
Synthesis of hypermodified adenosine derivatives as selective adenosine A3 receptor ligands
Upload
vanderbiltCategory
view
0download
0
of February 9, 2016.This information is current as
Signaling in Mast CellsAblation on Proinflammatory Adenosine
Adenosine Receptor Gene2BEffect of A
Blackburn, Italo Biaggioni and Igor FeoktistovSergey V. Novitskiy, Mikhail M. Dikov, Michael R. Sergey Ryzhov, Rinat Zaynagetdinov, Anna E. Goldstein,
http://www.jimmunol.org/content/180/11/7212doi: 10.4049/jimmunol.180.11.7212
2008; 180:7212-7220; ;J Immunol
Referenceshttp://www.jimmunol.org/content/180/11/7212.full#ref-list-1
, 33 of which you can access for free at: cites 61 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 9, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

Effect of A2B Adenosine Receptor Gene Ablation onProinflammatory Adenosine Signaling in Mast Cells1
Sergey Ryzhov,*§ Rinat Zaynagetdinov,*§ Anna E. Goldstein,†¶ Sergey V. Novitskiy,‡§
Mikhail M. Dikov,‡§ Michael R. Blackburn,� Italo Biaggioni,†§¶ and Igor Feoktistov2*§¶
Pharmacological studies suggest that A2B adenosine receptors mediate proinflammatory effects of adenosine in human mast cellsin part by up-regulating production of Th2 cytokines and angiogenic factors. This concept has been recently challenged by thefinding that mast cells cultured from bone marrow-derived mast cells (BMMCs) of A2B knockout mice display an enhanceddegranulation in response to Fc�RI stimulation. This finding was interpreted as evidence of anti-inflammatory functions of A2B
receptors and it was suggested that antagonists with inverse agonist activity could promote activation of mast cells. In this report,we demonstrate that genetic ablation of the A2B receptor protein has two distinct effects on BMMCs, one is the previously reportedenhancement of Ag-induced degranulation, which is unrelated to adenosine signaling; the other is the loss of adenosine signalingvia this receptor subtype that up-regulates IL-13 and vascular endothelial growth factor secretion. Genetic ablation of A2B
receptors had no effect on A3 adenosine receptor-dependent potentiation of Ag-induced degranulation in mouse BMMCs, butabrogated A2B adenosine receptor-dependent stimulation of IL-13 and vascular endothelial growth factor secretion. Adenosinereceptor antagonists MRS1706 and DPCPX with known inverse agonist activity at the A2B subtype inhibited IL-13 secretioninduced by the adenosine analog NECA, but did not mimic the enhanced Ag-induced degranulation observed in A2B knockoutBMMCs. Thus, our study confirmed the proinflammatory role of adenosine signaling via A2B receptors and the anti-inflammatoryactions of A2B antagonists in mouse BMMCs. The Journal of Immunology, 2008, 180: 7212–7220.
A denosine is an intermediate product in the metabolism ofATP. Adenosine exerts its action by binding to G pro-tein-coupled adenosine receptors. Four subtypes of
adenosine receptors have been cloned and classified as A1, A2A,A2B, and A3 receptors (1). Among adenosine receptors, the A2B
subtype has the lowest affinity to adenosine. A2B receptors arethought to remain silent under normal physiological conditionswhen adenosine concentrations are low and become active onlywhen extracellular adenosine levels rise as a result of inflamma-tion, ischemia, or cell damage (2).
There is growing evidence that adenosine plays an importantrole in chronic lung inflammation. Elevated concentrations ofadenosine are found in bronchoalveolar lavage fluid (3) and ex-haled breathe condensate (4) obtained from asthma patients. Stud-ies in adenosine deaminase (ADA)3-deficient mice, characterizedby elevated lung tissue levels of adenosine, strongly suggest a
causal association between adenosine and an inflammatory pheno-type (5, 6). These mice exhibit a lung phenotype with features oflung inflammation, bronchial hyperresponsiveness, enhanced mu-cus secretion, increased IgE synthesis, and elevated levels ofproinflammatory cytokines and angiogenic factors that could bereversed by lowering adenosine levels with exogenous ADA (5).Even more striking are the similarities in lung inflammatory phe-notypes found between ADA-deficient mice and mice overex-pressing IL-13 (7). The A2B adenosine receptor subtype appears toplay an important role in this model because pharmacological in-hibition of A2B receptors significantly reduced elevations in proin-flammatory cytokines as well as mediators of airway remodelinginduced by high adenosine levels in the lungs of ADA-deficientmice (8).
Mast cell activation has been long recognized to be crucial to thepathophysiology of asthma (9) and adenosine has been shown tomodulate mast cell functions (10). We have previously demon-strated that stimulation of A2B receptors in human mast cell lineHMC-1 increases production of proinflammatory Th2 cytokinesIL-4 and IL-13 (11, 12), as well as angiogenic factors IL-8 andvascular endothelial growth factor (VEGF) (13, 14). Furthermore,conditioned medium from these activated mast cells promoted cap-illary formation in an angiogenesis model (14) and induced IgEsynthesis in naive B lymphocytes (11). For this and other reasons,mast cell A2B adenosine receptors have been suggested to promotepulmonary inflammation and airway remodeling associated withasthma.
Contrary to this body of evidence, a recent report by Hua et al.(15) on a mouse phenotype resulting from deletion of A2B receptorgene suggested that the A2B receptor functions as a negative reg-ulator of mast cell activation. This conclusion was based on height-ened susceptibility of the A2B adenosine receptor knockout(A2BKO) mice to Ag-induced anaphylaxis and mast cell degran-ulation, which was attributed to the loss of purportedly constitutive
*Division of Cardiovascular Medicine, †Division of Clinical Pharmacology, ‡Divi-sion of Hematology/Oncology, and §Department of Medicine and ¶Department ofPharmacology, Vanderbilt University, Nashville, TN 37232; and �Department ofBiochemistry and Molecular Biology, University of Texas Medical School, Hous-ton TX 77030
Received for publication March 24, 2008. Accepted for publication March 24, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by Grants R01 HL070952 and R01 HL076306 from theNational Institutes of Health.2 Address correspondence and reprint requests Dr. Igor Feoktistov, Division of Car-diovascular Medicine, 360 Preston Research Building, Vanderbilt University, 2220Pierce Avenue, Nashville, TN 37232-6300. E-mail address: [email protected] Abbreviations used in this paper: ADA, adenosine deaminase; KO, knockout;BMMC, bone marrow-derived mast cell; HSA, human serum albumin; VEGF, vas-cular endothelial growth factor; WT, wild type.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

stimulation of adenylate cyclase by these receptors in mast cells.This results led the authors to propose that A2B antagonists withinverse agonist activity, binding to the same receptor site as ago-nists but exerting the opposite pharmacological effect, could en-hance Ag-induced mast cell degranulation and proinflammatorycytokine production. However, the effect of direct agonist stimu-lation of adenosine receptors was not explored in that study, and inthe absence of receptor stimulation, it is difficult to determinewhether the enhanced degranulation of A2BKO mast cells indeedreflects the loss of adenosine-dependent signaling functions of A2B
receptors or whether it could be due to the loss of a previouslyunrecognized function of the A2B receptor protein independent ofadenosine ligation. Furthermore, the study of Hua et al. (15) didnot address the previously suggested role of A2B receptors in stim-ulation of Th2 cytokines and angiogenic factors, nor did it sub-stantiate the claim of potential proinflammatory action of A2B an-tagonists on mast cells.
Because all these unresolved issues may have important impli-cations for the understanding of the role of A2B receptors in reg-ulation of inflammation and hence for development of new ther-apeutic approaches to diseases associated with inflammation, weaddressed these questions in the current work by studying the ef-fects of genetic A2B receptor ablation on adenosine receptor ago-nist-induced potentiation of mast cell degranulation, stimulation ofIL-13, and VEGF secretion and by comparing these effects of A2B
receptor gene disruption to the action of A2B receptor antagonistswith known inverse agonist activity.
Materials and MethodsReagents
NECA (5�-N-ethylcarboxamidoadenosine), CGS21680 (2-p(2-carboxy-ethyl)phenethylamino-5�-N-ethylcarboxamidoadenosine), DPCPX (8-cy-clopentyl-1,3-dipropylxanthine), and forskolin were purchased fromSigma-Aldrich. MRS1706 (N-(4-acetylphenyl)-2-(-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy) acetamide) was obtained fromTocris. Adenylate cyclase inhibitor 2�,5�-dideoxyadenosine, MEK1/2-spe-cific inhibitor UO126 (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenyl-thio) butadiene) and its inactive analog UO124 (1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene), selective inhibitor of p38 MAPK SB202190(4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole) andits inactive analog SB202474 (4-Ethyl-2(p-methoxyphenyl)-5-(4�-pyridyl)-1H-imidazole) were purchased from Calbiochem. DMSO was purchasedfrom Sigma-Aldrich. When used as a solvent, final DMSO concentrationsin all assays did not exceed 0.1% and the same DMSO concentrations wereused in vehicle controls.
Mice and bone marrow-derived mast cell (BMMC) culture
All studies were conducted in accordance with the Guide for the Care andUse of Laboratory Animals as adopted and promulgated by the NationalInstitutes of Health. Animal studies were reviewed and approved by theInstitutional Animal Care and Use Committee of Vanderbilt University(Nashville, TN). Six- to 8-week-old, age- and sex-matched mice were used.A2BKO mice were obtained from Deltagen and wild-type (WT) C57BL/6mice were purchased from Harlan World Headquarters. Genotyping pro-tocols for A2BKO have been previously described (16). All of the A2BKOmice used in these studies were backcrossed to the C57BL/6 genetic back-ground for 10 generations.
BMMCs were cultured as previously described (15). In brief, bone mar-row was flushed from the femurs and tibias of each mouse with RPMI 1640medium, supplemented with 2% FBS and 10 U/�l heparin. After centrif-ugation and washing with RPMI 1640, cells were resuspended at 0.5 � 106
cells/ml in RPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine, nonessential amino acids, 50 �M 2-ME, 1� antibiotic-antimy-cotic mixture (Sigma-Aldrich), 20 ng/ml mouse IL-3 (PeproTech), and 20ng/ml stem cell factor (United States Biologicals). Cells were maintainedin suspension culture under humidified atmosphere of air/CO2 (19:1) at37°C. The medium was renewed every week and 5- to 6-wk cultures wereused for experiments.
Expression of Fc�RI and c-kit on BMMC surface was determined byflow cytometry. After treatment with Fc Receptor Blocking reagent (Milte-
nyi Biotec), BMMCs (106 cells) were incubated with 10 �g/ml mouseanti-DNP IgE (clone SPE-7; Sigma-Aldrich) for 30 min on ice. After briefcentrifugation to remove unbound IgE, cells were resuspended and labeledusing FITC-conjugated anti-IgE (clone 23G3; eBioscience) and anti-CD117-PE (clone 2B8; BD Pharmingen) Abs for 20 min. Data acquisitionwas performed on a FACSCalibur flow cytometer (BD ImmunocytometrySystems) and the data were analyzed with WinList 5.0 software. Nonviablecells were excluded by using 7-aminoactinomycin D. Ag negativity wasdefined as having the same fluorescent intensity as the isotype control.
Human mast cells
Human mast cell line HMC-1 was a gift from Dr. J. H. Butterfield (MayoClinic, Rochester, MN). HMC-1 cells were maintained in suspension cul-ture at a density between 3 and 6 � 105 cells/ml by dilution with Iscove’smedium supplemented with 10% FBS, 2 mM glutamine, 1.2 mM �-thio-glycerol, and 1� antibiotic-antimycotic mixture. Cells were kept underhumidified atmosphere of air/CO2 (19:1) at 37°C.
Real-time RT-PCR
Real-time RT-PCR analysis was performed as previously described (17).Total RNA was isolated from cells using the RNeasy Mini kit (Qiagen).Real-time RT-PCR was conducted on ABI PRISM 7900HT Sequence De-tection System (PE Applied Biosystems). Primer pairs and 6-carboxy-flu-orescein-labeled probes for murine adenosine receptors and �-actin wereprovided by Applied Biosystems. RT-PCR using 1 �g of DNase-treatedtotal RNA was performed under conditions recommended by the manu-facturer. A standard curve for each amplicon was obtained using serialdilutions of total RNA. The results from triplicate PCR for a given gene ateach time point were used to determine mRNA quantity relative to thecorresponding standard curve. The relative mRNA quantity for a givengene measured from a single reverse transcription reaction was divided bythe value obtained for �-actin to correct for fluctuations in input RNAlevels and varying efficiencies of RT-PCR.
Measurement of cAMP accumulation
Cyclic AMP accumulation was measured as previously described (18).BMMCs (5 � 106 cells/ml) were preincubated in Tyrode’s buffer (150 mMNaCl, 2.7 mM KCl, 0.37 mM NaH2PO4, 1 mM MgSO4, 1 mM CaCl2, 5g/L D-glucose, 10 mM HEPES-NaOH (pH 7.4)) containing 1 U/ml ADAand the cAMP phosphodiesterase inhibitor papaverine (1 mM) for 15 minat 37°C. To test the effect of adenylate cyclase inhibition on cAMP accu-mulation, HMC-1 cells (2 � 106 cells/ml) were preincubated in the samebuffer containing increasing concentrations of 2�,5�-dideoxyadenosine for30 min at 37°C. Adenosine receptor agonists, antagonists, forskolin or theirvehicle (DMSO) were added to cells, and the incubation was allowed toproceed for 5 min at 37°C. The reaction was stopped by the addition of 1/5volume of 25% TCA. The extracts were washed five times with 10 volumesof water-saturated ether. Cyclic AMP concentrations were determined us-ing a cAMP assay kit (GE Healthcare).
Mast cell degranulation assay
Mast cell degranulation was determined by measuring �-hexosaminidaseactivity and histamine concentrations. BMMCs were incubated 1 h at 37°Cwith a monoclonal IgE directed against DNP-human serum albumin (DNP-HSA, clone SPE-7; Sigma-Aldrich) at a concentration of 10 �g/ml/106
cells in Tyrode’s buffer lacking Ca2�. After removal of unbound IgE bywashing twice with Tyrode’s buffer, BMMCs were resuspended at a con-centration of 106 cells/ml in Tyrode’s buffer containing 1 mM CaCl2 and1 U/ml ADA. Cells were incubated in the presence or absence of DNP-HSA (Sigma-Aldrich) and in the presence or absence of NECA at 37°C. Insome experiments, A2B antagonists or their vehicle were included in Ty-rode’s buffer at this and all prior steps (see Fig. 5B). Reactions were ter-minated after 20 min by placing tubes on ice. The release of �-hexosamini-dase was quantified in cell supernatants and pellets by the hydrolysis ofp-nitrophenyl-N-acetyl-�-D-glucosaminide (Sigma-Aldrich), as detailedelsewhere (19). Histamine concentrations were determined using an en-zyme immunoassay kit (Immunotech Histamine kit; Beckman Coulter).
Analysis of IL-6, IL-13, and VEGF secretion
When indicated, BMMCs were preincubated with anti-DNP-HSA as de-scribed. To determine IL-6 and IL-13 release, BMMCs (5 � 106 cells/ml)were incubated in the presence or absence of DNP-HSA, and in the pres-ence or absence of NECA and A2B receptor antagonists in RPMI 1640medium with 10% calf serum, 1� antibiotic-antimycotic mixture and 1U/ml ADA for 6 h at 37°C. To determine VEGF release, BMMCs wereincubated in the same medium for 16 h at 37°C.
7213The Journal of Immunology
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

HMC-1 cells were resuspended at a concentration of 2 � 106 cells/ml inIscove’s medium containing 2 mM glutamine, 1.2 mM �-thioglycerol, and1 U/ml ADA in the absence or presence of tested reagents. Reactions werestarted by addition of NECA or its vehicle and continued for 6 h underhumidified atmosphere of air/CO2 (19:1) at 37°C. IL-6, IL-13, and VEGFconcentrations in supernatants were measured using ELISA kits (R&DSystems).
To determine the effects of inhibitors of intracellular signaling path-ways, BMMCs or HMC-1 cells were preincubated in the presence of testedcompounds for 30 min at 37°C before addition of NECA.
Transfections and luciferase reporter assay
HMC-1 cells were transfected using Fugene 6 transfection reagent (Roche).A total of 2 �g of plasmid DNA (0.5 �g/�l) was mixed with 84 �l ofserum-free Iscove’s medium containing 6 �l of Fugene 6. After 20 min ofincubation at room temperature, the transfection mixture was added to 1 �106 cells suspended in 1 ml of growth medium. The ratio 10:1 was used forVEGF or IL-13 firefly luciferase reporter to control Renilla luciferase re-porter combinations. VEGF promoter-driven luciferase reporter, a fireflyluciferase reporter plasmid comprising 5� flanking �1005 to �379 bp ofthe human VEGF gene (20), was provided by Dr. G. L. Semenza (JohnsHopkins Hospital, Baltimore, MD). IL-13 promoter-driven luciferase re-porter pIL13-939/�48-luc, a firefly luciferase reporter plasmid comprising5� flanking �939 to �48 bp of the human IL-13 gene (21), was gifted byDrs. H. Young, O. Shimozato, and D. Derse (National Cancer Institute,Frederick, MD). A control constitutively active Renilla luciferase plasmidpRL-SV40 was purchased from Promega. Twenty-four hours after trans-fection, cells were incubated in the presence or absence of 10 �M NECAfor an additional 6 h. Reporter activity was then measured using a Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase reporteractivities were normalized against Renilla luciferase activities and ex-pressed as relative luciferase activities over basal (set as 1).
Evaluation of ERK and p38 MAPK activation
BMMCs were cultured in their growth medium without IL-3 and stem cellfactor for 24 h. Three hours before experiment, BMMCs and HMC-1 cellswere starved by culturing in their corresponding growth medium withoutserum. After starvation, cells were resuspended in Tyrode’s buffer at aconcentration of 2 � 107 cells/ml and stimulated with 10 �M NECA forthe indicated periods. Reactions were stopped by cell lysis in ice-coldRIPA buffer (Santa Cruz Biotechnology). Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (NEN LifeScience Products). Membranes were blocked and incubated consecutivelywith primary Ab and HRP-conjugated secondary Ab. Immunoreactive pro-teins were visualized by ECL reagents (NEN Life Science Products). Anti-phospho-ERK (sc-7976), anti-phospho-p38 (sc-7975), anti-ERK (sc-154),and anti-p38 (sc-7149) MAPKs were purchased from Santa CruzBiotechnology.
ResultsExpression of mast cell surface markers in A2BKO and WTBMMCs
Mast cells obtained from A2BKO mice were morphologically sim-ilar to those obtained from WT animals. After 5 wk in culture, over90% of A2BKO and WT BMMCs expressed both c-kit and Fc�RIon their surface (Fig. 1A). The expression levels of these mast cellsurface markers were the same in A2BKO and WT BMMCs asdetermined by flow cytometry (Fig. 1B).
Effect of adenosine A2B receptor gene ablation on adenosinereceptor mRNA expression in BMMCs
Real-time RT-PCR analysis of WT BMMCs revealed mRNA en-coding A2A, A2B, and A3 receptor subtypes (1.57 � 0.21%, 1.8 �0.27%, and 1.34 � 0.24% of �-actin, respectively) and no detect-able levels of A1 receptor transcripts (Fig. 1C). As expected, wedid not detect the expression of A2B receptor mRNA in A2BKOBMMCs. We also documented that A2B receptor gene ablation hadno significant effect on A2A and A3 receptor mRNA expression inA2BKO BMMCs (1.28 � 0.09% and 1.11 � 0.08% of �-actin,respectively).
Effect of adenosine A2B receptor gene ablation on cAMP levelsin BMMCs
Adenosine receptor subtypes were initially characterized by theireffects on adenylate cyclase activity; A2A and A2B receptors stim-ulate adenylate cyclase via coupling to Gs proteins, whereas A1
and A3 receptors inhibit stimulation of this enzyme via coupling toGi proteins (1). Therefore, we determined whether A2B receptorgene ablation has any effect on cAMP levels in BMMCs. As seenin Fig. 2A, comparative analysis revealed no significant differencein basal cAMP levels between BMMCs isolated from WT andA2BKO mice (0.602 � 0.210 and 0.625 � 0.127 pmol/106 cells,respectively). Similarly, no significant difference between WT andA2BKO BMMCs was found in cAMP accumulation induced withthe adenylate cyclase stimulator forskolin (10 �M). However,stimulation of adenosine receptors with the nonselective agonistNECA (10 �M) induced greater cAMP accumulation in WT cellscompared with A2BKO BMMCs (1.65 � 0.17 and 1.15 � 0.04pmol/106 cells, respectively; p � 0.05, n � 5 separate cell prep-arations). In contrast, we found no significant difference in cAMPaccumulation between WT and A2BKO BMMCs stimulated withthe selective A2A receptor agonist CGS21680 (1 �M). Thus,cAMP accumulation induced by activation of all adenosine recep-tors with NECA was significantly lower in A2BKO BMMCs due tothe lack of A2B receptors, but genetic ablation of A2B receptors hadno effect on basal cAMP levels and cAMP accumulation inducedby stimulation of A2A receptors with CGS21680 or by receptor-independent stimulation of adenylate cyclase with forskolin. Theselective A2B antagonist MRS1706 (100 nM) and the A1/A2B an-tagonist DPCPX (1 �M) had no significant effect on basal cAMPlevels in both WT and A2BKO cells. However, these antagonists
0
100
200
300
400 WTA2BKO
FcεRI
Fluo
resc
ense
inte
nsity
FcεRI
c-K
it
A WT A2BKO
A1 A2A A2B A30.0
0.5
1.0
1.5
2.0
2.5 WTA2BKO
ND ND ND
mR
NA
, %β
-act
in
CB
10 1 10 2 10 3 10 4
FL1-H
101
102
103
104
FL2-H
3.8% 93.8%
1.3% 1.1%
10 1 10 2 10 3 10 4
FL1-H
101
102
103
104
FL2-H
4.3% 93.9%
1.0% 0.8%
0
100
200
300
400 WTA2BKO
FcεRI c-Kit
Fluo
resc
ense
inte
nsity
FcεRI
c-K
it
A WT A2BKO
A1 A2A A2B A30.0
0.5
1.0
1.5
2.0
2.5 WTA2BKO
ND ND ND
mR
NA
, %β
-act
in
10 1 10 2 10 3 10 4
FL1-H
101
10 1 10 2 10 3 10 4
FL1-H
101
102
103
104
FL2-H
3.8% 93.8%
1.3% 1.1%
10 1 10 2 10 3 10 4
FL1-H
101
102
10 1 10 2 10 3 10 4
FL1-H
101
102
103
104
FL2-H
4.3% 93.9%
1.0% 0.8%
FIGURE 1. Expression of Fc�RI, c-kit and adenosine receptors in WTand A2BKO BMMCs. A, Representative cytofluorographic dot plots ofFc�RI and c-kit cell surface expression on mast cells derived from WT andA2BKO mice by culturing bone marrow cells in the presence of IL-3 andstem cell factor for 5 wk as described in Materials and Methods. B, Ex-pression levels of Fc�RI and c-kit on WT and A2BKO BMMCs are pre-sented as mean fluorescence intensity above isotype control. Values areexpressed as mean � SEM of five separate cell preparations. C, Real-timeRT-PCR analysis of mRNA encoding adenosine receptor subtypes wasperformed as described in Materials and Methods. Values are expressed asmean � SEM of five separate cell preparations. Nd, No transcriptsdetected.
7214 ADENOSINE A2B RECEPTORS IN MAST CELLS
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

significantly attenuated NECA-induced cAMP accumulation inWT but not in A2BKO BMMCs (Fig. 2B).
Effect of adenosine A2B receptor gene ablation onadenosine-dependent potentiation of BMMC degranulation
Stimulation of adenosine receptors per se does not produce the releaseof preformed mediators from BMMCs, but potentiates degranulationinduced by clustering Fc�RI (22). This adenosine action is mediatedvia A3 adenosine receptors because adenosine agonists do not poten-tiate Ag-induced degranulation of A3KO BMMCs (23). As seen inFig. 3A, basal release of �-hexosaminidase, a granule-associated en-zyme, from A2BKO BMMCs was similar to that observed in WTcells. Stimulation of adenosine receptors with 10 �M NECA had no
significant effect on basal �-hexosaminidase release, but potentiatedthe release induced by 10 ng/ml DNP-HSA from both A2BKO andWT cells. Our results show that Ag-dependent clustering of Fc�RIwith DNP-HSA produced greater degranulation of A2BKO BMMCscompared with WT cells as determined by release of �-hexosamini-dase (Fig. 3A) and histamine (data not shown). Ag-induced degran-ulation of A2BKO BMMCs was also greater in the presence of NECA(Fig. 3A). However, the degree of potentiation of DNP-HSA-induceddegranulation by NECA was virtually the same in both WT andA2BKO cells (1.6 � 0.1- and 1.7 � 0.1-fold, respectively) (Fig. 3B),suggesting that A2B receptors do not play a role in this adenosineaction on mouse mast cells.
Effect of adenosine A2B receptor gene ablation on IL-6, IL-13,and VEGF release from BMMCs
In addition to triggering the release of preformed mediators,Fc�RI-dependent stimulation of BMMCs induces synthesis andsecretion of several proinflammatory cytokines and cell growthfactors including IL-6, IL-13, and VEGF (24–26). As seen in Fig.4A, activation of WT BMMCs with increasing concentrations ofAg significantly increased IL-6 in culture medium from basal lev-els from 48 � 8 to 113 � 25 pg/ml at 5 ng/ml DNP-HSA, and to236 � 46 pg/ml at 10 ng/ml DNP-HSA ( p � 0.01, ANOVA withDunnett’s posttests, n � 5 separate cell preparations). However,stimulation of adenosine receptors with NECA had no significant
0
1
2 BasalForskolinNECACGS21680
WT A2BKO
*
cAM
P, p
mol
/106
cells
0
1
2BasalMRS1706DPCPXNECANECA+MRS1706NECA+DPCPX
WT A2BKO
**
cAM
P, p
mol
/106
cells
A
B
0
1
2 BasalForskolinNECACGS21680
WT A2BKO
*
cAM
P, p
mol
/106
cells
0
1
2BasalMRS1706DPCPXNECANECA+MRS1706NECA+DPCPX
WT A2BKO
**
cAM
P, p
mol
/106
cells
FIGURE 2. Intracellular cAMP levels in WT and A2BKO BMMCs. A,Cyclic AMP accumulation in WT and A2BKO mast cells incubated in thepresence of 1 �M forskolin, 10 �M NECA, 1 �M CGS21680 or their vehicle(Basal). Values are expressed as mean � SEM of five separate cell prepara-tions. �, p � 0.05, significant difference between corresponding WT andA2BKO values determined using unpaired two-tail t test. B, Effects of 100 nMMRS1706, 1 �M DPCPX, or their vehicle on cAMP accumulation in theabsence (MRS1706, DPCPX, or Basal, respectively) or presence(NECA�MRS1706, NECA�DPCPX, or NECA, respectively) of 10 �MNECA. Values are expressed as mean � SEM of five separate cell prepara-tions. �, p � 0.05, significant difference from vehicle in NECA-stimulated WTBMMCs determined using one-way ANOVA with Dunnett’s posttest.
0
10
20
30
40
50 BasalDNP-HSANECADNP-HSA+NECA
WT A2BKO
Deg
ranu
latio
n, %
A
WT A2BKO0.0
0.5
1.0
1.5
2.0
NE
CA
pot
entia
tion,
fold
B
0
10
20
30
40
50 BasalDNP-HSANECADNP-HSA+NECA
WT A2BKO
Deg
ranu
latio
n, %
A
WT A2BKO0.0
0.5
1.0
1.5
2.0
NE
CA
pot
entia
tion,
fold
B
FIGURE 3. Potentiation of Ag-induced degranulation by NECA in WTand A2BKO mast cells. A, Degranulation of WT and A2BKO BMMCs wasassessed by measuring �-hexosaminidase release from cells preincubatedwith anti-DNP IgE after stimulation with 10 ng/ml DNP-HSA Ag (DNP-HSA, DNP-HSA�NECA) or from nonstimulated cells (Basal, NECA) inthe absence (Basal, DNP-HSA) or presence (NECA, DNP-HSA�NECA)of 10 �M NECA. Values are expressed as mean percentage � SEM ofthree separate cell preparations. B, Potentiation of Ag-induced degranula-tion by NECA in WT and A2BKO BMMCs calculated as fold value fromdata presented in A.
0 5 10
0
100
200
300 A2BKO VehicleNECA
DNP-HSA, ng/ml0 5 10
0
100
200
300 WT
DNP-HSA, ng/ml
IL-6
, pg/
106 ce
lls
A B
0 5 10
0
100
200
300 A2BKO VehicleNECA
DNP-HSA, ng/ml0 5 10
0
100
200
300 WT
DNP-HSA, ng/ml
IL-6
, pg/
106 ce
lls
0 5 10
0
100
200
300
400
500WT
***
*
DNP-HSA, ng/ml
IL-1
3, p
g/10
6 cel
ls0 5 10
0
20
40
60
80WT
*
*
*
DNP-HSA, ng/mlV
EG
F, p
g/10
6 cells
0 5 10
0
100
200
300
400
500 VehicleNECAA2BKO
DNP-HSA, ng/ml
0 5 10
0
20
40
60
80A2BKO Vehicle
NECA
DNP-HSA, ng/ml
E F
DC
0 5 10
0
100
200
300
400
500WT
***
*
DNP-HSA, ng/ml
IL-1
3, p
g/10
6 cel
ls0 5 10
0
20
40
60
80WT
*
*
*
DNP-HSA, ng/mlV
EG
F, p
g/10
6 cells
0 5 10
0
100
200
300
400
500 VehicleNECAA2BKO
DNP-HSA, ng/ml
0 5 10
0
20
40
60
80A2BKO Vehicle
NECA
DNP-HSA, ng/ml
FIGURE 4. Effect of A2B receptor gene ablation on IL-6, IL-13, andVEGF secretion from BMMCs. WT (A, C, and E) and A2BKO (B, D, andF) BMMCs were preincubated with anti-DNP IgE and stimulated with 0,1, 5, or 10 ng/ml DNP-HSA in the presence of 10 �M NECA (F) or itsvehicle (E) for 6 h (A–D), or for 16 h (E and F). IL-6, IL-13, and VEGFwere measured in culture medium and expressed as picograms per 106
cells. Data are presented as mean � SEM of five separate cell preparations.�, p � 0.05 and ��, p � 0.01, statistical differences of secretion in thepresence of NECA from corresponding values obtained in the presence ofvehicle determined by unpaired two-tail t test.
7215The Journal of Immunology
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
effect on basal or Ag-induced IL-6 release. Similar levels of basaland Ag-induced IL-6 release and the lack of NECA stimulationwere observed in A2BKO BMMCs (Fig. 4B). Thus, our resultsindicate that adenosine receptors play no role in the regulation ofIL-6 secretion in BMMCs.
In contrast, stimulation of adenosine receptors with 10 �MNECA significantly increased IL-13 secretion in WT BMMCsfrom basal levels from 81 � 19 to 172 � 9 pg/ml in the absenceof Ag and from 280 � 51 to 468 � 50 pg/ml in the presence of 10ng/ml DNP-HSA (Fig. 4C). Because this effect of NECA was notseen in A2BKO cells (Fig. 4D), our data suggest that A2B receptorsmediate adenosine-dependent stimulation of IL-13 in BMMCs.
Similarly, stimulation of adenosine receptors with 10 �MNECA significantly increased VEGF secretion in WT BMMCs forbasal levels from 14 � 3 to 35 � 6 pg/ml in the absence of Ag andfrom 35 � 5 to 66 � 10 pg/ml in the presence of 10 ng/ml DNP-HSA (Fig. 4E). As in the case of IL-13 secretion, NECA had nosignificant effect on VEGF secretion in A2BKO BMMCs (Fig. 4F),indicating that adenosine-dependent release of both IL-13 andVEGF is regulated via A2B receptors.
Effects of inverse A2B receptor agonists on BMMCdegranulation
It has been recently speculated, although not tested, that A2B an-tagonists with inverse agonist activity could enhance Ag-induceddegranulation in BMMCs similarly to the effect of genetic A2B
receptor ablation (15). To test this hypothesis, we chose adenosinereceptor antagonists MRS1706 and DPCPX because they were re-cently characterized as inverse agonists at the A2B receptor (27).We initially confirmed that these compounds antagonize an A2B
receptor-mediated process in these cells. As seen in Fig. 5A, 1 �MDPCPX and 100 nM MRS1706 significantly inhibited NECA-in-duced IL-13 secretion by 87 � 8 and 53 � 8%, respectively ( p �0.01, ANOVA with Dunnett’s posttests, n � 5 separate cell prep-arations). However, these compounds used at the same concentra-tions had no effect on Ag-induced BMMC degranulation (Fig. 5B).
HMC-1 as a model to study A2B receptor-dependent regulationof mast cell cytokine and growth factor production
Although HMC-1 cells express very low levels of functionalFc�RI (28), this human mast cell line has proven useful to uncoverthe A2B receptor-dependent up-regulation of Th2 cytokines andangiogenic factors (11, 14), a process independent from Fc�RIstimulation as we show in the current study in BMMCs. Becausethe use of an established cell line for detailed analysis of intracel-lular signaling pathways could be advantageous due to availabilityof plentiful material and better reproducibility of results, we nextsought to determine whether HMC-1 can serve as an appropriatemodel to study A2B receptor-dependent regulation of mast cellcytokine and growth factor production, by comparing IL-13 andVEGF regulation in BMMCs and HMC-1 cells.
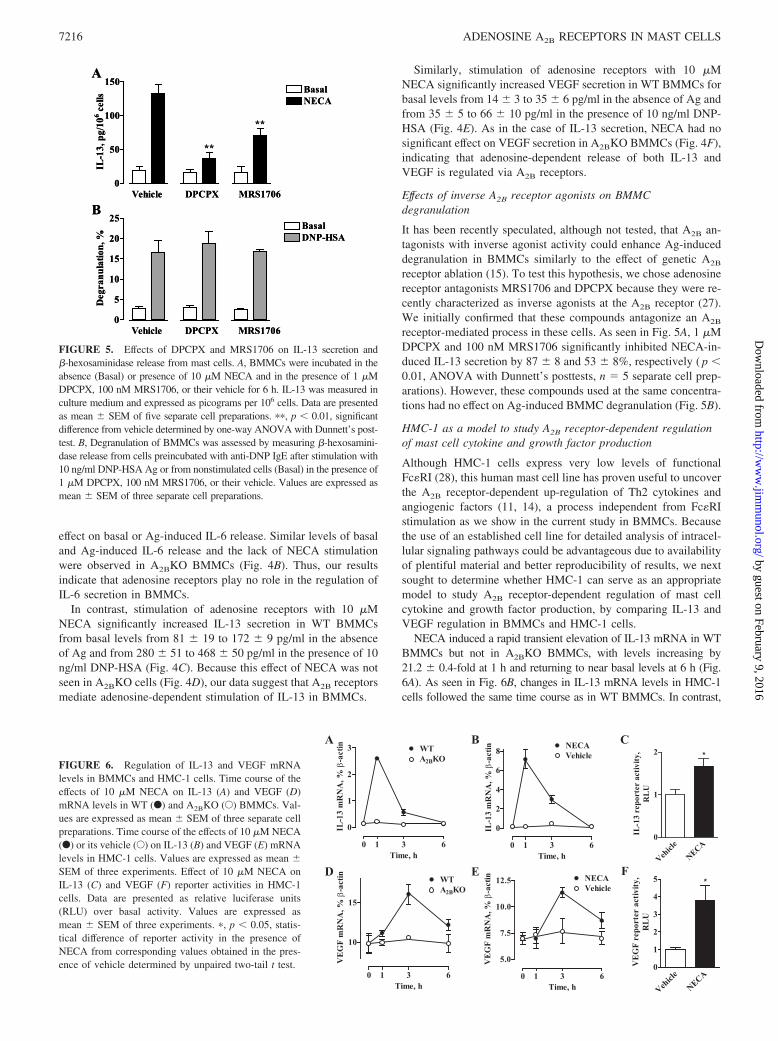
NECA induced a rapid transient elevation of IL-13 mRNA in WTBMMCs but not in A2BKO BMMCs, with levels increasing by21.2 � 0.4-fold at 1 h and returning to near basal levels at 6 h (Fig.6A). As seen in Fig. 6B, changes in IL-13 mRNA levels in HMC-1cells followed the same time course as in WT BMMCs. In contrast,
0
5
10
15
20
25Basal
Vehicle DPCPX MRS1706
DNP-HSA
Deg
ranu
latio
n, %
0
50
100
150BasalNECA
Vehicle DPCPX MRS1706
**
**
IL-1
3, p
g/10
6 cel
ls
B
A
0
5
10
15
20
25Basal
Vehicle DPCPX MRS1706
DNP-HSA
Deg
ranu
latio
n, %
0
50
100
150BasalNECA
Vehicle DPCPX MRS1706
**
**
IL-1
3, p
g/10
6 cel
ls
B
A
FIGURE 5. Effects of DPCPX and MRS1706 on IL-13 secretion and�-hexosaminidase release from mast cells. A, BMMCs were incubated in theabsence (Basal) or presence of 10 �M NECA and in the presence of 1 �MDPCPX, 100 nM MRS1706, or their vehicle for 6 h. IL-13 was measured inculture medium and expressed as picograms per 106 cells. Data are presentedas mean � SEM of five separate cell preparations. ��, p � 0.01, significantdifference from vehicle determined by one-way ANOVA with Dunnett’s post-test. B, Degranulation of BMMCs was assessed by measuring �-hexosamini-dase release from cells preincubated with anti-DNP IgE after stimulation with10 ng/ml DNP-HSA Ag or from nonstimulated cells (Basal) in the presence of1 �M DPCPX, 100 nM MRS1706, or their vehicle. Values are expressed asmean � SEM of three separate cell preparations.
Vehicl
e
NECA0
1
2 *
IL-1
3 re
port
er a
ctiv
ity,
RL
U
Vehicl
e
NECA0
1
2
3
4
5 *
VE
GF
repo
rter
act
ivity
,R
LU
0 1 3 6
0
1
2
3 WTA2BKO
Time, h
IL-1
3 m
RN
A, %
β-a
ctin
0 1 3 6
0
2
4
6
8 NECAVehicle
Time, h
IL-1
3 m
RN
A, %
β-a
ctin
0 1 3 6
10
15
WTA2BKO
Time, h
VE
GF
mR
NA
, %β
-act
in
0 1 3 6
5.0
7.5
10.0
12.5VehicleNECA
Time, h
VE
GF
mR
NA
, %β
-act
in
A B C
D E F
FIGURE 6. Regulation of IL-13 and VEGF mRNAlevels in BMMCs and HMC-1 cells. Time course of theeffects of 10 �M NECA on IL-13 (A) and VEGF (D)mRNA levels in WT (F) and A2BKO (E) BMMCs. Val-ues are expressed as mean � SEM of three separate cellpreparations. Time course of the effects of 10 �M NECA(F) or its vehicle (E) on IL-13 (B) and VEGF (E) mRNAlevels in HMC-1 cells. Values are expressed as mean �SEM of three experiments. Effect of 10 �M NECA onIL-13 (C) and VEGF (F) reporter activities in HMC-1cells. Data are presented as relative luciferase units(RLU) over basal activity. Values are expressed asmean � SEM of three experiments. �, p � 0.05, statis-tical difference of reporter activity in the presence ofNECA from corresponding values obtained in the pres-ence of vehicle determined by unpaired two-tail t test.
7216 ADENOSINE A2B RECEPTORS IN MAST CELLS
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

VEGF mRNA elevations were delayed in WT BMMCs peaking onlyat 3 h after stimulation with NECA (Fig. 6D). Yet again, NECA-induced changes in VEGF mRNA levels in HMC-1 followed exactlythe same time course as in WT BMMCs (Fig. 6E). Stimulation ofreporter activities with 10 �M NECA (Fig. 6, C and F) suggests thatregulation of IL-13 and VEGF mRNA levels via A2B adenosine re-ceptors occurs at least in part due to increase in their transcription.
Because stimulation of A2B receptors in both BMMCs andHMC-1 cells increases intracellular cAMP (29) (Fig. 2), we nextprobed whether elevations of cAMP concentrations induced byforskolin could mimic the effects of NECA on IL-13 and VEGFsecretion from these cells. We found that incubation of WT orA2BKO BMMCs with 100 �M forskolin had no effect on IL-13secretion (Fig. 7A), but stimulated VEGF release (Fig. 7C). Sim-ilarly, forskolin stimulated VEGF secretion, but not IL-13 secre-tion in HMC-1 cells (Fig. 7, B and D). However, inhibition ofcAMP production in HMC-1 cells with 100 �M 2�,5�-dideoxya-denosine (Fig. 7E) only partially suppressed NECA-inducedVEGF secretion (Fig. 7F), suggesting that cAMP-dependent
0
50
100
150 BasalNECAForskolin
WT A2BKO
**IL
-13,
pg/
106 c
ells
0
100
200 BasalNECAForskolin
**
IL-1
3, p
g/10
6 cel
ls
0
10
20
30
40
50 BasalNECAForskolin
WT A2BKO
VE
GF,
pg/
106 c
ells
** ** **
0.0
0.5
1.0
1.5
2.0
2.5BasalNECAForskolin**
*V
EGF,
ng/
106 c
ells
-6 -5 -4
0
50
100
Log [ddAdo], M-6 -5 -4
0
50
100
IL-13VEGF
Log [ddAdo], M
Rel
ease
stim
ulat
ion,
%
cAM
P St
imul
atio
n, %
A B
C D
E F
FIGURE 7. Effect of modulation of cAMP levels in BMMCs and HMC-1cells on IL-13 and VEGF secretion. Effects of 10 �M NECA and 100 �Mforskolin on IL-13 (A) and VEGF (C) secretion from WT and A2BKOBMMCs. Values are expressed as mean � SEM of three separate cell prep-arations. ��, p � 0.01, significant difference from basal shown by one-wayANOVA with Dunnett’s posttest. Effects of 10 �M NECA and 100 �Mforskolin on IL-13 (B) and VEGF (D) secretion from HMC-1 cells. Valuesare expressed as mean � SEM of three experiments. �, p � 0.05 and��, p � 0.01, significant differences from basal determined by one-wayANOVA with Dunnett’s posttest. Effect of adenylate cyclase inhibitor2�,5�-dideoxyadenosine (ddAdo) on cAMP accumulation (E) in HMC-1cells stimulated with 10 �M NECA. In the absence of 2�,5�-dideoxyade-nosine, 10 �M NECA increased cAMP levels from 3.2 � 0.6 to 19.4 � 2.9pmol/106 cells. Values are presented as mean percentage � SEM ofNECA-stimulated response in three experiments. F, Effect of 2�,5�-dideoxyadenosine on IL-13 (E) and VEGF (F) secretion from HMC-1 cellsstimulated with 10 �M NECA. Values are presented as mean percentage �SEM of NECA-stimulated response for three experiments.
FIGURE 8. Effect of inhibitors of MAPK pathways on NECA-depen-dent IL-13 and VEGF secretion from BMMCs and HMC-1 cells. A, West-ern blot analysis of phosphorylation of ERK and p38 MAPK (phospho-ERK and phospho-p38, respectively) in BMMCs and HMC-1 cells thatwere either not stimulated (�) or stimulated with 10 �M NECA for theindicated time period. Equal loading of proteins was verified by reprobingthe same blot with total ERK and p38 MAPK Abs (ERK and p38, respec-tively). Blots are representative of three experiments. Effects of MEK in-hibitor UO126 (F) and its inactive analog UO124 (E) on IL-13 (B) orVEGF (D) secretion from HMC-1 cells stimulated with 10 �M NECA.Values are presented as mean percentage � SEM of NECA-stimulatedresponse from three experiments. Effects of p38 inhibitor SB202190 (F)and its inactive analog SB202474 (E) on IL-13 (C) or VEGF (E) secretionfrom HMC-1 cells stimulated with 10 �M NECA. Values are presented asmean percentage � SEM of NECA-stimulated response from three exper-iments. F, Effects of MEK inhibitor UO126 (10 �M), p38 inhibitorSB202190 (10 �M), and their inactive analogs UO124 (10 �M) andSB202474 (10 �M), respectively, on IL-13 secretion from BMMCs stim-ulated with 10 �M NECA. Significant difference (p � 0.05) was deter-mined by one-way ANOVA. Data are expressed as the percentage ofNECA-stimulated response and presented as mean � SEM of three sepa-rate cell preparations. G, Effects of MEK inhibitor UO124 (10 �M), p38inhibitor SB202190 (10 �M), and their inactive analogs UO126 (10 �M)and SB202474 (10 �M) on VEGF secretion from BMMCs stimulated with10 �M NECA. Significant difference (p � 0.001) was determined by one-way ANOVA. Data are expressed as the percentage of NECA-stimulatedresponse and presented as mean � SEM of three separate cell preparations.
7217The Journal of Immunology
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

modulation of VEGF release cannot be solely responsible for theeffects of NECA.
We have previously shown that ERK and p38 MAPKs, but notcAMP-dependent pathways play an important role in regulation ofA2B receptor-mediated IL-8 production in HMC-1 cells (30). Sim-ilarly to HMC-1 cells, stimulation of BMMCs with 10 �M NECAinduced a rapid transient phosphorylation of ERK and p38 MAPKs(Fig. 8A). We determined that the inhibitors of ERK and p38MAPK pathways UO126 and SB202190, but not their inactiveanalogs UO124 and SB202474, respectively, effectively sup-pressed NECA-stimulated IL-13 and VEGF release from HMC-1cells in a concentration-dependent manner (Fig. 8, B–E). We foundthat these inhibitors of MAPK pathways, but not their inactiveanalogs used at the same concentrations (10 �M) also attenuatedNECA-stimulated IL-13 and VEGF release from BMMCs (Fig. 8,F and G). Taken together, our results demonstrate similarity incytokine regulation in both mast cells and suggest that HMC-1cells are an appropriate model for delineation of A2B receptor-linked intracellular signaling pathways leading to secretion of Th2cytokines and angiogenic factors.
DiscussionThe low-affinity A2B receptor is known to mediate proinflamma-tory effects of adenosine by up-regulating production of cytokinesand growth factors. This view has been supported by a large bodyof evidence provided by pharmacological analysis of adenosine-dependent cytokine and growth factor secretion in various cells,tissues, and organs (8, 11, 13, 18, 31–38). Pharmacological inhi-bition of A2B receptors significantly reduced elevations in proin-flammatory cytokines as well as mediators of airway remodelinginduced by high adenosine levels in the lungs of ADA-deficientmice (8). In the ragweed allergic mouse model, A2B antagonismplays an important role in inhibition of airway reactivity and in-flammation (39, 40). In agreement with data obtained in these an-imal models of pulmonary inflammation, stimulation of A2B re-ceptors in the human mast cell line HMC-1 was shown to inducesecretion of proinflammatory Th2 cytokines IL-4 and IL-13 (11,12), as well as angiogenic factors IL-8 and VEGF (13, 14).
A recent report by Hua et al. (15) disputed the proinflammatoryrole of A2B receptors in mast cells. Based on the finding in thisstudy of an enhanced Ag-induced mast cell degranulation in micelacking the A2B receptor, the authors inferred that this receptorsubtype normally limits the magnitude of responsiveness towardAg in WT mast cells. The authors postulated that A2B receptors areconstitutively active in mast cells and speculated that A2B antag-onists with inverse agonist activity would lower cAMP levelswithin the mast cell, potentially enhancing Ag-induced degranu-lation and proinflammatory cytokine production. However, nopharmacological data were presented to support this claim. In thecurrent study, we combined pharmacological and genetic ap-proaches to revisit these conclusions using exactly the same ex-perimental model of mouse BMMCs obtained from WT andA2BKO mice backcrossed onto the same C57BL/6 geneticbackground.
Our data confirmed that genetic A2B receptor ablation inBMMCs does not change the surface expression of Fc�RI or c-kitreceptors, nor does it affect the expression of mRNA encoding A2A
and A3 adenosine receptor subtypes present in these cells. In agree-ment with the report by Hua et al. (15), we observed an enhancedstimulation of Ag-induced degranulation in A2BKO BMMCs com-pared with WT cells. Notwithstanding the potential importance ofthis phenomenon, we thought that characterization of A2BKO BM-MCs would be incomplete without actual testing whether adeno-sine agonist-induced signaling has been changed in these cells.
Adenosine is known to potentiate Ag-induced degranulation ofmast cells. This effect of adenosine on preformed mediator releaseis mediated by A3 adenosine receptors in rodents (23, 41–43), butperhaps not in human (44) or canine (45) mast cells. We found thatan enhanced Ag-induced degranulation in A2BKO BMMCs wasobserved both in the absence and presence of the adenosine re-ceptor agonist NECA, but the degree of potentiation mediated viaA3 receptors was essentially the same as in WT cells. Thus, weconclude that genetic ablation of A2B receptors not only has nosignificant effect on the expression of A3 receptors, but also ontheir function in potentiating Ag-induced degranulation ofBMMCs.
In addition to release of preformed mediators from mast cellgranules, stimulation of mast cells with Ag results in de novosynthesis and secretion of proinflamatory cytokines and angio-genic factors (46). These events are regulated via distinct pathwaysthat can be activated independently (24, 47–50). Adenosine canexert differential effects on some of these processes via distinctadenosine receptor subtypes or may have no effect at all. In agree-ment with previous reports in BMMCs (24) and in human mastcells (11), we found no effect of stimulation of adenosine receptorswith NECA on IL-6 secretion in WT BMMCs. Consequentially,there was no significant effect of A2B receptor ablation on IL-6secretion either in resting or Ag-stimulated BMMCs. However,stimulation of A2B receptors in human mast cell line HMC-1 waspreviously shown to induce secretion of other proinflammatoryfactors, and particularly Th2 cytokines and angiogenic factors (11–14). Even though it has been argued that the proinflammatory func-tions of A2B receptors described in HMC-1 expressing low levelsof functional IgE receptors could be irrelevant to adenosine sig-naling in BMMCs (15), in this study we show for the first time thatA2B receptors stimulate secretion of these factors in BMMCs aswell. The adenosine agonist NECA stimulated IL-13 and VEGFsecretion only in WT BMMCs but not in A2BKO BMMCs. Con-trary to adenosine action on degranulation, which is only apparentin Ag-stimulated BMMCs, these effects did not require activationof Fc�RI because they were evident even in the absence of Ag. Wealso found that A2B receptor-dependent cytokine regulation wasidentical in BMMCs and HMC-1 cells, based on effects of cAMPmodulators and inhibitors of MAPK signaling pathways on IL-13and VEGF production. Thus, our data in mouse mast cells express-ing high levels of functional Fc�RI confirmed our previous findingin human mast cell line HMC-1 that stimulation of A2B adenosinereceptors results in secretion of Th2 cytokines and angiogenic fac-tors. These observations suggest that HMC-1 cells are appropriateas a model for delineation of A2B receptor-linked intracellular sig-naling pathways.
Our study has clearly demonstrated that genetic ablation of A2B
receptors has two different effects on BMMCs, one is the loss ofadenosine signaling mediated by this receptor that regulates secre-tion of IL-13 and VEGF, whereas the other is the adenosine-inde-pendent phenomenon of enhanced Ag-induced degranulation pre-viously described by Hua et al. (15). The latter phenomenon issimilar to an enhanced production of TNF-� observed in macro-phages obtained from A2BKO mice (51, 52). We have recentlydemonstrated that this enhanced basal and LPS-stimulated TNF-�secretion in A2BKO macrophages cannot be explained by the lossof adenosine signaling via A2B receptor (52). Likewise, our data inthe current study indicate that the enhanced Ag-induced degranu-lation in BMMCs obtained from A2BKO mice cannot be explainedby the loss of tonic stimulation of A2B receptors by endogenousadenosine. Because our experiments were conducted in thepresence of ADA, it is unlikely that endogenous adenosine could
7218 ADENOSINE A2B RECEPTORS IN MAST CELLS
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

accumulate in concentrations sufficient to stimulate this low-affin-ity receptor.
Acknowledging the implausibility of tonic stimulation of thelow-affinity A2B receptor by endogenous adenosine, Hua et al. (15)proposed the alternative explanation that A2B receptors are con-stitutively active in WT BMMCs even in the absence of an agonist.However, we determined that A2B receptors expressed in WT BM-MCs display no constitutive activity in regulation of cAMP. Ifthese receptors were indeed constitutively active in BMMCs, thenthey are expected to activate Gs proteins in the absence of agoniststimulation. Because the increased levels of free Gs� subunits areknown to enhance the responses to forskolin, this agent is oftenused to verify constitutive activity of Gs protein-coupled receptors(53). However, not only did we not find differences in basal cAMPlevels between BMMCs obtained from A2BKO and WT mice, butthere was no difference in forskolin-induced accumulation ofcAMP between mast cells expressing and lacking A2B receptors.In contrast, cAMP accumulation stimulated with the nonselectiveadenosine receptor agonist NECA, but not with the selective A2A
receptor agonist CGS21680, was significantly lower in A2BKOBMMCs compared with WT cells, reflecting the loss of adenosinesignaling via A2B receptors. Finally, the lack of constitutive A2B
receptor activity in BMMCs is supported by our experiments withinverse agonists, agents that bind to the same receptor binding siteas agonists but exert the opposite pharmacological effect. If thesereceptors were indeed constitutively active, then inverse agonistsare expected to repress their spontaneous activity (for a review, seeRef. 54). Recent studies have identified adenosine antagonistsZM241385, DPCPX, and MRS1706 as inverse agonists at A2B
receptors (27). We chose two structurally unrelated compounds,the selective A2B antagonist MRS1706 (55) and the A1/A2B an-tagonist DPCPX (56), to study A2B receptor signaling in BMMCs.Given the fact that these cells do not express A1 receptors (Fig.1C), we used DPCPX as an inverse agonist at A2B receptorscomplementary to MRS1706. We have demonstrated that bothMRS1706 and DPCPX inhibited cAMP accumulation mediatedby A2B receptor stimulation, but they had no significant effecton basal cAMP levels, further proving the lack of constitutiveA2B receptor activity in BMMCs. Contrary to the hypothesis ofHua et al. (15), these compounds had no effect on Ag-inducedmast cell degranulation, but they inhibited A2B-mediated IL-13secretion in BMMCs.
Our results, therefore, confirm the proinflammatory effects re-sulting from activation of A2B receptors. They also confirm thephenomenon described by Hua et al. (15) of exaggerated Ag-in-duced degranulation in BMMCs deficient of A2B receptors. Thisphenomenon, however, is unrelated to the adenosine signalingfunction of A2B receptors. An alternative and more likely expla-nation of this phenomenon could be that the A2B receptor proteininteracts with other signaling pathways unrelated to adenosine. Re-cent evidence suggests that adenosine receptors play a role in as-sembly of multiprotein signaling complexes (57–61). Rearrange-ment of proteins normally coupled to the A2B receptor as a resultof the A2BKO may affect Fc�RI-dependent regulation of degran-ulation. Future studies that define the full makeup of A2B receptorcomplexes with associated signaling components are needed tovalidate this hypothesis.
In conclusion, we report that adenosine receptors mediate dif-ferential regulation of murine mast cell functions depending on thereceptor subtype involved. Adenosine signaling that regulatesBMMC degranulation previously shown being mediated throughA3 receptors has no effect in resting cells, but potentiates the Ag-induced degranulation. In contrast, adenosine signaling throughA2B receptors stimulates secretion of Th2 cytokines and angio-
genic factors even in the absence of Ag. In this regard, our studyin BMMCs obtained from A2BKO and WT mice has confirmed theconcept drawn from previous pharmacological data that stimula-tion of A2B receptors with adenosine leads to proinflammatoryevents based on the observation that A2B receptors mediate ade-nosine-dependent secretion of IL-13 and VEGF. Our data argueagainst a constitutive activity of A2B receptors or their tonic acti-vation by endogenous adenosine in BMMCs purportedly limitingAg-stimulated mast cell degranulation. An enhancement in Fc�RI-mediated degranulation observed in A2BKO BMMCs is not di-rectly related to adenosine signaling through the A2B receptor, butmay represent a loss of other yet unidentified functions of thisprotein, or reflect a developmental adaptation to the global A2B
receptor gene ablation in mice.
DisclosuresDrs. I. Biaggioni and I. Feoktistov are inventors named onU.S. Patent No. 6,815,446 for “Selective antagonists of A2B adenosinereceptors,” which was licensed through the Vanderbilt University Officeof Technology Transfer and Enterprise Development (Nashville, TN) toCV Therapeutics (Palo Alto, CA) for the development of antiasthmaticdrugs. Drs. I. Biaggioni, I. Feoktistov, and M. R. Blackburn have beenrecipients of research funding from CV Therapeutics. The remainingauthors have no financial conflict of interest.
References1. Fredholm, B. B., A. P. Ijzerman, K. A. Jacobson, K. N. Klotz, and J. Linden.
2001. International Union of Pharmacology. XXV. Nomenclature and classifi-cation of adenosine receptors. Pharmacol. Rev. 53: 527–552.
2. Fredholm, B. B., E. Irenius, B. Kull, and G. Schulte. 2001. Comparison of thepotency of adenosine as an agonist at human adenosine receptors expressed inChinese hamster ovary cells. Biochem. Pharmacol. 61: 443–448.
3. Driver, A. G., C. A. Kukoly, S. Ali, and S. J. Mustafa. 1993. Adenosine inbronchoalveolar lavage fluid in asthma. Am. Rev. Respir. Dis. 148: 91–97.
4. Huszar, E., G. Vass, E. Vizi, Z. Csoma, E. Barat, V. G. Molnar, I. Herjavecz, andI. Horvath. 2002. Adenosine in exhaled breath condensate in healthy volunteersand in patients with asthma. Eur. Respir. J. 20: 1393–1398.
5. Blackburn, M. R., J. B. Volmer, J. L. Thrasher, H. Zhong, J. R. Crosby, J. J. Lee,and R. E. Kellems. 2000. Metabolic consequences of adenosine deaminase de-ficiency in mice are associated with defects in alveogenesis, pulmonary inflam-mation, and airway obstruction. J. Exp. Med. 192: 159–170.
6. Blackburn, M. R. 2003. Too much of a good thing: adenosine overload in aden-osine-deaminase-deficient mice. Trends Pharmacol. Sci. 24: 66–70.
7. Blackburn, M. R., C. G. Lee, H. W. Young, Z. Zhu, J. L. Chunn, M. J. Kang,S. K. Banerjee, and J. A. Elias. 2003. Adenosine mediates IL-13-induced inflam-mation and remodeling in the lung and interacts in an IL-13-adenosine amplifi-cation pathway. J. Clin. Invest. 112: 332–344.
8. Sun, C. X., H. Zhong, A. Mohsenin, E. Morschl, J. L. Chunn, J. G. Molina,L. Belardinelli, D. Zeng, and M. R. Blackburn. 2006. Role of A2B adenosinereceptor signaling in adenosine-dependent pulmonary inflammation and injury.J. Clin. Invest. 116: 2173–2182.
9. Bradding, P., A. F. Walls, and S. T. Holgate. 2006. The role of the mast cell inthe pathophysiology of asthma. J. Allergy Clin. Immunol. 117: 1277–1284.
10. Rorke, S., and S. T. Holgate. 2002. Targeting adenosine receptors: novel thera-peutic targets in asthma and chronic obstructive pulmonary disease.Am. J. Respir. Med. 1: 99–105.
11. Ryzhov, S., A. E. Goldstein, A. Matafonov, D. Zeng, I. Biaggioni, andI. Feoktistov. 2004. Adenosine-activated mast cells induce IgE synthesis by Blymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13with implications for asthma. J. Immunol. 172: 7726–7733.
12. Ryzhov, S., A. E. Goldstein, I. Biaggioni, and I. Feoktistov. 2006. Cross-talkbetween Gs- and Gq-coupled pathways in regulation of interleukin-4 by A2B
adenosine receptors in human mast cells. Mol. Pharmacol. 70: 727–735.13. Feoktistov, I., and I. Biaggioni. 1995. Adenosine A2B receptors evoke interleu-
kin-8 secretion in human mast cells: an enprofylline-sensitive mechanism withimplications for asthma. J. Clin. Invest. 96: 1979–1986.
14. Feoktistov, I., S. Ryzhov, A. E. Goldstein, and I. Biaggioni. 2003. Mast cell-mediated stimulation of angiogenesis: cooperative interaction between A2B andA3 adenosine receptors. Circ. Res. 92: 485–492.
15. Hua, X., M. Kovarova, K. D. Chason, M. Nguyen, B. H. Koller, and S. L. Tilley.2007. Enhanced mast cell activation in mice deficient in the A2b adenosine re-ceptor. J. Exp. Med. 204: 117–128.
16. Scoka, B., Z. H. Nemeth, L. Virag, P. Gergely, S. J. Leibovich, P. Pacher, C.-X.Sun, M. R. Blackburn, E. S. Vizi, E. A. Deitch, and G. Hasko. 2007. A2A aden-osine receptors and C/EBP� are crucially required for IL-10 production by mac-rophages exposed to E. coli. Blood 110: 2685–2695.
17. Ryzhov, S., J. L. McCaleb, A. E. Goldstein, I. Biaggioni, and I. Feoktistov. 2007.Role of adenosine receptors in the regulation of angiogenic factors and neovas-cularization in hypoxia. J. Pharmacol. Exp. Ther. 382: 565–572.
7219The Journal of Immunology
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

18. Feoktistov, I., A. E. Goldstein, S. Ryzhov, D. Zeng, L. Belardinelli,T. Voyno-Yasenetskaya, and I. Biaggioni. 2002. Differential expression of aden-osine receptors in human endothelial cells: role of A2B receptors in angiogenicfactor regulation. Circ. Res. 90: 531–538.
19. Schwartz, L. B., K. F. Austen, and S. I. Wasserman. 1979. Immunologic releaseof �-hexosaminidase and �-glucoronidase from peritoneal rat serosal mast cells.J. Immunol. 330: 393–411.
20. Forsythe, J. A., B. H. Jiang, N. V. Iyer, F. Agani, S. W. Leung, R. D. Koos, andG. L. Semenza. 1996. Activation of vascular endothelial growth factor gene tran-scription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16: 4604–4613.
21. Chung, H. K., H. A. Young, P. K. Goon, G. Heidecker, G. L. Princler,O. Shimozato, G. P. Taylor, C. R. Bangham, and D. Derse. 2003. Activation ofinterleukin-13 expression in T cells from HTLV-1-infected individuals and inchronically infected cell lines. Blood 102: 4130–4136.
22. Marquardt, D. L., L. L. Walker, and S. I. Wasserman. 1984. Adenosine receptorson mouse bone marrow-derived mast cells: functional significance and regulationby aminophylline. J. Immunol. 133: 932–937.
23. Salvatore, C. A., S. L. Tilley, A. M. Latour, D. S. Fletcher, B. H. Koller, andM. A. Jacobson. 2000. Disruption of the A3 adenosine receptor gene in mice andits effect on stimulated inflammatory cells. J. Biol. Chem. 275: 4429–4434.
24. Marquardt, D. L., J. L. Alongi, and L. L. Walker. 1996. The phosphatidylinositol3-kinase inhibitor wortmannin blocks mast cell exocytosis but not IL-6 produc-tion. J. Immunol. 156: 1942–1945.
25. Burd, P. R., W. C. Thompson, E. E. Max, and F. C. Mills. 1995. Activated mastcells produce interleukin 13. J. Exp. Med. 181: 1373–1380.
26. Boesiger, J., M. Tsai, M. Maurer, M. Yamaguchi, L. F. Brown, K. P. Claffey,H. F. Dvorak, and S. J. Galli. 1998. Mast cells can secrete vascular permeabilityfactor/vascular endothelial cell growth factor and exhibit enhanced release afterimmunoglobulin E-dependent upregulation of Fc� receptor I expression. J. Exp.Med. 188: 1135–1145.
27. Li, Q., K. Ye, C. C. Blad, H. den Dulk, J. Brouwer, A. P. Ijzerman, andM. W. Beukers. 2007. ZM241385, DPCPX, MRS1706 are inverse agonists withdifferent relative intrinsic efficacies on constitutively active mutants of the humanadenosine A2B receptor. J. Pharmacol. Exp. Ther. 320: 637–645.
28. Butterfield, J. H., D. Weiler, G. Dewald, and G. J. Gleich. 1988. Establishmentof an immature mast cell line from a patient with mast cell leukemia. LeukemiaRes. 12: 345–355.
29. Feoktistov, I., and I. Biaggioni. 1998. Pharmacological characterization of aden-osine A2B receptors: studies in human mast cells co-expressing A2A an A2B
adenosine receptor subtypes. Biochem. Pharmacol. 55: 627–633.30. Feoktistov, I., A. E. Goldstein, and I. Biaggioni. 1999. Role of p38 mitogen-
activated protein kinase and extracellular signal-regulated protein kinase kinasein adenosine A2B receptor-mediated interleukin-8 production in human mastcells. Mol. Pharmacol. 55: 726–735.
31. Fiebich, B. L., K. Biber, K. Guyfko, M. Berger, J. Bauer, and D. van Calker.1996. Adenosine A2b receptors mediate an increase in interleukin (IL)-6 mRNAand IL-6 protein synthesis in human astroglioma cells. J. Neurochem. 66:1426–1431.
32. Schwaninger, M., M. Neher, E. Viegas, A. Schneider, and M. Spranger. 1997.Stimulation of interleukin-6 secretion and gene transcription in primary astro-cytes by adenosine. J. Neurochem. 69: 1145–1150.
33. Zeng, D., T. Maa, U. Wang, I. Feoktistov, I. Biaggioni, and L. Belardinelli. 2003.Expression and function of A2B adenosine receptors in the U87MG tumor cells.Drug Dev. Res. 58: 405–411.
34. Rees, D. A., B. M. Lewis, M. D. Lewis, K. Francis, M. F. Scanlon, and J. Ham.2003. Adenosine-induced IL-6 expression in pituitary folliculostellate cells ismediated via A2b adenosine receptors coupled to PKC and p38 MAPK.Br. J. Pharmacol. 140: 764–772.
35. Zhong, H., L. Belardinelli, T. Maa, I. Feoktistov, I. Biaggioni, and D. Zeng. 2004.A2B adenosine receptors increase cytokine release by bronchial smooth musclecells. Am. J. Resp. Cell Mol. Biol. 30: 118–125.
36. Fiebich, B. L., R. S. Akundi, K. Biber, M. Hamke, C. Schmidt, R. D. Butcher,D. van Calker, and F. Willmroth. 2005. IL-6 expression induced by adenosineA2b receptor stimulation in U373 MG cells depends on p38 mitogen activatedkinase and protein kinase C. Neurochem. Int. 46: 501–512.
37. Zhong, H., L. Belardinelli, T. Maa, and D. Zeng. 2005. Synergy between A2B
adenosine receptors and hypoxia in activating human lung fibroblasts.Am. J. Resp. Cell Mol. Biol. 32: 2–8.
38. Evans, B. A., C. Elford, A. Pexa, K. Francis, A. C. Hughes, A. Deussen, andJ. Ham. 2006. Human osteoblast precursors produce extracellular adenosine,which modulates their secretion of IL-6 and osteoprotegerin. J. Bone Miner. Res.21: 228–236.
39. Fan, M., W. Qin, and S. J. Mustafa. 2003. Characterization of adenosine recep-tor(s) involved in adenosine-induced bronchoconstriction in an allergic mousemodel. Am. J. Physiol. 284: L1012–L1019.
40. Mustafa, S. J., A. Nadeem, M. Fan, H. Zhong, L. Belardinelli, and D. Zeng. 2007.Effect of a specific and selective A2B adenosine receptor antagonist on adenosine
agonist AMP and allergen-induced airway responsiveness and cellular influx in amouse model of asthma. J. Pharmacol. Exp. Ther. 320: 1246–1251.
41. Ramkumar, V., G. L. Stiles, M. A. Beaven, and H. Ali. 1993. The A3 adenosinereceptor is the unique adenosine receptor which facilitates release of allergicmediators in mast cells. J. Biol. Chem. 268: 16887–16890.
42. Reeves, J. J., C. A. Jones, M. J. Sheehan, C. J. Vardey, and C. J. Whelan. 1997.Adenosine A3 receptors promote degranulation of rat mast cells both in vitro andin vivo. Inflamm. Res. 46: 180–184.
43. Zhong, H., S. G. Shlykov, J. G. Molina, B. M. Sanborn, M. A. Jacobson,S. L. Tilley, and M. R. Blackburn. 2003. Activation of murine lung mast cells bythe adenosine A3 receptor. J. Immunol. 171: 338–345.
44. Walker, B. A., M. A. Jacobson, D. A. Knight, C. A. Salvatore, T. Weir, D. Zhou,and T. R. Bai. 1997. Adenosine A3 receptor expression and function in eosino-phils. Am. J. Resp. Cell Mol. Biol. 16: 531–537.
45. Auchampach, J. A., X. Jin, T. C. Wan, G. H. Caughey, and J. Linden. 1997.Canine mast cell adenosine receptors: Cloning and expression of the A3 receptorsand evidence that degranulation is mediated by the A2B receptor. Mol. Pharma-col. 52: 846–860.
46. Metcalfe, D. D., D. Baram, and Y. A. Mekori. 1997. Mast cells. Physiol. Rev. 77:1033–1079.
47. Arzubiaga, C., J. Morrow, L. J. Roberts, and I. Biaggioni. 1991. Neuropeptide Y,a putative cotransmitter in noradrenergic neurons, induces mast cell degranula-tion but not prostaglandin D2 release. J. Allergy Clin. Immunol. 87: 88–93.
48. Abdel-Majid, R. M., and J. S. Marshall. 2004. Prostaglandin E2 induces degran-ulation-independent production of vascular endothelial growth factor by humanmast cells. J. Immunol. 172: 1227–1236.
49. Nakayama, T., N. Mutsuga, L. Yao, and G. Tosato. 2006. Prostaglandin E2 pro-motes degranulation-independent release of MCP-1 from mast cells. J. LeukocyteBiol. 79: 95–104.
50. Qiao, H., M. V. Andrade, F. A. Lisboa, K. Morgan, and M. A. Beaven. 2006.Fc�R1 and Toll-like receptors mediate synergistic signals to markedly augmentproduction of inflammatory cytokines in murine mast cells. Blood 107: 610–618.
51. Yang, D., Y. Zhang, H. G. Nguyen, M. Koupenova, A. K. Chauhan, M. Makitalo,M. R. Jones, C. St. Hilaire, D. C. Seldin, P. Toselli, et al. 2006. The A2B aden-osine receptor protects against inflammation and excessive vascular adhesion.J. Clin. Invest. 116: 1913–1923.
52. Ryzhov, S., R. Zaynagetdinov, A. E. Goldstein, S. V. Novitskiy,M. R. Blackburn, I. Biaggioni, and I. Feoktistov. 2008. Effect of A2B adenosinereceptor gene ablation on adenosine-dependent regulation of pro-inflammatorycytokines. J. Pharmacol. Exp. Ther. 324: 694–700.
53. Alewijnse, A. E., M. J. Smit, M. S. Rodriguez Pena, D. Verzijl, H. Timmerman,and R. Leurs. 1997. Modulation of forskolin-mediated adenylyl cyclase activa-tion by constitutively active GS-coupled receptors. FEBS Lett. 419: 171–174.
54. Costa, T., and S. Cotecchia. 2005. Historical review: negative efficacy and theconstitutive activity of G-protein-coupled receptors. Trends Pharmacol. Sci. 26:618–624.
55. Kim, Y. C., X. Ji, N. Melman, J. Linden, and K. A. Jacobson. 2000. Anilidederivatives of an 8-phenylxanthine carboxylic congener are highly potent andselective antagonists at human A2B adenosine receptors. J. Med. Chem. 43:1165–1172.
56. Kreckler, L. M., T. C. Wan, Z. D. Ge, and J. A. Auchampach. 2006. Adenosineinhibits tumor necrosis factor-alpha release from mouse peritoneal macrophagesvia A2A and A2B but not the A3 adenosine receptor. J. Pharmacol. Exp. Ther.317: 172–180.
57. Gsandtner, I., C. Charalambous, E. Stefan, E. Ogris, M. Freissmuth, andJ. Zezula. 2005. Heterotrimeric G protein-independent signaling of a G protein-coupled receptor: direct binding of ARNO/cytohesin-2 to the carboxyl terminusof the A2A adenosine receptor is necessary for sustained activation of the ERK/MAP kinase pathway. J. Biol. Chem. 280: 31898–31905.
58. Milojevic, T., V. Reiterer, E. Stefan, V. M. Korkhov, M. M. Dorostkar, E. Ducza,E. Ogris, S. Boehm, M. Freissmuth, and C. Nanoff. 2006. The ubiquitin-specificprotease Usp4 regulates the cell surface level of the A2A receptor. Mol. Phar-macol. 69: 1083–1094.
59. Sun, C. N., H. C. Cheng, J. L. Chou, S. Y. Lee, Y. W. Lin, H. L. Lai, H. M. Chen,and Y. Chern. 2006. Rescue of p53 blockage by the A2A adenosine receptor viaa novel interacting protein, translin-associated protein X. Mol. Pharmacol. 70:454–466.
60. Sitaraman, S. V., L. Wang, M. Wong, M. Bruewer, M. Hobert, C. H. Yun,D. Merlin, and J. L. Madara. 2002. The adenosine 2b receptor is recruited to theplasma membrane and associates with E3KARP and Ezrin upon agonist stimu-lation. J. Biol. Chem. 277: 33188–33195.
61. Pacheco, R., J. M. Martinez-Navio, M. Lejeune, N. Climent, H. Oliva,J. M. Gatell, T. Gallart, J. Mallol, C. Lluis, and R. Franco. 2005. CD26, adenosinedeaminase, and adenosine receptors mediate costimulatory signals in the immu-nological synapse. Proc. Natl. Acad. Sci. USA 102: 9583–9588.
7220 ADENOSINE A2B RECEPTORS IN MAST CELLS
by guest on February 9, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
Copyright © 2022 FDOKUMEN

















![Corrigendum to “1-, 3- and 8-substituted-9-deazaxanthines as potent and selective antagonists at the human A2B adenosine receptor” [Bioorg. Med. Chem. 16 (2008) 2852–2869]](https://static.fdokumen.com/doc/165x107/63357c0ca1ced1126c0ad0ad/corrigendum-to-1-3-and-8-substituted-9-deazaxanthines-as-potent-and-selective.jpg)


