
Evaluation of a novel method based on amplification of DNA fragments surrounding rare restriction...
11
Evaluation of a novel method based on amplification of DNA fragments surrounding rare restriction sites (ADSRRS fingerprinting) for typing strains of vancomycin-resistant Enterococcus faecium Beata Krawczyk a , Krzysztof Lewandowski a , Marek Bronk b , Alfred Samet b , Przemysl aw Myjak c , Jo ´zef Kur a, * a Department of Microbiology, Technical University of Gdan ´sk, ul. G. Narutowicza 11/12, 80-952 Gdan ´sk, Poland b Department of Clinical Bacteriology, State Hospital No. 1, ul. De ˛binki 7, 80-211 Gdan ´sk, Poland c Institute of Maritime and Tropical Medicine, 81-519 Gdynia, ul. Powstania Styczniowego 9 B, Poland Received 17 April 2002; received in revised form 23 July 2002; accepted 6 September 2002 Abstract In the search for an effective DNA-typing technique for use in hospital epidemiology, the performance and convenience of a novel assay based on the fingerprinting of bacterial genomes by amplification of DNA fragments surrounding rare restriction sites (ADSRRS fingerprinting) was tested. A large number of vancomycin-resistant Enterococcus faecium (VREM) isolates from haematological ward patients of the Clinical Hospital in Gdan ´sk were examined. We found that ADSRRS fingerprinting analysis is a rapid method that offers good discriminatory power. The method demonstrated also excellent reproducibility. The usefulness of the ADSRRS fingerprinting method for molecular typing was compared with pulsed field gel electrophoresis (PFGE) method, which is currently considered the gold standard for molecular typing of isolates recovered from patients and the environment in the course of investigation and control of nosocomial outbreaks. Clustering of ADSRRS fingerprinting data matched pulsed field gel electrophoresis data. The features of ADSRRS fingerprinting technique is discussed in comparison with conventional methods. Data presented here demonstrate the complexity of the epidemiological situation concerning VREM that may occur in a single medical ward. D 2002 Elsevier Science B.V. All rights reserved. Keywords: Enterococcus faecium; PCR; PCR fingerprinting; PCR suppression; Vancomycin-resistant enterococci 1. Introduction Over the past 10 years, a number of Enterococcus strains with high-level inducible resistance to vanco- mycin have been identified, and the relative incidence of these strains has increased sharply in the last years. In addition, many reports of nosocomial outbreaks of infection with VRE have been published, especially from North America and Europe (Karanfil et al., 1992; Handwerger et al., 1993; Jordens et al., 1994; Montecalvo et al., 1994). Recently, the isolation of a vancomycin-resistant Enterococcus faecium (VREM) in Poland was reported (Samet et al., 1999; Hrynie- 0167-7012/02/$ - see front matter D 2002 Elsevier Science B.V. All rights reserved. PII:S0167-7012(02)00187-2 * Corresponding author. Tel./fax: +48-58-3471822. E-mail address: [email protected] (J. Kur). www.elsevier.com/locate/jmicmeth Journal of Microbiological Methods 52 (2003) 341 – 351
-
Upload
independent -
Category
Documents
-
view
7 -
download
0
Transcript of Evaluation of a novel method based on amplification of DNA fragments surrounding rare restriction...

Evaluation of a novel method based on amplification of DNA
fragments surrounding rare restriction sites (ADSRRS
fingerprinting) for typing strains of vancomycin-resistant
Enterococcus faecium
Beata Krawczyka, Krzysztof Lewandowskia, Marek Bronkb, Alfred Sametb,Przemyslaw Myjakc, Jozef Kura,*
aDepartment of Microbiology, Technical University of Gdansk, ul. G. Narutowicza 11/12, 80-952 Gdansk, PolandbDepartment of Clinical Bacteriology, State Hospital No. 1, ul. Debinki 7, 80-211 Gdansk, Poland
c Institute of Maritime and Tropical Medicine, 81-519 Gdynia, ul. Powstania Styczniowego 9 B, Poland
Received 17 April 2002; received in revised form 23 July 2002; accepted 6 September 2002
Abstract
In the search for an effective DNA-typing technique for use in hospital epidemiology, the performance and convenience of a
novel assay based on the fingerprinting of bacterial genomes by amplification of DNA fragments surrounding rare restriction
sites (ADSRRS fingerprinting) was tested. A large number of vancomycin-resistant Enterococcus faecium (VREM) isolates
from haematological ward patients of the Clinical Hospital in Gdansk were examined. We found that ADSRRS fingerprinting
analysis is a rapid method that offers good discriminatory power. The method demonstrated also excellent reproducibility. The
usefulness of the ADSRRS fingerprinting method for molecular typing was compared with pulsed field gel electrophoresis
(PFGE) method, which is currently considered the gold standard for molecular typing of isolates recovered from patients and
the environment in the course of investigation and control of nosocomial outbreaks. Clustering of ADSRRS fingerprinting data
matched pulsed field gel electrophoresis data.
The features of ADSRRS fingerprinting technique is discussed in comparison with conventional methods. Data presented
here demonstrate the complexity of the epidemiological situation concerning VREM that may occur in a single medical ward.
D 2002 Elsevier Science B.V. All rights reserved.
Keywords: Enterococcus faecium; PCR; PCR fingerprinting; PCR suppression; Vancomycin-resistant enterococci
1. Introduction
Over the past 10 years, a number of Enterococcus
strains with high-level inducible resistance to vanco-
mycin have been identified, and the relative incidence
of these strains has increased sharply in the last years.
In addition, many reports of nosocomial outbreaks of
infection with VRE have been published, especially
from North America and Europe (Karanfil et al.,
1992; Handwerger et al., 1993; Jordens et al., 1994;
Montecalvo et al., 1994). Recently, the isolation of a
vancomycin-resistant Enterococcus faecium (VREM)
in Poland was reported (Samet et al., 1999; Hrynie-
0167-7012/02/$ - see front matter D 2002 Elsevier Science B.V. All rights reserved.
PII: S0167 -7012 (02 )00187 -2
* Corresponding author. Tel./fax: +48-58-3471822.
E-mail address: [email protected] (J. Kur).
www.elsevier.com/locate/jmicmeth
Journal of Microbiological Methods 52 (2003) 341–351

wicz et al., 1999; Kawalec et al., 2000). The E.
faecium PCR-based specific diagnostic assay was
used for confirmation of the phenotype identification
of E. faecium (Cheng et al., 1997) and the multiplex
PCR-restriction fragment length polymorphism me-
thod for type of Van resistance (Patel et al., 1997). To
determine whether the isolates were epidemiologically
related, isolated strains were differentiated by PCR
fingerprinting method (Samet et al., 1999). The
applied PCR fingerprinting system allowed for the
differentiation of the clinical isolates from the Hae-
matological Unit. Two main strains (genotypes) were
identified. The PCR fingerprinting of VREM from the
Haematological Unit demonstrated only small genetic
heterogeneity among the isolates over 11 months, with
two strains being identified. These strains were genet-
ically closely related. In the present study, 100 VREM
strains (including 25 strains described previously;
Samet et al., 1999) within a duration of 36 months
(between January 1997 and December 1999) taken
from 100 patients were examined using a novel
fingerprinting method (ADSRRS fingerprinting)
described by Masny and Plucienniczak (2001). The
utility of the ADSRRS fingerprinting method was
evaluated with data obtained using pulsed field gel
electrophoresis (PFGE) method.
Genomic fingerprints are increasingly used to
study relationships at the intra- or even interspecific
level. The fingerprints are obtained by visualising
many parts of the genome. Differences in these finger-
prints between individuals are interpreted as genetic
distances. Obviously, the differences should reflect
variations in DNA rather than artifacts due to a non-
robust method. Furthermore, the method should pro-
vide the appropriate level of discriminatory power and
it should be relatively rapid and cheap, especially in
large-scale population genetic studies.
Macrorestriction analysis of genomic DNA fol-
lowed by pulsed field gel electrophoresis (PFGE)
has become the ‘‘gold standard’’ for molecular typing.
However, PFGE is limited in its resolving power
(Gerner-Smidt et al., 1998), and this contributes to
difficulties with gel-to-gel and interlaboratory repro-
ducibility (Van Belkum et al., 1998).
A variety of PCR-based methods for displaying
DNA sequence polymorphism have been developed.
Some of the methods such as RAPD (Williams et al.,
1990) and AFLP (Vos et al., 1995) do not require prior
knowledge of the DNA sequence. RAPD is generally
regarded to be less time consuming, while the AFLP
method is more robust (Mueller and Wolfenbarger,
1999; Pejic et al., 1998). RAPD allows detection of
DNA polymorphisms between strains of a species but
does not exhibit high rates of reproducibility. The
performance of RAPD is sensitive to many factors
such as selection of primers, magnesium concentra-
tion in the PCR buffers and the thermocycler used for
PCR (De Zoysa and Efstratiou, 1999). An amplified
restriction fragment polymorphism (AFLP) over-
comes many of the problems of RAPD. There are
three major steps in the AFLP procedure: (i) restric-
tion endonuclease digestion of genomic DNA and the
ligation of specific adapters; (ii) amplification of the
restriction fragments by PCR using primer pairs con-
taining common sequences of the adapter and one to
three arbitrary nucleotides; (iii) analysis of the ampli-
fied fragments using gel electrophoresis. The combi-
nation of different restriction enzymes and the choice
of selective nucleotides in the primers for PCR make
AFLP a useful system for molecular typing of micro-
organisms but requires the use of sequencing gels and
usually labeled primers because of the quantity of
simultaneously amplified DNA fragments.
Here, we show evaluation of a novel fingerprinting
method described by Masny and Plucienniczak (2001)
based on the fingerprinting of bacterial genomes by
amplification of DNA fragments surrounding rare
restriction sites (ADSRRS fingerprinting) for epide-
miological studies. This method is based on the
digestion of total bacterial DNA with two restriction
enzymes differing in cleavage frequency, ligation with
two different oligonucleotide adapters and suppres-
sion of PCR (Lukyanov et al., 1994; Diatchenko et al.,
1996; Shagin et al., 1999). PCR suppression allows
the amplification of only a limited subset of DNA
fragments, as only those with two different oligonu-
cleotides ligated at the ends of complementary DNA
strands are amplified in the PCR. The method does
not require prior knowledge of the sequence of the
analyzed DNA and generates a limited number of
DNA fragments, whose band pattern on the gel differs
between strains of a bacterial species. Furthermore,
the DNA fragments can be easily analyzed on poly-
acrylamide gels stained with ethidium bromide. We
have implemented this method using a set of clinical
vancomycin-resistant E. faecium (VREM) isolated
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351342

from Haematological Unit patients of the Clinical
Hospital in Gdansk.
2. Materials and methods
2.1. Isolates and patients
The first VREM isolate was cultured in the Uni-
versity Hospital in Gdansk from the urinary tract
infection of a patient located in the Haematological
Unit in December 1996. From January 1997 to Decem-
ber 1999, about 15,000 clinical samples were exam-
ined for the presence of E. faecium isolates and the
isolates were tested for the resistance to vancomycin
and teicoplanin. A total of 587 E. faecium isolates were
recovered and 344 of them were vancomycin- and
teicoplanin-resistant. A hundred isolates were chosen
for further examination by molecular typing methods
(one isolate from one patient). VREM were isolated
from blood (22), urine (13), stool (58), sputum (2), pus
(2), skin (1), vagina (1) and throat (1). Some clinical
data of patients colonised or infected with vancomy-
cin-resistant E. faecium are presented in Table 1.
2.2. DNA isolation
DNA isolations (from 1.5 ml of culture) were
carried out with the Genomic DNA Prep Plus (A&A
Biotechnology, Poland) according to the manufactures
procedure with minor modifications. For disruption of
enteroccocal cells, the incubation at 37 jC for 30 min,
before DNA isolation, with 2.5 mg of lysosyme
(Sigma-Aldrich Chemie, Steinheim, Germany) and
100 Ag lysostaphin (Sigma, St. Louis, MO, USA)
per 1 ml of TE buffer was applied. DNA quantities in
the samples were estimated by electrophoresis of 1 AlDNA solution on 1% agarose (Sigma) gels run
together with samples with known amounts of DNA
and subsequent ethidium bromide staining (Sigma-
Aldrich) in 0.5 mg/l solution for 10–15 min. The
DNA concentration range was from about 100 ng/Alto several hundred nanograms per microliter.
2.3. PCR
A PCR assay for identification of E. faecium and
primers used were the same as in Cheng et al. (1997),
with some modifications as described by Samet et al.
(1999). A multiplex PCR-restriction fragment length
polymorphism (MPCR/RFLP) assay for Van-type
identification of E. faecium isolates were carried out
according to Patel et al. (1997). Amplification prod-
ucts obtained with Van-specific primers were further
analysed by digestion with MspI restriction endonu-
clease (RFLP).
2.4. ADSRRS fingerprinting
Enterococcal DNA (100–500 ng) was digested
with a combination of two enzymes: XbaI (10 U/Al)(Sigma) and BglII (10 U/Al) (Sigma) for 2–3 h at 37
Table 1
Selected clinical details of patients infected or colonised with vancomycin-resistant E. faecium
Year of Age/sex/ Diagnosisa/ Source/no. of isolates
isolation no. of patient no. of patientInfection Colonisation
1997 19–54/F/14, 16–61/M/19 AML/14, CML/14,
ALL/4, NHL/1
blood/9, urine/2 stool/20, sputum/1, throat/1
1998 20–59/F/14, 20–77/M/18 AML/14, CML/7,
ALL/6, NHL/2, AA/3
blood/6, urine/6 stool/18, vagina/1, skin/1
1999 9–77/F/13, 4–52/M/22 AML/11, CML/12,
ALL/3, NHL/3, AA/1,
CABG/3, UTI/1, TR/1
blood/7, urine/5 stool/20, sputum/1, pus/2
1997–1999 9–77/F/41, 4–77/M/59 AML/39, CML/33, ALL/13,
NHL/6, AA/4, CABG/3,
UTI/1, TR/1
blood/22, urine/13,
pus/2
stool/58, sputum/2, throat/1,
vagina/1, skin/1
Total 100
a AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; ALL, acute lymphoid leukaemia; NHL, non-Hodgkin’s lymphoma;
AA, aplastic anaemia; CABG, coronary artery bypass graft; UTI, urinary tract infection; TR, trauma.
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351 343

jC in a total volume 50 Al of appropriate buffer. To
each of the digested samples, 20 Al 3 M NaAc, 150 AlTE buffer, 400 Al 96% ethanol and 3 Al glycogen (20
mg/ml) (Sigma) were added. Samples were vortexed,
mixed and incubated at � 20 jC for 60 min, then
centrifuged at 12,000� g for 10 min. Ethanol was
removed and the pellets were washed with 200 Al of70% ethanol, then centrifuged for 5 min at 12,000� g
and again ethanol was removed, and pellets were
dried for 20 min at room temperature.
Adapters were assembled from two oligonucleoti-
des (Table 2). Oligonucleotides were dissolved in
water to concentration of 20 pmol/Al (each oligonu-
cleotide), then heated at 90 jC in a water bath for 2 min
and subsequently left at room temperature for 10 min to
anneal. Appropriate adapters were ligated to the cor-
responding cohesive ends (Table 2 and Fig. 1). Dry
pellets of DNA digests were dissolved in a solution
consisting of 2 Al 10� ligation buffer (66 mM Tris–
HCl, pH 8.5, 6.6 mM MgCl2, 10 mM DTT, 66 AMATP), 1 Al (20 pmol each adapter) of the solution of the
adapters corresponding to the cohesive ends left by the
enzymes used for the prior digestion of the sample, 0.5
Al (1 U/Al) T4 DNA ligase (Epicentre, USA), 2 Al 50%PEG 4000 solution and water to 20 Al. The ligation
reactions were carried out at 16 jC for 2 h. After
ligation, 80 Al of TE buffer was added, and DNAwas
isopropanol precipitated according to standard proce-
dure. After centrifugation at 12,000� g for 10 min, the
DNA pellets were dissolved in 20 Al TE buffer.
The PCR reaction was carried out in a 50-Alreaction mixture containing 2 Al ligation solution, 5
Al PCR buffer (100 mM Tris–HCl, pH 8.8, 500 mM
KCl, 20 mM MgCl2, 1% Triton X-100), 5 Al of a
deoxynucleoside triphosphate (dNTP) mixture (con-
centration of each dNTP, 2.5 mM), 10 Al betaine (5 M
solution; Sigma), 1 Al (1U) of Taq polymerase
(Shark2, DNA Gdansk II, Poland) or 1 Al (1U) of
Pwo polymerase (DNA Gdansk II), 50 pM each RC
and FC primer (Table 2) and water to 50 Al. Thermal
cycl ing was performed in a Perkin-Elmer
GENEAMPR PCR System 2400 or Hot-Shot24 ther-
mal cycler (DNA Gdansk II). The following thermal
profile was applied: an initial cycle at 94 jC for 5 min
and then at 72 jC for 5 min for filling the ends of the
DNA fragments, followed by denaturation at 94 jCfor 5 min, then 19 cycles of 94 jC for 30 s, 62 jC for
30 s, 72 jC for 90 s, followed by 5 min at 72 jC. PCRproducts (10 of 50 Al) were electrophoresed on 6%
polyacrylamide gels with TBE buffer, stained in
ethidium bromide at 0.5 mg/l aqueous solution for
10–15 min and images of the gels were documented
by photographing using a White/Ultraviolet Trans-
illuminator. The patterns obtained from the electro-
pherograms were converted and analyzed using the
Quanty One software, version 4.3.1 (Bio-Rad, USA).
A UPGMA dendrogram was generated using Dice
correlation coefficient in the Quanty One software.
The reproducibility of the technique was examined by
performing three ADSRRS fingerprinting runs for
each isolate with three separate DNA extractions
and by using two different thermal cyclers (Perkin-
Elmer GENEAMPR PCR System 2400 or Hot-
Shot24 thermal cycler, DNA Gdansk II).
2.5. PFGE
PFGE was performed using Bio-Rad’s GenePath
Group 1 kits. A single colony from a 24-h isolate was
grown overnight in LB under aerobic conditions at 37
jC. Cells were collected and resuspended in cell
suspension buffer. The suspension was mixed with
Table 2
Adapters and PCR primers used in this study
Adaptersa and primers Nucleotides sequences
XbaI short adapter 5V-CTAGGTCGACGTT-3V3V-CAGCTGCAACCACCTACTTCC-5V
oligo-Xba helper,
oligo-Xba-lig
BglII long adapter 5V-GATCCGTCGACAACGGCGTTCCTTCGTCTACCATCC-3V3V-GCAGCTGTTGCCGCAAGGAAGCAGATGGTAGG-5V
oligo-Bgl helper,
oligo-Bgl-lig
XbaI short primer
(oligo-Xba-lig)
5V-CCTTCATCCACCAACGTCGAC-3V
BglII long primer 5V-GGATGGTAGACGAAGGAACGC-3Va Adapters are double stranded but only their constant parts (italic) are ligated, helper parts serve just to create dsDNA fragment and
protruding 5Vend.
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351344
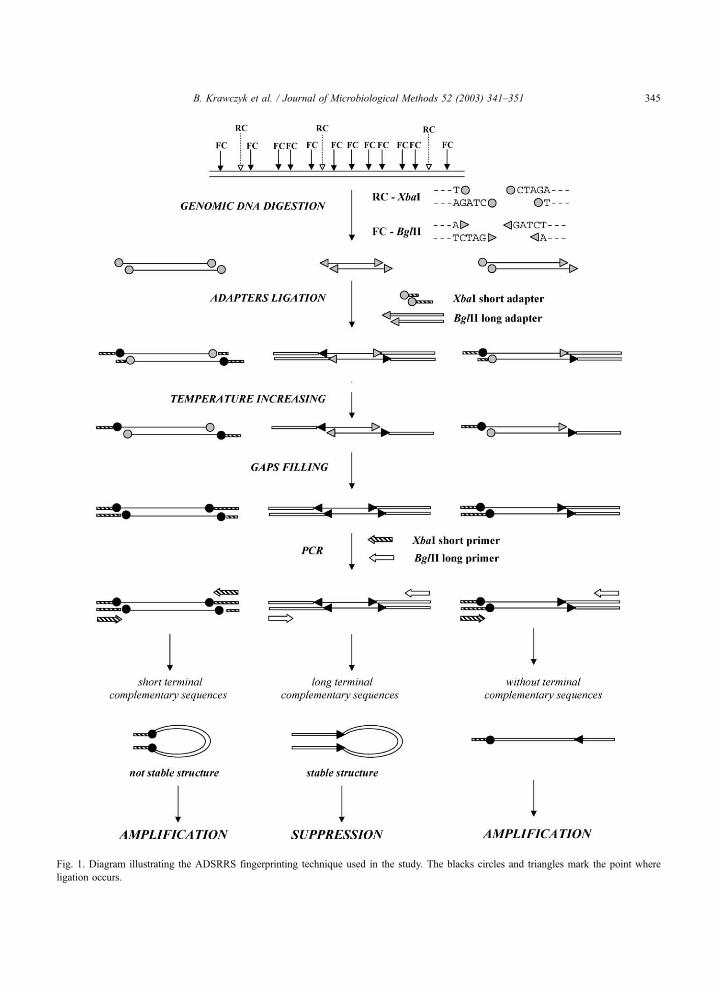
Fig. 1. Diagram illustrating the ADSRRS fingerprinting technique used in the study. The blacks circles and triangles mark the point where
ligation occurs.
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351 345

lysozyme–lysostaphin and embedding agarose, and
plugs were made. Plugs were placed in lyses buffer
with additional lysozyme–lysostaphin and incubated
for 3 h at 37 jC without agitation. The plugs were
washed, placed in proteinase K solution and incubated
overnight at 50 jC without agitation. Plugs were
washed four times with wash buffer for 30–60 min at
room temperature on a rocker.
DNA was digested overnight with 25 U of XbaI
(Sigma) at 37 jC. DNAwas separated on an agarose gel
using the GenePath instrument (based on the pulsed
field method of contoured clamped homogenous elec-
tric field). The run time was 20 h. Gels were stained
with ethidium bromide. The patterns obtained from the
electropherograms were converted and analyzed using
the Quanty One software, version 4.3.1 (Bio-Rad).
3. Results
3.1. PCR identification of E. faecium and Van-type
To confirm the phenotypic identification, the PCR
identification of E. faecium and Van-type of antibiotic
resistance was carried out. Using the EM1A and EM1B
primers (Cheng et al., 1997; Samet et al., 1999), a
specific 658-bp DNA product, upon PCR amplification
of DNA from all isolates identified as E. faecium by
standard biochemical assays, was identified (results not
shown). No amplification was observed with isolates
identified as E. faecalis or E. gallinarum. These results
confirmed that examined isolates in fact belong to E.
faecium. Next, the convenient multiplex PCR-restric-
tion fragment length polymorphism (MPCR/RFLP)
assay to detect and discriminate vanA, vanB and
vanC-1 genes according to Patel et al. (1997) was
applied. All examined clinical isolates, phenotypically
identified as vancomycin-resistant VanA-type of the E.
faecium, yielded the 885-bp amplicon, which is char-
acteristic for vanA and vanB genes. The amplified
DNAs from all isolates digested with MspI restriction
enzyme gave distinct electrophoretic patterns for vanA
gene (231, 184, 163, 133 and 131 bp restriction frag-
ments) (results not shown). These experiments con-
firmed that the isolates belong in fact to VanA-type of
the vancomycin resistance.
3.2. ADSRRS fingerprinting analysis
We applied the newly discovered technique for
fingerprinting of bacterial strains described by Masny
and PlCucienniczak (2001), with some minor modifica-
tions. Their proposal is to amplify exclusively DNA
fragments surrounding relatively rare nucleotide
sequences (e.g., rare restriction sites) and the PCR
suppression (SP PCR) phenomenon is the basis for
obtaining limited representation of the DNA fragments
that form the bacterial genome. The outline of that
method applied in our experiments is shown in Fig. 1.
A genomic total DNA is digested with two restriction
enzymes, rare (XbaI, RC) and frequent (BglII, FC)
cutters. Three kinds of DNA fragments—abundant,
sporadic and limited arise that are formed after diges-
tion with frequent, rare and both cutters at the same
time, respectively. The mixture of DNA fragments is
ligated with two different synthetic adapters (XbaI
short adapter and BglII long adapter). All 5Vends of
the most abundant DNA fragments produced by
digestion with a frequent cutter (FC) are modified by
joining the same synthetic oligonucleotide (oligo-Bgl-
lig). Similarly, sporadic fragments generated by diges-
tion with a rare cutter are modified by ligation of
oligonucleotide oligo-Xba-lig to both 5Vends of eachdsDNA fragment. After filling in of the modifying
oligonucleotides joined to the 5Vends with DNA
polymerase all single-stranded abundant and sporadic
DNA fragments have complementary sequences on
their 5Vand 3Vends and because of that, the proper
usage of suppression PCR (SP PCR) during amplifi-
cation of the genomic fragment mixture should elim-
inate the most and least abundant DNA fragments from
the mixture. However, fragments arising after diges-
Fig. 2. Panel A: ADSRRS fingerprints and UPGMA dendrogram of the vancomycin-resistant E. faecium strains (representative results). The
lane designated M contained molecular mass marker (1008, 883, 615, 517, 466 bp). Lanes 1–10: strains isolated in 1997 at the beginning of
outbreak (from urine—lane 1, from blood—lanes 6 and 9, from sputum—lane 3, from stool—lanes 2, 4, 5, 7, 8 and 10). The remaining lanes
show ADSRRS fingerprints of the sporadic strains representing patterns of the D, E, F, G and H groups. ADSRRS fingerprinting types are given
above each lane. The DNA fragments were electrophoresed in 8% polyacrylamide gel by using 1�Tris–borate EDTA running buffer at a field
strength of 8 V cm� 1. Panel B: PFGE profiles and UPGMA dendrogram of the E. faecium strains (representative results). Numbers refer to
strains shown in panel A. Chromosomal DNA was digested with XbaI, and the fragments were fractionated on a 0.9% agarose gel.
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351346

Fig. 2.
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351 347

Fig. 2 (continued).
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351348

tion with rare and frequent restriction enzymes at the
same time and consequently joined with two distinct
modifying oligonucleotides, oligo-Xba-lig oligo-Bgl-
lig, are amplified exponentially. After filling in of the
modifying oligonucleotides joined to the 5V ends with
DNA polymerase, single-stranded fragments do not
have complementary sequences on their 5V and 3Vendsand consequently are not susceptible to SP PCR.
The obtained ADSRRS fingerprinting patterns for
representative isolates are presented on the Fig. 2A.
Each pattern consists of approximately 20–25 frag-
ments in the size range of 50–2000 bp. ADSRRS
fingerprinting patterns of the 100 VREM found eight
unique profiles represented by A to H groups in Table 3
and Fig. 2A. Isolates of VREM recovered from patients
in the Haematological Unit in 1997 were typed into
ADSRRS fingerprinting groups A, B, C, D and E.
Groups A, B, C, D, F, G, H and groups A, B, C, G, H
were detected in 1998 and 1999, respectively. Strains
representing group E disappeared in 1998 and group D
in 1999. Groups F, G and H appeared later in 1998.
Group F is identified only in 1998 and is represented
by two isolates from stool. Two groups, A and B, were
markedly predominant, as these were represented
by 41% and 25%, respectively. Dice coefficients of
pattern similarity of the ADSRRS fingerprints revealed
high similarity of 0.83 only for the isolates of groups
A and B.
It was investigated next whether similar typing
results might be obtained by the use of different
restriction enzymes and oligonucleotide adapters.
ADSRRS fingerprinting analysis with NotI and StyI
enzymes and appropriate adapters exhibited the same
discriminatory power (results not shown).
3.3. ADSRRS fingerprinting validation
The validation of the ADSRRS fingerprinting
method for epidemiological investigation was based
on a set of 10 VREM isolates with comprehensive
epidemiological, microbiological and molecular typ-
ing data from previously described outbreak (Samet et
al., 1999), and eight isolates with no epidemiological
linkage (vancomycin-sensitive E. faecium isolates
from different hospitals). The applied PCR finger-
printing method enabled differentiation of two main
clones genetically closely related (Samet et al., 1999).
Analysis of the ADSRRS fingerprinting data obtained
with those VREM isolates correctly identified the
outbreak isolates and grouped them also into two
groups (results not shown). As expected, the unrelated
epidemiologically isolates show a high degree of
diversity with unique pattern of bands for each strain
(results not shown).
We also validated the reproducibility of the
ADSRRS fingerprinting method. All VREM isolates
were taken from pure culture to ADSRRS fingerprint
on three separate occasions to assess reproducibility
of the method. The resulting ADSRRS fingerprints
from each separate run produced identical profiles.
There was small variation in peak heights but this did
not result in the gain or loss of any information.
Table 3
E. faecium VREM typing by ADSRRS fingerprinting
Year of
isolation
Source/no.
of isolates
ADSRRS fingerprinting groups/
no. of isolates
A B C D E F G H
1997 Infection
blood/9 3 3 1 1 1
urine/2 2
Colonisation
stool/20 12 3 2 1 2
sputum/1 1
throat/1 1
1998 Infection
blood/6 3 2 1
urine/6 3 2 1
Colonisation
stool/18 12 2 1 2 1
vagina/1 1
skin/1 1
1999 Infection
blood/7 3 3 1
urine/5 1 3 1
Colonisation
stool/20 3 6 4 1 6
sputum/1 1
pus/2 2
1997–
1999
Infection
blood/22 9 6 4 1 1 1
urine/13 6 5 2
pus/2 2
Colonisation
stool/58 27 11 7 1 2 2 2 6
sputum/2 1 1
throat/1 1
vagina/1 1
skin/1 1
Total 100 41 25 12 4 4 2 5 7
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351 349

3.4. PFGE analysis
The molecular typing by PFGE found seven unique
profiles (represented by AB, C, D, E, F, G and H group
in Fig. 2B). Each pattern consists of approximately
12–17 fragments. Isolates of VREM recovered from
patients in 1997 were typed into PFGE groups AB, C,
D and E. The PFGE group AB is identified as groups
A and B by ADSRRS fingerprinting method. Groups
AB, C, D, F, G, H and groups AB, C, G, H were
detected in 1998 and 1999, respectively.
4. Discussion
PFGE, especially when combined with serotyping,
is currently considered the gold standard for molec-
ular typing of isolates recovered from patients and the
environment in the course of investigation and control
of nosocomial outbreaks. However, PFGE is time-
consuming and labor-intensive and can be performed
only in reference laboratories with skillful technicians.
Due to these drawbacks, PFGE is not an ideal typing
method for health departments undertaking routine
analysis of large numbers of isolates.
Here, we show for the first time the evaluation of a
novel fingerprinting method described by Masny and
Plucienniczak (2001) based on the fingerprinting of
bacterial genomes by amplification of DNA fragments
surrounding rare restriction sites (ADSRRS finger-
printing) for epidemiological studies. The high differ-
entiation power of the ADSRRS fingerprinting method
is shown on clinical strains of E. faecium. Fig. 2A
shows a high degree of diversity of the analyzed strains
representing each of eight identified groups (only
groups A and B are closely related). Identical results
were obtained in independent experiments performed
with DNA isolated from different cultures of one strain.
Results obtained in PCRs do not depend on the thermo-
stable DNA polymerase used (Taq polymerase or Pwo
polymerase) or on the thermal cycler used. The use of
betaine improves the PCR efficiency, especially for
high-molecular-weight DNA fragments. There are
several advantages of the ADSRRS fingerprinting
method: (i) the method does not require prior knowl-
edge of an analyzed sequence; (ii) results can be easily
analyzed even on polyacrylamide gels stained with
ethidium bromide; (iii) one set of adapters and enzymes
can be applied to analyze DNA from diverse species of
bacteria; (iv) PCR products can be directly isolated
from the polyacrylamide gel and subsequently se-
quenced.
We suggested based on this study that there is at
least a similar power of discrimination between the
present gold-standard PFGE and a novel method,
ADSRRS fingerprinting. Although the ADSRRS fin-
gerprinting method may appear to be more complex
than RAPD technique, we found it to be fast and
reproducible.
The ADSRRS fingerprinting method described
here (with the same restriction enzymes, adapters
and primers) was successfully used also for epidemio-
logical studies of Acinetobacter baumannii and Ser-
ratia marcescens outbreaks (in preparation).
Numerous isolations of VREM in the Haematolog-
ical Unit of the Clinical Hospital in Gdansk in 1997 to
1999 indicated that the first nosocomial outbreak of
VREM had occurred in the country (Samet et al.,
1999; Hryniewicz et al., 1999; Kawalec et al., 2000).
Several lines of evidence obtained in this study of
large number of isolates suggested that the VanA
phenotype was selected by most likely one or two
independent events within the ward enterococcal pop-
ulation. This was supported by the observation of the
dominant existence of the two closely related
ADSRRS fingerprinting groups (A and B). Only
minor differences in ADSRRS fingerprinting patterns
observed in both groups revealed the ongoing evolu-
tionary diversification process within their populations
(no difference was observed using PFGE method,
group AB). The outbreak in the ward was polyclonal,
as demonstrated by the diversity of the distinguished
E. faecium ADSRRS fingerprinting patterns. The
observed relatedness of some of the E. faecium iso-
lates (groups A and B) suggested, however, that clonal
spread has also occurred. It may be postulated that
originally a single variant of the Tn1546-like trans-
poson was spread among the nonrelated enterococcal
strains circulating in the ward (Kawalec et al., 2000).
The present report confirmed the results described
previously, where VREM isolated between December
1996 and October 1997 were analyzed using PCR
fingerprinting (RAPD) technique (Samet et al., 1999).
The applied PCR fingerprinting system distinguished
two main closely related genotypes of the clinical
isolates from Haematological Unit (groups A and B,
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351350

and AB identified by ADSRRS fingerprinting and
PFGE method, respectively) and demonstrated only
small genetic heterogeneity among the isolates over
11 months.
It is not certain from our studies whether the VanA-
type resistance genes may have been transferred
between strains of E. faecium that were present on
the unit or whether two strains of VREM may have
been introduced into Haematological Unit independ-
ently over a short period of time, either by the transfer
from other hospitals or from the community. Data
presented here demonstrate the complexity of the
epidemiological situation concerning VREM that
may occur in a single medical ward.
Acknowledgements
We wish to thank A. PlCucienniczak and A. Masny
for their helpful suggestions during this work. This
work was supported by grant nos. 6 PO5A 151 21 and
DOT 67/2001 from The State Committee for Scientific
Research (Poland).
References
Cheng, S., McCleskey, F.K., Gress, M.J., Petroziello, J.M., Liu, R.,
Namdari, H., Beninga, K., Salmen, A., DelVecchio, V.G., 1997.
A PCR assay for identification of Enterococcus faecium. J. Clin.
Microbiol. 35, 1248–1250.
De Zoysa, A.S., Efstratiou, A., 1999. PCR typing of Corynebacte-
rium diphtheriae by random amplification of polymorphic
DNA. J. Med. Microbiol. 48, 335–340.
Diatchenko, L., Lau, Y.F., Campbell, A.P., Chenchik, A., Moqa-
dam, F., Huang, B., Lukyanov, S., Lukyanov, K., Gurskaya, N.,
Sverdlov, E.D., Siebert, P.D., 1996. Suppression subtractive hy-
bridization: a method for generating differentially regulated or
tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci.
U. S. A. 93, 6025–6030.
Gerner-Smidt, P., Graves, L.M., Hunter, S., Swaninathan, B., 1998.
Computerized analysis of restriction fragment length polymor-
phism patterns: comparative evaluation of two commercial soft-
ware packages. J. Clin. Microbiol. 36, 1318–1323.
Handwerger, S., Raucher, B., Altarac, D., Monka, J., Marchinoe, S.,
Singh, K.V., Murray, B.E., Wolff, J., Walters, B., 1993. Noso-
comial outbreak due to Enterococcus faecium highly resistant to
vancomycin, penicillin, and gentamicin. Clin. Infect. Dis. 16,
750–755.
Hryniewicz, W., Szczypa, K., Bronk, M., Samet, A., Hellmann, A.,
Trzcinski, K., 1999. First report of vancomycin-resistant Enter-
ococcus faecium isolated in Poland. Clin. Microbiol. Infect. 5,
503–505.
Jordens, J.Z., Bates, J., Griffiths, D.T., 1994. Faecal carriage and
nosocomial spread of vancomycin-resistant Enterococcus faeci-
um. J. Antimicrob. Chemother. 34, 515–528.
Karanfil, L.V., Murphy, M., Josephson, A., Gaynes, R., Mandel, L.,
Hill, B.C., 1992. A cluster of vancomycin-resistant Enterococ-
cus faecium in an intensive care unit. Infect. Control Hosp.
Epidemiol. 13, 195–200.
Kawalec, M., Gniadkowski, M., Hryniewicz, W., 2000. Outbreak of
vancomycin-resistant enterococci in a hospital in Gdansk, Po-
land, due to horizontal transfer of different Tn1546-like trans-
poson variants and clonal spread of several strains. J. Clin.
Microbiol. 38, 3317–3322.
Lukyanov, S.A., Gurskaya, N.G., Lukyanov, K.A., Tarabykin, V.S.,
Sverdlov, E.D., 1994. Highly efficient subtractive hybridization
of cDNA. Bioorg. Khim. 20, 701–704.
Masny, A., Plucienniczak, A., 2001. Fingerprinting of bacterial
genomes by amplification of DNA fragments surrounding rare
restriction sites. BioTechniques 31, 930–936.
Montecalvo, M.A., Horowitz, H., Gedris, C., Carbonaro, C., Ten-
over, F.C., Issah, A., Cook, P., Wormser, G.P., 1994. Outbreak
of vancomycin-, ampicillin-, and aminoglycoside-resistant En-
terococcus faecium bacteremia in an adult oncology unit. Anti-
microb. Agents Chemother. 38, 1363–1367.
Mueller, U.G., Wolfenbarger, L.L., 1999. AFLP genotyping and
fingerprinting. Trends Ecol. Evol. 14, 389–394.
Patel, R., Uhl, J.R., Kohner, P., Hopkins, M.K., Cockerill III, F.R.,
1997. Multiplex PCR detection of vanA, vanB, vanC-1, and
vanC-2/3 genes in enterococci. J. Clin. Microbiol. 35, 703–707.
Pejic, I., Ajmone-Marsan, P., Morgante, M., Kozumplick, V., Cas-
tiglioni, P., Taramino, G., Motto, M., 1998. Comparative anal-
ysis of genetic similarity among maize inbred lines detected by
RFLPs, RAPDs, SSRs and AFLPs. Theor. Appl. Genet. 97,
1248–1255.
Samet, A., Bronk, M., Hellmann, A., Kur, J., 1999. Isolation and
epidemiological study of vancomycin-resistant Enterococcus
faecium from patients of a haematological unit in Poland. J.
Hosp. Infect. 41, 137–143.
Shagin, D.A., Lukyanov, K.A., Vagner, L.L., Matz, M.V., 1999.
Regulation of average length of complex PCR product. Nucleic
Acids Res. 27:e23.
Van Belkum, A., van Leeuwen, W., Kaufmann, M.E., Cookson, B.,
Forey, F., Etienne, J., Goering, R., Tenover, F., Steward, C.,
O’Brein, F., Grubb, W., Tassios, P., Legakis, N., Morvan, A.,
El Solh, N., de Ryck, R., Struelens, M., Salmenlinna, S., Vuo-
pio-Varkila, J., Kooistra, M., Talens, A., Witte, W., Verbrugh,
H., 1998. Assessment of resolution and intercenter reproduci-
bility of results of genotyping Staphylococcus aureus by pulsed-
field gel electrophoresis of SmaI macrorestriction fragments: a
multicenter study. J. Clin. Microbiol. 36, 1653–1659.
Vos, P., Hogers, R., Bleeker, M., Reijans, M., Vandelee, T.,
Hornes, M., Frijters, A., Pot, J., Peleman, J., Kuiper, M., Za-
beau, M., 1995. AFLP: a new technique for DNA fingerprint-
ing. Nucleic Acids Res. 23, 4407–4414.
Williams, J.G.K., Kubelik, A.R., Livak, K.J., Rafalski, J.A., Tin-
gey, S.V., 1990. DNA, polymorphisms amplified by arbitrary
primers are useful as genetic markers. Nucleic Acids Res. 18,
6531–6535.
B. Krawczyk et al. / Journal of Microbiological Methods 52 (2003) 341–351 351




















