
Electronic properties of atoms and molecules containing one and two negative muons
17
Electronic properties of atoms and molecules containing one and two negative muons F´ elix Moncada a , Daniel Cruz a , Andr´ es Reyes a a Departamento de Qu´ ımica, Universidad Nacional de Colombia, Av. Cra. 30 #45-03, Bogot´a, Colombia Abstract Any-Particle Molecular Orbital/Hartree-Fock (APMO/HF) calculations are performed for a variety of atoms and simple diatomic molecular systems containing one and two negative muons (μ). In these calculations electrons and muons are described quantum mechanically whereas nuclei are treated as point charges. Our results for atoms containing n=1,2 negative muons reveal that electronic properties such as electronic densities and ionization potentials shift to those of all-electron atoms with atomic numbers Z - n. In the case of diatomic molecules these muonic effects are more diverse ranging from transmutation of atomic properties to drastic changes in equilibrium geometries and energies. Keywords: Muonic Chemistry, Muonic Atoms, Muonic Molecules, Any Particle Molecular Orbital, Exotic Particles 1. Introduction The study of atomic and molecular systems containing either positrons and positive muons has received growing attention by the chemistry, atomic and molecular physics and material science communities. This is mainly due to the continuous advances in the experimental techniques for generating and manipulating these subatomic particles, to the unique properties displayed by these exotic systems and to their potential for scientific and technologi- cal applications [1–3]. The vast amount of knowledge accumulated over the years is such that positron and (positive) muon chemistry are nowadays well Email address: [email protected] (Andr´ es Reyes) Preprint submitted to Chemical Physics Letters January 26, 2015
-
Upload
uniamazonia -
Category
Documents
-
view
1 -
download
0
Transcript of Electronic properties of atoms and molecules containing one and two negative muons

Electronic properties of atoms and molecules containing
one and two negative muons
Felix Moncadaa, Daniel Cruza, Andres Reyesa
aDepartamento de Quımica, Universidad Nacional de Colombia,Av. Cra. 30 #45-03, Bogota, Colombia
Abstract
Any-Particle Molecular Orbital/Hartree-Fock (APMO/HF) calculations areperformed for a variety of atoms and simple diatomic molecular systemscontaining one and two negative muons (µ). In these calculations electronsand muons are described quantum mechanically whereas nuclei are treatedas point charges. Our results for atoms containing n=1,2 negative muonsreveal that electronic properties such as electronic densities and ionizationpotentials shift to those of all-electron atoms with atomic numbers Z−n. Inthe case of diatomic molecules these muonic effects are more diverse rangingfrom transmutation of atomic properties to drastic changes in equilibriumgeometries and energies.
Keywords: Muonic Chemistry, Muonic Atoms, Muonic Molecules, AnyParticle Molecular Orbital, Exotic Particles
1. Introduction
The study of atomic and molecular systems containing either positronsand positive muons has received growing attention by the chemistry, atomicand molecular physics and material science communities. This is mainly dueto the continuous advances in the experimental techniques for generating andmanipulating these subatomic particles, to the unique properties displayedby these exotic systems and to their potential for scientific and technologi-cal applications [1–3]. The vast amount of knowledge accumulated over theyears is such that positron and (positive) muon chemistry are nowadays well
Email address: [email protected] (Andres Reyes)
Preprint submitted to Chemical Physics Letters January 26, 2015

defined branches of research in nuclear chemistry [4, 5]. On the other hand,the exploration of systems containing negative muons (denoted as muons orµ in the rest of the document) has mainly attracted the physics commu-nity because of the potential of muons to catalyse nuclear fusion processes.[6–8]. A wide variety of related aspects have been investigated on muonicsystems: muon transfer and capture processes [6, 7, 9, 10], nuclear fusionreaction rates [6, 7, 11], quantum electrodynamics effects (e.g. Lamb shiftsand vacuum polarization) [12, 13], X-ray spectroscopy [14, 15] among oth-ers. In contrast, there is a limited amount of experimental studies on thechemical properties of atomic and molecular systems containing muons. Tothe best of our knowledge there are a number of µ-spin resonance studieson solids [16–20]. The lack of reports on these muonic species is certainly aconsequence of experimental difficulties associated to their preparation andmanipulation [5].
The landscape of muon chemistry drastically changed when Fleming andcowokers made a major breakthrough. In a series of recent papers [21–23]they reported the measurement of reaction rates for the 4He(µ) + H2 →4He(µ)H + H collisional process. Perhaps, the most surprising finding of theirresearch is that He(µ) chemically behaves as a heavy isotope of hydrogen.
Inspired by this finding, we carried out a series of calculations at theAny-Particle Molecular Orbital (APMO) level of theory for muonic atoms.These calculations allowed us to establish that the electronic properties of anatom mimic those of the neighboring all-electron atom with atomic numberZ-1 when one of its electrons is replaced by a muon [24].
Along these lines, two questions naturally arise: 1) Will the transmutationof electronic properties still be effective when two electrons are replaced bytwo muons? 2) Will this transmutation remain when molecules containingmounic atoms are formed?.
In this paper we have tried to answer these two questions. To do so,we have treated the atomic and molecular muonic systems of interest underthe APMO/HF approach. In this non-relativistic method both muons andelectrons are described quantum mechanically in a self-consistent field (SCF)manner. The electronic properties of atomic systems containing two muonsare analyzed in terms of their radial distributions and ionization potentials.The latter are calculated with the recently developed Any-Particle Propa-gator method [25]. In addition, the properties of small molecular systemssuch as H+
2 (µ), H2(µ), H2(2µ), [He(µ)]2, He(µ)H (the number of µ replacingelectrons is shown in parenthesis) are examined in terms of bond strengths
2

and distances and muonic and electronic densities.This paper is organized as follows. In section 2 we summarize the different
approximations utilized in this study. In section 3 we provide some compu-tational details. In section 4 we present the calculated electron and muonradial distributions and ionization potentials for bimuonic atoms and com-pare them with theoretical and experimental results for all-electron atoms.In addition, we present distances, energies, muonic and electronic distribu-tions of H+
2 (µ), H2(µ), H2(2µ), [He(µ)]2, He(µ)H. Finally, in section 5 weprovide some concluding remarks and perspectives.
2. Theoretical Aspects
The wave function and energy expressions of the APMO/HF method[26, 27] are presented below. This non-relativistic approach extends theelectronic HF method to any number and type of quantum species. Notethat APMO approach equations are related to those of multi-componentmolecular orbital (MCMO) [28] and nuclear orbital plus molecular orbital(NOMO) [29] methods.
2.1. APMO Hartree-Fock theory
The Hamiltonian in atomic units of a molecular system containing N e
quantum electrons, Nµ quantum muons and Nnuc nuclei is expressed in termsof kinetic and potential energy operators is
Htot =−Ne∑i
1
2∇2i −
Nµ∑i
1
2mµ
∇2i −
Nnuc∑A
1
2MA
∇2i
+Ne∑i
Ne∑j>i
1
rij+
Nµ∑i
Nµ∑j>i
1
rij+
Nnuc∑A
Nnuc∑B>A
ZAZBrAB
+Ne∑i
Nµ∑j
1
rij−
Ne∑i
Nnuc∑A
ZAriA−
Nµ∑i
Nnuc∑A
ZAriA
. (1)
where mµ = 206.8 and MA and ZA are the mass and the charge of theA nucleus respectively. We consider the nuclei as point charges under theBorn-Oppenheimer approximation (BOA). The resulting Born-OppenheimerHamiltonian HBO is equal to Htot neglecting the nuclear kinetic energy op-erators.
3

At the APMO/HF level, the wave function of the electron-muon system,Ψ0, is expressed as a product of single configurational wave functions
Ψ0 = Φe · Φµ. (2)
Because of their fermionic nature, electronic and muonic wavefunctions, Φe
and Φµ, are represented as Slater determinants of molecular orbitals (MO),ψi. These MOs are obtained solving the one particle APMO/HF equations
f e(i)ψei = εeiψei , i = 1, . . . , N e, (3)
fµ(i)ψµi = εµi ψµi , i = 1, . . . , Nµ. (4)
The above Fock operators f are written as
f e(i) =te(i) +Ne∑j
[Jej −Kej ] +
Nµ∑j
Jµj (5)
fµ(i) =tµ(i) +Nµ∑j
[Jµj −Kµj ] +
Ne∑j
Jej , (6)
where tα(i), Jαi and Kαi (α = e, µ) are the kinetic energy, Coulomb and
exchange operators for particle i of species α.By solving equations 3-4 iteratively converged MOs of electrons and
muons are obtained. These MOs are constructed as linear combinationsof gaussian type functions. As observed in equations 5-6, the effective fieldexperienced by one quantum species depends on the MOs of the other.
2.2. Finite Nuclear Mass Correction (FNMC)
It is well known that use of the BOA for muonic systems may lead tolarge errors in the calculation of total energies [30] because the mass of µ isabout 1/9 that of a proton. To correct for possible errors, we have utilizedthe finite nuclear mass correction (FNMC) proposed by Mohallem [31, 32].The extension of their method to the APMO equations for electrons andmuons is straightfoward. After moving from the laboratory reference frameto the molecular reference frame we arrive to the Hamiltonian [32], We havechanged eq. 7 and added a paragraph to the manuscript:
HFNMC = HBO +Nnuc∑A
PA
( Ne∑ij
∇i · ∇j
2MA
+Ne∑i
Nµ∑j
∇i · ∇j
MA
+Nµ∑ij
∇i · ∇j
2MA
)PA,
(7)
4

where PA is a projector operator onto the wavefunction subspace of muonicatom A. At HF level, muonic atoms wavefunctions are of the form of eq. 2.
The largest correction to the BO energy arises from the muon-muon diag-onal mass polarization terms, because of the muons larger mass. On the otherhand, the inclusion of electron-electron diagonal mass polarization terms isuseful to study nuclear mass effects on electronic properties [31]. In our cal-culations we have utilized the diagonal correction terms to the BOA energybecause in regular electronic structure calculations non-diagonal terms arenegligible far from conical intersections [33]. Therefore in this work we ne-glect muon-muon, electron-electron non-diagonal and muon-electron crossedterms from equation 7.
HFNMC = HBO +Nnuc∑A
PA
( Ne∑i
∇2i
2MA
+Nµ∑i
∇2i
2MA
)PA. (8)
At the APMO-HF level we arrive to a correction matrix, Qα, to each BOFock matrix [31],
FαFNMC = Fα + Qα. (9)
Electronic and muonic correction matrix elements QαijAB are related to the
corresponding kinetic energy matrix elements TαijAB,
QαijAB =
{mαMA
TαijAB if A = B
0 if A 6= B(10)
TαijAB =
⟨φαiA
∣∣∣∣− ∇2
2mα
∣∣∣∣φαjB⟩ , (11)
where φαiA is a basis function of species α centered on nucleus A. This correc-tion compares well with the common diagonal Born-Oppenheimer correction[31].
2.3. Generalized Propagator Theory
In this approach a corrected electronic ionization potential for the pth or-bital, ωep, is obtained from the orbital energy, εep, and a self energy correction,
Σe(2)pp (ωep),
ωep = εep + Σe(2)pp (ωep) (12)
5

Σe(2)pp (ωep) =
∑ae,re>se
|〈peae||rese〉|2
ωep + εea − εer − εes+
∑re,ae>be
|〈pere||aebe〉|2
ωep + εer − εea − εeb
+∑re
∑aµ,rµ
|〈peaµ|rerµ〉|2
ωep + εµa − εer − εµr
+∑ae
∑rµ,aµ
|〈perµ|aeaµ〉|2
ωep + εµr − εea − εµa, (13)
here a and b represent occupied molecular orbitals and r and s representvirtual ones. We will refer to this method as APMO/EP2 [25].
3. Computational details
All calculations presented in this article were performed with the LOWDINcomputational package [34]. In these calculations electrons and negativemuons were described as quantum particles at the APMO/HF level of the-ory including the FNMC and atomic nuclei were treated as point charges.The nuclear mass of the most abundant isotope was considered for each ele-ment. In all calculations even-tempered basis sets containing thirteen s-typeprimitive functions [24] were employed for muons and uncontracted primi-tives of the Dunning cc-pVTZ basis sets were employed for electrons [35–37].Preliminary calculations for atoms containing n muons revealed that lowertotal energies could be achieved by employing the electronic basis set of theatom with atomic number Z−n in place of that for atomic number Z. There-fore, we opted for using this choice of electronic basis sets in our calculations[24].
For diatomic molecules, we have solved the vibrational Schrodinger equa-tion for a Morse potential,
〈Ψ0 | HFNMC | Ψ0〉 ≈ De
(1− e−a(r−re)
)(14)[
− 1
2mred
d2
dr2+De
(1− e−a(r−re)
)]Ψvib = VZPEΨvib, (15)
where r is the variable corresponding to the internuclear distance and mred
is the reduced mass of the two nuclei. APMO/HF potential energy curveswere fitted to Morse potentials. De, a and re fitted values can be found insupporting information. Vibrational zero-point energies (VZPE) and averagebond distances 〈r〉 were calculated solving Eq. 15 [38].
6
0
100
200
300
400
500
600
10−4 10−3 10−2 10−1 1000.0
0.1
0.2
0.3
0.4
0.5
0.6m
uoni
c ra
dial
dis
trib
utio
n fu
nctio
n / a
.u.
elec
tron
ic r
adia
l dis
trib
utio
n fu
nctio
n / a
.u.
Distance / Å
µ in Li(2µ singlet)e in Li(2µ singlet)µ in Li(2µ triplet)e in Li(2µ triplet)
e in H
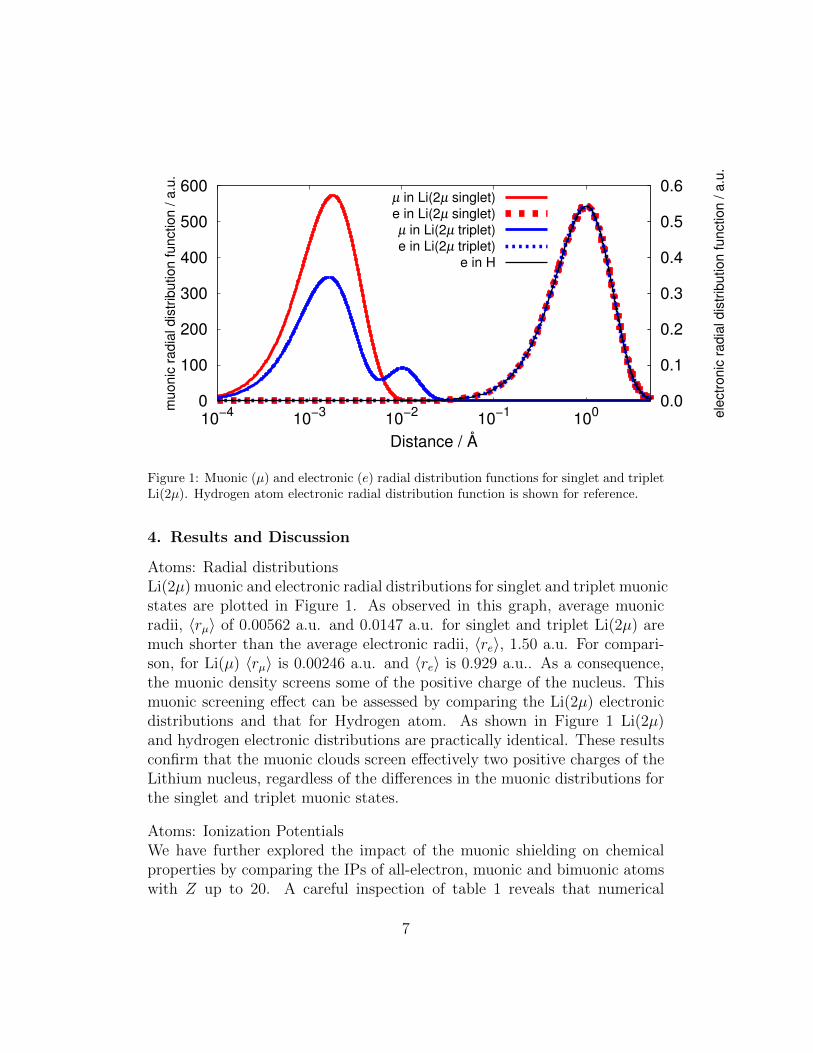
Figure 1: Muonic (µ) and electronic (e) radial distribution functions for singlet and tripletLi(2µ). Hydrogen atom electronic radial distribution function is shown for reference.
4. Results and Discussion
Atoms: Radial distributionsLi(2µ) muonic and electronic radial distributions for singlet and triplet muonicstates are plotted in Figure 1. As observed in this graph, average muonicradii, 〈rµ〉 of 0.00562 a.u. and 0.0147 a.u. for singlet and triplet Li(2µ) aremuch shorter than the average electronic radii, 〈re〉, 1.50 a.u. For compari-son, for Li(µ) 〈rµ〉 is 0.00246 a.u. and 〈re〉 is 0.929 a.u.. As a consequence,the muonic density screens some of the positive charge of the nucleus. Thismuonic screening effect can be assessed by comparing the Li(2µ) electronicdistributions and that for Hydrogen atom. As shown in Figure 1 Li(2µ)and hydrogen electronic distributions are practically identical. These resultsconfirm that the muonic clouds screen effectively two positive charges of theLithium nucleus, regardless of the differences in the muonic distributions forthe singlet and triplet muonic states.
Atoms: Ionization PotentialsWe have further explored the impact of the muonic shielding on chemicalproperties by comparing the IPs of all-electron, muonic and bimuonic atomswith Z up to 20. A careful inspection of table 1 reveals that numerical
7

Table 1: APMO-EP2 ionization potentials of all-electron, muonic and bimuonic (singlet,1Φµ, and triplet ,3Φµ, muonic states) atoms in eV.
No. Atom Exp APMO Muonic APMO Bimuonic APMO IPe- IPa IP atom IP atom 1Φµ 3Φµ
1 H 13.598 13.593 He(µ) 13.599 Li(2µ) 13.600 13.6022 He 24.587 24.546 Li(µ) 24.548 Be(2µ) 24.549 24.5533 Li 5.392 5.375 Be(µ) 5.375 B(2µ) 5.375 5.3764 Be 9.323 8.937 B(µ) 8.937 C(2µ) 8.937 8.9385 B 8.298 8.411 C(µ) 8.411 N(2µ) 8.412 8.4116 C 11.260 11.315 N(µ) 11.316 O(2µ) 11.316 11.3157 N 14.534 14.450 O(µ) 14.450 F(2µ) 14.451 14.4508 O 13.618 12.969 F(µ) 12.969 Ne(2µ) 12.969 12.9689 F 17.423 16.412 Ne(µ) 16.412 Na(2µ) 16.412 16.411
10 Ne 21.565 20.157 Na(µ) 20.157 Mg(2µ) 20.157 20.15611 Na 5.139 4.985 Mg(µ) 4.985 Al(2µ) 4.985 4.98612 Mg 7.646 7.355 Al(µ) 7.355 Si(2µ) 7.355 7.35613 Al 5.986 5.933 Si(µ) 5.933 P(2µ) 5.933 5.93314 Si 8.152 8.141 P(µ) 8.141 S(2µ) 8.141 8.14115 P 10.487 10.522 S(µ) 10.523 Cl(2µ) 10.523 10.52216 S 10.360 10.053 Cl(µ) 10.053 Ar(2µ) 10.053 10.05317 Cl 12.968 12.637 Ar(µ) 12.636 K(2µ) 12.636 12.63618 Ar 15.760 15.450 K(µ) 15.451 Ca(2µ) 15.451 15.451
a Experimental IPs taken from Ref. [39]
differences in IPs of all-electron, muonic and bimuonic atoms are only ashigh as 9 meV.
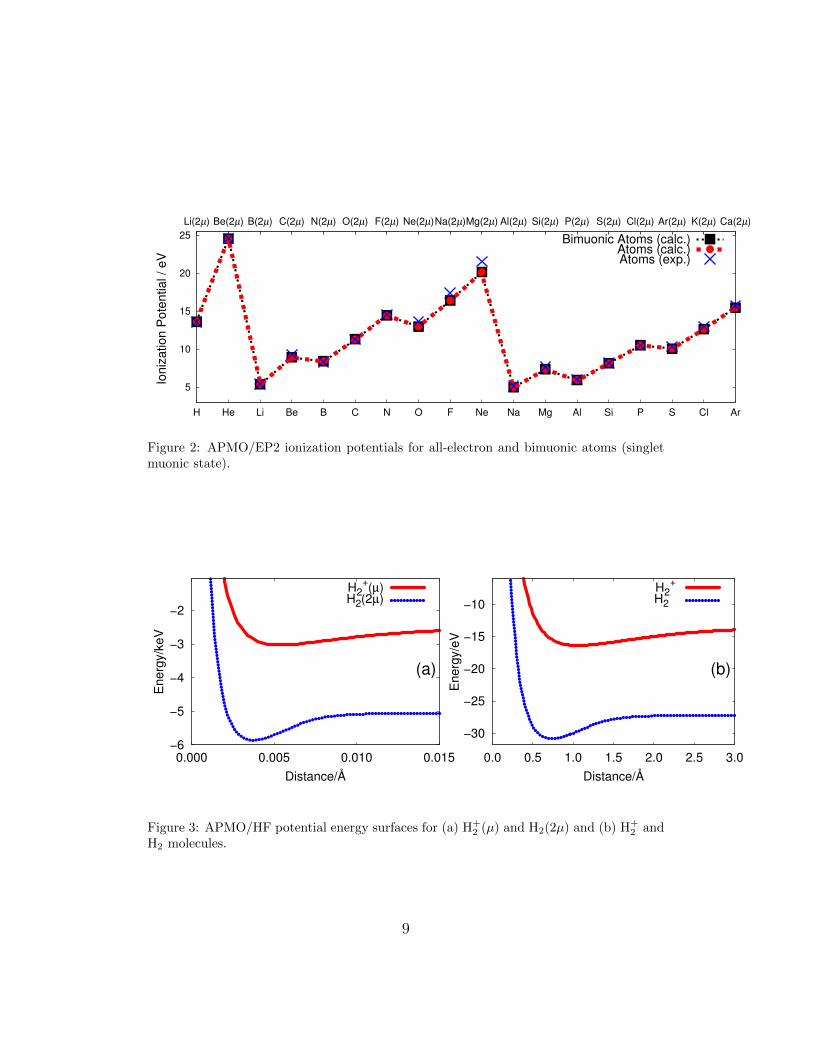
The IPs of bimuonic atoms are contrasted to calculated and experimen-tal IPs of all-electron atoms in Figure 2. It is brought to light that theIPs of muonic atoms match those of all-electron atoms after shifting themto the left by two atomic numbers. For instance, muonic noble gases areBe(2µ), Mg(2µ) and Ca(2µ); muonic alkaline metals are B(2µ) and Al(2µ);and muonic halogens are Na(2µ) and K(2µ).
Overall results presented so far confirm that by replacing two electrons ofan atom by two muons there is a transmutation of its electronic propertiesto those of an all-electron atom with atomic number Z-2.
8
5
10
15
20
25
H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar
Li(2µ) Be(2µ) B(2µ) C(2µ) N(2µ) O(2µ) F(2µ) Ne(2µ)Na(2µ)Mg(2µ) Al(2µ) Si(2µ) P(2µ) S(2µ) Cl(2µ) Ar(2µ) K(2µ) Ca(2µ)
Ioniz
ation P
ote
ntial / eV
Bimuonic Atoms (calc.)Atoms (calc.)Atoms (exp.)
Figure 2: APMO/EP2 ionization potentials for all-electron and bimuonic atoms (singletmuonic state).
−6
−5
−4
−3
−2
0.000 0.005 0.010 0.015
Ene
rgy/
keV
Distance/Å
H2+(µ)
H2(2µ)
−30
−25
−20
−15
−10
0.0 0.5 1.0 1.5 2.0 2.5 3.0
Ene
rgy/
eV
Distance/Å
(a) (b)
H2+
H2
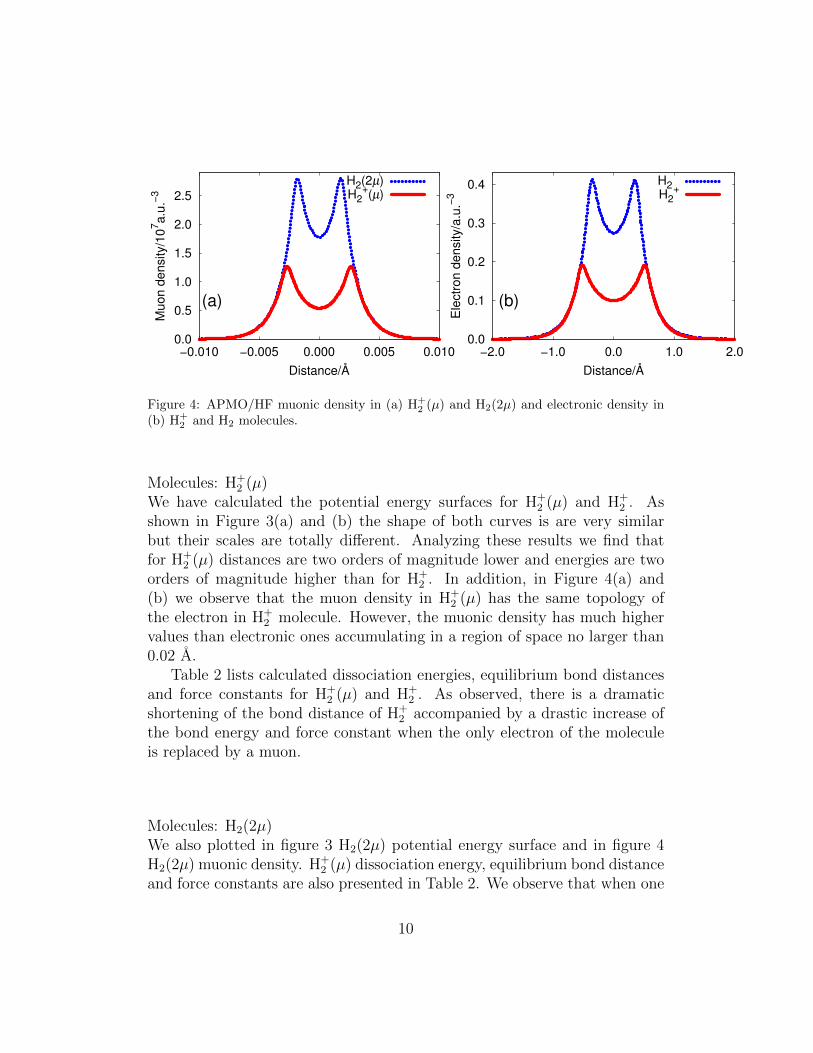
Figure 3: APMO/HF potential energy surfaces for (a) H+2 (µ) and H2(2µ) and (b) H+
2 andH2 molecules.
9
0.0
0.5
1.0
1.5
2.0
2.5
−0.010 −0.005 0.000 0.005 0.010
Muo
n de
nsity
/107 a.
u.−3
Distance/Å
H2(2µ)H2
+(µ)
0.0
0.1
0.2
0.3
0.4
−2.0 −1.0 0.0 1.0 2.0
Ele
ctro
n de
nsity
/a.u
.−3
Distance/Å
(b)(a)
H2 H2
+
Figure 4: APMO/HF muonic density in (a) H+2 (µ) and H2(2µ) and electronic density in
(b) H+2 and H2 molecules.
Molecules: H+2 (µ)
We have calculated the potential energy surfaces for H+2 (µ) and H+
2 . Asshown in Figure 3(a) and (b) the shape of both curves is are very similarbut their scales are totally different. Analyzing these results we find thatfor H+
2 (µ) distances are two orders of magnitude lower and energies are twoorders of magnitude higher than for H+
2 . In addition, in Figure 4(a) and(b) we observe that the muon density in H+
2 (µ) has the same topology ofthe electron in H+
2 molecule. However, the muonic density has much highervalues than electronic ones accumulating in a region of space no larger than0.02 A.
Table 2 lists calculated dissociation energies, equilibrium bond distancesand force constants for H+
2 (µ) and H+2 . As observed, there is a dramatic
shortening of the bond distance of H+2 accompanied by a drastic increase of
the bond energy and force constant when the only electron of the moleculeis replaced by a muon.
Molecules: H2(2µ)We also plotted in figure 3 H2(2µ) potential energy surface and in figure 4H2(2µ) muonic density. H+
2 (µ) dissociation energy, equilibrium bond distanceand force constants are also presented in Table 2. We observe that when one
10

Table 2: Force constants (ke), equilibrium bond distances (r0) and dissociation energiesD0 for small molecular systems containing muons. Results for H+
2 and H2 are presentedfor reference.
System ke/ eVA−2 r0/A D0/eVH+
2 10.1 1.086 2.647H2(µ)+ 7.26×107 8.635 ×10−3 260.9H2(2µ) 3.24×108 6.676 ×10−3 400.9
H2 39.8 0.760 3.359He(µ)H 40.1 0.755 3.414[He(µ)]2 40.2 0.747 3.492H2(µ) 7.260×107 8.635 ×10−3 260.9
extra µ is added to the H+2 (µ) complex there is an accumulation of muonic
density between the nuclei which makes the molecular bond even shorter (29%) and stronger (54 %) than that in H+
2 (µ).
Molecules: H2(µ)Unlike H2(2µ), the effects of adding an electron to H2(µ)+ are negligibleon the bond distance and force constant. These results can be understoodby analyzing the electronic density in H2(µ) plotted in Figure 5. Therewe observe that the electron orbits further apart from the H2(µ)+ complexbecause of its lighter mass. The accumulation of electronic density in theregion between nuclei is so small that its effects on the H-µ-H bond areinsignificant. In Figure 5 the electronic density of the hydrogen atom ispresented as a reference. It overlaps perfectly with the electronic density ofH2(µ). In an effort to unveil this result, we have to recall that the H2(µ)+
complex is so tightly packed that the extra electron on average feels theattraction of a positive charge such as that of a proton. As shown in theupper box in figure 5 differences in electronic densities only arise in theregion around the two nuclei.
Along these lines, we have calculated the electronic IP of H2(µ) (13.597eV) finding that it is practically identical to that of the hydrogen atom(13.593 eV). This result further confirms that the electron in H2(µ) feelsone net nuclear positive charge. All the above evidence allow us to concludethat the H2(µ) molecule will chemically behave as another heavier isotope ofhydrogen.
11

0.0
0.1
0.2
0.3
−2.0 −1.5 −1.0 −0.5 0.0 0.5 1.0 1.5 2.0
Ele
ctro
n de
nsity
/a.u
.−3
Distance/Å
H2(µ)H
0.284
0.286
0.288
−0.02 0.00 0.02
Figure 5: APMO/HF electronic density for H2(µ). For reference electronic density of aH atom is also displayed. The densities around the H+
2 (µ) moeity are zoomed in at theupper left corner box. H nuclei in H2(µ) are depicted as black dots.
The H2(µ)+ complex acting as a hydrogen nucleus has been reported instudies of collisional processes and resonant formation [41], hyperfine struc-ture calculation [42–44] and ortho-para transitions [45, 46]. However, noclear reference to chemical or electronic properties is given is these studies.
Results presented in Ref. [24] for He(µ) confirm the finding of Flemingand coworkers that He(µ) reacts as an isotope of Hydrogen [21–23]. Newresults presented above allow us to predict that Li(2µ) and H2(µ) will alsochemically behave as isotopes of hydrogen.
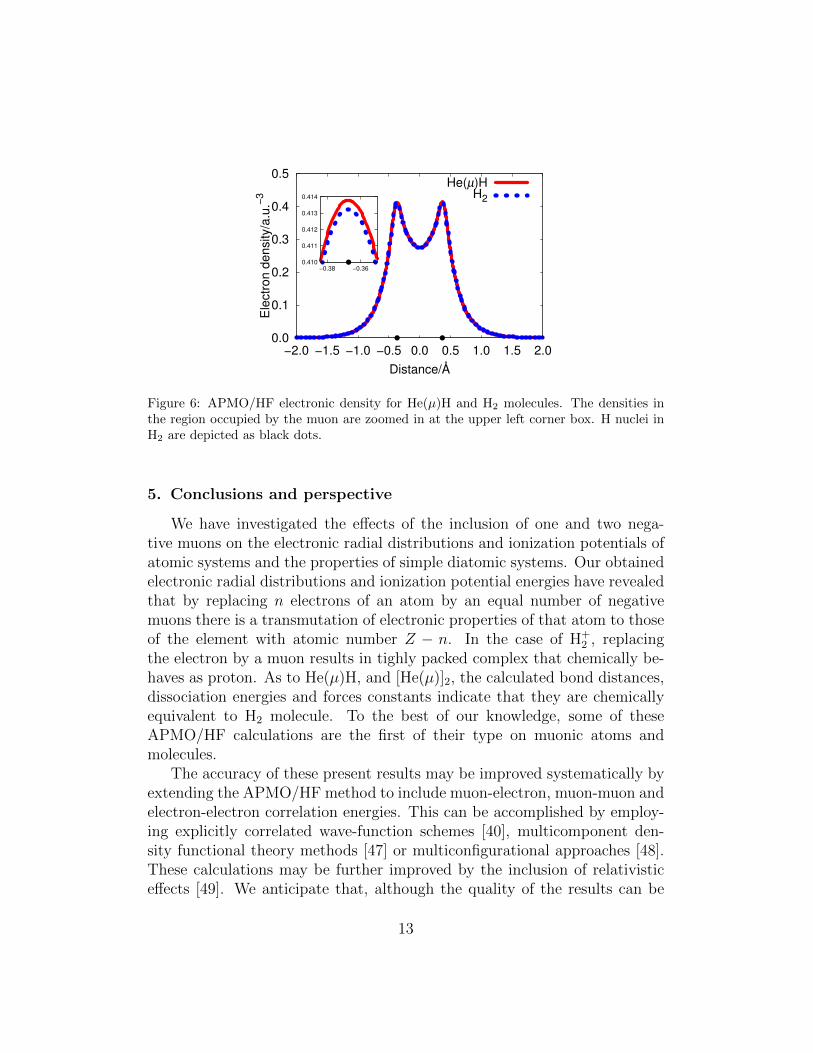
Molecules: He(µ)H and [He(µ)]2We now study the properties of He(µ)H which is the resulting product ofthe reaction of He(µ) and H2 [21–23]. The results for this molecule arecontrasted with those obtained for H2 and for the hypothetical [He(µ)]2 intable 2. As observed, the bond distance, force constant and dissociationenergies of He(µ)H, H2 and [He(µ)]2 are practically identical. In addition, wehave plotted in figure 6 the electronic densities of He(µ)H and H2 and foundthey overlap perfectly except for the region occupied by the muon. Theseresults allow us to conclude that He(µ)H, H2 and [He(µ)]2 are chemicallyequivalent.
12
0.0
0.1
0.2
0.3
0.4
0.5
−2.0 −1.5 −1.0 −0.5 0.0 0.5 1.0 1.5 2.0
Ele
ctro
n de
nsity
/a.u
.−3
Distance/Å
He(µ)HH2
0.410
0.411
0.412
0.413
0.414
−0.38 −0.36
Figure 6: APMO/HF electronic density for He(µ)H and H2 molecules. The densities inthe region occupied by the muon are zoomed in at the upper left corner box. H nuclei inH2 are depicted as black dots.
5. Conclusions and perspective
We have investigated the effects of the inclusion of one and two nega-tive muons on the electronic radial distributions and ionization potentials ofatomic systems and the properties of simple diatomic systems. Our obtainedelectronic radial distributions and ionization potential energies have revealedthat by replacing n electrons of an atom by an equal number of negativemuons there is a transmutation of electronic properties of that atom to thoseof the element with atomic number Z − n. In the case of H+
2 , replacingthe electron by a muon results in tighly packed complex that chemically be-haves as proton. As to He(µ)H, and [He(µ)]2, the calculated bond distances,dissociation energies and forces constants indicate that they are chemicallyequivalent to H2 molecule. To the best of our knowledge, some of theseAPMO/HF calculations are the first of their type on muonic atoms andmolecules.
The accuracy of these present results may be improved systematically byextending the APMO/HF method to include muon-electron, muon-muon andelectron-electron correlation energies. This can be accomplished by employ-ing explicitly correlated wave-function schemes [40], multicomponent den-sity functional theory methods [47] or multiconfigurational approaches [48].These calculations may be further improved by the inclusion of relativisticeffects [49]. We anticipate that, although the quality of the results can be
13

improved by employing one of the above methods, the qualitative featuresdiscovered in this study will be conserved. We are currently implementingsome of these approaches in the LOWDIN package.
ACKNOWLEDGEMENTSWe thank Jonathan Romero and Prof. Eduardo Chamorro for helpfuldiscussions. We gratefully acknowledge the nancial support of UniversidadNacional de Colombia (Quipu 201010016739).
References
[1] D. L. Bailey, D. W. Townsend, P. E. Valk, M. N. Maisey, PositronEmission Tomography: Basic Sciences, Springer, 2005.
[2] A. Dupasquier, A. Mills Jr., Positron Spectroscopy of Solids, IOSPress, 1995.
[3] A. G. E. Schenck, Muon Spin Rotation Spectroscopy Principles andApplications in Solid State Physics, Taylor & Francis, 1985.
[4] Y. C. Jean, P. E. Mallon, D. M. Schrader, Principles and Applicationsof Positron and Positronium Chemistry, World Scientific Publishing,2003.
[5] D. C. Walker, Muon and Muonium Chemistry, Cambridge UniversityPress, 2009.
[6] K. Nagamine, Introductory Muon Science, Cambridge UniversityPress, 2003.
[7] P. Froelich, Adv. Phys. 41 (1992) 405–508.
[8] K. Ishida, K. Nagamine, T. Matsuzaki, N. Kawamura, J. Phys. G 29(2003) 2043–2046.
[9] J. Cohen, Rep. Prog. Phys. 67 (2004) 1769–1819.
[10] J. Cohen, Phys. Rev. A 59 (1999) 1160–1169.
[11] R. Gheisari, Nucl. Instrum. Meth. A 634 (2011) 1–4.
[12] S. G. Karshenboim, Phys. Rep. 422 (2005) 1–63.
14

[13] R. Pohl, A. Antognini, F. Nez, F. Amaro, F. Biraben, J. Cardoso,D. Covita, A. Dax, S. Dhawan, L. Fernandes, Nature 466 (2010)213–216.
[14] D. Gotta, Prog. Part. Nucl. Phys. 52 (2004) 133–195.
[15] K. Ninomiya, T. U. Ito, W. Higemoto, M. Kita, A. Shinohara,T. Nagatomo, K. Kubo, P. Strasser, N. Kawamura, K. Shimomura,Y. Miyake, T. Miura, J. Korean Phys. Soc. 59 (2011) 2917–2920.
[16] T. Yamazaki, K. Nagamine, S. Nagamiya, O. Hashimoto, K. Sugimoto,K. Nakai, S. Kobayashi, Phys. Scr. 11 (1975) 133–139.
[17] K. Nagamine, Hyperfine Interact. 6 (1979) 347–355.
[18] T. Yamazaki, Hyperfine Interact. 8 (1981) 463–469.
[19] E. Torikai, K. Nagamine, K. Nishiyama, E. Hirose, P. Birrer,I. Tanaka, H. Kojima, S. Srinivas, T. Das, S. Maekawa, HyperfineInteract. 97-98 (1996) 387–394.
[20] T. Mamedov, D. Andrianov, D. Gerlach, K. Gritsai, V. Gorelkin,O. Cormann, J. Major, A. Stoikov, M. Shevchik, U. Zimmerman,JETP Lett. 71 (2000) 438–441.
[21] D. J. Arseneau, D. G. Fleming, O. Sukhorukov, J. H. Brewer, B. C.Garrett, D. G. Truhlar, Physica B 404 (2009) 946–949.
[22] D. Fleming, D. Arseneau, O. Sukhorukov, J. Brewer, S. Mielke,G. Schatz, B. Garrett, K. Peterson, D. Truhlar, Science 331 (2011)448–450.
[23] D. G. Fleming, D. J. Arseneau, O. Sukhorukov, J. H. Brewer, S. L.Mielke, D. G. Truhlar, G. C. Schatz, B. C. Garrett, K. A. Peterson, J.Chem. Phys. 135 (2011) 184310.
[24] F. Moncada, E. Posada, R. Flores-Moreno, A. Reyes, Chem. Phys. 400(2012) 103–107.
[25] J. Romero, E. Posada, R. Flores-Moreno, A. Reyes, J. Chem. Phys.137 (2012) 074105.
15

[26] S. Gonzalez, N. Aguirre, A. Reyes, Int. J. Quantum Chem. 108 (2008)1742–1749.
[27] S. Gonzalez, A. Reyes, Int. J. Quantum Chem. 110 (2010) 689–696.
[28] M. Tachikawa, K. Mori, H. Nakai, K. Iguchi, Chem. Phys. Lett. 290(1998) 437–442.
[29] H. Nakai, Int. J. Quantum Chem. 86 (2002) 511–517.
[30] S. Takahashi, K. Takatsuka, J. Chem. Phys. 124 (2006) 144101.
[31] C. P. Goncalves, J. R. Mohallem, Theor. Chem. Acc. 110 (2003)367–370.
[32] J. R. Mohallem, L. G. Diniz, A. S. Dutra, Chem. Phys. Lett. 501(2011) 575–579.
[33] N. C. Handy, A. M. Lee, Chem. Phys. Lett. 252 (1996) 425–430.
[34] R. Flores-Moreno, S. A. Gonzalez, N. F. Aguirre, E. F. Posada,J. Romero, F. Moncada, J. Charry, M. Diaz-Tinoco, K. Pineda-Urbina,A. Reyes, Lowdin: A general code for the treatment of any quantumparticle, 2012. URL:https://sites.google.com/site/lowdinproject/.
[35] T. H. Dunning, J. Chem. Phys. 90 (1989) 1007–1023.
[36] D. E. Woon, T. H. Dunning, J. Chem. Phys. 98 (1993) 1358–1371.
[37] D. E. Woon, T. H. Dunning, J. Chem. Phys. 100 (1994) 2975–2988.
[38] P. M. Morse, Phys. Rev. 34 (1929) 57–64.
[39] W. L. Jolly, Modern Inorganic Chemistry, 2nd ed., McGraw-Hill, 1991.
[40] M. Cafiero, S. Bubin, L. Adamowicz, Phys. Chem. Chem. Phys. 5(2003) 1491–1501.
[41] A. Scrinzi, P. Kammel, J. Zmeskal, J. Marton, M. P. Faifman,L. I.Ponomarev,T. A. Strizh, Nuc. Phys. A 47 (1993) 4961–4704.
16

[42] M. R. Harston, I. Shimamura, M. Kamimura, Phys. Rev. A 45 (1992)94–100.
[43] M. R. Harston,S. Hara, Y. Kino, I. Shimamura,H. Sato,M. Kamimura,Phys. Rev. A 56 (1997) 2685–2691.
[44] D. Bakalov, K. Bakalova, V. Korobov, H. J. Monkhorst,I. Shimamura,Phys. Rev. A 57 (1998) 3370–3375.
[45] D. D. Bakalov, M. P. Faifman, L. I, Ponomarev, S. I. Vinitsky, Nuc.Phys. A 384 (1982) 302–322.
[46] S. S. Gershtein, A. V. Luchunsky, Phys. Atomic Nuclei 65 (2002)102–108.
[47] T. Kreibich, E. Gross, Phys. Rev. Lett. 86 (2001) 2984–2987.
[48] T. Ishimoto, M. Tachikawa, U. Nagashima, J. Chem. Phys. 128 (2008)164118.
[49] B. Fricke, Phys. Rev. Lett. 30 (1973) 119–122.
17




















