
Contrats de plus de X $ totalisant plus de X $ par fournisseur
Upload
khangminh22Category
view
3download
0
© Stephan Hasse, 2021
Étude de l'implication de l'acide lysophosphatidique par la production de vésicules extracellulaires vasculaires
dans les dommages associés aux maladies rhumatismales auto-immunes systémiques
Thèse
Stephan Hasse
Doctorat en microbiologie-immunologie
Philosophiæ doctor (Ph. D.)
Québec, Canada

ii
Résumé
L’acide lysophosphatidique (LPA) est un lipide bioactif qui est formé dans le sang par
l’autotaxine. Le LPA est un médiateur important du système vasculaire, notamment par sa
modulation de l’immunité et de l’inflammation. Plusieurs espèces moléculaires de LPA
existent en fonction de leur acide gras. Les espèces moléculaires de LPA ont des affinités
différentes pour les récepteurs aux LPA. Il en résulte que les espèces moléculaires de LPA
peuvent avoir des effets différents, même si elles ciblent une même cellule.
Parmi ses nombreux effets, le LPA induit l’activation des plaquettes et est le seul activateur
endogène connu des globules rouges (GR). L’activation des plaquettes et des GR induit la
libération de vésicules extracellulaires (EV). Les EV de plaquettes (PEV) et les EV de GR
(REV) ont des effets pro-inflammatoires et sont des acteurs importants de la coagulation.
Le LPA est connu pour promouvoir la pathophysiologie de la polyarthrite rhumatoïde (PAR),
une maladie rhumatismale auto-immune systémique (MRAS). Les patients touchés par les
MRAS comme la PAR ou le lupus érythémateux disséminé (LED) présentent une
inflammation vasculaire importante et sont plus à même de développer des maladies
cardiovasculaires comme l’athérosclérose. Les maladies cardiovasculaires sont la première
cause de mortalité chez ces patients. Le LPA et les EV promeuvent tous deux l’inflammation
vasculaire et le développement de maladies cardiovasculaires. Ils sont également impliqués
dans la coagulation. L’hypothèse à l’origine des travaux de cette thèse est que le LPA via
l’activation des GR peut promouvoir l’inflammation vasculaire et participer aux dommages
vasculaires associés aux MRAS comme l’athérosclérose et la thrombose.
Dans cette thèse, nous avons d’abord étudié l’action des principales espèces moléculaires de
LPA trouvées dans le plasma sur les GR. Par des approches de cytométrie en flux à haute
sensibilité, nous avons montré que certaines espèces moléculaires de LPA induisent
l’exposition de la phosphatidylsérine (PS) par les GR et la libération de REV PS- et PS+
similaire à celles trouvées dans le plasma de patients LED. Cependant, d’autres espèces
moléculaires de LPA inhibent l’activation des GR. J’ai établi les principales voies de
signalisation impliquées dans l’activation et l’inhibition des GR. De plus, nous avons mis en

iii
évidence que, même si elle est possible dans le plasma, l’activation des GR par le LPA
dépend de son environnement.
Notre deuxième focus était centré les potentielles associations entre l’autotaxine et les EV
avec le risque de thrombose et le développement de l’athérosclérose chez des patients LED.
Nous avons montré que l’autotaxine n’était pas augmentée ni associée avec l’activité de la
maladie chez les patients LED. Et bien que les patients LED présentaient des quantités très
importantes de PEV et de REV, elles n’étaient pas associées avec l’activité de la maladie.
Cependant, les quantités de REV PS+ sont associées avec un risque plus élevé de thrombose
chez les patients SLE. De plus, le groupe de patients avec des quantités élevées de REV PS+
présentait également des concentration d’autotaxine plasmatique plus élevées.
Le travail présenté dans cette thèse approfondit la compréhension de l’effet du LPA sur
l’activation des GR et leur libération de REV. Il met également en évidence l’implication
potentielle du LPA et des REV dans les thromboses associées aux patients MRAS.

iv
Abstract
The lysophosphatidic acid (LPA) is a bioactive lipid which is formed by autotaxin in blood.
LPA is an important mediator in the vascular system mainly through its modulation of
immunity and inflammation. Several LPA species exist depending on the fatty acid. LPA
species varies in their affinity for the LPA receptors, which means that LPA species may
have different effects, even if they target a same cell.
Among its numerous biological actions, LPA induces platelet activation and is the only
known endogenous activator of red blood cells (RBCs). Both platelet and RBC activation
lead to the liberation of extracellular vesicles (EVs). Platelet EVs (PEVs) and RBC EVs
(REVs) are the two main populations of EVs found in blood. Both PEVs and REVs have
been described as pro-inflammatory mediators and are important actors of the coagulation.
LPA is a known promoter of the pathophysiology of rheumatoid arthritis (RA), a systemic
autoimmune rheumatic disease (SARD). Patients affected by SARDs such as RA and
systemic lupus erythematosus (SLE) present high vascular inflammation and are more prone
to develop cardiovascular diseases for instance atherosclerosis. Cardiovascular diseases are
the first cause of mortality for these patients. LPA and EVs are two mediators which
promotes vascular inflammation and the development of cardiovascular diseases. Also, both
are pro-coagulant factors. The hypothesis driving this thesis is that LPA through the
activation of RBCs promotes vascular inflammation and participate to the vascular damages
associated with MRAS patients such as atherosclerosis and thrombosis.
In this thesis, we first focused our interest to study the action of major blood LPA species on
RBCs. Through high sensitivity flow cytometry, we found that some LPA species induces
the exposition of phosphatidylserine (PS) by RBCs and the liberation of PS- and PS+ REVs
similar to those found in the plasma of LED patients. However, other species were inhibitors
of RBC activation. We have established the main LPA’s signaling pathways involved in the
activation and inhibition as well that even if it is possible in the plasma, RBC activation by
LPA is affected by the environment.

v
Our second focus was on the potential associations of autotaxin and EVs with thrombotic
risk and the development of atherosclerosis in SLE patients. We found that autotaxin were
not elevated in SLE patients nor associated with the disease activity. Even though, SLE
patients presented high quantities of PEVs and REVs, they were not associated with the
disease activity. However, we showed that the quantities of PS+ REVs were associated with
a higher risk of thrombosis in SLE patients. Moreover, the group of patients with high
quantities of PS+ REVs also presented higher quantities of plasmatic autotaxin.
The work presented in this thesis brings a better understanding of LPA impact on RBC
activation and REV liberation. It also highlights the potential implication of both LPA and
REVs in thrombosis associated with SARD patients.

vi
Table des matières
Résumé _________________________________________________________________ ii Abstract ________________________________________________________________ iv Table des matières ________________________________________________________ vi Liste des figures __________________________________________________________ ix Liste des tableaux _________________________________________________________ x
Liste des abréviations ______________________________________________________ xi Remerciements __________________________________________________________ xv Avant-propos __________________________________________________________ xvii Introduction _____________________________________________________________ 1
1 L’axe autotaxine / acide lysophosphatidique / lipide-phosphate phosphatase _____ 1 1.1 L’acide lysophosphatidique ______________________________________________________ 2
1.1.1 Structure, espèce et nomenclature _____________________________________________ 2 1.1.2 L’isolation et l’identification ________________________________________________ 3 1.1.3 L’acide lysophosphatidique chez les mammifères ________________________________ 4
1.2 Synthèse de l’acide lysophosphatidique _____________________________________________ 5 1.2.1 Synthèse intracellulaire _____________________________________________________ 6 1.2.2 Synthèse extracellulaire ____________________________________________________ 6
1.3 L’autotaxine __________________________________________________________________ 7 1.3.1 Isoformes _______________________________________________________________ 8 1.3.2 Structure et expression _____________________________________________________ 9 1.3.3 Régulation de l’expression _________________________________________________ 10
1.4 L’acide lysophosphatidique vasculaire _____________________________________________ 11 1.5 Signalisation dépendante de l’acide lysophosphatidique _______________________________ 14
1.5.1 La famille EDG des récepteurs couplés aux protéines G : LPA1, 2 et 3 ______________ 15 1.5.1.1 LPA1/Edg2 ________________________________________________________ 15 1.5.1.2 LPA2/Edg4 ________________________________________________________ 18 1.5.1.3 LPA3/Edg7 ________________________________________________________ 21
1.5.2 Récepteurs couplés aux protéines G de type non-EDG ___________________________ 24 1.5.2.1 LPA4/P2Y9 ________________________________________________________ 24 1.5.2.2 LPA5 _____________________________________________________________ 25 1.5.2.3 LPA6/P2Y5 ________________________________________________________ 27 1.5.2.4 GPR87 ____________________________________________________________ 28
1.5.3 Récepteurs non couplés à des protéines G _____________________________________ 28 1.5.3.1 TRPV1____________________________________________________________ 28 1.5.3.2 TREK-1/-2 _________________________________________________________ 29 1.5.3.3 PPARγ ____________________________________________________________ 29 1.5.3.4 Activation des cibles intracellulaires par le LPA ___________________________ 29
1.6 Régulation de l’activité du LPA : les lipide-phosphate phosphatases _____________________ 30 2 Vésicules extracellulaires _______________________________________________ 32
2.1 Diversité et formation des EV ___________________________________________________ 32 2.1.1 Les classes : exosomes, microvésicules, corps apoptotiques _______________________ 32 2.1.2 Isolation et étude _________________________________________________________ 35
2.2 Vésicules extracellulaires de plaquettes ____________________________________________ 37 2.2.1 Présentation de la plaquette ________________________________________________ 37 2.2.2 Description générale ______________________________________________________ 39 2.2.3 Fonctions_______________________________________________________________ 40
2.3 Vésicules extracellulaires de globules rouges _______________________________________ 40 2.3.1 Présentation des globules rouges ____________________________________________ 40 2.3.2 Description générale ______________________________________________________ 43 2.3.3 Fonctions_______________________________________________________________ 44
3 L’acide lysophosphatidique et les vésicules extracellulaires dans les maladies
rhumatismales auto-immunes systémiques _______________________________________ 45

vii
3.1 Polyarthrite rhumatoïde ________________________________________________________ 45 3.2 Lupus érythémateux disséminé __________________________________________________ 46 3.3 Comorbidité : athérosclérose ____________________________________________________ 48
4 Objectif _____________________________________________________________ 52 Chapitre 1 : Interplay between LPA2 and LPA3 in LPA-mediated phosphatidylserine cell
surface exposure and extracellular vesicles release by erythrocytes ________________ 53 1 Résumé ______________________________________________________________ 53 2 Abstract _____________________________________________________________ 55 3 Introduction __________________________________________________________ 56 4 Material and methods __________________________________________________ 57
4.1 Products ____________________________________________________________________ 57 4.2 Human plasma samples ________________________________________________________ 58 4.3 RBC isolation and activation ____________________________________________________ 58 4.4 Platelet and EV-free plasma preparation ___________________________________________ 59 4.5 RBC and REV labeling for flow cytometry _________________________________________ 59 4.6 Control for REV detection by flow cytometry _______________________________________ 60 4.7 Analysis and statistics _________________________________________________________ 60
5 Results ______________________________________________________________ 61 5.1 Detection of activated RBCs and REVs by flow cytometry _____________________________ 61 5.2 LPA species differentially activate RBCs. __________________________________________ 61 5.3 Characterization of RBC activation by LPA 18:1. ____________________________________ 62 5.4 LPA3 receptor induce RBC activation. ____________________________________________ 63 5.5 LPA2 receptor inhibits PS- REV formation. _________________________________________ 63 5.6 LPA 20:4 inhibits both RBC PS exposure and the production of PS- REVs. ________________ 64 5.7 RBC activation by LPA in physiological condition. __________________________________ 65
6 Discussion ___________________________________________________________ 66 7 References ___________________________________________________________ 69 8 Figures and legends ___________________________________________________ 74
Chapitre 2 : Plasma level of red blood cell-derived phosphatidylserine positive
extracellular vesicles are associated with thrombosis in systemic erythematous lupus
patients ________________________________________________________________ 83 1 Résumé ______________________________________________________________ 83 2 Abstract _____________________________________________________________ 85 3 Introduction __________________________________________________________ 86 4 Material and methods __________________________________________________ 87
4.1 SLE patients and healthy donors _________________________________________________ 87 4.2 SARD-BDB protocol __________________________________________________________ 87 4.3 Flow cytometry_______________________________________________________________ 88
4.3.1 Detection of platelet activation ______________________________________________ 88 4.3.2 Detection of plasmatic EVs ________________________________________________ 88
4.4 Autotaxin measurement ________________________________________________________ 89 4.5 Analysis and Statistics _________________________________________________________ 89
5 Results ______________________________________________________________ 89 5.1 Patient’s characteristics ________________________________________________________ 89 5.2 SLE patients present higher platelet activation and plasma EV levels at baseline ____________ 90 5.3 Prevalent and incident SLE patients show similar levels of plasma EVs and platelet activation. 90 5.4 Platelet activation is associated with the SLEDAI score in incident cases of SLE. ___________ 91 5.5 Higher PS+ REVs are associated with vascular damages in SLE patients. __________________ 91
6 Discussion ___________________________________________________________ 93 7 References ___________________________________________________________ 97 8 Figures, legends and tables ____________________________________________ 101
Discussion ____________________________________________________________ 110 1 Mise en contexte _____________________________________________________ 110

viii
2 Impact des limitations techniques dans l’analyse des vésicules extracellulaires __ 110 3 Résumé des travaux et discussion _______________________________________ 112
3.1 L’acide lysophosphatidique et les vésicules extracellulaires de globules rouges ____________ 112 3.2 Les vésicules extracellulaires de globules rouges dans le lupus érythémateux disséminée ____ 115
4 Perspectives _________________________________________________________ 117 Conclusion ____________________________________________________________ 119 Bibliographie __________________________________________________________ 121 Annexe I : Targeting the autotaxin - Lysophosphatidic acid receptor axis in cardiovascular
diseases _______________________________________________________________ 170 1 Abstract ____________________________________________________________ 171 2 Graphical abstract ___________________________________________________ 172 3 Lysophosphatidic acid and its receptors __________________________________ 173 4 LPA production pathways _____________________________________________ 175 5 The LPA-induced responses in cells of the cardiovascular system ____________ 176 6 The ATX-LPA axis in cardiovascular diseases ____________________________ 180 7 Targeted ATX-LPA therapy ___________________________________________ 184 8 Conclusions _________________________________________________________ 187 9 References __________________________________________________________ 187
Annexe II :Phosphatidylserine-specific phospholipase A1: A friend or the devil in disguise
_____________________________________________________________________ 196 1 Abstract ____________________________________________________________ 197 2 General introduction _________________________________________________ 198 3 Expression of PLA1A and lysoPS receptors in cells ________________________ 205 4 Expression of PLA1A in disease states ___________________________________ 207 5 Other enzymes regulating serine phospholipid metabolism in neural system ___ 216 6 Conclusions _________________________________________________________ 217 7 References __________________________________________________________ 217
Annexe III: Platelet-derived extracellular vesicles contain an active proteasome involved
in protein processing for antigen presentation via class I major histocompatibility
molecules _____________________________________________________________ 225 1 Abstract ____________________________________________________________ 227 2 Introduction _________________________________________________________ 228 3 Material and methods _________________________________________________ 229 4 Results _____________________________________________________________ 230 5 Discussion __________________________________________________________ 237 6 References: _________________________________________________________ 241 7 Figures _____________________________________________________________ 246

ix
Liste des figures
Introduction
Figure 1 : Structure biochimique du LPA. ................................................................... 2
Figure 2 : Sites d’hydrolyses des phospholipases de type A, B, C et D. ........................... 5
Figure 3 : Structure des isoformes d’autotaxine. .......................................................... 9
Figure 4 : Synthèse, signalisation et dégradation du LPA. ............................................13
Figure 5 : Récapitulatif du contenu trouvé dans les EV à l’exception des organelles. ......33
Figure 6 : Libération des différentes vésicules extracellulaires. ....................................34
Figure 7 : Les différentes méthodes d’isolation des EV. ..............................................35
Figure 8 : Interaction des globules rouges pour leur élimination par les macrophages. ....42
Figure 9 : Progression d’une plaque d’athérosclérose. .................................................50
Chapitre 1 :
Figure 1 : RBC activation and REV detection by high-sensitivity flow cytometry. .........74
Figure 2 : RBC activation by LPA varies depending of the fatty acid. ...........................75
Figure 3 : LPA induced RBC activation leads to two distinct REV populations. .............76
Figure 4 : LPA1/3 mediates RBC activation by LPA. ..................................................77
Figure 5 : LPA2 inhibits PS- REV production. ...........................................................78
Figure 6 : LPA 20:4 inhibits PS- REV production through LPA2 and PS exposure by RBCs.
...............................................................................................................................79
Figure 7 : LPA 18:1 induces PS+ REVs in platelet-free and EV-free plasma from healthy
donors. ....................................................................................................................80
Figure 8 : High plasmatic quantities of PS+ and PS- REV are present in SLE patients. ....81
Figure 9 : LPA signaling in RBCs. ............................................................................82
Chapitre 2 :
Figure 1 : High platelet activation and EV quantities are found in incident and prevalent SLE
patients. . ............................................................................................................... 105
Supplementary Figure 1 : Platelet activation and EV detection by high-sensitivity flow
cytometry. . ............................................................................................................ 107
Conclusion
Figure 1 : Récapitulatif des contributions des travaux de cette thèse. .......................... 120

x
Liste des tableaux
Chapitre 2 :
Table 1 : characteristics for SLE patients included in the study at baseline. ................. 101
Table 2 : High platelet activation and EV quantities are found in SLE patients at baseline.
............................................................................................................................. 103
Table 3 : Spearman correlation between our measurement and the total SLEDAI score for
SLE patients. .......................................................................................................... 103
Table 4 : Comparison of SLE patients with low and high PS+ REVs. .......................... 104

xi
Liste des abréviations
ACR American College of Rheumatology
ADN Acide désoxyribonucléique
ADP Adénosine diphosphate
AGPTA Acylglycerophosphate acyltransférase
ARF6 (ADP-ribosylation factor 6)
ARN Acide ribonucléique
ATP Adénosine triphosphate
ATX Autotaxine
CCL (chemokine CC ligands)
CD (cluster of differentiation)
CMH Complexe majeur d’histocompatibilité
CXCL (chemokine CXC ligands)
DAGK Diacylglycerols kinases
DLD (Deterministic lateral displacement)
Edg Gènes de différentiation endothéliales (endothelial differentiation gene)
EGF Facteurs de croissance épidermique (Epidermal Growth Factor)
EGFR Récepteur des EGF (EGF receptor)
ELISA (enzyme-linked immunosorbent assay)
ESCRT Complexe de tri endosomal nécessaire au transport (Endosomal Sorting
Complex Required for Transport)
EULAR European Alliance of Associations for Rheumatology (anciennement
EUropean League Against Rheumatism)
EV Vésicule extracellulaire (Extracellular Vesicle)

xii
Fc receptors Récepteurs aux fragments cristallisables
FGF Facteurs de croissance des fibroblastes (fibroblast growth factor)
GIPC Protéine d’interaction en C-terminus de type Gα (Galpha-interacting protein
C-terminus)
GPAT Glycerophosphates acyltransferases
GPR Récepteur couplé au protéine G (G Protein-Coupled Receptor)
GR Globules rouges
Ig Immunoglobuline
IL Interleukine
IP3 Inositol trisphosphate
ISEV Société internationale pour les vésicules extracellulaires (International
Society for Extracellular Vesicles)
LDL Lipoprotéine de basse densité (low density lipoprotein)
LED Lupus érythémateux disséminé
LPA Acide lysophosphatidique (lysophosphatidic acid)
LPAR Récepteur au LPA (LPA receptor)
LPP Lipide-phosphate phosphatases
Lyso-PS Lysophosphatidylsérine
MAGI-3 Guanylate kinase associé à la membrane avec une orientation inversé 3
(membrane-associated guanylate kinase with inverted orientation-3)
MAGK Monoacylglycerols kinases
MAP kinases (Mitogen-activated protein kinases)
MRAS Maladie rhumatismale auto-immune systémique
Nabs Auto-anticorps naturels (natural antibodies)

xiii
NFTA1 Facteur nucléaire des lymphocytes T activés 1 (Nuclear factor of activated T
cells 1)
NFκB Facteur nucléaire κ B (Nuclear factor κ B)
NHERF2 facteur 2 de régulation des échanges sodium hydrogène (sodium/hydrogen
exchanger regulatory factor 2)
oxLDL LDL oxydé (oxidized LDL)
PAR Polyarthrite rhumatoïde
PCR réaction de polymérisation en chaîne (Polymerase chain reaction)
PDZ (post synaptic density protein (PSD95), Drosophila disc large tumor
suppressor (Dlg1), and zonula occludens-1 protein (zo-1))
PEV Vésicules extracellulaires de plaquettes (platelet-derived EV)
PI3K Phosphoinositide 3-kinase
PKC Protéine kinase C
PLA/B/C/D Phospholipase de type A/B/C/D
PMA (Phorbol myristate acetate)
PPARγ récepteur intracellulaire activé par les proliférateurs de peroxysomes
(peroxisome proliferator-activated receptor gamma)
PRP Plasma riche en plaquettes
PS Phosphatidylsérine
RAB (RAS-related protein in brain)
REV EV de globule rouge (red blood cell EVs)
RhoA (Ras homolog family member A)
RhoGEF facteur d’échange de nucléotide guanine spécifique à RhoA (Rho-specific
guanine nucleotide exchange factors)
Rnase Ribonucléase

xiv
ROCK (Rho-associated protein kinase)
RT-PCR Transcription inverse PCR (reverse transcription-PCR)
SIMPLE (small integral membrane protein of lysosomes and late endosomes)
SIRPα (Signal regulatory protein α)
SLEDAI Indice d’activité de la maladie LED (SLE disease activity index)
SLICC Systemic Lupus International Collaborating Clinics
STAT3 Signal de transduction et d’activation de transcription 3 (Signal transducer
and activator of transcription 3)
TAP Protéines de transport associées à l’antigène (Transporters associated with
Antigen Processing)
TAZ (PDZ-binding motif)
TGF (Transforming growth factor)
TLR Récepteurs de type Toll (Toll-like receptors)
TNF Facteur de nécrose tumorale (Tumor necrosis factor)
TREK-1/-2 Canaux à ion de potassium apparenté TWIK-1 et -2 (TWIK related K+
channel-1 and -2)
TRIP6 protéine d’interaction au récepteur de la thyroïde 6 (thyroid receptor-
interacting protein 6)
TRPV1 Récepteur transitoire à potentiel vanilloïde 1 (transient receptor potential
vanilloid 1)
YAP (yes-associated protein 1)

xv
Remerciements
Je remercie mon directeur de thèse, le docteur Sylvain Bourgoin pour avoir m’avoir
accompagné tout au long de cette thèse. J’ai énormément apprécié notre relation de travail
que ce soit l’autonomie et la liberté qu’il m’a accordées pour gérer mes projets ou sa patience
et son acceptation de mes résultats négatifs.
Je remercie également l’ensemble des membres de notre laboratoire. Merci Lynn Davis, cela
a toujours été un plaisir de travailler et de discuter avec toi. Merci Chenqi Zhao, j’ai toujours
apprécié ta gentillesse et ton aide sur mes projets. Enfin merci Myriam pour ton esprit et ta
gentillesse ainsi que pour m’avoir permis mettre le point final à mes expériences.
Je remercie également l’ensemble des membres de l’équipe Boilard et Fernandez avec
lesquelles j’ai beaucoup interagi au cours de mon doctorat. Je pense tout particulièrement à
Isabelle Allaeys, Tania Lévesque, et Anne Zufferey. Vous avez été une source précieuse
d’aide et de conseil ainsi que de la bonne humeur dans nos rangées de travail. Mes
compétences ne seraient pas ce qu’elles sont sans votre apport.
Je remercie aussi les nombreux étudiants, post-doctorants et professionnels de recherche que
j’ai côtoyé. Je ne serai jamais suffisamment reconnaissant pour avoir croisé le chemin de
Geneviève, Julien, Anne, Pepito, Patate, Tania, Yann, Katerina, Oona, Aurélie, Marine,
Régis, Andréa, et Anthony. Vous avez tous été au long de ces cinq années une source de joie,
de conseil, d’encouragement et d’inspiration. Et, je suis sûr que vous continuerez de l’être.
Je ne permettrai pas d’oublier mes colocataires, Camille, Yann, Abde, Bibi et Guinness ainsi
que Mathieu et Laurence qui me permirent de me changer régulièrement les idées autour d’un
repas, d’un jeu de société ou d’un beigne.
Je remercie mes amis clermontois, Lucas, Loïc et Mazière pour ne citer qu’eux, qui malgré
leurs habitudes, ont eu la retenue et la délicatesse de ne pas me demander trop souvent quand
je finissais et ce que j’allais faire après. Merci à ma famille de m’avoir soutenu et d’avoir
compris la démarche qui nous a séparés par un océan. Et merci à mon frère de m’avoir
accueilli pour une partie de l’écriture de cette thèse.

xvi
Enfin, je ne remercierai jamais assez ma compagne qui, malgré la distance, m’a soutenu,
encouragé et a été une source de bien-être et de calme au quotidien. Ta compétence et ta
passion pour la science m’impressionne et me stimule pour améliorer mon travail. Merci.

xvii
Avant-propos
Le Dr Sylvain G Bourgoin a conçu et dirigé le projet de recherche. Il a participé à l’analyse
des données et corrigé les articles de cette thèse. Le Dr Éric Boilard a apporté son expertise
au projet de recherche. Le Dr Paul Fortin a contribué et à l’analyse des données obtenues sur
les échantillons de la biobanque MRAS du CHU de Québec – Université Laval.
J’ai conçu et réalisé les expériences, analysé et interprété les données, réalisé les analyses
statistiques et écrit les manuscrits des articles présentés aux chapitre 2 et 3 de ce document
en collaboration avec les auteurs mentionnés ci-dessous.
L’article qui constitue le chapitre 1, intitulé Interplay between LPA2 and LPA3 in LPA-
mediated phosphatidylserine cell surface exposure and extracellular vesicles release by
erythrocytes, a été publié dans le journal Biochemical Pharmacology en 2021:
Hasse S, Duchez AC, Fortin P, Boilard E, Bourgoin SG. (2021) Interplay between LPA2 and
LPA3 in LPA-mediated phosphatidylserine cell surface exposure and extracellular vesicles
release by erythrocytes. Biochem Pharmacol. 2021 Jun 30;192:114667. doi:
10.1016/j.bcp.2021.114667. Online ahead of print. PMID: 34216604
L’article qui constitue le chapitre 2, intitulé Plasma level of red blood cell-derived
phosphatidylserine positive extracellular vesicles are associated with thrombosis in
systemic erythematous lupus patients, a été soumis au journal Lupus Science & Medicine
pour publication (lupus-2021-000605).
Hasse S, Julien AS, Duchez AC, Chenqi Zhao C, Fortin P, Boilard E, Bourgoin SG.
Au cours de mon doctorat, j’ai collaboré à la rédaction de deux revues de littérature. La
première en tant que co-premier auteur qui est intitulée Targeting the autotaxin -
Lysophosphatidic acid receptor axis in cardiovascular diseases est présentée en
Annexe I :

xviii
Zhao Y, Hasse S, Zhao C, Bourgoin SG. (2019) Targeting the autotaxin - Lysophosphatidic
acid receptor axis in cardiovascular diseases. Biochem Pharmacol. 2019 Jun;164:74-81. doi:
10.1016/j.bcp.2019.03.035. Epub 2019 Mar 27. PMID: 30928673
La seconde, intitulée Phosphatidylserine-specific phospholipase A1: A friend or the devil in
disguise est présentée en Annexe II :
Zhao Y, Hasse S, Bourgoin SG. (2021) Phosphatidylserine-specific phospholipase A1: A
friend or the devil in disguise. Prog Lipid Res. 2021 Jun 22;83:101112. doi:
10.1016/j.plipres.2021.101112. Online ahead of print. PMID: 34166709
J’ai aussi collaboré significativement à plusieurs projets qui ne sont pas inclus dans cette
thèse. J’ai participé significativement à l’imagerie en microscopie à transmission ainsi qu’à
la rédaction et aux analyses statistiques dans cet article en Annexe III qui vient d’être publié
dans le journal Blood :
Marcoux G, Laroche A, Hasse S, Bellio M, Mbarik M, Tamagne M, Allaeys I, Zufferey A,
Lévesque T, Rebetz J, Karakeussian-Rimbaud A, Turgeon J, Bourgoin SG, Hamzeh-
Cognasse H, Cognasse F, Kapur R, Semple JW, Hebert MJ, Pirenne F, Overkleeft H, Florea
B, Dieude M, Vingert B and Boilard E. Platelet EVs contain an active proteasome involved
in protein processing for antigen presentation via MHC-I molecules. Blood. 2021 Jul
22:blood.2020009957. doi: 10.1182/blood.2020009957. Epub ahead of print. PMID:
34293122
J’ai également assisté significativement la réalisation d’expérience de l’article intitulé
Phospholipase A1 member A activates fibroblast-like synoviocytes through the autotax-
in-lysophosphatidic acid receptor axis, qui est en cours de révision par le journal
International Journal of Molecular Sciences.
Zhao Y, Hasse S, Vaillancourt M, Zhao C, Davis L, Boilard E, Fortin P, Di Battista J,
Poubelle PE, Bourgoin SG. Phospholipase A1 member A activates fibroblast-like
synoviocytes through the autotax-in-lysophosphatidic acid receptor axis. (ijms-1418430)
Seule la numérotation des titres a été changé par rapport aux versions publiées des articles.

1
Introduction
1 L’axe autotaxine / acide lysophosphatidique / lipide-
phosphate phosphatase
Les lipides qui présentent un groupement phosphate sont nommés phospholipides. Ils sont
essentiels aux structures membranaires ainsi qu’au transport des protéines. Bien que leur
identification et leur caractérisation datent du milieu du 19e siècle, l’identification d’une
activité biologique propre aux phospholipides ne débute que dans les années 1950. L’intérêt
pour l’acide lysophosphatidique (lysophosphatidic acid, LPA) découle du travail sur les
facteurs de Darmstoff1 et d’Arneil2. Vogt associe un facteur de stimulation des muscles lisses
qu’il nomme Darmstoff à des extraits de lipides d’intestins de mammifères et
d’amphibiens1,3. En 1963, Vogt montre les effets contractiles de différents lipides sur des
duodénums de lapins, dont celui du LPA1. Dans ces mêmes années, l’équipe d’Arneil met en
évidence un facteur vasoconstricteur dans le plasma humain qui bien que peu détectable lors
de la préparation du plasma, s’accumule quand il est entreposé à température ambiante2. De
plus, il montre par des approches de chromatographie et de traitement par des phospholipases
que ce facteur, dit d’Arneil, est proche de la lysophosphatidylcholine4. Il faut attendre
l’année 1979 pour que cette substance soit formellement identifiée comme le LPA5. Les
premiers travaux sur le LPA se concentrent sur son effet vasoconstricteur et sur les
plaquettes5,6. La découverte, au début des années 90, d’un récepteur au LPA va accélérer son
étude7. Depuis que l’importance du LPA a été établie dans des processus variés aussi bien
physiologiques, du développement embryonnaire, au recrutement lymphocytaire ou encore à
la pousse des cheveux que pathologiques comme dans le cancer, les maladies
cardiovasculaires ou les maladies rhumatismales auto-immunes systémiques (MRAS).

2
1.1 L’acide lysophosphatidique
1.1.1 Structure, espèce et nomenclature
Le LPA regroupe l’ensemble des lipides formés par un squelette de glycérol avec un
groupement phosphate en position sn-3 et un acide gras en position sn-1 ou 2 (Figure 1) et
fait partie de la famille des médiateurs lipidiques bioactifs. L’acide gras forme une queue
hydrophobe connectée par le glycérol à un groupement phosphate hydrophile.
Les LPA sont divisés en trois classes en fonction de la liaison de l’acide gras au squelette de
glycérol. Les trois classes de LPA possibles sont les acyl-, alkyl- ou alkenyl-, soit
respectivement une liaison ester, éther ou vinyl éther. Les LPA vont être ensuite subdivisés
en différentes espèces selon de la nature de la chaîne carbonée de l’acide gras présent,
considérant sa longueur et son nombre d’insaturations. La longueur de la chaine est
importante pour l’activité du LPA. Les LPA à chaine courte, en dessous de 14 carbones, ne
présentent pas d’activité biologique, contrairement aux LPA à chaine longue, de 14 à 26
carbones8. Enfin, un dernier élément est la position de l’acide gras sur le squelette de glycérol
qui est soit en position sn-1 ou sn-2. Les LPA avec l’acide gras en position sn-2 sont instables
et peuvent spontanément migrer en position sn-1 à un ratio de 1 pour 99. Ces trois éléments,
illustrés en Figure 1, modifient la conformation en 3 dimensions du LPA et donc son activité
biologique8,10.
Je vais utiliser la nomenclature suivante pour définir les espèces de LPA: la position de
l’acide gras sur le glycérol- la liaison entre le glycérol et l’acide gras- LPA la longueur : le
Figure 1: Structure biochimique du LPA. L’acide gras se présente le plus souvent sous forme acyl- en
position sn-1 du glycérol. Quand l’acide gras est présent sur la position sn-2 du glycérol il peut migrer
spontanément en position sn-1.

3
nombre d’insaturations de l’acide gras. Par exemple, un 1-acyl-LPA 18:1, est un LPA dont
l’acide gras est en position sn-1 du glycérol par une liaison acyl et à une chaîne de 18 carbones
qui contient une insaturation.
1.1.2 L’isolation et l’identification
L’isolation et l’extraction de lipides reposent sur leurs propriétés de solvatation. L’acide gras
des lipides est lipophile tandis que le groupement de tête est hydrophile. Donc plus l’acide
gras présente une longue chaine carbonée, plus le lipide sera soluble dans des solvants
organiques et insoluble dans l’eau. À l’inverse plus le groupement de tête est important, plus
le lipide sera soluble dans l’eau11. Les phospholipides, ce qui comprend le LPA, présentent
une solubilité intermédiaire dans la balance lipophile/hydrophile. Historiquement, l’isolation
et l’extraction des lipides avec une solubilité intermédiaire se font avec la méthode de Folch12
ou celle de Bligh et Dyer13. Ces deux méthodes reposent sur le potentiel de ces lipides à être
soluble dans un mélange de méthanol/chloroforme. Le mélange de l’échantillon avec le
méthanol, le chloroforme et une solution aqueuse va permettre l’obtention d’une phase
aqueuse et d’une phase méthanol/chloroforme qui contient les lipides. Ces deux méthodes
utilisent une solution aqueuse différente et effectuent la séparation de la phase organique et
aqueuse selon des techniques différentes. La méthode Bligh et Dyer utilise de l’eau et
l’obtention des 2 phases se fait par ajout d’eau et de chloroforme au mélange de
méthanol/chloroforme/eau13. La méthode Folch utilise du sérum physiologique comme
solution aqueuse et la séparation des 2 phases se fait sans ajout de solvant organique ou
aqueux12.
De nombreuses adaptations existent pour certaines applications, notamment pour des raisons
de sécurité et pour cibler certains glycérophospholipides difficilement isolables avec les
méthodes d’origine. Les modifications sont principalement l’utilisation de solvant moins
toxiques ou plus adapté à l’automatisation par exemple, ou servent à modifier les conditions
comme le pH ou la température. Une méthode à base de butanol a été développée pour
l’isolation et l’extraction du LPA. Les échantillons sont mélangés au 1-butanol et du 1-
butanol saturé en eau (2:1, v/v). Une fois formée, la phase organique contient le LPA14,15.
Cette méthode présente un taux de récupération supérieur à 95 %14.

4
Historiquement, l’étude des lipides se faisait par chromatographies sur couche mince puis à
l’aide de la chromatographie en phase liquide à haute performance. Actuellement, la
technique la plus répandue est la chromatographie en phase liquide à haute performance
associée à la spectrométrie de masse. Elle permet d’une part la détection des espèces
moléculaires de LPA et leur quantification relative14,16-18. D’autre part, la quantification
absolue du LPA total et de ses espèces est également possible à l’aide de standards internes16.
La manipulation des lipides peut engendrer des modifications qui vont affecter la nature des
espèces et des lipides identifiés. Pour limiter la perte, les modifications et la dégradation des
lipides au cours du processus d’isolation et d’identification, il est recommandé de travailler
avec de la verrerie et de limiter l’exposition à l’air et à la lumière16,18.
1.1.3 L’acide lysophosphatidique chez les mammifères
Les classes acyl-, alkyl- et alkenyl-LPA sont détectées dans les fluides biologiques humains19
et dans les tissus de rat20-22. Les acyl-LPA sont toutefois la forme prépondérante19-22. Les
formes acyl-LPA 16:0, 18:0, 18:1 et 20:4 sont les quatre espèces présentes en quantités
élevées dans les tissus de rat. La proportion de chacune de ces espèces varie significativement
d’un tissu à l’autre20,23. Les espèces saturées sont celles les plus abondantes avec, notamment,
les acyl-LPA 16:0 et 18:0 qui peuvent représenter jusqu’à 30% et 60%, respectivement du
LPA total détecté dans les tissus22.
Le LPA est trouvé dans le milieu extracellulaire24,25 et dans le milieu intracellulaire26 au
niveau du noyau27, du réticulum endoplasmique28 et des mitochondries29. L’activité
biologique du LPA est principalement médiée par le LPA extracellulaire et les récepteurs qui
y sont associés. Le LPA intracellulaire est avant tout un intermédiaire dans le métabolisme
des glycérophospholipides. Sa formation intracellulaire a aussi été décrite dans le contexte
d’inhibition de l’activité de l’acide phosphatidique. Cependant, l’activation d’un récepteur
intracellulaire par le LPA intracellulaire a été mis en évidence30,31 et l’accumulation de LPA
intracellulaire module la survie et la migration de cellules tumorales32. Une activité
biologique propre au LPA intracellulaire reste encore débattue33. Bien que le LPA ait un rôle
dans le milieu extra- et intracellulaire, il n’y a pas d’évidence qu’il soit transporté à travers

5
la membrane plasmique26. La synthèse du LPA extra- et intracellulaire est faite par des
mécanismes distincts.
1.2 Synthèse de l’acide lysophosphatidique
Différents mécanismes conduisent à la formation de LPA intracellulaire et extracellulaire. La
seule voie commune qui peut conduire à la production de LPA intracellulaire et
extracellulaire est l’oxydation de phospholipide sous l’action d’oxydants et de radicaux
libres. L’oxydation de lipoprotéine de base densité permet la production d’acyl- et d’alkyl-
LPA34,35. Les autres voies de synthèse du LPA sont spécifiques soit aux compartiments intra
ou extracellulaires et font intervenir diverses phospholipases.
Les phospholipases sont la classe d’enzyme capable d’hydrolyser certaine liaison ester dans
les lipides qui présentent un groupement phosphate. Il existe quatre classes de phospholipases
en fonction de la position de la liaison qu’elles hydrolysent (Figure 2)36. Les phospholipases
de type A (PLA) sont capables d’hydrolyser la liaison de l’acide gras position sn-1 ou sn-2
du squelette de glycérol et forment un acide gras et un lysophospholipide. Deux sous-classes
existent, les PLA1 qui hydrolysent la liaison en position sn-1 et les PLA2 qui hydrolysent la
liaison en position sn-2. Les phospholipases de type B (PLB) sont capables d’hydrolyser la
liaison entre les acides gras et le squelette de glycérol en position sn-1 et sn-2. Les
phospholipases de type C (PLC) et D (PLD) hydrolysent les liaisons de chaque côté du
groupement phosphate. Les PLC hydrolysent la liaison entre le groupement phosphate et le
squelette de glycérol. Enfin, les PLD hydrolysent la liaison entre le groupement phosphate et
le groupement de tête qui peut être une choline, une sérine ou une éthanolamine.
Figure 2 : Sites d’hydrolyses des phospholipases de type A, B, C et D.

6
1.2.1 Synthèse intracellulaire
Le LPA intracellulaire peut être formé selon trois substrats différents : le monoacylglycérol,
le glycérol-3-phosphate et l’acide phosphatidique (Figure 4, partie supérieure)37. Le LPA
peut être formé par l’hydrolyse d’un acide gras de l’acide phosphatidique par une PLA1/238.
L’acide phosphatidique est généré par l’hydrolyse du groupement de tête de phospholipides
membranaires par des PLD1/2 ou par l’ajout du groupement phosphate à un diacylglycérol
par une diacylglycérol kinase. La formation à partir du glycérol-3-phosphate se fait par
l’ajout d’un acide gras par des glycérol-3-phosphate acyltransférases39. Enfin, la formation
de LPA à partir du monoacylglycérol se fait par ajout d’un groupement phosphate en sn-3
par une monoacylglycérol kinase40.
La fonction principale du LPA intracellulaire est de servir d’intermédiaire au métabolisme
des glycérophospholipides. Il peut donc être rapidement pris en charge par des
lysophosphatases et des acyl glycerol-3-phosphate acyltransférases41 pour former
respectivement du glycérol-3-phosphate et de l’acide phosphatidique.
La famille des glycérol-3-phosphate acyltransférases (1 à 4) et les acyl glycérol-3-phosphates
acyltransférases sont localisées sur la membrane des mitochondries29,39 et du réticulum
endoplasmique28,39. La production du LPA est donc localisée à ces compartiments.
Cependant, le LPA peut être transporté entre différents compartiments intracellulaires par
son association avec la protéine de liaison cytosolique du LPA (cytosolic LPA-binding
protein)39. La protéine de transport est capable d’inhiber, dans la mitochondrie, ou de
stimuler, dans le réticulum endoplasmique, la production de LPA39.
1.2.2 Synthèse extracellulaire
À ce jour, deux mécanismes de production de LPA extracellulaire ont été décrits, soit à partir
d’acide phosphatidique, soit de phospholipides comme la phosphatidyl-sérine, -choline ou -
éthanolamine (Figure 4, partie du centre).
Des PLA1/2 peuvent hydrolyser un acide gras de l’acide phosphatidique pour former le LPA.
Ce mécanisme a été mis en évidence, dans les follicules pileux avec la PLA1 membranaire
spécifique pour l’acide phosphatidique, PA-PLA1α également appelé lipase H42-44. Il a aussi

7
été montré sur des vésicules extracellulaires (extracellular vesicles, EV) par des PLA2
secrétées45 et dans des lignées de cellules cancéreuses d’ovaire par des PLA2 membranaires46.
Le second mécanisme requiert l’hydrolyse d’un des acides gras de la phosphatidyl-sérine, -
choline ou -éthanolamine par des PLA1/2 pour former des lysophospholipides42,47. Le
groupement de tête des lysophospholipides est ensuite clivé par une PLD pour former le LPA.
L’activité de la PLD dans les milieux extracellulaires est assurée par l’autotaxine et constitue
la source majoritaire du LPA extracellulaire48,49.
Cette voie de synthèse utilise des sources diverses de substrats. Les phosphatidyl -sérines, -
cholines ou -éthanolamines peuvent être présentes dans les lipoprotéines50 et à la suite d’une
asymétrie de la membrane plasmique des cellules42,46,47 et de certaines EV45,51. Les EV
peuvent aussi être une source directe de lysophospholipides51. L’oxydation de lipoprotéines
génère également des lysophospholipides qui peuvent être transformés en LPA par
l’autotaxine52-54. Enfin, le milieu extracellulaire peut contenir des quantités importantes de
lysophospholipides comme c’est le cas dans le sang où la concentration en
lysophosphatidylcholine est de l’ordre de 140 µM chez l’humain55-57.
1.3 L’autotaxine
L’autotaxine (abrégée en ATX dans les figures et manuscrit) est une enzyme secrétée de la
famille des ectonucléotides pyrophosphatase/phosphodiestérase (ENPP) et peut être notée
ENPP2. Les modèles d’invalidation génique de l’autotaxine sont léthaux à cause de son rôle
essentiel dans le maintien des vaisseaux lors de la vasculogenèse embryonnaire58-60 et lors du
développement du système nerveux59,61. Un défaut d’autotaxine ou de son activité chez des
souris adultes n’induit aucune létalité ni phénotype visible62. En revanche, elle est impliquée
dans le développement de nombreuses pathologies dont l’obésité63,64, des cancers65,66, des
maladies pulmonaires67, cardiovasculaires68,69 et inflammatoires70. À ce jour, l’ensemble des
effets biologiques de l’autotaxine ont été associés à la production de LPA58-60.
L’autotaxine présente une double activité catalytique. D’une part elle peut hydrolyser des
groupements phosphates sur des nucléotides par son activité
pyrophosphatase/phosphodiestérase71. D’autre part, elle peut former du LPA et de la

8
sphingosine-1-phosphate à partir de lysophospholipide48,49 et de
sphingosylphosphorylcholine72, respectivement, par son activité de PLD (Figure 2).
L’affinité de l’autotaxine pour la lysophosphatidylcholine, un des principaux substrats pour
la formation du LPA, est trois fois plus forte que pour la sphingosylphosphorylcholine72 et
dix fois plus forte que pour les nucléotides49. L’activité PLD est donc son activité principale
et son affinité pour les lysophospholipides expliquent que les effets biologiques de
l’autotaxine sont associés avec la formation de LPA.
L’expression de l’autotaxine est détectée dans la quasi-totalité des tissus testés, à l’exception
des cellules musculaires lisses et des cellules endothéliales aortiques73-76. Les tissus adipeux
et lymphoïdes présentent une expression importante de l’autotaxine75,76. En effet, depuis qu’il
a été montré que l’expression de l’autotaxine dans le tissu adipeux affecte ses quantités
plasmatiques, le tissu adipeux est considéré comme une source majeure de l’autotaxine du
milieu extracellulaire76.
1.3.1 Isoformes
Le gène de l’autotaxine, noté ATX, est composé de 27 exons et 26 introns et est présent chez
l’humain sur la région chromosomiale 8q2473,77,78. La forme murine présente une homologie
de 93% et une structure similaire à la forme humaine73,79. De nombreux épissages alternatifs
du gène sont possibles et cinq isoformes ont été détectées chez l’humain, notées ATXα, β, γ,
δ et ε (Figure 3)73,74. Le gène ATX est fortement conservé dans l’évolution80 et les isoformes
α, β, γ sont présentes chez d’autres mammifères comme la souris et le rat73. L’ensemble de
ces isoformes ont des activités PLD et de pyrophosphatase/phosphodiestérase avec des
affinités similaires pour leurs substrats73,74. Les différentes isoformes se distinguent par leur
proportion et la localisation tissulaire de leur expression73. L’ATXβ est la plus représentée et
est considérée comme la forme canonique. Elle représente la forme la plus abondante dans
les tissus testés à l’exception du cerveau. C’est l’isoforme qui est la plus utilisée pour l’étude
de l’ATX73,74,81. ATXδ est la deuxième isoforme la plus fréquente et présente une distribution
similaire à ATXβ. ATXγ est l’isoforme majoritaire dans le cerveau, mais elle est peu détectée
dans les autres tissus73. ATXα et ATXε sont faiblement détectées dans les tissus73,74. En

9
revanche, ATXα est la seule isoforme capable de lier les héparanes sulfates ce qui lui permet
de localiser la production de LPA à la membrane plasmique81.82
1.3.2 Structure et expression
Toutes les isoformes de l’autotaxine ont la même structure et le même mécanisme de
sécrétion (Figure 4, partie supérieur droite)73,74. L’autotaxine est d’abord sous forme de
pré-pro-enzyme et partage la structure des ENPP1-3 soit en N-terminal un peptide signal,
avec domaine transmembranaire, suivi de 2 domaines somatomedin B, puis le domaine
catalytique, et se termine avec un domaine de type nucléase en C-terminal. Contrairement
aux ENPP1 et 3 dont le peptide signal est conservé et permet l’ancrage du domaine
transmembranaire à la membrane plasmique, le peptide signal de l’autotaxine contient un site
de clivage83. Une fois le peptide signal clivé, la forme pro-enzyme de l’autotaxine entre dans
la voie de sécrétion classique, soit dépendante du Golgi83,84. Les domaines somatomedin B
et de type nucléase sont nécessaires à cette sécrétion. Le domaine de type nucléase permet
son transport au Golgi85, où une glycosylation dans les domaines somatomedin B permet sa
sécrétion et son activité catalytique86. Avant d’être libérée dans le milieu extracellulaire, la
forme pro-enzyme de l’autotaxine est clivée davantage en N-terminal par une pro-protéine
convertase de type furine et perd son domaine transmembranaire83. La forme extracellulaire
active de l’autotaxine consiste en N-terminal de 2 domaines somatomedin B avec une
glycosylation suivie du domaine catalytique et enfin du domaine de type nucléase83,86,87.
Le domaine catalytique de l’autotaxine assure l’activité nucléotide
pyrophosphatase/phosphodiestérase et PLD sur un même site actif88,89. Bien que le domaine
de type nucléase ne porte pas d’activité catalytique, il est essentiel pour assurer la liaison des
Figure 3: Structure des isoformes d’autotaxine. ATX autotaxine, SP peptide signal, SMB domaines
somatomedin B, PDE domaine central catalytique phosphodiestérase, NUC domaine C-terminal similaire à
nucléase. (Adaptée de Perrakis et Moolenaar, 2014(82))

10
substrats et est nécessaire à l’activité du domaine catalytique90,91. Les produits de l’activité
catalytique de l’autotaxine, que sont le LPA et la sphingosine-1-phosphate, inhibent son
activité enzymatique83,87. Cette inhibition est de type mixte, c’est-à-dire que le LPA et la
sphingosine ne sont pas en compétition avec le substrat pour le site actif, mais se lient à un
deuxième site sur les domaines somatomedin B83,87.
Enfin, ce sont les domaines somatomedin B qui permettent la liaison de l’autotaxine aux
intégrines β1 et β387,92 et aux héparanes sulfates pour l’isoforme α81. Cela permet de localiser
la production de LPA à proximité des récepteurs aux LPA à la surface des cellules81,87,92.
1.3.3 Régulation de l’expression
L’expression du gène ATX est régulée au niveau épigénétique93,94, transcriptionnelle et post-
transcriptionnelle95. Sous le contrôle de nombreux facteurs de transcription, sa transcription
est stimulée par la famille de la protéine activatrice 1 (activating protein-1, AP-1) qui met en
jeu c-Jun96-98, la protéine de spécificité 1 (Specific protein 1, SP1)98, le facteur HOX1399, le
facteur nucléaire κ B (Nuclear factor κ B, NFκB)100-102, le facteur nucléaire des lymphocytes
T activés 1 (Nuclear factor of activated T cells 1, NFTA1)103, le signal de transduction et
d’activation de transcription 3 (Signal transducer and activator of transcription 3,
STAT3)104 ainsi que la β-caténine105. SP3 est le seul facteur de transcription identifié comme
un répresseur98. Enfin sa régulation post-transcriptionnelle est sous le contrôle de deux
protéines de liaison à l’ARN. L’antigène humain R (Human antigen R) permet la stabilisation
de l’ARN messager et stimule l’expression de l’ATX tandis que le facteur de liaison et
dégradation ARE/poly(U) 1 (ARE/poly(U)-binding/degradation factor 1) l’inhibe95.
Ces régulations de l’expression d’ATX sont mises en jeux par différents médiateurs
extracellulaires comme des facteurs de croissance ou encore des médiateurs pro-
inflammatoires. Les facteurs de croissance épidermique (Epidermal Growth Factor, EGF),
des fibroblastes (fibroblast growth factor, FGF)106 ainsi que le facteur de nécrose tumorale
(tumor necrosis factor, TNF)70,106,107 et l’interleukine (IL-) 6108 stimulent directement
l’expression d’ATX. L’activation des récepteurs de type Toll (Toll-like receptors, TLR) 3, 4
et 9 par, respectivement, des ARN doubles brins, des lipopolysaccharides et l’ADN,
stimulent également son expression de manière indirecte105,109. Les TLR induisent la

11
production d’interféron α et β qui activent le récepteur à l’interféron α/β 1 et stimulent
l’expression d’ATX109. Enfin, l’IL-1β a été rapportée comme un activateur106 et un
inhibiteur110,111 de son expression. Outre l’IL-1, le facteur de croissance transformant β106,
l’IL-4106 ainsi que les produits de l’autotaxine, soit le LPA et la sphingosine-1-phosphate107,
inhibent son expression.
1.4 L’acide lysophosphatidique vasculaire
Dans le compartiment vasculaire, qui est le compartiment d’intérêt des travaux de cette thèse,
le LPA est de forme acyl-LPA que ce soit dans le plasma ou dans le sérum humain, bien que
la présence d’alkyl-LPA ait été proposée dans certains contextes35,112-114. Les espèces acyl-
LPA 16:0, 18:0, 18:1, 18:2 et 20:4 sont parmi les espèces les plus représentées14. Cependant,
la proportion de chaque espèce varie du plasma au sérum. Dans le plasma, l’espèce la plus
représentée est l’acyl LPA 18:2 suivie du 18:1, 18:0, 16:0 et 20:414. Dans le sérum, l’acyl
LPA 20:4 et 18:2 sont trouvés en quantité similaire suivis de l’acyl LPA 18:1, 16:0 et 18:014.
Dans la vasculature, le LPA peut être associé avec l’albumine23,115 ou avec la gelsoline116,117.
L’association avec ces protéines protègent le LPA de la dégradation et peut moduler
positivement et négativement ses effets.
Le LPA vasculaire est formé directement dans le milieu extracellulaire par l’autotaxine qui
explique l’augmentation observée dans le sérum14,47,118. L’accumulation du LPA lors de la
préparation du sérum est associée avec la libération de l’autotaxine contenue dans les
granules des plaquettes119. L’activation plaquettaire est une importante source de manière
locale d’autotaxine119. L’autotaxine peut être associée avec différents acteurs vasculaires :
d’une part les cellules, notamment les plaquettes activées92,120 et les EV, et d’autre part avec
les lipoprotéines, où elle utilise les phospholipides oxydés comme substrat54,68. Le tissu
adipeux serait une source importante de l’autotaxine75,121. En effet, la perte d’expression de
l’autotaxine dans les adipocytes entraine la diminution de 40% des quantités de LPA
plasmatique dans des modèles murins75.
La quantité plasmatique de LPA chez les personnes saines est encore sujette à débat. En
fonction des études, elle varie en quantités de l’ordre de 0,1 µM122,123 jusqu’à atteindre
1 µM124,125 alors qu’un dernier groupe d’études détecte des quantités de l’ordre de

12
0,7 µM14,126,127. L’ensemble des études établissent que la concentration de LPA plasmatique
est plus élevées chez la femme que chez l’homme14,122,123,126. Enfin, une étude a associé
positivement les quantités de LPA plasmatique avec l’indice de masse corporelle126. En
situation pathologique, les quantités de LPA plasmatique peuvent atteindre jusqu’à
12 µM124,125,127.
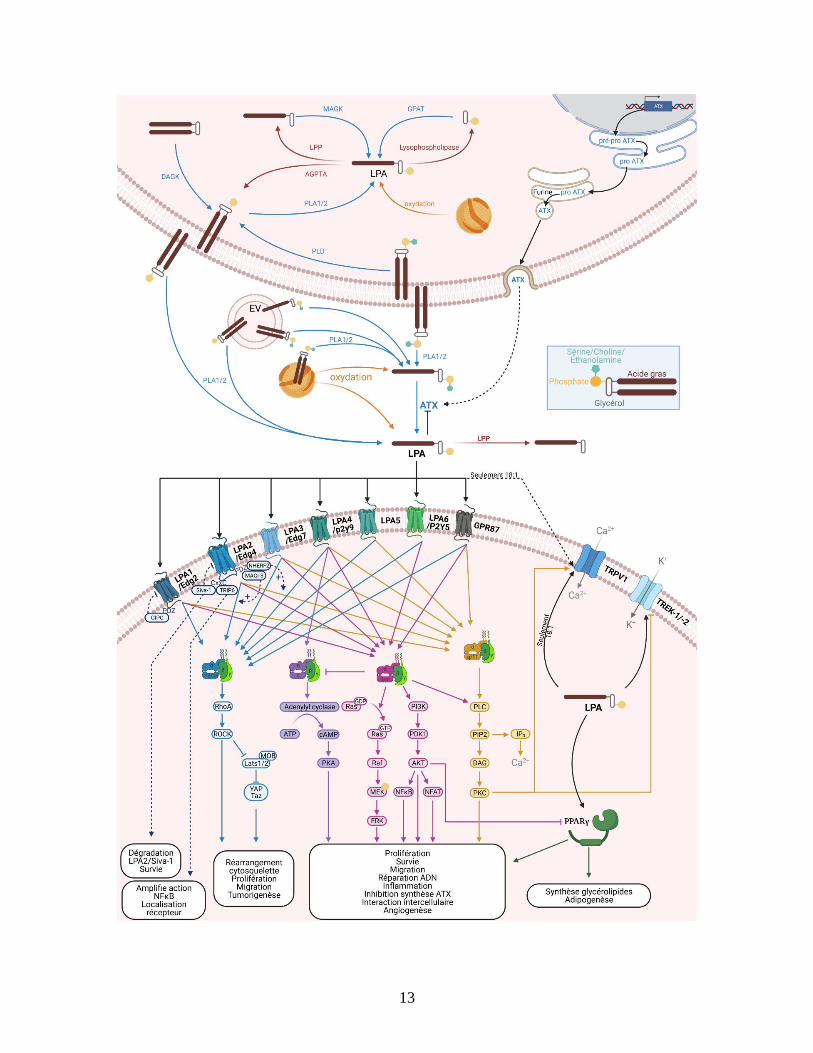
Figure 4: Synthèse, signalisation et dégradation du LPA. La partie supérieure gauche présente la synthèse
la synthèse intracellulaire du LPA par l’action des diacylglycerol kinases (DAGK); monoacylglycerol kinases
(MAGK); glycerophosphate acyltransférases (GPAT) et les phospholipases de type A et D (PLA1/2 et PLD).
Le LPA intracellulaire est dégradé par des phosphatases, des lipide-phosphate phosphatases (LPP) et par
l’acylglycerophosphate acyltransférase (AGPTA). La partie supérieure gauche présente la synthèse et la
sécrétion d’ATX. Le centre présente la synthèse extracellulaire du LPA par l’ATX et les PLA1/2 et sa
dégradation par les LPP. L’oxydation des lipoprotéines à faible densité peut également produire du LPA intra
et extracellulaire. La partie inférieure présente la signalisation induite du LPA sur les 7 récepteurs aux protéines
G (LPA1 à 6 et GPR87), le récepteur intracellulaire PPARγ et les canaux ioniques TRPV1, TREK1 et 2. La
figure a été créée à l’aide de BioRender.com.
13

14
1.5 Signalisation dépendante de l’acide lysophosphatidique
L’étude de la signalisation dépendante du LPA a débuté en 1996 avec la découverte du
premier récepteur au LPA et s’est étoffé au fil des ans7. Depuis, six récepteurs aux protéines
G sont considérés comme les cibles principales du LPA et ont été nommés récepteurs au LPA
1 à 6, notés LPA1-6 selon l’ordre de leur découverte7,128-132. Les LPA1-6 sont subdivisés en
deux familles, ceux dont l’expression est reliée aux gènes de différentiation endothéliales
(endothelial differentiation gene, Edg) et ceux qui sont proches des récepteurs purinergiques,
P2Y. En plus de ces récepteurs, le LPA peut activer le récepteur couplé aux protéines G 87
(G Protein-Coupled Receptor 87, GPR87)133,134. Enfin, il a été rapporté que le LPA active le
récepteur couplé aux protéines G P2Y10135. Cependant, une étude subséquente a identifié
non pas le LPA, mais la lysophosphatidylsérine (Lyso-PS) comme ligand de P2Y10136. Il n’y
a pas eu de publication additionnelle rapportant le LPA comme activateur du P2Y10.
Outre les récepteurs aux protéines G, le LPA est un ligand du récepteur intracellulaire activé
par les proliférateurs de peroxysomes (peroxisome proliferator-activated receptor gamma,
PPARγ)30, ainsi que de plusieurs canaux ioniques comme le récepteur transitoire à potentiel
vanilloïde 1 (transient receptor potential vanilloid 1, TRPV1)137 et les canaux à ion de
potassium apparenté TWIK-1 et -2 (TWIK related K+ channel-1 and -2, TREK-1/-2)138.
La signalisation du LPA a plusieurs niveaux de complexité. D’abord, il y a le patron
d’expression de chaque récepteur qui varie en fonction du tissu et du développement128,139.
Chaque récepteur peut s’associer avec différents médiateurs intracellulaires. Les récepteurs
aux protéines G activés par le LPA peuvent interagir avec quatre protéines G différentes
(Gα12/13, Gαq/11, Gαs et Gαi). Ensuite, les effets médiés par les récepteurs au LPA peuvent leur
être propres, partagés ou encore opposés à ceux des autres récepteurs au LPA. Enfin, les
récepteurs ont des affinités différentes pour les espèces moléculaires de LPA. Il en résulte
que les effets du LPA sur l’environnement cellulaire ne s’explique pas seulement en fonction
des récepteurs présents et de leur médiateurs intracellulaires associés mais également en
fonction des espèces moléculaires de LPA mis en jeux dans l’environnement extracellulaire.
La signalisation dépendante des récepteurs est récapitulée dans la partie inférieure de la
Figure 4.

15
1.5.1 La famille EDG des récepteurs couplés aux protéines G : LPA1, 2 et 3
1.5.1.1 LPA1/Edg2
En 1996, le LPA est identifié comme ligand du récepteur codé par le gène de la zone
ventriculaire 1 (ventricular zone gene-1)7. Le récepteur est nommé par la suite LPA1. LPA1
est codé par le gène LPAR1 dans la région chromosomique 9q31.3140. Il est le deuxième
membre de la famille des récepteurs couplés aux protéines G de type EDG. De ce fait, il
partage une homologie importante avec LPA2 et 3 qui sont également des récepteurs de la
famille EDG128,141. LPA1 est fortement exprimé dans le cerveau, le cœur et les intestins, mais
son expression est communément détectée dans d’autres tissus128,139. LPA1 est le récepteur
au LPA le plus étudié. Un de ses principaux rôles est le développement et le fonctionnement
du système nerveux142. Cependant, il régule aussi de nombreux mécanismes biologiques
comme la tumorigenèse143-145, l’ostéogenèse146,147, l’inflammation et l’immunité148,149 ou
encore le système vasculaire150-152.
Activation et signalisation
La position de l’acide gras sur le squelette de glycérol n’affecte pas l’interaction du LPA
avec LPA1, cependant les formes acyl-LPA présentent une affinité plus forte comparée aux
alkyl- ou alkeny-LPA8. Les espèces insaturées de LPA présentent une affinité plus forte avec
LPA1. Les espèces de LPA qui contiennent les acides gras 16:1, 18:1, 18:2, 18:3 et 20:4
présentent la plus grande affinité pour LPA1, viennent ensuite les formes saturées, d’abord
le LPA 16:0 et ensuite 18:08. LPA 12:0 et 14:0 sont capables d’activer le LPA1 mais
nécessitent de très forte concentrations8.
La signalisation de LPA1 repose sur son interaction avec les 3 protéines G, Gα12/13, Gαq/11, et
Gαi/o 8,153,154. Son association avec Gα12/13 permet l’activation de la signalisation dépendante
de RhoA/ROCK principalement impliquée dans le réarrangement du cytosquelette155. LPA1
active la voie de signalisation dépendante de la PLC et des PKC par son interaction avec
Gαq/11154. Cette voie permet notamment à LPA1 de réguler l’entrée d’ions à la membrane
cellulaire par l’activation du canal calcique TRPV1156,157 ou l’inhibition du canal potassium
TREK-1158. Enfin sous contrôle de LPA1, Gαi/o induit les signalisations dépendantes des

16
MAP kinases et de la PI3K153 ainsi que l’inhibition de l’adénylyl cyclase154. L’association de
LPA1 à Gαi/o permet notamment la transactivation du récepteur à l’EGFR159.
Le LPA1 présente des domaines PDZ en C-terminal qui est l’acronyme des trois premières
protéines où ce domaine a été mis en évidence160. Les domaines PDZ permettent l’interaction
de l’extrémité C-terminale avec des protéines spécifiques161. Seules deux protéines
interagissent avec LPA1 par les domaines PDZ, soit la protéine d’interaction en C-terminus
de type Gα (Galpha-interacting protein C-terminus, GIPC)162 et le facteur d’échange de
nucléotide guanine spécifique à RhoA (Rho-specific guanine nucleotide exchange factors,
RhoGEF)163. L’association avec GIPC induit la dégradation de LPA1 dans les endosomes et
ainsi inhibe sa signalisation du LPA1162,164. L’interaction avec RhoGEF stimule la
signalisation dépendante de RhoA163.
Principales fonctions
Dans le système nerveux, LPA1 est exprimé par les neurones centraux et périphériques ainsi
que par les cellules gliales comme les astrocytes, les cellules de Schwann ou les
oligodendrocytes165. LPA1 promeut leur migration, leur prolifération166 et leur différentiation
ainsi que la survie cellulaire pour les cellules de Schwann167 et les neurones168. Cependant,
LPA1 peut également induire l’apoptose des neurones par l’initiation de dysfonctionnements
mitochondriaux169. De plus, le LPA1 module les flux calciques neuronaux170,171, la
production de neurotransmetteurs170,172 ainsi que l’expression des gènes associés avec la
balance d’excitation et d’inhibition dans l’hippocampe173. Enfin, LPA1 est impliqué dans le
changement de morphologie154,165,174 et dans la formation de myéline167,175.
Il résulte que le modèle d’invalidation génique de LPA1 chez la souris entraine, entre autres,
une malformation de plusieurs régions du cerveau ainsi qu’une mortalité néonatale de 50 %
à cause d’un défaut dans le comportement d’allaitement142. Les études d’invalidations
géniques conditionnelles subséquentes ont mis en évidence un lien entre LPA1 avec
différents comportements comme l’anxiété173,176,177, la régulation des émotions173, la
consommation d’alcool173 ou de nourriture178 ou encore la mémoire spatiale176,177. Enfin, son
absence induit des symptômes similaires à ceux induits lors de la schizophrénie170,172,179.
Outre le développement et le comportement, LPA1 est impliqué dans la médiation de la

17
douleur. Dans la douleur neuropathique, qui est la douleur due à une blessure ou une maladie
du système de somesthésie, LPA1 participe à l’initiation et à l’amplification de la douleur
centrale et périphérique155,180,181. Plus récemment, LPA1 a été associé à la médiation de la
douleur d’origine inflammatoire182 et aux réponses anormales de douleurs dans le contexte
du diabète183. Enfin des études récentes s’intéressent à son rôle dans la progression de la
sclérose en plaque et de la sclérose latérale amyotrophique184,185.
En dehors de ses effets spécifiques au système nerveux, le LPA1 module la minéralisation
osseuse en stimulant sa formation186 et sa résorption146. Sa signalisation stimule la formation
osseuse par la différenciation des ostéoblastes en ostéocytes187-189. Elle induit aussi le
bourgeonnement de la membrane des ostéoblastes et par conséquent la libération d’EV190.
Bien qu’il augmente l’expression de médiateur anti-inflammatoire comme le suppresseur de
tumorigénicité 2 (suppression of tumorigenity 2)191, LPA1 induit également l’expression de
cytokines pro-inflammatoires par les ostéoblastes comme IL-6 et IL-8 qui promeuvent la
différenciation en ostéoclaste147,187,192. De plus, LPA1 stimule l’activité de résorption146.
LPA1 semble davantage favoriser la minéralisation de l’os en situation physiologique186,189.
Cependant dans des modèles pathologiques, son rôle dans la résorption est mis en avant146,147.
Similairement, LPA1 stimule la formation du cartilage en stimulant la prolifération des
chondrocytes et l’assemblage de fibronectine193,194, mais il semble lié à sa dégradation dans
l’arthrite rhumatoïde147,195. Cette différence peut venir de son implication dans
l’inflammation qui promeut l’activité des ostéoclastes et donc la dégradation osseuse comme
dans un modèle d’arthrite rhumatoïde147,192.
En effet LPA1 promeut l’inflammation par la production de nombreuses cytokines pro-
inflammatoires comme IL-1, IL-6, IL-8 et IL-17 par différents types cellulaires147-
149,187,192,196. Il favorise l’adoption d’un phénotype inflammatoire par les macrophages185 et
la différenciation des lymphocytes T en lymphocytes T auxiliaires149. Il induit également
l’expression des protéines d’adhésion des cellules endothéliales148,149 et le recrutement de
neutrophiles et de macrophages au site inflammatoire148,185,197,198. LPA1 a été associé avec
l’inflammation neuronale185,199, pulmonaire198, abdominale196, systémique196 ainsi que celle
dans la membrane synoviale qui contribue à la progression de l’arthrite
rhumatoïde147,149,195,200.

18
LPA1 promeut le développement de fibroses par son effet pro-inflammatoire201-203 et en
participant au recrutement et à la prolifération des fibroblastes150,204 ainsi qu’à la formation
de collagène198,203,205. Le LPA1 est essentielle pour le développement de la fibrose dans le
modèle de sclérodermie induit par la bléomycine206.
L’effet de LPA1 dans le recrutement de cellules immunitaires et dans la fibrose, est
partiellement médié par l’augmentation de la perméabilité des parois intestinales et
vasculaires150,207. Outre son action sur la perméabilité de l’endothélium vasculaire, LPA1
stimule le recrutement et la prolifération des cellules du muscle lisse vasculaire208,209 ce qui
lui permet d’induire la formation de néo-intima à la suite de dommages aux vaisseaux
sanguins209. LPA1 module le tonus vasculaire. L’activation du LPA1 des cellules
endothéliales induit la vasodilatation151,210 alors que son activation sur les cellules
musculaires lisses induit une vasoconstriction152.
Enfin, le LPA1 présente un rôle mixte dans la tumorigenèse. LPA1 peut stimuler la survie211,
la motilité212,213, l’invasion212,213, la prolifération cellulaire214 ou encore la formation de
métastases par différentes lignées cancéreuses213. Il peut aussi stimuler l’expression
d’oncogènes143,144, de facteurs de croissance215-217 et de cytokines215. De même, plusieurs
études impliquent LPA1 dans les mécanismes de résistance aux traitements anti-
cancéreux143,144. Cependant, de nombreuses lignées cancéreuses présentent une mutation
rendant LPA1 inactif ou réprimant son expression145. Il inhibe la progression de certaines
tumeurs par la répression de la motilité218,219 ou de l’expression de facteurs de croissance220.
1.5.1.2 LPA2/Edg4
LPA2 est codé par le gène LPAR2 situé dans la région chromosomique 19p13.11 chez
l’humain et présente une forte homologie avec les deux autres récepteurs de type EDG, LPA1
et LPA3128,141. L’expression de LPA2 est détectée dans de nombreux tissus mais à des
niveaux souvent plus faibles que LPA1128,139. Une forte expression de LPA2 est présente chez
les leucocytes et dans le tissu testiculaire128 et LPA2 est également retrouvé dans l’intestin et
le cerveau139,221. L’étude de LPA2 est principalement axée sur la protection de l’endothélium
intestinal, l’organisation du système nerveux et vasculaire ainsi que sur l’immunité. Enfin,

19
LPA2 stimule également la tumorigenèse ce qui explique pourquoi il est présent dans de
nombreuses lignées cancéreuses128,222-225.
Activation et signalisation
L’affinité des espèces de LPA pour LPA2 n’est pas affectée par la position de l’acide gras
sur le squelette de glycérol8. En revanche, les formes acyl-LPA sont privilégiées par rapport
aux formes alkyl- et alkenyl-LPA8. Les formes de LPA avec les acides gras 16:0, 16:1, 18:1,
18:2, 18:3 et 20:4 ont l’affinité la plus forte avec LPA2, vient ensuite LPA 14:0 puis LPA
18:0 et enfin LPA 12:0 qui active très faiblement le récepteur8.
LPA2 est capable de s’associer avec 3 protéines G, Gα12/13, Gαq/11, et Gαi/o154,226,227. Bien que
LPA2 puisse s’associer à Gα12/13 pour activer RhoA/ROCK227, LPA2 peut aussi activer cette
voie par son association avec Gαq/11 201,228,229. L’activation de la voie RhoA/ROCK par Gαq/11
permet à LPA2 d’induire l’expression de l’intégrine β6 et la transactivation de la signalisation
au TGF-β 201,228,229. Gαq/11 permet également de promouvoir l’activation de la PLC et
l’accumulation de DAG et d’IP3. L’accumulation d’IP3 permet à LPA2 d’induire la
mobilisation de calcium, pas seulement par son association avec Gαq/11, mais également avec
Gαi/o230,231. L’association de LPA2 avec Gαi/o permet également l’activation des voies des
MAP kinases et de PI3 kinase/AKT ainsi que la transactivation d’EGFR232-234.
LPA2 se différencie des autres récepteurs au LPA par un niveau de régulation supplémentaire
de son activité. Son extrémité C-terminale présente des domaines de liaison de CXXC235,236
et PDZ235,237. À l’aide des domaines PDZ, LPA2 interagit avec le facteur 2 de régulation des
échanges sodium hydrogène (sodium/hydrogen exchanger regulatory factor 2, NHERF2)236-
238, la guanylate kinase associée à la membrane avec une orientation inversée 3 (membrane-
associated guanylate kinase with inverted orientation-3, MAGI-3)238,239 et RhoGEF163. Son
interaction avec RhoGEF promeut l’activation de RhoA163. L’interaction de LPA2 avec
MAGI-3 promeut son association avec Gα12/13 tandis que NHERF2 stimule celle avec
Gαq/11238. MAGI-3 et NHERF2 sont en compétition pour LPA2 et donc ont un impact sur la
voie de signalisation induite suite à l’activation de LPA2238.

20
Outre les protéines qui interagissent avec ses domaines PDZ, LPA2 présente également des
motifs CXXC qui peuvent lier la protéine d’interaction au récepteur de la thyroïde 6 (thyroid
receptor-interacting protein 6, TRIP6)235,236,240 et le facteur inducteur d’apoptose Siva-
1236,241. Le recrutement de TRIP6 au LPA2 amplifie l’activation de NFκB dépendante de
LPA2240. Contrairement à TRIP6, Siva-1 réprime la signalisation dépendante de LPA2236,241.
Une fois lié à Siva-1, LPA2 est ubiquitinylé, ce qui conduit à la dégradation de LPA2 et de
Siva-1241.
Les interactions de LPA2 avec NHEFR2 and TRIP6 permettent également de localiser ses
effets dans l’environnement intracellulaire235,242,243. Cela permet, par exemple, d’orienter la
migration cellulaire en fonction d’un gradient de LPA242, de localiser l’effet de LPA2 au
cytosquelette235 ou proche d’effecteurs membranaires243.
Principales fonctions
Similairement à LPA1, LPA2 protège les progéniteurs de neurones contre l’apoptose244, mais
semble induire l’apoptose des neurones169. LPA2 participe aussi à la transduction neuronale
par la libération de glutamate et la mobilisation de calcium245. Enfin, LPA2 stimule la perte
de myéline après des blessures du système nerveux246.
LPA2 est impliqué dans le maintien de l’intégrité vasculaire et intestinale247-249. Il régule les
échanges de liquide par l’activation d’échangeur d’anion qui lui donne un effet anti-
diarrhéique234,250. Il induit la production de prostaglandines E2 qui sont impliquées dans la
protection des cellules gastriques contre l’environnement délétère de l’estomac248. Enfin, le
LPA2 stimule plusieurs mécanismes cellulaires impliqués dans la survie cellulaire et la
résistance à la radiation251-255. D’une part, LPA2 promeut la réparation de l’ADN252-254.
D’autre part, il diminue différents signaux apoptotiques. LPA2 inhibe la translocation de
BAX à la mitochondrie ainsi que la production de médiateurs pro-apoptotiques solubles254.
Enfin, l’activation de LPA2 induit la dégradation du facteur pro-apoptotique Siva-2 qui lie le
LPA2 activé241.
Au niveau vasculaire, LPA2 stimule la lymphangiogenèse et l’angiogenèse par l’induction
d’IL-8256,257. Il participe également au recrutement des cellules du muscle lisse en partenariat

21
avec LPA1 en réponse à une blessure de la paroi vasculaire258. Enfin, LPA2 inhibe la
différenciation des progéniteurs myéloïdes vers les voies de mégacaryocytes et érythrocytes
dans les étapes précoces de différenciation et uniquement vers le destin d’érythrocytes dans
les étages tardives259-261.
LPA2 impacte le milieu vasculaire et intestinal également de par son rôle dans l’immunité et
l’inflammation. Chez les macrophages, il stimule leur recrutement262, la production de
purine246, de cytokines pro-inflammatoires263 et des métalloprotéases matricielles264. La
signalisation du LPA2 chez les macrophages est associée à de l’inflammation dans le système
nerveux central ainsi qu’à de l’inflammation musculaire, intestinale et vasculaire262-264.
L’importance de LPA2 dans l’immunité a été la plus étudiée dans le contexte allergique. En
effet, LPA2 est impliqué dans l’allergie en réponse à une stimulation des muqueuses,
systémique ou des voies respiratoires265. Au niveau pulmonaire, LPA2 stimule le recrutement
des éosinophiles, des lymphocytes TH2 au poumon266,267. Il induit l’activation des cellules
dendritiques, lymphocytes T et la production de cytokines pro-inflammatoires265-269.
Outre ses effets pro-inflammatoires, LPA2 induit la production de cytokines pro-fibrosantes
et la transactivation du récepteur au TGFβ201,228,229,270. De plus, il induit le recrutement et la
différenciation de fibroblastes en myofibroblastes ainsi que l’accumulation de fibronectine,
d’actine et de collagène270. Ses effets promeuvent le développement de fibroses, notamment
pulmonaire228,270.
Enfin, LPA2 est fréquemment exprimé dans les cancers et supporte leur développement.
C’est notamment le cas pour les cancers de l’intestin222,271,272, du sein273,274 et des
ovaires233,275-277. Il stimule la transformation tumorale278,279, la prolifération219,240,273,280,281,
migration219,273,276,277 et l’invasion des cellules tumorales222,273,276,277. Récemment,
l’expression de LPA2 par les cellules cancéreuses est un facteur de
chimiorésistance223,224,227,282.
1.5.1.3 LPA3/Edg7
LPA3 est codé par le gène LPAR3 situé dans la région chromosomique 1p22.3 chez l’humain.
Il présente une forte homologie avec les récepteurs autres récepteurs de type EDG, LPA1 et

22
2129,141. Bien que trouvé dans de nombreux tissus, LPA3 est fortement exprimé au cerveau,
au cœur, aux testicules, à l’utérus, et aux poumons129,283-285. Les principaux domaines où le
rôle de LPA3 est étudié, sont la grossesse286,287, le maintien du système vasculaire259,288,289,
l’immunité148,290 ainsi que dans le cancer215,291.
Activation et signalisation
Le LPA3 a une affinité plus forte pour les espèces de LPA avec l’acide gras en position sn-2
et pour les formes insaturées8,129. LPA3 lie plus facilement d’abord les formes acyl-LPA puis
les alkyl- et enfin les alkeny-LPA8. Les formes de LPA avec les acides gras insaturées 18:1,
18:2 et 18:3 présentent la plus forte affinité avec LPA3 suivi du LPA 20:4 puis des formes
14:0, 16:0, 16:1 et 18:08. La forme LPA 12:0 est également capable d’activer LPA3, mais
seulement à de très fortes concentrations8.
LPA3 s’associe avec les protéines G, Gα12/13292,293, Gαq/11
283, et Gαi/o294. Quand LPA3 interagit
avec Gα12/13, il active la signalisation dépendante de RhoA, ROCK et YAP292,293. Par son
association avec Gαq/11, LPA3 peut activer la signalisation dépendante de la PLC154,219,295
ainsi que la mobilisation de calcium par l’activation de la PLC et la production d’IP3154,296.
Par l’activation de Gαi/o, LPA3 induit, d’une part, la signalisation dépendante de PI3K,
d’AKT et de NFκB219,256,297, qui permet notamment l’activation de la β-caténine297 et la voie
des MAP kinases105. D’autre part, l’interaction de LPA3 avec Gαi/o permet également
d’activer la voie de signalisation dépendante de la PLC, du DAG et des PKC qui est commune
avec Gαq/11297. Enfin, son association avec Gαi/o lui permet d’inhiber l’accumulation d’AMP
cyclique154. Les trois voies, dépendantes de Gα12/13, Gαq/11 et de Gαi/o, sont capables d’induire
la transactivation du récepteur à l’EGFR293,294,298. Enfin la signalisation dépendante de RhoA
et ROCK peut être activée par LPA3 de manière indépendante de Gα12/13219,296.
Principales fonctions
Similairement à LPA1, LPA3 est impliqué au cerveau dans la réponse aux douleurs
neuropathiques et anormales180,183,299,300. Les effets cellulaires qui ont été rapportés sont en
revanche bien plus limités que pour LPA1. LPA3 induit la formation chez les neurones de
neurites, soit un prolongement de celui-ci qui peut être un axone ou une dendrite154,300. Il peut

23
également induire la libération d’ATP par les macrophages du système nerveux, les
microglies301.
LPA3 a différentes fonctions au niveau vasculaire. D’une part, de manière opposée à LPA2,
LPA3 stimule l’érythropoïèse259,261,297. Il inhibe également la différenciation vers les
mégacaryocytes302. D’autre part, similairement à LPA2, LPA3 stimule l’angiogenèse et la
lymphangiogenèse par la production d’IL-8 et de VEGF256,288,298,303,304. Il est également
impliqué dans la réparation de la paroi vasculaire et du cœur après blessure par le recrutement
et la prolifération de cellules musculaires lisses et de cardiomyocytes209,289,293. Cependant les
effets de LPA3 sur les cardiomyocytes peuvent mener à une hypertrophie cardiaque305-307.
LPA3 permet le recrutement aux sites inflammatoires et l’activation de neutrophiles et de
monocytes ainsi que de ses formes différenciées que sont les macrophages et les cellules
dendritiques 105,197,290,308. Leur activation par LPA3 conduit à la libération de nombreux
médiateurs pro-inflammatoires comme le leucotriène B4, la prostaglandine E2 ou
CCL8105,197,290. Le recrutement des monocytes par LPA3 se fait par l’expression de cytokines
dont l’IL-1, CXCL8 (IL-8) et CCL2 ainsi que par l’expression de facteur d’adhésion par les
cellules endothéliales148. Enfin, LPA3 induit l’internalisation de lipoprotéines oxydées de
basse densité (oxidized low density lipoprotein, oxLDL) par les macrophages et donc leur
transformation en cellules spumeuses309,310. La motilité des cellules spumeuses est inhibée
par LPA3311.
Les modèles d’invalidation génique ont mis évidence que LPA3 est un acteur important dans
l’implantation des embryons dans l’endomètre utérin286,287. De même, chez l’humain, la
diminution de son expression dans les endomètres utérins est associée avec une baisse de la
fertilité à cause de soucis d’implantation des embryons312,313. LPA3 active la cyclooxygénase
2 et l’oxide nitrique synthase inductible314-316. Cela lui permet de stimuler la production des
prostaglandines E2 et I2286,315-317 et de transformer les cellules stromales en cellules
sécrétrices315. Cela permet à LPA3 de promouvoir l’implantation de l’embryon par le
développement de la vascularisation314,315 et la décidualisation, soit une modification de
l’endomètre permettant l’implantation de l’embryon306,314-316. Enfin, LPA3 est impliqué dans
les contractions utérines318,319.

24
LPA3 est fréquemment exprimé par les cellules cancéreuses215,275,320-323. LPA3 stimule la
migration et l’invasion de ces cellules144,215,219,292,321,323. De plus, il stimule leur survie,
notamment en réponse à des traitements, et leur prolifération cellulaire par l’inhibition de la
sénescence et l’expression de facteurs de croissance144,219,324,325. Cependant, l’absence
d’expression de LPA3 est associée à une mortalité plus forte dans le cancer du sein et lors de
métastases pulmonaires ou au cerveau291. De plus, des études mettent en évidence une
inhibition de la migration et de la survie ainsi que de l’angiogenèse282,326-328. Il en résulte que
l’implication de LPA3 dans la tumorigenèse est ambigüe.
1.5.2 Récepteurs couplés aux protéines G de type non-EDG
1.5.2.1 LPA4/P2Y9
LPA4 est le premier récepteur de type non-EDG identifié qui est codé par le gène LPAR4
présent dans la région chromosomique Xq21.1 chez l’humain. Il présente moins de 20 %
d’homologie avec les LPA1 à 3 mais est proche des LPA5 et 6132,141. LPA4 est fortement
exprimé dans les ovaires bien qu’une faible expression de LPA4 soit retrouvée dans de
nombreux tissus comme le cœur ou le thymus130,329. La signalisation de LPA4 a
principalement été étudiée dans le cancer330-332, l’ostéogenèse333,334, ainsi que la perméabilité
vasculaire335,336 et l’angiogenèse335,337-339.
Activation et signalisation
L’affinité des espèces de LPA pour LPA4 a été étudiée de manière limitée. Le LPA4 préfère
les LPA sous forme acyl- puis alkyl- et enfin alkenyl-LPA130. Parmi les espèces testées, le
LPA avec l’acide gras 18:1 présente l’affinité la plus forte suivie du LPA 18:0, puis de 16:0
et enfin 14:0130.
Une fois activé, le LPA4 est le seul récepteur au LPA capable de s’associer aux quatre
protéines G soit Gα12/13, Gαq/11, Gαs et Gαi/o 130,329. L’activation de LPA4 peut donc conduire à
l’accumulation d’AMP cyclique par son association avec Gαs 130,340. Son association avec
Gα12/13 active la signalisation RhoA/ROCK et YAP/TAZ 339,341. Quand LPA4 est associé à
Gαi/o, il met en jeu la signalisation ERK et PI3K278,330 et peut induire l’entrée de calcium dans
la cellule par l’activation de canaux cationiques329. Enfin LPA4 peut aussi induire l’activation

25
de la PLC et la mobilisation du calcium intracellulaire par son association avec Gαq/11329.
Étonnamment, LPA4 peut interagir avec Gαi/o et Gαs, alors que Gαi/o peut inhiber
l’accumulation d’AMP cyclique dépendante de Gαs. Cependant LPA4 privilégie l’interaction
avec Gαs à celle avec Gαi/o329. Enfin, la signalisation dépendante de LPA4 peut inhiber
l’activité de PPARγ342.
Principales fonctions
Bien que LPA4 inhibe la motilité cellulaire330-332 et promeut l’infiltration des leucocytes dans
les tumeurs341, son implication dans la tumorigenèse est contreversée. LPA4 stimule
également la formation d’invadopode343, et la transformation des cellules en cellules
cancéreuses278. LPA4 réprime la formation de tissu osseux en inhibant la différenciation des
ostéoblastes333,334.
Dans l’angiogenèse, LPA4 est impliqué dans le bourgeonnement de nouveaux vaisseaux par
le réarrangement du cytosquelette d’actine des cellules endothéliales335,338,339,341. De plus,
LPA4 renforce les jonctions adhérentes des cellules endothéliales vasculaires335 et stimule le
recrutement des cellules de la paroi vasculaire comme les cellules du muscle lisse et les
péricytes337. Il permet donc d’assurer le maintien et de moduler la perméabilité de la paroi
vasculaire335,336,341. Il est notamment impliqué dans la transmigration des leucocytes336,341.
Son action dans le maintien et le développement du système vasculaire est partiellement
médiée en partenariat avec LPA6339.
1.5.2.2 LPA5
LPA5 est codé par le gène LPAR5 présent dans la région chromosomique 12p13.31 chez
l’humain. Le LPA5 présente une homologie plus importante avec les LPA4 et 6 qu’avec les
récepteurs LPA1 à 3132,141. Son expression est principalement détectée dans le cœur, le
placenta, le cerveau et les intestins131 et dans certaines cellules immunitaires344,345. LPA5 est
impliqué dans les accidents ischémiques transitoires346,347, le comportement comme
l’anxiété348, la nociception348, l’immunité349-352 la régulation de la progression tumorale353.

26
Activation et signalisation
LPA5 présente une affinité plus forte avec des espèces de LPA sous ses formes alkyl- plutôt
qu’acyl-LPA112,354. Les espèces de LPA avec un acide gras 16:0, 18:1, 18:2, 18:3 et 20:4
présentent des affinités similaires pour LPA5. Seules les formes LPA 18:0 et 20:0 ont des
affinités plus faibles354. La farnesyl pyrophosphate et l’arachidonoyl-glycine sont également
des ligands de LPA5 même s’ils présentent des affinités plus faibles que le LPA354,355.
Les effets du LPA5 sont induits par son association avec deux protéines G. L’association à
la protéine Gα12/13 permet d’activer la signalisation RhoA/ROCK131. LPA5 interagit
également avec la protéine Gαq/11, ce qui induitl’activation de la PLC et des PKC131,356. Les
voies de signalisation en aval de LPA5 permettent l’activation de canaux ioniques 357,358 et la
transactivation du récepteur à l’EGFR357. Enfin la signalisation dépendante de LPA5 permet
l’accumulation d’AMP cyclique bien que LPA5 n’interagisse pas avec la protéine Gαs131.
Principales fonctions
La signalisation de LPA5 dans l’intestin et les reins permet l’absorption de liquides par
l’activation de l’échangeur d’ion sodium et potassium 3 356,357,359. Au cerveau, l’implication
de LPA5 dans la nociception et le comportement s’explique d’abord par son activation du
canal ionique TRPV1358. D’autre part, l’activation de LPA5 promeut un environnement
inflammatoire par l’activation des macrophages346,360, la différentiation de la microglie en
macrophage de type M346,361 et la production de cytokines346,352.
En dehors du cerveau, son action est plus ambiguë. LPA5 induit la sécrétion de cytokines
pro-inflammatoires chez les mastocytes362 et contrôle la voie d’activation des plaquettes
dépendante du LPA112,354,363. Cependant, LPA5 est décrit comme un activateur et un
inhibiteur de l’activation des macrophages351,360. Ses effets répresseurs de l’inflammation et
de l’immunité sont supportés également par son inhibition de la signalisation des récepteurs
des lymphocytes B349 et T350 et la libération de cytokines anti-inflammatoires351.
Enfin, dans le cancer, LPA5 diminue la chimiorésistance de différentes lignées
cancéreuses223,353,364 par l’inhibition de la migration353,365-368, de l’invasion332,369 et de la
survie cellulaire332,353. Bien que le LPA5 est majoritairement décrit comme un inhibiteur de

27
la tumorigenèse, plusieurs études ont rapporté que le LPA promeut la prolifération et la
motilité de certaine cellules cancéreuses et inhibe l’activité cytotoxique des lymphocytes T
CD8+ 350,370-372.
1.5.2.3 LPA6/P2Y5
LPA6, codé par le gène LPAR6 présent dans la région chromosomique 13q14 chez l’humain,
est le dernier ajout aux récepteurs au LPA. Il est un récepteur de la classe P2Y qui est
phylogénétiquement proche des récepteurs LPA4 et LPA5132,141. Bien que son étude soit
encore limitée, LPA6 est étudié dans les domaines classiques du LPA comme le
cancer331,332,373, l’immunité et l’homéostasie vasculaire336,351,374,375, mais également dans la
pousse de cheveux44,132.
Activation et signalisation
Le LPA6 a besoin de concentration de LPA plus élevée que les LPA1 à 5 pour être activé376.
Les formes alkyl- et acyl-LPA partagent des affinités similaires pour le LPA6376. De plus, il
est, avec LPA3, le seul récepteur au LPA à préférer les formes de LPA avec l’acide gras en
position sn-2 sur le squelette de glycérol376. LPA6 utilise du LPA formé au niveau des
follicules pileux, non par l’autotaxine, mais par la PA-PLA1α qui forme des 2-acyl-LPA43,44.
Enfin, l’espèce de LPA avec un acide gras 18:2 présente l’affinité la plus forte pour LPA6,
puis vient le LPA 18:1, ensuite 20:4 puis 16:0 après 18:0 et en dernier le LPA 14:0376.
LPA6 active la signalisation RhoA/ROCK par son association avec la protéine Gα12/13375,377
ainsi que les signalisations des MAPK et de la PI3K par la protéine Gαi377. Enfin,
l’accumulation d’AMP cyclique, qui est dépendante de la protéine Gαs, mise en évidence par
une première étude132, n’a pas été observé par deux autres équipes132,376. L’association de
LPA6 avec la protéine Gαs est donc incertaine. L’activation de LPA6 induit la transactivation
du récepteur au EGF44, module l’expression de cytokines351, la motilité331,332,373 et l’adhésion
cellulaire336,374,375,378.

28
Principales fonctions
LPA6 est un acteur majeur dans la différentiation et la maturation des follicules pileux43,44,379.
Sa mutation est notamment associée à la perte de cheveux chez l’humain44,132. Outre le
domaine capillaire, LPA6 participe la formation de nouveaux vaisseaux vasculaires339 et la
transmigration des lymphocytes336 bien que ce soit LPA4 qui soit déterminant dans ces
processus.
1.5.2.4 GPR87
Le LPA a été identifié comme le ligand du récepteur orphelin couplé aux protéines G
GPR87133. Les rares études sur le GPR87 se concentrent sur son rôle pro-tumorale dans le
cancer380-382.
Bien que l’activation de GPR87 par le LPA ait été mise en évidence, aucune étude décrivant
en détail son interaction avec les espèces de LPA ou ses voies de signalisation n’a été publiée.
Une seule étude a lié les effets du LPA induits par GPR87 à l’activation de la voie AKT134.
1.5.3 Récepteurs non couplés à des protéines G
1.5.3.1 TRPV1
TRPV1 est un canal calcique exprimé fortement dans les neurones383,384. Il est également
présent dans de nombreux autres tissus dont les tissus nerveux, vasculaires et certaines
cellules immunitaires384. TRPV1 est impliqué dans la thermoception, la nociception et
l’inflammation385,386. L’activation de TRPV1 par le LPA a été mis en évidence dans le
système nerveux pour la nociception chronique et aigue137,156,157,387,388 et pour les
démangeaisons358. L’activation de TRPV1 est également impliquée dans la vasoconstriction
dépendante au LPA389.
Le LPA active TRPV1 de manière indirecte dans les neurones du ganglion spinal par sa
signalisation dépendante de LPA1156,157 ou de LPA5358. Le LPA peut interagir directement
avec TRPV1 pour l’activer137,358,387. Parmi les espèces testées, seules les formes de acyl-,
alkyl-LPA 18:1 et leur analogue l’acide phosphatidique 18:1 cyclique peuvent interagir
directement avec TRPV1387. Le site de liaison du LPA est situé sur la partie intracellulaire

29
de TRPV1358,387. En revanche, le mécanisme par lequel le LPA intracellulaire active TREK-
1 et -2 est encore inconnu.
1.5.3.2 TREK-1/-2
TREK-1 et -2 sont des canaux à ion potassium de la famille des canaux potassium à 2 pores.
Ces canaux sont fortement exprimés dans les neurones et les cellules cardiaques. Ils sont
également détectés dans les poumons, le tractus intestinal, les reins et les testicules390. Cette
famille de récepteurs est impliquée dans la conduction de potassium de la membrane. Ces
récepteurs jouent un rôle neuroprotecteur390 et sont impliqués dans la nociception155.
Le LPA inhibent l’activité de TREK-1 et -2 de manière indirecte par la signalisation
dépendante du LPA1/3155,158. Cependant, de manière similaire à TRPV1, le LPA
intracellulaire peut activer TREK-1 et -2138.
1.5.3.3 PPARγ
Le PPARγ est un facteur de transcription de la famille des récepteurs nucléaires. Fortement
exprimé dans les tissus vasculaires et adipeux, PPARγ a été associé avec le métabolisme des
lipides, la régulation de la fonction endocrine et l’inflammation391,392. L’activation du PPARγ
par le LPA promeut, entre autres, le remodelage de la paroi vasculaire35,393, ainsi que la
transition de monocyte en macrophage394,395.
La signalisation du LPA dépendante de LPA1 et 3 est capable de moduler positivement et
négativement l’activité du PPARγ33,394,396-399. Outre cette action indirecte, le LPA active
également le PPARγ par interaction directe30,399. L’activation du PPARγ peut être
indépendante de l’ajout de LPA extracellulaire31,393,398, mais au contraire dépendre de la
synthèse de LPA intracellulaire31.
1.5.3.4 Activation des cibles intracellulaires par le LPA
Le mécanisme d’action du LPA sur ses cibles intracellulaires n’est pas encore complétement
décrit. Bien que le LPA peut moduler ces récepteurs par sa signalisation dépendante de ses
récepteurs membranaires, il reste à élucider les mécanismes d’interaction directe. Le LPA
active le PPARγ chez des levures transfectées en absence d’hormones et des récepteurs au

30
LPA30. Il pourrait donc être internalisé par un mécanisme encore inconnu et avoir une action
transcellulaire. En revanche, l’activation de TRPV1 par le LPA5 est abolie par l’inhibition
de la PLD intracellulaire nécessaire à la production de LPA intracellulaire358. Par ailleurs,
stimuler la voie de synthèse de LPA dépendant d’une glycerol-3-phosphate acyltransferase 1
permet l’activation du PPARγ31 et promeut la migration cellulaire32. Ces études suggèrent un
rôle de second messager pour le LPA intracellulaire.
1.6 Régulation de l’activité du LPA : les lipide-phosphate phosphatases
L’activité du LPA est principalement régulée par sa dégradation en monoacyl-glycérol à la
suite de l’hydrolyse du groupement phosphate par des lipides phosphate-phosphatases400,401.
Les monoacyl-glycérols n’ont pas d’activité biologique propre à l’exception du 2-
arachidonylglycérol qui peut être impliqué dans la signalisation des cannabinoïdes402.
Différentes lipides phosphate-phosphatases existent chez l’humain en fonction des tissus et
des compartiments cellulaires considérés, comme la LPA phosphatase dans les
mitochondries403,404 ou la phosphatase prostatique acide dans le liquide séminal405.
Cependant, les régulateurs majeurs des quantités de LPA extracellulaires sont les lipide-
phosphate phosphatases (LPP)400.
Les LPP, anciennement nommées phosphatases d’acide phosphatidique, sont des protéines
transmembranaires qui peuvent hydrolyser le groupement phosphate de certains lipides
phosphorylés que sont le LPA, la shingosine-1-phosphate, les acides phosphatidiques et les
céramides-1-phosphate. L’hydrolyse du groupement phosphate peut être fait par les LPP
quand ces lipides sont associés à de l’albumine ou sous forme de micelle401. Il existe 3 classes
distinctes de LPP, soit les LPP1, LPP2 et LPP3. Toutes les LPP sont des protéines avec 6
domaines transmembranaires et qui présentent 3 sites catalytiques406. Les 3 classes de LPP
partagent la même orientation avec les extrémités N- et C-terminales cytosoliques et leurs
sites catalytiques sont situées dans le milieu extracellulaire ou du bord de la lumière des
membrane intracellulaires406,407. Les 3 classes de LPP sont présentes à la membrane
plasmique et ont une activité catalytique dans le milieu extracellulaire400,408,409, même si elles
sont associées à des sous-domaines distincts de la membrane plasmique410,411. Elles peuvent
également être localisées sur des membranes intracellulaires. Les LPP3 sont détectées au

31
niveau du réticulum endoplasmique412, de vésicules cytoplasmiques et dans le compartiment
périnucléaire413 alors que les LPP2 sont détectées au Golgi414 et sur des vésicules
cytoplasmiques413. Les LPP1 ont été décrites à ce jour uniquement à la membrane
plasmique410,411, même si elle a des effets intracellulaires409. Les LPP peuvent former des
homo ou hétérodimères ce qui n’affecte pas leur activité, mais affecte leur localisation407.
Outre leur localisation cellulaire, les LPP varient dans leur patron d’expression tissulaire. Les
LPP1 et 3 sont détectées dans de nombreux tissu au contraire des LPP2.
À l’inverse de l’invalidation génique de LPP2 qui ne présente pas de phénotype415,
l’invalidation de LPP3 est létale au stade embryonnaire à cause d’un défaut de
vasculogenèse416, et celle de LPP1 à une incidence sur la morphologie, la reproduction et le
pelage des souris417. De plus, la LPP2 promeut l’avancée du cycle cellulaire418 alors qu’au
contraire les LPP1 et 3 inhibent la prolifération et la survie cellulaire419,420. Même si les LPP
partagent une même activité enzymatique, structure et orientation, elles ont, par leur
distribution tissulaire et cellulaire, des fonctions distinctes bien qu’il existe une certaine
redondance entre LPP1 et LPP3.
Des études d’invalidations géniques conditionnelles ont montré que LPP1 et 3 inhibent
l’action du LPA421,422 et régulent les quantités plasmatiques de LPA400. L’action des LPP sur
la signalisation du LPA se fait, d’une part, par la dégradation du LPA extracellulaire dont le
LPA plasmatique400,423, et d’autre part, en agissant sur la signalisation intracellulaire en aval
des récepteurs au LPA409,424. La localisation des LPP à des domaines membranaires
spécifiques permet de localiser la production du LPA à certains sites378. Les mécanismes
d’action des LPP sur la signalisation intracellulaire des récepteurs au LPA ne sont pas encore
décrits en détail. Cependant, une étude a associé l’inhibition intracellulaire de la signalisation
du LPA par les LPP avec la réduction des quantités intracellulaires d’acide phosphatidique
409. Cela est supporté par le fait que les LPP modulent les quantités d’acide phosphatidique
intracellulaire413 qui sont nécessaires au recrutement de certains médiateurs importants de la
signalisation dépendante des protéines G telles les protéines ras425 ou Raf-1426.

32
2 Vésicules extracellulaires
J’utilise le terme EV selon la définition de la Société internationale pour les vésicules
extracellulaires (ISEV) soit « terme générique pour les particules naturellement libérées par
des cellules, qui sont délimitées par une bicouche lipidique et qui ne peuvent pas se répliquer,
i.e. ne contiennent pas un noyau fonctionnel »427. Les EV sont conservées dans l’ensemble
des trois domaines du vivant soit archée, procaryote et eucaryote y compris chez les
végétaux428. Chez l’humain, elles sont trouvées dans l’ensemble des fluides biologiques
comme le plasma, la lymphe ou les liquides synoviaux429-431. Les principaux rôles associés
aux EV sont la communication intercellulaire, l’élimination de composé cellulaire, la
coagulation, la modulation de l’inflammation et de la réponse immune432,433. Les EV qui sont
impliquées dans la progression de différentes pathologies inflammatoires, auto-immunes et
dans le cancer, peuvent servir de biomarqueurs434-436. Enfin leur utilisation comme des
plateformes médicamenteuses ou vaccinales est également étudiée435-437.
2.1 Diversité et formation des EV
2.1.1 Les classes : exosomes, microvésicules, corps apoptotiques
Les EV sont libérées soit de manière passive ou active par les cellules. Le feuillet externe de
leur membrane présente des protéines comme des récepteurs, des molécules d’adhésion, ainsi
que des lipides, notamment la PS438-441 (Figure 5). Leur cytosol contient des protéines et des
acides nucléiques soit de l’ADN, des ARN messagers ou encore des microARN438,439,442
(Figure 5). Les EV peuvent également contenir des organelles comme les mitochondries ou
le protéasome439,442,443. 438
La composition de la membrane et le contenu du cytosol des EV dépend d’abord de la cellule
qui les produit, puis du stimulus et enfin du mécanisme qui conduit à leur formation. En effet
bien que la classification des EV soit encore débattue, leur subdivision en trois catégories
selon leur mécanisme de formation est fréquemment trouvée dans la littérature. Ces trois
subdivisions sont les corps apoptotiques, les microvésicules et les exosomes432(Figure 6).

33
Tout d’abord, les corps apoptotiques sont des fragments de cellules entrées en apoptose. Ce
sont des EV de grande taille, entre 500 et 5 000 nm qui peuvent contenir de l’ADN et des
organelles. Les corps apoptotiques transfèrent des protéines et de l’ADN, dont des oncogènes
à des cellules hôtes444,445 et sont également une source d’auto-antigènes446.
Les microvésicules, aussi appelées microparticules ou ectosomes, sont les EV produites par
un bourgeonnement de la membrane plasmique447. Les microvésicules ont une forme de
sphère ou de tubule448 et une taille comprise entre 100 et 1 000 nm, similaire aux bactéries et
à différents agrégats protéiques dont les complexes immuns449. Les plus grandes peuvent
contenir différentes organelles442. Le bourgeonnement des microvésicules a été associé avec
la perte de l’asymétrie de la membrane plasmique par la mobilisation de calcium et l’action
des flippases, floppases et scramblases450-452. Bien que l’exposition de la phosphatidylsérine
(PS) à la suite de l’asymétrie ne soit pas ubiquitaire, la PS reste fréquemment utilisée comme
un marqueur des microvésicules448,453. Leur formation peut faire intervenir la protéine
ARF6454 ainsi que des protéines de la famille du complexe de tri endosomal nécessaire au
Figure 5: Récapitulatif du contenu trouvé dans les EV à l’exception des organelles. (Colombo et al.,
2014(434))

34
transport (i.e. endosomal sorting complex required for transport ou ESCRT) bien qu’on
eût initialement restreint leurs fonctions à la production d’exosomes455-458.
Enfin les exosomes sont des EV formées dans les corps multivésiculaires puis libérées dans
le milieu extracellulaire par la fusion des corps multivésiculaires à la membrane plasmique459.
Les exosomes ont une taille similaire au virus entre 50 à 150 nm460. La principale voie de
formation des exosomes dans les corps multivésiculaires repose sur les protéines
ESCRT459,461. Des voies indépendantes des ESCRT existent aussi et font intervenir soit des
céramides, des tétraspanines ou les protéines dites SIMPLE (i.e. small integral membrane
protein of lysosomes and late endosomes)459,462-464. La fusion les corps multivésiculaires
avec la membrane plasmique est sous le contrôle de protéines de la famille RAB (i.e. RAS-
related protein in brain)459,465.
L’origine cellulaire et les mécanismes de production des EV permet d’obtenir une grande
hétérogénéité dans la composition des EV détectées dans les fluides biologiques429. Cette
hétérogénéité est accrue par la nature des stimuli à l’origine de la formation des EV qui
modifient le contenu des EV en cours de formation et donc leur fonction432. Il en résulte
qu’une même cellule peut, par exemple, libérer des EV pro- et anti-inflammatoires en
fonction du contexte cellulaire466-469. 470
Figure 6 : Libération des différentes vésicules extracellulaires. Les cellules non-apoptotiques peuvent libérer
des microvésicules par l’invagination de la membrane plasmique et former des exosomes dans les corps
multivésiculaires qui sont libérés après fusion des corps multivésiculaires à la membrane plasmique. Les
cellules apoptotiques subissent une fragmentation de la cellule pour former des corps apoptotiques. (D’après
Bian et al., 2019(462))

35
Les EV peuvent interagir avec les composants du milieu extracellulaire et avec des cellules
par l’intermédiaire des protéines et des lipides présents à leur surface. L’interaction des EV
avec une cellule peut activer différentes réponses cellulaires dont leur absorption.
L’absorption des EV est faite soit par endocytose, soit par fusion de la membrane de l’EV
avec celle de la cellule432,459. L’absorption de l’EV permet le transfert de son contenu à la
cellule hôte et provoque des réponses cellulaires variées. Ce transfert peut être une source de
métabolites, mais également de modification de la cellule hôte. Par exemple, la fusion des
membranes peut apporter de nouvelles protéines membranaires ou encore l’apport d’ARNm
ou interférent peut modifier l’expression protéique de la cellule hôte432,459.
2.1.2 Isolation et étude
Les EV partagent des caractéristiques physiques similaires à différents composés présents
dans les fluides biologiques que ce soient les complexes immuns, les lipoprotéines, des
agrégats protéiques ou encore des virus471. Cela, ajouté à l’absence de consensus pour les
différentes classes d’EV que ce soit pour leurs caractéristiques physiques ou des marqueurs
spécifiques, rend difficile l’isolement et l’étude des EV472.
Les approches d’isolation d’EV reposent sur des critères distincts qui sont la densité, la taille
et la composition de la surface des EV473(Figure 7). Il est également possible d’isoler les EV
par leur précipitation dans un polymère473(Figure 7). Bien que la combinaison de plusieurs
Figure 7 : Les différentes méthodes d’isolation des EV. L’isolation des EV peut être faite en fonction de la
densité, la taille, la composition de leur surface ou par précipitation de polymère. Les avancées dans les
techniques microfluidiques ont permis le développement de puce d’isolement d’EV. DLD, Deterministic lateral
displacement (D’après Wang et al., 2018(469))

36
approches soit généralement privilégiée, deux méthodes sont plus communément utilisées,
soit l’ultracentrifugation, avec ou sans un gradient de densité, et la chromatographie
d’exclusion de taille473. Considérant qu’aucune technique ne permette l’isolation spécifique
ou complète d’une population d’EV, que ce soit en fonction du type, (exosomes ou
microvésicules) ou d’une origine cellulaire par exemple, il reste possible d’enrichir une
population ciblée.
Leur composition est étudiée de manière ciblée par des puces ARN/ADN ou par PCR pour
les acides nucléiques et par immunobuvardages de type western et des méthodes immuno-
enzymatiques de type ELISA (i.e enzyme-linked immunosorbent assay) pour les
protéines474,475. Les études non ciblées se font par séquençage et par spectrométrie de
masse474,476,477.
Il est aussi possible d’étudier les EV de manière individuelle. L’observation de la structure
et de la taille des EV a d’abord été faite à l’aide de la microscopie électronique et
atomique448,478. La microscopie optique est limitée aux plus grandes EV puisque la résolution
est limitée à 400 nm478. Cependant, la microscopie optique à fluorescence reste utilisée pour
détecter ou suivre des EV dans des tissus ou des modèles animaux, ce qui peut être fait par
vidéomicroscopie454,479. La taille des EV peut aussi être mesurée par des techniques de
diffusion dynamique de la lumière478 ou par suivi individuel de particule478,480. Ces approches
présentent cependant des inconvénients, notamment le temps et la complexité d’analyse d’un
grand nombre d’EV ou de plusieurs marqueurs simultanément. L’utilisation d’un cytomètre
en flux, dit de haute sensibilité d’une résolution pour les évènements jusqu’à une taille de
100 nm permet de combler ces limitations par l’analyse rapide d’un grand nombre d’EV. Ces
cytomètres en flux permettent la distinction de différentes populations d’EV en fonction de
leur taille, mais aussi selon différents marqueurs de surface ou intracellulaires tel que la
présence d’organelle429,442,478. Cette approche s’est imposée dans l’étude des EV477 et sera
centrale dans les travaux présentés dans cette thèse.
Les EV trouvées dans de la circulation sanguine sont pour la majorité issues soit des
plaquettes, dites PEV, soit des GR, dites REV448,481,482. Puisque les travaux de cette thèse
s’intéressent plus spécifiquement à ces EV, elles seront décrites plus en détail.

37
2.2 Vésicules extracellulaires de plaquettes
2.2.1 Présentation de la plaquette
Les plaquettes sont les cellules les plus représentées avec les GR. Les plaquettes sont des
éléments du sang de l’ordre de 2 µm délimités par une membrane plasmique qui sont
produites par les mégacaryocytes. Elles ne contiennent pas de noyau et ne sont donc pas
capable de réplication483,484. Par conséquent, les plaquettes seraient conformes avec la
définition des EV faite par l’ISEV, citée précédemment. Cependant, par souci de clarté et
pour éviter un débat sur la nature des plaquettes, je vais, dans ce document, assimiler les
plaquettes à des cellules vasculaires. Les PEV correspondent donc aux vésicules libérées par
les plaquettes soit des exosomes ou des microvésicules de plaquettes.
Par le réarrangement des microtubules, les mégacaryocytes forment des protubérances qui
peuvent se subdiviser pour former plusieurs pro-plaquettes. Les mégacaryocytes étant situés
dans la moelle osseuse, il est nécessaire pour les protubérances qui contiennent les pro-
plaquettes de s’allonger à travers l’endothélium vasculaire pour atteindre la circulation
sanguine485,486. Une fois dans le flux sanguin, les pro-plaquettes sont scindées pour libérer
les plaquettes487,488. Au cours de l’élongation et de la formation des pro-plaquettes, le
mégacaryocyte fait la synthèse d’une grande quantité de protéines plaquettaires. Dans le
même temps, des protéines de membranes et des composants intracellulaires, dont des
organelles, vont être incorporés aux pro-plaquettes489. De fait, le contenu et la composition
membranaire des plaquettes reflète celui des mégacaryocytes. Elles présentent des organelles
incluant les mitochondries, les granules denses et α, le protéasome et les ribosomes.
D’ailleurs, les plaquettes sont capables de faire de la synthèse protéique grâce aux ARN
hérités des mégacaryocytes. Par l’intermédiaire de leurs granules, les plaquettes ont une
quantité importante de cytokines et chimiokines ainsi que de protéines membranaires. Une
partie des médiateurs présents dans les granules n’est pas transférée par le mégacaryocyte,
mais est acquise par internalisation d’EV490,491.
Le rôle premier des plaquettes est la coagulation. La présence de collagène ou du facteur de
von Willebrand induit l’activation des plaquettes492-495. Leur activation libère les granules α,
ce qui induit la présentation de protéines membranaires comme des intégrines à la surface

38
des plaquettes et la libération de protéines, comme l’autotaxine, le fibrinogène ou encore le
facteur de von Willebrand dans le milieu extracellulaire. Parmi les protéines membranaires
des plaquettes activées, des intégrines permettent l’agrégation des plaquettes entre elles ainsi
qu’avec d’autres cellules vasculaires comme les GR pour former un caillot496. En parallèle,
l’activation induit l’exposition de la PS à la surface des plaquettes, ce qui permet le
recrutement de cofacteurs de la coagulation497. Cela aboutit à la production de thrombine qui
stabilise le caillot par l’assemblage de filaments d’actine498. La thrombine et les médiateurs
libérés par les granules, amplifient l’activation plaquettaire et la synthèse de thrombine. Cela
permet de stabiliser le caillot498,499.
Le rôle des plaquettes n’est pas limité à l’initiation et à la promotion de la coagulation. Elles
présentent un nombre important de récepteurs qui peuvent induire leur activation.
L’ensemble des TLR exprimés chez l’humain, soit TLR1 à 10, y sont trouvés500. Les TLR
permettent la reconnaissance des motifs moléculaires associés soit aux dommages cellulaires,
des molécules d’origine endogène, soit aux pathogènes, alors des molécules d’origine
exogène. Le rôle de chaque TLR est dépendant de sa localisation et du motif qu’il reconnait.
Les TLR1, 2, 4, 5 et 6 sont présents à la membrane et reconnaissent différents types de
molécules de structures de micro-organismes501. Les TLR3, 7, 8 et 9 sont présents dans des
vésicules intracellulaires et reconnaissent des motifs d’acides nucléiques501. Également à leur
surface, plusieurs récepteurs aux fragments cristallisables des immunoglobulines (Ig) sont
présents et permettent de reconnaitre les IgA, E et G502-504. Elles présentent également des
récepteurs pour la thrombine505, l’ADP506, les prostaglandines506, différentes chimiokines507,
la sphingosine-1-phosphate508 et le LPA, en particulier LPA5 comme déjà présenté dans les
sections précédentes112,354,363. Il en résulte que les plaquettes peuvent être activées dans de
nombreux contextes inflammatoires et lors des réponses immunitaires innées et adaptatives.
En effet, les plaquettes activées sont capables de phagocytose, notamment de
pathogènes509,510, et elles libèrent des médiateurs pro-inflammatoires492,511,512. Ensuite, les
cytokines libérées et les intégrines présentes à la surface des plaquettes activées permettent
le recrutement et l’activation des cellules vasculaires dont les neutrophiles513,514, les
monocytes515, les cellules dendritiques515 et les lymphocytes516-519. L’activation des
lymphocytes T CD8+ par les plaquettes se fait lors de la présentation antigénique, par

39
l’intermédiaire du complexe majeur d’histocompatibilité de classe I (CMH I)520. Les
plaquettes contiennent un protéasome fonctionnel qui permet la production d’antigène pour
l’apprêtement du CMH I520-522. L’activation des plaquettes ne se limite pas uniquement à
stimuler la réponse immunitaire. Elles libèrent également des modulateurs de l’immunité
adaptative telle la sérotonine ou encore le facteur plaquettaire 4 qui inhibent respectivement
l’activation des cellules dendritiques523 ou des lymphocytes T524.
2.2.2 Description générale
Les PEV constituent la première population d’EV dans le sang et représentent plus de la
moitié des EV trouvées dans le sang448,481,482. Elles sont libérées de manière passive en
réponse aux pressions physiques exercées par le flot sanguin sur les plaquettes, dites
contrainte de cisaillement (i.e. shear stress)525 ou lors de leur entreposage suite à un
prélèvement sanguin526,527. Les EV peuvent également être produites de manière active par
l’activation des plaquettes en réponse à une multitude de signaux qui ont été brièvement
présentés dans la section précédente, dont la thrombine528 ou le LPA112,354,363. La libération
de PEV en réponse à la thrombine ou au collagène est associée à l’activation d’une
scramblase dépendante du calcium et à la perte d’asymétrie de la membrane plasmique452,529.
Les plaquettes présentent différentes protéines de surface dont des intégrines, la PS et le
CD41439. Ce dernier est souvent utilisé comme marqueur de l’origine plaquettaire des EV.
Les PEV contiennent une grande diversité de protéines intracellulaires dont des récepteurs,
des facteurs de transcription, de coagulation et des cytokines439. En plus des différentes
protéines, les PEV peuvent aussi contenir des microARN530,531. Enfin, elles peuvent aussi
contenir des organelles comme le protéasome ou les mitochondries442. Ces dernières peuvent
également être libérées par les plaquettes activées sans nécessairement être contenues dans
une vésicule, elles sont appelées alors mitochondries libres442,527. Les PEV présentent une
grande hétérogénéité de contenu429. D’une part, celles de grandes tailles ont une plus grande
probabilité d’avoir des mitochondries fonctionnelles et d’autre part les stimuli à l’origine de
la libération des PEV modulent leur contenu et donc leur activité429,468,532.

40
Les PEV sont éliminées de la circulation par phagocytose par les macrophages533 ou par
internalisation par les cellules endothéliales vasculaires si elles présentent de la PS534. Les
PEV négatives pour la PS seraient également drainées dans les vaisseaux lymphatiques535.
2.2.3 Fonctions
Les fonctions des PEV sont le reflet de celles des plaquettes, soit la coagulation466,467 et la
modulation de l’immunité. Les PEV peuvent adhérer à l’endothélium vasculaire endommagé
par leur fixation au collagène, au facteur de von Willebrand, au fibrinogène et aux
plaquettes466,536. De plus, certaines PEV peuvent exposer du facteur tissulaire et de la PS ce
qui permet le recrutement de différents facteurs de la coagulation et la production de
thrombine526,536,537. Cependant dans certaines conditions, les plaquettes libèrent également
des PEV avec des propriétés anticoagulantes468,469.
Dans l’immunité, les PEV ne sont pas seulement une source de médiateurs pro-
inflammatoires et de motifs de dommages associés aux dégâts cellulaires, mais peuvent
moduler l’activité de certaines cellules immunitaires439,442,538-540. Elles peuvent notamment
induire la différenciation de lymphocytes T régulateurs et aussi inhiber leur activité541,542.
Les PEV induisent également la différenciation et l’activation de lymphocyte B543,544.
Certaines sous-populations de PEV sont capables de présenter des antigènes et d’activer la
lymphoprolifération443,544. Les PEV ont donc un rôle dans la stimulation, mais également
dans l’inhibition de la réponse immune. Conséquence de leur petite taille, les PEV peuvent
diffuser dans les tissus et propager l’inflammation, comme c’est notamment le cas dans le
compartiment synovial de patients souffrant d’arthrite rhumatoïde447,545.
2.3 Vésicules extracellulaires de globules rouges
2.3.1 Présentation des globules rouges
Les GR sont des cellules sanguines les plus représentées avec des concentrations autour de
5x109 cellules/mL chez l’humain et représentent approximativement 45% du volume
sanguin. Leur fonction principale est d’assurer les échanges gazeux d’oxygène et de dioxyde
de carbone entre les poumons et les tissus.

41
Les globules rouges sont constitués de 1 à 3% de réticulocytes546. Les réticulocytes sont
formés dans la moelle osseuse par l’énucléation des érythroblastes, avant de rejoindre la
circulation sanguine. Ils ont une taille de 10 à 13 µm. Bien qu’ils ne présentent plus de noyau,
leur contenu faible en ARN messagers permet encore la synthèse de protéines nécessaires à
leur maturation547. Lors de la maturation, les réticulocytes produisent une importante quantité
d’hémoglobine, dont la synthèse débute plus tôt dans l’érythropoïèse. Cependant la
production d’hémoglobine est cette fois combinée à une dégradation des organelles, comme
les mitochondries, et également celles nécessaires à la synthèse protéique comme les
ribosomes548-551. Les réticulocytes présentent aussi une forte activité de RNase551,552 et il y a
également une dégradation ou une expulsion de protéines membranaires et intracellulaires,
soit par des autophagosomes553 ou bien par la production d’EV554-557. La maturation permet
d’une part la perte de nombreuses fonctions cellulaires des réticulocytes, mais aussi un
remodelage important de la membrane avec la perte de 20% de sa surface558-560. Ce
remodelage permet l’obtention de la forme biconcave qui leur confère une déformabilité
importante560,561. L’issue du processus de maturation aboutit aux érythrocytes.
Les érythrocytes constituent 97 à 99% des globules rouges546. Ce sont des cellules d’une
taille de 6 à 8 µm d’une forme biconcave qui contiennent de grande quantité d’hémoglobine
(plus de 90% de leur masse sèche) pour le transport d’oxygène et de dioxyde de carbone.
L’étape de maturation fait en sorte que les érythrocytes ne présentent ni noyau, ni organelles.
Il n’y a donc pas de synthèse de protéine chez les érythrocytes même s’ils contiennent encore
des ARN, notamment de petites tailles et des ARN interférents562-564. L’absence de
mitochondrie les empêche de consommer l’oxygène qu’ils transportent. Ils utilisent donc la
glycolyse comme source d’ATP560. Ils ont d’ailleurs une réserve importante d’ATP qui leur
permet de moduler leur forme ainsi que le tonus vasculaire par sa libération565. Enfin, leur
élimination de la circulation sanguine est faite après environ 120 jours par leur phagocytose
par des macrophages. Plusieurs mécanismes sont proposés (Figure 8). Le premier est que
BAND-3, la protéine transmembranaire la plus représentée à la membrane des érythrocytes,
soit ciblée par des auto-anticorps naturels (i.e. natural occurring antibodies)566. Le
deuxième reposerait sur la présence de CD47 à la membrane. CD47 interagit avec SIRPα
pour inhiber la phagocytose. Le vieillissement des érythrocytes est associé à une diminution
du CD47 membranaire567,568. Également, chez des érythrocytes vieillissants, l’interaction de
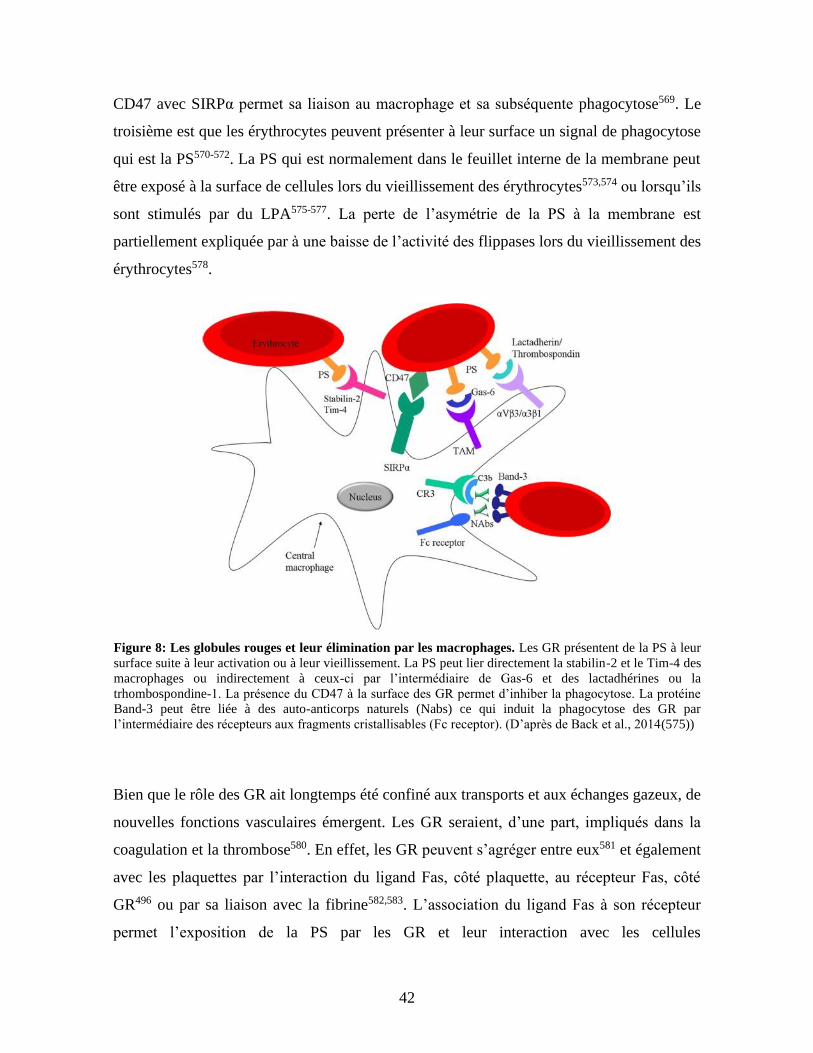
42
CD47 avec SIRPα permet sa liaison au macrophage et sa subséquente phagocytose569. Le
troisième est que les érythrocytes peuvent présenter à leur surface un signal de phagocytose
qui est la PS570-572. La PS qui est normalement dans le feuillet interne de la membrane peut
être exposé à la surface de cellules lors du vieillissement des érythrocytes573,574 ou lorsqu’ils
sont stimulés par du LPA575-577. La perte de l’asymétrie de la PS à la membrane est
partiellement expliquée par à une baisse de l’activité des flippases lors du vieillissement des
érythrocytes578. 579
Bien que le rôle des GR ait longtemps été confiné aux transports et aux échanges gazeux, de
nouvelles fonctions vasculaires émergent. Les GR seraient, d’une part, impliqués dans la
coagulation et la thrombose580. En effet, les GR peuvent s’agréger entre eux581 et également
avec les plaquettes par l’interaction du ligand Fas, côté plaquette, au récepteur Fas, côté
GR496 ou par sa liaison avec la fibrine582,583. L’association du ligand Fas à son récepteur
permet l’exposition de la PS par les GR et leur interaction avec les cellules
Figure 8: Les globules rouges et leur élimination par les macrophages. Les GR présentent de la PS à leur
surface suite à leur activation ou à leur vieillissement. La PS peut lier directement la stabilin-2 et le Tim-4 des
macrophages ou indirectement à ceux-ci par l’intermédiaire de Gas-6 et des lactadhérines ou la
trhombospondine-1. La présence du CD47 à la surface des GR permet d’inhiber la phagocytose. La protéine
Band-3 peut être liée à des auto-anticorps naturels (Nabs) ce qui induit la phagocytose des GR par
l’intermédiaire des récepteurs aux fragments cristallisables (Fc receptor). (D’après de Back et al., 2014(575))

43
endothéliales496,584. L’exposition de la PS permet également la génération de
thrombine496,584,585. Enfin, la présence de GR au sein du thrombus le stabilise et permet
d’accroitre la résistance de la fibrine à la lyse565,586,587.
D’autre part, les GR seraient un modulateur de l’inflammation vasculaire. Ils servent
d’entreposage vasculaire pour plusieurs cytokines et chimiokines588-590. Les cytokines
présentes chez les GR proviennent soit d’un stade plus précoce de l’érythropoïèse590, soit du
captage de cytokines circulantes par le récepteur aux antigènes et chimiokines Duffy (i.e.
Duffy antigen/chemokine receptor)588,591-593. Les cytokines et chimiokines stockées par les
GR peuvent être sécrétées par ces derniers et stimuler différentes cellules immunitaires dont
les neutrophiles et les lymphocytes T594,595. Outre les cytokines, les globules rouges
présentent le TLR9 qui permet la liaison de l’ADN CpG notamment l’ADN
mitochondriale596. Enfin, le CD235a et le récepteur Duffy permettent la liaison des GR à
plusieurs pathogènes et leur élimination lors de la phagocytose des GR par les
macrophages596-601.
2.3.2 Description générale
Les REV sont la deuxième population d’EV la plus abondante dans le système vasculaire
après celle d’origine plaquettaire448. Les REV peuvent présenter à leur surface le canal d’ion
Band 3, la PS, des tétraspanines et des glycoprotéines comme le CD235a, ou la glycophorine
A602. Cette dernière est notamment utilisée comme un marqueur spécifique des EV de GR.
Les REV contiennent notamment de l’hémoglobine et des ARN de petites tailles602.
La libération d’EV par les GR est souvent considérée comme un mécanisme passif en réponse
à des contraintes de cisaillement603,604, à leur vieillissement605,606 ou à leur stockage557.
Cependant, les GR libèrent également des EV de manière active au cours de leur maturation,
lors de l’hémolyse et en réponse à certains stimuli comme le LPA575,576. Le LPA est le seul
médiateur endogène connu qui peut activer la production de REV, mais des composés comme
le calcium ionophore ou le phorbol-12-myristate-13-acétate peuvent également
l’induire575,576. La production d’EV par les GR peut être faite de manière dépendante et
indépendante de la mobilisation intracellulaire de calcium575,576.

44
2.3.3 Fonctions
La production d’EV par les GR à plusieurs fonctions. Lors de la maturation, la libération
d’EV par la voie des exosomes ou par bourgeonnement, permet le remodelage de la
membrane du GR ou l’élimination de médiateurs intracellulaires tel le récepteur au facteur
tissulaire554-557,575. Ce mécanisme est également présent lors du vieillissement des GR
matures et des érythrocytes, qui perdent notamment de l’hémoglobine par la libération de
REV607.
Les REV agissent dans la continuité des fonctions des GR. Grâce à leur cargo en
hémoglobine, ils participent à la régulation des échanges gazeux par le captage de l’oxyde
d’azote circulant608-610 ou encore à la libération d’espèces réactives de l’oxygène611. Le
captage de l’oxyde d’azote par les REV inhibe la vasodilatation dépendante des cellules
endothéliales vasculaires608,609. La présentation de PS à leur surface permet également de lier
l’hème du milieu extracellulaire612.
Également, les REV sont capables d’initier et de promouvoir la coagulation441,613,614. Elles
sont d’une part une source mobilisable du facteur de von Willebrand615 et induisent
l’expression de protéines d’adhésion par cellules endothéliales615. Et d’autre part, elles
participent à la production de thrombine par l’activation du facteur XII441 ou par la cascade
de kallicréine616. De manière similaire aux GR et aux plaquettes, la production de thrombine
est initiée par l’exposition de la PS à la surface des REV, ce qui permet la liaison de cofacteur
de la coagulation ainsi que des inhibiteurs de la coagulation comme la protéine C
activée440,441.
Outre leur rôle dans la coagulation, elles participent à la modulation de l’immunité et de
l’inflammation. La génération de thrombine par les REV permet d‘activer le système du
complément617. Ils permettent également les interactions entre les plaquettes, les
neutrophiles, les lymphocytes T et les monocytes/macrophages614,618. Ces interactions
résultent en une production de nombreuses cytokines et chimiokines pro-
inflammatoires614,615,618 et également à l’activation des cellules endothéliales vasculaires615.
Sur les neutrophiles, les REV favorisent leur recrutement et leur « amorçage » (i.e.
priming)619,620. En parallèle à ces effets d’activation de l’immunité, les REV ont un potentiel

45
d’inhibition. En effet, leur internalisation inhibe l’activation et la survie des lymphocytes T621
et diminue la production de cytokines pro-inflammatoires pour les macrophages622.
3 L’acide lysophosphatidique et les vésicules extracellulaires
dans les maladies rhumatismales auto-immunes systémiques
3.1 Polyarthrite rhumatoïde
La polyarthrite rhumatoïde (PAR) est une maladie rhumatismales auto-immune systémique
avec une prévalence de 0,5 à 1% de la population et touche principalement les femmes qui
représente de 60 à 80% des cas623. La susceptibilité à développer l’PAR dépend en grande
partie de facteurs génétiques mais il existe de nombreux facteurs environnementaux comme
le tabagisme624. Le développement de la PAR se fait par l’activation et la propagation de la
réponse immunitaire adaptative contre des protéines modifiées du soi, notamment les
protéines citrullinées, ou d’anticorps appelés « facteur rhumatoïde »625. Cette première
réponse immunitaire ne suffit pas au déclenchement d’une PAR625. Un deuxième stress (i.e.
additional hit), comme la formation de complexes immuns ou l’activation du complément,
est nécessaire pour déclencher l’inflammation et le recrutement des cellules immunitaires au
niveau du tissu synovial de manière chronique. L’inflammation chronique du tissu synovial
conduit au cours du temps à la destruction du cartilage et de l’os. Les effets de la PAR ne se
limitent pas aux articulations et présentent des manifestations systémiques. Il existe
également une forme d’PAR dite séronégative où les patients ne présentent pas d’anticorps
dirigés contre des protéines du soi modifiées. Les patients atteint par une PAR sont
notamment plus à risque de développer des maladies cardiovasculaires, qui sont la première
cause de surmortalité des patients PAR par rapport à la population générale626-628. Les
patients PAR sont notamment sujet à un risque accru d’infarctus du myocarde et
d’évènements cérébrovasculaires plus élevés627,629. Ce risque a été associé à un
développement plus rapide de l’athérosclérose et à une instabilité accrue des plaques
d’athéroscléroses qui lors de leur rupture, entraine la formation d’un thrombus et le blocage
du flux sanguin627,629,630.
Le LPA a été montré comme un promoteur important de la pathophysiologie de la PAR dans
plusieurs modèles murins de PAR70,147,192. Chez l’humain, l’autotaxine est détectée dans le

46
liquide synoviale et son expression ainsi que celles de certains récepteurs au LPA, comme
LPA1, sont augmentés dans les tissus synoviaux, notamment les fibroblastes synoviaux de
type B70,149,192,631. Les fibroblastes synoviaux de type B contribuent à l’inflammation et à
l’hyperplasie du tissu synovial. Le LPA stimule la prolifération des synoviocytes de types B
et induit la libération de médiateurs pro-inflammatoires comme l’IL-6 et l’IL-8149,631,632 et de
métalloprotéases matricielles350. L’action du LPA ne se limite pas aux fibroblastes
synoviaux. Il stimule également le recrutement et l’activation de lymphocytes et de
macrophages dans le tissu synovial192,633. Enfin, il participe à la dégradation du cartilage et
de l’os par la formation et l’activation des ostéoclastes147,192.
L’inflammation dans les tissus synoviaux de patients PAR met également en jeux les EV.
Les patients présentent des quantités importantes d’EV d’origines diverses comme de
fibroblastes, de lymphocytes et de macrophages634-637. Le liquide synovial présente
également une infiltration d’EV de la circulation sanguine sous forme de PEV447,545,637,638.
Les PEV stimulent l’inflammation par l’apport d’IL-1α et β, ainsi que par l’induction de l’IL-
6 et l’IL-8 par les fibroblastes synoviaux447. Les PEV sont une source d’auto-antigènes, tel
que des protéines citrullinées ou de complexes immuns, et elles activent la réponse
inflammatoire lorsqu’elles sont internalisées par les neutrophiles545,639. Outre les PEV, les
EV du liquide synovial stimulent l’inflammation synoviale par la diffusion de microARN et
par l’apport de protéines citrullinées dans le tissu synoviale640-643. Les EV du liquide synovial
augmentent l’activation et la survie des lymphocytes636,641. Enfin, les EV des articulations
inflammées de patients PAR ont été proposées comme étant un facteur dans la coagulation
locale élevée et seraient impliquées dans les dépôts de fibrines visibles chez les patients
PAR644.
3.2 Lupus érythémateux disséminé
Le lupus érythémateux disséminé (LED) est une maladie rhumatismale auto-immune
systémique avec une prévalence qui varie de 19 à 159 par 100 000 personnes en fonction de
la zone géographique645. Le LED affecte principalement les femmes et, bien que le biais varie
fortement parmi les études, le ratio est considéré comme étant de 9 femmes pour 1
homme623,645.

47
Le LED est une pathologie complexe qui se caractérise par des atteintes à de multiples
organes et par un grand nombre de symptômes possibles. Parmi les manifestations les plus
fréquentes sont retrouvés les atteintes rénales, dite néprhite lupique, articulaires, cutanées ou
encore neurologiques645. Le fait que le LED puisse se présenter sous des formes très variées,
que ce soit sur des critères cliniques ou sérologiques, rend difficile son diagnostique. De plus,
le retard dans le diagnostique du LED augmente les risques dommages irréversibles aux
organes646.
Plusieurs systèmes de classification du LED ont été créés pour faciliter la conduite d’étude
clinique. Le premier a été fait par l’American College of Rheumatology (ACR) sur la base
de 11 critères cliniques et sérologiques647,648. Un patient doit présenter au moins 4 critères
pour être considéré comme atteint du LED. Un deuxième est fait par le Systemic Lupus
International Collaborating Clinics (SLICC) sur la base de 17 critères cliniques et
sérologiques648,649. Pour être considéré comme atteint du LED selon cette classification, un
patient a besoin de 4 critères, dont au minimum un clinique et un sérologique ou avoir une
atteinte rénale, la néphrite lupique, avec simultanément une mesure d’anticorps antinucléaire
ou contre l’ADN. Enfin, en 2019, l’ACR et l’European Alliance of Associations for
Rheumatology (EULAR) ont proposé une nouvelle classification basée sur 22 critères
cliniques et sérologiques648,650. Un patient est classifié comme atteint du LED s’il est positif
pour des anticorps antinucléaires et présente un score égal ou supérieur à 10. Cette nouvelle
classification présente une meilleure sensibilité, soit la probabilité que le test détecte la
maladie, que les classifications sur les critères ACR ou SLICC. Bien qu’elles puissent être
utilisées dans le cadre d’un diagnostique, ces classifications restent des outils pour aider la
recherche et n’ont pas une vocation à être couramment utilisé pour poser un diagnostic ou
pour décider d’un traitement.
La progression dans le temps du LED peut se faire de trois manières. D’abord la forme
quiescente, qui ne présente pas de symptôme clinique, mais qui est actif selon les critères
sérologiques, ensuite une forme active de manière chronique, et enfin une dernière forme
cyclique, qui alterne des poussées de la maladie avec des phases de rémission651. De plus, les
patients ont besoin d’un suivie important pour réduire, d’une part les risques de complication
liés à la pathologie et à son traitement, et d’autre part les dommages permanents aux organes.

48
Il est donc important d’évaluer l’activité de la maladie chez les patients645,651. Bien qu’il
existe plusieurs index, le plus utilisé est l’index SLEDAI pour systemic lupus erythematosus
disease index. Le SLEDAI est basé sur la présence de 24 critères qui inclus des atteintes de
9 organes différents lors des 10 jours précédents le test. Le SLEDAI permet l’obtention d’un
score qui augmente avec l’activité de la maladie652.
L’étiologie du LED reste encore floue mais implique des composantes génétiques et
environnementales645. Le LED est causé par une réponse de l’immunité innée et adaptative
contre des acides nucléiques ou des complexes protéiques contenant des acides nucléiques
originaires du soi, des phospholipides et des protéines mitochondriales653,654. La réponse
innée repose en grande partie sur la production d’interféron de type I par les cellules
dendritiques, les granulocytes et les neutrophiles655-657. La production d’interféron se fait
principalement en réponse à l’activation des TLR 7 et 9, respectivement par l’ARN simple
brin et par l’ADN CpG non méthylé657. Tandis que l’activation des lymphocytes B et T
conduit à la production de nombreux auto-anticorps et à la libération de cytokines658-661. Les
patients LED présentent des dommages aux organes en fonction de la nature locale de la
réponse immunitaire excessive aux auto-antigènes dans ces organes662-666. Similairement aux
patients RA, les patients affectés par le LED sont plus à risque que la population générale de
développer des maladies cardiovasculaires665,667-669. Les premières causes de mortalité chez
patients LED sont les maladies cardiovasculaires dont l’athérosclérose665,669.
Les patients lupiques présentent des quantités élevées d’EV circulantes. Similairement à leur
rôle dans la PAR, les EV stimulent l’inflammation principalement par la diffusion de
microARN670-672. Cela résulte à la libération augmentée de cytokines et chimiokines pro-
inflammatoires importantes dans la progression de la LED comme l’interféron-α, un
interféron de type I671-673. Elles sont également une source d’auto-antigènes674-676. Enfin, chez
les patients LED, les quantités de PEV sont associées avec l’épaississement de la paroi
vasculaire, un facteur de risque des accidents vasculaires674.
3.3 Comorbidité : athérosclérose
Les maladies cardiovasculaires représentent la première cause de mortalité au monde677-679.
L’athérosclérose est la principale cause des maladies cardiovasculaires678,679. L’inflammation

49
joue un rôle moteur dans la progression de l’athérosclérose ce qui explique de la trouver
comme facteur de comorbidité dans la PAR et le LED qui sont toutes deux des pathologies
associées à une forte inflammation systémique vasculaire627,629,630,665,669.
L’athérosclérose consiste en un épaississement de la paroi vasculaire par la formation d’un
noyaux fibreux et lipidique qui conduit à une réduction du flux sanguin. La progression de
l’athérosclérose se fait en trois phases (Figure 9). Les vaisseaux sanguins sont composés de
trois couches tissulaires, l’intima, la média et l’adventice, dans l’ordre d’éloignement à la
lumière du vaisseau. Dans la phase d’initiation, des lipoprotéines de la circulation
s’accumulent dans l’intima de la paroi vasculaire680. Cette accumulation permet la création
d’une plaque d’athérosclérose680. Au cours de la phase de progression, la plaque
d’athérosclérose se développe par la poursuite de l’accumulation de lipides et par le
recrutement de cellules. D’une part les cellules musculaires lisses de la média voient leur
prolifération et leur migration stimulées681. D’autre part, il y a également une infiltration par
des leucocytes, majoritairement des macrophages, mais également des lymphocytes T et des
neutrophiles682,683. Une fois présent dans la plaque d’athérosclérose les macrophages y
prolifèrent683. L’accumulation de lipides ne dépend plus uniquement des lipoprotéines
circulantes, mais également de l’intégration de cellules spumeuse dans la plaque. Les cellules
spumeuses sont des macrophages ou des cellules musculaires lisses gorgées de lipides684,685.
L’accumulation des lipides et le recrutement des cellules immunitaires dans la plaque
d’athérosclérose est facilité par l’activation de l’endothélium qui le rend plus perméable686.
Au fil des années les cellules présentes dans la plaque induisent la production de matrice
extracellulaire et accentuent la capture et la rétention des lipides678,681. Cette production
permet aussi la mise en place d’une chape fibreuse qui permet de stabiliser la plaque681,687.
Au cœur de la plaque se forme une accumulation de cellules mortes, de macrophages et de
cellules du muscle lisse, ainsi que de lipides qui est appelé noyau nécrotique688,689.

50
La progression de la plaque peut déboucher sur plusieurs complications, qui constituent la
dernière phase. D’une part, la plaque peut prendre trop d’espace dans la lumière du vaisseau
sanguin, ce qui va gêner le flux sanguin et peut mener à des ischémies lorsque l’apport en
oxygène devient insuffisant. D’autre part, si l’instabilité de la plaque est trop grande, une
rupture de la plaque peut se produire690-692. À la suite de cette rupture, le noyau nécrotique
va au contact de la circulation déclencher la coagulation et la formation d’un thrombus. La
formation de ce dernier peut bloquer le passage du sang686,690.
Figure 9: Progression d’une plaque d’athérosclérose. L’activation de l’endothélium augmente sa
perméabilité et induit la libération de cytokines pro-inflammatoires et l’expression de molécules d’adhésion.
La perméabilité de l’endothélium facilite l’accumulation de lipides tandis que les cytokines et les molécules
d’adhésion facilitent le recrutement des cellules immunitaires, principalement les macrophages.
L’inflammation au sein de la paroi vasculaire cause d’une part, le recrutement et la prolifération des cellules du
muscle lisse et d’autre part la formation de cellules spumeuses par internalisation de lipides par les macrophages
et les cellules du muscle lisse. Il en résulte un épaississement localisé de la paroi vasculaire. Le stress oxydatif
et l’hypoxie à l’intérieur de la plaque induit l’apoptose cellulaire et la formation d’un cœur nécrotique riche en
lipide. La plaque d’athérosclérose est protégée par la formation d’une chape fibreuse. Le développement de la
plaque résulte en une obstruction partielle de la lumière du vaisseau. La plaque peut rompre à cause d’un
amincissement de le chape fibreuse ou à cause d’une instabilité de la plaque. La rupture de celle-ci expose le
cœur nécrotique riche en lipides et en protéines matricielles aux plaquettes sanguines, ce qui induit la formation
d’un thrombus. (D’après Skeoch et Bruce, 2015(675))

51
Des modèles murins d’athérosclérose ont mis en évidence l’implication de l’autotaxine et de
certains récepteurs du LPA dans la progression de l’athérosclérose693,694. L’autotaxine
transportée par les lipoprotéines peut former du LPA lors de leur oxydation et favoriser
l’accumulation du LPA dans les plaques d’athérosclérose34,68,113. Le LPA participe à
l’inflammation et à l’hyperplasie de l’endothélium vasculaire par la libération de cytokines
pro-inflammatoires311,695-697. Il est également impliqué dans la formation de néo-intima par
la stimulation de la migration et de la prolifération des cellules endothéliales et du muscle
lisse311,698-700. Il augmente l’absorption de lipides par les macrophages ce qui permet leur
transformation en cellules spumeuses309,395,701. Le LPA inhibe la migration des cellules
spumeuses ce qui pourrait contribuer à leur rétention dans la plaque d’athérosclérose311.
Enfin, lors de la rupture de la plaque, le LPA et les fibres de collagène activent les plaquettes
enclenchant ainsi le processus de coagulation34,112,113.
Les plaques d’athérosclérose présentent différentes populations d’EV dont les REV702-705.
L’injection de certaines EV accélèrent la progression de l’athérosclérose dans un modèle
murin706. Les EV sont capables de moduler plusieurs mécanismes mis en jeu dans le
développement de l’athérosclérose. Tout d’abord, elles stimulent la migration et le
recrutement des lymphocytes et des monocytes/macrophages dans le la paroi vasculaire704,706-
709. Elles induisent également la production et la libération de cytokines et chimiokines pro-
inflammatoires702-704,706,710. D’autres EV sont impliquées dans le recrutement et la migration
de leucocytes. Les EV sont des apports de lipides aux macrophages et stimulent leur rétention
dans la plaque710-713. Cette modulation du contenu lipidique des macrophages favorise leur
transformation en cellules spumeuses711. Les EV sont également impliquées dans la
formation de néovascularisations et de microcalcifications au sein de la plaque
d’athérosclérose, ce qui la déstabilise714,715. Enfin le processus de coagulation qui se produit
lors d’une rupture de plaque implique les EV, comme cela a été décrit dans la section sur les
PEV et les REV.
Cependant, certaines populations d’EV ont des effets protecteurs contre l’athérosclérose.
Certaines EV protègent l’intégrité des cellules endothéliales716,717. D’autres peuvent inhiber
le recrutement et l’activation des monocytes et macrophages718-720. Enfin des EV peuvent
avoir des effets positifs et négatifs sur la progression des plaques comme c’est le cas pour les

52
EV de monocytes qui stimulent la formation de cellules spumeuses, mais inhibent
l’activation de lymphocytes T et la production de cytokines pro-inflammatoires711.
4 Objectif
Le LPA promeut le développement de plusieurs pathologies inflammatoires dont la PAR et
l’athérosclérose. Il est par ailleurs le seul médiateur endogène connu capable d’induire
l’activation des GR ce qui entraine notamment la libération de REV. De plus, un rôle des GR
dans la modulation de l’inflammation est proposé. Outre la capacité de stocker et libérer des
cytokines par les GR, leurs REV ont des activités pro-inflammatoires et stimulent la
coagulation. L’hypothèse au centre de cette thèse est que le LPA via l’activation des GR peut
promouvoir l’inflammation vasculaire et participer aux dommages vasculaires, comme
l’athérosclérose, associés aux maladies rhumatismales auto-immunes systémiques.
Notre premier objectif était d’évaluer le potentiel activateur de différentes espèces de LPA
sur les GR et d’examiner les voies de signalisation impliquées.
Un deuxième objectif était d’évaluer au sein de cohortes de patients atteints de maladies
rhumatismales auto-immunes systémiques si l’autotaxine et des niveaux élevés d’EV
sanguines étaient associés à un risque accru de thrombose et au développement de plaques
athéromateuses.
Ce projet permettrait donc d’approfondir l’implication du LPA et des EV sur les dysfonctions
du système vasculaire dans le contexte de maladies systémiques et auto-immunes telle l’PAR
et le LED.

53
Chapitre 1 : Interplay between LPA2 and LPA3 in LPA-
mediated phosphatidylserine cell surface exposure and
extracellular vesicles release by erythrocytes 1 Résumé
Un rôle plus large des globules rouges (GR) dans l’homéostasie vasculaire émerge,
notamment dans les évènements thrombotiques et dans l’inflammation. L’acide
lysophosphatidique (LPA) est le seul activateur connu des GR et induit l’exposition de
phosphatidylsérine et la libération de vésicules extracellulaires de GR (REV). Par des
approches de cytométrie en flux à haute sensibilité, nous avons étudié l’effet d’espèces
majeures de LPA plasmatique sur les GR. Trois de ces espèces induisent la présentation de
la PS et la libération de petites REV PS- et de grandes REV PS+ par l’activation du récepteur
LPA3. L’activation des GR est possible dans le plasma et libère des REV similaires à celles
trouvées dans le plasma de patients. Une quatrième espèce inhibe l’activation des GR par le
récepteur LPA2. Nos résultats suggèrent que les espèces de LPA présentent des activités
biologiques différentes chez les GR en fonction de l’activation des récepteurs LPA2 et/ou
LPA3.

54
Interplay between LPA2 and LPA3 in LPA-mediated phosphatidylserine cell surface
exposure and extracellular vesicles release by erythrocytes
Stephan Hasse1, Anne-Claire Duchez2, Paul Fortin2, Eric Boilard1, Sylvain G. Bourgoin1
1Centre de recherche du CHU de Québec-Université Laval, Centre ARThrite de l'Université
Laval, Département de microbiologie-infectiologie et d’immunologie, Université Laval,
Québec, QC, Canada G1V 4G2.
2Centre de recherche du CHU de Québec-Université Laval, Centre ARThrite de l'Université
Laval, Département de médecine, Faculté de médecine, Université Laval, QC, Canada G1V
4G2.
Corresponding author:
Sylvain G. Bourgoin, PhD
Centre de Recherche du Centre Hospitalier Universitaire de Québec
Faculté de Médecine de l’Université Laval
2705 Boul. Laurier, Québec, QC, Canada G1V 4G2
Phone: +1 (418) 525-4444, ext. 46136 Fax: (418) 654-2765
Key words: LPA, Erythrocytes, LPA receptors, Phosphatidylserine, Autoimmunity,
Extracellular Vesicles

55
2 Abstract
Evidence is growing for the role of red blood cells (RBCs) in vascular homeostasis, including
thrombogenic events and inflammation. Lysophosphatidic acid (LPA) is known to induce
phosphatidylserine (PS) exposure and the release of RBC Extracellular Vesicles (REVs).
Using high sensitivity flow cytometry, we examined the effects and the mechanisms by
which the LPA species commonly found in human plasma could activate RBCs. We report
that LPA 16:0, 18:0 and 18:1, but not LPA 20:4, induced PS exposure and the release of
small PS- and large PS+ REVs through LPA3 receptor signalling in RBCs. The release of
large PS+ REVs required higher concentrations of LPA. RBCs were not activated by LPA
20:4. Interestingly, blockade of LPA2 enhanced LPA-mediated PS- REV release in RBCs.
Furthermore, LPA receptor agonists and antagonists highlighted that LPA 20:4 inhibited
LPA3-dependent PS exposure and, through the LPA2 receptor, inhibited PS- REV
production. Activation of RBCs with LPA 18:1 in normal plasma stimulated the release of
PS- and PS+ REVs. REVs released in response to LPA were similar to those found in the
plasma of systemic lupus erythematosus patients. Our results suggest that LPA species
exhibit different biological activities in RBCs through targeting LPA2 and/or LPA3
receptors.

56
3 Introduction
Red blood cells (RBCs) have long been limited to their role as vehicles for gas exchanges
between blood and tissues and considered passive for other vascular processes such as
inflammation or coagulation. However, a lower concentration of RBCs in polycythemia vera
patients is associated with a lower number of thrombotic events and a lower death rate from
cardiovascular causes [1], thereby suggesting a role for RBCs in coagulation and thrombosis.
Furthermore, inhibition of RBC - platelet interaction protects from arterial thrombosis in
vivo[2]. In vitro experiment shows that interaction between RBCs and platelets contributes
to thrombin generation and thrombus formation[2]. A small subset of RBCs exposing
phosphatidylserine (PS) at their surface could contribute up to 40 % to the thrombin
generated in blood. PS exposition by RBCs promoted their binding to endothelial cells[3]
and could contribute to retinal vein occlusion[3]. In addition to the coagulation process,
RBCs constitute a storage site of vascular cytokines[4, 5] and are potential modulators of
blood cytokines and chemokines. Some such as IL-33 come from an earlier stage of RBC
development[6]. Others such as IL-8 are actively captured by RBCs through the Duffy
antigen receptor for chemokines[4]. Furthermore, RBCs can release cytokines in the milieu
without undergoing hemolysis[7]. Such RBC conditioned media could promote neutrophil
transmigration in vitro[8] and lymphocyte T survival and growth[7].
The release of extracellular vesicles (EVs) by RBCs also contributes to coagulation and
vascular inflammation. EVs are small vesicles from 30 nm up to 1 µm. EVs can originate
from the endosomal compartment before being released (exosomes) or through plasma
membrane budding of activated cells (microvesicles, ectosomes or microparticles). The
release of EVs containing membrane receptors by RBCs (REVs) is a mechanism associated
with a loss of functional responses during maturation of erythrocytes[9-11]. REVs are
capable of initiating and sustaining coagulation in vitro[12]. The increase of
hypercoagulation following REV injection in a mouse model of transfusion highlights their
role in coagulation processes[13]. REVs procoagulant activity is in part explained by the
upregulation of endothelial cell adhesion markers[14] and as a source of thrombin[15]
through the activation of factor XII or of the kallikrein cascade[12]. REVs could also have a
role in immunity, as suggested by the exacerbation of pro-inflammatory cytokine production

57
by REVs in lipopolysaccharide-induced inflammation[16]. REVs promote inflammation
further through thrombin generation[15, 16] and activation of the complement cascade[16].
With platelet EVs, REVs are the main EV population in the bloodstream[17]. Even if REV
release has long been considered passive in response to shear stress[18], aging[19, 20] or
storage[21], REV release can be induced in vitro by a mediator present in the plasma, the
lysophosphatidic acid (LPA)[22].
LPA is a bioactive lipid formed by a phosphate group on a glycerol backbone attached to a
fatty acid. LPA activity is mediated by six widely expressed LPA receptors (LPA1-6) and
atypical receptors such as the peroxisome proliferator-activated receptor γ[23]. The
functional responses to LPA depend on the pattern of LPA receptor expression and LPA
species affinity for the receptors[24, 25]. LPA is a critical mediator of the vascular system
homeostasis[26, 27]. LPA contributes to pro-thrombotic events and inflammatory diseases
such as atherosclerosis[28, 29]. LPA pro-inflammatory and pro-thrombotic functions are in
part explained by the activation of vascular cells such as platelets[30] and endothelial
cells[29]. It promotes platelet aggregation[30], cytokine production[29], and immune cell
recruitment[31]. However, knowledge on LPA impact on RBCs is limited. LPA is known to
have a dual role in the early steps of hematopoiesis. LPA3 promotes erythropoiesis at the
expense of megakaryopoiesis[32, 33], while LPA2 inhibits erythropoiesis[34]. LPA induces
the aggregation of mature RBCs[35], as well as the presentation of surface PS and the release
of pro-thrombotic REVs[22, 36].
Since RBCs and REVs have an active role in vascular physiology and LPA-mediated RBC
activation contributes to inflammation and coagulation, we studied the effect of common
blood LPA species on RBC PS exposure and EV release.
4 Material and methods
4.1 Products
LPA species (16:0, 18:0, 18:1, and 20:4), LPA3 agonist 2S-OMPT, and LPA1/3 antagonist
VPC 32183 were from Avanti Polar Lipids (Alabaster, Alabama, USA). LPA2 agonist GRI-
977143 and LPA2 antagonist H2L5186303 were from Tocris Bioscience (Bristol, UK).

58
4.2 Human plasma samples
Platelet-free plasma (PFP) samples, from systemic lupus erythematosus (SLE) patients and
age- and gender-matched healthy controls, were obtained from the Biobank and Repository
Data for Systemic Autoimmune Rheumatic Diseases of the CHU de Québec-Université
Laval Research Center. PFPs were prepared from blood samples using previously described
standardized procedures[37], which are presented in the section Platelet and EV-free plasma
preparation. All blood samples were obtained after informed consent. The study was
reviewed and approved by the ethics review board of the CHU de Québec-Université Laval
(Project # 2016-2558).
4.3 RBC isolation and activation
Whole blood was collected with sodium citrate as an anticoagulant from healthy anonymous
donors after obtaining informed consent. The blood was centrifuged at 282 g for 10 min to
separate the RBC pellet from plasma and leukocytes. The RBC pellet was collected and
washed thrice in phosphate-buffered saline (PBS: 1 mM KH2PO4, 154 mM NaCl, 3 mM
Na2HPO4; pH 7.4) unless otherwise stated. Washed RBCs were used right away at 108
cells/mL in PBS, when not stated otherwise, and were stimulated up to 2 h at 37 °C under
continuous agitation. In experiments looking at RBC activation in the presence of calcium or
albumin, washed RBCs in HEPES buffered physiological solution (HPS: 145 mM NaCl, 7.5
mM KCl, 10 mM glucose, 10 mM HEPES, pH 7.4 as previously described[36]) containing
2 mM CaCl2 (Sigma Aldrich, Oakville, ON, Canada) or 1 % lipid-free bovine serum albumin
(BSA, Sigma Aldrich, Oakville, ON, Canada) were stimulated with LPA 18:1 for 1 hour at
37 °C. Washed RBCs in EV-free and PFP (V/V), prepared as described in the section below,
were used to mimic physiological conditions. RBCs were centrifuged at 282 g for 10 min,
resuspended one last time in EV-free PFP (V/V), and stimulated with LPA 18:1 for 1 h and
24 h under rotating agitation at 37°C. PBS and HPS were prepared with chemical of
analytical grade obtain from Sigma Aldrich (Oakville, ON, Canada) and HEPES from Wisent
(St-Bruno, Québec, Canada).

59
4.4 Platelet and EV-free plasma preparation
PFP were prepared as previously described[37]. Blood from healthy donors was centrifuged
at 282 g for 10 min at room temperature (RT). The plasmatic fraction was centrifuged at 2
500 g for 20 min at RT. The supernatant was further centrifuged 3 times at 3 500 g for 5 min
at RT to obtain PFP. Finally, PFP was centrifuged at 100 000 g for 90 min at 18 °C to remove
all EVs. The plasma free of both platelets and EVs of 3 healthy donors were pooled and
stored at -20 °C before use.
4.5 RBC and REV labeling for flow cytometry
Following stimulation, 15 µl of stimulated RBCs at 108 cells/mL or 2 µl RBCs in PFP (V/V)
were collected and labeled with 3 µl anti-CD235a-PECy7 (BD Bioscience Canada,
Mississauga, ON, Canada), a marker of RBC, and 3 µl Annexin V FITC (BD Bioscience
Canada, Mississauga, ON, Canada) for PS detection, in 100 µl final of Annexin V binding
buffer (BD Bioscience Canada, Mississauga, ON, Canada) for 30 min in the dark at RT.
Labeling was stopped by adding 200 µl of Annexin V binding buffer and samples were
analyzed within 90 min to prevent labeling loss. PS exposure on RBCs and REVs were
analyzed using a high sensitivity flow cytometer BD Canto II Special Order Research Product
with a small particle option as previously described[38]. Performance tracking of high
sensitivity flow cytometry was done the day of use with BD cytometer setup and tracking
beads (BD Bioscience Canada, Mississauga, ON, Canada). REVs and PS+ REVs absolute
concentrations were determined using 2 µm APC polystyrene beads (BD Bioscience Canada,
Mississauga, ON, Canada) or 3 µm polystyrene beads (Polysciences, PA, USA) of known
concentration in each sample. The gating strategy for RBC activation and REV detection are
presented in Figure 1 A and B. The gate to discriminate the event of a size between 100 nm
and 1,000 nm was set using silica particles of known dimensions of 100, 500 and 1,000 nm
(Kisker Biotech GmbH & Co. Steinfurt, Germany) (Figure 1C). Voltages for the detection
of RBCs were set as follows: FSC at 160 Volt (V), SSC at 300 V, PECy7 at 450 V and FITC
at 500 V. A diode was used to optimize RBC detection. REVs were detected with the
following settings: FSC-PMT at 390 volts (V), SSC at 460 V, PECy7 at 500 V, FITC at 400
V and APC at 550 V. Detection of REVs in the plasma of SLE patients were done with the

60
following settings: 300 V, SSC at 300 V, PECy7 at 500 V, FITC at 500 V and APC at 500
V.
4.6 Control for REV detection by flow cytometry
To evaluate the sensitivity of REV detection, we performed serial dilution, triton treatment
and ultracentrifugation of REV samples as previously described[38]. RBCs were first
removed by centrifugation at 1 300 g for 10 min at RT, and the supernatants were then
centrifuged two more times at 3 500 g for 10 min at RT. EVs were destroyed using 0.05 %
Triton X-100 (Sigma Aldrich, Oakville, ON, Canada) for 1 h at 37 °C or removed by
centrifugation at 100 000 g for 1 h and 30 min at 18 °C. EV depletion data are express as a
percentage of the total number of EVs in untreated samples. Labeling in PBS with 50 µM
EDTA (Wisent, St-Bruno, Québec, Canada), instead of Annexin V buffer, verified the
specificity of Annexin V labeling. We also performed a coincidence test for the detection of
REVs to verify that our measurements were quantitative. Two-fold serial dilution of REV
samples were prepared and measured by high sensitivity flow cytometry (Figure 1F).
4.7 Analysis and statistics
Flow cytometry data were analyzed using FlowJo V10 software (FlowJo, LLC, OR, USA),
and statistical analyses performed using GraphPad Prism 7.0 software (GraphPad Software,
San Diego, USA). Since the number of repetitions for each experience is under 15, we only
used nonparametric tests for our statistical analysis. The strength of activation by LPA
species or LPA receptor agonists showed substantial variations between blood donors.
Therefore, when looking for increase or inhibition of REV production by RBCs, we mitigated
the variations by normalizing the data to the positive control. The Mann-Whitney test
assessed the difference between two experimental conditions. Multiple comparisons used a
two-way ANOVA with Dunnett's multiple comparison post-test to analyze RBC activation
in response to increasing concentrations of LPA and stimulation length. Statistical analyzes
used Kruskal-Wallis tests with Dunn’s multiple comparison post-test or Friedman's test with
Dunn's multiple comparison post-test. Post-test comparisons used the positive or negative
controls, and results were expressed as the mean ± standard error of the mean.

61
5 Results
5.1 Detection of activated RBCs and REVs by flow cytometry
We monitored PS exposure at the surface of RBCs as a marker of RBC activation. Events
positive for the RBC marker CD235a and Annexin V to detect PS were considered (Figure
1A). The gating for PS+ RBCs was validated using RBCs stimulated with calcium ionophore
as a positive control (Figure 1A). We also used the RBC marker CD235a to detect REVs.
Events of a size comprised between 100 nm and 1 000 nm and positive for the RBC marker
CD235a were considered as REVs (Figure 1B). We further discriminated the PS+ REVs
using Annexin V-FITC labeling (Figure 1B). The gate to discriminate the events between
100 nm and 1 000 nm were set with 100 nm, 500 nm and 1 000 nm beads (Figure 1C). To
validate the specificity of REV detection, we monitored the loss of signal for REVs in
supernatants of RBCs activated with LPA 18:1 after treatment with Triton X 100 or
ultracentrifugation at 100 000 g, which destroys the EV lipid bilayer and pellets REVs,
respectively. Triton X 100 treatment or ultracentrifugation reduced the REV populations by
at least 95% compared to untreated supernatants (Figure 1D). The data confirmed the
specificity of REV detection using our experimental approaches. Calcium-free medium
supplemented with EDTA abolished Annexin V labeling of REVs and validated the
specificity of PS detection by Annexin V at the surface of REVs (Figure 1E). Finally, serial
dilution of REV samples resulted in similar reduction of REV detection (Figure 1F).
Although, we detected fewer event, it did not affect the mean nor the median fluorescence of
the RBC marker, CD235a-PECy7, which confirmed that each event we measured is a single
EV and not a cluster of EVs (Figure 1F-G).
5.2 LPA species differentially activate RBCs.
Though LPA is known to induce PS exposure and EV release by RBCs, there is no report on
individual LPA species. We choose to evaluate 4 LPA species, i.e. LPA 16:0, 18:0, 18:1 and
LPA 20:4. It allowed us to have LPA with a long or short fatty acid, either unsaturated,
monounsaturated, or polyunsaturated. Furthermore, all 4 species are commonly found in the
plasma[39, 40] and they are also produced in large quantities following platelet activation[40,

62
41] and during lipoprotein oxidation[40]. We used a concentration of 10 µM of LPA species
to stimulate RBCs, as was done in a previous study of RBC activation by LPA[22].
LPA 16:0, 18:0, and 18:1 induced PS exposure by RBCs (Figure 2A) and REV release
(Figure 2B and C). Surprisingly, LPA 20:4 did not induce PS exposure (Figure 2A) nor
REV release (Figure 2B and C). Our results show a functional discrepancy between LPA
species. Further analysis of LPA-mediated RBC activation used LPA 18:1, since it is the
most potent species that we tested.
5.3 Characterization of RBC activation by LPA 18:1.
We assessed the kinetics of externalization of PS and REV release by RBCs incubated with
various concentrations of LPA 18:1. High concentrations of LPA, 10 µM, and 20 µM resulted
in rapid and transient PS exposure by RBCs (Figure 3A). Furthermore, LPA induced
significant REV accumulation for all concentrations tested except the lowest (Figure 3B).
REV release in the media is seen as soon as 2 min after stimulation and reaches a maximum
after 1 h (Figure 3B). Interestingly, low concentrations of LPA (2.5 µM) induced significant
REV accumulation (Figure 3B) but not PS exposure by RBCs (Figure 3A). Altogether the
data indicate that low concentrations of LPA 18:1 can induce the production of REVs by
RBCs in the absence of PS cell surface exposure.
Interestingly, PS- and PS+ REVs have distinct sizes. PS- REVs show relative size of about
100 nm. On the other hand, PS+ REVs have a size between 500 nm and 1 000 nm (Figure
3C). Using the same gating strategy, we detected similar smaller PS- and larger PS+ REV
subpopulations in PFPs of SLE patients (Figure 3D).
LPA 18:1 induced the release of PS+ REVs in a dose-dependent manner (Figure 3E). In
opposition, the production of PS- REVs reached a plateau with a concentration of 5 µM
(Figure 3F). RBCs released mainly PS- REVs when stimulated with 2.5 µM (87.3 % ± 5.5)
and 5 µM (85.7% ± 3.4) while PS+ REVs were produced at concentration of 10 µM (53.9 %
± 12.5) and 20 µM (90.2 % ± 3.4) (Figure 3G). Thus LPA 18:1, depending on the
concentration, induces the release of 2 distinct populations of REVs, small PS- and large PS+
possibly through two different mechanisms. To further understand the mechanisms leading

63
to RBC activation and the release of those two REV populations, we investigated whether
the production of those REVs was induced through the activation of LPA receptors.
5.4 LPA3 receptor induce RBC activation.
To our knowledge no published paper investigated the presence of LPA receptors on mature
RBCs and only two LPA receptors, LPA2 and 3, were reported on the precursors of RBCs[32,
33]. We first investigated the implication of the receptors LPA1 and LPA3 since both
receptors often mediate similar functions. Since no agonists for LPA1 are available, we
stimulated the RBCs with an agonist selective for LPA3, 2-OMPT. RBC stimulation with
2S-OMPT resulted in PS exposure and REV release (Figure 4A and B). As for RBC
activation with LPA 18:1, a low concentration of 2S-OMPT only induced the release of PS-
REVs (Figure 4B), and a high concentration of 2S OMPT led to significant accumulation of
both PS- and PS+ REVs (Figure 4C and D). To confirm that LPA activation of RBCs goes
through LPA3 (or LPA1), we stimulated RBCs with LPA 16:0, 18:0, and 18:1 in the presence
of VPC32183, an LPA1/3 receptor antagonist. VPC32183 reduced LPA 16:0-, 18:0-, and
18:1-induced cell surface exposure of PS and REV release by RBCs (Figure 4E and F).
Furthermore, VPC32183 strongly reduced the release of PS+ REVs in response to LPA 16:0,
18:0, and 18:1 (Figure 4G). VPC32183 strongly inhibited the release of PS- REVs in
response to LPA 16:0 and 18:0 or 18:1 to a lesser extent. Altogether these data suggest that
LPA-mediated RBC activation is dependent on LPA1/3 receptors.
5.5 LPA2 receptor inhibits PS- REV formation.
Knowing the complexity and redundancy of LPA signaling and the role played by LPA2 in
the regulation of erythropoiesis, we examined if LPA2 also contributes to LPA mediated
RBC activation and production of REVs. RBC incubation by GRI-977143, an LPA2 agonist,
did not induce RBC PS exposure (Figure 5A) nor the release of REVs (Figure 5B). We
further tested LPA2 implication in RBC activation by stimulating RBCs with LPA species
in the presence of H2L5186303, an LPA2 antagonist. H2L5186303 did not affect RBC PS
exposure induced by LPA 16:0, 18:0, and 18:1 (Figure 5C). Surprisingly, H2L5186303
enhanced the production of REVs induced by LPA 16:0 and 18:1 compared to the control
without H2L5186303, 199.2 % ± 88.0 (p=0.0350) and 157.3 % ± 59.5 (p=0.0450),

64
respectively (Figure 5D). Stimulation of RBCs with LPA 16:0 and 18:1 in the presence of
H2L5186303 only increased the release of PS- REVs (Figure 5F), but not that of PS+ REVs
(Figure 5E). To confirm the ability of LPA2 to inhibit PS- REV release, we induced the
release of REVs in the presence of 2S-OMPT, a selective LPA3 agonist, and the LPA2
agonist GRI-977143. As shown in Figure 5G, 10 µM of GRI-977143 reduced to 41.76 % ±
14.03 (p=0.0011) the production of PS- REVs. A higher concentration of GRI-977143 (20
µM) did not further inhibit the production of PS- REVs (Figure 5G), this might be due to
unknown off-target effects of high concentrations of the LPA2 antagonist. The data suggest
that LPA2 inhibits PS- REV release.
5.6 LPA 20:4 inhibits both RBC PS exposure and the production of PS-
REVs.
Since LPA2 has an inhibitory action, we assessed if LPA 20:4, which showed no activation
effect, could inhibit RBC activation. To address this point, we stimulated RBCs with 2S-
OMPT in the presence of LPA 20:4. Interestingly, 20µM LPA 20:4 reduced 2S-OMPT
mediated RBC PS exposure (11.30 % ± 8.81 versus 77.80 % ± 22.50; p=0.0107) (Figure 6A)
and strongly inhibited the production of REVs. Of note, LPA 20:4 had no impact on the
release of PS+ REVs (Figure 6B) but drastically reduced the production of PS- REVs induced
by 2S-OMPT (Figure 6C). Since PS+ REV accumulations were similar in the absence or
presence of LPA 20:4, we reasoned that LPA 20:4 mediated inhibition of PS- REV release
may be mediated by LPA2. To confirm this, we tried to rescue 2S-OMPT activation in the
presence of LPA 20:4 by adding the LPA2 antagonist, H2L5186303. The addition of
H2L5186303 enhanced in a dose-dependent manner LPA 20:4-mediated inhibition of PS cell
surface exposure induced by 2S-OMPT (Figure 6D). As expected, H2L5186303 at 30 µM
antagonized the inhibitory effect of LPA 20:4 on 2S-OMPT-mediated PS- REV release by
RBCs (Figure 6E). Together, these data support that LPA 20:4 inhibits the release of PS-
REVs through LPA2 signaling and inhibit PS exposure on RBCs through an LPA2
independent mechanism.

65
5.7 RBC activation by LPA in physiological condition.
All previous experiments used calcium- and albumin-free PBS. Given the presence of
albumin and millimolar concentration of calcium in the plasma, the vascular environment
likely impacts the ability of LPA to activate RBCs. Albumin binds numerous lipids including
lyso-phospholipids, thereby buffering the concentration of albumin-free LPA which prevents
the degradation of LPA by phosphatases and serves as a carrier to deliver this lipid to cells
and tissues [42, 43]. To evaluate the impact of albumin and calcium, two main components
of blood, on LPA-mediated effect on RBCs we used HEPES buffered physiological solution
(HPS) as described previously for RBC incubation in the presence of a high concentration of
calcium[36]. The strongest activator of RBCs LPA 18:1 was selected for this series of
experiments. LPA 18:1 induced PS externalization by RBCs and calcium enhanced LPA
18:1-mediated cell surface exposure of PS (Figure 7A). LPA 18:1 also stimulated the release
of PS+ (Figure 7B) and PS- REVs (Figure 7C). However, the release of PS+ REVs was lower
(Figure 7B), and that of PS- REVs strongly reduced in the presence of calcium (Figure 7C).
The addition of 1 % BSA to the medium supplemented with or without calcium abolished
LPA 18:1-mediated PS exposure by RBCs (Figure 7A) and the release of PS+ and PS- REVs
(Figure 7B and C).
Since major blood components impact LPA-mediated RBC activation, we investigated next
LPA effects in a closer physiological condition. We resuspended washed RBCs in an
equivalent volume of PFP- and EVs-free plasma to mimic the vascular compartment. In those
conditions, the stimulation with LPA 18:1 for 1 h or 24 h had no discernable impact on the
level of PS exposure by RBCs (Figure 7D). However, RBCs stimulated for 1 h with 20 µM
of LPA 18:1 showed a significant release of PS- REVs (Figure 7F) and higher quantities of
PS+ REVs albeit not significant (Figure 7E). After 24 h, the accumulation of PS+ REVs was
significantly higher for RBCs stimulated with 20 µM of LPA 18:1 (Figure 7E). REV
populations produced by RBCs stimulated with LPA 18:1 in EVs-free PFP, as characterized
by flow cytometry (Figure 7F), resemble those detected in the PFPs of SLE patients (Figure
3D). Interestingly, SLE patients showed higher plasmatic PS+ and PS- REV levels compared
to plasma of healthy controls (Figure 8A and B). Our data show that the REVs released by
RBCs in vitro can be observed in conditions that approximate to the vascular compartment.

66
6 Discussion
In this study, we showed that three LPA species, 16:0, 18:0, and 18:1, activate RBCs by
inducing PS exposure and the release of REVs. The REVs produced in response to LPA are
mainly small PS- EVs, but another population of larger PS+ EVs was also released. LPA-
mediated PS exposure and REV release as well were through the LPA3 receptor and possibly
LPA1. Among the LPA species tested, LPA 20:4 inhibited PS exposure on RBCs and the
release of small PS- REVs. Inhibition by LPA 20:4 of the release of the PS- REVs is through
the LPA2 receptors (Figure 9). Our data do not exclude the possibility that LPA 16:0 and
18:1 bind to LPA2 and contribute to limiting RBC activation through LPA3. Finally, we were
capable of reproducing part of RBC activation by LPA, including PS exposure and the release
of PS+ and PS- REVs, using conditions that mirror the vascular environment.
Even if we validated that LPA3 activates RBCs by inducing both PS exposure and REV
release using an agonist for LPA3 and an antagonist for LPA1 and LPA3, a role for LPA1
remains unclear. LPA species affinity for the LPA receptors is dependent on the fatty acid
chain[24, 25]. LPA 20:4 binding affinity for LPA3 has been reported among the lowest for
LPA species, whereas LPA 20:4 binding affinity for LPA1 is similar to LPA 16:0, 18:0 and
18:1[25]. A role for LPA1 in RBC activation is not excluded but could not be addressed in-
depth due to the lack of LPA1 selective agonists. Furthermore, even if LPA1 and LPA3
usually mediate similar biological functions, a role for LPA in promoting erythropoiesis has
been associated solely with LPA3[32]. To this day, no study showed an implication of LPA1
in RBC functions. An unambiguous approach to determine if LPA1 modulates the production
of REV and PS exposure by RBCs would require mouse knockout for LPA1.
LPA2 is known to inhibit to erythropoiesis[34]. In this study we provide evidence that
inhibition of small PS- REV release by LPA 20:4-activated RBCs depends on LPA2. Among
the species we tested, the one with the highest affinity for LPA2 is LPA 20:4[25], which is
in line with the effects of 20:4 reported in this study. However, LPA 16:0 and 18:1 could also
activate LPA2 but with a lower affinity than LPA 20:4[25]. Our data suggest that LPA2 can
mitigate LPA 16:0 or 18:1-mediated production of EVs by RBCs, as evidenced by the
increased production of EVs in the presence of an LPA2 antagonist. Other methods such as

67
silencing of LPA receptors are not amenable in mature RBCs. However, the observation that
LPA2 antagonist H2L5186303 can abolish the inhibitory effect of LPA 20:4 on LPA3
agonist-mediated production of EVs reinforces the hypothesis that signaling through LPA2
inhibits the release of small PS- EVs (Figure 9).
Increased inhibition of LPA 20:4-mediated PS exposure with H2L5186303 was unexpected
in experiments where RBCs were activated by the LPA2 agonist 2-S-OMPT. H2L518630 is
a competitive antagonist with a 50% inhibitory concentration (IC50) one hundred times
inferior for LPA2 than for LPA3. Competition between H2L518630 and 2-S-OMPT from
binding to LPA3 is unlikely since LPA 20:4-mediated inhibition of PS- REV release by 2-S-
OMPT is abolished by the LPA2 receptor antagonist. The inhibition of PS exposure by RBCs
seen with increasing concentration of H2L5186303 could be due to an unknown off-target
effect in the presence of LPA 20:4. Further, studies needed to unravel the mechanisms by
which LPA 20:4 modulates RBC PS exposure. Signaling through LPA5 is a pathway that
contributes to activation platelets, which share the same progenitor cell as RBCs[44].
Moreover, activation of LPA5 by 20:4 was reported[44]. The absence of available selective
agonists/antagonists for LPA5, LPA4, and LPA6 precludes further analysis of signaling
pathways involved in LPA 20:4 mediated inhibition of PS exposure in mature RBCs.
Depending on the study, the concentration of LPA in human plasma ranges from 50 nM to
1 µM[45-47], and up to 14 distinct LPA species are detected[45]. Most studies focus on 6
species: LPA 16:0, 18:0, 18:1, 18:2, 20:4 and 22:6; and with the following relative
abundance: LPA 18:2 > LPA 20:4 > LPA 16:0 ≥ LPA 18:1 = LPA 18:0 = LPA 22:6[39, 45-
47]. Platelet activation, a major driving force behind LPA increase in plasma, leads to LPA
16:0, 18:0, 18:1 and 20:4 accumulation with no significant changes in LPA 18:2 and 22:6
levels[40, 48, 49]. In the present study, we selected four LPA species (i.e. LPA 16:0, 18:0,
18:1 and 20:4) that are consistently detected in human plasma and produced upon platelet
activation or lipoprotein oxidation[40]. Since LPA 18:2 is the most prevalent species in
human plasma and the levels of LPA 22:6 and 18:2 are increased in acute coronary syndrome
patient[40], a future study of those LPA species on RBCs would be of interest.
An analysis of EVs present in human PFP by cryo-electronic microscopy reported that 95 %
of EVs in PFP were under 1 µm with an average diameter of 275 ± 150 nm[17]. Furthermore,

68
the same study found that only a fraction of PFP REVs were PS+[17]. Similarly, Zetasizer
analysis reported average size of 200 nm for REVs produced in vitro[36]. These findings
support our observations that RBCs release mainly small PS- EVs. However, confocal
microscopy and classic flow cytometry studies also highlighted the production of PS+
REVs[22, 36]. The previous study using classic flow cytometry reported that 98 % of REVs
produced in response to LPA were PS+[22]. Less than 1 % of EVs are detected using classic
flow cytometry as compared to cryo-electronic microscopy[17]. The main limitation of
classic flow cytometry is the detection of the small size vesicles released by cells. Our
findings suggest that the PS+ REVs detected in classic flow cytometry could apply only to
the largest REVs that only account for a small fraction of total REVs[17, 36].
We showed that plasma from healthy donors deprived of cells and EVs, LPA 18:1 was able
to induce the release of PS+ and PS- REVs. Since plasma components can alter the LPA
effects on RBCs, plasma composition may play a crucial role in inhibiting or promoting RBC
activation by LPA. Vascular inflammation is associated with chronic rheumatic
inflammatory diseases. PS+ and PS- REV levels are higher in pathologies with vascular
inflammation such as SLE. Further studies should aim at determining whether RBCs from
SLE and rheumatoid arthritis patients are more prone to activation by LPA or whether the
plasma of patients enhances LPA-mediate PS externalization and EV production by RBCs
from healthy controls.
Patients with rheumatoid arthritis and SLE suffer from comorbidities, with cardiovascular
diseases being the most preponderant factor[50, 51]. Cardiovascular dysfunctions
(atherosclerosis, coronary heart disease, stroke, heart failure) and thromboembolic events
correlated to systemic inflammation in rheumatoid arthritis and SLE[52]. REVs and PS
exposing RBCs were associated with activation of the blood coagulation cascade[3, 13] and
vascular inflammation[16, 53, 54]. Vascular LPA levels are higher in patients with
atherosclerosis[55] and those suffering from acute coronary syndrome[56, 57]. Plasma from
rheumatoid arthritis patients shows higher levels of autotaxin, the enzyme responsible for
LPA extracellular production[58]. Activated platelets are a significant source of LPA
including LPA 16:0, 18:0, 18:1, and 20:4[49]. The present study shows that some of those
species induce PS exposure and REV release by RBCs. Since both PS exposure and REVs

69
can promote coagulation and inflammation[13, 15, 16], RBC activation by LPA could be a
noteworthy mediator for the amplification of inflammation and the coagulation cascade.
Therefore, LPA capacity to modulate RBC physiology is a pathway worth investigating to
understand how RBCs contribute to vascular physiology in atherosclerosis or rheumatoid
arthritis.
7 References
[1] R. Marchioli, G. Finazzi, G. Specchia, R. Cacciola, R. Cavazzina, D. Cilloni, V. De
Stefano, E. Elli, A. Iurlo, R. Latagliata, F. Lunghi, M. Lunghi, R.M. Marfisi, P. Musto, A.
Masciulli, C. Musolino, N. Cascavilla, G. Quarta, M.L. Randi, D. Rapezzi, M. Ruggeri, E.
Rumi, A.R. Scortechini, S. Santini, M. Scarano, S. Siragusa, A. Spadea, A. Tieghi, E.
Angelucci, G. Visani, A.M. Vannucchi, T. Barbui, Cardiovascular events and intensity of
treatment in polycythemia vera, N Engl J Med 368(1) (2013) 22-33.
[2] C. Klatt, I. Krüger, S. Zey, K.J. Krott, M. Spelleken, N.S. Gowert, A. Oberhuber, L. Pfaff,
W. Lückstädt, K. Jurk, M. Schaller, H. Al-Hasani, J. Schrader, S. Massberg, K. Stark, H.
Schelzig, M. Kelm, M. Elvers, Platelet-RBC interaction mediated by FasL/FasR induces
procoagulant activity important for thrombosis, J Clin Invest 128(9) (2018) 3906-3925.
[3] M.P. Wautier, E. Héron, J. Picot, Y. Colin, O. Hermine, J.L. Wautier, Red blood cell
phosphatidylserine exposure is responsible for increased erythrocyte adhesion to
endothelium in central retinal vein occlusion, J Thromb Haemost 9(5) (2011) 1049-55.
[4] M.C. Szabo, K.S. Soo, A. Zlotnik, T.J. Schall, Chemokine class differences in binding to
the Duffy antigen-erythrocyte chemokine receptor, The Journal of biological chemistry
270(43) (1995) 25348-51.
[5] E. Karsten, E. Breen, B.R. Herbert, Red blood cells are dynamic reservoirs of cytokines,
Sci Rep 8(1) (2018) 3101.
[6] J. Wei, J. Zhao, V. Schrott, Y. Zhang, M. Gladwin, G. Bullock, Y. Zhao, Red Blood Cells
Store and Release Interleukin-33, J Investig Med 63(6) (2015) 806-10.
[7] R.F. Antunes, C. Brandão, M. Maia, F.A. Arosa, Red blood cells release factors with
growth and survival bioactivities for normal and leukemic T cells, Immunol Cell Biol 89(1)
(2011) 111-21.
[8] K. Fredriksson, J. Lundahl, L. Palmberg, D.J. Romberger, X.D. Liu, S.I. Rennard, C.M.
Skold, Red blood cells stimulate human lung fibroblasts to secrete interleukin-8,
Inflammation 27(2) (2003) 71-8.
[9] C. Harding, J. Heuser, P. Stahl, Endocytosis and intracellular processing of transferrin
and colloidal gold-transferrin in rat reticulocytes: demonstration of a pathway for receptor
shedding, Eur J Cell Biol 35(2) (1984) 256-63.
[10] B.T. Pan, K. Teng, C. Wu, M. Adam, R.M. Johnstone, Electron microscopic evidence
for externalization of the transferrin receptor in vesicular form in sheep reticulocytes, J Cell
Biol 101(3) (1985) 942-8.
[11] R.M. Johnstone, A. Mathew, A.B. Mason, K. Teng, Exosome formation during
maturation of mammalian and avian reticulocytes: evidence that exosome release is a major
route for externalization of obsolete membrane proteins, J Cell Physiol 147(1) (1991) 27-36.

70
[12] E.N. Lipets, O.A. Antonova, O.N. Shustova, K.V. Losenkova, A.V. Mazurov, F.I.
Ataullakhanov, Use of Thrombodynamics for revealing the participation of platelet,
erythrocyte, endothelial, and monocyte microparticles in coagulation activation and
propagation, PLoS One 15(5) (2020) e0227932.
[13] Y. Kim, B.T. Xia, A.D. Jung, A.L. Chang, W.A. Abplanalp, C.C. Caldwell, M.D.
Goodman, T.A. Pritts, Microparticles from stored red blood cells promote a hypercoagulable
state in a murine model of transfusion, Surgery 163(2) (2018) 423-429.
[14] M. Straat, M.E. van Hezel, A. Böing, A. Tuip-De Boer, N. Weber, R. Nieuwland, R.
van Bruggen, N.P. Juffermans, Monocyte-mediated activation of endothelial cells occurs
only after binding to extracellular vesicles from red blood cell products, a process mediated
by β-integrin, Transfusion 56(12) (2016) 3012-3020.
[15] P.E. Van Der Meijden, M. Van Schilfgaarde, R. Van Oerle, T. Renné, H. ten Cate, H.M.
Spronk, Platelet- and erythrocyte-derived microparticles trigger thrombin generation via
factor XIIa, J Thromb Haemost 10(7) (2012) 1355-62.
[16] D. Zecher, A. Cumpelik, J.A. Schifferli, Erythrocyte-derived microvesicles amplify
systemic inflammation by thrombin-dependent activation of complement, Arterioscler
Thromb Vasc Biol 34(2) (2014) 313-20.
[17] N. Arraud, R. Linares, S. Tan, C. Gounou, J.M. Pasquet, S. Mornet, A.R. Brisson,
Extracellular vesicles from blood plasma: determination of their morphology, size,
phenotype and concentration, J Thromb Haemost 12(5) (2014) 614-27.
[18] P. Sens, N. Gov, Force balance and membrane shedding at the red-blood-cell surface,
Phys Rev Lett 98(1) (2007) 018102.
[19] F.L. Willekens, J.M. Werre, Y.A. Groenen-Döpp, B. Roerdinkholder-Stoelwinder, B.
de Pauw, G.J. Bosman, Erythrocyte vesiculation: a self-protective mechanism?, Br J
Haematol 141(4) (2008) 549-56.
[20] G.J. Bosman, E. Lasonder, Y.A. Groenen-Döpp, F.L. Willekens, J.M. Werre, The
proteome of erythrocyte-derived microparticles from plasma: new clues for erythrocyte aging
and vesiculation, J Proteomics 76 Spec No. (2012) 203-10.
[21] P. Burger, E. Kostova, E. Bloem, P. Hilarius-Stokman, A.B. Meijer, T.K. van den Berg,
A.J. Verhoeven, D. de Korte, R. van Bruggen, Potassium leakage primes stored erythrocytes
for phosphatidylserine exposure and shedding of pro-coagulant vesicles, Br J Haematol
160(3) (2013) 377-86.
[22] S.M. Chung, O.N. Bae, K.M. Lim, J.Y. Noh, M.Y. Lee, Y.S. Jung, J.H. Chung,
Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure
and procoagulant microvesicle generation in human erythrocytes, Arterioscler Thromb Vasc
Biol 27(2) (2007) 414-21.
[23] G. Tigyi, Aiming drug discovery at lysophosphatidic acid targets, Br J Pharmacol 161(2)
(2010) 241-70.
[24] A. Tokumura, J. Sinomiya, S. Kishimoto, T. Tanaka, K. Kogure, T. Sugiura, K.
Satouchi, K. Waku, K. Fukuzawa, Human platelets respond differentially to lysophosphatidic
acids having a highly unsaturated fatty acyl group and alkyl ether-linked lysophosphatidic
acids, The Biochemical journal 365(Pt 3) (2002) 617-28.
[25] K. Bandoh, J. Aoki, A. Taira, M. Tsujimoto, H. Arai, K. Inoue, Lysophosphatidic acid
(LPA) receptors of the EDG family are differentially activated by LPA species. Structure-
activity relationship of cloned LPA receptors, FEBS Lett 478(1-2) (2000) 159-65.
[26] M. Tanaka, S. Okudaira, Y. Kishi, R. Ohkawa, S. Iseki, M. Ota, S. Noji, Y. Yatomi, J.
Aoki, H. Arai, Autotaxin stabilizes blood vessels and is required for embryonic vasculature

71
by producing lysophosphatidic acid, The Journal of biological chemistry 281(35) (2006)
25822-30.
[27] L.A. van Meeteren, P. Ruurs, C. Stortelers, P. Bouwman, M.A. van Rooijen, J.P.
Pradère, T.R. Pettit, M.J. Wakelam, J.S. Saulnier-Blache, C.L. Mummery, W.H. Moolenaar,
J. Jonkers, Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation
during development, Mol Cell Biol 26(13) (2006) 5015-22.
[28] M. Bot, S.C. de Jager, L. MacAleese, H.M. Lagraauw, T.J. van Berkel, P.H. Quax, J.
Kuiper, R.M. Heeren, E.A. Biessen, I. Bot, Lysophosphatidic acid triggers mast cell-driven
atherosclerotic plaque destabilization by increasing vascular inflammation, J Lipid Res 54(5)
(2013) 1265-74.
[29] Z. Zhou, P. Subramanian, G. Sevilmis, B. Globke, O. Soehnlein, E. Karshovska, R.
Megens, K. Heyll, J. Chun, J.S. Saulnier-Blache, M. Reinholz, M. van Zandvoort, C. Weber,
A. Schober, Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing
CXCL1 from the endothelium, Cell Metab 13(5) (2011) 592-600.
[30] N. Haserück, W. Erl, D. Pandey, G. Tigyi, P. Ohlmann, C. Ravanat, C. Gachet, W. Siess,
The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte
aggregate formation in whole blood: involvement of P2Y1 and P2Y12 receptors, Blood
103(7) (2004) 2585-92.
[31] C. Zhao, A. Sardella, J. Chun, P.E. Poubelle, M.J. Fernandes, S.G. Bourgoin, TNF-alpha
promotes LPA1- and LPA3-mediated recruitment of leukocytes in vivo through CXCR2
ligand chemokines, J Lipid Res 52(7) (2011) 1307-18.
[32] C.L. Chiang, S.S. Chen, S.J. Lee, K.C. Tsao, P.L. Chu, C.H. Wen, S.M. Hwang, C.L.
Yao, H. Lee, Lysophosphatidic acid induces erythropoiesis through activating
lysophosphatidic acid receptor 3, Stem Cells 29(11) (2011) 1763-73.
[33] K.H. Lin, M.W. Li, Y.C. Chang, Y.N. Lin, Y.H. Ho, W.C. Weng, C.J. Huang, B.E.
Chang, C.L. Yao, H. Lee, Activation of Lysophosphatidic Acid Receptor 3 Inhibits
Megakaryopoiesis in Human Hematopoietic Stem Cells and Zebrafish, Stem Cells Dev 27(3)
(2018) 216-224.
[34] K.H. Lin, Y.H. Ho, J.C. Chiang, M.W. Li, S.H. Lin, W.M. Chen, C.L. Chiang, Y.N. Lin,
Y.J. Yang, C.N. Chen, J. Lu, C.J. Huang, G. Tigyi, C.L. Yao, H. Lee, Pharmacological
activation of lysophosphatidic acid receptors regulates erythropoiesis, Sci Rep 6 (2016)
27050.
[35] L. Kaestner, P. Steffen, D.B. Nguyen, J. Wang, L. Wagner-Britz, A. Jung, C. Wagner,
I. Bernhardt, Lysophosphatidic acid induced red blood cell aggregation in vitro,
Bioelectrochemistry 87 (2012) 89-95.
[36] D.B. Nguyen, T.B. Ly, M.C. Wesseling, M. Hittinger, A. Torge, A. Devitt, Y. Perrie, I.
Bernhardt, Characterization of Microvesicles Released from Human Red Blood Cells, Cell
Physiol Biochem 38(3) (2016) 1085-99.
[37] I. Melki, I. Allaeys, N. Tessandier, T. Lévesque, N. Cloutier, A. Laroche, N. Vernoux,
Y. Becker, H. Benk-Fortin, A. Zufferey, E. Rollet-Labelle, M. Pouliot, G. Poirier, N. Patey,
C. Belleannee, D. Soulet, S.E. McKenzie, A. Brisson, M.E. Tremblay, C. Lood, P.R. Fortin,
E. Boilard, Platelets release mitochondrial antigens in systemic lupus erythematosus, Sci
Transl Med 13(581) (2021).
[38] G. Marcoux, A.C. Duchez, N. Cloutier, P. Provost, P.A. Nigrovic, E. Boilard, Revealing
the diversity of extracellular vesicles using high-dimensional flow cytometry analyses, Sci
Rep 6 (2016) 35928.

72
[39] D.L. Baker, D.M. Desiderio, D.D. Miller, B. Tolley, G.J. Tigyi, Direct quantitative
analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray
ionization liquid chromatography-mass spectrometry, Anal Biochem 292(2) (2001) 287-95.
[40] M. Kurano, A. Suzuki, A. Inoue, Y. Tokuhara, K. Kano, H. Matsumoto, K. Igarashi, R.
Ohkawa, K. Nakamura, T. Dohi, K. Miyauchi, H. Daida, K. Tsukamoto, H. Ikeda, J. Aoki,
Y. Yatomi, Possible involvement of minor lysophospholipids in the increase in plasma
lysophosphatidic acid in acute coronary syndrome, Arterioscler Thromb Vasc Biol 35(2)
(2015) 463-70.
[41] A.L. Bolen, A.P. Naren, S. Yarlagadda, S. Beranova-Giorgianni, L. Chen, D. Norman,
D.L. Baker, M.M. Rowland, M.D. Best, T. Sano, T. Tsukahara, K. Liliom, Y. Igarashi, G.
Tigyi, The phospholipase A1 activity of lysophospholipase A-I links platelet activation to
LPA production during blood coagulation, J Lipid Res 52(5) (2011) 958-70.
[42] A.E. Thumser, J.E. Voysey, D.C. Wilton, The binding of lysophospholipids to rat liver
fatty acid-binding protein and albumin, The Biochemical journal 301 ( Pt 3)(Pt 3) (1994)
801-6.
[43] G. Tigyi, R. Miledi, Lysophosphatidates bound to serum albumin activate membrane
currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells, The
Journal of biological chemistry 267(30) (1992) 21360-7.
[44] J.R. Williams, A.L. Khandoga, P. Goyal, J.I. Fells, D.H. Perygin, W. Siess, A.L. Parrill,
G. Tigyi, Y. Fujiwara, Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate
receptor indicates role in human platelet activation, The Journal of biological chemistry
284(25) (2009) 17304-19.
[45] K. Kano, H. Matsumoto, N. Kono, M. Kurano, Y. Yatomi, J. Aoki, Suppressing post-
collection lysophosphatidic acid (LPA) metabolism improves the precision of plasma LPA
quantification, J Lipid Res (2021) 100029.
[46] D.L. Baker, P. Morrison, B. Miller, C.A. Riely, B. Tolley, A.M. Westermann, J.M.
Bonfrer, E. Bais, W.H. Moolenaar, G. Tigyi, Plasma lysophosphatidic acid concentration and
ovarian cancer, Jama 287(23) (2002) 3081-2.
[47] B. Yang, L. Wang, X. Wan, Y. Li, X. Yu, Y. Qin, Y. Luo, F. Wang, O. Huang, Elevated
plasma levels of lysophosphatidic acid and aberrant expression of lysophosphatidic acid
receptors in adenomyosis, BMC Womens Health 17(1) (2017) 118.
[48] T. Sano, D. Baker, T. Virag, A. Wada, Y. Yatomi, T. Kobayashi, Y. Igarashi, G. Tigyi,
Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and
sphingosine 1-phosphate generation in blood, The Journal of biological chemistry 277(24)
(2002) 21197-206.
[49] J.M. Gerrard, P. Robinson, Identification of the molecular species of lysophosphatidic
acid produced when platelets are stimulated by thrombin, Biochim Biophys Acta 1001(3)
(1989) 282-5.
[50] F. Wolfe, D.M. Mitchell, J.T. Sibley, J.F. Fries, D.A. Bloch, C.A. Williams, P.W. Spitz,
M. Haga, S.M. Kleinheksel, M.A. Cathey, The mortality of rheumatoid arthritis, Arthritis
Rheum 37(4) (1994) 481-94.
[51] G. Thomas, J. Mancini, N. Jourde-Chiche, G. Sarlon, Z. Amoura, J.R. Harlé, E. Jougla,
L. Chiche, Mortality associated with systemic lupus erythematosus in France assessed by
multiple-cause-of-death analysis, Arthritis Rheumatol 66(9) (2014) 2503-11.
[52] M. Prasad, J. Hermann, S.E. Gabriel, C.M. Weyand, S. Mulvagh, R. Mankad, J.K. Oh,
E.L. Matteson, A. Lerman, Cardiorheumatology: cardiac involvement in systemic rheumatic
disease, Nat Rev Cardiol 12(3) (2015) 168-76.

73
[53] M. Straat, A.N. Böing, A. Tuip-De Boer, R. Nieuwland, N.P. Juffermans, Extracellular
Vesicles from Red Blood Cell Products Induce a Strong Pro-Inflammatory Host Response,
Dependent on Both Numbers and Storage Duration, Transfus Med Hemother 43(4) (2016)
302-305.
[54] R.M. Belizaire, P.S. Prakash, J.R. Richter, B.R. Robinson, M.J. Edwards, C.C. Caldwell,
A.B. Lentsch, T.A. Pritts, Microparticles from stored red blood cells activate neutrophils and
cause lung injury after hemorrhage and resuscitation, J Am Coll Surg 214(4) (2012) 648-55;
discussion 656-7.
[55] W. Siess, K.J. Zangl, M. Essler, M. Bauer, R. Brandl, C. Corrinth, R. Bittman, G. Tigyi,
M. Aepfelbacher, Lysophosphatidic acid mediates the rapid activation of platelets and
endothelial cells by mildly oxidized low density lipoprotein and accumulates in human
atherosclerotic lesions, Proc Natl Acad Sci U S A 96(12) (1999) 6931-6.
[56] T. Dohi, K. Miyauchi, R. Ohkawa, K. Nakamura, M. Kurano, T. Kishimoto, N.
Yanagisawa, M. Ogita, T. Miyazaki, A. Nishino, K. Yaginuma, H. Tamura, T. Kojima, K.
Yokoyama, T. Kurata, K. Shimada, H. Daida, Y. Yatomi, Increased lysophosphatidic acid
levels in culprit coronary arteries of patients with acute coronary syndrome, Atherosclerosis
229(1) (2013) 192-7.
[57] M. Kurano, K. Kano, T. Dohi, H. Matsumoto, K. Igarashi, M. Nishikawa, R. Ohkawa,
H. Ikeda, K. Miyauchi, H. Daida, J. Aoki, Y. Yatomi, Different origins of lysophospholipid
mediators between coronary and peripheral arteries in acute coronary syndrome, J Lipid Res
58(2) (2017) 433-442.
[58] I. Nikitopoulou, N. Oikonomou, E. Karouzakis, I. Sevastou, N. Nikolaidou-Katsaridou,
Z. Zhao, V. Mersinias, M. Armaka, Y. Xu, M. Masu, G.B. Mills, S. Gay, G. Kollias, V.
Aidinis, Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of
modeled arthritis, J Exp Med 209(5) (2012) 925-33.
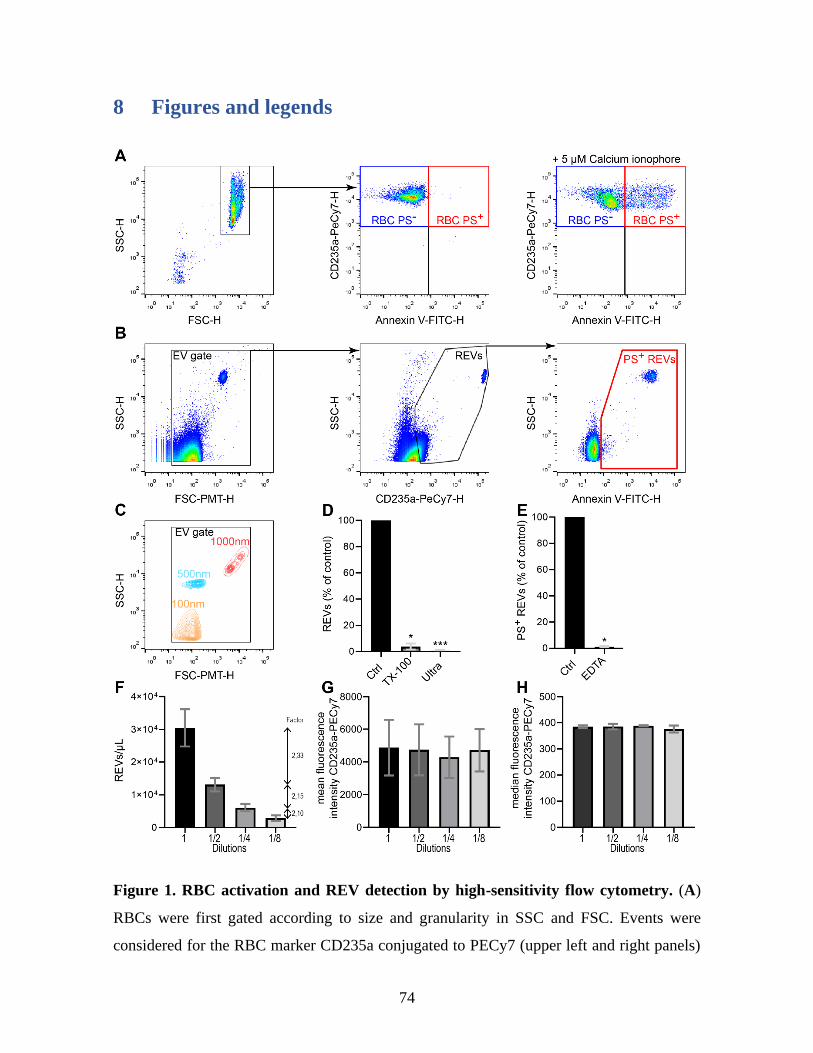
74
8 Figures and legends
Figure 1. RBC activation and REV detection by high-sensitivity flow cytometry. (A)
RBCs were first gated according to size and granularity in SSC and FSC. Events were
considered for the RBC marker CD235a conjugated to PECy7 (upper left and right panels)
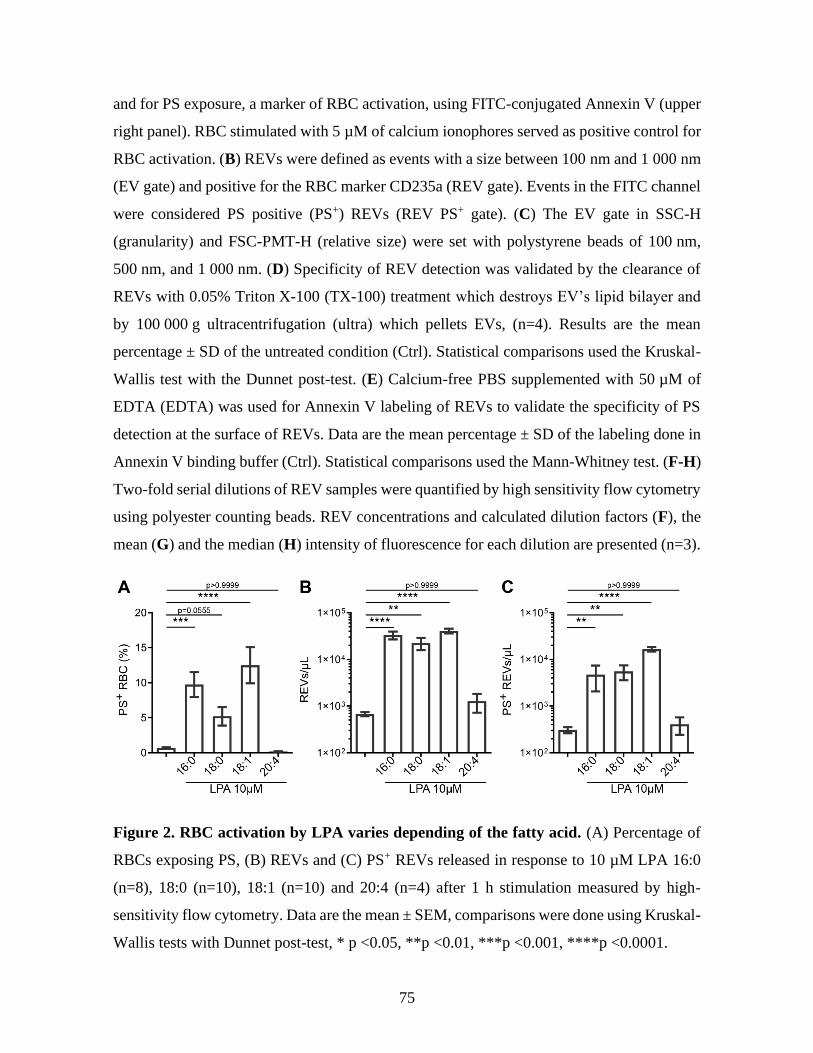
75
and for PS exposure, a marker of RBC activation, using FITC-conjugated Annexin V (upper
right panel). RBC stimulated with 5 µM of calcium ionophores served as positive control for
RBC activation. (B) REVs were defined as events with a size between 100 nm and 1 000 nm
(EV gate) and positive for the RBC marker CD235a (REV gate). Events in the FITC channel
were considered PS positive (PS+) REVs (REV PS+ gate). (C) The EV gate in SSC-H
(granularity) and FSC-PMT-H (relative size) were set with polystyrene beads of 100 nm,
500 nm, and 1 000 nm. (D) Specificity of REV detection was validated by the clearance of
REVs with 0.05% Triton X-100 (TX-100) treatment which destroys EV’s lipid bilayer and
by 100 000 g ultracentrifugation (ultra) which pellets EVs, (n=4). Results are the mean
percentage ± SD of the untreated condition (Ctrl). Statistical comparisons used the Kruskal-
Wallis test with the Dunnet post-test. (E) Calcium-free PBS supplemented with 50 µM of
EDTA (EDTA) was used for Annexin V labeling of REVs to validate the specificity of PS
detection at the surface of REVs. Data are the mean percentage ± SD of the labeling done in
Annexin V binding buffer (Ctrl). Statistical comparisons used the Mann-Whitney test. (F-H)
Two-fold serial dilutions of REV samples were quantified by high sensitivity flow cytometry
using polyester counting beads. REV concentrations and calculated dilution factors (F), the
mean (G) and the median (H) intensity of fluorescence for each dilution are presented (n=3).
Figure 2. RBC activation by LPA varies depending of the fatty acid. (A) Percentage of
RBCs exposing PS, (B) REVs and (C) PS+ REVs released in response to 10 µM LPA 16:0
(n=8), 18:0 (n=10), 18:1 (n=10) and 20:4 (n=4) after 1 h stimulation measured by high-
sensitivity flow cytometry. Data are the mean ± SEM, comparisons were done using Kruskal-
Wallis tests with Dunnet post-test, * p <0.05, **p <0.01, ***p <0.001, ****p <0.0001.
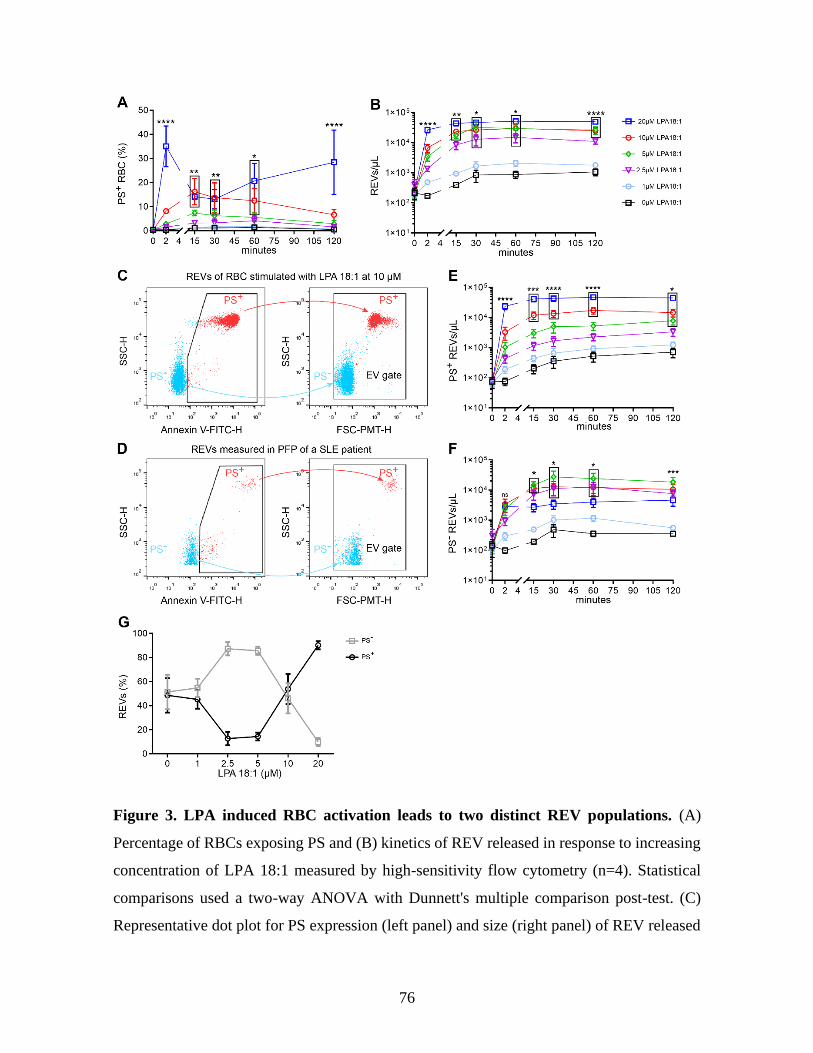
76
Figure 3. LPA induced RBC activation leads to two distinct REV populations. (A)
Percentage of RBCs exposing PS and (B) kinetics of REV released in response to increasing
concentration of LPA 18:1 measured by high-sensitivity flow cytometry (n=4). Statistical
comparisons used a two-way ANOVA with Dunnett's multiple comparison post-test. (C)
Representative dot plot for PS expression (left panel) and size (right panel) of REV released
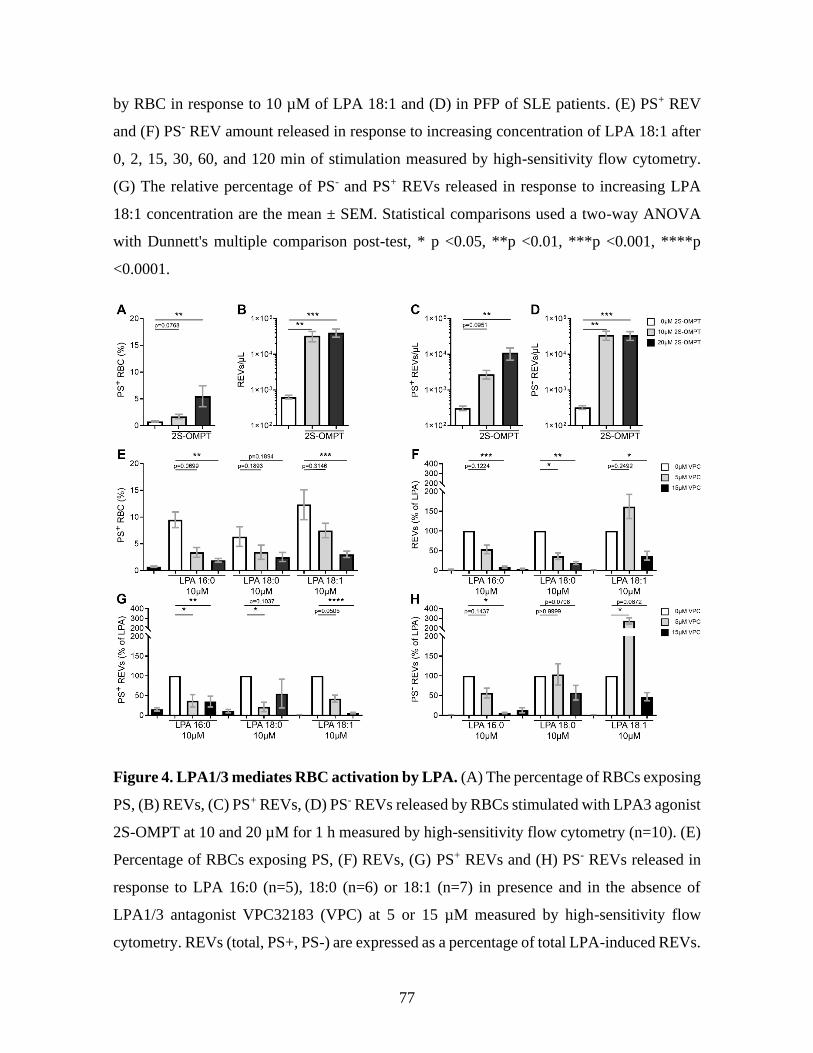
77
by RBC in response to 10 µM of LPA 18:1 and (D) in PFP of SLE patients. (E) PS+ REV
and (F) PS- REV amount released in response to increasing concentration of LPA 18:1 after
0, 2, 15, 30, 60, and 120 min of stimulation measured by high-sensitivity flow cytometry.
(G) The relative percentage of PS- and PS+ REVs released in response to increasing LPA
18:1 concentration are the mean ± SEM. Statistical comparisons used a two-way ANOVA
with Dunnett's multiple comparison post-test, * p <0.05, **p <0.01, ***p <0.001, ****p
<0.0001.
Figure 4. LPA1/3 mediates RBC activation by LPA. (A) The percentage of RBCs exposing
PS, (B) REVs, (C) PS+ REVs, (D) PS- REVs released by RBCs stimulated with LPA3 agonist
2S-OMPT at 10 and 20 µM for 1 h measured by high-sensitivity flow cytometry (n=10). (E)
Percentage of RBCs exposing PS, (F) REVs, (G) PS+ REVs and (H) PS- REVs released in
response to LPA 16:0 (n=5), 18:0 (n=6) or 18:1 (n=7) in presence and in the absence of
LPA1/3 antagonist VPC32183 (VPC) at 5 or 15 µM measured by high-sensitivity flow
cytometry. REVs (total, PS+, PS-) are expressed as a percentage of total LPA-induced REVs.
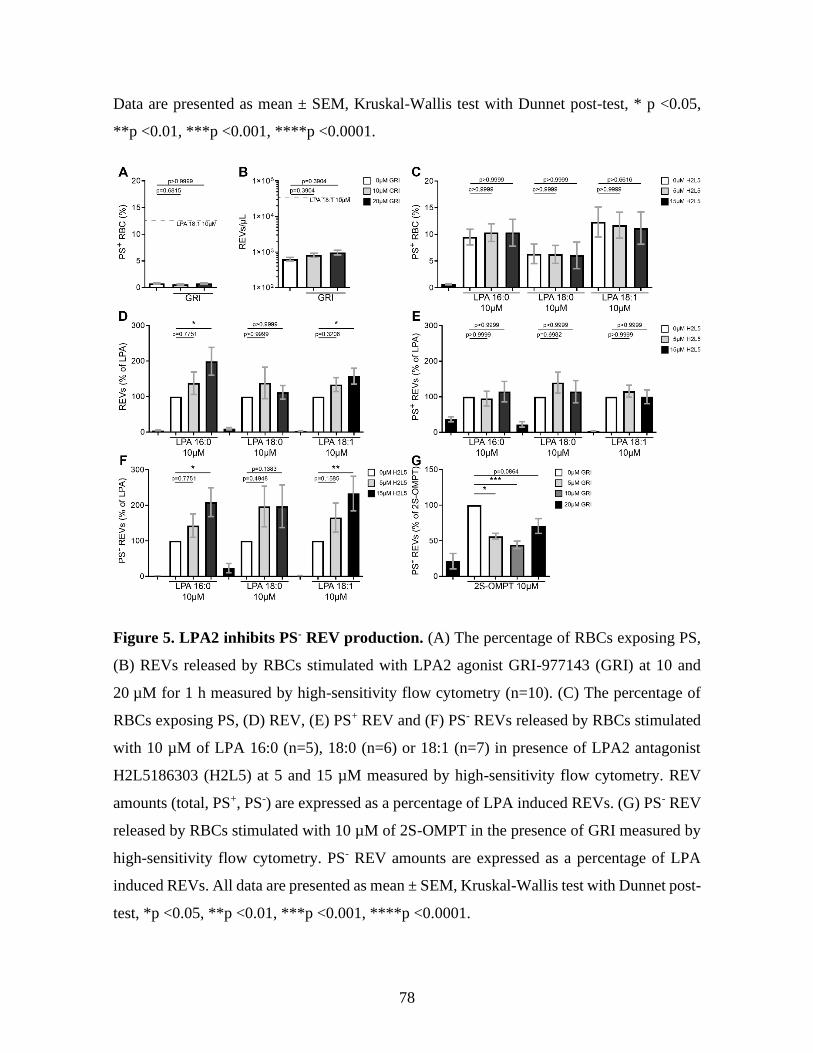
78
Data are presented as mean ± SEM, Kruskal-Wallis test with Dunnet post-test, * p <0.05,
**p <0.01, ***p <0.001, ****p <0.0001.
Figure 5. LPA2 inhibits PS- REV production. (A) The percentage of RBCs exposing PS,
(B) REVs released by RBCs stimulated with LPA2 agonist GRI-977143 (GRI) at 10 and
20 µM for 1 h measured by high-sensitivity flow cytometry (n=10). (C) The percentage of
RBCs exposing PS, (D) REV, (E) PS+ REV and (F) PS- REVs released by RBCs stimulated
with 10 µM of LPA 16:0 (n=5), 18:0 (n=6) or 18:1 (n=7) in presence of LPA2 antagonist
H2L5186303 (H2L5) at 5 and 15 µM measured by high-sensitivity flow cytometry. REV
amounts (total, PS+, PS-) are expressed as a percentage of LPA induced REVs. (G) PS- REV
released by RBCs stimulated with 10 µM of 2S-OMPT in the presence of GRI measured by
high-sensitivity flow cytometry. PS- REV amounts are expressed as a percentage of LPA
induced REVs. All data are presented as mean ± SEM, Kruskal-Wallis test with Dunnet post-
test, *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001.
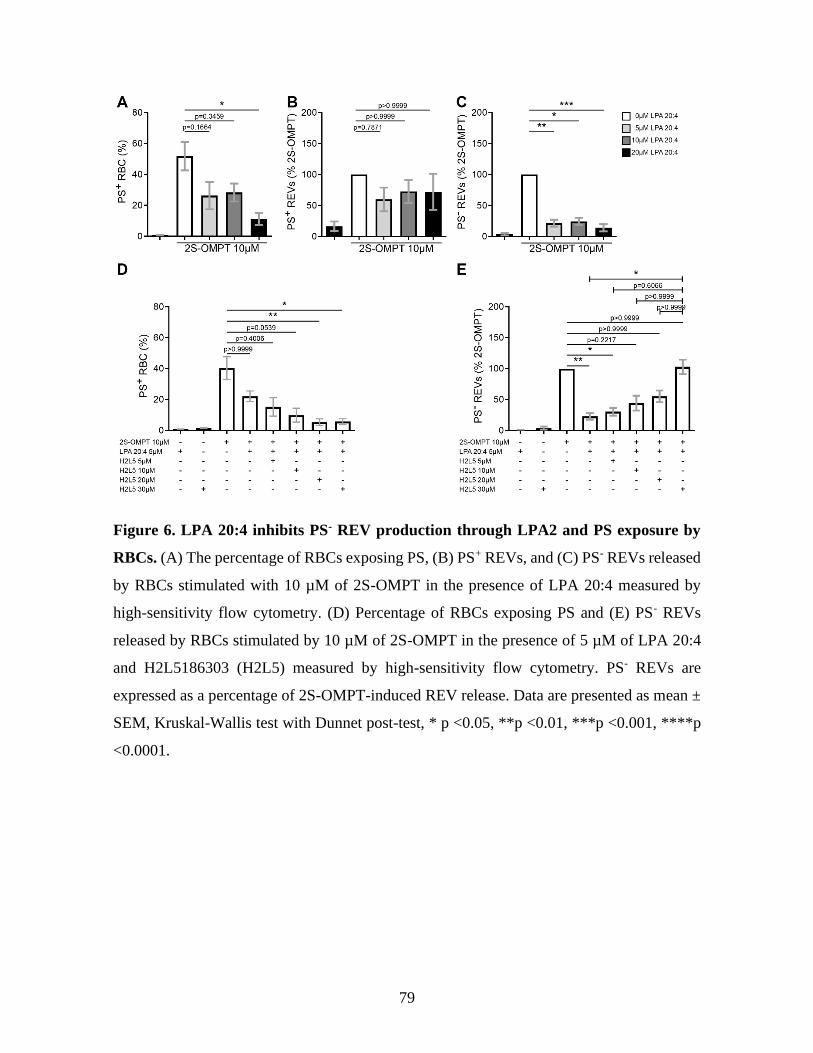
79
Figure 6. LPA 20:4 inhibits PS- REV production through LPA2 and PS exposure by
RBCs. (A) The percentage of RBCs exposing PS, (B) PS+ REVs, and (C) PS- REVs released
by RBCs stimulated with 10 µM of 2S-OMPT in the presence of LPA 20:4 measured by
high-sensitivity flow cytometry. (D) Percentage of RBCs exposing PS and (E) PS- REVs
released by RBCs stimulated by 10 µM of 2S-OMPT in the presence of 5 µM of LPA 20:4
and H2L5186303 (H2L5) measured by high-sensitivity flow cytometry. PS- REVs are
expressed as a percentage of 2S-OMPT-induced REV release. Data are presented as mean ±
SEM, Kruskal-Wallis test with Dunnet post-test, * p <0.05, **p <0.01, ***p <0.001, ****p
<0.0001.
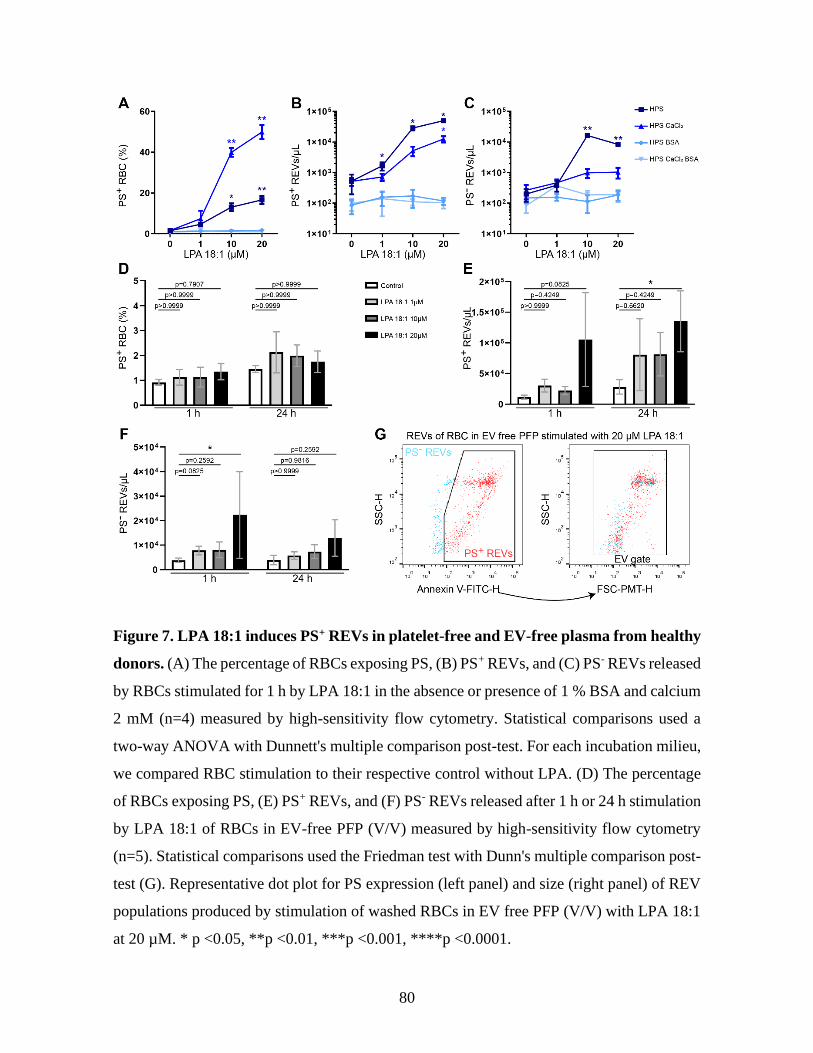
80
Figure 7. LPA 18:1 induces PS+ REVs in platelet-free and EV-free plasma from healthy
donors. (A) The percentage of RBCs exposing PS, (B) PS+ REVs, and (C) PS- REVs released
by RBCs stimulated for 1 h by LPA 18:1 in the absence or presence of 1 % BSA and calcium
2 mM (n=4) measured by high-sensitivity flow cytometry. Statistical comparisons used a
two-way ANOVA with Dunnett's multiple comparison post-test. For each incubation milieu,
we compared RBC stimulation to their respective control without LPA. (D) The percentage
of RBCs exposing PS, (E) PS+ REVs, and (F) PS- REVs released after 1 h or 24 h stimulation
by LPA 18:1 of RBCs in EV-free PFP (V/V) measured by high-sensitivity flow cytometry
(n=5). Statistical comparisons used the Friedman test with Dunn's multiple comparison post-
test (G). Representative dot plot for PS expression (left panel) and size (right panel) of REV
populations produced by stimulation of washed RBCs in EV free PFP (V/V) with LPA 18:1
at 20 µM. * p <0.05, **p <0.01, ***p <0.001, ****p <0.0001.
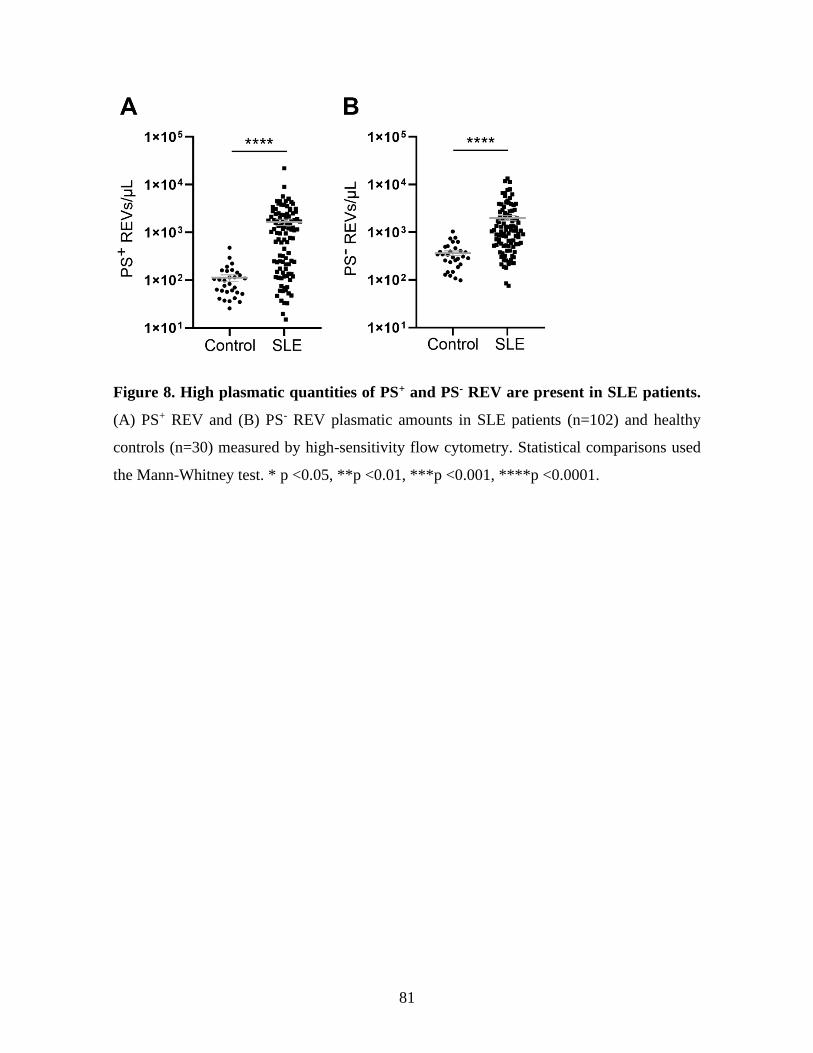
81
Figure 8. High plasmatic quantities of PS+ and PS- REV are present in SLE patients.
(A) PS+ REV and (B) PS- REV plasmatic amounts in SLE patients (n=102) and healthy
controls (n=30) measured by high-sensitivity flow cytometry. Statistical comparisons used
the Mann-Whitney test. * p <0.05, **p <0.01, ***p <0.001, ****p <0.0001.
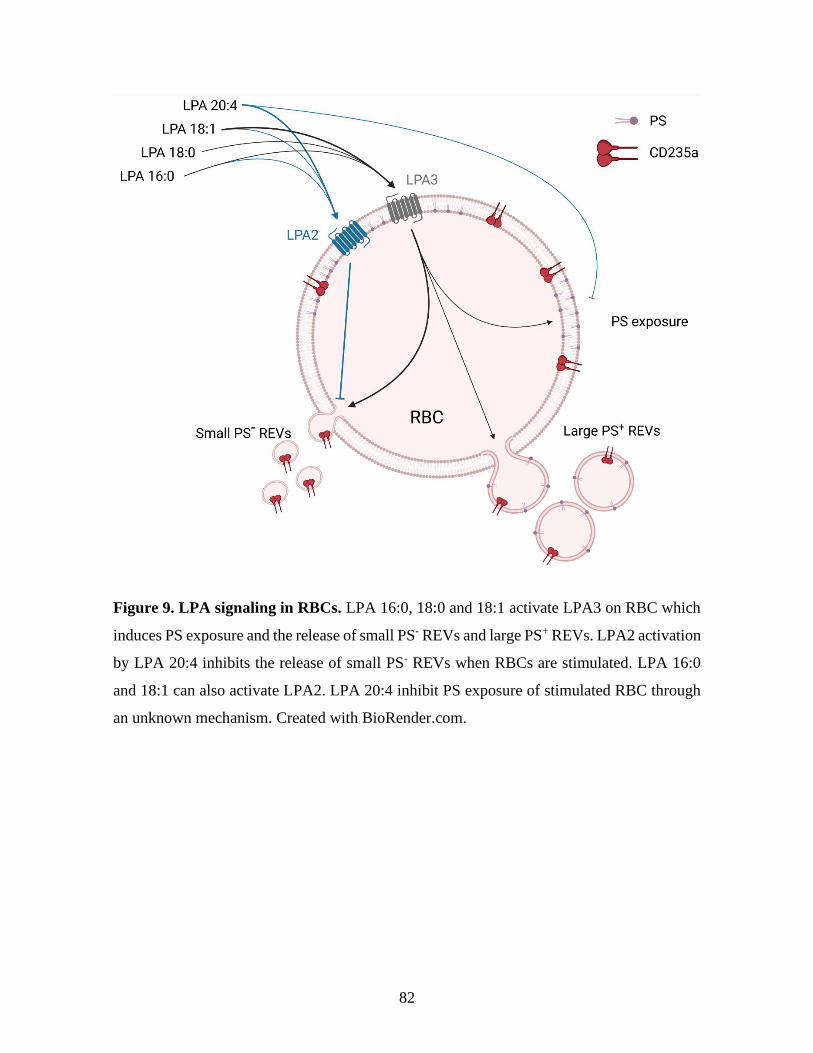
82
Figure 9. LPA signaling in RBCs. LPA 16:0, 18:0 and 18:1 activate LPA3 on RBC which
induces PS exposure and the release of small PS- REVs and large PS+ REVs. LPA2 activation
by LPA 20:4 inhibits the release of small PS- REVs when RBCs are stimulated. LPA 16:0
and 18:1 can also activate LPA2. LPA 20:4 inhibit PS exposure of stimulated RBC through
an unknown mechanism. Created with BioRender.com.

83
Chapitre 2 : Plasma level of red blood cell-derived
phosphatidylserine positive extracellular vesicles are
associated with thrombosis in systemic erythematous
lupus patients
1 Résumé
Les cellules activées libèrent des vésicules extracellulaires (EV). Les EV promeuvent la
coagulation et l’inflammation notamment en étant une source d’antigènes du soi. Les patients
atteints par le lupus érythémateux disséminé (LED) présentent un inflammation vasculaire
importante et ont un risque accru de développer des maladies cardiovasculaires. C’est
pourquoi nous pensions que les EV de plaquettes et de globules rouges (REV) pouvaient être
corrélées à l’activité de la maladie et aux dommages cardiovasculaires qui sont associés au
LED. Bien que les PEV et les REV soient augmentées chez les patients LED, elles ne sont
pas associées avec l’activité de la maladie. Cependant, la stratification de la cohorte en
fonction des REV positives pour la phosphatidylsérine a mis en évidence une incidence plus
importante de thrombose chez les patients qui en présentent de grandes quantités.

84
Red blood cell-derived phosphatidylserine positive extracellular vesicles are
associated with thrombosis in systemic erythematous lupus patients
Stephan Hasse1, Anne-Sophie Julien2, Anne-Claire Duchez1, Chenqi Zhao1, Eric Boilard1,
Paul Fortin3, Sylvain G. Bourgoin1
1Centre de recherche du CHU de Québec-Université Laval, Centre ARThrite de l'Université
Laval, Département de microbiologie-infectiologie et d’immunologie, Université Laval,
Québec, QC, Canada G1V 4G2.
2Département de mathématiques et statistique, Université Laval, QC, Canada G1V 4G2.
3Centre de recherche du CHU de Québec-Université Laval, Centre ARThrite de l'Université
Laval, Département de médecine, Faculté de médecine, Université Laval, QC, Canada G1V
4G2.
Short title: Extracellular vesicles promote thrombosis in SLE
Corresponding author:
Sylvain G. Bourgoin, PhD
Centre de Recherche du Centre Hospitalier Universitaire de Québec
Faculté de Médecine de l’Université Laval
2705 Boul. Laurier, Québec, QC, Canada G1V 4G2
Phone: +1 (418) 525-4444, ext. 46136 Fax: (418) 654-2765
Key words: Extracellular vesicles, ATX, CD62P, phosphatidylserine, platelets, erythrocytes,
lupus, thrombocytopenia, thrombosis

85
2 Abstract
Background. Extracellular vesicles (EVs) released by blood cells have pro-inflammation
and pro-coagulant action. Systemic lupus erythematosus (SLE) patients present high vascular
inflammation and are prone to develop cardiovascular diseases. Therefore, we postulated that
the EV populations found in blood, platelet EVs (PEVs) and red blood cell EVs (REVs) are
associated with SLE disease activity and SLE-associated cardiovascular accidents.
Method. We assessed ATX plasma levels by ELISA, the platelet activation markers PAC1
and CD62P, ATX bound to platelets, and the amounts of plasma PEVs and REVs by flow
cytometry in a cohort of 102 SLE patients, including 29 incident cases of SLE and 30
controls. Correlation analyses explored the associations with the clinical parameters.
Result. Platelet activation markers were increased in SLE patients compared to control, with
the marker CD62P associated with the SLEDAI. The incident cases show additional
associations between platelet markers (CD62P/ATX and PAC1/CD62P) and the SLEDAI.
SLE patients presented higher levels of PEVs, phosphatidylserine positive (PS+) PEVs,
REVs, and PS+ REVs, but there is no association with disease activity. When stratified
according to the plasma level of PS+ REVs, the group of SLE patients with a high level of
PS+ REVs presented a higher number of past thrombosis events and higher ATX levels.
Conclusion. Incident and prevalent forms of SLE cases present similar levels of platelet
activation markers, with CD62P correlating with disease activity. Though EVs are not
associated with disease activity, the incidence of thrombosis is higher in patients with a high
level of PS+ REVs.

86
3 Introduction
Systemic lupus erythematosus (SLE) is a Systemic Autoimmune Rheumatic Disease
(SARD). SLE patients present a wide range of clinical phenotypes. One characteristic of SLE
is a high vascular inflammation associated with damages in multiple organs [1]. Patients with
SLE have a higher risk of dying from cardiovascular diseases [2]. The vascular inflammation
associated with SLE development relates to an immune response to autoantigens [3, 4] and
a production of type I interferon [5-7].
Platelet activation also induces the liberation of extracellular vesicles [8]. Extracellular
vesicles (EVs) are small vesicles liberated by activated cells. The EVs includes exosomes
and microvesicles. The fusion of multivesicular bodies with the plasma membrane releases
the exosomes. Plasma membrane budding generates the microvesicles, which often occurs
after cell loss of the membrane phospholipid asymmetry. Some EVs formed after the loss of
the plasma membrane asymmetry presents the phosphatidylserine (PS) at their surface. EVs
from platelets (PEVs) and red blood cells (REVs) can, through the exposition of PS, recruit
mediators of the coagulation cascade and initiate the formation of blood cloth [9-11]. SLE
patients present higher levels of platelet activation in SLE is a source of autoantigen, notably
by the release of free mitochondria [8, 12]. SLE patients have increased circulating EVs,
notably from platelet origin [13, 14] and serve as a source for interferon-α and bind sites for
immune complexes [15-17]. The exosomes are also associated with enhanced pro-
inflammatory cytokine and chemokine production, notably type I interferon in SLE [15, 16,
18].
PEVs are the largest EV population in the blood, which are found in higher amounts in the
plasma SLE patients. However, besides that PEVs are a source of autoantigens and bind
immune complexes, there is little knowledge on their role in SLE [14, 19]. In some studies,
there was an association between PEVs and the SLE disease index (SLEDAI) [14], but no
association was reported in others [20]. However, PEVs were associated with the progression
of atherosclerosis in SLE patients through the thickening of the vascular wall [19].
Platelet activation also releases autotaxin (ATX), a phospholipase with pro-inflammatory
properties. ATX is associated with the pathophysiology of rheumatoid arthritis and

87
cardiovascular diseases [21-23]. ATX catalyzes the production of lysophosphatidic acid
(LPA), one of the few known activators of red blood cells which induce the liberation of
REVs [24, 25]. SLE are more at risk to suffer from cardiovascular diseases that are the
leading cause of death for SLE patients. EVs and platelet activation are factors in the
development of cardiovascular diseases and other autoimmune diseases. PEVs and REVs
have pro-inflammatory and pro-coagulant activities [9, 10, 26-28]. Since SLE patients have
a high level of plasma EVs, we focused our study on PEVs and REVs, the two most abundant
EV populations found in the blood, and the associated vascular events.
4 Material and methods
4.1 SLE patients and healthy donors
SARD-BDB (Systemic Autoimmune Rheumatic Disease biobank and database repository of
the CHU de Québec-Université Laval) recruited prevalent SLE patients with a disease
duration superior to 15 months and incident SLE patients with a disease duration equal or
under 15 months. A control group formed by 30 healthy donors, under no medication and
without known illness, was recruited (mean age 50±8 years, female 63.33%).
4.2 SARD-BDB protocol
The SARD-BDB provided the plasma and platelet-free plasma (PFP) from SLE patients. The
PFP was processed using previously described standardized protocols [12]. Patients included
in the study gave informed written consent according to the declaration of Helsinki. The
ethics review board of the CHU de Québec-Université Laval reviewed and validated the
study (Project # 2016-2558). SLE patients had to meet the American College of
Rheumatology (ACR) classification criteria for SLE revised in 1997 [29, 30].
Antiphospholipid syndrome (APS) was diagnosed according to the 2006 revised Sapporo
criteria [31]. Variables were collected at the time of the first visit to the SARD-BDB
including, sociodemographic variables, diseases characteristics, clinical variables, common
hematology tests, cardiovascular and thrombosis risk factors, and current use medication.

88
4.3 Flow cytometry
4.3.1 Detection of platelet activation
Five µL of PRP were incubated 30 min at room temperature in the dark with 3 µL of anti-
CD41-V450 (BD Bioscience Canada, Mississauga, ON, Canada), a marker of platelets, 15
µL of anti-CD62P-APC and 15 µL of anti-PAC1-FITC (BD Bioscience Canada,
Mississauga, ON, Canada) and anti-ATX (BD Bioscience Canada, Mississauga, ON,
Canada) in 100 µL of PBS. The samples were mixed with 400 µL of PBS to stop labelling
and analyzed using a high sensitivity flow cytometer BD Canto II Special Order Research
Product with the gating strategy described in supplementary figure 1A. The flow cytometer
settings were as follows: FSC at 300 V, SSC at 335 V, Pacific blue at 500 V, FITC at 500 V,
PE at 500 V and APC at 500 V.
4.3.2 Detection of plasmatic EVs
Five µL of PFP were incubated for 30 min at room temperature and in the dark with 3 µl
anti-CD41-V450 (BD Bioscience Canada, Mississauga, ON, Canada), a marker for platelet-
derived EVs, and with 3 µl anti-CD235a-PECy7 (BD Bioscience Canada, Mississauga, ON,
Canada) to label the REVs. To detect PS exposed on the outer membrane leaflet, we added
3 µl Annexin V FITC (BD Bioscience Canada, Mississauga, ON, Canada) to the plasma
samples in 100 µl final Annexin V binding buffer (BD Bioscience Canada, Mississauga, ON,
Canada). The samples were mixed with Annexin V binding buffer (200 µL) to stop labelling
and processed under 90 min on a high sensitivity flow cytometer BD Canto II Special Order
Research Product with a small particle option as previously described [32]. The
supplementary figure 1B shows the gating strategy for the detection of plasmatic PEVs and
REVs. Silica particles of 100, 500 and 1,000 nm (Kisker Biotech GmbH & Co. Steinfurt,
Germany) allowed to set up a gate differentiating the events of size between 100 to 1 000 nm
(Supp. Fig. 1B). The flow cytometer settings were as follows: FSC at 300 V, SSC at 300 V,
PECy7 at 500 V, FITC at 500 V and APC at 500 V. To determine the absolute amounts of
REVs and PEVs in the samples, we added known concentrations of 2 µm APC polystyrene
beads (BD Bioscience Canada, Mississauga, ON, Canada) or 3 µm polystyrene beads
(Polysciences, PA, USA).

89
Specificity of EV detection was validated by destroying EVs from the sample by Triton X-
100 treatment or pelleting EVs by a 100 000 g ultracentrifugation (Supp. Fig. 1D). Labelling
in EDTA-supplemented buffer and absence of annexin V buffer was used to validate the
specificity of Annexin V labelling (Supp. Fig. 1E). Finally, a coincidence test validated that
our measurements of PEV and REVs were quantitative (Supp. Fig. 1F and G).
Every day before monitoring platelets activation markers and plasma EVs, a test of
performance tracking of high sensitivity flow cytometry was done using BD cytometer setup
and tracking beads (BD Bioscience Canada, Mississauga, ON, Canada).
4.4 Autotaxin measurement
Plasmatic concentrations of autotaxin (ng/mL) were quantified using a Human ENPP-
2/Autotaxin Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) and following
the manufacturer instructions.
4.5 Analysis and Statistics
Flow cytometry data analysis used the FlowJo V10 software (FlowJo, LLC, OR, USA) and
statistical analysis with GraphPad Prism 7.0 software (GraphPad Software, San Diego, USA)
and SAS version 9.4 (SAS Institute Inc, Cary, North Carolina, USA). Comparisons between
groups used the Kruskal-Wallis tests with Dunn’s multiple comparison post-test or the
Wilcoxon Mann Whitney test for continuous variables, depending on the number of groups.
The Exact Pearson Chi-Square Test was used to compare groups for discrete variables.
Spearman’s correlation coefficient (rs) determined the association between continuous
variables. Only variables monitored at the first visit (baseline) were considered for the
analyses.
5 Results
5.1 Patient’s characteristics
The characteristics at baseline of the 102 SLE patients included in the cohort are presented
in Table 1. For 29 of 102 patients, the SLE diagnosis was 15 months or less before their

90
inclusion in the cohort. These patients that started their treatments recently and had not their
SLE under control were considered incident cases. The SLE prevalent cases were the patients
treated for more than 15 months. Women represented 83% of all SLE cases. About a quarter
of the patients had thrombocytopenia (23.5%). SLE patients with renal disorders and
antiphospholipid syndrome represented 24.5% and 15.7% of the SLE cohort, respectively.
The prevalence of thrombophilia in the SLE cohort was 11.8%. Furthermore, 30 patients had
atherosclerosis plaques, and 42 had no plaques (Table 1).
5.2 SLE patients present higher platelet activation and plasma EV levels at
baseline
When all the prevalent and the incident cases were considered at baseline, SLE patients
present a higher percentage of platelet with the activation marker PAC1 and CD62P by
comparison to healthy controls (Table 2). In addition, a higher number of platelets were
positive for the phospholipase ATX (Table 2). ATX is known to be secreted by and to bind
the integrins of activated platelets [33-35]. However, the total ATX plasma level was not
different between the SLE and the healthy group (Table 2). The plasma of SLE patients
shows high levels of platelet- and RBC-derived EVs, including those exposing PS at their
surface (Table 2). Furthermore, 79.3% of the plasma PEVs and 23.6% of the plasma REVs
were PS+ in the control group, and this proportion increased respectively to 87.8% and 45.2%
in the SLE cohort (Table 2).
5.3 Prevalent and incident SLE patients show similar levels of plasma EVs
and platelet activation.
We investigated the difference between recently diagnosed and established cases of SLE by
comparing the incident and prevalent cases (n=29 and n=73, respectively). The levels of
platelet activation markers and plasma EVs of the prevalent SLE were not significantly
different from those of the incident SLE cases (Fig. 1A & 1B). ATX plasma concentration
in healthy controls, prevalent and incident SLE cases are similar (Fig. 1C). However, plasma
EV levels in prevalent and incident cases were significantly different from those of the
control group (Fig. 1C & 1D). The incident SLE cases show no significant difference for the

91
platelet activation marker PAC1 or CD62P compared to healthy control (Fig. 1A, upper &
middle panels). In comparison to healthy patients, we report a significantly higher
percentage of platelets exposing both the activation markers PAC1 and CD62P (Fig. 1A,
lower panel). The prevalent and the incident SLE cases did not show a significant difference
for the platelet activation marker PAC1 compared to healthy control (Fig. 1A, upper panel).
In contrast, the amounts of CD62P positive (Fig. 1A, middle panel) and PAC1-CD62P
double-positive platelets (Fig. 1A, lower panel) were significantly different from the control
group. Compared to healthy controls, the levels of double-positive PAC1-ATX (Fig. 1B,
upper panel), double-positive CD62P-ATX (Fig. 1B, middle panel), and triple-positive
PAC1-CD62P-ATX platelets (Fig. 1B, lower panel) were significantly different with those
of prevalent but not those the incident SLE cases.
5.4 Platelet activation is associated with the SLEDAI score in incident cases
of SLE.
Despite significant increases in the incident and prevalent SLE groups, there was no
association between the EV populations and the SLEDAI score (Table 3). Only CDP62P, a
marker of platelet activation, was consistently associated with a higher SLEDAI score for the
incident and prevalent cases, or when we considered the whole SLE cohort patients
(Table 3). However, incident SLE cases show additional associations with platelet activation
markers as the levels of platelets highly positive for both CD62P and ATX or CD62P and
PAC1 are significantly associated with the SLEDAI score (Table 3).
5.5 Higher PS+ REVs are associated with vascular damages in SLE patients.
Incident and prevalent SLE patients did not present differences in plasmatic levels of ATX,
platelet activation and for both RBC and platelet EVs (Fig. 1A-E). Therefore, we choose not
to distinguish between the incident and prevalent SLE cases while analyzing the potential
clinical implications of high plasma levels of PS+ REVs. Based on the levels of PS+ REVs,
we divided the SLE cohort into two distinct groups (Fig. 1E). One group of patients had a
concentration of PS+ REVs similar to that of healthy controls. The second group of patients
showed a high plasma concentration of PS+ REVs. We applied a cut off at 1000 EV/µL to

92
distinguish the patients with a level of plasma PS+ REVs within the range of the healthy
controls from those with EV concentrations superior to the cut-off. The latter were considered
high plasma levels of PS+ REVs. Fifty-two SLE patients (51%) had a plasma PS+ REV
number over the cut-off level. The 50 SLE patients with a concentration of plasma PS+ REVs
lower than the cut off were considered “normal” for these biomarkers.
As anticipated, the SLE patients with a higher level of PS+ REVs also show a higher number
of REVs and PEVs (total and PS+) compared to those with “normal” concentrations of PS+
REVs (Table 4). Furthermore, the high PS+ REVs levels are associated with a lower platelet
count and high plasma level of plasma ATX (Table 4). Compared to patients with a normal
PS+ REV level, the plasma ATX amount was significantly higher in patients with elevated
PS+ REVs. The high PS+ REV counts were not associated with the disease duration or recent
disease onset but correlated with thrombophilia (Table 4). Only two patients with low plasma
PS+ REV levels (4%) had a history of venous thrombosis. Among patients with high PS+
REVs, 5 (10%) presented a history of venous thrombosis, 4 (8%) of arterial thrombosis and
one (2%) of microcirculation thrombosis. In this group of patients, we also highlight an
association between the plasma levels of PS+ REVs and the presence of autoantibodies
(Table 4). Surprisingly, patients with high amounts of PS+ REVs tend to have a lower
SLEDAI score, and antiphospholipid syndrome incidence tended to be higher than for
patients with low PS+ REV levels (Table 4).

93
6 Discussion
In this study, we report on a high plasma level of REVs in SLE patients. Furthermore, the
percentage of PS+ REVs in the plasma of SLE patients almost doubled compared to the
healthy group. The patients with a high level of plasma REVs also have elevated plasma
PEVs. There was no correlation between the EV levels, including those that are PS+, and the
SLEDAI. About half of the SLE patients had PS+ REV levels like the healthy controls.
However, a high level of PS+ REVs was positively associated with thrombocytopenia and a
higher incidence of thrombotic events.
A high level of platelet activation markers and plasma PEVs were reported previously in SLE
patients [36]. CD62P (P-selectin) is only present on the activated platelet following the fusion
of the α-granules with the plasma membrane [37]. The platelet activation marker CD62P (P-
selectin) is elevated in SLE patients and is positively associated with the SLEDAI score [36].
This study confirms the positive association between the number of CD62P+ platelets and
disease activity. The stratification of the incident and prevalent cases of SLE did not highlight
differences regarding the levels of plasma EVs, PS+ EVs, and platelet activation markers. In
incident SLE cases, the percentages of platelets positive for CD62P, CD62P/ATX or
PAC1/ATX double-positive, and PAC1/CD32P/ATX triple-positive, were not statistically
different from the healthy group. It may be due to the small number of newly diagnosed SLE
patients included in the present study. However, in the incident and prevalent cases, the
SLEDAI score correlated with platelet activation as monitored by the cell surface exposure
of CD62P. In incident SLE cases, we found other associations between the platelet activation
markers and the SLEDAI score. Those include the CD62P/ATX and CD62P/PAC1 double-
positive platelets. PAC1 monitor the activation of αIIbβ3 integrin complex in platelets [38,
39]. Of note, the ATX stored in α-granules and released upon platelet activation can bind the
platelet αIIbβ3 integrin [33-35]. It would suggest that the liberation of α-granule and the
activated form of platelets integrins contributes to the early phase of SLE disease progression.
While, at later stages, when the disease becomes chronic, only the liberation of the content
of α-granule is of importance.

94
It was possible to stratify the SLE patients based on the levels of plasma PS+ REVs. About
half of the patients had plasma levels of PS+ REVs below the threshold of 1 000 REVs/µL
found in healthy controls. The other half were SLE patients with high to very high levels of
PS+ REVs. The results suggest that a high amount of plasma PS+ REVs is associated with a
higher incidence of cardiovascular events. A longitudinal study on incident cases with no
antecedent thrombosis or cardiovascular diseases would confirm if SLE patients with a high
level of plasma PS+ REVs are more at risk of thrombosis. REVs exposing PS+ can recruit
different actors of the coagulation cascade and be a source for the generation of large
quantities of thrombin [10, 40]. REVs are also a source of the von Willebrand factor [41].
SLE patients with a high plasma level of PS+ REVs also show high amounts of plasma ATX.
ATX is the enzyme that produces LPA [42]. The binding of ATX to αIIbβ3 integrin of
activated platelets enhance its catalytic activity [33, 34]. Though we did not monitor the
plasma LPA levels, a role for LPA in RBC activation and production of PS+ REVs cannot be
excluded [24, 43].
Recent studies highlighted a possible role for phosphatidylserine-specific phospholipase A1
in SLE physiopathogenesis [44, 45]. Serum levels of phosphatidylserine-specific
phospholipase A1 are significantly higher in SLE patients with high disease activity. Besides,
patient treatment with immunosuppressive therapies lowered the amount of serum
phosphatidylserine-specific phospholipase A1 [44]. Serum phosphatidylserine-specific
phospholipase A1 and ATX are also higher in patients with lupus nephritis [45]. However,
there was an inverse correlation between the levels of serum ATX and disease activity [45].
Of note, we observed that patients with a normal plasma PS+ REV level tend to have a higher
SLEDAI and lower levels of plasma ATX. The role of ATX and phosphatidylserine-specific
phospholipase A1 in SLE pathophysiology is not known. Increased expression of
phosphatidylserine-specific phospholipase A1 is associated with many pathological
conditions, including autoimmune and cardiovascular diseases [46]. Phosphatidylserine-
specific phospholipase A1 can hydrolyse PS into lysoPS, and ATX can hydrolyze the lysoPS
into LPA [46]. On one side, long-chain lysoPS may contribute to immune cell activation,
including macrophages [47]. On the other side, stimulation of red blood cells with LPA
induces the liberation of REVs [24, 25]. LPA induces the production of REVs by red blood
cells in a concentration- and LPA species-dependent manner and through activation of

95
LPAR3 [48]. Low concentrations of LPA induce of the production of PS- REVs while the
production of PS+ REVs by red blood cells requires concentrations of LPA ≥ 5 µM [48].
Further studies should determine if the SLE patients with high plasma of PS+ REVs and ATX
also show elevated amounts of phosphatidylserine-specific phospholipase A1.
A higher percentage of patients with elevated amounts of plasma PS+ REV presented
abnormal quantities of glycoprotein and cardiolipin auto-antibodies. In SLE patients, higher
disease activity and clinical manifestations such as thrombosis were associated previously
with glycoprotein and cardiolipin antibody levels [49-53]. During our study, a change in the
coding variables occurred for cardiolipin and glycoprotein auto-antibodies. The changes may
have affected the validity of the statistical analysis between the group, especially for auto-
antibody variables.
Atherosclerosis is a lead cause of cardiovascular incidents and is a known comorbidity factor
in SLE [54-58]. PEVs exposing PS were associated with accelerated thickening of the intima-
media in SLE patients [19]. In addition, several EV populations promote the development of
atherosclerosis through the recruitment of immune cells in the vascular wall and the
production of cytokines[59-61]. Besides, through the uptake of EVs, vascular wall infiltrated
macrophages accumulate lipids and transform into foam cells [62-64]. Macrophages
phagocyte the PS+ EVs at a higher rate [65]. Therefore, even if PS+ REVs are not associated
with SLE progression, they still could be implicated in the progression of atherosclerosis and
thrombotic events associated with SLE. We did not dispose of sufficient measurements to
investigate a potential link between the levels of PS+ REVs and the carotid intima-media
thickness test. The overtime impacts of PS+ REVs and other blood cell-derived PS+ EVs on
the thickening of carotid intima-media of SLE patients would be worth investigating in SLE
patients.
In summary, SLE patients show a high number of plasma PEVs and REVs and high surface
exposure of the platelet activation markers PAC1 and CD62P compared to age- and gender-
matched controls. There are no significant differences between the incident and prevalent
cases of SLE. The levels of EVs do not correlate with the SLEDAI score. However, the
analyses of CD62P exposure on the platelet surface show an association with SLEDAI,
consistent with the documented association between this platelet activation marker and

96
disease activity. The plasma PS+ REVs segregate the patients into groups with normal and
high PS+ REV levels, respectively. The analyses show a higher incidence of
thrombocytopenia and thrombotic events in SLE patients with a high level of PS+ REVs.
Further studies are required to determine if the plasma level of PS+ REVs is a potential
biomarker for managing the cardiovascular risk of individuals with SLE.

97
7 References
1. Kaul, A., et al., Systemic lupus erythematosus. Nat Rev Dis Primers, 2016. 2: p.
16039.
2. Yurkovich, M., et al., Overall and cause-specific mortality in patients with systemic
lupus erythematosus: a meta-analysis of observational studies. Arthritis Care Res (Hoboken),
2014. 66(4): p. 608-16.
3. Zhu, H., et al., Autoantigen Microarray for High-throughput Autoantibody Profiling
in Systemic Lupus Erythematosus. Genomics Proteomics Bioinformatics, 2015. 13(4): p.
210-8.
4. Yaniv, G., et al., A volcanic explosion of autoantibodies in systemic lupus
erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun
Rev, 2015. 14(1): p. 75-9.
5. Crow, M.K., M. Olferiev, and K.A. Kirou, Targeting of type I interferon in systemic
autoimmune diseases. Transl Res, 2015. 165(2): p. 296-305.
6. Stetson, D.B., Endogenous retroelements and autoimmune disease. Curr Opin
Immunol, 2012. 24(6): p. 692-7.
7. Lövgren, T., et al., Induction of interferon-alpha production in plasmacytoid dendritic
cells by immune complexes containing nucleic acid released by necrotic or late apoptotic
cells and lupus IgG. Arthritis Rheum, 2004. 50(6): p. 1861-72.
8. Lood, C., et al., Decreased platelet size is associated with platelet activation and anti-
phospholipid syndrome in systemic lupus erythematosus. Rheumatology (Oxford), 2017.
56(3): p. 408-416.
9. Kim, Y., et al., Microparticles from stored red blood cells promote a hypercoagulable
state in a murine model of transfusion. Surgery, 2018. 163(2): p. 423-429.
10. Lipets, E.N., et al., Use of Thrombodynamics for revealing the participation of
platelet, erythrocyte, endothelial, and monocyte microparticles in coagulation activation and
propagation. PLoS One, 2020. 15(5): p. e0227932.
11. Tripisciano, C., et al., Different Potential of Extracellular Vesicles to Support
Thrombin Generation: Contributions of Phosphatidylserine, Tissue Factor, and Cellular
Origin. Sci Rep, 2017. 7(1): p. 6522.
12. Melki, I., et al., Platelets release mitochondrial antigens in systemic lupus
erythematosus. Sci Transl Med, 2021. 13(581).
13. Sellam, J., et al., Increased levels of circulating microparticles in primary Sjögren's
syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease
activity. Arthritis Res Ther, 2009. 11(5): p. R156.
14. López, P., et al., Circulating microparticle subpopulations in systemic lupus
erythematosus are affected by disease activity. Int J Cardiol, 2017. 236: p. 138-144.
15. Salvi, V., et al., Exosome-delivered microRNAs promote IFN-α secretion by human
plasmacytoid DCs via TLR7. JCI Insight, 2018. 3(10).
16. Kato, Y., et al., Apoptosis-derived membrane vesicles drive the cGAS-STING
pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum
Dis, 2018. 77(10): p. 1507-1515.

98
17. Dominguez-Gutierrez, P.R., et al., Positive correlation of STAT1 and miR-146a with
anemia in patients with systemic lupus erythematosus. J Clin Immunol, 2014. 34(2): p. 171-
80.
18. Dong, C., et al., Circulating Exosomes Derived-miR-146a from Systemic Lupus
Erythematosus Patients Regulates Senescence of Mesenchymal Stem Cells. Biomed Res Int,
2019. 2019: p. 6071308.
19. Fortin, P.R., et al., Distinct Subtypes of Microparticle-containing Immune Complexes
Are Associated with Disease Activity, Damage, and Carotid Intima-media Thickness in
Systemic Lupus Erythematosus. J Rheumatol, 2016. 43(11): p. 2019-2025.
20. Pereira, J., et al., Circulating platelet-derived microparticles in systemic lupus
erythematosus. Association with increased thrombin generation and procoagulant state.
Thromb Haemost, 2006. 95(1): p. 94-9.
21. Miyabe, Y., et al., Activation of fibroblast-like synoviocytes derived from rheumatoid
arthritis via lysophosphatidic acid-lysophosphatidic acid receptor 1 cascade. Arthritis Res
Ther, 2014. 16(5): p. 461.
22. Miyabe, Y., et al., Necessity of lysophosphatidic acid receptor 1 for development of
arthritis. Arthritis Rheum, 2013. 65(8): p. 2037-47.
23. Yang, L., et al., LPA receptor 4 deficiency attenuates experimental atherosclerosis. J
Lipid Res, 2019. 60(5): p. 972-980.
24. Nguyen, D.B., et al., Characterization of Microvesicles Released from Human Red
Blood Cells. Cell Physiol Biochem, 2016. 38(3): p. 1085-99.
25. Khandoga, A.L., et al., GPR92/LPA₅ lysophosphatidate receptor mediates
megakaryocytic cell shape change induced by human atherosclerotic plaques. Cardiovasc
Res, 2011. 90(1): p. 157-64.
26. Melki, I., et al., Platelet microvesicles in health and disease. Platelets, 2017. 28(3): p.
214-221.
27. Zecher, D., A. Cumpelik, and J.A. Schifferli, Erythrocyte-derived microvesicles
amplify systemic inflammation by thrombin-dependent activation of complement.
Arterioscler Thromb Vasc Biol, 2014. 34(2): p. 313-20.
28. Fischer, D., et al., Microparticles from stored red blood cells enhance procoagulant
and proinflammatory activity. Transfusion, 2017. 57(11): p. 2701-2711.
29. Hochberg, M.C., Updating the American College of Rheumatology revised criteria
for the classification of systemic lupus erythematosus. Arthritis Rheum, 1997. 40(9): p. 1725.
30. Tan, E.M., et al., The 1982 revised criteria for the classification of systemic lupus
erythematosus. Arthritis Rheum, 1982. 25(11): p. 1271-7.
31. Miyakis, S., et al., International consensus statement on an update of the classification
criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost, 2006. 4(2): p.
295-306.
32. Marcoux, G., et al., Revealing the diversity of extracellular vesicles using high-
dimensional flow cytometry analyses. Sci Rep, 2016. 6: p. 35928.
33. Fulkerson, Z., et al., Binding of autotaxin to integrins localizes lysophosphatidic acid
production to platelets and mammalian cells. J Biol Chem, 2011. 286(40): p. 34654-63.

99
34. Pamuklar, Z., et al., Autotaxin/lysopholipase D and lysophosphatidic acid regulate
murine hemostasis and thrombosis. J Biol Chem, 2009. 284(11): p. 7385-94.
35. Leblanc, R., et al., Interaction of platelet-derived autotaxin with tumor integrin αVβ3
controls metastasis of breast cancer cells to bone. Blood, 2014. 124(20): p. 3141-50.
36. Boilard, E., P. Blanco, and P.A. Nigrovic, Platelets: active players in the pathogenesis
of arthritis and SLE. Nat Rev Rheumatol, 2012. 8(9): p. 534-42.
37. Blair, P. and R. Flaumenhaft, Platelet alpha-granules: basic biology and clinical
correlates. Blood Rev, 2009. 23(4): p. 177-89.
38. Shattil, S.J., et al., Changes in the platelet membrane glycoprotein IIb.IIIa complex
during platelet activation. J Biol Chem, 1985. 260(20): p. 11107-14.
39. Lu, Q. and R.A. Malinauskas, Comparison of two platelet activation markers using
flow cytometry after in vitro shear stress exposure of whole human blood. Artif Organs, 2011.
35(2): p. 137-44.
40. Van Der Meijden, P.E., et al., Platelet- and erythrocyte-derived microparticles trigger
thrombin generation via factor XIIa. J Thromb Haemost, 2012. 10(7): p. 1355-62.
41. Straat, M., et al., Monocyte-mediated activation of endothelial cells occurs only after
binding to extracellular vesicles from red blood cell products, a process mediated by β-
integrin. Transfusion, 2016. 56(12): p. 3012-3020.
42. Tanaka, M., et al., Autotaxin stabilizes blood vessels and is required for embryonic
vasculature by producing lysophosphatidic acid. J Biol Chem, 2006. 281(35): p. 25822-30.
43. Chung, S.M., et al., Lysophosphatidic acid induces thrombogenic activity through
phosphatidylserine exposure and procoagulant microvesicle generation in human
erythrocytes. Arterioscler Thromb Vasc Biol, 2007. 27(2): p. 414-21.
44. Sawada, T., et al., Serum phosphatidylserine-specific phospholipase A(1) as a novel
biomarker for monitoring systemic lupus erythematosus disease activity. Int J Rheum Dis,
2019. 22(11): p. 2059-2066.
45. Iwata, Y., et al., Higher serum levels of autotaxin and phosphatidylserine-specific
phospholipase A(1) in patients with lupus nephritis. Int J Rheum Dis, 2021. 24(2): p. 231-
239.
46. Zhao, Y., S. Hasse, and S.G. Bourgoin, Phosphatidylserine-specific phospholipase
A1: A friend or the devil in disguise. Prog Lipid Res, 2021. 83: p. 101112.
47. Khandelwal, N., et al., Fatty acid chain length drives lysophosphatidylserine-
dependent immunological outputs. Cell Chem Biol, 2021. 28(8): p. 1169-1179.e6.
48. Hasse, S., et al., Interplay between LPA2 and LPA3 in LPA-mediated
phosphatidylserine cell surface exposure and extracellular vesicles release by erythrocytes.
Biochem Pharmacol, 2021. 192: p. 114667.
49. Samarkos, M., et al., Clinical significance of IgA anticardiolipin and anti-beta2-GP1
antibodies in patients with systemic lupus erythematosus and primary antiphospholipid
syndrome. Clin Rheumatol, 2006. 25(2): p. 199-204.
50. Sebastiani, G.D., et al., Anticardiolipin and anti-beta2GPI antibodies in a large series
of European patients with systemic lupus erythematosus. Prevalence and clinical
associations. European Concerted Action on the Immunogenetics of SLE. Scand J
Rheumatol, 1999. 28(6): p. 344-51.

100
51. Danowski, A., T.S. Kickler, and M. Petri, Anti-beta2-glycoprotein I: prevalence,
clinical correlations, and importance of persistent positivity in patients with antiphospholipid
syndrome and systemic lupus erythematosus. J Rheumatol, 2006. 33(9): p. 1775-9.
52. Shen, Y.M., et al., IgA antiphospholipid antibodies are an independent risk factor for
thromboses. Lupus, 2008. 17(11): p. 996-1003.
53. Sweiss, N.J., et al., IgA anti-beta2-glycoprotein I autoantibodies are associated with
an increased risk of thromboembolic events in patients with systemic lupus erythematosus.
PLoS One, 2010. 5(8): p. e12280.
54. Libby, P., et al., Atherosclerosis. Nat Rev Dis Primers, 2019. 5(1): p. 56.
55. Gustafsson, J.T., et al., Risk factors for cardiovascular mortality in patients with
systemic lupus erythematosus, a prospective cohort study. Arthritis Res Ther, 2012. 14(2):
p. R46.
56. Kaplan, M.J., Premature vascular damage in systemic lupus erythematosus.
Autoimmunity, 2009. 42(7): p. 580-6.
57. Urowitz, M.B., D. Ibañez, and D.D. Gladman, Atherosclerotic vascular events in a
single large lupus cohort: prevalence and risk factors. J Rheumatol, 2007. 34(1): p. 70-5.
58. Organization, W.H., Cardiovascular diseases (CVDs) Fact Sheet. 2017. 2017.
59. Fu, Z., et al., Oxidized low-density lipoprotein-induced microparticles promote
endothelial monocyte adhesion via intercellular adhesion molecule 1. Am J Physiol Cell
Physiol, 2017. 313(5): p. C567-c574.
60. Li, C., et al., Endothelial microparticles-mediated transfer of microRNA-19b
promotes atherosclerosis via activating perivascular adipose tissue inflammation in apoE(-/-
) mice. Biochem Biophys Res Commun, 2018. 495(2): p. 1922-1929.
61. Gao, W., et al., Exosomes derived from mature dendritic cells increase endothelial
inflammation and atherosclerosis via membrane TNF-α mediated NF-κB pathway. J Cell
Mol Med, 2016. 20(12): p. 2318-2327.
62. Nguyen, M.A., et al., Extracellular Vesicles Secreted by Atherogenic Macrophages
Transfer MicroRNA to Inhibit Cell Migration. Arterioscler Thromb Vasc Biol, 2018. 38(1):
p. 49-63.
63. Barberio, M.D., et al., Cholesterol efflux alterations in adolescent obesity: role of
adipose-derived extracellular vesical microRNAs. J Transl Med, 2019. 17(1): p. 232.
64. Keyel, P.A., et al., Coordinate stimulation of macrophages by microparticles and TLR
ligands induces foam cell formation. J Immunol, 2012. 189(9): p. 4621-9.
65. Matsumoto, A., et al., Phosphatidylserine-deficient small extracellular vesicle is a
major somatic cell-derived sEV subpopulation in blood. iScience, 2021. 24(8): p. 102839.

101
8 Figures, legends and tables
Table 1: characteristics for SLE patients included in the study at baseline.
Demographics mean±SD or % (n)
Age, years, n=102 49.98 ± 14.58
Gender, Female, n=102 83.33 (85)
BMI, n=101 25.49 ± 4.66
Disease duration, years, n=98 10.63 ± 12.07
Onset, n=102
Incident 28.43 (29)
Prevalent 71.57 (73)
APS, n=102 15.69 (16)
ACR criteria n=99 % (n)
Arthritis 77.45 (79)
Thrombocytopenia 23.53 (24)
Malar rash 25.49 (26)
Discoid rash 17.65 (18)
Hemolytic anemia 3.92 (4)
Renal disorder 24.51 (25)
Medication n=102 % (n)
NSAID/ Cox-II Inhibitors 24.51 (25)
Antipalutic drugs 75.49 (77)
Immunomodulators 84.31 (86)
Methotrexate 15.69 (16)
Biologic agents 6.86 (7)
Steroids 21.57 (22)
Other DMARD 27.45 (28)
Prednisone 20.59 (21)
Clinical characteristics mean±SD or % (n)
SLEDAI, n=99 3.07 ± 3.70
Platelet, 109/L, n=101 227.20 ± 74.91
MPV, fL, n=101 8.92 ± 6.10
Hemoglobin, g/L, n=101 129.34 ± 12.29
CRP, mg/L, n=82 4.47 ± 7.25
ESR, mm/hour, n=94 13.55 ± 16.55
Lupus anticoagulant, n=97 10.78 (11)
Anti-cardiolipine IGG, n=97 equivocal 1.96 (2)
abnormal 14.71 (13)
Anti-cardiolipine IGM, n=97 equivocal 0.98 (1)

102
abnormal 12.74 (12)
Glycoprotein IGG, n=97 abnormal 7.84 (8)
Glycoprotein IGM, n=97 abnormal 14.71 (15)
Cardiovascular damages mean±SD or % (n)
Thrombophilia, n=102 Venous 6.86 (7)
Arterial 3.92 (4)
Microcirculation 0.98 (1)
Plaque, n=72 29.41 (30)
CIMT, n=35 0.62 ± 0.12
ACR American college of rheumatology; APS antiphospholipid syndrome; BMI body mass index; CIMT carotid intima-media thickness; CRP C reactive protein; DMARD Disease Modifying Anti Rheumatic Drug; ESR erythrocyte sedimentation rate; MPV mean platelet volume; NSAID nonsteroidal anti-inflammatory drug; SLEDAI SLE disease activity index.
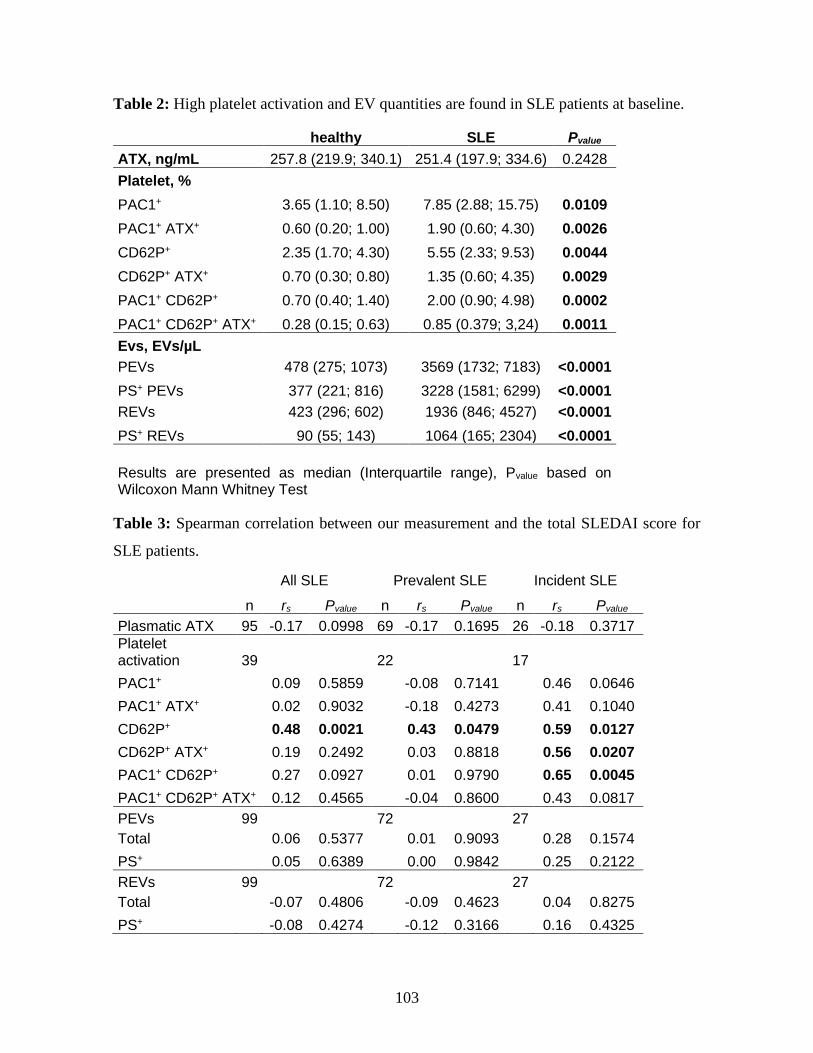
103
Table 2: High platelet activation and EV quantities are found in SLE patients at baseline.
healthy SLE Pvalue
ATX, ng/mL 257.8 (219.9; 340.1) 251.4 (197.9; 334.6) 0.2428
Platelet, %
PAC1+ 3.65 (1.10; 8.50) 7.85 (2.88; 15.75) 0.0109
PAC1+ ATX+ 0.60 (0.20; 1.00) 1.90 (0.60; 4.30) 0.0026
CD62P+ 2.35 (1.70; 4.30) 5.55 (2.33; 9.53) 0.0044
CD62P+ ATX+ 0.70 (0.30; 0.80) 1.35 (0.60; 4.35) 0.0029
PAC1+ CD62P+ 0.70 (0.40; 1.40) 2.00 (0.90; 4.98) 0.0002
PAC1+ CD62P+ ATX+ 0.28 (0.15; 0.63) 0.85 (0.379; 3,24) 0.0011
Evs, EVs/µL
PEVs 478 (275; 1073) 3569 (1732; 7183) <0.0001
PS+ PEVs 377 (221; 816) 3228 (1581; 6299) <0.0001
REVs 423 (296; 602) 1936 (846; 4527) <0.0001
PS+ REVs 90 (55; 143) 1064 (165; 2304) <0.0001
Results are presented as median (Interquartile range), Pvalue based on Wilcoxon Mann Whitney Test
Table 3: Spearman correlation between our measurement and the total SLEDAI score for
SLE patients.
All SLE Prevalent SLE Incident SLE
n rs Pvalue n rs Pvalue n rs Pvalue
Plasmatic ATX 95 -0.17 0.0998 69 -0.17 0.1695 26 -0.18 0.3717
Platelet activation 39 22 17
PAC1+ 0.09 0.5859 -0.08 0.7141 0.46 0.0646
PAC1+ ATX+ 0.02 0.9032 -0.18 0.4273 0.41 0.1040
CD62P+ 0.48 0.0021 0.43 0.0479 0.59 0.0127
CD62P+ ATX+ 0.19 0.2492 0.03 0.8818 0.56 0.0207
PAC1+ CD62P+ 0.27 0.0927 0.01 0.9790 0.65 0.0045
PAC1+ CD62P+ ATX+ 0.12 0.4565 -0.04 0.8600 0.43 0.0817
PEVs 99 72 27
Total 0.06 0.5377 0.01 0.9093 0.28 0.1574
PS+ 0.05 0.6389 0.00 0.9842 0.25 0.2122
REVs 99 72 27
Total -0.07 0.4806 -0.09 0.4623 0.04 0.8275
PS+ -0.08 0.4274 -0.12 0.3166 0.16 0.4325
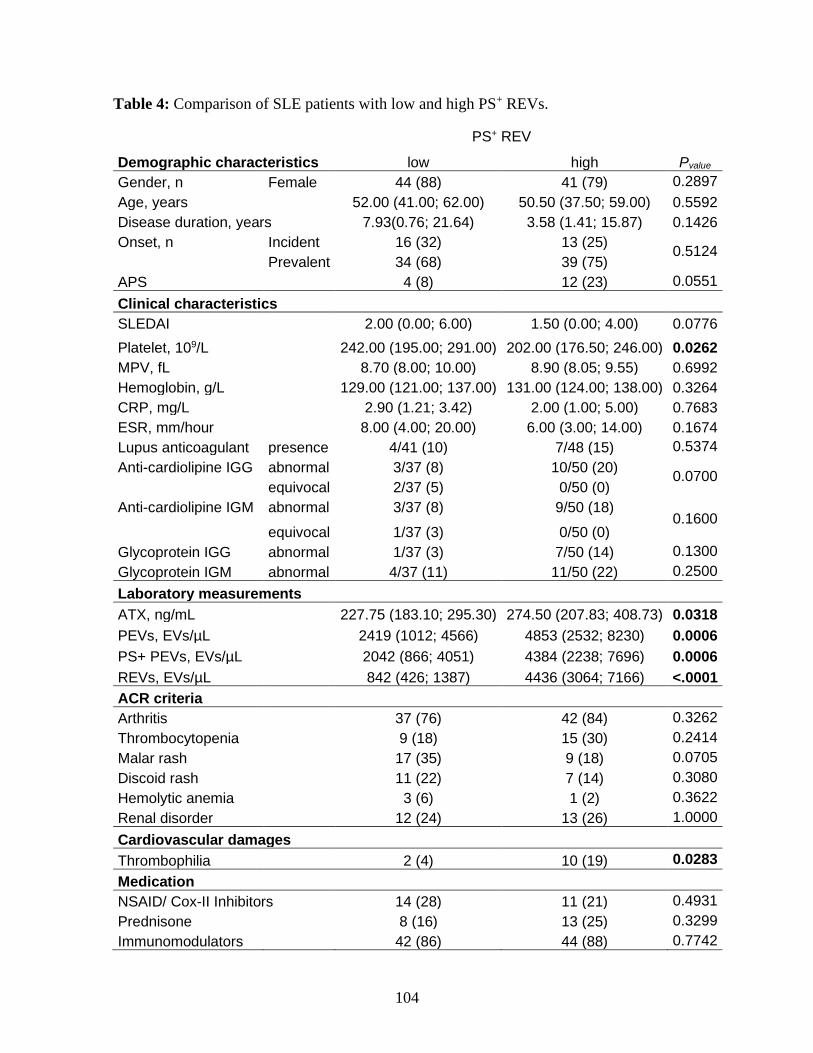
104
Table 4: Comparison of SLE patients with low and high PS+ REVs.
PS+ REV
Demographic characteristics low high Pvalue
Gender, n Female 44 (88) 41 (79) 0.2897
Age, years 52.00 (41.00; 62.00) 50.50 (37.50; 59.00) 0.5592
Disease duration, years 7.93(0.76; 21.64) 3.58 (1.41; 15.87) 0.1426
Onset, n Incident 16 (32) 13 (25) 0.5124
Prevalent 34 (68) 39 (75)
APS 4 (8) 12 (23) 0.0551
Clinical characteristics
SLEDAI 2.00 (0.00; 6.00) 1.50 (0.00; 4.00) 0.0776
Platelet, 109/L 242.00 (195.00; 291.00) 202.00 (176.50; 246.00) 0.0262
MPV, fL 8.70 (8.00; 10.00) 8.90 (8.05; 9.55) 0.6992
Hemoglobin, g/L 129.00 (121.00; 137.00) 131.00 (124.00; 138.00) 0.3264
CRP, mg/L 2.90 (1.21; 3.42) 2.00 (1.00; 5.00) 0.7683
ESR, mm/hour 8.00 (4.00; 20.00) 6.00 (3.00; 14.00) 0.1674
Lupus anticoagulant presence 4/41 (10) 7/48 (15) 0.5374
Anti-cardiolipine IGG abnormal 3/37 (8) 10/50 (20) 0.0700
equivocal 2/37 (5) 0/50 (0)
Anti-cardiolipine IGM abnormal 3/37 (8) 9/50 (18) 0.1600
equivocal 1/37 (3) 0/50 (0)
Glycoprotein IGG abnormal 1/37 (3) 7/50 (14) 0.1300
Glycoprotein IGM abnormal 4/37 (11) 11/50 (22) 0.2500
Laboratory measurements
ATX, ng/mL 227.75 (183.10; 295.30) 274.50 (207.83; 408.73) 0.0318
PEVs, EVs/µL 2419 (1012; 4566) 4853 (2532; 8230) 0.0006
PS+ PEVs, EVs/µL 2042 (866; 4051) 4384 (2238; 7696) 0.0006
REVs, EVs/µL 842 (426; 1387) 4436 (3064; 7166) <.0001
ACR criteria
Arthritis 37 (76) 42 (84) 0.3262
Thrombocytopenia 9 (18) 15 (30) 0.2414
Malar rash 17 (35) 9 (18) 0.0705
Discoid rash 11 (22) 7 (14) 0.3080
Hemolytic anemia 3 (6) 1 (2) 0.3622
Renal disorder 12 (24) 13 (26) 1.0000
Cardiovascular damages
Thrombophilia 2 (4) 10 (19) 0.0283
Medication
NSAID/ Cox-II Inhibitors 14 (28) 11 (21) 0.4931
Prednisone 8 (16) 13 (25) 0.3299
Immunomodulators 42 (86) 44 (88) 0.7742
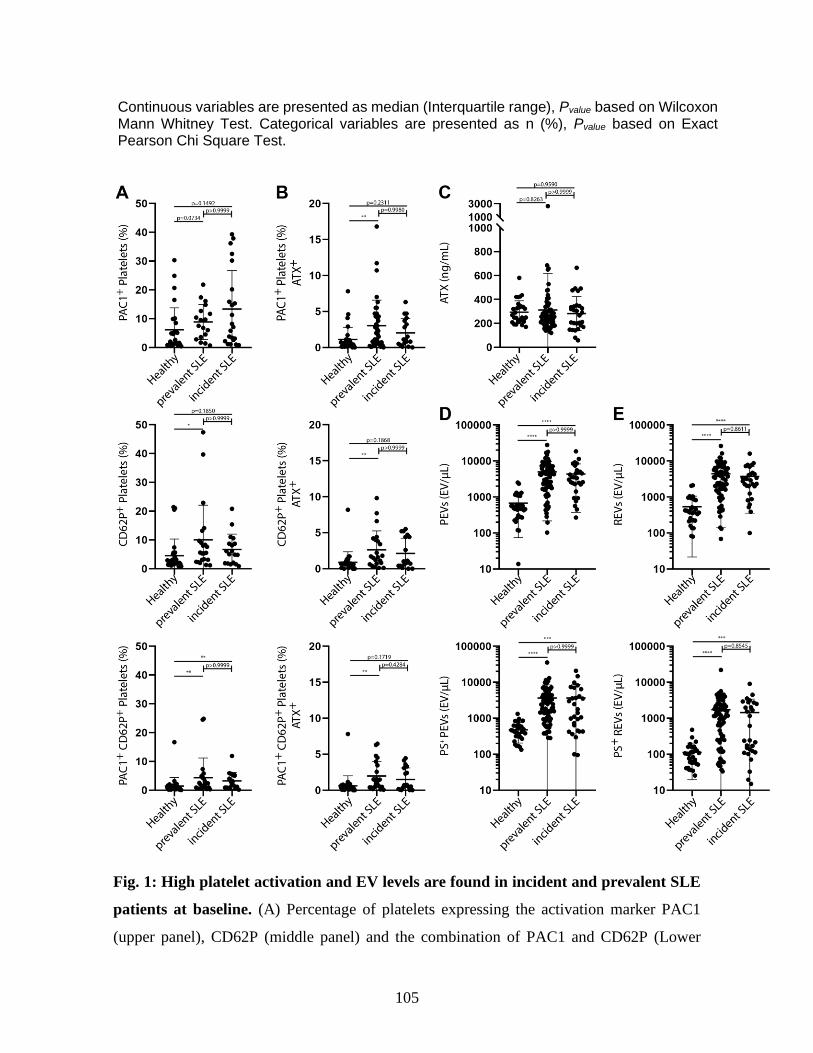
105
Continuous variables are presented as median (Interquartile range), Pvalue based on Wilcoxon Mann Whitney Test. Categorical variables are presented as n (%), Pvalue based on Exact Pearson Chi Square Test.
Fig. 1: High platelet activation and EV levels are found in incident and prevalent SLE
patients at baseline. (A) Percentage of platelets expressing the activation marker PAC1
(upper panel), CD62P (middle panel) and the combination of PAC1 and CD62P (Lower

106
panel) for the healthy (n=30), prevalent (n=22) and incident SLE (n=18) group. (B)
Percentage of platelets expressing ATX and the activation marker PAC1 (upper panel),
CD62P (middle panel) or the combination of PAC1 and CD62P (Lower panel) for the healthy
(n=30), prevalent (n=22) and incident SLE (n=18) group. (C) Plasma concentration of ATX
for the healthy (n=30), prevalent (n=70) and incident SLE (n=28) (D) Number of plasma
PEVs (upper panel) and PS+ PEVs (lower panel) for the healthy (n=30), prevalent (n=73)
and incident SLE (n=29) group. (E) Plasma REV (upper panel) and PS+ REV levels (lower
panel) for the healthy (n=30), prevalent (n=73) and incident SLE (n=29) group. Data are
presented as median with interquartile range, Kruskal-Wallis test with Dunnett post-test,
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
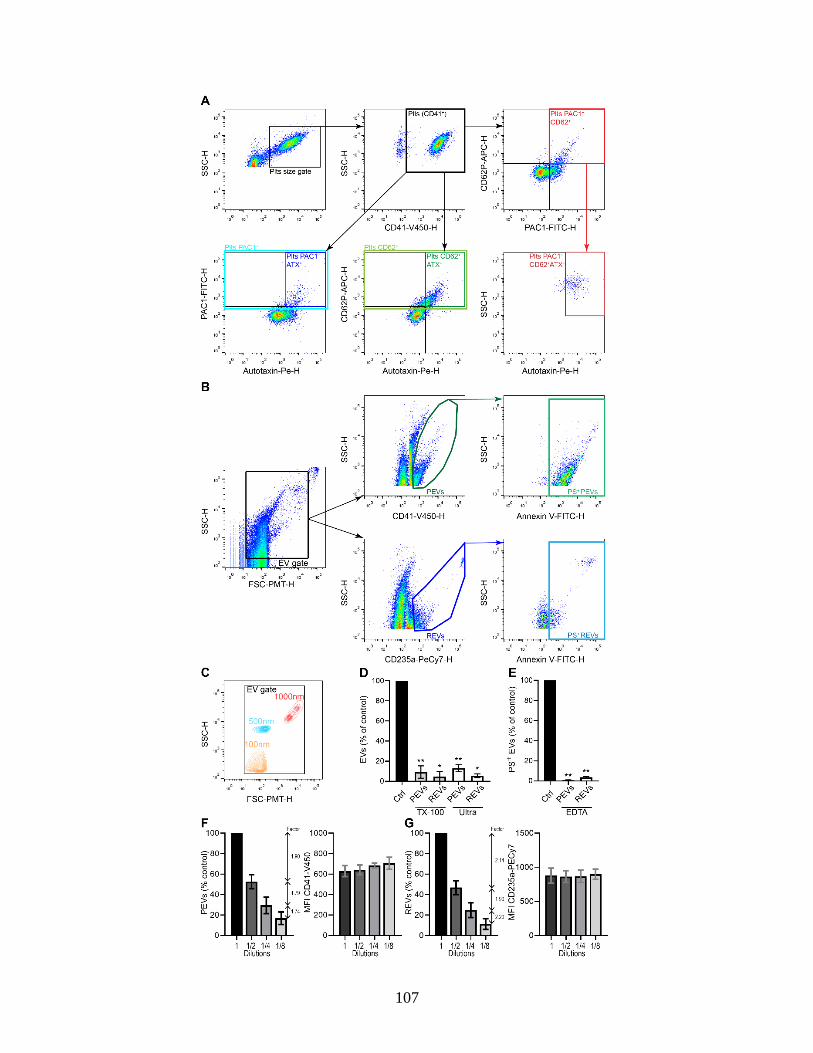
107

108
Supplementary Figure 1: Platelet activation and EV detection by high-sensitivity flow
cytometry. (A) Plasma samples of SLE patients were assessed by high-sensitivity flow
cytometry (BD Canto II Special Order Research Product). Platelet were first gated according
to size (SSC) and granularity (FSC), and then for labeling with V450 fluorochrome-
conjugated antibodies directed against CD41a (CD41-V450-H), a platelet marker. Platelets
where then analyzed for the expression of several activation markers, firstly FITC
fluorochrome-conjugated antibodies directed against PAC1 (PAC1-FITC-H) alone or in
combination with ATX-PE-H, the APC fluorochrome-conjugated antibodies directed against
CD62P (CD62P-FITC-H) alone or in combination with ATX-PE-H, and lastly the
combination of PAC1-FITC-H and CD62P-FITC-H. Platelets positive for both PAC1 and
CD62P activation markers and labeled with PE fluorochrome-conjugated antibodies directed
against ATX (ATX-PE-H) were monitored. (B) PFP samples of SLE patients were assessed
by high-sensitivity flow cytometry (BD Canto II Special Order Research Product). Events
positive for the EV gate set for relative sizes comprised between 100 nm and 1 000 nm on
SSC and FSC-photomultiplier tube (PMT). EVs positive for the expression of CD41-V450-
H were considered PEVs and EVs positive for the expression of PECy7 fluorochrome-
conjugated antibodies directed against CD235a (CD235a-PECy7-H), a marker of RBC, were
considered REVs. Finally, we assessed the fluorescence labelling of PEVs and REVs using
FITC-conjugated Annexin V which binds PS (Annexin V-FTIC-H). (C) The EV gate in SSC-
H (granularity) and FSC-PMT-H (relative size) were set with polystyrene beads of 100 nm,
500 nm, and 1 000 nm. (D) Specificity of PEV and REV detection was validated by the
clearance of PEVs and REVs with 0.05% Triton X-100 (TX-100) treatment (n=6) which
destroys EV’s lipid bilayer and by 100, 000g ultracentrifugation (ultra) which pellets EVs,
(n=3) which are presented as the percentage of the untreated control (Ctrl). Data show the
mean percentage ± SD. Each condition was compared to its untreated control using the paired
t test. (E) Calcium-free PBS supplemented with 50 µM of EDTA (EDTA) was used for
Annexin V labeling of PEVs and REVs to validate the specificity of PS detection at their
surface (n=6) which is presented as the percentage of untreated (Ctrl). Data show the mean
percentage ± SD. Statistical comparisons used the paired t test. (F) Two-fold serial dilutions
of PEV samples were quantified by high sensitivity flow cytometry using polyester counting
beads. PEV concentrations and calculated dilution factors (left panel), and the median (right

109
panel) intensity of fluorescence for each dilution are presented (n=3). Data are presented as
mean ± SD (G) Two-fold serial dilutions of REV samples were quantified by high sensitivity
flow cytometry using polyester counting beads. REV concentrations and calculated dilution
factors (left panel), and the median (right panel) intensity of fluorescence for each dilution
are presented (n=3). Data are presented as mean ± SD.

110
Discussion
1 Mise en contexte
Le LPA est un lipide bioactif avec un rôle important dans la physiologie vasculaire. Le LPA
est un médiateur pro-inflammatoire qui est associé avec la progression de pathologies
inflammatoires comme l’arthrite rhumatoïde, une maladie rhumatismale auto-immune, ou
encore comme l’athérosclérose. Le LPA est notamment associé à l’activation des plaquettes
et à l’activation de la cascade de la coagulation lors de la rupture de plaques
d’athéroscléroses. Outre les plaquettes, le LPA est également connu comme le seul activateur
endogène des GR.
L’activation de cellules induit la libération d’EV. Les EV sont de petites vésicules
membranaires libérées dans le milieu extracellulaire par les cellules. Elles sont notamment
impliquées dans la communication intercellulaire et dans la régulation de l’environnement
vasculaire avec des effets pro-inflammatoires et sur le processus de la coagulation.
Les travaux présentés dans cette thèse ont examiné la modulation de l’activité des GR
par le LPA et leur implication possible dans la promotion des dommages vasculaires
associés aux MRAS.
2 Impact des limitations techniques dans l’analyse des vésicules
extracellulaires
La recherche sur les EV est un champ d’investigation récent qui ne présente pas encore
d’approches standardisées que ce soit dans la nomenclature, l’isolation, la caractérisation,
l’entreposage ou encore l’analyse des EV. Bien que les recommandations de l’ISEV
permettent d’avoir de plus en plus d’études qui respectent des normes communes, il reste
encore de nombreuses considérations méthodologiques et expérimentales à résoudre427,471,721.
Ma thèse étant centrée sur l’analyse d’EV par cytométrie en flux à haute sensibilité, je me
suis heurté à plusieurs limitations et problèmes posés par cette approche. Je vais revenir sur
les plus significatifs.

111
L’analyse d’EV dans des échantillons obtenus sur de grandes périodes, lors d’études
longitudinales par exemple, se heurte à plusieurs limitations. L’évolution de la sensibilité de
l’appareil lors d’un bris peut modifier grandement les limites de détection de l’appareil. Cela
peut rendre difficile, voire impossible, l’inclusion de mesure prise avant et après le bris dans
une même étude, et cela même avec l’utilisation d’un contrôle interne et une quantification
absolue. Même sans bris, la puissance des lasers diminue au fur et à mesure de leur utilisation.
Cette diminution réduit progressivement la fluorescence détectée pour un même réglage. Si
cela n’est pas vérifié et pris en compte régulièrement, il y a un risque de perdre les populations
de EV qui sont proches de la limite de détection.
De plus, le comportement des lots d’anticorps peut également varier au cours de l’étude
même pour des cibles bien établies. Cela peut donc conduire à l’abandon de marqueur
d’intérêt en cours d’étude à cause d’une perte de sensibilité de l’anticorps pour sa cible sur
les EV ou par l’apparition d’agrégats d’anticorps détectés par le cytomètre en flux à haute
sensibilité qui parasitent la détection des EV ciblées.
Ces soucis sont apparus au cours de mes travaux de thèse. Dans une première situation,
l’utilisation de tubes d’un même lot, mais commandés à des temps différents sur un même
échantillon produisait des résultats différents bien que l’analyse soit effectuée en parallèle
avec le même protocole. Cependant, le même test sur des cellules conduisait à des résultats
similaires. Dans la deuxième situation, un changement de lot d’anticorps a résulté dans
l’apparition d’agrégats non spécifiques qui se superposaient avec nos populations d’intérêt.
Le test des anticorps sur des populations d’EV de grande taille et sur des cellules a montré
une même spécificité de l’anticorps par rapport au lot précédent. Dans les deux cas, les
compagnies nous ont communiqué qu’elles n’avaient pas modifié le protocole de production
des anticorps et que la validation des anticorps avait été faite sur des cellules. L’absence de
validation des anticorps sur des EV pose plusieurs problèmes. D’une part, cela augmente la
variabilité des mesures déjà importante avec cette approche et d’autre part limite la
reproduction de résultats et donc limite leur validation722,723.
Réaliser les mesures une fois sur l’ensemble des échantillons dans un laps de temps limité
permet de réduire l’impact de l’évolution du matériel et des anticorps utilisés. Cependant, si
le projet requiert l’obtention d’échantillon sur de grandes périodes comme cela était notre

112
cas, il est nécessaire d’entreposer les échantillons à analyser ce qui peut avoir un impact sur
les résultats. En effet, la préparation et l’entreposage des EV peuvent induire la perte des EV
les plus fragiles ou de forte densité ainsi que modifier leur membrane724,725. Il en résulte qu’en
fonction de la solution d’entreposage des EV et des cycles de congélation / décongélation, le
contenu en EV de l’échantillon peut augmenter ou diminuer427,724,726. De plus au cours du
temps, malgré des conditions adéquates d’entreposage la quantité d’EV diminue dans les
échantillons727. Enfin, l’effet de l’entreposage n’affecte pas les populations d’EV de manière
homogène728.
La modification des populations d’EV peut mener à des analyses biaisées. Les
problématiques liées à l’entreposage des EV sont également importantes lorsque les
échantillons sont analysés au fur et à mesure du projet, mais peuvent être limitées notamment
par l’analyse des EV directement dans les échantillons frais, soit sans entreposage. Plusieurs
études conseillent cette approche pour les analyses dans les liquides biologiques427,724.
3 Résumé des travaux et discussion
3.1 L’acide lysophosphatidique et les vésicules extracellulaires de globules
rouges
Bien qu’il soit déjà connu, que le LPA active les GR par l’induction de la présentation de PS
à leur surface ainsi que par la libération d’EV positives pour la PS, les mécanismes ont été
peu étudiés575,576. L’externalisation de la PS par les GR en réponse au LPA est associée à la
mobilisation de calcium intracellulaire et à l’activation de la PKC. Les deux protéines G,
Gαq/11, et Gαi/o, peuvent être associées aux récepteurs au LPA et sont capables d’activer la
PKC. Par ailleurs, aucune étude n’avait étudié la possible hétérogénéité d’effet des
différentes espèces moléculaires de LPA sur l’activation des GR. Au vu de la littérature
limitée sur l’activation des GR par le LPA, nous avons voulu évaluer l’impact des espèces
moléculaires de LPA sur les GR, en particulier celles qui sont les plus abondantes dans la
circulation. Nous avons donc privilégié quatre espèces de LPA qui d’une part représentaient
la diversité des espèces moléculaires du LPA (saturé, mono-insaturé et poly-insaturé).
D’autre part, ces espèces étaient des formes majeures dans le plasma d’individu sain, mais
étaient produites lors de l’activation plaquettaire et lors de la préparation de sérum14,47,118,119.

113
Nous avons montré que l’activation des GR par le LPA varie en fonction de l’espèce
moléculaire avec notamment le LPA 20:4 qui ne présente aucun effet activateur. Et
contrairement aux études précédentes, nous avons montré que l’activation des GR par le LPA
ne débouche pas uniquement sur la libération de REV PS+, mais également sur la libération
de plus petites REV négatives pour la PS575,576. Cela s’explique en partie par la sensibilité de
la technique que nous avons utilisée qui permet de détecter des EV de petites tailles
(~ 100 nm). De plus, nous avons identifié un nouvel effet du LPA, il peut aussi inhiber
l’activation des GR. Enfin, nous avons été les premiers à associer l’effet du LPA sur les GR
matures à l’activation des récepteurs LPA2 et LPA3.
Notre étude de l’effet des espèces moléculaires de LPA sur les GR s’est heurtée à plusieurs
limitations. L’expression des récepteurs au LPA, a été mise en évidence dans les progéniteurs
myéloïdes communs729 et certains comme le LPA2 et les LPA3 sont impliqués dans la
régulation de l’érythropoïèse260,261,297. Cependant, leur présence chez les GR matures reste
encore inconnue. Des travaux antérieurs dans le laboratoire sur différentes cellules connues
pour exprimer les récepteurs au LPA ont montré que les anticorps disponibles contre les
récepteurs aux LPA ne permettaient pas la détection par immuno-buvardage de leur
expression basale. De plus, bien que les GR matures présentent des ARN messagers, la
maturation des globules rouges après l’énucléation présente une forte activité de dégradation
de l’ARN547,551,564 et l’hémoglobine contenue par les GR altère l’efficacité de la transcription
et de l’amplification des ARN par la technique de PCR730,731. Ces deux raisons pourraient
expliquer pourquoi nous n’avons pas pu détecter d’ARN messager pour les récepteurs au
LPA par des approches de RT-PCR. Il en résulte que nous n’avons pas pu valider la présence
des récepteurs au LPA autrement qu’avec des approches fonctionnelles soit par l’utilisation
d’agonistes et d’antagonistes. La disponibilité d’agonistes et d’antagonistes sélectifs pour les
récepteurs au LPA a limité le nombre de récepteurs que nous avons pu étudier. Il serait donc
intéressant d’approfondir les mécanismes d’action du LPA sur les GR à partir de modèles
murins ou par la différenciation in vitro de progéniteurs de GR. Ces modèles permettraient
notamment de supprimer ou de moduler l’expression de manière certaine des récepteurs au
LPA à des étapes précoces de l’érythropoïèse et ainsi s’assurer de leur absence sur les GR
matures.

114
Bien que nous ayons montré que l’activation des GR par le LPA est possible dans le plasma,
il n’y a pas encore d’estimations fiables des concentrations de LPA présente en circulation.
En effet, les études chez des sujets sains font état de concentrations allant de 50 nM à 1 µM
et jusqu’à 12 µM dans des situations pathologiques14,124,125,127,732. Il n’existe pas de protocole
standardisé pour le dosage du LPA plasmatique et il peut être synthétisé ou dégradé lors de
la manipulation d’échantillon sanguin. Les techniques d’extractions du LPA pour également
produire du LPA de façon artéfactuelle. Cela explique la grande variabilité entre les études
et empêche une estimation précise des concentrations du LPA plasmatique. De plus, la demi-
vie du LPA est courte et sa production peut être faite proche de son site d’action suite à la
liaison de l’autotaxine aux intégrines ou aux héparanes sulfates présents à la surface des
cellules. La concentration locale de LPA pourrait donc fortement varier de celle mesurée
dans la circulation générale, mais avoir un plus grand impact dans l’activation des cellules
vasculaires.
Nous avons aussi montré que les différentes espèces moléculaires de LPA n’ont pas toutes le
même potentiel activateur avec certaines qui montrent uniquement un effet inhibiteur pour
des concentrations physiologiques. L’activation des GR par la concentration de LPA
plasmatique pourrait donc dépendre de l’équilibre entre les espèces activatrices et inhibitrices
et pas seulement de la concentration totale de LPA.
De plus, nous avons montré que l’environnement module l’activation dépendante du LPA. Il
reste donc difficile à évaluer si l’activation par le LPA des GR est possible et est impliquée
dans des situations physiologiques ou pathologiques. Même si l’activation des GR n’a été
montrée que pour le LPA, ceux-ci peuvent être activés par d’autres voies signalétiques. En
effet, les GR expriment notamment le TLR9 qui peuvent lier de l’ADN mitochondrial596.
L’activation du TLR9 pourrait constituer un autre signal activateur que le LPA. Il serait donc
aussi intéressant de voir l’impact d’autres signaux activateurs des GR sur l’activation
dépendante du LPA. Stimuler les GR avec du PFP issu de patients souffrant de différentes
pathologies permettrait d’étudier plus en détail la potentiel implication du LPA plasmatique
dans les processus pathologiques.
Enfin, selon mes observations, le LPA induit deux populations de REV. Les REV de grande
taille PS+ ont déjà été associées dans des études antérieures à la coagulation et à

115
l’inflammation441,614. Cependant, l’études fonctionnelles des REV utilisent des REV
provenant de l’entreposage de GR441,613,614. Le stimulus à l’origine de la production des EV
est connu pour moduler leur composition et leur fonction429,468,532. Il serait donc important de
valider les effets biologiques des REV PS+ et PS- induit par le LPA dans l’inflammation et la
coagulation. De plus, les EV PS+ et PS- ont des demi-vies et une bio-distributions
différentes733. Les REV PS+ et les REV PS- pourraient donc jouer des rôles différents dans
l’environnement vasculaire.
3.2 Les vésicules extracellulaires de globules rouges dans le lupus
érythémateux disséminée
Contrairement à notre hypothèse, les quantités plasmatiques d’autotaxine chez les patients
LED étaient similaires à celles des individus sains, et celles-ci n’étaient pas associées avec
l’activité de la maladie. De plus, les quantités de PEV et de REV des patients LED étaient
élevées bien que non associées à l’activité de la maladie. Bien que les études s’accordent sur
l’augmentation des PEV chez les patients LED, leur association avec le SLEDAI est encore
débattue734,735. Nos résultats corroborent donc la publication qui n’associe pas les PEV avec
l’activité de la maladie735. Les quantités de REV PS+ nous ont permis de stratifier la cohorte
de patient LED en deux groupes, celui avec des quantités similaires aux individus sains et
l’autre qui présente des quantités élevées de EV plasmatique. Les patients LED avec des
quantités élevées de REV PS+ sont plus à risque d’avoir un historique de thrombose.
Les REV participent à la coagulation notamment par l’apport de thrombine et du facteur de
von Willebrand441,613,615,616. Ils sont aussi capables d’initier la coagulation dans certaines
conditions expérimentales441. Le recrutement des médiateurs de la cascade de coagulation
qui conduit à la production de thrombine par les REV est favorisé par la présence de la PS+
à leur surface440,441. Les quantités élevées de REV PS+ pourraient donc faciliter la coagulation
chez les patients LED et conférer une susceptibilité au développement de thromboses.
D’autre part, les EV sont associées à plusieurs étapes du développement de l’athérosclérose
qui la première cause des évènements thrombotiques677-679, notamment par l’apport de lipide
aux macrophages qui se transforment en cellules spumeuses710-713. En effet, les EV qui
présentent la PS sont plus sujets à leur internalisation par les macrophages733. Des quantités

116
importantes de REV PS+ pourraient donc également stimuler le développement de
l’athérosclérose chez les patients LED par leur phagocytose par les macrophages.
Les patients LED qui ont des quantités élevées de REV PS+ présentent des concentrations
d’autotaxine plus élevées que les patients avec des quantités faibles de REV PS+. Les
quantités d’autotaxine pourraient ainsi avoir un impact sur les concentrations de LPA
plasmatiques et avoir des effets sur les cellules sanguines. L’activation des GR par le LPA
est une source potentielle de REV PS+. De plus, le LPA stimule la progression de plusieurs
étapes de l’athérosclérose qui est la cause majeure des évènements thrombotiques677-679. En
effet, le LPA stimule l’infiltration et l’activation des macrophages dans la paroi vasculaire et
leur différenciation en cellules de spumeuses par accumulation de lipides309,395,701. Il en
résulte un environnement pro-inflammatoire qui peut attirer d’autres cellules incluant des
neutrophiles. De plus lors de la rupture des plaques, le LPA est un des signaux d’activation
des plaquettes et promeut la coagulation et la formation d’un caillot thrombotique34,112,113. Ce
serait en accord avec notre observation que les patients avec des quantités élevées de REV
PS+ présentent également des concentrations de plaquettes circulantes plus faibles que le
groupe avec des quantités normales de REV PS+. Donc même si l’autotaxine ne semble pas
associée au développement du LED, elle pourrait tout de même être impliquée dans le
développement de l’athérosclérose et dans la formation de thromboses associées avec le
LED.
Les quantités élevées de REV PS+ pourraient également servir de source locale de
phospholipides pour la production de lyso-PS par des phospholipases capables d’utiliser les
phospholipides présents sur la membrane d’EV45,51. Cela est supporté par une étude récente
qui a fait état de quantité plus élevé de la PLA1 spécifique de la PS dans le sérum de patients
LED et que les quantités de la PLA1 spécifique de la PS étaient associées avec le SLEDAI736.
Les lyso-PS pourraient ensuite servir de précurseur pour la production de LPA par
l’autotaxine mais pourraient également présenter une activité biologique qui leur est propre,
notamment par l’intermédiaire de ses trois récepteurs couplés aux protéines G, P2Y10,
GPR34, et GPR174136,737. Ces récepteurs sont exprimés par les cellules hématopoïétiques. Le
lyso-PS à des effets répresseurs sur l’inflammation en inhibant la prolifération des
lymphocytes T738,739. Cependant il peut également promouvoir l’inflammation par

117
l’inhibition de la différenciation des lymphocytes T en lymphocytes régulateurs738,
l’activation du TLR2737,740, la libération d’histamine par les mastocytes, ainsi qu’en facilitant
la phagocytose et la libération de cytokines pro-inflammatoires par les macrophages737,741,742.
Les REV PS+ pourraient potentiellement avoir un rôle pro-inflammatoire et promouvoir
l’athérosclérose en étant une source de phospholipides pour la production de LPA et de lyso-
PS. En effet, l’inflammation pourrait augmenter la perméabilité vasculaire et facilité
l’extravasation des EV dans les tissus vasculaires.
Bien que notre étude n’ait pas montré d’augmentation des quantités plasmatiques
d’autotaxine, une augmentation de l’autotaxine sérique a déjà été rapportée pour des patients
LED avec des atteintes aux reins (néphrite lupique)743. L’autotaxine est sécrétée dans le
milieu extracellulaire et peut être trouvée sous forme libre, mais également associée à
différents transporteurs comme les plaquettes activées et les lipoprotéines. Les patients LED
présentent également une proportion de plaquettes activées et de plaquettes activées positives
pour l’autotaxine plus importante que les contrôles sains. Nos mesures ont été faites sur du
plasma sans plaquette et donc ne prend pas en considération l’autotaxine associée aux
plaquettes. Nos mesures pourraient donc sous-estimer les quantités d’autotaxine présentes
dans la circulation.
4 Perspectives
Les travaux de cette thèse ont souligné la complexité de la signalisation du LPA qui permet
des effets différents sur une même cellule en fonction de l’espèce moléculaire utilisée. De
plus, les différentes espèces moléculaires de LPA ont des concentrations différentes dans le
plasma et ne fluctuent pas de manière homogène, par exemple lors de l’activation des
plaquettes14,25,127. Cependant, l’étude des effets du LPA est encore souvent réalisé avec une
seule espèce moléculaire, dont l'idendité n'est pas toujours précisée 112,144,213,575. Il serait donc
important d’étudier l’effet de chaque espèce moléculaire majeure de LPA vasculaire sur les
cellules vasculaires comme les plaquettes ou les cellules endothéliales ainsi que dans les
fonctions vasculaires que ce soit l’inflammation ou la coagulation. Ces études seraient
bénéfiques pour une meilleure compréhension du rôle du LPA en situation pathologique.

118
Nous avons établi que certaines espèces moléculaires de LPA induisaient la libération de
deux populations de REV, les PS- et PS+. Il serait important d’une part de vérifié si elles sont
également pro-coagulantes et pro-inflammatoire à l’instar des REV issues de l’entreposage
de GR576,613,614,617,618. D’autre part, le LPA et les EV sont associées au développement de
l’athérosclérose et il est connu que les REV sont retrouvées dans les plaques 705. Il serait donc
pertinent d’évaluer les effets des REV induit par le LPA sur les cellules et fonctions
impliquées dans la progression des plaques d’athérosclérose comme les cellules
endothéliales, les cellules musculaires lisses ou les macrophages.
Nous avons observé que le groupe de patients LED avec des quantités élevées de REV PS+
avait une incidence de thrombose plus élevée. La construction de notre étude ne permet pas
de définir si les quantités importantes REV PS+ puissent être un facteur de risque ou
simplement une conséquence de la thrombose. Il serait donc nécessaire de compléter ce
travail avec une étude longitudinale pour définir si les quantités importantes de REV PS+ sont
un facteur de risque du développement de thromboses. Ce type d’étude sur plusieurs années
ou décennies nécessiterait une large cohorte de patients pour assurer qu’à son terme, le
nombre de patients suivis soit suffisant pour chaque groupe et qu’il y ait un nombre suffisant
de cas de thrombose pour tirer une conclusion.
Sachant que les REV sont impliquées dans la coagulation et peuvent induire un état d’hyper
coagulabilité dans des modèles murins de transfusion441,613,614, il serait à propos d’examiner
si de grandes quantités de REV PS+ dans le plasma de patients LED facilitent le
déclenchement de la coagulation par rapport à des plasmas de patients LED avec peu de REV
PS+ ou issus de donneurs sains. D’autre part, il serait intéressant d’établir si cette association,
entre la thrombose et les REV PS+, est retrouvée dans d’autres MRAS, notamment dans la
PAR. Également, les PEV ont déjà été associées à l’épaississement de la paroi vasculaire qui
est une conséquence de l’athérosclérose674. Puisque l’athérosclérose est une cause majeure
de thrombose, il serait pertinent d’évaluer une potentielle association entre les REV PS+ avec
l’épaississement de la paroi vasculaire chez les patients LED.
Enfin, les patients LED avec des quantités importantes de REV PS+ présentaient aussi des
concentrations d’autotaxine plasmatique plus forte que les patients LED avec de faibles
quantités de REV PS+. Il serait pertinent de mesurer les quantités de LPA présentes pour

119
chaque groupe. Cela permettrait d’une part d’évaluer si l’augmentation des concentrations
d’autotaxine se reflètent bien par une augmentation de LPA. Et d’autre part, cela permettrait
de déterminer les espèces moléculaires de LPA et d’étudier une potentielle association de ces
dernières avec les quantités de REV.
Conclusion
Les travaux présentés dans cette thèse ont permis la description des effets des espèces
vasculaires majeures de LPA sur l’activation des GR. Ces travaux ont mis en évidence des
effets activateurs et inhibiteurs du LPA sur l’exposition de PS par les GR et la production de
REV. Enfin, ces travaux ont permis, à notre connaissance, la première association d’une
population de REV, les PS+, à une situation pathologique qu’est la thrombose. Les
contributions de cette thèse sont soulignées dans le schéma récapitulatif (Figure 10).
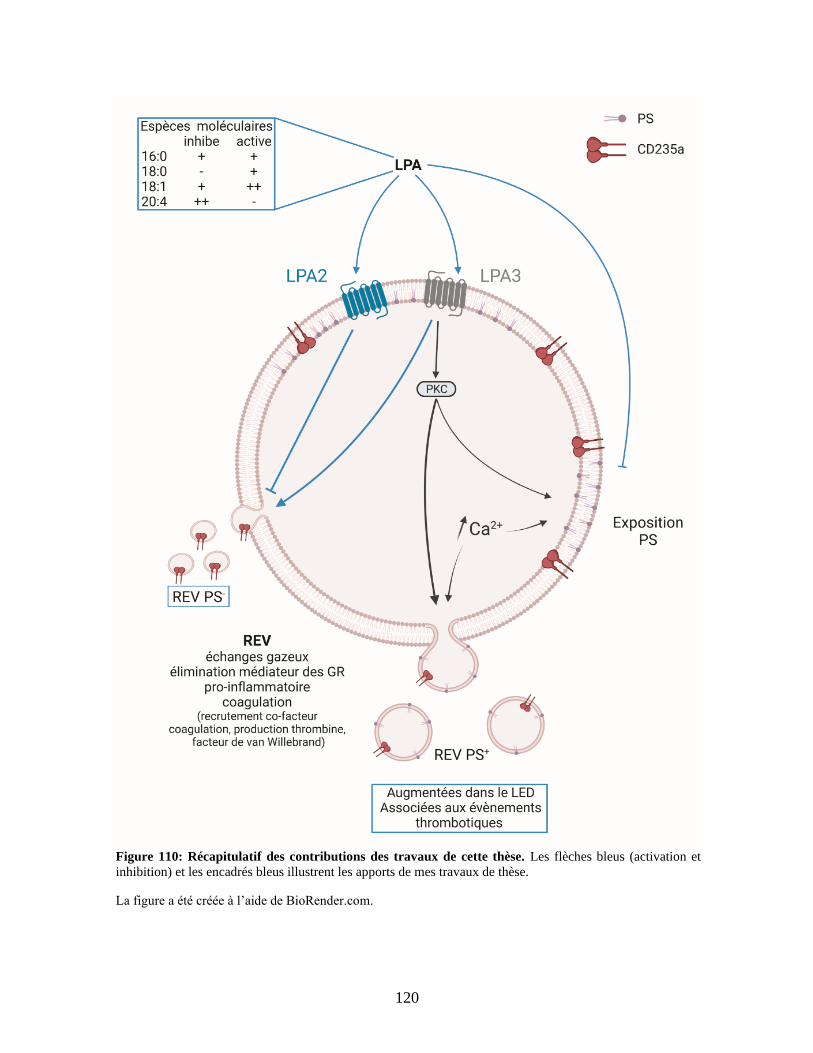
120
Figure 110: Récapitulatif des contributions des travaux de cette thèse. Les flèches bleus (activation et
inhibition) et les encadrés bleus illustrent les apports de mes travaux de thèse.
La figure a été créée à l’aide de BioRender.com.

121
Bibliographie
1. Vogt W. Pharamacologically active acidic phospholipids and glycolipids. Biochem
Pharmacol. 1963;12:415-420.
2. Arneil GC, Dekanski JB. Excess of vasopressor activity in plasma of nephritic
children with hypertension. Lancet. 1954;267(6850):1204-1207.
3. Vogt W. Pharmacologically active lipidsoluble acids of natural occurrence. Nature.
1957;179(4554):300-304; passim.
4. Khairallah PA, Page IH. A vasopressor lipid in incubated plasma. Am J Physiol.
1960;199:341-345.
5. Schumacher KA, Classen HG, Spath M. Platelet aggregation evoked in vitro and in
vivo by phosphatidic acids and lysoderivatives: identity with substances in aged serum
(DAS). Thromb Haemost. 1979;42(2):631-640.
6. Tokumura A, Fukuzawa K, Tsukatani H. Effects of synthetic and natural
lysophosphatidic acids on the arterial blood pressure of different animal species. Lipids.
1978;13(8):572-574.
7. Hecht JH, Weiner JA, Post SR, Chun J. Ventricular zone gene-1 (vzg-1) encodes a
lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral
cortex. J Cell Biol. 1996;135(4):1071-1083.
8. Bandoh K, Aoki J, Taira A, Tsujimoto M, Arai H, Inoue K. Lysophosphatidic acid
(LPA) receptors of the EDG family are differentially activated by LPA species. Structure-
activity relationship of cloned LPA receptors. FEBS Lett. 2000;478(1-2):159-165.
9. Pluckthun A, Dennis EA. Acyl and phosphoryl migration in lysophospholipids:
importance in phospholipid synthesis and phospholipase specificity. Biochemistry.
1982;21(8):1743-1750.
10. Tokumura A, Sinomiya J, Kishimoto S, et al. Human platelets respond differentially
to lysophosphatidic acids having a highly unsaturated fatty acyl group and alkyl ether-linked
lysophosphatidic acids. Biochem J. 2002;365(Pt 3):617-628.
11. Triebl A, Trotzmuller M, Eberl A, Hanel P, Hartler J, Kofeler HC. Quantitation of
phosphatidic acid and lysophosphatidic acid molecular species using hydrophilic interaction
liquid chromatography coupled to electrospray ionization high resolution mass spectrometry.
J Chromatogr A. 2014;1347:104-110.
12. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and
purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497-509.
13. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J
Biochem Physiol. 1959;37(8):911-917.
14. Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi GJ. Direct quantitative analysis
of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization
liquid chromatography-mass spectrometry. Anal Biochem. 2001;292(2):287-295.
15. Scherer M, Schmitz G, Liebisch G. Simultaneous quantification of cardiolipin,
bis(monoacylglycero)phosphate and their precursors by hydrophilic interaction LC-MS/MS
including correction of isotopic overlap. Anal Chem. 2010;82(21):8794-8799.

122
16. Peterson BL, Cummings BS. A review of chromatographic methods for the
assessment of phospholipids in biological samples. Biomed Chromatogr. 2006;20(3):227-
243.
17. Khoury S, Canlet C, Lacroix MZ, Berdeaux O, Jouhet J, Bertrand-Michel J.
Quantification of Lipids: Model, Reality, and Compromise. Biomolecules. 2018;8(4).
18. Fuchs B, Suss R, Teuber K, Eibisch M, Schiller J. Lipid analysis by thin-layer
chromatography--a review of the current state. J Chromatogr A. 2011;1218(19):2754-2774.
19. Xiao YJ, Schwartz B, Washington M, et al. Electrospray ionization mass
spectrometry analysis of lysophospholipids in human ascitic fluids: comparison of the
lysophospholipid contents in malignant vs nonmalignant ascitic fluids. Anal Biochem.
2001;290(2):302-313.
20. Sugiura T, Nakane S, Kishimoto S, et al. Occurrence of lysophosphatidic acid and its
alkyl ether-linked analog in rat brain and comparison of their biological activities toward
cultured neural cells. Biochim Biophys Acta. 1999;1440(2-3):194-204.
21. Liliom K, Guan Z, Tseng JL, Desiderio DM, Tigyi G, Watsky MA. Growth factor-
like phospholipids generated after corneal injury. Am J Physiol. 1998;274(4):C1065-1074.
22. Das AK, Hajra AK. Quantification, characterization and fatty acid composition of
lysophosphatidic acid in different rat tissues. Lipids. 1989;24(4):329-333.
23. Thumser AE, Voysey JE, Wilton DC. The binding of lysophospholipids to rat liver
fatty acid-binding protein and albumin. Biochem J. 1994;301 ( Pt 3):801-806.
24. Gellett AM, Kharel Y, Sunkara M, Morris AJ, Lynch KR. Biosynthesis of alkyl
lysophosphatidic acid by diacylglycerol kinases. Biochem Biophys Res Commun.
2012;422(4):758-763.
25. Bolen AL, Naren AP, Yarlagadda S, et al. The phospholipase A1 activity of
lysophospholipase A-I links platelet activation to LPA production during blood coagulation.
J Lipid Res. 2011;52(5):958-970.
26. Watson SP, McConnell RT, Lapetina EG. Decanoyl lysophosphatidic acid induces
platelet aggregation through an extracellular action. Evidence against a second messenger
role for lysophosphatidic acid. Biochem J. 1985;232(1):61-66.
27. Baker RR, Chang HY. Lysophosphatidic acid, alkylglycerophosphate and
alkylacetylglycerophosphate increase the neuronal nuclear acetylation of 1-acyl
lysophosphatidyl choline by inhibition of lysophospholipase. Mol Cell Biochem. 1999;198(1-
2):47-55.
28. Das AK, Horie S, Hajra AK. Biosynthesis of glycerolipid precursors in rat liver
peroxisomes and their transport and conversion to phosphatidate in the endoplasmic
reticulum. J Biol Chem. 1992;267(14):9724-9730.
29. Haldar D, Lipfert L. Export of mitochondrially synthesized lysophosphatidic acid. J
Biol Chem. 1990;265(19):11014-11016.
30. McIntyre TM, Pontsler AV, Silva AR, et al. Identification of an intracellular receptor
for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl
Acad Sci U S A. 2003;100(1):131-136.
31. Stapleton CM, Mashek DG, Wang S, et al. Lysophosphatidic acid activates
peroxisome proliferator activated receptor-gamma in CHO cells that over-express glycerol
3-phosphate acyltransferase-1. PLoS One. 2011;6(4):e18932.
32. Marchan R, Buttner B, Lambert J, et al. Glycerol-3-phosphate Acyltransferase 1
Promotes Tumor Cell Migration and Poor Survival in Ovarian Carcinoma. Cancer Res.
2017;77(17):4589-4601.

123
33. Simon MF, Daviaud D, Pradere JP, et al. Lysophosphatidic acid inhibits adipocyte
differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of
peroxisome proliferator-activated receptor gamma2. J Biol Chem. 2005;280(15):14656-
14662.
34. Siess W, Zangl KJ, Essler M, et al. Lysophosphatidic acid mediates the rapid
activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and
accumulates in human atherosclerotic lesions. Proc Natl Acad Sci U S A. 1999;96(12):6931-
6936.
35. Zhang C, Baker DL, Yasuda S, et al. Lysophosphatidic acid induces neointima
formation through PPARgamma activation. J Exp Med. 2004;199(6):763-774.
36. Aloulou A, Rahier R, Arhab Y, Noiriel A, Abousalham A. Phospholipases: An
Overview. Methods Mol Biol. 2018;1835:69-105.
37. Yu J, Loh K, Song ZY, Yang HQ, Zhang Y, Lin S. Update on glycerol-3-phosphate
acyltransferases: the roles in the development of insulin resistance. Nutr Diabetes.
2018;8(1):34.
38. Gerrard JM, Robinson P. Identification of the molecular species of lysophosphatidic
acid produced when platelets are stimulated by thrombin. Biochim Biophys Acta.
1989;1001(3):282-285.
39. Vancura A, Haldar D. Regulation of mitochondrial and microsomal phospholipid
synthesis by liver fatty acid-binding protein. J Biol Chem. 1992;267(20):14353-14359.
40. Simpson CM, Itabe H, Reynolds CN, King WC, Glomset JA. Swiss 3T3 cells
preferentially incorporate sn-2-arachidonoyl monoacylglycerol into sn-1-stearoyl-2-
arachidonoyl phosphatidylinositol. J Biol Chem. 1991;266(24):15902-15909.
41. Vancura A, Carroll MA, Haldar D. A lysophosphatidic acid-binding cytosolic protein
stimulates mitochondrial glycerophosphate acyltransferase. Biochem Biophys Res Commun.
1991;175(1):339-343.
42. Sonoda H, Aoki J, Hiramatsu T, et al. A novel phosphatidic acid-selective
phospholipase A1 that produces lysophosphatidic acid. J Biol Chem. 2002;277(37):34254-
34263.
43. Yoshimasu T, Kanazawa N, Kambe N, Nakamura M, Furukawa F. Identification of
736T>A mutation of lipase H in Japanese siblings with autosomal recessive woolly hair. J
Dermatol. 2011;38(9):900-904.
44. Inoue A, Arima N, Ishiguro J, Prestwich GD, Arai H, Aoki J. LPA-producing enzyme
PA-PLA(1)alpha regulates hair follicle development by modulating EGFR signalling. EMBO
J. 2011;30(20):4248-4260.
45. Fourcade O, Simon MF, Viode C, et al. Secretory phospholipase A2 generates the
novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated
cells. Cell. 1995;80(6):919-927.
46. Eder AM, Sasagawa T, Mao M, Aoki J, Mills GB. Constitutive and lysophosphatidic
acid (LPA)-induced LPA production: role of phospholipase D and phospholipase A2. Clin
Cancer Res. 2000;6(6):2482-2491.
47. Sano T, Baker D, Virag T, et al. Multiple mechanisms linked to platelet activation
result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J Biol
Chem. 2002;277(24):21197-21206.
48. Umezu-Goto M, Kishi Y, Taira A, et al. Autotaxin has lysophospholipase D activity
leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol.
2002;158(2):227-233.

124
49. Tokumura A, Majima E, Kariya Y, et al. Identification of human plasma
lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a
multifunctional phosphodiesterase. J Biol Chem. 2002;277(42):39436-39442.
50. Bergmark C, Dewan A, Orsoni A, et al. A novel function of lipoprotein [a] as a
preferential carrier of oxidized phospholipids in human plasma. J Lipid Res.
2008;49(10):2230-2239.
51. Jethwa SA, Leah EJ, Zhang Q, et al. Exosomes bind to autotaxin and act as a
physiological delivery mechanism to stimulate LPA receptor signalling in cells. J Cell Sci.
2016;129(20):3948-3957.
52. Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification
of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of
low density lipoprotein phospholipids. Proc Natl Acad Sci U S A. 1984;81(12):3883-3887.
53. Choi J, Zhang W, Gu X, et al. Lysophosphatidylcholine is generated by spontaneous
deacylation of oxidized phospholipids. Chem Res Toxicol. 2011;24(1):111-118.
54. Nsaibia MJ, Mahmut A, Boulanger MC, et al. Autotaxin interacts with lipoprotein(a)
and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients
with coronary artery disease. J Intern Med. 2016;280(5):509-517.
55. Rabini RA, Galassi R, Fumelli P, et al. Reduced Na(+)-K(+)-ATPase activity and
plasma lysophosphatidylcholine concentrations in diabetic patients. Diabetes.
1994;43(7):915-919.
56. Okita M, Gaudette DC, Mills GB, Holub BJ. Elevated levels and altered fatty acid
composition of plasma lysophosphatidylcholine(lysoPC) in ovarian cancer patients. Int J
Cancer. 1997;71(1):31-34.
57. Sasagawa T, Suzuki K, Shiota T, Kondo T, Okita M. The significance of plasma
lysophospholipids in patients with renal failure on hemodialysis. J Nutr Sci Vitaminol
(Tokyo). 1998;44(6):809-818.
58. Tanaka M, Okudaira S, Kishi Y, et al. Autotaxin stabilizes blood vessels and is
required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem.
2006;281(35):25822-25830.
59. van Meeteren LA, Ruurs P, Stortelers C, et al. Autotaxin, a secreted
lysophospholipase D, is essential for blood vessel formation during development. Mol Cell
Biol. 2006;26(13):5015-5022.
60. Ferry G, Giganti A, Coge F, Bertaux F, Thiam K, Boutin JA. Functional invalidation
of the autotaxin gene by a single amino acid mutation in mouse is lethal. FEBS Lett.
2007;581(18):3572-3578.
61. Fotopoulou S, Oikonomou N, Grigorieva E, et al. ATX expression and LPA
signalling are vital for the development of the nervous system. Dev Biol. 2010;339(2):451-
464.
62. Katsifa A, Kaffe E, Nikolaidou-Katsaridou N, et al. The Bulk of Autotaxin Activity
Is Dispensable for Adult Mouse Life. PLoS One. 2015;10(11):e0143083.
63. D'Souza K, Paramel GV, Kienesberger PC. Lysophosphatidic Acid Signaling in
Obesity and Insulin Resistance. Nutrients. 2018;10(4).
64. Nishimura S, Nagasaki M, Okudaira S, et al. ENPP2 contributes to adipose tissue
expansion and insulin resistance in diet-induced obesity. Diabetes. 2014;63(12):4154-4164.
65. Euer N, Schwirzke M, Evtimova V, et al. Identification of genes associated with
metastasis of mammary carcinoma in metastatic versus non-metastatic cell lines. Anticancer
Res. 2002;22(2A):733-740.

125
66. Brisbin AG, Asmann YW, Song H, et al. Meta-analysis of 8q24 for seven cancers
reveals a locus between NOV and ENPP2 associated with cancer development. BMC Med
Genet. 2011;12:156.
67. Oikonomou N, Mouratis MA, Tzouvelekis A, et al. Pulmonary autotaxin expression
contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol.
2012;47(5):566-574.
68. Bouchareb R, Mahmut A, Nsaibia MJ, et al. Autotaxin Derived From Lipoprotein(a)
and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve.
Circulation. 2015;132(8):677-690.
69. Dohi T, Miyauchi K, Ohkawa R, et al. Increased lysophosphatidic acid levels in
culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis.
2013;229(1):192-197.
70. Nikitopoulou I, Oikonomou N, Karouzakis E, et al. Autotaxin expression from
synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J Exp Med.
2012;209(5):925-933.
71. Clair T, Lee HY, Liotta LA, Stracke ML. Autotaxin is an exoenzyme possessing 5'-
nucleotide phosphodiesterase/ATP pyrophosphatase and ATPase activities. J Biol Chem.
1997;272(2):996-1001.
72. Clair T, Aoki J, Koh E, et al. Autotaxin hydrolyzes sphingosylphosphorylcholine to
produce the regulator of migration, sphingosine-1-phosphate. Cancer Res.
2003;63(17):5446-5453.
73. Giganti A, Rodriguez M, Fould B, et al. Murine and human autotaxin alpha, beta, and
gamma isoforms: gene organization, tissue distribution, and biochemical characterization. J
Biol Chem. 2008;283(12):7776-7789.
74. Hashimoto T, Okudaira S, Igarashi K, Hama K, Yatomi Y, Aoki J. Identification and
biochemical characterization of a novel autotaxin isoform, ATXdelta, with a four-amino acid
deletion. J Biochem. 2012;151(1):89-97.
75. Dusaulcy R, Rancoule C, Gres S, et al. Adipose-specific disruption of autotaxin
enhances nutritional fattening and reduces plasma lysophosphatidic acid. J Lipid Res.
2011;52(6):1247-1255.
76. Kanda H, Newton R, Klein R, Morita Y, Gunn MD, Rosen SD. Autotaxin, an
ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into
secondary lymphoid organs. Nat Immunol. 2008;9(4):415-423.
77. Kawagoe H, Soma O, Goji J, et al. Molecular cloning and chromosomal assignment
of the human brain-type phosphodiesterase I/nucleotide pyrophosphatase gene (PDNP2).
Genomics. 1995;30(2):380-384.
78. Lee HY, Murata J, Clair T, et al. Cloning, chromosomal localization, and tissue
expression of autotaxin from human teratocarcinoma cells. Biochem Biophys Res Commun.
1996;218(3):714-719.
79. Nishimasu H, Okudaira S, Hama K, et al. Crystal structure of autotaxin and insight
into GPCR activation by lipid mediators. Nat Struct Mol Biol. 2011;18(2):205-212.
80. Bollen M, Gijsbers R, Ceulemans H, Stalmans W, Stefan C. Nucleotide
pyrophosphatases/phosphodiesterases on the move. Crit Rev Biochem Mol Biol.
2000;35(6):393-432.
81. Houben AJ, van Wijk XM, van Meeteren LA, et al. The polybasic insertion in
autotaxin alpha confers specific binding to heparin and cell surface heparan sulfate
proteoglycans. J Biol Chem. 2013;288(1):510-519.

126
82. Perrakis A, Moolenaar WH. Autotaxin: structure-function and signaling. J Lipid Res.
2014;55(6):1010-1018.
83. van Meeteren LA, Ruurs P, Christodoulou E, et al. Inhibition of autotaxin by
lysophosphatidic acid and sphingosine 1-phosphate. J Biol Chem. 2005;280(22):21155-
21161.
84. Jansen S, Stefan C, Creemers JW, et al. Proteolytic maturation and activation of
autotaxin (NPP2), a secreted metastasis-enhancing lysophospholipase D. J Cell Sci.
2005;118(Pt 14):3081-3089.
85. Jansen S, Andries M, Derua R, Waelkens E, Bollen M. Domain interplay mediated
by an essential disulfide linkage is critical for the activity and secretion of the metastasis-
promoting enzyme autotaxin. J Biol Chem. 2009;284(21):14296-14302.
86. Jansen S, Callewaert N, Dewerte I, Andries M, Ceulemans H, Bollen M. An essential
oligomannosidic glycan chain in the catalytic domain of autotaxin, a secreted
lysophospholipase-D. J Biol Chem. 2007;282(15):11084-11091.
87. Hausmann J, Kamtekar S, Christodoulou E, et al. Structural basis of substrate
discrimination and integrin binding by autotaxin. Nat Struct Mol Biol. 2011;18(2):198-204.
88. Gijsbers R, Aoki J, Arai H, Bollen M. The hydrolysis of lysophospholipids and
nucleotides by autotaxin (NPP2) involves a single catalytic site. FEBS Lett. 2003;538(1-
3):60-64.
89. Koh E, Clair T, Woodhouse EC, Schiffmann E, Liotta L, Stracke M. Site-directed
mutations in the tumor-associated cytokine, autotaxin, eliminate nucleotide
phosphodiesterase, lysophospholipase D, and motogenic activities. Cancer Res.
2003;63(9):2042-2045.
90. Gijsbers R, Ceulemans H, Bollen M. Functional characterization of the non-catalytic
ectodomains of the nucleotide pyrophosphatase/phosphodiesterase NPP1. Biochem J.
2003;371(Pt 2):321-330.
91. Cimpean A, Stefan C, Gijsbers R, Stalmans W, Bollen M. Substrate-specifying
determinants of the nucleotide pyrophosphatases/phosphodiesterases NPP1 and NPP2.
Biochem J. 2004;381(Pt 1):71-77.
92. Pamuklar Z, Federico L, Liu S, et al. Autotaxin/lysopholipase D and lysophosphatidic
acid regulate murine hemostasis and thrombosis. J Biol Chem. 2009;284(11):7385-7394.
93. Parris TZ, Kovacs A, Hajizadeh S, et al. Frequent MYC coamplification and DNA
hypomethylation of multiple genes on 8q in 8p11-p12-amplified breast carcinomas.
Oncogenesis. 2014;3:e95.
94. Li S, Wang B, Xu Y, Zhang J. Autotaxin is induced by TSA through HDAC3 and
HDAC7 inhibition and antagonizes the TSA-induced cell apoptosis. Mol Cancer.
2011;10:18.
95. Sun S, Zhang X, Lyu L, Li X, Yao S, Zhang J. Autotaxin Expression Is Regulated at
the Post-transcriptional Level by the RNA-binding Proteins HuR and AUF1. J Biol Chem.
2016;291(50):25823-25836.
96. Black EJ, Clair T, Delrow J, Neiman P, Gillespie DA. Microarray analysis identifies
Autotaxin, a tumour cell motility and angiogenic factor with lysophospholipase D activity,
as a specific target of cell transformation by v-Jun. Oncogene. 2004;23(13):2357-2366.
97. Sioletic S, Czaplinski J, Hu L, et al. c-Jun promotes cell migration and drives
expression of the motility factor ENPP2 in soft tissue sarcomas. J Pathol. 2014;234(2):190-
202.

127
98. Farina AR, Cappabianca L, Ruggeri P, et al. Constitutive autotaxin transcription by
Nmyc-amplified and non-amplified neuroblastoma cells is regulated by a novel AP-1 and
SP-mediated mechanism and abrogated by curcumin. FEBS Lett. 2012;586(20):3681-3691.
99. Williams TM, Williams ME, Kuick R, et al. Candidate downstream regulated genes
of HOX group 13 transcription factors with and without monomeric DNA binding capability.
Dev Biol. 2005;279(2):462-480.
100. Corcoran DL, Feingold E, Dominick J, et al. Footer: a quantitative comparative
genomics method for efficient recognition of cis-regulatory elements. Genome Res.
2005;15(6):840-847.
101. Lovas A, Weidemann A, Albrecht D, Wiechert L, Weih D, Weih F. p100 Deficiency
is insufficient for full activation of the alternative NF-kappaB pathway: TNF cooperates with
p52-RelB in target gene transcription. PLoS One. 2012;7(8):e42741.
102. Cloney DL, Gray RW, Bruns ME, et al. Intestinal vitamin D-dependent calbindin-
D9k and alkaline phosphatase in spontaneously hypertensive rats. Am J Physiol. 1991;260(5
Pt 1):G691-697.
103. Braeuer RR, Zigler M, Kamiya T, et al. Galectin-3 contributes to melanoma growth
and metastasis via regulation of NFAT1 and autotaxin. Cancer Res. 2012;72(22):5757-5766.
104. Azare J, Doane A, Leslie K, et al. Stat3 mediates expression of autotaxin in breast
cancer. PLoS One. 2011;6(11):e27851.
105. Li S, Xiong C, Zhang J. ATX and LPA receptor 3 are coordinately up-regulated in
lipopolysaccharide-stimulated THP-1 cells through PKR and SPK1-mediated pathways.
FEBS Lett. 2012;586(6):792-797.
106. Wu JM, Xu Y, Skill NJ, et al. Autotaxin expression and its connection with the TNF-
alpha-NF-kappaB axis in human hepatocellular carcinoma. Mol Cancer. 2010;9:71.
107. Benesch MG, Zhao YY, Curtis JM, McMullen TP, Brindley DN. Regulation of
autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J
Lipid Res. 2015;56(6):1134-1144.
108. Castelino FV, Bain G, Pace VA, et al. An Autotaxin/Lysophosphatidic
Acid/Interleukin-6 Amplification Loop Drives Scleroderma Fibrosis. Arthritis Rheumatol.
2016;68(12):2964-2974.
109. Song J, Guan M, Zhao Z, Zhang J. Type I Interferons Function as Autocrine and
Paracrine Factors to Induce Autotaxin in Response to TLR Activation. PLoS One.
2015;10(8):e0136629.
110. Kehlen A, Englert N, Seifert A, et al. Expression, regulation and function of autotaxin
in thyroid carcinomas. Int J Cancer. 2004;109(6):833-838.
111. Kehlen A, Lauterbach R, Santos AN, et al. IL-1 beta- and IL-4-induced down-
regulation of autotaxin mRNA and PC-1 in fibroblast-like synoviocytes of patients with
rheumatoid arthritis (RA). Clin Exp Immunol. 2001;123(1):147-154.
112. Khandoga AL, Pandey D, Welsch U, Brandl R, Siess W. GPR92/LPA(5)
lysophosphatidate receptor mediates megakaryocytic cell shape change induced by human
atherosclerotic plaques. Cardiovasc Res. 2011;90(1):157-164.
113. Rother E, Brandl R, Baker DL, et al. Subtype-selective antagonists of
lysophosphatidic Acid receptors inhibit platelet activation triggered by the lipid core of
atherosclerotic plaques. Circulation. 2003;108(6):741-747.
114. Haseruck N, Erl W, Pandey D, et al. The plaque lipid lysophosphatidic acid stimulates
platelet activation and platelet-monocyte aggregate formation in whole blood: involvement
of P2Y1 and P2Y12 receptors. Blood. 2004;103(7):2585-2592.

128
115. Tigyi G, Miledi R. Lysophosphatidates bound to serum albumin activate membrane
currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J Biol
Chem. 1992;267(30):21360-21367.
116. Goetzl EJ, Lee H, Azuma T, Stossel TP, Turck CW, Karliner JS. Gelsolin binding
and cellular presentation of lysophosphatidic acid. J Biol Chem. 2000;275(19):14573-14578.
117. Mintzer E, Sargsyan H, Bittman R. Lysophosphatidic acid and lipopolysaccharide
bind to the PIP2-binding domain of gelsolin. Biochim Biophys Acta. 2006;1758(1):85-89.
118. Yagi T, Shoaib M, Kuschner C, et al. Challenges and Inconsistencies in Using
Lysophosphatidic Acid as a Biomarker for Ovarian Cancer. Cancers (Basel). 2019;11(4).
119. Leblanc R, Lee SC, David M, et al. Interaction of platelet-derived autotaxin with
tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood.
2014;124(20):3141-3150.
120. Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS. Binding of autotaxin
to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J
Biol Chem. 2011;286(40):34654-34663.
121. Smyth SS, Mueller P, Yang F, Brandon JA, Morris AJ. Arguing the case for the
autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the
development and complications of atherosclerosis. Arterioscler Thromb Vasc Biol.
2014;34(3):479-486.
122. Hosogaya S, Yatomi Y, Nakamura K, et al. Measurement of plasma lysophosphatidic
acid concentration in healthy subjects: strong correlation with lysophospholipase D activity.
Ann Clin Biochem. 2008;45(Pt 4):364-368.
123. Garcia-Marchena N, Pizarro N, Pavon FJ, et al. Potential association of plasma
lysophosphatidic acid (LPA) species with cognitive impairment in abstinent alcohol use
disorders outpatients. Sci Rep. 2020;10(1):17163.
124. Sedlakova I, Vavrova J, Tosner J, Hanousek L. Lysophosphatidic acid (LPA)-a
perspective marker in ovarian cancer. Tumour Biol. 2011;32(2):311-316.
125. Zhang YJ, Cao LY, Fu ZZ, Wang YJ, Wang GX, Gu T. Clinical significance of
plasma lysophosphatidic acid levels in the differential diagnosis of ovarian cancer. J Cancer
Res Ther. 2015;11(2):375-380.
126. Michalczyk A, Budkowska M, Dolegowska B, Chlubek D, Safranow K.
Lysophosphatidic acid plasma concentrations in healthy subjects: circadian rhythm and
associations with demographic, anthropometric and biochemical parameters. Lipids Health
Dis. 2017;16(1):140.
127. Baker DL, Morrison P, Miller B, et al. Plasma lysophosphatidic acid concentration
and ovarian cancer. JAMA. 2002;287(23):3081-3082.
128. An S, Bleu T, Hallmark OG, Goetzl EJ. Characterization of a novel subtype of human
G protein-coupled receptor for lysophosphatidic acid. J Biol Chem. 1998;273(14):7906-
7910.
129. Bandoh K, Aoki J, Hosono H, et al. Molecular cloning and characterization of a novel
human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem.
1999;274(39):27776-27785.
130. Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-
coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol
Chem. 2003;278(28):25600-25606.

129
131. Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a new G12/13- and Gq-
coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J Biol Chem.
2006;281(33):23589-23597.
132. Pasternack SM, von Kugelgen I, Al Aboud K, et al. G protein-coupled receptor P2Y5
and its ligand LPA are involved in maintenance of human hair growth. Nat Genet.
2008;40(3):329-334.
133. Tabata K, Baba K, Shiraishi A, Ito M, Fujita N. The orphan GPCR GPR87 was
deorphanized and shown to be a lysophosphatidic acid receptor. Biochem Biophys Res
Commun. 2007;363(3):861-866.
134. Ochiai S, Furuta D, Sugita K, Taniura H, Fujita N. GPR87 mediates lysophosphatidic
acid-induced colony dispersal in A431 cells. Eur J Pharmacol. 2013;715(1-3):15-20.
135. Murakami M, Shiraishi A, Tabata K, Fujita N. Identification of the orphan GPCR,
P2Y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor.
Biochem Biophys Res Commun. 2008;371(4):707-712.
136. Inoue A, Ishiguro J, Kitamura H, et al. TGFalpha shedding assay: an accurate and
versatile method for detecting GPCR activation. Nat Methods. 2012;9(10):1021-1029.
137. Nieto-Posadas A, Picazo-Juarez G, Llorente I, et al. Lysophosphatidic acid directly
activates TRPV1 through a C-terminal binding site. Nat Chem Biol. 2011;8(1):78-85.
138. Chemin J, Patel A, Duprat F, Zanzouri M, Lazdunski M, Honore E. Lysophosphatidic
acid-operated K+ channels. J Biol Chem. 2005;280(6):4415-4421.
139. Ohuchi H, Hamada A, Matsuda H, et al. Expression patterns of the lysophospholipid
receptor genes during mouse early development. Dev Dyn. 2008;237(11):3280-3294.
140. Contos JJ, Chun J. Complete cDNA sequence, genomic structure, and chromosomal
localization of the LPA receptor gene, lpA1/vzg-1/Gpcr26. Genomics. 1998;51(3):364-378.
141. Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature
review: IUPHAR Review 8. Br J Pharmacol. 2014;171(15):3575-3594.
142. Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1
lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A.
2000;97(24):13384-13389.
143. Venkatraman G, Benesch MG, Tang X, Dewald J, McMullen TP, Brindley DN.
Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved
in drug resistance and oxidative stress responses: implications for cancer treatment. FASEB
J. 2015;29(3):772-785.
144. Fukushima K, Takahashi K, Yamasaki E, et al. Lysophosphatidic acid signaling via
LPA1 and LPA3 regulates cellular functions during tumor progression in pancreatic cancer
cells. Exp Cell Res. 2017;352(1):139-145.
145. Obo Y, Yamada T, Furukawa M, et al. Frequent mutations of lysophosphatidic acid
receptor-1 gene in rat liver tumors. Mutat Res. 2009;660(1-2):47-50.
146. David M, Machuca-Gayet I, Kikuta J, et al. Lysophosphatidic acid receptor type 1
(LPA1) plays a functional role in osteoclast differentiation and bone resorption activity. J
Biol Chem. 2014;289(10):6551-6564.
147. Orosa B, Garcia S, Martinez P, Gonzalez A, Gomez-Reino JJ, Conde C.
Lysophosphatidic acid receptor inhibition as a new multipronged treatment for rheumatoid
arthritis. Ann Rheum Dis. 2014;73(1):298-305.
148. Lin CI, Chen CN, Lin PW, Chang KJ, Hsieh FJ, Lee H. Lysophosphatidic acid
regulates inflammation-related genes in human endothelial cells through LPA1 and LPA3.
Biochem Biophys Res Commun. 2007;363(4):1001-1008.

130
149. Miyabe Y, Miyabe C, Iwai Y, et al. Activation of fibroblast-like synoviocytes derived
from rheumatoid arthritis via lysophosphatidic acid-lysophosphatidic acid receptor 1
cascade. Arthritis Res Ther. 2014;16(5):461.
150. Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1
links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak.
Nat Med. 2008;14(1):45-54.
151. Ruisanchez E, Dancs P, Kerek M, et al. Lysophosphatidic acid induces vasodilation
mediated by LPA1 receptors, phospholipase C, and endothelial nitric oxide synthase. FASEB
J. 2014;28(2):880-890.
152. Dancs PT, Ruisanchez E, Balogh A, et al. LPA1 receptor-mediated thromboxane A2
release is responsible for lysophosphatidic acid-induced vascular smooth muscle contraction.
FASEB J. 2017;31(4):1547-1555.
153. Gomez-Larrauri A, Gangoiti P, Presa N, et al. Phosphatidic Acid Stimulates Myoblast
Proliferation through Interaction with LPA1 and LPA2 Receptors. Int J Mol Sci. 2021;22(3).
154. Ishii I, Contos JJ, Fukushima N, Chun J. Functional comparisons of the
lysophosphatidic acid receptors, LP(A1)/VZG-1/EDG-2, LP(A2)/EDG-4, and LP(A3)/EDG-
7 in neuronal cell lines using a retrovirus expression system. Mol Pharmacol.
2000;58(5):895-902.
155. Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic
pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10(7):712-718.
156. Pan HL, Zhang YQ, Zhao ZQ. Involvement of lysophosphatidic acid in bone cancer
pain by potentiation of TRPV1 via PKCepsilon pathway in dorsal root ganglion neurons. Mol
Pain. 2010;6:85.
157. Robering JW, Gebhardt L, Wolf K, Kuhn H, Kremer AE, Fischer MJM.
Lysophosphatidic acid activates satellite glia cells and Schwann cells. Glia. 2019;67(5):999-
1012.
158. Cohen A, Sagron R, Somech E, Segal-Hayoun Y, Zilberberg N. Pain-associated
signals, acidosis and lysophosphatidic acid, modulate the neuronal K(2P)2.1 channel. Mol
Cell Neurosci. 2009;40(3):382-389.
159. Komachi M, Damirin A, Malchinkhuu E, et al. Signaling pathways involved in DNA
synthesis and migration in response to lysophosphatidic acid and low-density lipoprotein in
coronary artery smooth muscle cells. Vascul Pharmacol. 2009;50(5-6):178-184.
160. Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between
NMDA receptor subunits and the postsynaptic density protein PSD-95. Science.
1995;269(5231):1737-1740.
161. Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and
modification. Cell Commun Signal. 2010;8:8.
162. Varsano T, Taupin V, Guo L, Baterina OY, Jr., Farquhar MG. The PDZ protein GIPC
regulates trafficking of the LPA1 receptor from APPL signaling endosomes and attenuates
the cell's response to LPA. PLoS One. 2012;7(11):e49227.
163. Yamada T, Ohoka Y, Kogo M, Inagaki S. Physical and functional interactions of the
lysophosphatidic acid receptors with PDZ domain-containing Rho guanine nucleotide
exchange factors (RhoGEFs). J Biol Chem. 2005;280(19):19358-19363.
164. Shano S, Hatanaka K, Ninose S, Moriyama R, Tsujiuchi T, Fukushima N. A
lysophosphatidic acid receptor lacking the PDZ-binding domain is constitutively active and
stimulates cell proliferation. Biochim Biophys Acta. 2008;1783(5):748-759.

131
165. Weiner JA, Fukushima N, Contos JJ, Scherer SS, Chun J. Regulation of Schwann cell
morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. J Neurosci.
2001;21(18):7069-7078.
166. Shano S, Moriyama R, Chun J, Fukushima N. Lysophosphatidic acid stimulates
astrocyte proliferation through LPA1. Neurochem Int. 2008;52(1-2):216-220.
167. Weiner JA, Chun J. Schwann cell survival mediated by the signaling phospholipid
lysophosphatidic acid. Proc Natl Acad Sci U S A. 1999;96(9):5233-5238.
168. Olianas MC, Dedoni S, Onali P. Inhibition of TNF-alpha-induced neuronal apoptosis
by antidepressants acting through the lysophosphatidic acid receptor LPA1. Apoptosis.
2019;24(5-6):478-498.
169. Zhang J, Li Y, Wang C, et al. Lysophosphatidic Acid Induces Apoptosis of PC12
Cells Through LPA1 Receptor/LPA2 Receptor/MAPK Signaling Pathway. Front Mol
Neurosci. 2020;13:16.
170. Musazzi L, Di Daniel E, Maycox P, Racagni G, Popoli M. Abnormalities in
alpha/beta-CaMKII and related mechanisms suggest synaptic dysfunction in hippocampus of
LPA1 receptor knockout mice. Int J Neuropsychopharmacol. 2011;14(7):941-953.
171. Moller T, Contos JJ, Musante DB, Chun J, Ransom BR. Expression and function of
lysophosphatidic acid receptors in cultured rodent microglial cells. J Biol Chem.
2001;276(28):25946-25952.
172. Harrison SM, Reavill C, Brown G, et al. LPA1 receptor-deficient mice have
phenotypic changes observed in psychiatric disease. Mol Cell Neurosci. 2003;24(4):1170-
1179.
173. Moreno-Fernandez RD, Rosell-Valle C, Bacq A, et al. LPA1 receptor and chronic
stress: Effects on behaviour and the genes involved in the hippocampal excitatory/inhibitory
balance. Neuropharmacology. 2020;164:107896.
174. Anliker B, Choi JW, Lin ME, et al. Lysophosphatidic acid (LPA) and its receptor,
LPA1 , influence embryonic schwann cell migration, myelination, and cell-to-axon
segregation. Glia. 2013;61(12):2009-2022.
175. Weiner JA, Hecht JH, Chun J. Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-
2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain.
J Comp Neurol. 1998;398(4):587-598.
176. Castilla-Ortega E, Sanchez-Lopez J, Hoyo-Becerra C, et al. Exploratory, anxiety and
spatial memory impairments are dissociated in mice lacking the LPA1 receptor. Neurobiol
Learn Mem. 2010;94(1):73-82.
177. Santin LJ, Bilbao A, Pedraza C, et al. Behavioral phenotype of maLPA1-null mice:
increased anxiety-like behavior and spatial memory deficits. Genes Brain Behav.
2009;8(8):772-784.
178. Dusaulcy R, Daviaud D, Pradere JP, Gres S, Valet P, Saulnier-Blache JS. Altered
food consumption in mice lacking lysophosphatidic acid receptor-1. J Physiol Biochem.
2009;65(4):345-350.
179. Roberts C, Winter P, Shilliam CS, et al. Neurochemical changes in LPA1 receptor
deficient mice--a putative model of schizophrenia. Neurochem Res. 2005;30(3):371-377.
180. Uchida H, Nagai J, Ueda H. Lysophosphatidic acid and its receptors LPA1 and LPA3
mediate paclitaxel-induced neuropathic pain in mice. Mol Pain. 2014;10:71.
181. Rivera RR, Lin ME, Bornhop EC, Chun J. Conditional Lpar1 gene targeting identifies
cell types mediating neuropathic pain. FASEB J. 2020;34(7):8833-8842.

132
182. Srikanth M, Chew WS, Hind T, et al. Lysophosphatidic acid and its receptor LPA1
mediate carrageenan induced inflammatory pain in mice. Eur J Pharmacol. 2018;841:49-56.
183. Ueda H, Neyama H, Matsushita Y. Lysophosphatidic Acid Receptor 1- and 3-
Mediated Hyperalgesia and Hypoalgesia in Diabetic Neuropathic Pain Models in Mice.
Cells. 2020;9(8).
184. Gento-Caro A, Vilches-Herrando E, Garcia-Morales V, et al. Interfering with
lysophosphatidic acid receptor edg2/lpa1 signalling slows down disease progression in
SOD1-G93A transgenic mice. Neuropathol Appl Neurobiol. 2021.
185. Fransson J, Gomez-Conde AI, Romero-Imbroda J, et al. Activation of Macrophages
by Lysophosphatidic Acid through the Lysophosphatidic Acid Receptor 1 as a Novel
Mechanism in Multiple Sclerosis Pathogenesis. Mol Neurobiol. 2021;58(2):470-482.
186. Gennero I, Laurencin-Dalicieux S, Conte-Auriol F, et al. Absence of the
lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased
bone mass. Bone. 2011;49(3):395-403.
187. Aki Y, Kondo A, Nakamura H, Togari A. Lysophosphatidic acid-stimulated
interleukin-6 and -8 synthesis through LPA1 receptors on human osteoblasts. Arch Oral Biol.
2008;53(3):207-213.
188. Yu ZL, Li DQ, Huang XY, et al. Lysophosphatidic acid upregulates connective tissue
growth factor expression in osteoblasts through the GPCR/PKC and PKA pathways. Int J
Mol Med. 2016;37(2):468-474.
189. Alioli CA, Demesmay L, Laurencin-Dalacieux S, et al. Expression of the type 1
lysophosphatidic acid receptor in osteoblastic cell lineage controls both bone mineralization
and osteocyte specification. Biochim Biophys Acta Mol Cell Biol Lipids.
2020;1865(8):158715.
190. Panupinthu N, Zhao L, Possmayer F, Ke HZ, Sims SM, Dixon SJ. P2X7 nucleotide
receptors mediate blebbing in osteoblasts through a pathway involving lysophosphatidic
acid. J Biol Chem. 2007;282(5):3403-3412.
191. Waters KM, Tan R, Genetos DC, Verma S, Yellowley CE, Karin NJ. DNA
microarray analysis reveals a role for lysophosphatidic acid in the regulation of anti-
inflammatory genes in MC3T3-E1 cells. Bone. 2007;41(5):833-841.
192. Miyabe Y, Miyabe C, Iwai Y, et al. Necessity of lysophosphatidic acid receptor 1 for
development of arthritis. Arthritis Rheum. 2013;65(8):2037-2047.
193. Nishioka T, Arima N, Kano K, et al. ATX-LPA1 axis contributes to proliferation of
chondrocytes by regulating fibronectin assembly leading to proper cartilage formation. Sci
Rep. 2016;6:23433.
194. Hurst-Kennedy J, Boyan BD, Schwartz Z. Lysophosphatidic acid signaling promotes
proliferation, differentiation, and cell survival in rat growth plate chondrocytes. Biochim
Biophys Acta. 2009;1793(5):836-846.
195. Mizuno K, Komiya M, Okuyama K, Imada K, Sato T. Lysophosphatidic Acid
Augments the Gene Expression and Production of Matrix Metalloproteinases-1 and -3 in
Human Synovial Fibroblasts in Vitro. Biol Pharm Bull. 2021;44(1):131-135.
196. Zhao J, Wei J, Weathington N, et al. Lysophosphatidic acid receptor 1 antagonist
ki16425 blunts abdominal and systemic inflammation in a mouse model of peritoneal sepsis.
Transl Res. 2015;166(1):80-88.
197. Zhao C, Sardella A, Chun J, Poubelle PE, Fernandes MJ, Bourgoin SG. TNF-alpha
promotes LPA1- and LPA3-mediated recruitment of leukocytes in vivo through CXCR2
ligand chemokines. J Lipid Res. 2011;52(7):1307-1318.

133
198. Chen X, Walther FJ, van Boxtel R, et al. Deficiency or inhibition of lysophosphatidic
acid receptor 1 protects against hyperoxia-induced lung injury in neonatal rats. Acta Physiol
(Oxf). 2016;216(3):358-375.
199. Gao L, Shi H, Sherchan P, et al. Inhibition of lysophosphatidic acid receptor 1
attenuates neuroinflammation via PGE2/EP2/NOX2 signalling and improves the outcome of
intracerebral haemorrhage in mice. Brain Behav Immun. 2021;91:615-626.
200. Wang H, Tu S, Yang S, et al. Berberine Modulates LPA Function to Inhibit the
Proliferation and Inflammation of FLS-RA via p38/ERK MAPK Pathway Mediated by
LPA1. Evid Based Complement Alternat Med. 2019;2019:2580207.
201. Geng H, Lan R, Liu Y, et al. Proximal tubule LPA1 and LPA2 receptors use divergent
signaling pathways to additively increase profibrotic cytokine secretion. Am J Physiol Renal
Physiol. 2021;320(3):F359-F374.
202. Ohashi T, Yamamoto T. Antifibrotic effect of lysophosphatidic acid receptors LPA1
and LPA3 antagonist on experimental murine scleroderma induced by bleomycin. Exp
Dermatol. 2015;24(9):698-702.
203. Ledein L, Leger B, Dees C, et al. Translational engagement of lysophosphatidic acid
receptor 1 in skin fibrosis: from dermal fibroblasts of patients with scleroderma to tight skin
1 mouse. Br J Pharmacol. 2020;177(18):4296-4309.
204. Olianas MC, Dedoni S, Onali P. Antidepressants activate the lysophosphatidic acid
receptor LPA(1) to induce insulin-like growth factor-I receptor transactivation, stimulation
of ERK1/2 signaling and cell proliferation in CHO-K1 fibroblasts. Biochem Pharmacol.
2015;95(4):311-323.
205. Pradere JP, Klein J, Gres S, et al. LPA1 receptor activation promotes renal interstitial
fibrosis. J Am Soc Nephrol. 2007;18(12):3110-3118.
206. Castelino FV, Seiders J, Bain G, et al. Amelioration of dermal fibrosis by genetic
deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model
of scleroderma. Arthritis Rheum. 2011;63(5):1405-1415.
207. Lin S, Han Y, Jenkin K, et al. Lysophosphatidic Acid Receptor 1 Is Important for
Intestinal Epithelial Barrier Function and Susceptibility to Colitis. Am J Pathol.
2018;188(2):353-366.
208. Bao L, Qi J, Wang YW, et al. The atherogenic actions of LPC on vascular smooth
muscle cells and its LPA receptor mediated mechanism. Biochem Biophys Res Commun.
2018;503(3):1911-1918.
209. Subramanian P, Karshovska E, Reinhard P, et al. Lysophosphatidic acid receptors
LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in
neointima formation. Circ Res. 2010;107(1):96-105.
210. Chabowski DS, Kadlec AO, Ait-Aissa K, et al. Lysophosphatidic acid acts on LPA1
receptor to increase H2 O2 during flow-induced dilation in human adipose arterioles. Br J
Pharmacol. 2018;175(22):4266-4280.
211. Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, Johnson LR.
Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and
chemotherapy-induced apoptosis. Gastroenterology. 2002;123(1):206-216.
212. Fisher KE, Pop A, Koh W, Anthis NJ, Saunders WB, Davis GE. Tumor cell invasion
of collagen matrices requires coordinate lipid agonist-induced G-protein and membrane-type
matrix metalloproteinase-1-dependent signaling. Mol Cancer. 2006;5:69.
213. Shi W, Zhang C, Ning Z, et al. CMTM8 as an LPA1-associated partner mediates
lysophosphatidic acid-induced pancreatic cancer metastasis. Ann Transl Med. 2021;9(1):42.

134
214. Amaral RF, Geraldo LHM, Einicker-Lamas M, TCLS ES, Mendes F, Lima FRS.
Microglial lysophosphatidic acid promotes glioblastoma proliferation and migration via
LPA1 receptor. J Neurochem. 2021;156(4):499-512.
215. Yu S, Murph MM, Lu Y, et al. Lysophosphatidic acid receptors determine
tumorigenicity and aggressiveness of ovarian cancer cells. J Natl Cancer Inst.
2008;100(22):1630-1642.
216. Kitayoshi M, Kato K, Tanabe E, et al. Enhancement of endothelial cell migration by
constitutively active LPA(1)-expressing tumor cells. Biochem Biophys Res Commun.
2012;422(2):339-343.
217. Shida D, Fang X, Kordula T, et al. Cross-talk between LPA1 and epidermal growth
factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer
cell motility and invasion. Cancer Res. 2008;68(16):6569-6577.
218. Kato K, Yoshikawa K, Tanabe E, et al. Opposite roles of LPA1 and LPA3 on cell
motile and invasive activities of pancreatic cancer cells. Tumour Biol. 2012;33(5):1739-
1744.
219. Hayashi M, Okabe K, Kato K, et al. Differential function of lysophosphatidic acid
receptors in cell proliferation and migration of neuroblastoma cells. Cancer Lett.
2012;316(1):91-96.
220. Windischhofer W, Huber E, Rossmann C, et al. LPA-induced suppression of periostin
in human osteosarcoma cells is mediated by the LPA(1)/Egr-1 axis. Biochimie.
2012;94(9):1997-2005.
221. Contos JJ, Chun J. Genomic characterization of the lysophosphatidic acid receptor
gene, lp(A2)/Edg4, and identification of a frameshift mutation in a previously characterized
cDNA. Genomics. 2000;64(2):155-169.
222. Zhang Z, Zhu X. Clinical Significance of Lysophosphatidic Acid Receptor-2 (LPA2)
and Kruppel-Like Factor 5 (KLF5) Protein Expression Detected by Tissue Microarray in
Gastric Adenocarcinoma. Med Sci Monit. 2019;25:4705-4715.
223. Ishimoto K, Minami A, Minami K, Ueda N, Tsujiuchi T. Different effects of
lysophosphatidic acid receptor-2 (LPA2) and LPA5 on the regulation of chemoresistance in
colon cancer cells. J Recept Signal Transduct Res. 2021;41(1):93-98.
224. Minami K, Ueda N, Ishimoto K, Tsujiuchi T. Lysophosphatidic acid receptor-2
(LPA2)-mediated signaling enhances chemoresistance in melanoma cells treated with
anticancer drugs. Mol Cell Biochem. 2020;469(1-2):89-95.
225. Komachi M, Tomura H, Malchinkhuu E, et al. LPA1 receptors mediate stimulation,
whereas LPA2 receptors mediate inhibition, of migration of pancreatic cancer cells in
response to lysophosphatidic acid and malignant ascites. Carcinogenesis. 2009;30(3):457-
465.
226. Han SG, Baek SI, Son TJ, Lee H, Kim NH, Yu YG. Preparation of functional human
lysophosphatidic acid receptor 2 using a P9( *) expression system and an amphipathic
polymer and investigation of its in vitro binding preference to Galpha proteins. Biochem
Biophys Res Commun. 2017;487(1):103-108.
227. Minami K, Ueda N, Ishimoto K, et al. Cooperation of G12/13 and Gi proteins via
lysophosphatidic acid receptor-2 (LPA2) signaling enhances cancer cell survival to cisplatin.
Biochem Biophys Res Commun. 2020;532(3):427-432.
228. Xu MY, Porte J, Knox AJ, et al. Lysophosphatidic acid induces alphavbeta6 integrin-
mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am
J Pathol. 2009;174(4):1264-1279.

135
229. Geng H, Lan R, Singha PK, et al. Lysophosphatidic acid increases proximal tubule
cell secretion of profibrotic cytokines PDGF-B and CTGF through LPA2- and Galphaq-
mediated Rho and alphavbeta6 integrin-dependent activation of TGF-beta. Am J Pathol.
2012;181(4):1236-1249.
230. Oh YS, Jo NW, Choi JW, et al. NHERF2 specifically interacts with LPA2 receptor
and defines the specificity and efficiency of receptor-mediated phospholipase C-beta3
activation. Mol Cell Biol. 2004;24(11):5069-5079.
231. An S, Bleu T, Zheng Y, Goetzl EJ. Recombinant human G protein-coupled
lysophosphatidic acid receptors mediate intracellular calcium mobilization. Mol Pharmacol.
1998;54(5):881-888.
232. Wang GL, Wen ZQ, Xu WP, Wang ZY, Du XL, Wang F. Inhibition of
lysophosphatidic acid receptor-2 expression by RNA interference decreases lysophosphatidic
acid-induced urokinase plasminogen activator activation, cell invasion, and migration in
ovarian cancer SKOV-3 cells. Croat Med J. 2008;49(2):175-181.
233. Chen SU, Chou CH, Lee H, Ho CH, Lin CW, Yang YS. Lysophosphatidic acid up-
regulates expression of interleukin-8 and -6 in granulosa-lutein cells through its receptors
and nuclear factor-kappaB dependent pathways: implications for angiogenesis of corpus
luteum and ovarian hyperstimulation syndrome. J Clin Endocrinol Metab. 2008;93(3):935-
943.
234. Singla A, Kumar A, Priyamvada S, et al. LPA stimulates intestinal DRA gene
transcription via LPA2 receptor, PI3K/AKT, and c-Fos-dependent pathway. Am J Physiol
Gastrointest Liver Physiol. 2012;302(6):G618-627.
235. Xu J, Lai YJ, Lin WC, Lin FT. TRIP6 enhances lysophosphatidic acid-induced cell
migration by interacting with the lysophosphatidic acid 2 receptor. J Biol Chem.
2004;279(11):10459-10468.
236. E S, Lai YJ, Tsukahara R, et al. Lysophosphatidic acid 2 receptor-mediated
supramolecular complex formation regulates its antiapoptotic effect. J Biol Chem.
2009;284(21):14558-14571.
237. Holcomb J, Jiang Y, Lu G, et al. Structural insights into PDZ-mediated interaction of
NHERF2 and LPA(2), a cellular event implicated in CFTR channel regulation. Biochem
Biophys Res Commun. 2014;446(1):399-403.
238. Lee SJ, Ritter SL, Zhang H, Shim H, Hall RA, Yun CC. MAGI-3 competes with
NHERF-2 to negatively regulate LPA2 receptor signaling in colon cancer cells.
Gastroenterology. 2011;140(3):924-934.
239. Zhang H, Wang D, Sun H, Hall RA, Yun CC. MAGI-3 regulates LPA-induced
activation of Erk and RhoA. Cell Signal. 2007;19(2):261-268.
240. Lin FT, Lin VY, Lin VT, Lin WC. TRIP6 antagonizes the recruitment of A20 and
CYLD to TRAF6 to promote the LPA2 receptor-mediated TRAF6 activation. Cell Discov.
2016;2.
241. Lin FT, Lai YJ, Makarova N, Tigyi G, Lin WC. The lysophosphatidic acid 2 receptor
mediates down-regulation of Siva-1 to promote cell survival. J Biol Chem.
2007;282(52):37759-37769.
242. Ren A, Moon C, Zhang W, et al. Asymmetrical macromolecular complex formation
of lysophosphatidic acid receptor 2 (LPA2) mediates gradient sensing in fibroblasts. J Biol
Chem. 2014;289(52):35757-35769.

136
243. Zhang W, Penmatsa H, Ren A, et al. Functional regulation of cystic fibrosis
transmembrane conductance regulator-containing macromolecular complexes: a small-
molecule inhibitor approach. Biochem J. 2011;435(2):451-462.
244. Sun Y, Nam JS, Han DH, et al. Lysophosphatidic acid induces upregulation of Mcl-
1 and protects apoptosis in a PTX-dependent manner in H19-7 cells. Cell Signal.
2010;22(3):484-494.
245. Vogt J, Kirischuk S, Unichenko P, et al. Synaptic Phospholipid Signaling Modulates
Axon Outgrowth via Glutamate-dependent Ca2+-mediated Molecular Pathways. Cereb
Cortex. 2017;27(1):131-145.
246. Lopez-Serrano C, Santos-Nogueira E, Francos-Quijorna I, Coll-Miro M, Chun J,
Lopez-Vales R. Lysophosphatidic acid receptor type 2 activation contributes to secondary
damage after spinal cord injury in mice. Brain Behav Immun. 2019;76:258-267.
247. Tanaka T, Morito K, Kinoshita M, et al. Orally administered phosphatidic acids and
lysophosphatidic acids ameliorate aspirin-induced stomach mucosal injury in mice. Dig Dis
Sci. 2013;58(4):950-958.
248. Tanaka T, Ohmoto M, Morito K, et al. Type 2 lysophosphatidic acid receptor in
gastric surface mucous cells: Possible implication of prostaglandin E2 production.
Biofactors. 2014;40(3):355-361.
249. Deng W, Shuyu E, Tsukahara R, et al. The lysophosphatidic acid type 2 receptor is
required for protection against radiation-induced intestinal injury. Gastroenterology.
2007;132(5):1834-1851.
250. Singla A, Dwivedi A, Saksena S, et al. Mechanisms of lysophosphatidic acid (LPA)
mediated stimulation of intestinal apical Cl-/OH- exchange. Am J Physiol Gastrointest Liver
Physiol. 2010;298(2):G182-189.
251. Kuo B, Szabo E, Lee SC, et al. The LPA2 receptor agonist Radioprotectin-1 spares
Lgr5-positive intestinal stem cells from radiation injury in murine enteroids. Cell Signal.
2018;51:23-33.
252. Balogh A, Shimizu Y, Lee SC, et al. The autotaxin-LPA2 GPCR axis is modulated
by gamma-irradiation and facilitates DNA damage repair. Cell Signal. 2015;27(9):1751-
1762.
253. Patil R, Szabo E, Fells JI, et al. Combined mitigation of the gastrointestinal and
hematopoietic acute radiation syndromes by an LPA2 receptor-specific nonlipid agonist.
Chem Biol. 2015;22(2):206-216.
254. Kiss GN, Lee SC, Fells JI, et al. Mitigation of radiation injury by selective stimulation
of the LPA(2) receptor. Biochim Biophys Acta. 2013;1831(1):117-125.
255. Schleicher SM, Thotala DK, Linkous AG, et al. Autotaxin and LPA receptors
represent potential molecular targets for the radiosensitization of murine glioma through
effects on tumor vasculature. PLoS One. 2011;6(7):e22182.
256. Chen RJ, Chen SU, Chou CH, Lin MC. Lysophosphatidic acid receptor 2/3-mediated
IL-8-dependent angiogenesis in cervical cancer cells. Int J Cancer. 2012;131(4):789-802.
257. Mu H, Calderone TL, Davies MA, et al. Lysophosphatidic acid induces
lymphangiogenesis and IL-8 production in vitro in human lymphatic endothelial cells. Am J
Pathol. 2012;180(5):2170-2181.
258. Panchatcharam M, Miriyala S, Yang F, et al. Lysophosphatidic acid receptors 1 and
2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res.
2008;103(6):662-670.

137
259. Ho YH, Yao CL, Lin KH, et al. Opposing regulation of megakaryopoiesis by LPA
receptors 2 and 3 in K562 human erythroleukemia cells. Biochim Biophys Acta.
2015;1851(2):172-183.
260. Chiang JC, Chen WM, Lin KH, et al. Lysophosphatidic acid receptors 2 and 3
regulate erythropoiesis at different hematopoietic stages. Biochim Biophys Acta Mol Cell Biol
Lipids. 2021;1866(1):158818.
261. Lin KH, Ho YH, Chiang JC, et al. Pharmacological activation of lysophosphatidic
acid receptors regulates erythropoiesis. Sci Rep. 2016;6:27050.
262. Puigdomenech-Poch M, Martinez-Muriana A, Andres-Benito P, Ferrer I, Chun J,
Lopez-Vales R. Dual Role of Lysophosphatidic Acid Receptor 2 (LPA2) in Amyotrophic
Lateral Sclerosis. Front Cell Neurosci. 2021;15:600872.
263. Wang Z, Shi W, Tian D, et al. Autotaxin stimulates LPA2 receptor in macrophages
and exacerbates dextran sulfate sodium-induced acute colitis. J Mol Med (Berl).
2020;98(12):1781-1794.
264. Gu C, Wang F, Zhao Z, Wang H, Cong X, Chen X. Lysophosphatidic Acid Is
Associated with Atherosclerotic Plaque Instability by Regulating NF-kappaB Dependent
Matrix Metalloproteinase-9 Expression via LPA2 in Macrophages. Front Physiol.
2017;8:266.
265. Emo J, Meednu N, Chapman TJ, et al. Lpa2 is a negative regulator of both dendritic
cell activation and murine models of allergic lung inflammation. J Immunol.
2012;188(8):3784-3790.
266. Kondo M, Tezuka T, Ogawa H, et al. Lysophosphatidic Acid Regulates the
Differentiation of Th2 Cells and Its Antagonist Suppresses Allergic Airway Inflammation.
Int Arch Allergy Immunol. 2021;182(1):1-13.
267. Zhao Y, Tong J, He D, et al. Role of lysophosphatidic acid receptor LPA2 in the
development of allergic airway inflammation in a murine model of asthma. Respir Res.
2009;10:114.
268. Park GY, Lee YG, Berdyshev E, et al. Autotaxin production of lysophosphatidic acid
mediates allergic asthmatic inflammation. Am J Respir Crit Care Med. 2013;188(8):928-940.
269. Knowlden SA, Hillman SE, Chapman TJ, et al. Novel Inhibitory Effect of a
Lysophosphatidic Acid 2 Agonist on Allergen-Driven Airway Inflammation. Am J Respir
Cell Mol Biol. 2016;54(3):402-409.
270. Huang LS, Fu P, Patel P, et al. Lysophosphatidic acid receptor-2 deficiency confers
protection against bleomycin-induced lung injury and fibrosis in mice. Am J Respir Cell Mol
Biol. 2013;49(6):912-922.
271. Lin S, Wang D, Iyer S, et al. The absence of LPA2 attenuates tumor formation in an
experimental model of colitis-associated cancer. Gastroenterology. 2009;136(5):1711-1720.
272. Yamashita H, Kitayama J, Shida D, et al. Differential expression of lysophosphatidic
acid receptor-2 in intestinal and diffuse type gastric cancer. J Surg Oncol. 2006;93(1):30-35.
273. Li M, Xiao D, Zhang J, et al. Expression of LPA2 is associated with poor prognosis
in human breast cancer and regulates HIF-1alpha expression and breast cancer cell growth.
Oncol Rep. 2016;36(6):3479-3487.
274. Chen M, Towers LN, O'Connor KL. LPA2 (EDG4) mediates Rho-dependent
chemotaxis with lower efficacy than LPA1 (EDG2) in breast carcinoma cells. Am J Physiol
Cell Physiol. 2007;292(5):C1927-1933.
275. Hu YL, Albanese C, Pestell RG, Jaffe RB. Dual mechanisms for lysophosphatidic
acid stimulation of human ovarian carcinoma cells. J Natl Cancer Inst. 2003;95(10):733-740.

138
276. Jeong KJ, Park SY, Seo JH, et al. Lysophosphatidic acid receptor 2 and Gi/Src
pathway mediate cell motility through cyclooxygenase 2 expression in CAOV-3 ovarian
cancer cells. Exp Mol Med. 2008;40(6):607-616.
277. Park J, Jang JH, Oh S, et al. LPA-induced migration of ovarian cancer cells requires
activation of ERM proteins via LPA1 and LPA2. Cell Signal. 2018;44:138-147.
278. Taghavi P, Verhoeven E, Jacobs JJ, et al. In vitro genetic screen identifies a
cooperative role for LPA signaling and c-Myc in cell transformation. Oncogene.
2008;27(54):6806-6816.
279. Kortlever RM, Brummelkamp TR, van Meeteren LA, Moolenaar WH, Bernards R.
Suppression of the p53-dependent replicative senescence response by lysophosphatidic acid
signaling. Mol Cancer Res. 2008;6(9):1452-1460.
280. No YR, Lee SJ, Kumar A, Yun CC. HIF1alpha-Induced by Lysophosphatidic Acid Is
Stabilized via Interaction with MIF and CSN5. PLoS One. 2015;10(9):e0137513.
281. Yun CC, Sun H, Wang D, et al. LPA2 receptor mediates mitogenic signals in human
colon cancer cells. Am J Physiol Cell Physiol. 2005;289(1):C2-11.
282. Ueda N, Minami K, Ishimoto K, Tsujiuchi T. Effects of lysophosphatidic acid (LPA)
receptor-2 (LPA2) and LPA3 on the regulation of chemoresistance to anticancer drug in lung
cancer cells. Cell Signal. 2020;69:109551.
283. Im DS, Heise CE, Harding MA, et al. Molecular cloning and characterization of a
lysophosphatidic acid receptor, Edg-7, expressed in prostate. Mol Pharmacol.
2000;57(4):753-759.
284. Contos JJ, Chun J. The mouse lp(A3)/Edg7 lysophosphatidic acid receptor gene:
genomic structure, chromosomal localization, and expression pattern. Gene.
2001;267(2):243-253.
285. Ye X, Herr DR, Diao H, Rivera R, Chun J. Unique uterine localization and regulation
may differentiate LPA3 from other lysophospholipid receptors for its role in embryo
implantation. Fertil Steril. 2011;95(6):2107-2113, 2113 e2101-2104.
286. Ye X, Hama K, Contos JJ, et al. LPA3-mediated lysophosphatidic acid signalling in
embryo implantation and spacing. Nature. 2005;435(7038):104-108.
287. Hama K, Aoki J, Inoue A, et al. Embryo spacing and implantation timing are
differentially regulated by LPA3-mediated lysophosphatidic acid signaling in mice. Biol
Reprod. 2007;77(6):954-959.
288. Wei H, Wang F, Wang X, et al. Lysophosphatidic acid promotes secretion of VEGF
by increasing expression of 150-kD Oxygen-regulated protein (ORP150) in mesenchymal
stem cells. Biochim Biophys Acta. 2013;1831(8):1426-1434.
289. Wang F, Liu S, Pei J, et al. LPA3-mediated lysophosphatidic acid signaling promotes
postnatal heart regeneration in mice. Theranostics. 2020;10(24):10892-10907.
290. D'Aquilio F, Procaccini M, Izzi V, et al. Activatory properties of lysophosphatidic
acid on human THP-1 cells. Inflammation. 2005;29(4-6):129-140.
291. Shim SJ, Shin E, Lee CS, Koo JS. The expressions of autotaxin-lysophosphatidate
signaling-related proteins in metastatic breast cancer. Int J Clin Exp Pathol.
2019;12(8):2920-2930.
292. Cai H, Xu Y. The role of LPA and YAP signaling in long-term migration of human
ovarian cancer cells. Cell Commun Signal. 2013;11(1):31.
293. Zuo C, Li X, Huang J, et al. Osteoglycin attenuates cardiac fibrosis by suppressing
cardiac myofibroblast proliferation and migration through antagonizing lysophosphatidic

139
acid 3/matrix metalloproteinase 2/epidermal growth factor receptor signalling. Cardiovasc
Res. 2018;114(5):703-712.
294. Hernandez M, Barrero MJ, Crespo MS, Nieto ML. Lysophosphatidic acid inhibits
Ca2+ signaling in response to epidermal growth factor receptor stimulation in human
astrocytoma cells by a mechanism involving phospholipase C(gamma) and a G(alphai)
protein. J Neurochem. 2000;75(4):1575-1582.
295. Lin CC, Lin CE, Lin YC, et al. Lysophosphatidic acid induces reactive oxygen species
generation by activating protein kinase C in PC-3 human prostate cancer cells. Biochem
Biophys Res Commun. 2013;440(4):564-569.
296. Sriwai W, Zhou H, Murthy KS. G(q)-dependent signalling by the lysophosphatidic
acid receptor LPA(3) in gastric smooth muscle: reciprocal regulation of MYPT1
phosphorylation by Rho kinase and cAMP-independent PKA. Biochem J. 2008;411(3):543-
551.
297. Chiang CL, Chen SS, Lee SJ, et al. Lysophosphatidic acid induces erythropoiesis
through activating lysophosphatidic acid receptor 3. Stem Cells. 2011;29(11):1763-1773.
298. Lin CI, Chen CN, Huang MT, et al. Lysophosphatidic acid upregulates vascular
endothelial growth factor-C and tube formation in human endothelial cells through LPA(1/3),
COX-2, and NF-kappaB activation- and EGFR transactivation-dependent mechanisms. Cell
Signal. 2008;20(10):1804-1814.
299. Ma L, Uchida H, Nagai J, et al. Lysophosphatidic acid-3 receptor-mediated feed-
forward production of lysophosphatidic acid: an initiator of nerve injury-induced neuropathic
pain. Mol Pain. 2009;5:64.
300. Furuta D, Yamane M, Tsujiuchi T, Moriyama R, Fukushima N. Lysophosphatidic
acid induces neurite branch formation through LPA3. Mol Cell Neurosci. 2012;50(1):21-34.
301. Fujita R, Ma Y, Ueda H. Lysophosphatidic acid-induced membrane ruffling and
brain-derived neurotrophic factor gene expression are mediated by ATP release in primary
microglia. J Neurochem. 2008;107(1):152-160.
302. Lin KH, Li MW, Chang YC, et al. Activation of Lysophosphatidic Acid Receptor 3
Inhibits Megakaryopoiesis in Human Hematopoietic Stem Cells and Zebrafish. Stem Cells
Dev. 2018;27(3):216-224.
303. Saatian B, Zhao Y, He D, et al. Transcriptional regulation of lysophosphatidic acid-
induced interleukin-8 expression and secretion by p38 MAPK and JNK in human bronchial
epithelial cells. Biochem J. 2006;393(Pt 3):657-668.
304. Rivera-Lopez CM, Tucker AL, Lynch KR. Lysophosphatidic acid (LPA) and
angiogenesis. Angiogenesis. 2008;11(3):301-310.
305. Yang J, Nie Y, Wang F, et al. Reciprocal regulation of miR-23a and lysophosphatidic
acid receptor signaling in cardiomyocyte hypertrophy. Biochim Biophys Acta.
2013;1831(8):1386-1394.
306. Yang J, Xu J, Han X, et al. Lysophosphatidic Acid Is Associated With Cardiac
Dysfunction and Hypertrophy by Suppressing Autophagy via the LPA3/AKT/mTOR
Pathway. Front Physiol. 2018;9:1315.
307. Chen J, Chen Y, Zhu W, et al. Specific LPA receptor subtype mediation of LPA-
induced hypertrophy of cardiac myocytes and involvement of Akt and NFkappaB signal
pathways. J Cell Biochem. 2008;103(6):1718-1731.
308. Chan LC, Peters W, Xu Y, Chun J, Farese RV, Jr., Cases S. LPA3 receptor mediates
chemotaxis of immature murine dendritic cells to unsaturated lysophosphatidic acid (LPA).
J Leukoc Biol. 2007;82(5):1193-1200.

140
309. Chang CL, Hsu HY, Lin HY, Chiang W, Lee H. Lysophosphatidic acid-induced
oxidized low-density lipoprotein uptake is class A scavenger receptor-dependent in
macrophages. Prostaglandins Other Lipid Mediat. 2008;87(1-4):20-25.
310. Chen L, Zhang J, Deng X, et al. Lysophosphatidic acid directly induces macrophage-
derived foam cell formation by blocking the expression of SRBI. Biochem Biophys Res
Commun. 2017;491(3):587-594.
311. Chen L, Zhang J, Yang X, Liu Y, Deng X, Yu C. Lysophosphatidic acid decreased
macrophage foam cell migration correlated with downregulation of fucosyltransferase 8 via
HNF1alpha. Atherosclerosis. 2019;290:19-30.
312. Wei Q, St Clair JB, Fu T, Stratton P, Nieman LK. Reduced expression of biomarkers
associated with the implantation window in women with endometriosis. Fertil Steril.
2009;91(5):1686-1691.
313. Achache H, Tsafrir A, Prus D, Reich R, Revel A. Defective endometrial prostaglandin
synthesis identified in patients with repeated implantation failure undergoing in vitro
fertilization. Fertil Steril. 2010;94(4):1271-1278.
314. Beltrame JS, Sordelli MS, Canumil VA, Franchi AM, Ribeiro ML. Lysophosphatidic
acid-triggered pathways promote the acquisition of trophoblast endovascular phenotype in
vitro. J Cell Biochem. 2018;119(1):758-772.
315. Inoue S, Tanabe E, Shibata A, et al. Ethionine regulates cell motile activity through
LPA receptor-3 in liver epithelial WB-F344 cells. Mol Cell Biochem. 2013;383(1-2):173-
177.
316. Sordelli MS, Beltrame JS, Cella M, et al. Interaction between lysophosphatidic acid,
prostaglandins and the endocannabinoid system during the window of implantation in the rat
uterus. PLoS One. 2012;7(9):e46059.
317. Shah BH, Catt KJ. Roles of LPA3 and COX-2 in implantation. Trends Endocrinol
Metab. 2005;16(9):397-399.
318. Chen Q, Zhang Y, Peng H, et al. Transient {beta}2-adrenoceptor activation confers
pregnancy loss by disrupting embryo spacing at implantation. J Biol Chem.
2011;286(6):4349-4356.
319. Markiewicz W, Kaminska K, Bogacki M, Maslanka T, Jaroszewski J. Participation
of analogues of lysophosphatidic acid (LPA): oleoyl-sn-glycero-3-phosphate (L-alpha-LPA)
and 1-oleoyl-2-O-methyl-rac-glycerophosphothionate (OMPT) in uterine smooth muscle
contractility of the pregnant pigs. Pol J Vet Sci. 2012;15(4):635-643.
320. Si J, Su Y, Wang Y, Yan YL, Tang YL. Expressions of lysophosphatidic acid
receptors in the development of human ovarian carcinoma. Int J Clin Exp Med.
2015;8(10):17880-17890.
321. Okabe K, Hayashi M, Kato K, et al. Lysophosphatidic acid receptor-3 increases
tumorigenicity and aggressiveness of rat hepatoma RH7777 cells. Mol Carcinog.
2013;52(4):247-254.
322. Nakamoto T, Yasuda K, Yasuhara M, et al. Expression of the endothelial cell
differentiation gene 7 (EDG-7), a lysophosphatidic acid receptor, in ovarian tumor. J Obstet
Gynaecol Res. 2005;31(4):344-351.
323. Sun K, Cai H, Duan X, et al. Aberrant expression and potential therapeutic target of
lysophosphatidic acid receptor 3 in triple-negative breast cancers. Clin Exp Med.
2015;15(3):371-380.

141
324. Fukui R, Kato K, Okabe K, et al. Enhancement of Drug Resistance by
Lysophosphatidic Acid Receptor-3 in Mouse Mammary Tumor FM3A Cells. J Toxicol
Pathol. 2012;25(3):225-228.
325. Lin CE, Chen SU, Lin CC, et al. Lysophosphatidic acid enhances vascular endothelial
growth factor-C expression in human prostate cancer PC-3 cells. PLoS One.
2012;7(7):e41096.
326. Minami K, Ueda N, Ishimoto K, et al. Roles of endothelial cells in the regulation of
cell motility via lysophosphatidic acid receptor-2 (LPA2) and LPA3 in osteosarcoma cells.
Exp Mol Pathol. 2021;118:104596.
327. Tanabe E, Kitayoshi M, Hirane M, et al. Downregulation of activation factors of
endothelia and fibroblasts via lysophosphatidic acid signaling in a mouse lung cancer LL/2
cell line. J Recept Signal Transduct Res. 2013;33(5):286-290.
328. Fukui R, Tanabe E, Kitayoshi M, Yoshikawa K, Fukushima N, Tsujiuchi T. Negative
regulation of cell motile and invasive activities by lysophosphatidic acid receptor-3 in colon
cancer HCT116 cells. Tumour Biol. 2012;33(6):1899-1905.
329. Lee CW, Rivera R, Dubin AE, Chun J. LPA(4)/GPR23 is a lysophosphatidic acid
(LPA) receptor utilizing G(s)-, G(q)/G(i)-mediated calcium signaling and G(12/13)-mediated
Rho activation. J Biol Chem. 2007;282(7):4310-4317.
330. Lee Z, Cheng CT, Zhang H, et al. Role of LPA4/p2y9/GPR23 in negative regulation
of cell motility. Mol Biol Cell. 2008;19(12):5435-5445.
331. Takahashi K, Fukushima K, Onishi Y, et al. Lysophosphatidic acid (LPA) signaling
via LPA4 and LPA6 negatively regulates cell motile activities of colon cancer cells. Biochem
Biophys Res Commun. 2017;483(1):652-657.
332. Ishii S, Hirane M, Fukushima K, Tomimatsu A, Fukushima N, Tsujiuchi T. Diverse
effects of LPA4, LPA5 and LPA6 on the activation of tumor progression in pancreatic cancer
cells. Biochem Biophys Res Commun. 2015;461(1):59-64.
333. Liu YB, Kharode Y, Bodine PV, Yaworsky PJ, Robinson JA, Billiard J. LPA induces
osteoblast differentiation through interplay of two receptors: LPA1 and LPA4. J Cell
Biochem. 2010;109(4):794-800.
334. Salles JP, Laurencin-Dalicieux S, Conte-Auriol F, Briand-Mesange F, Gennero I.
Bone defects in LPA receptor genetically modified mice. Biochim Biophys Acta.
2013;1831(1):93-98.
335. Takara K, Eino D, Ando K, et al. Lysophosphatidic Acid Receptor 4 Activation
Augments Drug Delivery in Tumors by Tightening Endothelial Cell-Cell Contact. Cell Rep.
2017;20(9):2072-2086.
336. Hata E, Sasaki N, Takeda A, et al. Lysophosphatidic acid receptors LPA4 and LPA6
differentially promote lymphocyte transmigration across high endothelial venules in lymph
nodes. Int Immunol. 2016;28(6):283-292.
337. Sumida H, Noguchi K, Kihara Y, et al. LPA4 regulates blood and lymphatic vessel
formation during mouse embryogenesis. Blood. 2010;116(23):5060-5070.
338. Yukiura H, Hama K, Nakanaga K, et al. Autotaxin regulates vascular development
via multiple lysophosphatidic acid (LPA) receptors in zebrafish. J Biol Chem.
2011;286(51):43972-43983.
339. Yasuda D, Kobayashi D, Akahoshi N, et al. Lysophosphatidic acid-induced
YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4.
J Clin Invest. 2019;129(10):4332-4349.

142
340. Rhee HJ, Nam JS, Sun Y, et al. Lysophosphatidic acid stimulates cAMP accumulation
and cAMP response element-binding protein phosphorylation in immortalized hippocampal
progenitor cells. Neuroreport. 2006;17(5):523-526.
341. Eino D, Tsukada Y, Naito H, et al. LPA4-Mediated Vascular Network Formation
Increases the Efficacy of Anti-PD-1 Therapy against Brain Tumors. Cancer Res.
2018;78(23):6607-6620.
342. Yanagida K, Igarashi H, Yasuda D, et al. The Galpha12/13-coupled receptor LPA4
limits proper adipose tissue expansion and remodeling in diet-induced obesity. JCI Insight.
2018;3(24).
343. Harper K, Arsenault D, Boulay-Jean S, Lauzier A, Lucien F, Dubois CM. Autotaxin
promotes cancer invasion via the lysophosphatidic acid receptor 4: participation of the cyclic
AMP/EPAC/Rac1 signaling pathway in invadopodia formation. Cancer Res.
2010;70(11):4634-4643.
344. Kotarsky K, Boketoft A, Bristulf J, et al. Lysophosphatidic acid binds to and activates
GPR92, a G protein-coupled receptor highly expressed in gastrointestinal lymphocytes. J
Pharmacol Exp Ther. 2006;318(2):619-628.
345. Amisten S, Braun OO, Bengtsson A, Erlinge D. Gene expression profiling for the
identification of G-protein coupled receptors in human platelets. Thromb Res.
2008;122(1):47-57.
346. Sapkota A, Lee CH, Park SJ, Choi JW. Lysophosphatidic Acid Receptor 5 Plays a
Pathogenic Role in Brain Damage after Focal Cerebral Ischemia by Modulating
Neuroinflammatory Responses. Cells. 2020;9(6).
347. Sapkota A, Park SJ, Choi JW. Receptor for Advanced Glycation End Products Is
Involved in LPA5-Mediated Brain Damage after a Transient Ischemic Stroke. Life (Basel).
2021;11(2).
348. Callaerts-Vegh Z, Leo S, Vermaercke B, Meert T, D'Hooge R. LPA5 receptor plays
a role in pain sensitivity, emotional exploration and reversal learning. Genes Brain Behav.
2012;11(8):1009-1019.
349. Hu J, Oda SK, Shotts K, et al. Lysophosphatidic acid receptor 5 inhibits B cell antigen
receptor signaling and antibody response. J Immunol. 2014;193(1):85-95.
350. Mathew D, Kremer KN, Strauch P, Tigyi G, Pelanda R, Torres RM. LPA5 Is an
Inhibitory Receptor That Suppresses CD8 T-Cell Cytotoxic Function via Disruption of Early
TCR Signaling. Front Immunol. 2019;10:1159.
351. Ciesielska A, Hromada-Judycka A, Ziemlinska E, Kwiatkowska K. Lysophosphatidic
acid up-regulates IL-10 production to inhibit TNF-alpha synthesis in Mvarphis stimulated
with LPS. J Leukoc Biol. 2019;106(6):1285-1301.
352. Plastira I, Joshi L, Bernhart E, et al. Small-Molecule Lysophosphatidic Acid Receptor
5 (LPAR5) Antagonists: Versatile Pharmacological Tools to Regulate Inflammatory
Signaling in BV-2 Microglia Cells. Front Cell Neurosci. 2019;13:531.
353. Minami K, Ueda N, Maeda H, Ishimoto K, Otagaki S, Tsujiuchi T. Modulation of
chemoresistance by lysophosphatidic acid (LPA) signaling through LPA5 in melanoma cells
treated with anticancer drugs. Biochem Biophys Res Commun. 2019;517(2):359-363.
354. Williams JR, Khandoga AL, Goyal P, et al. Unique ligand selectivity of the
GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J Biol
Chem. 2009;284(25):17304-17319.

143
355. Oh DY, Yoon JM, Moon MJ, et al. Identification of farnesyl pyrophosphate and N-
arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283(30):21054-
21064.
356. Cha B, Chen T, Sarker R, et al. Lysophosphatidic acid stimulation of NHE3
exocytosis in polarized epithelial cells occurs with release from NHERF2 via ERK-PLC-
PKCdelta signaling. Am J Physiol Cell Physiol. 2014;307(1):C55-65.
357. Yoo BK, He P, Lee SJ, Yun CC. Lysophosphatidic acid 5 receptor induces activation
of Na(+)/H(+) exchanger 3 via apical epidermal growth factor receptor in intestinal epithelial
cells. Am J Physiol Cell Physiol. 2011;301(5):C1008-1016.
358. Kittaka H, Uchida K, Fukuta N, Tominaga M. Lysophosphatidic acid-induced itch is
mediated by signalling of LPA5 receptor, phospholipase D and TRPA1/TRPV1. J Physiol.
2017;595(8):2681-2698.
359. Jenkin KA, He P, Yun CC. Expression of lysophosphatidic acid receptor 5 is
necessary for the regulation of intestinal Na(+)/H(+) exchanger 3 by lysophosphatidic acid
in vivo. Am J Physiol Gastrointest Liver Physiol. 2018;315(4):G433-G442.
360. Gaire BP, Lee CH, Kim W, Sapkota A, Lee DY, Choi JW. Lysophosphatidic Acid
Receptor 5 Contributes to Imiquimod-Induced Psoriasis-Like Lesions through NLRP3
Inflammasome Activation in Macrophages. Cells. 2020;9(8).
361. Plastira I, Bernhart E, Goeritzer M, et al. 1-Oleyl-lysophosphatidic acid (LPA)
promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype.
J Neuroinflammation. 2016;13(1):205.
362. Lundequist A, Boyce JA. LPA5 is abundantly expressed by human mast cells and
important for lysophosphatidic acid induced MIP-1beta release. PLoS One.
2011;6(3):e18192.
363. Khandoga AL, Fujiwara Y, Goyal P, et al. Lysophosphatidic acid-induced platelet
shape change revealed through LPA(1-5) receptor-selective probes and albumin. Platelets.
2008;19(6):415-427.
364. Minami K, Ueda N, Ishimoto K, Tsujiuchi T. LPA5-mediated signaling induced by
endothelial cells and anticancer drug regulates cellular functions of osteosarcoma cells. Exp
Cell Res. 2020;388(1):111813.
365. Jongsma M, Matas-Rico E, Rzadkowski A, Jalink K, Moolenaar WH. LPA is a
chemorepellent for B16 melanoma cells: action through the cAMP-elevating LPA5 receptor.
PLoS One. 2011;6(12):e29260.
366. Dong Y, Hirane M, Araki M, Fukushima N, Tsujiuchi T. Lysophosphatidic acid
receptor-5 negatively regulates cellular responses in mouse fibroblast 3T3 cells. Biochem
Biophys Res Commun. 2014;446(2):585-589.
367. Dong Y, Hirane M, Araki M, Fukushima N, Honoki K, Tsujiuchi T. Lysophosphatidic
acid receptor-5 negatively regulates cell motile and invasive activities of human sarcoma cell
lines. Mol Cell Biochem. 2014;393(1-2):17-22.
368. Araki M, Kitayoshi M, Dong Y, et al. Inhibitory effects of lysophosphatidic acid
receptor-5 on cellular functions of sarcoma cells. Growth Factors. 2014;32(3-4):117-122.
369. Lee SC, Fujiwara Y, Tigyi GJ. Uncovering unique roles of LPA receptors in the tumor
microenvironment. Receptors Clin Investig. 2015;2(1).
370. Lee SC, Fujiwara Y, Liu J, et al. Autotaxin and LPA1 and LPA5 receptors exert
disparate functions in tumor cells versus the host tissue microenvironment in melanoma
invasion and metastasis. Mol Cancer Res. 2015;13(1):174-185.

144
371. Hidaka M, Nishihara M, Tokumura A. Three lysophosphatidic acids with a distinct
long chain moiety differently affect cell differentiation of human colon epithelial cells to
goblet cells. Life Sci. 2018;197:73-79.
372. Zhao WJ, Zhu LL, Yang WQ, et al. LPAR5 promotes thyroid carcinoma cell
proliferation and migration by activating class IA PI3K catalytic subunit p110beta. Cancer
Sci. 2021;112(4):1624-1632.
373. Takahashi K, Fukushima K, Otagaki S, et al. Effects of LPA1 and LPA6 on the
regulation of colony formation activity in colon cancer cells treated with anticancer drugs. J
Recept Signal Transduct Res. 2018;38(1):71-75.
374. Ren Y, Guo L, Tang X, et al. Comparing the differential effects of LPA on the barrier
function of human pulmonary endothelial cells. Microvasc Res. 2013;85:59-67.
375. Masago K, Kihara Y, Yanagida K, et al. Lysophosphatidic acid receptor, LPA6,
regulates endothelial blood-brain barrier function: Implication for hepatic encephalopathy.
Biochem Biophys Res Commun. 2018;501(4):1048-1054.
376. Yanagida K, Masago K, Nakanishi H, et al. Identification and characterization of a
novel lysophosphatidic acid receptor, p2y5/LPA6. J Biol Chem. 2009;284(26):17731-17741.
377. Lee M, Choi S, Hallden G, Yo SJ, Schichnes D, Aponte GW. P2Y5 is a G(alpha)i,
G(alpha)12/13 G protein-coupled receptor activated by lysophosphatidic acid that reduces
intestinal cell adhesion. Am J Physiol Gastrointest Liver Physiol. 2009;297(4):G641-654.
378. Yukiura H, Kano K, Kise R, Inoue A, Aoki J. LPP3 localizes LPA6 signalling to non-
contact sites in endothelial cells. J Cell Sci. 2015;128(21):3871-3877.
379. Hayashi R, Inoue A, Suga Y, Aoki J, Shimomura Y. Analysis of unique mutations in
the LPAR6 gene identified in a Japanese family with autosomal recessive woolly
hair/hypotrichosis: Establishment of a useful assay system for LPA6. J Dermatol Sci.
2015;78(3):197-205.
380. Okazoe H, Zhang X, Liu D, et al. Expression and role of GPR87 in urothelial
carcinoma of the bladder. Int J Mol Sci. 2013;14(6):12367-12379.
381. Wang L, Zhou W, Zhong Y, et al. Overexpression of G protein-coupled receptor
GPR87 promotes pancreatic cancer aggressiveness and activates NF-kappaB signaling
pathway. Mol Cancer. 2017;16(1):61.
382. Glatt S, Halbauer D, Heindl S, et al. hGPR87 contributes to viability of human tumor
cells. Int J Cancer. 2008;122(9):2008-2016.
383. Sanchez JF, Krause JE, Cortright DN. The distribution and regulation of vanilloid
receptor VR1 and VR1 5' splice variant RNA expression in rat. Neuroscience.
2001;107(3):373-381.
384. Toth A, Czikora A, Pasztor ET, et al. Vanilloid receptor-1 (TRPV1) expression and
function in the vasculature of the rat. J Histochem Cytochem. 2014;62(2):129-144.
385. Morales-Lazaro SL, Rosenbaum T. A painful link between the TRPV1 channel and
lysophosphatidic acid. Life Sci. 2015;125:15-24.
386. Bujak JK, Kosmala D, Szopa IM, Majchrzak K, Bednarczyk P. Inflammation, Cancer
and Immunity-Implication of TRPV1 Channel. Front Oncol. 2019;9:1087.
387. Morales-Lazaro SL, Serrano-Flores B, Llorente I, et al. Structural determinants of the
transient receptor potential 1 (TRPV1) channel activation by phospholipid analogs. J Biol
Chem. 2014;289(35):24079-24090.
388. Tigyi G. Lipids: LPA activates TRPV1--and it hurts. Nat Chem Biol. 2011;8(1):22-
23.

145
389. Phan TX, Ton HT, Gulyas H, et al. TRPV1 expressed throughout the arterial
circulation regulates vasoconstriction and blood pressure. J Physiol. 2020;598(24):5639-
5659.
390. Hernandez-Araiza I, Morales-Lazaro SL, Canul-Sanchez JA, Islas LD, Rosenbaum
T. Role of lysophosphatidic acid in ion channel function and disease. J Neurophysiol.
2018;120(3):1198-1211.
391. Marx N, Duez H, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors
and atherogenesis: regulators of gene expression in vascular cells. Circ Res.
2004;94(9):1168-1178.
392. Sharma AM, Staels B. Review: Peroxisome proliferator-activated receptor gamma
and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose
metabolism. J Clin Endocrinol Metab. 2007;92(2):386-395.
393. Cheng Y, Makarova N, Tsukahara R, et al. Lysophosphatidic acid-induced arterial
wall remodeling: requirement of PPARgamma but not LPA1 or LPA2 GPCR. Cell Signal.
2009;21(12):1874-1884.
394. Gustin C, Van Steenbrugge M, Raes M. LPA modulates monocyte migration directly
and via LPA-stimulated endothelial cells. Am J Physiol Cell Physiol. 2008;295(4):C905-914.
395. Ray R, Rai V. Lysophosphatidic acid converts monocytes into macrophages in both
mice and humans. Blood. 2017;129(9):1177-1183.
396. Bagga S, Price KS, Lin DA, Friend DS, Austen KF, Boyce JA. Lysophosphatidic acid
accelerates the development of human mast cells. Blood. 2004;104(13):4080-4087.
397. Rodway HA, Hunt AN, Kohler JA, Postle AD, Lillycrop KA. Lysophosphatidic acid
attenuates the cytotoxic effects and degree of peroxisome proliferator-activated receptor
gamma activation induced by 15-deoxyDelta12,14-prostaglandin J2 in neuroblastoma cells.
Biochem J. 2004;382(Pt 1):83-91.
398. Murch O, Collin M, Thiemermann C. Lysophosphatidic acid reduces the organ injury
caused by endotoxemia-a role for G-protein-coupled receptors and peroxisome proliferator-
activated receptor-gamma. Shock. 2007;27(1):48-54.
399. Tsukahara T, Yamagishi S, Matsuda Y, Haniu H. Lysophosphatidic acid signaling
regulates the KLF9-PPARgamma axis in human induced pluripotent stem cell-derived
neurons. Biochem Biophys Res Commun. 2017;491(1):223-227.
400. Tomsig JL, Snyder AH, Berdyshev EV, et al. Lipid phosphate phosphohydrolase type
1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J. 2009;419(3):611-
618.
401. Roberts R, Sciorra VA, Morris AJ. Human type 2 phosphatidic acid
phosphohydrolases. Substrate specificity of the type 2a, 2b, and 2c enzymes and cell surface
activity of the 2a isoform. J Biol Chem. 1998;273(34):22059-22067.
402. van der Bend RL, de Widt J, van Corven EJ, Moolenaar WH, van Blitterswijk WJ.
Metabolic conversion of the biologically active phospholipid, lysophosphatidic acid, in
fibroblasts. Biochim Biophys Acta. 1992;1125(1):110-112.
403. Li J, Dong Y, Lu X, et al. Crystal structures and biochemical studies of human
lysophosphatidic acid phosphatase type 6. Protein Cell. 2013;4(7):548-561.
404. Hiroyama M, Takenawa T. Isolation of a cDNA encoding human lysophosphatidic
acid phosphatase that is involved in the regulation of mitochondrial lipid biosynthesis. J Biol
Chem. 1999;274(41):29172-29180.

146
405. Tanaka M, Kishi Y, Takanezawa Y, Kakehi Y, Aoki J, Arai H. Prostatic acid
phosphatase degrades lysophosphatidic acid in seminal plasma. FEBS Lett. 2004;571(1-
3):197-204.
406. Zhang QX, Pilquil CS, Dewald J, Berthiaume LG, Brindley DN. Identification of
structurally important domains of lipid phosphate phosphatase-1: implications for its sites of
action. Biochem J. 2000;345 Pt 2:181-184.
407. Long JS, Pyne NJ, Pyne S. Lipid phosphate phosphatases form homo- and hetero-
oligomers: catalytic competency, subcellular distribution and function. Biochem J.
2008;411(2):371-377.
408. Ishikawa T, Kai M, Wada I, Kanoh H. Cell surface activities of the human type 2b
phosphatidic acid phosphatase. J Biochem. 2000;127(4):645-651.
409. Alderton F, Darroch P, Sambi B, et al. G-protein-coupled receptor stimulation of the
p42/p44 mitogen-activated protein kinase pathway is attenuated by lipid phosphate
phosphatases 1, 1a, and 2 in human embryonic kidney 293 cells. J Biol Chem.
2001;276(16):13452-13460.
410. Jia YJ, Kai M, Wada I, Sakane F, Kanoh H. Differential localization of lipid
phosphate phosphatases 1 and 3 to cell surface subdomains in polarized MDCK cells. FEBS
Lett. 2003;552(2-3):240-246.
411. Kai M, Sakane F, Jia YJ, Imai S, Yasuda S, Kanoh H. Lipid phosphate phosphatases
1 and 3 are localized in distinct lipid rafts. J Biochem. 2006;140(5):677-686.
412. Barila D, Plateroti M, Nobili F, et al. The Dri 42 gene, whose expression is up-
regulated during epithelial differentiation, encodes a novel endoplasmic reticulum resident
transmembrane protein. J Biol Chem. 1996;271(47):29928-29936.
413. Long J, Darroch P, Wan KF, et al. Regulation of cell survival by lipid phosphate
phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1-
phosphate pools. Biochem J. 2005;391(Pt 1):25-32.
414. Kai M, Wada I, Imai S, Sakane F, Kanoh H. Cloning and characterization of two
human isozymes of Mg2+-independent phosphatidic acid phosphatase. J Biol Chem.
1997;272(39):24572-24578.
415. Zhang N, Sundberg JP, Gridley T. Mice mutant for Ppap2c, a homolog of the germ
cell migration regulator wunen, are viable and fertile. Genesis. 2000;27(4):137-140.
416. Escalante-Alcalde D, Hernandez L, Le Stunff H, et al. The lipid phosphatase LPP3
regulates extra-embryonic vasculogenesis and axis patterning. Development.
2003;130(19):4623-4637.
417. Yue J, Yokoyama K, Balazs L, et al. Mice with transgenic overexpression of lipid
phosphate phosphatase-1 display multiple organotypic deficits without alteration in
circulating lysophosphatidate level. Cell Signal. 2004;16(3):385-399.
418. Morris KE, Schang LM, Brindley DN. Lipid phosphate phosphatase-2 activity
regulates S-phase entry of the cell cycle in Rat2 fibroblasts. J Biol Chem. 2006;281(14):9297-
9306.
419. Tang X, Benesch MG, Dewald J, et al. Lipid phosphate phosphatase-1 expression in
cancer cells attenuates tumor growth and metastasis in mice. J Lipid Res. 2014;55(11):2389-
2400.
420. Tanyi JL, Morris AJ, Wolf JK, et al. The human lipid phosphate phosphatase-3
decreases the growth, survival, and tumorigenesis of ovarian cancer cells: validation of the
lysophosphatidic acid signaling cascade as a target for therapy in ovarian cancer. Cancer Res.
2003;63(5):1073-1082.

147
421. Panchatcharam M, Salous AK, Brandon J, et al. Mice with targeted inactivation of
ppap2b in endothelial and hematopoietic cells display enhanced vascular inflammation and
permeability. Arterioscler Thromb Vasc Biol. 2014;34(4):837-845.
422. Panchatcharam M, Miriyala S, Salous A, et al. Lipid phosphate phosphatase 3
negatively regulates smooth muscle cell phenotypic modulation to limit intimal hyperplasia.
Arterioscler Thromb Vasc Biol. 2013;33(1):52-59.
423. Hooks SB, Santos WL, Im DS, Heise CE, Macdonald TL, Lynch KR.
Lysophosphatidic acid-induced mitogenesis is regulated by lipid phosphate phosphatases and
is Edg-receptor independent. J Biol Chem. 2001;276(7):4611-4621.
424. Pilquil C, Dewald J, Cherney A, et al. Lipid phosphate phosphatase-1 regulates
lysophosphatidate-induced fibroblast migration by controlling phospholipase D2-dependent
phosphatidate generation. J Biol Chem. 2006;281(50):38418-38429.
425. Tsai MH, Yu CL, Wei FS, Stacey DW. The effect of GTPase activating protein upon
ras is inhibited by mitogenically responsive lipids. Science. 1989;243(4890):522-526.
426. Rizzo MA, Shome K, Watkins SC, Romero G. The recruitment of Raf-1 to
membranes is mediated by direct interaction with phosphatidic acid and is independent of
association with Ras. J Biol Chem. 2000;275(31):23911-23918.
427. Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of
extracellular vesicles 2018 (MISEV2018): a position statement of the International Society
for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles.
2018;7(1):1535750.
428. Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and
archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun.
2012;80(6):1948-1957.
429. Marcoux G, Duchez AC, Cloutier N, Provost P, Nigrovic PA, Boilard E. Revealing
the diversity of extracellular vesicles using high-dimensional flow cytometry analyses. Sci
Rep. 2016;6:35928.
430. Tessandier N, Melki I, Cloutier N, et al. Platelets Disseminate Extracellular Vesicles
in Lymph in Rheumatoid Arthritis. Arterioscler Thromb Vasc Biol. 2020;40(4):929-942.
431. Foers AD, Chatfield S, Dagley LF, et al. Enrichment of extracellular vesicles from
human synovial fluid using size exclusion chromatography. J Extracell Vesicles.
2018;7(1):1490145.
432. Yanez-Mo M, Siljander PR, Andreu Z, et al. Biological properties of extracellular
vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
433. Tetta C, Ghigo E, Silengo L, Deregibus MC, Camussi G. Extracellular vesicles as an
emerging mechanism of cell-to-cell communication. Endocrine. 2013;44(1):11-19.
434. Slomka A, Urban SK, Lukacs-Kornek V, Zekanowska E, Kornek M. Large
Extracellular Vesicles: Have We Found the Holy Grail of Inflammation? Front Immunol.
2018;9:2723.
435. Tian J, Casella G, Zhang Y, Rostami A, Li X. Potential roles of extracellular vesicles
in the pathophysiology, diagnosis, and treatment of autoimmune diseases. Int J Biol Sci.
2020;16(4):620-632.
436. Urbanelli L, Buratta S, Tancini B, et al. The Role of Extracellular Vesicles in Viral
Infection and Transmission. Vaccines (Basel). 2019;7(3).
437. de Jong OG, Kooijmans SAA, Murphy DE, et al. Drug Delivery with Extracellular
Vesicles: From Imagination to Innovation. Acc Chem Res. 2019;52(7):1761-1770.

148
438. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions
of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255-289.
439. Aatonen M, Gronholm M, Siljander PR. Platelet-derived microvesicles: multitalented
participants in intercellular communication. Semin Thromb Hemost. 2012;38(1):102-113.
440. Koshiar RL, Somajo S, Norstrom E, Dahlback B. Erythrocyte-derived microparticles
supporting activated protein C-mediated regulation of blood coagulation. PLoS One.
2014;9(8):e104200.
441. Lipets EN, Antonova OA, Shustova ON, Losenkova KV, Mazurov AV,
Ataullakhanov FI. Use of Thrombodynamics for revealing the participation of platelet,
erythrocyte, endothelial, and monocyte microparticles in coagulation activation and
propagation. PLoS One. 2020;15(5):e0227932.
442. Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria serving
as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation.
Blood. 2014;124(14):2173-2183.
443. Marcoux G, Laroche A, Hasse S, et al. Platelet EVs contain an active proteasome
involved in protein processing for antigen presentation via MHC-I molecules. Blood. 2021.
444. Holmgren L, Szeles A, Rajnavolgyi E, et al. Horizontal transfer of DNA by the uptake
of apoptotic bodies. Blood. 1999;93(11):3956-3963.
445. Singh P, Goel H, Husain M, et al. Tubular cell HIV-entry through apoptosed CD4 T
cells: a novel pathway. Virology. 2012;434(1):68-77.
446. Turiak L, Misjak P, Szabo TG, et al. Proteomic characterization of thymocyte-derived
microvesicles and apoptotic bodies in BALB/c mice. J Proteomics. 2011;74(10):2025-2033.
447. Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis
via collagen-dependent microparticle production. Science. 2010;327(5965):580-583.
448. Arraud N, Linares R, Tan S, et al. Extracellular vesicles from blood plasma:
determination of their morphology, size, phenotype and concentration. J Thromb Haemost.
2014;12(5):614-627.
449. Gyorgy B, Modos K, Pallinger E, et al. Detection and isolation of cell-derived
microparticles are compromised by protein complexes resulting from shared biophysical
parameters. Blood. 2011;117(4):e39-48.
450. Baroni M, Pizzirani C, Pinotti M, et al. Stimulation of P2 (P2X7) receptors in human
dendritic cells induces the release of tissue factor-bearing microparticles. FASEB J.
2007;21(8):1926-1933.
451. Beer KB, Rivas-Castillo J, Kuhn K, et al. Extracellular vesicle budding is inhibited
by redundant regulators of TAT-5 flippase localization and phospholipid asymmetry. Proc
Natl Acad Sci U S A. 2018;115(6):E1127-E1136.
452. Fujii T, Sakata A, Nishimura S, Eto K, Nagata S. TMEM16F is required for
phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc
Natl Acad Sci U S A. 2015;112(41):12800-12805.
453. Linares R, Tan S, Gounou C, Brisson AR. Imaging and Quantification of
Extracellular Vesicles by Transmission Electron Microscopy. Methods Mol Biol.
2017;1545:43-54.
454. Muralidharan-Chari V, Clancy J, Plou C, et al. ARF6-regulated shedding of tumor
cell-derived plasma membrane microvesicles. Curr Biol. 2009;19(22):1875-1885.
455. Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV
Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol.
2006;172(6):923-935.

149
456. Fang Y, Wu N, Gan X, Yan W, Morrell JC, Gould SJ. Higher-order oligomerization
targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol. 2007;5(6):e158.
457. Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q. Formation and release of arrestin domain-
containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment
of TSG101 protein. Proc Natl Acad Sci U S A. 2012;109(11):4146-4151.
458. Hurley JH. ESCRTs are everywhere. EMBO J. 2015;34(19):2398-2407.
459. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular
vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213-228.
460. Gyorgy B, Szabo TG, Pasztoi M, et al. Membrane vesicles, current state-of-the-art:
emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68(16):2667-2688.
461. Thery C, Boussac M, Veron P, et al. Proteomic analysis of dendritic cell-derived
exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol.
2001;166(12):7309-7318.
462. Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles
into multivesicular endosomes. Science. 2008;319(5867):1244-1247.
463. Nazarenko I, Rana S, Baumann A, et al. Cell surface tetraspanin Tspan8 contributes
to molecular pathways of exosome-induced endothelial cell activation. Cancer Res.
2010;70(4):1668-1678.
464. Zhu H, Guariglia S, Yu RY, et al. Mutation of SIMPLE in Charcot-Marie-Tooth 1C
alters production of exosomes. Mol Biol Cell. 2013;24(11):1619-1637, S1611-1613.
465. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol.
2009;10(8):513-525.
466. Zubairova LD, Nabiullina RM, Nagaswami C, et al. Circulating Microparticles Alter
Formation, Structure, and Properties of Fibrin Clots. Sci Rep. 2015;5:17611.
467. Sinauridze EI, Kireev DA, Popenko NY, et al. Platelet microparticle membranes have
50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb
Haemost. 2007;97(3):425-434.
468. Tans G, Rosing J, Thomassen MC, Heeb MJ, Zwaal RF, Griffin JH. Comparison of
anticoagulant and procoagulant activities of stimulated platelets and platelet-derived
microparticles. Blood. 1991;77(12):2641-2648.
469. Berckmans RJ, Nieuwland R, Boing AN, Romijn FP, Hack CE, Sturk A. Cell-derived
microparticles circulate in healthy humans and support low grade thrombin generation.
Thromb Haemost. 2001;85(4):639-646.
470. Bian X, Ma K, Zhang C, Fu X. Therapeutic angiogenesis using stem cell-derived
extracellular vesicles: an emerging approach for treatment of ischemic diseases. Stem Cell
Res Ther. 2019;10(1):158.
471. Lotvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for
definition of extracellular vesicles and their functions: a position statement from the
International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913.
472. Witwer KW, Buzas EI, Bemis LT, et al. Standardization of sample collection,
isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2.
473. Wang W, Luo J, Wang S. Recent Progress in Isolation and Detection of Extracellular
Vesicles for Cancer Diagnostics. Adv Healthc Mater. 2018;7(20):e1800484.
474. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated
transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells.
Nat Cell Biol. 2007;9(6):654-659.

150
475. Ueda K, Ishikawa N, Tatsuguchi A, Saichi N, Fujii R, Nakagawa H. Antibody-
coupled monolithic silica microtips for highthroughput molecular profiling of circulating
exosomes. Sci Rep. 2014;4:6232.
476. Li J, He X, Deng Y, Yang C. An Update on Isolation Methods for Proteomic Studies
of Extracellular Vesicles in Biofluids. Molecules. 2019;24(19).
477. Gardiner C, Di Vizio D, Sahoo S, et al. Techniques used for the isolation and
characterization of extracellular vesicles: results of a worldwide survey. J Extracell Vesicles.
2016;5:32945.
478. Szatanek R, Baj-Krzyworzeka M, Zimoch J, Lekka M, Siedlar M, Baran J. The
Methods of Choice for Extracellular Vesicles (EVs) Characterization. Int J Mol Sci.
2017;18(6).
479. Di Vizio D, Morello M, Dudley AC, et al. Large oncosomes in human prostate cancer
tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;181(5):1573-
1584.
480. Dragovic RA, Gardiner C, Brooks AS, et al. Sizing and phenotyping of cellular
vesicles using Nanoparticle Tracking Analysis. Nanomedicine. 2011;7(6):780-788.
481. Benameur T, Osman A, Parray A, Ait Hssain A, Munusamy S, Agouni A. Molecular
Mechanisms Underpinning Microparticle-Mediated Cellular Injury in Cardiovascular
Complications Associated with Diabetes. Oxid Med Cell Longev. 2019;2019:6475187.
482. Flaumenhaft R, Dilks JR, Richardson J, et al. Megakaryocyte-derived microparticles:
direct visualization and distinction from platelet-derived microparticles. Blood.
2009;113(5):1112-1121.
483. Patel SR, Hartwig JH, Italiano JE, Jr. The biogenesis of platelets from megakaryocyte
proplatelets. J Clin Invest. 2005;115(12):3348-3354.
484. Thon JN, Italiano JE. Platelets: production, morphology and ultrastructure. Handb
Exp Pharmacol. 2012(210):3-22.
485. Cramer EM, Norol F, Guichard J, et al. Ultrastructure of platelet formation by human
megakaryocytes cultured with the Mpl ligand. Blood. 1997;89(7):2336-2346.
486. Schulze H, Shivdasani RA. Molecular mechanisms of megakaryocyte differentiation.
Semin Thromb Hemost. 2004;30(4):389-398.
487. Sabri S, Foudi A, Boukour S, et al. Deficiency in the Wiskott-Aldrich protein induces
premature proplatelet formation and platelet production in the bone marrow compartment.
Blood. 2006;108(1):134-140.
488. Jiang J, Woulfe DS, Papoutsakis ET. Shear enhances thrombopoiesis and formation
of microparticles that induce megakaryocytic differentiation of stem cells. Blood.
2014;124(13):2094-2103.
489. Richardson JL, Shivdasani RA, Boers C, Hartwig JH, Italiano JE, Jr. Mechanisms of
organelle transport and capture along proplatelets during platelet production. Blood.
2005;106(13):4066-4075.
490. Lopez-Vilchez I, Diaz-Ricart M, Galan AM, et al. Internalization of Tissue Factor-
Rich Microvesicles by Platelets Occurs Independently of GPIIb-IIIa, and Involves CD36
Receptor, Serotonin Transporter and Cytoskeletal Assembly. J Cell Biochem.
2016;117(2):448-457.
491. Beikmann BS, Tomlinson ID, Rosenthal SJ, Andrews AM. Serotonin uptake is
largely mediated by platelets versus lymphocytes in peripheral blood cells. ACS Chem
Neurosci. 2013;4(1):161-170.

151
492. Cloutier N, Allaeys I, Marcoux G, et al. Platelets release pathogenic serotonin and
return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S
A. 2018;115(7):E1550-E1559.
493. Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC. Glycoprotein VI-
dependent and -independent pathways of thrombus formation in vivo. Blood.
2006;107(10):3902-3906.
494. Massberg S, Gawaz M, Gruner S, et al. A crucial role of glycoprotein VI for platelet
recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197(1):41-49.
495. Dubois C, Panicot-Dubois L, Gainor JF, Furie BC, Furie B. Thrombin-initiated
platelet activation in vivo is vWF independent during thrombus formation in a laser injury
model. J Clin Invest. 2007;117(4):953-960.
496. Klatt C, Kruger I, Zey S, et al. Platelet-RBC interaction mediated by FasL/FasR
induces procoagulant activity important for thrombosis. J Clin Invest. 2018;128(9):3906-
3925.
497. Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood
coagulation. Prog Lipid Res. 2003;42(5):423-438.
498. Jackson SP, Nesbitt WS, Kulkarni S. Signaling events underlying thrombus
formation. J Thromb Haemost. 2003;1(7):1602-1612.
499. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med.
2008;359(9):938-949.
500. Koupenova M, Mick E, Mikhalev E, Benjamin EJ, Tanriverdi K, Freedman JE. Sex
differences in platelet toll-like receptors and their association with cardiovascular risk
factors. Arterioscler Thromb Vasc Biol. 2015;35(4):1030-1037.
501. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors
in infection and immunity. Immunity. 2011;34(5):637-650.
502. Qian K, Xie F, Gibson AW, Edberg JC, Kimberly RP, Wu J. Functional expression
of IgA receptor FcalphaRI on human platelets. J Leukoc Biol. 2008;84(6):1492-1500.
503. Hasegawa S, Pawankar R, Suzuki K, et al. Functional expression of the high affinity
receptor for IgE (FcepsilonRI) in human platelets and its' intracellular expression in human
megakaryocytes. Blood. 1999;93(8):2543-2551.
504. King M, McDermott P, Schreiber AD. Characterization of the Fc gamma receptor on
human platelets. Cell Immunol. 1990;128(2):462-479.
505. Xu WF, Andersen H, Whitmore TE, et al. Cloning and characterization of human
protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95(12):6642-6646.
506. Hollopeter G, Jantzen HM, Vincent D, et al. Identification of the platelet ADP
receptor targeted by antithrombotic drugs. Nature. 2001;409(6817):202-207.
507. Clemetson KJ, Clemetson JM, Proudfoot AE, Power CA, Baggiolini M, Wells TN.
Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human
platelets. Blood. 2000;96(13):4046-4054.
508. Motohashi K, Shibata S, Ozaki Y, Yatomi Y, Igarashi Y. Identification of
lysophospholipid receptors in human platelets: the relation of two agonists, lysophosphatidic
acid and sphingosine 1-phosphate. FEBS Lett. 2000;468(2-3):189-193.
509. Male R, Vannier WE, Baldeschwieler JD. Phagocytosis of liposomes by human
platelets. Proc Natl Acad Sci U S A. 1992;89(19):9191-9195.
510. White JG. Platelets are covercytes, not phagocytes: uptake of bacteria involves
channels of the open canalicular system. Platelets. 2005;16(2):121-131.

152
511. von Hundelshausen P, Schmitt MM. Platelets and their chemokines in
atherosclerosis-clinical applications. Front Physiol. 2014;5:294.
512. Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates.
Blood Rev. 2009;23(4):177-189.
513. Ehlers R, Ustinov V, Chen Z, et al. Targeting platelet-leukocyte interactions:
identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein
Ibalpha. J Exp Med. 2003;198(7):1077-1088.
514. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular
traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463-469.
515. Han P, Hanlon D, Arshad N, et al. Platelet P-selectin initiates cross-presentation and
dendritic cell differentiation in blood monocytes. Sci Adv. 2020;6(11):eaaz1580.
516. Silva-Cardoso SC, Affandi AJ, Spel L, et al. CXCL4 Exposure Potentiates TLR-
Driven Polarization of Human Monocyte-Derived Dendritic Cells and Increases Stimulation
of T Cells. J Immunol. 2017;199(1):253-262.
517. Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to
antigen-presenting cells: a potential bridge between injury and immune activation. Exp
Hematol. 2004;32(2):135-139.
518. Buchner K, Henn V, Grafe M, de Boer OJ, Becker AE, Kroczek RA. CD40 ligand is
selectively expressed on CD4+ T cells and platelets: implications for CD40-CD40L
signalling in atherosclerosis. J Pathol. 2003;201(2):288-295.
519. Mudd JC, Panigrahi S, Kyi B, et al. Inflammatory Function of CX3CR1+ CD8+ T
Cells in Treated HIV Infection Is Modulated by Platelet Interactions. J Infect Dis.
2016;214(12):1808-1816.
520. Chapman LM, Aggrey AA, Field DJ, et al. Platelets present antigen in the context of
MHC class I. J Immunol. 2012;189(2):916-923.
521. Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization
of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J
Proteomics. 2014;101:130-140.
522. Klockenbusch C, Walsh GM, Brown LM, et al. Global proteome analysis identifies
active immunoproteasome subunits in human platelets. Mol Cell Proteomics.
2014;13(12):3308-3319.
523. Katoh N, Soga F, Nara T, et al. Effect of serotonin on the differentiation of human
monocytes into dendritic cells. Clin Exp Immunol. 2006;146(2):354-361.
524. Fleischer J, Grage-Griebenow E, Kasper B, et al. Platelet factor 4 inhibits
proliferation and cytokine release of activated human T cells. J Immunol. 2002;169(2):770-
777.
525. Reininger AJ, Heijnen HF, Schumann H, Specht HM, Schramm W, Ruggeri ZM.
Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under
high shear stress. Blood. 2006;107(9):3537-3545.
526. Keuren JF, Magdeleyns EJ, Govers-Riemslag JW, Lindhout T, Curvers J. Effects of
storage-induced platelet microparticles on the initiation and propagation phase of blood
coagulation. Br J Haematol. 2006;134(3):307-313.
527. Marcoux G, Duchez AC, Rousseau M, et al. Microparticle and mitochondrial release
during extended storage of different types of platelet concentrates. Platelets. 2017;28(3):272-
280.
528. Sims PJ, Wiedmer T, Esmon CT, Weiss HJ, Shattil SJ. Assembly of the platelet
prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies

153
in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem.
1989;264(29):17049-17057.
529. Bevers EM, Comfurius P, Zwaal RF. Changes in membrane phospholipid distribution
during platelet activation. Biochim Biophys Acta. 1983;736(1):57-66.
530. Laffont B, Corduan A, Ple H, et al. Activated platelets can deliver mRNA regulatory
Ago2*microRNA complexes to endothelial cells via microparticles. Blood.
2013;122(2):253-261.
531. Laffont B, Corduan A, Rousseau M, et al. Platelet microparticles reprogram
macrophage gene expression and function. Thromb Haemost. 2016;115(2):311-323.
532. Perez-Pujol S, Marker PH, Key NS. Platelet microparticles are heterogeneous and
highly dependent on the activation mechanism: studies using a new digital flow cytometer.
Cytometry A. 2007;71(1):38-45.
533. Dasgupta SK, Abdel-Monem H, Niravath P, et al. Lactadherin and clearance of
platelet-derived microvesicles. Blood. 2009;113(6):1332-1339.
534. Dasgupta SK, Le A, Chavakis T, Rumbaut RE, Thiagarajan P. Developmental
endothelial locus-1 (Del-1) mediates clearance of platelet microparticles by the endothelium.
Circulation. 2012;125(13):1664-1672.
535. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular
vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell
Vesicles. 2016;5:31427.
536. Keuren JF, Magdeleyns EJ, Bennaghmouch A, Bevers EM, Curvers J, Lindhout T.
Microparticles adhere to collagen type I, fibrinogen, von Willebrand factor and surface
immobilised platelets at physiological shear rates. Br J Haematol. 2007;138(4):527-533.
537. Hoffman M, Monroe DM, Roberts HR. Coagulation factor IXa binding to activated
platelets and platelet-derived microparticles: a flow cytometric study. Thromb Haemost.
1992;68(1):74-78.
538. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and
disease. Platelets. 2017;28(3):214-221.
539. Nomura S, Okamae F, Abe M, et al. Platelets expressing P-selectin and platelet-
derived microparticles in stored platelet concentrates bind to PSGL-1 on filtrated leukocytes.
Clin Appl Thromb Hemost. 2000;6(4):213-221.
540. Marcoux G, Magron A, Sut C, et al. Platelet-derived extracellular vesicles convey
mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse
reactions. Transfusion. 2019;59(7):2403-2414.
541. Sadallah S, Amicarella F, Eken C, Iezzi G, Schifferli JA. Ectosomes released by
platelets induce differentiation of CD4+T cells into T regulatory cells. Thromb Haemost.
2014;112(6):1219-1229.
542. Dinkla S, van Cranenbroek B, van der Heijden WA, et al. Platelet microparticles
inhibit IL-17 production by regulatory T cells through P-selectin. Blood. 2016;127(16):1976-
1986.
543. Yari F, Motefaker M, Nikougoftar M, Khayati Z. Interaction of Platelet-Derived
Microparticles with a Human B-Lymphoblast Cell Line: A Clue for the Immunologic
Function of the Microparticles. Transfus Med Hemother. 2018;45(1):55-61.
544. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-
mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-
derived membrane vesicles. Blood. 2008;111(10):5028-5036.

154
545. Cloutier N, Tan S, Boudreau LH, et al. The exposure of autoantigens by
microparticles underlies the formation of potent inflammatory components: the
microparticle-associated immune complexes. EMBO Mol Med. 2013;5(2):235-249.
546. Deiss A, Kurth D. Circulating reticulocytes in normal adults as determined y the new
methylene blue method. Am J Clin Pathol. 1970;53(4):481-484.
547. Lee E, Choi HS, Hwang JH, Hoh JK, Cho YH, Baek EJ. The RNA in reticulocytes is
not just debris: it is necessary for the final stages of erythrocyte formation. Blood Cells Mol
Dis. 2014;53(1-2):1-10.
548. Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic
maturation of erythroid cells. Nature. 2008;454(7201):232-235.
549. Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for
mitochondrial clearance. EMBO Rep. 2010;11(1):45-51.
550. Grullich C, Duvoisin RM, Wiedmann M, van Leyen K. Inhibition of 15-lipoxygenase
leads to delayed organelle degradation in the reticulocyte. FEBS Lett. 2001;489(1):51-54.
551. Burka ER. RNase activity in erythroid cell lysates. J Clin Invest. 1969;48(9):1724-
1732.
552. Valentine WN, Fink K, Paglia DE, Harris SR, Adams WS. Hereditary hemolytic
anemia with human erythrocyte pyrimidine 5'-nucleotidase deficiency. J Clin Invest.
1974;54(4):866-879.
553. Griffiths RE, Kupzig S, Cogan N, et al. Maturing reticulocytes internalize plasma
membrane in glycophorin A-containing vesicles that fuse with autophagosomes before
exocytosis. Blood. 2012;119(26):6296-6306.
554. Harding C, Heuser J, Stahl P. Endocytosis and intracellular processing of transferrin
and colloidal gold-transferrin in rat reticulocytes: demonstration of a pathway for receptor
shedding. Eur J Cell Biol. 1984;35(2):256-263.
555. Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for
externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol.
1985;101(3):942-948.
556. Johnstone RM, Mathew A, Mason AB, Teng K. Exosome formation during
maturation of mammalian and avian reticulocytes: evidence that exosome release is a major
route for externalization of obsolete membrane proteins. J Cell Physiol. 1991;147(1):27-36.
557. Burger P, Kostova E, Bloem E, et al. Potassium leakage primes stored erythrocytes
for phosphatidylserine exposure and shedding of pro-coagulant vesicles. Br J Haematol.
2013;160(3):377-386.
558. Waugh RE, McKenney JB, Bauserman RG, Brooks DM, Valeri CR, Snyder LM.
Surface area and volume changes during maturation of reticulocytes in the circulation of the
baboon. J Lab Clin Med. 1997;129(5):527-535.
559. Come SE, Shohet SB, Robinson SH. Surface remodelling of reticulocytes produced
in response to erythroid stress. Nat New Biol. 1972;236(66):157-158.
560. Kirby BS, Hanna G, Hendargo HC, McMahon TJ. Restoration of intracellular ATP
production in banked red blood cells improves inducible ATP export and suppresses RBC-
endothelial adhesion. Am J Physiol Heart Circ Physiol. 2014;307(12):H1737-1744.
561. Li H, Yang J, Chu TT, et al. Cytoskeleton Remodeling Induces Membrane Stiffness
and Stability Changes of Maturing Reticulocytes. Biophys J. 2018;114(8):2014-2023.
562. Rathjen T, Nicol C, McConkey G, Dalmay T. Analysis of short RNAs in the malaria
parasite and its red blood cell host. FEBS Lett. 2006;580(22):5185-5188.

155
563. Chen SY, Wang Y, Telen MJ, Chi JT. The genomic analysis of erythrocyte
microRNA expression in sickle cell diseases. PLoS One. 2008;3(6):e2360.
564. Doss JF, Corcoran DL, Jima DD, Telen MJ, Dave SS, Chi JT. A comprehensive joint
analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genomics.
2015;16:952.
565. van der Spuy WJ, Pretorius E. Interaction of red blood cells adjacent to and within a
thrombus in experimental cerebral ischaemia. Thromb Res. 2013;132(6):718-723.
566. Hornig R, Lutz HU. Band 3 protein clustering on human erythrocytes promotes
binding of naturally occurring anti-band 3 and anti-spectrin antibodies. Exp Gerontol.
2000;35(8):1025-1044.
567. Fossati-Jimack L, Azeredo da Silveira S, Moll T, et al. Selective increase of
autoimmune epitope expression on aged erythrocytes in mice: implications in anti-
erythrocyte autoimmune responses. J Autoimmun. 2002;18(1):17-25.
568. Anniss AM, Sparrow RL. Expression of CD47 (integrin-associated protein) decreases
on red blood cells during storage. Transfus Apher Sci. 2002;27(3):233-238.
569. Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R. CD47
functions as a molecular switch for erythrocyte phagocytosis. Blood. 2012;119(23):5512-
5521.
570. Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA. Phosphatidylserine receptor
is required for clearance of apoptotic cells. Science. 2003;302(5650):1560-1563.
571. Kobayashi N, Karisola P, Pena-Cruz V, et al. TIM-1 and TIM-4 glycoproteins bind
phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927-940.
572. Park SY, Jung MY, Kim HJ, et al. Rapid cell corpse clearance by stabilin-2, a
membrane phosphatidylserine receptor. Cell Death Differ. 2008;15(1):192-201.
573. Bosman GJ, Cluitmans JC, Groenen YA, Werre JM, Willekens FL, Novotny VM.
Susceptibility to hyperosmotic stress-induced phosphatidylserine exposure increases during
red blood cell storage. Transfusion. 2011;51(5):1072-1078.
574. Boas FE, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in
red cell aging and in hemolytic anemia. Proc Natl Acad Sci U S A. 1998;95(6):3077-3081.
575. Nguyen DB, Ly TB, Wesseling MC, et al. Characterization of Microvesicles Released
from Human Red Blood Cells. Cell Physiol Biochem. 2016;38(3):1085-1099.
576. Chung SM, Bae ON, Lim KM, et al. Lysophosphatidic acid induces thrombogenic
activity through phosphatidylserine exposure and procoagulant microvesicle generation in
human erythrocytes. Arterioscler Thromb Vasc Biol. 2007;27(2):414-421.
577. Nguyen DB, Wagner-Britz L, Maia S, et al. Regulation of phosphatidylserine
exposure in red blood cells. Cell Physiol Biochem. 2011;28(5):847-856.
578. Seki M, Arashiki N, Takakuwa Y, Nitta K, Nakamura F. Reduction in flippase
activity contributes to surface presentation of phosphatidylserine in human senescent
erythrocytes. J Cell Mol Med. 2020;24(23):13991-14000.
579. de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R. Of
macrophages and red blood cells; a complex love story. Front Physiol. 2014;5:9.
580. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of
treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33.
581. Yu FT, Armstrong JK, Tripette J, Meiselman HJ, Cloutier G. A local increase in red
blood cell aggregation can trigger deep vein thrombosis: evidence based on quantitative
cellular ultrasound imaging. J Thromb Haemost. 2011;9(3):481-488.

156
582. Lominadze D, Dean WL. Involvement of fibrinogen specific binding in erythrocyte
aggregation. FEBS Lett. 2002;517(1-3):41-44.
583. Carvalho FA, Connell S, Miltenberger-Miltenyi G, et al. Atomic force microscopy-
based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano.
2010;4(8):4609-4620.
584. Wautier MP, Heron E, Picot J, Colin Y, Hermine O, Wautier JL. Red blood cell
phosphatidylserine exposure is responsible for increased erythrocyte adhesion to
endothelium in central retinal vein occlusion. J Thromb Haemost. 2011;9(5):1049-1055.
585. Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood
coagulation: the erythrocyte contribution to thrombin generation. Blood. 2012;120(18):3837-
3845.
586. Wohner N, Sotonyi P, Machovich R, et al. Lytic resistance of fibrin containing red
blood cells. Arterioscler Thromb Vasc Biol. 2011;31(10):2306-2313.
587. Choi MH, Park GH, Lee JS, et al. Erythrocyte Fraction Within Retrieved Thrombi
Contributes to Thrombolytic Response in Acute Ischemic Stroke. Stroke. 2018;49(3):652-
659.
588. Szabo MC, Soo KS, Zlotnik A, Schall TJ. Chemokine class differences in binding to
the Duffy antigen-erythrocyte chemokine receptor. J Biol Chem. 1995;270(43):25348-
25351.
589. Karsten E, Breen E, Herbert BR. Red blood cells are dynamic reservoirs of cytokines.
Sci Rep. 2018;8(1):3101.
590. Wei J, Zhao J, Schrott V, et al. Red Blood Cells Store and Release Interleukin-33. J
Investig Med. 2015;63(6):806-810.
591. Mannu F, Arese P, Cappellini MD, et al. Role of hemichrome binding to erythrocyte
membrane in the generation of band-3 alterations in beta-thalassemia intermedia
erythrocytes. Blood. 1995;86(5):2014-2020.
592. Neote K, Darbonne W, Ogez J, Horuk R, Schall TJ. Identification of a promiscuous
inflammatory peptide receptor on the surface of red blood cells. J Biol Chem.
1993;268(17):12247-12249.
593. Lee JS, Wurfel MM, Matute-Bello G, et al. The Duffy antigen modifies systemic and
local tissue chemokine responses following lipopolysaccharide stimulation. J Immunol.
2006;177(11):8086-8094.
594. Fredriksson K, Lundahl J, Palmberg L, et al. Red blood cells stimulate human lung
fibroblasts to secrete interleukin-8. Inflammation. 2003;27(2):71-78.
595. Antunes RF, Brandao C, Maia M, Arosa FA. Red blood cells release factors with
growth and survival bioactivities for normal and leukemic T cells. Immunol Cell Biol.
2011;89(1):111-121.
596. Hotz MJ, Qing D, Shashaty MGS, et al. Red Blood Cells Homeostatically Bind
Mitochondrial DNA through TLR9 to Maintain Quiescence and to Prevent Lung Injury. Am
J Respir Crit Care Med. 2018;197(4):470-480.
597. Horuk R, Chitnis CE, Darbonne WC, et al. A receptor for the malarial parasite
Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261(5125):1182-
1184.
598. McHenry AM, Adams JH. The crystal structure of P. knowlesi DBPalpha DBL
domain and its implications for immune evasion. Trends Biochem Sci. 2006;31(9):487-491.

157
599. Asher DR, Cerny AM, Finberg RW. The erythrocyte viral trap: transgenic expression
of viral receptor on erythrocytes attenuates coxsackievirus B infection. Proc Natl Acad Sci
U S A. 2005;102(36):12897-12902.
600. Beck Z, Brown BK, Wieczorek L, et al. Human erythrocytes selectively bind and
enrich infectious HIV-1 virions. PLoS One. 2009;4(12):e8297.
601. He W, Neil S, Kulkarni H, et al. Duffy antigen receptor for chemokines mediates
trans-infection of HIV-1 from red blood cells to target cells and affects HIV-AIDS
susceptibility. Cell Host Microbe. 2008;4(1):52-62.
602. Thangaraju K, Neerukonda SN, Katneni U, Buehler PW. Extracellular Vesicles from
Red Blood Cells and Their Evolving Roles in Health, Coagulopathy and Therapy. Int J Mol
Sci. 2020;22(1).
603. Sens P, Gov N. Force balance and membrane shedding at the red-blood-cell surface.
Phys Rev Lett. 2007;98(1):018102.
604. Buerck JP, Burke DK, Schmidtke DW, Snyder TA, Papavassiliou DV, O'Rear EA.
Production of erythrocyte microparticles in a sub-hemolytic environment. J Artif Organs.
2021;24(2):135-145.
605. Bosman GJ, Lasonder E, Groenen-Dopp YA, Willekens FL, Werre JM. The proteome
of erythrocyte-derived microparticles from plasma: new clues for erythrocyte aging and
vesiculation. J Proteomics. 2012;76 Spec No.:203-210.
606. Willekens FL, Werre JM, Groenen-Dopp YA, Roerdinkholder-Stoelwinder B, de
Pauw B, Bosman GJ. Erythrocyte vesiculation: a self-protective mechanism? Br J Haematol.
2008;141(4):549-556.
607. Willekens FL, Roerdinkholder-Stoelwinder B, Groenen-Dopp YA, et al. Hemoglobin
loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood.
2003;101(2):747-751.
608. Donadee C, Raat NJ, Kanias T, et al. Nitric oxide scavenging by red blood cell
microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion.
Circulation. 2011;124(4):465-476.
609. Liu C, Zhao W, Christ GJ, Gladwin MT, Kim-Shapiro DB. Nitric oxide scavenging
by red cell microparticles. Free Radic Biol Med. 2013;65:1164-1173.
610. Poisson J, Tanguy M, Davy H, et al. Erythrocyte-derived microvesicles induce
arterial spasms in JAK2V617F myeloproliferative neoplasm. J Clin Invest.
2020;130(5):2630-2643.
611. Jank H, Salzer U. Vesicles generated during storage of red blood cells enhance the
generation of radical oxygen species in activated neutrophils. ScientificWorldJournal.
2011;11:173-185.
612. Camus SM, De Moraes JA, Bonnin P, et al. Circulating cell membrane microparticles
transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood.
2015;125(24):3805-3814.
613. Kim Y, Xia BT, Jung AD, et al. Microparticles from stored red blood cells promote
a hypercoagulable state in a murine model of transfusion. Surgery. 2018;163(2):423-429.
614. Fischer D, Büssow J, Meybohm P, et al. Microparticles from stored red blood cells
enhance procoagulant and proinflammatory activity. Transfusion. 2017;57(11):2701-2711.
615. Straat M, van Hezel ME, Boing A, et al. Monocyte-mediated activation of endothelial
cells occurs only after binding to extracellular vesicles from red blood cell products, a process
mediated by beta-integrin. Transfusion. 2016;56(12):3012-3020.

158
616. Van Der Meijden PE, Van Schilfgaarde M, Van Oerle R, Renné T, ten Cate H, Spronk
HM. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor
XIIa. J Thromb Haemost. 2012;10(7):1355-1362.
617. Zecher D, Cumpelik A, Schifferli JA. Erythrocyte-derived microvesicles amplify
systemic inflammation by thrombin-dependent activation of complement. Arterioscler
Thromb Vasc Biol. 2014;34(2):313-320.
618. Danesh A, Inglis HC, Jackman RP, et al. Exosomes from red blood cell units bind to
monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood.
2014;123(5):687-696.
619. Cardo LJ, Wilder D, Salata J. Neutrophil priming, caused by cell membranes and
microvesicles in packed red blood cell units, is abrogated by leukocyte depletion at
collection. Transfus Apher Sci. 2008;38(2):117-125.
620. Belizaire RM, Prakash PS, Richter JR, et al. Microparticles from stored red blood
cells activate neutrophils and cause lung injury after hemorrhage and resuscitation. J Am Coll
Surg. 2012;214(4):648-655; discussion 656-647.
621. Nazimek K, Bustos-Moran E, Blas-Rus N, et al. Syngeneic red blood cell-induced
extracellular vesicles suppress delayed-type hypersensitivity to self-antigens in mice. Clin
Exp Allergy. 2019;49(11):1487-1499.
622. Sadallah S, Eken C, Schifferli JA. Erythrocyte-derived ectosomes have
immunosuppressive properties. J Leukoc Biol. 2008;84(5):1316-1325.
623. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front
Neuroendocrinol. 2014;35(3):347-369.
624. Smolen JS, Aletaha D, Barton A, et al. Rheumatoid arthritis. Nat Rev Dis Primers.
2018;4:18001.
625. Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to
clinical synovitis. Nat Rev Rheumatol. 2012;8(10):573-586.
626. Radner H, Lesperance T, Accortt NA, Solomon DH. Incidence and Prevalence of
Cardiovascular Risk Factors Among Patients With Rheumatoid Arthritis, Psoriasis, or
Psoriatic Arthritis. Arthritis Care Res (Hoboken). 2017;69(10):1510-1518.
627. Aubry MC, Maradit-Kremers H, Reinalda MS, Crowson CS, Edwards WD, Gabriel
SE. Differences in atherosclerotic coronary heart disease between subjects with and without
rheumatoid arthritis. J Rheumatol. 2007;34(5):937-942.
628. Sparks JA, Chang SC, Liao KP, et al. Rheumatoid Arthritis and Mortality Among
Women During 36 Years of Prospective Follow-Up: Results From the Nurses' Health Study.
Arthritis Care Res (Hoboken). 2016;68(6):753-762.
629. Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident
cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational
studies. Ann Rheum Dis. 2012;71(9):1524-1529.
630. Mason JC, Libby P. Cardiovascular disease in patients with chronic inflammation:
mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur
Heart J. 2015;36(8):482-489c.
631. Zhao C, Fernandes MJ, Prestwich GD, et al. Regulation of lysophosphatidic acid
receptor expression and function in human synoviocytes: implications for rheumatoid
arthritis? Mol Pharmacol. 2008;73(2):587-600.
632. Zhao C, Hui W, Fernandes MJ, Poubelle PE, Bourgoin SG. Lysophosphatidic acid-
induced IL-8 secretion involves MSK1 and MSK2 mediated activation of CREB1 in human
fibroblast-like synoviocytes. Biochem Pharmacol. 2014;90(1):62-72.

159
633. Hui W, Zhao C, Bourgoin SG. LPA Promotes T Cell Recruitment through Synthesis
of CXCL13. Mediators Inflamm. 2015;2015:248492.
634. Pasztoi M, Sodar B, Misjak P, et al. The recently identified hexosaminidase D enzyme
substantially contributes to the elevated hexosaminidase activity in rheumatoid arthritis.
Immunol Lett. 2013;149(1-2):71-76.
635. Messer L, Alsaleh G, Freyssinet JM, et al. Microparticle-induced release of B-
lymphocyte regulators by rheumatoid synoviocytes. Arthritis Res Ther. 2009;11(2):R40.
636. Zhang HG, Liu C, Su K, et al. A membrane form of TNF-alpha presented by
exosomes delays T cell activation-induced cell death. J Immunol. 2006;176(12):7385-7393.
637. Michael BNR, Kommoju V, Kavadichanda Ganapathy C, Negi VS. Characterization
of cell-derived microparticles in synovial fluid and plasma of patients with rheumatoid
arthritis. Rheumatol Int. 2019;39(8):1377-1387.
638. Burbano C, Rojas M, Munoz-Vahos C, et al. Extracellular vesicles are associated with
the systemic inflammation of patients with seropositive rheumatoid arthritis. Sci Rep.
2018;8(1):17917.
639. Duchez AC, Boudreau LH, Naika GS, et al. Platelet microparticles are internalized in
neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-
IIA. Proc Natl Acad Sci U S A. 2015;112(27):E3564-3573.
640. Stanczyk J, Pedrioli DM, Brentano F, et al. Altered expression of MicroRNA in
synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum.
2008;58(4):1001-1009.
641. Takamura Y, Aoki W, Satomura A, Shibasaki S, Ueda M. Small RNAs detected in
exosomes derived from the MH7A synovial fibroblast cell line with TNF-alpha stimulation.
PLoS One. 2018;13(8):e0201851.
642. Skriner K, Adolph K, Jungblut PR, Burmester GR. Association of citrullinated
proteins with synovial exosomes. Arthritis Rheum. 2006;54(12):3809-3814.
643. Foers AD, Garnham AL, Chatfield S, et al. Extracellular Vesicles in Synovial Fluid
from Rheumatoid Arthritis Patients Contain miRNAs with Capacity to Modulate
Inflammation. Int J Mol Sci. 2021;22(9).
644. Berckmans RJ, Nieuwland R, Tak PP, et al. Cell-derived microparticles in synovial
fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-
dependent mechanism. Arthritis Rheum. 2002;46(11):2857-2866.
645. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis
Primers. 2016;2:16039.
646. Faurschou M, Starklint H, Halberg P, Jacobsen S. Prognostic factors in lupus
nephritis: diagnostic and therapeutic delay increases the risk of terminal renal failure. J
Rheumatol. 2006;33(8):1563-1569.
647. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of
systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271-1277.
648. Aringer M, Leuchten N, Johnson SR. New Criteria for Lupus. Curr Rheumatol Rep.
2020;22(6):18.
649. Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the Systemic
Lupus International Collaborating Clinics classification criteria for systemic lupus
erythematosus. Arthritis Rheum. 2012;64(8):2677-2686.
650. Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against
Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus
Erythematosus. Arthritis Rheumatol. 2019;71(9):1400-1412.

160
651. Ceccarelli F, Perricone C, Massaro L, et al. Assessment of disease activity in Systemic
Lupus Erythematosus: Lights and shadows. Autoimmun Rev. 2015;14(7):601-608.
652. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the
SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies
in SLE. Arthritis Rheum. 1992;35(6):630-640.
653. Zhu H, Luo H, Yan M, Zuo X, Li QZ. Autoantigen Microarray for High-throughput
Autoantibody Profiling in Systemic Lupus Erythematosus. Genomics Proteomics
Bioinformatics. 2015;13(4):210-218.
654. Yaniv G, Twig G, Shor DB, et al. A volcanic explosion of autoantibodies in systemic
lupus erythematosus: a diversity of 180 different antibodies found in SLE patients.
Autoimmun Rev. 2015;14(1):75-79.
655. Crow MK, Olferiev M, Kirou KA. Targeting of type I interferon in systemic
autoimmune diseases. Transl Res. 2015;165(2):296-305.
656. Stetson DB. Endogenous retroelements and autoimmune disease. Curr Opin
Immunol. 2012;24(6):692-697.
657. Lövgren T, Eloranta ML, Båve U, Alm GV, Rönnblom L. Induction of interferon-
alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic
acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum.
2004;50(6):1861-1872.
658. Ettinger R, Sims GP, Robbins R, et al. IL-21 and BAFF/BLyS synergize in
stimulating plasma cell differentiation from a unique population of human splenic memory
B cells. J Immunol. 2007;178(5):2872-2882.
659. Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic
lupus erythematosus lymphocytes. J Clin Invest. 1996;98(3):826-837.
660. Simpson N, Gatenby PA, Wilson A, et al. Expansion of circulating T cells resembling
follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus
erythematosus. Arthritis Rheum. 2010;62(1):234-244.
661. McKinney EF, Lyons PA, Carr EJ, et al. A CD8+ T cell transcription signature
predicts prognosis in autoimmune disease. Nat Med. 2010;16(5):586-591, 581p following
591.
662. Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and
kidney damage in autoimmune glomerulonephritis. Science. 1998;279(5353):1052-1054.
663. Knight JS, Kaplan MJ. Lupus neutrophils: 'NET' gain in understanding lupus
pathogenesis. Curr Opin Rheumatol. 2012;24(5):441-450.
664. Deng GM, Tsokos GC. Pathogenesis and targeted treatment of skin injury in SLE.
Nat Rev Rheumatol. 2015;11(11):663-669.
665. Kaplan MJ. Premature vascular damage in systemic lupus erythematosus.
Autoimmunity. 2009;42(7):580-586.
666. McMahon M, Grossman J, Skaggs B, et al. Dysfunctional proinflammatory high-
density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus
erythematosus. Arthritis Rheum. 2009;60(8):2428-2437.
667. Yurkovich M, Vostretsova K, Chen W, Aviña-Zubieta JA. Overall and cause-specific
mortality in patients with systemic lupus erythematosus: a meta-analysis of observational
studies. Arthritis Care Res (Hoboken). 2014;66(4):608-616.
668. Gustafsson JT, Simard JF, Gunnarsson I, et al. Risk factors for cardiovascular
mortality in patients with systemic lupus erythematosus, a prospective cohort study. Arthritis
Res Ther. 2012;14(2):R46.

161
669. Urowitz MB, Ibañez D, Gladman DD. Atherosclerotic vascular events in a single
large lupus cohort: prevalence and risk factors. J Rheumatol. 2007;34(1):70-75.
670. Dong C, Zhou Q, Fu T, et al. Circulating Exosomes Derived-miR-146a from Systemic
Lupus Erythematosus Patients Regulates Senescence of Mesenchymal Stem Cells. Biomed
Res Int. 2019;2019:6071308.
671. Salvi V, Gianello V, Busatto S, et al. Exosome-delivered microRNAs promote IFN-
alpha secretion by human plasmacytoid DCs via TLR7. JCI Insight. 2018;3(10).
672. Kato Y, Park J, Takamatsu H, et al. Apoptosis-derived membrane vesicles drive the
cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus.
Ann Rheum Dis. 2018;77(10):1507-1515.
673. Dominguez-Gutierrez PR, Ceribelli A, Satoh M, Sobel ES, Reeves WH, Chan EK.
Positive correlation of STAT1 and miR-146a with anemia in patients with systemic lupus
erythematosus. J Clin Immunol. 2014;34(2):171-180.
674. Fortin PR, Cloutier N, Bissonnette V, et al. Distinct Subtypes of Microparticle-
containing Immune Complexes Are Associated with Disease Activity, Damage, and Carotid
Intima-media Thickness in Systemic Lupus Erythematosus. J Rheumatol. 2016;43(11):2019-
2025.
675. Ullal AJ, Reich CF, 3rd, Clowse M, et al. Microparticles as antigenic targets of
antibodies to DNA and nucleosomes in systemic lupus erythematosus. J Autoimmun.
2011;36(3-4):173-180.
676. Nielsen CT, Østergaard O, Stener L, et al. Increased IgG on cell-derived plasma
microparticles in systemic lupus erythematosus is associated with autoantibodies and
complement activation. Arthritis Rheum. 2012;64(4):1227-1236.
677. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017
Update: A Report From the American Heart Association. Circulation. 2017;135(10):e146-
e603.
678. Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers.
2019;5(1):56.
679. Organization WH. Cardiovascular diseases (CVDs) Fact Sheet; 2017.
680. Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause
atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical
studies. A consensus statement from the European Atherosclerosis Society Consensus Panel.
Eur Heart J. 2017;38(32):2459-2472.
681. Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis.
Circ Res. 2016;118(4):692-702.
682. Wanschel A, Seibert T, Hewing B, et al. Neuroimmune guidance cue Semaphorin 3E
is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler
Thromb Vasc Biol. 2013;33(5):886-893.
683. Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional
macrophage accumulation in atherosclerosis. Nat Med. 2013;19(9):1166-1172.
684. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA.
Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-
like cells in human atherosclerosis. Circulation. 2014;129(15):1551-1559.
685. Singh A, Sen P. Lipid droplet: A functionally active organelle in monocyte to
macrophage differentiation and its inflammatory properties. Biochim Biophys Acta Mol Cell
Biol Lipids. 2021;1866(10):158981.

162
686. Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: is it all about
inflammation? Nat Rev Rheumatol. 2015;11(7):390-400.
687. Huang H, Virmani R, Younis H, Burke AP, Kamm RD, Lee RT. The impact of
calcification on the biomechanical stability of atherosclerotic plaques. Circulation.
2001;103(8):1051-1056.
688. Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis
induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition
of phagocytosis. Circ Res. 2010;106(2):363-372.
689. Geng YJ, Libby P. Evidence for apoptosis in advanced human atheroma.
Colocalization with interleukin-1 beta-converting enzyme. Am J Pathol. 1995;147(2):251-
266.
690. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and
rupture. Circ Res. 2014;114(12):1852-1866.
691. Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively
and negatively regulate interstitial collagen gene expression in human vascular smooth
muscle cells. Arterioscler Thromb. 1991;11(5):1223-1230.
692. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix
metalloproteinases and matrix degrading activity in vulnerable regions of human
atherosclerotic plaques. J Clin Invest. 1994;94(6):2493-2503.
693. Kritikou E, van Puijvelde GH, van der Heijden T, et al. Inhibition of lysophosphatidic
acid receptors 1 and 3 attenuates atherosclerosis development in LDL-receptor deficient
mice. Sci Rep. 2016;6:37585.
694. Yang L, Kraemer M, Fang XF, et al. LPA receptor 4 deficiency attenuates
experimental atherosclerosis. J Lipid Res. 2019;60(5):972-980.
695. Chang CL, Lin ME, Hsu HY, et al. Lysophosphatidic acid-induced interleukin-1 beta
expression is mediated through Gi/Rho and the generation of reactive oxygen species in
macrophages. J Biomed Sci. 2008;15(3):357-363.
696. Zhou Z, Subramanian P, Sevilmis G, et al. Lipoprotein-derived lysophosphatidic acid
promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab.
2011;13(5):592-600.
697. Tripathi H, Al-Darraji A, Abo-Aly M, et al. Autotaxin inhibition reduces cardiac
inflammation and mitigates adverse cardiac remodeling after myocardial infarction. J Mol
Cell Cardiol. 2020;149:95-114.
698. Panetti TS, Nowlen J, Mosher DF. Sphingosine-1-phosphate and lysophosphatidic
acid stimulate endothelial cell migration. Arterioscler Thromb Vasc Biol. 2000;20(4):1013-
1019.
699. Kim J, Keys JR, Eckhart AD. Vascular smooth muscle migration and proliferation in
response to lysophosphatidic acid (LPA) is mediated by LPA receptors coupling to Gq. Cell
Signal. 2006;18(10):1695-1701.
700. Gaaya A, Poirier O, Mougenot N, et al. Plasticity-related gene-1 inhibits
lysophosphatidic acid-induced vascular smooth muscle cell migration and proliferation and
prevents neointima formation. Am J Physiol Cell Physiol. 2012;303(10):C1104-1114.
701. An D, Hao F, Zhang F, et al. CD14 is a key mediator of both lysophosphatidic acid
and lipopolysaccharide induction of foam cell formation. J Biol Chem. 2017;292(35):14391-
14400.
702. Canault M, Leroyer AS, Peiretti F, et al. Microparticles of human atherosclerotic
plaques enhance the shedding of the tumor necrosis factor-alpha converting

163
enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am
J Pathol. 2007;171(5):1713-1723.
703. Gao W, Liu H, Yuan J, et al. Exosomes derived from mature dendritic cells increase
endothelial inflammation and atherosclerosis via membrane TNF-alpha mediated NF-
kappaB pathway. J Cell Mol Med. 2016;20(12):2318-2327.
704. Li C, Li S, Zhang F, et al. Endothelial microparticles-mediated transfer of microRNA-
19b promotes atherosclerosis via activating perivascular adipose tissue inflammation in
apoE(-/-) mice. Biochem Biophys Res Commun. 2018;495(2):1922-1929.
705. Leroyer AS, Isobe H, Lesèche G, et al. Cellular origins and thrombogenic activity of
microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol.
2007;49(7):772-777.
706. Hoyer FF, Giesen MK, Nunes Franca C, Lutjohann D, Nickenig G, Werner N.
Monocytic microparticles promote atherogenesis by modulating inflammatory cells in mice.
J Cell Mol Med. 2012;16(11):2777-2788.
707. Fu Z, Zhou E, Wang X, et al. Oxidized low-density lipoprotein-induced
microparticles promote endothelial monocyte adhesion via intercellular adhesion molecule
1. Am J Physiol Cell Physiol. 2017;313(5):C567-c574.
708. Rautou PE, Leroyer AS, Ramkhelawon B, et al. Microparticles from human
atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and
transendothelial migration. Circ Res. 2011;108(3):335-343.
709. Wadey RM, Connolly KD, Mathew D, Walters G, Rees DA, James PE. Inflammatory
adipocyte-derived extracellular vesicles promote leukocyte attachment to vascular
endothelial cells. Atherosclerosis. 2019;283:19-27.
710. Zakharova L, Svetlova M, Fomina AF. T cell exosomes induce cholesterol
accumulation in human monocytes via phosphatidylserine receptor. J Cell Physiol.
2007;212(1):174-181.
711. Keyel PA, Tkacheva OA, Larregina AT, Salter RD. Coordinate stimulation of
macrophages by microparticles and TLR ligands induces foam cell formation. J Immunol.
2012;189(9):4621-4629.
712. Nguyen MA, Karunakaran D, Geoffrion M, et al. Extracellular Vesicles Secreted by
Atherogenic Macrophages Transfer MicroRNA to Inhibit Cell Migration. Arterioscler
Thromb Vasc Biol. 2018;38(1):49-63.
713. Barberio MD, Kasselman LJ, Playford MP, et al. Cholesterol efflux alterations in
adolescent obesity: role of adipose-derived extracellular vesical microRNAs. J Transl Med.
2019;17(1):232.
714. Wang F, Chen FF, Shang YY, et al. Insulin resistance adipocyte-derived exosomes
aggravate atherosclerosis by increasing vasa vasorum angiogenesis in diabetic ApoE(-/-)
mice. Int J Cardiol. 2018;265:181-187.
715. Hutcheson JD, Goettsch C, Bertazzo S, et al. Genesis and growth of extracellular-
vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15(3):335-
343.
716. Zernecke A, Bidzhekov K, Noels H, et al. Delivery of microRNA-126 by apoptotic
bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2(100):ra81.
717. Jansen F, Yang X, Hoelscher M, et al. Endothelial microparticle-mediated transfer of
MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in
glucose-damaged endothelial microparticles. Circulation. 2013;128(18):2026-2038.

164
718. Li J, Tan M, Xiang Q, Zhou Z, Yan H. Thrombin-activated platelet-derived exosomes
regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-
inflammation response. Thromb Res. 2017;154:96-105.
719. Njock MS, Cheng HS, Dang LT, et al. Endothelial cells suppress monocyte activation
through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood.
2015;125(20):3202-3212.
720. Huang C, Han J, Wu Y, et al. Exosomal MALAT1 derived from oxidized low-density
lipoprotein-treated endothelial cells promotes M2 macrophage polarization. Mol Med Rep.
2018;18(1):509-515.
721. Poupardin R, Wolf M, Strunk D. Adherence to minimal experimental requirements
for defining extracellular vesicles and their functions. Adv Drug Deliv Rev.
2021;176:113872.
722. van der Pol E, Sturk A, van Leeuwen T, Nieuwland R, Coumans F. Standardization
of extracellular vesicle measurements by flow cytometry through vesicle diameter
approximation. J Thromb Haemost. 2018;16(6):1236-1245.
723. Lacroix R, Judicone C, Mooberry M, Boucekine M, Key NS, Dignat-George F.
Standardization of pre-analytical variables in plasma microparticle determination: results of
the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J
Thromb Haemost. 2013.
724. Yuana Y, Böing AN, Grootemaat AE, et al. Handling and storage of human body
fluids for analysis of extracellular vesicles. J Extracell Vesicles. 2015;4:29260.
725. Lőrincz Á M, Timár CI, Marosvári KA, et al. Effect of storage on physical and
functional properties of extracellular vesicles derived from neutrophilic granulocytes. J
Extracell Vesicles. 2014;3:25465.
726. Bæk R, Søndergaard EK, Varming K, Jørgensen MM. The impact of various
preanalytical treatments on the phenotype of small extracellular vesicles in blood analyzed
by protein microarray. J Immunol Methods. 2016;438:11-20.
727. Kriebardis AG, Antonelou MH, Georgatzakou HT, Tzounakas VL, Stamoulis KE,
Papassideri IS. Microparticles variability in fresh frozen plasma: preparation protocol and
storage time effects. Blood Transfus. 2016;14(2):228-237.
728. Trummer A, De Rop C, Tiede A, Ganser A, Eisert R. Recovery and composition of
microparticles after snap-freezing depends on thawing temperature. Blood Coagul
Fibrinolysis. 2009;20(1):52-56.
729. Evseenko D, Latour B, Richardson W, et al. Lysophosphatidic acid mediates myeloid
differentiation within the human bone marrow microenvironment. PLoS One.
2013;8(5):e63718.
730. Sidstedt M, Hedman J, Romsos EL, et al. Inhibition mechanisms of hemoglobin,
immunoglobulin G, and whole blood in digital and real-time PCR. Anal Bioanal Chem.
2018;410(10):2569-2583.
731. Al-Soud WA, Rådström P. Purification and characterization of PCR-inhibitory
components in blood cells. J Clin Microbiol. 2001;39(2):485-493.
732. Kano K, Matsumoto H, Kono N, Kurano M, Yatomi Y, Aoki J. Suppressing
postcollection lysophosphatidic acid metabolism improves the precision of plasma LPA
quantification. J Lipid Res. 2021;62:100029.
733. Matsumoto A, Takahashi Y, Ogata K, et al. Phosphatidylserine-deficient small
extracellular vesicle is a major somatic cell-derived sEV subpopulation in blood. iScience.
2021;24(8):102839.

165
734. López P, Rodríguez-Carrio J, Martínez-Zapico A, Caminal-Montero L, Suárez A.
Circulating microparticle subpopulations in systemic lupus erythematosus are affected by
disease activity. Int J Cardiol. 2017;236:138-144.
735. Pereira J, Alfaro G, Goycoolea M, et al. Circulating platelet-derived microparticles in
systemic lupus erythematosus. Association with increased thrombin generation and
procoagulant state. Thromb Haemost. 2006;95(1):94-99.
736. Sawada T, Kurano M, Shirai H, et al. Serum phosphatidylserine-specific
phospholipase A1 as a novel biomarker for monitoring systemic lupus erythematosus disease
activity. Int J Rheum Dis. 2019;22(11):2059-2066.
737. Shanbhag K, Mhetre A, Khandelwal N, Kamat SS. The Lysophosphatidylserines-An
Emerging Class of Signalling Lysophospholipids. J Membr Biol. 2020;253(5):381-397.
738. Barnes MJ, Li CM, Xu Y, An J, Huang Y, Cyster JG. The lysophosphatidylserine
receptor GPR174 constrains regulatory T cell development and function. J Exp Med.
2015;212(7):1011-1020.
739. Barnes MJ, Cyster JG. Lysophosphatidylserine suppression of T-cell activation via
GPR174 requires Gαs proteins. Immunol Cell Biol. 2018;96(4):439-445.
740. van der Kleij D, Latz E, Brouwers JF, et al. A novel host-parasite lipid cross-talk.
Schistosomal lyso-phosphatidylserine activates toll-like receptor 2 and affects immune
polarization. J Biol Chem. 2002;277(50):48122-48129.
741. Kamat SS, Camara K, Parsons WH, et al. Immunomodulatory
lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat Chem Biol.
2015;11(2):164-171.
742. Frasch SC, Bratton DL. Emerging roles for lysophosphatidylserine in resolution of
inflammation. Prog Lipid Res. 2012;51(3):199-207.
743. Iwata Y, Kitajima S, Yamahana J, et al. Higher serum levels of autotaxin and
phosphatidylserine-specific phospholipase A1 in patients with lupus nephritis. Int J Rheum
Dis. 2021;24(2):231-239.
744. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med.
2007;357(24):2482-2494.
745. Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of Innate Immune Responses
by Platelets. Front Immunol. 2019;10:1320.
746. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune cell
interactions in thrombo-inflammation. Res Pract Thromb Haemost. 2020;4(1):23-35.
747. Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat
Rev Immunol. 2011;11(4):264-274.
748. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets
as immune and inflammatory cells. Blood. 2014;123(18):2759-2767.
749. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with
inflammation. J Immunol. 2015;194(12):5579-5587.
750. Cunin P, Nigrovic PA. Megakaryocytes as immune cells. J Leukoc Biol.
2019;105(6):1111-1121.
751. Garraud O, Cognasse F. Are Platelets Cells? And if Yes, are They Immune Cells?
Front Immunol. 2015;6:70.
752. Opperman CM, Sishi BJ. Tumor necrosis factor alpha stimulates p62 accumulation
and enhances proteasome activity independently of ROS. Cell Biol Toxicol. 2015;31(2):83-
94.

166
753. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser
B Phys Biol Sci. 2009;85(1):12-36.
754. Dieude M, Bell C, Turgeon J, et al. The 20S proteasome core, active within apoptotic
exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl
Med. 2015;7(318):318ra200.
755. Shi DS, Smith MC, Campbell RA, et al. Proteasome function is required for platelet
production. J Clin Invest. 2014;124(9):3757-3766.
756. Murai K, Kowata S, Shimoyama T, et al. Bortezomib induces thrombocytopenia by
the inhibition of proplatelet formation of megakaryocytes. Eur J Haematol. 2014;93(4):290-
296.
757. Nayak MK, Kulkarni PP, Dash D. Regulatory role of proteasome in determination of
platelet life span. J Biol Chem. 2013;288(10):6826-6834.
758. Grundler K, Rotter R, Tilley S, et al. The proteasome regulates collagen-induced
platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb Res.
2016;148:15-22.
759. Nayak MK, Kumar K, Dash D. Regulation of proteasome activity in activated human
platelets. Cell Calcium. 2011;49(4):226-232.
760. Grundler Groterhorst K, Mannell H, Pircher J, Kraemer BF. Platelet Proteasome
Activity and Metabolism Is Upregulated during Bacterial Sepsis. Int J Mol Sci. 2019;20(23).
761. Gupta N, Li W, Willard B, Silverstein RL, McIntyre TM. Proteasome proteolysis
supports stimulated platelet function and thrombosis. Arterioscler Thromb Vasc Biol.
2014;34(1):160-168.
762. Colberg L, Cammann C, Greinacher A, Seifert U. Structure and function of the
ubiquitin-proteasome system in platelets. J Thromb Haemost. 2020.
763. Yukawa M, Sakon M, Kambayashi J, et al. Purification and characterization of
endogenous protein activator of human platelet proteasome. J Biochem. 1993;114(3):317-
323.
764. Yukawa M, Sakon M, Kambayashi J, et al. Proteasome and its novel endogeneous
activator in human platelets. Biochem Biophys Res Commun. 1991;178(1):256-262.
765. Boegel S, Lower M, Bukur T, Sorn P, Castle JC, Sahin U. HLA and proteasome
expression body map. BMC Med Genomics. 2018;11(1):36.
766. Semple JW, Speck ER, Milev YP, Blanchette V, Freedman J. Indirect allorecognition
of platelets by T helper cells during platelet transfusions correlates with anti-major
histocompatibility complex antibody and cytotoxic T lymphocyte formation. Blood.
1995;86(2):805-812.
767. Zufferey A, Speck ER, Machlus KR, et al. Mature murine megakaryocytes present
antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv.
2017;1(20):1773-1785.
768. Iannacone M, Sitia G, Isogawa M, et al. Platelets mediate cytotoxic T lymphocyte-
induced liver damage. Nat Med. 2005;11(11):1167-1169.
769. Verschoor A, Neuenhahn M, Navarini AA, et al. A platelet-mediated system for
shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb
and complement C3. Nat Immunol. 2011;12(12):1194-1201.
770. Maouia A, Rebetz J, Kapur R, Semple JW. The Immune Nature of Platelets Revisited.
Transfus Med Rev. 2020.
771. Marcoux G, Laroche A, Espinoza Romero J, Boilard E. Role of platelets and
megakaryocytes in adaptive immunity. Platelets. 2020:1-12.

167
772. Pariser DN, Hilt ZT, Ture SK, et al. Lung megakaryocytes are immune modulatory
cells. J Clin Invest. 2020.
773. Puhm F, Boilard E, Machlus KR. Platelet Extracellular Vesicles: Beyond the Blood.
Arterioscler Thromb Vasc Biol. 2020:ATVBAHA120314644.
774. Siljander PR. Platelet-derived microparticles - an updated perspective. Thromb Res.
2011;127 Suppl 2:S30-33.
775. Burnouf T, Chou ML, Goubran H, Cognasse F, Garraud O, Seghatchian J. An
overview of the role of microparticles/microvesicles in blood components: Are they
clinically beneficial or harmful? Transfus Apher Sci. 2015;53(2):137-145.
776. Gyorgy B, Szabo TG, Turiak L, et al. Improved flow cytometric assessment reveals
distinct microvesicle (cell-derived microparticle) signatures in joint diseases. PLoS One.
2012;7(11):e49726.
777. Tessandier N, Melki I, Cloutier N, et al. Platelets Disseminate Extracellular Vesicles
in Lymph in Rheumatoid Arthritis. Arterioscler Thromb Vasc Biol.
2020:ATVBAHA119313698.
778. French SL, Butov KR, Allaeys I, et al. Platelet-derived extracellular vesicles infiltrate
and modify the bone marrow during inflammation. Blood Adv. 2020;4(13):3011-3023.
779. Verdoes M, Florea BI, Menendez-Benito V, et al. A fluorescent broad-spectrum
proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol.
2006;13(11):1217-1226.
780. Raz V, Raz Y, Paniagua-Soriano G, et al. Proteasomal activity-based probes mark
protein homeostasis in muscles. J Cachexia Sarcopenia Muscle. 2017;8(5):798-807.
781. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release
two types of membrane vesicles: microvesicles by surface shedding and exosomes derived
from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94(11):3791-
3799.
782. Semple JW, Rebetz J, Kapur R. Transfusion-associated circulatory overload and
transfusion-related acute lung injury. Blood. 2019;133(17):1840-1853.
783. Kapur R, Kim M, Rebetz J, et al. Gastrointestinal microbiota contributes to the
development of murine transfusion-related acute lung injury. Blood Adv. 2018;2(13):1651-
1663.
784. Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect against
transfusion-related acute lung injury via IL-10. Blood. 2017;129(18):2557-2569.
785. McKenzie SE, Taylor SM, Malladi P, et al. The role of the human Fc receptor Fc
gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol.
1999;162(7):4311-4318.
786. Melki I, Allaeys I, Tessandier N, et al. Platelets release mitochondrial antigens in
systemic lupus erythematosus. Sci Transl Med. 2021;13(581).
787. Angenieux C, Dupuis A, Gachet C, de la Salle H, Maitre B. Cell surface expression
of HLA I molecules as a marker of young platelets. J Thromb Haemost. 2019;17(9):1511-
1521.
788. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME,
Corona-de la Pena NA, Salazar MI. Dengue Virus Induces the Release of sCD40L and
Changes in Levels of Membranal CD42b and CD40L Molecules in Human Platelets. Viruses.
2018;10(7).

168
789. Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization,
quantitation, and in situ detection of specific peptide-MHC class I complexes using a
monoclonal antibody. Immunity. 1997;6(6):715-726.
790. Rand ML, Wang H, Bang KW, Packham MA, Freedman J. Rapid clearance of
procoagulant platelet-derived microparticles from the circulation of rabbits. J Thromb
Haemost. 2006;4(7):1621-1623.
791. Rank A, Nieuwland R, Crispin A, et al. Clearance of platelet microparticles in vivo.
Platelets. 2011;22(2):111-116.
792. Lefrancais E, Ortiz-Munoz G, Caudrillier A, et al. The lung is a site of platelet
biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105-109.
793. Campbell RA, Schwertz H, Hottz ED, et al. Human megakaryocytes possess intrinsic
antiviral immunity through regulated induction of IFITM3. Blood. 2019;133(19):2013-2026.
794. Princiotta MF, Finzi D, Qian SB, et al. Quantitating protein synthesis, degradation,
and endogenous antigen processing. Immunity. 2003;18(3):343-354.
795. Sixt SU, Dahlmann B. Extracellular, circulating proteasomes and ubiquitin -
incidence and relevance. Biochim Biophys Acta. 2008;1782(12):817-823.
796. Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet
microparticle proteome. J Proteome Res. 2005;4(5):1516-1521.
797. Dean WL, Lee MJ, Cummins TD, Schultz DJ, Powell DW. Proteomic and functional
characterisation of platelet microparticle size classes. Thromb Haemost. 2009;102(4):711-
718.
798. Capriotti AL, Caruso G, Cavaliere C, Piovesana S, Samperi R, Laganà A. Proteomic
characterization of human platelet-derived microparticles. Anal Chim Acta. 2013;776:57-63.
799. Benaroudj N, Tarcsa E, Cascio P, Goldberg AL. The unfolding of substrates and
ubiquitin-independent protein degradation by proteasomes. Biochimie. 2001;83(3-4):311-
318.
800. Chen Z, Larregina AT, Morelli AE. Impact of extracellular vesicles on innate
immunity. Curr Opin Organ Transplant. 2019;24(6):670-678.
801. Lindenbergh MFS, Stoorvogel W. Antigen Presentation by Extracellular Vesicles
from Professional Antigen-Presenting Cells. Annu Rev Immunol. 2018;36:435-459.
802. Lindenbergh MFS, Wubbolts R, Borg EGF, van 't Veld EM, Boes M, Stoorvogel W.
Dendritic cells release exosomes together with phagocytosed pathogen; potential
implications for the role of exosomes in antigen presentation. J Extracell Vesicles.
2020;9(1):1798606.
803. Federici C, Shahaj E, Cecchetti S, et al. Natural-Killer-Derived Extracellular
Vesicles: Immune Sensors and Interactors. Front Immunol. 2020;11:262.
804. Zeng F, Morelli AE. Extracellular vesicle-mediated MHC cross-dressing in immune
homeostasis, transplantation, infectious diseases, and cancer. Semin Immunopathol.
2018;40(5):477-490.
805. Noulsri E. Effects of Cell-Derived Microparticles on Immune Cells and Potential
Implications in Clinical Medicine. Lab Med. 2020.
806. Gitz E, Pollitt AY, Gitz-Francois JJ, et al. CLEC-2 expression is maintained on
activated platelets and on platelet microparticles. Blood. 2014;124(14):2262-2270.
807. Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue viruses
infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis.
2019;13(11):e0007837.

169
808. Becker Y, Marcoux G, Allaeys I, et al. Autoantibodies in Systemic Lupus
Erythematosus Target Mitochondrial RNA. Front Immunol. 2019;10:1026.
809. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights
into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21(5):298-312.
810. Sabanovic B, Piva F, Cecati M, Giulietti M. Promising Extracellular Vesicle-Based
Vaccines against Viruses, Including SARS-CoV-2. Biology (Basel). 2021;10(2).
811. Bliss CM, Parsons AJ, Nachbagauer R, et al. Targeting Antigen to the Surface of EVs
Improves the. Mol Ther Methods Clin Dev. 2020;16:108-125.
812. Baker GR, Sullam PM, Levin J. A simple, fluorescent method to internally label
platelets suitable for physiological measurements. Am J Hematol. 1997;56(1):17-25.
813. Blessinger SA, Tran JQ, Jackman RP, et al. Immunodeficient mice are better for
modeling the transfusion of human blood components than wild-type mice. PLoS One.
2020;15(7):e0237106.

170
Annexe I : Targeting the autotaxin - Lysophosphatidic
acid receptor axis in cardiovascular diseases
Yang Zhao 1 , Stephan Hasse 1 , Chenqi Zhao 2 , Sylvain G Bourgoin 3
Affiliations :
1 Centre de Recherche du Centre Hospitalier Universitaire de Québec - Université Laval,
Canada; Département de microbiologie, infectiologie et immunologie, Faculté de Médecine,
Université Laval, Québec, QC G1V4G2, Canada.
2 Centre de Recherche du Centre Hospitalier Universitaire de Québec - Université Laval,
Canada.
3 Centre de Recherche du Centre Hospitalier Universitaire de Québec - Université Laval,
Canada; Département de microbiologie, infectiologie et immunologie, Faculté de Médecine,
Université Laval, Québec, QC G1V4G2, Canada. Electronic address:
Keywords: Lysophosphatidic acid, Autotaxin, Atherosclerosis, Inflammation, Vascular
remodelling

171
1 Abstract
Lysophosphatidic acid (LPA) is a well-characterized bioactive lipid mediator, which is
involved in development, physiology, and pathological processes of the cardiovascular
system. LPA can be produced both inside cells and in biological fluids. The majority of
extracellular LPA is produced locally by the secreted lysophospholipase D, autotaxin (ATX),
through its binding to various β integrins or heparin sulfate on cell surface and hydrolyzing
various lysophospholipids. LPA initiates cellular signalling pathways upon binding to and
activation of its G protein-coupled receptors (LPA1-6). LPA has potent effects on various
blood cells and vascular cells involved in the development of cardiovascular diseases such
as atherosclerosis and aortic valve sclerosis. LPA signalling drives cell migration and
proliferation, cytokine production, thrombosis, fibrosis, as well as angiogenesis. For instance,
LPA promotes activation and aggregation of platelets through LPA5, increases expression of
adhesion molecules in endothelial cells, and enhances expression of tissue factor in vascular
smooth muscle cells. Furthermore, LPA induces differentiation of monocytes into
macrophages and stimulates oxidized low-density lipoproteins (oxLDLs) uptake by
macrophages to form foam cells during formation of atherosclerotic lesions through LPA1-
3. This review summarizes recent findings of the roles played by ATX, LPA and LPA
receptors (LPARs) in atherosclerosis and calcific aortic valve disease. Targeting the ATX-
LPAR axis may have potential applications for treatment of patients suffering from various
cardiovascular diseases.

172
2 Graphical abstract
LPA initiates cellular signalling pathways through LPA1-6 expressed by blood cells and
vascular cells, mediating the development of atherosclerosis. The ATX-LPA axis can be
targeted for potential treatment of CVDs.

173
3 Lysophosphatidic acid and its receptors
Lysophosphatidic acid (LPA) is a bioactive lipid mediator required for the maintenance of
homeostasis in multiple physiological functions and pathological processes. LPA possesses
a glycerol backbone with an aliphatic fatty acid chain attached at sn-1 or sn-2 position
(Fig. 1). The sn-1 and the sn-2 positions are predominantly occupied by saturated and
unsaturated fatty acids, respectively [1]. The subclasses of LPA include the acyl-, alkyl-, and
alkenyl-LPA species (Fig. 1). Among them, acyl-LPA species are the most abundant
glycerophospholipid species [2]. The length and number of the fatty acid chain unsaturation
determine the diversity of molecular species of acyl-LPA. Common acyl-LPA species
include 16:0, 18:0, 18:1, 18:2, 18:3, 20:4, 20:5, and 22:6. Additionally, sn-2 acyl-LPA
species are unstable and easily undergo acyl chain migration to produce sn-1 acyl-LPA [1].
LPA biological activities rely at least in part on the fatty acid position on the glycerol
backbone, length, and degree of saturation [2].
Fig. 1. LPA structures. Basic structure of LPA includes a glycerol backbone, an aliphatic
acid chain, and a phosphate moiety. LPA comprises the acyl-, alkyl-, and alkenyl-LPA
species. Acyl-LPA can also be divided into 1-acyl and 2-acyl LPA according to the sn
position of the acyl chain.
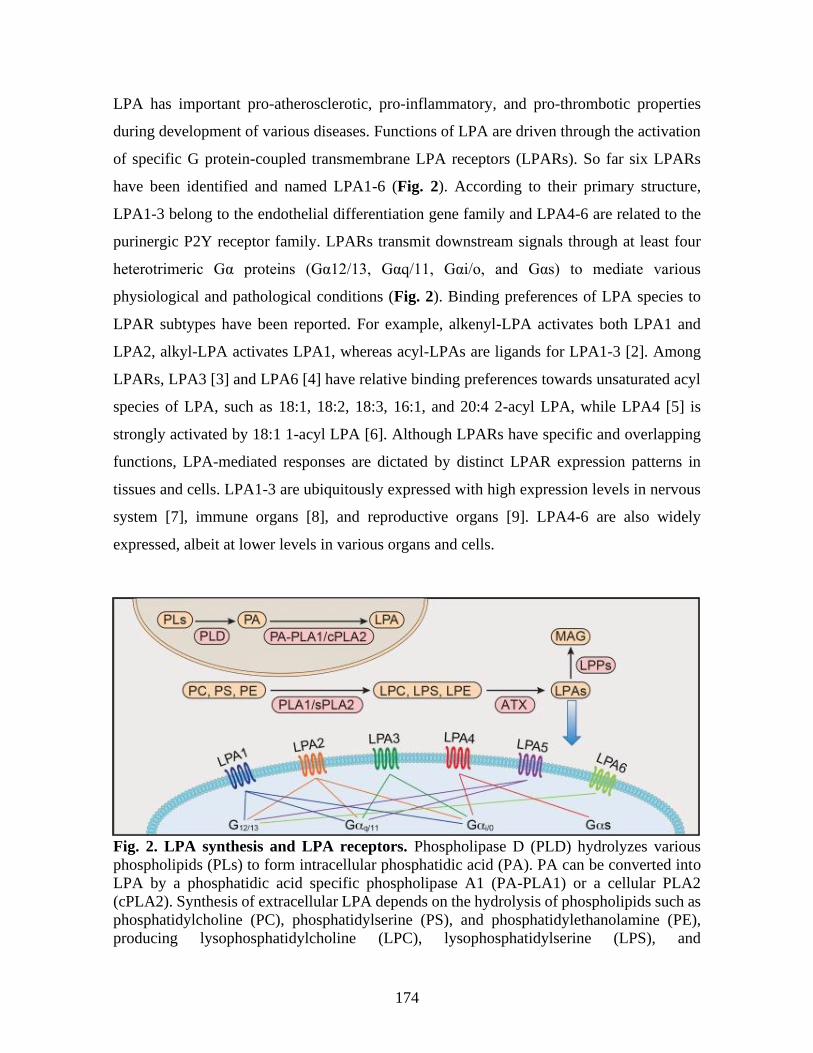
174
LPA has important pro-atherosclerotic, pro-inflammatory, and pro-thrombotic properties
during development of various diseases. Functions of LPA are driven through the activation
of specific G protein-coupled transmembrane LPA receptors (LPARs). So far six LPARs
have been identified and named LPA1-6 (Fig. 2). According to their primary structure,
LPA1-3 belong to the endothelial differentiation gene family and LPA4-6 are related to the
purinergic P2Y receptor family. LPARs transmit downstream signals through at least four
heterotrimeric Gα proteins (Gα12/13, Gαq/11, Gαi/o, and Gαs) to mediate various
physiological and pathological conditions (Fig. 2). Binding preferences of LPA species to
LPAR subtypes have been reported. For example, alkenyl-LPA activates both LPA1 and
LPA2, alkyl-LPA activates LPA1, whereas acyl-LPAs are ligands for LPA1-3 [2]. Among
LPARs, LPA3 [3] and LPA6 [4] have relative binding preferences towards unsaturated acyl
species of LPA, such as 18:1, 18:2, 18:3, 16:1, and 20:4 2-acyl LPA, while LPA4 [5] is
strongly activated by 18:1 1-acyl LPA [6]. Although LPARs have specific and overlapping
functions, LPA-mediated responses are dictated by distinct LPAR expression patterns in
tissues and cells. LPA1-3 are ubiquitously expressed with high expression levels in nervous
system [7], immune organs [8], and reproductive organs [9]. LPA4-6 are also widely
expressed, albeit at lower levels in various organs and cells.
Fig. 2. LPA synthesis and LPA receptors. Phospholipase D (PLD) hydrolyzes various
phospholipids (PLs) to form intracellular phosphatidic acid (PA). PA can be converted into
LPA by a phosphatidic acid specific phospholipase A1 (PA-PLA1) or a cellular PLA2
(cPLA2). Synthesis of extracellular LPA depends on the hydrolysis of phospholipids such as
phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidylethanolamine (PE),
producing lysophosphatidylcholine (LPC), lysophosphatidylserine (LPS), and

175
lysophosphatidylethanolamine (LPE) by phospholipases A1 (PLA1) or secreted
phospholipase A2 (sPLA2), respectively. Lysophospholipids are subsequently hydrolysed by
ATX to produce LPA. LPA is rapidly metabolized into monoacylglycerol (MAG) by the
ecto-activities of lipid phosphate phosphatases (LPPs). LPA induces various physiological
and pathophysiological responses through binding to and activation of six LPA receptors
(LPA1 to LPA6), which transmit downstream signals through at least four heterotrimeric Gα
proteins (Gα12/13, Gαq/11, Gαi/o, and Gαs).
4 LPA production pathways
Intracellular produced LPA works not only as a precursor or an intermediate in the synthesis
of cell phospholipids, but it can also serve as an intracellular signalling molecule. LPA can
be produced inside cells through sequential hydrolysis of phosphatidylcholine by a
phospholipase D (PLD) and a phospholipase A (PLA) [10] (Fig. 2). In this pathway, PLD
produced phosphatidic acid in the inner layer of cell membrane is subsequently deacylated
into LPA by a phosphatidic acid specific PLA1 [10].
LPA produced in the circulation acts in an autacoid way. Autotaxin (ATX) is responsible for
the production of extracellular LPA using various lysophospholipids as substrates (Fig. 2),
with plasma lysophosphatidylcholine (LPC) being the most abundant one. ATX, a member
of the nucleotide pyrophophatase/phosphodiesterase protein family, was originally isolated
from melanoma cells and characterized as a cell motility factor [11]. ATX is an enzyme with
unique lysophospholipase D activity that cleaves the choline group of LPC to produce LPA.
Structure of ATX consists of two N-terminal somatomedin B-like domains, a central catalytic
phosphodiester domain, and a C-terminal nuclease-like domain; these domains form a
hydrophobic channel containing the lysophospholipid-binding pocket [12]. The N-terminal
somatomedin B-like domain of ATX can bind to β integrins to access cell surface
lysophospholipids and to locally produce LPA [12].
There are three isoforms of ATX, namely ATX-α, -β, and -γ. ATX has a broad tissue
expression such as in brain, kidney, and lymphoid organs; and ATX can be produced locally
by a plethora of cell types [13]. ATX expression can be increased by growth factors, such as
fibroblast and epidermal growth factor, bone morphogenetic protein 2 (BMP-2) and the Wnt-
1 signalling pathway, but be inhibited by TGF-β and cytokines, such as interleukin-1 (IL-1),

176
IL-4, and IFN-γ [14]. In the vascular compartment, ATX-β is the most abundant ATX
isoform responsible for synthesizing plasma LPA [15]. Although the relative cellular origins
of plasma ATX are still uncertain, adipose tissues are a major source of ATX [16].
Megakaryocytes, the cells responsible for the production of platelets, do not express ATX
mRNA. But the enzyme was nevertheless found stored in α-granules of resting platelets and
was secreted upon platelet activation [17].
Plasma LPA was originally reported to be produced by activated platelets, especially under
pathological conditions such as inflammation and atherosclerosis [18]. During the
coagulation process, activated platelets release a large amount of lysophospholipids, which
are subsequently converted to 18:2 and 20:4 LPA by ATX [1]. Recently, a novel
phospholipase purified from thrombin activated human platelets, the acyl-protein
thioesterase 1 also known as lysophospholipase A1, was reported to possess PLA1 activity
[1]. Lysophospholipase A1 can generate sn-2-esterified LPC, which can be converted into
LPA by ATX. Almost half of serum LPA is produced in a platelet-dependent pathway
according to previous studies [10]. Furthermore, LPA can be generated in a cell-free system,
by cell-derived microparticles (MPs) [19]. Extracellular LPA is metabolized by the ecto-
activities of cell-associated lipid phosphate phosphatases (LPPs), which are responsible for
the rapid turnover of plasma LPA [20] and for maintaining the physiological concentration
of extracellular LPA in the low µM range.
5 The LPA-induced responses in cells of the cardiovascular
system
5.1. LPA and platelets
Research findings on LPARs expression in blood cells/vascular bed cells and LPA-induced
functional responses in context of cardiovascular diseases (CVDs) are summarized in Table
1. ATX has been reported to bind β1 and β3 integrins [12]. One of the consequences of
platelet activation is the release of ATX stored in α-granules [17]. ATX binding to activated
platelet β3 integrins promotes LPA production and LPA-dependent responses [12]. LPA
induces platelet shape change [21] and activation [22]. In blood, LPA-activated platelets form
platelet aggregates and platelet-monocyte aggregates [21], [23]. LPA also promotes the
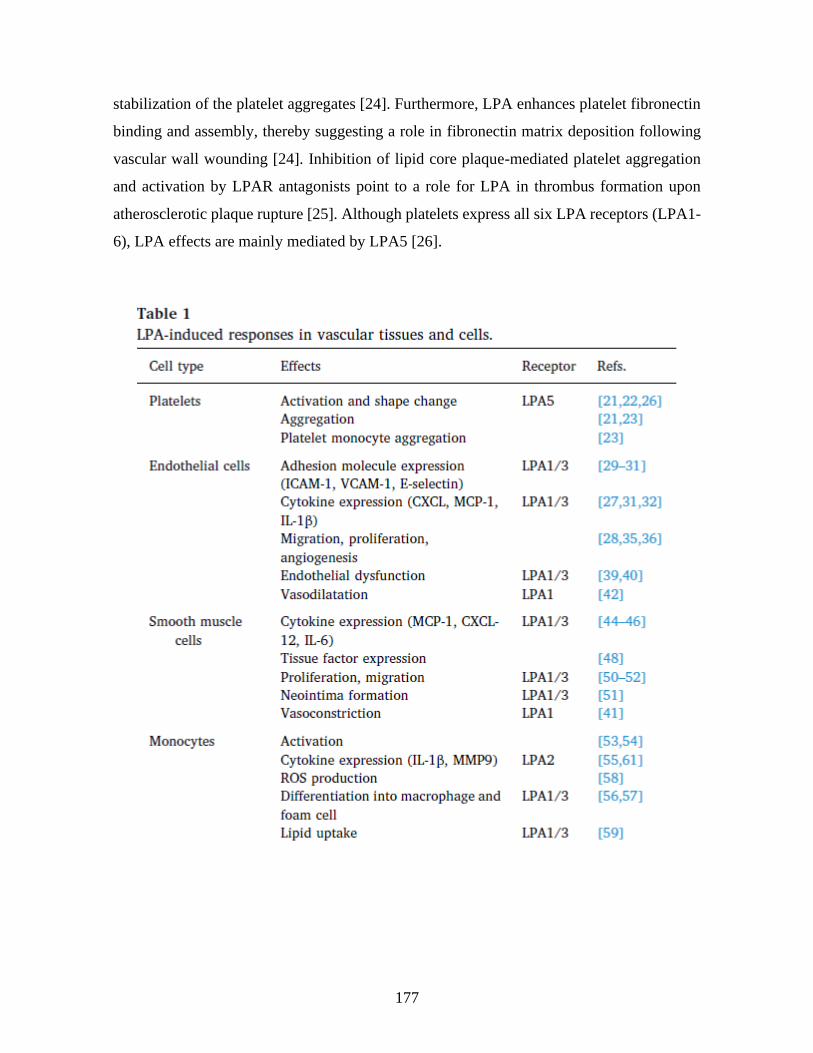
177
stabilization of the platelet aggregates [24]. Furthermore, LPA enhances platelet fibronectin
binding and assembly, thereby suggesting a role in fibronectin matrix deposition following
vascular wall wounding [24]. Inhibition of lipid core plaque-mediated platelet aggregation
and activation by LPAR antagonists point to a role for LPA in thrombus formation upon
atherosclerotic plaque rupture [25]. Although platelets express all six LPA receptors (LPA1-
6), LPA effects are mainly mediated by LPA5 [26].

178
5.2. LPA and endothelial cells
LPA contributes to the regulation of three endothelial cell-dependent processes: leukocyte
recruitment, angiogenesis, and vascular functions (Table 1). Silencing of LPA1 and LPA3 or
blocking those receptors with the selective antagonist Ki16425 attenuated LPA-induced
endothelial cell functional responses including chemokine C-X-C motif ligand 1 (CXCL1)
production [27], migration and proliferation [28]. LPA promotes leukocyte interaction with
the vascular wall through activation of LPA1/3 expressed on endothelial cells. Adhesion
proteins play a crucial role in leukocyte adhesion and migration across the vascular wall.
LPA increases the expression of the vascular cell adhesion molecules ICAM-1, VCAM-1,
and E-selectin [29], [30], [31]. LPA-mediated production of CXCL1 [27], IL-8 and monocyte
chemoattractant protein 1 (MCP-1) in endothelial cells also stimulates leukocyte adhesion to
the vascular wall [32]. IL-8 and MCP-1 are important mediators of leukocyte adhesion to the
endothelium under flow conditions [33]. In addition to these chemokines, LPA also induces
expression and secretion of the pro-inflammatory cytokine IL-1 in endothelial cells [31].
Studies using a mouse model of atherosclerosis suggested that LPA-mediated leukocyte
recruitment contributes to the initiation and the progression of atherosclerosis [27].
In vitro assays showed that LPA stimulated endothelial cell proliferation, migration, as well
as tube formation [28], [34]. LPA-induced cell migration, proliferation and tube formation
all contribute to angiogenesis, leading to new blood vessel formation from the existing
vasculature. LPA-induced angiogenesis depends on upregulation of the vascular endothelial
growth factor expression [35] and concomitant suppression of the angiogenesis repressor
CD36 expression in endothelial cells [36]. Taken together, these results suggest that the pro-
angiogenic properties of LPA contribute to development of imperfect intimal microvessels
commonly found in atherosclerotic plaques [37]. Those intimal microvessels are important
risk factors of plaque vulnerability since they are a source of intraplaque atherogenic lipids
and a cause for intraplaque haemorrhages [38]. Thus, LPA participates in atherosclerotic
plaque development and instability in part through its pro-angiogenic action and its ability to
recruit inflammatory cells.
Endothelial cell dysfunction is a characteristic of CVDs that is crucial to the initiation and
development of atherosclerosis. LPA influences some of endothelial cell functions that affect

179
the structure and the properties of the vascular endothelium. LPA stimulation decreases
endothelial cell confluence [39] and increases their motility [40]. Measurement of hydraulic
permeability in rat vessels showed increased permeability of the vascular endothelium in
response to LPA [39]. LPA also induces the expression of matrix metalloproteinase 2 (MMP-
2) in endothelial cells [40]. MMP-2 contributes to remodelling of the extracellular matrix.
Therefore, LPA could alter vessel functions and increase vessel leakage through upregulation
of MMP-2. LPA impact on vessel function is not limited to modulation of vascular
endothelium permeability, it also impacts the vascular tone by inducing vasodilatation or
vasoconstriction [41], [42]. Injection of LPA in the lumen of isolated murine aortae resulted
in nitric oxide synthase dependent vasodilation. Genetic depletion of the nitric oxide synthase
or the mechanical removal of endothelial cells resulted in vasoconstriction [42]. LPA was
shown to induced smooth muscle cell contraction through liberation of thromboxane A2
which results in vasoconstriction [41]. In both studies, the use of an antagonist of LPA1/3,
Ki16425, and genetic deletion of LPA1 abolished LPA-induced vasoconstriction and
vasodilation [41], [42].
5.3. LPA and smooth muscle cells
Smooth muscle cells are a major cell type in the hyperplasic vascular lesions seen in
atherosclerosis [43]. LPA administered to mice and rat was reported to increase neointima
formation [44]. LPA-induced neointima formation is partly mediated through the recruitment
of progenitor cells and their differentiation into vascular smooth muscle cells. Similar as in
endothelial cells, LPA-mediated activation of smooth muscle cells induces the expression
and the release of several cytokines and chemokines such as MCP-1 [45], CXCL12 [44] and
IL-6 [46], which promote leukocyte recruitment and inflammation. The LPA1/3 antagonist
Ki16425 inhibits LPA-induced cytokine production and neointima formation as well (Table
1 and [44]). A transcriptomics study only detected the expression of LPA1 mRNA in human
aortic smooth muscle cells [47], thereby suggesting that the effects of LPA in these cells were
mediated by LPA1. LPA also increases the production of tissue factor [48], which is found
at high level in the atherosclerotic plaque and is an important initiator of atherothrombosis
[49].

180
LPA stimulated smooth muscle cell proliferation and migration in vitro [50], [51] and in vivo
[51] in part through the induction of early growth response gene-1 expression and the
secretion of IL-6 [52]. In addition, smooth muscle cell growth was inhibited by an inhibitor
of NADPH oxidase, indicating a role for reactive oxygen species in LPA-dependent smooth
muscle cell proliferation [45].
5.4. LPA and monocytes
LPA also modulates monocyte recruitment and activation [53], [54] through the production
of reactive oxygen species, and the release of arachidonic acid and IL-1β [55]. LPA is an
important initiator of monocyte differentiation into macrophage [56] and formation of foam
cells [57], [58]. The LPA-dependent differentiation was associated with the inhibition of
high-density lipoprotein receptor SRB1 expression and generation of reactive oxygen species
[57], [58]. In atherosclerotic plaques, LPA enhances uptake of oxidized phospholipid by
macrophages [59]. Lipid uptake [59] and foam cell formation [57] are likely mediated
through LPA1 and/or LPA3. LPA may contribute to macrophage accumulation into
atherosclerotic plaques by inhibiting reverse transmigration across the endothelial layer [60].
Furthermore, LPA stimulates MMP-9 expression in THP-1 derived macrophages through
LPA2 [61]. MMP-9 also accelerates remodelling of the extracellular matrix of the artery wall,
resulting in progression and instability of the atherosclerotic plaque [61].
6 The ATX-LPA axis in cardiovascular diseases
ATX as well as the LPA-induced functional responses have been extensively studied in
various pathological conditions. Comprehensive reviews on the roles of lysophospholipids
in the development of atherosclerosis and other CVDs have been published previously [62],
[63]. Here we reviewed the most recent findings on the roles played by the ATX-LPA axis
to the development of cardiovascular pathologies such as atherosclerosis and calcific aortic
valve disease.
6.1. The ATX-LPA axis in atherosclerosis
LPA plays an important pro-thrombotic role and contributes to the development of
atherosclerotic plaques. Atherosclerosis is a slowly progressing arterial disease that is

181
characterized by inflammatory and regenerative processes, resulting in vascular remodelling
and formation of atherosclerotic plaques. Although plasma LPA level is in low µM range,
unsaturated long chain acyl-LPA species can be predominantly accumulated in the
atherosclerotic lipid-rich core [22]. In atherosclerosis, LPA can be generated locally by
oxidization of low-density lipoprotein (oxLDL) [22]. OxLDL are transporters of oxidized
lipids and ATX in human plasma [64]. These oxidized lipids and ATX participate to the
production of LPA in atherosclerotic lesions, which subsequently promote several
pathological processes of atherosclerosis [22]. In addition to oxLDL-derived LPA, ATX
originated from various cell types also produces LPA within the atherosclerotic lesions.
Plasma ATX originating from adipose tissues together with ATX secreted by vascular cells,
such as endothelial cells [65], smooth muscle cells [47] and macrophages [66], can bind to
activated β1 and β3 cell integrins [12]. ATX binding to integrins increases its catalytic
activity and contributes to localized LPA production [12]. Furthermore, ATX also binds to
exosomes [67]. When incubated with phospholipases in vitro, cell-derived MPs are capable
of producing LPA [19]. During development of atherosclerosis, vascular inflammation
results in increased endothelial permeability, allowing circulating MPs in the blood to diffuse
within the vascular wall [68], [69]. In addition to oxLDL, higher levels of MPs originated
from vascular cells are accumulated at the atherosclerotic plaque and could contribute to
intra-plaque LPA production [70]. Whether MPs contribute to the ATX-dependent LPA
production in CVDs is yet to be confirmed.
The primary location of newly generated LPA at the cell surface can be in proximity to its
receptors [71]. On the other hand, inflammation associated with atherosclerotic lesions and
vascular injuries can increase the expression of ATX and LPARs. The enhanced LPA
production subsequently promotes neointima formation, which worsens the atherosclerotic
lesions [72]. A mouse model showed that exogenously administered LPA can increase
atherosclerotic plaque burden through LPA1/3 [27]. Extracellular LPA levels can be
decreased by LPP3, which localizes to the plasma membrane and serves as a negative
regulator of LPA signalling through its dephosphorylation catalyzed function. Enhanced
LPP3 expression in animal models was shown to decrease circulating LPA level [73]. In
atherosclerosis, alterations of LPP3 expression in monocytes and vascular wall cells are
closely related to circulating LPA levels [74]. LPP3 was therefore suggested to be involved

182
in preventing the development of atherosclerosis, stabilizing atherosclerotic plaque, and
reducing the risks of complication associated with atherosclerosis [74]. Strategies to
stimulate LPP activity or protein expression could be envision for future treatments of CVDs.
Within the digestive tract, lysophospholipids coming from the diet are hydrolysed by ATX
to produce LPA in the intestinal lumen [75]. Intestinal LPC and LPA are absorbed and
transported into the plasma. These lysophospholipids, especially species with unsaturated
fatty acids such as arachidonic acid, can contribute to increased risks of CVDs by promoting
systemic inflammation and cell dysfunctions [76]. Atherosclerosis plaques in diabetic
patients are more vulnerable than those in non-diabetic patients with similar size of plaques.
One possibility is that carotid atheroma plaques of diabetic patients have a higher proportion
of polyunsaturated phospholipids such as 2-arachidonoyl-lysophosphatidylcholine [77]. This
lysophospholipid can be subsequently hydrolysed by ATX to produce C20:4 LPA, thereby
contributing to inflammation and to decreased atherosclerotic plaque stability. In addition,
ATX-derived LPA is also involved in mediating blood and vascular cell activation after
plaque rupture, further contributing to the progression and the complication of
atherosclerosis. LPA also serves as endogenous toll like receptor 4 ligand to activate NF-κB.
NF-κB signalling contributes to the development of atherosclerosis and the formation of
unstable plaques through enhanced inflammatory cytokine production and MMP-9
expression [78].
Acute coronary syndrome (ACS) is a life-threatening complication of atherosclerosis. In
ACS, most of the infarction is due to the formation of an occluding thrombus on the surface
of the atherosclerotic plaque. LPA has been suggested to increase the susceptibility of
atherosclerosis and its complications. For instance, a cross-sectional study of consecutive
patients showed a significant relationship between plasma LPA concentrations, especially
long chain unsaturated LPA species, and ACS [79]. Indeed, increased LPA levels were
detected in ACS patients, which were associated with ATX and platelet activation [79]. ATX
mass and enzymatic activity in the plasma from patients with coronary artery disease (CAD)
were associated with a higher risk of concurrent calcific aortic valve stenosis [80]. LPA levels
were higher in the plasma samples collected from coronary arteries than that from peripheral
arteries [81]. Elevated plasma LPA levels can be derived from different sources, to name a

183
few, ATX-mediated production of 18:2 LPA, the platelet-related production of 20:4 LPA,
and other pathways, which might include not only LPC but also
lysophosphatidylethanolamine and lysophosphatidylglycerol as sources of 22:6 LPA [82].
Indeed, CAD susceptibility was also linked to the PPAP2B gene, which encodes the
expression of LPP3 and negatively regulates plasma LPA level [74]. A genome-wide
association study identified 13 novel loci harboring one or more single nucleotide
polymorphisms associated with CAD, and reported that among these 13 novel loci, PPAP2B
displayed risk allele frequencies at 0.91 that are highly associated with the risk of CAD [74].
Taken together, those studies suggest that ATX-LPA axis contributes to the pathogenesis of
ACS.
6.2. The ATX-LPA axis in calcific aortic valve diseases
Calcific aortic valve stenosis (CAVS) is the most common chronic and multifactorial
valvular disorder among the calcific aortic valve diseases (CAVDs). Pathological changes
associated with CAVD include progressive fibrosis, large mineral deposits in the lipid-rich
area of aortic valve, leading to gradual obstruction of the aortic valve orifice. Fibrosis and
valve mineralization are two intertwine factors that play crucial roles in the pathological
hemodynamic changes of CAVDs [83]. Mendelian randomization studies revealed a
significant association between the development of CAVD and the lipoprotein(a) gene
variant re10455872 in these patients [84], [85], [86]. CAVD patients with high lipoprotein
levels were at higher risk to develop aortic valve stenosis. Lipoproteins were reported to be
transporters of oxidized phospholipids and ATX, which accumulate not only in
atherosclerotic plaques but also in the mineralized aortic valves [64]. Plasma lipoprotein-
associated PLA2 is expressed in platelets [87] and macrophages [88]. Mineralized aortic
valve tissues express high levels of lipoprotein-associated PLA2 [88]. This phospholipase
and ATX both contribute to LPA production from plasma lipoproteins, promoting a pro-
inflammatory condition that drives mineralization of the aortic valve. LPA derived from
oxLDL was shown to promote aortic valve mineralization through the LPA1-RhoA/ROCK-
NF-κB signalling pathway [87]. In human CAVS tissues, LPA1 mRNA was increased by
1.5-fold compared to those in control non-mineralized aortic valves [87]. Activation of LPA1

184
upregulates the expression of BMP2, a powerful morphogen signal that drives the osteogenic
program [88].
In addition to lipoproteins transported ATX, human explanted mineralized aortic valves
express high levels of ATX compared to non-mineralized valves. A 60% increase in ATX
enzymatic activity and a 1.5-fold increase in the levels of LPA in human mineralized aortic
valves were reported, respectively [64]. In a mouse model of CAVS, LPA promoted
inflammation and an osteogenic response through enhanced secretion of IL-6 and expression
of BMP2 [64]. Mass spectrophotometry analyses confirmed the enhanced LPA levels,
especially the unsaturated LPA species, in human aortic valve leaflets of CAVS patients [89].
A recent study revealed that the aggregation of platelets to endothelium-denudated aortic
valves contributes to the mineralization process of aortic valve through production of LPA
[90]. Platelets derived adenosine diphosphate can induce the release of ATX by valve
interstitial cells through P2RY1 receptors [90]. In turn, ATX binding to glycoprotein IIb/IIIa
(also known as integrin αIIbβ3) of platelets can promote the production of LPA during this
osteogenic process [90]. Both ex vivo and in vitro studies suggested that down regulation of
the LPP3 gene can promote the mineralization of aortic valves [91]. Medical interventions
targeting ATX and LPA1 have been suggested for treatment of CAVD.
7 Targeted ATX-LPA therapy
So far, no treatment targeting the ATX-LPA axis in CVDs has been fully developed.
However, pharmacological approaches targeting this pathway in other pathological
conditions, such as pulmonary and liver fibrosis, have been suggested (summarized below
and in Table 2). These examples of therapeutic approaches could potentially be developed
for the treatment of CVDs.
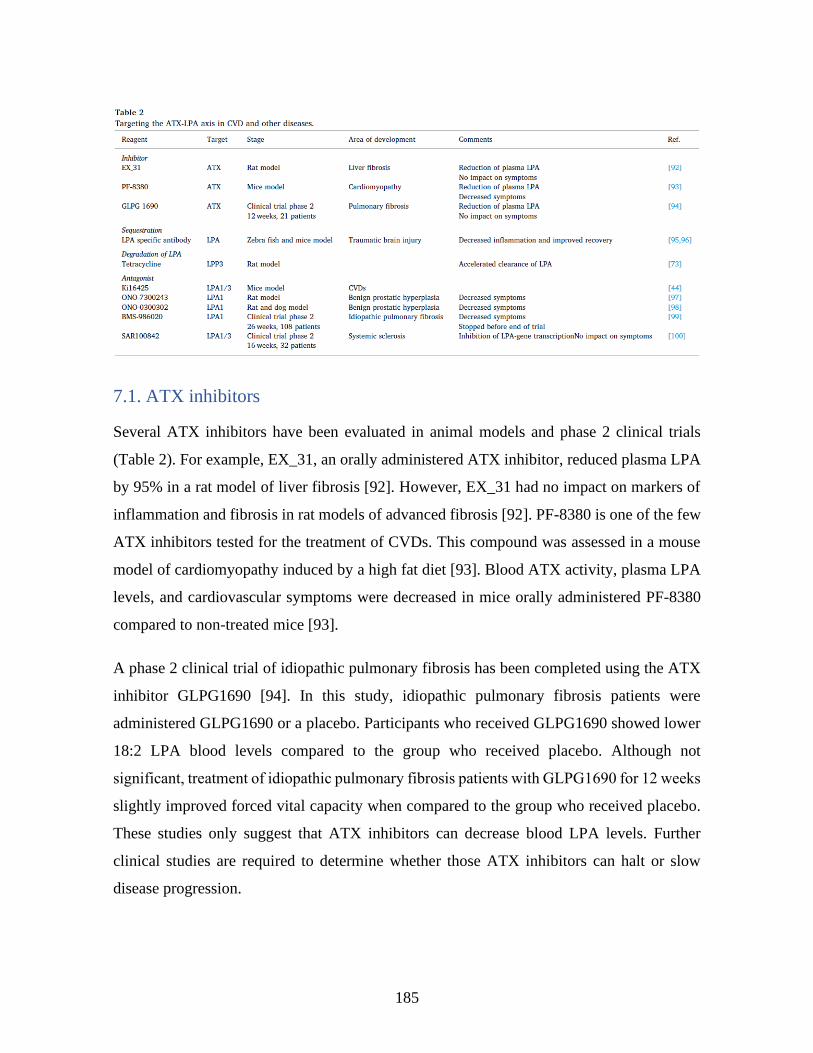
185
7.1. ATX inhibitors
Several ATX inhibitors have been evaluated in animal models and phase 2 clinical trials
(Table 2). For example, EX_31, an orally administered ATX inhibitor, reduced plasma LPA
by 95% in a rat model of liver fibrosis [92]. However, EX_31 had no impact on markers of
inflammation and fibrosis in rat models of advanced fibrosis [92]. PF-8380 is one of the few
ATX inhibitors tested for the treatment of CVDs. This compound was assessed in a mouse
model of cardiomyopathy induced by a high fat diet [93]. Blood ATX activity, plasma LPA
levels, and cardiovascular symptoms were decreased in mice orally administered PF-8380
compared to non-treated mice [93].
A phase 2 clinical trial of idiopathic pulmonary fibrosis has been completed using the ATX
inhibitor GLPG1690 [94]. In this study, idiopathic pulmonary fibrosis patients were
administered GLPG1690 or a placebo. Participants who received GLPG1690 showed lower
18:2 LPA blood levels compared to the group who received placebo. Although not
significant, treatment of idiopathic pulmonary fibrosis patients with GLPG1690 for 12 weeks
slightly improved forced vital capacity when compared to the group who received placebo.
These studies only suggest that ATX inhibitors can decrease blood LPA levels. Further
clinical studies are required to determine whether those ATX inhibitors can halt or slow
disease progression.

186
7.2. LPA sequestration
Systemic injection of anti-LPA antibodies was shown to reduce inflammation and
neurodegeneration in a zebrafish and in a mouse model of spinal cord lesion [95]. Mice
administered anti-LPA antibodies showed improved motor functions after spinal cord lesion
compared to non-treated mice [95]. LPA antibodies also diminished brain tissue damage and
inflammation in a mouse model of traumatic brain injury [96]. These studies suggest that
LPA antibody could be used to minimize injury-mediated neurone death and its associated
neurological dysfunctions.
7.3. LPA degradation
A genome-wide study highlighted a relation between CADs and the PPAP2B gene coding
for LPP3 [74]. Tetracyclines were shown in vitro to increase LPP1 and LPP3 cell surface
expression in malignant and non-malignant cell lines [73]. Interestingly, doxycycline
administration to rats accelerated the clearance of LPA from the circulation [73]. Enhancing
LPA degradation by vascular cells is a potential therapeutic option to reduce atherosclerotic
plaque development. Further studies are required to seriously evaluate the merits of this
strategy.
7.4. LPA receptors
The LPA1/3 antagonist Ki16425 has been used to study LPA signalling in CVDs [44] and
LPA-mediated responses in vascular wall cells [55]. So far Ki16425 has been used only in
preclinical studies. Other selective LPA1 antagonists such as ONO-7300243 [97] and ONO-
0300302 [98] have been tested for the treatment of benign prostatic hyperplasia and been
reported to reduce LPA-mediated increase in intraurethral pressure in rats and dogs.
Other LPAR antagonists have been evaluated in clinical trials. The LPA1 antagonist BMS-
986020 was used in a phase 2 clinical trial for the treatment of idiopathic pulmonary fibrosis
[99]. Patients receiving BMS-986020 showed a slower rate in respiratory capability decline
compared to the group administered placebo. However, the study was terminated before
completion due to serious side effects. On the other hand, a phase 2 clinical trials with another
LPA1/3 antagonist SAR100842 has been completed for the treatment of systemic sclerosis,

187
a disease characterized by skin fibrosis [100]. Transcription of LPA-induced genes
(plasminogen activator inhibitor-1, Wnt-2, and sFRP-4) was attenuated in skin samples of
patients administered SAR100842. No significant clinical improvement was achieved
compared to the control group [100]. However, it should be highlighted that the phase 2
clinical trials mentioned above have several limitations including the duration of the
treatments and the size of the cohorts (Table 2). Further studies are required to assess the
beneficial effects of those compounds for the treatment of fibrotic diseases.
8 Conclusions
Taking together, emerging evidence shows that the ATX-LPA axis is involved in CVD
development through: (1) production of pro-inflammatory cytokines and mediators, (2)
neointima formation, (3) immune cells recruitment, and (4) oxidized phospholipids uptake.
All those actions are involved in and associated to atherosclerosis development and increased
risk of atherothrombosis. Therefore, ATX and LPARs, especially LPA1, are potential targets
to mitigate the development of CVDs. Since the ATX-LPA axis is druggable, future studies
should evaluate whether ATX inhibitors and LPAR antagonists represent promising
strategies for preventing CVDs such as atherosclerosis and CAVD.
Acknowledgements
This project was supported by a research grant from the Canadian Institutes for Health
Research (MOP-142210). YZ is supported by the scholarship from China Scholarship
Council (CSC).
Disclosure statement
The authors have declared no conflicts of interest.
9 References
[1] A.L. Bolen, A.P. Naren, S. Yarlagadda, S. Beranova-Giorgianni, L. Chen, D. Norman,
D.L. Baker, M.M. Rowland, M.D. Best, T. Sano, T. Tsukahara, K. Liliom, Y. Igarashi, G.
Tigyi, The phospholipase A1 activity of lysophospholipase A-I links platelet activation to
LPA production during blood coagulation, J. Lipid Res. 52(5) (2011) 958-70.
[2] A. Tokumura, J. Sinomiya, S. Kishimoto, T. Tanaka, K. Kogure, T. Sugiura, K. Satouchi,
K. Waku, K. Fukuzawa, Human platelets respond differentially to lysophosphatidic acids

188
having a highly unsaturated fatty acyl group and alkyl ether-linked lysophosphatidic acids,
Biochem. J. 365(Pt 3) (2002) 617-28.
[3] K. Bandoh, J. Aoki, A. Taira, M. Tsujimoto, H. Arai, K. Inoue, Lysophosphatidic acid
(LPA) receptors of the EDG family are differentially activated by LPA species. Structure-
activity relationship of cloned LPA receptors, FEBS Lett. 478(1-2) (2000) 159-65.
[4] K. Yanagida, K. Masago, H. Nakanishi, Y. Kihara, F. Hamano, Y. Tajima, R. Taguchi,
T. Shimizu, S. Ishii, Identification and characterization of a novel lysophosphatidic acid
receptor, p2y5/LPA6, J. Biol. Chem. 284(26) (2009) 17731-41.
[5] K. Noguchi, S. Ishii, T. Shimizu, Identification of p2y9/GPR23 as a novel G protein-
coupled receptor for lysophosphatidic acid, structurally distant from the Edg family, J. Biol.
Chem. 278(28) (2003) 25600-6.
[6] S. Okudaira, H. Yukiura, J. Aoki, Biological roles of lysophosphatidic acid signaling
through its production by autotaxin, Biochimie 92(6) (2010) 698-706.
[7] J.H. Hecht, J.A. Weiner, S.R. Post, J. Chun, Ventricular zone gene-1 (vzg-1) encodes a
lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral
cortex, J. Cell Biol. 135(4) (1996) 1071-83.
[8] S. An, T. Bleu, O.G. Hallmark, E.J. Goetzl, Characterization of a novel subtype of human
G protein-coupled receptor for lysophosphatidic acid, J. Biol. Chem. 273(14) (1998) 7906-
10.
[9] K. Bandoh, J. Aoki, H. Hosono, S. Kobayashi, T. Kobayashi, K. Murakami-Murofushi,
M. Tsujimoto, H. Arai, K. Inoue, Molecular cloning and characterization of a novel human
G-protein-coupled receptor, EDG7, for lysophosphatidic acid, J. Biol. Chem. 274(39) (1999)
27776-85.
[10] J. Aoki, A. Inoue, S. Okudaira, Two pathways for lysophosphatidic acid production,
Biochim. Biophys. Acta 1781(9) (2008) 513-8.
[11] M.L. Stracke, H.C. Krutzsch, E.J. Unsworth, A. Arestad, V. Cioce, E. Schiffmann, L.A.
Liotta, Identification, purification, and partial sequence analysis of autotaxin, a novel
motility-stimulating protein, J. Biol. Chem. 267(4) (1992) 2524-9.
[12] Z. Fulkerson, T. Wu, M. Sunkara, C.V. Kooi, A.J. Morris, S.S. Smyth, Binding of
autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian
cells, J. Biol. Chem. 286(40) (2011) 34654-63.
[13] S. Emoto, M. Kurano, K. Kano, K. Matsusaki, H. Yamashita, M. Nishikawa, K. Igarashi,
H. Ikeda, J. Aoki, J. Kitayama, Y. Yatomi, Analysis of glycero-lysophospholipids in gastric
cancerous ascites, J. Lipid Res. 58(4) (2017) 763-771.
[14] L.A. van Meeteren, W.H. Moolenaar, Regulation and biological activities of the
autotaxin-LPA axis, Prog Lipid Res 46(2) (2007) 145-60.
[15] A. Tokumura, E. Majima, Y. Kariya, K. Tominaga, K. Kogure, K. Yasuda, K.
Fukuzawa, Identification of human plasma lysophospholipase D, a lysophosphatidic acid-
producing enzyme, as autotaxin, a multifunctional phosphodiesterase, J. Biol. Chem. 277(42)
(2002) 39436-42.
[16] S.S. Smyth, P. Mueller, F. Yang, J.A. Brandon, A.J. Morris, Arguing the case for the
autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the
development and complications of atherosclerosis, Arterioscler Thromb Vasc Biol 34(3)
(2014) 479-86.
[17] R. Leblanc, S.C. Lee, M. David, J.C. Bordet, D.D. Norman, R. Patil, D. Miller, D. Sahay,
J. Ribeiro, P. Clezardin, G.J. Tigyi, O. Peyruchaud, Interaction of platelet-derived autotaxin

189
with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone, Blood
124(20) (2014) 3141-50.
[18] J. Aoki, A. Taira, Y. Takanezawa, Y. Kishi, K. Hama, T. Kishimoto, K. Mizuno, K.
Saku, R. Taguchi, H. Arai, Serum lysophosphatidic acid is produced through diverse
phospholipase pathways, J. Biol. Chem. 277(50) (2002) 48737-44.
[19] O. Fourcade, M.F. Simon, C. Viode, N. Rugani, F. Leballe, A. Ragab, B. Fournie, L.
Sarda, H. Chap, Secretory phospholipase A2 generates the novel lipid mediator
lysophosphatidic acid in membrane microvesicles shed from activated cells, Cell 80(6)
(1995) 919-27.
[20] J.L. Tomsig, A.H. Snyder, E.V. Berdyshev, A. Skobeleva, C. Mataya, V. Natarajan,
D.N. Brindley, K.R. Lynch, Lipid phosphate phosphohydrolase type 1 (LPP1) degrades
extracellular lysophosphatidic acid in vivo, Biochem. J. 419(3) (2009) 611-8.
[21] M. Retzer, M. Essler, Lysophosphatidic acid-induced platelet shape change proceeds
via Rho/Rho kinase-mediated myosin light-chain and moesin phosphorylation, Cell. Signal.
12(9-10) (2000) 645-8.
[22] W. Siess, K.J. Zangl, M. Essler, M. Bauer, R. Brandl, C. Corrinth, R. Bittman, G. Tigyi,
M. Aepfelbacher, Lysophosphatidic acid mediates the rapid activation of platelets and
endothelial cells by mildly oxidized low density lipoprotein and accumulates in human
atherosclerotic lesions, Proc Natl Acad Sci U S A 96(12) (1999) 6931-6.
[23] N. Haseruck, W. Erl, D. Pandey, G. Tigyi, P. Ohlmann, C. Ravanat, C. Gachet, W. Siess,
The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte
aggregate formation in whole blood: involvement of P2Y1 and P2Y12 receptors, Blood
103(7) (2004) 2585-92.
[24] O.E. Olorundare, O. Peyruchaud, R.M. Albrecht, D.F. Mosher, Assembly of a
fibronectin matrix by adherent platelets stimulated by lysophosphatidic acid and other
agonists, Blood 98(1) (2001) 117-24.
[25] E. Rother, R. Brandl, D.L. Baker, P. Goyal, H. Gebhard, G. Tigyi, W. Siess, Subtype-
selective antagonists of lysophosphatidic Acid receptors inhibit platelet activation triggered
by the lipid core of atherosclerotic plaques, Circulation 108(6) (2003) 741-7.
[26] J.R. Williams, A.L. Khandoga, P. Goyal, J.I. Fells, D.H. Perygin, W. Siess, A.L. Parrill,
G. Tigyi, Y. Fujiwara, Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate
receptor indicates role in human platelet activation, J. Biol. Chem. 284(25) (2009) 17304-19.
[27] Z. Zhou, P. Subramanian, G. Sevilmis, B. Globke, O. Soehnlein, E. Karshovska, R.
Megens, K. Heyll, J. Chun, J.S. Saulnier-Blache, M. Reinholz, M. van Zandvoort, C. Weber,
A. Schober, Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing
CXCL1 from the endothelium, Cell Metab 13(5) (2011) 592-600.
[28] C.I. Lin, C.N. Chen, M.T. Huang, S.J. Lee, C.H. Lin, C.C. Chang, H. Lee,
Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation
in human endothelial cells through LPA(1/3), COX-2, and NF-kappaB activation- and EGFR
transactivation-dependent mechanisms, Cell. Signal. 20(10) (2008) 1804-14.
[29] H. Shimada, L.E. Rajagopalan, Rho kinase-2 activation in human endothelial cells drives
lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65,
J. Biol. Chem. 285(17) (2010) 12536-42.
[30] C. Rizza, N. Leitinger, J. Yue, D.J. Fischer, D.A. Wang, P.T. Shih, H. Lee, G. Tigyi,
J.A. Berliner, Lysophosphatidic acid as a regulator of endothelial/leukocyte interaction, Lab.
Invest. 79(10) (1999) 1227-35.

190
[31] C.I. Lin, C.N. Chen, P.W. Lin, K.J. Chang, F.J. Hsieh, H. Lee, Lysophosphatidic acid
regulates inflammation-related genes in human endothelial cells through LPA1 and LPA3,
Biochem. Biophys. Res. Commun. 363(4) (2007) 1001-8.
[32] C.I. Lin, C.N. Chen, J.H. Chen, H. Lee, Lysophospholipids increase IL-8 and MCP-1
expressions in human umbilical cord vein endothelial cells through an IL-1-dependent
mechanism, J. Cell. Biochem. 99(4) (2006) 1216-32.
[33] R.E. Gerszten, E.A. Garcia-Zepeda, Y.C. Lim, M. Yoshida, H.A. Ding, M.A. Gimbrone,
Jr., A.D. Luster, F.W. Luscinskas, A. Rosenzweig, MCP-1 and IL-8 trigger firm adhesion of
monocytes to vascular endothelium under flow conditions, Nature 398(6729) (1999) 718-23.
[34] H. Lee, E.J. Goetzl, S. An, Lysophosphatidic acid and sphingosine 1-phosphate
stimulate endothelial cell wound healing, Am. J. Physiol. Cell Physiol. 278(3) (2000) C612-
8.
[35] C.I. Lin, C.N. Chen, M.T. Huang, S.J. Lee, C.H. Lin, C.C. Chang, H. Lee,
Lysophosphatidic acid up-regulates vascular endothelial growth factor-C and lymphatic
marker expressions in human endothelial cells, Cell. Mol. Life Sci. 65(17) (2008) 2740-51.
[36] B. Ren, J. Hale, S. Srikanthan, R.L. Silverstein, Lysophosphatidic acid suppresses
endothelial cell CD36 expression and promotes angiogenesis via a PKD-1-dependent
signaling pathway, Blood 117(22) (2011) 6036-45.
[37] C.M. Rivera-Lopez, A.L. Tucker, K.R. Lynch, Lysophosphatidic acid (LPA) and
angiogenesis, Angiogenesis 11(3) (2008) 301-10.
[38] R. Di Stefano, F. Felice, A. Balbarini, Angiogenesis as risk factor for plaque
vulnerability, Curr. Pharm. Des. 15(10) (2009) 1095-106.
[39] N.A. Neidlinger, S.K. Larkin, A. Bhagat, G.P. Victorino, F.A. Kuypers, Hydrolysis of
phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates
lysophosphatidic acid and results in vascular dysfunction, J. Biol. Chem. 281(2) (2006) 775-
81.
[40] W.T. Wu, C.N. Chen, C.I. Lin, J.H. Chen, H. Lee, Lysophospholipids enhance matrix
metalloproteinase-2 expression in human endothelial cells, Endocrinology 146(8) (2005)
3387-400.
[41] P.T. Dancs, E. Ruisanchez, A. Balogh, C.R. Panta, Z. Miklos, R.M. Nusing, J. Aoki, J.
Chun, S. Offermanns, G. Tigyi, Z. Benyo, LPA1 receptor-mediated thromboxane A2 release
is responsible for lysophosphatidic acid-induced vascular smooth muscle contraction,
FASEB J. 31(4) (2017) 1547-1555.
[42] E. Ruisanchez, P. Dancs, M. Kerek, T. Nemeth, B. Farago, A. Balogh, R. Patil, B.L.
Jennings, K. Liliom, K.U. Malik, A.V. Smrcka, G. Tigyi, Z. Benyo, Lysophosphatidic acid
induces vasodilation mediated by LPA1 receptors, phospholipase C, and endothelial nitric
oxide synthase, FASEB J. 28(2) (2014) 880-90.
[43] L. Jonasson, J. Holm, O. Skalli, G. Bondjers, G.K. Hansson, Regional accumulations of
T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque,
Arteriosclerosis 6(2) (1986) 131-8.
[44] P. Subramanian, E. Karshovska, P. Reinhard, R.T. Megens, Z. Zhou, S. Akhtar, U.
Schumann, X. Li, M. van Zandvoort, C. Ludin, C. Weber, A. Schober, Lysophosphatidic acid
receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell
recruitment in neointima formation, Circ Res 107(1) (2010) 96-105.
[45] U. Kaneyuki, S. Ueda, S. Yamagishi, S. Kato, T. Fujimura, R. Shibata, A. Hayashida, J.
Yoshimura, M. Kojiro, K. Oshima, S. Okuda, Pitavastatin inhibits lysophosphatidic acid-
induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth

191
muscle cells by suppressing Rac-1-mediated reactive oxygen species generation, Vascul
Pharmacol 46(4) (2007) 286-92.
[46] F. Hao, M. Tan, D.D. Wu, X. Xu, M.Z. Cui, LPA induces IL-6 secretion from aortic
smooth muscle cells via an LPA1-regulated, PKC-dependent, and p38alpha-mediated
pathway, Am. J. Physiol. Heart Circ. Physiol. 298(3) (2010) H974-83.
[47] L. Bao, J. Qi, Y.W. Wang, Q. Xi, T. Tserennadmid, P.F. Zhao, J. Qi, A. Damirin, The
atherogenic actions of LPC on vascular smooth muscle cells and its LPA receptor mediated
mechanism, Biochem. Biophys. Res. Commun. 503(3) (2018) 1911-1918.
[48] M.Z. Cui, G. Zhao, A.L. Winokur, E. Laag, J.R. Bydash, M.S. Penn, G.M. Chisolm, X.
Xu, Lysophosphatidic acid induction of tissue factor expression in aortic smooth muscle
cells, Arterioscler Thromb Vasc Biol 23(2) (2003) 224-30.
[49] M.B. Taubman, L. Wang, C. Miller, The role of smooth muscle derived tissue factor in
mediating thrombosis and arterial injury, Thromb Res 122 Suppl 1 (2008) S78-81.
[50] J. Kim, J.R. Keys, A.D. Eckhart, Vascular smooth muscle migration and proliferation
in response to lysophosphatidic acid (LPA) is mediated by LPA receptors coupling to Gq,
Cell. Signal. 18(10) (2006) 1695-701.
[51] A. Gaaya, O. Poirier, N. Mougenot, T. Hery, F. Atassi, A. Marchand, J.S. Saulnier-
Blache, J. Amour, J. Vogt, A.M. Lompre, F. Soubrier, S. Nadaud, Plasticity-related gene-1
inhibits lysophosphatidic acid-induced vascular smooth muscle cell migration and
proliferation and prevents neointima formation, Am. J. Physiol. Cell Physiol. 303(10) (2012)
C1104-14.
[52] M.Z. Cui, E. Laag, L. Sun, M. Tan, G. Zhao, X. Xu, Lysophosphatidic acid induces
early growth response gene 1 expression in vascular smooth muscle cells: CRE and SRE
mediate the transcription, Arterioscler Thromb Vasc Biol 26(5) (2006) 1029-35.
[53] M. Fueller, D.A. Wang, G. Tigyi, W. Siess, Activation of human monocytic cells by
lysophosphatidic acid and sphingosine-1-phosphate, Cell. Signal. 15(4) (2003) 367-75.
[54] F. D'Aquilio, M. Procaccini, V. Izzi, V. Chiurchiu, V. Giambra, F. Carotenuto, P. Di
Nardo, P.M. Baldini, Activatory properties of lysophosphatidic acid on human THP-1 cells,
Inflammation 30(5) (2007) 167-77.
[55] C.L. Chang, M.E. Lin, H.Y. Hsu, C.L. Yao, S.M. Hwang, C.Y. Pan, C.Y. Hsu, H. Lee,
Lysophosphatidic acid-induced interleukin-1 beta expression is mediated through Gi/Rho
and the generation of reactive oxygen species in macrophages, J. Biomed. Sci. 15(3) (2008)
357-63.
[56] R. Ray, V. Rai, Lysophosphatidic acid converts monocytes into macrophages in both
mice and humans, Blood 129(9) (2017) 1177-1183.
[57] L. Chen, J. Zhang, X. Deng, Y. Liu, X. Yang, Q. Wu, C. Yu, Lysophosphatidic acid
directly induces macrophage-derived foam cell formation by blocking the expression of
SRBI, Biochem. Biophys. Res. Commun. 491(3) (2017) 587-594.
[58] S.S. Barbieri, S. Eligini, M. Brambilla, E. Tremoli, S. Colli, Reactive oxygen species
mediate cyclooxygenase-2 induction during monocyte to macrophage differentiation: critical
role of NADPH oxidase, Cardiovasc Res 60(1) (2003) 187-97.
[59] C.L. Chang, H.Y. Hsu, H.Y. Lin, W. Chiang, H. Lee, Lysophosphatidic acid-induced
oxidized low-density lipoprotein uptake is class A scavenger receptor-dependent in
macrophages, Prostaglandins Other Lipid Mediat 87(1-4) (2008) 20-5.
[60] J. Llodra, V. Angeli, J. Liu, E. Trogan, E.A. Fisher, G.J. Randolph, Emigration of
monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not
progressive, plaques, Proc Natl Acad Sci U S A 101(32) (2004) 11779-84.

192
[61] C. Gu, F. Wang, Z. Zhao, H. Wang, X. Cong, X. Chen, Lysophosphatidic Acid Is
Associated with Atherosclerotic Plaque Instability by Regulating NF-κB Dependent Matrix
Metalloproteinase-9 Expression via LPA(2) in Macrophages, Front Physiol 8 (2017) 266.
[62] A. Abdel-Latif, P.M. Heron, A.J. Morris, S.S. Smyth, Lysophospholipids in coronary
artery and chronic ischemic heart disease, Curr Opin Lipidol 26(5) (2015) 432-7.
[63] A. Schober, W. Siess, Lysophosphatidic acid in atherosclerotic diseases, Br. J.
Pharmacol. 167(3) (2012) 465-82.
[64] E. Im, R. Motiejunaite, J. Aranda, E.Y. Park, L. Federico, T.I. Kim, T. Clair, M.L.
Stracke, S. Smyth, A. Kazlauskas, Phospholipase Cgamma activation drives increased
production of autotaxin in endothelial cells and lysophosphatidic acid-dependent regression,
Mol. Cell. Biol. 30(10) (2010) 2401-10.
[65] S. Li, J. Zhang, Lipopolysaccharide induces autotaxin expression in human monocytic
THP-1 cells, Biochem. Biophys. Res. Commun. 378(2) (2009) 264-8.
[66] S.A. Jethwa, E.J. Leah, Q. Zhang, N.A. Bright, D. Oxley, M.D. Bootman, S.A. Rudge,
M.J. Wakelam, Exosomes bind to autotaxin and act as a physiological delivery mechanism
to stimulate LPA receptor signalling in cells, J. Cell Sci. 129(20) (2016) 3948-3957.
[67] N. Cloutier, A. Pare, R.W. Farndale, H.R. Schumacher, P.A. Nigrovic, S. Lacroix, E.
Boilard, Platelets can enhance vascular permeability, Blood 120(6) (2012) 1334-43.
[68] E. Boilard, P.A. Nigrovic, K. Larabee, G.F. Watts, J.S. Coblyn, M.E. Weinblatt, E.M.
Massarotti, E. Remold-O'Donnell, R.W. Farndale, J. Ware, D.M. Lee, Platelets amplify
inflammation in arthritis via collagen-dependent microparticle production, Science
327(5965) (2010) 580-3.
[69] A.S. Leroyer, H. Isobe, G. Leseche, Y. Castier, M. Wassef, Z. Mallat, B.R. Binder, A.
Tedgui, C.M. Boulanger, Cellular origins and thrombogenic activity of microparticles
isolated from human atherosclerotic plaques, J. Am. Coll. Cardiol. 49(7) (2007) 772-7.
[70] Z. Pamuklar, L. Federico, S. Liu, M. Umezu-Goto, A. Dong, M. Panchatcharam, Z.
Fulkerson, E. Berdyshev, V. Natarajan, X. Fang, L.A. van Meeteren, W.H. Moolenaar, G.B.
Mills, A.J. Morris, S.S. Smyth, Autotaxin/lysopholipase D and lysophosphatidic acid
regulate murine hemostasis and thrombosis, J. Biol. Chem. 284(11) (2009) 7385-94.
[71] C. Zhang, D.L. Baker, S. Yasuda, N. Makarova, L. Balazs, L.R. Johnson, G.K. Marathe,
T.M. McIntyre, Y. Xu, G.D. Prestwich, H.S. Byun, R. Bittman, G. Tigyi, Lysophosphatidic
acid induces neointima formation through PPARgamma activation, J. Exp. Med. 199(6)
(2004) 763-74.
[72] X. Tang, Y.Y. Zhao, J. Dewald, J.M. Curtis, D.N. Brindley, Tetracyclines increase lipid
phosphate phosphatase expression on plasma membranes and turnover of plasma
lysophosphatidate, J. Lipid Res. 57(4) (2016) 597-606.
[73] H. Schunkert, I.R. Konig, S. Kathiresan, M.P. Reilly, T.L. Assimes, H. Holm, M. Preuss,
A.F. Stewart, M. Barbalic, C. Gieger, D. Absher, Z. Aherrahrou, H. Allayee, D. Altshuler,
S.S. Anand, K. Andersen, J.L. Anderson, D. Ardissino, S.G. Ball, A.J. Balmforth, T.A.
Barnes, D.M. Becker, L.C. Becker, K. Berger, J.C. Bis, S.M. Boekholdt, E. Boerwinkle, P.S.
Braund, M.J. Brown, M.S. Burnett, I. Buysschaert, J.F. Carlquist, L. Chen, S. Cichon, V.
Codd, R.W. Davies, G. Dedoussis, A. Dehghan, S. Demissie, J.M. Devaney, P. Diemert, R.
Do, A. Doering, S. Eifert, N.E. Mokhtari, S.G. Ellis, R. Elosua, J.C. Engert, S.E. Epstein, U.
de Faire, M. Fischer, A.R. Folsom, J. Freyer, B. Gigante, D. Girelli, S. Gretarsdottir, V.
Gudnason, J.R. Gulcher, E. Halperin, N. Hammond, S.L. Hazen, A. Hofman, B.D. Horne, T.
Illig, C. Iribarren, G.T. Jones, J.W. Jukema, M.A. Kaiser, L.M. Kaplan, J.J. Kastelein, K.T.
Khaw, J.W. Knowles, G. Kolovou, A. Kong, R. Laaksonen, D. Lambrechts, K. Leander, G.

193
Lettre, M. Li, W. Lieb, C. Loley, A.J. Lotery, P.M. Mannucci, S. Maouche, N. Martinelli,
P.P. McKeown, C. Meisinger, T. Meitinger, O. Melander, P.A. Merlini, V. Mooser, T.
Morgan, T.W. Muhleisen, J.B. Muhlestein, T. Munzel, K. Musunuru, J. Nahrstaedt, C.P.
Nelson, M.M. Nothen, O. Olivieri, R.S. Patel, C.C. Patterson, A. Peters, F. Peyvandi, L. Qu,
A.A. Quyyumi, D.J. Rader, L.S. Rallidis, C. Rice, F.R. Rosendaal, D. Rubin, V. Salomaa,
M.L. Sampietro, M.S. Sandhu, E. Schadt, A. Schafer, A. Schillert, S. Schreiber, J.
Schrezenmeir, S.M. Schwartz, D.S. Siscovick, M. Sivananthan, S. Sivapalaratnam, A. Smith,
T.B. Smith, J.D. Snoep, N. Soranzo, J.A. Spertus, K. Stark, K. Stirrups, M. Stoll, W.H. Tang,
S. Tennstedt, G. Thorgeirsson, G. Thorleifsson, M. Tomaszewski, A.G. Uitterlinden, A.M.
van Rij, B.F. Voight, N.J. Wareham, G.A. Wells, H.E. Wichmann, P.S. Wild, C. Willenborg,
J.C. Witteman, B.J. Wright, S. Ye, T. Zeller, A. Ziegler, F. Cambien, A.H. Goodall, L.A.
Cupples, T. Quertermous, W. Marz, C. Hengstenberg, S. Blankenberg, W.H. Ouwehand,
A.S. Hall, P. Deloukas, J.R. Thompson, K. Stefansson, R. Roberts, U. Thorsteinsdottir, C.J.
O'Donnell, R. McPherson, J. Erdmann, N.J. Samani, Large-scale association analysis
identifies 13 new susceptibility loci for coronary artery disease, Nat. Genet. 43(4) (2011)
333-8.
[74] M. Navab, G. Hough, G.M. Buga, F. Su, A.C. Wagner, D. Meriwether, A.
Chattopadhyay, F. Gao, V. Grijalva, J.S. Danciger, B.J. Van Lenten, E. Org, A.J. Lusis, C.
Pan, G.M. Anantharamaiah, R. Farias-Eisner, S.S. Smyth, S.T. Reddy, A.M. Fogelman,
Transgenic 6F tomatoes act on the small intestine to prevent systemic inflammation and
dyslipidemia caused by Western diet and intestinally derived lysophosphatidic acid, J. Lipid
Res. 54(12) (2013) 3403-18.
[75] M. Navab, A. Chattopadhyay, G. Hough, D. Meriwether, S.I. Fogelman, A.C. Wagner,
V. Grijalva, F. Su, G.M. Anantharamaiah, L.H. Hwang, K.F. Faull, S.T. Reddy, A.M.
Fogelman, Source and role of intestinally derived lysophosphatidic acid in dyslipidemia and
atherosclerosis, J. Lipid Res. 56(4) (2015) 871-87.
[76] L. Menegaut, D. Masson, N. Abello, D. Denimal, C. Truntzer, P. Ducoroy, L. Lagrost,
J.P. Pais de Barros, A. Athias, J.M. Petit, L. Martin, E. Steinmetz, B. Kretz, Specific
enrichment of 2-arachidonoyl-lysophosphatidylcholine in carotid atheroma plaque from type
2 diabetic patients, Atherosclerosis 251 (2016) 339-347.
[77] Z.B. Zhou, B. Yang, X. Li, H. Liu, G. Lei, Lysophosphatidic Acid Promotes Expression
and Activation of Matrix Metalloproteinase 9 (MMP9) in THP-1 Cells via Toll-Like
Receptor 4/Nuclear Factor-kappaB (TLR4/NF-kappaB) Signaling Pathway, Med Sci Monit
24 (2018) 4861-4868.
[78] T. Dohi, K. Miyauchi, R. Ohkawa, K. Nakamura, M. Kurano, T. Kishimoto, N.
Yanagisawa, M. Ogita, T. Miyazaki, A. Nishino, K. Yaginuma, H. Tamura, T. Kojima, K.
Yokoyama, T. Kurata, K. Shimada, H. Daida, Y. Yatomi, Increased lysophosphatidic acid
levels in culprit coronary arteries of patients with acute coronary syndrome, Atherosclerosis
229(1) (2013) 192-7.
[79] M.J. Nsaibia, A. Mahmut, M.C. Boulanger, B.J. Arsenault, R. Bouchareb, S. Simard,
J.L. Witztum, M.A. Clavel, P. Pibarot, Y. Bosse, S. Tsimikas, P. Mathieu, Autotaxin interacts
with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve
stenosis in patients with coronary artery disease, J Intern Med 280(5) (2016) 509-517.
[80] M. Kurano, K. Kano, T. Dohi, H. Matsumoto, K. Igarashi, M. Nishikawa, R. Ohkawa,
H. Ikeda, K. Miyauchi, H. Daida, J. Aoki, Y. Yatomi, Different origins of lysophospholipid
mediators between coronary and peripheral arteries in acute coronary syndrome, J. Lipid Res.
58(2) (2017) 433-442.

194
[81] M. Kurano, A. Suzuki, A. Inoue, Y. Tokuhara, K. Kano, H. Matsumoto, K. Igarashi, R.
Ohkawa, K. Nakamura, T. Dohi, K. Miyauchi, H. Daida, K. Tsukamoto, H. Ikeda, J. Aoki,
Y. Yatomi, Possible involvement of minor lysophospholipids in the increase in plasma
lysophosphatidic acid in acute coronary syndrome, Arterioscler Thromb Vasc Biol 35(2)
(2015) 463-70.
[82] N. Cote, A. Mahmut, D. Fournier, M.C. Boulanger, C. Couture, J.P. Despres, S. Trahan,
Y. Bosse, S. Page, P. Pibarot, P. Mathieu, Angiotensin receptor blockers are associated with
reduced fibrosis and interleukin-6 expression in calcific aortic valve disease, Pathobiology
81(1) (2014) 15-24.
[83] G. Thanassoulis, C.Y. Campbell, D.S. Owens, J.G. Smith, A.V. Smith, G.M. Peloso,
K.F. Kerr, S. Pechlivanis, M.J. Budoff, T.B. Harris, R. Malhotra, K.D. O'Brien, P.R.
Kamstrup, B.G. Nordestgaard, A. Tybjaerg-Hansen, M.A. Allison, T. Aspelund, M.H.
Criqui, S.R. Heckbert, S.J. Hwang, Y. Liu, M. Sjogren, J. van der Pals, H. Kalsch, T.W.
Muhleisen, M.M. Nothen, L.A. Cupples, M. Caslake, E. Di Angelantonio, J. Danesh, J.I.
Rotter, S. Sigurdsson, Q. Wong, R. Erbel, S. Kathiresan, O. Melander, V. Gudnason, C.J.
O'Donnell, W.S. Post, Genetic associations with valvular calcification and aortic stenosis, N
Engl J Med 368(6) (2013) 503-12.
[84] P.R. Kamstrup, A. Tybjaerg-Hansen, B.G. Nordestgaard, Elevated lipoprotein(a) and
risk of aortic valve stenosis in the general population, J. Am. Coll. Cardiol. 63(5) (2014) 470-
7.
[85] B.J. Arsenault, S.M. Boekholdt, M.P. Dube, E. Rheaume, N.J. Wareham, K.T. Khaw,
M.S. Sandhu, J.C. Tardif, Lipoprotein(a) levels, genotype, and incident aortic valve stenosis:
a prospective Mendelian randomization study and replication in a case-control cohort, Circ
Cardiovasc Genet 7(3) (2014) 304-10.
[86] R. Bouchareb, A. Mahmut, M.J. Nsaibia, M.C. Boulanger, A. Dahou, J.L. Lepine, M.H.
Laflamme, F. Hadji, C. Couture, S. Trahan, S. Page, Y. Bosse, P. Pibarot, C.A. Scipione, R.
Romagnuolo, M.L. Koschinsky, B.J. Arsenault, A. Marette, P. Mathieu, Autotaxin Derived
From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization
of the Aortic Valve, Circulation 132(8) (2015) 677-90.
[87] M.J. Nsaibia, M.C. Boulanger, R. Bouchareb, G. Mkannez, K. Le Quang, F. Hadji, D.
Argaud, A. Dahou, Y. Bosse, M.L. Koschinsky, P. Pibarot, B.J. Arsenault, A. Marette, P.
Mathieu, OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve
stenosis through a LPAR1-RhoA-NF-kappaB pathway, Cardiovasc Res 113(11) (2017)
1351-1363.
[88] A. Mahmut, M.C. Boulanger, D. El Husseini, D. Fournier, R. Bouchareb, J.P. Despres,
P. Pibarot, Y. Bosse, P. Mathieu, Elevated expression of lipoprotein-associated
phospholipase A2 in calcific aortic valve disease: implications for valve mineralization, J.
Am. Coll. Cardiol. 63(5) (2014) 460-9.
[89] M. Torzewski, A. Ravandi, C. Yeang, A. Edel, R. Bhindi, S. Kath, L. Twardowski, J.
Schmid, X. Yang, U.F.W. Franke, J.L. Witztum, S. Tsimikas, Lipoprotein(a) Associated
Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve
Stenosis, JACC Basic Transl Sci 2(3) (2017) 229-240.
[90] R. Bouchareb, M.C. Boulanger, L. Tastet, G. Mkannez, M.J. Nsaibia, F. Hadji, A.
Dahou, Y. Messadeq, B.J. Arsenault, P. Pibarot, Y. Bosse, A. Marette, P. Mathieu, Activated
platelets promote an osteogenic programme and the progression of calcific aortic valve
stenosis, Eur Heart J (2018). doi: 10.1093/eurheartj/ehy696

195
[91] G. Mkannez, V. Gagne-Ouellet, M. Jalloul Nsaibia, M.C. Boulanger, M. Rosa, D.
Argaud, F. Hadji, N. Gaudreault, G. Rheaume, L. Bouchard, Y. Bosse, P. Mathieu, DNA
methylation of a PLPP3 MIR transposon-based enhancer promotes an osteogenic programme
in calcific aortic valve disease, Cardiovasc Res 114(11) (2018) 1525-1535.
[92] M. Baader, T. Bretschneider, A. Broermann, J.F. Rippmann, B. Stierstorfer, C.A.
Kuttruff, M. Mark, Characterization of the properties of a selective, orally bioavailable
autotaxin inhibitor in preclinical models of advanced stages of liver fibrosis, Br. J.
Pharmacol. 175(4) (2018) 693-707.
[93] J. Weng, S. Jiang, L. Ding, Y. Xu, X. Zhu, P. Jin, Autotaxin/lysophosphatidic acid
signaling mediates obesity-related cardiomyopathy in mice and human subjects, J. Cell. Mol.
Med. 23(2) (2019) 1050-1058.
[94] T.M. Maher, E.M. van der Aar, O. Van de Steen, L. Allamassey, J. Desrivot, S. Dupont,
L. Fagard, P. Ford, A. Fieuw, W. Wuyts, Safety, tolerability, pharmacokinetics, and
pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary
fibrosis (FLORA): a phase 2a randomised placebo-controlled trial, Lancet Respir Med 6(8)
(2018) 627-635.
[95] Y. Goldshmit, R. Matteo, T. Sztal, F. Ellett, F. Frisca, K. Moreno, D. Crombie, G.J.
Lieschke, P.D. Currie, R.A. Sabbadini, A. Pebay, Blockage of lysophosphatidic acid
signaling improves spinal cord injury outcomes, Am J Pathol 181(3) (2012) 978-92.
[96] P.J. Crack, M. Zhang, M.C. Morganti-Kossmann, A.J. Morris, J.M. Wojciak, J.K.
Fleming, I. Karve, D. Wright, M. Sashindranath, Y. Goldshmit, A. Conquest, M. Daglas,
L.A. Johnston, R.L. Medcalf, R.A. Sabbadini, A. Pebay, Anti-lysophosphatidic acid
antibodies improve traumatic brain injury outcomes, J Neuroinflammation 11 (2014) 37.
[97] M. Terakado, H. Suzuki, K. Hashimura, M. Tanaka, H. Ueda, H. Kohno, T. Fujimoto,
H. Saga, S. Nakade, H. Habashita, Y. Takaoka, T. Seko, Discovery of ONO-7300243 from
a Novel Class of Lysophosphatidic Acid Receptor 1 Antagonists: From Hit to Lead, ACS
Med Chem Lett 7(10) (2016) 913-918.
[98] M. Terakado, H. Suzuki, K. Hashimura, M. Tanaka, H. Ueda, K. Hirai, M. Asada, M.
Ikura, N. Matsunaga, H. Saga, K. Shinozaki, N. Karakawa, Y. Takada, M. Minami, H.
Egashira, Y. Sugiura, M. Yamada, S. Nakade, Y. Takaoka, Discovery of a Slow Tight
Binding LPA1 Antagonist (ONO-0300302) for the Treatment of Benign Prostatic
Hyperplasia, ACS Med Chem Lett 8(12) (2017) 1281-1286.
[99] S.M. Palmer, L. Snyder, J.L. Todd, B. Soule, R. Christian, K. Anstrom, Y. Luo, R.
Gagnon, G. Rosen, Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-
986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic
Pulmonary Fibrosis, Chest 154(5) (2018) 1061-1069.
[100] Y. Allanore, O. Distler, A. Jagerschmidt, S. Illiano, L. Ledein, E. Boitier, I. Agueusop,
C.P. Denton, D. Khanna, Lysophosphatidic Acid Receptor 1 Antagonist SAR100842 for
Patients With Diffuse Cutaneous Systemic Sclerosis: A Double-Blind, Randomized, Eight-
Week Placebo-Controlled Study Followed by a Sixteen-Week Open-Label Extension Study,
Arthritis Rheumatol 70(10) (2018) 1634-1643.

196
Annexe II :Phosphatidylserine-specific phospholipase
A1: A friend or the devil in disguise
Yang Zhao 1 , Stephan Hasse 1 , Sylvain G Bourgoin 2
Affiliations :
1 Centre de recherche du CHU de Québec-Université Laval, Centre ARThrite de l'Université
Laval, Département de microbiologie-infectiologie et d'immunologie, Université Laval,
Québec, G1V 4G2, Canada.
2 Centre de recherche du CHU de Québec-Université Laval, Centre ARThrite de l'Université
Laval, Département de microbiologie-infectiologie et d'immunologie, Université Laval,
Québec, G1V 4G2, Canada. Electronic address: [email protected].
Keywords: Phosphatidylserine, Lysophosphatidylserine, PLA1A, Lysophosphatidylserine
receptors, Autoimmunity, Cancer

197
1 Abstract
Various human tissues and cells express phospholipase A1 member A (PLA1A), including
the liver, lung, prostate gland, and immune cells. The enzyme belongs to the pancreatic lipase
family. PLA1A specifically hydrolyzes sn-1 fatty acid of phosphatidylserine (PS) or 1-acyl-
lysophosphatidylserine (1-acyl-lysoPS). PS externalized by activated cells or apoptotic cells
or extracellular vesicles is a potential source of substrate for the production of unsaturated
lysoPS species by PLA1A. Maturation and functions of many immune cells, such as T cells,
dendritic cells, macrophages, and mast cells, can be regulated by PLA1A and lysoPS. Several
lysoPS receptors, including GPR34, GPR174 and P2Y10, have been identified. High serum
levels and high PLA1A expression are associated with autoimmune disorders such as Graves'
disease and systemic lupus erythematosus. Increased expression of PLA1A is associated with
metastatic melanomas. PLA1A may contribute to cardiometabolic disorders through
mediating cholesterol transportation and producing lysoPS. Furthermore, PLA1A is
necessary for hepatitis C virus assembly and can play a role in the antivirus innate immune
response. This review summarizes recent findings on PLA1A expression, lysoPS and lysoPS
receptors in autoimmune disorders, cancers, cardiometabolic disorders, antivirus immune
responses, as well as regulations of immune cells.

198
2 General introduction
Sato et al. named the enzyme phosphatidylserine-specific phospholipase A1 (PS-PLA1) in
1997 [1]. PS-PLA1 specifically hydrolyzes sn-1 fatty acid of phosphatidylserine (PS) or
lysophosphatidylserine (lysoPS) [1]. PS-PLA1 has the name phospholipase A1 member A
(PLA1A) in databases. We will use the official HGCN gene nomenclature PLA1A
throughout the review.
Activated rat platelets release two types of lipases having phospholipase A2 (PLA2) and
lysoPS specific lysophospholipase A activities [2]. These lipases come from dense granules
or α-granules rather than the lysosomal compartments [2]. The lysophospholipase activity
hydrolyzes both 1-acyl- and 2-acyl-lysoPS, but no other lysophospholipids [2]. At the same
time, Higashi et al. also partially purified a lysophospholipase from thrombin-activated rat
platelets [3]. The lysophospholipase activity was towards 1-acyl-sn-glycerol-3-phospho-l-
serine but none other lysophospholipids [3]. Yokoyama et al. later reported that platelets
released one protein with phospholipase A1 and lysophospholipase activities [4]. This
enzyme worked with PLA2 to hydrolyze platelet phospholipids during blood clotting [4].
The cDNA sequences encoding rat and human PLA1A were published in 1997 [1,5]. Purified
PLA1A has an apparent molecular weight of 55-kDa on SDS-polyacrylamide gel
electrophoresis [1]. They assessed the enzyme activity towards PS and lysoPS and
highlighted the structural similarity between PLA1A and other mammalian lipases [1]. Nagai
et al. mapped the human PLA1A gene to chromosome 3q13.13–13.2 [6] and PLA1A was
identified as a gene previously named nmd [5]. The murine ortholog of Pla1a localizes to
mouse chromosome 16 [7].
Following PLA1A purification, several studies have assessed its biological functions. For
instance, Aoki et al. reported that the addition of recombinant PLA1A to activated rat
platelets accelerated lysophosphatidic acid (LPA) production, thereby suggesting that
PLA1A could contribute to serum LPA level during blood clotting [8]. PLA1A proteins from
rat, human, and mouse are highly conserved [7]. PLA1A molecular structure, expression, and
hydrolytic activities have been reviewed previously [[9], [10], [11], [12]] and will not be
discussed in depth in this review. There is a growing number of publications on the putative

199
roles of PLA1A in disease states or its cellular functions. The involvements of
lysophospholipids and the potential roles of PLA1A in several diseases, such as acute
coronary syndrome (ACS), atherosclerosis, and gastric cancer, have been reviewed recently
[13]. This review will focus on more recent studies concerning the characterization of
PLA1A, expression in cells, and putative functions in various diseases, including cancer and
autoimmunity.
2.1. PLA1A belongs to the pancreatic lipase family
The pancreatic lipase family consists of six members: PLA1A, membrane-associated
phosphatidic acid-selective phospholipase A1a (mPA-PLA1a), mPA-PLA1b, hepatic lipase,
endothelial lipase, and pancreatic lipase-related protein 2. They are essentially extracellular
PLA1s [14,15]. PLA1A, mPA-PLA1a, and mPA-PLA1b consist of a subfamily with distinct
molecular features (a short lid domain and a deleted β9 domain) that distinguish them from
other lipases [15]. Extracellular PLA1s have no sequence homologies to intracellular PLA1s
and exhibit distinct functions [10]. We will not discuss the structures and roles of intracellular
PLA1s in this review.
The domain structure of pancreatic lipase family members determines their substrate
specificity and the ability to hydrolyze triglyceride and phospholipids. Crucial roles of lid,
β5, and β9 loops of pancreatic lipases in choosing substrate have been reviewed [9,12,14].
Fig. 1 shows the substrate specificity of the pancreatic lipases. Once the lipase contacts its
substrate, the lid undergoes a conformational change and adopts an open conformation with
β9 [9]. This conformational change allows the hydrophobic interaction with substrate acyl
chains and the ligand docking in the catalytic site [14]. Lipases are inactive in aqueous
solutions because the catalytic triad is obstructed by the lid [16].

200
Fig. 1. Substrate preference of pancreatic lipase family members. PLA1A, mPA-PLA1a and
b consist of a subfamily exhibiting phospholipase activity towards PLs. PL and LPL can
hydrolyze TG. EL, PLRP2 and HL can hydrolyze both TG and PLs. PLs, phospholipids; TG,
triglyceride; PLA1A, phospholipase A1 member A; mPA-PLA1, membrane-associated
phosphatidic acid-selective phospholipase A1; EL, endothelial lipase; PLRP2, pancreatic
lipase-related protein 2; HL, hepatic lipase; PL, pancreatic lipase; LPL, lipoprotein lipase.
All pancreatic lipase family members have quite a distinct affinity to heparan sulfate
proteoglycans (HSPGs). PLA1A affinity to heparin was also reported [3,15]. Membrane-
bound PLA1A can hydrolyze externalized PS and be internalized into living mammalian cells
through binding to HSPGs. There is no specific inhibitor of PLA1A. Like phospholipases
with a catalytic serine, PLA1A is sensitive to inhibition by diisopropyl fluorophosphate [1].
There are five transcript variants in homo sapiens encoding for different isoforms of PLA1A,
NM_015900.4 (variant 1), NM_001206960.2 (variant 2), NM_001206961.2 (variant 3),
NM_001293225.2 (variant 4), and NR_120610.2 (variant 5, non-coding) (https://www-ncbi-
nlm-nih-gov.acces.bibl.ulaval.ca/gene/51365). Variant 1 represents the longest transcript
encoding the full-length protein. Isoform 2 has the same N- and C- terminal but is shorter
than the full-length PLA1A, whereas isoform 3 and 4 both have a shorter N-terminus.
Among the two PLA1A mRNAs characterized in vivo, the larger one encodes the full-length
PLA1A, and the second mRNA encodes a truncated form named PLA1AΔC [12]. PLA1AΔC
lacks two-thirds of the C-terminal domain, including the β5 loop and basic residues [12],
which eliminates enzyme ability to hydrolyze PS in liposomes but retains its
lysophospholipase activity towards 1-acyl-lysoPS in solution [6,12]. As for other lipases, the
β5 loop of PLA1A is likely required for interfacial binding to membrane leaflet [12,14]. The
N-terminal domain, which is conserved in both PLA1A and PLA1AΔC, encompasses the
catalytic triad and the heparin-binding site, thereby suggesting that both isoforms have
similar affinity to HSPGs [6]. PLA1A and PLA1AΔC are expressed in various human organs,

201
tissues, and cells, with PLA1AΔC representing about 10–20% of total PLA1A [6].
PLA1AΔC and PLA1A can synergistically induce lipid signaling through hydrolyzing PS
exposed on damaged or activated cell surface and control the level of lysoPS [12].
2.2. Expression patterns of PLA1A
In rats, PLA1A is expressed in platelets, hearts, and lungs [10]. Rat platelets can release
PLA1A upon activation. Unlike rat, mouse and human platelets poorly express PLA1A [10].
Several human tissues express PLA1A
(https://gtexportal.org/home/gene/PLA1A#spliceVizBlock), including muscle, kidney, small
intestine, spleen, placenta, and testis, with the highest expression in the liver and prostate
gland [5,6]. Human fibroblasts, keratinocytes, melanomas, HepG2 and HeLa cells express
the PLA1A mRNA [6]. The sources of PLA1A in human serum have remained elusive [17].
One liver cancer study revealed that the serum PLA1A level was closely related to the PLA1A
mRNA level in non-hepatocellular carcinoma (HCC) tissues, indicating that the liver might
be a source of circulating PLA1A [18].
In 2010, Nakamura et al. generated anti-human PLA1A monoclonal antibodies [17]. Using
a novel enzyme immunoassay, they reported that the serum level of PLA1A in healthy
subjects was 33.8 ± 16.6 μg/L [17]. The concentration was significantly higher in men (13.8–
80.6 μg/L) than in women (12.1–68.8 μg/L), with no correlation between the age of the
subjects [17]. Furthermore, there was no association between serum PLA1A level and other
laboratory tests, such as total IgE concentration, platelet and leukocyte counts, tumor
markers, etc. [17]. Expression levels of PLA1A in pathophysiological conditions will be
reviewed in following paragraphs (summarized in Table 1).
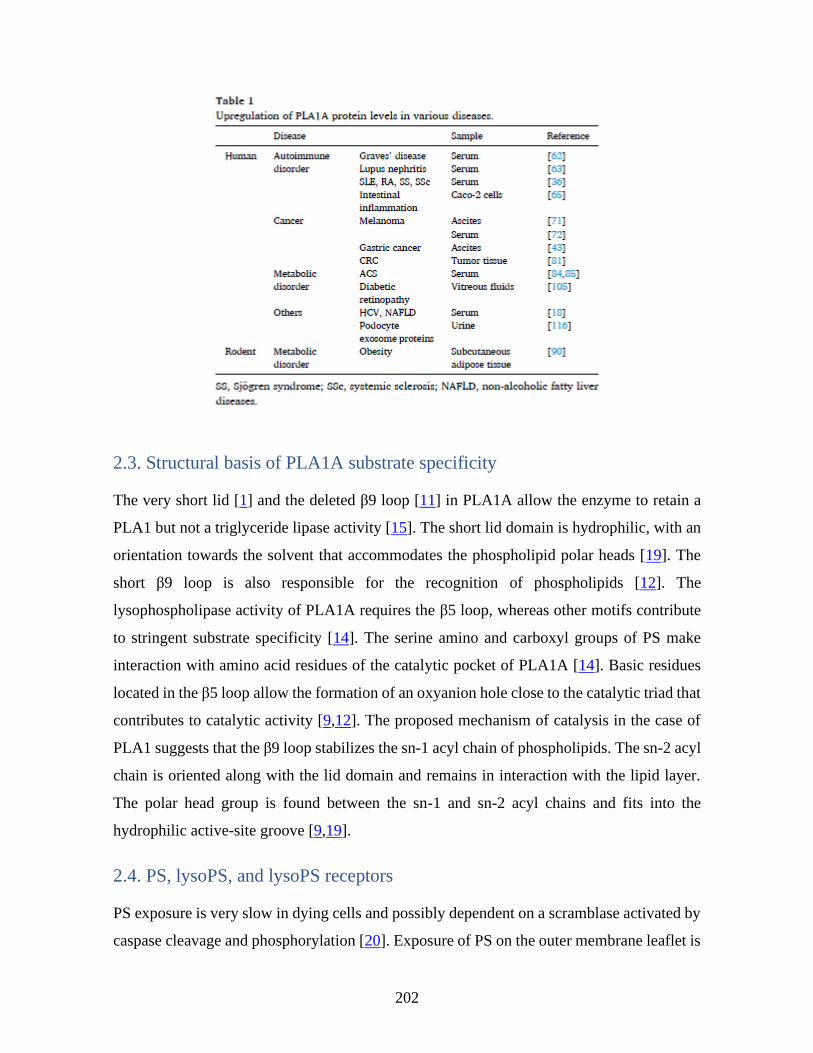
202
2.3. Structural basis of PLA1A substrate specificity
The very short lid [1] and the deleted β9 loop [11] in PLA1A allow the enzyme to retain a
PLA1 but not a triglyceride lipase activity [15]. The short lid domain is hydrophilic, with an
orientation towards the solvent that accommodates the phospholipid polar heads [19]. The
short β9 loop is also responsible for the recognition of phospholipids [12]. The
lysophospholipase activity of PLA1A requires the β5 loop, whereas other motifs contribute
to stringent substrate specificity [14]. The serine amino and carboxyl groups of PS make
interaction with amino acid residues of the catalytic pocket of PLA1A [14]. Basic residues
located in the β5 loop allow the formation of an oxyanion hole close to the catalytic triad that
contributes to catalytic activity [9,12]. The proposed mechanism of catalysis in the case of
PLA1 suggests that the β9 loop stabilizes the sn-1 acyl chain of phospholipids. The sn-2 acyl
chain is oriented along with the lid domain and remains in interaction with the lipid layer.
The polar head group is found between the sn-1 and sn-2 acyl chains and fits into the
hydrophilic active-site groove [9,19].
2.4. PS, lysoPS, and lysoPS receptors
PS exposure is very slow in dying cells and possibly dependent on a scramblase activated by
caspase cleavage and phosphorylation [20]. Exposure of PS on the outer membrane leaflet is

203
the gold marker of cells undergoing apoptosis and is an eat-me signal for the clearance of
dying cells by macrophages (Mφ) [21], a process called efferocytosis. The engulfment of
apoptotic cells does not induce inflammation but promotes the secretion of anti-inflammatory
cytokines (IL-10 and TGFβ) and decreases the production of TNFα, IL-1β and IL-12 [[22],
[23], [24]]. PS functions as an immunosuppressive mediator for silent clearance of apoptotic
debris by Mφ.
The exposure of PS on the cell surface can occur in the absence of apoptosis. In that case,
cell surface exposure of PS is rapid and reversible, thereby preventing the engulfment of
viable PS exposing cells by Mφ and dendritic cells (DCs) [25,26]. The externalization of PS
has been reported in viable monocytes [27], activated mast cells [27,28], CD8+ T cells [29],
regulatory B cells [30], cancer cells and cancer cell-derived extracellular vesicles (EVs)
[31,32]. PS externalization on activated platelets is required for microparticles release and
plays a critical role in the recruitment and the activation of clotting factors with gamma-
carboxyglutamic domains [33,34]. Platelet-derived microparticles harboring IgG are
positively correlated with systemic lupus erythematosus (SLE) disease activity and vascular
damage [35]. EVs can be derived by various cell types and serve as cargos for the delivery
of proteins, lipids, and RNA to the target cells. In platelet-free plasma of SLE patients, the
amounts of EVs derived by platelets and red blood cells were significantly increased,
respectively (unpublished data). Among these EVs, the PS+ EVs were increased in SLE
patients compared to healthy controls (unpublished data). Besides, PLA1A level is increased
in active SLE patients [36]. Thus, PLA1A might impact immune regulatory processes and
inflammation through the production of 2-acyl-lysoPS converted from PS exposed at the cell
surface of activated cells, apoptotic cells, or various types of EVs (Fig. 2 and [[37], [38],
[39]]).
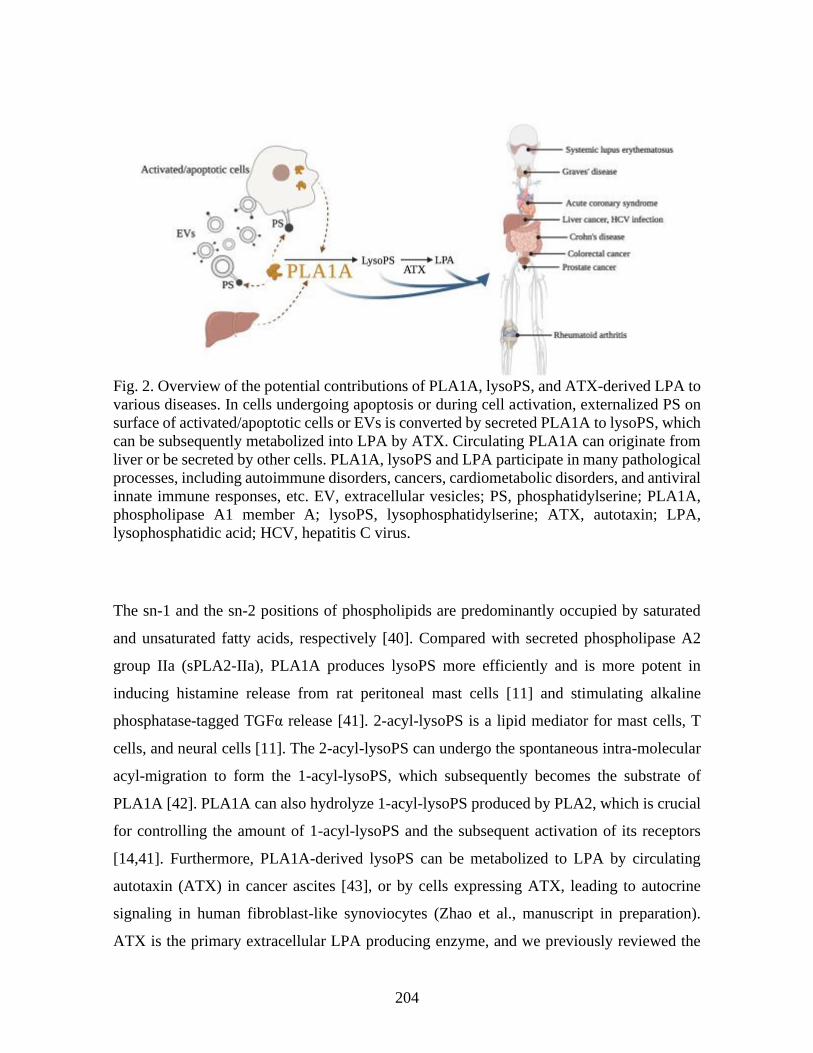
204
Fig. 2. Overview of the potential contributions of PLA1A, lysoPS, and ATX-derived LPA to
various diseases. In cells undergoing apoptosis or during cell activation, externalized PS on
surface of activated/apoptotic cells or EVs is converted by secreted PLA1A to lysoPS, which
can be subsequently metabolized into LPA by ATX. Circulating PLA1A can originate from
liver or be secreted by other cells. PLA1A, lysoPS and LPA participate in many pathological
processes, including autoimmune disorders, cancers, cardiometabolic disorders, and antiviral
innate immune responses, etc. EV, extracellular vesicles; PS, phosphatidylserine; PLA1A,
phospholipase A1 member A; lysoPS, lysophosphatidylserine; ATX, autotaxin; LPA,
lysophosphatidic acid; HCV, hepatitis C virus.
The sn-1 and the sn-2 positions of phospholipids are predominantly occupied by saturated
and unsaturated fatty acids, respectively [40]. Compared with secreted phospholipase A2
group IIa (sPLA2-IIa), PLA1A produces lysoPS more efficiently and is more potent in
inducing histamine release from rat peritoneal mast cells [11] and stimulating alkaline
phosphatase-tagged TGFα release [41]. 2-acyl-lysoPS is a lipid mediator for mast cells, T
cells, and neural cells [11]. The 2-acyl-lysoPS can undergo the spontaneous intra-molecular
acyl-migration to form the 1-acyl-lysoPS, which subsequently becomes the substrate of
PLA1A [42]. PLA1A can also hydrolyze 1-acyl-lysoPS produced by PLA2, which is crucial
for controlling the amount of 1-acyl-lysoPS and the subsequent activation of its receptors
[14,41]. Furthermore, PLA1A-derived lysoPS can be metabolized to LPA by circulating
autotaxin (ATX) in cancer ascites [43], or by cells expressing ATX, leading to autocrine
signaling in human fibroblast-like synoviocytes (Zhao et al., manuscript in preparation).
ATX is the primary extracellular LPA producing enzyme, and we previously reviewed the

205
roles and involvements of ATX-LPA receptor axis [44]. LPA receptors have ligand
selectivity and can be activated differentially by the LPA species [45]. LysoPS receptors also
show preference for lysoPS species. Several G protein-coupled receptors for lysoPS were
identified, including GPR34 (LPS1), P2Y10 (LPS2), A630033H20 (LPS2L), and GPR174
(LPS3) [46,47]. A630033H20 is expressed in mouse but is a pseudo-gene in human [46].
GPR34 prefers lysoPS with an unsaturated fatty acid at the sn-2 position like those produced
by PLA1A [41]. GPR174 is highly activated by 16:1 lysoPS and P2Y10 can be activated
similarly by 16:0 and 18:1 lysoPS [48]. We will discuss the functions of lysoPS and its
receptors in the following paragraphs.
3 Expression of PLA1A and lysoPS receptors in cells
3.1. Immune cells
Treg cells and memory CD4+ T cells, at a much lower level, express PLA1A
(https://www.proteinatlas.org/ENSG00000144837-PLA1A). A few reports suggested that
PLA1A and lysoPS receptors could be involved in immune cell rep rogramming, maturation,
and modulation of immune cell functional responses [38,39,49].
Unsaturated PS could inhibit mitogen-induced T cell activation, and serum PLA1A might
enhance the inhibitory effects through the generation of unsaturated lysoPS [38,39]. Of note,
among the tumor-infiltrating lymphocytes, there was a high enrichment of PLA1A mRNA in
tumor-promoting CD8+ T cells (31.1-fold) compared to native peripheral blood CD8+ T cells
[49]. Whether high levels of PLA1A expression are associated with reduced CD8+ T cell-
dependent cytotoxicity and poor prognostic survival are yet to establish. LysoPS species
(C18:0 > C18:1 > C16:0) and PLA1A transcripts were abundant in lymphoid tissues (spleen,
thymus) [39]. LysoPS suppressed T cell proliferation, Treg generation and homeostasis
through activation of GPR174, and the subsequent elevation of intracellular cAMP levels
[39,50]. GPR174 weakened the capacity of Treg to suppress the functions of Th1 subsets and
controlled tissue-specific immune responses [51]. TGFβ signals suppressed the expression
of GPR174 by Treg [51]. The blockade of GPR174 might be a potential treatment option for
autoimmune disorders [39].

206
Human and mouse DCs were reported to express PLA1A [39,52]. Treg cells can interfere
with the induction of mature DCs [52]. In immature Treg-DCs, which drive CD4+ T cells
polarization towards a regulatory phenotype, PLA1A was among the most downregulated
genes compared to mature DCs [52].
The TLR4 agonist lipopolysaccharide (LPS) enhanced PLA1A mRNA expression in human
THP-1 derived Mφ [53]. Immunosuppressive agents, such as corticosteroids, prednisolone,
6α-methylprednisolone, dexamethasone, and beclomethasone reduced LPS-mediated PLA1A
expression in THP-1 derived Mφ [53]. In LPS-induced peritoneal Mφ from microsomal
prostaglandin E synthase-1 knockout mice, the expression of Pla1a was modulated [54].
Kawamoto et al. found that nerve growth factor (NGF) could stimulate the release of
histamine from rat peritoneal mast cells incubated with activated rat platelets [55]. NGF
antibodies or inhibition of NGF receptor tyrosine kinase activity can completely block
histamine release by rat peritoneal mast cells [55]. Furthermore, histamine release by rat
peritoneal mast cells incubated with PS+ erythrocytes and NGF required the presence of
PLA1A, thereby suggesting a role for PLA1A-derived lysoPS in mast cell activation [55].
GPR34 is a highly expressed functional lysoPS receptor in rat mast cells [56]. PLA1A-
mediated production of 2-acyl-lysoPS may also contribute to histamine release by rat
peritoneal mast cells in response to cross-linking of the high-affinity IgE receptor, known as
FcεRI [57]. Histamine release was blocked by heparin, suggesting that PLA1A activity
required binding to cellular HSPGs [57].
3.2. Other cells
LysoPS was showed to promote the NGF-induced neural differentiation of PC12 cells [58].
NGF and lysoPS stimulated the growth of PC12 cells and enhanced neurite length [58]. The
cells developed only 1–2 neurites instead of the multipolar appearance induced by NGF alone
[58]. LysoPS was showed to stimulate intracellular calcium increase and chemotactic
migration in U87 human glioma cells [59] and L2071 mouse fibroblasts [60]. These effects
were dependent on the activation of PI3K, p38 MAPK, and JNK pathways [59,60]. A
pertussis toxin-insensitive but phospholipase C-dependent cascade was involved in the
intracellular calcium increase [60]. Pertussis toxin-sensitive chemotactic migration was

207
mediated through the PI3K and ERK pathways [60]. The lysoPS receptors expressed by
L2071 fibroblasts were not characterized.
4 Expression of PLA1A in disease states
Whether PLA1A has beneficial or harmful functions in healthy and disease states remains to
be determined. Under physiological conditions, the PS in the inner cell membrane layer is
not accessible to secreted PLA1A [11,12]. However, in cells undergoing apoptosis or during
cell activation, externalized PS can be converted by secreted PLA1A to lysoPS, which
potentially induces lysoPS receptor-dependent functional responses in various cell types
[11,12]. The PLA1A chromosome locus is not associated with human diseases [11]. However,
high PLA1A expression levels in melanoma cells or subtypes of prostate cancers,
autoimmune disorders such as SLE and Graves' disease suggest a role for PLA1A in the
regulation of tumor growth and autoimmunity [5,36,61,62]. The following paragraphs will
review the expression of PLA1A and the lysoPS receptors in autoimmune disorders, cancers,
cardiometabolic disorders, virus immunomodulation, and other diseases (summarized in
Table 1, Table 2, Table 3 and Fig. 2).
4.1. Autoimmune disorders
Serum PLA1A level was significantly higher in SLE patients compared to healthy controls
and patients with other systemic autoimmune rheumatic diseases, such as active rheumatoid
arthritis (RA), Sjögren syndrome, and systemic sclerosis [63]. Serum PLA1A level
significantly correlated with SLE disease activity index [36]. Patients with lupus nephritis
and diabetic nephropathy had similarly elevated plasma levels of PLA1A [63]. In the cohort
of patients with lupus nephritis, serum level of PLA1A was not correlated with activity index
or chronicity index, proteinuria, kidney survival, SLE disease activity index, and other
clinical laboratory data such as anti-double-stranded DNA antibody and complement proteins
(C3 and C4) [63]. Disease treatment reduced serum PLA1A levels [36,63]. However, serum
PLA1A level inversely correlated with the daily dose of prednisolone [63]. We have
compared plasma PLA1A levels between early diagnosed RA patients and a cohort of SLE
patients with age- and gender-matched controls. PLA1A levels were higher in early

208
diagnosed RA patients and SLE patients, with no sex-based differences. PLA1A levels in
synovial fluids from patients with RA and psoriatic arthritis were higher than those from
osteoarthritis and gout patients (Zhao et al., manuscript in preparation). Therefore, we do not
exclude that the levels of serum/plasma PLA1A in systemic autoimmune rheumatic diseases
would correlate with high systemic inflammation levels or tissue damages [36].
Graves' disease, also known as Basedow's disease, is an autoimmune disorder characterized
by overactive thyroid and the presence of antibody against the thyroid stimulating hormone
receptor [64]. Serum PLA1A level was higher in patients with Graves' disease and strongly
correlated with thyroid hormone levels [62]. Treatment with anti-thyroid reagents can lower
serum PLA1A levels, suggesting a possible link between PLA1A and the inflammation in
chronic autoimmune thyroiditis [62]. Changes in serum PLA1A levels also occurred in
patients with subacute thyroiditis or silent thyroiditis [62].
Intestinal inflammation modulates the expression of PLA1A and lysoPS receptors [65].
PLA1A expression was increased in inflamed ulcerated mucosa from Crohn's disease, even
in their noninflamed macroscopically normal-looking mucosa [66]. The PLA1A mRNA,
protein, and lipase activity were all enhanced in primary intestinal microvascular endothelial
cells and human colonic Caco-2 epithelial cells treated with inflammatory stimulations, such
as TNF-α, IL-1β, and LPS [65]. Besides, expression of all lysoPS receptors (GPR34,
GPR174, P2RY10) increased in Caco-2 cells after inflammatory stimulation [65].
The serum of systemic sclerosis patients showed an elevated level of PLA1A [36]. Serum
PLA1A is possibly associated with low-grade systemic inflammation. PLA1A may
contribute to progressive skin fibrosis in systemic sclerosis patients, as the expression of
PLA1A gene was increased 2.63-fold in response to the TLR3 agonist poly(I:C) [67].
Expression of PLA1A is elevated in various diseased tissues and is possibly induced in
response to inflammatory stimuli [57]. By hydrolyzing PS, PLA1A might contribute to
inflammation or interfere with processes involved in inflammation-resolution mechanisms
through coving the recognition of apoptotic cells/debris by other immune cells, such as Mφ
and DCs, leading to the development of autoimmunity [36,62]. The roles played by PLA1A
in the pathophysiology of many diseases remain to be established. Whether PLA1A is a
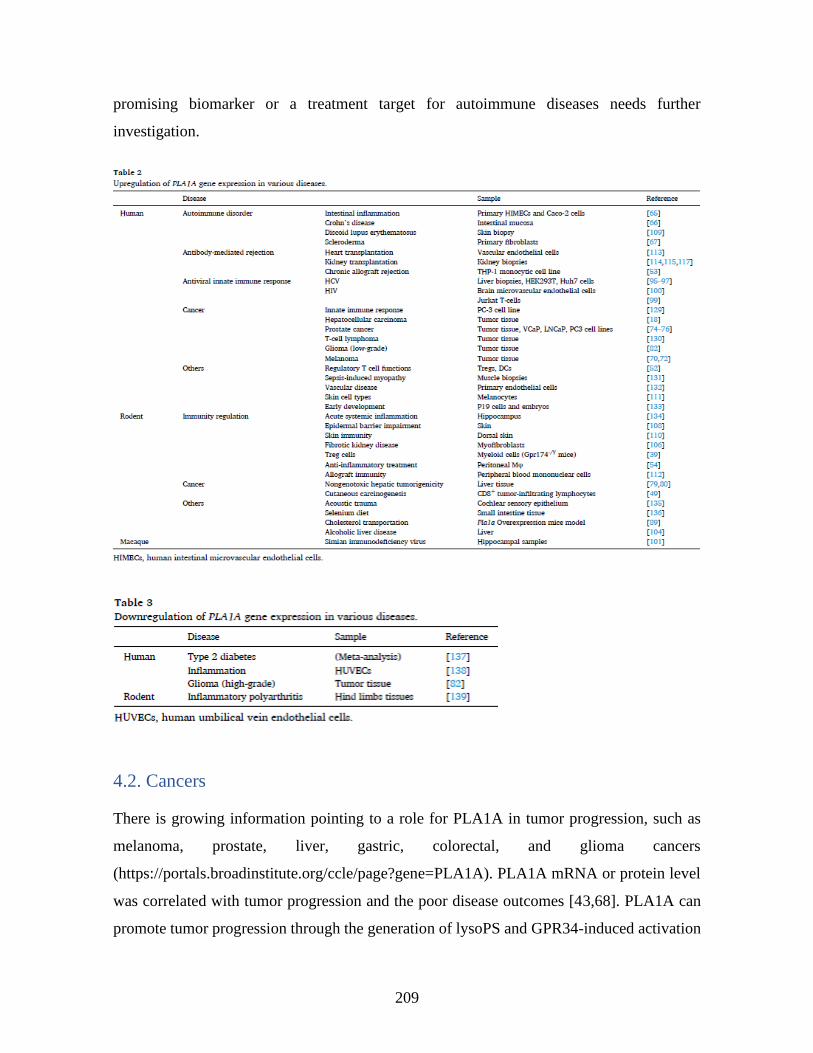
209
promising biomarker or a treatment target for autoimmune diseases needs further
investigation.
4.2. Cancers
There is growing information pointing to a role for PLA1A in tumor progression, such as
melanoma, prostate, liver, gastric, colorectal, and glioma cancers
(https://portals.broadinstitute.org/ccle/page?gene=PLA1A). PLA1A mRNA or protein level
was correlated with tumor progression and the poor disease outcomes [43,68]. PLA1A can
promote tumor progression through the generation of lysoPS and GPR34-induced activation

210
of the PI3K/Akt pathway [68] or through ATX-mediated conversion of lysoPS to LPA [43],
a lipid mediator associated with cancer progression and metastasis [69].
PLA1A was highly expressed in poorly-metastatic melanoma cell lines [5,70], suggesting a
link to cancer cell fate. Of note, the levels of serum PLA1A and that of ATX were
significantly higher in melanoma subjects and correlated with the clinical stages in females
[71]. In a recent study, increased PLA1A expression was positively correlated to disease
severity and routine diagnostic markers of metastatic melanoma [72].
Transmembrane serine protease 2/erythroblast transformation-specific transcription factor
(TMPRSS2/ERG) gene fusion events play a role in the initiation and progression of prostate
cancer [73]. The chromatin and gene expression are distinct between TMPRSS2/ERG positive
and non-TMPRSS2/ERG prostate cancers in the regions proximal to the PLA1A gene [74],
which is one of the target genes regulated by ERG [61,73]. Poly ADP-ribose polymerase 1
and the catalytic subunit of DNA protein kinase were required for ERG mediated
transcriptional activities, and the expression of PLA1A in VCaP cells decreased following
siRNA knockdown of ERG or incubation with small molecule inhibitors of poly ADP-ribose
polymerase 1 and catalytic subunit of DNA protein kinase [73,75,76]. However,
overexpression of various ΔERG constructs had no significant effect on PLA1A mRNA
expression [75]. In VCaP cells, the vitamin D receptor agonists induced expression of ERG
target genes but had mixed outcomes on PLA1A expression [77]. In PCa cells, PLA1A gene
expression was not induced in response to stimulation with androgen [78].
Elevated levels of PLA1A mRNA were detected in HCC tissues [18] and were associated
with higher serum PLA1A levels when compared to healthy controls, but no relationship
with clinical parameters was observed [18]. However, elevated serum PLA1A level
correlated to background PLA1A mRNA expression in normal tissues adjacent to the tumors,
but not in the HCC tissues [18]. The relationship between high serum PLA1A and hepatic
enzyme levels suggested that liver tissue injury could contribute to serum PLA1A in HCC
patients [18]. High GPR34 expression in HCC tissues correlated with poorly differentiated
HCC tumors [18]. Pla1a is part of a signature gene set modulated by nongenotoxic hepatic
tumorigens in rats, including peroxisome proliferator-activated receptor agonists and steroid
hormones [79]. Pla1a expression was increased in response to hepatotoxic agents such as

211
pirinixic acid, nafenopin, or phenobarbital, and was decreased by chloroform, pravastatin, or
methapyrilene [80].
In ascites of gastric cancer patients, the levels of lysoPS and PLA1A were positively
correlated [43]. Although ascites from gastric tumor patients and control cirrhosis patients
expressed similar amounts of PLA1A, higher levels of C18:0 and C18:1 lysoPS were
observed in the gastric tumor group [43].
PLA1A and lysoPS have different effects on colorectal cancer (CRC) cell growth and tumor
metastasis [68,81]. PLA1A expression was associated with tumor invasion, hematogenous
metastasis, and poor disease-free survival in CRC patients [81]. LysoPS failed to stimulate
CRC cell proliferation, but it stimulated tumor cell migration through GPR34 and PI3K/Akt
pathway [68]. These results possibly correlate with different expression patterns of lysoPS
receptors among cancer cell lines. For instance, CRC cell lines expressing only GPR34 were
less sensitive to high dose lysoPS-mediated inhibition of proliferation than cell lines
expressing all lysoPS receptors [68].
Differential expression of phospholipases and lysophospholipases correlated with alterated
glycerophosphocholine lipid metabolism in gliomas [82]. Expression of PLA1A was
increased in low-grade glioma but reduced in high-grade glioma compared to normal brain
tissues [82]. The distinct expression of PLA1A and other phospholipase genes (PLA2G4A
and LYPLA1) in low- and high-grade could help to grade astrocytomas [82].
Altogether the studies suggest a role for PLA1A in tumor invasion and metastasis through
GPR34-induced signaling [68,81]. The roles of PLA1A should take the tumor
microenvironment into account [43,69,71]. Surface exposure of PS, the release of PS-
positive EVs by tumor cells, and PLA1A can negatively impact tumor immunity and tumor-
killing through lysoPS-mediated suppression of immune cell functions and lysoPS
conversion into LPA by ATX [[37], [38], [39],69,83].
4.3. Cardiometabolic disorders
PLA1A might participate in ACS pathogenesis [13]. High PLA1A levels in serum samples
from culprit coronary arteries of patients with ACS significantly correlated with C18:0 and

212
18:1 lysoPS levels [84]. The monitoring of plasma serotonin levels in 141 consecutive
patients undergoing coronary angiography emphasized that serotonin, a biomarker of platelet
activation, was significantly associated with plasma lysoPS level in the ACS group [85].
Aspirin intake was without effect on plasma lysoPS levels, but plasma LPA and other
lysophospholipid levels were lower in patients who had taken aspirin regularly [85].
Correlation between serum PLA1A and plasma lysoPS levels was observed only in the ACS
group [85]. There was no significant difference in serum PLA1A level among the patients
with normal coronary arteries, stable angina pectoris, and ACS groups, thereby suggesting
that PLA1A might only contribute to plasma lysoPS during ACS [85].
In rats, PLA1A is released by activated platelets since it is only detected at high levels in the
serum but not in the plasma [10]. During blood coagulation, PS+ platelet-derived EVs provide
a catalytic surface for tenase and prothrombinase complexes [4]. Besides, these platelet-
derived EVs expose the PS substrate to the extracellular phospholipases, such as PLA1A and
sPLA2, which are responsible for lysoPS production [4]. LysoPS might activate platelets and
serotonin release through the P2Y10 receptor [13]. Incubation of washed activated rat
platelets reduced PS content but enhanced lysoPS accumulation, which was inhibited
partially by a PLA2 inhibitor [4]. Besides, blood clotting time was decreased, and thrombin
formation was increased, thereby suggesting a role for PLA1A in blood clotting [4]. Platelet
activation also initiated an upsurge in polyunsaturated (18:2 and 20:4) LPA production [40].
Incubation of activated rat platelets with recombinant PLA1A, sPLA2-IIa, and ATX induced
a drastic decrease in PS and an increase in LPA, indicating that PLA1A works with sPLA2
for producing lysophospholipids including various lysoPS species [4,8].
Evidence suggests an independent association between plasma triglyceride concentrations
and increased atherosclerosis risk [86]. In addition to its immunomodulatory functions,
lysoPS may contribute to atherosclerosis by influencing Mφ and platelet functions [13]. A
mouse insertional lipid defect (lpd) mutation led to hypertriglyceridemia and a fatty liver
phenotype [7]. Although PLA1A has homology to triglyceride lipases, the mouse Pla1a gene
is distinct from the lpd locus [7]. Lpdl, another lipase gene in the lpd locus, and its human
homolog LPDL have high sequence homology with PLA1A [86]. Nine DNA polymorphisms
in PLA1A were identified in a Caucasian population [87]. Although the sample size studied

213
was small (10 patients) to draw a valid conclusion, DNA sequence comparison of Caucasian
descents with hypertriglyceridemia and clinically normal subjects revealed no association
between PLA1A polymorphisms and hypertriglyceridemia [87].
Removal of excess cholesterol from cells and the capacity of high-density lipoproteins to
transport cholesterol for elimination by the liver is thought to be atheroprotective [88].
PLA1A likely participates in the high-density lipoprotein metabolism in vivo [89].
Overexpression of Pla1a in human apoA-I transgenic C57BL/6 mice led to increases in serum
phospholipids/apoA-I ratio and cholesterol efflux [89]. The high high-density lipoprotein
cholesterol levels and phospholipids/apoA-I ratio were associated with enhanced cholesterol
efflux via the scavenger receptor class BI and reduced efflux via the ATP-binding cassette
transporter 1, respectively [89].
Mesenchymal stromal cells contribute to the homeostasis of many organs. Proteomic
analyses of proteins secreted by mesenchymal stromal cells isolated from bone marrows,
visceral and subcutaneous adipose tissues only identified PLA1A in mesenchymal stromal
cells from tissues of high-fat but not normal diet-fed mice [90]. Thus, obesity can modify the
secretome content of MSCs [90]. Future studies using Pla1a knockout mice will establish
whether PLA1A plays a role in atherosclerosis, cardiometabolic syndromes, and obesity.
4.4. Antiviral innate immune responses
Several phospholipases contribute to hepatitis C virus (HCV) replication [[91], [92], [93],
[94], [95]]. Serum levels of PLA1A were higher in HCC patients with HCV-related liver
injury than in those with hepatitis B virus or non-hepatitis B virus, non-HCV-related liver
diseases [18]. PLA1A expression was upregulated by HCV infection [95], and PLA1A levels
were higher in the liver from HCV-infected patients [96]. In cells, PLA1A showed a reticular
pattern reminiscent of the endoplasmic reticulum that was not disturbed by HCV infection
[95]. PLA1A facilitated viral assembly through direct binding to HCV E2, NS2, and NS5A
proteins, leading to stabilization of the NS2-E2 and the NS2/NS5A complexes during
infection [95,96]. An amino acid motif essential for viral RNA replication in the C-terminal
domain I of NS5A drove the interaction with PLA1A [96]. The reduced expression of PLA1A
by siRNA silencing resulted in reduced HCV replication, decline in intracellular HCV RNA

214
level and viral proteins expression [95]. Though the impact of catalytically inactive PLA1A
was not tested on HCV replication, the effect of PLA1A silencing was reversed by addition
of lysoPS [95]. Furthermore, PLA1A might participate in the antiviral immune response
through TANK-binding kinase 1-mediated signaling [97]. PLA1A modulated the recruitment
of TANK-binding kinase 1 and its interactions with interferon regulatory factor 3 and
mitochondrial antiviral signaling proteins [97]. The knockdown of PLA1A reduced
recruitment of TANK-binding kinase 1 and interferon regulatory factor 3 to mitochondria,
leading to mitochondria morphology changes and inhibition of signaling for type I interferon
production [97].
HIV-1 Tat protein released by infected cells can affect bystander uninfected T cells functions,
contributing to HIV pathogenesis [98,99]. PLA1A mRNA was enhanced in Jurkat T cells [99]
and human brain microvascular endothelial cells exposed to Tat protein [100]. In the
cerebrospinal fluid of rhesus macaques with simian immunodeficiency virus-infected central
nervous system, fatty acids and phospholipids (C18:2, C16:0, and C18:0) were increased
[101]. These increases in lipid metabolites were concomitant with enhanced expression of
PLA1A (9.3-fold) and PLA2G4C (6.4-fold) in the hippocampus [101].
4.5. Other diseases
In patients with non-alcoholic fatty liver diseases, high serum PLA1A levels were reported
[18]. Non-alcoholic fatty liver diseases are common liver diseases. Multiple factors can
contribute to oxidative-stress-induced liver fibrosis, such as viruses, alcohol, high-fat diet,
obesity, insulin resistance, and gut-derived LPS [102,103]. LPS administered to rats
differently modulated numerous genes in the liver, including a 24.3-fold increase in Pla1a
expression compared to pair-fed animals, but in the alcohol-fed rats, LPS-mediated
expression of Pla1a was suppressed [104].
Vitreous eye fluids from proliferative diabetic retinopathy patients expressed higher PLA1A
compared to nondiabetic controls [105]. PLA1A might contribute to the development of
proliferative diabetic retinopathy through the synthesis of bioactive lysophospholipids [105].
Increased expression of PLA1A seems to be associated with fibrosis in multiple forms of
chronic disease, including liver, eyes, and kidneys [18,105,106].

215
Treatment of skin diseases includes the use of all-trans retinoic acid, but the all-trans retinoic
acid adverse effects on the epidermal barrier function limit its use [107]. In mouse skin
treated with all-trans retinoic acid, corneocytes and keratinocytes contained abnormal lipid
droplets in the cytoplasm, which is related to a hyperproliferative state [108]. Pla1a is one of
the upregulated epithelial barrier-associated genes in the all-trans retinoic acid-treated mouse
skin [108]. Besides Pla1a, several other phospholipases were either upregulated (Pla2g4e)
or downregulated (Pla2g2e and Lypla) [108]. PLA1A is highly expressed in skin samples of
discoid lupus patients compared to psoriasis patient samples, suggesting that specific
pathological traits regulate its expression [109]. Exposure of mouse skin to Staphylococcus
aureus strongly induced Pla1a transcripts [110]. Among human skin cells, PLA1A is only
expressed in melanocytes but not in keratinocytes or fibroblasts [111]. PLA1A expression in
diseased or infected skins might be related to recruitment of T-cell subsets expressing PLA1A
in skin lesions [49,109], stimulation of skin fibroblasts by TLR3 microbial ligands [67], or
TLR4 ligand-induced PLA1A expression in tissue-resident Mφ [53]. Further studies are
required to understand the roles played by PLA1A in skin immunity, inflammation, and
repair.
Pla1a was the most upregulated gene in peripheral blood cells isolated from long-term
surviving rats following allogeneic heart transplant compared to syngeneic graft
transplantation [112]. Increased expression of PLA1A was also observed in human biopsies
undergoing antibody-mediated rejection after heart transplantation [113]. In biopsies from
patients post kidney transplantation, elevated PLA1A expression in renal proximal tubule
epithelial cells and INFγ-stimulated endothelial cells during antibody-mediated rejection
have been reported [114,115]. INFγ-activated renal epithelial cells were likely the principal
source of PLA1A transcripts in acute allograft rejection [115]. Expression of PLA1A
correlated with histological lesions [115]. Urinary exosomes were released into the
extracellular environment by podocyte epithelial cells [116]. Proteomics analysis identified
PLA1A in enriched podocyte vesicles isolated from human urine [116]. The assessment of
PLA1A expression can help diagnose antibody-mediated rejection after organ transplantation,
possibly predicting future failure [115,117].

216
5 Other enzymes regulating serine phospholipid metabolism in
neural system
ABHD (α/β-hydrolase domain) containing-protein family plays a crucial role in lipid
metabolism. Mutations of the ABHD protein family members led to inherited inborn lipid
metabolic disorders [118]. Among the ABHD family members, ABHD16A and ABHD12
play crucial roles in the metabolism of serine phospholipids in mammalian brains as the
foremost PS lipase [119] and lysoPS lipase [120], respectively. Deficiencies of ABHD16A
(PS production) and ABHD12 (PS degradation) are associated with metabolic syndrome and
inflammatory neurodegenerative disease, respectively [[118], [119], [120]]. Mutation in
ABHD12 causes the neurodegenerative disorder PHARC, which means polyneuropathy,
hearing loss, ataxia, retinitis pigmentosa, and cataract [121]. The brain of ABHD12−/− mice
displayed at a younger age (2–6 months) a massive increase of long-chain lysoPS, which
acted as a TLR2 agonist in the microglia [120]. Activation of microglial TLR2 was
responsible for the subsequent age-dependent proinflammatory responses and neural death
in the ABHD12−/− mouse model, which was associated with auditory and motor defects
[120]. Both ABHD16A and ABHD12 are localized in the endoplasmic reticulum in the
central nervous system, especially in the cerebellum that is the most atrophic brain region in
PHARC patients [122]. ABHD16A [123] and ABHD12 [121,124] also exhibited
monoacylglycerol lipase activity towards 2-arachidonoylglycerol, responsible for neural
pain. Thus, ABHD16A and ABHD12 were responsible for the intracellular serine
phospholipids metabolism, and the ABHD16A-ABHD12-lysoPS pathway was assessed as
an emerging lysophospholipid signaling network for neuro-immunological disorders,
indicating their potential therapeutic relevance [119]. It is unknown that the conversion of
lysoPS to LPA by ATX in the brain contributes to neurodegenerative diseases.
DO264, a selective inhibitor of ABHD12, elevated the lysoPS level in mice brain and induced
manifestations similar to PHARC [125]. In contrast, a reversible inhibitor of ABHD16A (12-
thiazole abietanes) inhibiting the lipase activity towards PS reduced the lysoPS level and
brain inflammation [126]. GPR34 is expressed in microglial cells [127,128]. Deletion of
GPR34 impaired glial cell morphology, functional responses such as phagocytosis [127], and

217
the production of inflammatory cytokines involved in neuropathic pain [128]. Altogether, the
available information suggests that the altered metabolism of PS lipids and lysoPS-mediated
signaling in the brain can play a role in the pathogenesis of neurodegenerative disorders.
6 Conclusions
There is growing evidence suggesting roles for PLA1A in many pathological conditions,
including autoimmune disorders, cancers, cardiometabolic disorders, antiviral innate
immune responses, and other diseases. Elevated PLA1A expression and high PLA1A protein
levels are associated with various pathologies. Increased PLA1A expression and release in
tissues and biological fluids result in hydrolysis of surface exposed PS. Production of high
level of lysoPS might subsequently contribute to disease development through lysoPS
receptor activation. However, the roles of PLA1A in disease mechanism remain to be
established. Future studies will determine whether PLA1A is a valuable clinical biomarker
for disease diagnosis or a drug target.
Acknowledgments
This work was supported by the Canadian Institutes for Health Research (MOP-142210). YZ
was the recipient of a scholarship from the China Scholarship Council (CSC).
7 References
[1] Sato T, et al. Serine phospholipid-specific phospholipase A that is secreted from activated
platelets. A new member of the lipase family. J. Biol. Chem. 1997;272(4):2192–8.
[2] Horigome K, et al. Selective release of phospholipase A2 and lysophosphatidylserine-
specific lysophospholipase from rat platelets. J. Biochem.1987;101(1):53–61.
[3] Higashi S, et al. Purification and characterization of lysophospholipase released from rat
platelets. J. Biochem. 1988;103(3):442–7.
[4] Yokoyama K, Kudo I, Inoue K. Phospholipid degradation in rat calcium ionophore-
activated platelets is catalyzed mainly by two discrete secretory phospholipase As. J.
Biochem. 1995;117(6):1280–7.
[5] van Groningen JJ, et al. nmd, a novel gene differentially expressed in human melanoma
cell lines, encodes a new atypical member of the enzyme family of lipases. FEBS Lett.
1997;404(1):82–6.
[6] Nagai Y, et al. An alternative splicing form of phosphatidylserine-specific phospholipase
A1 that exhibits lysophosphatidylserine-specific lysophospholipase activity in humans. J.
Biol. Chem. 1999;274(16):11053–9.

218
[7] Wen XY, et al. Murine phosphatidylserine-specific phospholipase A1 (Ps-pla1) maps to
chromosome 16 but is distinct from the lpd (lipid defect) locus. Mamm. Genome
2001;12(2):129–32.
[8] Aoki J, et al. Serum lysophosphatidic acid is produced through diverse phospholipase
pathways. J. Biol. Chem. 2002;277(50):48737–44.
[9] Carri`ere F, et al. Structural basis for the substrate selectivity of pancreatic lipases and
some related proteins. Biochim. Biophys. Acta 1998;1376(3):417–32.
[10] Aoki J, et al. Structure and function of extracellular phospholipase A1 belonging to the
pancreatic lipase gene family. Biochimie 2007;89(2):197–204.
[11] Aoki J, et al. Structure and function of phosphatidylserine-specific phospholipase A1.
Biochim. Biophys. Acta 2002;1582(1–3):26–32.
[12] Aloulou A, et al. Exploring the specific features of interfacial enzymology based on
lipase studies. Biochim. Biophys. Acta 2006;1761(9):995–1013.
[13] Yatomi Y, et al. Lysophospholipids in laboratory medicine. Proc. Jpn. Acad. Ser. B
Phys. Biol. Sci. 2018;94(10):373–89.
[14] Arima N, et al. Surface loops of extracellular phospholipase A(1) determine both
substrate specificity and preference for lysophospholipids. J. Lipid Res. 2012;53(3):513–21.
[15] Hiramatsu T, et al. Biochemical and molecular characterization of two phosphatidic
acid-selective phospholipase A1s, mPA-PLA1alpha and mPAPLA1beta. J. Biol. Chem.
2003;278(49):49438–47.
[16] Winkler FK, D’Arcy A, Hunziker W. Structure of human pancreatic lipase. Nature
1990;343(6260):771–4.
[17] Nakamura K, et al. A novel enzyme immunoassay for the determination of
phosphatidylserine-specific phospholipase A(1) in human serum samples. Clin. Chim. Acta
2010;411(15–16):1090–4.
[18] Uranbileg B, et al. Possible involvement of PS-PLA1 and lysophosphatidylserine
receptor (LPS1) in hepatocellular carcinoma. Sci. Rep. 2020;10(1):2659.
[19] Withers-Martinez C, et al. A pancreatic lipase with a phospholipase A1 activity: crystal
structure of a chimeric pancreatic lipase-related protein 2 from guinea pig. Structure
1996;4(11):1363–74.
[20] Sakuragi T, Kosako H, Nagata S. Phosphorylation-mediated activation of mouse Xkr8
scramblase for phosphatidylserine exposure. Proc. Natl. Acad. Sci. U. S. A.
2019;116(8):2907–12.
[21] Schroit AJ, Madsen JW, Tanaka Y. In vivo recognition and clearance of red blood cells
containing phosphatidylserine in their plasma membranes. J. Biol. Chem.1985;260(8):5131–
8.
[22] Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of
apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin.
Invest. 2002;109(1):41–50.
[23] Voll RE, et al. Immunosuppressive effects of apoptotic cells. Nature 1997;390
(6658):350–1.
[24] Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression
of proinflammatory macrophage transcription is coupled directly to specific recognition. J.
Immunol. 2004;172(2):880–9.
[25] Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable
cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108(48):19246–51.

219
[26] Elliott JI, et al. Membrane phosphatidylserine distribution as a non-apoptotic signalling
mechanism in lymphocytes. Nat. Cell Biol. 2005;7(8):808–16.
[27] Appelt U, et al. Viable, apoptotic and necrotic monocytes expose phosphatidylserine:
cooperative binding of the ligand Annexin V to dying but not viable cells and implications
for PS-dependent clearance. Cell Death Differ. 2005;12(2):194–6.
[28] Smrz D, Draberova L, Draber P. Non-apoptotic phosphatidylserine externalization
induced by engagement of glycosylphosphatidylinositol-anchored proteins. J. Biol. Chem.
2007;282(14):10487–97.
[29] Fischer K, et al. Antigen recognition induces phosphatidylserine exposure on the cell
surface of human CD8+ T cells. Blood 2006;108(13):4094–101.
[30] Audo R, et al. Phosphatidylserine outer layer translocation is implicated in IL-10
secretion by human regulatory B cells. PLoS One 2017;12(1):e0169755.
[31] Riedl S, et al. In search of a novel target - phosphatidylserine exposed by nonapoptotic
tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys.
Acta 2011;1808(11):2638–45.
[32] Sharma R, et al. Detection of phosphatidylserine-positive exosomes for the diagnosis of
early-stage malignancies. Br. J. Cancer 2017;117(4):545–52.
[33] Yang H, et al. TMEM16F forms a Ca2+ activated cation channel required for lipid
scrambling in platelets during blood coagulation. Cell 2012;151(1):111–22.
[34] Fujii T, et al. TMEM16F is required for phosphatidylserine exposure and microparticle
release in activated mouse platelets. Proc. Natl. Acad. Sci. U. S. A. 2015;112(41):12800–5.
[35] Fortin PR, et al. Distinct subtypes of microparticle-containing immune complexes are
associated with disease activity, damage, and carotid intima-media thickness in systemic
lupus erythematosus. J. Rheumatol. 2016;43(11):2019–25.
[36] Sawada T, et al. Serum phosphatidylserine-specific phospholipase A(1) as a novel
biomarker for monitoring systemic lupus erythematosus disease activity. Int. J.Rheum. Dis.
2019;22(11):2059–66.
[37] Birge RB, et al. Phosphatidylserine is a global immunosuppressive signal in
efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–78.
[38] Bellini F, Bruni A. Role of a serum phospholipase A1 in the phosphatidylserineinduced
T cell inhibition. FEBS Lett. 1993;316(1):1–4.
[39] Barnes MJ, et al. The lysophosphatidylserine receptor GPR174 constrains regulatory T
cell development and function. J. Exp. Med. 2015;212(7):1011–20.
[40] Bolen AL, et al. The phospholipase A1 activity of lysophospholipase A-I links platelet
activation to LPA production during blood coagulation. J. Lipid Res.2011;52(5):958–70.
[41] Kitamura H, et al. GPR34 is a receptor for lysophosphatidylserine with a fatty acid at
the sn-2 position. J. Biochem. 2012;151(5):511–8.
[42] Plückthun A, Dennis EA. Acyl and phosphoryl migration in lysophospholipids:
importance in phospholipid synthesis and phospholipase specificity. Biochemistry
1982;21(8):1743–50.
[43] Emoto S, et al. Analysis of glycero-lysophospholipids in gastric cancerous ascites. J.
Lipid Res. 2017;58(4):763–71.
[44] Zhao Y, et al. Targeting the autotaxin - Lysophosphatidic acid receptor axis in
cardiovascular diseases. Biochem. Pharmacol. 2019;164:74–81.
[45] Bandoh K, et al. Lysophosphatidic acid (LPA) receptors of the EDG family are
differentially activated by LPA species. Structure-activity relationship of cloned LPA
receptors. FEBS Lett. 2000;478(1–2):159–65.

220
[46] Inoue A, et al. TGFalpha shedding assay: an accurate and versatile method for detecting
GPCR activation. Nat. Methods 2012;9(10):1021–9.
[47] Kihara Y, et al. Lysophospholipid receptor nomenclature review: IUPHAR Review 8.
Br. J. Pharmacol. 2014;171(15):3575–94.
[48] Ikubo M, et al. Structure-activity relationships of lysophosphatidylserine analogs as
agonists of G-protein-coupled receptors GPR34, P2Y10, and GPR174. J. Med. Chem.
2015;58(10):4204–19.
[49] Kwong BY, et al. Molecular analysis of tumor-promoting CD8+ T cells in two stage
cutaneous chemical carcinogenesis. J. Invest. Dermatol. 2010;130(6):1726–36.
[50] Barnes MJ, Cyster JG. Lysophosphatidylserine suppression of T-cell activation via
GPR174 requires Galphas proteins. Immunol. Cell Biol. 2018;96(4):439–45.
[51] Konkel JE, et al. Transforming growth factor-beta signaling in regulatory T cells
controls T Helper-17 cells and tissue-specific immune responses. Immunity 2017;46(4):660–
74.
[52] Mavin E, et al. Human regulatory T cells mediate transcriptional modulation ofdendritic
cell function. J. Immunol. 2017;198(1):138–46.
[53] Hosono H, et al. Expression of phosphatidylserine-specific phospholipase A(1) mRNA
in human THP-1-derived macrophages. Cell Transplant. 2010;19(6):759–64.
[54] Idborg H, et al. A9. 3 Deletion of mPGES-1 affects fatty acid composition and
eicosanoid profiles in mice. Ann. Rheum. Dis. 2013;72(Suppl. 1):A65.
[55] Kawamoto K, et al. Nerve growth factor activates mast cells through the collaborative
interaction with lysophosphatidylserine expressed on the membrane surface of activated
platelets. J. Immunol. 2002;168(12):6412–9.
[56] Sugo T, et al. Identification of a lysophosphatidylserine receptor on mast cells. Biochem.
Biophys. Res. Commun. 2006;341(4):1078–87.
[57] Hosono H, et al. Phosphatidylserine-specific phospholipase A1 stimulates histamine
release from rat peritoneal mast cells through production of 2-acyl-1- lysophosphatidylserine.
J. Biol. Chem. 2001;276(32):29664–70.
[58] Lourenssen S, Blennerhassett MG. Lysophosphatidylserine potentiates nerve growth
factor-induced differentiation of PC12 cells. Neurosci. Lett. 1998;248(2):77–80.
[59] Lee SY, et al. Lysophosphatidylserine stimulates chemotactic migration in U87 human
glioma cells. Biochem. Biophys. Res. Commun. 2008;374(1):147–51.
[60] Park KS, et al. Lysophosphatidylserine stimulates L2071 mouse fibroblast chemotactic
migration via a process involving pertussis toxin-sensitive trimeric Gproteins. Mol.
Pharmacol. 2006;69(3):1066–73.
[61] Tomlins SA, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia
2008;10(2):177–88.
[62] Nakawatari K, et al. Elevated phosphatidylserine-specific phospholipase A1 level in
hyperthyroidism. Clin. Chim. Acta 2020;503:99–106.
[63] Iwata Y, et al. Higher serum levels of autotaxin and phosphatidylserine-specific
phospholipase A(1) in patients with lupus nephritis. Int. J. Rheum. Dis. 2021;24(2):231–9.
[64] Sugenoya A, et al. Correlation between thyrotropin-displacing activity and human
thyroid-stimulating activity by immunoglobulins from patients with Graves’ disease and
other thyroid disorders. J. Clin. Endocrinol. Metab. 1979;48(3):398–402.
[65] Tepasse P-R, et al. Tu1780-regulated expression of phosphatidylserine-specific
phospholipase A1 and lysophosphatidylserine receptors in human intestinal inflammation.
Gastroenterology 2018;154(6):S-1017.

221
[66] Hong SN, et al. RNA-seq reveals transcriptomic differences in inflamed and
noninflamed intestinal mucosa of Crohn’s disease patients compared with normal mucosa of
healthy controls. Inflamm. Bowel Dis. 2017;23(7):1098–108.
[67] Fang F, et al. A synthetic TLR3 ligand mitigates profibrotic fibroblast responses by
inducing autocrine IFN signaling. J. Immunol. 2013;191(6):2956–66.
[68] Iida Y, et al. Lysophosphatidylserine stimulates chemotactic migration of colorectal
cancer cells through GPR34 and PI3K/Akt pathway. Anticancer Res.2014;34(10):5465–72.
[69] Lee SC, et al. Regulation of tumor immunity by lysophosphatidic acid. Cancers (Basel)
2020;12(5):1202.
[70] Liu W, Peng Y, Tobin DJ. A new 12-gene diagnostic biomarker signature of melanoma
revealed by integrated microarray analysis. PeerJ. 2013;1:e49.
[71] Kurano M, et al. Association between serum autotaxin or phosphatidylserinespecific
phospholipase A1 levels and melanoma. J. Dermatol. 2018;45(5):571–9.
[72] Yang G, et al. PLA1A expression as a diagnostic marker of BRAF-mutant metastasis in
melanoma cancer. Sci. Rep. 2021;11(1):6056.
[73] Wang J, et al. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG
fusion gene transcripts. Cancer Res. 2008;68(20):8516–24.
[74] Kron KJ, et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates
NOTCH signaling in primary prostate cancer. Nat. Genet. 2017;49(9):1336–45.
[75] Paulo P, et al. Molecular subtyping of primary prostate cancer reveals specific and
shared target genes of different ETS rearrangements. Neoplasia 2012;14(7):600–11.
[76] Brenner JC, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase
in ETS gene fusion-positive prostate cancer. Cancer Cell 2011;19(5):664–78.
[77] Washington MN, Weigel NL. 1{alpha},25-Dihydroxyvitamin D3 inhibits growth of
VCaP prostate cancer cells despite inducing the growth-promoting TMPRSS2:ERG gene
fusion. Endocrinology 2010;151(4):1409–17.
[78] Goodwin JF, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer
progression and metastasis. Cancer Cell 2015;28(1):97–113.
[79] Fielden MR, Brennan R, Gollub J. A gene expression biomarker provides early
prediction and mechanistic assessment of hepatic tumor induction by nongenotoxic
chemicals. Toxicol. Sci. 2007;99(1):90–100.
[80] Fielden MR, et al. Development and evaluation of a genomic signature for the prediction
and mechanistic assessment of nongenotoxic hepatocarcinogens in the rat. Toxicol. Sci.
2011;124(1):54–74.
[81] Iida Y, et al. Phosphatidylserine-specific phospholipase A1 (PS-PLA1) expression in
colorectal cancer correlates with tumor invasion and hematogenous metastasis. Anticancer
Res. 2015;35(3):1459–64.
[82] Righi V, et al. 1H HR-MAS and genomic analysis of human tumor biopsies discriminate
between high and low grade astrocytomas. NMR Biomed. 2009;22(6):629–37.
[83] Tang X, Benesch MGK, Brindley DN. Role of the autotaxin-lysophosphatidate axis in
the development of resistance to cancer therapy. Biochim. Biophys. Acta Mol. Cell Biol.
Lipids 1865;2020(8):158716.
[84] Kurano M, et al. Different origins of lysophospholipid mediators between coronary and
peripheral arteries in acute coronary syndrome. J. Lipid Res. 2017;58(2):433–42.
[85] Kurano M, et al. Blood levels of serotonin are specifically correlated with plasma
lysophosphatidylserine among the glycero-lysophospholipids. BBA Clin. 2015;4:92–8.

222
[86] Wen XY, et al. Identification of a novel lipase gene mutated in lpd mice with
hypertriglyceridemia and associated with dyslipidemia in humans. Hum. Mol. Genet.
2003;12(10):1131–43.
[87] Wang J, et al. Polymorphisms in the gene encoding phosphatidylserine-specific
phospholipase A1 (PSPLA1). J. Hum. Genet. 2002;47(11):611–3.
[88] Kudinov VA, et al. High-density lipoproteins as homeostatic nanoparticles of blood
plasma. Int. J. Mol. Sci. 2020;21(22):8737.
[89] Yancey PG, et al. In vivo modulation of HDL phospholipid has opposing effects on SR-
BI- and ABCA1-mediated cholesterol efflux. J. Lipid Res. 2004;45(2):337–46.
[90] Ayaz-Guner S, et al. A comparative study on normal and obese mice indicates that the
secretome of mesenchymal stromal cells is influenced by tissue environment and
physiopathological conditions. Cell Commun. Sign. 2020;18(1):118.
[91] Xu S, et al. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus
replication and assembly. J. Virol. 2012;86(23):13025–37.
[92] Li X, et al. Hepatocyte nuclear factor 4α and downstream secreted phospholipase A2
GXIIB regulate production of infectious hepatitis C virus. J. Virol. 2014;88(1):612–27.
[93] Farquhar MJ, et al. Autotaxin-lysophosphatidic acid receptor signalling regulates
hepatitis C virus replication. J. Hepatol. 2017;66(5):919–29.
[94] Menzel N, et al. MAP-kinase regulated cytosolic phospholipase A2 activity is essential
for production of infectious hepatitis C virus particles. PLoS Pathog.2012;8(7):e1002829.
[95] Guo M, et al. Phosphatidylserine-specific phospholipase A1 involved in hepatitis C virus
assembly through NS2 complex formation. J. Virol. 2015;89(4):2367–77.
[96] Yang Q, et al. Phosphatidylserine-specific phospholipase A1 is the critical bridge for
hepatitis C virus assembly. Virol. Sin. 2019;34(5):521–37.
[97] Gao X, et al. PLA1A participates in the antiviral innate immune response by facilitating
the recruitment of TANK-binding kinase 1 to mitochondria. J. Innate Immun.
2018;10(4):315–27.
[98] Ajasin D, Eugenin EA. HIV-1 tat: role in bystander toxicity. Front. Cell. Infect.
Microbiol. 2020;10:61.
[99] Liao W, et al. Combined metabonomic and quantitative real-time PCR analyses reveal
systems metabolic changes in Jurkat T-cells treated with HIV-1 Tat protein. J. Proteome Res.
2012;11(11):5109–23.
[100] Woollard SM, et al. Differential effects of Tat proteins derived from HIV-1 subtypes
B and recombinant CRF02_AG on human brain microvascular endothelial cells: implications
for blood-brain barrier dysfunction. J. Cereb. Blood Flow Metab. 2014;34(6):1047–59.
[101] Wikoff WR, et al. Metabolomic analysis of the cerebrospinal fluid reveals changes in
phospholipase expression in the CNS of SIV-infected macaques. J. Clin. Invest.
2008;118(7):2661–9.
[102] Ferro D, et al. New insights into the pathogenesis of non-alcoholic fatty liver disease:
gut-derived lipopolysaccharides and oxidative stress. Nutrients 2020;12 (9):2762.
[103] Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of nonalcoholic
fatty liver disease (NAFLD). Metabolism 2016;65(8):1038–48.
[104] Deaciuc IV, et al. Microarray gene analysis of the liver in a rat model of chronic,
voluntary alcohol intake. Alcohol 2004;32(2):113–27.
[105] Abu El-Asrar AM, et al. Expression of bioactive lysophospholipids and processing
enzymes in the vitreous from patients with proliferative diabetic retinopathy. Lipids Health
Dis. 2014;13:187.

223
[106] Gomez IG, et al. TWEAK-Fn14 signaling activates myofibroblasts to drive progression
of fibrotic kidney disease. J. Am. Soc. Nephrol. 2016;27(12):3639–52.
[107] Ale SI, Laugier JP, Maibach HI. Differential irritant skin responses to tandem
application of topical retinoic acid and sodium lauryl sulphate: II. Effect of time between first
and second exposure. Br. J. Dermatol. 1997;137(2):226–33.
[108] Li J, Li Q, Geng S. All-trans retinoic acid alters the expression of the tight junction
proteins Claudin-1 and -4 and epidermal barrier function-associated genes in the epidermis.
Int. J. Mol. Med. 2019;43(4):1789–805.
[109] Jabbari A, et al. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed
by transcriptomic comparison with psoriasis. J. Invest. Dermatol. 2014;134(1):87–95.
[110] Harris TA, et al. Resistin-like molecule α provides vitamin-A-dependent antimicrobial
protection in the skin. Cell Host Microbe 2019;25(6):777–88. e8.
[111] Reemann P, et al. Melanocytes in the skin–comparative whole transcriptome analysis
of main skin cell types. PLoS One 2014;9(12):e115717.
[112] Lu H, et al. Identification of alterations in gene expression in rat recipients with long-
term-surviving cardiac grafts. Transplant. Proc. 2002;34(7):2729–31.
[113] Loupy A, et al. Gene expression profiling for the identification and classification of
antibody-mediated heart rejection. Circulation 2017;135(10):917–35.
[114] Hidalgo LG, et al. NK cell transcripts and NK cells in kidney biopsies from patients
with donor-specific antibodies: evidence for NK cell involvement in antibodymediated
rejection. Am. J. Transplant. 2010;10(8):1812–22.
[115] Venner JM, et al. The molecular landscape of antibody-mediated kidney transplant
rejection: evidence for NK involvement through CD16a Fc receptors. Am. J. Transplant.
2015;15(5):1336–48.
[116] Prunotto M, et al. Proteomic analysis of podocyte exosome-enriched fraction from
normal human urine. J. Proteome 2013;82:193–229.
[117] Sellar´es J, et al. Molecular diagnosis of antibody-mediated rejection in human kidney
transplants. Am. J. Transplant. 2013;13(4):971–83.
[118] Xu J, et al. Sequence analysis and structure prediction of ABHD16A and the roles of
the ABHD family members in human disease. Open Biol. 2018;8(5):180017.
[119] Kamat SS, et al. Immunomodulatory lysophosphatidylserines are regulated by
ABHD16A and ABHD12 interplay. Nat. Chem. Biol. 2015;11(2):164–71.
[120] Blankman JL, et al. ABHD12 controls brain lysophosphatidylserine pathways that are
deregulated in a murine model of the neurodegenerative disease PHARC. Proc. Natl. Acad.
Sci. U. S. A. 2013;110(4):1500–5.
[121] Fiskerstrand T, et al. Mutations in ABHD12 cause the neurodegenerative disease
PHARC: an inborn error of endocannabinoid metabolism. Am. J. Hum. Genet.
2010;87(3):410–7.
[122] Singh S, Joshi A, Kamat SS. Mapping the neuroanatomy of ABHD16A, ABHD12, and
lysophosphatidylserines provides new insights into the pathophysiology of the human
neurological disorder PHARC. Biochemistry 2020;59(24):2299–311.
[123] Savinainen JR, et al. Biochemical and pharmacological characterization of the human
lymphocyte antigen B-associated transcript 5 (BAT5/ABHD16A). PLoS One
2014;9(10):e109869.
[124] Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that
hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007;14(12):1347–56.

224
[125] Ogasawara D, et al. Selective blockade of the lyso-PS lipase ABHD12 stimulates
immune responses in vivo. Nat. Chem. Biol. 2018;14(12):1099–108.
[126] Ahonen TJ, et al. Discovery of 12-thiazole abietanes as selective inhibitors of the
human metabolic serine hydrolase hABHD16A. ACS Med. Chem. Lett. 2018;9(12):1269–
73.
[127] Preissler J, et al. Altered microglial phagocytosis in GPR34-deficient mice. Glia
2015;63(2):206–15.
[128] Sayo A, et al. GPR34 in spinal microglia exacerbates neuropathic pain in mice. J.
Neuroinflammation 2019;16(1):82.
[129] Spivack K, et al. Enhancement of transgene expression by the β-catenin inhibitor
iCRT14. Plasmid 2021;114:102556.
[130] Xiong J, et al. Dysregulated choline metabolism in T-cell lymphoma: role of choline
kinase-α and therapeutic targeting. Blood Cancer J. 2015;5(3):287.
[131] Ning YL, et al. Bioinformatics analysis identifies hub genes and molecular pathways
involved in sepsis-induced myopathy. Med. Sci. Monit. 2020;26:e919665.
[132] Xu S. Transcriptome profiling in systems vascular medicine. Front. Pharmacol.
2017;8:563.
[133] Silvestri C, et al. Genome-wide identification of Smad/Foxh1 targets reveals a role for
Foxh1 in retinoic acid regulation and forebrain development. Dev. Cell 2008; 14(3):411–23.
[134] Czapski GA, et al. Acute systemic inflammatory response alters transcription profile
of genes related to immune response and Ca(2+) homeostasis inHippocampus; relevance to
neurodegenerative disorders. Int. J. Mol. Sci. 2020;21(21):7838.
[135] Yang S, et al. Immune defense is the primary function associated with the differentially
expressed genes in the cochlea following acoustic trauma. Hear. Res. 2016;333:283–94.
[136] Barger JL, et al. Gene expression profiling reveals differential effects of sodium
selenite, selenomethionine, and yeast-derived selenium in the mouse. Genes Nutr.
2012;7(2):155–65.
[137] Mencucci MV, et al. Integrative transcriptomic analysis of pancreatic islets from
patients with prediabetes/type 2 diabetes. Diabetes Metab. Res. Rev. 2021;37(1):e3359.
[138] Chan B, Sukhatme VP. Suppression of Tie-1 in endothelial cells in vitro induces a
change in the genome-wide expression profile reflecting an inflammatory function. FEBS
Lett. 2009;583(6):1023–8.
[139] Karagianni N, et al. An integrative transcriptome analysis framework for drug efficacy
and similarity reveals drug-specific signatures of anti-TNF treatment in a mouse model of
inflammatory polyarthritis. PLoS Comput. Biol. 2019;15(5):e1006933.

225
Annexe III: Platelet-derived extracellular vesicles contain
an active proteasome involved in protein processing
for antigen presentation via class I major
histocompatibility molecules
Genevieve Marcoux1,2, Audrée Laroche1,2, Stephan Hasse1,2, Marie Bellio1,2,
Maroua Mbarik1,2, Marie Tamagne3,4,5, Isabelle Allaeys1,2, Anne Zufferey1,2, Tania
Lévesque1,2, Johan Rebetz6, Annie Karakeussian-Rimbaud7,8, Julie Turgeon7,8,
Sylvain G. Bourgoin1,2, Hind Hamzeh-Cognasse9, Fabrice Cognasse9,10, Rick
Kapur11, John W. Semple6,12, Marie-Josée Hébert7,8, France Pirenne3,4,5, Herman S.
Overkleeft12, Bogdan I. Florea12, Mélanie Dieude7,8,14, Benoit Vingert3,4,5, Eric
Boilard1,2,8.
Affiliations :
1 Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval,
Québec, QC, Canada.
2 Centre de Recherche Arthrite, Faculté de Médecine de l'Université Laval, Québec, QC,
Canada.
3 Univ Paris Est Créteil, INSERM, IMRB, F-94010 Créteil, France
4 Etablissement Français du Sang, Ivry sur Seine, F-94200, France
5 Laboratory of Excellence GR-Ex, Paris, France
6 Division of Hematology and Transfusion Medicine, Lund University, Lund, Sweden
7 Research Centre, Centre hospitalier de l'Université de Montréal (CRCHUM), Montréal,
Québec, Canada.
8 Canadian Donation and Transplantation Research Program, Edmonton, Alberta, Canada
9 Université de Lyon, Université Jean Monnet, INSERM U1059, Saint-Etienne, France
10 Établissement Français du Sang Auvergne-Rhône-Alpes, Saint-Etienne, France.
11 Sanquin Research, Department of Experimental Immunohematology, Amsterdam and
Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, the
Netherlands.

226
12 Departments of Pharmacology and Medicine, University of Toronto, Toronto, Canada
13 Gorlaeus Laboratories, Leiden Institute of Chemistry and Netherlands Proteomics Centre,
Leiden, The Netherlands.
14 Département Microbiologie, Infectiologie et Immunologie, Faculté de Médecine,
Université de Montréal, Montréal, Québec, Canada.
Keywords: platelet, extracellular vesicles, proteasome, MHC-I, antigen
presentation, immunity

227
1 Abstract
In addition to their hemostatic role, platelets play a significant role in immunity. Once
activated, platelets release extracellular vesicles (EVs) formed by budding of their
cytoplasmic membranes. Because of their heterogeneity, platelet EVs (PEVs) are thought to
perform diverse functions. It is unknown, however, whether the proteasome is transferred
from platelets to PEVs or whether its function is retained. We hypothesized that functional
protein processing and antigen presentation machinery is transferred to PEVs by activated
platelets. Using molecular and functional assays, we show that the active 20S proteasome is
enriched in PEVs along with MHC-I and lymphocyte costimulatory molecules (CD40L and
OX40L). Proteasome-containing PEVs were identified in healthy donor blood, but did not
increase in platelet concentrates that caused adverse transfusion reactions. They were,
however, augmented after immune complex injections in mice. The complete biodistribution
of murine PEVs following injection into mice revealed that they could principally reach
lymphoid organs such as spleen and lymph nodes, in addition to the bone marrow, and to a
lesser extent liver and lungs. The PEV proteasome processed exogenous ovalbumin (OVA)
and loaded its antigenic peptide onto MHC-I molecules which promoted OVA-specific
CD8+ T lymphocyte proliferation. These results suggest that PEVs contribute to adaptive
immunity through cross-presentation of antigens and have privileged access to immune cells
through the lymphatic system, a tissue location that is inaccessible to platelets.

228
2 Introduction
Platelets are the second most abundant lineage in the blood and are best known for
their role in hemostasis.744 Platelets are small fragments produced by the large
multinucleated megakaryocyte in the bone marrow. They bear receptors that permit
recruitment of immune cells and carry an extensive set of immune and inflammatory
molecules (e.g. cytokines/chemokines, lipid mediators, hormones) stored in their
granules, cytoplasm, or synthesized by mRNA translation following platelet
activation. Thus, while platelets may mount an innate immune response against
injury, which is critical to combat pathogen invasion, organ and tissue damage may
also favor platelet activation and inflammation in chronic inflammatory diseases.745-
751
Albeit anucleate, the platelet cytoplasm includes numerous molecules comprising
the proteasome, which are transferred from megakaryocytes to their progeny. The
proteasome is a high molecular weight cylindrical protein complex through which
unwanted or damaged proteins are degraded.752,753 The central complex part, called
the 20S proteasome, is made up of twenty-eight distinct subunits,522 comprising the
three catalytic subunits necessary for the degradation of proteins into peptides of
three to fifteen amino acids in length.522,754 Proteasome activity in megakaryocytes
is required for platelet production755,756 and in platelets, the proteasome regulates
platelet lifespan,757 activation758-760 and the release of PEVs.761,762 The platelet
proteasome can hydrolyze proteins into smaller peptides,522,763,764 thereby enabling
peptide loading onto the platelet major histocompatibility complex (MHC) class I
molecules (MHC-I).765-767 Components of the peptide loading complex are also
expressed in platelets and are found in close proximity with MHC-I during platelet
activation.520,521 As platelets can efficiently form an immunological synapse with T-
lymphocytes to activate lymphocyte proliferation,520,768,769 they are known to fulfill
roles in cross-presentation of antigens in adaptive immunity. In a similar manner,
megakaryocytes cross-present antigens to CD8 T-lymphocytes, thereby suggesting
that they may also play a dual role in innate and adaptive immunity.767,770-772

229
Extracellular vesicles, produced in abundance by platelets, are small (up to 1µm in
diameter) membrane-bound vesicles released from the plasma membrane or
endosomal compartments of activated cells. Platelet EVs are heterogeneous in
terms of surface molecules and content (e.g. nucleic acids, lipids, transcription
factors, enzymes, mitochondria) and as such, may play diverse functions beyond
hemostasis.538,773,774 For instance, PEVs convey mitochondrial components that are
associated with inflammation and adverse transfusion reactions (ATRs).442,540,775
Despite the fact that platelets are restricted to the blood circulation, PEVs can cross
tissue barriers and enter synovial fluid,447,776 lymph535,777 and bone marrow778 where
they can deliver platelet-derived molecules and modulate target cells.773 For
instance, PEVs promote the formation of germinal centers and the production of IgG
by B-cells.543,544 They also interact with and modulate regulatory T cell differentiation
and activity.541,542 Thus, PEVs may be able to transport platelet-derived molecules
relevant to adaptive immunity into lymphoid organs. However, it is unknown whether
the proteasome and the molecules necessary for antigen presentation are also
transferred during the budding of PEVs. In this study, we evaluated whether
functional protein processing and antigen presentation machinery is transferred to
PEVs by activated platelets.
3 Material and methods
More details are presented in supplemental methods.
Labelling of murine platelets, DCs and PEVs
Platelets were isolated from C57BL/6J mice by retro-orbital or cardiac puncture in 200µL
ACD, 350µL Tyrode’s buffer pH 6.5. Whole blood was centrifuged at 600xg for 3min and
then at 400xg for 2min to remove red blood cells. Supernatant was spun at 1,300xg for 5 min
and the platelet-containing pellet was gently resuspended in 600µL Tyrode’s buffer pH 7.4.
Platelets were either left nonactivated or activated with thrombin (0.1U/mL) after addition of
5 mM of calcium for 90 min at RT (time based on kinetics of CD41+ Proteasome+ PEV

230
release shown in Supplementary figure 3C). Platelet EVs were obtained by two rounds of
centrifugation of stimulated platelets at 1,300xg for 5min at RT. Either activated platelets,
EVs or DCs were pulsed with 100µg/mL OVA protein (Sigma-Aldrich), 200µg/mL of OVA
peptide (SIINFEKL [Invivogen]) or left unpulsed for 4h at RT. These conditions were either
left unlabelled for lymphoproliferation and intracellular staining experiments or labelled for
Hs-FCM experiments.
Five µL of PEVs or platelet suspensions were labelled with 250nM LWA300 proteasome
probe in a total volume of 100μL for 90min at 30°C. Samples were then incubated with the
following antibodies for 30min at RT prior to dilution in Annexin V binding buffer and
analysis by Hs-FCM: BUV395 anti-CD41, BV650 anti-CD62p, BUV395 anti-CD41,
BV650 anti-CD62p, BV711 Annexin V, BV421 anti-OX40L, BUV737 anti-CD154 (all BD
Biosciences), PeCy7 anti-CD40, PeCy7 anti-MHC-I (AF6-88.5) and PE anti MHC-I bound
to OVA peptide (25D1.16) (all from Biolegend).
4 Results
PEVs contain functional proteasome
Following platelet activation by thrombin, remnant platelets were eliminated by
centrifugation and larger EVs were isolated by a second high-speed centrifugation
(18Kxg fraction). The supernatant obtained was further centrifuged at 100,000g and
smaller EVs (likely exosomes) were obtained from this pellet. We found that 98.10.5
% of proteins were retrieved in the larger EV 18Kxg fraction. Immunoblotting
confirmed that human PEVs from this fraction were enriched in proteasome 20S α
subunit, in addition to mitochondria (indicated by TOM20 expression) and CD41, but
lacked TSG101 (putative marker of exosomes) (Figure 1A-E). Using platelets as a
positive control, we assessed proteasome function in these PEVs. Proteasome-
associated trypsin-, caspase- and chymotrypsin-like activities were detectable in
platelets and significatively increased in the PEV fraction, but were undetectable
after treatment with epoxomicin, a proteasome inhibitor (Figure 1F). Visualization of
immunogold-labelled proteasome 20S α subunit by transmission electron
microscopy confirmed the presence of proteasomes in PEVs (Figure 1G). These

231
data suggest the catalytically active proteasome was transferred to PEVs upon their
release from platelets.
LWA300 is a conjugate between epoxomicin and BODIPY FL fluorophore that
generates an activity-based, plasma membrane-permeable inhibitor that can identify
the proteasome in cells.779,780 Using LWA300, we detected and quantified active
proteasome-containing PEVs directly in the platelet secretome.779,780 High-sensitivity
flow cytometry (Hs-FCM) confirmed PEV heterogeneity following platelet activation
by thrombin (Figure 1H). Approximately 16.66.5% of the larger (i.e. 500–900nm)
PEVs781 contained proteasome whereas smaller vesicles (i.e. less than 500nm) had
no detectable proteasome (Figure 1H and Supplementary figure 1). The detection
specificity of proteasome-containing PEVs by hs-FCM was confirmed using a
combination of controls. We confirmed efficient competition of the LWA300 probe by
unlabelled epoxomicin, and we determined the particulate nature and membrane
moiety of proteasome-containing PEVs, as they were respectively pelleted by
ultracentrifugation and sensitive to detergent treatment (Figure 1I-J). Confocal
microscopic visualization of platelets as positive controls, and PEVs from thrombin-
activated platelets labelled with LWA300 revealed that both platelets and a
subpopulation of PEVs contained active proteasome (Figure 1K).
Hs-FCM was further used to characterize proteasome-containing PEVs in terms of
surface markers and mitochondrial content. Approximately half of the proteasome-
containing PEVs exposed phosphatidylserine while the vast majority expressed
surface P-selectin (Supplementary figure 2A). Furthermore, 68.37.8% of the
proteasome-containing PEVs also contained mitochondria (Supplementary
figure 2A). Investigation of the mechanisms underlying release of active
proteasome-positive PEVs revealed that the total number of PEVs (with and without
proteasomes) were significantly reduced in the presence of actin inhibitors
(cytochalasins B, D, E and latrunculin A) but not by the tubulin polymerization
inhibitor nocodazole (Supplementary figure 2B). Proteasome release in PEVs was
not unique to thrombin stimulation as ADP, cross-linked collagen related peptide

232
(CRP-XL) and heat-aggregated IgG (HA-IgG) also triggered release of proteasome-
containing PEVs (Supplementary figure 2C).
Identification of proteasome-containing PEVs under physiological and
pathological conditions
The presence of proteasome-containing PEVs was assessed under conditions
conducive to platelet activation and PEV release. A mean of 1.82×106
(range:1.13×105 to 8.11×106, n=6) proteasome-containing PEVs/mL were detected
by hs-FCM in the blood of healthy individuals, which corresponded to 2.61.8% of
the total PEVs in blood. PEVs were quantified in platelet concentrates (PCs) known
to have caused ATRs and compared with control PCs that did not induce ATRs.
Given the reported increase in mitochondria-containing PEVs in ATRs,442,540 we also
determined their levels. High levels of proteasome-containing PEVs were found in
all tested PCs (Figure 2A) but the concentrations of proteasome-containing PEVs
(with or without mitochondria) were not significantly elevated in PCs that induced
ATR (Figure 2A). In contrast, compared with controls, the concentrations of
mitochondria-containing PEVs were increased in ATR-associated PCs, consistent
with prior findings.442,540
Transfusion-related acute lung injury (TRALI) is a potentially lethal adverse reaction
that can result from transfusion of PCs.782 Thus, we quantified proteasome-
containing PEVs in murine bronchoalveolar lavages in an inducible TRALI
model.783,784 Proteasome-containing PEVs were detected in bronchoalveolar
lavages from both TRALI and control mice (Figure 2B), however, no significant
difference was observed between the two groups (Figure 2B). This suggests that
proteasome-containing PEVs are not increased during lung inflammation in this
model and therefore may not participate to acute inflammation that characterizes the
pathogenesis.
Our in vitro investigations pointed to the high potency of immune complexes (HA-
IgG) in generating proteasome-containing PEVs (Supplementary figure 2C).
Although mice lack FcγRIIA, this is the only Fcγ receptor expressed by human

233
platelets that is capable of responding to immune complexes.785 Recent findings
indicate that circulating immune complexes stimulate the release of mitochondria-
containing PEVs in mice expressing the FcγRIIA transgene.492,786 Compared with
diluent injected control mice, there were significantly elevated levels of proteasome-
containing PEVs in plasma of mice with immune-complexes challenge (Figure 2C).
These findings confirmed that proteasome-containing PEVs are present under
various physiological and pathological conditions.
Protein processing by proteasome-containing PEVs
In order to study proteasome function in PEVs, we investigated its ability to process
proteins into smaller peptides by assessing their successful loading into the antigen-
binding groove of MHC-I molecules. We confirmed the expression of MHC-I on
resting and thrombin-activated murine platelets and verified whether MHC-I is
maintained on PEVs present in the platelet secretome. We found that washed resting
platelets did not express MHC-I on their surface (Figure 3A-B), however, thrombin
activation led to a significant increase in surface MHC-I expression (Figure 3A-B),
consistent with the reported presence of this molecule in α-granules and its release
upon activation.520,521,787,788 A small proportion (0.930.13%) of the spontaneously
released PEVs expressed MHC-I, but this proportion significantly increased upon
platelet activation with thrombin (means of 4.640.98%).
To determine whether PEV MHC-I can indeed load small peptides, we pulsed PEVs
present in the platelet secretome with the ovalbumin (OVA) peptide SIINFEKL and
monitored its association with MHC-I molecules using the 25D1.16 monoclonal
antibody, which specifically recognizes MHC-I/SIINFEKL complexes.789 Similarly
with platelets, PEVs loaded the SIINFEKL peptide onto their MHC-I molecules
(Figure 3C-D). Native OVA was also efficiently processed by platelets and the
SIINFEKL peptide was loaded in MHC-I (Figure 3C-E), consistent with prior work.520
We found that an average of 2.53±0.74% of CD41+ PEVs pulsed with the peptide
and 1.83±0.24% of CD41+ PEVs pulsed with OVA (n=18) were positive for 25D.1.16.
Of interest, incubation of native OVA with PEVs resulted in proteolysis of the former
and retrieval of the SIINFEKL peptide from MHC-I molecules expressed by the

234
PEVs. Taken together, the data show that PEVs can process native proteins into
smaller peptides thereby enabling antigen presentation through MHC-I.
Proteasome-containing PEVs can reach lymphoid organs and circulate through
the lymphatic system
Intravenously injected PEVs have a limited circulation time in human blood, ranging
from 10 min to hours depending on studies.790,791 It is unclear, however, whether
they can reach lymphoid organs. Fluorescently labelled PEVs generated from
activated mouse platelets were intravenously injected into mice and their presence
in blood and different organs was monitored. We could identify free PEVs (unbound
to cells) for up to 2 minutes in blood (Figure 4A and Supplementary figure 4A-C).
PEVs in blood were also mainly found bound to platelets and to leukocytes, mainly
Ly6G+ neutrophils, and to a lesser extent lymphocytes, but were mostly undetectable
by 60min (Figure 4A). Screening of individual PEVs in whole tissue sections in
different organs identified spleen and lymph nodes (popliteal and inguinal) as
primary targets, followed by liver, bone marrow, lungs, and kidneys, while none were
found in brain (Figure 4B-C and Supplementary figure 4D-E). Moreover,
aggregates of PEVs (i.e. larger than 1µm2 and up to 541µm2) were mainly observed
in spleen (mean size 2.84±0.16µm2), popliteal (4.16±0.40µm2) and inguinal
(4.08±0.30µm2) lymph nodes, followed by bone marrow (2.75±0.15µm2), lung
(33.51±7.15µm2) and liver (3.40±0.21µm2). This may reflect their accumulation into
smaller vessels or the internalization of numerous PEVs within single cellular
recipients in these organs (Figure 4B-C and Supplementary figure 4D-E).
Platelet EVs can circulate through the lymphatic system and the levels of PEVs in
lymph are increased in mouse models of atherosclerosis and autoimmune
inflammatory arthritis.535,773,777 Using the lymph from mice, we evaluated whether
PEVs were associated with proteasome and MHC-I molecules. We found that a
fraction of the PEVs in lymph expressed MHC-I (11.22.2%) and contained an active
proteasome (12.03.9%). Remarkably, a detectable proportion (1.60.7%) of the
lymph PEVs contained both proteasome and MHC-I molecules (Figure 4D-E) and

235
this was significant given the substantial number of PEVs in lymph (mean of
2.5×107/mL in mice777).
Proteasome-containing PEVs express lymphocyte co-stimulatory molecules
Efficient stimulation of adaptive immunity requires both recognition of the antigen-
MHC-I complexes by the T-cell receptor (TCR) and the activity of co-stimulatory
molecules. We evaluated whether platelets or proteasome-containing PEVs loaded
with SIINFEKL expressed co-stimulatory molecules in addition to other known PEV
markers displayed by CD41+ Proteasome+ EVs. Compared with PEVs that had
undetectable SIINFEKL loading, both platelets and PEVs present in the platelet
secretome loaded with SIINFEKL (25D1.16-positive) expressed higher levels of
proteasome (Figure 5). Moreover, in contrast to thrombin-activated platelets, where
phosphatidylserine expression is increased when loaded with SIINFEKL, both PEVs
bearing SIINFEKL and those negative for SIINFEKL expressed similar levels of
phosphatidylserine (Figure 5). Furthermore, both platelets and SIINFEKL-bearing
PEVs expressed higher levels of P-selectin, and the co-stimulatory
molecules CD40L, CD40 and OX40L (Figure 5). Thus, among the different subtypes
of PEVs, those with a higher density of antigen–MHC-I complexes show more
abundant expression of lymphocytes co-stimulatory molecules and bear a higher
content of active proteasome.
Proteasome-containing PEVs can support antigen-specific T cell activation
T cells isolated from OT-1 mice100 were co-incubated for 18h with PEVs present in
the platelet secretome that were either pulsed or not with the SIINFEKL peptide or
native OVA. Dendritic cells and platelets were treated similarly as positive controls
and for comparison (Figure 6A). The T cells (CD3+CD8+) were then washed and the
expression of CD40, OX40, IL-2 and IFN-γ was evaluated to assess T-cell activation.
Compared with DCs and platelets, PEVs could induce a significant release of IFN-γ
when pulsed with the OVA peptide, whereas native OVA led to an increase in IFN-γ

236
but did not reach statistical significance (Figure 6B). Moreover, DCs and, to a lesser
extent, platelets and PEVs were only capable of inducing significant CD40
expression by T lymphocytes previously pulsed with the OVA peptide (Figure 6C).
In contrast, OX40 and IL-2 expression were not induced by DCs, platelets or PEVs
under these experimental conditions (Figure 6D-6E).
Whether PEVs could stimulate T cell proliferation, a hallmark response by the
lymphocyte antigen-MHC-I complex was evaluated. T cells from OT-1 mice were
labelled with CFSE to monitor cellular division and co-incubated for 5 days with either
DCs, activated platelets or PEVs, which were either pulsed or not with either
SIINFEKL or native OVA (Figure 7A). Lymphoproliferation would be represented by
a decrease in the mean fluorescence intensity histogram, i.e. a dilution of CFSE
fluorescence. (Figure 7B).
As expected, we found that the proportion of lymphoproliferative cells was
significantly higher when OT-1 T lymphocytes were incubated with peptide- or native
OVA-pulsed DCs or activated platelets (Figure 7B-C). Of particular note is that PEVs
also supported T cell proliferation when pulsed with either the SIINFEKL or native
OVA (Figure 7B-C). In addition, when PEVs present in the platelet secretome were
removed from pulsed conditions by ultracentrifugation, no proliferation was
observed, confirming that the pulsed proteins alone, or the platelet secretome devoid
of PEVs, cannot support proliferation (Figure 7D). Furthermore, inhibition of PEV
proteasome by epoxomicin before pulsing with native OVA inhibited the ability of
PEV to induce T cell proliferation (Figure 7E left panel). The effect was directed
toward PEV proteasome, as addition of epoxomicin prior to peptide pulsing at the
same concentration used on DCs did not inhibit proliferation (Figure 7E right
panel). Thus, PEVs are capable of proteosome-dependent processing of native
proteins, thereby enabling peptide loading onto MHC-I. Platelet EVs express co-
stimulatory molecules, and their interaction with T lymphocytes promotes
lymphocyte cytokine production and proliferation.

237
5 Discussion
Megakaryocytes and platelets are emerging as active players in innate and adaptive
immunity.750,770,771 The platelet’s role in immunity is mainly confined to the blood
circulation, while megakaryocytes are localized in bone marrow and lungs. The latter
location potentially provides the megakaryocyte with more direct access to airborne
pathogens and allergens.772,792,793 In contrast, PEVs can additionally disseminate
into organs and tissues and this may be possibly due to their small dimensions and
the presence of unique surface molecules. In this study, we found that the
proteasome and the necessary machinery to process and present antigens to CD8+
T cells are packaged into PEVs by platelets. Thus, PEVs may extend the immune
functions played by platelets and megakaryocytes outside the confines of the blood.
Platelet EVs are heterogeneous in terms of surface molecules and their platelet-
derived content. The presence of mitochondria within PEVs is well
documented,442,527,540 but it was unknown whether other organelles were also
transferred from the platelet. The proteasome is much more abundant than
mitochondria, at around 800,000 copies per cell794 in contrast to approximately 3–7
mitochondria per platelet.442 Further investigation will be necessary to determine if
the presence of multiple organelles within a single vesicle is the result of a specific
sorting mechanism, or because those vesicles are larger and may have more
storage capacity. Nonetheless, we observed that the release of proteasome-
containing PEVs requires cytoskeleton remodeling via intact actin microfilament
dynamics and that a broad array of platelet agonists induce the release of
proteasome-containing PEVs.442
The presence of an extracellular proteasome has already been documented in
normal human blood, and elevated levels have been found in patients suffering from
autoimmune diseases, sepsis or trauma.795 Moreover, the 20S proteasome core is
present and active within EVs derived from apoptotic endothelial cells and regulates
tertiary lymphoid structure formation, autoantibody production and graft rejection
following transplantation.754 While some evidence supports that a circulating

238
extracellular proteasome may be transported by EVs, we show that EVs of platelet
origin, among the most abundant EVs in blood, do contain the proteasome. We
further suggest, based on our characterization of these EVs, that platelet
microvesicles, not exosomes, contain the proteasome. Consistent with this, mass
spectrometry analysis of the human PEV proteome identified numerous proteasomal
subunits.796-798 These include subunits of the 20S catalytic core and
immunoproteasome subunit (PSMB8), subunits of the 11S and 19S regulator and
the 26S proteasome.796-798 Moreover, with calnexin, calreticulin, ERP57 and ERP29,
other members of the ubiquitin-proteasome pathway were identified in PEVs such
as members of the E1 and E2 ubiquitin-conjugating enzyme family.796,798
Considering the presence of these proteins and the fact that intact ovalbumin needs
to be ubiquitinated for degradation by the proteasome,799 it points to the occurrence
of functional ubiquitination in PEVs. To our knowledge, there is no evidence of
protein TAP-1 and TAP-2 (related to TAP transporter) presence in PEVs. Further
investigations are required to see if the TAP transporter is present in PEVs and/or if
the processing pathway of the antigen differs in extracellular vesicles since there is
no reported endoplasmic reticulum in PEV. Thus, while proteomic data points to
ubiquitin-proteasome system proteins in PEVs, the present work unequivocally
demonstrates its presence and documents that the extracellular proteasome in
PEVs is functional and can contribute to antigen processing.
We used complementary approaches and developed a Hs-FCM-based assay to
detect active proteasome at the single EV level, thereby permitting quantification and
assessment of other molecules expressed by the EVs. In particular, the proteasome-
containing PEVs also expressed MHC-I and co-stimulatory molecules, which
enabled lymphocyte activation/proliferation and cytokine generation. These findings
demonstrate a novel and potentially important role for PEVs in adaptive immunity.
While our work suggests that PEVs may be involved in adaptive immunity through
antigen presentation, it does not necessarily exclude that other cells may release
proteasome-containing EVs capable of playing this role. Indeed, EVs derived from
DCs, B and T lymphocytes, macrophages and NK cells can perform cross-
presentation, suggesting that they also contain the necessary antigen

239
processing machinery.800-805 Further studies will be necessary to determine the
impact and the importance of PEVs as antigen presenting elements.
We identified proteasome-containing PEVs in the blood of healthy donors. As most
PEVs in blood under healthy conditions are suggested to originate from
megakaryocytes,482,806 the latter may also constitutively release proteasome-
containing EVs. Moreover, we found that numerous stimuli of human platelets, as
well as in vivo stimulation of mouse platelets could induce release of proteasomes
in PEVs, suggesting that proteasome release is at least conserved in both humans
and mice and takes place via platelet activation. Furthermore, platelets can actively
induce immunity against the Plasmodium berghei parasite520 and megakaryocytes
can be infected by Dengue virus807 and can also phagocytose E. coli.772 Given their
small size, intact microorganisms may not necessarily be present inside PEVs, but
PEVs might process cytosolic microbial proteins derived from intact
platelets/megakaryocytes that lack the ability to enter the lymphatic system. Thus,
PEVs may be implicated in immune surveillance and might contribute to presentation
of microbial antigens within lymph tissues. Future studies are however needed to
determine whether exposure to PEVs suffices to establish immunity in vivo, such as
to less immunodominant antigens than OVA, or whether co-stimulation by
inflammation or infection are needed to establish sustained immune response.
Self-antigens may also be presented by PEVs. Mitochondria were identified in a
proportion of the proteasome-containing PEVs, and although prior studies showed
that mitochondria-containing PEVs are rare in lymph (0.41±0.25% (n=4) of the PEVs
in mouse lymph contain mitochondria)777 in comparisons to proteasome-containing
PEVs 13.61±4.27% (n=6), these proportions might be augmented in certain
diseases. It would be interesting to determine whether PEVs contribute to the
formation of mitochondrial autoantibodies that are described in autoimmune
diseases, such as systemic lupus erythematosus.808 Furthermore, the presence of
proteasome-containing PEVs in platelet concentrates was not associated with
increased risks of ATR or TRALI in a mouse model. It remains to be verified whether
the presentation of platelet antigens (e.g. CD41 or CD61) by PEVs from PCs might

240
contribute to generation of anti-platelet immunity in transfused recipients, although
this has been shown with megakaryocytes.56 It is also not excluded that PEV might
also participate in other immune responses such as autoantibody production or in
tissue remodeling, or if they could be used a platform for cell-based
vaccines.434,436,437 The cross-presentation of PEVs presented here may allow for
new therapeutic possibilities such as in anti-tumor or anti-viral immunity or to induce
cytotoxic immunity by vaccination.809 For example, PEVs have already been
proposed as antigen carriers for vaccination,810,811 and our results suggest that these
types of PEVs are also endowed with cross-priming properties that offer new
prophylactic or therapeutic vaccination.
Human platelets injected into WT mice circulate less than 2h, in contrast to mouse
platelets transfused into mice that can circulate for several days.812,813 We thus used
mouse PEVs in our transfusion experiments, and yet most were undetectable from
the blood circulation after 15min, pointing to their rapid uptake in surrounding tissues.
In blood, the main absolute cellular target was the platelets, mostly because platelets
outnumber leukocytes, which might suggest that PEVs might recycle molecules back
to platelets. PEVs were also found in bone marrow, consistent with recent findings
that pointed to their role in the stimulation of megakaryocyte biogenesis.778 The main
organs that were targeted were the lymphoid organs. Our findings in mouse lymph
revealed that proteasome-containing PEVs can circulate in the lymphatic system,
potentially explaining their accumulation in lymphoid organs following intravenous
injection. This access to the lymphatic system by proteasome-containing PEVs may
reveal a new immune route for PEVs to reach lymphoid organs or infected tissues.
Our study highlights the diversity of PEVs and supports the concept that different
subtypes of PEVs may play different roles depending on their cargo and tissue
distribution.
Acknowledgments
We are grateful to the blood donors and patients who participated in this study. We
acknowledge the generous technical help provided by Nicolas Tessandier and
Carolanne Gélinas. We are thankful to Julie-Christine Lévesque from the Cytometry

241
and Microscopy platform (CHU de Quebec) and Richard Janvier from the
Microscopy platform (Université Laval).
6 References:
1. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med.
2007;357(24):2482-2494.
2. Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of Innate Immune Responses
by Platelets. Front Immunol. 2019;10:1320.
3. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune cell
interactions in thrombo-inflammation. Res Pract Thromb Haemost. 2020;4(1):23-35.
4. Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat
Rev Immunol. 2011;11(4):264-274.
5. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets
as immune and inflammatory cells. Blood. 2014;123(18):2759-2767.
6. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with
inflammation. J Immunol. 2015;194(12):5579-5587.
7. Cunin P, Nigrovic PA. Megakaryocytes as immune cells. J Leukoc Biol.
2019;105(6):1111-1121.
8. Garraud O, Cognasse F. Are Platelets Cells? And if Yes, are They Immune Cells?
Front Immunol. 2015;6:70.
9. Opperman CM, Sishi BJ. Tumor necrosis factor alpha stimulates p62 accumulation
and enhances proteasome activity independently of ROS. Cell Biol Toxicol. 2015;31(2):83-
94.
10. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser
B Phys Biol Sci. 2009;85(1):12-36.
11. Klockenbusch C, Walsh GM, Brown LM, et al. Global proteome analysis identifies
active immunoproteasome subunits in human platelets. Mol Cell Proteomics.
2014;13(12):3308-3319.
12. Dieude M, Bell C, Turgeon J, et al. The 20S proteasome core, active within apoptotic
exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl
Med. 2015;7(318):318ra200.
13. Shi DS, Smith MC, Campbell RA, et al. Proteasome function is required for platelet
production. J Clin Invest. 2014;124(9):3757-3766.
14. Murai K, Kowata S, Shimoyama T, et al. Bortezomib induces thrombocytopenia by
the inhibition of proplatelet formation of megakaryocytes. Eur J Haematol. 2014;93(4):290-
296.
15. Nayak MK, Kulkarni PP, Dash D. Regulatory role of proteasome in determination of
platelet life span. J Biol Chem. 2013;288(10):6826-6834.
16. Grundler K, Rotter R, Tilley S, et al. The proteasome regulates collagen-induced
platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb Res.
2016;148:15-22.
17. Nayak MK, Kumar K, Dash D. Regulation of proteasome activity in activated human
platelets. Cell Calcium. 2011;49(4):226-232.
18. Grundler Groterhorst K, Mannell H, Pircher J, Kraemer BF. Platelet Proteasome
Activity and Metabolism Is Upregulated during Bacterial Sepsis. Int J Mol Sci. 2019;20(23).

242
19. Gupta N, Li W, Willard B, Silverstein RL, McIntyre TM. Proteasome proteolysis
supports stimulated platelet function and thrombosis. Arterioscler Thromb Vasc Biol.
2014;34(1):160-168.
20. Colberg L, Cammann C, Greinacher A, Seifert U. Structure and function of the
ubiquitin-proteasome system in platelets. J Thromb Haemost. 2020.
21. Yukawa M, Sakon M, Kambayashi J, et al. Purification and characterization of
endogenous protein activator of human platelet proteasome. J Biochem. 1993;114(3):317-
323.
22. Yukawa M, Sakon M, Kambayashi J, et al. Proteasome and its novel endogeneous
activator in human platelets. Biochem Biophys Res Commun. 1991;178(1):256-262.
23. Boegel S, Lower M, Bukur T, Sorn P, Castle JC, Sahin U. HLA and proteasome
expression body map. BMC Med Genomics. 2018;11(1):36.
24. Semple JW, Speck ER, Milev YP, Blanchette V, Freedman J. Indirect allorecognition
of platelets by T helper cells during platelet transfusions correlates with anti-major
histocompatibility complex antibody and cytotoxic T lymphocyte formation. Blood.
1995;86(2):805-812.
25. Zufferey A, Speck ER, Machlus KR, et al. Mature murine megakaryocytes present
antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv.
2017;1(20):1773-1785.
26. Chapman LM, Aggrey AA, Field DJ, et al. Platelets present antigen in the context of
MHC class I. J Immunol. 2012;189(2):916-923.
27. Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization
of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J
Proteomics. 2014;101:130-140.
28. Iannacone M, Sitia G, Isogawa M, et al. Platelets mediate cytotoxic T lymphocyte-
induced liver damage. Nat Med. 2005;11(11):1167-1169.
29. Verschoor A, Neuenhahn M, Navarini AA, et al. A platelet-mediated system for
shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb
and complement C3. Nat Immunol. 2011;12(12):1194-1201.
30. Maouia A, Rebetz J, Kapur R, Semple JW. The Immune Nature of Platelets Revisited.
Transfus Med Rev. 2020.
31. Marcoux G, Laroche A, Espinoza Romero J, Boilard E. Role of platelets and
megakaryocytes in adaptive immunity. Platelets. 2020:1-12.
32. Pariser DN, Hilt ZT, Ture SK, et al. Lung megakaryocytes are immune modulatory
cells. J Clin Invest. 2020.
33. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and
disease. Platelets. 2017;28(3):214-221.
34. Puhm F, Boilard E, Machlus KR. Platelet Extracellular Vesicles: Beyond the Blood.
Arterioscler Thromb Vasc Biol. 2020:ATVBAHA120314644.
35. Siljander PR. Platelet-derived microparticles - an updated perspective. Thromb Res.
2011;127 Suppl 2:S30-33.
36. Marcoux G, Magron A, Sut C, et al. Platelet-derived extracellular vesicles convey
mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse
reactions. Transfusion. 2019;59(7):2403-2414.
37. Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria serving
as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation.
Blood. 2014;124(14):2173-2183.

243
38. Burnouf T, Chou ML, Goubran H, Cognasse F, Garraud O, Seghatchian J. An
overview of the role of microparticles/microvesicles in blood components: Are they
clinically beneficial or harmful? Transfus Apher Sci. 2015;53(2):137-145.
39. Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis
via collagen-dependent microparticle production. Science. 2010;327(5965):580-583.
40. Gyorgy B, Szabo TG, Turiak L, et al. Improved flow cytometric assessment reveals
distinct microvesicle (cell-derived microparticle) signatures in joint diseases. PLoS One.
2012;7(11):e49726.
41. Tessandier N, Melki I, Cloutier N, et al. Platelets Disseminate Extracellular Vesicles
in Lymph in Rheumatoid Arthritis. Arterioscler Thromb Vasc Biol.
2020:ATVBAHA119313698.
42. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular
vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell
Vesicles. 2016;5:31427.
43. French SL, Butov KR, Allaeys I, et al. Platelet-derived extracellular vesicles infiltrate
and modify the bone marrow during inflammation. Blood Adv. 2020;4(13):3011-3023.
44. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-
mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-
derived membrane vesicles. Blood. 2008;111(10):5028-5036.
45. Yari F, Motefaker M, Nikougoftar M, Khayati Z. Interaction of Platelet-Derived
Microparticles with a Human B-Lymphoblast Cell Line: A Clue for the Immunologic
Function of the Microparticles. Transfus Med Hemother. 2018;45(1):55-61.
46. Sadallah S, Amicarella F, Eken C, Iezzi G, Schifferli JA. Ectosomes released by
platelets induce differentiation of CD4+T cells into T regulatory cells. Thromb Haemost.
2014;112(6):1219-1229.
47. Dinkla S, van Cranenbroek B, van der Heijden WA, et al. Platelet microparticles
inhibit IL-17 production by regulatory T cells through P-selectin. Blood. 2016;127(16):1976-
1986.
48. Verdoes M, Florea BI, Menendez-Benito V, et al. A fluorescent broad-spectrum
proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol.
2006;13(11):1217-1226.
49. Raz V, Raz Y, Paniagua-Soriano G, et al. Proteasomal activity-based probes mark
protein homeostasis in muscles. J Cachexia Sarcopenia Muscle. 2017;8(5):798-807.
50. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release
two types of membrane vesicles: microvesicles by surface shedding and exosomes derived
from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94(11):3791-
3799.
51. Semple JW, Rebetz J, Kapur R. Transfusion-associated circulatory overload and
transfusion-related acute lung injury. Blood. 2019;133(17):1840-1853.
52. Kapur R, Kim M, Rebetz J, et al. Gastrointestinal microbiota contributes to the
development of murine transfusion-related acute lung injury. Blood Adv. 2018;2(13):1651-
1663.
53. Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect against
transfusion-related acute lung injury via IL-10. Blood. 2017;129(18):2557-2569.
54. McKenzie SE, Taylor SM, Malladi P, et al. The role of the human Fc receptor Fc
gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol.
1999;162(7):4311-4318.

244
55. Cloutier N, Allaeys I, Marcoux G, et al. Platelets release pathogenic serotonin and
return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S
A. 2018;115(7):E1550-E1559.
56. Melki I, Allaeys I, Tessandier N, et al. Platelets release mitochondrial antigens in
systemic lupus erythematosus. Sci Transl Med. 2021;13(581).
57. Angenieux C, Dupuis A, Gachet C, de la Salle H, Maitre B. Cell surface expression
of HLA I molecules as a marker of young platelets. J Thromb Haemost. 2019;17(9):1511-
1521.
58. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME,
Corona-de la Pena NA, Salazar MI. Dengue Virus Induces the Release of sCD40L and
Changes in Levels of Membranal CD42b and CD40L Molecules in Human Platelets. Viruses.
2018;10(7).
59. Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization,
quantitation, and in situ detection of specific peptide-MHC class I complexes using a
monoclonal antibody. Immunity. 1997;6(6):715-726.
60. Rand ML, Wang H, Bang KW, Packham MA, Freedman J. Rapid clearance of
procoagulant platelet-derived microparticles from the circulation of rabbits. J Thromb
Haemost. 2006;4(7):1621-1623.
61. Rank A, Nieuwland R, Crispin A, et al. Clearance of platelet microparticles in vivo.
Platelets. 2011;22(2):111-116.
62. Lefrancais E, Ortiz-Munoz G, Caudrillier A, et al. The lung is a site of platelet
biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105-109.
63. Campbell RA, Schwertz H, Hottz ED, et al. Human megakaryocytes possess intrinsic
antiviral immunity through regulated induction of IFITM3. Blood. 2019;133(19):2013-2026.
64. Marcoux G, Duchez AC, Rousseau M, et al. Microparticle and mitochondrial release
during extended storage of different types of platelet concentrates. Platelets. 2017;28(3):272-
280.
65. Princiotta MF, Finzi D, Qian SB, et al. Quantitating protein synthesis, degradation,
and endogenous antigen processing. Immunity. 2003;18(3):343-354.
66. Sixt SU, Dahlmann B. Extracellular, circulating proteasomes and ubiquitin -
incidence and relevance. Biochim Biophys Acta. 2008;1782(12):817-823.
67. Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet
microparticle proteome. J Proteome Res. 2005;4(5):1516-1521.
68. Dean WL, Lee MJ, Cummins TD, Schultz DJ, Powell DW. Proteomic and functional
characterisation of platelet microparticle size classes. Thromb Haemost. 2009;102(4):711-
718.
69. Capriotti AL, Caruso G, Cavaliere C, Piovesana S, Samperi R, Laganà A. Proteomic
characterization of human platelet-derived microparticles. Anal Chim Acta. 2013;776:57-63.
70. Benaroudj N, Tarcsa E, Cascio P, Goldberg AL. The unfolding of substrates and
ubiquitin-independent protein degradation by proteasomes. Biochimie. 2001;83(3-4):311-
318.
71. Chen Z, Larregina AT, Morelli AE. Impact of extracellular vesicles on innate
immunity. Curr Opin Organ Transplant. 2019;24(6):670-678.
72. Lindenbergh MFS, Stoorvogel W. Antigen Presentation by Extracellular Vesicles
from Professional Antigen-Presenting Cells. Annu Rev Immunol. 2018;36:435-459.
73. Lindenbergh MFS, Wubbolts R, Borg EGF, van 't Veld EM, Boes M, Stoorvogel W.
Dendritic cells release exosomes together with phagocytosed pathogen; potential

245
implications for the role of exosomes in antigen presentation. J Extracell Vesicles.
2020;9(1):1798606.
74. Federici C, Shahaj E, Cecchetti S, et al. Natural-Killer-Derived Extracellular
Vesicles: Immune Sensors and Interactors. Front Immunol. 2020;11:262.
75. Zeng F, Morelli AE. Extracellular vesicle-mediated MHC cross-dressing in immune
homeostasis, transplantation, infectious diseases, and cancer. Semin Immunopathol.
2018;40(5):477-490.
76. Noulsri E. Effects of Cell-Derived Microparticles on Immune Cells and Potential
Implications in Clinical Medicine. Lab Med. 2020.
77. Flaumenhaft R, Dilks JR, Richardson J, et al. Megakaryocyte-derived microparticles:
direct visualization and distinction from platelet-derived microparticles. Blood.
2009;113(5):1112-1121.
78. Gitz E, Pollitt AY, Gitz-Francois JJ, et al. CLEC-2 expression is maintained on
activated platelets and on platelet microparticles. Blood. 2014;124(14):2262-2270.
79. Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue viruses
infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis.
2019;13(11):e0007837.
80. Becker Y, Marcoux G, Allaeys I, et al. Autoantibodies in Systemic Lupus
Erythematosus Target Mitochondrial RNA. Front Immunol. 2019;10:1026.
81. Słomka A, Urban SK, Lukacs-Kornek V, Żekanowska E, Kornek M. Large
Extracellular Vesicles: Have We Found the Holy Grail of Inflammation? Front Immunol.
2018;9:2723.
82. de Jong OG, Kooijmans SAA, Murphy DE, et al. Drug Delivery with Extracellular
Vesicles: From Imagination to Innovation. Acc Chem Res. 2019;52(7):1761-1770.
83. Urbanelli L, Buratta S, Tancini B, et al. The Role of Extracellular Vesicles in Viral
Infection and Transmission. Vaccines (Basel). 2019;7(3).
84. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights
into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21(5):298-312.
85. Sabanovic B, Piva F, Cecati M, Giulietti M. Promising Extracellular Vesicle-Based
Vaccines against Viruses, Including SARS-CoV-2. Biology (Basel). 2021;10(2).
86. Bliss CM, Parsons AJ, Nachbagauer R, et al. Targeting Antigen to the Surface of EVs
Improves the. Mol Ther Methods Clin Dev. 2020;16:108-125.
87. Baker GR, Sullam PM, Levin J. A simple, fluorescent method to internally label
platelets suitable for physiological measurements. Am J Hematol. 1997;56(1):17-25.
88. Blessinger SA, Tran JQ, Jackman RP, et al. Immunodeficient mice are better for
modeling the transfusion of human blood components than wild-type mice. PLoS One.
2020;15(7):e0237106.
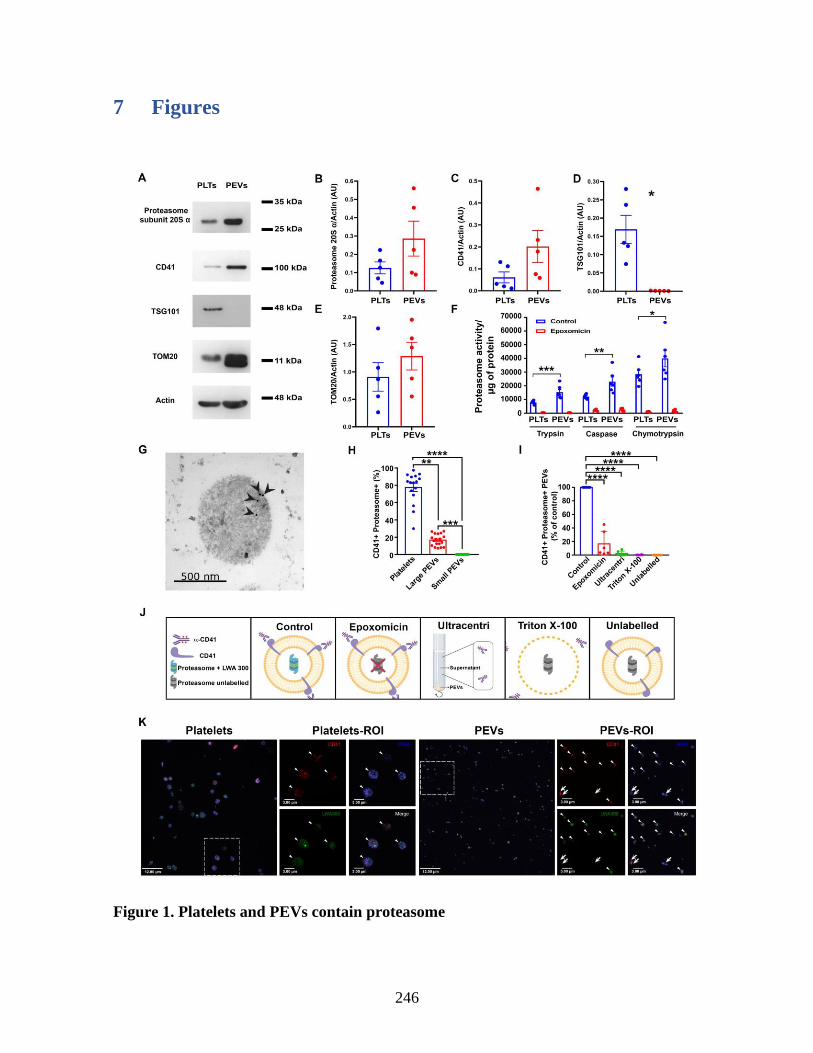
246
7 Figures
Figure 1. Platelets and PEVs contain proteasome
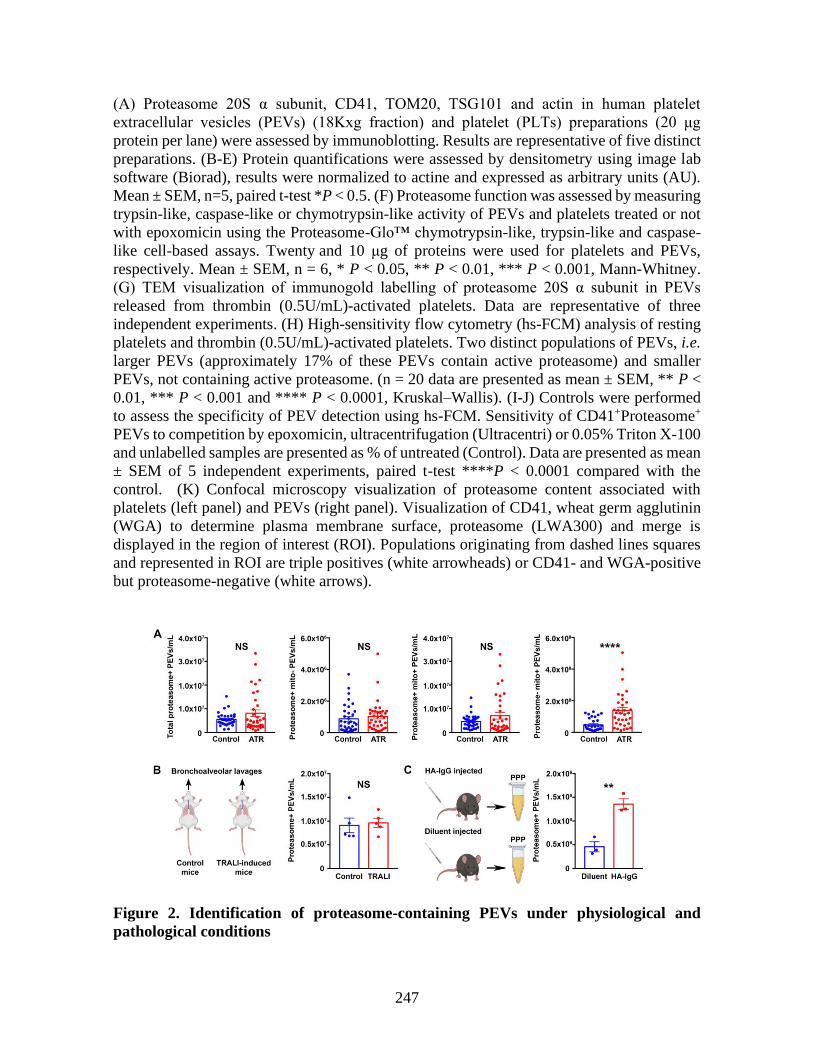
247
(A) Proteasome 20S α subunit, CD41, TOM20, TSG101 and actin in human platelet
extracellular vesicles (PEVs) (18Kxg fraction) and platelet (PLTs) preparations (20 μg
protein per lane) were assessed by immunoblotting. Results are representative of five distinct
preparations. (B-E) Protein quantifications were assessed by densitometry using image lab
software (Biorad), results were normalized to actine and expressed as arbitrary units (AU).
Mean ± SEM, n=5, paired t-test *P < 0.5. (F) Proteasome function was assessed by measuring
trypsin-like, caspase-like or chymotrypsin-like activity of PEVs and platelets treated or not
with epoxomicin using the Proteasome-Glo™ chymotrypsin-like, trypsin-like and caspase-
like cell-based assays. Twenty and 10 μg of proteins were used for platelets and PEVs,
respectively. Mean ± SEM, n = 6, * P < 0.05, ** P < 0.01, *** P < 0.001, Mann-Whitney.
(G) TEM visualization of immunogold labelling of proteasome 20S α subunit in PEVs
released from thrombin (0.5U/mL)-activated platelets. Data are representative of three
independent experiments. (H) High-sensitivity flow cytometry (hs-FCM) analysis of resting
platelets and thrombin (0.5U/mL)-activated platelets. Two distinct populations of PEVs, i.e.
larger PEVs (approximately 17% of these PEVs contain active proteasome) and smaller
PEVs, not containing active proteasome. (n = 20 data are presented as mean ± SEM, ** P <
0.01, *** P < 0.001 and **** P < 0.0001, Kruskal–Wallis). (I-J) Controls were performed
to assess the specificity of PEV detection using hs-FCM. Sensitivity of CD41+Proteasome+
PEVs to competition by epoxomicin, ultracentrifugation (Ultracentri) or 0.05% Triton X-100
and unlabelled samples are presented as % of untreated (Control). Data are presented as mean
± SEM of 5 independent experiments, paired t-test ****P < 0.0001 compared with the
control. (K) Confocal microscopy visualization of proteasome content associated with
platelets (left panel) and PEVs (right panel). Visualization of CD41, wheat germ agglutinin
(WGA) to determine plasma membrane surface, proteasome (LWA300) and merge is
displayed in the region of interest (ROI). Populations originating from dashed lines squares
and represented in ROI are triple positives (white arrowheads) or CD41- and WGA-positive
but proteasome-negative (white arrows).
Figure 2. Identification of proteasome-containing PEVs under physiological and
pathological conditions

248
(A) Proteasome-containing PEVs detected by hs-FCM are found in PFP from platelet
concentrates that have caused adverse transfusion reaction (ATR) in recipients and in control
concentrates that did not induce ATR. The total number of proteasome-containing PEVs
(containing or not mitochondria (mito)), proteasome+mito-PEVs or proteasome+mito+PEVs
does not significantly differ between control and ATR, while proteasome- mito+ PEVs are
increased in ATR (no adverse reaction group [n = 33] vs. adverse reaction group [n = 34]
matched in terms of storage duration; data are presented as mean ± SEM, NS non-significant,
**** P < 0.0001, Student's t-test). (B) Proteasome-containing PEVs detected by hs-FCM are
found in bronchoalveolar lavages from mice after induction of transfusion related acute lung
injury (TRALI) with 34-1-2s and AF6-88.5.5.3 antibody and in control mice (n = 5, data are
presented as mean ± SEM, NS non-significant, Student's t-test). (C) Proteasome-containing
PEVs are detected at significantly higher levels in mice 1-hour post i.v. injection of HA-IgG
vs. control (diluent) mice. (n = 3, **P< 0.01, data are presented as mean ± SEM, Student’s t-
test)
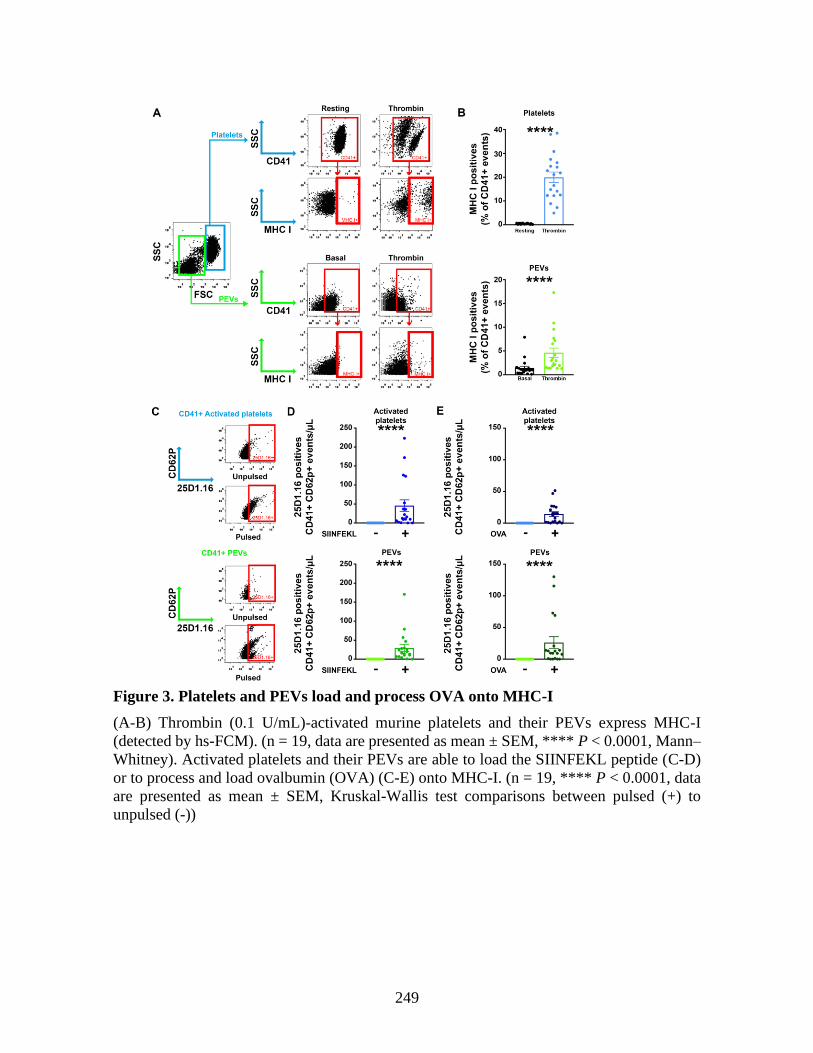
249
Figure 3. Platelets and PEVs load and process OVA onto MHC-I
(A-B) Thrombin (0.1 U/mL)-activated murine platelets and their PEVs express MHC-I
(detected by hs-FCM). (n = 19, data are presented as mean ± SEM, **** P < 0.0001, Mann–
Whitney). Activated platelets and their PEVs are able to load the SIINFEKL peptide (C-D)
or to process and load ovalbumin (OVA) (C-E) onto MHC-I. (n = 19, **** P < 0.0001, data
are presented as mean ± SEM, Kruskal-Wallis test comparisons between pulsed (+) to
unpulsed (-))
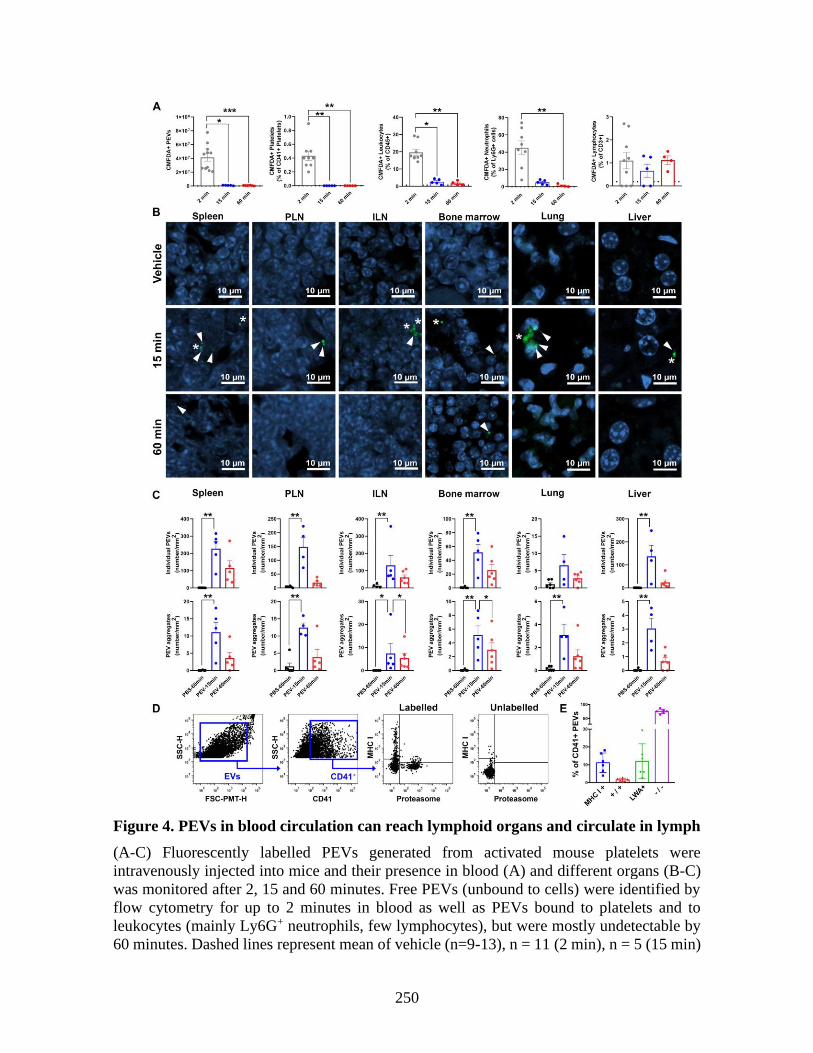
250
Figure 4. PEVs in blood circulation can reach lymphoid organs and circulate in lymph
(A-C) Fluorescently labelled PEVs generated from activated mouse platelets were
intravenously injected into mice and their presence in blood (A) and different organs (B-C)
was monitored after 2, 15 and 60 minutes. Free PEVs (unbound to cells) were identified by
flow cytometry for up to 2 minutes in blood as well as PEVs bound to platelets and to
leukocytes (mainly Ly6G+ neutrophils, few lymphocytes), but were mostly undetectable by
60 minutes. Dashed lines represent mean of vehicle (n=9-13), n = 11 (2 min), n = 5 (15 min)

251
and n = 6 (60 min), data are presented as mean ± SEM, * P < 0.05, ** P < 0.01, *** P <
0.001, Kruskal-Wallis). (B) Representative images of CMFDA-labelled (Green) individual
PEVs (White arrowhead) and PEV aggregates (* white asterisk) in whole tissue sections
(Spleen, popliteal LN (PLN), inguinal LN (ILN), bone marrow, lungs and liver) at 15 and 60
min by confocal microscopy, nuclei (Hoeschst 3342) are in blue. Results are representative
of observations made in 5-6 mice per group. (C) PEVs and aggregates were quantified using
5 different sections for lymph nodes (PLN and ILN) (representing a total surface of at least
1.5mm2), 8 zones of 500,000 µm2 each, randomly assigned on 2 different sections for femurs
(total surface of 4 mm2) and 10 zones of 500,000 µm2 each, randomly assigned on 2 different
sections for lungs, spleen, kidneys and brain and 1 section for liver (total surface of 5 mm2)
using Zen 3.3 software. (n=6 (PBS 60min), 5 (15 min) and 6 (60 min, data are presented as
mean ± SEM, * P < 0.05, ** P < 0.01, Kruskal-Wallis). (D-E) PEVs in lymph were detected
by hs-FCM. (D) Gating strategy to analyze expression of MHC-I and proteasome (LWA300)
on CD41+ EVs in lymph and representative dot plot of labelled and unlabelled (CD41 only)
lymph. (E) Expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph was
determined. +/+ double positive and −/− double negative for MHC-I and proteasome. (n = 6,
data are presented as mean ± SEM).
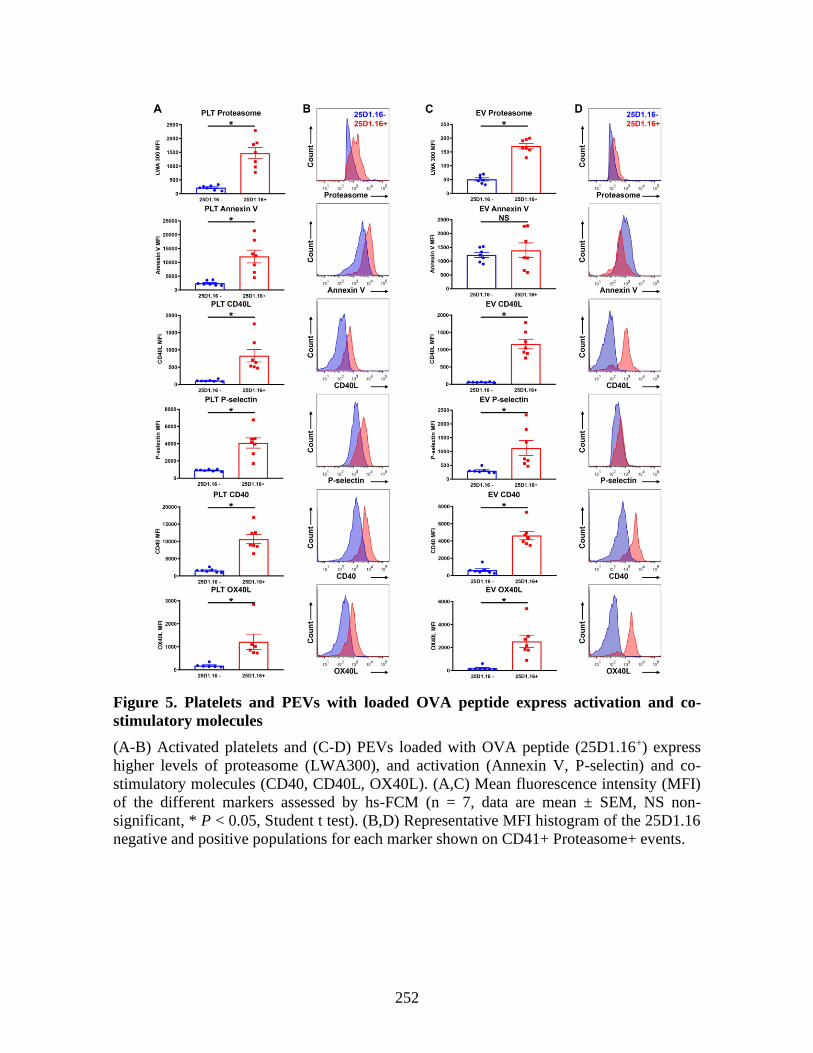
252
Figure 5. Platelets and PEVs with loaded OVA peptide express activation and co-
stimulatory molecules
(A-B) Activated platelets and (C-D) PEVs loaded with OVA peptide (25D1.16+) express
higher levels of proteasome (LWA300), and activation (Annexin V, P-selectin) and co-
stimulatory molecules (CD40, CD40L, OX40L). (A,C) Mean fluorescence intensity (MFI)
of the different markers assessed by hs-FCM (n = 7, data are mean ± SEM, NS non-
significant, * P < 0.05, Student t test). (B,D) Representative MFI histogram of the 25D1.16
negative and positive populations for each marker shown on CD41+ Proteasome+ events.
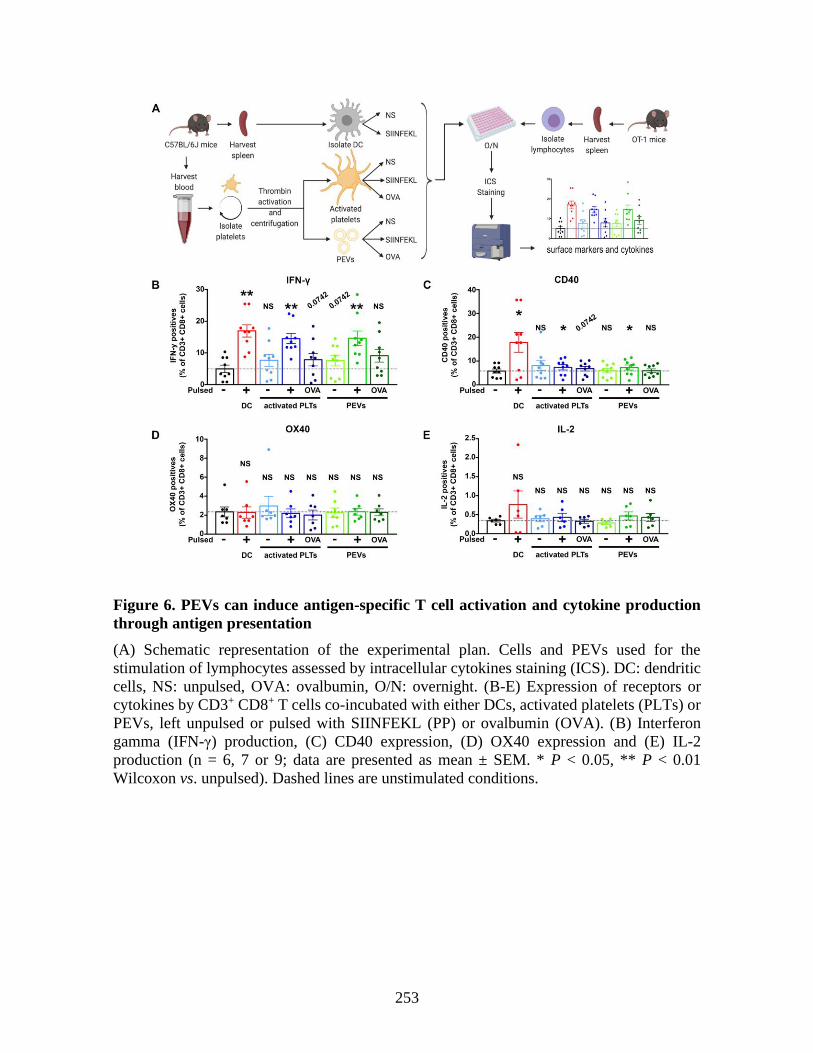
253
Figure 6. PEVs can induce antigen-specific T cell activation and cytokine production
through antigen presentation
(A) Schematic representation of the experimental plan. Cells and PEVs used for the
stimulation of lymphocytes assessed by intracellular cytokines staining (ICS). DC: dendritic
cells, NS: unpulsed, OVA: ovalbumin, O/N: overnight. (B-E) Expression of receptors or
cytokines by CD3+ CD8+ T cells co-incubated with either DCs, activated platelets (PLTs) or
PEVs, left unpulsed or pulsed with SIINFEKL (PP) or ovalbumin (OVA). (B) Interferon
gamma (IFN-γ) production, (C) CD40 expression, (D) OX40 expression and (E) IL-2
production (n = 6, 7 or 9; data are presented as mean ± SEM. * P < 0.05, ** P < 0.01
Wilcoxon vs. unpulsed). Dashed lines are unstimulated conditions.
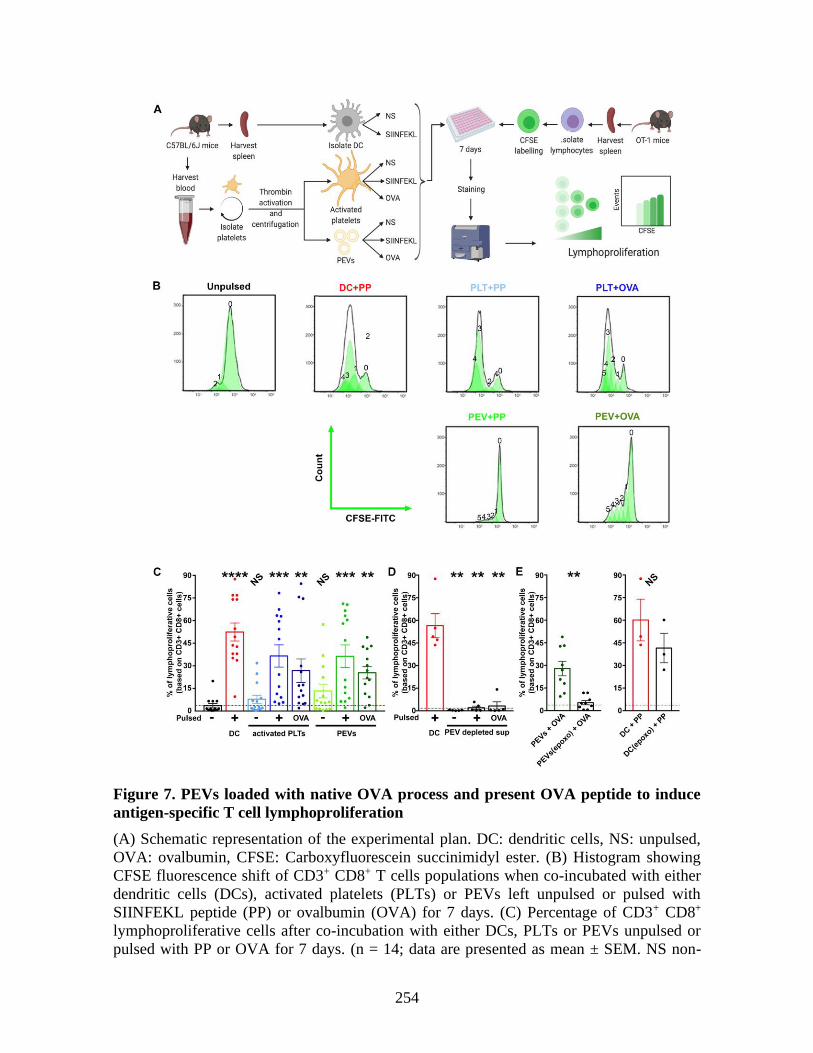
254
Figure 7. PEVs loaded with native OVA process and present OVA peptide to induce
antigen-specific T cell lymphoproliferation
(A) Schematic representation of the experimental plan. DC: dendritic cells, NS: unpulsed,
OVA: ovalbumin, CFSE: Carboxyfluorescein succinimidyl ester. (B) Histogram showing
CFSE fluorescence shift of CD3+ CD8+ T cells populations when co-incubated with either
dendritic cells (DCs), activated platelets (PLTs) or PEVs left unpulsed or pulsed with
SIINFEKL peptide (PP) or ovalbumin (OVA) for 7 days. (C) Percentage of CD3+ CD8+
lymphoproliferative cells after co-incubation with either DCs, PLTs or PEVs unpulsed or
pulsed with PP or OVA for 7 days. (n = 14; data are presented as mean ± SEM. NS non-

255
significant, ** P < 0.01, *** P < 0.001, ****P < 0.0001, Friedman test followed by Dunn’s
post-test for multiple comparisons to unpulsed). (D) Percentage of CD3+ CD8+
lymphoproliferative cells after 7 days co-incubation with either PP pulsed DCs or supernatant
(surn) depleted of PEVs by ultracentrifugation, left unpulsed or pulsed with PP or OVA.
(n=5; data are presented as mean ± SEM, ** P < 0.01, Mann-Whitney vs. unpulsed). (E)
Proportion of CD3+ CD8+ lymphoproliferative cells after 7 days co-incubation with OVA-
pulsed PEVs treated or not with epoxomicin (epoxo) for 2 hours and PP-pulsed DCs (DC +
PP) treated or not with epoxomicin (epoxo). (n = 9 for PEVs and n = 3 for DC; data are mean
± SEM, NS non-significant, ** P < 0.01, Wilcoxon). Dashed lines are unstimulated
conditions.
Copyright © 2022 FDOKUMEN




















