-restricted enhancersfound in several genes important for Treg cell function. Analysis of DNA sequences atFoxp3 binding sites identified a forkhead motif only in a small subset of these DNA regionssuggesting cofactor contribution. High-resolution digital footprinting analysis revealedsimilar footprints in Foxp3 expressing Treg cells and Foxp3- negative CD4+ T cells forseveral Foxp3 cofactors supporting the notion that Foxp3 functions through pre-existingenhancers. Moreover, a related transcription factor Foxo1 appeared to serve as a predecessorat many Foxp3-binding loci in precursor cells and its displacement in Treg cells by Foxp3resulted in downregulation of proximal genes.
Thus, Foxp3 does not substantially change the accessible chromatin landscape but ratherbinds at previously established enhancers with cofactors already present and establishes theTreg cell transcriptional and functional programs likely by modification of transcriptionalactivity of these enhancers and by recruiting additional nuclear factors. These results suggestthat late-acting lineage specification transcription factors like Foxp3 can establishfunctionality and define identity of their corresponding cell type by exploiting a subset of
Samstein et al. Page 2
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

enhancers pre-established in precursor cells and maintained in a poised state by a distinct setof cofactors and predecessors.
ResultsA similar enhancer landscape in Foxp3-positive Treg and Foxp3-negative CD4+ T cells
To test whether Treg cells exhibit a unique set of enhancers supporting their distinctfunction and phenotype we employed DNase-seq, which affords the most reliableassessment of genome-wide chromatin accessibility and regulatory element activity(Thurman et al., 2012). Thus, we isolated nuclei from FACS sorted purified Foxp3+Treg andFoxp3−CD4+ T cells and subjected them to DNase I digestion followed by high-throughputsequencing to find DNase hypersensitive sites (DHSs; Figures 1A, S1). We identified over100,000 DNase I hypersensitive sites in Treg and CD4+Foxp3− T cells each correspondingto the most accessible regions in the genome (Figure 1B). Unexpectedly, the location andthe extent of accessibility of over 99% of DHSs was similar between naïve CD4+ T cells andTreg cells implying that very few alterations in chromatin accessibility occur duringdifferentiation of Treg cells from their precursors (Figures 1B, S1). DHSs are found atexons, promoters, CTCF-bound elements, and enhancers. Indeed, comparison of ChIP-seqdatasets for a variety of histone marks vs. DNase-seq datasets showed extremely strongcorrelation between DHSs and histone marks that are traditionally associated with enhancersincluding H3K4me1 (ENCODE Consortium, 2012).
While most DHSs were comparable in precursor CD4+ T cells and Treg cells, a small, butlikely important subset of DHSs (<1% of all DHSs; 679 total) were found in Treg cells, butnot in naïve Foxp3−CD4+ T cells. These new sites were located near or within many Tregcell characteristic genes including Foxp3, Ctla4, and Helios (Figure 1B). In addition to thosesites newly accessible in Treg cells, 250 DHSs prominent in naïve Foxp3−CD4+ T cellswere markedly diminished in Treg cells. The observed differences in chromatin accessibilitywere associated with directionally consistent changes in the nearest gene’s expression(Figure 1D). In contrast to the nearly identical DHS landscape observed in Treg cells andtheir precursors, the genome-wide DHS landscape of B cells, a sister lineage of T cells, wasmarkedly different from that of either T cell subsets (Figure 1C). Thus, these results suggestthat Treg cells utilize predominantly T-cell specific enhancers present in precursor Foxp3-negative cells and very few novel enhancers emerge in Treg cells.
Foxp3 binds predominantly to pre-accessible chromatin sitesThe observation that very few regulatory elements are accessible only in Treg cells raisedthe question as to whether Foxp3 binds preferentially to sites of accessible chromatin thatare Treg-specific or to sites that are pre-accessible in Foxp3-negative precursor cells. Toaddress this question we performed ChIP-seq analysis of the genome-wide binding sites ofFoxp3 in purified Treg cells. Foxp3 was bound at over 2800 sites across the genomeassociated with more than 1400 genes (Figures 2A and S2). Foxp3 binding sites were highlyenriched in the promoters and first introns of genes (p<10−77 and p<10−300, respectively;binomial test), as were DHSs (p<10−300 and p<10−300, respectively; binomial test),consistent with direct Foxp3 mediated transcriptional regulation of gene expression viabinding to promoters and enhancers (Figure 2B, S3). Foxp3 bound loci were also highlyenriched for genes differentially expressed in Treg cells relative to precursor CD4+ T cells(Figure 2C). We confirmed association of Foxp3 with up and down regulation of geneexpression by stratifying the data across both Foxp3 peak ranks and the magnitude of geneexpression changes (Figure S2). Foxp3 binding correlated with both up- and down-regulatedtranscripts in Treg cells known to be involved in T cell activation and regulation by GOontology.
Samstein et al. Page 3
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

We next identified regulatory elements that are bound by Foxp3 and are similarly ordifferentially accessible between Foxp3−CD4+ T cells and Treg cells. To do this, we cross-referenced DNase-seq read counts with Foxp3 binding sites (Figure 2D). Consistent with theobserved broad distribution of Foxp3 binding sites within the genome and overall similarchromatin architecture of Treg cells and their precursors, 98% of Foxp3 binding sites wereobserved to be already accessible, i.e. DHS positive in CD4+ T cells (Figure 2E). Theremaining Foxp3 binding sites corresponded to Treg-specific enhancers including thepreviously identified CNS2 enhancer within the Foxp3 locus (Figure 2D) responsible for theauto-regulatory feed-forward loop for stabilizing Foxp3 expression (Zheng et al., 2010). Amuch larger proportion of Foxp3 binding was observed at T-cell specific DHS sites incontrast to Treg specific sites (29% T-cell specific, 2% Treg specific; Figures 2E,F). Uponcross-referencing our DHS-seq dataset and the recent analysis of histone H3 modifications(Wei et al., 2009) we observed significant overlap between the increased presence ofH3K4me3 marks and newly accessible DHSs and between H3K27me3 marks and decreasedchromatin accessibility (Figure S2L).
These results indicated that Foxp3 does not dramatically alter the chromatin accessibilitylandscape. Instead, it binds primarily to already accessible enhancers and promoters in orderto coordinate the lineage-specific gene expression program of Treg cells consistent with theidea that Foxp3 exploits chromatin features established during differentiation of Tregprecursor cells.
Foxp3 binds through a network of cofactorsGiven that Foxp3 predominantly binds to chromatin that is accessible in Foxp3−CD4+ Tcells, we next wanted to know if proteins occupying these sites in precursor cells mightserve as cofactors to facilitate Foxp3 recruitment to, or function at these sites. Foxp3 isknown to physically interact with several transcription factors. To explore if any of thesefactors could associate with Foxp3 binding sites we examined Foxp3 binding regions for thepresence of known and novel motifs. Significant enrichment for canonical ETS, RUNX,CNOT, and forkhead (FKHD) motifs (as defined by TRANSFAC and JASPAR) accountedfor 58% of Foxp3 occupied sites (p < 10−270, comparison to flanking region; Fisher’s exacttest), whereas the remaining peaks contained weaker motifs (Figure 3A, 3B, S4). As Foxp3contains a forkhead domain that can bind to the canonical FKHD motif (Koh et al., 2009), itwas surprising that ETS and RUNX motifs were substantially more enriched at Foxp3binding sites than the FKHD motif (Figure 3B). These motifs were also enriched in theFoxp3 peaks with the highest rank by read count (Figure 3C). Furthermore, ETS and RUNXmotifs were enriched at Foxp3 bound loci relative to genome-wide DHS sites (Figure S4Band S4C). It is noteworthy that our recent mass-spectrometric analysis identified ETS familymembers, Cnot3, and Runx1 and its cofactor Cbf-β as components of Foxp3 transcriptionalcomplexes (Rudra et al., submitted).
Next, we employed ChIP-seq to test whether several transcription factors, whose DNAbinding motifs were enriched at sites occupied by Foxp3, were bound to these sites. Theseexperiments confirmed binding of ETS family members Ets-1 and Elf-1 as well as Runx/Cbf-β to Foxp3 occupied loci (Fig 3D); the binding was specifically enriched at Foxp3 siteswith corresponding motifs (Figure 3E). These results suggested that Foxp3 and severaltranscription factors co-occupy a large portion of Foxp3 binding sites in Treg cells andraised the possibility that these cofactors are associated with the maintenance of preformedaccessible sites in precursor cells and facilitate Foxp3 recruitment to these regulatoryelements.
Samstein et al. Page 4
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Foxp3 binding loci are bound by cofactors in precursor cellsTo better understand the role of DNA-binding cofactors in precursor cells, we wanted todetermine if Foxp3 factors were directly associated with DNA already in naïve Foxp3−CD4+
T cells. Thus, we examined deeply sequenced DNase-seq libraries (~400 million reads each)for DHSs that contained cofactor motifs preferentially protected from DNase I cleavage,suggesting direct protein binding (Hesselberth et al., 2009). These digital genomic footprints(DGFs) of Foxp3 cofactors allowed genome-wide identification of specific protein-protectedDNA footprints and provide evidence for specific DNA-protein interactions in situ.
Genome-wide DGFs containing a FKHD motif sorted by locus accessibility were observedto be enriched for Foxp3 binding sites (Figure 4A and S5). To gain further insights intothese particular protein-DNA interactions the number of reads at individual nucleotides ofaccessible regions of the genome were aligned to the same motif and a corresponding mapof contacts was observed consistent with conservation of the nucleotides and previouslypublished crystal structures (Tahirov et al., 2001; Bandukwala et al., 2011). Footprints ofdistinct sizes and patterns were found for cofactor motifs including ETS, RUNX, and FKHD(Figure 4B).
We next wanted to know if these motifs were preferentially protected in Treg vs. Foxp3−
CD4+ cells and whether Foxp3 presence was correlated with alterations of the footprint. Todetermine whether cofactors binding to these motifs were pre-bound to regulatory DNAelements prior to Treg cell differentiation and expression of Foxp3, DGFs were analyzed inprecursor Foxp3−CD4+ cells (Figure 4B). Interestingly, similar footprints were observed forthe Foxp3 binding sites containing ETS and RUNX as well as FKHD motifs in Treg cellsand corresponding sites in Foxp3−CD4+ T cells suggesting that this group of Foxp3 bindingsites is pre-bound and protected in precursor cells. Consistent with the results of digitalfootprinting, binding of Runx/Cbf-β and Ets family members to corresponding sites inprecursor Foxp3−CD4+ cells was confirmed by ChIP-seq experiments, suggesting thatFoxp3-independent cofactor binding was significantly enriched at sites of Foxp3 peaks whenthe corresponding cofactor motifs were present (Figure 4C). Furthermore, Elf1 and Foxp3co-bound loci showed highly similar quantitative binding patterns of Elf1 in Treg andFoxp3−CD4+ cells (Figure 4D). Thus, at a large portion of its overall binding sites within theTreg cell genome Foxp3 binds indirectly likely via interactions with its cofactors pre-boundin precursor cells or directly via substantially weaker FKHD motif(s).
Foxo1 acts as a “predecessor” and is displaced by Foxp3As Foxp3 binding sites are predominantly accessible in precursor cells and infrequentalterations of DNase footprints in Treg cells were observed, we hypothesized that Foxp3cofactors might maintain the enhancer chromatin state to allow for Foxp3 binding duringTreg cell lineage specification. Similar DGFs observed at Foxp3 binding sites containingFKHD motif in both Treg and Foxp3−CD4+ T cells pointed to the possibility that anotherforkhead transcription family member could serve as a “predecessor” at Foxp3 binding sitesin Foxp3− CD4+ cells. Foxp3 in Treg cells could then displace this putative factor in acompetitive manner leading to changes in gene expression. We considered the possibilitythat Foxo1, a forkhead TF family member, might serve a role of a Foxp3 predecessor boundto these sites in precursor cells because Foxo1 has been implicated in regulation of geneexpression in both effector T cells and in Treg cells (Ouyang et al., 2009; 2010). Consistentwith this idea ChIP-seq analysis of Foxo1 binding showed that in Foxp3−CD4+ T cellsFoxo1 was bound to sites that were occupied by Foxp3 in Treg cells (Figure 5A).Furthermore, in Treg cells Foxo1 binding was preferentially decreased at Foxp3 bound, butnot at unbound sites in comparison to Foxp3−CD4+ cells (Figure 5B). We also excluded thescenario that the observed decrease of Foxo1 at Foxp3 bound sites in Treg cells is due to an
Samstein et al. Page 5
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

overall decrease in chromatin accessibility since Foxp3-bound Foxo1 sites were on averageincreased in accessibility (Figure S6).
Given that Foxo1 acts as a transcriptional regulator, its displacement could be an importantcomponent of the Foxp3-mediated program of gene repression. A corollary to thishypothesis is that corresponding genes in precursor cells should be either repressed or de-repressed and exhibit diminished or increased expression, respectively, in comparison toTreg cells, where Foxp3 displaces its putative FKHD-containing predecessor, Foxo1. Thus,we examined Foxp3 dependent gene expression changes by transcriptional profiling of Tregcells and their precursors expressing a functional (Foxp3GFP) and null Foxp3 reporter allele(Foxp3GFPKO), respectively. We found that Foxp3 bound gene loci with a decreased Foxo1occupancy were significantly down-regulated in Treg cells in contrast to Foxp3 unboundgenes or Foxp3 bound sites that have increased Foxo1 (Figure 5C).
The observed gene repression associated with displacement of Foxo1 by Foxp3 highlightsits functional importance and implicates Foxo1 as a functionally relevant predecessor atFoxp3 direct binding sites in precursor cells. On a more general level, displacement of atranscriptional regulator by a structurally related TF with a similar DNA binding domainspecificity represents a mode for implementation of gene expression programming duringcellular differentiation.
The majority of “Treg-specific” enhancers are established in a Foxp3 independent mannerupon T cell activation
We next wished to understand how apparently Treg-specific enhancers are established. Wefound that while an overwhelming majority of Foxp3 binding sites exhibited an openchromatin state in precursor CD4+ cells, the 679 loci identified by DNase-seq as newlyaccessible in Treg cells were enriched in genes known to be critical for Treg cell function.Additionally, Foxp3 binding to these new DHSs in Treg cells was significantly enriched incomparison to all DNase accessible loci genome-wide (6% and 2%, respectively).
We wished to examine if Foxp3 facilitates establishment of these new sites throughrecruitment of chromatin remodelers or if remodeling of these sites might precede Foxp3expression and occur independently of it. To address these questions we first analyzed DNAsequence motifs present at Treg-specific DHSs. We found that they were highly enriched forthe AP-1 septamer motif (Figure 6A) and the enrichment was prevalent in those that weredirectly bound by Foxp3. AP-1 and its binding partner NFAT are activated in T cells uponTCR signaling (Macian, 2005). Furthermore, thymic and extrathymic Treg celldifferentiation and Foxp3 induction requires TCR stimulation by self or non-self antigens(Josefowicz et al., 2012). Thus, the presence of the AP-1 motif and necessity of TCRsignaling for Treg cell differentiation suggests that TCR signaling may be playing animportant role in the establishment of these enhancers in the Treg cell lineage even prior toFoxp3 expression. In contrast to AP-1 motif enrichment at Foxp3-bound sites newlyaccessible in Treg cells, sites with Treg cell-specific diminished accessibility were enrichedfor the RUNX motif along with a HMG motif associated with Lef1 and Tcf7 (Figure 6A).
In order to explore the role of TCR signaling in establishing Treg-specific accessiblechromatin loci, we performed DNase-seq analysis of chromatin state in activatedFoxp3−CD4+ T cells. For these experiments, activated Foxp3−CD4+ T cells were FACSpurified from Foxp3DTR mice after human diphtheria toxin receptor (DTR)-expressing Tregcells were ablated upon DT injection. Massive T cell activation observed upon Treg cellablation is dependent upon stimulation of TCR by self and environmental antigens (Kim etal., 2007). The vast majority of seemingly Treg-specific enhancers (>75%) acquired anincrease in chromatin accessibility in activated cells suggesting that TCR dependent T cell
Samstein et al. Page 6
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

activation without Foxp3 expression is sufficient to confer accessibility to these sites (Figure6B,C). Accordingly, genes associated with Treg-specific DHSs were largely up-regulated inTreg “wannabe” cells expressing Foxp3GFPKO allele relative to Foxp3−CD4+ T cells (Figure6D)(Gavin et al., 2007). These cells most likely received TCR signals and represent directTreg cell precursors, reflected by Foxp3 locus activation, but lack of Foxp3 proteinexpression and suppressor function.
The Foxp3-independent expression of genes containing “Treg-specific” DHSs enriched forthe AP-1 motif and chromatin accessibility at these sites in activated, but not resting Foxp3−
CD4+ T cells suggested that activation of AP-1 most likely in cooperation with NFAT mayaccount for these features. We next examined whether the impairment in NFAT activationupon ablation of calcineurin B1 in Foxp3CreCnb1fl/fl Treg cells disproportionally affectedthe genes containing “Treg-specific” DHSs. In the absence of calcineurin signaling, aprerequisite for NFAT activation and translocation to the nucleus, these genes hadsignificantly decreased expression, particularly those that both contained an AP-1 motif andwere Foxp3 bound (Figure 6E). Importantly, loss of NFAT activation in Treg cells led to aloss of their function and severe autoimmunity (data not shown).
The remaining subset of Foxp3 bound enhancers present in Treg cells, but not in precursorFoxp3−CD4+ or activated CD4+ T cells were found in a subset of genes encoding Ccr6,Lrrc32 (GARP), Foxp3, Itgb8, HOPX, Alk1, and PHLPP proteins whose functions in Tregcells include regulation of Foxp3 and TGF-β expression and activation, cell trafficking, andAkt signaling (Figure S7). Treg-specific DHS loci that are not more accessible in activatedFoxp3− CD4+ T cells had markedly less AP-1 motif enrichment. In accordance with thepresence of Treg-cell specific Foxp3-bound enhancers in these loci, these genes areexpressed in a Foxp3- dependent manner in Treg cells.
In addition to the aforementioned genes with a known role in Treg cells, genes containingTreg-specific DHS sites included genes whose role in Treg cells is currently unknown.Together these genes may play an important role in Treg cell function under homeostaticand inflammatory conditions. Consistent with the latter idea, we found overlaps betweenTreg-specific DHS sites and several single nucleotide polymorphisms (SNPs) identified ingenome-wide association studies as associated with a variety of autoimmune andinflammatory diseases (Figure S7). These include Il10 and Lrrc32 in ulcerative colitis, Ccr6in rheumatoid arthritis and Rhoh in Graves’ disease (Stahl et al., 2010; Anderson et al.,2011; Chu et al., 2011). These results highlight the power of the datasets generated bycombined DHS-seq and Foxp3 ChIP-seq analyses and might offer potential insights into therole of Treg cells in these diseases.
Taken together, our experiments suggest that over 98% of sites bound by Foxp3 in Tregcells are accessible in their precursors and are occupied by Foxp3 cofactors or Foxo1serving as a Foxp3 “placeholder” and that TCR signaling is responsible for Foxp3-independent establishment of the remaining minor “Treg-specific” subset of Foxp3-boundenhancers. Thus, Foxp3 controls Treg cell differentiation and function by modulating geneexpression upon binding to these preexistent enhancers without profound alterations inchromatin accessibility and enhancer repertoire.
DiscussionCell type specific gene expression and functional features of differentiated cells areestablished by genetically defined programs of specification, which employ epigeneticcontrol, transcriptional and post-transcriptional regulation. Recent genome-wide studies ofhistone modifications indicate that cell identity is largely defined by permissive or
Samstein et al. Page 7
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

repressive chromatin features present at enhancers (Heintzman et al., 2009; Mercer et al.,2011). We examined the late differentiation process of Treg lineage specification resultingfrom Foxp3 expression. Treg cell differentiation is known to occur at a late stage ofthymocyte maturation and beyond the thymus as mature naïve Foxp3-negative CD4+ T cellsacquire Foxp3 expression upon TCR stimulation under particular conditions that give rise toextrathymic Treg cells (Josefowicz et al., 2012). Our analysis of genome-wide DNase-seqand Foxp3 ChIP-seq datasets demonstrates that >99% of Foxp3 bound enhancers areaccessible in precursor Foxp3-negative CD4+ resting or activated T cells. While there maybe subtle undetected changes in chromatin accessibility imparted by Foxp3, DNase-seqtechnology offers the most accurate means of genome-wide enhancer analysis currentlyavailable (Boyle et al., 2008).
Interestingly, the lack of alterations in chromatin accessibility imparted by Foxp3 contrastswith early cellular differentiation processes, which are thought to rely on epigeneticmodifications of chromatin at regulatory gene loci that can persist even after removal of theinitiating factor (Cavalli and Paro, 1999). The establishment of heritable cell lineage-specificenhancer repertoires is critical for early tissue development (Cirillo et al., 2002; Heinz et al.,2010; Natoli, 2010; Mercer et al., 2011). Unlike early differentiation defined by lineage-specification factors, gene expression programs induced by some extracellular cues aredriven by ligand- or signaling-dependent mobilization of latent transcription factors, whichbind to preconditioned enhancers to alter gene expression without substantially altering thechromatin landscape (John et al., 2011). It must be noted that certain extracellular stimulisuch as LPS that result in drastic changes in gene expression may be associated withconsiderable changes in chromatin states (Ghisletti et al., 2010; Smale, 2010).
Thus, features of late cellular differentiation exemplified by Foxp3-dependent generation ofTreg cells are similar to those of responses triggered by extracellular stimuli likeglucocorticoid steroid receptor ligand response in that both are associated with minimalchromatin remodeling (Biddie et al., 2011). Importantly, these results can explain thepreviously demonstrated need for continuous expression of Foxp3 for the maintenance ofsuppressive function and phenotypic features in fully differentiated Treg cells (Williams andRudensky, 2007). This observation may also extend to the reported requirement forcontinuous expression for Pax5 in mature B cells, which may also be acting throughmodulation of preexisting enhancers instead of de novo heritable alterations in chromatinstate (Cobaleda et al., 2007).
We also observed that while the vast majority of enhancers were accessible in precursorCD4+ cells, 2% of Foxp3 binding sites appeared to be accessible in a Treg-specific mannerand were found at important Treg cell signature gene loci. However, a large majority ofthese sites emerged in activated T cells in a Foxp3 independent manner. These sites werehighly enriched for a motif bound by AP-1, which is particularly interesting since AP-1forms protein complexes with NFAT to jointly control expression of numerous genesdownstream of TCR signaling (Macian, 2005). Thus, we propose that TCR activation drivenmobilization of AP-1 likely in cooperation with NFAT facilitates chromatin remodeling atthese “Treg-specific” enhancers in precursor cells and prepares sites for Foxp3 binding (Fig.7c). Furthermore, X-ray crystallographic analysis has shown that Foxp3 forms proteincomplexes with NFAT and is thought to displace AP-1 in the NFAT complex in its DNAbound form (Wu et al., 2006), which suggests that Foxp3 might replace AP-1 at activation-dependent enhancers in Treg cells. Consistent with this idea we found altered expression ofgenes containing these enhancers in Treg cells subjected to ablation of a conditional Cnb1allele resulting in impaired NFAT activation. In conjunction with a recent study showingthat AP-1 maintains open chromatin, our data point to a general role for AP-1 as a pioneerfactor capable of establishing an accessible chromatin state at regulatory elements in
Samstein et al. Page 8
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

response to extracellular stimuli in diverse biological contexts including both cellulardifferentiation and activation (Biddie et al., 2011). We also note that T cell activation is ableto substantially remodel chromatin at multiple functionally important gene loci in addition toTCR response elements which were previously characterized as primed to be pre-accessiblefor rapid T cell response (Barski et al., 2009).
Since Foxp3 seemed to not alter chromatin accessibility directly, we wished to betterunderstand alternative mechanisms for how it implements the Treg cell lineage geneexpression program. Analysis of sequence patterns showed that a canonical FKHD motifwas present only in a minority of Foxp3 sites. In contrast, a majority of sites containedmotifs for Foxp3 cofactors, including members of ETS and RUNX families of nuclearfactors. Thus, Foxp3 may interact with DNA in large part indirectly, through protein-proteininteractions. This result is consistent with the observed loss-of-function mutations of Foxp3in the oligomerization leucine zipper domain in IPEX (immunodysregulation,polyendocrinopathy, enteropathy, X-linked) syndrome patients, which disrupts protein-protein interactions (Wildin et al., 2002; Lopes et al., 2006; van der Vliet and Nieuwenhuis,2007). This is also consistent with Foxp3 DNA binding properties revealed by EMSA andcrystallization studies (Koh et al., 2009; Bandukwala et al., 2011) and the large size ofFoxp3 protein complexes, which contain Runx/Cbf-β and ETS family members among othercofactors (Rudra et al., submitted). Also, both Ets and Runx/Cbf-β play important roles inTreg cell differentiation and function that can now be explained in part by their cooperationwith Foxp3 at its binding sites (Rudra et al., 2009; Mouly et al., 2010). Digital footprintingsuggested that in precursor cells these cofactors are already present at sites of Foxp3 bindingprior to expression of Foxp3 and this binding in precursor cells was confirmed by ChIP.Thus, Foxp3 may be recruited to these sites with prebound cofactors leading toconformational perturbation or recruitment of other factors and changes in gene expression(Fig. 7a).
In addition to co-binding and directly interacting cofactors, our results suggested a novelmode of transcriptional regulation through structural homologs of transcription factorsacting as predecessors or placeholders. Analysis of Foxo1 and Foxp3 ChIP-seq and DNase-seq datasets indicated that Foxp3 in Treg cells displaced another family member Foxo1,which was bound to FKHD motif-containing enhancers in precursor cells (Fig. 7b).Importantly, transcripts of genes associated with these sites were predominantly down-regulated genome-wide in a Foxp3- dependent manner. Previously, a swap of structurallyunrelated transcription factors at enhancers has been shown to cause a change in geneexpression (Sun et al., 2002; Reichard et al., 2007). Thus, while Foxo1 and its relativeFoxo3 enhance Treg cell function, possibly in part through Foxp3 induction (Ouyang et al.,2009; 2010), our results suggest an additional role for Foxo family members in establishingor maintaining enhancers for Foxp3. This mode of Foxp3 function raises an excitingpossibility that members of a given family of transcription factors with distinct DNAbinding specificity prepare or preserve enhancers in precursor cells during differentiation (oractivation) to pass them on to another member of the same family, which throughdislodgement of the predecessor imparts transcriptional activation or repression. This ideawas supported by an earlier observation that the forkhead transcription factor familymember FoxD3 binds at an enhancer within the Alb1 locus in embryonic stem cells andserves as a “placeholder” for FoxA1 and FoxA2 binding, which in turn promote chromatinremodeling and binding of GATA family members during definitive endoderm andhepatocyte differentiation (Xu et al., 2009). This “evolutionary” model, where a factorexploits highly similar structural domains for alternative means of transcriptional regulation,offers an intuitive mechanism for maintenance of enhancers in an “open” chromatin stateand its engagement by distinct members of different families of transcription factors.
Samstein et al. Page 9
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Finally, our datasets of Foxp3 targets and Treg cell-specific changes in enhancer repertoireidentified a number of novel regulatory targets, which may inform further exploration ofmechanisms and functions of Treg cells in human diseases. The potential importance ofthese regulatory elements and genes was suggested by the overlap of these datasets withSNPs associated with a variety of clinical inflammatory conditions.
In summation, our results suggest several modes for “opportunistic” control of Treg celldifferentiation and function by Foxp3 through a network of preformed enhancers andcofactors operating in precursor cells. Foxp3 exploits the pre-existing enhancer landscape bybinding to its cofactor-occupied enhancers accessible in precursor cells, by displacement ofits putative predecessor forkhead family member Foxo1, and by binding to TCR stimulationdependent, but Foxp3-independent enhancers established during Treg cell differentiation.Thus, in contrast to early cellular differentiation characterized by alteration of chromatinaccessibility at key enhancers, late differentiation relies overwhelmingly on a set ofenhancers established during developmental history of precursor cells and on a minor set ofenhancers established in precursor cells in response to extracellular cues promotingdifferentiation of a given cell type. However, given that even in early development, cellsrespond to extracellular cues from adjacent accessory cells and soluble ligand gradients, itmay very well be the case that the mechanisms we describe here are also extensivelyemployed during early developmental processes.
Experimental ProceduresMice and cell sorting
Foxp3GFPKO, Foxp3GFP-DTR, Foxp3YFP-Cre mice were previously described (Gavin et al.,2007; Kim et al., 2007; Rubtsov et al., 2008). All the mice were bred and housed in thespecific pathogen-free animal facility at the Memorial Sloan-Kettering Cancer Center andused in accordance with institutional guidelines. Activated Foxp3-CD4+ T cells were sortedfrom spleens and lymph nodes of mice 10 days after two administrations of i.p. diphtheriatoxin (DT) (Sigma). CD4 T cells were isolated by CD4 Dynabeads positive selection(Invitrogen) followed by sorting using an Aria2 flow cytometer (BD Biosciences).
Chromatin immunoprecipitation of Foxp3 and its cofactorsChromatin immunoprecipitation was performed as previously described (Zheng et al., 2007).Briefly, nuclei were isolated and lysed in 0.2% SDS followed by sonication to fragmentDNA to 200–300 bp fragments (Branson). The chromatin was then incubated overnight withthe appropriate antibody (polyclonal rabbit Foxp3 antibody (Zheng et al., 2007), Ets1, Elf1,and Cbf-β antibodies (all Santa Cruz clones C-20, C-20, FL-182). Precipitated chromatinwas then washed, de-cross-linked, digested with proteinase K, and DNA was isolated usingQiagen PCR purification kit or phenol-chloroform extraction. ChIP was validated by qPCRfor known targets of corresponding transcription factors. Sequencing libraries were preparedby ChIP-seq DNA prep kit and sequenced using a Genome Analyzer IIx or HiSeq(Illumina).
Analysis of ChIP-seqDNA reads generated in DNase-seq and Foxp3 ChIP-seq experiments were aligned to theUCSC mm9 genome using Bowtie allowing for 2 mismatches. Only uniquely aligning readswere analyzed. For ChIP experiments, all reads starting at an identical position werecompressed to single reads to remove monoclonal reads. ChIP-seq peaks were called usingSPP and peak-height was determined by number of reads that aligned to a 200bp windowaround peak center after strand-specific 75nt shift. Peaks with high input-control signal wereexcluded from subsequent analysis. This included peaks with input RPM greater than a
Samstein et al. Page 10
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

threshold of 0.5RPM and peaks that were not enriched relative to input (p < 0.001) asdetermined by a Poisson distribution using a local estimate (200bp) of λ. Several total peakestimation methods were examined and many analyses were done using peak rank instead ofdiscretized peak calls (Figure S2). The final number of peaks for Foxp3 was determined bythe overlap in top 5000 peaks in both replicates, resulting in 2886 peaks. Peaks wereassigned to genes by proximity to gene body defined by transcription start and end sites.When comparing multiple ChIP-seq experiments, peak heights were quantile normalized toaccount for potential differences in experimental data quality. When plotting tracks formultiple ChIP-seq experiments for a single transcription factor, a multiplicative factor wasused to normalize for enrichment i.e. differing numbers of tags in peaks.
Chromatin accessibility and DNase footprintingTreg and resting and activated Foxp3− T cells as well as B cells were assayed for chromatinaccessibility using DNase-seq as described elsewhere (John et al., 2011). Briefly, intactnuclei were treated with DNase I, DNA was isolated following nuclear lysis, and fragmentssized 300–1000bp were sequenced. DNase-seq data shown in scatter plots is in units ofreads per million (RPM). Data were incremented by 0.3 (to smooth and avoid zeros), log-transformed, capped at 50RPM, and quantile normalized. DNase-seq peaks were calledusing the HotSpot algorithm (FDR 1%) (Sabo et al., 2004).
Differential accessibility was determined by an asymmetric cutoff for up- and down-regulation of accessibility based on an empirical 5% FDR derived from replicate-to-replicatedifferences and consistency in replicates (Figure S1A, B). These loci were then confirmedindependently when comparing an additional replicate of Treg DNase-seq to two replicatesof CD4 DNase-seq (Figure S1C). This analysis revealed differential sites that were rankedhighly by rank expectation and negative binomial dispersion approaches (data not shown)(Robinson et al., 2010; Thomas et al., 2012).
Footprints were aggregated into heatmaps by aligning read start sites to specific motifinstances (Figure S5). De novo footprints at individual loci were identified using previouslydescribed methods (Hesselberth et al., 2009; Neph et al., 2012). Conservation acrossplacental mammals was analyzed using phyloP downloaded from UCSC genome browser.
Motif analysisFor analysis of transcription factor binding motif enrichment, motifs were taken fromJASPAR and TRANSFAC, whereas de novo motifs (AP-1 and HMG) were found byMDscan, cERMIT, and MEME motif discovery tools (Liu et al., 2002). To determine motifpresence at a ChIP-seq or DNase-seq peak, motifs were scanned within 100bp regionsaround peak centers. Motif scores were defined as the log likelihood ratio of observingnucleotide frequency defined by the PSSM compared to genome-wide nucleotidefrequencies. Motifs were considered present when the maximum PSSM score was greaterthan that found in 90% of the maximum scores of the flanking regions. This can beconsidered an empirical p-value of 0.1 on the distribution of maximum PSSM score over agiven window. While this threshold may seem insufficiently stringent, when we examinedregions considered positive for the motif, we found that nearly all sites (>90%) containedcanonical 6mer binding motifs for the FKHD, RUNX, and ETS motifs. The few sites thatlacked canonical sites had additional flanking nucleotides that increase statisticalassociation.
Gene Expression ProfilingGFP+ and GFP− CD4 T cells were isolated by CD4 positive selection (Invitrogen) followedby cell sorting using an Aria2 flow cytometer (BD Biosciences). To ensure high purity cells
Samstein et al. Page 11
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

were sorted through consecutive rounds of sorting to attain >99% purity. Cells wereresuspended in Trizol and RNA was isolated according to manufacturer instructions. cDNAlibraries were amplified and hybridized to Affymetrix MOE 430 2.0 chips. Arrays werenormalized using RMA and differentially expression was estimated using the limmapackage in Bioconductor. Genes were considered differentially expressed if they had a q-value <0.05 after Benjamini- Hochberg FDR estimation. Significance of differences in geneexpression between sets of genes (e.g. those associated with regions of increased/decreasedchromatin accessibility in Figure 1) was determined by a two-sample Kolmogorov-Smirnov(KS) test, where the background distribution was change in all expressed genes unlessspecified otherwise by dashed line emphasizing the two cumulative curves being compared.
Data accessibilityAll data are being deposited in the relevant public databases. Gene expression data will beuploaded to the GEO database and sequencing experiments to the SRA and ENCODEarchives.
Supplementary MaterialRefer to Web version on PubMed Central for supplementary material.
AcknowledgmentsWe would like to thank D. Rudra and A. Chaudhry for helpful discussions. This work was supported by NIHDK091968 grant, MSTP grant GM07739 (both R.M.S) and NIH 5R37AI034206 grant (A.Y.R.). A.Y.R. is aninvestigator with the Howard Hughes Medical Institute.
ReferencesAnderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P,
Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci,increasing the number of confirmed associations to 47. Nat Genet. 2011; 43:246–252. [PubMed:21297633]
Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, Stroud JC, Benoist C, Mathis D,Rao A, et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory Tcells. Immunity. 2011; 34:479–491. [PubMed: 21458306]
Barski A, Jothi R, Cuddapah S, Cui K, Roh T-Y, Schones DE, Zhao K. Chromatin poises miRNA- andprotein-coding genes for expression. Genome Res. 2009; 19:1742–1751. [PubMed: 19713549]
Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung M-H, TrumpS, Lightman SL, et al. Transcription factor AP1 potentiates chromatin accessibility andglucocorticoid receptor binding. Mol. Cell. 2011; 43:145–155. [PubMed: 21726817]
Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. [PubMed: 18243105]
Cavalli G, Paro R. Epigenetic inheritance of active chromatin after removal of the main transactivator.Science. 1999; 286:955–958. [PubMed: 10542150]
Chu X, Pan C-M, Zhao S-X, Liang J, Gao G-Q, Zhang X-M, Yuan G-Y, Li C-G, Xue L-Q, Shen M, etal. A genome-wide association study identifies two new risk loci for Graves' disease. Nat Genet.2011; 43:897–901. [PubMed: 21841780]
Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin byearly developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell. 2002; 9:279–289.[PubMed: 11864602]
Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function.Nat Immunol. 2007; 8:463–470. [PubMed: 17440452]
Samstein et al. Page 12
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Consortium TEP. Initial Analysis of the Encyclopedia of DNA Elements in the Human Genome.Nature. 2012 In press.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function ofCD4+CD25+ regulatory T cells. Nat Immunol. 2003; 4:330–336. [PubMed: 12612578]
Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007; 445:771–775. [PubMed:17220874]
Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A,Wei C-L, et al. Identification and characterization of enhancers controlling the inflammatory geneexpression program in macrophages. Immunity. 2010; 32:317–328. [PubMed: 20206554]
Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK,Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific geneexpression. Nature. 2009; 459:108–112. [PubMed: 19295514]
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK.Simple combinations of lineage-determining transcription factors prime cis-regulatory elementsrequired for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [PubMed: 20513432]
Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, KuehnMS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomicfootprinting. Nat. Methods. 2009; 6:283–289. [PubMed: 19305407]
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factorFoxp3. Science. 2003; 299:1057–1061. [PubMed: 12522256]
John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, Hager GL, StamatoyannopoulosJA. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet.2011; 43:264–268. [PubMed: 21258342]
Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance.Immunity. 2009; 30:616–625. [PubMed: 19464984]
Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation andFunction. Annu Rev Immunol. 2012
Khattri R, Cox T, Yasayko S-A, Ramsdell F. An essential role for Scurfin in CD4+CD25+ Tregulatory cells. Nat Immunol. 2003; 4:337–342. [PubMed: 12612581]
Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunitythroughout the lifespan of mice. Nat Immunol. 2007; 8:191–197. [PubMed: 17136045]
Koh KP, Sundrud MS, Rao A. Domain requirements and sequence specificity of DNA binding for theforkhead transcription factor FOXP3. PLoS ONE. 2009; 4:e8109. [PubMed: 19956618]
Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation.Cell. 2010; 140:845–858. [PubMed: 20303875]
Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications tochromatin-immunoprecipitation microarray experiments. Nat Biotechnol. 2002; 20:835–839.[PubMed: 12101404]
Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, Ziegler SF. Analysis ofFOXP3 reveals multiple domains required for its function as a transcriptional repressor. JImmunol. 2006; 177:3133–3142. [PubMed: 16920951]
Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol.2005; 5:472–484. [PubMed: 15928679]
Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T,Glass CK, Murre C. Multilineage priming of enhancer repertoires precedes commitment to the Band myeloid cell lineages in hematopoietic progenitors. Immunity. 2011; 35:413–425. [PubMed:21903424]
Mouly E, Chemin K, Nguyen HV, Chopin M, Mesnard L, Leite-de-Moraes M, Burlen-defranoux O,Bandeira A, Bories J-C. The Ets-1 transcription factor controls the development and function ofnatural regulatory T cells. J Exp Med. 2010; 207:2113–2125. [PubMed: 20855499]
Natoli G. Maintaining cell identity through global control of genomic organization. Immunity. 2010;33:12–24. [PubMed: 20643336]
Samstein et al. Page 13
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Neph S, Vierstra J, Stergachis AB, Reynolds A, Stamatoyannopoulos J. An expansive humanregulatory lexicon encoded in transcription factor footprints. Nature. 2012 In press.
Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J,Rolfe PA, Gifford DK, et al. Control of pancreas and liver gene expression by HNF transcriptionfactors. Science. 2004; 303:1378–1381. [PubMed: 14988562]
Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factorFoxo1 in control of T cell homeostasis and tolerance. Immunity. 2009; 30:358–371. [PubMed:19285438]
Ouyang W, Beckett O, Ma Q, Paik J-H, DePinho RA, Li MO. Foxo proteins cooperatively control thedifferentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010; 11:618–627. [PubMed:20467422]
Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of thetranscriptional repressor BACH1. Nucleic Acids Research. 2007; 35:7074–7086. [PubMed:17942419]
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expressionanalysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [PubMed: 19910308]
Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability ofthe regulatory T cell lineage in vivo. Science. 2010; 329:1667–1671. [PubMed: 20929851]
Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A,Henderson WR, et al. Regulatory T cell-derived interleukin-10 limits inflammation atenvironmental interfaces. Immunity. 2008; 28:546–558. [PubMed: 18387831]
Rudra D, Egawa T, Chong MMW, Treuting P, Littman DR, Rudensky AY. Runx-CBFbeta complexescontrol expression of the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2009;10:1170–1177. [PubMed: 19767756]
Sabo PJ, Hawrylycz M, Wallace JC, Humbert R, Yu M, Shafer A, Kawamoto J, Hall R, Mack J,Dorschner MO, et al. Discovery of functional noncoding elements by digital analysis of chromatinstructure. Proc Natl Acad Sci U S A. 2004; 101:16837–16842. [PubMed: 15550541]
Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010; 140:833–844.[PubMed: 20303874]
Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FAS,Zhernakova A, Hinks A, et al. Genome-wide association study meta-analysis identifies seven newrheumatoid arthritis risk loci. Nat Genet. 2010; 42:508–514. [PubMed: 20453842]
Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S,Slam J, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene.Embo. J. 2002; 21:5216. [PubMed: 12356737]
Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, KumasakaT, Yamamoto M, et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runtdomain and its allosteric control by CBFbeta. Cell. 2001; 104:755–767. [PubMed: 11257229]
Thomas S, Neph S, Reynolds A, Stamatoyannopoulos J. Quantifying the significant differencesbetween genomic datasets and rank expectation. 2012
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB,Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012In press.
van der Vliet HJJ, Nieuwenhuis EE. IPEX as a result of mutations in FOXP3. Clin. Dev. Immunol.2007; 2007:89017. [PubMed: 18317533]
Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh T-Y, Watford WT, et al. Globalmapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fatedetermination of differentiating CD4+ T cells. Immunity. 2009; 30:155–167. [PubMed: 19144320]
Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of theimmunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J. Med.Genet. 2002; 39:537–545. [PubMed: 12161590]
Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in matureregulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007; 8:277–284.[PubMed: 17220892]
Samstein et al. Page 14
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, ZieglerSF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. [PubMed: 16873067]
Xu J, Watts JA, Pope SD, Gadue P, Kamps M, Plath K, Zaret KS, Smale ST. Transcriptionalcompetence and the active marking of tissue-specific enhancers by defined transcription factors inembryonic and induced pluripotent stem cells. Genes Dev. 2009; 23:2824–2838. [PubMed:20008934]
Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010; 463:808–812.[PubMed: 20072126]
Zheng Y, Josefowicz SZ, Kas A, Chu T-T, Gavin MA, Rudensky AY. Genome-wide analysis ofFoxp3 target genes in developing and mature regulatory T cells. Nature. 2007; 445:936–940.[PubMed: 17237761]
Samstein et al. Page 15
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Research Highlights
• Treg cell specification factor Foxp3 binds to pre-accessible enhancers
• Foxp3 binds to sites occupied by cofactors in precursor cells
• Structural homolog Foxo1 serves as a Foxp3 predecessor in precursor cells
• TCR activation confers accessibility to the majority of Treg-specific enhancers
Samstein et al. Page 16
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
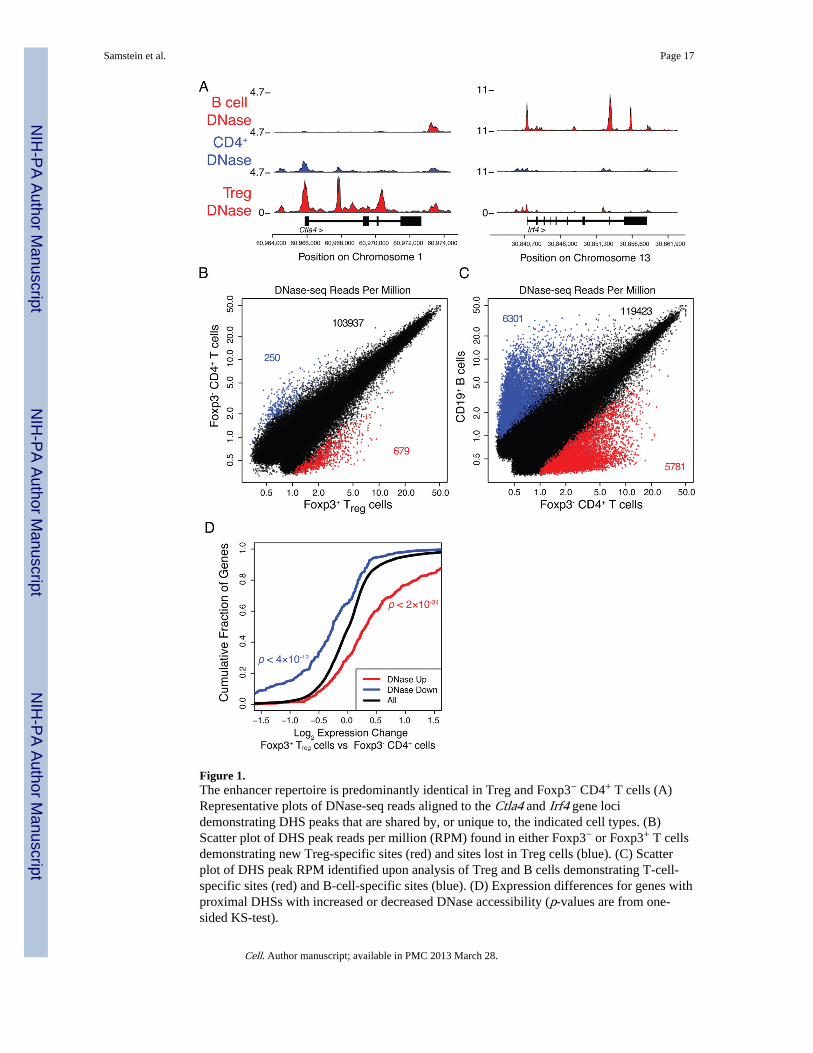
Figure 1.The enhancer repertoire is predominantly identical in Treg and Foxp3− CD4+ T cells (A)Representative plots of DNase-seq reads aligned to the Ctla4 and Irf4 gene locidemonstrating DHS peaks that are shared by, or unique to, the indicated cell types. (B)Scatter plot of DHS peak reads per million (RPM) found in either Foxp3− or Foxp3+ T cellsdemonstrating new Treg-specific sites (red) and sites lost in Treg cells (blue). (C) Scatterplot of DHS peak RPM identified upon analysis of Treg and B cells demonstrating T-cell-specific sites (red) and B-cell-specific sites (blue). (D) Expression differences for genes withproximal DHSs with increased or decreased DNase accessibility (p-values are from one-sided KS-test).
Samstein et al. Page 17
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
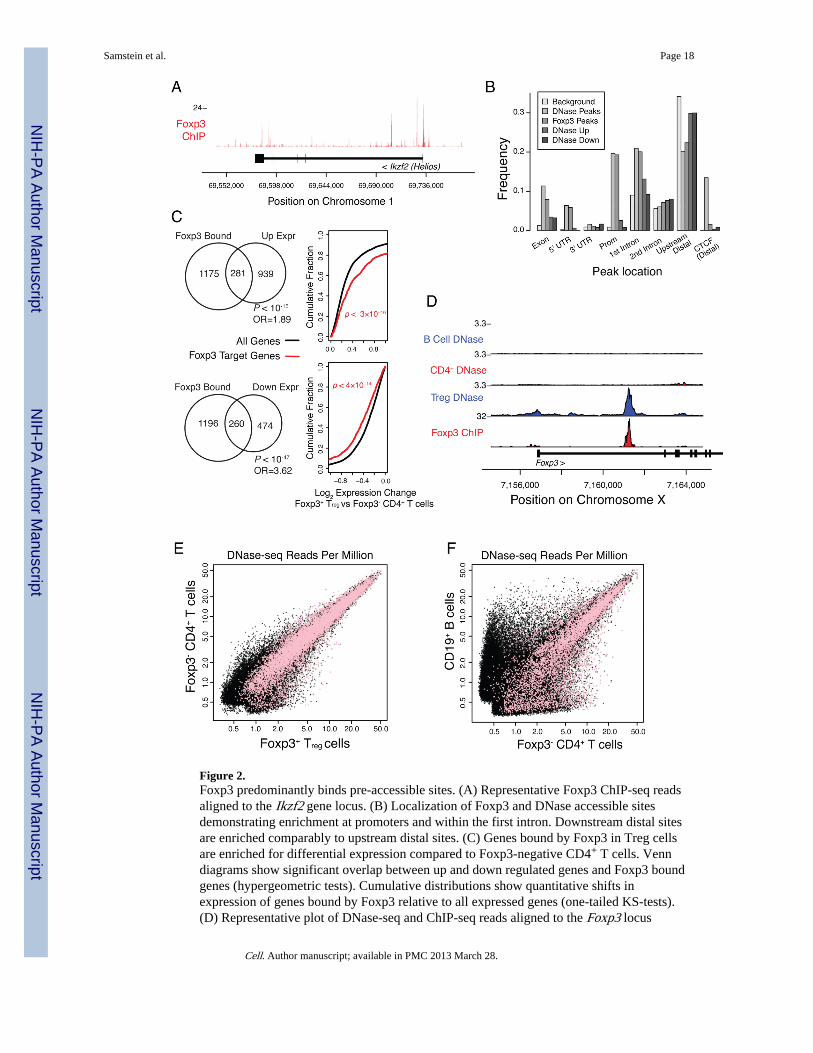
Figure 2.Foxp3 predominantly binds pre-accessible sites. (A) Representative Foxp3 ChIP-seq readsaligned to the Ikzf2 gene locus. (B) Localization of Foxp3 and DNase accessible sitesdemonstrating enrichment at promoters and within the first intron. Downstream distal sitesare enriched comparably to upstream distal sites. (C) Genes bound by Foxp3 in Treg cellsare enriched for differential expression compared to Foxp3-negative CD4+ T cells. Venndiagrams show significant overlap between up and down regulated genes and Foxp3 boundgenes (hypergeometric tests). Cumulative distributions show quantitative shifts inexpression of genes bound by Foxp3 relative to all expressed genes (one-tailed KS-tests).(D) Representative plot of DNase-seq and ChIP-seq reads aligned to the Foxp3 locus
Samstein et al. Page 18
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

illustrating Treg-specific DHS corresponding to a Foxp3 binding site. (E, F) Foxp3 bindingsite accessibility is largely similar in Foxp3+ Treg and Foxp3− CD4+ cells. Scatter plot of allDHS peaks (black) and those containing Foxp3 bound sites (pink) in the indicated cellpopulations.
Samstein et al. Page 19
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
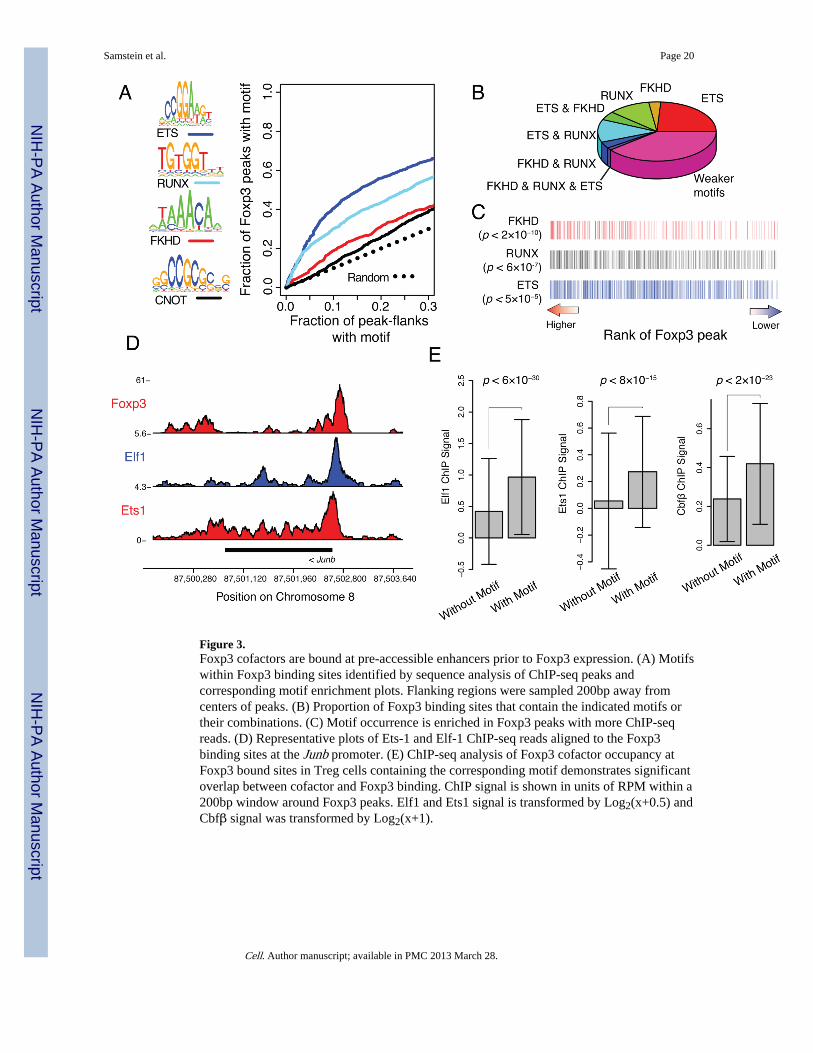
Figure 3.Foxp3 cofactors are bound at pre-accessible enhancers prior to Foxp3 expression. (A) Motifswithin Foxp3 binding sites identified by sequence analysis of ChIP-seq peaks andcorresponding motif enrichment plots. Flanking regions were sampled 200bp away fromcenters of peaks. (B) Proportion of Foxp3 binding sites that contain the indicated motifs ortheir combinations. (C) Motif occurrence is enriched in Foxp3 peaks with more ChIP-seqreads. (D) Representative plots of Ets-1 and Elf-1 ChIP-seq reads aligned to the Foxp3binding sites at the Junb promoter. (E) ChIP-seq analysis of Foxp3 cofactor occupancy atFoxp3 bound sites in Treg cells containing the corresponding motif demonstrates significantoverlap between cofactor and Foxp3 binding. ChIP signal is shown in units of RPM within a200bp window around Foxp3 peaks. Elf1 and Ets1 signal is transformed by Log2(x+0.5) andCbfβ signal was transformed by Log2(x+1).
Samstein et al. Page 20
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
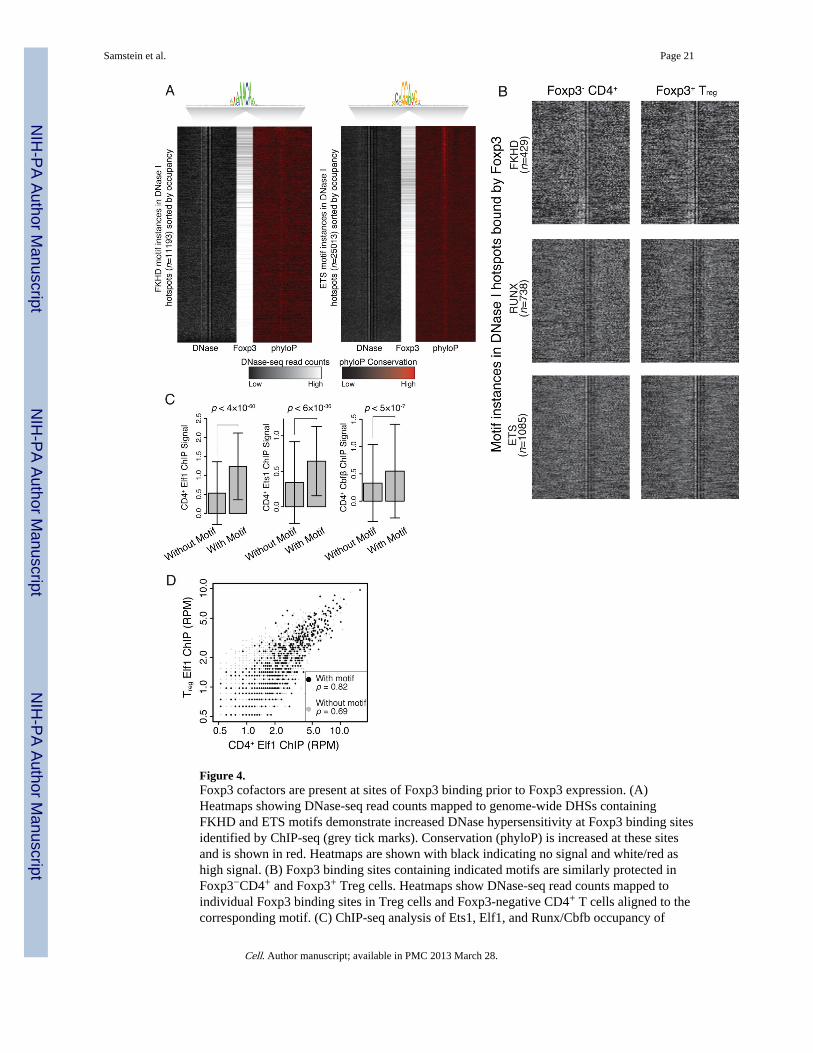
Figure 4.Foxp3 cofactors are present at sites of Foxp3 binding prior to Foxp3 expression. (A)Heatmaps showing DNase-seq read counts mapped to genome-wide DHSs containingFKHD and ETS motifs demonstrate increased DNase hypersensitivity at Foxp3 binding sitesidentified by ChIP-seq (grey tick marks). Conservation (phyloP) is increased at these sitesand is shown in red. Heatmaps are shown with black indicating no signal and white/red ashigh signal. (B) Foxp3 binding sites containing indicated motifs are similarly protected inFoxp3−CD4+ and Foxp3+ Treg cells. Heatmaps show DNase-seq read counts mapped toindividual Foxp3 binding sites in Treg cells and Foxp3-negative CD4+ T cells aligned to thecorresponding motif. (C) ChIP-seq analysis of Ets1, Elf1, and Runx/Cbfb occupancy of
Samstein et al. Page 21
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Foxp3-bound sites containing ETS and RUNX motifs, respectively, in Foxp3-negativeCD4+ T cells. Units and transforms are same as described in Figure 3 legend. (D) Elf1binding at Foxp3-bound sites in both Foxp3-negative CD4+ T cells and Treg cells revealedby ChIP-seq analysis. Spearman’s rank correlation is greater for Foxp3 binding sites withETS motif than without. Data is incremented by 0.5RPM to increase dynamic range.
Samstein et al. Page 22
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
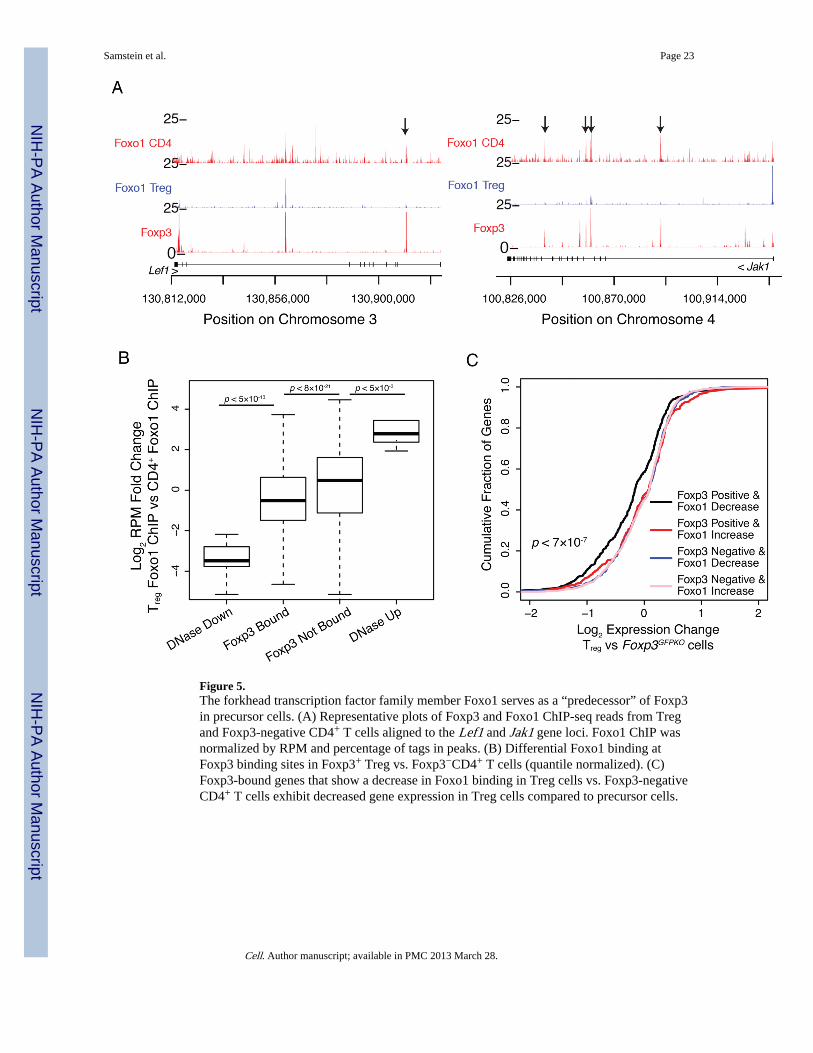
Figure 5.The forkhead transcription factor family member Foxo1 serves as a “predecessor” of Foxp3in precursor cells. (A) Representative plots of Foxp3 and Foxo1 ChIP-seq reads from Tregand Foxp3-negative CD4+ T cells aligned to the Lef1 and Jak1 gene loci. Foxo1 ChIP wasnormalized by RPM and percentage of tags in peaks. (B) Differential Foxo1 binding atFoxp3 binding sites in Foxp3+ Treg vs. Foxp3−CD4+ T cells (quantile normalized). (C)Foxp3-bound genes that show a decrease in Foxo1 binding in Treg cells vs. Foxp3-negativeCD4+ T cells exhibit decreased gene expression in Treg cells compared to precursor cells.
Samstein et al. Page 23
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
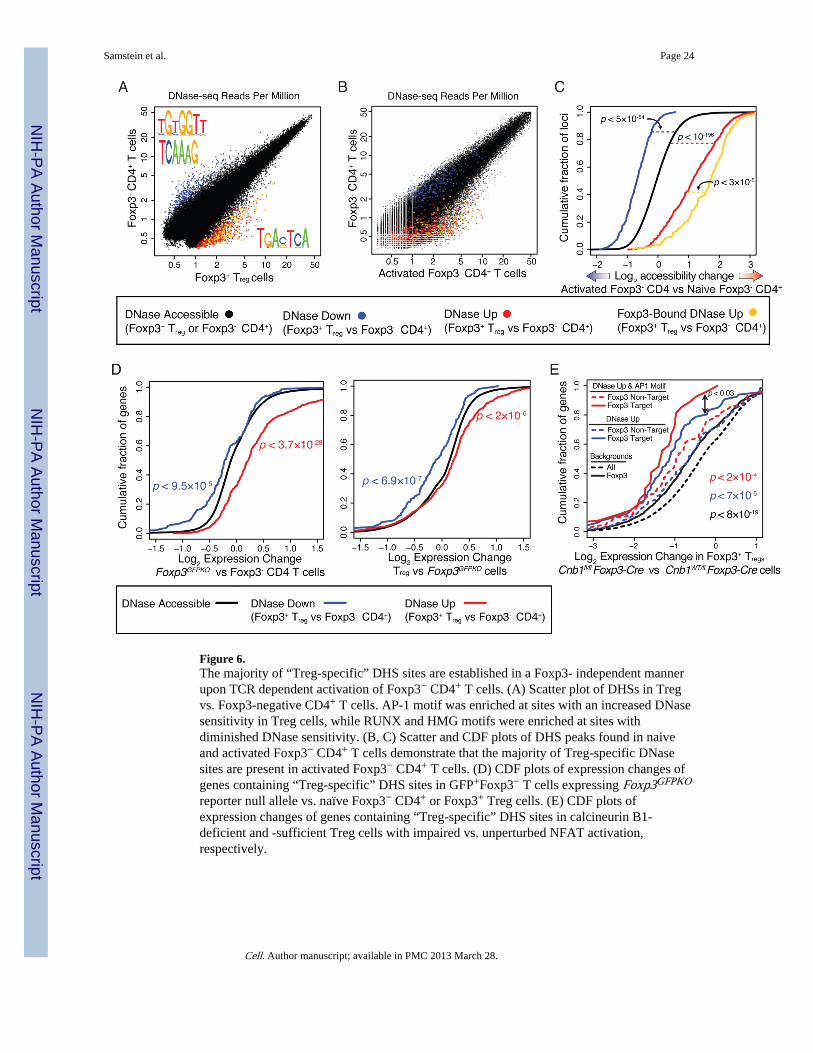
Figure 6.The majority of “Treg-specific” DHS sites are established in a Foxp3- independent mannerupon TCR dependent activation of Foxp3− CD4+ T cells. (A) Scatter plot of DHSs in Tregvs. Foxp3-negative CD4+ T cells. AP-1 motif was enriched at sites with an increased DNasesensitivity in Treg cells, while RUNX and HMG motifs were enriched at sites withdiminished DNase sensitivity. (B, C) Scatter and CDF plots of DHS peaks found in naiveand activated Foxp3− CD4+ T cells demonstrate that the majority of Treg-specific DNasesites are present in activated Foxp3− CD4+ T cells. (D) CDF plots of expression changes ofgenes containing “Treg-specific” DHS sites in GFP+Foxp3− T cells expressing Foxp3GFPKO
reporter null allele vs. naïve Foxp3− CD4+ or Foxp3+ Treg cells. (E) CDF plots ofexpression changes of genes containing “Treg-specific” DHS sites in calcineurin B1-deficient and -sufficient Treg cells with impaired vs. unperturbed NFAT activation,respectively.
Samstein et al. Page 24
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
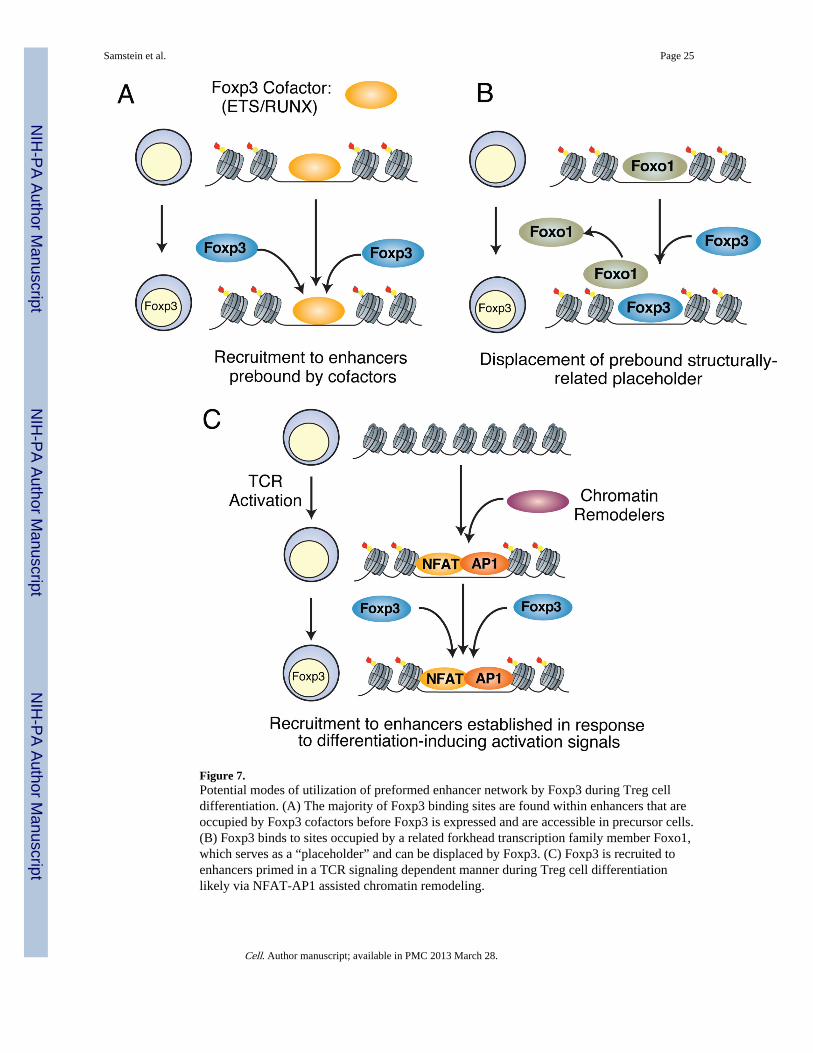
Figure 7.Potential modes of utilization of preformed enhancer network by Foxp3 during Treg celldifferentiation. (A) The majority of Foxp3 binding sites are found within enhancers that areoccupied by Foxp3 cofactors before Foxp3 is expressed and are accessible in precursor cells.(B) Foxp3 binds to sites occupied by a related forkhead transcription family member Foxo1,which serves as a “placeholder” and can be displaced by Foxp3. (C) Foxp3 is recruited toenhancers primed in a TCR signaling dependent manner during Treg cell differentiationlikely via NFAT-AP1 assisted chromatin remodeling.
Samstein et al. Page 25
Cell. Author manuscript; available in PMC 2013 March 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Copyright © 2022 FDOKUMEN




















