
In Situ Building of a Nanoprobe Based on Fluorescent Carbon Dots for Methylmercury Detection

ORIGINAL PAPER
Eicosanoid Signaling and Vascular Dysfunction: Methylmercury-Induced Phospholipase D Activation in Vascular Endothelial Cells
Shariq I. Sherwani • Sheila Pabon • Rishi B. Patel • Muzzammil M. Sayyid •
Thomas Hagele • Sainath R. Kotha • Ulysses J. Magalang •
Krishna R. Maddipati • Narasimham L. Parinandi
Published online: 22 October 2011
� Springer Science+Business Media, LLC 2011
Abstract Mercury, especially methylmercury (MeHg), is
implicated in the etiology of cardiovascular diseases. Ear-
lier, we have reported that MeHg induces phospholipase D
(PLD) activation through oxidative stress and thiol-redox
alteration. Hence, we investigated the mechanism of the
MeHg-induced PLD activation through the upstream reg-
ulation by phospholipase A2 (PLA2) and lipid oxygenases
such as cyclooxygenase (COX) and lipoxygenase (LOX) in
the bovine pulmonary artery endothelial cells (BPAECs).
Our results showed that MeHg significantly activated both
PLA2 (release of [3H]arachidonic acid, AA) and PLD
(formation of [32P]phosphatidylbutanol) in BPAECs in
dose- (0–10 lM) and time-dependent (0–60 min) fashion.
The cPLA2-specific inhibitor, arachidonyl trifluoromethyl
ketone (AACOCF3), significantly attenuated the MeHg-
induced [3H]AA release in ECs. MeHg-induced PLD
activation was also inhibited by AACOCF3 and the COX-
and LOX-specific inhibitors. MeHg also induced the for-
mation of COX- and LOX-catalyzed eicosanoids in ECs.
MeHg-induced cytotoxicity (based on lactate dehydroge-
nase release) was protected by PLA2-, COX-, and LOX-
specific inhibitors and 1-butanol, the PLD-generated PA
quencher. For the first time, our studies showed that MeHg
activated PLD in vascular ECs through the upstream action
of cPLA2 and the COX- and LOX-generated eicosanoids.
These results offered insights into the mechanism(s) of the
MeHg-mediated vascular endothelial cell lipid signaling as
an underlying cause of mercury-induced cardiovascular
diseases.
Keywords Cyclooxygenase � Lipoxygenase �Phospholipase D � Eicosanoid signaling �Phospholipase A2 � Vascular endothelial cells � PLD
Introduction
Mercury (Hg), a highly toxic heavy metal, is a potent
environmental pollutant and has been established to cause
neurotoxicity, cytotoxicity, and immunotoxicity in humans,
but appears to play no known physiological role [1–3].
Mercury-containing dental amalgam fillings have been in
use in dental care worldwide and mercury leaching from
the dental implants in the mouth has been attributed to the
adverse health effects in humans [4]. Mercury also arises as
a toxicant in the industrial environments, contributing to
the occupational disorders/diseases such as respiratory and
lung diseases, cardiovascular diseases, musculoskeletal
disorders, and nervous system diseases among transport
workers [5]. Inorganic form of mercury, which is toxic to
many organisms, undergoes biomethlyation into the highly
toxic form, methylmercury (MeHg) [6]. Dietary con-
sumption of fish has been shown as a major source of
environmental mercury (especially MeHg) in humans that
could lead to suppression of the beneficial effects of
omega-3 fatty acids on the coronary artery disease [7, 8].
Overall, humans are exposed to all these forms of mercury
S. I. Sherwani � S. Pabon � R. B. Patel �M. M. Sayyid � T. Hagele � S. R. Kotha �U. J. Magalang � N. L. Parinandi (&)
Lipidomics, Lipid Signaling, and Vasculotoxicity Laboratory,
Division of Pulmonary, Allergy, Critical Care, and Sleep
Medicine, Department of Internal Medicine, Dorothy M. Davis
Heart and Lung Research Institute, The Ohio State University
College of Medicine, 473 W. 12th Avenue, Columbus,
OH 43210, USA
e-mail: [email protected]
K. R. Maddipati
Department of Pathology, Wayne State University
School of Medicine, Detroit, MI 48202, USA
123
Cell Biochem Biophys (2013) 67:317–329
DOI 10.1007/s12013-011-9304-3

through accidents, environmental pollution, food contami-
nation, dental care, preventive medical practices, industrial
and agricultural operations, and occupational exposures
[7].
Increased exposure to mercury has been correlated to
risk of cardiovascular disease in humans [9]. The role of
mercury toxicity as a possible risk factor in cardiovascular
disease has been discussed [10]. Reports have been made
on the toxic effects of metals in several diseases among
humans including the vascular diseases [9]. Elevated body
levels of mercury, due to fish consumption by humans,
have been hypothesized as a risk factor in coronary heart
disease [11]. Increased levels of urinary mercury have been
shown to be associated with elevated cholesterol levels in
humans and mercury has been suggested as a risk factor of
myocardial infarction, coronary disease, and cardiovascular
disease [12]. Levels of mercury in toenail and urine sam-
ples have been found to be directly correlated with the
increased risk of myocardial infarction, and coronary heart
disease [13]. MeHg has been reported to cause hyperten-
sion in rats [13]. MeHg has also been shown to generate
reactive oxygen species (ROS) in several systems including
the vascular ECs, which can lead to cellular oxidative
stress [14, 15]. Despite the existence of a correlation
between mercury and cardiovascular diseases, there
remains a void on the role of vascular endothelial cells
(ECs) in the mechanism of mercury-induced cardiovascu-
lar diseases. Vascular endothelium is known to play a
pivotal role in the structure and function of the blood vessel
and maintains the homeostasis of the circulatory system
and the body in general [16]. Endothelial dysfunction has
been associated with vascular leak and the breakdown of
the cardiovascular system [17]. Hence, the mercury-
induced cardiovascular diseases may conceivably be a
result of the toxic effects of mercury on the vascular
endothelium.
Mammalian phospholipases are enzymes which specifi-
cally hydrolyze the membrane phospholipids and generate
bioactive lipid second messengers, which play a vital role in
cell signaling and regulation of the cellular functions [16,
18, 19]. One such enzyme, PLA2, is crucial in regulating the
cellular signaling cascades involving the formation and
repair of the phospholipid membrane and generation of the
inflammatory lipid metabolites [19, 20]. In addition to
functioning as a housekeeping enzyme [21], PLA2 hydro-
lyzes the membrane phospholipid at the sn-2 position,
releasing the free unsaturated fatty acid (arachidonic acid,
AA) from the membrane phospholipid and forming the
lysophospholipid [19, 20, 22]. The PLA2-released AA is a
preferred substrate for the lipid oxygenases including the
cyclooxygenases (COXs) and lipoxygenases (LOXs),
which catalyze the formation of bioactive AA metabolites
(eicosanoids) such as prostaglandins and leukotrienes [20].
Generation of the eicosanoids is tightly regulated by PLA2
action and they have been identified as crucial players in the
inflammatory cascades [22]. Another important member of
the phospholipase family, phospholipase D (PLD), is a
ubiquitous lipid signaling enzyme present in all mammalian
cells, and acts exclusively on the substrate phosphatidyl-
choline (PC) [19]. PLD hydrolyzes PC to generate choline
and phosphatidic acid (PA), which is subsequently metab-
olized to either 1,2-diacylglycerol (DAG) by phosphatidate
phosphohydrolase or lysophosphatidic acid (LPA) by
PLA1/PLA2 [18, 19, 23]. These PLD-derived lipid media-
tors have been shown to play vital roles in cell signaling and
signal transduction [16, 19, 22–25].
Earlier, we have reported that MeHg induces activation
of both PLA2 and PLD in the vascular ECs [15, 26–28].
However, the interdependent regulation of these two
phospholipases in the vascular ECs under mercury expo-
sure is yet to be shown. In order to define the underlying
bioactive lipid-mediated mechanism of mercury-induced
vascular dysfunction(s), we hypothesized that MeHg would
induce cytotoxicity in the vascular ECs through the inter-
active signaling of PLA2, COX, LOX, and PLD. Our
results demonstrated that MeHg induced the activation of
PLD and generation of the bioactive lipid, PA, through the
upstream activation of PLA2 and formation of COX- and
LOX-catalyzed eicosanoids, which led to the MeHg-
induced cytotoxicity in the well-established bovine pul-
monary artery ECs (BPAECs).
Materials and Methods
Materials
Bovine pulmonary artery endothelial cells (passage 2)
were purchased from Cell Applications, Inc. (San Diego,
CA). Antibiotic–antimycotic (10,000 units/ml penicillin,
10,000 lg/ml streptomycin, 25 lg/ml amphoteracin B),
fetal bovine serum (FBS), trypsin, and nonessential amino
acids were obtained from Gibco Invitrogen Co. (Grand
Island, NY). Phosphate-free Dulbecco’s modified Eagle’s
medium (DMEM), minimum essential medium (MEM),
MeHg chloride, and lactate dehydrogenase (LDH) cyto-
toxicity assay kits were obtained from Sigma Chemical Co.
(St. Louis, MO). [32P]orthophosphate (carrier-free) was
obtained from New England Nuclear (Wilmington, DE).
Phosphatidylbutanol (PBt) was purchased from Avanti
Polar Lipids (Alabaster, AL). Endothelial cell growth fac-
tor was obtained from Upstate Biotechnology (Lake Placid,
NY). [3H]arachidonic acid (AA) was acquired from
American Radiolabeled Chemicals, Inc. (St. Louis, MO).
Arachidonyl trifluoromethyl ketone (AACOCF3), BEL,
ibuprofen, cinnamyl-3,4-dihydroxy-a-cynanocinnamate
318 Cell Biochem Biophys (2013) 67:317–329
123

(CDC), 5,8,11-eicosatriynoic acid (ETI), 5,8,11,14-eico-
satetraynoic acid (ETYA), baicalein, caffeic acid, aspirin,
and indomethacin were obtained from Cayman Chemical
Co. (Ann Arbor, MI). Primary antibodies for cPLA2 and
phosphoserine-cPLA2, raised in rabbit, were obtained from
Cell Signaling Technology, Inc (Danvers, MA). Secondary
antibody (Anti-Rabbit IgG) and anti-b-actin antibody were
obtained from Amersham Biosciences (Piscataway, NJ).
Cell Culture
Bovine pulmonary artery endothelial cells were cultured in
MEM supplemented with 10% FBS, 1% nonessential
amino acids, 100 units/ml antibiotics (penicillin and
streptomycin) and 5 lg/ml endothelial growth factor
according to our previously published procedures [15, 16].
Cells in culture were maintained at 37�C in a humidified
environment of 95% air–5% CO2 and grown to contact-
inhibited monolayers with typical cobblestone morphol-
ogy. When confluence was reached, cells were trypsinized
and sub-cultured in sterile 35 or 17.5-mm tissue culture
dishes. Confluent cells were observed for cobblestone
morphology under light microscope and stained positive
for factor VIII. All experiments were conducted between 2
and 10 passages (75–80% confluence).
LDH Assay of Cytotoxicity
Cytotoxicity in BPAECs was determined by spectropho-
tometric determination of the extent of release of lactate
dehydrogenase (LDH) from cells according to our previ-
ously published methods [17, 26, 27]. BPAECs grown in
17.5-mm dishes were pretreated with MEM alone or MEM
containing the selected pharmacological inhibitors for 1 h
and then treated with MEM alone or MEM containing
MeHg for the desired lengths of time. At the end of
treatment, the medium was collected and the LDH released
into the medium was determined according to the manu-
facturer’s recommendations (Sigma Chemical Co.,
St. Louis, MO).
Ecosanoid Determination by Liquid Chromatography-
Mass Spectrometry (LC-MS)
Samples (media) were spiked with 10 ng of 8-iso prosta-
glandin F2a-d4 and 15(S)-HETE-d8 as internal standards and
mixed well. SEP-Pak C18 cartridges (100 mg adsorbent;
Waters Corporation, MA) were equilibrated with 1 ml each
of methanol followed by water. The internal standard sup-
plemented samples were applied to the conditioned C18
cartridges and the cartridges were washed with 5 ml of water
followed by 5 ml of hexane. Eicosanoids were eluted with
500 ll of ethyl acetate. The eluate was dried under nitrogen
and reconstituted in methanol:25 mM aqueous ammonium
acetate (1:1). The extracted and reconstituted sample was
subjected to HPLC on a Luna C18 column (2 9 150 mm,
3 l, Phenomenex) eluted with methanol:13 mM aqueous
ammonium acetate:acetonitrile gradient at a flow rate of
0.2 ml/min. Initial composition of the solvent gradient
50:45:5 (CH3OH:aq.NH4OCOCH3:CH3CN) was changed to
90:5:5 in 30 min and the final conditions were maintained for
5 min. The eluent was monitored for the eicosanoids by mass
spectrometer (QuattroLC; Micromass, UK) in the negative
ion mode under the following conditions: Source block:
120�C, Desolvation: 350�C, and Collision gas pressure:
3.2 9 10-4 mbar. Multiple Reaction Monitoring (MRM)
was used to detect and quantify the entire range of eicosa-
noids using this method and the complete list of transitions
are published elsewhere [29]. MRM transitions for the
detected eicosanoids were as follows (m/z): 8-iso PGF2a-d4:
357 ? 197, 8-iso PGF2a: 353 ? 193, PGE2: 351 ? 271,
15(S)-HETE-d8: 327 ? 226, 15(S)-HETE: 319 ? 219,
12(S)-HETE: 391 ? 179, and 5(S)-HETE: 319 ? 115.
Cone voltage (CV) and collision energies (CE) used for
MRM of the detected eicosanoids and the corresponding
deuterated standards were as follows: 8-iso PGF2a—40 V,
25 eV; prostaglandin E2—25 V, 15 eV; and for all HE-
TEs—25 V, 14 eV. Under these conditions, the retention
times for the detected eicosanoids were as follows:
8-iso prostaglandin F2a—9.8 min, PGE2—8.4 min, 15(S)-
HETE—26.3 min, 12(S)-HETE—27.5 min, and 5(S)-HETE—
29.2 min.
Phospholipase D activation in intact ECs: PLD activity in
BPAECs was determined according to our previously pub-
lished procedures [15, 16, 19]. BPAECs in 35-mm dishes
(5 9 105 cells/dish) were labeled with [32P]orthophosphate
(5 lCi/ml) in DMEM phosphate-free medium containing
2% (v/v) fetal bovine serum for 6–14 h. Cells were washed
with MEM and incubated at 37�C in 1 ml of MEM con-
taining 0.05% (v/v) 1-butanol in absence and presence of the
desired concentrations of MeHg for different lengths of time
under a humidified 95% air–5% CO2 atmosphere. In some
experiments, wherever required, ECs were pre-treated for
1 h with the selected pharmacological inhibitors prior to
exposure to MeHg for 30 min. The incubations were termi-
nated by addition of 1 ml methanol:HCl (100:1 v/v). Lipids
were extracted essentially according to the method of Bligh
and Dyer as described previously [15, 28]. [32P]-labeled
phosphatidylbutanol ([32P]PBt), formed from PLD activa-
tion and transphosphatidylation reaction as an index of PLD
activity in intact cells, was separated by thin-layer chroma-
tography (TLC). Radioactivity associated with the [32P]PBt
was quantified by liquid scintillation counting and data were
expressed as DPM normalized to 106 counts in the total
cellular lipid extract.
Cell Biochem Biophys (2013) 67:317–329 319
123
[3H]AA Labeling and Assay of PLA2 Activity
The activity of PLA2 was assayed according to our previ-
ously published procedure [19, 26, 27]. BPAECs in 35-mm
dishes (70% confluence) were labeled for 12 h with 1 ml of
the medium containing 0.5 lCi/ml of [3H]AA following
which they were washed and treated with MEM alone or
MEM-containing MeHg (1–10 lM) for the desired lengths
of time. Following the treatment of cells, [3H]AA released
into the medium was measured in the Packard Tricarb 2900
TR liquid scintillation counter. PLA2 activity was expres-
sed as DPM of [3H]AA released/dish.
SDS-PAGE and Western Blotting
Preparation of cell lysates, separation of proteins by
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
(SDS-PAGE), and Western blotting were done according
to our previously published procedures [16]. Cell lysates
containing equal amounts of proteins (40 lg) were sub-
jected to SDS-PAGE on 12% gels, transferred on to
polyvinylidene difluoride (PVDF) membranes, and then
subjected to overnight immunoblotting with either anti-
cPLA2 or anti-phosphoserine-cPLA2 (1:1000 dilution)
rabbit polyclonal antibodies at 4�C. The membranes were
then washed three times with TBST and incubated for
1–2 h at room temperature with horseradish peroxidase-
conjugated goat anti-rabbit secondary antibody (1:2000
dilution). The immunoblots were then developed on the
film with the enhanced chemiluminescence (ECL) reagents
according to the manufacturer’s recommendations. The
intensities of protein bands developed on the film were
quantified by digital densitometric analysis.
Statistical Analysis of Data
All experiments were done in triplicates and data were
expressed as mean ± standard deviation (SD). One-way
analysis of variance (ANOVA) and pair wise multiple
statistical comparisons were done by Dunnett’s method
with P \ 0.05 indicating significance.
Results
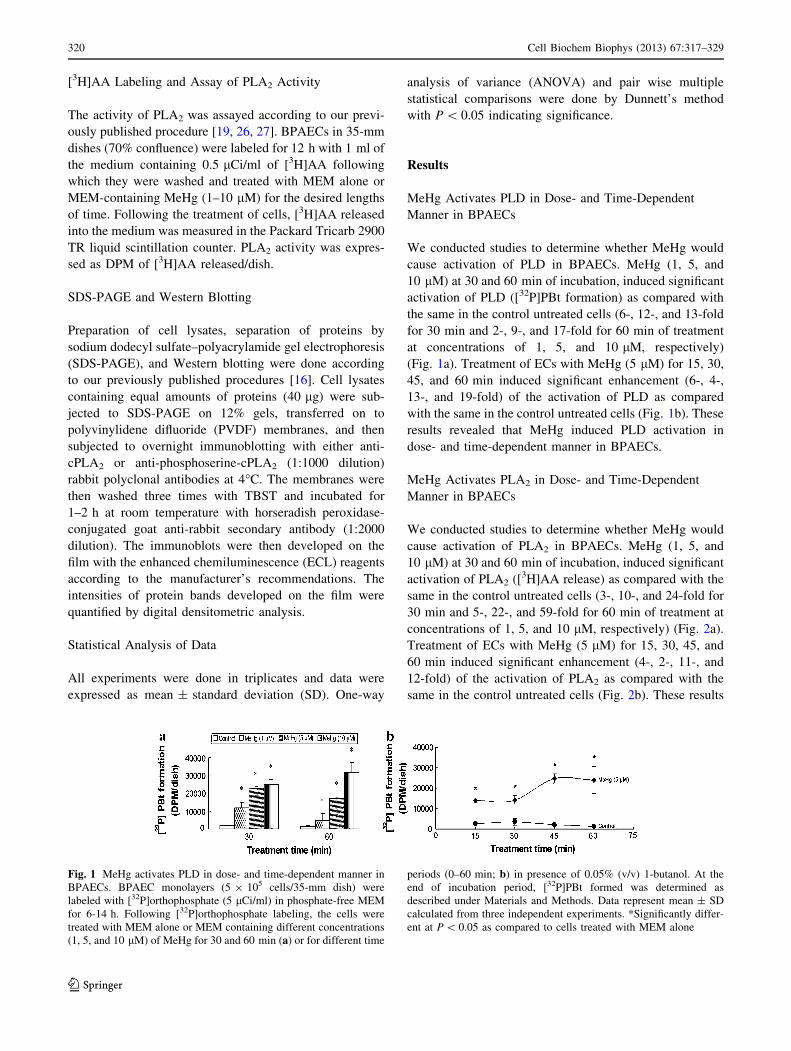
MeHg Activates PLD in Dose- and Time-Dependent
Manner in BPAECs
We conducted studies to determine whether MeHg would
cause activation of PLD in BPAECs. MeHg (1, 5, and
10 lM) at 30 and 60 min of incubation, induced significant
activation of PLD ([32P]PBt formation) as compared with
the same in the control untreated cells (6-, 12-, and 13-fold
for 30 min and 2-, 9-, and 17-fold for 60 min of treatment
at concentrations of 1, 5, and 10 lM, respectively)
(Fig. 1a). Treatment of ECs with MeHg (5 lM) for 15, 30,
45, and 60 min induced significant enhancement (6-, 4-,
13-, and 19-fold) of the activation of PLD as compared
with the same in the control untreated cells (Fig. 1b). These
results revealed that MeHg induced PLD activation in
dose- and time-dependent manner in BPAECs.
MeHg Activates PLA2 in Dose- and Time-Dependent
Manner in BPAECs
We conducted studies to determine whether MeHg would
cause activation of PLA2 in BPAECs. MeHg (1, 5, and
10 lM) at 30 and 60 min of incubation, induced significant
activation of PLA2 ([3H]AA release) as compared with the
same in the control untreated cells (3-, 10-, and 24-fold for
30 min and 5-, 22-, and 59-fold for 60 min of treatment at
concentrations of 1, 5, and 10 lM, respectively) (Fig. 2a).
Treatment of ECs with MeHg (5 lM) for 15, 30, 45, and
60 min induced significant enhancement (4-, 2-, 11-, and
12-fold) of the activation of PLA2 as compared with the
same in the control untreated cells (Fig. 2b). These results
Fig. 1 MeHg activates PLD in dose- and time-dependent manner in
BPAECs. BPAEC monolayers (5 9 105 cells/35-mm dish) were
labeled with [32P]orthophosphate (5 lCi/ml) in phosphate-free MEM
for 6-14 h. Following [32P]orthophosphate labeling, the cells were
treated with MEM alone or MEM containing different concentrations
(1, 5, and 10 lM) of MeHg for 30 and 60 min (a) or for different time
periods (0–60 min; b) in presence of 0.05% (v/v) 1-butanol. At the
end of incubation period, [32P]PBt formed was determined as
described under Materials and Methods. Data represent mean ± SD
calculated from three independent experiments. *Significantly differ-
ent at P \ 0.05 as compared to cells treated with MEM alone
320 Cell Biochem Biophys (2013) 67:317–329
123

revealed that MeHg-induced PLA2 activation in dose- and
time-dependent manner in BPAECs.
PLA2-Specific Inhibitors Attenuate MeHg-Induced
PLD Activation in BPAECs
Earlier experiments of the current study showed that MeHg
caused the activation of both PLA2 and PLD. Therefore,
we investigated whether PLA2 activation would be
upstream of PLD activation in ECs under MeHg exposure.
In order to establish the role of PLA2 activation in the
MeHg-induced PLD activation in BPAECs, the well-
established PLA2-specific inhibitors (AACOCF3 for cPLA2
and BEL for iPLA2) were used. Cells were pre-treated for
1 h with MEM alone or MEM containing the chosen PLA2-
specific inhibitor (1, 3, and 5 lM) and then treated for
30 min with MeHg (5 lM). AACOCF3 caused effective
and significant attenuation of the MeHg-induced PLD
activation in BPAECs (58, 78, and 76% of inhibition at
concentrations of 1, 3, and 5 lM, respectively) (Fig. 3a).
BEL also offered effective and significant attenuation of
the MeHg-induced PLD activation in BPAECs (44, 82, and
69% of inhibition at concentrations of 1, 3, and 5 lM,
respectively) (Fig. 3b). These results revealed that the
PLA2-specific inhibitors effectively attenuated the MeHg-
induced PLD activation in BPAECs, suggesting the
involvement of upstream cPLA2 activation in the MeHg-
induced activation of PLD in BPAECs.
COX-Specific Inhibitors Attenuate MeHg-Induced PLD
Activation in BPAECs
As the earlier experiment of this study showed that MeHg-
induced PLA2 activation and caused the release of AA, we
further investigated whether the COX-generated eicosa-
noids would be involved in the MeHg-induced PLD
activation in BPAECs. In order to establish the role of
COX-generated eicosanoids in the MeHg-induced PLD
activation in BPAECs, here, the well-established general-
COX inhibitors (ibuprofen and CDC) were utilized. Cells
were pre-treated for 1 h with MEM alone or MEM con-
taining the chosen COX inhibitors (100 and 300 lM for
ibuprofen and 10 and 25 lM for CDC) and then treated for
30 min with MeHg (5 lM). Ibuprofen caused effective and
significant attenuation of the MeHg-induced PLD activa-
tion in BPAECs (9 and 30% at inhibition for concentrations
Fig. 2 MeHg activates PLA2 in dose- and time-dependent manner in
BPAECs. BPAEC monolayers (5 9 105 cells/35-mm dish) were
labeled with [3H]AA (5 lCi) in complete EC medium for 12 h.
Following [3H]AA labeling, the cells were treated with MEM alone or
MEM containing different concentrations (1, 5, and 10 lM) of MeHg
for 30 and 60 min (a) or for different time periods (0–60 min; b). At
the end of incubation, [3H]AA released into the medium was
determined as described under ‘‘Materials and Methods’’ section.
Data represent mean ± SD calculated from three independent
experiments. *Significantly different at P \ 0.05 as compared to
cells treated with MEM alone
Fig. 3 PLA2-specific inhibitors attenuate MeHg-induced PLD acti-
vation in BPAECs. BPAEC monolayers (5 9 105 cells/35-mm dish)
were labeled with [32P]orthophosphate (5 lCi/ml) in phosphate-free
MEM for 6–14 h. Following [32P]orthophosphate labeling, the cells
were pretreated for 1 h with MEM alone or MEM containing
AACOCF3 (1, 5, and 10 lM; a) or BEL (1, 5, and 10 lM; b) and then
subjected to treatment with MEM alone or MEM containing MeHg
(5 lM) for 30 min in presence of 0.05% (v/v) 1-butanol. At the end of
incubation, [32P]PBt formed was determined as described under
‘‘Materials and Methods’’ section. Data represent mean ± SD of three
independent experiments. *Significantly different at P \ 0.05 as
compared to cells treated with MEM alone. **Significantly different
at P \ 0.05 as compared to cells treated with MEM-containing MeHg
alone
Cell Biochem Biophys (2013) 67:317–329 321
123

of 100 and 300 lM, respectively) (Fig. 4a). CDC also
offered effective and significant attenuation of the MeHg-
induced PLD activation in BPAECs (28 and 51% of inhi-
bition at concentrations of 10 and 25 lM, respectively)
(Fig. 4b). These results revealed that the general-COX
inhibitors effectively attenuated the MeHg-induced PLD
activation in BPAECs, suggesting the involvement of
COX-generated eicosanoids in the MeHg-induced
upstream activation of PLD in BPAECs.
General-LOX Inhibitors Attenuate MeHg-Induced PLD
Activation in BPAECs
Earlier experiments of this study revealed that the COX-
generated eicosanoids were involved in the MeHg-induced
PLD activation, and therefore we further investigated to
determine whether the related LOX-generated eicosanoids
would be involved in the MeHg-induced PLD activation in
BPAECs. In order to establish the role of LOX-generated
eicosanoids in the MeHg-induced PLD activation in
BPAECs, the well-established general-LOX inhibitors (ETI
and ETYA) were used. Cells were pre-treated for 1 h with
MEM alone or MEM containing the chosen LOX inhibitors
(10, 50, and 100 lM) and then treated for 30 min with
MeHg (5 lM). ETI caused effective and significant atten-
uation of the MeHg-induced PLD activation in BPAECs
(54, 79, and 77% of inhibition at concentrations of 10, 50,
and 100 lM, respectively) (Fig. 5a). ETYA also offered
effective and significant attenuation of the MeHg-induced
PLD activation in BPAECs (1, 27, and 52% of inhibition at
concentrations of 10, 50, and 100 lM, respectively)
(Fig. 5b). These results revealed that the general-LOX
inhibitors effectively attenuated the MeHg-induced PLD
activation in BPAECs, suggesting the involvement of the
LOX-generated eicosanoids in the MeHg-induced
upstream activation of PLD in BPAECs.
12-LOX-Specific Inhibitors Attenuate MeHg-Induced
PLD Activation in BPAECs
Earlier experiments of this study revealed that the LOX-
generated eicosanoids were involved in the MeHg-induced
PLD activation, and therefore we further investigated to
demonstrate whether the 12-LOX-generated eicosanoids
would be responsible for the MeHg-induced PLD
activation in BPAECs. In order to establish the role of
Fig. 4 COX-specific inhibitors attenuate MeHg-induced PLD acti-
vation in BPAECs. BPAEC monolayers (5 9 105 cells/35-mm dish)
were labeled with [32P]orthophosphate (5 lCi/ml) in phosphate-free
MEM for 6–14 h. Following [32P]orthophosphate labeling, the cells
were pretreated for 1 h with MEM alone or MEM containing
ibuprofen (100 and 300 lM; a) or CDC (5, 10, and 25 lM; b) and
then subjected to treatment with MEM alone or MEM containing
MeHg (5 lM) for 30 min in presence of 0.05% (v/v) 1-butanol. At the
end of incubation, [32P]PBt formed was determined as described
under Materials and Methods. Data represent mean ± SD of three
independent experiments. *Significantly different at P \ 0.05 as
compared to cells treated with MEM alone. **Significantly different
at P \ 0.05 as compared to cells treated with MEM containing MeHg
alone
Fig. 5 LOX-specific inhibitors attenuate MeHg-induced PLD acti-
vation in BPAECs. BPAEC monolayers (5 9 105 cells/35-mm dish)
were labeled with [32P]orthophosphate (5 lCi/ml) in phosphate-free
MEM for 6–14 h. Following [32P]orthophosphate labeling, the cells
were pretreated for 1 h with MEM alone or MEM containing ETI (10,
50, and 100 lM; a) or ETYA (10, 50, and 100 lM; b) and then
subjected to treatment with MEM alone or MEM-containing MeHg
(5 lM) for 30 min in presence of 0.05% (v/v) 1-butanol. At the end of
incubation, [32P]PBt formed was determined as described under
‘‘Materials and Methods’’ sectiont at P \ 0.05 as compared to cells
treated with MEM alone. **Significantly different at P \ 0.05 as
compared to cells treated with MEM-containing MeHg alone
322 Cell Biochem Biophys (2013) 67:317–329
123

12-LOX-generated eicosanoids in the MeHg-induced PLD
activation in BPAECs, here, the well-established 12-LOX
inhibitors (baicalein and caffeic acid) were used. Cells
were pre-treated for 1 h with MEM alone or MEM con-
taining the chosen 12-LOX-specific inhibitors (5 lM for
baicalein and 10, 50, and 100 lM for caffeic acid) and then
treated for 30 min with MeHg (5 lM). Baicalein caused
effective and significant attenuation of the MeHg-induced
PLD activation in BPAECs (87% of inhibition for 5 lM)
(Fig. 6a). Caffeic acid also offered effective and significant
attenuation of the MeHg-induced PLD activation in
BPAECs (66, 75, and 79% of inhibition at concentrations
of 20, 50, and 100 lM, respectively) (Fig. 6b). These
results revealed that the 12-LOX-specific inhibitors sig-
nificantly attenuated the MeHg-induced PLD activation in
BPAECs, suggesting major involvement of 12-LOX-gen-
erated eicosanoids in the MeHg-induced activation of PLD
in BPAECs.
MeHg Induces COX- and LOX-Catalyzed Formation
of Eicosanoids in BPAECs
Earlier in this study, the involvement of PLA2 activity and
the COX- and LOX-generated eicosanoids were shown to
be involved in the MeHg-induced PLD activation in
BPAECs. Therefore, we further determined the extent of
COX- and LOX-generated eicosanoids in BPAECs fol-
lowing MeHg exposure. Treatment of ECs with MeHg
(5 lM) for 15, 30, and 60 min induced significant increase
(27-, 31-, and 48-fold) in PGE2 release as compared with
Fig. 6 12-LOX-specific inhibitors attenuate MeHg-induced PLD
activation in BPAECs. BPAEC monolayers (5 9 105 cells/35-mm
dish) were labeled with [32P]orthophosphate (5 lCi/ml) in phosphate-
free MEM for 6–14 h. Following [32P]orthophosphate labeling, the
cells were pretreated for 1 h with MEM alone or MEM containing
baicalein (5 lM; a) or caffeic acid (20, 50, and 100 lM; b) and then
subjected to treatment with MEM alone or MEM-containing MeHg
(5 lM) for 30 min in presence of 0.05% (v/v) 1-butanol. At the end of
incubation, [32P]PBt formed was determined as described under
‘‘Materials and Methods’’ section. Data represent mean ± SD of three
independent experiments. *Significantly different at P \ 0.05 as
compared to cells treated with MEM alone. **Significantly different
at P \ 0.05 as compared to cells treated with MEM-containing MeHg
alone
Fig. 7 MeHg induces COX- and LOX-catalyzed formation of
eicosanoids in BPAECs. BPAEC monolayers (5 9 105 cells/35-mm
dish) were treated with MEM alone or MEM containing different
concentrations (1, 5, and 10 lM) of MeHg for desired amount of time
and the formation of PGE2 (a), 5-HETE (b), 12-HETE (c), and
15-HETE (d) was measured as described under ‘‘Materials and
Methods’’ section. Data represent mean ± SD calculated from three
independent experiments. *Significantly different at P \ 0.05 as
compared to cells treated with MEM alone
Cell Biochem Biophys (2013) 67:317–329 323
123

the same in the control untreated cells (Fig. 7a). MeHg (1,
5, 10 lM), at 30 min of incubation, induced significant
5-HETE formation (2-, 1.5-, and 1.1-fold) as compared
with the same in the control untreated cells (Fig. 2a).
MeHg (5 lM) at 15, 30, and 60 min of treatment induced
significant increase in the 12-HETE formation (1.4-, 1.3-,
and 1.4-fold) as compared with the same in the control
untreated cells (Fig. 7c). MeHg (5 lM) at 15, 30, and
60 min of treatment induced significant increase in the
15-HETE formation (13-, 13-, and 12-fold) as compared
with the same in the control untreated cells (Fig. 7d). These
results revealed that MeHg enhanced the extent of forma-
tion of the COX- and LOX-generated eicosanoids in
BPAECs.
COX- and LOX-Specific Inhibitors Attenuate MeHg-
Induced LDH Release in BPAECs
Earlier in this study, MeHg was shown to activate PLA2
and generate eicosanoids in BPAECs. Our earlier studies
have also shown that cPLA2 inhibitor (AACOCF3) and
iPLA2 inhibitor (BEL) attenuate MeHg-induced cytotox-
icity in BPAECs [26, 27]. Therefore, here, we investigated
to demonstrate whether the formation of PA and the sub-
sequent COX- and LOX-generated eicosanoids would
be responsible for the MeHg-induced cytotoxicity in
BPAECs. In order to establish the role of COX- and LOX-
generated eicosanoids in the MeHg-induced cytotoxicity in
BPAECs, the well-established general-COX and LOX
inhibitors (indomethacin and ETYA) were used. Cells were
pre-treated for 1 h with MEM alone or MEM containing
the chosen inhibitors (100 lM) and then treated for 30 min
with MeHg (5 lM). Indomethacin offered effective and
significant attenuation of the MeHg-induced LDH leak in
BPAECs (45% of inhibition at a concentration of 100 lM)
(Fig. 8a). ETYA also offered effective and significant
attenuation of the MeHg-induced LDH leak in BPAECs
(73% of inhibition at a concentration of 100 lM) (Fig. 8b).
These results revealed that the COX- and LOX-specific
inhibitors effectively attenuated the MeHg-induced LDH
leak in BPAECs, suggesting the involvement of COX- and
LOX-generated eicosanoids in the MeHg-induced cyto-
toxicity in BPAECs.
PLA2-Specific Inhibitor and 1-Butanol, the Quencher
of PLD-Generated PA, Attenuate MeHg-Induced LDH
Release in BPAECs
Earlier in the study, MeHg was shown to induce cytotox-
icity which was reversed by the COX- and LOX-specific
inhibitors in BPAECs. In order to further understand the
role of the PLA2 and PLD in the MeHg-induced cytotox-
icity, the protective effects of well-established general
PLA2 inhibitor (quinacrine, 2 lM) and 1-butanol (2%), the
PLD-generated PA quencher were used. Cells were pre-
treated for 1 h with MEM alone or MEM containing the
chosen inhibitors and then treated for 30 min with MeHg
(10 lM). Quinacrine offered effective and significant
attenuation of the MeHg-induced LDH leak in BPAECs
(33% of inhibition at a concentration of 2 lM) (Fig. 9a).
1-Butanol also offered effective and significant attenuation
of the MeHg-induced LDH leak in BPAECs (92% of
inhibition at a concentration of 2%) (Fig. 9b). These results
revealed that PLA2 inhibition and quenching of the PLD-
generated PA effectively attenuated the MeHg-induced
LDH leak in BPAECs, suggesting the involvement of PLA2
and PLD activation in the MeHg-induced cytotoxicity in
BPAECs.
MeHg Induces Serine Phosphorylation of cPLA2
in BPAECs
As the earlier experiment of this study demonstrated the
MeHg-induced activation of cPLA2 in BPAECs, we
investigated whether the enzyme activation would be
associated with the serine phosphorylation of cPLA2. SDS-
PAGE and Western blot analysis of proteins revealed that
MeHg (5–25 lM) induced serine phosphorylation in a
Fig. 8 COX- and LOX-specific inhibitors attenuate MeHg-induced
LDH release in BPAECs. BPAEC monolayers (2.5 9 105 cells/17.5-
mm dish) were pretreated for 1 h with MEM alone or MEM
containing indomethacin (100 lM; a) or ETYA (100 lM; b) and then
subjected to treatment with MEM alone or MEM-containing MeHg
(5 lM) for 60 min. At the end of incubation period, release of LDH
into the medium was determined spectrophotometrically as described
under ‘‘Materials and Methods’’ section. Data represent mean ± SD
calculated from three independent experiments. *Significantly differ-
ent at P \ 0.05 as compared to cells treated with MEM alone.
**Significantly different at P \ 0.05 as compared to cells treated with
MEM containing MeHg alone
324 Cell Biochem Biophys (2013) 67:317–329
123

dose- and time-dependent manner (0–30 min) in BPAECs.
MeHg-induced serine phosphorylation of cPLA2 was
maximum at 10 lM dose (Fig. 10a). In addition, the extent
of serine phosphorylation of cPLA2 was the highest at
5 min of incubation and then declined from 5 to 30 min
(Fig. 10b) in BPAECs as compared to the same in the
control untreated cells. From these results, it was evident
that MeHg induced serine phosphorylation of cPLA2 in a
dose- and time-dependent fashion in BPAECs (Fig. 10).
Discussion
The results of this study revealed that the MeHg-induced
activation of PLD in BPAECs was regulated upstream by
the activation of PLA2 and eicosanoids generated by COXs
and LOXs. Furthermore, the results showed that the MeHg-
induced cytotoxicity in BPAECs was mediated by the
bioactive lipids generated by PLA2, COXs, LOXs, and
PLD. Overall, this study demonstrated that the bioactive
lipid signaling cascades, operated by the phospholipases
and lipid oxygenases, regulated the MeHg-induced vascu-
lar EC cytotoxicity.
Endothelium of the blood vessels plays a fundamental
role in the structure and function of the vasculature and
maintains the homeostasis of the circulation. Cellular
membrane phospholipids are crucial for the structure and
function of the living cells. Phospholipases including PLA2
and PLD are essential enzymes in the housekeeping of cells
as well as in the generation of bioactive lipid messengers
which play a vital role in cellular signaling [22, 30]. Hence,
it is conceivable to surmise that MeHg exerts its toxic
effects on the vascular endothelium leading to the modu-
lation or alteration of the functions of the blood vessel.
Phospholipase A2 is an important membrane phospho-
lipid-hydrolyzing enzyme which releases the unsaturated
fatty acid (typically AA) esterified at the sn-2 position of
the phospholipid [22]. Lipid oxygenases, including the
COXs and LOXs, utilize the PLA2-released free AA as the
substrate and convert it into potent bioactive AA metabo-
lites (eicosanoids) including the prostaglandins and leu-
kotrienes [20]. COX-1 and COX-2, the two isoforms of
mammalian COX convert AA into prostaglandin H2
(PGH2), which acts as a precursor for further metabolic
conversion into thromboxane A2 (TXA2), prostacyclin
(PGI2), and PGE2 in the vascular ECs [19]. LOXs are of
three main types viz., 5-LOX, 12-LOX, and 15-LOX which
convert the free AA into the hydroperoxyeicosatetraenoic
acids (5-HPETE, 12-HPETE, and 15-HPETE), which are
further reduced to the hydroxyeicosatetraenoic acids
(5-HETE, 12-HETE, and 15-HETE) by the cellular gluta-
thione peroxidase [19]. The HPETEs act as precursors for
the LOX-generated leukotrienes. The eicosanoids, gener-
ated by COXs and LOXs, have been established to play
crucial roles in the inflammatory cascades and their for-
mation is tightly regulated by the activity of PLA2 [22].
The LOX-derived eicosanoids are potent bioactive lipid
signaling molecules in mammalian cells including the
vascular ECs [19]. Both PLA2 and the eicosanoids are
emerging as critical players in cardiovascular diseases [30].
Hence, the unregulated activation of PLA2, mediated by
certain agonists (e.g., environmental toxicants), can
endanger the vascular endothelial structure and function,
and ultimately the blood vessel function.
Mammalian cells contain three major classes of PLA2:
(i) cytosolic calcium-dependent PLA2 (cPLA2), (ii) intra-
cellular calcium-independent PLA2 (iPLA2), and (iii)
secretory calcium-dependent PLA2 (sPLA2) [30]. Several
agonists have been identified that activate PLA2 in differ-
ent systems in vitro and in vivo [20]. Regulation of PLA2 is
apparently complex. The regulation of cPLA2 activation
has been extensively investigated, wherein the mitogen-
activated protein kinases (MAPKs), protein kinase A
(PKA), and protein kinase C (PKC) have been shown to
play important roles [20]. Results of this study revealed
that MeHg induced the serine phosphorylation of cPLA2 in
BPAECs, possibly through the upstream regulation by
MAPKs. On the other hand, the regulation of iPLA2 and
Fig. 9 PLA2-specific inhibitor and 1-butanol, the quencher of PLD-
generated PA, attenuate MeHg-induced LDH release in BPAECs.
BPAEC monolayers (2.5 9 105 cells/17.5-mm dish) were pretreated
for 1 h with MEM alone or MEM containing quinacrine (2 lM; a) or
1-butanol (2%; b) and then subjected to treatment with MEM alone or
MEM-containing MeHg (10 lM) for 60 min. At the end of incubation
period, LDH in the medium was determined spectrophotometrically
as described under ‘‘Materials and Methods’’ section. Data represent
mean ± SD calculated from three independent experiments. *Signif-
icantly different at P \ 0.05 as compared to cells treated with MEM
alone. **Significantly different at P \ 0.05 as compared to cells
treated with MEM containing MeHg alone
Cell Biochem Biophys (2013) 67:317–329 325
123

sPLA2 is not known in detail. Studies reveal that lipid
peroxidation activates sPLA2 [31]. ROS have been shown
to cause the activation of iPLA2 leading to the release of
AA in macrophages [32]. Hydrogen peroxide has been
reported to cause the release of AA in astrocytes that is
mediated by the activation of cPLA2 and iPLA2 [33].
Overall, these reports corroborate that PLA2 activity in
mammalian cells is regulated by signaling cascades, ROS,
and oxidative stress. Earlier, we have shown that MeHg
causes the formation of ROS, induces oxidative stress, and
activates PLA2 activity through ROS production and thiol
depletion in BPAECs [15, 26, 27]. As observed in this
study, cPLA2 activation by MeHg in BPAECs could be
mediated by the ROS generation, oxidative stress, and
thiol-redox alteration. Needless to mention, the involve-
ment of other signaling kinases such as the MAPKs, PKA,
and PKC in the regulation of MeHg-induced cPLA2 acti-
vation in the vascular ECs is not ruled out.
MeHg has been reported to cause toxicity in astrocytes
and neurons that is mediated by ROS, oxidative stress, and
loss of GSH [34, 35]. Mepacrine, a well-known PLA2
inhibitor, has been shown to protect against the MeHg-
induced cytotoxicity in the cerebellar granule cells, sug-
gesting the role of PLA2 activation in the MeHg-induced
neurotoxicity [36]. Earlier, we have also shown that inor-
ganic mercury- and MeHg-induced cytotoxicity in
BPAECs is protected by the cPLA2 and iPLA2 inhibitors
and inorganic mercury induces the formation of eicosa-
noids in BPAECs [26, 27], suggesting the role of PLA2 in
the mercury-induced cytotoxicity in the vascular ECs.
Along these lines, the results of the current study also
demonstrated that quinacrine attenuated the MeHg-induced
cytotoxicity in BPAECs. Thus, the findings of this
study reinforced the role of PLA2 in the mediation of
MeHg-induced cytotoxicity in the vascular ECs. Activation
of cPLA2, and probably iPLA2, might have contributed to
the cytotoxicity of MeHg through the formation and action
of eicosanoids generated by COXs and LOXs as the eico-
sanoids (prostaglandins, HPETEs, and leukotrienes) are
known as potent bioactive lipid mediators. This was further
supported by the results of this study that MeHg induced the
formation of PGE2 and 5-, 12-, and 15-HPETEs and the
COX- and LOX-specific inhibitors offered protection
against the MeHg cytotoxicity in BPAECs. Eicosanoids,
including the COX-generated prostanoids (prostaglandins,
thromboxane, and prostacyclin), are emerging as the
important mediators of inflammation in vascular endothelial
dysfunction and atherosclerosis [37, 38].
In mammalian cells, two predominant isoforms of PLD,
namely PLD1 and PLD2, have been identified, cloned, and
characterized [16, 19]. Vascular ECs have also been
reported to contain both PLD1 and PLD2 [16]. Distinct
cofactors including Arf, Rho, Cdc42, phosphatidylinositol
4,5-bisphosphate, and detergents have been reported to
cause PLD activation in vitro specific to the isoform [19].
ROS and oxidants have been shown to activate PLD in the
cultured cellular systems including the vascular ECs [16].
Signaling kinases such as the p38 MAPK, extracellular
signal-regulated kinases (ERKs), and Src kinases have
been identified to play a role in the regulation of the oxi-
dant-mediated activation of PLD in BPAECs [16]. Also,
the activation of PLD by several agonists has been
observed to be regulated by cellular calcium, PKC, het-
erotrimeric G proteins, small molecular weight G proteins,
protein tyrosine kinases, and protein tyrosine phosphatases
[16]. Earlier, we have shown that the MeHg-induced PLD
Fig. 10 MeHg induces serine phosphorylation of cPLA2 in BPAECs.
BPAECs monolayers (5 9 105 cells/35-mm dish) were subjected to
treatment with MEM alone or MEM containing MeHg (5, 10, and
25 lM) for 30 min (a) or for different time periods (0–30 min; b). At
the end of incubation period, cPLA2 and phosphoserine-cPLA2 were
probed using Western blotting as described under Materials and
Methods. The intensities of the blots were quantified. Data represent
mean ± SD calculated from three independent experiments. *Signif-
icantly different at P \ 0.05 as compared to cells treated with MEM
alone
326 Cell Biochem Biophys (2013) 67:317–329
123

activation in BPAECs is regulated by calcium and the
thiol-redox perturbation being associated with the ROS
generation [15, 28]. Therefore, calcium, ROS, and the
thiol-redox perturbation could have contributed to the
MeHg-induced PLD activation that was observed in this
study.
PGF2a-stimulated activation of PLD in MC3T3-E1 cells
has been observed to be associated with DAG formation
[39]. G proteins have been reported to be involved in the
PGF2a-induced PLD activation in the osteoblast-like cells
[40]. Another COX-generated eicosanoid, PGD2, has been
shown to activate PLD in the osteoblast-like cells through
the calcium/calmodulin signaling [41]. Earlier, we have
shown that the calcium/calmodulin signaling regulates the
MeHg-induced activation of PLD in BPAECs [28]. Thus, it
could be surmised, from the results of the current study,
that eicosanoids might be the possible bioactive lipids in
mediating the MeHg-induced activation of PLD in
BPAECs through calcium/calmodulin signaling. Linoleic
acid hydroperoxide, generated by the soybean LOX,
has been shown to activate PLD in BPAECs [42]. PGF2a-
induced PLD activation in rat luteal cells has been shown to
be attenuated by the LOX-specific inhibitors such as
nordihydroguaeretic acid and ETYA, suggesting the role of
LOX therein [43]. Earlier, we have also reported that the
vitamin C-induced activation of PLD in the lung micro-
vascular ECs is attenuated by the COX- and LOX-specific
inhibitors, suggesting the regulation of prooxidant-induced
PLD activation by COXs and LOXs [19]. The results of this
study were in agreement with these reports, and revealed
that both COX- and LOX-generated eicosanoids, derived
from the PLA2-released AA from the membrane phospho-
lipids, appeared as key regulators in the MeHg-induced
PLD activation in BPAECs (Scheme 1). In addition, the
results of the current study demonstrated that PLD-gener-
ated PA also played a crucial role in the MeHg-induced
cytotoxicity in BPAECs as revealed from the experiments
where the primary alcohol, 1-butanol, protected the MeHg-
induced cytotoxicity by quenching the PLD-generated PA
and rendering it physiologically inactive.
The roles of phospholipases and COXs in the vascular
diseases and ischemic injury of the tissue are becoming
increasingly evident [44, 45]. The three important isoforms
of LOX (5-LOX, 12-LOX, and 15-LOX) have been shown
to be associated with several human diseases including the
myocardial diseases [46–49]. The role of PLD in vascular
disorders is becoming increasingly evident [16]. MeHg is
an established heavy metal toxicant that depletes the cel-
lular thiols and causes the generation of ROS [15]. As
demonstrated in this study, the activation and cross-talk
among PLA2, COXs, LOXs, and PLD to generate the
potent bioactive lipid messengers that caused the cytotox-
icity in the cultured vascular ECs under MeHg exposure,
offered a possible lipid signaling mechanism of the mer-
cury-induced vascular diseases.
Acknowledgments This work was supported by the funding from
the International Academy of Oral Medicine and Toxicology (IA-
OMT), Davis Heart and Lung Research Institute, the Division of
Pulmonary, Allergy, Critical Care, and Sleep Medicine, and the
National Institutes of Health (HL 093463).
References
1. Clarkson, T. W., Magos, L., & Myers, G. J. (2003). The toxi-
cology of mercury–current exposures and clinical manifestations.
New England Journal of Medicine, 349(18), 1731–1737.
2. Bolger, P. M., & Schwetz, B. A. (2002). Mercury and health. New
England Journal of Medicine, 347(22), 1735–1736.
3. Mutter, J., Naumann, J., Schneider, R., Walach, H., & Haley, B.
(2005). Mercury and autism: Accelerating evidence? Neuroen-
docrinology Letters, 26(5), 439–446.
4. Mutter, J., Naumann, J., Sadaghiani, C., Walach, H., & Drasch,
G. (2004). Amalgam studies: Disregarding basic principles of
mercury toxicity. International Journal of Hygiene and Envi-
ronmental Health, 207(4), 391–397.
5. Ustinaviciene, R., Obelenis, V., & Ereminas, D. (2004). Occu-
pational health problems in modern work environment. Medicina
(Kaunas), 40(9), 897–904.
6. Renneberg, A. J., & Dudas, M. J. (2001). Transformations of
elemental mercury to inorganic and organic forms in mercury
and hydrocarbon co-contaminated soils. Chemosphere, 45(6–7),
1103–1109.
7. Clarkson, T. W. (2002). The three modern faces of mercury.
Environmental Health Perspectives, 110(Suppl 1), 11–23.
Scheme 1 MeHg induces PLA2 activation through ROS production
and decrease in thiols leading to the enhanced release of arachidonic
acid (AA) from membrane phospholipids. The AA is subsequently
converted into eicosanoids such as PGE2 by COX and HPETEs by
LOX. These eicosanoids are involved in the down-stream activation
of PLD in ECs. MeHg induces activation of PLD through the
upstream redox-dependent activation of PLA2 and subsequent
formation of COX- and LOX-catalyzed eicosanoids (PGE2 and
HPETEs). The activated PLD leads to the formation of the bioactive
lipid, phosphatidic acid, which is involved in the MeHg-induced EC
cytotoxicity
Cell Biochem Biophys (2013) 67:317–329 327
123

8. Landmark, K., & Aursnes, I. (2004). Mercury, fish, fish oil and
the risk of cardiovascular disease. Tidsskrift for den Norske
Laegeforening, 124(2), 198–200.
9. Nash, R. A. (2005). Metals in medicine. Alternative Therapies in
Health and Medicine, 11(4), 18–25.
10. Kostka, B. (1991). Toxicity of mercury compounds as a possible
risk factor for cardiovascular diseases. British Journal of Indus-
trial Medicine, 48(12), 845.
11. Yoshizawa, K., Rimm, E. B., Morris, J. S., Spate, V. L., Hsieh,
C. C., Speigelman, D., et al. (2002). Mercury and the risk of cor-
onary heart disease in men. New England Journal of Medicine,
347(22), 1755–1760.
12. Kim, D. S., Lee, E. H., Yu, S. D., Cha, J. H., & Ahn, S. C. (2005).
Heavy metal as risk factor of cardiovascular disease—an analysis
of blood lead and urinary mercury. Journal of Preventive Medi-
cine and Public Health, 38(4), 401–407.
13. Guallar, E., Sanz-Gallardo, M. I., van’t Veer, P., Bode, P.,
Aro, A., Gomez-Aracena, J., et al. (2002). Mercury, fish oils, and
the risk of myocardial infarction. New England Journal of
Medicine, 347(22), 1747–1754.
14. Wolf, M. B., & Baynes, J. W. (2007). Cadmium and mercury
cause an oxidative stress-induced endothelial dysfunction. Bio-
Metals, 20(1), 73–81.
15. Hagele, T. J., Mazerik, J. N., Gregory, A., Kaufman, B., Maga-
lang, U., Kuppusamy, M. L., et al. (2007). Mercury activates
vascular endothelial cell phospholipase D through thiols and
oxidative stress. The International Journal of Toxicology, 26(1),
57–69.
16. Varadharaj, S., Steinhour, E., Hunter, M. G., Watkins, T., Baran,
C. P., Magalang, U., et al. (2006). Vitamin C-induced activation
of phospholipase D in lung microvascular endothelial cells:
Regulation by MAP kinases. Cellular Signalling, 18(9),
1396–1407.
17. Sliman, S. M., Eubank, T. D., Kotha, S. R., Kuppusamy, M. L.,
Sherwani, S. I., Butler, E. S., et al. (2009). Hyperglycemic oxo-
aldehyde, glyoxal, causes barrier dysfunction, cytoskeletal alter-
ations, and inhibition of angiogenesis in vascular endothelial
cells: Aminoguanidine protection. Molecular and Cellular Bio-
chemistry, 333(1–2), 9–26.
18. Brindley, D. N., & Waggoner, D. W. (1996). Phosphatidate
phosphohydrolase and signal transduction. Chemistry and Phys-
ics of Lipids, 80(1–2), 45–57.
19. Steinhour, E., Sherwani, S. I., Mazerik, J. N., Ciapala, V.,
O’Connor Butler, E., Cruff, J. P., et al. (2008). Redox-active
antioxidant modulation of lipid signaling in vascular endothelial
cells: Vitamin C induces activation of phospholipase D through
phospholipase A2, lipoxygenase, and cyclooxygenase. Molecular
and Cellular Biochemistry, 315(1–2), 97–112.
20. Chakraborti, S. (2003). Phospholipase A(2) isoforms: A per-
spective. Cellular Signalling, 15(7), 637–665.
21. Balsinde, J., Winstead, M. V., & Dennis, E. A. (2000). Phos-
pholipase A2 regulation of acachidonic acid mobilization. FEBS
Letters, 531, 2–6.
22. Dennis, E. A., Rhee, S. G., Billah, M. M., & Hannun, Y. A.
(1991). Role of phospholipase in generating lipid second mes-
sengers in signal transduction. FASEB Journal, 5(7), 2068–2077.
23. Exton, J. H. (1997). New developments in phospholipase D.
Journal of Biological Chemistry, 272(25), 15579–15582.
24. Natarajan, V. (1995). Oxidants and signal transduction in vas-
cular endothelium. Journal of Laboratory and Clinical Medicine,
125(1), 26–37.
25. Singer, W. D., Brown, H. A., Jiang, X., & Sternweis, P. C.
(1996). Regulation of phospholipase D by protein kinase C is
synergistic with ADP-ribosylation factor and independent of
protein kinase activity. Journal of Biological Chemistry, 271(8),
4504–4510.
26. Mazerik, J. N., Hagele, T., Sherwani, S., Ciapala, V., Butler, S.,
Kuppusamy, M. L., et al. (2007). Phospholipase A2 activation
regulates cytotoxicity of methylmercury in vascular endothelial
cells. The International Journal of Toxicology, 26(6), 553–569.
27. Mazerik, J. N., Mikkilineni, H., Kuppusamy, V. A., Steinhour, E.,
Peltz, A., Marsh, C. B., et al. (2007). Mercury activates phos-
pholipase a(2) and induces formation of arachidonic acid
metabolites in vascular endothelial cells. Toxicology Mechanisms
and Methods, 17(9), 541–557.
28. Peltz, A., Sherwani, S. I., Kotha, S. R., Mazerik, J. N., O’Connor
Butler, E. S., Kuppusamy, M. L., et al. (2009). Calcium and
calmodulin regulate mercury-induced phospholipase D activation
in vascular endothelial cells. The International Journal of Toxi-
cology, 28(3), 190–206.
29. Murphy, R. C., Barkley, R. M., Zemski Berry, K., Hankin, J.,
Harrison, K., Johnson, C., et al. (2005). Electrospray ionization
and tandem mass spectrometry of eicosanoids. Analytical Bio-
chemistry, 346(1), 1–42.
30. Lambert, I. H., Pedersen, S. F., & Poulsen, K. A. (2006). Acti-
vation of PLA2 isoforms by cell swelling and ischaemia/hypoxia.
Acta physiologica (Oxford), 187(1–2), 75–85.
31. Nigam, S., & Schewe, T. (2000). Phospholipase A(2)s and lipid
peroxidation. Biochimica et Biophysica Acta, 1488(1–2), 167–
181.
32. Martinez, J., & Moreno, J. J. (2001). Role of Ca2?-independent
phospholipase A2 on arachidonic acid release induced by reactive
oxygen species. Archives of Biochemistry and Biophysics, 392(2),
257–262.
33. Xu, J., Yu, S., Sun, A. Y., & Sun, G. Y. (2003). Oxidant-mediated
AA release from astrocytes involves cPLA(2) and iPLA(2). Free
Radical Biology and Medicine, 34(12), 1531–1543.
34. Shanker, G., & Aschner, M. (2001). Identification and charac-
terization of uptake systems for cystine and cysteine in cultured
astrocytes and neurons: Evidence for methylmercury-targeted
disruption of astrocyte transport. Journal of Neuroscience
Research, 66(5), 998–1002.
35. Shanker, G., Syversen, T., Aschner, J. L., & Aschner, M. (2005).
Modulatory effect of glutathione status and antioxidants on
methylmercury-induced free radical formation in primary cul-
tures of cerebral astrocytes. Brain research. Molecular brain
research, 137(1–2), 11–22.
36. Verity, M. A., Sarafian, T., Pacifici, E. H., & Sevanian, A. (1994).
Phospholipase A2 stimulation by methyl mercury in neuron
culture. Journal of Neurochemistry, 62(2), 414–705.
37. Reiss, A. B., & Edelman, S. D. (2006). Recent insights into the
role of prostanoids in atherosclerotic vascular disease. Current
Vascular Pharmacology, 4(4), 395–408.
38. Bogatcheva, N. V., Sergeeva, M. G., Dudek, S. M., & Verin, A. D.
(2005). Arachidonic acid cascade in endothelial pathobiology.
Microvascular Research, 69(3), 107–127.
39. Sugiyama, T., Sakai, T., Nozawa, Y., & Oka, N. (1994). Pros-
taglandin F2 alpha-stimulated phospholipase D activation in
osteoblast-like MC3T3-E1 cells: Involvement in sustained 1,2-
diacylglycerol production. Biochemical Journal, 298(Pt 2),
479–484.
40. Kozawa, O., Suzuki, A., Kotoyori, J., Tokuda, H., Watanabe, Y.,
Ito, Y., et al. (1994). Prostaglandin F2 alpha activates phospho-
lipase D independently from activation of protein kinase C in
osteoblast-like cells. Journal of Cellular Biochemistry, 55(3),
373–379.
41. Imamura, Y., Kozawa, O., Suzuki, A., Watanabe, Y., Saito, H., &
Oiso, Y. (1995). Mechanism of phospholipase D activation
induced by prostaglandin D2 in osteoblast-like cells: Function of
Ca2?/calmodulin. Cellular Signalling, 7(1), 45–51.
42. Natarajan, V., Taher, M. M., Roehm, B., Parinandi, N. L., Sch-
mid, H. H., Kiss, Z., et al. (1993). Activation of endothelial cell
328 Cell Biochem Biophys (2013) 67:317–329
123

phospholipase D by hydrogen peroxide and fatty acid hydroper-
oxide. The Journal of Biological Chemistry, 268(2), 930–937.
43. Yamamoto, H., Endo, T., Kiya, T., Goto, T., Sagae, S., Ito, E.,
et al. (1995). Activation of phospholipase D by prostaglandin F2
alpha in rat luteal cells and effects of inhibitors of arachidonic
acid metabolism. Prostaglandins, 50(4), 201–211.
44. Hurt-Camejo, E., Camejo, G., Peilot, H., Oorni, K., & Kovanen,
P. (2001). Phospholipase A(2) in vascular disease. Circulation
Research, 89(4), 298–304.
45. Phillis, J. W., & O’Regan, M. H. (2003). The role of phospho-
lipases, cyclooxygenases, and lipoxygenases in cerebral ische-
mic/traumatic injuries. Critical Reviews in Neurobiology, 15(1),
61–90.
46. Avis, I. M., Jett, M., Boyle, T., Vos, M. D., Moody, T., Treston, A.
M., et al. (1996). Growth control of lung cancer by interruption of
5-lipoxygenase-mediated growth factor signaling. Journal of
Clinical Investigation, 97(3), 806–813.
47. Steele, V. E., Holmes, C. A., Hawk, E. T., Kopelovich, L., Lubet,
R. A., Crowell, J. A., et al. (1999). Lipoxygenase inhibitors as
potential cancer chemopreventives. Cancer Epidemiology, Bio-
markers and Prevention, 8(5), 467–483.
48. Tang, D. G., Chen, Y. Q., & Honn, K. V. (1996). Arachidonate
lipoxygenases as essential regulators of cell survival and apop-
tosis. Proceedings of the National Academy of Sciences, United
States of America, 93(11), 5241–5246.
49. Whitman, S., Gezginci, M., Timmermann, B. N., & Holman, T.
R. (2002). Structure-activity relationship studies of nord-
ihydroguaiaretic acid inhibitors toward soybean, 12-human, and
15-human lipoxygenase. Journal of Medicinal Chemistry, 45(12),
2659–2661.
Cell Biochem Biophys (2013) 67:317–329 329
123
Copyright © 2022 FDOKUMEN




















