
Polycyclic aromatic hydrocarbons in coal combustion flue gas under electron beam irradiation

This article was downloaded by:[Reza, Joel]On: 23 August 2007Access Details: [subscription number 781496450]Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Polycyclic Aromatic CompoundsPublication details, including instructions for authors and subscription information:http://www.informaworld.com/smpp/title~content=t713647615
EXTRACTION OF POLYCYCLIC AROMATICHYDROCARBONS FROM SOIL USING WATERUNDER SUBCRITICAL CONDITIONS
Online Publication Date: 01 August 2007To cite this Article: Moreno, Erick, Reza, Joel and Trejo, Arturo (2007)'EXTRACTION OF POLYCYCLIC AROMATIC HYDROCARBONS FROM SOILUSING WATER UNDER SUBCRITICAL CONDITIONS', Polycyclic AromaticCompounds, 27:4, 239 - 260To link to this article: DOI: 10.1080/10406630701462916URL: http://dx.doi.org/10.1080/10406630701462916
PLEASE SCROLL DOWN FOR ARTICLE
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article maybe used for research, teaching and private study purposes. Any substantial or systematic reproduction,re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expresslyforbidden.
The publisher does not give any warranty express or implied or make any representation that the contents will becomplete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should beindependently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings,demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with orarising out of the use of this material.
© Taylor and Francis 2007

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Polycyclic Aromatic Compounds, 27: 239–260, 2007
Copyright c© 2007 Taylor & Francis Group, LLC
ISSN: 1040-6638 print / 1563-5333 online
DOI: 10.1080/10406630701462916
EXTRACTION OF POLYCYCLIC AROMATICHYDROCARBONS FROM SOIL USING WATERUNDER SUBCRITICAL CONDITIONS
Erick MorenoJoel RezaArturo TrejoInstituto Mexicano del Petroleo, Programa de IngenierıaMolecular, Area de Investigacion en Termofısica, MexicoD. F., Mexico
Water under subcritical conditions (250◦C, 4100 kPa) wasused to extract polycyclic aromatic hydrocarbons (PAHs)from a Mexican hydrocarbon-contaminated soil. Extractionwas carried out by continuously circulating the solventthrough the sample contained in an extraction cell. Afterextraction, the non-extracted PAHs were eluted from the soiland then analyzed by high performance liquidchromatography. The elution and subsequent analysis werecarried out by directly coupling the extraction cell, througha switching valve system, to a liquid chromatograph. Theelution and analysis process takes place in a closed systemthat allows a fast, precise, and highly sensitivequantification of the studied compounds. Additionalextraction experiments with spiked samples (silicon dioxideand nonpolluted soil) were carried out in order to asses theperformance of the method. Extraction recoveries rangedfrom 95 to 100% with an average uncertainty of ±1.2%.
Received 18 March 2007; accepted 17 May 2007.This work was supported by Instituto Mexicano del Petroleo under Research Projects D.00406
and I.00348. E. M. thanks the Consejo Nacional de Ciencia y Tecnologıa (CONACyT-Mexico) fora research assistantship. The authors thank Mr. M. A. Avila-Chavez for carrying out the Soxhletextractions and for providing the soil samples.
Address correspondence to Joel Reza or Arturo Trejo. Instituto Mexicano del Petroleo, Pro-grama de Ingenierıa Molecular, Area de Investigacion en Termofısica, Eje Central Lazaro CardenasNorte 152. 07730. Mexico D. F., Mexico. E-mail: [email protected] (J. Reza); [email protected](A. Trejo).
239

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
240 E. Moreno et al.
In some cases it was possible to quantitatively extract PAHsat concentrations as high as 8522 mg·kg−1 .
Keywords Anthracene, 9,10-dihydroanthracene, silicon dioxide, soil,subcritical water extraction
INTRODUCTION
Subcritical water can be considered as water at pressure (P) and tem-perature (t) conditions below its gas-liquid critical point (P= 22.064MPa, t = 373.99◦C) (1). More specifically it is water heated at temper-atures between 100 and 373◦C and at a pressure high enough to keep itin the liquid state. Under these conditions the polarity (as measured bythe dielectric constant, ε), density, viscosity, and surface tension of wa-ter can be dramatically lowered by simply raising its temperature (2–4),thus yielding a solvent whose physicochemical properties can be widelyand easily adjustable as a function of temperature.
As a result of the change in the properties of water, as a functionof temperature, subcritical water has become a subject of growing sci-entific interest because of its potential application in processes such asthe extraction and degradation of pollutants (5–14), as a medium forthe development of chemical reactions (15–18), and as an eluant forchromatographic separations at elevated temperatures (19–22).
The extraction of polycyclic aromatic compounds (PAHs) in soil andother solid matrixes using water under subcritical conditions has beenextensively studied and improved over the years (14, 23–28). As a re-sult, it has been established that PAHs can be quantitatively extractedfrom highly contaminated soils at relatively mild conditions (250◦C,5 MPa, water flow rate ranging from 0.1 to 1.1 cm3·min−1, extractiontime 15 min) (23). At 250◦C the extraction efficiencies have very lit-tle dependence on pressure. On the other hand, additional increases intemperature do not increase the PAHs recoveries significantly (23).
It has been reported that it is possible to achieve quantitative extrac-tions of PAHs from soil at temperatures as low as 150◦C, especiallyin the presence of surfactants that increase the extractability of thesecompounds (5 MPa, 10 to 15 cm3, 3.0 cm3·min−1; surfactant concen-tration (sodium dodecyl sulfate): 0.05 mol·L−1) (25). It is important totake into account that as the molecular mass of the PAHs increases, theirextraction rates at lower temperatures decrease dramatically (23). Thus,in some cases it is recommended to use a higher temperature (∼275◦C)to improve the PAHs removal, especially of those hydrocarbons with amolecular mass of 252 and 276 (14).

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 241
Once the extraction is completed, the extracted solid phase and theextraction solvent are analyzed in order to determine the efficiency ofthe extraction process. The extracted solid phase usually requires a com-plementary extraction step (Soxhlet or sonication) prior to its quanti-tative analysis. In the same way, the analysis of the solvent containingthe extracted compounds commonly involves an additional treatmentand preparation of the sample. This treatment frequently involves a sol-vent trapping process, through which the water used in the extractionis percolated through an organic solvent, allowing the extracted com-pounds to partition into the solvent. Unfortunately, solvent trapping hassome intrinsic disadvantages. For example, for some analytes quantita-tive transference from the water to the organic solvent does not occurwhen this simple percolation technique is used (23). In addition, the or-ganic solvent could be evaporated during the process. These two factorsmay affect the experimental results by lowering the obtained recoveries(23). On the other hand, the sensitivity of the analysis could also be lim-ited because only a small amount of sample is analyzed. To circumventthese inconveniences the use of a sorbent trapping process has been pro-posed, in which the analytes dissolved in the liquid phase are adsorbedinto a solid-phase trap (24, 28). The trap is then connected to a chro-matographic system and the solutes are desorbed and analyzed throughan appropriate mobile phase. The desorption and analysis take place ina closed system and usually no sample pretreatment is required. Sincethe total mass of the collected solutes can be analyzed, the sensitivityof the analysis method is higher than that related to the solvent trappingtechnique (24).
In this work we present the results obtained after extracting a hy-drocarbon-contaminated soil using water under subcritical conditions.The primary aim of the work was to apply an experimental apparatusand method for extracting (using water under subcritical conditions) low-polarity organic pollutants occurring in hydrocarbon-contaminated Mex-ican soils. This work is part of a research program developed in our labo-ratory focused on the application of efficient extraction processes for thetreatment of highly polluted solid matrixes related to the oil industry (29).
The experimental apparatus presented was custom-built in our labo-ratory taking into account the operational characteristics as well as theimprovements and advantages of several devices previously describedin the literature (23–25, 28, 30, 31). Additionally, we included some im-provements in the analysis process that we applied during the study ofthe water solubility of PAHs as a function of temperature (32, 33).
The studied hydrocarbon-contaminated soil was collected at a hydro-carbons distribution facility (gasoline and other similar light distillates),

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
242 E. Moreno et al.
similar to many other distribution facilities throughout our country. Wewere interested in identifying and quantifying the 16 PAHs consid-ered priority pollutants by the United States Environmental ProtectionAgency (EPA). To determine the repeatability of the experimental resultscomplementary extractions were carried out on spiked solid matrixes(silicon dioxide and nonpolluted soil).
EXPERIMENTAL
Chemicals
The studied PAHs, anthracene and 9,10-dihydroanthracene, were ob-tained from Chem Service (West Chester, PA) with stated purities of99.0% and 98%, respectively, and were used without further purifica-tion. Acetonitrile (HPLC-grade) was obtained from EM Science (GibbsTown, NJ). Water used in the experiments was type 1 reagent froman Ultrapure water system, model Easypure RF (Barnstead Thermo-line, Dubuque, IA). Other reagents used in this work were suppliedby Aldrich (Milwaukee, WI) with the following stated purity; toluene(PRA grade), 99.8%; hexane (for GC), ≥99.0%; methanol (PRA grade),≥99.9%; sodium sulfate (anhydrous), ≥99.0%; and silicon dioxide, 99%(50–70 mesh).
Solid Matrixes and Sample Preparation
Two samples, a nonpolluted soil and a historically hydrocarbon-contaminated soil, were used for the extraction experiments. The non-polluted soil was sampled at the City of Tuxpan, Veracruz (Mexico). Thehydrocarbon-contaminated soil was collected at a hydrocarbons distri-bution facility located 5 km from the City of Tuxpan, Veracruz (Mexico).Both soil samples were dried, sieved to a size smaller than 1 mm, andmechanically mixed to ensure homogeneity. Table 1 gives some detailsof the features of the two soil samples.
TABLE 1. Features of the studied soil samples
mass%
Soil Sand Silt Clay Organic content pHa
Nonpolluted 88.9 8.0 3.1 3.01 7.9Hydrocarbon-contaminated 94.9 2.0 3.1 9.14 4.8
a1:2 soil:water (w/v).

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 243
To prepare the spiked samples, each solid matrix was placed directlyinto the extraction cell. This operation was carried out keeping closedthe lower end of the cell, while the upper end was kept open. After care-fully weighing the amount of the loaded matrix, the spiking compoundswere dissolved in acetonitrile and then were added to the solid ma-trix as a solution by a plunger-operated pipette (Transferpette c©, model704 174). The solutions of PAHs in acetonitrile were prepared with sucha concentration that in all cases the volume of the solution used for spik-ing the solid matrix was lower than 150 μl. By using this reduced volumeof solution any leak of sample through the lower end of the extractioncell was avoided. Once the addition of the solution was completed theextraction cell was kept open for about 1 h within a fumes hood in orderto allow the evaporation of acetonitrile. After this time, the upper end ofthe cell was closed, and the extraction cell was ready to be adapted intothe extraction apparatus.
Jimenez-Carmona and Luque de Castro (34) reported that the use ofa spiking procedure similar to the one described above gives the sameresults as those obtained through the application of the more traditionalprocedure in which a given mass of the solid matrix is mixed with asolution (in an appropriate and volatile solvent) of the analyte understudy and the solvent is subsequently evaporated to dryness in a rotaryevaporator. The spiking procedure used in this work is simpler and fasterthan the one that involves the use of a rotary evaporator. In addition, itreduces the steps related to the handling of harmful substances such asthe PAHs.
Experimental Apparatus and Procedure
The extraction apparatus used in this work was custom built in ourlaboratory. A schematic diagram of the apparatus is presented in Figure 1.
The main component of the extraction system was the extraction cell.This cell was constructed in stainless steel (2.0 × 0.2 cm i.d.) and fittedwith 2.0-μm fritted disks (stainless steel) and zero dead volume reducingunions at each end. This cell was packed with the solid matrixes to beextracted. The inlet of the loaded extraction cell was connected to aLabAlliance Series III isocratic pump (LabAlliance; State College, PA)through a small-bore stainless steel tubing; 1/16 in o.d. × 0.010 in i.d.(1.6 mm o.d. × 0.3 mm i.d.) and placed inside the oven of a Varian 3700gas chromatograph (GC).
The isocratic pump was used to provide pressurized water through thesolid matrix during the extraction experiments.

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
244 E. Moreno et al.
FIGURE 1. Subcritical water extraction apparatus. (1) Isocratic pump, (2)1.5 m-coil, (3) oven, (4) 5 m-preheating coil, (5) extraction cell, (6)thermometer, (7) cooling bath, (8) 1.5 m-cooling coil, and (9) outlet valve.
As shown in Figure 1, a 5 m-preheating coil (stainless steel; 1/16 ino.d. × 0.010 in i.d.) was adapted in the system previous to the extractioncell. This coil was adapted with the aim of ensuring that the water en-tering the extraction cell was adequately homogenized at the operatingtemperature. In addition, a 1.5 m-coil (stainless steel; 1/16 in o.d × 0.010in i.d.) was placed in-line after the pump in order to protect the pumpfrom the heat radiating from the oven and to prevent any possible solutemigration back to the pump.
Pressure in the system (read at the pump LED) was maintained byadjusting an outlet valve (Whitey, model SS-22RS4). To protect the outletvalve from the flow of water at high temperature leaving the oven, theaqueous phase was cooled by passing it through a 1.5-m cooling coil(stainless steel, 1/16 in o.d × 0.010 in i.d.) placed in a cooling bath(Haake, model A80) maintained at 4◦C.
The thermometer used to register the temperature during the extractionexperiments was a Systemteknik AB digital thermometer, model S1220(Lindigo, Sweden), adapted with a platinum resistance sensor, calibratedby comparison with a standard platinum resistance thermometer whose

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 245
calibration was traceable to the NIST (U.S.). The platinum resistancesensor was directly adapted to the external surface of the extractioncell.
To carry out an extraction experiment, a given mass (from 0.042 to0.095 g) of sample (either spiked or aged environmental matrixes) wasloaded and weighted into the extraction cell. After loading the sample,the extraction cell was connected to the extraction apparatus (Figure 1).Then, deionized water (previously purged for 3 h with nitrogen to re-move dissolved oxygen) was pumped through the extraction cell at asolvent flow rate of 0.2 cm3·min−1. The extraction cell was mountedvertically in the GC oven with water flowing from top to bottom. Thisarrangement was adopted to allow that the extracted compounds wereimmediately swept from the cell. The outlet valve was closed and thesystem was pressurized to 17.2 MPa to check for possible leaks of thesystem. When it was verified that no water leaks were present the pres-sure was reduced to around 6.9 MPa and the temperature was increasedat a rate of 20◦C·min−1, up to the desired extraction temperature. Atthe same time the pressure of the system was regulated and adjustedto the desired value (either 4.1 or 7.3 MPa) through the outlet valve.During the extraction process, the pump was set in a constant-flowrate mode. It has been reported that a temperature of 250◦C is ade-quate to achieve good recoveries in the extraction of PAHs (even high-molecular-mass PAHs) from several solid matrixes, including soil (23,27, 35). Thus, all experiments in this work were carried out at suchtemperature.
The cooled water flow leaving the cooling coil was collected andcarefully weighted at the end of the extraction process.
High-performance Liquid Chromatography Analysis
Once the extraction process concluded, the extraction cell was coupledto a liquid chromatograph by using a switching valve system, as shownin Figure 2.
The main components of the switching valves system were a 9725iRheodyne injector (I) (Rohnert Park, CA), two 7000 Rheodyne switchingvalves (A, B) (Rohnert Park, CA), and a constant volume loop (26.0 ±0.3 μl). A modified version of the switching valve system was previouslyused to quantify PAHs during the determination of their solubility inwater (32, 33).
The injector (I) was used for the injection of calibration solutions of thestudied compounds during the chromatographic calibration process. Theswitching valve (A), with the extraction cell at the position corresponding

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
246 E. Moreno et al.
FIGURE 2. Coupling of the extraction cell to the high-performance liquidchromatograph during the analytical step. (1) HPLC pump, (2) constantvolume loop, (3) extraction cell, (4) analytical column, (5) photo diode arraydetector, (6) fluorescence detector, and (7) data acquisition.
to the injection loop was installed after the injector. This valve al-lowed the on-line elution of the solute present in the extraction celltoward the analytical column. The switching valve (B) was incorporatedinto the system to avoid the unnecessary de-equilibration of the ana-lytical column during the cleaning process that followed the analysis ofsome dirty real samples. A 25.0 × 0.46 cm i.d. analytical column packedwith a bonded C18 stationary phase (Spherisorb S5 ODS-2; Waters As-sociates, Milford, MA) was used in all the analyses. All lines in the setupshown in Figure 2 were made of small-bore stainless steel tubing; 1/16in o.d. × 0.007 in (1.6 mm o.d. × 0.18 mm i.d.).
During the analysis step, the switching valve system was held as shownin Figure 2. In this arrangement, an appropriate mobile phase (usually amixture of acetonitrile and water, in a volume ratio of 70:30) entered theinjector through port 2 and exited the injector through port 3.
Then, the mobile phase provided by a Waters pump (solvent deliverysystem, model 626, adapted with a 600S controller) reached valve Athrough port 2 and was routed through the extraction cell in order toexhaustively elute all the adsorbed compounds. The mobile phase leftvalve A through port 3 and then was directed toward valve B. Afterthe mobile phase entered the valve B (through port 2), flowed towardthe analytical column through port 1, and then to the detectors. After

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 247
separation, the desorbed compounds were simultaneously detected by aphotodiode array detector, PDA (model 996, Waters, Milford, MA), anda fluorescence detector (model 474; waters, Milford, MA). Detectionof PAHs in the PDA detector was carried out at 254 nm. For detectinganthracene and 9,10-dihydroanthracene in the fluorescence detector, theexcitation and emission wavelengths (λexcitation, λemission) were set at 260nm and 380 nm, and at 250 nm and 280 nm, respectively.
All analyses were carried out isocratically with a 70:30 (v/v) acetoni-trile:water mixture.
In order to corroborate the exhaustive desorption of the remainingcompounds from the treated solid matrixes (after subcritical water ex-traction) the following experiment was carried out. Once the desorption-analysis process was completed the process was repeated; that is, themobile phase was flowed once again through the solid matrix. In allcases, and for all the studied solid matrixes, no PAHs were detected afterthis second desorption process.
The quantification of the PAHs in the studied solid matrixes was car-ried out by an external 6 to 7 point calibration. Calibration solutions ofknown concentration of each PAH (dissolved in acetonitrile) were pre-pared gravimetrically by dilution from a more concentrated (standard)solution of the PAH, also prepared in acetonitrile. In all the calibrationexperiments only fresh solutions were used to avoid errors caused by theadsorption of PAHs onto the glassware surfaces.
The standard deviation (Sx0 ) of a concentration, x0, calculated fromthe generated calibration curve was estimated according to the methodreported by Miller and Miller (36). The confidence limits for each deter-mined concentration was calculated as x0 ± tα/2(n−2)Sx0 where tα/2(n−2)is the t of Student with (n −2) degrees of freedom and a confidence level(α) of 95%. In addition, n represents the number of data points that wasused to generate the calibration curve.
Limits of detection and quantification were determined by serial dilu-tion and analysis of the standard solution of each PAH. The results fromthese analyses were then used to generate calibration curves in a diluteand narrow concentration region. The limits of detection and quantifi-cation were calculated from the statistical parameters derived from thecalibration curves according with the approach reported by Miller andMiller (36).
During the chromatographic calibration each calibration solution waseluted through the extraction cell packed with silicon dioxide in orderto take into account the possible band broadening effects occurring dur-ing the transfer of the extracted compounds from the extraction to theanalytical column.

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
248 E. Moreno et al.
The efficiency of the extraction process was assessed by determiningthe fraction extracted, E A (37), of the studied compounds:
E A = Q A/Q′A (1)
where Q A is the mass of the solute A extracted and Q′A is the total mass
of A in the solid matrix at the beginning of the extraction. The fractionextracted is also known as the recovery factor, and may be expressed asa percentage, % EA (37).
To determine Q A and Q′A the extraction cell (containing the extracted
or the nonextracted sample) was coupled to the switching valves system(Figure 2), and then the compounds in the solid matrix were desorbed andanalyzed through an appropriate mobile phase. The use of the switch-ing valves system eliminates the need for a liquid-liquid post extractionstep. The determination of Q A and Q′
A values for each extraction ex-periment was carried out by triplicate. In some cases, the analysis of theextracted spiked matrixes produced a chromatogram in which there wasno evidence of the presence of the studied PAHs. In these cases Q A wascalculated taking into account the limit of quantification of the studiedcompound.
Uncertainties in Q A and Q′A generated as a result of the uncertainties
in the chromatographic analysis and the other operational steps involvedin the experimental determinations were adequately considered to deter-mine the uncertainty of E A.
The above described desorption-analysis system has several intrin-sic advantages, for example, since the desorption and analysis processtakes place in a closed system, it avoids the external handling of thesolid sample, thus minimizing the possibility of solute losses (e.g. due toadsorptive phenomena) or the sample contamination by external impuri-ties. The system also minimizes the contact of the operator with potentialharmful compounds. In addition, since the total mass of the solutes in thesolid matrix can be analyzed the sensitivity of the analysis is enhanced.Through the use of the described system and the application of appro-priate statistical criteria (36), the uncertainty of the fraction extracted inthe experiments was reduced to a relatively low interval ranging from±0.9% to ±1.3%.
Other Analyses
To determine the PAHs that were present in the studied soil samplesboth the nonpolluted soil and the hydrocarbon-contaminated soil were

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 249
extracted according to the EPA method 3540 C (triplicate experiments,16-h Soxhlet extraction, with a 1:6 (v/v) toluene:methanol mixture) (38).The obtained extracts were then analyzed by high-performance liquidchromatography (UV and fluorescence detection). It must be pointedout that the extraction process was carried out with the toluene:methanolmixture because it has been established that this mixture is one of the bestsolvents for extracting PAHs (Soxhlet extraction) from solid matrixes ascomplex as soot and sediment (39).
In addition, the hexane extractable material, HEM, of the hydrocarbon-contaminated soil sample was also determined using EPA method 9071B(40).
RESULTS AND DISCUSSION
After Soxhlet extraction and HPLC analysis no PAHs were detectedin the nonpolluted soil. On the other hand, anthracene was the onlyPAH (from the EPA Priority Pollutant PAHs list) that could be identified(based on both UV spectra analyses and injection of pure polycyclicaromatic compound standards) in the hydrocarbon-contaminated soil.Anthracene was detected at a concentration of (28.1 ± 7.2) mg·kg−1.Thus, to determine the repeatability of the experimental results comple-mentary extractions were carried out on solid matrixes (silicon dioxideand nonpolluted soil) spiked with anthracene. Even though the deter-mined concentration of anthracene in the hydrocarbon-contaminatedsoil was low we were also interested in studying the extraction pro-cess of higher concentrations of PAHs from the solid matrixes. Thisinterest was the result of the fact that only few works reported in liter-ature are focused on studying the extraction of relatively high concen-trations of PAHs. Hawthorne et al. (26) reported the extraction of PAHsfrom soil (subcritical water at 250◦C and 5 MPa) with a total concentra-tion of 6936 mg·kg−1. Thus, we were interested in studying the extrac-tion of PAHs at concentrations higher than 7000 mg·kg−1. We tried tospike a nonpolluted soil sample with a high concentration of anthracene(>7000 mg·kg−1). Unfortunately, as a result of the limited solubilityof anthracene in acetonitrile (the solvent used to prepare the spikingsolutions) it was not possible to prepare such spiked sample. By consid-ering that 9,10-dihydroanthracene has a higher solubility in water thananthracene (32, 33) it was assumed that 9,10-dihydroanthracene wouldhave a higher solubility in acetonitrile than anthracene. In fact, using9,10-dihydroanthracene as the spiking compound it was possible to pre-pare spiked samples of non-polluted soil with a concentration of PAHas high as 8522 mg·kg−1. This was the reason for studying anthrace and

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
250 E. Moreno et al.
TABLE 2. Limit of detection and limit of quantification of PAHs usingfluorescence detection
Limit of λexcitation,PAH Limit of detection/g quantification/g λemission/nm
Anthracene 3.23 × 10−12 1.08 × 10−11 260, 3809,10-Dihydroanthracene 9.06 × 10−10 3.02 × 10−9 250, 280
9,10-dihydroanthrace (as well as some of their binary mixtures) in thespiking studies.
Limit of Detection and Limit of Quantification
Fluorescence detection is more sensitive than UV detection for theanalysis of several PAHs (41). In our study we found that the fluores-cence detector was more sensitive than the PDA detector for analyzinganthracene and 9,10-dihydroanthracene. Thus, the fluorescence detectorwas mainly used for quantification purposes. Table 2 shows the limit ofdetection and the limit of quantification of the two studied PAHs usingthe fluorescence detector. Nonetheless, the PDA detector was also usedin all the analyses.
Extraction of Spiked Samples
To evaluate the performance of the experimental apparatus and methodseveral samples of silicon dioxide and nonpolluted soil were spiked withanthracene, 9,10-dihydroanthracene, and some of their binary mixtures,and subsequently extracted with subcritical water. The results obtainedin the different extraction experiments are presented in Table 3.
Several experiments were carried out to determine the efficiency ofthe process for extracting the studied PAHs, spiked (as pure components)onto silicon dioxide. As observed in Table 3, both anthracene and 9,10-dihydroanthracene were quantitatively extracted from the solid phase,with water at 250◦C and 4140 kPa. An increase in the extraction pres-sure, up to 7300 kPa, did not have a significant effect on the extractedfraction of the considered PAHs. Furthermore, Hawthorne et al. (23) havedemonstrated that the water flow rate has little effect on the recoveriesof low molecular mass PAHs extracted with subcritical water. Thus, atemperature of 250◦C, a pressure of around 4140 kPa, and a water flow of0.2 cm3·min−1 were selected as the extraction conditions for all the restof the experiments. The extraction experiments with the PAHs spikedas pure compounds were repeated between three and seven times. As

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
TA
BL
E3
.E
xtra
ctio
nef
ficie
ncy
ofPA
Hs
from
spik
edsi
licon
diox
ide
and
nonp
ollu
ted
soil
usin
gw
ater
unde
rsu
bcri
tical
cond
ition
s
Silic
ondi
oxid
em
atri
xsp
iked
with
PAH
s
Ext
ract
ion
cond
ition
sA
nthr
acen
e9,
10-D
ihyd
roan
thra
cene
t/◦ C
250.
76±
0.02
250.
42±
0.02
250.
56±
0.02
250.
69±
0.02
P/k
Pa41
40±
9173
03±
161
4147
±93
7391
±18
3M
ass
ofso
lven
ta/g
18.5
507
±0.
0003
19.8
973
±0.
0003
19.0
079
±0.
0003
18.9
896
±0.
0003
Spik
edco
ncen
trat
ionb
/mg·k
g−1
47.7
4±
0.01
47.7
4±
0.01
——
——
3115
±2
3115
±2
Ext
ract
ion
effic
ienc
yof
PAH
s
EA
(ant
hrac
ene)
/%10
0.0
±0.
9c10
0.0
±1.
2c—
—E
A(9
10-d
ihyd
roan
thra
cene
)/%—
—10
0.0
±1.
0c10
0.0
±0.
9c
Silic
ondi
oxid
em
atri
xsp
iked
with
PAH
sm
ixtu
res
Ext
ract
ion
cond
ition
sM
ixtu
reA
dM
ixtu
reB
dM
ixtu
reC
d
t/◦ C
250.
04±
0.02
250.
27±
0.02
250.
87±
0.02
P/k
Pa41
03±
8741
33±
9141
25±
91M
ass
ofso
lven
ta/g
19.7
458
±0.
0002
18.7
642
±0.
0002
19.6
322
±0.
0001
Spik
edco
ncen
trat
ionb
/mg·k
g−1
Ant
hrac
ene
34.9
5±
0.03
2.40
6±
0.00
14.
477
±0.
003
9,10
-Dih
ydro
anth
race
ne76
7.0
±0.
12.
352
±0.
001
0.20
07±
0.00
02
(Con
tinue
don
next
page
)
251

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
TA
BL
E3
.E
xtra
ctio
nef
ficie
ncy
ofPA
Hs
from
spik
edsi
licon
diox
ide
and
nonp
ollu
ted
soil
usin
gw
ater
unde
rsu
bcri
tical
cond
ition
s(C
ontin
ued)
Ext
ract
ion
Effi
cien
cyof
PAH
s
EA
(ant
hrac
ene)
/%99
.8±
1.3e
99.7
±1.
3e99
.3±
1.3e
EA
(910
-dih
ydro
anth
race
ne)/%
100.
0±
1.2e
99.6
±1.
3e95
.1±
1.3e
Non
pollu
ted
soil
spik
edw
ithPA
Hs
Ext
ract
ion
cond
ition
s9,
10-D
ihyd
roan
thra
cene
Mix
ture
Dd
t/◦ C
250.
57±
0.02
250.
60±
0.02
P/k
Pa41
57±
109
4141
±97
Mas
sof
solv
enta
/g19
.888
3±
0.00
0220
.197
3±
0.00
02Sp
iked
conc
entr
atio
nb/m
g·kg−1
Ant
hrac
ene
—2.
67±
0.01
9,10
-Dih
ydro
anth
race
ne85
22±
4350
.7±
0.1
Ext
ract
ion
Effi
cien
cyof
PAH
s
EA
(ant
hrac
ene)
/%—
99.8
±1.
0e
EA
(910
-dih
ydro
anth
race
ne)/%
100.
0±
1.3e
100.
0±
1.2e
aM
ass
ofso
lven
tuse
din
the
extr
actio
nex
peri
men
t.bC
once
ntra
tion
ofth
ePA
Hs
spik
edin
the
solid
mat
rix
(mg
ofPA
Hpe
rkg
ofsi
licon
diox
ide
orpe
rkg
ofso
il).
c Ave
rage
ofse
ven
inde
pend
entd
eter
min
atio
ns.
dM
ixtu
res
A,B
,Can
dD
are
bina
rym
ixtu
res
ofan
thra
cene
+9,1
0-di
hydr
oant
hrac
ene;
the
mas
sfr
actio
nof
anth
race
nein
each
mix
ture
was
4.36
%(m
ixtu
reA
),50
.57%
(mix
ture
B),
91.7
5%(m
ixtu
reC
),an
d5.
27%
(mix
ture
D).
e Ave
rage
ofth
ree
inde
pend
entd
eter
min
atio
ns.I
nal
lthe
expe
rim
ents
the
aver
age
mas
sof
solid
mat
rix
load
edin
toth
eco
lum
nw
as0.
0950
g.
252

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 253
observed in Table 3, the uncertainty in the efficiency of the extractionprocess of the PAHs from the spiked samples ranged between 0.9 and1.3%.
When PAHs were spiked onto silicon dioxide as binary mixtures, theextracted fraction of each compound was also high. Table 3 shows thatthe extracted fraction of anthracene, spiked onto silicon dioxide (both aspure compound and in the binary mixture), ranged from 99.3 to 100.0%,with and average uncertainty of ±1.2%. For 9,10-dihydroanthrace theextracted fraction varied between 95.1 and 100.0% (average uncertaintyof ±1.2%). As observed in Table 3, high recovery factors were alsoobtained when PAHs were spiked onto the nonpolluted soil matrix, evenwhen the concentration of the spiked compounds was as high as 8522mg·kg−1.
By taking into account the results above described it was establishedthat both the extraction apparatus and experimental method worked ade-quately. Thus, they were employed to study the extraction of PAHs fromthe real world samples.
The use of spiked samples to determine extraction efficiencies ofhydrophobic compounds, from heterogeneous environmental solid ma-trixes, may not be a reliable mean for representing the extraction behaviorof environmentally aged matrixes (23, 42). This result derives from thefact that spiked and environmentally aged compounds may not be sit-uated on the same binding sites or exposed to the same physical andchemical interactions. In addition, since the spiking process usually in-volves the use of a solvent to spike the analyte onto the solid matrix,the solvent may affect the chemical integrity of the sample, thus ren-dering a sample that is not comparable with the sample containing theenvironmentally aged compounds (23, 42).
Thus, it must be pointed out again that the study performed in this workon spiked samples was carried out only with the purpose of evaluatingthe performance of the experimental apparatus and method.
Extraction of Real World Samples
The real world sample studied in this work was a hydrocarbon-contam-inated soil obtained at a hydrocarbons distribution facility. Its hexaneextractable material content (wet weight basis), as determined by EPAmethod 9071B, was 118450 ± 6740 mg·kg−1 (triplicate determination).
The hydrocarbon-contaminated soil was also Soxhlet extracted (16-h,1:6 (v/v) toluene:methanol mixture) (38), and the extract was analyzed byhigh-performance liquid chromatography (fluorescence detection) to de-tect and quantify the PAHs of interest. As previously stated anthracene

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
254 E. Moreno et al.
TABLE 4. Extraction conditions and fraction of anthracene extracted fromthe hydrocarbon-contaminated soil
Extraction Conditions
t /◦C 250.86 ± 0.02P/kPa 4207 ± 103Mass of the soil extracted/g 0.0498Mass of solvent/g 19.8883 ± 0.0002Solvent flow/cm3·min−1 0.2
Concentration of Components in the Soil Matrix
HEM (wet weight basis)a /mg·kg−1 118 450 ± 6740Anthraceneb/mg·kg−1 28.1 ± 7.2
Extraction efficiency
E A(anthracene)/% 100.0 ± 1.3
aHexane extractable material, as determined by EPA method 9071B (triplicate determination).bDetermined from HPLC analysis (fluorescence detection) after Soxhlet extraction (16-h, 1:6
(v/v) toluene:methanol mixture; average of triplicate determinations).
was the only priority pollutant PAH detected in the soil sample. It isimportant to mention that in addition to anthracene many other com-pounds were also detected in the extract; unfortunately their identitycould not be established.
The hydrocarbon-contaminated soil was extracted with subcriticalwater at the conditions presented in Table 4. As observed, the extractionof anthracene from the environmentally aged matrix was also quantitative(E A = 100.0 ± 1.3%).
The compounds contained in a sample of 0.0429 g of hydrocarbon-contaminated soil were desorbed employing a 70:30 (v/v) acetonitrile:water mixture. The desorption process was carried out passing the sol-vent mixture through the soil (contained in the extraction cell) at a flowrate of 1.0 cm3·min−1. The desorbed compounds were analyzed em-ploying fluorescence detection with the following program in the ex-citation and emission wavelengths: from 0 to 10 min λexcitation = 250nm, λemission = 280 nm; then, from 10 min to the end of the analysis(60 min) λexcitation = 260 nm and λemission = 380 nm. As a result ofthe large number and high concentration of desorbed compounds a sat-uration of the detector signal was observed (0 to 1000 AU). In order toavoid systematic errors in subsequent analyses, it was necessary to makea deep and exhaustive cleaning of the chromatographic system after theabove described desorption experiments.
Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 255
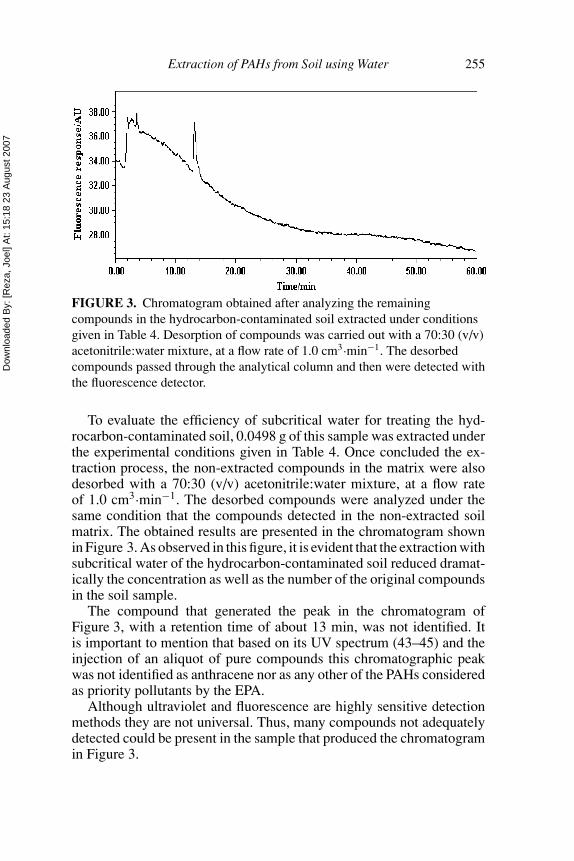
FIGURE 3. Chromatogram obtained after analyzing the remainingcompounds in the hydrocarbon-contaminated soil extracted under conditionsgiven in Table 4. Desorption of compounds was carried out with a 70:30 (v/v)acetonitrile:water mixture, at a flow rate of 1.0 cm3·min−1. The desorbedcompounds passed through the analytical column and then were detected withthe fluorescence detector.
To evaluate the efficiency of subcritical water for treating the hyd-rocarbon-contaminated soil, 0.0498 g of this sample was extracted underthe experimental conditions given in Table 4. Once concluded the ex-traction process, the non-extracted compounds in the matrix were alsodesorbed with a 70:30 (v/v) acetonitrile:water mixture, at a flow rateof 1.0 cm3·min−1. The desorbed compounds were analyzed under thesame condition that the compounds detected in the non-extracted soilmatrix. The obtained results are presented in the chromatogram shownin Figure 3. As observed in this figure, it is evident that the extraction withsubcritical water of the hydrocarbon-contaminated soil reduced dramat-ically the concentration as well as the number of the original compoundsin the soil sample.
The compound that generated the peak in the chromatogram ofFigure 3, with a retention time of about 13 min, was not identified. Itis important to mention that based on its UV spectrum (43–45) and theinjection of an aliquot of pure compounds this chromatographic peakwas not identified as anthracene nor as any other of the PAHs consideredas priority pollutants by the EPA.
Although ultraviolet and fluorescence are highly sensitive detectionmethods they are not universal. Thus, many compounds not adequatelydetected could be present in the sample that produced the chromatogramin Figure 3.

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
256 E. Moreno et al.
FIGURE 4. Desorption profile of the natural components contained in asample of 0.0421 g of the nonpolluted soil, with a 70:30 (v/v)acetonitrile:water mixture, at a flow rate of 1.0 cm3·min−1, and fluorescencedetection: (a) Original soil sample; (b) Soil sample extracted with water atsubcritical conditions (t = 250◦C, P = 4140 kPa).
A sample of the nonpolluted soil (0.0421 g) was also extracted withwater (t = 250.76 ± 0.02◦C, P = 4140 ± 91 kPa, solvent flow= 0.2 cm3·min−1, mass of solvent = 19.8987 ± 0.0002g; duplicated experiments).Chromatograms in Figure 4 present the results obtained in the analysisof the solid matrix before and after the extraction process. None of thecompounds that generated the peaks of chromatograms in Figure 4 wereidentified. However, as can be observed, the extraction process resultedin a large reduction in the number and concentration of many of thecomponents in the nonpolluted soil.

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 257
The selectivity of water under subcritical conditions for extractingnatural matrix components (especially for soils and sediments) is muchlower than that of solvents such as supercritical carbon dioxide (26).However, despite this apparent disadvantage some studies demonstratethat after treating a PAH-contaminated soil with subcritical water, thesoil could support plant growth after extraction (14).
CONCLUSIONS
Water under subcritical conditions was used for extracting organicpollutants from a hydrocarbon-contaminated Mexican soil. The deter-mination of the efficiency of the extraction process was carried out usingan analysis system that avoids the external handling of the solid matrixand allows minimizing the possibility of solute losses or sample contam-ination. In addition, the sensitivity of the analysis method is enhanced.Through the use of the method of analysis reported in this work theuncertainty of the fraction extracted was reduced to a relatively low in-terval ranging from ±0.9% to ±1.3%. The experimental apparatus andmethod presented are appropriate for quantitatively extracting PAHs atconcentrations as high as 8522 mg·kg−1.
REFERENCES
1. J. Kestin, J. V. Sengers, B. Kamgar-Parsi, and J. M. H. Levelt Sengers, Thermo-physical Properties of Fluid H2O, J. Phys. Chem. Ref. Data 13 (1984):175–183.
2. M. Uematsu and E. U. Franck, Static Dielectric Constant of Water and Steam,J. Phys. Chem. Ref. Data 9 (1980):1291–1305.
3. N. B. Vargaftik, B. N. Volkov, and L. D. Voljak, International Tables of the SurfaceTension of Water, J. Phys. Chem. Ref. Data 12 (1983):817–820.
4. J. V. Sengers and B. Kagmar-Parsi, Representative Equation for the Viscosity ofWater Substance, J. Phys. Chem. Ref. Data 13 (1984):185–205.
5. T. Gungoren, M. Saglam, M. Yuksel, H. Madenoglu, R. Isler, I. H. Metecan, A. R.Ozkan, and L. Ballice, Near-critical and Supercritical Fluid Extraction of IndustrialSewage Sludge, Ind. Eng. Chem. Res. 46 (2007):1051–1057.
6. Y. Yang and F. Hildebrand, Phenanthrene Degradation in Subcritical Water, Anal.Chim. Acta. 555 (2006):364–369.
7. A. A. Dadkhah and A. Akgerman, Hot Water Extraction with In Situ Wet Oxidation:Kinetics of PAHs Removal from Soil, J. Hazard. Mater. B. 137 (2006):518–526.
8. S. Hashimoto, K. Watanabe, K. Nose, and M. Morita, Remediation of Soil Contami-nated with Dioxins by Subcritical Water Extraction, Chemosphere 54 (2004):89–96.
9. S. B. Hawthorne and A. Kubatova, Hot (Subcritical) water Extraction. In Sam-pling and Sample Preparation for Field and Laboratory, J. Pawliszyn, Ed.: 2002,(Amsterdam: Elsevier). 587–608

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
258 E. Moreno et al.
10. A. Kubatova, A. J. M. Lagadec, and A. Hawthorne, Dechlorination of Lindane,Dieldrin, Tetrachloroethane, Trichloroethane and PVC in Subcritical Water, Envi-ron. Sci. Technol. 36 (2002):1337–1343.
11. J. Kronholm, S. Hunthala, H. Haario, and M. -L. Riekkola, Oxidation of 4-chloro-3-methylphenol in Pressurized Hot Water in Liquid and Vapor Phases, Adv. Environ.Res. 6 (2002):199–206.
12. L. Ramos, E. Kristenson, and U. A.Th. Brinkman, Current use of Pressurized LiquidExtraction and Subcritical Water Extraction in Environmental Analysis, J. Chro-matogr. A 975 (2002):3–29.
13. S. B. Hawthorne, A. J. M. Lagadec, D. Kalderis, A. V. Lilke, and D. J. Miller,Pilot-scale Destruction of TNT, RDX, and HMX on Contaminated Soils UsingSubcritical Water, Environ. Sci. Technol. 34 (2000):3224–3228.
14. A. J. M. Lagadec, D. J. Miller, A. V. Lilke, and S. B. Hawthorne, Pilot-scale Sub-critical Water Remediation of Polycyclic Aromatic Hydrocarbon- and Pesticide-contaminated Soil, Environ. Sci. Technol. 34 (2000):1542–1548.
15. H. C. Hailes, Reaction Solvent Selection: The Potential of Water as a Solvent forOrganic Transformations, Org. Process. Res. Dev. 11 (2007):114–120.
16. N. Akiya and P. E. Savage, Roles of Water for Chemical Reactions in High-temperature Water, Chem. Rev. 102 (2002):2725–2750.
17. A. R. Katritzky, S. M. Allin, and M. Siskin, Aquathermolysis: Reactions or OrganicCompounds with Superheated Water, Acc. Chem. Res. 29 (1996):399–406.
18. M. Siskin and A. R. Katritzky, Reactivity of Organic Compounds in Hot Water:Geochemical and Technological Implications, Science 254 (1991):231–237.
19. M. O. Fogwill and K. B. Thurbide, Rapid Column Heating Method for SubcriticalWater Chromatography, J. Chromatogr. A 1139 (2007):199–205.
20. P. He and Y. Yang, Studies on the Long-term Thermal Stability of StationaryPhases in Subcritical Water Chromatography, J. Chromatogr. A 989 (2003):55–63.
21. Y. Yang, L. J. Lamm, P. He, and T. Kondo, Temperature Effect on Peak Width andColumn Efficiency in Subcritical Water Chromatography, J. Chromatogr. Sci. 40(2002):107–112.
22. D. J. Miller and S. B. Hawthorne, Subcritical Water Chromatography with FlameIonization Detection, Anal. Chem. 69 (1997):623–627.
23. S. B. Hawthorne, Y. Yang, and D. J. Miller, Extraction of Organic Pollutantsfrom Environmental Solids with Sub- and Supercritical Water, Anal. Chem. 66(1994):2912–2920.
24. Y. Yang and B. Li, Subcritical Water Extraction Coupled to High-performanceLiquid Chromatography, Anal. Chem. 71 (1999):1491–1495.
25. V. Fernandez-Perez and M. D. Luque de Castro, Micelle Formation for Improvementof Continuous Subcritical Water Extraction of Polycyclic Aromatic Hydrocarbonsin Soil Prior to High Performance Liquid Chromatography-fluorescence Detection,J. Chromatogr. A 902 (2000):357–367.
26. S. B. Hawthorne, C. B. Grabanski, E. Martin, and D. J. Miller, Comparison ofSoxhlet Extraction, Pressurized Liquid Extraction, Supercritical Fluid Extractionand Subcritical Water Extraction for Environmental Solids: Recovery, Selectivityand Effects on Sample Matrix, J. Chromatogr. A 892 (2000):421–433.

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
Extraction of PAHs from Soil using Water 259
27. S. B. Hawthorne, S. Trembley, C. L. Moniot, C. B. Grabanski, and D. J. Miller,Static Subcritical Water Extraction with Simultaneous Solid-phase Extraction forDetermining Polycyclic Aromatic Hydrocarbons on Environmental Solids, J. Chro-matogr. A 886 (2000):237–244.
28. T. Hyotylainen, T. Andersson, K. Hartonen, K. Kuosmanen, and M.-L. Riekkola,Pressurized Hot Water Extraction Coupled On-line with LC-GC: Determination ofPolycyclic Hydrocarbons in Sediment, Anal. Chem. 72 (2000):3070–3076.
29. M. A. Avila-Chavez, R. Eustaquio-Rincon, J. Reza, and A. Trejo, Extraction ofHydrocarbons from Crude Oil Tank Bottom Sludges Using Supercritical Ethane,Sep. Sci. Technol. 42 (2007):2329–2347.
30. B. Li, Y. Yang, Y. Gan, C. D. Eaton, P. He, and A. D. Jones, On-line Coupling ofSubcritical Water Extraction with High-performance Liquid Chromatography viaSolid Phase Trapping, J. Chromatogr. A 873 (2000):175–184.
31. M. D. Johnson, W. Huang, Z. Dang, and W. J. Weber Jr., A Distributed Reactiv-ity Model for Sorption by Soils and Sediments. 12. Effects of Subcritical WaterExtraction and Alterations of Soil Organic Matter on Sorption Equilibria, Environ.Sci. Technol. 33 (1999):1657–1663.
32. J. Reza, A. Trejo, and L. E. Vera-Avila, Determination of the Temperature Depen-dence of Water Solubilities of Polycyclic Aromatic Hydrocarbons by a GeneratorColumn on Line-solid Phase Extraction Liquid Chromatographic Method, Chemo-sphere 47 (2002):933–945.
33. J. Reza and A. Trejo, Temperature Dependence of the Infinite Dilution ActivityCoefficient and Henry’s Law Constant of Polycyclic Aromatic Hydrocarbons inWater, Chemosphere 56 (2004):537–547.
34. M. M. Jimenez-Carmona and M. D. Luque de Castro, An Approach for MonitoringSubcritical Water Extraction Kinetics of Fluorescent Analytes, Anal. Chim. Acta.342 (1997):215–221.
35. K. J. Hageman, L. Mazeas, C. B. Grabanski, D. J. Miller, and S. B. Hawthorne,Coupled Subcritical Water Extraction with Solid-phase Microextraction for Deter-mining Semivolatile Organics in Environmental Solids, Anal. Chem. 68 (1996):3892–3898.
36. J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry.(Harlow, England: Pearson Prentice Hall (2005)):107–149.
37. IUPAC, Compendium of Chemical Terminology, 2nd ed. http://www.iupac.org/goldbook/F02503.pdf (1997) (accessed March 7, 2007).
38. United States Environmental Protection Agency, Method 3540C. Soxhlet Extrac-tion. Revision 3. http://www.epa.gov/epaoswer/hazwaste/test/pdfs/3540c.pdf (1996)(accessed March 7, 2007).
39. M. T. O. Jonker and A. A. Koelmans, Extraction of Polycyclic Aromatic Hydro-carbons from Soot and Sediment: Solvent Evaluation and Implications for SorptionMechanism, Environ. Sci. Technol. 36 (2002):4107–4113.
40. United States Environmental Protection Agency. Method 9071B, n-Hexane Ex-tractable Material (HEM) for Sludge, Sediment, and Soil Samples. Revision 2.http://www.epa.gov/epaoswer/hazwaste/test/pdfs/9071b.pdf (1998) (accessed March7, 2007).

Dow
nloa
ded
By:
[Rez
a, J
oel]
At:
15:1
8 23
Aug
ust 2
007
260 E. Moreno et al.
41. B. H. Chen, C. Y. Wang, and C. P. Chiu, Evaluation of Analysis of PolycyclicAromatic Hydrocarbons in Meat Products by Liquid Chromatography, J. Agric.Food Chem. 44 (1996):2244–2251.
42. M. Burford, S. B. Hawthorne, and D. J. Miller, Extraction Rates of Spiked versusNative PAHs from Heterogeneous Environmental Samples using Supercritical FluidExtraction and Sonication in Methylene Chloride, Anal. Chem. 65 (1993):1497–1505.
43. K. Jinno, Polycyclic Aromatic Hydrocarbons (PAHs) Data Base in Alpha-betical Order. http://chrom.tutms.tut.ac.jp/JINNO/DATABASE/00alphabet.html(accessed March 7, 2007).
44. Sadtler, The Sadtler Standard Spectra. Ultraviolet. Philapelphia. Sadtler ResearchLaboratories (UV Spectra number 256, 265, 269, 272, 281, 335, 351, 394, 594,2114, 3052, 3631, 6290) (1966).
45. E. Clar, Polycyclic Hydrocarbons Volume 2 (London: Academic Press) (1964).
Copyright © 2022 FDOKUMEN




















