
Epsilon Protein Kinase C Mediated Ischemic Tolerance Requires Activation of the Extracellular...
10
Epsilon Protein Kinase C Mediated Ischemic Tolerance Requires Activation of the Extracellular Regulated Kinase Pathway in the Organotypic Hippocampal Slice *Christian Lange-Asschenfeldt, *Ami P. Raval, *Kunjan R. Dave, †Daria Mochly-Rosen, *Thomas J. Sick, and *Miguel A. Pérez-Pinzón From the *Cerebral Vascular Disease Research Center, Department of Neurology and Neuroscience, University of Miami School of Medicine, Miami, Florida, U.S.A.; and the †Department of Molecular Pharmacology, Stanford University School of Medicine, Stanford, California, U.S.A. Summary: Ischemic preconditioning (IPC) promotes brain tolerance against subsequent ischemic insults. Using the orga- notypic hippocampal slice culture, we conducted the present study to investigate (1) the role of adenosine A 1 receptor (A 1 AR) activation in IPC induction, (2) whether epsilon protein kinase C (PKC) activation after IPC is mediated by the phos- phoinositol pathway, and (3) whether PKC protection is me- diated by the extracellular signal-regulated kinase (ERK) path- way. Our results demonstrate that activation of A 1 AR emulated IPC, whereas blockade of the A 1 AR during IPC diminished neuroprotection. The neuroprotection promoted by the A 1 AR was also reduced by the PKC antagonist. To determine wheth- er PKC activation in IPC and A 1 AR preconditioning is me- diated by activation of the phosphoinositol pathway, we incu- bated slices undergoing IPC or adenosine treatment with a phosphoinositol phospholipase C inhibitor. In both cases, pre- conditioning neuroprotection was significantly attenuated. To further characterize the subsequent signal transduction pathway that ensues after PKC activation, mitogen-activated protein kinase kinase was blocked during IPC and pharmacologic pre- conditioning (PPC) (with PKC, NMDA, or A 1 AR agonists). This treatment significantly attenuated IPC- and PPC-induced neuroprotection. In conclusion, we demonstrate that PKC ac- tivation after IPC/PPC is essential for neuroprotection against oxygen/glucose deprivation in organotypic slice cultures and that the ERK pathway is downstream to PKC. Key Words: Metabolism—In vitro culture—Adenosine receptors— Anoxia—Tolerance—Signal transduction. It is now well-established that brief ischemic/anoxic episodes may render brain, heart, and other tissues tol- erant against more severe subsequent ischemic insults, which is a phenomenon that is referred to as ischemic preconditioning (IPC). Robust IPC neuroprotection has been studied extensively for more than a decade in a variety of in vivo and in vitro models (Kato et al., 1992; Kitagawa et al., 1990; Schurr et al., 1986). The specific triggering mechanisms leading to this state of tolerance still remain unknown. However, stud- ies from different laboratories suggest that IPC-mediated neuroprotection is achieved by activation of NMDA (Best et al., 1996; Gonzalez-Zulueta et al., 2000; Kato et al., 1992; Pringle et al., 1996, 1997; Raval et al., 2003) and the adenosine A 1 receptor (A 1 AR) (Blondeau et al., 2000; Cohen et al., 2000; Di-Capua et al., 2003; Heur- teaux et al., 1995; Hiraide et al., 2001; Perez-Pinzon et al., 1996; Reshef et al., 2000). Although the NMDA pathway is better understood, the adenosine pathway has been less characterized. Be- cause both NMDA and A 1 receptors have different ef- fects in the CA1 region of the hippocampus, namely post-synaptic excitation (NMDA) and pre-synaptic inhi- bition (A 1 AR), the signal transduction pathway that en- sues after activation of these two receptors after IPC remains undefined. Recent evidence demonstrates that a common signal transduction pathway for these two dis- parate receptors involves the translocation of PKC in organotypic hippocampal slices (Raval et al., 2003) and primary rat neuronal cultures (Di-Capua et al., 2003). Received August 19, 2003; final version received December 16, 2003; accepted January 15, 2004. C.L-A. and A.P.R. contributed equally to this study. Supported by PHS grants NS34773, NS05820, NS38276 and AHA 0225227B Florida/Puerto Rico affiliate. Address correspondence and reprint requests to Miguel A. Pérez- Pinzón, Department of Neurology (D4-5), P.O. Box 016960, University of Miami School of Medicine, Miami, FL 33101; e-mail: [email protected]. Journal of Cerebral Blood Flow & Metabolism 24:636–645 © 2004 The International Society for Cerebral Blood Flow and Metabolism Published by Lippincott Williams & Wilkins, Baltimore 636 DOI: 10.1097/01.WCB.0000121235.42748.BF
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Epsilon Protein Kinase C Mediated Ischemic Tolerance Requires Activation of the Extracellular...

Epsilon Protein Kinase C Mediated Ischemic Tolerance RequiresActivation of the Extracellular Regulated Kinase Pathway in the
Organotypic Hippocampal Slice
*Christian Lange-Asschenfeldt, *Ami P. Raval, *Kunjan R. Dave, †Daria Mochly-Rosen,*Thomas J. Sick, and *Miguel A. Pérez-Pinzón
From the *Cerebral Vascular Disease Research Center, Department of Neurology and Neuroscience, University of Miami Schoolof Medicine, Miami, Florida, U.S.A.; and the †Department of Molecular Pharmacology, Stanford University School of Medicine,
Stanford, California, U.S.A.
Summary: Ischemic preconditioning (IPC) promotes braintolerance against subsequent ischemic insults. Using the orga-notypic hippocampal slice culture, we conducted the presentstudy to investigate (1) the role of adenosine A1 receptor(A1AR) activation in IPC induction, (2) whether epsilon proteinkinase C (�PKC) activation after IPC is mediated by the phos-phoinositol pathway, and (3) whether �PKC protection is me-diated by the extracellular signal-regulated kinase (ERK) path-way. Our results demonstrate that activation of A1AR emulatedIPC, whereas blockade of the A1AR during IPC diminishedneuroprotection. The neuroprotection promoted by the A1ARwas also reduced by the �PKC antagonist. To determine wheth-er �PKC activation in IPC and A1AR preconditioning is me-diated by activation of the phosphoinositol pathway, we incu-
bated slices undergoing IPC or adenosine treatment with aphosphoinositol phospholipase C inhibitor. In both cases, pre-conditioning neuroprotection was significantly attenuated. Tofurther characterize the subsequent signal transduction pathwaythat ensues after �PKC activation, mitogen-activated proteinkinase kinase was blocked during IPC and pharmacologic pre-conditioning (PPC) (with �PKC, NMDA, or A1AR agonists).This treatment significantly attenuated IPC- and PPC-inducedneuroprotection. In conclusion, we demonstrate that �PKC ac-tivation after IPC/PPC is essential for neuroprotection againstoxygen/glucose deprivation in organotypic slice cultures andthat the ERK pathway is downstream to �PKC. Key Words:Metabolism—In vitro culture—Adenosine receptors—Anoxia—Tolerance—Signal transduction.
It is now well-established that brief ischemic/anoxicepisodes may render brain, heart, and other tissues tol-erant against more severe subsequent ischemic insults,which is a phenomenon that is referred to as ischemicpreconditioning (IPC). Robust IPC neuroprotection hasbeen studied extensively for more than a decade in avariety of in vivo and in vitro models (Kato et al., 1992;Kitagawa et al., 1990; Schurr et al., 1986).
The specific triggering mechanisms leading to thisstate of tolerance still remain unknown. However, stud-ies from different laboratories suggest that IPC-mediated
neuroprotection is achieved by activation of NMDA(Best et al., 1996; Gonzalez-Zulueta et al., 2000; Kato etal., 1992; Pringle et al., 1996, 1997; Raval et al., 2003)and the adenosine A1 receptor (A1AR) (Blondeau et al.,2000; Cohen et al., 2000; Di-Capua et al., 2003; Heur-teaux et al., 1995; Hiraide et al., 2001; Perez-Pinzon etal., 1996; Reshef et al., 2000).
Although the NMDA pathway is better understood,the adenosine pathway has been less characterized. Be-cause both NMDA and A1 receptors have different ef-fects in the CA1 region of the hippocampus, namelypost-synaptic excitation (NMDA) and pre-synaptic inhi-bition (A1AR), the signal transduction pathway that en-sues after activation of these two receptors after IPCremains undefined. Recent evidence demonstrates that acommon signal transduction pathway for these two dis-parate receptors involves the translocation of �PKC inorganotypic hippocampal slices (Raval et al., 2003) andprimary rat neuronal cultures (Di-Capua et al., 2003).
Received August 19, 2003; final version received December 16,2003; accepted January 15, 2004.
C.L-A. and A.P.R. contributed equally to this study.Supported by PHS grants NS34773, NS05820, NS38276 and AHA
0225227B Florida/Puerto Rico affiliate.Address correspondence and reprint requests to Miguel A. Pérez-
Pinzón, Department of Neurology (D4-5), P.O. Box 016960, Universityof Miami School of Medicine, Miami, FL 33101; e-mail:[email protected].
Journal of Cerebral Blood Flow & Metabolism24:636–645 © 2004 The International Society for Cerebral Blood Flow and MetabolismPublished by Lippincott Williams & Wilkins, Baltimore
636 DOI: 10.1097/01.WCB.0000121235.42748.BF

It is also necessary to define the signal transductionpathway that ensues after �PKC activation. Previousstudies in the heart demonstrated that activation of �PKCafter IPC both in vivo and in vitro protects against isch-emia via activation of extracellular regulated kinase(ERK 1/2) and c-Jun N-terminal kinase (JNK) membersof the mitogen-activated protein kinase (MAPK) family(Li et al., 2000; Ping et al., 1999a,b; Punn et al., 2000).In cortical neurons, the signal transduction pathway afterIPC includes the activation of the p21 (RAS)/ERK path-way (Gonzalez-Zulueta et al., 2000). However, it re-mains to be determined whether �PKC and theRAS/ERK pathway are linked together or are two dif-ferent pathways that promote IPC tolerance in the brain.
The goals of the present study are (1) to elucidatewhether A1AR activation is key in the organotypic hip-pocampal slice model of IPC, (2) to determine whether�PKC translocation because of IPC and adenosine recep-tor activation is mediated by the phosphoinositol path-way, and (3) to determine whether there is a link between�PKC translocation and ERK in preconditioning medi-ated neuroprotection or whether these two pathways areindependent of each other.
MATERIALS AND METHODS
Preparation of organotypic slice culturesAll protocols were approved by the Animal Care and Use
Committee of the University of Miami. Neonatal (9–11 daysold) Sprague-Dawley rats were anesthetized by intraperitonealinjection of ketamine (1.0 mg/pup). Animals were decapitated,and the brains were quickly removed. Organotypic hippocam-pal slice cultures were prepared as described previously (Ravalet al., 2003; Xu et al., 2002). In summary, transverse slices (400�m) were dissected from the hippocampi and placed in Gey’sBalanced Salt Solution (Sigma, St. Louis, MO, U.S.A.) supple-mented with 6.5 mg/mL glucose at 4°C. After 1 hour, two sliceswere placed onto one 30-mm diameter membrane insert (Mil-licell-CM, Millipore, Bedford, MA, U.S.A.), and inserts weretransferred to six-well culture plates with 1 mL of culture me-dium per well. Culture medium consisted of 50% MinimumEssential Medium, 25% Hank’s Balanced Salt Solution, 25%Heat Inactivated Horse Serum (Sigma) supplemented with 6.5mg/mL glucose and 1 mM glutamine. Slice cultures were in-cubated (equilibrated at 36°C, 95% O2, 5% CO2, humidity100%) for 14 to 15 days before experiments were performed.
Induction of ischemiaOur ischemia and preconditioning protocols have been de-
fined in previous studies (Raval et al., 2003; Xu et al., 2002).For oxygen/glucose deprivation (OGD), slices were washedthree times with aglycemic Hank’s Balanced Salt Solution(AHBSS) (pH 7.4) of the following constitution: CaCl2 · 2H2O1.26 mM, KCl 5.37 mM, KH2PO4 0.44 mM, MgCl2 0.49 mM,MgSO4 · 7H2O 0.41 mM, NaCl 136.9 mM, NaHCO3 4.17 mM,Na2HPO4 · 7H2O 0.34 mM, and sucrose 15 mM (Sigma). Theslices were then transferred into an airtight chamber, which wasequilibrated with 95% N2/5% CO2 gas (preheated to 37°C andwater saturated) that was blown through the chamber for 5minutes (4 L/min) to achieve anoxic conditions. Then thechamber was sealed and remained incubated for 10 minutes
(for a total of 15 minutes for preconditioning) or 35 minutes(for a total of 40 minutes for ischemic insult). After OGD,slices were placed back in the incubator in plates containing thenormal culture medium.
Assessment of neuronal cell deathTo determine the extent of neuronal damage, we used the
propidium iodide (PI) method (Raval et al., 2003; Xu et al.,2002). Slices were incubated in culture medium supplementedwith 2 �g/mL PI (Sigma) for 1 hour. Images were taken usingan inverted fluorescence microscope (Olympus IX 50),equipped with a light intensifying SPOT CCD camera (Diag-nostic Instruments Inc., Sterling Heights, MI, U.S.A.) andSPOT Advanced software. Images of the cultured slices weretaken as follows: (1) as baseline before the preconditioningprocedure, (2) 24 hours after the preconditioning, (3) 24 hoursafter the test ischemic insult to assess ischemic damage, and (4)24 hours after NMDA treatment to assess maximum damage toneuronal cells. The hippocampal CA1 subfield was chosen asregion of interest, and quantification was performed usingScion Image software. The percentage of relative optical inten-sity (ROI) served as an index of neuronal cell death. Relativecell death was calculated from each ROI as follows: Relative %cell death � (Fexp − Fmin)/(Fmax − Fmin) × 100, where Fexpis the fluorescence of the test condition, Fmax is maximumfluorescence (100 �M NMDA treatment for 1 hour), and Fminis background fluorescence (before preconditioning or OGD).In all groups, experiments (except Western blot analysis) wereterminated by superfusing slices with an overdose of NMDA(100 �M) 24 hours after the end of the experiments to deter-mine total number of neurons using the PI technique (Fmaxabove).
Cell fractionation and Western blot analysisTo determine whether �PKC isozyme was translocated after
IPC in organotypic slices, we used Western blot analysis. Thismethod is adapted from one that was described previously(Mackay and Mochly-Rosen, 2001) with some modifications(Raval et al., 2003). Hippocampal organotypic slices were fro-zen in liquid nitrogen at different time intervals and stored at−80°C until the analysis. At the time of Western blot analysisthe hippocampal organotypic slice cultures were separatedfrom the supporting membrane. For one sample, 32 slices werepooled together. Thus, for Western blot analysis, n � 1 rep-resents 32 slices. Also, each slice was obtained from differentpups. The pooled slices were washed twice with cold phosphatebuffered saline (PBS). Slices were pelleted at 1,000×g and PBSwas removed. The pellet was resuspended in 400 �L cell lysisbuffer (4 mM ATP, 100 mM KCl, 10 mM imidazole, 2 mMEGTA, 1 mM MgCl2, 20% glycerol, 0.05% Triton X-100, 17�g/mL PMSF, 20 �g/mL soybean trypsin inhibitor, 25 �g/mLleupeptin, and 25 �g/mL aprotinin). The suspended slices werehomogenized using all glass homogenizer. The homogenatewas then centrifuged at 4°C at 1000×g for 10 minutes. Thesupernatant (soluble fraction) was carefully taken off and re-centrifuged at 16,000×g for 15 minutes to get rid of any con-taminating pellet material. The initial pellet was resuspended in250 �L cell lysis buffer containing 1% Triton X-100 and wasextracted on ice for 60 minutes. Samples were centrifuged at16,000×g for 15 minutes. The supernatant is the particulatefraction. Soluble and particulate fractions were analyzed forprotein content by the Bradford’s assay and 40 �g protein fromeach fraction separated by 12% SDS-PAGE. Each group con-sisted of four samples, and each sample was prepared from 32slices. Protein was transferred to Immobilon-P (Millipore, MA,U.S.A.) membrane and incubated with the primary antibody
ISCHEMIC PRECONDITIONING AND �PKC SIGNALING 637
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

anti-�PKC (Calbiochem, La Jolla, CA, U.S.A.) (1: 500). Im-munoreactivity was detected using enhanced chemilumines-cence (ECL Western blotting detection kit, Amershampharma-cia biotech, UK). Western blot images were digitized at 8-bitprecision by means of a charge-coupled-device-based (CCD)camera (8–12 bit, Xillix Technologies Corp., Vancouver,Canada) equipped with a 55-mm Micro-Nikkor lens (Nikon,Japan). The camera was interfaced to an advanced image-analysis system (MCID Model M2, Imaging Research, Inc., St.Catherines, Ont., Canada). The digitized immunoblots weresubjected to densitometric analysis using MCID software.
Peptide preparation and deliveryThe �V1–2 (�PKC inhibitor, amino acids 14–21
[EAVSLKPT]) (Gray et al., 1997) and ��RACK (�PKC acti-vator, amino acids 85–92 [HDAPIGYD]) (Dorn et al., 1999)peptides were synthesized at Stanford’s Protein and NucleicAcid facility and conjugated to Tat (carrier peptide, aminoacids 47–57 [YGRKKRRQRRR]) (Schwarze et al., 1999) via acysteine-cysteine bond at the N termini, as previously described(Chen et al., 2001). These peptides are conjugated via disulfidebond to short cell permeable peptides (Souroujon and Mochly-Rosen, 1998). Our previous study clearly showed efficacy ofthese peptides in the organotypic slice cultures (Raval et al.,2003).
Experimental designThe organotypic slices were divided into five major groups:
Group I: Sham. Slices were incubated for 15 minutes in HBSSsolution supplied with an equimolar concentration of glucoseinstead of sucrose (Sham-IPC) and were subsequently trans-ferred back to regular media. After 48 hours, the same pro-cedure was performed for an extended duration of 40 min-utes (sham OGD-ischemia).
Group II: Test ischemia. Sham IPC was induced as in Group Ifollowed after 48 hours by test ischemia (40 minutes ofOGD).
Group III: IPC. Slices underwent IPC (15 minutes of OGD)and, 48 hours later, test ischemia (40 minutes of OGD).
Group IV: Pharmacologic preconditioning (PPC) by selectivepharmacologic agents hypothesized to emulate IPC. Slicesunderwent PPC and, 48 hours later, test ischemia (40 min-utes of OGD)
Group V: Pharmacologic blockade of IPC.
IPC and PPC experiments were performed in the presence ofinhibitors of (A) �PKC, (B) phospholipase C (PLC), and (C)mitogen-activated protein kinase kinase (MAPKK) during andafter the preconditioning procedure. The slices were subjectedto test ischemia 48 hours later.
For Western blot analysis of �PKC translocation, slices werefrozen 30 minutes after IPC, 30 minutes after R-PIA, and 60minutes after ��RACK (an �PKC agonist) treatments.
To provide better controls in these experiments, all six-wellplates used contained at least one well for the sham, IPC, andischemia groups. In addition, for statistical purposes, each in-sert had two slices obtained from two different pups. Thusevery n � 1 (one slice) represents a different animal.
ChemicalsR-PIA (N6-(R)-Phenylisopropyladenosine) (A1AR agonist),
DPCPX (1,3 Dipropyl-8-cyclopentylxanthine) (A1AR antago-nist), DMPX (3,7-Dimethyl-1-propargylxanthine) (A2AR an-tagonist), U-73122 (1-(6-[(17�)]-3-Methoxyestra-1, 3,5 [10]-trien-17-yl) amino] hexyl)-1H-pyrrole-2, 5-dione) (PI-PLCinhibitor), and PD-98059 (2–2-Amino-3-methoxyphenyl)-4H-
1-benzopyran-4-one) (MAPKK inhibitor) were all purchasedfrom Sigma, St. Louis, MO, U.S.A.
Statistical analysisThe results are expressed as mean ± SD. Statistical signifi-
cance was determined with an ANOVA test, followed by aBonferroni’s post hoc test.
RESULTS
Confirming our previous findings, IPC was neuropro-tective against test ischemia in hippocampal organotypicslices.
To test whether IPC in hippocampal organotypic slicewas mediated by the A1AR, slices were superfused with10 �M of the selective A1AR antagonist DPCPX for theduration of IPC. This treatment diminished the neuro-protection afforded by IPC (P < 0.05). PI fluorescencevalues after the test ischemic insult were 55.39 ± 16.71%(n � 17) for untreated slices, 22.64 ± 19.32% (n � 12)after IPC, and 50.98 ± 17.23% (n � 12) after combinedIPC and DPCPX treatment (Fig. 1A).
As control experiments, we compared PI fluorescencevalues between sham IPC, IPC (followed by sham isch-emia), and IPC + DPCPX (followed by sham ischemia).PI fluorescence values for sham IPC, IPC, and IPC +DPCPX were determined at 24 and 48 hours. Further-more, in these experiments, PI fluorescence values weredetermined in the same groups 24 hours after sham isch-emia, which represents 72 hours after the initial insult. PIfluorescence values 24, 48, and 72 hours after sham IPCwere 3.15 ± 0.78, 3.33 ± 1.47, and 4.11 ± 0.75 (n � 4),respectively. PI fluorescence values 24, 48, and 72 hoursafter IPC were 2.97 ± 1.16, 3.06 ± 1.37, and 4.29 ± 1.24,respectively (n � 4). PI fluorescence values 24, 48, and72 hours after IPC + DPCPX were 3.38 ± 2.18, 3.97 ±2.53, and 5.37 ± 3.32, respectively (n � 5). No signifi-cant differences in PI fluorescence values were foundbetween sham IPC, IPC (followed by sham ischemia),and IPC + DPCPX groups at any of these times.
To further confirm the role of A1AR during IPC, weexamined whether IPC could be emulated by the selec-tive A1AR agonist R-PIA. Slices were superfused withR-PIA (at two different concentrations, 50 nM and 100�M) for 15 minutes, and 48 hours later, the test ischemicinsult was induced. Only pre-treatment with 100 �MR-PIA significantly reduced PI fluorescence in hippo-campal slices after test ischemia (R-PIA, 25.17 ±17.52%, n � 18, vs. test ischemia, 55.39 ± 16.71%, n �17) (P < 0.001) (Fig. 1A). 50 nM R-PIA concentrationwas not effective in reducing PI fluorescence after testischemia (62.89 ± 31.81, n � 10). PI fluorescence values24, 48, and 72 hours after R-PIA treatment alone were3.06 ± 0.60, 2.56 ± 1.15, and 4.96 ± 2.31, respectively(n � 6). These values were not significantly differentthan shams or IPC (followed by sham ischemia) groups.
C. LANGE-ASSCHENFELDT ET AL.638
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

Because 100 �M of R-PIA might also activate theA2AR, we performed an additional set of experiments todetermine whether this receptor was involved in the R-PIA induced preconditioning. Slices were subjected topharmacologic preconditioning with 100 �M R-PIA inpresence of the selective A2AR antagonist DMPX (10�M) for 15 minutes, and 48 hours later, test ischemiawas performed. Compared with ischemia (68.06 ± 16.47,n � 5), both R-PIA treatment alone (30.89 ± 17.47%,n � 5) and R-PIA + DMPX (39.46 ± 21.92%, n � 4)exhibited reduction of PI-fluorescence (Fig. 1B) (P <0.05). There was no significant difference in PI-fluorescence between the R-PIA and the R-PIA + DMPXgroups. These results suggest that IPC in rat organotypichippocampal slices requires A1AR activation. However,we cannot rule out the putative role of the A2AR in the
ischemic or R-PIA-induced preconditioning neuropro-tection in the organotypic hippocampal slices. As controlexperiments, we determined PI fluorescence after R-PIAand DMPX treatment alone. PI fluorescence values 24,48, and 72 hours after R-PIA + DMPX were 2.23 ± 1.33,3.27 ± 1.46, and 4.49 ± 2.16, respectively (n � 4). Thesevalues were not significantly different from shams orIPC (followed by sham ischemia) groups.
To test the hypothesis of whether A1AR induced neu-roprotection is mediated by activation/translocation of�PKC in the organotypic hippocampal slice, as we pre-viously reported for IPC- and NMDA-induced precondi-tioning (Raval et al., 2003), we inhibited �PKC with�V1–2 (20 nM, dosage determined in Raval et al., 2003)during R-PIA (100 �M) induced preconditioning, and 48hours later, test ischemia was induced. Neuroprotectionpromoted by R-PIA (20.23 ± 13.17%, n � 8) was sig-nificantly reduced in presence of the �PKC antagonist(52.46 ± 20.04%, n � 7) (P < 0.01), indicating an es-sential role of �PKC in A1AR induced ischemic toler-ance (Fig. 2).
Activation of �PKC is typically determined by trans-location of �PKC from the soluble fraction to particulatefraction (Dorn et al., 1999). To determine that all pre-conditioning treatments promoted the translocation of�PKC, we performed Western blot analysis in slices thatunderwent IPC or PPC (Fig. 3). When compared with
FIG. 1. (A) PI fluorescence values measured in the CA1 pyra-midal cells in rat organotypic hippocampal slices 1 day afterOGD. The selective A1AR antagonist DPCPX reduced IPC-induced neuroprotection. Selective A1AR agonist R-PIA emu-lated IPC. (B) A2AR did not significantly contribute to R-PIA me-diated ischemic tolerance; the selective A2AR antagonist DMPXwas superfused during the R-PIA-induced preconditioning 48hours before a test ischemic insult. Significance as comparedwith ischemia, aP < 0.05, and IPC + DPCPX, bP < 0.05. PI,propidium iodide; OGD, oxygen/glucose deprivation; A1 AR,adenosine A1 receptor; IPC, ischemic preconditioning; A2 AR,adenosine A2 receptor.
FIG. 2. The A1AR-induced ischemic tolerance was mediated byactivation of �PKC in rat organotypic hippocampal slices. Neuro-protection promoted by the selective A1AR agonist R-PIA wasreduced when the selective �PKC antagonist peptide �V1–2 wasadministered simultaneously. Significance as compared withischemia, aP < 0.01. A1 AR, adenosine A1 receptor; �PKC, epsi-lon protein kinase C.
ISCHEMIC PRECONDITIONING AND �PKC SIGNALING 639
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004
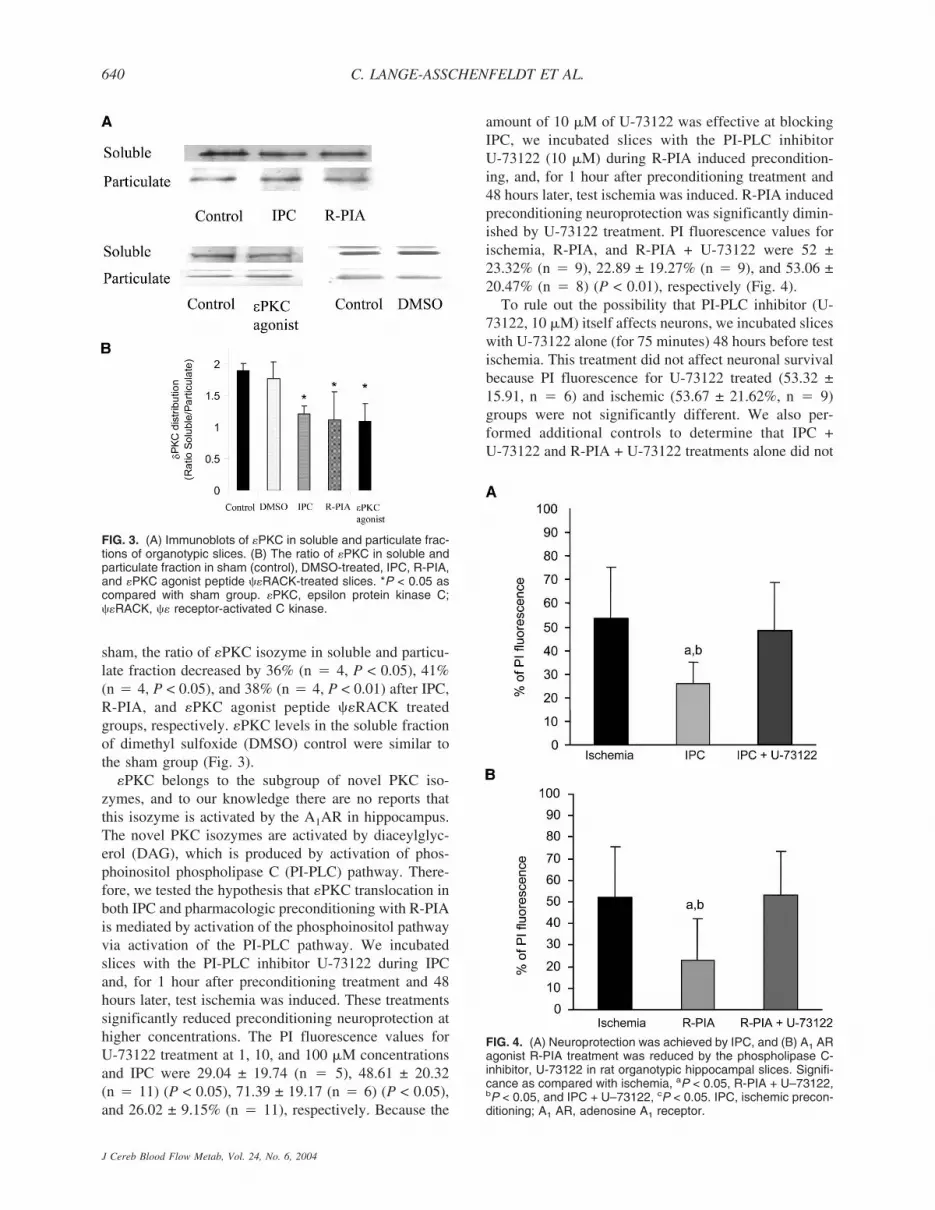
sham, the ratio of �PKC isozyme in soluble and particu-late fraction decreased by 36% (n � 4, P < 0.05), 41%(n � 4, P < 0.05), and 38% (n � 4, P < 0.01) after IPC,R-PIA, and �PKC agonist peptide ��RACK treatedgroups, respectively. �PKC levels in the soluble fractionof dimethyl sulfoxide (DMSO) control were similar tothe sham group (Fig. 3).
�PKC belongs to the subgroup of novel PKC iso-zymes, and to our knowledge there are no reports thatthis isozyme is activated by the A1AR in hippocampus.The novel PKC isozymes are activated by diaceylglyc-erol (DAG), which is produced by activation of phos-phoinositol phospholipase C (PI-PLC) pathway. There-fore, we tested the hypothesis that �PKC translocation inboth IPC and pharmacologic preconditioning with R-PIAis mediated by activation of the phosphoinositol pathwayvia activation of the PI-PLC pathway. We incubatedslices with the PI-PLC inhibitor U-73122 during IPCand, for 1 hour after preconditioning treatment and 48hours later, test ischemia was induced. These treatmentssignificantly reduced preconditioning neuroprotection athigher concentrations. The PI fluorescence values forU-73122 treatment at 1, 10, and 100 �M concentrationsand IPC were 29.04 ± 19.74 (n � 5), 48.61 ± 20.32(n � 11) (P < 0.05), 71.39 ± 19.17 (n � 6) (P < 0.05),and 26.02 ± 9.15% (n � 11), respectively. Because the
amount of 10 �M of U-73122 was effective at blockingIPC, we incubated slices with the PI-PLC inhibitorU-73122 (10 �M) during R-PIA induced precondition-ing, and, for 1 hour after preconditioning treatment and48 hours later, test ischemia was induced. R-PIA inducedpreconditioning neuroprotection was significantly dimin-ished by U-73122 treatment. PI fluorescence values forischemia, R-PIA, and R-PIA + U-73122 were 52 ±23.32% (n � 9), 22.89 ± 19.27% (n � 9), and 53.06 ±20.47% (n � 8) (P < 0.01), respectively (Fig. 4).
To rule out the possibility that PI-PLC inhibitor (U-73122, 10 �M) itself affects neurons, we incubated sliceswith U-73122 alone (for 75 minutes) 48 hours before testischemia. This treatment did not affect neuronal survivalbecause PI fluorescence for U-73122 treated (53.32 ±15.91, n � 6) and ischemic (53.67 ± 21.62%, n � 9)groups were not significantly different. We also per-formed additional controls to determine that IPC +U-73122 and R-PIA + U-73122 treatments alone did not
FIG. 3. (A) Immunoblots of �PKC in soluble and particulate frac-tions of organotypic slices. (B) The ratio of �PKC in soluble andparticulate fraction in sham (control), DMSO-treated, IPC, R-PIA,and �PKC agonist peptide ��RACK-treated slices. *P < 0.05 ascompared with sham group. �PKC, epsilon protein kinase C;��RACK, �� receptor-activated C kinase.
FIG. 4. (A) Neuroprotection was achieved by IPC, and (B) A1 ARagonist R-PIA treatment was reduced by the phospholipase C-inhibitor, U-73122 in rat organotypic hippocampal slices. Signifi-cance as compared with ischemia, aP < 0.05, R-PIA + U–73122,bP < 0.05, and IPC + U–73122, cP < 0.05. IPC, ischemic precon-ditioning; A1 AR, adenosine A1 receptor.
C. LANGE-ASSCHENFELDT ET AL.640
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

promote any significant cell death. PI fluorescence val-ues 24, 48, and 72 hours after IPC and U-73122 were3.16 ± 1.82, 3.53 ± 2.02, and 4.68 ± 1.56, respectively(n � 6). PI fluorescence values 24, 48, and 72 hoursafter R-PIA and U-73122 were 3.00 ± 0.72, 3.48 ± 1.05,and 5.23 ± 2.45, respectively (n � 6). These values werenot significantly different from sham or IPC (followedby sham ischemia) groups.
Because ERK pathway has been implicated in IPCneuroprotection in cortical culture cells (Gonzalez-Zulutea et al., 2000; Shamloo et al., 1999) and PKCappears to be an upstream signaling pathway to ERK, wetested the hypothesis that there is a link between trans-location of �PKC and activation of ERK after IPC. Forthat purpose, ERK activation was blocked with the mi-togen-activated protein kinase (MAPKK) inhibitor (PD-98059). Inhibition of MAPKK by PD-98059 is a goodapproach to inhibit the ERK pathway because PD-98059
prevented activation of MAPKK and subsequent phos-phorylation of ERK both in vitro and in intact cells (Dud-ley et al., 1995). The organotypic slices were incubatedwith PD-98059 (20, 50, and 100 �M) during IPC, and 48hours later, test ischemia was induced. The PI fluores-cence values for different concentrations of the MAPKKinhibitor were 66.14 ± 15.90% (n � 10), 68.54 ± 17.26(n � 7), and 69.94 ± 27.24% (n � 8) for 20, 50, and 100�M of PD-98059 treatment, respectively. All concentra-tions reduced IPC-induced neuroprotection (P < 0.001).The treatment at 20 �M concentration was used for fur-ther studies because this low dosage is advantageous inreducing the effects of PD-98059 on NMDA receptors(Pereira et al., 2002). The PI fluorescence values were30.11 ± 9.68% (n � 5), 66.14 ± 15.90% (n � 10), and61.50 ± 11.83% (n � 13) for IPC, IPC + PD-98059treated, and test ischemic groups, respectively (Fig. 5A).As control experiments, we determined that IPC +
FIG. 5. (A) The selective MAPKK antagonist PD-98059 reduced neuroprotection when superfused during IPC in rat organotypic hippo-campal slices; significance as compared with ischemia, aP < 0.001, and IPC + PD-98059, bP < 0.001. (B) The selective MAPKK antagonistPD-98059 reduced ��RACK-mediated neuroprotection; significance as compared with ischemia (aP < 0.001 and ��RACK + PD-98059,bP < 0.001. (C) Pharmacologic neuroprotection afforded by NMDA was reduced by selective MAPKK antagonist PD-98059; significanceas compared with ischemia, aP < 0.001, and NMDA + PD-98059, bP < 0.001. (D) The selective MAPKK antagonist PD-98059 reducedR-PIA mediated neuroprotection when superfused for 15 minutes 48 hours before test ischemic insult; significance as compared withischemia, aP < 0.001, and R-PIA + PD-98059, bP < 0.001. MAPKK, mitogen-activated protein kinase kinase; IPC, ischemic precondi-tioning; ��RACK, �� receptor-activated C kinase.
ISCHEMIC PRECONDITIONING AND �PKC SIGNALING 641
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

PD-98059 treatment alone did not promote any signifi-cant cell death. PI fluorescence values 24, 48, and 72hours after IPC + PD-98059 were 2.07 ± 1.28, 3.58 ±2.20, and 3.93 ± 2.32, respectively (n � 5). These valueswere not significantly different from sham or IPC (fol-lowed by sham ischemia) groups.
To further confirm the link between �PKC transloca-tion and the ERK pathway in IPC-induced neuroprotec-tion, we blocked ERK activation with PD-98059 (20�M) when IPC was emulated by superfusion of the�PKC agonist in organotypic slices. The PI fluorescencevalues for �PKC agonist treatment and �PKC agonisttreatment along with MAPKK inhibitor were 25.01 ±11.57% (n � 8) and 57.00 ± 21.68% (n � 10) (P <0.001), respectively. These results indicate the involve-ment of ERK pathway downstream of �PKC transloca-tion (Fig. 5B).
We have previously demonstrated that the NMDA re-ceptor mediates IPC neuroprotection via �PKC translo-cation (Raval et al., 2003). In the next two experiments,we tested whether the ERK pathway is involved inNMDA and A1AR mediated preconditioning. In the firstexperiment, ERK activation was blocked with PD-98059during NMDA-induced preconditioning. This treatmentsignificantly attenuated the NMDA induced neuroprotec-tion. The PI fluorescence values of NMDA precondition-ing and NMDA + PD-98059 treated groups were 28.07 ±5.52% (n � 4) and 49.08 ± 9.68% (n � 8), respectively(P < 0.001) (Fig. 5C). In the second experiment, ERKactivation was blocked during treatment with R-PIA. ThePI fluorescence values of R-PIA preconditioning and R-PIA + PD-98059 treated groups were 38.24 ± 4.13%(n � 4) and 74.42 ± 13.92% (n � 14), respectively(P < 0.001) (Fig. 5D).
DISCUSSION
The present study demonstrated that the A1AR is in-volved in the triggering phase of IPC in the organotypichippocampal slice preparation. We previously demon-strated that IPC requires activation of NMDA receptorsin the same model of ischemic preconditioning (Raval etal., 2003). Both findings are supported by previous stud-ies in other models of cerebral ischemia. For example, invitro studies from cortical cell cultures (Gonzalez-Zulueta et al., 2000; Grabb and Choi, 1999; Tauskela etal., 1999, 2003) and in vivo studies of global cerebralischemia by blocking of NMDA receptors during isch-emic preconditioning demonstrated the importance ofNMDA receptor activation in IPC neuroprotection (Bondet al., 1999; Kato et al., 1992). Additional evidence sup-porting the role of NMDA receptors emerged from stud-ies on cortical spreading depression induced precondi-tioning (Douen et al., 2000; Kawahara et al., 1997:Kobayashi et al., 1995; Kunkler and Kraig, 1998; Mat-
sushima et al., 1996). The role of A1AR was also dem-onstrated in vitro in primary neuronal cultures (Di-Capuaet al., 2003; Reshef et al., 2000) and in vivo during globalcerebral ischemia (Heurteaux et al., 1995).
A key finding in the current study is that both trigger-ing factors (i.e., NMDA and A1AR) promote IPC neu-roprotection in the hippocampal CA1 region by a similarsignal transduction pathway. Furthermore, to our knowl-edge for the first time, we also demonstrate that bothreceptors activate �PKC in hippocampus. A puzzle thatemerges from these findings is that both receptors exertopposite actions in the CA1 subregion of the hippocam-pus. For example, the A1AR mediates inhibition of syn-aptic potentiation in the CA1 region of hippocampus byreducing Ca+2 influx through voltage gated channels andreducing pre-synaptic neurotransmitter release (Abbrac-chio and Cattabeni, 1999; Kato et al., 1999; Thompson etal., 1993). In contrast, NMDA receptors in the hippo-campal CA1 region are associated with influx of Ca+2
and increase excitability post-synaptically (Purves et al.,2001). Thus a quandary emerges from the disparate ac-tions of these two receptors in this region of the hippo-campus. How do pre- and post-synaptic terminals cross-talk to promote ischemic tolerance in all hippocampalcells? Further studies in our laboratory are underwayto define putative cross-talk messengers between pre-and post-synaptic terminals in the CA1 region of thehippocampus.
In the present study, we also investigated whetherthese two disparate receptors activate �PKC duringIPC. �PKC belongs to the subgroup of novel, Ca+2-independent PKC-isozymes that are activated by bindingof diacylglycerol (DAG), a second messenger generatedby the phosphoinositol pathway (Lester et al., 2000). It iswell known that NMDA receptor activation promotesCa+2 influx, which in turn can activate phospholipase C(PLC), which in turn generates DAG (Nishizuka, 1992).Raval and colleagues (2003) demonstrate that the DAGanalogue OAG (oleoylacetyl glycerol) emulated IPCneuroprotection. In the present study, we further cor-roborate this pathway when ischemic tolerance was re-duced by PLC inhibition (Fig. 4B). Furthermore, in theheart, it was demonstrated that protection against isch-emia by A1AR-induced preconditioning was linked toincreased PLC activity and DAG turnover (Parsons et al.,2000). These data suggest that IPC in the brain triggeredby A1AR activation is also coupled to stimulation of thephosphoinositol pathway and subsequent �PKC signal-ing. These findings suggest that PLC may be a commonpathway in both pre- and post-synaptic terminals in theCA1 region of the hippocampus.
Taking into account the results of our previous studyin which �PKC also played a key role in NMDA-inducedpreconditioning (Raval et al, 2003), we conjectured thatsignals from different receptors may converge at �PKC,
C. LANGE-ASSCHENFELDT ET AL.642
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

underscoring once more the importance of this isozymefor preconditioning neuroprotection. It is important tonote that other PKC isozymes may play a role in IPC aswell. Translocation of other PKC isozymes, namely �, �,and � after IPC has been reported in the brain (Kurkinenet al., 2001; Shamloo and Wieloch, 1999). However, wecould not get translocation of �PKC or �PKC after IPCin this particular model of IPC (organotypic hippocampalslice cultures) (Raval et al., 2003). It will be importantfor future studies to define whether other PKC isozymesplay any role in IPC by using pharmacologic blockers asthe ones described in the current study or transgenicspecies where different PKC isozymes have beenknocked down.
Because not much is known about the downstreamsignals that ensue after �PKC translocation, we furtherinvestigated whether the ERK pathway was involved in�PKC-induced neuroprotection. Previous studies in theheart demonstrated that activation of �PKC both in vivoand in vitro protected against ischemia via activation ofERK 1/2 and JNK members of the MAPK family (Li etal., 2000; Ping et al., 1999a,b; Punn et al., 2000). Ac-cording to Shizukuda and Buttrick (2001), �PKC medi-ated ERK 1/2 regulation in adult rat ventricular myo-cytes. Furthermore, �PKC was implicated in thedownstream activation of ERK 1/2 during IPC in theheart (Ping et al., 1999a,b). These results support a pu-tative link between �PKC translocation and ERK 1/2activation leading to neuroprotection after IPC.
Although the effector pathways after activation of�PKC-ERK pathways remain undefined in the brain, anessential feature of the activation of �PKC during IPC isits subcellular redistribution and binding with its recep-tor-activated C kinase (RACKs) (Dorn et al., 1999; Kraftand Anderson, 1983; Mochly-Rosen, 1995; Ping et al.,1997, 1999). Subsequent to translocation to specificsites, PKC isozymes exert their effect by phosphorylat-ing specific target proteins. This translocation to the sub-cellular compartments was considered to be an importantmechanism for �PKC to direct downstream signalingcascades and induce protection (Dorn, 1999). One of thesubcellular sites thought to play a role in the induction ofneuroprotection is the mitochondria. Ischemic precondi-tioning has been shown to preserve mitochondrial func-tion in the late window of IPC (Dave et al., 2001; Fryeret al., 2000). There is lack of direct evidence suggestingtranslocation of �PKC to brain mitochondria; however,formation of mitochondrial �PKC-ERK 1/2 modulescoupled to the inactivation of BAD, a pro-apoptotic mol-ecule was demonstrated in the heart (Baines et al., 2002).ERK 1/2 was also reported to phosphorylate the pro-apoptotic protein BAD and dissociate it from BCL-XL.This balance was postulated to prevent apoptosis andenhance cell survival (Scheid et al., 1999). In the brain,similar pro-survival influence after the phosphorylation
of ERK 1/2 has been reported (Gonzalez-Zulueta et al.,2000; Shamloo et al., 1999). Further studies will be re-quired to determine whether �PKC + ERK 1/2 interactsin the same manner in the hippocampus after IPC.
Other pathways involved in �PKC + ERK 1/2-inducedneuroprotection after IPC may involve expression ofneuroprotective genes. For example, activation of ERK1/2 results in phosphorylation of downstream targets in-cluding p90rsk (Wood et al., 1992), as well as severaltranscription factors (Treisman, 1996).
CONCLUSION
We demonstrate here that the ischemic tolerance in-duced by IPC and two of its key triggering factors, theadenosine A1 and NMDA receptors, was mediated by�PKC-ERK pathway via PLC activation. Future studiesare required to define the exact subcellular sites targetedby this signal transduction pathway that leads to thisnaturally intrinsic state of ischemic tolerance in the brain.
REFERENCES
Abbracchio MP, Cattabeni F (1999) Brain adenosine receptors as tar-gets for therapeutic intervention in neurodegenerative diseases.Ann N Y Acad Sci 890:79–92
Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM,Bolli R, Ping P (2002) Mitochondrial PK Cepsilon and MAPKform signaling modules in the murine heart: enhanced mitochon-drial PKC epsilon-MAPK interactions and differential MAPK ac-tivation in PKCepsilon-induced cardioprotection. Circ Res90(4):390–397
Best N, Sunsstrom LE, Mitchell J, Wheal HV (1996) Pre-exposure tosubtoxic levels prevents kainic acid lesions in organotypic hippo-campal slice cultures: effects of kainic acid on parvalbumin-immunoreactive neurons and expression of heat shock protein-72following induction of tolerance. Eur J Neurosci 8:1209–1219
Blondeau N, Plamondon H, Richelme C, Heurteaux C, Lazdunski M(2000) K(ATP) channel openers, adenosine agonists and epilepticpreconditioning are stress signals inducing hippocampal neuropro-tection. Neuroscience 100:465–474
Bond A, Lodge D, Hicks CA, Ward MA, O’Neill MJ (1999) NMDAreceptor antagonism, but not AMPA receptor antagonism attenu-ates induced ischemic tolerance in the gerbil hippocampus. Eur JPharmacol 380:91–99
Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, CavallaroG, Banci L, Guo Y, Bolli R, Dorn GW 2nd, Mochly-Rosen D(2001). Opposing cardioprotective actions and parallel hypertro-phic effects of delta PKC and epsilon PKC. Proc Natl Acad SciU S A 98(20):11114–11119
Cohen MV, Baines CP, Downey JM (2000) Ischemic preconditioning:From adenosine receptor to KATP channel. Annu Rev Physiol62:79–109
Dave KR, Saul I, Busto R, Ginsberg MD, Sick TJ, Perez-Pinzon MA(2001) Ischemic preconditioning preserves mitochondrial functionafter global cerebral ischemia in rat hippocampus. J Cereb BloodFlow Metab 21(12):1401–1410
Di-Capua N, Sperling O, Zoref-Shani E (2003) Protein kinase C-epsilon is involved in the adenosine-activated signal transductionpathway conferring protection against ischemia-reperfusion injuryin primary rat neuronal cultures. J Neurochem 84:409–412
Dorn GW 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ,Csukai M, Wu G, Lorenz JN, Mochly-Rosen D (1999) Sustained invivo cardiac protection by a rationally designed peptide that causesepsilon protein kinase C translocation. Proc Natl Acad Sci U S A96(22):12798–12803
Douen AG, Akiyama K, Hogan MJ, Wang F, Dong L, Chow AK,
ISCHEMIC PRECONDITIONING AND �PKC SIGNALING 643
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

Hakim A (2000) Preconditioning with cortical spreading depres-sion decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2from rat cerebal cortex plasma membranes. J Neurochem 75:812–818
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR (1995) Asynthetic inhibitor of the mitogen-activated protein kinase cascade.Proc Natl Acad Sci U S A 92(17):7686–7689
Fryer RM, Eells JT, Hsu AK, Henry MM, Gross GJ (2000) Ischemicpreconditioning in rats: role of mitochondrial K(ATP) channel inpreservation of mitochondrial function. Am J Physiol Heart CircPhysiol 8(1):H305–312
Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF,Parada LF, Dawson TM, Dawson VL (2000) Requirement fornitric oxide activation of p21 (ras)/extracellular regulated kinase inneuronal ischemic preconditioning. Proc Natl Acad Sci U S A97(1):436–441
Grabb MC, Choi DW (1999) Ischemic tolerance in murine cortical cellculture: Critical role for NMDA receptors. J Neurosci 19:1657–1662
Gray MO, Karliner JS, Mochly-Rosen D (1997) A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytesfrom hypoxia-induced cell death. J Biol Chem 272(49):30945–30951
Heurteaux C, Lauritzen I, Widmann C, Lazdunski M (1995) Essentialrole of adenosine, adenosine A1 receptors, and ATP-sensitive K+
channels in cerebral ischemic preconditioning. Proc Natl Acad SciU S A 92:4666–4670
Hiraide T, Katsura K, Muramatsu H, Asano G, Katayama Y (2001)Adenosine receptor antagonists cancelled the ischemic tolerancephenomenon in gerbil. Brain Res 910(1–2): 94–98
Kato H, Araki T, Murase K, Kogure K (1992) Induction of tolerance toischemia: alterations in second-messenger systems in the gerbilhippocampus. Brain Res Bull 29(5):559–565
Kato K, Li ST, Zorumski CF (1999) Modulation of long-term poten-tiation induction in the hippocampus by N-methyl-D-aspartate-mediated presynaptic inhibition. Neuroscience 92:1261–1272
Kawahara N, Croll SD, Wiegand SJ, Klatzo I (1997) Cortical spreadingdepression induces long-term alterations of BDNF levels in cortexand hippocampus distinct from lesion effects: implications forischemic tolerance. Neurosci Res 29:37–47
Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M,Handa N, Fukunaga R, Kimura K, Mikoshiba K (1990) ‘Ischemictolerance’ phenomenon found in the brain. Brain Res 528(1):21–24
Kobayashi S, Harris VA, Welsh FA (1995) Spreading depression in-duces tolerance of cortical neurons to ischemia in rat brain. J CerebBlood Flow Metab 15(5):721–727
Kraft AS, Anderson WB (1983) Phorbol esters increase the amount ofCa2+, phospholipid-dependent protein kinase associated withplasma membrane. Nature 301(5901):621–623
Kunkler PE, Kraig RP (1998) Calcium waves precede electrophysi-ological changes of spreading depression in hippocampal organcultures. J Neurosci 18(9):3416–3425
Kurkinen K, Busto R, Goldsteins G, Koistinaho J, Perez-Pinzon MA(2001) Isoform-specific membrane translocation of protein kinaseC after ischemic preconditioning. Neurochem Res. 26(10):1139–1144
Lester JW, Hofmann PA (2000) Role for PKC in the adenosine-induceddecrease in shortening velocity of rat ventricular myocytes. Am JPhysiol Heart Circ Physiol 279(6):H2685–2693
Li RC, Ping P, Zhang J, Wead WB, Cao X, Gao J, Zheng Y, Huang S,Han J, Bolli R (2000) PKCepsilon modulates NF-kappaB andAP-1 via mitogen-activated protein kinases in adult rabbit cardio-myocytes. Am J Physiol Heart Circ Physiol 279(4):H1679–1689
Mackay K, Mochly-Rosen D (2001) Arachidonic acid protects neonatalrat cardiac myocytes from ischaemic injury through epsilon proteinkinase C. Cardiovasc Res 50:65–74
Matsushima K, Hogan MJ, Hakim AM (1996) Cortical spreading de-pression protects against subsequent focal cerebral ischemia inrats. J Cereb Blood Flow Metab 16(2):221–226
Mochly-Rosen D (1995) Localization of protein kinases by anchoring
proteins: a theme in signal transduction. Science 268 (5208):247–251
Nishizuka Y (1992) Intracellular signaling by hydrolysis of phospho-lipids and activation of protein kinase C. Science 258(5082):607–614
Parsons M, Young L, Lee JE, Jacobson KA, Liang BT (2000) Distinctcardioprotective effects of adenosine mediated by differential cou-pling of receptor subtypes to phospholipases C and D. FASEB J14(10):1423–1431
Pereira DB, Carvalho AP, Duarte CB (2002) Non-specific effects of theMEK inhibitors PD098,059 and U0126 on glutamate release fromhippocampal synaptosomes. Neuropharmacol 42(1):9–19
Pérez-Pinzón MA, Mumford PL, Rosenthal M, Sick TJ (1996) Anoxicpreconditioning in hippocampal slices: role of adenosine. Neuro-science 75:687–694
Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y,Clerk A, Sugden P, Han J, Bolli R (1999a) PKC-dependent acti-vation of p46/p54 JNKs during ischemic preconditioning in con-scious rabbits. Am J Physiol 277(5 Pt 2):H1771–H1785
Ping P, Zhang J, Cao X, Li RC, Kong D, Tang XL, Qiu Y, Manchi-kalapudi S, Auchampach JA, Black RG, Bolli R (1999b) PKC-dependent activation of p44/p42 MAPKs during myocardial isch-emia-reperfusion in conscious rabbits. Am J Physiol 276(5 Pt2):H1468–1481
Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R(1997) Ischemic preconditioning induces selective translocation ofprotein kinase C isoforms epsilon and eta in the heart of consciousrabbits without subcellular redistribution of total protein kinase Cactivity. Circ Res 81(3):404–414
Pringle AK, Benham CD, Sim L, Kennedy J, Iannotti F, Sundstrom LE(1996) Selective N-type calcium channel antagonist omega cono-toxin MVIIA is neuroprotective against hypoxic neurodegenera-tion in organotypic hippocampal-slice cultures. Stroke 27:2124–2130
Pringle AK, Iannotti F, Wilde GJ, Chad JE, Seeley PJ, Sundstrom LE(1997) Neuroprotection by both NMDA and non-NMDA receptorantagonists in in vitro ischemia. Brain Res 755:36–46
Punn A, Mockridge JW, Farooqui S, Marber MS, Heads RJ (2000)Sustained activation of p42/p44 mitogen-activated protein kinaseduring recovery from simulated ischaemia mediates adaptive cy-toprotection in cardiomyocytes. Biochem J 3:891–899
Purves D, Augustine G, Fitzpatrick D, Katz LC, LaMantia AS, McNa-mara JO, Williams SM (2001) Neurotransmitter receptors and theireffects. In: Neuroscience. Sinauer:Sunderland, MA, pp 141–163
Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Pérez-Pinzón MA(2003) �PKC is required for the induction of tolerance by ischemicand NMDA-mediated preconditioning in the organotypic hippo-campal slice. J Neurosci 23(2):384–391
Reshef A, Sperling O, Zoref-Shani E (2000) The adenosine-inducedmechanism for the acquisition of ischemic tolerance in primary ratneuronal cultures. Pharmacol Ther 87(2–3):151–159
Scheid MP, Schubert KM, Duronio V (1999) Regulation of bad phos-phorylation and association with Bcl-x(L) by the MAPK/Erk ki-nase. J Biol Chem 274(43):31108–31113
Schurr A, Reid KH, Tseng MT, West C, Rigor BM (1986) Adaptationof adult brain tissue to anoxia and hypoxia in vitro. Brain Res374(2):244–248
Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF (1999) In vivoprotein transduction: delivery of a biologically active protein intothe mouse. Science 285(5433):1569–1572
Shamloo M, Rytter A, Wieloch T (1999) Activation of the extracellularsignal-regulated protein kinase cascade in the hippocampal CA1region in a rat model of global cerebral ischemic preconditioning.Neuroscience 93(1):81–88
Shamloo M, Wieloch T (1999) Rapid decline in protein kinaseCgamma levels in the synaptosomal fraction of rat hippocampusafter ischemic preconditioning. Neuroreport 10(5):931–935
Shizukuda Y, Buttrick PM (2001) Protein kinase C(epsilon) modulatesapoptosis induced by beta-adrenergic stimulation in adult rat ven-tricular myocytes via extracellular signal-regulated kinase (ERK)activity. J Mol Cell Cardiol 33(10):1791–1803
C. LANGE-ASSCHENFELDT ET AL.644
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004

Souroujon MC, Mochly-Rosen D (1998) Peptide modulators of pro-tein-protein interactions in intracellular signaling. Nature Biotech-nol 16:919–924
Tauskela JS, Brunette E, Monette R, Comas T, Morley P (2003) Pre-conditioning of cortical neurons by oxygen-glucose deprivation:Tolerance induction through abbreviated neurotoxic signaling. AmJ Physiol Cell Physiol 285(4):C899–911
Tauskela JS, Chakravarthy BR, Murray CL, Wang Y, Comas T, HoganM, Hakim A, Morley P (1999) Evidence from cultured rat corticalneurons of differences in the mechanism of ischemic precondition-ing of brain and heart. Brain Res 827(1–2):143–151
Thompson SM, Capogna M, Scanziani M (1993) Presynaptic inhibitionin the hippocampus. Trends Neurosci 16(6):222–227
Treisman R (1996) Regulation of transcription by MAP kinase cas-cades. Curr Opin Cell Biol 8(2):205–215
Wood KW, Sarnecki C, Roberts TM, Blenis J (1992) ras mediatesnerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell68(6):1041–1050
Xu GP, Dave KR, Vivero R, Schmidt-Kastner R, Sick TJ, Perez-PinzonMA (2002) Improvement in neuronal survival after ischemic precon-ditioning in hippocampal slice cultures. Brain Res 952(2):153–158
ISCHEMIC PRECONDITIONING AND �PKC SIGNALING 645
J Cereb Blood Flow Metab, Vol. 24, No. 6, 2004



















