
Enzymatic biofuel cell based on anode and cathode powered by ethanol
7
Biosensors and Bioelectronics, 2008 Cite this article as : A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766. DOI:org/10.1016/j.bios.2008.06.048 Full printed journal version is available at: http://www.sciencedirect.com/science/article/pii/S0956566308003357 RESEARCH ARTICLE (Advanced authors version) A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766. DOI:org/10.1016/j.bios.2008.06.048 Enzymatic biofuel cell based on anode and cathode powered by ethanol Arunas Ramanavicius 1∗ , Asta Kausaite 1,2 , Almira Ramanaviciene 1,2 , 1 Center of Nanotechnology and Material Science, Faculty of Chemistry, Vilnius University, Naugarduko 24, 03225 Vilnius, Lithuania 2 Laboratory of Immunoanalysis and Nanotechnology, Institute of Immunology of Vilnius University, Moletu pl. 29, 08409 Vilnius, Lithuania Abstract Enzymatic biofuel cell based on enzyme modified anode and cathode electrodes are both powered by ethanol and operate at ambient temperature is described. The anode of the presented biofuel cell was based on immobilized quino- hemoprotein-alcohol dehydrogenase (QH-ADH), while the cathode on coimmobilized alcohol oxidase (AOx) and microperoxidase (MP-8). Two enzymes AOx and MP-8 acted in the consecutive mode and were applied in the design of the biofuel cell cathode. The ability of QHADH to transfer electrons directly towards the carbon-based electrode and the ability of MP-8 to accept electrons directly from the same type of electrodes was exploited in this biofuel cell design. Direct electron transfer (DET) to/from enzymes was the basis for generating an electric potential between the anode and cathode. Application of immobilized enzymes and the harvesting of the same type of fuel at both electrodes (cathode and anode) avoided the compartmentization of enzymatic biofuel cell. The maximal open circuit potential of the biofuel cell was 240 mV. c 2008 Elsevier B.V. All rights reserved. Keywords: Enzymatic fuel cell, Fuel cell, Biofuel cell, PQQ, Alcohol dehydrogenase, Alcohol oxidase, Microperoxidase.
Transcript of Enzymatic biofuel cell based on anode and cathode powered by ethanol

Biosensors and Bioelectronics, 2008
Cite this article as : A. Ramanavicius A. Kausaite-Minkstimiene, A.
Ramanaviciene, Enzymatic biofuel cell based on anode and cathode powered by
ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
Full printed journal version is available at:
http://www.sciencedirect.com/science/article/pii/S0956566308003357
RESEARCH ARTICLE
(Advanced authors version)
A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
Enzymatic biofuel cell based on anode and cathode powered by
ethanol
Arunas Ramanavicius1∗, Asta Kausaite
1,2, Almira Ramanaviciene
1,2,
1 Center of Nanotechnology and Material Science, Faculty of Chemistry, Vilnius University, Naugarduko 24, 03225 Vilnius,
Lithuania
2 Laboratory of Immunoanalysis and Nanotechnology, Institute of Immunology of Vilnius University, Moletu pl. 29, 08409 Vilnius,
Lithuania
Abstract
Enzymatic biofuel cell based on enzyme modified anode and cathode electrodes are both powered by ethanol and
operate at ambient temperature is described. The anode of the presented biofuel cell was based on immobilized quino-
hemoprotein-alcohol dehydrogenase (QH-ADH), while the cathode on coimmobilized alcohol oxidase (AOx) and
microperoxidase (MP-8). Two enzymes AOx and MP-8 acted in the consecutive mode and were applied in the design of
the biofuel cell cathode. The ability of QHADH to transfer electrons directly towards the carbon-based electrode and
the ability of MP-8 to accept electrons directly from the same type of electrodes was exploited in this biofuel cell
design. Direct electron transfer (DET) to/from enzymes was the basis for generating an electric potential between the
anode and cathode. Application of immobilized enzymes and the harvesting of the same type of fuel at both electrodes
(cathode and anode) avoided the compartmentization of enzymatic biofuel cell. The maximal open circuit potential of
the biofuel cell was 240 mV.
c 2008 Elsevier B.V. All rights reserved.
Keywords: Enzymatic fuel cell, Fuel cell, Biofuel cell, PQQ, Alcohol dehydrogenase, Alcohol oxidase, Microperoxidase.

A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
1. Introduction
The production of electrical power from low-cost
biofuels is an important challenge in the energetics, since
biofuels are renewable, sustainable, offer lower
greenhouse-gas emissions and reduce the demand for
common fuel sources, these are of special interest of
modern-energetics (Pizzariello et al., 2002). As the
majority of organic substrates undergo combustion with
the evolution of energy, the biocatalyzed oxidation of
organic substrates by oxygen at electrode interfaces
provides a means for the conversion of the chemical
energy to an electrical one (Willner et al., 1998a).
Significant concentrations of ethanol and other alcohols
are often present in the various biological substances
(Lim and Wang, 2003). Basic fermentation processes
might be applied to increase concentration of alcohols in
these substances. The chemical energy of abundant
biological substrates including polyhydroxylic
(Arechederra et al., 2007) and monohydroxylic alcohols
(Ikeda and Kano, 2001; Ramanavicius et al., 2005)may
be converted into electrical current by biofuel cells. The
advantage of biofuel cells when compared to standard
fuel cells is that biofuel cells are able to operate at a low
substrate concentration which can be even at the
micromolar level (Pizzariello et al., 2002). In the future
biofuel cells can be used as alternative energy supply
sources for biosensors and for other bio electronic
devices (Willner et al., 2001). These cells may also be
used as power sources of implantable devices (Barton et
al., 2004). The combination of bioelectronics with
nanotechnology allows integration of biofuel cells within
the operating devices, while the nanotechnology offers
novel perspectives for the miniaturization of
bioelectronic devices and the increase of their efficiency
(Willner et al., 2001; Wang et al., 2005). Microorganisms
and/or enzymes are catalysts that are able to convert
chemical energy to electrical energy (Katz et al., 1999),
for this reason they are important subjects of biofuel cells
(Bullen et al., 2006). The most efficient biofuel mediators
based system for re-oxidation of its active site. It was
demonstrated that both types of alcohol harvesting
enzymes could be applied in the design of biofuel cells,
however usually they are applied for designing of biofuel
cell anode. The majority of oxidases and dehydrogenases
require the application of red-ox mediators to establish
direct electron transfer (DET) with electrode
(Habermuller et al., 2000; Malinauskas et al., 2004).
Among the other types of enzymes, some
pyrroloquinoline quinine (PQQ)-dependent red-ox
enzymes were also employed for the construction of
biofuel cells (Ikeda and Kano, 2003). However the
majority of PQQ-dependent enzymes are unable to
transfer electrons without additional red-ox mediators
(Lapenaite et al., 2006; Habermuller et al., 2000). The
detailed characterization of the interfacial electron
transfer rates is essential in the construction of biofuel
cells, since the enzyme and electronic conductors are
foreign components in respect of one to the other, that
leads to a lack of electric current between them. The
modification of PQQ-dependent enzymes with covalently
attached red-ox mediators can be applied to facilitate
electron transfer from active site of enzyme towards
electrode (Laurinavicius et al., 2004). An alternative to
this is to apply DET-able enzymes (Gorton et al., 1999).
The red-ox enzymes containing heme-c are very
promising in this context (Freire et al., 2003;
Ramanavicius et al., 2006a). Quino-hemoprotein-alcohol
dehydrogenase from Gluconobacter sp. 33 (QH-ADH)
demonstrates DET toward glassy carbon (Ikeda et al.,
1993), other forms of carbon (Razumiene et al., 2002),
gold (Ikeda et al., 1993) and conducting polymer
polypyrrole (Ramanavicius et al., 1999). Another class of
enzymes, heme-c containing peroxidases, are able for
direct electron transfer and also are very promising for
construction biofuel cell cathode (Willner et al., 1998a,b;
Ferapontova and Gorton, 2001). Direct electrochemistry
of microperoxidases with a gold electrode (Willner et al.,
1998a), carbon electrodes (Ruzgas et al., 1996), along
with platinum electrodes modified with carbon nanotubes
(Wang et al., 2005),were investigated and exploited in
the design of biofuel cell cathode. However, DET-based
enzymatic biofuel cell utilizing the same substrate at both
electrodes is still a challenge. The aim of this study was
to design basic, non-compartmentalized, mediator free
biofuel cell based on enzymes exhibiting direct
bioelectrocatalysis and able to convert the chemical
energy of biological substrate – ethanol – at both cathode
and anode. cell designs allow operation’s without
compartments dividing membranes (Chen et al., 2001;
Ramanavicius et al., 2005). It allows applications of
biofuel cells as portable power sources. Biofuel cells
utilize biocatalysts for the conversion of chemical energy
to electrical energy (Katz et al., 1999; Chen et al., 2001;
Mano et al., 2002, 2003). The methodology based on the
application of purified redox enzymes for the targeted
oxidation and reduction of specific fuel and oxidizer
substrates at the electrode supports and the generation of
the electrical current output is used for the development
of biofuel cells (Chen et al., 2001). There are known
several red-ox enzymes able gain electrons from various
alcohols: methanol (Zhang et al., 2006a,b), ethanol
(Ramanavicius et al., 2006a), glycerol (Lapenaite et al.,
2006) and other alcohols (Ivnitski et al., 2006). Alcohol
oxidases (AOxs) utilizes oxygen as natural electron
acceptor thus producing hydrogen peroxide
(Ramanavicius et al., 2006b; Malinauskas et al., 2005).
Alcohol dehydrogenases are using NAD/NADH or
artificial red-ox mediators based system for re-oxidation
of its active site. It was demonstrated that both types of
alcohol harvesting enzymes could be applied in the
design of biofuel cells, however usually they are applied
for designing of biofuel cell anode. The majority of
oxidases and dehydrogenases require the application of
red-ox mediators to establish direct electron transfer
(DET) with electrode (Habermuller et al., 2000;
Malinauskas et al., 2004). Among the other types of
enzymes, some pyrroloquinoline quinine (PQQ)-
dependent red-ox enzymes were also employed for the
construction of biofuel cells (Ikeda and Kano, 2003).
However the majority of PQQ-dependent enzymes are
unable to transfer electrons without additional red-ox
mediators (Lapenaite et al., 2006; Habermuller et al.,
2000). The detailed characterization of the interfacial
electron transfer rates is essential in the construction of
biofuel cells, since the enzyme and electronic conductors
are foreign components in respect of one to the other, that
leads to a lack of electric current between them. The

A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
modification of PQQ-dependent enzymes with covalently
attached red-ox mediators can be applied to facilitate
electron transfer from active site of enzyme towards
electrode (Laurinavicius et al., 2004). An alternative to
this is to apply DET-able enzymes (Gorton et al., 1999).
The red-ox enzymes containing heme-c are very
promising in this context (Freire et al., 2003;
Ramanavicius et al., 2006a). Quino-hemoprotein-alcohol
dehydrogenase from Gluconobacter sp. 33 (QH-ADH)
demonstrates DET toward glassy carbon (Ikeda et al.,
1993), other forms of carbon (Razumiene et al., 2002),
gold (Ikeda et al., 1993) and conducting polymer
polypyrrolen (Ramanavicius et al., 1999). Another class
of enzymes, heme-c containing peroxidases, are able for
direct electron transfer and also are very promising for
construction biofuel cell cathode (Willner et al., 1998a,b;
Ferapontova and Gorton, 2001). Direct electrochemistry
of microperoxidases with a gold electrode (Willner et al.,
1998a), carbon electrodes (Ruzgas et al., 1996), along
with platinum electrodes modified with carbon nanotubes
(Wang et al., 2005),were investigated and exploited in
the design of biofuel cell cathode. However, DET-based
enzymatic biofuel cell utilizing the same substrate at both
electrodes is still a challenge. The aim of this study was
to design basic, non-compartmentalized, mediator free
biofuel cell based on enzymes exhibiting direct
bioelectrocatalysis and able to convert the chemical
energy of biological substrate – ethanol – at both cathode
and anode.
2. Experimental
2.1 Chemicals
Alcohol dehydrogenase from Gluconobacter sp. 33 (E.C.
1.1.99.8) was isolated and purified at the Institute of
Biochemistry (Vilnius, Lithuania). The enzyme had an
activity of 171 U/ml and 7.6mg/ml concentration of
proteins. Microperoxidase-8 (MP-8) from horse heart 250
U/mg; AOx from Pichia pastoris (E.C.1.1.3.13), 50
U/ml, 25% glutaraldehyde and 96% ethanol were
purchased from Sigma (Berlin, Germany). Carbon rod
electrodes “Ultra F purity” 3mm in diameter obtained
from Ultra Carbon Division of Carbon USA, RAVEN-M
were used. Carbon rod electrodes were sealed into epoxy
to prevent the contact of electrode side surface with the
solution.
2.2 Preparation of electrodes
Graphite electrodes modified with enzymes were
prepared as follows: (i) rods of spectroscopic graphite
were cut, and polished on fine emery paper, followed by
rinsing the electrode surface with ethanol and water.
Electrodes were dried at room temperature before coating
with enzyme; (ii) enzyme solutions were deposited on the
electrode surface and the coated electrodes were kept
for 20 h over the 5% solution of glutaraldehyde at 4 ◦C in
the closed vessel as it was previously described
(Ramanavicius, 2007). In preparing the anode, 6µl of
QH-ADH solution was thoroughly distributed on the
surface of the electrode and kept over glutaraldehyde
solution. In preparing the cathode, 3µl of MP-8 solution
(10mg/ml) was thoroughly distributed over the electrode
and dried for 30 min. Afterwards, 3µl of AOx solution
(10mg/ml) was deposited and the electrode was kept over
glutaraldehyde. Control carbon electrode modified with
MP-8 was prepared by deposition of 3µl ofMP-8 solution
(10mg/ml) and cross-linked by glutaraldehyde using the
same cross-linking protocol.
Fig. 1. Configuration of biofuel cell, using ethanol as a
fuel for cathode and anode.
2.3. Electrochemical measurements
All electrochemical measurements were performed
by potentiostat–galvanostat PGSTAT 30 using
specialized software GPES3 v3,2 Echochemie/Autolab
(Utrech, Netherlands). Two-electrode circuit was applied
for registration of electrical potential between electrodes.
Some potentiometric measurements were performed at
open circuit; during some other experiments the resistors
were switched into the external circuit (Fig. 1) to imitate
circuit-load on biofuel cell.
3. Results and discussion
Current developments in nanotechnology enables serious
thoughts about nano-devices and even nano-robots, but in
many cases such systems require miniature power
sources (Chen et al., 2001) otherwise such devices will
be very limited in function and application. Major idea of
the work presented here was to apply environmentally
friendly and/or biodegradable materials. This concept
does not allow us to apply any soluble red-ox mediators
or hazardous red-ox polymers. Direct electron transfer
exhibiting hemoproteins (Gorton et al., 1999) were
selected for this study to avoid application of any
additional environmentally and/or biologically hazardous
red-ox materials. Enzymes MP-8 and QH-ADH were
selected for the design of biofuel cell as the catalysts.
These enzymes are able to transduce chemical energy
into electrical one and to transfer electrical current
directly to the carbon electrodes. Carbon is selected as a
matrix-material, because various carbon forms are very
often applied in various nanotechnological applications
(Malinauskas et al., 2005). Moreover carbon is especially
suitable for application in bioelectronic devices since it
supports sufficient electron transfer due to proper enzyme
orientation (Shleev et al., 2005). The QH-ADH is
comprised of three subunits (Fre’bortova et al., 1998).
The ability of QH-ADH to transfer electrons directly
towards the electrodes was employed for generation of
anodic current in amperometric biosensors
(Ramanavicius et al., 1999) and later this process was

A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
applied in biofuel cell harvesting ethanol and glucose as
biofuels (Ramanavicius et al., 2005). The anode of the
constructed enzymatic fuel cell was based on
immobilized QH-ADH, which oxidized ethanol as the
anode fuel. The cooperative action of the enzyme-
integrated prosthetic groups pyrroloquinoline quinone
and hemes-c were assumed to allow direct electron-
transfer from the enzyme’s catalytic site to the carbon
electrode. The electrical contact between QH-ADH and
carbon electrode was efficient and the electrons extracted
from ethanol (1) were directly transferred to carbon
surface (Fig. 1, anode reaction). This phenomenon has
been exploited in anodes of other biofuel cells
(Ramanavicius et al., 2005; Treu et al., 2006). This
enzyme becomes attractive because of the possible
improvements when applying other carbon-based
nanomaterials, including carbon nanotubes (Treu et al., in
press).
Fig. 2. Open circuit potentials of: (1) QH-ADH functionalized
anode as a function of ethanol concentration; (2)MP-8/AOx functionalized cathode as a function of ethanol concentration;
(3) MP-8/AOx functionalized cathode as function of hydrogen
peroxide concentration; (4) QH-ADH functionalized anode and MP-8/AOx functionalized cathode-based biofuel cell as a
function of ethanol concentration. Investigations performed in
50mM Na acetate solution, pH 6.0, with 100mM KCl at open circuit.
The open circuit potential of here studied QH-ADH-
based anode was approximately −125mV (Fig. 2, curve
1). Anode reaction:
Ethanol QH−-→ADHEthanal+2H+ + 2e− (1)
The cathode reaction is also very important during the
creation of biofuel cells since it can significantly increase
electric potential and current of biofuel cell. Various
strategies were applied to maximize the performance of
cathodes in biofuel cells and the application of laccases
seems the most popular for this purpose (Barton et al.,
2001; Tarasevich et al., 1979; Tayhas et al., 1999), it was
demonstrated that some carbon-based nanomaterials may
significantly improve the current of laccase-based biofuel
cell cathode (Gallaway et al., 2008) and negative
influence of some halide ions could be eliminated by
proper orientation of laccases (Vaz-Dominguez et al.,
2008). Advantageous properties of laccases including
major DET aspects were in details reviewed (Shleev et
al., 2005), however some alternative ways including
application of heme-c-dependent enzymes could be also
applied to design cathodes of biofuel cells (Bullen et al.,
2006). In this respect, previously we have demonstrated
that glucose oxidase might be principally applied in the
design of biofuel cell cathode if combined with
microperoxidase-8 (Ramanavicius et al., 2005).
Hydrogen peroxide, which is produced by oxidase, is a
strong oxidizer. However electrochemical reduction of
hydrogen peroxide at the carbon electrode occurs with
very high over-potential, which may be reduced if the
carbon surface is modified by peroxidase activity
exhibiting enzymes (Ruzgas et al., 1996). From the
variety of peroxidases we have selected the smallest one,
MP-8, because this enzyme is very stable what is
determined by basic structure of this microperoxidases.
Although, this biocatalyst was expected to have the most
efficient carbon surface covering due to lowest molecular
weight (Adams, 1991). The influence of an efficient
surface covering was demonstrated by investigation of
several carbon electrode-covering strategies. The layered
design of the cathode based on two separated layers of
MP-8 and AOx was selected for this study, because the
investigations showed that in the case when pure MP-8
layer was deposited as first layer on these carbon
electrodes, then higher potential and higher current
density of the cathode are achieved in comparison with
the cathode based on mixed MP-8 and AOx. This type of
a layered design guaranties a higher surface-density of
MP-8, which is directly deposited on the electrode.
Hydrogen peroxide was formed in the reaction catalyzed
by AOx. Then hydrogen peroxide was used as a substrate
for MP-8, and electrons from this reaction were
transferred from the electrode to the active site of MP-8,
which was acting as a reducer of hydrogen peroxide (Fig.
1, cathode interface reaction). Due to the consecutive
action of AOx and MP-8 immobilized on the cathode,
chemical energy of ethanol oxidation by dissolved
oxygen was converted into electrical current. Cathode
interface reactions:
Ethanol + O2 + H2O−AO→xEthanal+H2O2 (2)
H2O2 + 2H+M−→P-82H2O − 2e− (3)
The potential generated by MP-8/AOx cathode in the
presence of different ethanol concentrations was
examined (Fig. 2, curve 2). The slope of the second curve
(Fig. 2, curve 2) seems much lower if compared with that
achieved by QH-ADH-based anode (Fig. 2, curve 1).
This effect might be based on diffusion of ethanol and
hydrogen peroxide, which was formed during the action
of AOx. From the potential values of MP-8/AOx
modified electrode at 2mM and higher hydrogen

A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
peroxide concentrations (Fig. 2, curve 3), the theoretical
limit of the MP-8 modified electrode potential is
estimated to be 150mV. The maximal voltage generated
by MP-8/AOx modified cathode was close to that
generated by biofuel cell cathode based on MP-11
modified gold cathode (Willner et al., 1998a). The
cathode potential exceeded 125mV if 5mM and higher
ethanol concentrations were added into the cell (Fig. 2,
curve 1). The maximal open circuit potential of the
complete biofuel cell was 240mVat ethanol
concentrations of 20mMand higher (Fig. 2, curve 4).
Experimental results indicates, that the open circuit
potential in this type of measurements is dependent on
substrate concentration because of some limitations in
measurement: (i) the “open circuit” is not ideal, despite
of very high resistance of measuring device (300M_), the
resistance of circuit cathode/solution/anode was just
1±0.2M_, because of conductivity of used buffer
solution, for this reason the measured potential at least
partially depends on enzymatic reaction rate, which is
highly influenced by concentration of substrate—ethanol.
Fig. 3. The pH dependence of open circuit potential for: (1) MP-
8 functionalized cathode in presence 20mMof hydrogen peroxide; (2)QH-ADH functionalized anode in presence 20mM
of ethanol; (3) MP-8/AOx functionalized cathode in presence
20mM of ethanol and (4) biofuel cell based on MP-8/AOx functionalized cathode and QH-ADH functionalized anode in
the presence 20mMof ethanol. Investigations performed at an
open circuit in 50mMof sodium acetate with accordingly adjusted pH, 100mM of potassium chloride.
The MP-8 modified electrode may be effectively applied
in a broad pH diapason: between pH 3 and pH 7 (Fig. 3,
curve 1). However, the QH-ADH modified anode (Fig. 3,
curve 2) and layered MP-8/AOx modified cathode (Fig.
3, curve 3) are much more dependent on pH level. The
result of this is a “sharp” dependence of electric potential
on pH (Fig. 3, curve 4). On the other hand the developed
biofuel cell is able to operate in the physiological pH
range. It is important for possible application of biofuel
cell in vivo (Tsujimura et al., 2001). To enable better
operation of biofuel cell in vivo, some enzymes might be
exchanged into ones harvesting glucose (Malinauskas et
al., 2004) and/or glycerol (Lapenaite et al., 2006). There
are many reasons why pH is affecting open circuit
potentials, e.g.: (i) there are significant differences in
thermodynamics of enzymatic red-ox reactions and in
thermodynamics intrinsic/extrinsic electron transfer
reactions at different pH; (ii) significant differences in
enzymatic activity, because conformation of enzyme is
pH dependent and protonated/deprotonated groups might
be crucial in catalytic site of enzyme; (iii) extrinsic
electron transfer might be dependent on enzyme
conformation, etc. The stability of biofuel cell was
examined at working state in presence of 2mM of ethanol
at the loading resistance of 1M_ as
Fig. 4. Open circuit potentials for: (1) MP-8/AOx functionalized
cathode and (2) QH-ADH functionalized anode as a function of
glucose concentration. Investigations performed at an open circuit in 50mMof sodium acetate, pH6.0, containing 100mM of
potassium chloride and 75mM of ethanol.
Fig. 5. Current–voltage behavior of: (1) complete biofuel cell
based on both MP- 8/AOx cathode and QH-ADH anode; (2) QH-ADH anode vs. unmodified carbon electrode; (3) MP-
8/AOx cathode vs. unmodified carbon electrode. All
measurements were performed in 25mM of ethanol.
a function of time in the presence of ethanol. The half-
life of the biofuel cell was found to be 26 h. Significant
improvement in stability of biofuel cell was detected if it
is compared with the results published (Willner et al.,
1998a) where the cathode was based on MP-11 while the
anode on GOx “wired” by PQQ derivatives assembled on
the gold electrode. The “increased” stability of the
biofuel cell might be based on application of unmodified
enzymes when compared with enzymes treated under
harsh conditions due to immobilization via self-
assembled monolayers or enzyme “wiring” techniques
applied in other studies (Willner et al., 1998a). On the
other hand, the structure of MP-8 is very basic, which
makes this enzyme more stable if comparing it with the
higher molecular weight enzyme—MP-11 used in
previous studies. However in this study used AOx was
found to be relatively unstable; for this reason AOx
negatively influenced stability of biofuel cell cathode.
The least expensive alcohol sources, for example natural
fermentation products, typically contain a lot of various
sugars. In order to study performance of biofuel cells in
an environment containing sugars the effectiveness of
MP-8/AOx (Fig. 4, curve 1) and QH-ADH (Fig. 4, curve
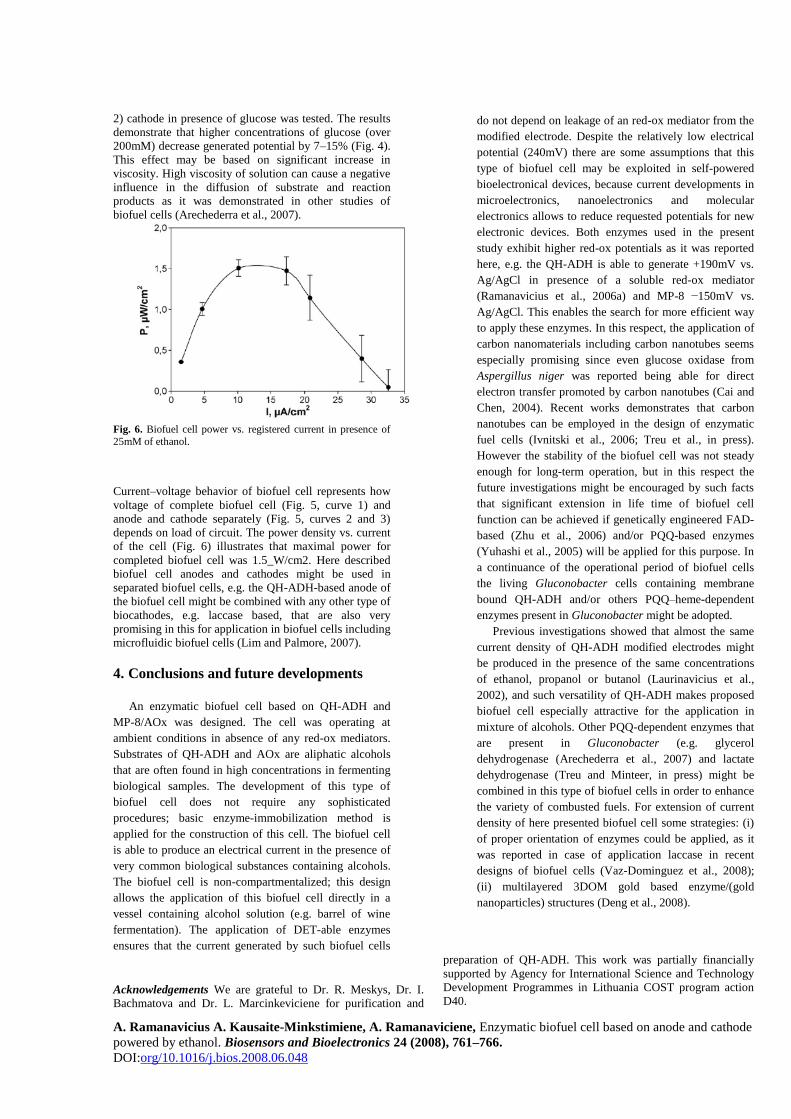
A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
2) cathode in presence of glucose was tested. The results
demonstrate that higher concentrations of glucose (over
200mM) decrease generated potential by 7–15% (Fig. 4).
This effect may be based on significant increase in
viscosity. High viscosity of solution can cause a negative
influence in the diffusion of substrate and reaction
products as it was demonstrated in other studies of
biofuel cells (Arechederra et al., 2007).
Fig. 6. Biofuel cell power vs. registered current in presence of
25mM of ethanol.
Current–voltage behavior of biofuel cell represents how
voltage of complete biofuel cell (Fig. 5, curve 1) and
anode and cathode separately (Fig. 5, curves 2 and 3)
depends on load of circuit. The power density vs. current
of the cell (Fig. 6) illustrates that maximal power for
completed biofuel cell was 1.5_W/cm2. Here described
biofuel cell anodes and cathodes might be used in
separated biofuel cells, e.g. the QH-ADH-based anode of
the biofuel cell might be combined with any other type of
biocathodes, e.g. laccase based, that are also very
promising in this for application in biofuel cells including
microfluidic biofuel cells (Lim and Palmore, 2007).
4. Conclusions and future developments
An enzymatic biofuel cell based on QH-ADH and
MP-8/AOx was designed. The cell was operating at
ambient conditions in absence of any red-ox mediators.
Substrates of QH-ADH and AOx are aliphatic alcohols
that are often found in high concentrations in fermenting
biological samples. The development of this type of
biofuel cell does not require any sophisticated
procedures; basic enzyme-immobilization method is
applied for the construction of this cell. The biofuel cell
is able to produce an electrical current in the presence of
very common biological substances containing alcohols.
The biofuel cell is non-compartmentalized; this design
allows the application of this biofuel cell directly in a
vessel containing alcohol solution (e.g. barrel of wine
fermentation). The application of DET-able enzymes
ensures that the current generated by such biofuel cells
do not depend on leakage of an red-ox mediator from the
modified electrode. Despite the relatively low electrical
potential (240mV) there are some assumptions that this
type of biofuel cell may be exploited in self-powered
bioelectronical devices, because current developments in
microelectronics, nanoelectronics and molecular
electronics allows to reduce requested potentials for new
electronic devices. Both enzymes used in the present
study exhibit higher red-ox potentials as it was reported
here, e.g. the QH-ADH is able to generate +190mV vs.
Ag/AgCl in presence of a soluble red-ox mediator
(Ramanavicius et al., 2006a) and MP-8 −150mV vs.
Ag/AgCl. This enables the search for more efficient way
to apply these enzymes. In this respect, the application of
carbon nanomaterials including carbon nanotubes seems
especially promising since even glucose oxidase from
Aspergillus niger was reported being able for direct
electron transfer promoted by carbon nanotubes (Cai and
Chen, 2004). Recent works demonstrates that carbon
nanotubes can be employed in the design of enzymatic
fuel cells (Ivnitski et al., 2006; Treu et al., in press).
However the stability of the biofuel cell was not steady
enough for long-term operation, but in this respect the
future investigations might be encouraged by such facts
that significant extension in life time of biofuel cell
function can be achieved if genetically engineered FAD-
based (Zhu et al., 2006) and/or PQQ-based enzymes
(Yuhashi et al., 2005) will be applied for this purpose. In
a continuance of the operational period of biofuel cells
the living Gluconobacter cells containing membrane
bound QH-ADH and/or others PQQ–heme-dependent
enzymes present in Gluconobacter might be adopted.
Previous investigations showed that almost the same
current density of QH-ADH modified electrodes might
be produced in the presence of the same concentrations
of ethanol, propanol or butanol (Laurinavicius et al.,
2002), and such versatility of QH-ADH makes proposed
biofuel cell especially attractive for the application in
mixture of alcohols. Other PQQ-dependent enzymes that
are present in Gluconobacter (e.g. glycerol
dehydrogenase (Arechederra et al., 2007) and lactate
dehydrogenase (Treu and Minteer, in press) might be
combined in this type of biofuel cells in order to enhance
the variety of combusted fuels. For extension of current
density of here presented biofuel cell some strategies: (i)
of proper orientation of enzymes could be applied, as it
was reported in case of application laccase in recent
designs of biofuel cells (Vaz-Dominguez et al., 2008);
(ii) multilayered 3DOM gold based enzyme/(gold
nanoparticles) structures (Deng et al., 2008).
Acknowledgements We are grateful to Dr. R. Meskys, Dr. I.
Bachmatova and Dr. L. Marcinkeviciene for purification and
preparation of QH-ADH. This work was partially financially
supported by Agency for International Science and Technology
Development Programmes in Lithuania COST program action
D40.

A. Ramanavicius A. Kausaite-Minkstimiene, A. Ramanaviciene, Enzymatic biofuel cell based on anode and cathode
powered by ethanol. Biosensors and Bioelectronics 24 (2008), 761–766.
DOI:org/10.1016/j.bios.2008.06.048
References Adams, P.A., 1991. In: Everse, J., Everse, K.E., Grisham, M.B.
(Eds.), Peroxidases in Chemistry and Biology, vol. 2. CRC
Press, Boca Raton, pp. 171–200 (Chapter 7).
Arechederra, R.L., Treu, B.L., Minteer, S.D., 2007. J. Power
Sources 173, 156–161.
Barton, S.C., Gallaway, J., Atanasov, P., 2004. Chem. Rev. 104,
4867–4886.
Barton, S.C., Kim, H.H., Binyamin, G., Zhang, Y., Heller, A.,
2001. J. Am. Chem. Soc. 123, 5802–5803. Bullen, R.A., Arnot, T.C., Lakeman, J.B., Walsh, F.C., 2006.
Biosens. Bioelectron. 21, 2015–2045.
Cai, C., Chen, J., 2004. Anal. Biochem. 332, 75–83. Chen, T., Barton, S.C., Binyamin, G., Gao, Z.Q., Zhang, Y.C.,
Kim, H.H., Heller, A., 2001. J. Am. Chem. Soc. 123, 8630–
8631.
Deng, L., Wang, F., Chen, H., Shang, L., Wang, L., Wang, T.,
Dong, S., 2008. Biosens. Bioelectron. 24, 329–333.
Ferapontova, E., Gorton, L., 2001. Electrochem. Commun. 3,
767–774.
Fre’bortova, J., Matsushita, K., Arata, H.,Adachi, O., 1998.
Biochim. Biophys.Acta 1363, 24–34.
Freire, R.S., Pessoa, C.A., Mello, L.D.,Kubota, L.D., 2003. J.
Braz. Chem. Soc. 14, 230–243.
Gallaway, J., Wheeldon, R., Rincon, I., Atanassov, P., Banta, S.,
Barton, S.C., 2008. Biosens. Bioelectron. 23, 1229–1235. Gorton, L., Lindgren, A., Larsson, T., Munteanu, F.D., Ruzgas,
T., Gazaryan, I., 1999. Anal. Chim. Acta 400, 91–108.
Habermüller, K., Ramanavicius, A., Laurinavicius, V.,
Schuhmann,W., 2000. Electroanalysis 12, 1383–1389.
Ikeda, T., Kano, K., 2001. Rev. J. Biosci. Bioeng. 92, 9–18.
Ikeda, T., Kano, K., 2003. Biochim. Biophys. Acta 1647, 121–
126.
Ikeda, T., Kobayashi, D., Matsuma, F., Sagara, T., Niki, K.,
1993. J. Electroanal. Chem. 361, 221–228.
Ivnitski, D., Branch, B., Atanassov, P., Apblett, Ch., 2006.
Electrochem. Commun. 8, 1204–1210.
Ivnitski, D., Branch, B., Atanassov, P., Apblett, Ch., 2006.
Electrochem. Commun. 8, 1204–1210. Katz, E., Willner, I., Kotlyar, A.B., 1999. J. Electroanal. Chem.
479, 64–68.
Lapenaite, I., Ramanaviciene, A., Ramanavicius, A., 2006. Crit.
Rev. Anal. Chem. 36, 13–25.
Lapenaite, I., Ramanaviciene, A., Ramanavicius, A., 2006. Crit.
Rev. Anal. Chem. 36, 13–25.
Laurinavicius, V., Razumiene, J., Kurtinaitiene, B., Lapenaite,
I., Bachmatova, I., Marcinkeviciene, L., Meskys, R.,
Ramanavicius, A., 2002. Bioelectrochemistry 55, 29–32. Laurinavicius, V., Razumiene, J., Ramanavicius, A., Ryabov,
D., 2004. Biosens. Bioelectron. 20, 1217–1222.
Lim, C.,Wang, C.Y., 2003. J. Power Sources 113, 145–150.
Lim, K.G., Palmore, G.T.R., 2007. Biosens. Bioelectron. 22,
941–947. Malinauskas, A., Kuzmarskyte, J., Meskys, R., Ramanavicius,
A., 2004. Sens. Actuator B 100, 387–394.
Malinauskas, A., Kuzmarskyte, J., Meskys, R., Ramanavicius,
A., 2004. Sens. Actuator B 100, 387–394.
Malinauskas, A., Malinauskiene, J., Ramanavicius, A., 2005.
Nanotechnology 16, 51–63.
Mano, N., Mao, F., Heller, A., 2002. J. Am. Chem. Soc. 124,
12962–12963.
Mano, N., Mao, F., Heller, A., 2003. J. Am. Chem. Soc. 125,
6588–6594.
Pizzariello, A., Stred’ansky, M., Miertuˇs, S., 2002.
Bioelectrochemistry 56, 99–105.
Pizzariello, A., Stred’ansky, M., Miertuˇs, S., 2002.
Bioelectrochemistry 56, 99–105.
Ramanavicius, A., 2007. Anal. Bional. Chem. 387, 1899–1906.
Ramanavicius, A., Habermuller, K., Csoregi, E., Laurinavicius,
V., Schuhmann, W., 1999. Anal. Chem. 71, 3581–3586.
Ramanavicius, A., Kausaite, A., Ramanaviciene, A., 2005.
Biosens. Bioelectron. 20,1962–1967.
Ramanavicius, A., Kausaite, A., Ramanaviciene, A., 2006a.
Sens. Actuator B 113, 435–444.
Ramanavicius, A., Ramanaviciene, A., Malinauskas, A., 2006b.
Electrochim. Acta 51, 6025–6037.
Razumiene, J., Niculescu, M., Ramanavicius, A., Laurinavicius,
V., Csoregi, E., 2002. Electroanalysis 14, 1–7.
Ruzgas, T., Csoregi, E., Emneus, J., Gorton, L., Marko-Varga,
G., 1996. Anal. Chim. Acta 330, 123–138.
Shleev, S., Tkac, J., Christenson, A., Ruzgas, T., Yaropolov,
A.,Whittaker, J., Gorton, L., 2005. Biosens. Bioelectron. 20,
2157–2554.
Tarasevich, M.R., Yaropolov, A.I., Bogdanovskaya, V.A.,
Varfolomeev, S.D., 1979. Bioelectrochem. Bioenerg. 6, 393–
403.
Tayhas, G., Palmore, R., Kim, H.H., 1999. J. Electroanal. Chem.
464, 110–117. Treu, B.L., Anderson, D., Behrmann, A., Minteer, S.D., 2006.
Abstracts Papers of The American Chemical Society. pp. 231. Treu, B.L., Arechederra R., Minteer, S.D., in press. J. Nanosci.
Nanotechnol. Treu, B.L., Minteer, S.D., in press. Bioelectrochemistry.
doi:10.1016/j.bioelectrochem. 2008.07.005.
Tsujimura, S., Fujita, M., Tatsumi, H., Kano, K., Ikeda, T.,
2001. Phys. Chem. Chem. Phys. 3, 1331–1335. Vaz-Dominguez, C., Campuzano, S., Rüdiger, O., Pita, M.,
Gorbacheva, M., Shleev, S., Fernandez, V.M., De Lacey, A.L.,
2008. Biosens. Bioelectron. 24, 531–537.
Wang, M., Zhao, F., Liu, Y., Dong, S., 2005. Biosens.
Bioelectron. 21, 159–166.
Willner, I., Arad, G., Katz, E., 1998a. Bioelectrochem.
Bioenerg. 44, 209–214.
Willner, I., Katz, E., Buckmann, A.F., 2001. J. Am. Chem. Soc.
123, 10752–10753.
Willner, I., Katz, E., Patolsky, F., Buckmann, A.F., 1998b. J.
Chem. Soc., Perkin Trans. 2, 1817–1822.
Yuhashi, N., Tomiyama, M., Okuda, J., Igarashi, S., Ikebukuro,
K., Sode, K., 2005. Biosens. Bioelectron. 20, 2145–2150. Zhang, E., Xu, W., Diao, G., Shuang, C., 2006a. J. Power
Sources 161, 820–825.
Zhang, X.C., Ranta, A., Halme, A., 2006b. Biosens. Bioelectron.
21, 2052–2057.
Zhu, Z., Momeu, C., Zakhartsev, M., Schwaneberg, U., 2006.
Biosens. Bioelectron. 21, 2046–2051.
Some other related references 1. Ramanavicius A., Ramanaviciene A. Hemoproteins in design of
biofuel cells. Fuel cells 2009, 9, 25–36. 2. Krikstolaityte V., Barrantes A., Ramanavicius A., Arnebrant T.,
Shleev S., Ruzgas T., Bioelectrocatalytic reduction of oxygen at gold
nanoparticles modified with laccase Bioelectrochemistry. Bioelectrochemistry 2014, 95, 1-6.
3. V. Krikstolaityte, Y. Oztekin, J. Kuliesius, A. Ramanaviciene,
Z.Yazicigil, M. Ersoz, A. Okumus, A. Kausaite-Minkstimiene, Z. Kilic, A.O. Solak, A. Makaraviciute, A. Ramanavicius, Biofuel Cell Based on
Anode and Cathode Modified by Glucose Oxidase. Electroanalysis 2014
(in press). DOI:10.1002/elan.201300482 4. T. Semashko, R. Mikhailova, A. Ramanaviciene, A. Ramanavicius
Specificity of Glucose Oxidase from Penicillium Funiculosum 46.1
Towards Some Redox Mediators Applied Biochemistry and Biotechnology 2013, 171, 1739–1749.




















