
Role of the Polarity Determinant Crumbs in Suppressing Mammalian Epithelial Tumor Progression
Upload
independentCategory
view
0download
0
Fock space multireference coupled-cluster theory for general single determinant reference functions
John F. Stanton and Rodney J. Bartlett Quantum Theory Project, Departments of Chemistry and Physics, University of Florida, Gainesville, Florida 32611
C. Magnus L. Rittby Department of Physics, Texas Christian University, Fort Worth, Texas 76129
(Received 11 June 1992; accepted 10 July 1992)
The technique of Fock space multireference coupled-cluster (FSMRCC) theory is applied for the first time to problems involving a high-spin open-shell ground state. Explicit spin-orbital equations applicable to any single determinant reference state are presented and some computational aspects of FSMRCC are discussed. The method is illustrated by two applications in which calculations are limited to single and double excitation operators (FSMRCCSD). First, several basis sets and choices of open-shell reference function are used to calculate selected ionization potentials of 0,. The FSMRCCSD results obtained with a large generally contracted basis set are uniformly within 0.1 eV of experiment. In ad- dition, FSMRCCSD is applied to a study of symmetry breaking in the 3A2 state of CO,, a classic multireference problem. The force constant for asymmetric distortion is shown to be predicted correctly as positive, unlike ordinary single-reference CCSD which predicts a double-minimum potential. The results of this paper suggest that the open-shell reference FSMRCC approach has wide applicability for the solution of chemical problems, particularly when significant nondynamic electron correlation effects are present.
I. INTRODUCTION
The Fock space multireference coupled cluster (FSM- RCC ) approach *-I* offers a convenient formalism for the calculation of transition energies between an arbitrarily chosen ground state and several final states which, in gen- eral, contain a different number of electrons. Operation- ally, the Fock space is partitioned into “sectors” denoted by an ordered pair of integers [(m,n)] which indicate the number of electrons which have been added to and re- moved from the system, respectively. Hence, the one- dimensional (0,O) sector is associated with the ground state, the (0,l) sector with singly ionized states, the ( 1,O) sector with electron attached states, the ( I,1 ) sector with singly excited states; etc. In the FSMRCC approach, the wave function for states in each (m,n) sector depends upon all of the (p,q); p<m, q<n, which recommends a hierarchical solution. For computational reasons, most FSMRCC calculations reported in the literature have been limited to sectors of relatively low valence rank (m fnG2).
The current FSMRCC method derives from the normal-ordered wave operator ansatz of Lindgren,13 but approached in a hierarchical strategy due to Mukhejee’(c) which allows the wave operator to be well defined. All previous formulations and applications of this method have been based on a closed-shell ground state. Such an ap- proach has a number of advantages, principally in the spin- adapted nature of the final state solutions. Although this point has received surprisingly little attention in the liter- ature, final state wave functions obtained in FSMRCC cal- culations which utilize a closed-shell ground state are rig- orous eigenfunctions of the S2 operator.14 Indeed, a recent
derivation of a spin-adapted coupled-cluster method via the techniques of unitary group theory ultimately led to the FSMRCC approach. ”
Nevertheless, the FSMRCC formalism may be applied to arbitrary (closed- or open-shell) ground states built on a single determinant starting point. Although such a rec- ommendation has been made previously,‘6 no calculations using open-shell ground states have yet been reported in the literature. While the spin-adapted nature of the solu- tions is lost when an open-shell reference state is used, such an approach has a number of attractive features. For ex- ample, the (0,l) sector of Fock space with respect to the (triplet) ground state of molecular oxygen contains both quartet and doublet ionized states. The considerable suc- cess” of high-spin open-shell CC calculations based on unrestricted” open-shell restricted” and quasirestricted” Hartree-Fock (UHF, ROHF, and QRHF, respectively) determinants suggests that the high-spin states of 0: (all of the quartets and the most stable 211 state) should be well characterized by an open-shell reference FSMRCC ap- proach. While it is true that one could also study the quar- tet states using a closed-shell reference via higher valence rank sectors [e.g., the (0,3) sector with respect to O”,-1, these calculations would be considerably more complicated and suffer from a less suitable orbital and correlation de- scription of the final state. Furthermore, due to the nature of the FSMRCC wave operator described in the following section, a certain class of triple and quadruple excitations relative to the Oi- ground state would be required to in- clude effects already included in calculations based on O,, when the latter is limited to only single and double excita- tion operators (FSMRCCSD) . Hence, the open-shell
5560 J. Chem. Phys. 97 (a), 15 October 1992 0021-9606/92/205560-08$006.00 0 1992 American Institute of Physics Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ground state approach offers a convenient means for ex- tending the scope of systems which can be treated easily with FSMRCC theory. The primary purposes of the present paper are to report an implementation of a general spin-orbital FSMRCC method and discuss its use by means of a few representative examples. In this report, discussion is limited to an implementation for the (0,l) sector, although generalization to other sectors is straight- forward.
II. THEORY
A. Derivation of the FSMRCC equations
In the following, we present a derivation of the FSM- RCC equations appropriate for the (0,l) sector of Fock space and document the final equations in spin-orbital form. It is not our intention to provide a rigorous founda- tion for FSMRCC theory, as a number of formal presen- tations can be found in the literature.*’ Rather, the inten- tion of this section is to provide the reader with a clear description of this approach as applied to the (0,l) sector, stressing similarities with more familiar single-reference CC techniques when appropriate. In the following, we ad- here to the usual convention that orbital labels ij,k,... refer to occupied orbitals, while a,&~,... refer to unoccupied or- bitals. The generic indices p,q,r,... are reserved for orbitals which may be either occupied or unoccupied. One should note that this convention leads to some ambiguities in dis- cussing FSMRCC methods since a number of different ref- erence determinants with varying occupation schemes are used. Therefore, we adopt the additional convention that these levels refer strictly to the occupation scheme in the ground state reference function.
In the FSMRCC approach, reference wave functions for each sector are given by specific linear combinations of Slater determinants
t@= F cljh (1)
where (pi represent the set of “model determinants” that span the reference space for the problem. The one-electron orbitals are partitioned into three categories-core, va- lence, and virtual. The core orbitals are occupied and the virtual orbitals are unoccupied in all determinants, while the valence orbitals may be occupied or unoccupied. In the (0,l) sector, all of the valence orbitals are occupied in the ground state reference function. The full set or any subset of these orbitals serves as an appropriate valence space. The model determinants for the (0, 1 > sector are then de- fined as those which can be obtained by deleting a single electron from the set of valence orbitals.
Exact wave functions are generated by application of an exponential wave operator
Y’i= f-l&. (2)
This wave operator is valence universal in the sense that it serves to map model functions in all sectors into the cor- responding exact wave functions. In the (0,l) case consid- ered here, R functions as the wave operator for both the
ground and ionized states. The definition of creation and annihilation operators is based on the orbital occupancies in the ground state reference function, which generally leads to hole and particle annihilation operators. Such terms are absent from the ground state [(O,O) sector] wave operator and their presence leads to the possibility of con- tractions among elements of the wave operator belonging to different sectors. To avoid this undesirable feature which severely complicates the structure of the equations, Lindgren”” introduced the normal-ordered wave operator
a= [exp(X> I, (3)
x=p,o, +x(0,‘) +xw + . . ., (4)
where the square brackets denote normal ordering. Terms in the Xtm*“’ sector contain m particle annihilation opera- tors (a) and n hole annihilation (i+ ) operators, and are partitioned into single excitation, double excitation, etc. operators in the usual way
x(m,n) =x;mn) +xhn) +-@d). . . . (5)
X(‘*‘) is simply the familiar excitation operator used in single-reference CC methods (usually symbolized as T) . The X(‘*‘) operators are given by
(6)
where the reader should note the presence of the hole an- nihilation operator (I+) in the definition. To simplify no- tation, we will express X(oPo) as T and X(‘*‘) as S in the following discussion:
To derive the working equations of the FSMRCC method, we begin with the Schrbdinger equation
HQIlj~“‘>=E,d21@‘>. (7)
In practice, one usually works directly with the model de- terminants, leading to the system of equations
Hi2Icp)=E9’QlI), (8)
where @ represents a vector of the determinants {&$2. * *}, and the matrix of coefficients C in Eq. ( 1) diagonalizes 8”. Considerable computational simplification is achieved through use of the canonically transformed Hamiltonian K
H=exp( - T)H exp( T) (9)
expressed in normal-ordered form
(10) where IO) represents the ground state single determinant reference function. In second-quantized notation, HN can be written as
rrN= C ~~~CP+q}+a~~rscp+q+sr)+... . (11) w
Note that .i?,v includes three- and higher-body terms and therefore has a different structure than the untransformed Hamiltonian, which contains only one- and two-particle operators. Explicit spin-orbital expressions for all of the
Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory 5561
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992 Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

-- (Q’~HN~~Q)-(Q’IiilQ)(Qlii-‘tiNii~Q)=O.
(17)
5562 Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory
one- and two-body matrix elements of EN needed for (0,l) sector calculations are given in Eqs.J 17)-( 24) of Ref. 21. Elements of the three-body part of HN are also needed for ionization @ential calculations.6(c)
Using HN, we may rewrite the Schriidinger equation as
E&Q)=eilQ), (12)
where
In the literature, Eq. (17) is frequently called the Bloch equation. Both terms individually contain both connected and disconnected parts, but it can be shown that the dis- connected components cancel one another, demonstrating the size extensivity of the FSMRCC approach.8
G=exp(S). (13)
The energy matrix g now represents the difference be- tween the final state energies and the CC energy of the ground state. It may be evaluated by application of the inverse wave operator on the left followed by projection onto the model determinants
If S is limited to one- and two-body operators, Eqs. ( 14) and ( 17) are equivalent to the following spin-orbital expressions, 23 which are the working equations for the (0,l) sector FSMRCCSD calculations presented in Sec. III
E$= -3,+ c SpL- 1 &%m (QIHeffIQ)~(QI~-‘H~~IQ)=~. (14)
The matrix $ is also known as the effective Hamiltonian for the model space of the (0,l) sector, since diagonaliza- tion gives directly the ionization potentials of the system under consideration. Of course, the potential energy sur- face and properties of the final states may also be evaluated by simply adding the ionization potentials- to the ground stateenergy, which is equivalent to using Hand KI instead of HN and fi in the equations above. This should be con- trasted with propagator methods which only provide well- defined energy differences. Note that one could also invoke intermediate normalization [(<p I fi I @) = l] thereby dis- pensing with the need to apply ai-’ on the left, since only the lead term (unity) can contribute to 8’. However, Mukherjee t(s) has pointed out that the assumption of in- termediate normalization for incomplete model spaces in higher sectors of Fock space can lead to a breakdown of the size-extensivity property. For this reason, we retain the 6-l term in our equations, even though it makes no dif- ference for the (0,l) [and ( l,O)] sector.
In order to obtain equations for excitation amplitudes in CC theory, one usually projects the Schrodinger equa- tion onto the complementary space (the space of all deter- minants which are not included in the model space) and sets the result to zero.22 In the single reference case, this task is carried out most easily by first applying a-’ on the left prior to projection, leading to the equation
-- m’ me
+; c -$zJTmJeJ,, (18)
O=FKit - C SK,,FmPif + C S~m9~~-~ C 42 yn7n,i’e In’ me mm
()= yu,Ka- C SK,,Wm~~,~-P-(ij) C eymj m' m
+ C $Fae--P- (ij) C $$rma,ie+2 C $$rmnjj
e me mn
+i m& $-$~(4lef) - C $+WSL M
where P- (ij) is an antisymmetrization operator. In the equations above, the occupied indices ijk*.* have been di- vided into three categories. Capitalized indices refer to va- lence orbitals, while the primed indices are reserved for core orbitals. The remaining occupied indices may repre- sent either valence or core orbitals.
(&$‘. 1 exp( - T)H exp( T) IO) =0, (15)
where 4$‘.‘.’ represents the complementary space determi- nants. The quantity exp( - T)H exp( T) can be expanded in terms of nested commutators (the Hausdorlf expan- sion), which immediately demonstrates that the method contains only connected diagrams and is therefore size ex- tensive. However, in the FSMRCC method, the normal- ordered ansatz for 6 complicates the form of the inverse wave operator (it is not simply [exp( -S)]} and it is less straightforward to show the extensivity of the method. To avoid consideration of fii-‘, one simply projects the SchrS- dinger equation directly onto (@’ I, leading to
Following the spirit of the Msller-Plesset partitioning technique, the one-body terms of the transformed Hamil- tonian (3) are separated into diagonal and off-diagonal parts. Contributions from the diagonal elements are moved to the left-hand side of the equations, thereby providing an initial guess for the s amplitudes. These equations are then iterated to convergence using standard reduced subspace methods to accelerate the process.24 When a solution is obtained, the eigenvalues of He’ provide the ionization po- tentials of the system.
(QrI~,iilQ)-~(QrIiilQ)=O, (16)
or
A particularly important feature of FSMRCC theory should be addressed at this point. Observe that the one- body S, excitation operator simply mixes the model space functions with other determinants which are contained in the (0,l) sector of Fock space. If the valence space in- cludes all orbitals which are occupied in the reference func- tion, there are no S, amplitudes, since information regard- ing the mixing of the model space determinants is then solely carried by the eigenvectors of pff. Hence, there is a redundancy between the functions of the St amplitudes and the coefficients C of Eq. ( 1). Indeed, it can be shown
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992
(19)
(20)
Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

that the eigenvalues and final state wave functions which satisfy the FSMRCC equations for the (0,l) and ( 1,O) sectors in a reduced model space are identical to those which would be obtained if all occupied orbitals in the reference state were chosen to be valence levels.2f It is therefore incorrect to assume that ( 1) use of a larger va- lence space leads to a more complete treatment and better results and (2) the magnitude of the off-diagonal elements of gff provides an indication of the amount of multirefer- ence character present in the system. The latter assumption is only appropriate if the full valence space is chosen, since both the eigenvectors of H’* and the S, amplitudes must be examined if a reduced space is used (see Ref. 26 for a particularly simple proof of this property).
The invariance of FSMRCC results with respect to the choice of valence space for ionization potential and elec- tron affinity calculations is important for chemical appli- cations, since all determinants in each sector are treated on an equalfooting, regardless of the choice of valence orbitals. Consequently, for computational reasons, one can choose a small valence space (even one which contains only a single determinant) without compromising the balanced nature of the method. The FSMRCC approach is therefore well suited for situations in which more than one model deter- minant is important, as illustrated by its application to the 3,4z state of CO, presented later in this paper. We note in passing that the open-shell FSMRCC method described here is not particularly appropriate for the study of low- spin final states, and no such results are reported here. The reason for this is that low-spin systems by definition re- quire a multideterminantal zeroth-order description which is not of the type handled automatically by these methods. In all cases, at least one of these determinants lies outside the (0,l) sector defined by the high-spin open-shell ground state and is therefore not treated in the balanced manner discussed above. FSMRCC techniques can indeed be ap- plied to such systems, but sectors of valence rank two or higher must be used.
8. Computational considerations
The spin-orbital-based FSMRCC method described in the previous section has been incorporated into the ACES II quantum chemistry package. 27 All operator products listed in Eqs. (18)~(20) have been coded as matrix multiplica- tions, resulting in a fully vectorized program for these cal- culations. In this section, we address some computational aspects of FSMRCC calculations-a subject which has largely been ignored in the literature-and limit our con- sideration to the (0,l) sector in the FSMRCCSD (S=S, +S2) approximation. In the following, n refers to the number of orbitals which are occupied in the ground state reference function and N refers to the number of unoccu- pied orbitals. The number of valence orbitals is denoted by n,. Assuming that N>n, which is nearly always the case in production calculations, the most computationally de- manding step of a (0,l) sector calculation is the fifth term in Eq. (20), which scales as n, n3N2. The cost of these calculations is directly proportional to the dimension of the valence space and it is therefore advantageous to work in
TABLE I. Computational details for FSMRCCSD ionization potential calculations using both open-shell and closed-shell reference states. The Nz ground state calculation used an RHF reference function, while an UHF-CCSD ground state was used for Oa. Both calculations used the PVQZ correlation consistent basis set ( 110 basis functions). Timings are in seconds and were obtained on a Cray-YMP supercomputer with the ACES II program system.
Computational step N2 02
Integral evaluation 128 127 Self-consistent field (SCF) calculation 10 18 Integral transformation 10 34 Molecular orbital (MO) integral sorting 12 30 Ground state CCSD equations 25 61 CCSD iterations 11 11 FSMRCCSD calculation 3 7 FSMRCCSD iterations 12 11 Number of roots obtained 4 4 Total CPU time 188 277
the smallest subspace which contains the desired number of solutions. An analysis of the computational require- ments of ground-state CCSD calculations for both open- and closed-shell references has been presented elsewhere.28 The most demanding step of these calculations in the N)n case is the “particle-particle ladder” term, which scales as n2+. Hence, the cost ratio of a FSMRCCSD ionization potential calculation to the ground state CCSD scales as -nnLJN2. The incremental cost of the FSMRCCSD (0,l) sector calculation therefore tends towards zero as the size of the basis is increased. In practice, the ground-state CC calculation is typically at least an order of magnitude more expensive than the FSMRCCSD step.
Exploitation of molecular symmetry is handled easily in FSMRCCSD calculations. In ACES II, the direct product decomposition scheme of Ref. 28 is used for both ground- state and FSMRCC calculations. This leads formally to a computational savings which scales with the square of the order of the molecular point group, although our imple- mentation is currently limited to Abelian groups. Finally, we remark that all four-index quantities required for (0,l) sector calculations contain at most two virtual indices. Consequently, these calculations do not require a signifi- cant amount of core memory, and full in-core algorithms for all products can almost always be used.
In Table I, we present timings and some computational details for FSMRCCSD calculations carried out for a closed-shell ground state (the N2 molecule) and an open- shell ground state (the 0, molecule). In both cases, the solution of the (0,l) sector equations requires a negligible amount of computer time [1.6% and 2.5% of the total central processing unit (CPU) time for N, and O,, respec- tively]. Furthermore, little difficulty is encountered in con- verging the FSMRCCSD equations, as the number of iter- ations required to achieve convergence is similar to that for the ground-state calculation in both examples. In ACES II, convergence acceleration is provided by the reduced linear equation (RLE) method of Purvis and Bartlett,24 where elements of the error matrix are defined by
Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory 5563
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992 Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
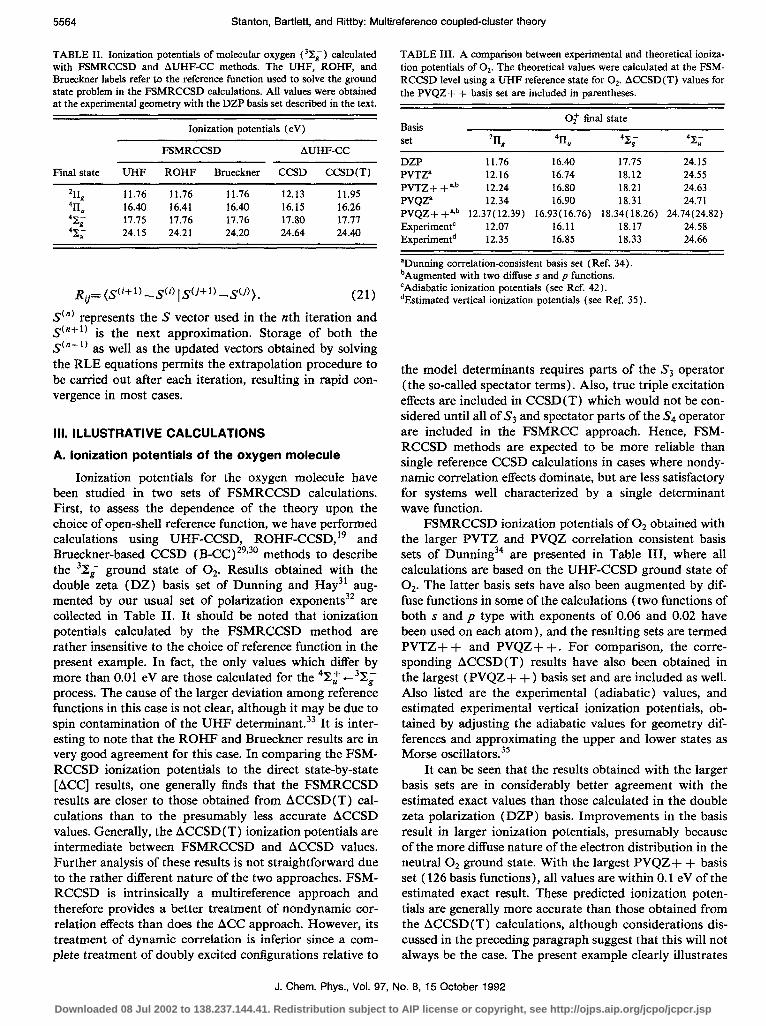
TABLE II. Ionization potentials of molecular oxygen (32,) calculated with FSMRCCSD and AUHF-CC methods. The UHF, ROHF, and Brueckner labels refer to the reference function used to solve the ground state problem in the FSMRCCSD calculations. All values were obtained at the experimental geometry with the DZP basis set described in the text.
Ionization potentials (eV)
Final state
% 4b ‘Xi 42;
FSMRCCSD AUHF-CC
UHF ROHF Brueckner CCSD CCSD(T)
11.76 11.76 11.76 12.13 11.95 16.40 16.41 16.40 16.15 16.26 Il.75 17.16 17.76 17.80 17.77 24.15 24.21 24.20 24.64 24.40
R,= (s(i+ 1) -s(i) Is(j+l)-s(i)). (21)
S(*) represents the S vector used in the nth iteration and SC”+‘) is the next approximation. Storage of both the ,!?+I) as well as the updated vectors obtained by solving the RLE equations permits the extrapolation procedure to be carried out after each iteration, resulting in rapid con- vergence in most cases.
Ill. ILLUSTRATIVE CALCULATIONS
A. Ionization potentials of the oxygen molecule
Ionization potentials for the oxygen molecule have been studied in two sets of FSMRCCSD calculations. First, to assess the dependence of the theory upon the choice of open-shell reference function, we have performed calculations using UHF-CCSD, ROHF-CCSD,19 and Brueckner-based CCSD (B-CC) 29v30 methods to describe the 3X;a ground state of Oz. Results obtained with the double zeta (DZ) basis set of Dunning and Hay31 aug- mented by our usual set of polarization exponents3* are collected in Table II. It should be noted that ionization potentials calculated by the FSMRCCSD method are rather insensitive to the choice of reference function in the present example. In fact, the only values which differ by more than 0.01 eV are those calculated for the 48,’ e38, process. The cause of the larger deviation among reference functions in this case is not clear, although it may be due to spin contamination of the UHF determinant.33 It is inter- esting to note that the ROHF and Brueckner results are in very good agreement for this case. In comparing the FSM- RCCSD ionization potentials to the direct state-by-state [ACC] results, one generally finds that the FSMRCCSD results are closer to those obtained from ACCSD (T) cal- culations than to the presumably less accurate ACCSD values. Generally, the ACCSD (T) ionization potentials are intermediate between FSMRCCSD and ACCSD values. Further analysis of these results is not straightforward due to the rather different nature of the two approaches. FSM- RCCSD is intrinsically a multireference approach and therefore provides a better treatment of nondynamic cor- relation effects than does the ACC approach. However, its treatment of dynamic correlation is inferior since a com- plete treatment of doubly excited configurations relative to
TABLE III. A comparison between experimental and theoretical ioniza- tion potentials of 0,. The theoretical values were calculated at the FSM- RCCSD level using a UHF reference state for 0s. ACCSD(T) values for the PVQZ+ + basis set are included in parentheses.
Basis set 2ns
0: final state
4K 42,
DZP 11.76 16.40 17.75 24.15 PVTZ* 12.16 16.74 18.12 24.55 pVTz+ +ab 12.24 16.80 18.21 24.63 PVQZ’ 12.34 16.90 18.31 24.71 PVQZ+ +4b 12.37( 12.39) 16.93( 16.76) 18.34( 18.26) 24.74(24.82) ExperimentC 12.07 16.11 18.17 24.58 Experimentd 12.35 16.85 18.33 24.66
5564 Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory
‘Dunning correlation-consistent basis set (Ref. 34). bAugmented with two diffuse s and p functions. ‘Adiabatic ionization potentials (see Ref. 42). dEstimated vertical ionization potentials (see Ref. 35).
the model determinants requires parts of the S3 operator (the so-called spectator terms). Also, true triple excitation effects are included in CCSD(T) which would not be con- sidered until all of S3 and spectator parts of the S4 operator are included in the FSMRCC approach. Hence, FSM- RCCSD methods are expected to be more reliable than single reference CCSD calculations in cases where nondy- namic correlation effects dominate, but are less satisfactory for systems well characterized by a single determinant wave function.
FSMRCCSD ionization potentials of O2 obtained with the larger PVTZ and PVQZ correlation consistent basis sets of Dunning34 are presented in Table III, where all calculations are based on the UHF-CCSD ground state of 0,. The latter basis sets have also been augmented by dif- fuse functions in some of the calculations (two functions of both s and p type with exponents of 0.06 and 0.02 have been used on each atom), and the resulting sets are termed PVTZ + + and PVQZ+ +. For comparison, the corre- sponding ACCSD(T) results have also been obtained in the largest (PVQZ f + ) basis set and are included as well. Also listed are the experimental (adiabatic) values, and estimated experimental vertical ionization potentials, ob- tained by adjusting the adiabatic values for geometry dif- ferences and approximating the upper and lower states as Morse oscillators.35
It can be seen that the results obtained with the larger basis sets are in considerably better agreement with the estimated exact values than those calculated in the double zeta polarization (DZP) basis. Improvements in the basis result in larger ionization potentials, presumably because of the more diffuse nature of the electron distribution in the neutral O2 ground state. With the largest PVQZ+ + basis set ( 126 basis functions), all values are within 0.1 eV of the estimated exact result. These predicted ionization poten- tials are generally more accurate than those obtained from the ACCSD( T) calculations, although considerations dis- cussed in the preceding paragraph suggest that this will not always be the case. The present example clearly illustrates
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992
Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

the suitability of the FSMRCCSD method for calculating ionization potential of open-shell molecules.
B. Application to the 3A2 state of COP
One class of problems which is particularly appropri- ate for FSMRCC theory involves cases where two different Hartree-Fock configurations lie close together at a sym- metrical geometry, but are prevented from interacting be- cause of symmetry. Certain modes of distortion lower the symmetry to a point group in which the two quasidegen- erate configurations can interact. This typically leads to symmetry breaking in Hartree-Fock approaches and pre- sents a delicate theoretical problem. Characterizing the curvature of the potential energy surface with respect to these displacements requires a treatment which is capable of describing the single-reference symmetrical and intrin- sically multireference unsymmetrical configurations in a reasonably balanced manner. It is not even straightforward to obtain a potential energy surface with the proper ana- lytic behavior, since any treatment of electron correlation (short of a full configuration interaction approach) based on a broken-symmetry Hartree-Fock reference state will lead to a potential energy surface with an unphysical cusp at the high symmetry point. To circumvent this difficulty, a number of techniques have been used including multiref- erence configuration interaction ( MRCI),36 CC calcula- tions based on QRHF37$38 and Brueckner3’ orbitals, and FSMRCC calculations.39 Limited experience has shown that all of these techniques can be applied successfully to these problems, although some treatment of triple excita- tions appears to be required when the QRHF-CC and B- CC approaches are used.40
In the following, we present the first application of open-shell reference state FSMRCC methods to such a problem. The 3,42 excited state of carbon dioxide was shown to suffer from severe symmetry breaking problems a decade ago.41 In this state of CO*, which is nonlinear, the electronic configurations
(core) 3u~2b~4uf3b~kz: lb;la24b;6ul
and
(core) 3u:2b$u:3b$: lb la24b26a 1 2 2 1
mix strongly in the C, region of the potential surface where the lb1 and la2 orbitals both transform as a”. This example is ideally suited to the open-shell reference FSMRCCSD method since the important configurations lie in the (0,l) sector with respect to the *A1 state of the CO, anion, which is well described by the determinant
2 2 2 2 2 2 2 2 (core)3u,2b24u,3b25a,1b,la24b26ul.
To study this problem, we have calculated the potential energy surface with respect to asymmetric (b2) distortion of the nuclear framework, using the optimized C2, struc- ture of Englebrecht and Liu4i as the reference geometry and our standard DZP basis set.31732 The results of these calculations are displayed graphically in Fig. 1, where the FSMRCCSD potential curve is compared with those from
,,n’ CO, (34) b, stretching potential
-2.m -1.54 -1.00 -0M 0.00 0.93 LW l.F.0 zmlo-’ AR (4
FIG. 1. Asymmetric (9) stretching potentials for the ‘A, excited state of carbon dioxide calculated with QRHF-CC and FSMRCCSD ap- proaches. All calculations were performed with the DZP basis set dis- cussed in the text at the minimum energy geometry of Ref. 41. The QRHF-CC calculations used the RHF orbitals of CO:-, while the FSM- RCCSD results were based on the UHF-CCSD solution for CO;.
CCSD and CCSD( T) calculations based on a QRHF ref- erence function constructed from the CO:- RHF orbitals. Although the QRHF-CCSD method predicts a broken- symmetry equilibrium geometry for the 3A2 state of CO,, both the theoretically more complete QRHF-CCSD (T) method and the FSMRCCSD calculations predict a sym- metrical structure. The latter results agree qualitatively with those of Englebrecht and Liu,41 who showed that all improvements in theory tended to favor the symmetric form. Their most accurate MRCI results predicted a po- tential similar in curvature to those obtained in the QRHF- CCSD (T) and FSMRCCSD calculations presented here. The close correspondence between the QRHF-CCSD (T) and FSMRCCSD results is encouraging, as it suggests that much of the improvement introduced by including the T3 operator in the QRHF-CC approach is already present in the more economical FSMRCCSD method. This pattern is similar to that observed in studies of the ground *Ai po- tential surface of the NO3 radica1.30137*39 The present results clearly demonstrate the utility of the FSMRCC approach for the study of systems which have significant nondynam- ical electron correlation effects and further illustrate that these calculations need not be based on closed-shell ground states.
IV. CONCLUSIONS
When applicable, FSMRCC offers one of the most powerful and computationally efficient approaches for the study of open-shell molecules, particularly when nondy- namic correlation effects are important. The principal ad- vantage of the method lies in its ability to provide a bal- anced description of several electronic configurations, thereby overcoming the bias of single-reference techniques towards a particular determinant. While FSMRCC calcu- lations based on a closed-shell ground state have the addi- tional advantage of providing spin-adapted solutions, the results presented in this paper suggest that use of a high- spin open-shell ground state does not compromise the
Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory 6565
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992 Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

5566 Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory
quality of the results. This is an important point because use of open-shell reference functions significantly expands the practical range of application of these methods. In the past, FSMRCC studies have been limited to systems which differ from a closed-shell ground state by at most two elec- tron processes. Such calculations are restricted to the study of doublet states via the rank one sectors of Fock space, and singlet and triplet states via the ( 1,l ), (0,2) and (2,O) sectors. However, the use of high-spin open-shell ground states has no such limitation, as illustrated by our study of the 3A2 excited state of CO, and quartet states of 0, using only the (0,l) sector. While it is true that any state of a molecule can be treated by FSMRCC methods in some sector of Fock space, all methods limited to the S=S, +Sz approximation become intrinsically less accurate as the rank of the sector is increased, since some single and dou- ble excitation effects relative to the model space determi- nants are carried by the S3 and higher-order spectator terms. Another consideration is that the orbitals used in these calculations are not particularly appropriate for a state far removed from the ground state. Hence, it is not clear that results obtained for a triplet state via a two- electron process from a closed-shell ground state will be generally superior to those obtained from (0,l) or ( l,O> sector calculations using a doublet ground state, even though the latter solutions are not spin adapted. Addi- tional experience with the open-shell reference state FSM- RCCSD method must be acquired before such questions can be addressed.
In this paper, we have presented the first application of FSMRCC methods to problems based on an open-shell ground state, and have demonstrated that the FSMRCC approach provides reliable results in these cases. In addi- tion, we have attempted to provide a clear description of the method as it applies to the calculation of ionization potentials and the study of final state potential energy sur- faces, describing in some detail the derivation of the FSM- RCC equations as well as many of the computational as- pects of these calculations. Finally, the applicability of FSMRCC methods to chemical problems has been dis- cussed, emphasizing the suitability of these techniques for studying problems which involve strong nondynamical correlation effects. It is our hope that the availability of the FSMRCC method in the ACES II program system will lead to increased application of these approaches to the solution of chemical problems.
Phys. 80,5058 (1984); (d) A. Haque and D. Mukherjee, Pramana 23, 651 (1984); (e) D. Sinha, S. Mukhopdhyay, and D. Mukherjee, Chem. Phys. Lett. 129,369 (1986); (f) D. Sinha, S. K. Mukhopadhyay, M. D. Prasad, and D. Mukherjee ibid. 125,213 (1986); (g) R. Chaudhuri, D. Sinha, and D. Mukherjee, ibid. 163, 165 (1989).
*(a) I. Lindgren, Int. J. Quantum Chem. Symp. 12, 33 ( 1978); (b) I. Lindgren, Phys. Ser. 32, 291 (1985); (c) 32, 611 (1985).
‘I. Lindgren and D. Mukherjee, Phys. Rep. 151, 93 (1987). 4(a) A. Haque and U. Kaldor, Chem. Phys. Lett. 117, 347 (1985); (b)
120, 261 (1985); (c) U. Kaldor and A. Haque, ibid. 128, 45 (1985); (c) U. Kaldor, J. Comput. Chem. 8, 448 (1987); J. Chem. Phys. 87, 467 (1987); Phys. Rev. A 38, 6013 (1988).
‘(a) L. 2. Stolarczyk and H. J. Monkhorst, Phys. Rev. A 32, 725 (1985); (b) 32,743 (1985); (c) 37, 1908 (1987); (d) 37, 1926 (1987).
‘(a) S. Pal, M. Rittby, and R. J. Bartlett, J. Chem. Phys. 88, 4357 (1987); (b) S. Pal, M. Rittby, R. J. Bartlett, D. Sinha, and D. Mukher- jee, Chem. Phys. Lett. 137, 273 (1987); (c) S. Pal, M. Rittby, and R. J. Bartlett, ibid. 150, 212 (1989); (d) M. Rittby, S. Pal, and R. J. Bartlett, ibid. 90, 3214 (1989); (e) J. D. Watts, M. Rittby, and R. J. Bartlett, J. Am. Chem. Sot. 111, 4155 (1989); (f) J. F. Stanton, M. Rittby, R. J. Bartlett, and D. W. Toohey, J. Phys. Chem. 95, 2107 (1991).
‘B. Jeziorski and J. Paldus, J. Chem. Phys. 90, 2714 (1989). 8D. Mukherjee and S. Pal, Adv. Quantum Chem. 20, 292 (1989). 9U. Kaldor, Theor. Chim. Acta 80,427 ( 1991). “D. Mukhopadhyay, S. Mukhopadhyay, R. Chaudhuri, and D. Mukher-
jee, Theor. Chim. Acta 80, 441 ( 1991). “C. M. L. Rittby and R. J. Bartlett, Theor. Chim. Acta 80, 469 (1991). ‘*M Barysz, H. J. Monkhorst, and L. Z. Stolarczyk, Theor. Chim. Acta
80; 483 (1991). 13See I. Lindgren and Morrison, Atomic Many-Body Theory (Springer,
Berlin, 1982), pp. 398-404 for a pedagogical discussion. 14Here we make a distinction between rigorous eigenfunctions of the S2
operator [S’I Y) =s(s+- 1) 1 Y)] and projected eigenfunctions, which sat- isfy the property (OlS2 1 Y) =s(s+ 1). Only the latter condition is sat- isfied by CC methods based on a spin-eigenfunction reference, such as ROHF and QRHF (see Ref. 19) determinants.
“J. S. Battle (unpublished research). 16R. J. Bartlett, Theor. Chim. Acta 80, 71 (1991). “For a review of CC methods which discuss a number of open-shell
applications, see R. J. Bartlett, J. Phys. Chem. 93, 1697 ( 1989). “R. J. Bartlett and G. D. Purvis, Int. J. Quantum Chem. 14,561 (1978);
J. A. Pople, R. Krishnan, H. B. Schlegel, and J. S. Binkley ibid. 15, 545 (1978) (CCD); G. D. Purvis and R. J. Bartlett, J. Chem. Phys. 76, 7918 (1982) (CCSD); J. D. Watts and R. J. Bartlett, ibid. 93, 6104 (1990) (CCSDT).
19MvI. Rittby and R. J. Bartlett, J. Phys. Chem. 92, 3033 (1988). ‘OFor reviews, see Refs. 7-l 1. 2’J. Gauss, J. F. Stanton, and R. J. Bartlett, J. Chem. Phys. 95, 2623
(1991).
ACKNOWLEDGMENTS
This work was supported by the Office of Naval Re- search. We would also like to thank the Ohio Supercom- puter Center for its continued support of the ACES II pro- gram system and providing the computer time used for the calculations presented here. In addition, discussions with R. P. Mattie of the Quantum Theory Project helped to shape aspects of this work.
’ (a) D. Mukherjee, R. K. Moitra, and A. Mukhopadhyay, Mol. Phys. 33,955 (1977); (b) A. Mukhopadhyay, R. K. Moitra, and D. Mukher- jee, J. Phys. B 12, 1 (1979); (c) A. Haque and D. Mukherjee, J. Chern.
22See, e.g., R. J. Bartlett, Annu. Rev. Phys. Chem. 32, 359 ( 1981). 23 For the (0,l) sector discussed here, only one of the three-body terms of
E,v contributes to the equations [the penultimate term of Eq. (20)]. Computationally, the strategy used to evaluatethis contribution diiers from the method of contracting elements of HN with the s amplitudes used for all other terms. Instead, the s and t amplitude terms are com- bined and the result is contracted with the integrals. Although this approach requires simultaneous storage of the t and s amplitudes, this does not represent any sort of limitation. An exactly analogous strategy is used in CC gradient calculations via the .Y? intermediates [see Eqs. (25) and (26) of Ref. 211.
“G. D. Purvis and R. J. Bartlett, J. Chem. Phys. 75, 1284 (1981). “L. Meissner and R. J. Bartlett, J. Chem. Phys. 94, 6670 (1989). 26 R. P. Mattie, J. F. Stanton, and R. J. Bartlett (to be published). 27AC~ II, an ab initio program system, authored by J. F. Stanton, J.
Gauss, J. D. Watts, W. J. Lauderdale, and R. J. Bartlett, Quantum Theory Project, University of Florida, 1991. ACES II also contains mod- ified versions of the MOLECULE Gaussian integral program (written by J. Almlof and P. R. Taylor) and the ABACUS integral derivative pro- gram (written by T. Helgaker, H. J. A. Jensen, P. R. Taylor, and P. Jorgensen).
**J. F. Stanton, J. Gauss, and R. J. Bartlett, J. Chem. Phys. 94, 4334 (1991).
29R. A. Chiles and C. E. Dykstra, J. Chem. Phys. 74,4544 (1981); L. Z.
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992
Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

Stanton, Bartlett, and Rittby: Multireference coupled-cluster theory 5567
Stolarczyk and H. J. Monkhorst, Int. J. Quantum Chem. Symp. 18, 267 (1984); N. C. Handy, J. A. Pople, M. Head-Gordon, and G. W. Trucks, Chem. Phys. Lett. 164, 185 (1989).
MJ. F. Stanton, J. Gauss, and R. J. Bartlett, J. Chem. Phys. 97, 5554 (1992).
“T. H. Dunning, J. Chem. Phys. 58, 2823 (1970). “L. T. Redmon, G. D. Purvis, and R. J. Bartlett, I. Am. Chem. Sot. 101,
2856 (1979). “The UHF spin multiplicity is 3.029 ((S’) =2.044), while values from
the projected CCSD multiplicity (calculated from (0 1 S* (Y,&) are 3.001 and 2.002, respectively. The theory for calculating spin multiplic- ities at the CC level is presented in G. D. Purvis, H. Sekino, and R. J. Bartlett, Collect. Czech. Chem. Commun. 53, 2205 (1988).
“T. H. Dunning, J. Chem. Phys. 90, 1007 (1989). 3J Parameters for the Morse potentials were obtained from the tables of K.
P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules (Van Nostrand-Reinhold, New York, 1979).
36See, e.g., C. F. Jackels and E. R. Davidson, J. Chem. Phys. 65, 2941 (1976); E. R. Davidson and W. D. Borden, J. Phys. Chem. 87, 4783 (1983); A. D. McLean, B. H. Lengsfeld, J. Pacansky, and Y. Ellinger,
J. Chem. Phys. 83, 3567 (1985); and references therein. 37J F. Stanton, J. Gauss, and R. J. Bartlett, J. Chem. Phys. 94, 4084
(1991). ‘8J. D. Watts, J. F. Stanton, J. Gauss, and R. J. Bartlett, J. Chem. Phys.
94, 4320 (1991). 39U. Kaldor, Chem. Phys. Lett. 185, 131 (1991). 40This point is made because of a trend noted in the study of these
problems in which the QRHF-CCSD and B-CCD methods tend to favor the asymmetrical structures, while QRHF-CCSD(T) and B- CCD(T) follow the behavior of FSMRCCSD calculations in favoring the symmetrical forms. However, the suitability of the (T) energy ap- proximation for Brueckner and QRHF references has not been studied in detail, and it is therefore not clear how well these results would agree with more rigorous QRHF-CCSDT and B-CCDT calculations.
41 L. Englebrecht and B. Liu, J. Chem. Phys. 78, 3907 (1983). 42The ionization potentials were taken from the following sources: R. G.
Tonkyn, J. W. Winniczek, and M. G. White, J. Chem. Phys. 91, 6632 (1989) (‘II,); E. Nishitani, I. Tanaka, K. Tanaka, T. Kato, and I. Koyano, ibid. 81, 3429 (1984) (4n,); R. M. Holmes and G. V. Marr, J. Phys. B 13, 945 (1980) (4X:,). The value for the ‘Z; state was calculated from the tables of Ref. 35.
J. Chem. Phys., Vol. 97, No. 8, 15 October 1992 Downloaded 08 Jul 2002 to 138.237.144.41. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
Copyright © 2022 FDOKUMEN




















