
Effects of glutamate, quisqualate, and N-methyl-d-aspartate in neonatal brain
7
EXPERIMENTAL NEUROLOGY 111,362-368 (1991) Effects of Glutamate, Quisqualate, and N-Methybaspartate in Neonatal Brain RICHARD S. K. YOUNG,**? OGNEN A. C. PETROFF,~ WILLIAM J. AQUILA,* AND JOHNNIE YATES* Departments of *Pediatrics and ?Neurology, Yale University School of Medicine, New Haven, Connecticut 06510 The intracerebral injection of the excitotoxins, gluta- mate (GLU), or its analogues, quisqualic acid (QA) and iv-methyl-D-aspartate (NMDA), produces neuropatho- logic changes which resemble those induced by hyp- oxic-ischemic injury. We employed proton magnetic resonance spectroscopy to investigate the acute bio- chemical changes which follow injection of these excito- toxins in the neonatal rat brain. Aspartate and GLU increased in animals injected with GLU or NMDA. Ala- nine, glycine, and taurine increased with all three exci- totoxins. There was no decrease in phosphocreatine (PCr) or glucose and only a modest increase in lactate after excitotoxin injection, but there was substantial change in these metabolites after hypoxia. GABA rose only after hypoxic-ischemic injury. Although NMDA and QA produced morphological changes which resem- bled those following hypoxic-ischemic injury, the ef- fect of these excitotoxins on levels of PCr, glucose, and excitatory and inhibitory amino acids was considerably different. 0 1991 Academic Press, Inc. INTRODUCTION It is widely believed that glutamate (GLU) is involved in the pathogenesis of brain damage due to hypoxia and ischemia (1,4,7,18). Three principal types of GLU ago- nists are recognized and classified according to their re- ceptors: kainate (KA), quisqualate (QA), and N- methyl-D-asparate (NMDA) (1, 4, 7, 18). Intracerebral injection of these three excitotoxins in the neonate pro- duces a spectrum of injury ranging from severe necrosis with NMDA (12) to moderate damage with QA (19,25) to no injury with KA (5). The mechanism by which GLU and other excitotoxic drugs cause brain damage is uncertain. Several investi- gators have demonstrated that excitotoxic injury is as- sociated with the fall in phosphocreatine and glucose and the rise in lactate (5, 9, 17). However, it is not known whether the disruption in high energy phos- phates and glucose is etiologically relevant or, rather, an epiphenomenon to excitotoxic injury. It is also hypothe- sized that a low energy state may contribute to gluta- mate neurotoxicity by reducing glutamate uptake func- tion (4). The purpose of this study was to test the hypothesis that the pathological effects of the endogenous amino acid, GLU, resemble those of the GLU analogues, QA and NMDA, in the developing nervous system. We also wished to test the hypothesis that changes in brain en- ergy state and carbohydrate metabolism induced by in- tracerebral administration of GLU, QA, and NMDA are similar to those produced by hypoxia-ischemia. METHODS Experimental Procedure Sprague-Dawley rats (7 days old, both sexes) were assigned to one of five groups: control, GLU, QA, NMDA, or hypoxia-ischemia. For the intracerebral in- jections, each animal was briefly immobilized in a form- fitting plaster mold. The dose of GLU, QA, or NMDA was neutralized with Tris and stereotactically injected percutaneously into the right striatum using a Hamilton syringe (coordinates: 2 mm posterior, 3 mm lateral, and 4 mm inferior to the bregma) (19). An LD,, dose of exci- totoxin (3.5 pmol GLU, 100 nmol QA, and 10 nmol NMDA) was used in these studies because it resulted in an intermediate degree of mortality. Doses of GLU (22), QA (19), and NMDA (12) of approximate equivalence have also been used in other laboratories. Littermate controls for the pathological experiments received an equivalent volume of Tris intracerebrally. A volume of 5 ~1 was employed as described by Wolf and Keilhoff (22) to dilute the excitotoxin and allow slower resorption. Preliminary studies showed no pathological changes re- sulting from intracerebral injection of l-10 PL of Tris buffer. Animals assigned to the hypoxia-ischemia group un- derwent unilateral carotid artery ligation and 3.5 h of hypoxia (gas mixture of 8% 0,/92% N,) as described (19, 24). After this exposure to hypoxia, animals were either immediately sacrificed for metabolic determina- tions (see below) or returned to their dams until perfu- sion-fixation for neuropathological examination. 0014-4686/91 $3.00 Copyright 0 1991 by Academic Press, Inc. All rights of reproduction in any form reserved. 362
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Effects of glutamate, quisqualate, and N-methyl-d-aspartate in neonatal brain

EXPERIMENTAL NEUROLOGY 111,362-368 (1991)
Effects of Glutamate, Quisqualate, and N-Methybaspartate in Neonatal Brain
RICHARD S. K. YOUNG,**? OGNEN A. C. PETROFF,~ WILLIAM J. AQUILA,* AND JOHNNIE YATES*
Departments of *Pediatrics and ?Neurology, Yale University School of Medicine, New Haven, Connecticut 06510
The intracerebral injection of the excitotoxins, gluta- mate (GLU), or its analogues, quisqualic acid (QA) and iv-methyl-D-aspartate (NMDA), produces neuropatho- logic changes which resemble those induced by hyp- oxic-ischemic injury. We employed proton magnetic resonance spectroscopy to investigate the acute bio- chemical changes which follow injection of these excito- toxins in the neonatal rat brain. Aspartate and GLU increased in animals injected with GLU or NMDA. Ala- nine, glycine, and taurine increased with all three exci- totoxins. There was no decrease in phosphocreatine (PCr) or glucose and only a modest increase in lactate after excitotoxin injection, but there was substantial change in these metabolites after hypoxia. GABA rose only after hypoxic-ischemic injury. Although NMDA and QA produced morphological changes which resem- bled those following hypoxic-ischemic injury, the ef- fect of these excitotoxins on levels of PCr, glucose, and excitatory and inhibitory amino acids was considerably different. 0 1991 Academic Press, Inc.
INTRODUCTION
It is widely believed that glutamate (GLU) is involved in the pathogenesis of brain damage due to hypoxia and ischemia (1,4,7,18). Three principal types of GLU ago- nists are recognized and classified according to their re- ceptors: kainate (KA), quisqualate (QA), and N- methyl-D-asparate (NMDA) (1, 4, 7, 18). Intracerebral injection of these three excitotoxins in the neonate pro- duces a spectrum of injury ranging from severe necrosis with NMDA (12) to moderate damage with QA (19,25) to no injury with KA (5).
The mechanism by which GLU and other excitotoxic drugs cause brain damage is uncertain. Several investi- gators have demonstrated that excitotoxic injury is as- sociated with the fall in phosphocreatine and glucose and the rise in lactate (5, 9, 17). However, it is not known whether the disruption in high energy phos- phates and glucose is etiologically relevant or, rather, an epiphenomenon to excitotoxic injury. It is also hypothe- sized that a low energy state may contribute to gluta-
mate neurotoxicity by reducing glutamate uptake func- tion (4).
The purpose of this study was to test the hypothesis that the pathological effects of the endogenous amino acid, GLU, resemble those of the GLU analogues, QA and NMDA, in the developing nervous system. We also wished to test the hypothesis that changes in brain en- ergy state and carbohydrate metabolism induced by in- tracerebral administration of GLU, QA, and NMDA are similar to those produced by hypoxia-ischemia.
METHODS
Experimental Procedure
Sprague-Dawley rats (7 days old, both sexes) were assigned to one of five groups: control, GLU, QA, NMDA, or hypoxia-ischemia. For the intracerebral in- jections, each animal was briefly immobilized in a form- fitting plaster mold. The dose of GLU, QA, or NMDA was neutralized with Tris and stereotactically injected percutaneously into the right striatum using a Hamilton syringe (coordinates: 2 mm posterior, 3 mm lateral, and 4 mm inferior to the bregma) (19). An LD,, dose of exci- totoxin (3.5 pmol GLU, 100 nmol QA, and 10 nmol NMDA) was used in these studies because it resulted in an intermediate degree of mortality. Doses of GLU (22), QA (19), and NMDA (12) of approximate equivalence have also been used in other laboratories. Littermate controls for the pathological experiments received an equivalent volume of Tris intracerebrally. A volume of 5 ~1 was employed as described by Wolf and Keilhoff (22) to dilute the excitotoxin and allow slower resorption. Preliminary studies showed no pathological changes re- sulting from intracerebral injection of l-10 PL of Tris buffer.
Animals assigned to the hypoxia-ischemia group un- derwent unilateral carotid artery ligation and 3.5 h of hypoxia (gas mixture of 8% 0,/92% N,) as described (19, 24). After this exposure to hypoxia, animals were either immediately sacrificed for metabolic determina- tions (see below) or returned to their dams until perfu- sion-fixation for neuropathological examination.
0014-4686/91 $3.00 Copyright 0 1991 by Academic Press, Inc. All rights of reproduction in any form reserved.
362

GLUTAMATE AGONISTS IN NEONATAL BRAIN 363
Metabolic Determinations
Whole brain metabolites were determined 3 h after injection of excitotoxin or Tris because studies by Retz and Coyle (17) showed significant alteration of brain metabolites as early as 30 min after injection of excito- toxin. For the metabolic studies, the animals were decap- itated and their heads immediately immersed in liquid N,. Tissue fixation by this method occurs rapidly in the neonatal brain and there is satisfactory preservation of energy charge in control animals. The right hemisphere was dissected, pulverized in liquid N,, extracted, lyophi- lized, and analyzed with proton magnetic resonance spectroscopy (MRS) as described (16,23). A total of 23 animals was utilized for the metabolic determinations (control, 4; GLU, 4; QA, 5; NMDA, 4; hypoxia-ischemia, 6). Data were analyzed by the Student t test with Bon- ferroni correction for multiple comparisons.
Neuropathological Studies
Neuropathologic examination was carried out 4 days after exposure to excitotoxin, to Tris, or to hypoxia- ischemia. After anesthetization with pentobarbital, ani- mals were perfuse-fixed through the aorta with a solu- tion of formalin as described (24,25). Neuronal damage was assessed in cerebral cortex, striatum, hippocampus, and cerebral white matter by two observers. A semi- quantitative rating scale was utilized as previously de- scribed (24). Neuropathologic damage was graded: 0, no damage seen; 1, a few cells damaged; 2, many cells dam- aged; 3, extensive area of necrosis. A total of 46 animals was injected and perfused for neuropathological exami- nation (GLU, 12; &A, 20; NMDA, 8; Tris, 6). The dam- age scores were analyzed by a randomized analysis of variance.
RESULTS
Behavioral Changes
All animals injected with GLU, QA, or NMDA exhib- ited clinical seizure consisting of sustained flexion or extension of the extremities and tail, stereotypic cir- cling, intermittent oral cyanosis, and rhythmic shaking of the head and neck which persisted for several hours.
Neuropathological Findings
Injection of GLU produced significantly less neuro- pathologic damage than did QA or NMDA (Table 1; Figs. 1 and 2). Linear disruption of the cerebral cortex occurred as a result of the needle artifact (Fig. 2A). A small area of cystic necrosis was present at the site of injection. Ventricular dilatation was minimal. Damage
to the germinal matrix, striatum, or periventricular white matter was not seen. Structures posterior to the injection site such as hippocampus and thalamus were unaffected.
Damage from QA was considerably greater than that seen with GLU. Surrounding the injection site was an area of necrosis (Fig. 2B) involving both neurons and glia. Damage was most prominent in striatum, extend- ing to the deeper layers of cerebral cortex, the adjacent periventricular white matter, the septum, and the ger- minal matrix. Microscopic examination of the damaged areas showed polymorphonuclear leukocytic infiltrate, karrhyorhexis, and “dark cell change” (shrunken, dark neurons with dispersed Nissl substance) (20). At the pe- riphery of the injection site, selective injury of neurons without damage to glia was evident in the CA1 and CA3-4 regions of hippocampus (Fig. 2C).
Damage was significantly greater in the NMDA-in- jetted animals than in the GLU- and QA-injected ani- mals. Prominent injury of the paraventricular struc- tures, septum, striatum, and germinal matrix was evi- dent, involving both neurons and glia (Fig. 2D). Ventriculomegaly was extreme and there was necrosis of all layers of the cerebral cortex and underlying peri- ventricular white matter (Fig. 2E). Damage extended from the injection site posteriorly to involve the cere- bral cortex and periventricular white matter overlying the thalamus. The hippocampus was destroyed almost in its entirety (Fig. 2E), excepting the inferior aspect of the fascia dentata. The area damaged by NMDA corre- sponded to the territory supplied by the middle cerebral artery and had gross similarity to the pattern of injury seen in animals subjected to hypoxia-ischemia (Fig. 2F), with sparing only of the territory nourished by the posterior and anterior cerebral arteries.
Biochemical Alterations
Analysis of brain extracts showed lactate and alanine doubled with GLU, QA, and NMDA, but rose sevenfold with hypoxia. Brain glucose fell with hypoxia, but nearly doubled with GLU injection. The decrease in phosphocreatine (PCr) during hypoxia approached sig- nificance (0.05 < P < 0.1). GLU levels doubled in GLU- and NMDA-injected animals. In these two groups, aspartate (ASP), GABA, and glutamine rose to varying degrees. With hypoxia, GABA doubled, while ASP was reduced by one-half. Succinate increased with hypoxia and GLU injection. Glycine and taurine increased mark- edly with ail stresses. Changes in NAA and acetate after excitotoxin injection appeared to be reciprocal with the greatest drop occurring after QA. Changes in P-hy- droxybutyrate, inositol, and choline varied with the in- sult; GLU appeared to produce the greatest changes.
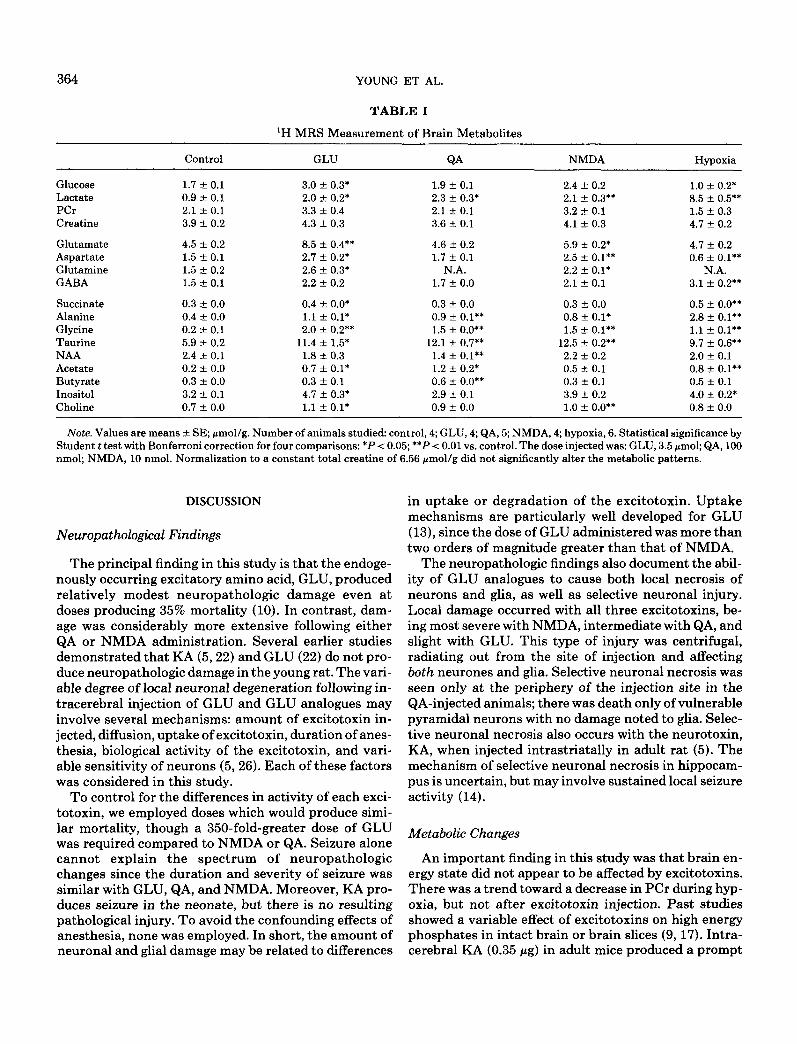
364 YOUNG ET AL.
TABLE 1
‘H MRS Measurement of Brain Metabolites
Glucose Lactate PCr Creatine
Glutamate Aspartate Glutamine GABA
Succinate Alanine Glycine Taurine NAA Acetate Butyrate Inositol Choline
Control GLU
1.7 * 0.1 3.0 + 0.3* 0.9 -t 0.1 2.0 zlz 0.2* 2.1 * 0.1 3.3 k 0.4 3.9 -t 0.2 4.3 iz 0.3
4.5 -t 0.2 8.5 + 0.4** 1.5 k 0.1 2.7 xk 0.2* 1.5 + 0.2 2.6 4 0.3* 1.5 -+ 0.1 2.2 r 0.2
0.3 2 0.0 0.4 + o.o* 0.4 + 0.0 1.1 * 0.1* 0.2 * 0.1 2.0 + 0.2** 5.9 -t 0.2 11.4 + 1.5* 2.4 + 0.1 1.8 + 0.3 0.2 2 0.0 0.7 f 0.1* 0.3 z!Y 0.0 0.3 f 0.1 3.2 -+ 0.1 4.7 + 0.3* 0.7 + 0.0 1.1 -c 0.1*
QA
1.9 f 0.1 2.3 +- 0.3* 2.1 f 0.1 3.6 + 0.1
4.6 + 0.2 1.7 + 0.1
N.A. 1.7 + 0.0
0.3 * 0.0 0.9 f 0.1** 1.5 * o-o**
12.1 + 0.7** 1.4 + 0.1** 1.2 +- 0.2* 0.6 + O.O** 2.9 -t 0.1 0.9 + 0.0
NMDA
2.4 zk 0.2 2.1 +- 0.3** 3.2 -t 0.1 4.1 + 0.3
5.9 +- 0.2* 2.5 f 0.1** 2.2 + 0.1* 2.1 + 0.1
0.3 t 0.0 0.8 + O.l* 1.5 r 0.1**
12.5 -+ 0.2** 2.2 k 0.2 0.5 rt 0.1 0.3 -e 0.1 3.9 rt 0.2 1.0 -t o.o**
Hypoxia
1.0 + 0.2* 8.5 f 0.5** 1.5 -+ 0.3 4.7 f 0.2
4.7 * 0.2 0.6 iz O.l**
N.A. 3.1 + 0.2**
0.5 +- o.o** 2.8 f O.l** 1.1 + 0.1** 9.7 zk 0.6** 2.0 + 0.1 0.8 + O.l** 0.5 rt 0.1 4.0 + 0.2* 0.8 + 0.0
Note. Values are means + SE; pmollg. Number of animals studied: control, 4; GLU, 4; QA, 5; NMDA, 4; hypoxia, 6. Statistical significance by Student t test with Bonferroni correction for four comparisons: *P < 0.05; **P < 0.01 vs. control. The dose injectedwas: GLU, 3.5 pmol; QA, 100 nmol; NMDA, 10 nmol. Normalization to a constant total creatine of 6.56 pmol/g did not significantly alter the metabolic patterns.
DISCUSSION
Neuropathological Findings
The principal finding in this study is that the endoge- nously occurring excitatory amino acid, GLU, produced relatively modest neuropathologic damage even at doses producing 35% mortality (10). In contrast, dam- age was considerably more extensive following either QA or NMDA administration. Several earlier studies demonstrated that KA (5,22) and GLU (22) do not pro- duce neuropathologic damage in the young rat. The vari- able degree of local neuronal degeneration following in- tracerebral injection of GLU and GLU analogues may involve several mechanisms: amount of excitotoxin in- jected, diffusion, uptake of excitotoxin, duration of anes- thesia, biological activity of the excitotoxin, and vari- able sensitivity of neurons (526). Each of these factors was considered in this study.
To control for the differences in activity of each exci- totoxin, we employed doses which would produce simi- lar mortality, though a 350-fold-greater dose of GLU was required compared to NMDA or QA. Seizure alone cannot explain the spectrum of neuropathologic changes since the duration and severity of seizure was similar with GLU, QA, and NMDA. Moreover, KA pro- duces seizure in the neonate, but there is no resulting pathological injury. To avoid the confounding effects of anesthesia, none was employed. In short, the amount of neuronal and glial damage may be related to differences
in uptake or degradation of the excitotoxin. Uptake mechanisms are particularly well developed for GLU (13), since the dose of GLU administered was more than two orders of magnitude greater than that of NMDA.
The neuropathologic findings also document the abil- ity of GLU analogues to cause both local necrosis of neurons and glia, as well as selective neuronal injury. Local damage occurred with all three excitotoxins, be- ing most severe with NMDA, intermediate with QA, and slight with GLU. This type of injury was centrifugal, radiating out from the site of injection and affecting both neurones and glia. Selective neuronal necrosis was seen only at the periphery of the injection site in the QA-injected animals; there was death only of vulnerable pyramidal neurons with no damage noted to glia. Selec- tive neuronal necrosis also occurs with the neurotoxin, KA, when injected intrastriatally in adult rat (5). The mechanism of selective neuronal necrosis in hippocam- pus is uncertain, but may involve sustained local seizure activity (14).
Metabolic Changes
An important finding in this study was that brain en- ergy state did not appear to be affected by excitotoxins. There was a trend toward a decrease in PCr during hyp- oxia, but not after excitotoxin injection. Past studies showed a variable effect of excitotoxins on high energy phosphates in intact brain or brain slices (9,17). Intra- cerebral KA (0.35 pg) in adult mice produced a prompt

GLUTAMATE AGONISTS IN NEONATAL BRAIN 365
CEREBRAL CORTEX STRIATUM
"1 -
NMDA Glutamate
HIPPOCAMPUS
"1
NMDA Glutamate I
NMDA QA Glutamate
PERIVENTRICULAR WHITE MATTER
NMDA Glutamate
FIG. 1. Neuropathologic damage. Damage to striatum, cerebral cortex, hippocampus, and white matter was significantly greater in NMDA-injected animals than in GLU- or QA-injected animals. Injury to the hippocampus was significantly greater in both NMDA- and QA-injected animals than in the GLU-injected animals. Neuropathologic damage was scored: 0, no damage seen; 1, a few cells damaged; 2, many cells damaged, 3, extensive area of necrosis. The total number of animals studied was 40 (GLU, 12; QA, 20; NMDA, 8). Analysis of variance: *P c 0.05 vs NMDA, #P < 0.05 vs QA.
and sustained decrease of PCr and ATP of approxi- mately one-third of control values. A decrease in PCr also occurred in neonatal brain slices incubated with NMDA (9). ATP fell in cerebellar slices exposed to O.l- 1 mM KA, but not to 1 n-144 NMDA or ibotenate (15).
This study also disclosed that GLU, QA, and NMDA produced increases in brain lactate and alanine. An in- crease in lactate of a similar magnitude has been seen following intracerebral injection of KA in adult mice (17) and following exposure of neonatal brain slices to NMDA (9). However, this modest rise in lactate is far less than that seen with hypoxia, &hernia, or status epilepticus (16, 23). For example, animals in our hyp- oxia-ischemia group had an 800% increase in both lac- tate and alanine and a 25% reduction in PCr. During bicuculline-induced status epilepticus in neonatal dog, there are increases in alanine, creatine, P-hydroxybu- tyrate, lactate, GABA, and succinate; decreases in glu- cose, PCr, and ASP (23); and no change in glycine, ace- tate, ATP, or GLU. Kainic acid seizure in the adult rat produced a decrease in GLU, ASP, glutamine, and glu- cose and an increase in GABA.
GLU and NMDA injection produced a rise in GLU with subsequent increase in its metabolites, GABA and
glutamine. ASP rose in parallel suggesting diversion of oxaloacetate from the Krebs cycle, transamination with GLU, to yield ASP and a-ketoglutarate, with entry of a-ketoglutarate into the Krebs cycle. Increased extra- cellular GLU caused by injection or enhanced release should promote uptake by glia and increase glutamine via the action of glutamine synthetase (13). Similarly, enhanced neuronal depolarization could increase GABA production and release. Other laboratories have observed a doubling of ASP and GLU levels in cerebel- lar slices incubated with either KA or NMDA (15).
Curiously, though the metabolic changes were greater with the injection of GLU, the tissue damage was far more extensive in all regions with NMDA. Taurine in- creased markedly with both excitotoxic and hypoxic in- jury. The fivefold increase in glycine in GLU, QA, and NMDA animals remains unexplained. It appeared to be inversely correlated with the degree of tissue damage, with the largest increases occurring with GLU injection and a lesser degree of increase with NMDA and hyp- oxia.
It is important to note that these metabolic analyses were performed on extracts of whole brain which repre- sent primarily cytosolic content. Experiments are

366 YOUNG ET AL.
F
FIG. 2. (A) Glutamate. A needle artifact shows the site of injection (thin arrow) of the GLU into the striatum (broad arrow). Mild ventricular dilatation is present. Damage to septum, striatum, periventricular white matter, and cerebral cortex is slight. H and E, X5. (B) Quisqualic Acid. Extensive damage is present in striatum, adjacent periventricular white matter, and deeper layers of cerebral cortex. Damage to the germinal matrix (arrow) is also seen. H and E, x5. (C) Quisqualic acid. Selective neuronal damage with sparing of adjacent white matter is demonstrated by necrosis of pyramidal cells throughout the CAl, CA3, and CA4 areas of hippocampus (arrowheads) with sparing of the adjacent periventricular white matter. H and E, x2.5. (D) N-Methyl-D-aspartate. There is virtually complete necrosis of cerebral cortex and periventricular white matter. The striatum, septum, and choroid plexus are atrophic. Marked ventriculomegaly is present. H and E, X10. (E) IV-Methyl-n-aspartate. Cystic encephalomalacia is present in the territory supplied by the middle cerebral artery with relative sparing of tissue in the region supplied by the anterior and posterior cerebral arteries (thin arrows). The hippocampus is necrotic with sparing of only the inferior aspect of the fascia dentata (wide arrow). H and E, X10. (F) Hypoxia-ischemia. Extensive necrosis is present in the territory supplied by the middle cerebral artery. The hippocampal formation is necrotic in its entirety, There is sparing of the regions supplied by the anterior and posterior cerebral arteries (arrows). Note the resemblance of the pattern of infarction following hypoxia-ischemia to the areas damaged by intracerebral injection of NMDA. H and E, X10.

GLUTAMATE AGONISTS IN NEONATAL BRAIN 367
currently underway in our laboratory using in vivo cere- bral microdialysis to measure excitatory and inhibitory amino acids in the extracellular space (11).
Differences between Mature and Developing Animals
A notable finding in this study was that the LD,, of GLU produced minimal injury in the developing brain, whereas GLU causes extensive damage in mature brain (20). This is remarkable because synaptosomal high af- finity uptake of endogenous L-GLU increases several- fold during development in the rat (9). This study also confirms that NMDA and QA produce extensive tissue injury in the developing animal (12, 19, 25), whereas they are relatively less injurious to adult brain.
The potential mechanisms underlying the differential sensitivity of young and mature animals include differ- ences in concentration of excitatory amino acids, recep- tor sites, uptake mechanisms, and afferent input. The postnatal number of GLU binding sites is low and rap- idly increases during development, whereas adult stria- turn is enriched in NMDA receptors (12). QA may be particularly toxic to striatum of l-week-old rats because of a preponderance of QA receptor subtype (12,19). The present data show that GLU agonists stimulating NMDA receptors are more potent.
Potential Therapies
It is expected that our understanding of the toxicity of excitatory amino acids will be enhanced by the ability to measure concentrations of excitatory and inhibitory amino acids in the extracellular space using in vivo mi- crodialysis (2,ll). Microdialysis measurements show in- creases in extracellular fluid concentrations of GLU and ASP both during hypoxic-ischemic injury and during seizure in the juvenile rabbit (11). Preliminary in vivo studies in our laboratory indicate that the noncompeti- tive NMDA receptor antagonist, MK-801, attenuates the increase in both excitatory and inhibitory amino acids during hypoxia and during seizure (21). Studies in this and other laboratories have shown that excitatory amino acid antagonists such as MK-801 and kynurenic acid can reduce hypoxic (6,8) as well as excitotoxic (12) neuronal injury. We are currently conducting studies in our laboratory which will test the hypothesis that atten- uating the rise in GLU during hypoxia will improve neuropathological outcome.
ACKNOWLEDGMENTS
We thank Dr. Jung Kim for reviewing the neuropathologic material and Peter Demou for assisting with the magnetic resonance suectros-
copy. These studies were supported by NIH Grants NS ROl-24605 (R.S.K.Y.) and NS ROl-21708 (O.A.C.P.).
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
REFERENCES
AUER, R. N., AND B. K. SIESJO. 1988. Biological differences be- tween ischemia, hypoglycemia, and epilepsy. Ann. Neural. 54: 699-707.
BENVENISTE, H., J. DRJZJER, A. SCHOUSBOE, AND N. H. DIEMER. 1984. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral isch- emia monitored by intracerebral microdialysis. J. Neurochem. 43: 1369-1374.
CAMPOCHIARO, P., AND J. T. COYLE. 1978. Ontogenetic develop- ment of kainate neurotoxicity: Correlates with glutamatergic in- nervation. Proc. Natl. Acad. Sci. USA 75: 2025-2029.
CHOI, D. W., M. MAULUCCI GEDDE, AND A. KRIEGSTEIN. 1987. Glutamate neurotoxicity in cortical cell culture. J. Neurosei. 7: 357-368.
COYLE, J. T. 1989. Neurotoxic action of kainic acid. J. Neuro- them. 41: l-11.
FORD, L. M., P. R. SANBERG, A. B. NORMAN, AND H. FOGELSON. 1989. MK-801 prevents hippocampal neurodegeneration in neo- natal hypoxic-ischemic rats. Arch. Neural. 46: 1090-1096.
GREENAMYRE, J. T. 1986. The role of glutamate in neurotrans- mission and in neurologic disease. Arch. Neural. 43: 1058-1063.
HA~ORI, H., A. M. MORIN, P. H. SCHWARTZ, D. G. FUJIKAWA, AND C. G. WASTERLAIN. 1989. Posthypoxic treatment with MK- 801 reduces hypoxic-ischemic damage in the neonatal rat. Neu- rology 39: 713-718.
JACQUIN, T., G. FORTIN, C. PASQUIER, B. GILLET, J. C. BELOEIL, AND J. CHAMPAGNAT. 1988. Metabolic acidosis induced by N- methyl-D-aspartate in brain slices of the neonatal rat: ‘iP and ‘H magnetic resonance spectroscopy. Neurosci. Lett. 92: 285-290.
JOHNSTON, G. A. R. 1972. Convulsions induced in 10 day old rats by intraperitoneal injection of monosodium glutamate and re- lated excitant amino acids. Biochem. Pharmacol. 22: 137.
LEY, E., R. S. K. YOUNG, M. J. DURING, AND W. J. AQUILA. 1990. Serial in uiuo microdialysis measurements of neurotransmitter concentrations during hypoxia and seizure. Pediatr. Res. 27: 345A.
MCDONALD, J. W., F. S. SILVERSTEIN, D. CARDONA, C. HUDSON, R. CHEN, AND M. V. JOHNSTON. 1990. Systemic administration of MK-801 protects against N-methyl-D-aspartate and quisqua- late-mediated neurotoxicity in perinatal rats. Neuroscience 36: 589-599.
MCILWAIN, H., AND H. S. BACHELARD. 1985. Biochemistry and the Central Nervous System, pp. 168-178. Churchill Livingstone, London.
MENINI, C., B. S. MELDRUM, D. RICHE, C. SILVA-COMTE, AND J. M. STUTZMANN. 1980. Sustained limbic seizures induced by intraamygdaloid kainic acid in the baboon. Ann. Neurol. 8: 501- 509.
NICKLAS, W. J., B. KRESPAN, AND S. BERL. 1980. Effect of kain- ate on ATP levels and glutamate metabolism in cerebellar slices. Eur. J. Pharmacol. 62: 209-213.
PETROFF, 0. A. C., T. OGINO, AND J. R. ALGER. 1988. High resolu- tion proton magnetic resonance spectroscopy of rabbit brain: Regional metabolite levels and postmortem changes. J. Neuro- them. 51: 163-171.
RETZ, K. C., AND J. T. COYLE. 1982. Effects of kainic acid on high

368 YOUNG ET AL.
18.
19.
20.
21.
22.
energy metabolites in the mouse striatum. J. Neurochem. 38: 196-203.
ROTHMAN, S. M., AND J. W. OLNEY. 1986. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann. Neurol. 19: 105-111.
SILVERSTEIN, F., R. CHEN, AND M. V. JOHNSTON. 1986. The glu- tamate analogue quisqualic acid is neurotoxic in striatum and hippocampus of immature rat brain. Neurosci. Lett. 71: 13-18. SLOVITER, R. S., AND D. W. DEMPSTER. 1985. Epileptic brain damage is replicated qualitatively in the rat hippocampus by central injection of glutamate or aspartate but not by GABA or acetylcholine. Bruin Res. Bull. 15: 39-60.
TENDLER, D. A., R. S. K. YOUNG, M. J. DURING, AND W. J. AQUILA. 1990. MK-801 Reduces Excitatory Amino Acid Levels. Presented at Research Biologicals, Inc., Symposium, Natick, MA, October 8,199O.
WOLF, G., AND G. KEILHOFF. 1984. Kainate and glutamate neuro-
toxicity in dependence on the postnatal development with spe- cial reference to hippocampal neurons. Deu. Brain Res. 14: 15- 21.
23. YOUNG, R. S. K., B. E. COWAN, 0. A. C. PETROFF, E. NOVOTNY, S. L. DUNHAM, AND R. W. BRIGGS. 1987. In vivo 31P and ‘H nuclear magnetic resonance study of hypoglycemia during neo- natal seizure. Ann. Neurol. 22: 622-628.
24. YOUNG, R. S. K., T. OLENGINSKI, S. K. YAGEL, AND J. TOWFIGHI. 1983. The effect of graded hypothermia on hypoxic-ischemic brain damage: A neuropathologic study in the neonatal rat. Stroke 14: 929-934.
25. YOUNG, R. S. K., 0. A. C. PETROFF, E. J. NOVOTNY, AND M. WONG. 1990. Neonatal excitotoxic brain injury. Dev. Neurosci. 12:210-220.
26. ZACZEK, R., S. SIMONTON, AND J. T. COYLE. 1980. Local and distant neuronal degeneration following intrastriatal injection of kainic acid. J. Neuropathol. Exp. Neurol. 39: 245-264.




















