
Comparison and differentiation of potyvirus isolates and ...
Upload
independentCategory
view
0download
0
7) 431–440www.elsevier.com/locate/yviro
Virology 364 (200
Entry inhibitor-based microbicides are active in vitro against HIV-1 isolatesfrom multiple genetic subtypes
Thomas J. Ketas a, Susan M. Schader a,1, Juan Zurita a, Esther Teo a, Victoria Polonis b, Min Lu c,Per Johan Klasse a, John P. Moore a,⁎
a Dept. of Microbiology and Immunology, Weill Medical College of Cornell University, 1300 York Avenue, Box 62, New York, NY 10021, USAb Division of Retrovirology, Walter Reed Army Institute of Research, Washington, DC 20307, USAc Dept. of Biochemistry, Weill Medical College of Cornell University, New York, NY 10021, USA
Received 18 January 2007; returned to author for revision 26 February 2007; accepted 2 March 2007Available online 10 April 2007
Abstract
Inhibitors of viral entry are under consideration as topical microbicides to prevent HIV-1 sexual transmission. Small molecules targeting HIV-1gp120 (BMS-378806) or CCR5 (CMPD167), and a peptide fusion inhibitor (C52L), each blocks vaginal infection of macaques by a SHIV. Amicrobicide, however, must be active against multiple HIV-1 variants. We therefore tested BMS-C (a BMS-378806 derivative), CMPD167, C52Land the CXCR4 ligand AMD3465, alone and in combination, against 25 primary R5, 12 X4 and 7 R5X4 isolates from subtypes A–G. At highconcentrations (0.1–1 μM), the replication of most R5 isolates in human donor lymphocytes was inhibited by >90%. At lower concentrations,double and triple combinations were more effective than individual inhibitors. Similar results were obtained with X4 viruses when AMD3465 wassubstituted for CMPD167. The R5X4 viruses were inhibited by combining AMD3465 with CMPD167, or by the coreceptor-independentcompounds. Thus, combining entry inhibitors may improve microbicide effectiveness.© 2007 Published by Elsevier Inc.
Keywords: HIV-1; Microbicide; Clades; Entry inhibitors; R5 X4 tropism
Introduction
In the continuing absence of an effective vaccine, the use of atopical microbicide represents a credible alternative method toreduce the sexual transmission of human immunodeficiencyvirus type 1 (HIV-1). The microbicide concept remains to bevalidated, in that no compounds have yet been shown to preventHIV-1 transmission in efficacy trials (Dhawan and Mayer, 2006;Klasse et al., 2006; Lee et al., 2003; Shattock and Moore, 2003).Several efficacy trials of early generation microbicide candi-dates, mostly polyanion-based, are due to be completed in thenext few years. Provided these microbicides are used regularly,it is possible that one or more of them will show significantefficacy, although trials of one polyanion, cellulose sulphate,
⁎ Corresponding author.E-mail address: [email protected] (J.P. Moore).
1 Present address: Lady Davis Institute, McGill University, Montreal, Quebec,Canada.
0042-6822/$ - see front matter © 2007 Published by Elsevier Inc.doi:10.1016/j.virol.2007.03.001
were recently abandoned due to an increased rate of infectionsin the active arm. If the other polyanions are also found not to beprotective, the focus will fall on the next generation ofmicrobicide candidates that are in pre-clinical development orearly-stage clinical trials (Dhawan and Mayer, 2006; Klasse etal., 2006; Lederman et al., 2006; Shattock and Moore, 2003).
Among such next-generation concepts are entry inhibitors,compounds that inhibit the fusion of HIV-1 with its target cellsby specifically interfering with virus–receptor interactions orthe subsequent stages of the entry process. Several differententry inhibitors have now been shown to be effective at pre-venting SHIV infection of macaques by the vaginal and/orrectal routes (Dhawan and Mayer, 2006; Klasse et al., 2006;Lederman et al., 2006). One significant problem that will needto be overcome by an entry-inhibitor-based microbicide,indeed by any microbicide, is the global sequence diversityof HIV-1 (McCutchan, 2006; Walker and Korber, 2001). As isnow well appreciated, HIV-1 is an extraordinarily variablevirus, its diversity exceeding other viral pathogens' by orders

432 T.J. Ketas et al. / Virology 364 (2007) 431–440
of magnitude. This factor affects the design and performanceof HIV-1 vaccines, and it is just as relevant an issue formicrobicide development; any product that could prevent thetransmission of only a small subset of the viruses its usersencountered would not be of much practical value.
The breadth of protection against diverse HIV-1 strainscannot be readily gauged in the macaque model, because only avery few SHIVs are available for microbicide testing (Klasse etal., 2006; Lederman et al., 2006). Tissue-culture studies must,therefore, suffice. It is possible to assess the breadth of reactivityof vaccine-induced antibodies or neutralizing monoclonalantibodies (MAbs) in vitro by measuring their ability to inhibitthe infection of target cells by viruses from multiple differentgenetic subtypes (Binley et al., 2004; Brown et al., 2005; Burtonet al., 2004; Li et al., 2005; Moore and Burton, 2004). Aneutralizing antibody is, of course, an entry inhibitor, one thatworks by binding to the viral envelope glycoproteins (Burton etal., 2000; Klasse and Sattentau, 2002). Hence the same orsimilar virus test panels used for neutralization studies can beused to gauge the breadth of activity of candidate microbicidesin vitro.
We have, therefore, assembled a multi-subtype HIV-1 testpanel and used it to study a selection of different entryinhibitors, including compounds we have previously shown tobe effective at preventing SHIV infection of macaques aftervaginal challenge (Veazey et al., 2005). The test compounds arethe small-molecule CCR5 inhibitor CMPD167, the gp120-binding attachment inhibitor BMS-C, the gp41-targeted fusioninhibitory peptide C52L, and the small-molecule CXCR4inhibitor AMD3465. We have tested each of these inhibitorsas individual agents, and we have also evaluated their per-formance in pair-wise and triple combinations. The rationale fortesting inhibitor combinations is that the use of severalcompounds with different, complementary mechanisms ofaction is one possible answer to the problems posed by HIV-1diversity (Veazey et al., 2005). The chances of a virus beingsimultaneously resistant to three compounds is obviously lessthan to any single inhibitor, a principle well established fromclinical experience with drug-based therapies for HIV-1infection (Yeni et al., 2004). Furthermore, the combination ofCXCR4- and CCR5-specific inhibitors may be useful forcountering the transmission of dual-tropic viruses since,although X4 viruses are rarely transmitted, this can occur(Dhawan and Mayer, 2006; Gupta and Klasse, 2006; Klasseet al., 2006; Margolis and Shattock, 2006; Moore et al., 2004).
Results
Entry inhibitor microbicide candidates and virus test panels
We have previously shown that the small-molecule CCR5inhibitor CMPD167, the gp120-binding small-molecule attach-ment inhibitor BMS-378806 and the gp41–peptide fusioninhibitor C52L, alone and in combination, can protect rhesusmacaques against infection by the R5 virus SHIV-162P3 whenused as vaginal microbicides (Veazey et al., 2005). BMS-C is amember of the same structural family of compounds as BMS-
378806 but has greater potency in vitro, so we selected it forfurther study. AMD3465 is a small-molecule inhibitor thatbinds to CXCR4 (Hatse et al., 2005). We are now evaluating itin the macaque model for protection against the vaginaltransmission of viruses that use CXCR4 for entry into primarycells in vitro.
In the present study, we have explored the activities of eachof these compounds against HIV-1 primary isolates fromdifferent genetic subtypes in vitro, to gain insights into theirpractical utility as topical microbicides. Based on the knownproperties of these inhibitors, CMPD167 should only be activeagainst R5 viruses and AMD3465 only against X4 viruses,whereas the inhibitory effects of BMS-C and C52L should becoreceptor-independent (Guo et al., 2003; Matthews et al.,2004). Some of the test viruses were derived from a test panelput together for evaluating the breadth of activity of vaccine-induced neutralizing antibodies (Brown et al., 2005). Otherswere isolated from individuals in the relatively early stages ofinfection, so might be representative of viruses that expand inthe new host post-transmission (Rusert et al., 2005).
The test viruses were grouped by coreceptor usage: R5, X4or dual-tropic (R5+X4 or R5X4). In total, we used 25 R5viruses, 12 X4 viruses and 7 dual-tropic viruses, from 7 geneticsubtypes or circulating recombinant forms (CRFs) (Table 1).We placed more emphasis on the R5 virus test panel becauseviruses with this phenotype are the most commonly transmittedsuccessfully (Lederman et al., 2006; Moore et al., 2004).However, although X4 viruses are rarely transmitted, it ispossible that a microbicide might have to counter viruses of thisphenotype, particularly if the transmitter has been infected forseveral years, allowing the evolution of CXCR4-using strains(Dhawan and Mayer, 2006; Gupta and Klasse, 2006; Klasse etal., 2006; Margolis and Shattock, 2006; Moore et al., 2004).Isolates that use only CXCR4 are rare compared to ones thatcontain mixtures of viruses with either phenotype (R5+X4)and/or clonal viruses that can use both coreceptors (R5X4)(Brumme et al., 2005; Moyle et al., 2005). A caveat is that themost commonly used assays for viral phenotype are based oncell lines such as U87/CD4-CCR5 and U87/CD4-CXCR4(Brumme et al., 2005; Moyle et al., 2005), and coreceptor usagepatterns on cell lines and on primary cells are not alwayscorrelated (Yi et al., 2005; Zhang and Moore, 1999; Zhang etal., 1998). We therefore tested the entry inhibitors only againstdual-tropic isolates that we had confirmed could use both CCR5and CXCR4 to enter primary CD4+ T-cells, by showing thatthey were sensitive to a combination of the CCR5-specificinhibitor CMPD167 with a CXCR4-specific inhibitor(AMD3465 or AMD3100) (Zhang and Moore, 1999; Zhanget al., 1998) (and data not shown). These isolates probablycontain mixtures of R5 and X4 viruses and, perhaps, alsogenuinely dual-tropic viruses able to use either coreceptor.
Evaluation of entry inhibitors, alone and in combination,against primary isolates from multiple genetic subtypes
Each inhibitor was initially tested against each virus in aPBMC-based replication assay at four different concentrations
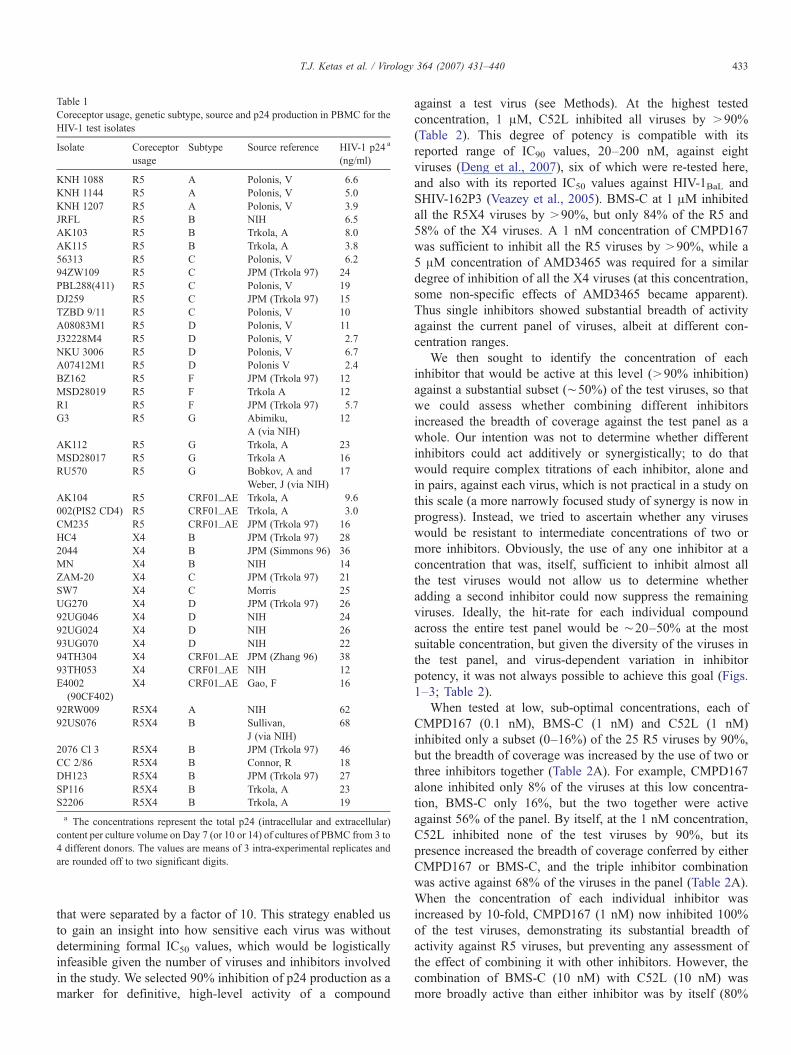
Table 1Coreceptor usage, genetic subtype, source and p24 production in PBMC for theHIV-1 test isolates
Isolate Coreceptorusage
Subtype Source reference HIV-1 p24 a
(ng/ml)
KNH 1088 R5 A Polonis, V 6.6KNH 1144 R5 A Polonis, V 5.0KNH 1207 R5 A Polonis, V 3.9JRFL R5 B NIH 6.5AK103 R5 B Trkola, A 8.0AK115 R5 B Trkola, A 3.856313 R5 C Polonis, V 6.294ZW109 R5 C JPM (Trkola 97) 24PBL288(411) R5 C Polonis, V 19DJ259 R5 C JPM (Trkola 97) 15TZBD 9/11 R5 C Polonis, V 10A08083M1 R5 D Polonis, V 11J32228M4 R5 D Polonis, V 2.7NKU 3006 R5 D Polonis, V 6.7A07412M1 R5 D Polonis V 2.4BZ162 R5 F JPM (Trkola 97) 12MSD28019 R5 F Trkola A 12R1 R5 F JPM (Trkola 97) 5.7G3 R5 G Abimiku,
A (via NIH)12
AK112 R5 G Trkola, A 23MSD28017 R5 G Trkola A 16RU570 R5 G Bobkov, A and
Weber, J (via NIH)17
AK104 R5 CRF01_AE Trkola, A 9.6002(PIS2 CD4) R5 CRF01_AE Trkola, A 3.0CM235 R5 CRF01_AE JPM (Trkola 97) 16HC4 X4 B JPM (Trkola 97) 282044 X4 B JPM (Simmons 96) 36MN X4 B NIH 14ZAM-20 X4 C JPM (Trkola 97) 21SW7 X4 C Morris 25UG270 X4 D JPM (Trkola 97) 2692UG046 X4 D NIH 2492UG024 X4 D NIH 2693UG070 X4 D NIH 2294TH304 X4 CRF01_AE JPM (Zhang 96) 3893TH053 X4 CRF01_AE NIH 12E4002
(90CF402)X4 CRF01_AE Gao, F 16
92RW009 R5X4 A NIH 6292US076 R5X4 B Sullivan,
J (via NIH)68
2076 Cl 3 R5X4 B JPM (Trkola 97) 46CC 2/86 R5X4 B Connor, R 18DH123 R5X4 B JPM (Trkola 97) 27SP116 R5X4 B Trkola, A 23S2206 R5X4 B Trkola, A 19
a The concentrations represent the total p24 (intracellular and extracellular)content per culture volume on Day 7 (or 10 or 14) of cultures of PBMC from 3 to4 different donors. The values are means of 3 intra-experimental replicates andare rounded off to two significant digits.
433T.J. Ketas et al. / Virology 364 (2007) 431–440
that were separated by a factor of 10. This strategy enabled usto gain an insight into how sensitive each virus was withoutdetermining formal IC50 values, which would be logisticallyinfeasible given the number of viruses and inhibitors involvedin the study. We selected 90% inhibition of p24 production as amarker for definitive, high-level activity of a compound
against a test virus (see Methods). At the highest testedconcentration, 1 μM, C52L inhibited all viruses by >90%(Table 2). This degree of potency is compatible with itsreported range of IC90 values, 20–200 nM, against eightviruses (Deng et al., 2007), six of which were re-tested here,and also with its reported IC50 values against HIV-1BaL andSHIV-162P3 (Veazey et al., 2005). BMS-C at 1 μM inhibitedall the R5X4 viruses by >90%, but only 84% of the R5 and58% of the X4 viruses. A 1 nM concentration of CMPD167was sufficient to inhibit all the R5 viruses by >90%, while a5 μM concentration of AMD3465 was required for a similardegree of inhibition of all the X4 viruses (at this concentration,some non-specific effects of AMD3465 became apparent).Thus single inhibitors showed substantial breadth of activityagainst the current panel of viruses, albeit at different con-centration ranges.
We then sought to identify the concentration of eachinhibitor that would be active at this level (>90% inhibition)against a substantial subset (∼50%) of the test viruses, so thatwe could assess whether combining different inhibitorsincreased the breadth of coverage against the test panel as awhole. Our intention was not to determine whether differentinhibitors could act additively or synergistically; to do thatwould require complex titrations of each inhibitor, alone andin pairs, against each virus, which is not practical in a study onthis scale (a more narrowly focused study of synergy is now inprogress). Instead, we tried to ascertain whether any viruseswould be resistant to intermediate concentrations of two ormore inhibitors. Obviously, the use of any one inhibitor at aconcentration that was, itself, sufficient to inhibit almost allthe test viruses would not allow us to determine whetheradding a second inhibitor could now suppress the remainingviruses. Ideally, the hit-rate for each individual compoundacross the entire test panel would be ∼20–50% at the mostsuitable concentration, but given the diversity of the viruses inthe test panel, and virus-dependent variation in inhibitorpotency, it was not always possible to achieve this goal (Figs.1–3; Table 2).
When tested at low, sub-optimal concentrations, each ofCMPD167 (0.1 nM), BMS-C (1 nM) and C52L (1 nM)inhibited only a subset (0–16%) of the 25 R5 viruses by 90%,but the breadth of coverage was increased by the use of two orthree inhibitors together (Table 2A). For example, CMPD167alone inhibited only 8% of the viruses at this low concentra-tion, BMS-C only 16%, but the two together were activeagainst 56% of the panel. By itself, at the 1 nM concentration,C52L inhibited none of the test viruses by 90%, but itspresence increased the breadth of coverage conferred by eitherCMPD167 or BMS-C, and the triple inhibitor combinationwas active against 68% of the viruses in the panel (Table 2A).When the concentration of each individual inhibitor wasincreased by 10-fold, CMPD167 (1 nM) now inhibited 100%of the test viruses, demonstrating its substantial breadth ofactivity against R5 viruses, but preventing any assessment ofthe effect of combining it with other inhibitors. However, thecombination of BMS-C (10 nM) with C52L (10 nM) wasmore broadly active than either inhibitor was by itself (80%
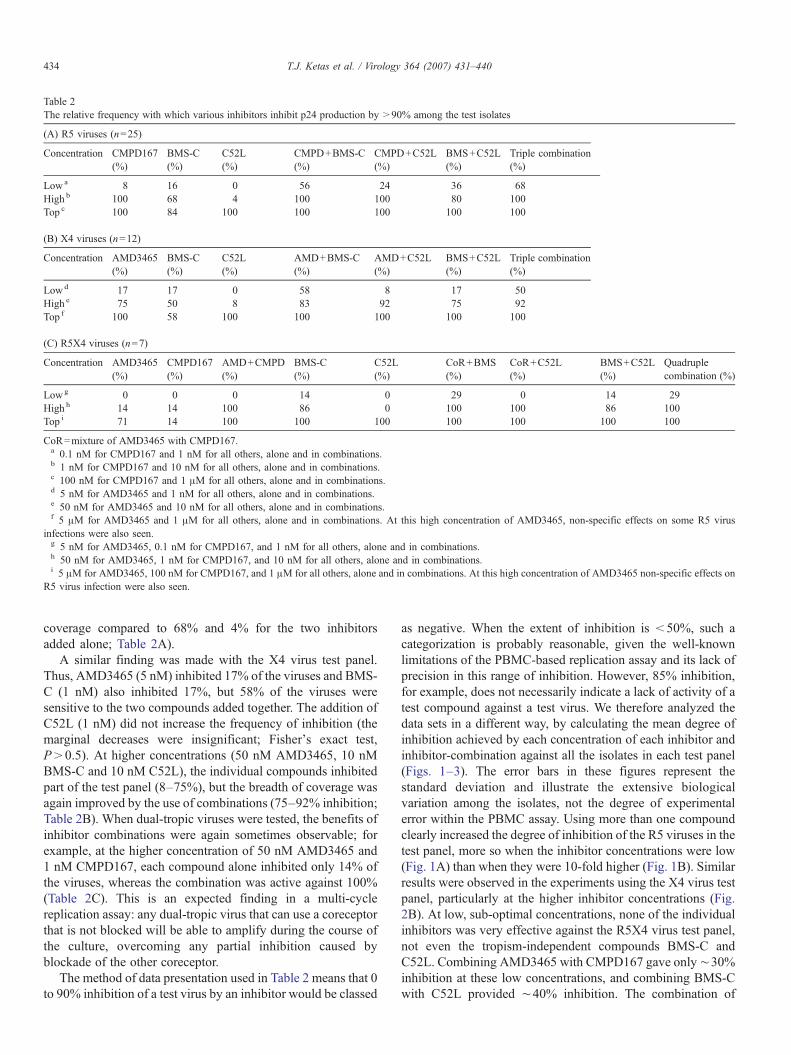
Table 2The relative frequency with which various inhibitors inhibit p24 production by >90% among the test isolates
(A) R5 viruses (n=25)
Concentration CMPD167(%)
BMS-C(%)
C52L(%)
CMPD+BMS-C(%)
CMPD+C52L(%)
BMS+C52L(%)
Triple combination(%)
Lowa 8 16 0 56 24 36 68High b 100 68 4 100 100 80 100Top c 100 84 100 100 100 100 100
(B) X4 viruses (n=12)
Concentration AMD3465(%)
BMS-C(%)
C52L(%)
AMD+BMS-C(%)
AMD+C52L(%)
BMS+C52L(%)
Triple combination(%)
Lowd 17 17 0 58 8 17 50High e 75 50 8 83 92 75 92Top f 100 58 100 100 100 100 100
(C) R5X4 viruses (n=7)
Concentration AMD3465(%)
CMPD167(%)
AMD+CMPD(%)
BMS-C(%)
C52L(%)
CoR+BMS(%)
CoR+C52L(%)
BMS+C52L(%)
Quadruplecombination (%)
Lowg 0 0 0 14 0 29 0 14 29High h 14 14 100 86 0 100 100 86 100Top i 71 14 100 100 100 100 100 100 100
CoR=mixture of AMD3465 with CMPD167.a 0.1 nM for CMPD167 and 1 nM for all others, alone and in combinations.b 1 nM for CMPD167 and 10 nM for all others, alone and in combinations.c 100 nM for CMPD167 and 1 μM for all others, alone and in combinations.d 5 nM for AMD3465 and 1 nM for all others, alone and in combinations.e 50 nM for AMD3465 and 10 nM for all others, alone and in combinations.f 5 μM for AMD3465 and 1 μM for all others, alone and in combinations. At this high concentration of AMD3465, non-specific effects on some R5 virus
infections were also seen.g 5 nM for AMD3465, 0.1 nM for CMPD167, and 1 nM for all others, alone and in combinations.h 50 nM for AMD3465, 1 nM for CMPD167, and 10 nM for all others, alone and in combinations.i 5 μM for AMD3465, 100 nM for CMPD167, and 1 μM for all others, alone and in combinations. At this high concentration of AMD3465 non-specific effects on
R5 virus infection were also seen.
434 T.J. Ketas et al. / Virology 364 (2007) 431–440
coverage compared to 68% and 4% for the two inhibitorsadded alone; Table 2A).
A similar finding was made with the X4 virus test panel.Thus, AMD3465 (5 nM) inhibited 17% of the viruses and BMS-C (1 nM) also inhibited 17%, but 58% of the viruses weresensitive to the two compounds added together. The addition ofC52L (1 nM) did not increase the frequency of inhibition (themarginal decreases were insignificant; Fisher's exact test,P>0.5). At higher concentrations (50 nM AMD3465, 10 nMBMS-C and 10 nM C52L), the individual compounds inhibitedpart of the test panel (8–75%), but the breadth of coverage wasagain improved by the use of combinations (75–92% inhibition;Table 2B). When dual-tropic viruses were tested, the benefits ofinhibitor combinations were again sometimes observable; forexample, at the higher concentration of 50 nM AMD3465 and1 nM CMPD167, each compound alone inhibited only 14% ofthe viruses, whereas the combination was active against 100%(Table 2C). This is an expected finding in a multi-cyclereplication assay: any dual-tropic virus that can use a coreceptorthat is not blocked will be able to amplify during the course ofthe culture, overcoming any partial inhibition caused byblockade of the other coreceptor.
The method of data presentation used in Table 2 means that 0to 90% inhibition of a test virus by an inhibitor would be classed
as negative. When the extent of inhibition is <50%, such acategorization is probably reasonable, given the well-knownlimitations of the PBMC-based replication assay and its lack ofprecision in this range of inhibition. However, 85% inhibition,for example, does not necessarily indicate a lack of activity of atest compound against a test virus. We therefore analyzed thedata sets in a different way, by calculating the mean degree ofinhibition achieved by each concentration of each inhibitor andinhibitor-combination against all the isolates in each test panel(Figs. 1–3). The error bars in these figures represent thestandard deviation and illustrate the extensive biologicalvariation among the isolates, not the degree of experimentalerror within the PBMC assay. Using more than one compoundclearly increased the degree of inhibition of the R5 viruses in thetest panel, more so when the inhibitor concentrations were low(Fig. 1A) than when they were 10-fold higher (Fig. 1B). Similarresults were observed in the experiments using the X4 virus testpanel, particularly at the higher inhibitor concentrations (Fig.2B). At low, sub-optimal concentrations, none of the individualinhibitors was very effective against the R5X4 virus test panel,not even the tropism-independent compounds BMS-C andC52L. Combining AMD3465 with CMPD167 gave only∼30%inhibition at these low concentrations, and combining BMS-Cwith C52L provided ∼40% inhibition. The combination of
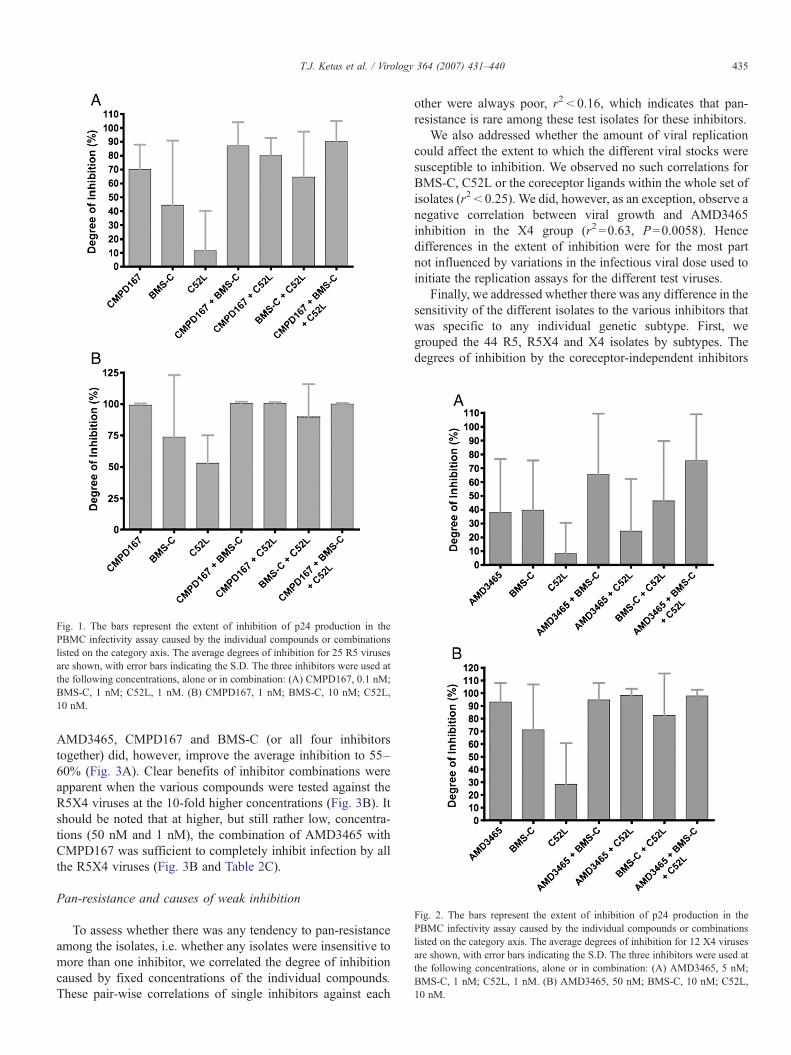
Fig. 2. The bars represent the extent of inhibition of p24 production in thePBMC infectivity assay caused by the individual compounds or combinationslisted on the category axis. The average degrees of inhibition for 12 X4 virusesare shown, with error bars indicating the S.D. The three inhibitors were used atthe following concentrations, alone or in combination: (A) AMD3465, 5 nM;BMS-C, 1 nM; C52L, 1 nM. (B) AMD3465, 50 nM; BMS-C, 10 nM; C52L,10 nM.
Fig. 1. The bars represent the extent of inhibition of p24 production in thePBMC infectivity assay caused by the individual compounds or combinationslisted on the category axis. The average degrees of inhibition for 25 R5 virusesare shown, with error bars indicating the S.D. The three inhibitors were used atthe following concentrations, alone or in combination: (A) CMPD167, 0.1 nM;BMS-C, 1 nM; C52L, 1 nM. (B) CMPD167, 1 nM; BMS-C, 10 nM; C52L,10 nM.
435T.J. Ketas et al. / Virology 364 (2007) 431–440
AMD3465, CMPD167 and BMS-C (or all four inhibitorstogether) did, however, improve the average inhibition to 55–60% (Fig. 3A). Clear benefits of inhibitor combinations wereapparent when the various compounds were tested against theR5X4 viruses at the 10-fold higher concentrations (Fig. 3B). Itshould be noted that at higher, but still rather low, concentra-tions (50 nM and 1 nM), the combination of AMD3465 withCMPD167 was sufficient to completely inhibit infection by allthe R5X4 viruses (Fig. 3B and Table 2C).
Pan-resistance and causes of weak inhibition
To assess whether there was any tendency to pan-resistanceamong the isolates, i.e. whether any isolates were insensitive tomore than one inhibitor, we correlated the degree of inhibitioncaused by fixed concentrations of the individual compounds.These pair-wise correlations of single inhibitors against each
other were always poor, r2<0.16, which indicates that pan-resistance is rare among these test isolates for these inhibitors.
We also addressed whether the amount of viral replicationcould affect the extent to which the different viral stocks weresusceptible to inhibition. We observed no such correlations forBMS-C, C52L or the coreceptor ligands within the whole set ofisolates (r2<0.25). We did, however, as an exception, observe anegative correlation between viral growth and AMD3465inhibition in the X4 group (r2 =0.63, P=0.0058). Hencedifferences in the extent of inhibition were for the most partnot influenced by variations in the infectious viral dose used toinitiate the replication assays for the different test viruses.
Finally, we addressed whether there was any difference in thesensitivity of the different isolates to the various inhibitors thatwas specific to any individual genetic subtype. First, wegrouped the 44 R5, R5X4 and X4 isolates by subtypes. Thedegrees of inhibition by the coreceptor-independent inhibitors
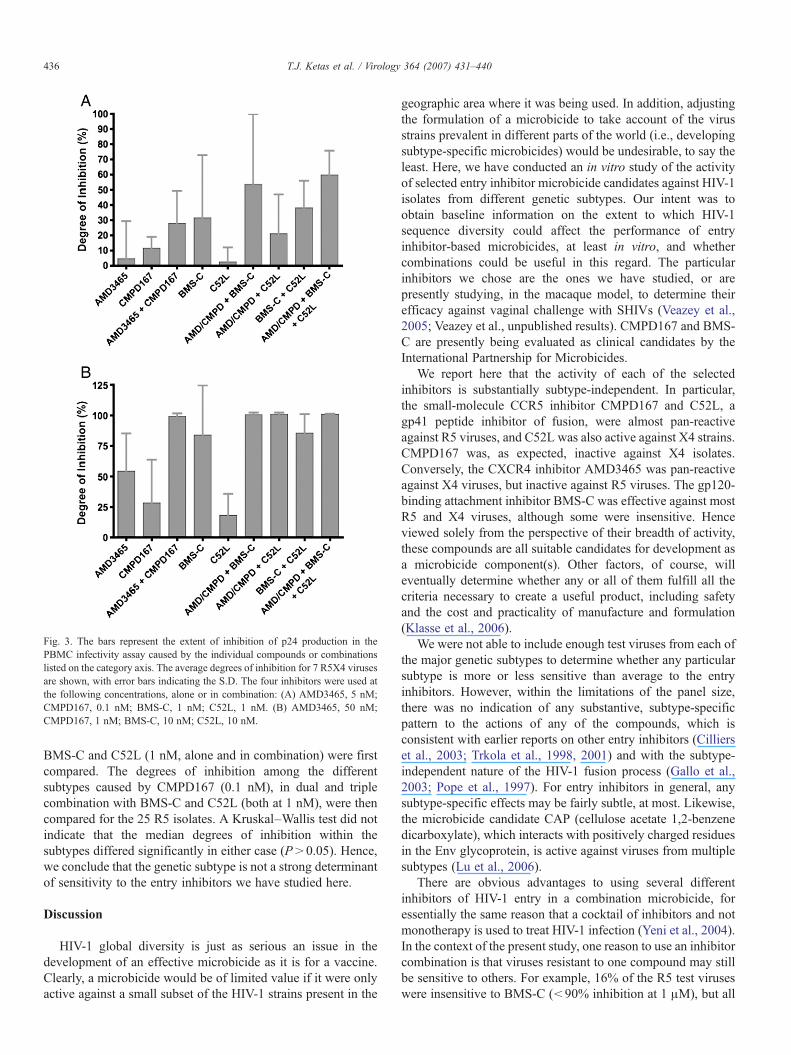
Fig. 3. The bars represent the extent of inhibition of p24 production in thePBMC infectivity assay caused by the individual compounds or combinationslisted on the category axis. The average degrees of inhibition for 7 R5X4 virusesare shown, with error bars indicating the S.D. The four inhibitors were used atthe following concentrations, alone or in combination: (A) AMD3465, 5 nM;CMPD167, 0.1 nM; BMS-C, 1 nM; C52L, 1 nM. (B) AMD3465, 50 nM;CMPD167, 1 nM; BMS-C, 10 nM; C52L, 10 nM.
436 T.J. Ketas et al. / Virology 364 (2007) 431–440
BMS-C and C52L (1 nM, alone and in combination) were firstcompared. The degrees of inhibition among the differentsubtypes caused by CMPD167 (0.1 nM), in dual and triplecombination with BMS-C and C52L (both at 1 nM), were thencompared for the 25 R5 isolates. A Kruskal–Wallis test did notindicate that the median degrees of inhibition within thesubtypes differed significantly in either case (P>0.05). Hence,we conclude that the genetic subtype is not a strong determinantof sensitivity to the entry inhibitors we have studied here.
Discussion
HIV-1 global diversity is just as serious an issue in thedevelopment of an effective microbicide as it is for a vaccine.Clearly, a microbicide would be of limited value if it were onlyactive against a small subset of the HIV-1 strains present in the
geographic area where it was being used. In addition, adjustingthe formulation of a microbicide to take account of the virusstrains prevalent in different parts of the world (i.e., developingsubtype-specific microbicides) would be undesirable, to say theleast. Here, we have conducted an in vitro study of the activityof selected entry inhibitor microbicide candidates against HIV-1isolates from different genetic subtypes. Our intent was toobtain baseline information on the extent to which HIV-1sequence diversity could affect the performance of entryinhibitor-based microbicides, at least in vitro, and whethercombinations could be useful in this regard. The particularinhibitors we chose are the ones we have studied, or arepresently studying, in the macaque model, to determine theirefficacy against vaginal challenge with SHIVs (Veazey et al.,2005; Veazey et al., unpublished results). CMPD167 and BMS-C are presently being evaluated as clinical candidates by theInternational Partnership for Microbicides.
We report here that the activity of each of the selectedinhibitors is substantially subtype-independent. In particular,the small-molecule CCR5 inhibitor CMPD167 and C52L, agp41 peptide inhibitor of fusion, were almost pan-reactiveagainst R5 viruses, and C52L was also active against X4 strains.CMPD167 was, as expected, inactive against X4 isolates.Conversely, the CXCR4 inhibitor AMD3465 was pan-reactiveagainst X4 viruses, but inactive against R5 viruses. The gp120-binding attachment inhibitor BMS-C was effective against mostR5 and X4 viruses, although some were insensitive. Henceviewed solely from the perspective of their breadth of activity,these compounds are all suitable candidates for development asa microbicide component(s). Other factors, of course, willeventually determine whether any or all of them fulfill all thecriteria necessary to create a useful product, including safetyand the cost and practicality of manufacture and formulation(Klasse et al., 2006).
We were not able to include enough test viruses from each ofthe major genetic subtypes to determine whether any particularsubtype is more or less sensitive than average to the entryinhibitors. However, within the limitations of the panel size,there was no indication of any substantive, subtype-specificpattern to the actions of any of the compounds, which isconsistent with earlier reports on other entry inhibitors (Cillierset al., 2003; Trkola et al., 1998, 2001) and with the subtype-independent nature of the HIV-1 fusion process (Gallo et al.,2003; Pope et al., 1997). For entry inhibitors in general, anysubtype-specific effects may be fairly subtle, at most. Likewise,the microbicide candidate CAP (cellulose acetate 1,2-benzenedicarboxylate), which interacts with positively charged residuesin the Env glycoprotein, is active against viruses from multiplesubtypes (Lu et al., 2006).
There are obvious advantages to using several differentinhibitors of HIV-1 entry in a combination microbicide, foressentially the same reason that a cocktail of inhibitors and notmonotherapy is used to treat HIV-1 infection (Yeni et al., 2004).In the context of the present study, one reason to use an inhibitorcombination is that viruses resistant to one compound may stillbe sensitive to others. For example, 16% of the R5 test viruseswere insensitive to BMS-C (<90% inhibition at 1 μM), but all

437T.J. Ketas et al. / Virology 364 (2007) 431–440
of the BMS-C-resistant viruses were inhibited by CMPD167and/or C52L. No virus from among the 44 tested wasinsensitive to two of the different categories of inhibitor at thehighest concentrations (0.1–5 μM excluding, of course, theexpected ineffectiveness of the CCR5 inhibitor against X4viruses and the CXCR4 inhibitor against R5 viruses). Thus in acombination of CMPD167, C52L and BMS-C, at least two ofthe inhibitors, and usually all three, were active against the R5viruses in the test panel. Moreover, combining a CCR5 inhibitorwith a CXCR4 inhibitor, or with coreceptor-independentinhibitors, is one solution to the problem of the raretransmissions of CXCR4-using viruses (Dhawan and Mayer,2006; Lederman et al., 2006; Margolis and Shattock, 2006;Moore et al., 2004).
All of the above conclusions are, of course, conditional onthe concentrations of the test compounds, because theiractivities are dose-dependent and we used different concentra-tions in the nM to μM range in various experiments. In themacaque model, the inhibitors are applied at much higher, mM,concentrations when they provide protection against vaginalchallenge with SHIV-162P3 (Veazey et al., 2005). Although thelocal inhibitor concentrations at the site of virus deposition arenot known, it seems unlikely that they will be as low as the nMrange, at least initially (Klasse et al., 2006). The localconcentrations will dissipate with time, so eventually thelower, less active range will be reached. What inhibitorconcentrations must be present within the vagina to protectwomen, and for how long, are not yet known, of course.
A second advantage of the use of inhibitor combinations isthe potential for synergy; that is, the mutually reinforcingactivity of two different compounds with complementarymechanisms of action. We did not, in this study, performformal synergy analyses, but we have previously reportedsynergy between CMPD167 and both C52L and BMS-378806against SHIV-162P3 replication in vitro (Veazey et al., 2005).There are also several other reports of synergy between CCR5inhibitors similar to CMPD167 and gp41 peptide fusioninhibitors similar to C52L (Tremblay et al., 2002,2005a,2005b). In the context of a microbicide formulation forhuman use, synergy would act to reduce the amount of eachinhibitor required to provide an effective blockade to HIV-1transmission, a useful bonus to combination-based strategies.
There are two obvious limitations to this study: the choicesof the test viruses and the target cells used in the infectioninhibition assay. The viruses were almost all isolated from theperipheral blood of HIV-1-infected people, some relativelyearly in infection, others several years later. How precisely suchviruses mimic those present in genital fluids that a microbicidemust block from establishing a new infection remains to bedetermined, because such viruses are unavailable (Klasse et al.,2006). The same considerations apply, of course, to the designof virus test panels for the evaluation of MAb-based HIV-1vaccines (Binley et al., 2004; Brown et al., 2005; Li et al., 2005;Mascola et al., 2005; Rusert et al., 2005). One possibleapproach is to use viruses isolated relatively early after HIV-1infection of a new host, and we included such examples in ourR5 test panel. However, viruses that expand in the host may
differ phenotypically from those present in semen or vaginalfluids (Klasse et al., 2006). Until test panels are assembled thatinclude the latter categories of HIV-1 variants, it cannot bedetermined whether this issue is truly relevant. We suspect,however, that any such influence on microbicide efficacy invitro will not be substantive enough to alter the broadconclusions we have drawn here.
The second principal limitation to this study is our use ofPBMC as the target cell for HIV-1 replication. Given the scaleof the study (44 test viruses and 4 inhibitors, alone and incombination) we had little choice but to work with PBMC.However, these cells are not the immediate target cells forsexual transmission of HIV-1 (Gupta and Klasse, 2006; Haase,2005; Miller et al., 2005). Thus, in principle, using PBMC couldaffect our conclusions, but again probably only to a minorextent. Firstly, several of the same compounds we have shownto be active against HIV-1 infection of PBMC also inhibitSHIV-162P3 vaginal transmission to macaques, so they must beactive against whatever cells are infected early in the infectionprocess (Veazey et al., 2005). Secondly, the principal cell type inwhich HIV-1 replicates in activated PBMC is the CD4+ T-cell.Such cells are present at early sites of virus replication aftervaginal transmission of SIVs or SHIVs, as are others such as themacrophage and dendritic cell (Gupta and Klasse, 2006; Haase,2005; Hladik et al., 2007; Miller et al., 2005). Small-moleculeCCR5 inhibitors and gp41-based fusion peptides have pre-viously been shown to inhibit HIV-1 infection of both these celltypes in vitro, so we would not expect too much difference fromour present results if we had used other cell types (Ketas et al.,2003a,2003b; Willey et al., 2005). In broad terms, themechanism of HIV-1 entry, and hence of its inhibition, isindependent of the target cell in vitro (Gallo et al., 2003).Nonetheless, it will be essential to also perform the present typeof study using other in vitro experimental systems, such ascervical tissue explants and dendritic cells, to see whether anyissues arise that are not visible in studies that use PBMC.Whether any in vitro system can always predict the activity ofan inhibitor against virus transmission in vivo, in a modelsystem or the real world, of course remains to be determined.
Using more than one entry inhibitor might also reduce thedevelopment of resistance (Olson and Maddon, 2003). Mostwomen in the developing world are unaware of their HIV-1status, so it is probable that, outside a clinical trial setting,microbicides would be used by infected women. It is unclearwhether any topical agent will cause resistant variants to evolvesystemically, but inhibitor absorption into the body could resultin sub-therapeutic levels that select for resistance, perhaps atlocal sites close to where the inhibitor is applied. This concernmay apply to conventional antiviral drugs that are now beingused widely for treating HIV-1 infection, something that doesnot yet apply to entry inhibitors. There is an argument that, inhigh-incidence areas like sub-Saharan Africa, entry inhibitorsmight best be reserved for the prevention of HIV-1 transmission,to avoid any possible impact of resistance development on theefficacy of therapy.
Overall, our results support the development of microbicideformulations based on the use of several different inhibitors of

438 T.J. Ketas et al. / Virology 364 (2007) 431–440
HIV-1 attachment and entry. If such a microbicide can bedeveloped successfully, its efficacy may not be influenced toodrastically by subtype-dependent HIV-1 sequence variation. Wedo not seek to trivialize the problem of HIV-1 sequencediversity, which is and will always remain formidable(McCutchan, 2006; Walker and Korber, 2001). However,carefully chosen entry inhibitors, if used in combination, maybe effective at blocking HIV-1 transmission irrespective of theviral subtype.
Methods
Entry inhibitors and MAbs
The small-molecule CCR5 inhibitor CMPD167 and thegp41-peptide fusion inhibitor C52L have been describedpreviously, including evaluations of their activity againstSHIV transmission to the rhesus macaque after vaginal admi-nistration (Veazey et al., 2005; Deng et al., 2007). BMS-C is amore potent member of the same structural family ofcompounds as the gp120-binding attachment inhibitor BMS-378806 (Guo et al., 2003). AMD3465 is a small-moleculeinhibitor that binds to CXCR4 (Hatse et al., 2005).
HIV-1 isolates
The isolates of defined coreceptor usage (R5, X4, R5X4) andgenetic subtypes used in this study have all been describedpreviously (see Table 1).
Preparation of PBMC and use in HIV-1 infection assay
Peripheral blood mononuclear cells (PBMC) were preparedfrom leukopacks obtained from the New York Blood Center(New York, NY). The PBMC were cultured in lymphocytemedium (LM) [RPMI 1640 supplemented with 10% fetalbovine serum (FBS), 2 mM L-glutamine (all media fromCellgro, Herndon, VA), and 100 U/ml IL-2 (NIH AIDS Re-search and Reference Reagent Program, contributed by Hoff-man-LaRoche, Inc.)]. Equal parts of each PBMC culture werestimulated with either 5 μg/ml phytohemagglutinin (PHA;Sigma, St. Louis, MO) or with supernatant from the OKT3hybridoma (anti-CD3 stimulation). After 3 days, an equalnumber of cells from the two different stimulation cultures werecombined. PHA and OKT3 were removed by washing. Titrationmedium (TM, which is LM containing 100 μg/ml penicillin/streptomycin) was added for the remainder of the culture period(Ketas et al., 2003b). The PBMC were seeded at 100 μl per wellinto a 96-well plate (Becton Dickinson) at 1.4×106 cells/ml inTM before addition of a serially diluted inhibitor immediatelyprior to inoculation of the test virus (100 TCID50/well). Thecultures were then continued with both virus and inhibitorspresent. HIV-1 replication was measured using an in-house p24antigen ELISA on Day 7, then again on Day 10 or 14 if viralreplication was insufficient. Residual p24 from the input viruswas measured and subtracted in order to calculate total net p24production (intracellular and extracellular) in the test wells.
Inhibition was expressed relative to p24 production in thecontrol wells (no inhibitor, defined as 0% inhibition).
Statistical and mathematical analysis
The effect of the various inhibitors on the replication of apanel of HIV-1 test isolates in PBMC is presented in twodifferent ways: (1) as the relative frequency of inhibition abovea defined cut-off value; and (2) as the average degree ofinhibition.
In the first method, the cut-off value for calculating thefrequency of inhibition was selected to be 90% reduction of p24production compared with control cultures (i.e., no inhibitorpresent). The rationale for this cut-off is that a 90% reduction inthe infectivity of the inocula commonly used for experimentaltransmission of challenge viruses to macaques should providesignificant protection from infection (Klasse et al., 2006).Within each virus test panel, the number of isolates inhibited by>90% is divided by the total number of isolates in the panel; theresulting relative frequency is presented as a percentage. Therationale here is that frequencies based on a high cut-off arelikely to be the most important from the perspective of thepractical development of an intervention strategy such as amicrobicide. In other words, what proportion of the divergentviruses an inhibitor might encounter when used as a microbicidecould it be expected to be effective against?
Because the above method of data presentation obscures anydifferences in partial inhibition that occur below the cut-offvalue (i.e., from 0 to 90% inhibition of p24 production), we alsocalculated the average degree of inhibition (±standard devia-tion) for all the isolates in each test panel. This strategy providesa complementary means of gauging the increase in inhibitionresulting from combining two or more inhibitors. Biologically,this is relevant to the question: When the concentrations ofindividual inhibitors at mucosal sites fall below what is requiredfor protection (e.g. in mucosal microenvironments, and overtime), will their use in combinations still be able to counter theinfectious inoculum?
In addition, to assess whether any test isolates were pan-resistant to all the relevant inhibitors (as opposed to beingresistant to individual ones), we performed pair-wise correla-tions of the degree of inhibition by fixed concentrations of eachinhibitor. Finally, to address whether variation in the extent towhich different test isolates replicated caused the extent ofinhibition by a test compound also to vary, we correlated theextent of inhibition with the amount of p24 produced in theabsence of an inhibitor. These correlations were performed andthe r2 values were calculated using Prism (GraphPad). Thejustification for performing this analysis by correlations, ratherthan linear regression, is that only experimental variables, andno controlled parameters, were plotted against each other.
We addressed whether there was any genetic subtype-specificeffect of the various inhibitors (alone and combined) bycomparing the degrees of inhibition at fixed, intermediateconcentrations, in a Kruskal–Wallis one-way analysis ofvariance by ranks. This non-parametric test was chosen becausethe sample numbers were sometimes too small to evaluate

439T.J. Ketas et al. / Virology 364 (2007) 431–440
Gaussian distributions. When the Kruskal–Wallis test indicatedthat there were no overall discrepancies in rank sums among thegroups (p>0.05), Dunn's post-test was not done. All statisticalanalyses were performed in Prism (GraphPad).
Acno leden ts
We thank Marty Springer and Jacqueline Fine (MerckResearch Labs) for the provision of CMPD167 and advice on itsuse, Richard Colonno (Bristol Myers Squibb, Inc.) forproviding BMS-C and advising how to use it, Gary Bridger(AnorMED, Inc.) for providing AMD3465 and advice on itsuse, and Alexandra Trkola for the provision of HIV-1 isolatesfrom acutely infected individuals. We appreciate receivingadvice from Robin Shattock. This work was supported by NIHgrant U19 AI 65413, and by an Unrestricted Infectious DiseasesAward from the Bristol-Myers Squibb Foundation to J.P.M. TheDepartment of Microbiology and Immunology at the WeillMedical College gratefully acknowledges the support of theWilliam Randolph Hearst Foundation.
Reerences
Binley, J.M., Wrin, T., Korber, B., Zwick, M.B., Wang, M., Chappey, C.,Stiegler, G., Kunert, R., Zolla-Pazner, S., Katinger, H., Petropoulos, C.J.,Burton, D.R., 2004. Comprehensive cross-clade neutralization analysis of apanel of anti-human immunodeficiency virus type 1 monoclonal antibodies.J. Virol. 78 (23), 13232–13252.
Brown, B.K., Darden, J.M., Tovanabutra, S., Oblander, T., Frost, J., Sanders-Buell, E., de Souza, M.S., Birx, D.L., McCutchan, F.E., Polonis, V.R., 2005.Biologic and genetic characterization of a panel of 60 human immunode-ficiency virus type 1 isolates, representing clades A, B, C, D, CRF01_AE,and CRF02_AG, for the development and assessment of candidate vaccines.J. Virol. 79 (10), 6089–6101.
Brumme, Z.L., Goodrich, J., Mayer, H.B., Brumme, C.J., Henrick, B.M.,Wynhoven, B., Asselin, J.J., Cheung, P.K., Hogg, R.S., Montaner, J.S.,Harrigan, P.R., 2005. Molecular and clinical epidemiology of CXCR4-usingHIV-1 in a large population of antiretroviral-naive individuals. J. Infect. Dis.192 (3), 466–474.
Burton, D.R., Williamson, R.A., Parren, P.W., 2000. Antibody and virus:binding and neutralization. Virology 270 (1), 1–3.
Burton, D.R., Desrosiers, R.C., Doms, R.W., Koff, W.C., Kwong, P.D., Moore,J.P., Nabel, G.J., Sodroski, J., Wilson, I.A., Wyatt, R.T., 2004. HIV vaccinedesign and the neutralizing antibody problem. Nat. Immunol. 5 (3), 233–236.
Cilliers, T., Nhlapo, J., Coetzer, M., Orlovic, D., Ketas, T., Olson, W.C., Moore,J.P., Trkola, A., Morris, L., 2003. The CCR5 and CXCR4 coreceptors areboth used by human immunodeficiency virus type 1 primary isolates fromsubtype C. J. Virol. 77 (7), 4449–4456.
Deng, Y., Zheng, Q., Ketas, T.A., Moore, J.P., Lu, M., 2007. Protein design of abacterially expressed HIV-1 gp41 fusion inhibitor. Biochemistry [Electronicpublication ahead of print].
Dhawan, D., Mayer, K.H., 2006. Microbicides to prevent HIV transmission:overcoming obstacles to chemical barrier protection. J. Infect. Dis. 193 (1),36–44.
Gallo, S.A., Finnegan, C.M., Viard, M., Raviv, Y., Dimitrov, A., Rawat, S.S.,Puri, A., Durell, S., Blumenthal, R., 2003. The HIV Env-mediated fusionreaction. Biochim. Biophys. Acta 1614 (1), 36–50.
Guo, Q., Ho, H.T., Dicker, I., Fan, L., Zhou, N., Friborg, J., Wang, T.,McAuliffe, B.V., Wang, H.G., Rose, R.E., Fang, H., Scarnati, H.T., Langley,D.R., Meanwell, N.A., Abraham, R., Colonno, R.J., Lin, P.F., 2003.Biochemical and genetic characterizations of a novel human immunodefi-ciency virus type 1 inhibitor that blocks gp120–CD4 interactions. J. Virol.77 (19), 10528–10536.
Gupta, K., Klasse, P.J., 2006. How do viral and host factors modulate the sexualtransmission of HIV? Can transmission be blocked? PLoS Med. 3 (2), e79,181–185.
Haase, A.T., 2005. Perils at mucosal front lines for HIVand SIVand their hosts.Nat. Rev., Immunol. 5 (10), 783–792.
Hatse, S., Princen, K., De Clercq, E., Rosenkilde, M.M., Schwartz, T.W.,Hernandez-Abad, P.E., Skerlj, R.T., Bridger, G.J., Schols, D., 2005.AMD3465, a monomacrocyclic CXCR4 antagonist and potent HIV entryinhibitor. Biochem. Pharmacol. 70 (5), 752–761.
Hladik, F., Sakchalathorn, P., Ballweber, L., Lentz, G., Fialkow, M.,Eschenbach, D., McElrath, M.J., 2007. Initial events in establishing vaginalentry and infection by human immunodeficiency virus type-1. Immunity 26(2), 257–270.
Ketas, T.J., Frank, I., Klasse, P.J., Sullivan, B.M., Gardner, J.P., Spenlehauer, C.,Nesin, M., Olson, W.C., Moore, J.P., Pope, M., 2003a. Human immuno-deficiency virus type 1 attachment, coreceptor, and fusion inhibitors areactive against both direct and trans infection of primary cells. J. Virol. 77 (4),2762–2767.
Ketas, T.J., Klasse, P.J., Spenlehauer, C., Nesin, M., Frank, I., Pope, M., Strizki,J.M., Reyes, G.R., Baroudy, B.M., Moore, J.P., 2003b. Entry inhibitorsSCH-C, RANTES, and T-20 block HIV type 1 replication in multiple celltypes. AIDS Res. Hum. Retroviruses 19 (3), 177–186.
Klasse, P.J., Sattentau, Q.J., 2002. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J. Gen. Virol. 83 (9), 2091–2108.
Klasse, P.J., Shattock, R.J., Moore, J.P., 2006. Which topical microbicides forblocking HIV-1 transmission will work in the real world? PLoS Med. 3 (9),1501–1507.
Lederman, M.M., Offord, R.E., Hartley, O., 2006. Microbicides and othertopical strategies to prevent vaginal transmission of HIV. Nat. Immunol.Rev. 6 (5), 371–382.
Lee, M.T., Coburn, G.A., McClure, M.O., Cullen, B.R., 2003. Inhibition ofhuman immunodeficiency virus type 1 replication in primary macrophagesby using Tat- or CCR5-specific small interfering RNAs expressed from alentivirus vector. J. Virol. 77 (22), 11964–11972.
Li, M., Gao, F., Mascola, J.R., Stamatatos, L., Polonis, V.R., Koutsoukos, M.,Voss, G., Goepfert, P., Gilbert, P., Greene, K.M., Bilska, M., Kothe, D.L.,Salazar-Gonzalez, J.F., Wei, X., Decker, J.M., Hahn, B.H., Montefiori, D.C.,2005. Human immunodeficiency virus type 1 env clones from acute andearly subtype B infections for standardized assessments of vaccine-elicitedneutralizing antibodies. J. Virol. 79 (16), 10108–10125.
Lu, H., Zhao, Q., Wallace, G., Liu, S., He, Y., Shattock, R., Neurath, A.R., Jiang,B.S., 2006. Cellulose acetate 1,2-benzenedicarboxylate inhibits infection bycell-free and cell-associated primary HIV-1 isolates. AIDS Res. Hum.Retroviruses 22 (5), 411–418.
Margolis, L., Shattock, R., 2006. Selective transmission of CCR5-utilizingHIV-1: the gatekeeper problem resolved? Nat. Rev., Microbiol. 4 (4),312–317.
Mascola, J.R., D'Souza, P., Gilbert, P., Hahn, B.H., Haigwood, N.L., Morris, L.,Petropoulos, C.J., Polonis, V.R., Sarzotti, M., Montefiori, D.C., 2005.Recommendations for the design and use of standard virus panels to assessneutralizing antibody responses elicited by candidate human immunodefi-ciency virus type 1 vaccines. J. Virol. 79 (16), 10103–10107.
Matthews, T., Salgo, M., Greenberg, M., Chung, J., DeMasi, R., Bolognesi, D.,2004. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into hostCD4 lymphocytes. Nat. Rev., Drug Discov. 3 (3), 215–225.
McCutchan, F.E., 2006. Global epidemiology of HIV. J. Med. Virol. 78(Suppl. 1), S7–S12.
Miller, C.J., Li, Q., Abel, K., Kim, E.Y., Ma, Z.M., Wietgrefe, S., La Franco-Scheuch, L., Compton, L., Duan, L., Shore, M.D., Zupancic, M., Busch, M.,Carlis, J., Wolinsky, S., Haase, A.T., 2005. Propagation and dissemination ofinfection after vaginal transmission of simian immunodeficiency virus. J.Virol. 79 (14), 9217–9227.
Moore, J.P., Burton, D.R., 2004. Urgently needed: a filter for the HIV-1 vaccinepipeline. Nat. Med. 10 (8), 769–771.
Moore, J.P., Kitchen, S.G., Pugach, P., Zack, J.A., 2004. The CCR5 and CXCR4coreceptors – central to understanding the transmission and pathogenesis ofhuman immunodeficiency virus type 1 infection. AIDS Res. Hum.Retroviruses 20 (1), 111–126.

440 T.J. Ketas et al. / Virology 364 (2007) 431–440
Moyle, G.J., Wildfire, A., Mandalia, S., Mayer, H., Goodrich, J., Whitcomb, J.,Gazzard, B.G., 2005. Epidemiology and predictive factors for chemokinereceptor use in HIV-1 infection. J. Infect. Dis. 191 (6), 866–872.
Olson, W.C., Maddon, P.J., 2003. Resistance to HIV-1 entry inhibitors. Curr.Drug Targets, Infect. Disord. 3 (4), 283–294.
Pope, M., Frankel, S.S., Mascola, J.R., Trkola, A., Isdell, F., Birx, D.L., Burke,D.S., Ho, D.D., Moore, J.P., 1997. Human immunodeficiency virus type 1strains of subtypes B and E replicate in cutaneous dendritic cell-T-cellmixtures without displaying subtype-specific tropism. J. Virol. 71 (10),8001–8007.
Rusert, P., Kuster, H., Joos, B., Misselwitz, B., Gujer, C., Leemann, C., Fischer,M., Stiegler, G., Katinger, H., Olson, W.C., Weber, R., Aceto, L., Gunthard,H.F., Trkola, A., 2005. Virus isolates during acute and chronic humanimmunodeficiency virus type 1 infection show distinct patterns of sensitivityto entry inhibitors. J. Virol. 79 (13), 8454–8469.
Shattock, R.J., Moore, J.P., 2003. Inhibiting sexual transmission of HIV-1infection. Nat. Rev., Microbiol. 1 (1), 25–34.
Tremblay, C.L., Giguel, F., Kollmann, C., Guan, Y., Chou, T.C., Baroudy, B.M.,Hirsch, M.S., 2002. Anti-human immunodeficiency virus interactions ofSCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agentsin vitro. Antimicrob. Agents Chemother. 46 (5), 1336–1339.
Tremblay, C.L., Giguel, F., Chou, T.C., Dong, H., Takashima, K., Hirsch, M.S.,2005a. TAK-652, a novel CCR5 inhibitor, has favourable drug interactionswith other antiretrovirals in vitro. Antiviral Ther. 10 (8), 967–968.
Tremblay, C.L., Giguel, F., Guan, Y., Chou, T.C., Takashima, K., Hirsch, M.S.,2005b. TAK-220, a novel small-molecule CCR5 antagonist, has favorableanti-human immunodeficiency virus interactions with other antiretrovirals invitro. Antimicrob. Agents Chemother. 49 (8), 3483–3485.
Trkola, A., Ketas, T., Kewalramani, V.N., Endorf, F., Binley, J.M., Katinger, H.,Robinson, J., Littman, D.R., Moore, J.P., 1998. Neutralization sensitivity ofhuman immunodeficiency virus type 1 primary isolates to antibodies andCD4-based reagents is independent of coreceptor usage. J. Virol. 72 (3),1876–1885.
Trkola, A., Ketas, T.J., Nagashima, K.A., Zhao, L., Cilliers, T., Morris, L.,Moore, J.P., Maddon, P.J., Olson, W.C., 2001. Potent, broad-spectruminhibition of human immunodeficiency virus type 1 by the CCR5 mono-clonal antibody PRO 140. J. Virol. 75 (2), 579–588.
Veazey, R.S., Klasse, P.J., Schader, S.M., Hu, Q., Ketas, T.J., Lu, M.,Marx, P.A., Dufour, J., Colonno, R.J., Shattock, R.J., Springer, M.S.,Moore, J.P., 2005. Protection of macaques from vaginal SHIV challengeby vaginally delivered inhibitors of virus–cell fusion. Nature 438(7064), 99–102.
Walker, B.D., Korber, B.T., 2001. Immune control of HIV: the obstacles of HLAand viral diversity. Nat. Immunol. 2 (6), 473–475.
Willey, S., Peters, P.J., Sullivan, W.M., Dorr, P., Perros, M., Clapham, P.R.,2005. Inhibition of CCR5-mediated infection by diverse R5 and R5X4HIV and SIV isolates using novel small molecule inhibitors of CCR5:effects of viral diversity, target cell and receptor density. Antiviral Res.68 (2), 96–108.
Yeni, P.G., Hammer, S.M., Hirsch, M.S., Saag, M.S., Schechter, M., Carpenter,C.C., Fischl, M.A., Gatell, J.M., Gazzard, B.G., Jacobsen, D.M.,Katzenstein, D.A., Montaner, J.S., Richman, D.D., Schooley, R.T.,Thompson, M.A., Vella, S., Volberding, P.A., 2004. Treatment for adultHIV infection: 2004 recommendations of the International AIDS Society—USA Panel. JAMA 292 (2), 251–265.
Yi, Y., Shaheen, F., Collman, R.G., 2005. Preferential use of CXCR4 by R5X4human immunodeficiency virus type 1 isolates for infection of primarylymphocytes. J. Virol. 79 (3), 1480–1486.
Zhang, Y.J., Moore, J.P., 1999. Will multiple coreceptors need to be targeted byinhibitors of human immunodeficiency virus type 1 entry? J. Virol. 73 (4),3443–3448.
Zhang, Y.J., Dragic, T., Cao, Y., Kostrikis, L., Kwon, D.S., Littman, D.R.,KewalRamani, V.N., Moore, J.P., 1998. Use of coreceptors other thanCCR5 by non-syncytium-inducing adult and pediatric isolates ofhuman immunodeficiency virus type 1 is rare in vitro. J. Virol. 72 (11),9337–9344.
Copyright © 2022 FDOKUMEN




















