
Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind,...
10
ARTHRITIS & RHEUMATISM Vol. 60, No. 2, February 2009, pp 335–344 DOI 10.1002/art.24266 © 2009, American College of Rheumatology Evaluation of the Efficacy and Safety of Pamapimod, a p38 MAP Kinase Inhibitor, in a Double-Blind, Methotrexate-Controlled Study of Patients With Active Rheumatoid Arthritis Stanley B. Cohen, 1 Tien-Tsai Cheng, 2 Vishala Chindalore, 3 Nemanja Damjanov, 4 Ruben Burgos-Vargas, 5 Patricia DeLora, 6 Kathleen Zimany, 6 Helen Travers, 7 and John P. Caulfield 8 Objective. To determine the efficacy and safety of pamapimod (a selective inhibitor of the -isoform of p38 MAP kinase) as monotherapy in comparison with methotrexate (MTX) treatment in adult patients with active rheumatoid arthritis (RA). Methods. Patients were randomly assigned to 1 of 4 treatment groups and received 12 weeks of double- blind treatment. One group received MTX (7.5 mg/week with planned escalation to 20 mg/week), and 3 groups received pamapimod (50, 150, or 300 mg) once daily. The primary efficacy end point was the proportion of patients meeting the American College of Rheumatology 20% improvement criteria (achieving an ACR20 re- sponse) at 12 weeks. Secondary end points included ACR50 and ACR70 responses, change from baseline in the Disease Activity Score in 28 joints (DAS28), cate- gorical analyses of DAS28/European League Against Rheumatism response, and change from baseline in each parameter of the ACR core set of measures. Safety monitoring included recording of adverse events (AEs), laboratory testing, immunology assessments, adminis- tration of electrocardiograms, and assessment of vital signs. Results. Patients assigned to receive MTX and pamapimod had similar demographics and baseline characteristics. At week 12, fewer patients taking pam- apimod had an ACR20 response (23%, 18%, and 31% in the 50-, 150-, and 300-mg groups, respectively) com- pared with patients taking MTX (45%). Secondary efficacy end points showed a similar pattern. AEs were typically characterized as mild and included infections, skin disorders, and dizziness. Pamapimod was generally well tolerated, but the 300-mg dose appeared to be more toxic than either the 2 lower doses or MTX. Conclusion. The present results showed that pam- apimod was not as effective as MTX in the treatment of active RA. Parenteral biologic therapies that selectively neu- tralize proinflammatory cytokines such as tumor necro- sis factor (TNF) and interleukin-1 (IL-1) or their receptors, such as the IL-6 receptor, substantially im- prove signs and symptoms of rheumatoid arthritis (RA), reduce the number of swollen and tender joints, and ClinicalTrials.gov identifier: NCT00303563. Supported by Hoffman-La Roche, Nutley, New Jersey. 1 Stanley B. Cohen, MD: Metroplex Clinical Research, Dallas, Texas; 2 Tien-Tsai Cheng, MD: Chang Gung Memorial Hospital– Kaohsiung Medical Center, and Chang Gung University College of Medicine, Kaohsiung, Taiwan; 3 Vishala Chindalore, MD: Pinnacle Research Group, Anniston, Alabama; 4 Nemanja Damjanov, MD, PhD: Institut za Reumatologiju, Belgrade University School of Med- icine, Belgrade, Serbia and Montenegro; 5 Ruben Burgos-Vargas, MD: Clı ´nica para el Diagnostico y Tratamiento de las Enfermedades Reuma ´ticas and Hospital General de Me ´xico, Mexico City, Mexico; 6 Patricia DeLora, BA, Kathleen Zimany, MS: Hoffman-La Roche, Nutley, New Jersey; 7 Helen Travers, PhD: Roche Products Ltd., Welwyn Garden City, UK; 8 John P. Caulfield, MD: Roche Palo Alto, LLC, Palo Alto, California. Dr. Cohen has served as a clinical investigator and research consultant for Genentech, Biogen Idec, Roche, Procter & Gamble, Pfizer, Centocor, Amgen, Scios, and Wyeth-Ayerst, and has received consulting and/or speaking fees from these companies (less than $10,000 each). Dr. Chindalore has received speaking fees from Roche, Pfizer, and GlaxoSmithKline (less than $10,000 each). Dr. Burgos- Vargas has received consulting fees, speaking fees, and/or honoraria from Roche, Schering-Plough, Wyeth, and Abbott (less than $10,000 each). Ms DeLora owns stock or stock options in Hoffman-La Roche. Dr. Caulfield owns stock or stock options in Roche. Address correspondence and reprint requests to Stanley B. Cohen, MD, Metroplex Clinical Research Center, 5939 Harry Hines Boulevard, Suite 441, Dallas, TX 75235. E-mail: [email protected]. Submitted for publication June 6, 2008; accepted in revised form October 22, 2008. 335
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind,...

ARTHRITIS & RHEUMATISMVol. 60, No. 2, February 2009, pp 335–344DOI 10.1002/art.24266© 2009, American College of Rheumatology
Evaluation of the Efficacy and Safety of Pamapimod,a p38 MAP Kinase Inhibitor, in a Double-Blind,Methotrexate-Controlled Study of Patients With
Active Rheumatoid Arthritis
Stanley B. Cohen,1 Tien-Tsai Cheng,2 Vishala Chindalore,3 Nemanja Damjanov,4
Ruben Burgos-Vargas,5 Patricia DeLora,6 Kathleen Zimany,6 Helen Travers,7
and John P. Caulfield8
Objective. To determine the efficacy and safety ofpamapimod (a selective inhibitor of the �-isoform ofp38 MAP kinase) as monotherapy in comparison withmethotrexate (MTX) treatment in adult patients withactive rheumatoid arthritis (RA).
Methods. Patients were randomly assigned to 1 of4 treatment groups and received 12 weeks of double-blind treatment. One group received MTX (7.5 mg/weekwith planned escalation to 20 mg/week), and 3 groupsreceived pamapimod (50, 150, or 300 mg) once daily.
The primary efficacy end point was the proportion ofpatients meeting the American College of Rheumatology20% improvement criteria (achieving an ACR20 re-sponse) at 12 weeks. Secondary end points includedACR50 and ACR70 responses, change from baseline inthe Disease Activity Score in 28 joints (DAS28), cate-gorical analyses of DAS28/European League AgainstRheumatism response, and change from baseline ineach parameter of the ACR core set of measures. Safetymonitoring included recording of adverse events (AEs),laboratory testing, immunology assessments, adminis-tration of electrocardiograms, and assessment of vitalsigns.
Results. Patients assigned to receive MTX andpamapimod had similar demographics and baselinecharacteristics. At week 12, fewer patients taking pam-apimod had an ACR20 response (23%, 18%, and 31% inthe 50-, 150-, and 300-mg groups, respectively) com-pared with patients taking MTX (45%). Secondaryefficacy end points showed a similar pattern. AEs weretypically characterized as mild and included infections,skin disorders, and dizziness. Pamapimod was generallywell tolerated, but the 300-mg dose appeared to be moretoxic than either the 2 lower doses or MTX.
Conclusion. The present results showed that pam-apimod was not as effective as MTX in the treatment ofactive RA.
Parenteral biologic therapies that selectively neu-tralize proinflammatory cytokines such as tumor necro-sis factor � (TNF�) and interleukin-1� (IL-1�) or theirreceptors, such as the IL-6 receptor, substantially im-prove signs and symptoms of rheumatoid arthritis (RA),reduce the number of swollen and tender joints, and
ClinicalTrials.gov identifier: NCT00303563.Supported by Hoffman-La Roche, Nutley, New Jersey.1Stanley B. Cohen, MD: Metroplex Clinical Research, Dallas,
Texas; 2Tien-Tsai Cheng, MD: Chang Gung Memorial Hospital–Kaohsiung Medical Center, and Chang Gung University College ofMedicine, Kaohsiung, Taiwan; 3Vishala Chindalore, MD: PinnacleResearch Group, Anniston, Alabama; 4Nemanja Damjanov, MD,PhD: Institut za Reumatologiju, Belgrade University School of Med-icine, Belgrade, Serbia and Montenegro; 5Ruben Burgos-Vargas, MD:Clınica para el Diagnostico y Tratamiento de las EnfermedadesReumaticas and Hospital General de Mexico, Mexico City, Mexico;6Patricia DeLora, BA, Kathleen Zimany, MS: Hoffman-La Roche,Nutley, New Jersey; 7Helen Travers, PhD: Roche Products Ltd.,Welwyn Garden City, UK; 8John P. Caulfield, MD: Roche Palo Alto,LLC, Palo Alto, California.
Dr. Cohen has served as a clinical investigator and researchconsultant for Genentech, Biogen Idec, Roche, Procter & Gamble,Pfizer, Centocor, Amgen, Scios, and Wyeth-Ayerst, and has receivedconsulting and/or speaking fees from these companies (less than$10,000 each). Dr. Chindalore has received speaking fees from Roche,Pfizer, and GlaxoSmithKline (less than $10,000 each). Dr. Burgos-Vargas has received consulting fees, speaking fees, and/or honorariafrom Roche, Schering-Plough, Wyeth, and Abbott (less than $10,000each). Ms DeLora owns stock or stock options in Hoffman-La Roche.Dr. Caulfield owns stock or stock options in Roche.
Address correspondence and reprint requests to Stanley B.Cohen, MD, Metroplex Clinical Research Center, 5939 Harry HinesBoulevard, Suite 441, Dallas, TX 75235. E-mail: [email protected].
Submitted for publication June 6, 2008; accepted in revisedform October 22, 2008.
335

slow radiographic progression of erosive disease. How-ever, there are no marketed orally active, safe, andeffective agents that act primarily to inhibit TNF� orIL-1�, despite attempts to develop such compounds(1–3).
The p38� MAP kinase (MAPK) has been a majormolecular target for the development of an anti–proinflammatory cytokine small-molecule oral therapyfor RA. The �-isoform is an enzyme important to theintracellular signaling pathway for the generation ofTNF� or IL-1�. Multiple extracellular stimuli, includingstress signals (lipopolysaccharide), osmotic or heatshock, and proinflammatory cytokines (TNF� or IL-1�),stimulate the p38 pathway (4). The p38� MAPK alsoregulates the expression of cyclooxygenase 2, the en-zyme that regulates prostanoids (e.g., prostaglandin E2)in inflammation (5). Activation of the p38� pathwaycauses both transcriptional and translational modulationof gene expression of TNF� and IL-1� that is both celltype and signal specific. Inhibitors of p38� block theproduction of TNF� and IL-1� in monocytes and inanimal models of arthritis (6). The evidence for a role ofthe other 3 isoforms, �, �, and �, in TNF generation isnot as strong as that for the �-isoform, although the �and � isoforms have been localized to the rheumatoidsynovium (7–9).
Pamapimod is a novel small molecule of thepyridopyrimidine class that selectively inhibits the�-isoform of p38 MAPK (10). Its mean � SD 50%inhibition concentration against recombinant p38� is0.014 � 0.002 �M, while that against TNF� release fromcultured monocytic leukemia-derived THP-1 cells is0.025 � 0.001 �M, and that against IL-1� productionfrom human whole blood is 0.10 � 0.03 �M. In a varietyof in vitro and in vivo preclinical models, pamapimodinhibited TNF�, IL-1�, and IL-6 production; it alsoreduced inflammation and provided joint protection incollagen-induced arthritis in mice. In studies in healthyvolunteers, pamapimod was well tolerated and wasassociated with mild dizziness, mild infections (such asfolliculitis, bronchitis, and urinary tract infections), andmild, reversible elevations of creatinine phosphokinase(CPK) (Zhang X, Huang Y, Caulfield JP: unpublisheddata). Therefore, phase II development of pamapimodas an oral agent to treat RA was initiated. In the studyreported here (PA18604), patients not currently receiv-ing methotrexate (MTX) were given either pamapimodonce daily at 1 of 3 dosage levels or MTX weekly withappropriate increase of the MTX dosage. In a compan-ion study (PA18439), patients with an incomplete re-sponse to MTX were given once-daily or twice-daily
dosing regimens of pamapimod or were given placebo(Alten RE, Zerbini C, Jeka S, Irazoque F, Khatib F,Emery P, et al: unpublished observations).
PATIENTS AND METHODS
Patients. Eligible patients were age �18 years, hadactive RA according to the 1987 revised criteria of the Amer-ican College of Rheumatology (ACR; formerly, the AmericanRheumatism Association) (11), received antiinflammatorytherapy for RA on an outpatient basis, had �6 swollen joints(of 66 joints assessed) and �8 tender joints (of 68 jointsassessed), and had a high-sensitivity C-reactive protein(hsCRP) level �0.6 mg/dl or an erythrocyte sedimentation rate(ESR) �28 mm/hour or morning stiffness �45 minutes. Pa-tients were excluded if they were unable to get out of bedwithout assistance or required use of a wheelchair, had ahistory of or current inflammatory joint disease other than RA,or had a rheumatic autoimmune disease other than RA orsignificant systemic involvement secondary to RA; Sjogren’ssyndrome with RA was allowed. Other exclusion criteriaincluded discontinuation of previous MTX therapy due toclinically important toxicity or insufficient efficacy, previoustreatment with alkylating agents, evidence of serious uncon-trolled disease, history or evidence of tuberculosis, activeinfections, history of recurrent bacterial infections, history ofhereditary or acquired immune deficiency disorder, pregnancyor breast-feeding, or neuropathies or other painful conditionsthat had the potential to interfere with pain evaluations.
Treatment with disease-modifying antirheumatic drugs(DMARDs) and biologic agents had to be stopped prior tobaseline as follows: infliximab and adalimumab, 8 weeks;leflunomide, 8 weeks, or after cholestyramine, 4 weeks; intra-articular or parenteral corticosteroids, 4 weeks; sulfasalazine,azathioprine, cyclosporine, D-penicillamine, hydroxychloro-quine, chloroquine, and gold, 3 weeks; etanercept and anak-inra, 2 weeks.
Study protocol. This 12-week, double-blind, double-dummy, parallel-group, active-controlled study was conductedat 61 centers in the US, Canada, Mexico, Europe, SouthAfrica, and Taiwan. All participating sites received approvalfrom their governing institutional review board (or equivalent),all patients provided informed written consent, and the studywas performed in accordance with the Declaration of Helsinki.A list of investigators who recruited patients for this study isshown in Appendix A.
At screening, medical history was recorded, and infor-mation was collected regarding swollen and tender joints andduration of morning stiffness. Vital signs and physical mea-surements were obtained, and a complete physical examina-tion, electrocardiography (EKG), chest radiography, tubercu-lin skin test, clinical laboratory tests (hematology, serumchemistry, nonfasting lipid panel, and urinalysis), and a serumpregnancy test (as required) were performed. Blood sampleswere obtained to test for hepatitis B surface antigen andhepatitis C virus and to determine the hsCRP level and ESR.
Patients were randomly assigned (1:1:1:1) to 1 of 4treatment regimens in a blinded manner and stratified bygeographic region and previous MTX exposure. The firstgroup received 50 mg pamapimod once daily (two 25-mg
336 COHEN ET AL

tablets) plus MTX placebo weekly. The second group received150 mg pamapimod once daily (two 75-mg tablets) plus MTXplacebo weekly. The third group received 300 mg pamapimodonce daily (two 150-mg tablets) plus MTX placebo weekly. Thefourth group received placebo pamapimod once daily (twoplacebo tablets) plus MTX weekly in increasing doses. Patientsrandomly assigned to the MTX group received MTX orallyonce a week, commencing at 7.5 mg (three 2.5-mg tablets) onday 1, which was increased to 15 mg weekly by week 4; afurther increase to 20 mg weekly was an option at week 8,depending on whether the patient had painful and swollenjoints and did not have signs of significant MTX-relatedtoxicity. Of those in the MTX group who completed 12 weeksof dosing, 71% were taking 15 mg/week and 29% were taking20 mg/week at the end of the study. The mean cumulative doseof MTX was 156.5 mg. In the 3 pamapimod groups, MTXplacebo was escalated to achieve similar nominal mean cumu-lative doses as in the MTX group, namely, 155.1 mg for the50-mg group, 152.4 mg for the 150-mg group, and 142.4 mg forthe 300-mg group. MTX dosage reductions were permitted forpatients who experienced possible MTX-related side effects,and dosage adjustments were performed in the same mannerfor both active and placebo forms of MTX, due to the blindednature of the study.
In addition to the study drug, patients were to receivefolic acid (5 mg weekly) during the study. Oral corticosteroids(�10 mg/day prednisone or equivalent) and nonsteroidalantiinflammatory drugs were permitted if the dosage wasstable for �4 weeks and �2 weeks, respectively, prior tobaseline and maintained during the study. The followingtreatments were prohibited during the study: DMARDs, MTX(except as part of protocol-defined study therapy), prednisoneor equivalent at a dosage of �10 mg/day, intraarticular orparenteral corticosteroids, biologic agents, cell-depleting ther-apies, alkylating agents (including cyclophosphamide), anyinvestigational agent other than pamapimod, immunizations,and immunoabsorption columns.
Efficacy parameters were assessed at baseline and thenroutinely throughout the 12 weeks of treatment according tothe following schedule. The swollen and tender joint countsand the Stanford Health Assessment Questionnaire disabilityindex (HAQ DI) score (12) were obtained at weeks 2, 4, 8, and12. An assessment of fatigue using the fatigue subscale of theFunctional Assessment of Chronic Illness Therapy question-naire (FACIT-F) (13) was performed at weeks 4 and 12. Allother efficacy assessments (patient’s and physician’s globalassessment of disease activity, patient’s assessment of pain,hsCRP level, ESR, and morning stiffness) were performed atweeks 1, 2, 4, 8, and 12.
Safety assessments, including EKGs and vital signs,were monitored routinely. Safety laboratory testing (hematol-ogy, blood chemistry including CPK level, lipid panel, andurinalysis) was performed at screening and baseline and atweeks 1, 2, 4, 8, and 12. In addition, immunology laboratorytesting for rheumatoid factor (RF), anti–cyclic citrullinatedpeptide (anti-CCP), lymphocyte subtype analysis (CD4�,CD3�, CD8�, and CD19� cells were sorted), anti–double-stranded DNA (anti-dsDNA), antitetanus titer, IgG, IgA, andIgM was performed at baseline and at week 12.
All adverse events (AEs) encountered during the studyand through 4 weeks following discontinuation of study treat-
ment were recorded. AEs were monitored until they stabilizedor returned to baseline status. For each AE, the investigatorassessed and recorded the intensity (as mild, moderate, orsevere), relationship to the study drug (unrelated or remotely,possibly, or probably related), action taken (i.e., whether atreatment was given or a procedure performed and whetherthe study drug was discontinued or the dose adjusted), andoutcome. A serious AE was defined as any experience thatresulted in death, was life-threatening, required hospitalizationor prolonged an existing hospitalization, resulted in a persis-tent or significant disability, resulted in a congenital anomaly/birth defect, or required intervention to prevent one or moreof the outcomes listed above.
Clinical assessments. The primary efficacy end pointwas the proportion of patients meeting the ACR 20% improve-ment criteria (achieving an ACR20 response) at week 12 (14),defined as a 20% improvement from baseline in swollen jointcount and tender joint count and in at least 3 of the following5 variables: HAQ DI score, patient’s global assessment ofdisease activity, patient’s global assessment of pain, physician’sglobal assessment of disease activity, and level of an acute-phase reactant (primarily CRP; ESR was used only if CRP wasmissing). Secondary end points included ACR50 and ACR70responses (50% and 70% improvement, respectively, frombaseline in the parameters used to define an ACR20 re-sponse), change from baseline in the Disease Activity Score in28 joints (DAS28) (15), categorical analyses of DAS28/European League Against Rheumatism response (16), andchange from baseline in each parameter of the ACR core setof measures (17) (swollen joint count, tender joint count,patient’s and physician’s global assessment of disease activity,patient’s assessment of pain, HAQ DI score, hsCRP level,and ESR). Additional secondary end points included assess-ments of the FACIT-F score and the severity and duration ofmorning stiffness.
Statistical analysis. Pamapimod and MTX were ex-pected to have similar efficacy levels, with an assumed ACR20response rate of 50–70% based on historical data. For aresponse rate of 70%, a sample size of 50 patients for a giventreatment group would provide a 90% confidence interval(90% CI) with a margin of error of �10%. For a response rateof 50%, the same sample size would provide a 90% CI with amargin of error of �12%.
Efficacy analyses were based on the intent-to-treat(ITT) population, defined as all patients who received at least1 dose of double-blind study medication. Patients who prema-turely withdrew from the study for any reason, or for whom anassessment was not performed for any reason, were included inthe ITT population. Analysis of safety data included allrandomized patients who received at least 1 dose of studymedication and had at least 1 safety assessment.
For the primary efficacy analysis, no formal hypothesistesting was performed. Descriptive statistics, including CIestimates, were used to evaluate differences among treatmentgroups. Patients with missing ACR20 response values at week12 (including patients who had withdrawn from the study priorto week 12) were classified as nonresponders for the purposeof the analysis. The primary efficacy variable (ACR20 responseat week 12) was analyzed using the Cochran-Mantel-Haenszeltest, with region and previous MTX exposure as stratificationfactors.
PAMAPIMOD VERSUS MTX IN PATIENTS WITH ACTIVE RA 337

For the secondary efficacy analyses, results for cate-gorical parameters were analyzed as described for the primaryefficacy analysis. Patients with missing ACR50 and ACR70response values were considered nonresponders for the week-12analyses. For all other categorical variables, the observed datawere used. Results for continuous parameters were summa-rized using descriptive statistics.
RESULTS
Patient characteristics. Study data were collectedbetween February 9, 2006 and June 21, 2007. Of the 271patients screened, 204 were enrolled; 53 were random-ized to receive MTX and 151 to receive pamapimod (52,51, and 48 patients to the 50-, 150-, and 300-mg groups,respectively). Table 1 summarizes the demographics andbaseline characteristics of the 4 treatment groups. Themajority of patients were women, and the mean age ofthe patients ranged from 47.3 years to 51.4 years acrossthe treatment groups. At baseline, the majority ofpatients tested positive for RF and anti-CCP, 88% ofpatients reported having morning stiffness of �45 min-utes, and 47% had CRP values that were �1.0 mg/dl.Data on duration of disease were not captured. Fifty-six
percent of patients had reported prior use of DMARDs(mean of 2) for treatment of RA, 37% of patients hadprior exposure to MTX, and 2–8% of patients had usedan anti-TNF medication. At baseline, approximately halfof the patients were using corticosteroids at a dosageequivalent to �10 mg prednisone once daily. The 4treatment groups were generally similar at baseline withregard to the ACR core set (17) characteristics ofswollen and tender joint counts, HAQ DI score, patient-assessed pain, and patient and physician globally as-sessed disease activity (Table 1).
All patients received at least 1 dose of the studydrug and were included in both the ITT and safetyanalysis populations. Overall, 81% of patients completed12 weeks of treatment; 15.1% of patients in the MTXgroup and 19.9% of patients in the combined pamapi-mod groups discontinued study treatment prematurely(Table 2). Reasons for discontinuation were attributedto AEs, lack of efficacy, and refusal of continued treat-ment (which included noncompliance and withdrawal ofconsent). The rates of and reasons for discontinuationwere similar between the MTX group and the 2 lower-
Table 1. Baseline characteristics of the patients*
CharacteristicMTX
(n � 53)
Pamapimod
50 mgonce daily(n � 52)
150 mgonce daily(n � 51)
300 mgonce daily(n � 48)
Age, years 47.3 � 12.4 51.4 � 11.5 50.7 � 12.4 50.2 � 12.8Female, no. (%) 44 (83) 45 (87) 43 (84) 40 (83)Swollen joint count (of 66 joints) 15.5 � 8.4 17.5 � 9.2 15.7 � 9.5 19.2 � 10.9Tender joint count (of 68 joints) 28.5 � 14.6 27.0 � 12.6 27.0 � 15.2 28.2 � 14.7RF positive, no. (%) 34 (64) 37 (71) 33 (67) 27 (57)RF, IU/ml 154.4 � 282.4 308.4 � 650.3 235.0 � 411.3 142.6 � 248.0Anti-CCP positive, no. (%) 32 (60) 36 (69) 38 (75) 33 (69)CRP level, mg/dl 1.62 � 1.91 2.04 � 2.41 1.95 � 2.10 2.58 � 2.95ESR, mm/hour 39.7 � 22.4 47.8 � 33.7 44.3 � 25.1 42.0 � 22.8Morning stiffness �45 minutes, no. (%) 47 (89) 46 (88) 45 (90) 42 (88)DAS28 using ESR 6.40 � 0.90 6.42 � 0.97 6.30 � 1.03 6.61 � 1.00HAQ DI score 1.36 � 0.65 1.33 � 0.74 1.37 � 0.75 1.58 � 0.74FACIT-F score 28.1 � 11.1 29.9 � 11.0 30.4 � 11.3 26.6 � 11.2Patient’s global assessment of disease activity,
0–100-mm VAS62.4 � 26.7 55.6 � 24.2 58.6 � 24.8 63.6 � 23.9
Physician’s global assessment of disease activity,0–100-mm VAS
58.1 � 20.8 56.8 � 19.5 54.4 � 20.4 59.9 � 21.2
Patient’s assessment of pain, 0–100-mm VAS 53.6 � 25.0 49.8 � 19.5 52.2 � 19.8 55.7 � 24.3Use of corticosteroids, no. (%) 25 (47) 28 (54) 21 (41) 25 (52)Previous exposure to MTX, no. (%) 21 (40) 22 (42) 16 (31) 16 (33)Previous use of DMARDs, no. (%) 29 (55) 32 (62) 28 (55) 25 (52)Number of previous DMARDs 2.4 � 1.2 1.8 � 0.9 2.0 � 1.2 2.2 � 1.1Previous use of anti-TNF agents, no. (%) 4 (8) 3 (6) 1 (2) 3 (6)
* Except where indicated otherwise, values are the mean � SD. MTX � methotrexate; RF � rheumatoid factor; anti-CCP � anti–cyclic citrullinatedpeptide; CRP � C-reactive protein; ESR � erythrocyte sedimentation rate; DAS28 � Disease Activity Score in 28 joints; HAQ DI � HealthAssessment Questionnaire disability index; FACIT-F � Functional Assessment of Chronic Illness Therapy–Fatigue; VAS � visual analog scale;DMARDs � disease-modifying antirheumatic drugs; anti-TNF � anti–tumor necrosis factor.
338 COHEN ET AL
dose pamapimod groups. The incidence of discontinua-tion due to an AE was highest in the 300-mg pamapimodgroup (with a rate of 21%, compared with 6–8% in theother 3 groups).
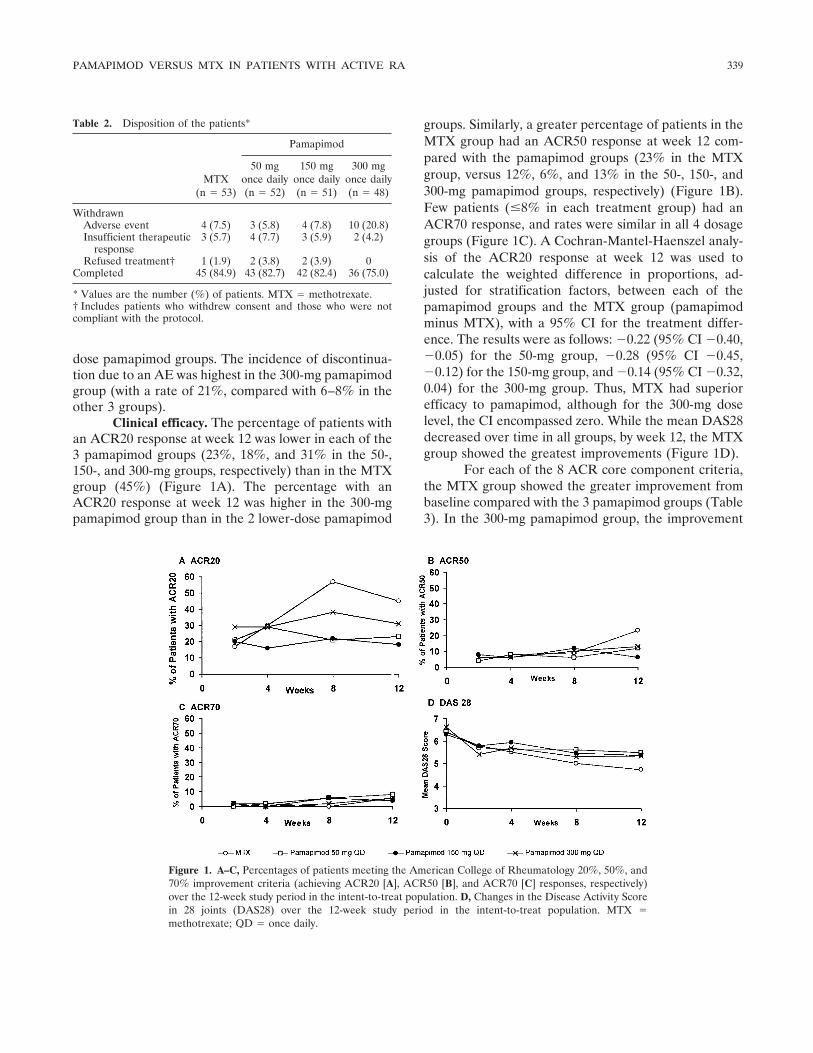
Clinical efficacy. The percentage of patients withan ACR20 response at week 12 was lower in each of the3 pamapimod groups (23%, 18%, and 31% in the 50-,150-, and 300-mg groups, respectively) than in the MTXgroup (45%) (Figure 1A). The percentage with anACR20 response at week 12 was higher in the 300-mgpamapimod group than in the 2 lower-dose pamapimod
groups. Similarly, a greater percentage of patients in theMTX group had an ACR50 response at week 12 com-pared with the pamapimod groups (23% in the MTXgroup, versus 12%, 6%, and 13% in the 50-, 150-, and300-mg pamapimod groups, respectively) (Figure 1B).Few patients (�8% in each treatment group) had anACR70 response, and rates were similar in all 4 dosagegroups (Figure 1C). A Cochran-Mantel-Haenszel analy-sis of the ACR20 response at week 12 was used tocalculate the weighted difference in proportions, ad-justed for stratification factors, between each of thepamapimod groups and the MTX group (pamapimodminus MTX), with a 95% CI for the treatment differ-ence. The results were as follows: �0.22 (95% CI �0.40,�0.05) for the 50-mg group, �0.28 (95% CI �0.45,�0.12) for the 150-mg group, and �0.14 (95% CI �0.32,0.04) for the 300-mg group. Thus, MTX had superiorefficacy to pamapimod, although for the 300-mg doselevel, the CI encompassed zero. While the mean DAS28decreased over time in all groups, by week 12, the MTXgroup showed the greatest improvements (Figure 1D).
For each of the 8 ACR core component criteria,the MTX group showed the greater improvement frombaseline compared with the 3 pamapimod groups (Table3). In the 300-mg pamapimod group, the improvement
Table 2. Disposition of the patients*
MTX(n � 53)
Pamapimod
50 mgonce daily(n � 52)
150 mgonce daily(n � 51)
300 mgonce daily(n � 48)
WithdrawnAdverse event 4 (7.5) 3 (5.8) 4 (7.8) 10 (20.8)Insufficient therapeutic
response3 (5.7) 4 (7.7) 3 (5.9) 2 (4.2)
Refused treatment† 1 (1.9) 2 (3.8) 2 (3.9) 0Completed 45 (84.9) 43 (82.7) 42 (82.4) 36 (75.0)
* Values are the number (%) of patients. MTX � methotrexate.† Includes patients who withdrew consent and those who were notcompliant with the protocol.
Figure 1. A–C, Percentages of patients meeting the American College of Rheumatology 20%, 50%, and70% improvement criteria (achieving ACR20 [A], ACR50 [B], and ACR70 [C] responses, respectively)over the 12-week study period in the intent-to-treat population. D, Changes in the Disease Activity Scorein 28 joints (DAS28) over the 12-week study period in the intent-to-treat population. MTX �methotrexate; QD � once daily.
PAMAPIMOD VERSUS MTX IN PATIENTS WITH ACTIVE RA 339
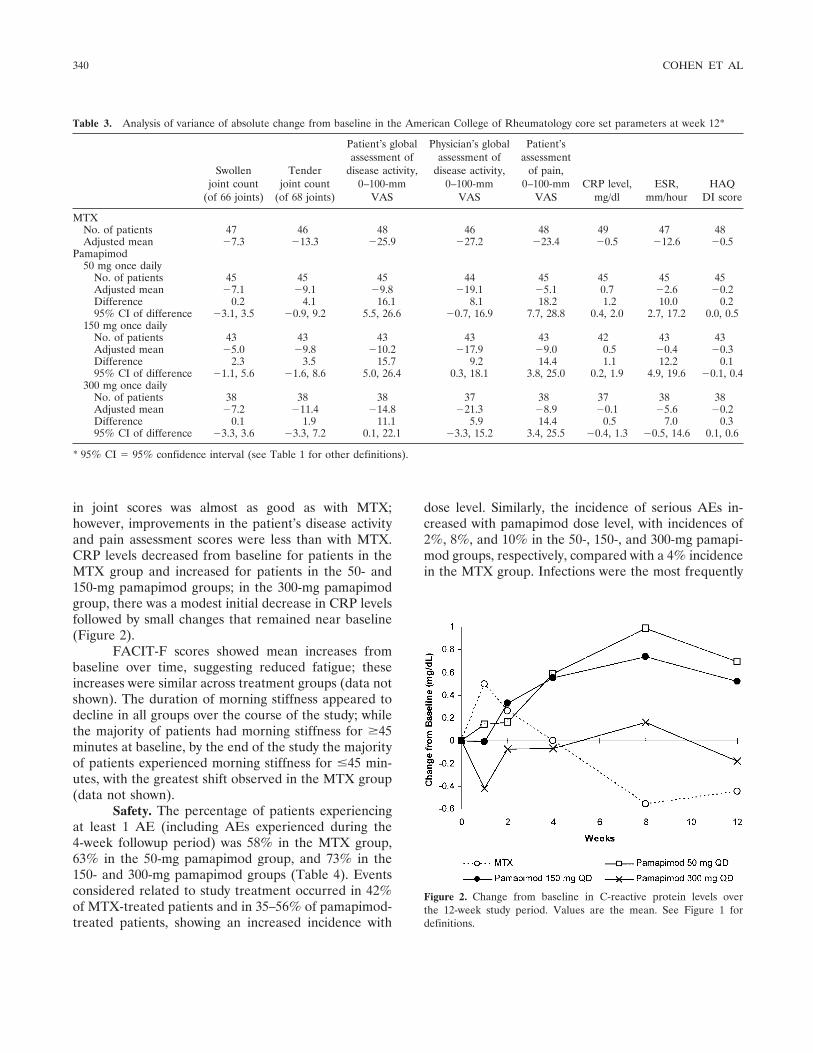
in joint scores was almost as good as with MTX;however, improvements in the patient’s disease activityand pain assessment scores were less than with MTX.CRP levels decreased from baseline for patients in theMTX group and increased for patients in the 50- and150-mg pamapimod groups; in the 300-mg pamapimodgroup, there was a modest initial decrease in CRP levelsfollowed by small changes that remained near baseline(Figure 2).
FACIT-F scores showed mean increases frombaseline over time, suggesting reduced fatigue; theseincreases were similar across treatment groups (data notshown). The duration of morning stiffness appeared todecline in all groups over the course of the study; whilethe majority of patients had morning stiffness for �45minutes at baseline, by the end of the study the majorityof patients experienced morning stiffness for �45 min-utes, with the greatest shift observed in the MTX group(data not shown).
Safety. The percentage of patients experiencingat least 1 AE (including AEs experienced during the4-week followup period) was 58% in the MTX group,63% in the 50-mg pamapimod group, and 73% in the150- and 300-mg pamapimod groups (Table 4). Eventsconsidered related to study treatment occurred in 42%of MTX-treated patients and in 35–56% of pamapimod-treated patients, showing an increased incidence with
dose level. Similarly, the incidence of serious AEs in-creased with pamapimod dose level, with incidences of2%, 8%, and 10% in the 50-, 150-, and 300-mg pamapi-mod groups, respectively, compared with a 4% incidencein the MTX group. Infections were the most frequently
Table 3. Analysis of variance of absolute change from baseline in the American College of Rheumatology core set parameters at week 12*
Swollenjoint count
(of 66 joints)
Tenderjoint count
(of 68 joints)
Patient’s globalassessment of
disease activity,0–100-mm
VAS
Physician’s globalassessment of
disease activity,0–100-mm
VAS
Patient’sassessment
of pain,0–100-mm
VASCRP level,
mg/dlESR,
mm/hourHAQ
DI score
MTXNo. of patients 47 46 48 46 48 49 47 48Adjusted mean �7.3 �13.3 �25.9 �27.2 �23.4 �0.5 �12.6 �0.5
Pamapimod50 mg once daily
No. of patients 45 45 45 44 45 45 45 45Adjusted mean �7.1 �9.1 �9.8 �19.1 �5.1 0.7 �2.6 �0.2Difference 0.2 4.1 16.1 8.1 18.2 1.2 10.0 0.295% CI of difference �3.1, 3.5 �0.9, 9.2 5.5, 26.6 �0.7, 16.9 7.7, 28.8 0.4, 2.0 2.7, 17.2 0.0, 0.5
150 mg once dailyNo. of patients 43 43 43 43 43 42 43 43Adjusted mean �5.0 �9.8 �10.2 �17.9 �9.0 0.5 �0.4 �0.3Difference 2.3 3.5 15.7 9.2 14.4 1.1 12.2 0.195% CI of difference �1.1, 5.6 �1.6, 8.6 5.0, 26.4 0.3, 18.1 3.8, 25.0 0.2, 1.9 4.9, 19.6 �0.1, 0.4
300 mg once dailyNo. of patients 38 38 38 37 38 37 38 38Adjusted mean �7.2 �11.4 �14.8 �21.3 �8.9 �0.1 �5.6 �0.2Difference 0.1 1.9 11.1 5.9 14.4 0.5 7.0 0.395% CI of difference �3.3, 3.6 �3.3, 7.2 0.1, 22.1 �3.3, 15.2 3.4, 25.5 �0.4, 1.3 �0.5, 14.6 0.1, 0.6
* 95% CI � 95% confidence interval (see Table 1 for other definitions).
Figure 2. Change from baseline in C-reactive protein levels overthe 12-week study period. Values are the mean. See Figure 1 fordefinitions.
340 COHEN ET AL

reported serious AEs, occurring in 2%, 2%, and 6% ofpatients in the 50-, 150-, and 300-mg pamapimod groups,respectively, with 0% in the MTX group. Of theseinfections, 3 were considered by the investigator to bepossibly or probably related to the study drug: sialadeni-tis (50-mg group), pneumonia (300-mg group), andgastrointestinal (GI) infection (300-mg group). Oneadditional serious AE, GI hemorrhage in the 150-mgpamapimod group, was considered related to the studydrug. There were no deaths reported during the 12-weekstudy or 4-week followup period.
Withdrawals from the study due to an AE weresimilar between the MTX group and the 2 lower-dosepamapimod groups (6–8%); in contrast, the rate wassubstantially higher in the 300-mg pamapimod group(21%). In the MTX group, AEs that led to treatmentwithdrawal included elevated liver enzyme levels (3patients) and dizziness (1 patient). In contrast, in the300-mg pamapimod group, AEs that led to withdrawalincluded infection (1 patient), skin disorders (2 pa-tients), elevated liver enzyme levels (3 patients), GIdisorders (3 patients), and RA flare (1 patient).
AEs involving infections or skin disorders as wellas dizziness and increased hepatic enzymes occurredwith greater frequency in pamapimod-treated patientsthan in MTX-treated patients (Table 4). Nausea oc-curred more frequently in the MTX group than in thepamapimod groups (11% of MTX-treated patients,compared with 3% of all pamapimod-treated patients).
The proportion of patients experiencing skindisorders trended upward with increasing pamapimoddosage, and while the type of skin disorders varied, themost commonly reported were rash and acne. Skinlesions were predominantly on the face and torso andconsisted of maculopapular lesions with or withoutpustules. The majority of the skin disorders resolvedwhile patients continued pamapimod treatment, andonly 2 patients receiving pamapimod discontinued treat-ment because of the dermatitis.
Dizziness appeared to be associated with pamapi-mod, and the incidence increased with dosage level; only1 patient (in the 150-mg pamapimod group) withdrewfrom the study due to dizziness. In the pamapimodgroups, dizziness generally occurred soon after treat-
Table 4. Summary of adverse events*
MTX(n � 53)
Pamapimod
50 mgonce daily(n � 52)
150 mgonce daily(n � 51)
300 mgonce daily(n � 48)
Any adverse event 31 (58) 33 (63) 37 (73) 35 (73)Serious adverse event† 2 (4) 1 (2) 4 (8) 5 (10)Related serious adverse events‡ – 1 (2) 1 (2) 2 (4)Adverse event leading to withdrawal 4 (8) 3 (6) 4 (8) 10 (21)Adverse events with �5% incidence
Infections and infestations(system organ class)
13 (25) 12 (23) 14 (27) 17 (35)
Skin and subcutaneous tissuedisorders (system organ class)
2 (4) 6 (12) 4 (8) 9 (19)
Dizziness 3 (6) 4 (8) 5 (10) 7 (15)Hepatic enzyme increased 2 (4) – 2 (4) 6 (13)Upper respiratory tract infection 3 (6) 2 (4) 1 (2) 4 (8)Nausea 6 (11) – 5 (10) –Rheumatoid arthritis 4 (8) 2 (4) 1 (2) 3 (6)Constipation 1 (2) 1 (2) 2 (4) 4 (8)Urinary tract infection 3 (6) 2 (4) 1 (2) 3 (6)Diarrhea 1 (2) 2 (4) 3 (6) 1 (2)Vertigo – 1 (2) 2 (4) 3 (6)Headache 3 (6) 1 (2) 1 (2) 2 (4)Pyrexia 1 (2) 1 (2) 3 (6) 1 (2)Fatigue 2 (4) 1 (2) – 3 (6)Bronchitis 4 (8) 1 (2) 1 (2) –Cough 3 (6) 2 (4) – –Insomnia 3 (6) – 1 (2) –
* Values are the number (%) of patients. MTX � methotrexate.† Defined in Patients and Methods.‡ Considered by the investigator to be remotely, possibly, or probably related to the study drug.
PAMAPIMOD VERSUS MTX IN PATIENTS WITH ACTIVE RA 341

ment was started (ranging from 1 day to 12 days) andgenerally persisted for �2 weeks in the lower-dosegroups and for �2 weeks through the duration of thestudy for patients in the 300-mg group.
Several patients had normal values on laboratorymeasures at baseline but had at least 1 value outside thenormal limits following the start of MTX or pamapimodtreatment. These laboratory abnormalities includedneutrophilia, defined as �9.25 � 109 cells/liter and a�20% increase from baseline at any postdose time point(13% of patients in the MTX group, 20% in the 50-mgpamapimod group, 12% in the 150-mg pamapimodgroup, and 25% in the 300-mg pamapimod group),hematuria (8% of patients in the MTX group, 18% inthe 50-mg pamapimod group, 10% in the 150-mg pam-apimod group, and 15% in the 300-mg pamapimodgroup), and leukocyturia (9% of patients in the MTXgroup, 8% in the 50-mg pamapimod group, 6% in the150-mg pamapimod group, and 13% in the 300-mgpamapimod group). Hematuria and leukocyturia weredefined on a scale of 1–4�; values �1� and increasedby �2� over baseline were recorded as abnormal.
Changes in laboratory parameters were observedin all 4 treatment groups. Notably, the MTX group andthe 300-mg pamapimod group showed changes frombaseline to the end of treatment in levels of alanineaminotransferase (ALT), aspartate aminotransferase(AST), and CPK. Substantially elevated ALT and/orAST values (i.e., values both �2 times the upper limit ofnormal [ULN] and �50% over baseline) occurred in 6MTX-treated patients (11%) and in a total of 12pamapimod-treated patients (8%) (3 patients in the150-mg group [6%] and 9 patients in the 300-mg group[19%]). In both MTX- and pamapimod-treated patients,elevated values generally occurred within 2 months afterstarting the study treatment.
Within the MTX group, 3 patients with normalALT and AST values at baseline discontinued studytreatment due to elevations of both ALT (4.2, 14.3, and4.0 times the ULN) and AST (2.9, 3.3, and 3.8 timesthe ULN). In all 3 patients, the elevated liver enzymelevels resolved without sequelae within 1 month fol-lowing discontinuation of MTX. Similarly, within thepamapimod-treated groups, 3 patients, all in the 300-mggroup and with normal values at baseline, discontinuedthe study drug due to elevated liver enzyme levels; all 3patients had elevated levels of ALT (3.3, 3.5, and 16.4times the ULN), and 2 of the patients had high levelsof AST (5.0 and 7.9 times the ULN). All events wereconsidered possibly or probably related to the studydrug. In 2 patients, these AEs resolved without sequelae
within 1 month following discontinuation of pamapi-mod; in the third patient, the event was consideredongoing at the time of last contact (30 days afterdiscontinuation of pamapimod). Patients with elevatedALT and AST values had concurrent total bilirubinvalues that remained within the normal range (0–17�moles/liter); there were no values �15.6 �moles/liter.
Elevations of CPK levels, ranging from 2.9-foldto 9.4-fold the ULN, were observed in 3 MTX-treatedpatients (6%) and in 4 pamapimod-treated patients(3%); of the latter patients, 3 in the 150-mg group hadvalues ranging from 2.0-fold to 6.2-fold the ULN, and 1in the 300-mg group had a value of 2.2-fold the ULN.Two patients, 1 in the MTX group and 1 in the 150-mgpamapimod group, each had 2 reports of elevated CPKvalues measured �30 days apart; all other patients hadonly a single episode. There was no trend in time of firstonset of elevated CPK levels; the range was 10–58 daysfollowing the start of treatment. No patients reportedsymptoms of muscle pain or weakness, and there wereno treatment discontinuations due to elevated CPKlevels.
There were no changes in RF, anti-CCP, anti-dsDNA, or immunoglobulin concentrations, lymphocytesubtypes, or antitetanus titer between baseline and 12weeks in any treatment group. There were no clinicallysignificant changes in EKG findings or vital signs duringthe study for any treatment group.
DISCUSSION
This 12-week study demonstrated that treatmentwith 50-, 150-, and 300-mg once-daily doses of pamapi-mod, a novel p38 MAPK inhibitor, resulted in lowerfrequencies of response according to the ACR improve-ment criteria and the DAS28, compared with MTXstarted at 7.5 mg/week and increased to 15 mg/week. The2 lower doses of pamapimod yielded similar frequenciesof ACR improvement response, and while the frequencyof this response with the 300-mg dose was higher thanthat with the lower doses, it was well below the fre-quency of ACR improvement response with MTX.Pamapimod was generally well tolerated, but the overallside effect profile suggested that the 300-mg dose wasmore toxic than the 2 lower doses or MTX.
The results of this study are important, since thisis the first large randomized clinical trial evaluating ap38 MAPK inhibitor for which efficacy and safety resultshave been reported. Several p38 inhibitors have beeninvestigated in the clinic. In terms of efficacy, VX-745was shown to result in a significantly better ACR20
342 COHEN ET AL

response than placebo in a phase II study, but develop-ment was stopped because of preclinical toxicity findings(18). Doramapimod (BIRB796), showed no efficacy inCrohn’s disease (19). Scio-469 was efficacious in therelief of dental pain (20) and has been tested in phaseIIa and IIb trials, but the results are unreported.
Why did pamapimod fail to show efficacy? Invitro, pamapimod selectively inhibits p38� MAPK andinhibits the production of TNF, IL-1, and IL-6 in bothhuman and rodent cell cultures. Pamapimod also inhib-its the in vitro release of constitutively expressed TNFfrom primary synovial explant cultures, with potencysimilar to that found in other in vitro assays (10). Thedrop in CRP levels in the first 2 weeks with pamapimodat the 300-mg dose suggests that at this dosage level,pamapimod may have suppressed proinflammatory cy-tokine pathways in the patients studied. However, at thelower dosage levels, pamapimod failed to suppress CRP,and the dosage levels were probably too low for thiseffect to occur. The weakening of CRP inhibition seenafter 2 weeks in the 300-mg pamapimod group suggeststhat inflammatory pathways may have been up-regulatedin response to the p38� blockade. Candidates for p38�bypass might include other p38 isoforms or NF-�B,ERK, activator protein 1, and/or JNK pathways. Asubstantial early decline in CRP levels with subsequentelevation was also seen with BIRB796 in Crohn’s disease(19). There was no decrease in plasma exposures ofpamapimod at any dosage level (data not shown), sug-gesting that metabolism did not account for the lack ofeffect. The AE profile seen with the 300-mg pamapimoddose (i.e., worse than that with MTX) mitigates againsttesting higher once-daily dosing levels for long-termtreatment of RA.
The dominant side effects seen in this trial areconsistent with preclinical toxicities and side effectsreported with other compounds. Pamapimod causedan increase in infections that were generally mild andprimarily of the upper respiratory tract and naso-pharynx, skin lesions usually described as folliculitis oracneiform rash, dizziness or vertigo, and GI AEs. Infec-tions are often seen with immunomodulatory compounds.The skin lesions may be infectious in origin, and p38 hasbeen implicated in maintenance of skin barrier function(21). However, skin lesions have been reported withother kinase inhibitors (22), so the precise mechanism isnot clear. Skin lesions (23) and dizziness (24) have alsobeen described with other p38 inhibitors. Since p38� isfound in the cerebellum (25), the maximumconcentration–related dizziness may be caused by inhi-bition of the molecular target. Finally, GI AEs have
been reported with other p38 inhibitors in smallerstudies and may be a target-related class toxicity perhapsassociated with infection or changes in gut flora. Eleva-tion of transaminase levels has occurred with p38 inhib-itors as well (19). Preclinical data suggest that p38 is alsofound in myocardium, developing tissues, skeletal mus-cle, and hematopoietic cells as well as in the immunesystem, liver, and central nervous system (26). However,there is no clear correlation between AEs in humans andthe first 4 of these tissues. At this point the p38 class sideeffect profile in RA patients appears to be dizziness, skinlesions, infections, GI AEs, and transaminase elevations.
While the results of this study are disappointing,over time the p38 inhibitors have become progressivelymore potent and selective (27). In addition, studies areongoing in neuropathic pain, chronic obstructive pulmo-nary disease, atherosclerosis, and oncologic indicationsas well as in RA (28). Different selectivity and phar-macokinetic profiles of other p38 inhibitors may offersome advantage over pamapimod. Alternatively, RAmay be the wrong indication for p38 inhibitors, as sepsiswas for the anti-TNF biologic agents. Additional studieswill be needed to define the clinical utility of this newclass of immunomodulators.
AUTHOR CONTRIBUTIONS
Dr. Cohen had full access to all of the data in the study andtakes responsibility for the integrity of the data and the accuracy of thedata analysis.Study design. Cohen, Caulfield.Acquisition of data. Cheng, Chindalore, Damjanov, Burgos-Vargas,DeLora, Zimany.Analysis and interpretation of data. Cohen, Damjanov, DeLora,Zimany, Travers, Caulfield.Manuscript preparation. Cohen, Chindalore, Damjanov, Burgos-Vargas, DeLora, Zimany, Travers, Caulfield, D. M. Lidgate (non-author; Roche), S. Peng (nonauthor; Roche).Statistical analysis. DeLora, Travers, M. Rabbia (nonauthor; Roche).Medical monitoring. S. Wax (nonauthor; Roche).Programming. J. Chen (nonauthor; Roche), X. Y. Lue (nonauthor;Roche).
ROLE OF THE STUDY SPONSOR
The study was funded by Hoffman-La Roche (Nutley, NJ)and managed by Roche personnel at several locations (Nutley, NJ;Welwyn Garden City, UK; and Palo Alto, CA). The study wasdesigned by clinicians and scientists at Roche with input from theinvestigators. Data were collected by the investigators and entered intoa database that was maintained by Roche. The data were analyzed byRoche statisticians and programmers. Dr. Caulfield and Dr. Cohenwrote a draft of the manuscript with assistance from a medical writer.All authors reviewed, offered revisions, and approved the content ofthe manuscript before submission. All authors agreed to submit thefinal version of the manuscript.
PAMAPIMOD VERSUS MTX IN PATIENTS WITH ACTIVE RA 343

REFERENCES
1. Goldstein DM, Gabriel T. Pathway to the clinic: inhibition of p38MAP kinase. A review of ten chemotypes selected for develop-ment. Curr Top Med Chem 2005;5:1017–29.
2. Gracie JA, Leung BP, McInnes IB. Novel pathways that regulatetumor necrosis factor-� production in rheumatoid arthritis. CurrOpin Rheumatol 2002;14:270–5.
3. Salituro FG, Germann UA, Wilson KP, Bemis GW, Fox T, Su MS.Inhibitors of p38 MAP kinase: therapeutic intervention incytokine-mediated diseases. Curr Med Chem 1999;6:807–23.
4. Herlaar E, Brown Z. p38 MAPK signalling cascades in inflamma-tory disease. Mol Med Today 1999;5:439–47.
5. Guan Z, Buckman SY, Pentland AP, Templeton DJ, MorrisonAR. Induction of cyclooxygenase-2 by the activated MEKK13SEK1/MKK43p38 mitogen-activated protein kinase pathway.J Biol Chem 1998;273:12901–8.
6. Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors—mechanisms and therapeuticpotentials. Pharmacol Ther 1999;82:389–97.
7. Hale KK, Trollinger D, Rihanek M, Manthey CL. Differentialexpression and activation of p38 mitogen-activated protein kinase�, �, �, and � in inflammatory cell lineages. J Immunol 1999;162:4246–52.
8. Schett G, Tohidast-Akrad M, Smolen JS, Schmid BJ, Steiner CW,Bitzan P, et al. Activation, differential localization, and regula-tion of the stress-activated protein kinases, extracellular signal–regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoidarthritis. Arthritis Rheum 2000;43:2501–12.
9. Korb A, Tohidast-Akrad M, Cetin E, Axmann R, Smolen J, SchettG. Differential tissue expression and activation of p38 MAPK �, �,�, and � isoforms in rheumatoid arthritis. Arthritis Rheum 2006;54:2745–56.
10. Hill RJ, Dabbagh K, Phippard D, Li C, Suttmann RT, Welch M,et al. Pamapimod, a novel p38 MAP kinase inhibitor: preclinicalanalysis of efficacy and selectivity. J Pharmacol Exp Ther 2008.E-pub ahead of print.
11. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF,Cooper NS, et al. The American Rheumatism Association 1987revised criteria for the classification of rheumatoid arthritis.Arthritis Rheum 1988;31:315–24.
12. Fries JF, Spitz P, Kraines RG, Holman HR. Measurement ofpatient outcome in arthritis. Arthritis Rheum 1980;23:137–45.
13. Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, GroberJ. Validation of the Functional Assessment of Chronic IllnessTherapy Fatigue Scale relative to other instrumentation in patientswith rheumatoid arthritis. J Rheumatol 2005;32:811–9.
14. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D,Goldsmith C, et al. American College of Rheumatology prelimi-nary definition of improvement in rheumatoid arthritis. ArthritisRheum 1995;38:727–35.
15. Prevoo ML, van ’t Hof MA, Kuper HH, van Leeuwen MA, van dePutte LB, van Riel PL. Modified disease activity scores thatinclude twenty-eight–joint counts: development and validation in aprospective longitudinal study of patients with rheumatoid arthri-tis. Arthritis Rheum 1995;38:44–8.
16. Van Gestel AM, Prevoo ML, van ’t Hof MA, van Rijswijk MH, vande Putte LB, van Riel PL. Development and validation of theEuropean League Against Rheumatism response criteria for rheu-matoid arthritis: comparison with the preliminary American Col-lege of Rheumatology and the World Health Organization/International League Against Rheumatism criteria. ArthritisRheum 1996;39:34–40.
17. Felson DT, Anderson JJ, Boers M, Bombardier C, Chernoff M,
Fried B, et al. The American College of Rheumatology prelimi-nary core set of disease activity measures for rheumatoid arthritisclinical trials. Arthritis Rheum 1993;36:729–40.
18. Weisman M, Furst D, Schiff M, Kauffman R, Merica E, Martin-Munley S, et al. A double-blind, placebo-controlled trial of VX-745, an oral p38 mitogen activated protein kinase (MAPK)inhibitor, in patients with rheumatoid arthritis (RA) [abstract].Presented at the 2002 annual European Congress of Rheumatol-ogy; 2002 June 12–15; Stockholm, Sweden. Abstract FRI0018.URL: http://www.eular.org.
19. Schreiber S, Feagan B, D’Haens G, Colombel JF, Geboes K,Yurcov M, et al, BIRB 796 Study Group. Oral p38 mitogen-activated protein kinase inhibition with BIRB 796 for activeCrohn’s disease: a randomized, double-blind, placebo-controlledtrial. Clin Gastroenterol Hepatol 2006;4:325–34.
20. Tong SE, Daniels SE, Nontano T, Chang S, Desjardins P. Scio-469, a novel p38� MAPK inhibitor, provides efficacy in acutepost-surgical dental pain. Clin Pharmacol Ther 2004;75:3.
21. Kobayashi H, Aiba S, Yoshino Y, Tagami H. Acute cutaneousbarrier disruption activates epidermal p44/42 and p38 mitogen-activated protein kinases in human and hairless guinea pig skin.Exp Dermatol 2003;12:734–46.
22. Laffitte E, Saurat JH. Kinase inhibitor-induced pustules. Derma-tology 2005;211:305–6.
23. Ding C. Drug evaluation: VX-702, a MAP kinase inhibitor forrheumatoid arthritis and acute coronary syndrome. Curr OpinInvestig Drugs 2006;7:1020–5.
24. Nikas SN, Drosos AA. SCIO-469 Scios Inc. Curr Opin InvestigDrugs 2004;5:1205–12.
25. Lee SH, Park J, Che Y, Han PL, Lee JK. Constitutive activity anddifferential localization of p38� and p38� MAPKs in adult mousebrain. J Neurosci Res 2000;60:623–31.
26. Dambach DM. Potential adverse effects associated with inhibitionof p38�/� MAP kinases. Curr Top Med Chem 2005;5:929–39.
27. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE,Campbell BT, et al. A quantitative analysis of kinase inhibitorselectivity. Nat Biotechnol 2008;26:127–32.
28. Clinical Trials.gov. A service of the US National Institutes ofHealth. URL: http://clinicaltrials.gov.
APPENDIX A: INVESTIGATORS
Investigators who recruited patients for this study were asfollows: Canada: A. Bookman (Toronto), D. Sholter (Edmonton);Croatia: D. Martinovic Kaliterna (Split), S. Novak (Rijeka); CzechRepublic: S. Augustinova (Ceske Budejovice), J. Vencovsky (Prague),P. Vitek (Zlin), R. Zahora (Terezin); France: C. Jorgensen (Montpel-lier), M. Nguyen (Paris); Italy: C. M. Montecucco (Pavia), B. Seriolo(Genoa), G. Valesini (Rome); Mexico: H. Avila-Armengol (Guadala-jara), R. Burgos-Vargas (Mexico City), C. Ramos-Remus (Guadala-jara); Romania: G. Mirea (Brascov), C. Tanasescu (Bucuresti); Serbia/Montenegro: N. Damjanov (Belgrade); South Africa: F. Khatib(Johannesburg), D. Nel (Pretoria), B. J. van Rensburg (Bloemfontein);Spain: F. Blanco (La Corunda), S. Marsal (Barcelona), R. Queiro(Oviedo); Taiwan: T.-T. Cheng (Kaohsiung), S. Luo (Taoyuan);United States: J. Anderson (Wichita, KS), R. Capps (Knoxville, TN),V. Chindalore (Anniston, AL), S. B. Cohen (Dallas, TX), K. Colburn(Loma Linda, CA), G. M. Eisenberg (Morton Grove, IL), J. Habros(Scottsdale, AZ), T. Harrington (Danville, PA), H. Holt (Memphis,TN), S. Ishaq (Covington, LA), D. Kirby (Belmont, NC), M. J. Maricic(Tucson, AZ), C. Matejicka (Lancaster, PA), D. Radin (Stamford,CT), G. Roane (Charleston, SC), D. Sandoval (Bend, OR), N. Strani-ero (South Bend, IN), M. Thurmond Anderle (Amarillo, TX),C. Young (Escondido, CA).
344 COHEN ET AL




















