
New generations of boron-doped diamond structures by delta ...
Upload
independentCategory
view
0download
0
Chemosphere 87 (2012) 1126–1133
Contents lists available at SciVerse ScienceDirect
Chemosphere
journal homepage: www.elsevier .com/locate /chemosphere
Electrochemical incineration of sulfanilic acid at a boron-doped diamond anode
Abdellatif El-Ghenymy, Conchita Arias, Pere Lluís Cabot, Francesc Centellas, José Antonio Garrido,Rosa María Rodríguez, Enric Brillas ⇑Laboratori d’Electroquímica dels Materials i del Medi Ambient, Departament de Química Física, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1-11,08028 Barcelona, Spain
a r t i c l e i n f o
Article history:Received 13 December 2011Received in revised form 1 February 2012Accepted 2 February 2012Available online 25 February 2012
Keywords:Anodic oxidationBoron-doped diamondOxidation intermediatesSulfanilic acidWater treatment
0045-6535/$ - see front matter � 2012 Elsevier Ltd. Adoi:10.1016/j.chemosphere.2012.02.006
⇑ Corresponding author. Tel.: +34 934021223; fax:E-mail address: [email protected] (E. Brillas).
a b s t r a c t
The anodic oxidation of sulfanilic acid solutions has been studied in acidic medium using a divided cellwith a boron-doped diamond (BDD) anode and a stainless steel cathode. Overall mineralization wasachieved under all experimental conditions tested due to the efficient destruction of sulfanilic acid andall its by-products with hydroxyl radicals generated at the BDD anode from water oxidation. The alter-native use of an undivided cell with the same electrodes gave rise to the coating of the cathode with poly-meric compounds, thus preventing the complete electrochemical incineration of sulfanilic acid. Thesolutions treated in the anodic compartment of the divided cell were degraded at similar rate underpH regulation within the pH interval 2.0–6.0. The mineralization current efficiency was enhanced whenthe applied current decreased and the initial substrate concentration increased. The decay of sulfanilicacid was followed by reversed-phase HPLC, showing a pseudo first-order kinetics. Hydroquinone andp-benzoquinone were identified as aromatic intermediates by gas chromatography-mass spectrometryand/or reversed-phase HPLC. Maleic, acetic, formic, oxalic and oxamic acids were detected as generatedcarboxylic acids by ion-exclusion HPLC. Ionic chromatographic analysis of electrolyzed solutions revealedthat the N content of sulfanilic acid was mainly released as NHþ4 ion and in much smaller proportion asNO�3 ion.
� 2012 Elsevier Ltd. All rights reserved.
1. Introduction react with most organics yielding their mineralization (Flox et al.,
The contamination of water bodies by man-made organicchemicals is a critical issue that recent water framework directivesfrom all countries are trying to address for achieving good waterquality status and healthy ecosystems. A large variety of chemicals,dyes, pesticides and drugs are found as pollutants in industrialwastewaters, as well as at low contents of lg L�1 in soils, surfacewaters, ground waters and even drinking waters (Martínez-Huitleand Brillas, 2008). Although physicochemical methods and biore-mediation are the most extended technologies for the removal ofcolor and organic load from the contaminated effluents, advancedoxidation processes (AOPs) are gaining attention because they canshorten the treatments and be coupled to renewable energysources to enhance their performance and decrease the cost(Andreozzi et al., 1999; Cañizares et al., 2008; Sirés and Brillas,2012). AOPs are chemical, photochemical, photocatalytic and elec-trochemical methods involving the in situ generation of �OH asmain oxidizing agent (Boye et al., 2002; Pera-Titus et al., 2004;Panizza and Cerisola, 2009). The �OH radical has so high standardreduction potential (E� = 2.80 V vs SHE) that can non-selectively
ll rights reserved.
+34 934021231.
2005; Özcan et al., 2008).Electro-oxidation or anodic oxidation (AO) is the most popular
electrochemical AOP for water remediation. In this procedure,the organic matter contained in a contaminated solution is oxi-dized by direct charge transfer at the anode (M) and reaction withphysisorbed hydroxyl radicals (M(�OH)) formed from water oxida-tion by reaction (1) (Martínez-Huitle and Ferro, 2006):
MþH2O!Mð�OHÞ þHþ þ e� ð1Þ
However, the effectiveness of AO depends largely on the anodematerial. When active anodes like Pt, IrO2 and RuO2 are utilized,the corresponding M(�OH) is oxidized to a chemisorbed ‘‘superox-ide’’ species with weaker oxidizing power only allowing the electro-chemical conversion of organics to carboxylic acids (Gao et al., 2009;Liu et al., 2009; Brillas et al., 2010). In contrast, the physisorbedM(�OH) radicals become stable in non-active anodes like PbO2 andboron-doped diamond (BDD) causing the electrochemical incinera-tion of organics (Ammar et al., 2006; Lei et al., 2010; Zhao et al.,2010). Among these anodes, BDD electrodes are preferred for AO be-cause they interact very weakly with �OH resulting in a much greaterO2-overvoltage than other anodes and in an enhancement of organicremoval with reactive BDD(�OH) (Panizza and Cerisola, 2007, 2009).Thus, BDD is potent enough to mineralize aromatic pollutants fromwaters (Marselli et al., 2003; Carvalho et al., 2007; Sáez et al., 2007;

A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133 1127
Santos et al., 2010) and their generated carboxylic acids (Cañizareset al., 2003; Garcia-Segura and Brillas, 2011) and possesses muchhigher oxidation power than other common anodes such as Pt (Siréset al., 2006; Hamza et al., 2009; Liu et al., 2009) and PbO2 (Sirés et al.,2008; Flox et al., 2009). Nevertheless, most of AO treatments withBDD are performed with undivided cells discarding the possiblereduction of organics at the cathode, a process that can be veryimportant for sulfonated compounds (Hastie et al., 2006).
Sulfanilic acid or 4-aminobenzenesulfonic acid is widely used inthe production of sulfonamide pharmaceuticals, pesticides, sulfo-nated azo dyes, dye mordants, species and food pigments. It is toxicand carcinogenic. Its high water solubility favors its accumulationin rivers and surface waters when it is produced, for example, fromthe reduction of sulfonated azo dyes under anaerobic conditions(Coughlin et al., 2003; Yemashova et al., 2004; Tan et al., 2005),although it can be very slowly biodegraded by different bacterialstrains under aerobic conditions (Perei et al., 2001; Singh et al.,2006; Wang et al., 2009; Ming et al., 2011). It has been reportedthe slow destruction of sulfanilic acid by ozonation (Gul et al.,1999), as well as by TiO2 photocatalysis (Hu et al., 2007) and Fen-ton reagent (Marciocha et al., 2009) when it is formed as interme-diate in the degradation of the antimicrobial sulfamethoxazole andby AO (Carvalho et al., 2007) and electro-Fenton (Hammami et al.,2008) in the removal of Acid Orange 7. Recently, Santos et al.(2010) described the almost total mineralization of aniline andseveral sulfonated derivatives, including sulfanilic acid, at theirnatural pH using AO with an undivided BDD/stainless steel (SS)cell. However, they discarded the possible formation of cathodicby-products, which can affect significantly the oxidation powerof BDD for the removal of sulfanilic acid.
This paper presents an extensive and careful study on the AO ofsulfanilic acid solutions in the anodic compartment of a dividedBDD/SS tank reactor. The influence of pH, applied current and sub-strate concentration on the degradation rate, mineralization de-gree and mineralization current efficiency (MCE) was examined.The kinetics of sulfanilic acid decay was monitored by HPLC. Stableintermediates were identified by gas chromatography-massspectrometry (GC–MS) and the evolution of aromatic by-products,generated carboxylic acids and released inorganic nitrogen ionswas followed by different LC techniques. From these results, aplausible reaction sequence for sulfanilic acid mineralizationinvolving measured intermediates is proposed.
2. Experimental
2.1. Chemicals
Sulfanilic acid (>99% purity) of reagent grade supplied by Sigma–Aldrich was used without further purification. Oxalic, oxamic,maleic, acetic and formic acids were of reagent grade purchasedfrom Merck and Panreac. Anhydrous sodium sulfate, used as back-ground electrolyte, and sulfuric acid and sodium hydroxide, usedto regulate the solution pH, were of analytical grade supplied byMerck. Solutions were prepared with high-purity water obtainedfrom a Millipore Milli-Q system with resistivity >18 MX cm at25 �C. Organic solvents and other chemicals used were either ofHPLC or analytical grade purchased from Merck, Fluka and Avocado.
2.2. Electrolytic system
A scheme of the system used for the AO treatment of sulfanilicacid is depicted in Fig. 1. The electrolyses were conducted in adivided and cylindrical tank reactor of 150 mL capacity with a dou-ble jacket in which external thermostated water recirculated tomaintain the solution temperature using a Thermo Electron
Corporation HAAKE DC 10 thermostat. Since the efficiency of AO isenhanced with increasing temperature (Panizza and Cerisola,2009), all experiments were performed at 35 �C, which is the max-imum temperature allowed by the open tank reactor with insignif-icant water evaporation during prolonged electrolysis. The anodiccompartment contained the treated solution and a 3 cm2 BDDthin-film electrode provided by Adamant Technologies. The catho-dic compartment consisted of a glass tube with its bottom sealedwith a glass filter separator (porosity number 2) in contact withthe treated solution. It was filled with 10 mL of the same back-ground electrolyte where a SS (AISI 304) wire was immersed ascathode. All the assays were performed at constant current providedwith an Amel 2053 potentiostat–galvanostat and the potential dif-ference between electrodes was measured with a Demestres 6005digital multimeter. Some experiments were also performed with asimilar undivided tank reactor placing both, the same BDD anodeand a SS sheet cathode of 3 cm2 area in 100 mL of the treated solu-tion with an interelectrode gap of 1 cm. Before the use of the BDDanode in the electrolytic trials, it was polarized in the undivided cellcontaining a 0.05 M Na2SO4 solution at 300 mA for 60 min to re-move the impurities of its surface.
Solutions of 100 mL of 240 mg L�1 sulfanilic acid (100 mg L�1 ofdissolved organic carbon (DOC)) with 0.5 M Na2SO4 in the pH range2.0–6.0, initially adjusted with H2SO4, were degraded in the anodiccompartment of the divided cell by AO at 200 mA. The influence ofapplied current between 50 and 450 mA and substrate concentra-tion between 240 and 2530 mg L�1 on the oxidation power of theprocess at pH 3.0 was examined. This pH value was chosen becauseit is close to the natural pH of 3.23 of sulfanilic acid solutions(Santos et al., 2010). In all the trials, the solution was stirred witha magnetic bar at 700 rpm to ensure its mixing and the transport ofreactants towards/from the electrodes. Since the treated solutionsin the anodic compartment were rapidly acidified, their pH wascontinuously regulated to their initial values by adding small vol-umes of 0.1 M NaOH.
2.3. Instruments and analytical procedures
The solution pH was determined on a Crison GLP 22 pH-meter.The aliquots withdrawn from electrolyzed solutions were filteredwith 0.45 lm PTFE filters purchased from Whatman before analy-sis. The mineralization of sulfanilic acid solutions was monitoredfrom their DOC decay, measured with a Shimadzu VCSN TOC ana-lyzer. Reproducible DOC values with an accuracy of ±1% werefound by injecting 50 lL aliquots to the analyzer. From these data,the MCE value for each trial at current I (A) and time t (h) was thenestimated from Eq. (2) (Skoumal et al., 2008):
MCEð%Þ ¼nF V sDðDOCÞexp
4:32� 107 mI t� 100 ð2Þ
where F is the Faraday constant (96,487 C mol�1), Vs is the solutionvolume (L), D(DOC)exp is the experimental DOC decay (mg L�1),4.32 � 107 is an homogenization factor (3600 s h�1 � 12000 mgmol�1) and m is the number of carbon atoms of sulfanilic acid(6 atoms). The number of electrons (n) consumed per molecule wastaken as 28 assuming that sulfanilic acid is completely mineralizedto carbon dioxide and sulfate and ammonium ions from reaction (3):
C6H7NO3Sþ 13H2O! 6CO2 þ NHþ4 þ SO2�4 þ 29Hþ þ 28e� ð3Þ
The formation of sulfate ion is expected from the oxidationbehavior of sulfonated compounds (Panizza and Cerisola, 2007;Hammami et al., 2008; Borràs et al., 2011). However, only a partialconversion of the initial nitrogen into NHþ4 ion was detected, as willbe discussed below.
The kinetics for sulfanilic acid decay and the evolution of aromaticintermediates were followed by reversed-phase HPLC using a

− +
V
Thermostat
Power supply
Multimeter
Magnetic stirrer
Magnetic bar
Water inlet
Water outlet
Stainless steel cathode
Boron-doped diamond anode
Cathodic compartment
Anodic compartment
Glass filter separator
Fig. 1. Sketch of the experimental set-up used for the anodic oxidation (AO) of sulfanilic acid solutions with a divided boron-doped diamond (BDD)/stainless steel (SS) cell.
1128 A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133
Shimadzu LC-10 ADvp fitted with a Thermo Electron CorporationHypersil ODS 5 lm, 150 mm� 3 mm (id), column at room tempera-ture and coupled with a Shimadzu ACvp photodiode array detectorset at k = 249 nm for sulfanilic acid, k = 289 nm for hydroquinoneand k = 246 nm for p-benzoquinone. The mobile phase was a 5:95(v/v) acetonitrile/water (phosphate buffer of pH 3) mixture at0.6 mL min�1. Generated carboxylic acids were detected and quanti-fied by ion-exclusion HPLC with a Waters 600 LC fitted with a Bio-Rad Aminex HPX 87H, 300 mm� 7.8 mm (id), column at 35 �C andcoupled with a Waters 996 photodiode array detector selected atk = 210 nm, using 4 mM H2SO4 at 0.6 mL min�1 as mobile phase. TheNHþ4 concentration in electrolyzed solutions was determined by ionicchromatography with a Shimadzu 10 Avp LC fitted with a Shodex ICYK-421, 125 mm� 4.6 mm (id), cation column at 40 �C coupled witha Shimadzu CDD 10 Avp conductivity detector. The mobile phase wasa 5.0 mM tartaric acid, 2.0 mM dipicolinic acid, 24.2 mM boric acid and1.50 mM crown ether solution at 1.0 mL min�1. The NO�3 content wasobtained with the above LC system fitted with a Shim-Pack IC-A1S,100 mm� 4.6 mm (id), anion column at 40 �C using a 2.5 mM phthalicacid and 2.4 mM tris(hydroxymethyl)aminomethane solution at1.5 mL min�1 as mobile phase.
To identify the stable intermediates formed during sulfanilicacid degradation, the organic components of short-time electro-lyzed solutions were extracted three times with dichloromethaneusing 15 mL each. The resulting organic solution was dried overNa2SO4, filtered and rotavaporated to reduce its volume up toabout 1 mL for its further analysis by GC–MS. This technique wascarried out with a Hewlett–Packard system composed of a HP5890 Series II GC fitted with a polar HP-Innowax 0.25 lm,30 m � 0.25 mm (id), column and a HP 5989A MS in electron im-pact mode at 70 eV. The temperature ramp was 50 �C for 3 min,5 �C min�1 up to 260 �C and hold time 20 min. The temperatureof the injector and detector was 240 and 260 �C, respectively. Themass spectra were identified through a NIFT05 data library.
3. Results and discussion
3.1. Effect of cathodic by-products and pH on sulfanilic acidmineralization
A first assay was performed by electrolyzing 100 mL of a240 mg L�1 sulfanilic acid solution in 0.05 M Na2SO4 at natural
pH of 3.2, 200 mA and 35 �C using an undivided BDD/SS cell(similar to that of Fig. 1 but without the cathodic compartment)with electrodes of 3 cm2 apparent area. While its DOC decayed rap-idly and the solution pH became slightly acidified, it was observedthe progressive formation of a black coating on the cathode, whichcan be related to polymeric compounds generated from the catho-dic reduction of sulfanilic acid and/or its intermediates. This meansthat the electrochemical incineration of the substrate by theBDD(�OH) radicals produced at the anode from reaction (1) cannotbe correctly assessed using an undivided cell because part of theorganic matter is removed via a reduction process at the cathodewithout being mineralized.
From the above considerations, the AO behavior of sulfanilicacid solutions was investigated in the anodic compartment of thedivided BDD/SS tank reactor shown in Fig. 1, where organics aredestroyed under the action of BDD(�OH). Nevertheless, the glass fil-ter separator between the anodic and cathodic compartments ofthe divided cell originated so large potential penalty that no highcurrents could be applied from the 30 V power supply for low con-tents of Na2SO4 like 0.05 M. To solve this problem, the conductivityof the treated solution was increased using 0.5 M Na2SO4 as back-ground electrolyte. Note that no significant change in the oxidationpower of BDD on the organic matter is expected in AO by raisingthe Na2SO4 content up to 0.5 M, since greater amount of back-ground electrolyte only increases the conductivity with the con-comitant decay in ohmic drop of the solution (Flox et al., 2006).Another problem arose from the protons released during the waterdischarge to O2 at the BDD anode initiated by reaction (1) and fol-lowed by reaction (4) (Skoumal et al., 2008; Panizza and Cerisola,2009):
2BDDð�OHÞ ! 2BDDþ O2ðgÞ þ 2Hþ þ 2e� ð4Þ
Since these anodically generated protons are not neutralized bytheir consumption (or OH� production) at the cathode, the solutionof the anodic compartment was continuously acidified. This wassolved by regulating the solution pH at its initial value by additionof 0.1 M NaOH.
In view of the need of pH regulation when a sulfanilic acid solu-tion is treated in a divided cell, a series of trials were made to clar-ify the role of this parameter over the oxidation power of BDD.Fig. 2 depicts a very rapid and quite similar DOC abatement for a240 mg L�1 substrate solution with 0.5 M Na2SO4 in the pH range2.0–6.0 operating at 200 mA and 35 �C. This trend indicates the
0
20
40
60
80
100
120
0 60 120 180 240 300 360 420
DO
C /
mg
L-1
Time / min
0
10
20
30
40
0 3 6 9 12 15
% M
CE
Q / A h L-1
(a)
0
20
40
60
80
100
120(b)
(c)
DO
C /
mg
L-1
Fig. 3. DOC abatement with (a) electrolysis time and (b) consumed specific chargefor 100 mL of a 240 mg L�1 sulfanilic acid solution in 0.5 M Na2SO4 at pH 3.0 and35 �C by means of AO in the anodic compartment of a divided BDD/SS cell. Applied
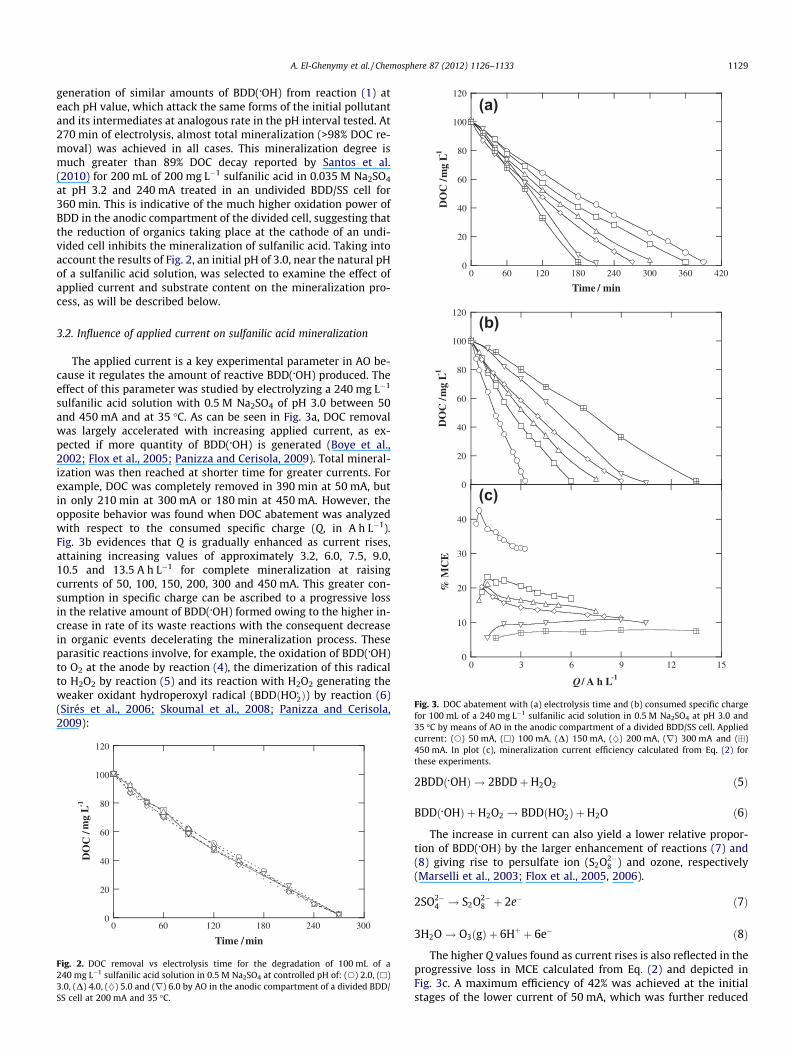
A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133 1129
generation of similar amounts of BDD(�OH) from reaction (1) ateach pH value, which attack the same forms of the initial pollutantand its intermediates at analogous rate in the pH interval tested. At270 min of electrolysis, almost total mineralization (>98% DOC re-moval) was achieved in all cases. This mineralization degree ismuch greater than 89% DOC decay reported by Santos et al.(2010) for 200 mL of 200 mg L�1 sulfanilic acid in 0.035 M Na2SO4
at pH 3.2 and 240 mA treated in an undivided BDD/SS cell for360 min. This is indicative of the much higher oxidation power ofBDD in the anodic compartment of the divided cell, suggesting thatthe reduction of organics taking place at the cathode of an undi-vided cell inhibits the mineralization of sulfanilic acid. Taking intoaccount the results of Fig. 2, an initial pH of 3.0, near the natural pHof a sulfanilic acid solution, was selected to examine the effect ofapplied current and substrate content on the mineralization pro-cess, as will be described below.
3.2. Influence of applied current on sulfanilic acid mineralization
The applied current is a key experimental parameter in AO be-cause it regulates the amount of reactive BDD(�OH) produced. Theeffect of this parameter was studied by electrolyzing a 240 mg L�1
sulfanilic acid solution with 0.5 M Na2SO4 of pH 3.0 between 50and 450 mA and at 35 �C. As can be seen in Fig. 3a, DOC removalwas largely accelerated with increasing applied current, as ex-pected if more quantity of BDD(�OH) is generated (Boye et al.,2002; Flox et al., 2005; Panizza and Cerisola, 2009). Total mineral-ization was then reached at shorter time for greater currents. Forexample, DOC was completely removed in 390 min at 50 mA, butin only 210 min at 300 mA or 180 min at 450 mA. However, theopposite behavior was found when DOC abatement was analyzedwith respect to the consumed specific charge (Q, in A h L�1).Fig. 3b evidences that Q is gradually enhanced as current rises,attaining increasing values of approximately 3.2, 6.0, 7.5, 9.0,10.5 and 13.5 A h L�1 for complete mineralization at raisingcurrents of 50, 100, 150, 200, 300 and 450 mA. This greater con-sumption in specific charge can be ascribed to a progressive lossin the relative amount of BDD(�OH) formed owing to the higher in-crease in rate of its waste reactions with the consequent decreasein organic events decelerating the mineralization process. Theseparasitic reactions involve, for example, the oxidation of BDD(�OH)to O2 at the anode by reaction (4), the dimerization of this radicalto H2O2 by reaction (5) and its reaction with H2O2 generating theweaker oxidant hydroperoxyl radical (BDDðHO�2Þ) by reaction (6)(Sirés et al., 2006; Skoumal et al., 2008; Panizza and Cerisola,2009):
0
20
40
60
80
100
120
0 60 120 180 240 300
Time / min
DO
C /
mg
L-1
Fig. 2. DOC removal vs electrolysis time for the degradation of 100 mL of a240 mg L�1 sulfanilic acid solution in 0.5 M Na2SO4 at controlled pH of: (s) 2.0, (h)3.0, (D) 4.0, (}) 5.0 and (r) 6.0 by AO in the anodic compartment of a divided BDD/SS cell at 200 mA and 35 �C.
current: (s) 50 mA, (h) 100 mA, (D) 150 mA, (}) 200 mA, (r) 300 mA and (�)450 mA. In plot (c), mineralization current efficiency calculated from Eq. (2) forthese experiments.
2BDDð�OHÞ ! 2BDDþH2O2 ð5Þ
BDDð�OHÞ þH2O2 ! BDDðHO�2Þ þH2O ð6Þ
The increase in current can also yield a lower relative propor-tion of BDD(�OH) by the larger enhancement of reactions (7) and(8) giving rise to persulfate ion (S2O2�
8 ) and ozone, respectively(Marselli et al., 2003; Flox et al., 2005, 2006).
2SO2�4 ! S2O2�
8 þ 2e� ð7Þ
3H2O! O3ðgÞ þ 6Hþ þ 6e� ð8Þ
The higher Q values found as current rises is also reflected in theprogressive loss in MCE calculated from Eq. (2) and depicted inFig. 3c. A maximum efficiency of 42% was achieved at the initialstages of the lower current of 50 mA, which was further reduced

1130 A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133
to 31% at the end of the treatment. Fig. 3c also shows the same ten-dency, but with decreasing MCE values, operating up to 200 mA. Incontrast, the efficiency at higher currents remained practically con-stant, achieving the lowest value of 7% at 450 mA. These findingssuggest that the degradation of sulfanilic acid until 200 mA yieldsby-products that are more difficultly oxidized with BDD(�OH) atlong electrolysis time, whereas the application of higher currentsproduces sufficient amount of this oxidant to quickly destroy suchspecies, leading to a constant efficiency during the AO treatment.
3.3. Effect of sulfanilic acid concentration on the AO behavior
The influence of sulfanilic acid concentration on its mineraliza-tion process was also checked. To do this, solutions containing be-tween 240 and 2530 mg L�1 of this compound and 0.5 M Na2SO4 ofpH 3.0 were comparatively degraded by AO at 200 mA and 35 �C.Fig. 4a evidences the high oxidation ability of the BDD anode in thisprocedure because all tested solutions were completely mineral-ized by prolonging the electrolysis time as the substrate contentincreased. Thus, the time needed for total mineralization variedfrom 270 min at 240 mg L�1 to 600 min at 2530 mg L�1. Thisbehavior can be simply accounted for by the slower destructionof more organic matter by a similar quantity of BDD(�OH) formedat the same current. Nevertheless, at a given time higher amountof DOC was removed at greater substrate content, making the pro-cess more efficient since it is mass-transfer controlled at the highcurrent of 200 mA (Flox et al., 2005). For example, at 240 minDOC was reduced to 97, 167, 233, 300, 357, 378, 397, 440 and462 mg L�1 starting from 240, 480, 720, 960, 1200, 1440, 1730,1970 and 2530 mg L�1 of sulfanilic acid, respectively. This trendsuggests that the parasitic reactions of electrogenerated BDD(�OH),like reactions (4)–(6), become slower since a larger proportion of
0
200
400
600
800
1000
1200
DO
C /
mg
L-1
0
10
20
30
40
50
60
0 120 240 360 480 600 720
Time / min
% M
CE
(a)
(b)
Fig. 4. Change of (a) DOC and (b) mineralization current efficiency calculated fromEq. (2) with electrolysis time for the AO of 100 mL of sulfanilic acid solutions in0.5 M Na2SO4 at pH 3.0 in the anodic compartment of a divided BDD/SS cell at200 mA and 35 �C. Substrate concentration: (r) 240 mg L�1, (�) 480 mg L�1, (})720 mg L�1, (N) 960 mg L�1, (D) 1200 mg L�1, (j) 1440 mg L�1, (h) 1730 mg L�1,(d) 1970 mg L�1 and (s) 2530 mg L�1.
this radical destroys greater amounts of organics, thus enhancingtheir mineralization.
Fig. 4b depicts the expected increase in MCE with raising sulfa-nilic acid concentration calculated from Eq. (2) for the experimentsof Fig. 4a. The highest MCE obtained in these trials was as high as66%, attained after 360 min of the AO treatment of 2530 mg L�1,confirming the great oxidation power of this method to mineralizehigh contents of this compound. Fig. 4b also shows that for concen-trations >480 mg L�1 the efficiency presents a maximal that isshifted to longer times with increasing substrate content. This ten-dency suggests that from the first stages of each process to its max-imum efficiency, an increasing amount of intermediates is oxidizedto CO2 after more time as greater organic matter is present in solu-tion. In contrast, at long electrolysis times, the efficiency decaysbecause of the formation of by-products that are more hardly oxi-dized with BDD(�OH), as pointed out above.
3.4. Kinetics for sulfanilic acid decay
The kinetics for the reaction between sulfanilic acid and electro-generated BDD(�OH) was followed by reversed-phase HPLC, where itdisplayed a well-defined peak at retention time (tr) of 1.48 min.Fig. 5a presents the concentration decay of this compound for a240 mg L�1 substrate solution in 0.5 M Na2SO4 of pH 3.0 at currentsranging between 100 and 450 mA. As can be seen, sulfanilic acid wascompletely removed more rapidly at higher current owing to thegeneration of more amount of BDD(�OH) from reaction (1). Compar-ison of these results with those of Fig. 3a corroborates that at eachcurrent the initial compound disappears at much shorter time thanthat required for its total mineralization, indicating the formation ofoxidation by-products that are more difficultly destroyed at theBDD anode. The above concentration decays were analyzed usingkinetic equations related to simple reaction orders and excellent lin-ear correlations were only found for a pseudo first-order reaction, asshown in the inset panel of Fig. 5a. From this analysis, an increasingpseudo first-order rate constant (k1) of 3.4 � 10�4 s�1 (R2 = 0.989) at100 mA, 4.3 � 10�4 s�1 (R2 = 0.990) at 200 mA, 6.2 � 10�4 s�1
(R2 = 0.988) at 300 mA and 8.6 � 10�4 s�1 (R2 = 0.983) at 450 mAwas obtained. This behavior suggests the attack of sulfanilic acidwith a constant amount of BDD(�OH) at each applied current. How-ever, k1 only rises 2.5-fold when the current increases 4.5 times,indicating a loss of relative proportion of oxidant BDD(�OH) by thelarger enhancement of its waste reactions (4)–(6).
The effect of sulfanilic acid concentration on its decay kinetics isexemplified in Fig. 5b for three solutions of pH 3.0 treated at200 mA. These results show that the BDD anode was potent enoughto completely remove the higher content of 2530 mg L�1 in450 min, a time lower than 600 min needed for the total mineraliza-tion of its intermediates (see Fig. 4a). The inset panel of Fig. 5b pre-sents the good straight lines obtained if sulfanilic acid verifies apseudo first-order reaction, with decreasing k1 values of4.3 � 10�4 s�1 (R2 = 0.990) at 240 mg L�1, 1.5 � 10�4 s�1 (R2 =0.989) at 1200 mg L�1 and 8.4 � 10�5 s�1 (R2 = 0.980) at2530 mg L�1. Although k1 decreases with increasing substrate con-tent, an inspection of Fig. 5b indicates, for example at 60 min of elec-trolysis, the concomitant destruction of more concentration ofsulfanilic acid, as expected if greater amounts of generatedBDD(�OH) attack this compound.
3.5. Detection and evolution of intermediates
Several solutions of 240 mg L�1 of sulfanilic acid with 0.5 MNa2SO4 of pH 3.0 were electrolyzed by AO for 10 min to detectthe primary aromatic intermediates by GC–MS. The mass spectraonly revealed the formation of hydroquinone [m/e = 110 (100,M+)] at tr = 44.1 min, coming from the hydroxylation of the initial

0
400
800
1200
1600
2000
2400
2800
0 60 120 180 240 300 360 420 480 540
Time / min
0
50
100
150
200
250
0 20 40 60 80 100 120 140 160
[Sul
fani
lic a
cid]
/ m
g L-1
0.0
0.5
1.0
1.5
2.0
2.5
0 20 40 60 80 100 120ln
(C
0 / C
)
Time / min
0.0
0.5
1.0
1.5
2.0
0 60 120 180 240 300
ln (
C0
/ C )
Time / min
(a)
(b)
[Sul
fani
lic a
cid]
/ m
g L-1
Fig. 5. Effect of the applied current and substrate content on the decay of sulfanilicacid concentration by AO for 100 mL of a solution with 0.5 M Na2SO4 at pH 3.0 and35 �C in the anodic compartment of a divided BDD/SS cell. (a) Substrate concen-tration: 240 mg L�1; current: (h) 100 mA, (}) 200 mA, (r) 300 mA and (�) 450 mA.(b) Sulfanilic acid concentration: (r) 240 mg L�1, (D) 1200 mg L�1 and (s)2530 mg L�1; current: 200 mA. The inset panels present the corresponding kineticanalysis assuming that sulfanilic acid follows a pseudo first-order reaction.
0
1
2
3
4
0 60 120 180 240 300
0 60 120 180 240 300 360 420
Time / min
[Ino
rgan
ic n
itro
gen
ions
] / m
g L-1
Con
cent
rati
on/ m
g L-1
(b) 0.00
0.04
0.08
0.12
0.16(a)
(c)
0
2
4
6
8
10
12
14
16
Fig. 6. Time course of the concentration of: (a) p-benzoquinone and (b) ultimatecarboxylic acids detected during the AO treatment of 100 mL of a 240 mg L�1
sulfanilic acid solution in 0.5 M Na2SO4 at pH 3.0 in the anodic compartment of adivided BDD/SS cell at 200 mA and 35 �C. (c) Evolution of released inorganicnitrogen ions during the similar degradation of the same solution, but with 0.05 MNa2SO4 and at 50 mA. Compound: (r) p-benzoquinone, (s) oxalic acid, (h) oxamicacid, (D) NHþ4 ion and (}) NO�3 ion.
A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133 1131
compound. Reversed-phase chromatograms recorded during theabove electrolyses exhibited peaks associated with hydroquinoneat tr = 3.5 min and its oxidation derivative p-benzoquinone attr = 5.3 min. Both aromatics were unequivocally identified by com-paring their retention times and UV–Vis spectra, measured on thephotodiode array detector, with those of pure standards. Hydroqui-none was rapidly oxidized because only traces were found for50 min of electrolysis. In contrast, p-benzoquinone was compara-tively accumulated in larger extent, attaining a maximum amountof 0.15 mg L�1 in solution about 60 min of AO treatment, as can beseen in Fig. 6a. This by-product persisted up to 135 min, a timemuch longer than 80 min needed for the total disappearance of sul-fanilic acid (see Fig. 5a), indicating that p-benzoquinone possessesa much lower reactivity with BDD(�OH).
A different behavior was detected for generated carboxylic acids.Ion-exclusion chromatograms of the above electrolyzed solutiondisplayed defined peaks related to oxalic (tr = 6.9 min), maleic(tr = 8.2 min), oxamic (tr = 9.4 min), formic (tr = 13.7 min) and acetic(tr = 15.6 min) acids. Maleic and acetic acids are expected to beformed from the cleavage of the benzenic ring of intermediates, fur-ther being transformed into oxalic and formic acids (Marselli et al.,2003; Flox et al., 2006; Brillas et al., 2010). Oxamic acid could thenbe generated from the breaking of aromatic moieties containing a –NH2 group. Note that oxalic, oxamic and formic acids are ultimatecarboxylic acids that are directly oxidized to CO2 (Skoumal et al.,2008; Garcia-Segura and Brillas, 2011).
Maleic, acetic and formic acids were poorly accumulated(<0.8 mg L�1), quickly disappearing from the medium between 90and 180 min of treatment. In contrast, Fig. 6b evidences that oxalic
and oxamic acids were totally removed at 270 min, time in whichthe solution was completely mineralized (see Fig. 3a). Oxalic acidwas continuously accumulated to 4.2 mg L�1 at 180 min, where-upon it was rapidly removed, whereas oxamic acid reached an al-most quasi-steady content near 1 mg L�1 for 180 min before itsfast disappearance. A simple mass balance at 210 min revealed thatthe concentration of both acids only corresponded to 1.1 mg L�1 ofDOC, a value much lower than 17.5 mg L�1 DOC found for the trea-ted solution (see Fig. 3a). This indicates that a higher amount ofother undetected recalcitrant organics is present in solution at longelectrolysis time.
The mineralization of N-compounds by AO is expected to beaccompanied by the release of inorganic ions like NO�3 and NHþ4(Flox et al., 2005; Ammar et al., 2006; Hammami et al., 2008).The generation of both ions during the degradation of 240 mg L�1
sulfanilic acid (19.4 mg L�1 of N) at pH 3.0 was confirmed by ionic

Fig. 7. Proposed reaction sequence for the electrochemical incineration of sulfanilic acid at a BDD anode.
1132 A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133
chromatography, without detecting the presence of other inor-ganic nitrogen ions such as NO�2 . However, this assay was madewith 0.05 M Na2SO4 because higher contents of this salt causedthe overlapping of the chromatographic peak of Na+ or SO2�
4 ionwith that of NHþ4 or NO�3 ion, respectively. The use of this low con-centration of background electrolyte then limited the applied cur-rent to 50 mA corresponding to the maximum potential differenceof 30 V that was able to provide the power supply utilized. Fig. 6cshows that under these electrolytic conditions, NHþ4 ion was accu-mulated in a much larger extent than NO�3 ion, as proposed in reac-tion (3). At 360 min of electrolysis, the solution contained14.4 mg L�1 of the former ion (58% of initial N) and 1.8 mg L�1 ofthe latter one (2% of initial N). The fact that only a 60% of initialN is converted into inorganic ions at such time when about 91%of DOC has been removed from the solution (see Fig. 3a) suggeststhat a large part of the initial N content remain in the recalcitrantby-products of the solution (9% of initial DOC) or is lost as volatileN-compounds, probably NxOy species.
3.6. Proposed reaction sequence for sulfanilic acid mineralization
On the basis of by-products detected, the reaction sequence ofFig. 7 is proposed for the electrochemical incineration of sulfanilicacid at a BDD anode. In this pathway, the BDD(�OH) radical gener-ated from reaction (1) is considered the oxidizing agent of organics.
The sequence is initiated by the attack of this oxidant on theC(1)- and C(4)-positions of sulfanilic acid yielding the dihydroxyl-ated derivative hydroquinone with loss of SO2�
4 and primordiallyNHþ4 ions. Hydroquinone is subsequently oxidized to p-benzoqui-none, which is degraded to a mixture of carboxylic acids likemaleic and acetic from the cleavage of its benzenic ring. Theseacids can also be produced from the parallel destruction of unde-tected intermediates formed from other reactions of the substratewith BDD(�OH). The loss of SO2�
4 ion alone from sulfanilic acid givesrise to N-derivatives that generate oxamic acid, which is then min-eralized losing NHþ4 and NO�3 ions (Garcia-Segura and Brillas,2011). Further oxidation of maleic and acetic acids yields the ulti-mate oxalic and formic acids that are directly transformed intoCO2.
4. Conclusions
It has been demonstrated that solutions of sulfanilic acid inacidic medium were completely mineralized in the anodic com-partment of a divided BDD/SS tank reactor by the oxidation actionof BDD(�OH) generated from water oxidation by reaction (1). Incontrast, the use of an undivided BDD/SS cell did not allow the totalmineralization of this compound since the cathode was coated bypolymeric compounds generated from the reduction of sulfanilicacid and/or its intermediates. A similar degradation rate was found

A. El-Ghenymy et al. / Chemosphere 87 (2012) 1126–1133 1133
in the pH range 2.0–6.0 using the divided cell, indicating the oxida-tion of the same forms of the initial pollutant and its intermediatesat analogous rate in all pH values tested. The increase in currentenhanced the mineralization process, but with the consumptionof more specific charge and the drop of mineralization current effi-ciency by the larger acceleration of parasitic reactions of BDD(�OH).Higher contents of sulfanilic acid yielded a lower percentage ofDOC removal with a greater efficiency due to the inhibition ofnon-oxidizing reactions of BDD(�OH). Sulfanilic acid was removedfollowing a pseudo first-order kinetics, with a rate constant that in-creased with increasing the applied current and with decreasingthe initial substrate content. Hydroquinone and p-benzoquinonewere identified as aromatic intermediates and maleic, acetic, for-mic, oxalic and oxamic acids were detected as generated carboxylicacids. The initial N was mainly released as NHþ4 ion and in muchsmaller proportion as NO�3 ion.
Acknowledgments
The authors are grateful to MICINN (Ministerio de Ciencia eInnovación, Spain) by financial support under Project CTQ2010-16164/BQU, co-financed with FEDER funds, and the grant givento A. El Ghenymy to do this work.
References
Ammar, S., Abdelhedi, R., Flox, C., Arias, C., Brillas, E., 2006. Electrochemicaldegradation of the dye indigo carmine at boron-doped diamond anode forwastewaters remediation. Environ. Chem. Lett. 4, 229–233.
Andreozzi, R., Caprio, V., Insola, A., Marotta, R., 1999. Advanced oxidation processes(AOP) for water purification and recovery. Catal. Today 53, 51–59.
Boye, B., Michaud, P.A., Marselli, B., Dieng, M.M., Brillas, E., Comninellis, Ch., 2002.Anodic oxidation of 4-chlorophenoxyacetic acid on synthetic boron-dopeddiamond electrodes. New Diamond Front. Carbon Technol. 12, 63–72.
Borràs, N., Arias, C., Oliver, R., Brillas, E., 2011. Mineralization of desmetryne byelectrochemical advanced oxidation processes using a boron-doped diamondanode and an oxygen-diffusion cathode. Chemosphere 85, 1167–1175.
Brillas, E., Garcia-Segura, S., Skoumal, M., Arias, C., 2010. Electrochemicalincineration of diclofenac in neutral aqueous medium by anodic oxidationusing Pt and boron-doped diamond anodes. Chemosphere 79, 605–612.
Cañizares, P., García-Gómez, J., Lobato, J., Rodrigo, M.A., 2003. Electrochemicaloxidation of aqueous carboxylic acid wastes using diamond thin-filmelectrodes. Ind. Eng. Chem. Res. 42, 956–962.
Cañizares, P., Paz, R., Sáez, C., Rodrigo, M.A., 2008. Electrochemical oxidation ofalcohols and carboxylic acids with diamond anodes. A comparison with otheradvanced oxidation processes. Electrochim. Acta 53, 2144–2153.
Carvalho, C., Fernandes, A., Lopes, A., Pinheiro, H., Gonçalves, I., 2007.Electrochemical degradation applied to the metabolites of Acid Orange 7anaerobic biotreatment. Chemosphere 67, 1316–1324.
Coughlin, M.F., Kinkle, B.K., Bishop, P.L., 2003. High performance degradation of azodye Acid Orange 7 and sulfanilic acid in a laboratory scale reactor after seedingwith cultured bacterial strains. Water Res. 37, 2757–2763.
Flox, C., Arias, C., Brillas, E., Savall, A., Groenen-Serrano, K., 2009. Electrochemicalincineration of cresols: a comparative study between PbO2 and boron-dopeddiamond anodes. Chemosphere 74, 1340–1347.
Flox, C., Cabot, P.L., Centellas, F., Garrido, J.A., Rodríguez, R.M., Arias, C., Brillas, E.,2006. Electrochemical combustion of herbicide mecoprop in aqueous mediumusing a flow reactor with a boron-doped diamond anode. Chemosphere 64,892–902.
Flox, C., Garrido, J.A., Rodríguez, R.M., Centellas, F., Cabot, P.L., Arias, C., Brillas, E.,2005. Degradation of 4,6-dinitro-o-cresol from water by anodic oxidation witha boron-doped diamond electrode. Electrochim. Acta 50, 3685–3692.
Gao, J., Zhao, G., Liu, M., Li, D., 2009. Mechanism of enhanced electrochemicaloxidation of 2,4-dichlorophenoxyacetic acid with in situ microwave activatedboron-doped diamond and platinum anodes. J. Phys. Chem. A 113, 10466–10473.
Garcia-Segura, S., Brillas, E., 2011. Mineralization of the recalcitrant oxalic andoxamic acids by electrochemical advanced oxidation processes using a boron-doped diamond anode. Water Res. 45, 2975–2984.
Gul, S., Serindag, O., Boztepe, H., 1999. Effects of ozonation on COD elimination ofsubstituted aromatic compounds in aqueous solution. Turk. J. Chem. 23, 21–26.
Hammami, S., Bellakhal, N., Oturan, N., Oturan, M.A., Dachraoui, M., 2008.Degradation of Acid Orange 7 by electrochemically generated �OH radicals inacidic aqueous medium using a boron-doped diamond or platinum anode: amechanistic study. Chemosphere 73, 678–684.
Hamza, M., Abdelhedi, R., Brillas, E., Sirés, I., 2009. Comparative electrochemicaldegradation of the triphenylmethane dye Methyl Violet with boron-dopeddiamond and Pt anodes. J. Electroanal. Chem. 627, 41–50.
Hastie, J., Bejan, D., Teutli-Leon, M., Bunce, N.J., 2006. Electrochemical methods fordegradation of Orange II (sodium 4-(2-hydroxy-1-naphthylazo)benzenesulfonate). Ind. Eng. Chem. Res. 45, 4898–4904.
Hu, L., Flanders, P.M., Miller, P.L., Strathmann, T.J., 2007. Oxidation ofsulfamethoxazole and related antimicrobial agents by TiO2 photocatalysis.Water Res. 41, 2621–2626.
Lei, Y., Zhao, G., Zhang, Y., Liu, M., Liu, L., Lv, B., Gao, J., 2010. Highly efficient andmild electrochemical incineration: Mechanism and kinetic process of refractoryaromatic hydrocarbon pollutants on superhydrophobic PbO2 anode. Environ.Sci. Technol. 44, 7921–7927.
Liu, L., Zhao, G., Wu, M., Lei, Y., Geng, R., 2009. Electrochemical degradation ofchlorobenzene on boron-doped diamond and platinum electrodes. J. Hazard.Mater. 168, 179–186.
Marciocha, D., Kalka, J., Turek-Szytow, J., Wiszniowski, J., Surmacz-Gorska, J., 2009.Oxidation of sulfamethoxazole by UVA radiation and modified Fenton reagent:Toxicity and biodegradability of by-products. Water Sci. Technol. 60, 2555–2562.
Marselli, B., García-Gomez, J., Michaud, P.A., Rodrigo, M.A., Comninellis, Ch., 2003.Electrogeneration of hydroxyl radicals on boron-doped diamond electrodes. J.Electrochem. Soc. 150, D79–D83.
Martínez-Huitle, C.A., Brillas, E., 2008. Electrochemical alternatives for drinkingwater disinfection. Angew. Chem. Int. Ed. 47, 1998–2005.
Martínez-Huitle, C.A., Ferro, S., 2006. Electrochemical oxidation of organicpollutants for the wastewater treatment: direct and indirect processes. Chem.Soc. Rev. 35, 1324–1340.
Ming, G.H., Shafinaz, S., Zaharah, I., Adibah, Y., 2011. Biodegradation of 4-aminobenzenesulfonate by Ralstonia sp. PBA and Hydrogenophaga sp. PBCisolated from textile wastewater treatment plant. Chemosphere 82, 507–513.
Özcan, A., S�ahin, Y., Savas� Koparal, A., Oturan, M.A., 2008. Propham mineralizationin aqueous medium by anodic oxidation using boron-doped diamond anode:influence of experimental parameters on degradation kinetics andmineralization efficiency. Water Res. 42, 2889–2898.
Panizza, M., Cerisola, G., 2007. Electrocatalytic materials for the electrochemicaloxidation of synthetic dyes. Appl. Catal. B: Environ. 75, 95–101.
Panizza, M., Cerisola, G., 2009. Direct and mediated anodic oxidation of organicpollutants. Chem. Rev. 109, 6541–6569.
Pera-Titus, M., García-Molina, V., Baños, M.A., Giménez, J., Esplugas, S., 2004.Degradation of chlorophenols by means of advanced oxidation processes: ageneral review. Appl. Catal. B: Environ. 47, 219–256.
Perei, K., Rakhely, G., Kiss, I., Polyak, B., Kovacs, K.L., 2001. Biodegradation ofsulfanilic acid by Pseudomonas paucimobilis. Appl. Microbiol. Biotechnol. 55,101–107.
Sáez, C., Panizza, M., Rodrigo, M.A., Cerisola, G., 2007. Electrochemical incinerationof dyes using a boron-doped diamond anode. J. Chem. Technol. Biotechnol. 82,575–581.
Santos, V., Diogo, J., Pacheco, M.J.A., Ciríaco, L., Morão, A., Lopes, A., 2010.Electrochemical degradation of sulfonated amines on Si/BDD electrodes.Chemosphere 79, 637–645.
Singh, P., Mishra, L.C., Pandey, A., Iyengar, L., 2006. Degradation of 4-aminobenzenesulfonate by alginate encapsulated cells of Agrobacterium sp.PNS-1. Bioresour. Technol. 97, 1655–1659.
Sirés, I., Brillas, E., 2012. Remediation of water pollution caused by pharmaceuticalresidues based on electrochemical separation and degradation technologies: areview. Environ. Int. 40, 212–229.
Sirés, I., Brillas, E., Cerisola, G., Panizza, M., 2008. Comparative depollution ofmecoprop aqueous solutions by electrochemical incineration using BDD andPbO2 as high oxidation power anodes. J. Electroanal. Chem. 613, 151–159.
Sirés, I., Cabot, P.L., Centellas, F., Garrido, J.A., Rodríguez, R.M., Arias, C., Brillas, E.,2006. Electrochemical degradation of clofibric acid in water by anodicoxidation. Comparative study with platinum and boron-doped diamondelectrodes. Electrochim. Acta 52, 75–85.
Skoumal, M., Arias, C., Cabot, P.L., Centellas, F., Garrido, J.A., Rodríguez, R.M., Brillas,E., 2008. Mineralization of the biocide chloroxylenol by electrochemicaladvanced oxidation processes. Chemosphere 71, 1718–1729.
Tan, N.C.G., van Leeuwen, A., van Voorthuizen, E.M., Slenders, P., Prenafeta-Boldu,F.X., Temmink, H., Lettinga, G., Field, J.A., 2005. Fate and biodegradability ofsulfonated aromatic amines. Biodegradation 16, 527–537.
Wang, Y.Q., Zhang, J.S., Zhou, J.T., Zhang, Z.P., 2009. Biodegradation of4aminobenzenesulfonate by a novel Pannonibacter sp. W1 isolated fromactivated sludge. J. Hazard. Mater. 169, 1163–1167.
Yemashova, N., Telegina, A., Kotova, I., Netrusov, A., Kalyuzhnyi, S., 2004.Decolorization and partial degradation of selected azo dyes by methanogenicsludge. Appl. Biochem. Biotechnol. 119, 31–40.
Zhao, G., Zhang, Y., Lei, Y., Lv, B., Gao, J., Zhang, Y., Li, D., 2010. Fabrication andelectrochemical treatment application of a novel lead dioxide anode withsuperhydrophobic surfaces, high oxygen evolution potential, and oxidationcapability. Environ. Sci. Technol. 44, 1754–1759.
Copyright © 2022 FDOKUMEN




















