
Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute...
25
©2004 FASEB The FASEB Journal express article 10.1096/fj.04-2178fje. Published online October 26, 2004. Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct Felicity N. E. Gavins,* Ahmad M. Kamal,* Michele D'Amico, † Sonia M. Oliani, ‡ and Mauro Perretti* *Centre of Biochemical Pharmacology, The William Harvey Research Institute, London EC1M 6BQ, United Kingdom; † Department of Experimental Medicine, Second University of Naples, 80138 Naples, Italy; and ‡ Department of Biology, IBILCE–UNESP, São Paulo, Brazil Corresponding author: Mauro Perretti, The William Harvey Research Institute, Bart’s and the London, Queen Mary School of Medicine and Dentistry, Charterhouse Square, London EC1M 6BQ, UK. E-mail: [email protected] ABSTRACT Recent interest in the annexin 1 field has come from the notion that specific G-protein-coupled receptors, members of the formyl-peptide receptor (FPR) family, appear to mediate the anti- inflammatory actions of this endogenous mediator. Administration of the annexin 1 N-terminal derived peptide Ac2-26 to mice after 25 min ischemia significantly attenuated the extent of acute myocardial injury as assessed 60 min postreperfusion. Evident at the dose of 1 mg/kg (~9 nmol per animal), peptide Ac2-26 cardioprotection was intact in FPR null mice. Similarly, peptide Ac2-26 inhibition of specific markers of heart injury (specifically myeloperoxidase activity, CXC chemokine KC contents, and endogenous annexin 1 protein expression) was virtually identical in heart samples collected from wild-type and FPR null mice. Mouse myocardium expressed the mRNA for FPR and the structurally related lipoxin A 4 receptor, termed ALX; thus, comparable equimolar doses of two ALX agonists (W peptide and a stable lipoxin A 4 analog) exerted cardioprotection in wild-type and FPR null mice to an equal extent. Curiously, marked (>95%) blood neutropenia produced by an anti-mouse neutrophil serum did not modify the extent of acute heart injury, whereas it prevented the protection afforded by peptide Ac2-26. Thus, this study sheds light on the receptor mechanism(s) mediating annexin 1-induced cardioprotection and shows a pivotal role for ALX and circulating neutrophil, whereas it excludes any functional involvement of mouse FPR. These mechanistic data can help in developing novel therapeutics for acute cardioprotection. Key words: FPR • ischemia/reperfusion • PMN he literature shows contrasting data on the role played by circulating leukocytes (predominantly polymorphonuclear leukocytes or PMN) in myocardial ischemia/reperfusion (I/R) damage. Indeed, whereas initial observations indicated that active PMN promoted reperfusion injury in the heart, subsequent work failed to provide T Page 1 of 25 (page number not for citation purposes)
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute...

©2004 FASEB
The FASEB Journal express article 10.1096/fj.04-2178fje. Published online October 26, 2004.
Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct
Felicity N. E. Gavins,* Ahmad M. Kamal,* Michele D'Amico,† Sonia M. Oliani,‡ and Mauro Perretti* *Centre of Biochemical Pharmacology, The William Harvey Research Institute, London EC1M 6BQ, United Kingdom; †Department of Experimental Medicine, Second University of Naples, 80138 Naples, Italy; and ‡Department of Biology, IBILCE–UNESP, São Paulo, Brazil Corresponding author: Mauro Perretti, The William Harvey Research Institute, Bart’s and the London, Queen Mary School of Medicine and Dentistry, Charterhouse Square, London EC1M 6BQ, UK. E-mail: [email protected]
ABSTRACT
Recent interest in the annexin 1 field has come from the notion that specific G-protein-coupled receptors, members of the formyl-peptide receptor (FPR) family, appear to mediate the anti-inflammatory actions of this endogenous mediator. Administration of the annexin 1 N-terminal derived peptide Ac2-26 to mice after 25 min ischemia significantly attenuated the extent of acute myocardial injury as assessed 60 min postreperfusion. Evident at the dose of 1 mg/kg (~9 nmol per animal), peptide Ac2-26 cardioprotection was intact in FPR null mice. Similarly, peptide Ac2-26 inhibition of specific markers of heart injury (specifically myeloperoxidase activity, CXC chemokine KC contents, and endogenous annexin 1 protein expression) was virtually identical in heart samples collected from wild-type and FPR null mice. Mouse myocardium expressed the mRNA for FPR and the structurally related lipoxin A4 receptor, termed ALX; thus, comparable equimolar doses of two ALX agonists (W peptide and a stable lipoxin A4 analog) exerted cardioprotection in wild-type and FPR null mice to an equal extent. Curiously, marked (>95%) blood neutropenia produced by an anti-mouse neutrophil serum did not modify the extent of acute heart injury, whereas it prevented the protection afforded by peptide Ac2-26. Thus, this study sheds light on the receptor mechanism(s) mediating annexin 1-induced cardioprotection and shows a pivotal role for ALX and circulating neutrophil, whereas it excludes any functional involvement of mouse FPR. These mechanistic data can help in developing novel therapeutics for acute cardioprotection.
Key words: FPR • ischemia/reperfusion • PMN
he literature shows contrasting data on the role played by circulating leukocytes (predominantly polymorphonuclear leukocytes or PMN) in myocardial ischemia/reperfusion (I/R) damage. Indeed, whereas initial observations indicated that
active PMN promoted reperfusion injury in the heart, subsequent work failed to provide T
Page 1 of 25(page number not for citation purposes)

conclusive evidence (for recent commentaries, see refs 1, 2). It is now generally accepted that activation of circulating PMN and subsequent infiltration into the myocardial tissue have a causal link in I/R-induced heart tissue damage (3–8). These processes are part of the local inflammatory processes initiated in the ischemic and postischemic heart, which all contribute to the remodeling process (3). Nonetheless, many other inflammatory mediators generated in the early phases of the I/R response contribute to the early reperfusion damage (9), including radical species, cytokines (10–17), and chemokines (8, 18).
We have recently reported on the ability of exogenously administered annexin 1 (ANX-A1) to markedly attenuate I/R injury in the rat heart. Treatment of rats with human recombinant ANX-A1 (8) or with its N-terminal derived peptide Ac2-26 (19) inhibited the acute injury measured 2 h postreperfusion by >50%. This cardioprotective effect was associated with reduced tissue levels of proinflammatory chemokines and cytokines and appeared related to a reduced degree of tissue infiltration by blood-borne PMN (8, 19), as determined using histological analyses and by measuring myelo-peroxidase activity.
ANX-A1, originally described as a glucocorticoid-inducible protein (20) involved in the early feedback inhibition of the hypothalamus-pituitary adrenal axis (21), exerts inhibitory effects on the process of leukocyte extravasation (22, 23). Intravital microscopy studies have highlighted ANX-A1-dependent alterations in the degree of PMN recruitment, with a particular efficacy in blocking the transition between leukocyte adhesion to leukocyte emigration (or diapedesis) (24). Data obtained in these initial models of innate inflammation have subsequently been supported by models in which I/R-mediated tissue recruitment of blood borne cells was determined (25).
Recent interest to the ANX-A1 field has come from the notion that specific G-protein-coupled receptors, members of the formyl-peptide receptor (FPR) family, might mediate its protective action. An initial in vitro study has highlighted the role of FPR in the anti-migratory effects of ANX-A1-derived peptides (26); however, a subsequent study detected direct interaction between endogenous and exogenous ANX-A1 with another member of the FPR family, the lipoxin A4 receptor or ALX (27). Importantly, the ANX-A1/ALX interaction was also supported by binding experiments. However, the distinct role that these two, and possibly more, receptors of this family plays in mediating specific ANX-A1 effects in inflammation and tissue injury is unclear. We have used genetically deficient mice (28) to observe a partial involvement of mouse FPR in the anti-migratory actions of ANX-A1 and its peptides in a model of peritonitis (29). However, when I/R-induced leukocyte endothelium interaction in the mouse mesenteric microcirculation was monitored, it was found to be highly susceptible to the inhibition afforded by peptide Ac2-26 and to be partly independent from FPR (30). This aspect was significantly evident when the leukocyte detachment phenomenon was monitored, a response that was unaltered in FPR knockout (KO) animals and mimicked by a lipoxin A4 stable analog (30), a known agonist at mouse ALX (31). Thus, it seems that a functional link between ANX-A1 peptides and ALX is maintained in the murine systems and its receptors. However, no studies have yet determined the potential receptor involvement in the cardioprotective effects of ANX-A1 and its peptides.
The present study used a combined genetic and pharmacological approach to gain information on the receptor mediating ANX-A1 cardioprotection. The data obtained indicated a lack of involvement of mouse FPR in the inhibition that peptide Ac2-26 affords on mouse acute
Page 2 of 25(page number not for citation purposes)

myocardial I/R injury, despite the receptor being present in the heart tissue, and rather indicate a possible role for mouse ALX.
MATERIALS AND METHODS
Animals
A colony of FPR KO mice (28), backcrossed with C57Bl/6 for six generations, was bred on site, with breeding pairs being kindly donated by Drs. P. M. Murphy and J.-L. Gao (Laboratory of Host Defenses, NIAID, NIH, Bethesda, MD). FPR KO and C57Bl/6 mice (purchased from B&K Universal Ltd., Hull, UK), used as wild-type (WT) controls, were maintained on a standard chow pellet diet with tap water ad libitum using a 12 h light/dark cycle. Animal experimental work was performed according to Home Office regulations (Guidance on the Operation of Animals, Scientific Procedures Act, 1986).
In vivo I/R injury of the mouse heart
Surgical procedure
The mouse MIR surgical protocol and infarct size determination were performed similar as recently described (18). Animals were anesthetized with diazepam (6 mg/kg sc) and Hypnorm™ (0.7 mg/kg fentanyl citrate and 20 mg/kg fluanisone im), and a tracheotomy was performed for artificial ventilation (Harvard Apparatus, AH 40-1000). The tidal volume of the respirator was set at 1 ml/min, with the rate set at 100 strokes/min, and was supplemented with 100% oxygen. Core body temperature was maintained at 37°C with the use of a rectal thermometer attached to a heating pad. The right carotid artery was cannulated with PE-10 tubing, and the mean arterial blood pressure (MABP) and heart rate (HR) were continuously recorded by a MacLab system (AD Instruments, Chalgrove, UK). HR was automatically calculated from the blood pressure. A rectal thermometer was inserted, and the mice were kept at a constant body temperature of 37-38°C by a homeothermic blanket.
After completion of the surgical procedures, mice were allowed to stabilize for 30 min before ligation of the left anterior descending coronary artery (LADCA) was performed using a 6/0 silk suture (attached to a 10 mm micropoint reverse cutting needle; Ethicon, W593 7/0 BV1, Edinburgh, UK). Ischemia was confirmed by the appearance of hypokinesis and pallor distal to the occlusion and a fall in MABP. After a 25 min period of myocardial ischemia, the clip was removed so that the tension on the ligature was released and reperfusion (visually confirmed) occurred for 60 min.
Measurement of area at risk and infarct size
At the end of the reperfusion period, LADCA was re-occluded and Evans blue dye (1.5 ml of a 1% w/v solution in PBS; Sigma-Aldrich, Poole, UK) was injected into the carotid artery catheter to delineate the area at risk (AAR). The heart was removed, and the left ventricle (LV) was excised and weighed. After this procedure, the heart was sectioned transversely into five sections with one section being made at the site of ligature, and each section was weighed. Sections of the ventricle above the site of the ligature were uniformly completely blue. Sections of the ventricle from the level of the ligature to the apex that were not blue (defining the AAR) were then
Page 3 of 25(page number not for citation purposes)

incubated in 1.5% w/v triphenyltetrazolium chloride in PBS (TTC; Sigma-Aldrich). After TTC staining, viable myocardium stains brick red and the infarct appears pale white (see Fig. 1). The area of infarction for each slice was determined by computerized planimetry using an image analysis software program (NIH 1.57 Image Software, NIH).
Color enhancement was used to accentuate the differences between areas stained in blue, red, or that remain pale. The size of infarction (IS) was determined by the following equation:
IS = (A1 – WT1) + (A2 – WT2) + (A3 – WT3) + (A4 – WT4) + (A5 – WT5)
where A is percent area of infarction by planimetry from subscripted numbers 1-5 representing section and WT is weight of the same numbered sections. Percentage of infarcted LV is obtained from the equation (WT of infarction/WT of LV) – 100, as described previously (32).
Drug treatment
The ANX-A1 mimetic peptide Ac2-26 (Ac-AMVSEFLKQAWFIENEEQEYVQTVK) was purchased from Tocris Cookson (Bristol, UK). The dose range used (0.3-1 mg/kg corresponding to 13-30 µg per mouse; MW, 3065) was chosen from previous studies (19, 30). Doses of the antagonist Boc2 (N-t-butyloxy-carbonyl-Phe-DLeu-Phe-DLeu-Phe) were also based on these previous studies, and it was obtained from ICN Pharmaceuticals Ltd. (Basingstoke, UK). The high affinity ALX agonist W peptide (Trp-Lys-Tyr-Met-Val-DMet or WKYMVm; MW, 856) was synthesized by the Advanced Biotechnology Centre (Imperial College School of Medicine, London, UK) by using solid-phase stepwise synthesis. Purity was >95% as assessed by HPLC and capillary electrophoresis (data supplied by the manufacturer). W peptide was administered at doses of 5-10 µg per mouse (0.15-0.3 mg/kg), since equimolar to the active doses of peptide Ac2-26. The lipoxin A4 analog 15-epi-16-(parafluoro)-phenoxy lipoxin A4 methyl ester (ATLa-ME) was a kind gift of Dr. C. N. Serhan (Harvard Medical School, Boston, MA) and was given at the dose of 5 µg per mouse (equivalent to 11 nmol). In all cases, animals received compounds or saline (100 µl) at the beginning of reperfusion phase.
In vitro procedures
RT-PCR
Total RNA was isolated from heart tissue using RNeasy spin columns (Qiagen, Crawley, West Sussex) using a modified protocol for heart tissue according to the instructions of the manufacturer. Briefly, heart tissue samples (30-50 mg) were homogenized in RLT buffer and treated with 10 mg/ml proteinase K (10 min at 55°C). Total RNA yield was determined spectrophotmetrically, and 2 µg RNA were reverse transcribed using 2 µg oligo(dT)15 primer (Promega, Southampton, UK), 10 units AMV reverse transcriptase, 40 units RNase inhibitor (Promega), and 1.25 mM each dNTP for 20 min at 42°C. The resultant cDNA was used for PCR using murine FPR (1095 bp), ALX (1055 bp), or GAPDH (363 bp) primers. The primers for FPR and ALX were designed to amplify the entire cDNA and have been already reported (30). PCR was performed on 5 µl of the reverse transcribed cDNA in the presence of 25 pmol of each primer, 1.5 mM MgCl2, 0.2 mM each dNTP, and 2.5 units Taq polymerase (Invitrogen, Paisley, UK). Samples were denatured for 5 min at 94°C followed by 94°C for 1 min, 60°C for 1 min,
Page 4 of 25(page number not for citation purposes)

and 72°C for 1 min for a total of 35 cycles. A final extension step at 72°C for 10 min was also included. All RT-PCR was performed on a TouchDownTM thermal cycler (Thermo Electron Corporation, Basingstoke, UK). Samples were visualized on 1% agarose gels containing ethidium bromide, and gel images were analyzed by densitometry using NIH Image 1.63.
Histology
Preparation of the tissue sections for histological analysis was similar to a previously published protocol (19). Briefly, animals (WT or FPR KO) subjected to sham operation, I/R plus vehicle, or I/R plus peptide Ac2-26 (1 mg/kg) at the start of reperfusion, were killed by cervical dislocation at the end of the experimental protocol. Hearts were promptly removed, and the AAR was determined and snap-frozen in liquid nitrogen. Transversal (from apex-base axis) sections (10 µm) from two randomly chosen small tissue fragments derived from the AAR were cut. Tissue fragments were fixed in 4% paraformaldehyde and 0.5% glutaraldehyde, 0.1% sodium cacodylate buffer (pH 7.4), for 24 h at 4°C; then washed in sodium cacodylate and dehydrated through graded percentages of ethanol; and embedded in LRGold (London Resin Co., Reading, Berkshire, UK). Sections (0.5 µm thick) were stained with 1% toludine blue in 1% borax solution (TAAB, UK) before examination on an Axioskop 2-mot plus Zeiss microscope (Carl Zeiss, Jena, Germany).
Western blot analysis
In separate experiments, sham or infarcted hearts were infused with saline to remove excess blood and the left ventricles were homogenized in 50 mM Tris-HCl (pH 7.2), containing 1 µM leupeptin, 1 µM pepstatin A, 200 µM phenyl-methylsulfonylfluoride, and total protein concentration was determined according to Bradford. To detect ANX-A1, protein extracts (30 µg) were loaded onto a 10% SDS-PAGE for electrophoresis together with the appropriate molecular weight markers and transferred to ECL Hybond nitrocellulose membrane. Reversible protein staining of the membranes with 0.1% Ponceau S in 5% acetic acid was used to verify even protein transfer. Membranes were incubated overnight in 5% nonfat dry milk together with a rabbit polyclonal antibody raised against full-length human ANX-A1 (1:1000; kindly donated by Dr E Solito, Imperial College, London) in phosphate buffered saline with 0.1% Tween 20 (PBST). This was followed by 30 min washing with PBST and incubation for 60 min at room temperature with peroxidase-conjugated goat anti-mouse IgG (1:2000), and immunoreactive proteins were detected using an enhanced chemiluminescence ECL kit (Amersham, UK). Relative band intensity was quantified using NIH image software 1.63.
Measurement of PMN influx and activation
Selected heart samples were obtained from other WT or FPR KO mice subjected to sham operation, I/R and I/R + peptide Ac2-26 as described above. Samples were minced and homogenized in a phosphate buffer containing 0.5% hexadecyl trimethylammonium bromide (HTAB) detergent. PMN accumulation was measured by comparing MPO activity in tissue extracts with that measured from extracts prepared from known numbers of mouse neutrophils (elicited in the peritoneal cavity of other mice by injection of thioglycollate). For the enzymatic reaction, a mixture HTAB buffer (25 µl), K-Blue substrate® (tetramethylbenzidine; 100 µl; Sigma-Aldrich, Poole, UK), and samples (25 µl) were added to a 96-well plate and optical
Page 5 of 25(page number not for citation purposes)

density (OD) readings at 620 nm were taken at 5 min intervals for 30 min, using GENios (TECAN, Reading, UK). Values were plotted, and a reaction rate (OD/time) was measured from the initial slope of the curve. A calibration curve was then produced, with the rate of reaction plotted against the number of PMN in the standard samples. This was used to convert reaction rates to number of neutrophils for the heart sample homogenates. Levels of the CXC chemokine KC (CXCL1) in heart tissue extracts were determined with an enzyme linked immunoabsorbant assay (Quantikine™ immunoassay kit; R&D Systems; sensitivity 0.001 pg/ml).
Statistical analysis
All values are means ± SE of, with number (n) of animals per group where stated. Statistical analysis for the intravital microscopy studies was assessed either by Student’s t test (2 groups) or by one-way ANOVA followed by Bonferroni post-hoc test (>2 groups). Differences among groups in in vitro experiments were determined by the Mann-Whitney test. In all cases, a probability value of P < 0.05 was considered significant.
RESULTS
Peptide Ac2-26 protects against acute myocardial I/R in WT and FPR KO
Initially, we tested whether peptide Ac2-26 retained its cardioprotective properties in a murine model of I/R-induced myocardial damage. When administered at a dose of 1 mg/kg (corresponding ~30 µg or 9 nmol/mouse) immediately after LADCA reopening, peptide Ac2-26 afforded significant cardioprotection (Fig. 1). Figure 1A illustrates the visible changes in coloration seen in representative tissue slices, highlighting the ischemic area (which did not retain staining; Fig. 1b), and the reversibility of this effect with peptide Ac2-26 (Fig. 1c) that brings staining grossly back to sham tissue (Fig. 1a). Whereas administration of peptide Ac2-26 did not modify AAR values (Fig. 1B), significant attenuation of myocardial tissue damage was measured, with an ~40% reduction (Fig. 1C). The lower dose of 0.3 mg/kg peptide Ac2-26 (corresponding to 10 µg or 3 nmol/mouse) did not produce significant protection. In line with the study in the rat (19), the Boc2 antagonist abrogated the inhibitory effect of 1 mg/kg peptide Ac2-26 (Fig. 1C). More importantly, gene deficiency in mouse FPR did not affect the cardioprotection afforded by peptide Ac2-26 (Fig. 2). Administered at the dose of 1 mg/kg, peptide Ac2-26 markedly attenuated tissue damage, as measured 60 min postreperfusion. The antagonist Boc2 fully prevented the cardioprotective effect of peptide Ac2-26 even in FPR KO mice (Fig. 2B). In either genotype, none of the treatments modified the AAR values (Figs. 1B and 2A)
Occlusion and subsequent reperfusion of LADCA (control vehicle) did not significantly modify MABP, HR, or PRI with respect to sham-operated mice, except the expected modest reduction in HR seen at the end of the ischemic period in WT and FPR KO (Tables 1 and 2, respectively). Treatment of animals with peptide Ac2-26 alone or together with Boc2 did not modify any of these figures (Tables 1 and 2).
These protective actions of peptide Ac2-26 were also confirmed at the histological level (Fig. 3). Classical alignment of cardiac fibers was seen in heart tissue samples taken from sham WT or FPR KO mouse (Fig. 3A–B). I/R produced marked damage easily detectable by loss of fiber
Page 6 of 25(page number not for citation purposes)

organization and mitochondrial swelling (Fig. 3C–D). Administered at the beginning of reperfusion, peptide Ac2-26 afforded protection, with an evident proportion of the ischemic tissue retaining its integrity (Fig. 3E–F). In line with the colorimetric and quantitative analyses (Figs. 1 and 2), the effect of peptide Ac2-26 was visibly identical in WT and FPR KO mice.
Peptide Ac2-26-mediated cardioprotection and markers of tissue inflammation
Myocardial injury was associated with accumulation of PMN as detected by measuring MPO activity into the AAR. Figure 4A shows this increase in equivalent PMN numbers in the infarcted heart with respect to the values measured in hearts collected from sham-operated animals. Administration of the protective dose of peptide Ac2-26 (1 mg/kg) produced significant attenuation in these values, hence in the number of PMN, as measured 60 min postreperfusion (Fig. 4A). The effect was similar in WT and KO mice, with calculated net inhibitions of 55 and 45%, respectively (not significant between the 2 genotypes).
Histological assessment of the infarcted myocardium showed the presence of leukocytes in the blood vessels of the myocardial tissue. Leukocytes were clearly detected both on the endothelium of myocardial blood vessel as well as into the perivascular connective tissue, selectively after the I/R procedure (Fig. 4B) but not in the heart of sham-operated animals (not shown). Treatment of mice with 1 mg/kg peptide Ac2-26 reduced the extent of leukocyte recruitment: Fig. 4C shows a representative vessel in which some intravascular, but few extravasated, cells were seen. These photographs refer to heart tissue collected from WT mice, but similar results were obtained in FPR KO mice (data not shown).
Changes in the extent of leukocyte interaction with myocardial vessels were associated with parallele changes in the tissue content of the CXC chemokine KC. The results obtained mirrored those referring to MPO activity, though treatment with peptide Ac2-26 practically abolished I/R-induced increase in KC levels in either WT or FPR KO mice (Fig. 4D).
Finally, endogenous ANX-A1 expression was also monitored by Western blotting analysis. Whereas sham-operated hearts did not express much of the protein, leukocyte infiltration associated with I/R increased ANX-A1 protein content to detectable levels (Fig. 5A). Both ANX-A1 isoforms were detected, with molecular masses of 37 and 33 kDa; Fig. 5A shows a representative blot with different tissue samples prepared from WT mice, however, similar results were obtained for FPR KO mice (not shown). Myocardial ANX-A1 protein expression was increased after I/R and significantly attenuated by peptide Ac2-26 administration at LADCA re-opening (Fig. 5A and B). The effect of peptide Ac2-26 was evident on both isoforms and in either mouse genotype, with the exception of the intact protein (37 kDa) in FPR KO mice (Fig. 5B).
Mouse ALX and circulating PMN
Finally, we monitored mouse ALX expression in the heart of WT and FPR KO mice. Genomic DNA analyses confirmed the expression of the correct genes in both mouse genotypes, with clear evidence of the construct used to disrupt the FPR gene in KO animals (Fig. 6A). mRNA for both receptors was also detected in naïve myocardial samples. Figure 6A, right panel, shows a representative RT-PCR from a WT mouse; interestingly, there was an evident increase in ALX
Page 7 of 25(page number not for citation purposes)

message after I/R, though analysis of few distinct samples showed that this was not a consistent result (Fig. 6B).
Then, we tested the effect of ALX agonists W peptide (33) and ATLa-ME (34). Injected at doses of 5 and 10 µg per mouse (corresponding to 5.5 and 11 nmol/mouse, or 0.15-0.3 mg/kg), W peptide displayed a dose-dependent cardioprotective effect with similar degree of protection in WT and FPR KO mice (Fig. 6C). The lipoxin A4 analog also afforded cardioprotection. In FPR KO mice administered at the dose of 5 µg per mouse immediately after LADCA re-opening, ATLa-ME reduced myocardial infarct (expressed as %AAR) to 20 ± 2 from the 54 ± 2 values measured in vehicle-treated mice (n=4, P<0.05). MABP, HR, and PRI values were measured for both W peptide and ATLa-ME groups, and they were in line with those reported in Tables 1 and 2 (data not shown).
In the last set of experiments, we tested an anti-neutrophil serum RB68C5. Pretreatment of WT mice with the anti-neutrophil serum selectively reduced the number of circulating neutrophils by >90%, without significantly altering the values for mononuclear cells (Table 3). However, RB68C5 did not affect the extent of acute myocardial damage as measured 60 min postreperfusion (Fig. 7B) nor did it affect the AAR (Fig. 7A). Interestingly, peptide Ac2-26 cardioprotective effect was no longer detected in neutropenic mice (Fig. 7B).
DISCUSSION
This study describes the protective effect of the ANX-A1 mimetic peptide Ac2-26 in a recently developed murine model of acute myocardial I/R-induced damage and provides conclusive evidence on the receptor(s)/mechanism(s) involved. Using an integrated transgenic and pharmacological approach, we could exclude any involvement of FPR in the protection afforded by peptide Ac2-26, whereas a less convincing involvement of blood-borne PMN is also apparent. Finally, indications for a functional involvement of ALX were obtained.
Two studies of myocardial I/R injury in the rat have described the ability of ANX-A1 and its peptido-mimetics to afford cardioprotection (8, 19); however, the search for the potential receptor target mediating these effects has been so far elusive. Using the Boc2 antagonist, also used in the present study, we proposed an involvement of a receptor of the FPR family (19). A breakthrough study of Walther et al. (26) proposed the existence of a functional interaction between ANX-A1 N-terminal peptides and FPR, raising a novel interest in the ANX-A1 field. The possibility of identifying the molecular receptor(s) responsible for the biological actions of this protein and its peptido-mimetic represents a major advancement and could have therapeutic impact to capitalize on over 20 yr of investigations on this anti-inflammatory mediator. However, further analyses of a series of in vitro and in vivo experimentations suggested that FPR was not the sole or the major ANX-A1 receptor. Lack of binding data showing direct interaction with FPR (26) and observation of different profiles of effect between ANX-A1 and formylated peptide (35) have led to some confusion in the field. In the present study, postischemic treatment with peptide Ac2-26 produced a dose-dependent cardioprotection associated with a marked reduction in local markers of tissue inflammation, including MPO activity (36) and levels of the CXC chemokine KC and of endogenous ANX-A1 (19, 37). These findings are similar to those obtained in the rat model, though development of the mouse model allowed us the use FPR KO mice. These animals appear to resemble their WT counterparts apart from the fact that they
Page 8 of 25(page number not for citation purposes)

display higher susceptibility to experimental infection (28). The inhibitory and protective actions of peptide Ac2-26 on I/R-induced heart injury were intact in mice deficient for FPR, clearly indicating the lack of involvement of this receptor in this specific action of this anti-inflammatory compound.
In line with our previous study (19), peptide Ac2-26 cardioprotection was sensitive to the FPR antagonist Boc2 (38). However, we now know that derivatives bearing the bulky tert-butyloxycarbonyl group interact also with other receptors of the family (30). At least five distinct receptors of the FPR family have been identified in the mouse (39). Having established the lack of involvement of FPR, in the next part of the study we focused on mouse ALX. This selection was supported by two notions: first, ANX-A1 and its peptide compete for lipoxin A4 binding to human ALX (27); and, second, as for ANX-A1, this lipid agonist activates mouse ALX to down-regulate PMN functions and trafficking (31, 40). Mouse heart tissue samples expressed ALX mRNA (as they did for FPR mRNA). The same is true for mouse PMN in which we confirmed ALX expression both at the mRNA and protein level (30). Relevantly, Boc2 blocks ALX-mediated functions, as detected in WT and FPR KO (30).
These molecular analyses were then supported by a functional study of another agonist at this class of receptors. Originally identified as a more potent agonist at human ALX than FPR (41, 42), W peptide has then been found to be a potent activator also of mouse FPR (33). In our hands, administration of W peptide immediately after re-opening of LADCA produced a dose-dependent cardioprotective effect at doses comparable to those required for peptide Ac2-26. Importantly, W peptide cardioprotection was fully retained also in FPR KO mice. Altogether these data indicate that a Boc2-sensitive receptor, different from FPR and likely to be ALX, mediates the cardioprotective actions of the ANX-A1 mimetic in acute myocardial I/R-induced injury. However, due to multiplicity of murine receptors present in the FPR family (39, 43), some precaution must be taken and we cannot exclude that other members might be mediating at least part of these cardioprotective actions. In the human system, for instance, W peptide is also a potent agonist for human FPR-like 2 receptor (44) and the same has recently been reported for peptide Ac2-26 (45). The involvement of mouse ALX in the present experimental system, though, is reinforced by the inhibition obtained with the selective ALX agonist ALTa-ME (34). Analysis of the data obtained with peptide Ac2-26, W peptide, and ATLa-ME indicates comparable potencies and efficacies for all these ALX agonists.
Comparative analysis of ANX-A1 physio-pharmacological effects suggests that distinct receptors, likely all belonging to the FPR family, mediate the actions of this anti-inflammatory mediator in a tissue specific fashion. Using a peritonitis model, we could highlight a fundamental role for mouse FPR in mediating the anti-migratory properties of peptide Ac2-26, but this was only partially true for full-length ANX-A1 (29). However, direct observation of the leukocyte-endothelium interaction by intravital microscopy analysis of postischemic mesenteric venules demonstrated that a significant proportion of the anti-adhesive effect of peptide Ac2-26 was unaltered in FPR KO mice. We now show that FPR is not involved in acute myocardial protection afforded by peptide Ac2-26 in the mouse model; the same may be applicable to the effects of these peptides on the isolated pituitary (46). Thus, in analogy to other mediators, it appears that different ANX-A1 receptors mediate specific inhibitory actions in different tissue sites and organs. It is also noteworthy that the ANX-A1 system is particularly potent at inhibition of ischemia- (47) and I/R-mediated vascular (25, 30) and myocardial (8, 19) inflammation.
Page 9 of 25(page number not for citation purposes)

The final part of the study yielded, perhaps, the most intriguing and thought-provoking observation. The issue of the causal role of the blood-borne PMN in acute myocardial I/R injury has created disparate views and conflicting experimental data (see Introduction for a succinct summary). Previously, based on histological analysis and MPO measurements, we have linked the acute (1-2 h) cardioprotective effect of ANX-A1 and derived peptides to local inhibition of PMN adhesion and recruitment (8, 19). Here we have used an anti-PMN serum known to cause prolonged neutropenia (48) to investigate the functional relationship between ANX-A1 and PMN. Administered at the dose of 150 µg, antibody RB68C5 reduced circulating PMN by >90%, as measured at the 24 h time point. This value is in line with those obtained in other studies (48–50). Importantly, the acute damage provoked in the myocardium by I/R was not affected by treatment with antibody RB68C5. However, and intriguingly, the cardioprotective effect of peptide Ac2-26 was lost in mice pretreated with RB68C5. Thus, it seems that circulating PMN play an ancillary, important but not exclusive, role in the damage associated with acute myocardial I/R injury. Another explanation, at least at the theoretical level, is that blood PMN play a protective, rather than detrimental, role in this acute model. However, even with this alternative hypothesis, not supported by the fact that ANX-A1 and peptide Ac2-26 have long been known to inhibit PMN adhesion and trafficking (9, 22, 51), RB68C5 should have increased infarct size, whereas the antibody was inactive when given on its own. It is noteworthy that a similar dichotomy of behavior has been reported for inducible nitric oxide synthase and the acute cardioprotection mediated by statins. In mice null for inducible nitric oxide synthase, myocardial I/R injury was not different from WT animals however the protective properties of simvastatin were no longer evident. Thus, nitric oxide derived from the inducible enzyme is functionally involved only in the cardioprotective effect of the pharmacological agent, whereas it did not affect the pathological process per se. The uncertain and perhaps ancillary role of circulating PMN discussed above could be at the basis of the discrepancy reported in the literature (1, 2; see also Introduction). It is also possible that other leukocyte types might play an accessory or singular role in this mouse model, an example being the T lymphocyte (53–55). To conclude, this study has investigated the receptor mechanism(s) responsible for the acute cardioprotection afforded by peptide Ac2-26. The data obtained show the lack of involvement of FPR, though expressed in the myocardium, and suggest a potential role for mouse ALX. It is likely that ALX-bearing PMN might be the target, in view of the functional data produced with antibody RB68C5. It cannot be excluded that the adherent/migrated PMN might produce other ALX agonists, e.g., lipoxin A4 or endogenous ANX-A1, which in itself might act on the myocardial ALX. Finally, in the context of cardioprotection and ANX-A1-derived peptides, it must be highlighted that, though these mediators were inactive in the rat isolated heart (Langerdoff) preparation (8), in vitro efficacy of peptide Ac2-26 on rat cardio-myocytes has also been reported, though using much longer incubation times (24 vs. 1-2 h) (56). It is therefore possible that multiple mechanisms closely related to cell type as well as the postinjury time examined might be modulated by ANX-A1 and its mimetics. Investigation of the ultimate effect of the ANX-A1 system on activation may lead to the design of novel therapeutics with potential efficacy on I/R-mediated injury including myocardial infarct.
Page 10 of 25(page number not for citation purposes)

ACKNOWLEDGMENTS
This study was funded by the British Heart Foundation (Ph.D. studentship FS/2000076). M. Perretti is a Senior Fellow of the Arthritis Research Campaign UK. We thank Drs. Phil M. Murphy and J.-L. Gao (National Institute of Allergy and Infectious Diseases, NIH) for the supply of breeding pairs of FPR KO mice. We are also grateful to Dr. D. Neil Granger (Louisiana State University, Health Science Center, Shreveport, LA) for advice and the supply of antibody RB68C5.
REFERENCES
1. Baxter, G. F. (2002) The neutrophil as a mediator of myocardial ischemia-reperfusion injury: time to move on. Basic Res. Cardiol. 97, 268–275
2. Lefer, D. J. (2002) Do neutrophils contribute to myocardial reperfusion injury? Basic Res. Cardiol. 97, 263–267
3. Frangogiannis, N. G., Smith, C. W., and Entman, M. L. (2002) The inflammatory response in myocardial infarction. Cardiovasc. Res. 53, 31–47
4. Jones, S. P., Trocha, S. D., Strange, M. B., Granger, D. N., Kevil, C. G., Bullard, D. C., and Lefer, D. J. (2000) Leukocyte and endothelial cell adhesion molecules in a chronic murine model of myocardial reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 279, H2196–H2201
5. Jones, S. P., Girod, W. G., Palazzo, A. J., Granger, D. N., Grisham, M. B., Jourd'Heuil, D., Huang, P. L., and Lefer, D. J. (1999) Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am. J. Physiol. 276, H1567-1573
6. Lefer, A. M., Campbell, B., Scalia, R., and Lefer, D. J. (1998) Synergism between platelets and neutrophils in provoking cardiac dysfunction after ischemia and reperfusion: role of selectins. Circulation 98, 1322–1328
7. Dewald, O., Frangogiannis, N. G., Zoerlein, M., Duerr, G. D., Klemm, C., Knuefermann, P., Taffet, G., Michael, L. H., Crapo, J. D., Welz, A., et al. (2003) Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc. Natl. Acad. Sci. USA 100, 2700–2705
8. D'Amico, M., Di Filippo, C., La, M., Solito, E., McLean, P. G., Flower, R. J., Oliani, S. M., and Perretti, M. (2000) Lipocortin 1 reduces myocardial ischaemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 14, 1867–1869
9. La, M., Tailor, A., D'Amico, M., Flower, R. J., and Perretti, M. (2001) Analysis of the protection afforded by annexin 1 in ischaemia-reperfusion injury: focus on neutrophil recruitment. Eur. J. Pharmacol. 429, 263–278
10. Latini, R., Bianchi, M., Correale, E., Dinarello, C. A., Fantuzzi, G., Fresco, C., Maggioni, A. P., Mengozzi, M., Romano, S., Shapiro, L., et al. (1994) Cytokines in acute myocardial
Page 11 of 25(page number not for citation purposes)

infarction: selective increase in circulating tumor necrosis factor, its soluble receptor, and interleukin-1 receptor antagonist. J. Cardiovasc. Pharmacol. 23, 1–6
11. Blum, A. (1996) Interleukin-1 in acute myocardial infarction. Lancet 347, 56
12. Maulik, N., Engelman, R. M., Wei, Z., Lu, D., Rousou, J. A., and Das, D. K. (1993) Interleukin-1 alpha preconditioning reduces myocardial ischemia reperfusion injury. Circulation 88, II387–II394
13. Pannitteri, G., Marino, B., Campa, P. P., Martucci, R., Testa, U., and Peschle, C. (1997) Interleukins 6 and 8 as mediators of acute phase response in acute myocardial infarction. Am. J. Cardiol. 80, 622–625
14. Abe, Y., Kawakami, M., Kuroki, M., Yamamoto, T., Fujii, M., Kobayashi, H., Yaginuma, T., Kashii, A., Saito, M., and Matsushima, K. (1993) Transient rise in serum interleukin-8 concentration during acute myocardial infarction. Br. Heart J. 70, 132–134
15. Anguera, I., Miranda-Guardiola, F., Bosch, X., Filella, X., Sitges, M., Marin, J. L., Betriu, A., and Sanz, G. (2002) Elevation of serum levels of the anti-inflammatory cytokine interleukin-10 and decreased risk of coronary events in patients with unstable angina. Am. Heart J. 144, 811–817
16. de Lemos, J. A., Morrow, D. A., Sabatine, M. S., Murphy, S. A., Gibson, C. M., Antman, E. M., McCabe, C. H., Cannon, C. P., and Braunwald, E. (2003) Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation 107, 690–695
17. Mallat, Z., Henry, P., Fressonnet, R., Alouani, S., Scoazec, A., Beaufils, P., Chvatchko, Y., and Tedgui, A. (2002) Increased plasma concentrations of interleukin-18 in acute coronary syndromes. Heart 88, 467–469
18. Di Filippo, C., Rossi, F., Rossi, S., and M, D. A. (2004) Cannabinoid CB2 receptor activation reduces mouse myocardial ischemia-reperfusion injury: involvement of cytokine/chemokines and PMN. J. Leukoc. Biol. 75, 453-459
19. La, M., D'Amico, M., Bandiera, S., Di Filippo, C., Oliani, S. M., Gavins, F. N., Flower, R. J., and Perretti, M. (2001) Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. FASEB J. 15, 2247–2256
20. Flower, R. J. (1988) Lipocortin and the mechanism of action of the glucocorticoids. Br. J. Pharmacol. 94, 987–1015
21. John, C. D., Christian, H. C., Morris, J. F., Flower, R. J., Solito, E., and Buckingham, J. C. (2004) Annexin 1 and the regulation of endocrine function. Trends Endocrinol. Metab. 15, 103–109
22. Perretti, M., Ahluwalia, A., Harris, J. G., Goulding, N. J., and Flower, R. J. (1993) Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in
Page 12 of 25(page number not for citation purposes)

the mouse: a qualitative comparison with an anti-CD11b monoclonal antibody. J. Immunol. 151, 4306–4314
23. Perretti, M., and Gavins, F. N. (2003) Annexin 1: an endogenous anti-inflammatory protein. News Physiol. Sci. 18, 60–64
24. Lim, L. H., Solito, E., Russo-Marie, F., Flower, R. J., and Perretti, M. (1998) Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc. Natl. Acad. Sci. USA 95, 14535–14539
25. Cuzzocrea, S., Tailor, A., Zingarelli, B., Salzman, A. L., Flower, R. J., Szab¢, C., and Perretti, M. (1997) Lipocortin 1 protects against splanchnic artery occlusion and reperfusion injury by affecting neutrophil migration. J. Immunol. 159, 5089-5097
26. Walther, A., Riehemann, K., and Gerke, V. (2000) A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 5, 831–840
27. Perretti, M., Chiang, N., La, M., Fierro, I. M., Marullo, S., Getting, S. J., Solito, E., and Serhan, C. N. (2002) Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 8, 1296–1302
28. Gao, J. L., Lee, E. J., and Murphy, P. M. (1999) Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J. Exp. Med. 189, 657–662
29. Perretti, M., Getting, S. J., Solito, E., Murphy, P. M., and Gao, J. L. (2001) Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of annexin 1 and its mimetics. Am. J. Pathol. 158, 1969–1973
30. Gavins, F. N., Yona, S., Kamal, A. M., Flower, R. J., and Perretti, M. (2003) Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 101, 4140–4147
31. Takano, T., Fiore, S., Maddox, J. F., Brady, H. R., Petasis, N. A., and Serhan, C. N. (1997) Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti- inflammatory receptors. J. Exp. Med. 185, 1693–1704
32. Michael, L. H., Entman, M. L., Hartley, C. J., Youker, K. A., Zhu, J., Hall, S. R., Hawkins, H. K., Berens, K., and Ballantyne, C. M. (1995) Myocardial ischemia and reperfusion: a murine model. Am. J. Physiol. 269, H2147–H2154
33. He, R., Tan, L., Browning, D. D., Wang, J. M., and Ye, R. D. (2000) The synthetic peptide Trp-Lys-Tyr-Met-Val-D-Met is a potent chemotactic agonist for mouse formyl peptide receptor. J. Immunol. 165, 4598–4605
Page 13 of 25(page number not for citation purposes)

34. Schottelius, A. J., Giesen, C., Asadullah, K., Fierro, I. M., Colgan, S. P., Bauman, J., Guilford, W., Perez, H. D., and Parkinson, J. F. (2002) An aspirin-triggered lipoxin A4 stable analog displays a unique topical anti-inflammatory profile. J. Immunol. 169, 7063–7070
35. Solito, E., Kamal, A. M., Russo-Marie, F., Buckingham, J. C., Marullo, S., and Perretti, M. (2003) A novel calcium-dependent pro-apoptotic effect of annexin 1 on human neutrophils. FASEB J. 17, 1544–1546
36. Mullane, K. M., Kraemer, R., and Smith, B. (1985) Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J. Pharmacol. Meth. 14, 157–163
37. Vergnolle, N., Coméra, C., and Buéno, L. (1995) Annexin 1 is overexpressed and specifically secreted during experimentally induced colitis in rats. Eur. J. Biochem. 232, 603–610
38. Prossnitz, E. R., and Ye, R. D. (1997) The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol. Ther. 74, 73–102
39. Le, Y., Murphy, P. M., and Wang, J. M. (2002) Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548
40. Chiang, N., Fierro, I. M., Gronert, K., and Serhan, C. N. (2000) Activation of lipoxin A4 receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191, 1197–1208
41. Le, Y., Gong, W., Li, B., Dunlop, N. M., Shen, W., Su, S. B., Ye, R. D., and Wang, J. M. (1999) Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation. J. Immunol. 163, 6777–6784
42. Dahlgren, C., Christophe, T., Boulay, F., Madianos, P. N., Rabiet, M. J., and Karlsson, A. (2000) The synthetic chemoattractant Trp-Lys-Tyr-Met-Val-DMet activates neutrophils preferentially through the lipoxin A4 receptor. Blood 95, 1810–1818
43. Vaughn, M. W., Proske, R. J., and Haviland, D. L. (2002) Identification, cloning, and functional characterization of a murine lipoxin A4 receptor homologue gene. J. Immunol. 169, 3363–3369
44. Christophe, T., Karlsson, A., Dugave, C., Rabiet, M. J., Boulay, F., and Dahlgren, C. (2001) The synthetic peptide Trp-Lys-Tyr-Met-Val-Met-NH2 specifically activates neutrophils through FPRL1/lipoxin A4 receptors and is an agonist for the orphan monocyte-expressed chemoattractant receptor FPRL2. J. Biol. Chem. 276, 21585–21593
45. Ernst, S., Lange, C., Wilbers, A., Goebeler, V., Gerke, V., and Rescher, U. (2004) An annexin 1 N-terminal Peptide activates leukocytes by triggering different members of the formyl Peptide receptor family. J. Immunol. 172, 7669–7676
Page 14 of 25(page number not for citation purposes)

46. John, C., Perretti, M., Solito, E., Morris, J. F., Flower, R. J., and Buckingham, J. C. (2001) Does annexin 1 act via the FPR receptor? J. Endocrinol. 169, P150
47. Relton, J. K., Strijbos, P. J. L. M., O'Shaughnessy, C. T., Carey, F., Forder, R. A., Tilders, F. J., and Rothwell, N. J. (1991) Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. J. Exp. Med. 174, 305–310
48. Stokes, K. Y., Clanton, E. C., Bowles, K. S., Fuseler, J. W., Chervenak, D., Chervenak, R., Jennings, S. R., and Granger, D. N. (2002) The role of T-lymphocytes in hypercholesterolemia-induced leukocyte-endothelial interactions. Microcirculation 9, 407–417
49. Bonder, C. S., Ajuebor, M. N., Zbytnuik, L. D., Kubes, P., and Swain, M. G. (2004) Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J. Immunol. 172, 45–53
50. Cerwinka, W. H., Cooper, D., Krieglstein, C. F., Ross, C. R., McCord, J. M., and Granger, D. N. (2003) Superoxide mediates endotoxin-induced platelet-endothelial cell adhesion in intestinal venules. Am. J. Physiol. Heart Circ. Physiol. 284, H535–H541
51. Mancuso, F., Flower, R. J., and Perretti, M. (1995) Leukocyte transmigration, but not rolling or adhesion, is selectively inhibited by dexamethasone in the hamster post-capillary venule. Involvement of endogenous lipocortin 1. J. Immunol. 155, 377–386
52. Scalia, R., Gooszen, M. E., Jones, S. P., Hoffmeyer, M., Rimmer, D. M., III, Trocha, S. D., Huang, P. L., Smith, M. B., Lefer, A. M., and Lefer, D. J. (2001) Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation 103, 2598–2603
53. Lim, Y. C., Garcia-Cardena, G., Allport, J. R., Zervoglos, M., Connolly, A. J., Gimbrone, M. A., Jr., and Luscinskas, F. W. (2003) Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am. J. Pathol. 162, 1591–1601
54. Varda-Bloom, N., Leor, J., Ohad, D. G., Hasin, Y., Amar, M., Fixler, R., Battler, A., Eldar, M., and Hasin, D. (2000) Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J. Mol. Cell. Cardiol. 32, 2141–2149
55. Spagnoli, L. G., Bonanno, E., Mauriello, A., Palmieri, G., Partenzi, A., Sangiorgi, G., and Crea, F. (2002) Multicentric inflammation in epicardial coronary arteries of patients dying of acute myocardial infarction. J. Am. Coll. Cardiol. 40, 1579–1588
56. Ritchie, R. H., Sun, X., Bilszta, J. L., Gulluyan, L. M., and Dusting, G. J. (2003) Cardioprotective actions of an N-terminal fragment of annexin-1 in rat myocardium in vitro. Eur. J. Pharmacol. 461, 171–179
Received May 18, 2004; accepted September 16, 2004.
Page 15 of 25(page number not for citation purposes)

Table 1 Cardiovascular parameters of WT mice subjected to 25 min of myocardial occlusion and 60 min reperfusiona
Experimental Groups
Pre-occlusion -5 min
Occlusion Reperfusion
0 min 60 min Sham-operated MABP 49 ± 4 53 ± 4 46 ± 5 44 ± 4 HR 212 ± 54 261 ± 24 203 ± 20 253 ± 18 PRI 12 ± 3 15 ± 1 9 ± 2 10 ± 2 PBS MABP 66 ± 5 56 ± 8 57 ± 5 44 ± 8* HR 213 ± 52 191 ± 50 240 ± 80 354 ± 77 PRI 14 ± 4 14 ± 3 15 ± 4 16 ± 5 Ac2-26 (1 mg/kg) MABP 58 ± 5 55 ± 3 51 ± 3 42 ± 8 HR 280 ± 58 213 ± 38 287 ± 34 274 ± 54 PRI 17 ± 4 11 ± 2 15 ± 2 13 ± 4 Ac2-26 (0.3 mg/kg) MABP 66 ± 6 62 ± 5 58 ± 5 51 ± 7 HR 292 ± 61 270 ± 81 284 ± 99 408 ± 44* PRI 20 ± 5 18 ± 6 22 ± 4 24 ± 3 Boc2 MABP 72 ± 5 74 ± 6 64 ± 6 54 ± 6* HR 294 ± 61 370 ± 21 284 ± 99 408 ± 44* PRI 20 ± 4 27 ± 2 22 ± 3 20 ± 3 Ac2-26 + Boc2 MABP 62 ± 9 70 ± 5 62 ± 5 53 ± 5 HR 324 ± 32 371 ± 33 356 ± 35 314 ± 34 PRI 21 ± 4 26 ± 3 22 ± 3 17 ± 3 aMAP (mmHg), HR (beats/min), and PRI (mmHg/min/103) recorded in mice before coronary occlusion (-5 min), at occlusion (0 min), at start of reperfusion (+25 min), and 60 min after reperfusion. Animals were treated at beginning of reperfusion with either peptide Ac2-26 (0.3 and 1 mg/kg), Boc2 (0.4 mg/kg), Ac2-26 (1 mg/kg) + Boc2 (0.4 mg/kg), or PBS. Data are means ± SE of 6 mice/group; *P < 0.05 vs. respective pre-occlusion value.
Page 16 of 25(page number not for citation purposes)

Table 2 Cardiovascular parameters of FPR KO mice subjected to 25 min of myocardial occlusion and 60 min reperfusion1
Experimental Groups Pre-occlusion
-5 min Occlusion
0 min Reperfusion
+25 min +60 min Sham-operated MABP 52 ± 3 52 ± 5 48 ± 5 46 ± 4 HR 306 ± 18 331 ± 25 342 ± 39 354 ± 28 PRI 16 ± 1 17 ± 1 16 ± 1 16 ± 1 PBS MABP 65 ± 9 55 ± 8 63 ± 5 48 ± 8 HR 386 ± 47 387 ± 31 381 ± 35 364 ± 22 PRI 26 ± 5 23 ± 4 22 ± 2 18 ± 1 Ac2-26 MABP 60 ± 8 59 ± 12 55 ± 5 43 ± 6 HR 382 ± 27 358 ± 60 432 ± 18 342 ± 19 PRI 23 ± 3 24 ± 3 24 ± 2 15 ± 2* Ac2-26 + Boc2 MABP 45 ± 10 44 ± 4 42 ± 1 37 ± 5 HR 317 ± 23 364 ± 35 352 ± 25 315 ± 20 PRI 15 ± 3 16 ± 2 15 ± 1 10 ± 1 aMAP (mmHg), HR (beats/min), and PRI (mmHg/min/103) recorded in mice before coronary occlusion (-5 min), at occlusion (0 min), at start of reperfusion (+25 min), and +60 min after reperfusion. Animals were treated at beginning of reperfusion (25 min) with either Ac2-26 (1 mg/kg), Ac2-26 (1 mg/kg) + Boc2 (0.4 mg/kg), or PBS. Data are means ± SE of 6 mice/group; *P < 0.05 vs. respective pre-occlusion value.
Page 17 of 25(page number not for citation purposes)
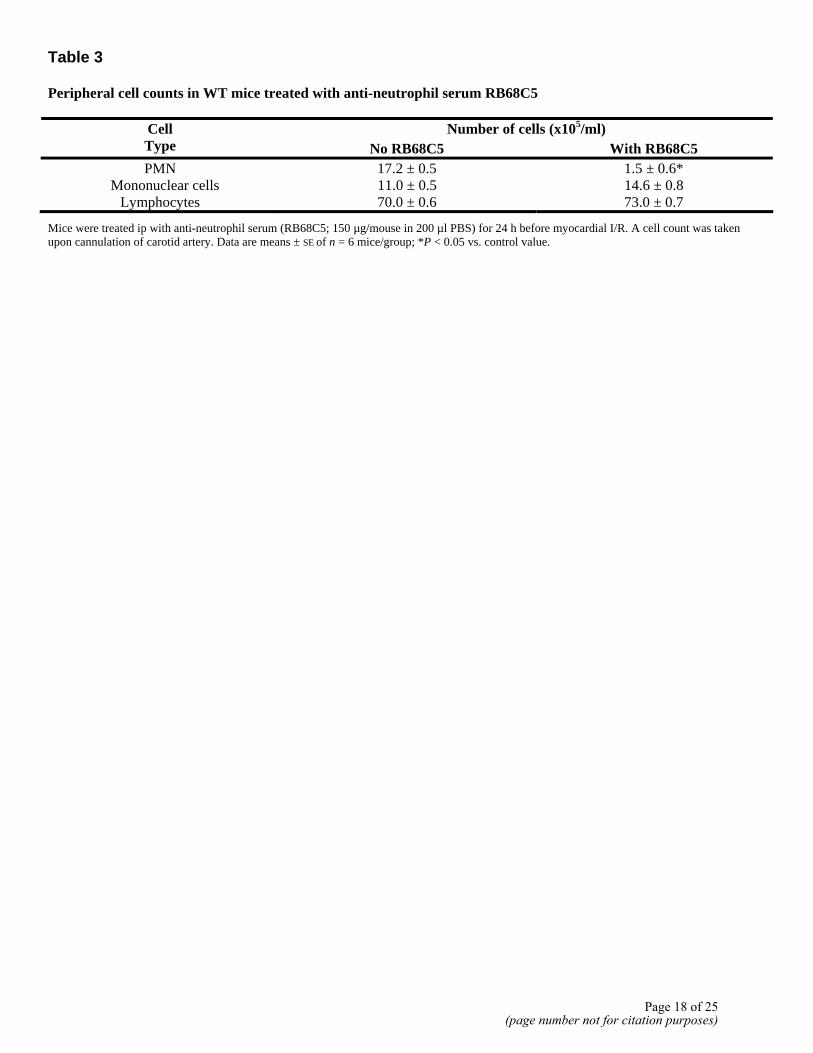
Table 3 Peripheral cell counts in WT mice treated with anti-neutrophil serum RB68C5
Number of cells (x105/ml) Cell Type No RB68C5 With RB68C5 PMN 17.2 ± 0.5 1.5 ± 0.6*
Mononuclear cells 11.0 ± 0.5 14.6 ± 0.8 Lymphocytes 70.0 ± 0.6 73.0 ± 0.7
Mice were treated ip with anti-neutrophil serum (RB68C5; 150 µg/mouse in 200 µl PBS) for 24 h before myocardial I/R. A cell count was taken upon cannulation of carotid artery. Data are means ± SE of n = 6 mice/group; *P < 0.05 vs. control value.
Page 18 of 25(page number not for citation purposes)

Fig. 1
Figure 1. Cardioprotective effect of peptide Ac2-26 in WT mice. Mice were subjected to 25 min ischaemia followed by 60 min reperfusion, by occlusion, s described in Materials and Methods. PBS vehicle, peptide Ac2-26 alone, or with Boc2 (1 mg/kg peptide + 0.4 mg/kg Boc2) were administered immediately after reperfusion. A) Representative photographs showing vital heart tissue (blue staining) and damage (white areas) produced by I/R procedure: left, sham heart; middle, heart subject to I/R; right, I/R + peptide Ac2-26 (1 mg/kg). B) Area at risk (AAR). C) Myocardial infarct expressed as percentage of AAR. Data are means ± SE of n = 6 mice/group. *P < 0.05.
Page 19 of 25(page number not for citation purposes)

Fig. 2
Figure 2. Cardioprotective effect of peptide Ac2-26 in FPR KO mice. Mice were subjected to I/R and compound treatment as described for WT in Fig. 1. A) AAR. B) Myocardial infarct expressed as percentage of AAR. Data are means ± SE of n = 6 mice/group. *P < 0.05.
Page 20 of 25(page number not for citation purposes)

Fig. 3
Figure 3. Histological examination of heart tissues. Myocardial sections (0.5 µm) were visualized after staining with Toluidine blue. Typical morphological presentation of fibers (f) in myocardium of a sham WT (A) and FPR KO (B) mouse. This alignment of fibers (f) is clearly altered after I/R (25 min+60 min) both in WT (C) and FPR KO (D) mice. Quantitative data in Figs. 1 and 2 corroborated this lack of histological difference. Administration of 1 mg/kg peptide Ac2-26 immediately after re-opening of LADCA affords at least partial protection in both WT (E) and FPR KO (F) mice. Micrographs are of similar 5 analyses performed on tissue samples collected from different mice. Bar = 10 µm.
Page 21 of 25(page number not for citation purposes)

Fig. 4
Figure 4. Modulation of I/R-induced markers of local inflammation by peptide Ac2-26. WT and FPR KO mice were subjected to I/R in the absence (PBS) or presence of peptide Ac2-26 (1 mg/kg iv) treatment immediately after re-opening of LADCA. A) Values for MPO activity expressed as number of PMN. B) Micrograph of a representative section obtained from a WT mouse at end of I/R, in which blood-borne leukocytes are visible both in vessel (v) and in perivascular tissue (arrow). C) In tissues treated with peptide Ac2-26, leukocytes were more restrained within myocardial vessel (v). Bar = 10 µm. D) Values for tissue content of KC. Data are means ± SE of n = 6 mice/goup. #P < 0.05 vs. sham and *P < 0.05 vs. PBS treatment.
Page 22 of 25(page number not for citation purposes)

Fig. 5
Figure 5. Modulation of endogenous ANX-A1 expression by I/R and peptide Ac2-26. Heart samples were obtained from animals treated as in Fig. 1 and Fig. 2. A) Representative Western blot analysis of endogenous annexin 1 expression in WT mice subjected to sham (S), I/R alone, or I/R + 1 mg/kg peptide Ac2-26 (P). Two preparations of mouse PMN were used as control. Blots were stained for ANX-A1 and then stripped and stained for tubulin to monitor protein loading. Densitometric analysis of ANX-A1 in WT and FPR KO is shown for both 37 kDa isoform (B) and cleaved 34 kDA isoform (C). Values in B and C are means ± SE of n = 6 mice/group. *P < 0.05.
Page 23 of 25(page number not for citation purposes)

Fig. 6
Figure 6. Molecular and functional analysis for ALX. A) Hearts were removed from WT and FPR KO mice and analyzed for genomic FPR and ALX expression (left panel) or for mRNA for each receptor by RT-PCR (right panel; WT only). The latter analysis was repeated on hearts collected at end of I/R procedure. B) Cumulative densitometric analysis of heart samples collected from 3 mice for ALX mRNA expression. C) Selective ALX agonist W peptide immediately after LADCA re-opening in WT or FPR KO mice. Data are means ± SE of n = 6 mice/group; *P < 0.05 vs. vehicle (Dose 0).
Page 24 of 25(page number not for citation purposes)

Fig. 7
Figure 7. Analysis of effects of anti-neutrophil serum RB68C5. WT mice were treated with RB68C5 (150 µg/mouse in 200 µl) 24 h before I/R. Peptide Ac2-26 (1 mg/kg iv) was administered immediately after LADCA re-opening. A) AAR. B) Myocardial infarct expressed as percentage of AAR. Data are mean ± SE of n = 6 mice/group. *P < 0.05 vs. PBS group.
Page 25 of 25(page number not for citation purposes)




















