
Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity...
21
©2013 Landes Bioscience. Do not distribute. BASIC RESEARCH PAPER www.landesbioscience.com Autophagy 1 Autophagy 9:11, 1–21; November 2013; © 2013 Landes Bioscience BASIC RESEARCH PAPER Introduction Cadmium (Cd) is an extremely toxic environmental and occupational contaminant, arising primarily from batteries, the food chain, and cigarette smoke. 1,2 Given its high rate of release into the environment, Cd levels in humans are likely to increase in the coming years and to lead to a higher incidence of Cd-related diseases. Among the affected organs, the liver is critically damaged by both acute and chronic exposure to Cd. 3,4 Several studies have demonstrated that mitochondria are primary intracellular targets the context of Cd hepatotoxicity. 5-7 Recently, mitochondrial loss has been revealed to play a prominent role in the observed mitochondrial dysfunction. Growing evidence indicates that Cd exposure leads to mitochondrial loss in liver cells, reflected by a decrease in both mitochondrial DNA copy number and in the expression of components of the mitochondrial respiratory chain. 8-12 However, the mechanism by which Cd induces mitochondrial loss during Cd-induced hepatotoxicity is not fully understood. Identifying the mechanism that underlies Cd-induced mitochondrial reduction during hepatotoxicity is an important area of clinical research, and the present results offer insight into new mitochondrial targets for therapeutic intervention in Cd-affected populations. Submitted: 11/08/2012; Revised: 07/01/2013; Accepted: 07/08/2013 http://dx.doi.org/10.4161/auto.25665 *Correspondence to: Zhou Zhou; Email: [email protected]; Zhengping Yu; Email: [email protected] Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium Huifeng Pi, 1 Shangcheng Xu, 1,† Lei Zhang, 1 Pan Guo, 1 Yuming Li, 2 Jia Xie, 1 Li Tian, 1 Mindi He, 1 Yonghui Lu, 1 Min Li, 1 Yanwen Zhang, 1 Min Zhong, 1 Yang Xiang, 3 Linhong Deng, 4 Zhou Zhou 1, * Zhengping Yu 1, * 1 Department of Occupational Health; Third Military Medical University; Chongqing, China; 2 Institute of Hepatobiliary Surgery; XinQiao Hospital; Third Military Medical University; Chongqing, China; 3 Medical Research Center; Southwest Hospital; Third Military Medical University; Chongqing, China; 4 Key Laboratory of Biorheological Science and Technology; Ministry of Education; Bioengineering College; Chongqing University; Chongqing, China † These authors contributed equally to this work. Keywords: cadmium, mitochondrial loss,mitophagy, DNM1L, mitochondrial fission, hepatotoxicity Abbreviations: ACTB, actin, beta; AL, autolysosome; AP, autophagosome; ATG5, autophagy-related 5; Baf A1, bafilomycin A 1 ; Cd, cadmium; CdCl 2 , cadmium chloride; COX4I1, cytochrome c oxidase subunit IV isoform 1; DNM1L, dynamin 1-like; FIS1, fission 1 (mitochondrial outer membrane) homolog (S. cerevisiae); LC3, microtubule-associated protein 1 light chain 3; MFN1, mitofusin 1; MFN2, mitofusin 2; NAO, 10-N-nonyl-acridine orange; OPA1, optic atrophy 1 (autosomal dominant); PARK2, parkinson protein 2, E3 ubiquitin protein ligase (parkin); PINK1, PTEN-induced putative kinase 1; PPARGC1A, peroxisome proliferator-activated receptor gamma, coactivator 1 alpha; SQSTM1/p62, sequestosome 1; TFAM, transcription factor A, mitochondrial How cadmium (Cd) induces mitochondrial loss in the context of its hepatotoxic effects remains enigmatic. The purpose of the study was to investigate whether mitophagy contributes to mitochondrial loss in cadmium-induced hepatotoxicity and to determine the potential mechanism. In normal human liver L02 cells, we observed that Cd treatment led to a significant increase in LC3-II formation, the number of GFP-LC3 puncta and lysosomal colocalization with mitochondria. These results were associated with mitochondrial loss and bioenergetic deficit. Additionally, the abrogation of excessive mitophagy by ATG5 siRNA treatment efficiently suppressed the mitochondrial loss and cytotoxicity of Cd. Before overactivating mitophagy, Cd induced excessive mitochondrial fragmentation as a result of increasing dynamin 1-like (DNM1L) expression and enhancing the DNM1L mitochondrial translocation. Moreover, reversing the excessive mitochondrial fragmentation via the administration of DNM1L siRNA significantly inhibited the observed overactivation of mitophagy in Cd-induced hepatotoxicity. Notably, the selective DNM1L inhibitor Mdivi-1 blocked abnormal mitophagy and subsequently ameliorated Cd-induced hepatotoxicity in vivo. Together, our data indicated that Cd induces mitochondrial loss via the overactivation of mitophagy following DNM1L-dependent mitochondrial fragmentation. The balanced activity of DNM1L and mitophagy signaling may be a potential therapeutic approach to treat Cd-induced hepatotoxicity.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity...

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
Basic ReseaRch PaPeR
www.landesbioscience.com autophagy 1
autophagy 9:11, 1–21; November 2013; © 2013 Landes Bioscience
Basic ReseaRch PaPeR
Introduction
Cadmium (Cd) is an extremely toxic environmental and occupational contaminant, arising primarily from batteries, the food chain, and cigarette smoke.1,2 Given its high rate of release into the environment, Cd levels in humans are likely to increase in the coming years and to lead to a higher incidence of Cd-related diseases. Among the affected organs, the liver is critically damaged by both acute and chronic exposure to Cd.3,4 Several studies have demonstrated that mitochondria are primary intracellular targets the context of Cd hepatotoxicity.5-7 Recently, mitochondrial loss has
been revealed to play a prominent role in the observed mitochondrial dysfunction. Growing evidence indicates that Cd exposure leads to mitochondrial loss in liver cells, reflected by a decrease in both mitochondrial DNA copy number and in the expression of components of the mitochondrial respiratory chain.8-12 However, the mechanism by which Cd induces mitochondrial loss during Cd-induced hepatotoxicity is not fully understood. Identifying the mechanism that underlies Cd-induced mitochondrial reduction during hepatotoxicity is an important area of clinical research, and the present results offer insight into new mitochondrial targets for therapeutic intervention in Cd-affected populations.
Submitted: 11/08/2012; Revised: 07/01/2013; Accepted: 07/08/2013http://dx.doi.org/10.4161/auto.25665*Correspondence to: Zhou Zhou; Email: [email protected]; Zhengping Yu; Email: [email protected]
Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy
in the hepatotoxicity of cadmiumhuifeng Pi,1 shangcheng Xu,1,† Lei Zhang,1 Pan Guo,1 Yuming Li,2 Jia Xie,1 Li Tian,1 Mindi he,1 Yonghui Lu,1 Min Li,1
Yanwen Zhang,1 Min Zhong,1 Yang Xiang,3 Linhong Deng,4 Zhou Zhou1,* Zhengping Yu1,*
1Department of Occupational health; Third Military Medical University; chongqing, china; 2institute of hepatobiliary surgery; XinQiao hospital; Third Military Medical University; chongqing, china; 3Medical Research center; southwest hospital; Third Military Medical University; chongqing, china; 4Key Laboratory of Biorheological science
and Technology; Ministry of education; Bioengineering college; chongqing University; chongqing, china
†These authors contributed equally to this work.
Keywords: cadmium, mitochondrial loss,mitophagy, DNM1L, mitochondrial fission, hepatotoxicity
Abbreviations: ACTB, actin, beta; AL, autolysosome; AP, autophagosome; ATG5, autophagy-related 5; Baf A1, bafilomycin A1;
Cd, cadmium; CdCl2, cadmium chloride; COX4I1, cytochrome c oxidase subunit IV isoform 1; DNM1L, dynamin 1-like;
FIS1, fission 1 (mitochondrial outer membrane) homolog (S. cerevisiae); LC3, microtubule-associated protein 1 light chain 3; MFN1, mitofusin 1; MFN2, mitofusin 2; NAO, 10-N-nonyl-acridine orange; OPA1, optic atrophy 1 (autosomal dominant);
PARK2, parkinson protein 2, E3 ubiquitin protein ligase (parkin); PINK1, PTEN-induced putative kinase 1; PPARGC1A, peroxisome proliferator-activated receptor gamma, coactivator 1 alpha; SQSTM1/p62, sequestosome 1;
TFAM, transcription factor A, mitochondrial
how cadmium (cd) induces mitochondrial loss in the context of its hepatotoxic effects remains enigmatic. The purpose of the study was to investigate whether mitophagy contributes to mitochondrial loss in cadmium-induced hepatotoxicity and to determine the potential mechanism. in normal human liver L02 cells, we observed that cd treatment led to a significant increase in Lc3-ii formation, the number of GFP-Lc3 puncta and lysosomal colocalization with mitochondria. These results were associated with mitochondrial loss and bioenergetic deficit. additionally, the abrogation of excessive mitophagy by ATG5 siRNa treatment efficiently suppressed the mitochondrial loss and cytotoxicity of cd. Before overactivating mitophagy, cd induced excessive mitochondrial fragmentation as a result of increasing dynamin 1-like (DNM1L) expression and enhancing the DNM1L mitochondrial translocation. Moreover, reversing the excessive mitochondrial fragmentation via the administration of DNM1L siRNa significantly inhibited the observed overactivation of mitophagy in cd-induced hepatotoxicity. Notably, the selective DNM1L inhibitor Mdivi-1 blocked abnormal mitophagy and subsequently ameliorated cd-induced hepatotoxicity in vivo. Together, our data indicated that cd induces mitochondrial loss via the overactivation of mitophagy following DNM1L-dependent mitochondrial fragmentation. The balanced activity of DNM1L and mitophagy signaling may be a potential therapeutic approach to treat cd-induced hepatotoxicity.
©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
2 autophagy Volume 9 issue 11
Autophagy, an evolutionarily conserved pathway, plays a crucial role in degrading a wide variety of cellular components, such as peroxisomes, the endoplasmic reticulum, and mitochondria. The specific process that clears the mitochondria from the cell through autophagy has been termed “mitophagy.”13 The mitochondria are the primary sites for reactive oxygen species formation, and they suffer more oxidative damage than other cellular organelles, thereby easily inducing the autophagic removal of defective mitochondria. In certain circumstances, mitophagy plays a benef icial role in eliminating damaged and unhealthy mitochondria and maintaining the quality of the organelle.14 In contrast, massive and persistent mitophagy creates the overactivation of intracellular mitochondrial degradation machinery. In particular, increased mitochondrial degradation may contribute to a bioenergetic deficit and cell death.15-18
Mitochondria have recently been considered dynamic organelles that continuously fuse and fragment during the
cell cycle. These processes are regulated by mitochondrial fission/fusion proteins, such as dynamin 1-like (DNM1L).19 A delicate balance between fusion and fission events is responsible for the morphology of the mitochondrial network within cells. Mitochondrial dynamic alterations are associated with metabolism, cell development, and cell death.20,21 Recent studies have indicated that the fusion/ fission balance plays an essential role in autophagosome biogenesis.22 Mitophagy targets small, round mitochondria for digestion and elimination, whereas mitochondrial elongation can protect the mitochondria from autophagic degradation and can also sustain the ATP levels.15,16,23
In the present study, we proposed an intriguing mechanism whereby cadmium-induced hepatotoxicity may arise from the loss of mitochondria due to the overactivation of mitophagy. Notably, we confirmed the roles of abnormal mitochondrial dynamics in Cd-induced mitophagy.
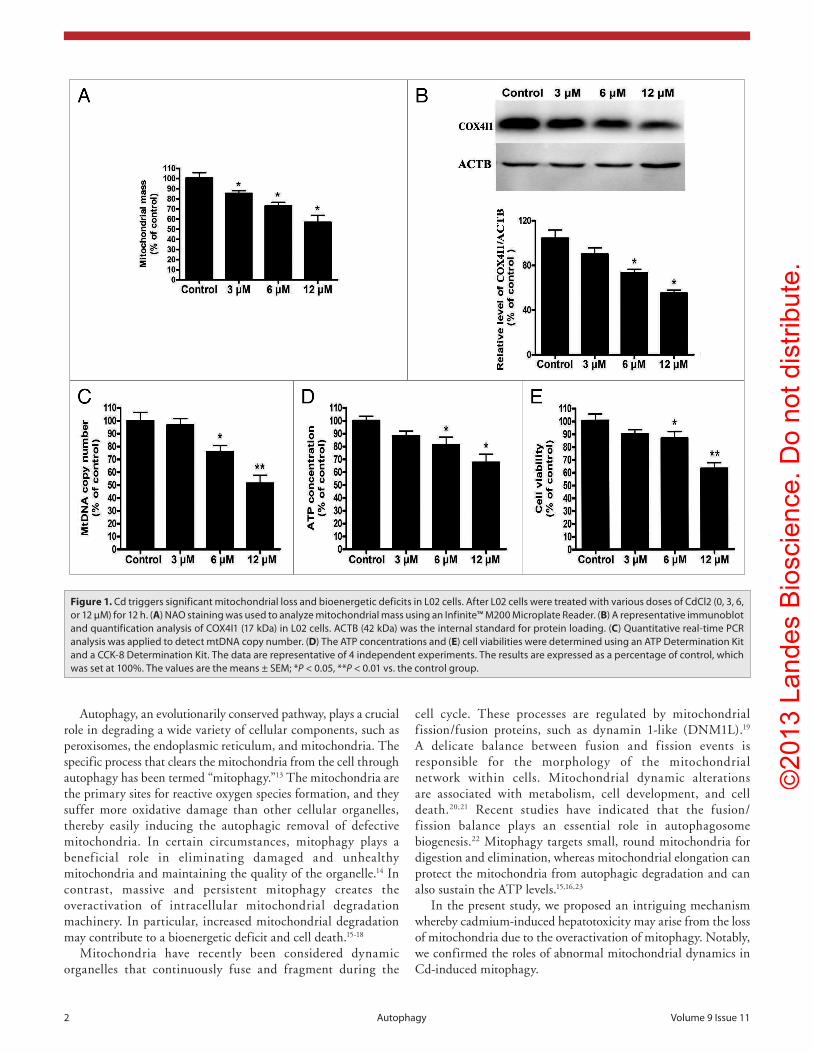
Figure 1. cd triggers significant mitochondrial loss and bioenergetic deficits in L02 cells. after L02 cells were treated with various doses of cdcl2 (0, 3, 6, or 12 μM) for 12 h. (A) NaO staining was used to analyze mitochondrial mass using an infinite™ M200 Microplate Reader. (B) a representative immunoblot and quantification analysis of cOX4i1 (17 kDa) in L02 cells. acTB (42 kDa) was the internal standard for protein loading. (C) Quantitative real-time PcR analysis was applied to detect mtDNa copy number. (D) The aTP concentrations and (E) cell viabilities were determined using an aTP Determination Kit and a ccK-8 Determination Kit. The data are representative of 4 independent experiments. The results are expressed as a percentage of control, which was set at 100%. The values are the means ± seM; *P < 0.05, **P < 0.01 vs. the control group.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 3
Results
Cd-induced mitochondrial loss and bioenergetic deficits in L02 cells
To determine the effects of Cd on the mitochondrial loss in liver cells, we stained L02 cells with the mitochondria-specific chemical NAO. We observed that the treatment of L02 cells with 3 μM, 6 μM, or 12 μM cadmium chloride (CdCl
2) for 12 h
caused 18%, 28%, and 39% reductions in the mitochondrial mass, respectively (Fig. 1A). Furthermore, the levels of the COX4I1 subunit (a protein constituent of the mitochondrial respiratory chain) and mtDNA copy number decreased in a dose-dependent manner (Fig. 1B and C). In line with the observed mitochondrial loss, the ATP level was decreased by 28% and 33% following the 6 μM and 12 μM CdCl
2 treatments, respectively
(Fig. 1D). As expected, cell viability decreased in a similar pattern (Fig. 1E). These results indicate that Cd activates significant degradation of mitochondria while exerting its hepatotoxicity.
Cd targets autolysosomes to mitochondriaTo better understand the mechanisms that underlie the
observed reduction in the number of mitochondria, we first evaluated the increase in mitophagy, which may contribute to mitochondrial loss.24,25 The conversion of the cellular protein LC3-I to LC3-II is a critical hallmark in the autophagy process. As shown in Figure 2A, CdCl
2 induced a dose-dependent increase
in the LC3-II/LC3-I ratio. The EM studies revealed an increased number of autophagic vacuoles in the CdCl
2-treated cells
compared with the nontreated control cells, supporting the hypothesis that Cd induces autophagy in L02 cells (Fig. 2B). We further transfected L02 cells with GFP-LC3 prior to treatment with 12 μM CdCl
2, and 3-dimension (3-D) surface rendering of
Z-stacks exhibited that CdCl2 significantly increased the
colocalization of GFP-LC3 puncta with mitochondria compared with vehicle-treated cells (cells with 10 or more mitochondria-containing APs, 6% in the control vs. 42% in CdCl
2-treated cells)
(Fig. 2C and E; Fig. S1A and S1C). Moreover, we observed the potential colocalization of mitochondria and autolysosomes by confocal microscopy. As shown in Figure 2D and F, Figure S1B and S1D, 3-D surface rendering of the Z-stacks revealed that the green and red fluorescence signals did not greatly overlap in the majority of the control cells. In the culture that was treated with 12 μM CdCl
2, however, the number of cells with visible
colocalization substantially increased (48% of the cells with 10 or more mitochondria-containing ALs). However, the increase in the number of mitochondria-containing APs could ref lect either increased autophagosome formation due to enhanced autophagic activity or an accumulation of mitochondria-containing APs due to impairment in the downstream degradation pathway.26 We used several approaches to investigate whether Cd influences autophagic f lux in L02 cells. We first examined the change of SQSTM1 protein levels. This protein is selectively incorporated into autophagosomes through direct binding to LC3 and is efficiently degraded by autophagy.27 We observed an evident decrease in SQSTM1 protein levels in L02 cells that were treated with CdCl
2
in a dose-dependent manner, confirming intact autophagic flux in the CdCl
2-treated cell lines (Fig. 3A). Second, we used
bafilomycin A1 (Baf A1), which is an inhibitor of the lysosomal
V-ATPase and which causes an accumulation of autophagosomes due to a defect in the fusion between autophagosomes and lysosomes.28 We observed that the presence of 10 nM Baf A1 significantly increased the percentage of cells exhibiting GFP-LC3 positive autophagosomes in L02 cells that were treated with 12 μM CdCl
2 (Fig. 3B). These results suggested that the increase in
autophagosome number was due to increased formation rather than an inhibition of fusion with lysosomes. Another possible reason for the observed decrease in the number of mitochondria is impairment of mitochondrial biogenesis. Mitochondrial biogenesis is regulated by PPARGC1A and TFAM, which are responsible for initiating the synthesis of mtDNA-encoded respiratory chain proteins, such as the complex IV subunit.29,30 However, in the present study, the expression of the transcription factor, PPARGC1A and that of its downstream target TFAM were unaffected by CdCl
2 treatment (Fig. 3C).
The overactivation of mitophagy contributes to Cd-induced mitochondrial loss and cell death
To address whether Cd-induced mitophagy was involved in Cd-induced mitochondrial loss and hepatotoxicity, we suppressed mitophagy using siRNA against ATG5. As expected, the increase in mitochondria-containing APs was significantly attenuated by ATG5 siRNA following exposure to 12 μM CdCl
2 for 12 h
(Fig. 4A and B). In particular, ATG5 siRNA treatment efficiently prevented the CdCl
2-treated cells against a loss of mitochondrial
mass and inhibited the decrease in both the levels of the mitochondrial protein COX4I1 and mtDNA copy number (Fig. 4C–E). These effects were followed by partial improvement in both the ATP level and cell survival (Fig. 4F and G). Consistent with the above result, limiting the abnormal mitophagic pathway may restore the mitochondrial numbers and protect against Cd-induced cell death.
Cd-induced mitochondrial fragmentation via enhanced DNM1L expression and translocation into mitochondria in L02 cells
As the mitochondrial fusion/fission balance has a close relationship with mitophagy,31 we next examined the effects of Cd on mitochondrial dynamics. The mitochondria displayed large and tubular morphology in the control L02 cells. However, CdCl
2
induced a significant dose-dependent increase in the number of fragmented mitochondria with ring-shaped structures as well as an increase in the number of small-sized mitochondria in the EM images (Fig. 5A and C; Fig. 2B). Notably, time-lapse microscopy revealed that this change in mitochondrial morphology occurred early as 3 h following treatment with Cd (Fig. 5B and D). Various proteins responsible for fusion/fission balance include OPA1, MFN1, MFN2, DNM1L, and FIS1.19 In our study, accompanied with the changes of mitochondria morphology, only DNM1L protein levels were significantly increased in the CdCl
2-treated
L02 cells, and no significant changes was detected in the levels of the other mitochondrial fusion/fission proteins (Fig. 6A). Furthermore, DNM1L was observed to translocate to the mitochondria following exposure to 12 μM CdCl
2 for 12 h. CdCl
2
induced an increase in the number of cells with a punctuate distribution of DNM1L, and western blot analysis revealed an

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
4 autophagy Volume 9 issue 11
Figure 2A and B. cd triggers mitophagy in L02 cells. (A) a representative immunoblot and quantification analysis of Lc3 protein levels (17 kDa) in L02 cells. acTB (42 kDa) was the internal standard for protein loading. (B) electron microscopy revealed an increased number of aVs.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 5
Figure 2C–F. cd triggers mitophagy in L02 cells. (C) To visualize the mitochondria-containing aP formations, the colocalization of GFP-Lc3 with TOMM20 determined in L02 cells. Red: TOMM20, green: GFP-Lc3, orange-yellow: Merge. The orange-yellow puncta were counted as mitochondria-containing aPs. (D) To visualize the colocalization between mitochondria and autolysosomes, the colocalization of LysoTracker Red with MitoTracker Green in L02 cells was analyzed. Red: LysoTracker Red, green: MitoTracker Green, orange-yellow: Merge. The orange-yellow puncta were counted as mitochondria-containing aLs. (E) Quantification analysis of mitochondria-containing aPs. (F) Quantification analysis of mitochondria-containing aLs. a minimum of 50 cells were analyzed for each experiment. The results are expressed as a percentage of control, which was set at 100%. The values are the means ± seM; *P < 0.05, **P < 0.01 vs. the control group. arrows indicate the colocalization dots.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
6 autophagy Volume 9 issue 11
increase in DNM1L levels in the mitochondrial sub-fractions (Fig. 6B and C). The formation of ring-shape structures and the increase in DNM1L expression suggested that Cd caused abnormal mitochondrial dynamics in L02 cells.
DNM1L-dependent mitochondrial fragmentation was critical for Cd-induced mitophagy and mitochondrial loss in L02 cells
To further explore the relationship between abnormal mitochondrial dynamics and the overactivation of mitophagy in Cd-induced hepatotoxicity, time-course studies were performed to assay the expression of LC3-II/ LC3-I and DNM1L, respectively. As a result, the LC3-II/LC3-I ratio was significantly increased only over a 6 h period in L02 cells, whereas DNM1L levels were increased as early as 3 h following exposure (Fig. 7A and B), These results suggest that Cd-induced mitochondrial
fragmentation occurs much earlier than Cd-induced mitophagy. To confirm this result, we transfected L02 cells with DNM1L siRNA plasmids (Fig. 8A). Following the restoration of mitochondrial dynamics, the LC3-II/LC3-I ratio significantly decreased by 1.5 fold in DNM1L-deficient cells compared with the control cells that were treated with 12 μM CdCl
2 for 12 h
(Fig. 8D). Consistent with the LC3-II/LC3-I immunoblot results, a significant decrease in the formation of mitochondria-containing APs in the DNM1L-deficient cells was observed after 12 h of CdCl
2 treatment as well as the results of cell cultures were
pretreated for 30 min with Mdivi-1 (10 µM) before being challenged with 12 μM Cd (Fig. 8E; Fig.S3). Moreover, DNM1L knockdown substantially increased the mitochondrial mass (Fig. 8F). However, ATG5 siRNA treatment neither prevented the CdCl
2-induced upregulation of DNM1L nor abrogated the
Figure 3. cd does not inhibit autophagic flux in L02 cells. (A) a representative immunoblot and quantification analysis of the sQsTM1 (75 kDa) protein levels in L02 cells. acTB (42 kDa) was the internal standard for protein loading. (B) The L02 cells were transfected with a GFP-Lc3 plasmid and exposed to 12 μM cdcl2 and 10 nM bafilomycin a1 alone or together for 12 h. The formation of GFP-Lc3 puncta was examined using confocal microscopy and was quantified. (C) The western blot analyses for the expression of PPaRGc1a and TFaM protein. acTB (42 kDa) was the internal standard for protein loading. The data are representative of 4 independent experiments. The results are expressed as a percentage of control, which was set at 100%. The values are the means ± seM; *P < 0.05 vs. the control group; #P < 0.05 vs. the cdcl2 group.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 7
Figure 4A and B. ATG5 siRNa treatment reverses mitochondrial loss by inhibiting mitophagy. (A) a representative immunoblot of aTG5 protein levels (55 kDa) in L02 cells following ATG5 silencing using a commercial vector. acTB (42 kDa) was the internal standard for protein loading. (B) The colocalization of GFP-Lc3 with TOMM20 was determined in L02 cells to visualize the mitochondria-containing aP formations. Red: TOMM20, green: GFP-Lc3, orange-yellow: Merge. The orange-yellow puncta were counted as mitochondria-containing aPs. a minimum of 50 cells were analyzed for each experiment.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
8 autophagy Volume 9 issue 11
mitochondrial morphological alterations that were observed with confocal microscopy (Fig. 4B; Fig. 8C). These results suggest that Cd-induced mitochondrial fragmentation requires the core mitochondrial fission machinery and that mitochondrial fission is required for mitophagy during Cd-induced hepatotoxicity.
Mdivi-1 suppresses mitophagy and minimizes Cd induced-hepatotoxicity in C57BL/6 mice
To determine the role of DNM1lL-dependent mitochondrial fragmentation in vivo, we examined the effects of Mdivi-1 in a
mouse model. Mdivi-1 is an effective inhibitor of mitochondrial division that functions by selectively inhibiting DNM1L.32 Using EM, we confirmed that Mdivi-1 partially suppressed the Cd-induced mitochondrial fragmentation in hepatic cells from 49% to 23% (Fig. 9A). Western blot analysis revealed a decrease in DNM1L levels in the mitochondrial subfractions of liver tissues in Mdivi-1-treated mice (Fig. 9B). The expression of mitophagy markers, such as LC3 and the mitochondrial marker COX4I1, were normalized in the Mdivi-1-treated mice compared with the
Figure 4C–G. ATG5 siRNa treatment reverses mitochondrial loss by inhibiting mitophagy. (C) a representative immunoblot of cOX4i1 (17 kDa) pro-tein levels in L02 cells silenced using a commercial vector. acTB (42 kDa) was the internal standard for protein loading. (E) Quantitative real-time PcR analysis was applied to detect mtDNa copy number. (D) The mitochondrial mass, (F) aTP concentrations, and (G) cell viability were analyzed using an infinite™ M200 Microplate Reader. The data are representative of 4 independent experiments. The results were expressed as a percentage of control, which was set at 100%. The values are the means ± seM; *P < 0.05 vs. the control group; #P < 0.05, ##P < 0.01versus the cdcl2 group. arrows indicate the colocalization dots.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 9
Figure 5. cd exposure disrupts mitochondrial morphology in L02 cells. The specific fluores-cence probe MitoTracker Red cMXRos (red) was used to detect alterations in mitochondrial morphology. (A and C) after the L02 cells were treated with different doses of cdcl2, repre-sentative changes in the mito-chondrial morphology were detected using confocal micros-copy. (B and D) Representative individual mitochondria were tracked using time-lapse micros-copy upon treatment of the L02 cells with 12 μM cdcl2. The data are representative of 3 indepen-dent experiments. The results were expressed as a percent-age of control, which was set at 100%. The values are the means ± seM; *P < 0.05; **P < 0.01 vs. the control group.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
10 autophagy Volume 9 issue 11
Figure 6. changes in the levels of mitochondrial fusion/fission proteins and an increase in the mitochondrial localization of DNM1L following cd treat-ment. (A) a representative immunoblot and quantification analysis of DNM1L (84 kDa), Fis1 (17 kDa), MFN1 (86 kDa), MFN2 (86 kDa) and OPa1 (112 kDa) protein levels in L02 cells. acTB (42 kDa) was the internal standard for protein loading. The cells were treated with different doses of cdcl2 for 12 h. (B) confocal scanning microscope images indicated increased mitochondrial localization of DNM1L following treatment of the L02 cells with cdcl2. Red: MitoTracker Red cMXRos, green: DNM1L. (C) Representative immunoblots and quantification analysis of the DNM1L protein levels (84 kDa) that were observed in the mitochondrial fraction. cOX4i1 (17 kDa) was the internal standard for the mitochondrial protein loading. The data are representative of 4 independent experiments. The results were expressed as a percentage of control, which was set at 100%. The values are the means ± seM; *P < 0.05; **P < 0.01 vs. the control group.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 11
CdCl2-treated controls (Fig. 9C and D). Moreover, the levels of
mitochondrial LC3-II rapidly decreased, and the levels of mitochondrial SQSTM1 significantly increased in the Mdivi-1-treated mice (Fig. 9E and F). Furthermore, ATP levels were evaluated to determine the effect of Mdivi-1 on CdCl
2-induced
liver dysfunction. We observed that inhibiting mitochondrial fragmentation with Mdivi-1 aided in the amelioration of the CdCl
2-induced bioenergetic deficit (Fig. 9G). These results
suggest that inhibiting DNM1L-dependent mitochondrial fragmentation with Mdivi-1 blocks abnormal mitophagy, restores the number of mitochondria and aids in the amelioration of CdCl
2-induced hepatotoxicity in vivo.
Discussion
Mitochondrial loss is a major outcome of numerous human mitochondrial diseases, such as diabetes, cardiovascular diseases, and neurodegenerative disorders.33-35 Mitochondrial loss is also tightly linked to Cd-related diseases,9-12 however, the precise mechanism for these effects are unclear. In this study, we provided evidence that Cd induced significant mitochondrial loss through the overactivation of mitophagy during hepatotoxicity. The inhibition of excessive mitophagy may restore mitochondrial mass, compensate for the reduced function of the respiratory chain and maintain overall ATP production. Notably, we demonstrated that Cd induced mitophagy through irreversible mitochondrial fragmentation in a DNM1L-dependent manner. The silencing of DNM1L expression eff iciently prevented mitochondrial fragmentation, thereby attenuating the increased mitophagy that occurs in Cd-induced mitochondrial loss in vitro. Lastly, pharmacological DNM1L inhibition with Mdivi-1 attenuated the observed abnormal mitophagy and protected against Cd-induced hepatotoxicity in vivo. To the best of our knowledge, this study is the first to investigate the mechanism of Cd hepatotoxicity with a focus on (1) the roles of mitophagy in Cd-induced mitochondrial loss and (2) the relationship between abnormal mitochondrial dynamics and the impairment of mitophagy both in vitro and vivo.
Mitophagy is a mechanism of mitochondrial degradation that occurs under various stress conditions and has been widely observed in multiple mitochondrial dysfunction-induced diseases, such as 6-OHDA and MPP+ toxicity.36-38 Evidence indicates that mitophagy governs the mitochondrial number and that mitochondrial loss is the consequence of autophagy-mediated degradation.39 Here, we initially demonstrated the induction of mitophagy in the Cd-exposed L02 cells. Cd-induced mitophagy was directly observed by examination of the formation of mitochondria-containing APs and mitochondria-containing ALs.40 Moreover, Cd-induced increases in the number of GFP-LC3 puncta resulted from an increase in autophagic flux rather than an inhibition of autophagosome degradation. Notably, a critical link between the induction of mitophagy and mitochondrial loss is supported by the ATG5 interference experiments. The suppression of mitophagy protected the mitochondria from removal and reversed the mitochondrial loss. This finding provides evidence that mitophagy is a cause rather than a consequence of mitochondrial loss.
Currently, the role of mitophagy in cell death remains controversial.23,41,42 It is postulated that mitophagy emerges as a quality control mechanism, which is responsible for the elimination of damaged mitochondria. In hepatocytes, the inhibition of autophagy sensitizes the cells to apoptosis presumably due to the cell’s failure to remove permeabilized mitochondria.43
Figure 7. Time-course analysis for the expression of Lc3 and DNM1L in cd-treated L02 cells. (A) a representative immunoblot and quantification analysis of the Lc3 protein levels (17 kDa) in L02 cells. (B) a representa-tive immunoblot and quantification analysis of DNM1L protein levels (84 kDa). acTB (42 kDa) was the internal standard for protein loading. The cells were treated with 12 µM cdcl2 for various periods of time (0, 3, 6, 12, or 24 h). The data are representative of 4 independent experiments. The results are expressed as a percentage of control, which was set at 100%. The values are means ± seM; *P < 0.05 vs. the control group.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
12 autophagy Volume 9 issue 11
Figure 8A–C. DNM1L siRNa blocks mitochondrial fragmentation and mitophagy in cd-treated L02 cells. (A) a representative immunoblot of the DNM1L protein levels (84 kDa) in L02 cells in which DNM1L was silenced using a commercial vector. acTB (42 kDa) was the internal standard for protein loading. (B) after the L02 cells were treated with 12 μM cdcl2 for 12 h, MitoTracker Red cMXRos (red) was used to detect the effects of the DNM1L silencing vehicle (green) on mitochondrial fragmentation. (C) a representative immunoblot of the DNM1L protein levels (84 kDa) in L02 cells in which ATG5 was silenced. acTB (42 kDa) was the internal standard for protein loading.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 13

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
14 autophagy Volume 9 issue 11
However, mitophagy can have fatal consequences.23,44-46 If too many mitochondria are removed by increased mitophagy, cell viability cannot be maintained. Mitophagy has also been observed as a form of cell death regardless of whether caspases are activated. Furthermore, partially blocking mitochondrial autophagic degradation was observed to prevent decreased ATP levels and reversed cell death during Cd treatment. Consistent with our results, the suppression of mitophagy partially restored the ATP pool and protected against cell death during SNCA/α-synuclein-induced neurotoxicity.16 This result suggests that a significant degree of mitophagy is specifically caused by Cd treatment and may be an important event that leads to the decrease of mitochondrial mass, subsequent ATP depletion and cell death during Cd treatment. These data also provide new evidence as to the mechanism by which Cd exposure inhibits the respiratory chain reaction, reduces the ATP concentration and induces autophagic cell death.47,48 Indeed, recent data showed that mitophagic cell death occurs due to extensive mitochondrial damage.49 Cd may directly or indirectly damage mitochondria, thereby inducing the autophagic removal of defective mitochondria. Here, we also cannot exclude that mitophagy activation is a secondary response to Cd-induced mitochondrial damage. In turn, reduced mitochondrial function might further exacerbate the production of mitophagy, which could induce further mitochondrial loss. This vicious cycle might proceed until cell death occurred. That may be why mitophagy inhibition is protective in an initial phase, whereas in a later stage (48 h) the accumulation of damaged mitochondria induced by cadmium would promote in any case cell death (Fig. S4).
Mitochondria are dynamic remodeling organelles that continuously undergo fusion and fission.50 Mitochondrial morphological dynamics are linked to the regulation of many specific cell functions, such as apoptosis regulation,51-53 Ca2+-transfer45 and mitochondrial quality control.46 Very recently, mitochondrial morphological changes were revealed to participate in autophagosome biogenesis, but mitochondrial morphology is not modified during autophagy.54,55 Here, time-course analyses and DNM1L silencing indicated that Cd-induced mitochondrial fragmentation occurred earlier than Cd-induced mitophagy, and could be efficiently prevented by DNM1L silencing. However, Cd treatment still led to increased mitochondrial fragmentation in ATG5-deficient cells. Therefore, mitochondrial fragmentation precedes Cd-induced mitophagy, but autophagosomal machinery
is not a prerequisite for Cd-induced abnormal mitochondrial dynamics. Consistent with this observation, excessive mitochondrial fission caused by overexpression of Fis1 or Dnm1l reduces the mitochondrial number by activating mitochondrial autophagy.56 By contrast, the inhibition of mitochondrial fission also impairs mitophagy, suggesting an essential role for the shape of mitochondria in this process.22,23 Taken together, these data suggest that an imbalance of mitochondrial dynamics is intimately involved in Cd-induced mitophagy in L02 cells.
However, it remains unclear why mitochondrial fragmentation precedes mitophagy during cadmium treatment. Mitochondria may play an important role in providing membrane for the formation of the autophagosome. An individual mitochondrial averages 5 μm in length, whereas the autophagosomes have a diameter of approximately 1 μm. Therefore, only fragmented, short mitochondria can fit inside the autophagosomes.57,58 Additionally, only shorter, but not longer, mitochondria may have an exposed “eat me” signal that makes them susceptible to the autophagosome. Furthermore, mitochondrial elongation can cause cells to maximize the efficiency of energy conversion and ATP production to be excluded from autophagic degradation.55,59
Mitochondrial fragmentation is a highly regulated process that is mediated by a defined set of protein factors. DNM1L is a large GTPase of the dynamin family and is a key player in mitochondrial fragmentation activation.60,61 Here, we observed that Cd-induced mitochondrial fragmentation results in an increase in DNM1L levels in a dose-dependent manner. Moreover, most of the DNM1L in treated cells is recruited to the mitochondria. Notably, mitochondrial fission and specifically fragmentation, is required for the progression of mitophagy, and DNM1L assembles into rings around the mitochondria and constricts their membranes, thereby breaking down the organelles.23,62-64 Importantly, we further demonstrated that the pharmacological blockade of mitochondrial fission with Mdivi-1 can attenuate the observed abnormal mitophagy in vivo. Mdivi-1, a known potent DNM1L inhibitor, has demonstrated great therapeutic promise in a wide array of disease models in vivo.65,66 Our findings suggest (1) that mitochondrial fragmentation is a novel mechanism that contributes to overactivation of mitophagy during Cd-induced hepatotoxicity, and (2) that the manipulation of DNM1L can decrease Cd-induced hepatotoxicity in an animal model. PINK1 acts through a signal transduction cascade to promote PARK2 to ubiquitinate particular cytoplasmic targets of the mitochondrial
Figure 8D–F (See previous page). DNML1 siRNa blocks mitochondrial fragmentation and mitophagy in cd-treated L02 cells. (D) Representative immu-noblot and quantification analysis of Lc3 (17 kDa) levels in DNM1L-RNai-treated L02 cells following treatment with 12 μM cdcl2 for 12 h. acTB (42 kDa) was the internal standard for protein loading. (E) To examine the effect of DNM1L reduction on cdcl2-induced mitophagy, L02 cells were cotransfected with an empty/negative control vehicle. Two days following transfection, the cells were treated with 12 μM cdcl2 for 12 h, fixed, and stained with TOMM20 and an anti-Lc3 antibody. The number of mitochondria-containing aPs was quantified. a minimum of 50 cells were analyzed for each experi-ment. Red: TOMM20, green: DNM1L, white: Lc3. (F) Quantification of mitochondrial mass in DNM1L RNai-treated L02 cells following treatment with 12 μM cdcl2 for 12 h. The results were obtained from 4 independent experiments. The values are the means ± seM; *P < 0.05; **P < 0.01 vs. the control group; #P < 0.05; ##P < 0.01 vs. the cdcl2 group. arrows indicate the colocalization dots.
Figure 9A–C (See opposite page). Mdivi-1 suppresses cd-induced mitophagy in vivo. (A) 2D eM analysis of mitochondrial fragmentation in liver tis-sues. eM micrographs and quantification of elongated (> 2 μm) mitochondria in a control and cdcl2-injured hepatic cell. a minimum of 50 cells from 8 animals were evaluated. (B) Mdivi-1 inhibits the translocation of DNM1L following cd treatment in vivo. Representative immunoblots and quantification analyses of DNM1L protein levels (86 kDa) in the mitochondrial fraction of liver tissues. cOX4i1 (17 kDa) was the internal standard for protein loading. Representative immunoblots and quantification analyses of Lc3 (17 kDa) (C) and cOX4i1 (17 kDa) protein levels in liver tissues.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 15

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
16 autophagy Volume 9 issue 11

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 17
morphogenesis machinery.67,68 DNM1L is one possible target of the PINK1–PARK2 pathway. DNM1L ubiquitination by PARK2 could promote DNM1L to assemble with mitochondria to activate fission. The relationship between DNM1L and the PINK1–PARK2 pathway in Cd-induced hepatotoxicity would be clarified in further studies.
Taken together, these results demonstrated that overactivated mitophagy may be the primary contributing factor underlying mitochondrial loss in the Cd exposure model, resulting in cellular energy depletion and cell death. In particular, our data suggest that DNM1L-dependent mitochondrial fragmentation plays a critical role in mediating these effects. The present study provides novel findings regarding the balanced activity of DNM1L and mitophagy signaling by Cd. Furthermore, these results may contribute to a better understanding of Cd-mediated hepatotoxicity.
Materials and Methods
Cell culture and treatmentThe human normal liver cell line, L02, was purchased from
the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China). The L02 cells were cultured in RPMI 1640 medium (Invitrogen, 11875) that was supplemented with 5% heat-inactivated FCS (HyClone, SV30087.02) and 1% (v/v) penicillin/streptomycin (Sigma, P4333) in a 5% CO
2 humidified atmosphere
at 37 °C. At 80% confluence, the L02 cells were treated with CdCl
2 (Sigma, 439800) at different concentrations (0, 3, 6,
12 μM) for 12 h or with 12 μM CdCl2 for various periods (0, 3,
6, 12, 24 h). The CdCl2 was dissolved in distilled deionized water
to produce a 12 mM stock solution, which was then used for the serial dilutions the cell culture medium prior to application.
Mitochondrial mass determinationThe mitochondrial mass was measured using 10-N-nonyl-
acridine orange (NAO) (Invitrogen, A1372) according to the manufacturer’s instructions. The L02 cells were seeded at a final concentration of 0.5 × 104 cells/well in a 96-well tissue culture plate. The cells were then treated with 5 μM NAO for 30 min. After the cells were washed twice, the emitted fluorescence was measured at 530 nm following excitation at 485 nm using in an Infinite® 200 Microplate Reader (Tecan, M200). The experiment was repeated 4 times, and the cellular fluorescence intensity was expressed as the fold change relative to the level that was observed in the control cells.
Real-time PCR analysis to detect mtDNA copy numbermtDNA copy number was analyzed using the iQ5 Real-Time
PCR Detection System (Bio-Rad) with the SYBR Green I detection method, as previously described.69 The mtDNA probe was designed to detect MT-CO2 (mitochondrially encoded cytochrome c oxidase II).70 The mtDNA probes were
(5´- C A A AC C TAC G C C A A A ATC C A-3´ ) a nd (5´-GAAATGAATG AGCCTACAGA-3´); and the GAPDH probes were (5´-TGACAACAGC CTCAAGAT-3´) and (5´- GAGTCCTTCC ACGATACC-3´).
ATP content determinationThe ATP levels were measured using an ATP Determination
Kit (Beyotime Company, S0026). Briefly, liver tissue (1:10 wt/vol) or cells (106 cells/ml) were resuspended a reaction buffer that contained 1 mM dithiothreitol, 0.5 mM luciferin, and 12.5 μg/ml luciferase. After gentle mixing of the solutions, the intensity readings for the above mixtures measured using an Infinite® 200 Microplate Reader (Tecan, M200). The ATP concentration in the samples was calculated using an ATP standard curve. The cellular ATP level was expressed as the percentage of the level that was observed in the control groups.
Cell viability assayCell viability was analyzed using a Cell Counting Kit-8 (CCK-
8) (Dojindo Laboratories, CK04) according to the manufacturer’s protocol. Briefly, 0.5 × 104 cells were seeded onto 96-well plates. Following CdCl
2 treatment, 10 μl CCK-8 was added to each well.
The cells were then incubated at 37 °C for 2 h. Following incubation, the OD value at 450 nm was determined using an Infinite® 200 Microplate Reader (Tecan, M200).
Plasmids and transfectionA siRNA that was targeted to human DNM1L was designed
by the Invitrogen Corporation and was cloned into the pcDNA™6.2-GW/EmGFPmiR vector; the negative controls were also provided by the Invitrogen Corporation. The plasmid expressing GFP-LC3 was a generous gift from Dr J Lian.71 The plasmids were transfected into L02 cells using Optimum-Minimum Essential Medium with Lipofectamine 2000 (Invitrogen, 11668027).
RNA interference of ATG5The L02 cells (1 × 105) were transfected with either 100 nmol/L
ATG5-targeting small interfering RNA (Santa Cruz, sc41445) or a control nonspecific siRNA (Santa Cruz, sc37007). 48 h following transfection, the cells were exposed 12 μM CdCl
2 for
12 h. The cells were then collected and processed for immunoblotting of ATG5 and for immunof luorescence microscopy of mitochondria- containing autophagosomes.
Confocal laser microscopyTo measure the changes in the mitochondrial morphology in
L02 cells, the CdCl2-exposed cells were treated with 100 nM
MitoTracker Red CMXRos probe (Invitrogen, M7512) for 30 min at 37 °C according to the manufacturer’s instructions. After 2 washes with PBS, the live cells were visualized under a Leica confocal laser scanning microscope (Leica TCS SP5). Consistent with previous studies,19,51,65,72 the fragmented mitochondria were characterized as shortened, punctate, and occasionally rounded organelles, whereas filamentous mitochondria displayed a
Figure 9D–G (See opposite page). Mdivi-1 suppresses cd-induced mitophagy in vivo. (D) acTB (42 kDa) was the internal standard for protein loading. Mdivi-1 inhibits mitochondrial Lc3-ii expression and increases the levels of mitochondrial sQsTM1. Representative immunoblots and quantification analyses of Lc3 (17 kDa) (E) and sQsTM1 (75 kDa) protein levels (F) in the mitochondrial fraction of liver tissues. cOX4i1 (17 kDa) was the internal standard for protein loading. (G) The aTP concentrations in the mouse liver were determined using an aTP Determination Kit. The aTP levels were expressed as a percentage of control, which was set at 100%. The values are the means ± seM, *P < 0.05 vs. the control group; #P < 0.05 vs. the cdcl2 group.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
18 autophagy Volume 9 issue 11
thread-like tubular structure. The classification of mitochondria was performed as follows: fragmented mitochondria (< 0.5 μm), intermediate mitochondria (0.5 to 5 μm), and tubular mitochondria (> 5 μm). The cells that exhibited 70% fragmented or intermediate mitochondria were scored as cells with fragmented mitochondria. Several random fields of cells (≥ 100 cells per dish) from each sample were evaluated for mitochondrial morphology.
For the analysis of DNM1L mitochondrial translocation, the CdCl
2-exposed cells were fixed with 4% (w/v) paraformaldehyde
in PBS at room temperature for 20 min following incubation with MitoTracker Red. The cells were then permeabilized with 0.5% Triton X-100 in PBS for 15 min at room temperature. The cells were blocked with 10% FBS at 37 °C for 30 min after 3 washes with PBS. Subsequently, the cells were incubated with a mouse monoclonal antibody against human DNM1L (Santa Cruz, sc271583) at a 1:50 dilution overnight at 4 °C. The cells were then incubated with an Alexa Fluor® 488 goat anti-mouse IgG (H+L) antibody (Invitrogen, A11001) at a 1:100 dilution for 1 h at 37 °C. The cells were mounted in mounting medium and visualized under a Leica confocal laser scanning microscope (Leica TCS SP5), as previously described.70
To assess autophagy, the cells that overexpressed GFP-LC3 were examined at 63× magnification, and autophagic cells were classified as those with diffuse GFP-LC3 fluorescence or as cells with > 20 GFP-LC3 puncta/cell.73 For the quantitation of number of mitochondrial autophagosomes in single cells using three-dimensional reconstruction, 0.5μm thick Z-stacks of 20 to 25 slices of at least 50 representative cells per condition in 4 separate experiments. To visua lize mitochondria-containing autophagosome (AP) formations, the cells (1 × 105) were transfected with the GFP-LC3 plasmid for 48 h after being cultured in 6-well plates. Following transfection, the cells were incubated with rabbit anti-TOMM20 antibody (Sigma, HPA011562,) to examine the colocalization of GFP-LC3 positive autophagosomes with the mitochondria, and this colocalization was used as a measure of mitophagy.73 To visualize mitochondria-containing autolysosome (AL) formation, the colocalization of LysoTracker Red (LTR; Beyotime Company, C1046) with mitochondria was assessed from fluorescent confocal images of cells that were treated with 75 nM red-fluorescing LTR and 200 nM green-f luorescing MitoTracker Green-labeled (Beyotime Company, C1048) mitochondria, as previously described.74 The cells were treated with either vehicle control or 12 μM CdCl
2 for
12 h, and then imaged using a Zeiss confocal laser scanning microscope (Carl Zeiss, LSM 510 Meta Confocal Laser Scanning Microscope). The three-dimensional projections were generated using LSM Image Examiner software. Cells with 10 or more mitochondria-containing APs or ALs were determined as “cells positive for mitophagy.”
For the analysis of mitochondria-containing APs in the DNM1L-deficient cells, the L02 cells (1 × 105) were transfected human DNM1L siRNA and vehicle. Forty-eight hours following transfection, the cells were exposed 12 μM CdCl
2 for 12 h. The
CdCl2-exposed cells were fixed with 4% (w/v) paraformaldehyde
in PBS at room temperature for 20 min and then permeabilized with 0.5% Triton X-100 in PBS for 15 min at room temperature.
The cells were blocked with 10% FBS at 37 °C for 30 min after 3 washes with PBS and then incubated with a mouse monoclonal to antibody against TOMM20 (Abcam, ab56783,) at a 1:100 dilution overnight at 4 °C. The cells were then incubated with an Alexa Fluor® 568 Donkey Anti-Mouse IgG (H+L) antibody (Invitrogen, A10037) at a 1:100 dilution for 1 h at 37 °C. The samples were subsequently incubated with a rabbit polyclonal antibody against LC3B (Sigma, L7543) at a 1:100 dilution overnight at 4 °C. Lastly, the cells were then incubated with an Alexa Fluor® 647 goat anti-rabbit IgG (H+L) antibody (Invitrogen, A21244) at a 1:100 dilution for 1 h at 37 °C, and were imaged using a Zeiss confocal laser scanning microscope(Carl Zeiss, LSM 510 Meta Confocal Laser Scanning Microscope).
Time-lapse microscopyThe L02 cells (1 × 105) were seeded onto glass-bottomed dishes.
After the cells were exposed to 12 μM CdCl2, the mitochondria
were labeled with the 100 nM MitoTracker Red CMXRos probe for 30 min at 37 °C according to the manufacturer’s instructions. After the cells were washed twice in PBS, the individual mitochondria were tracked in real time using a Leica confocal laser scanning microscope (Leica TCS SP5). MitoTracker Red was monitored at an excitation wavelength of 579 nm and an emission wavelength of 599 nm. The images were acquired every 10 min, as previously described.53 During the experiments, the cells were placed in a well-equipped live imaging station with a controlled temperature of 37 °C and a humidified atmosphere that consisted of 95% air and 5% CO
2. Several random fields of cells (30 cells
per dish) from each sample were evaluated for mitochondrial morphology.
Animal treatmentThirty-three adult (8 week old, 20 to 21 g), wild-type mice
(WT, C57BL6/J) were purchased from the Experimental Animal Center of the Third Military Medical University (Chongqing, China). The mice were housed at 25 °C under standard conditions with a 12 h light-dark cycle. For the cadmium treatment experiments, randomly selected mice were injected intraperitoneally (i.p.) with 1 mg/kg (n = 8) CdCl
2 for 7 d. The
CdCl2 was dissolved in 0.9% physiological saline. To test the
effects of Mdivi-1(Enzo Life Sciences, BML-CM127), the mice were injected (i.p.) with 50 mg/kg (n = 8) Mdivi-1 1 h prior to CdCl
2 treatment, as previously described.65 The Mdivi-1 was
dissolved in DMSO; therefore, additional randomly selected mice were injected intraperitoneally with 0.9% physiological saline and/or DMSO as controls. The mice were sacrificed by decapitation 24 h following the final injection, and the livers were harvested for the analysis. All of the procedures were approved by the Third Military Medical University for Accreditation of Laboratory Animal Care.
Transmission electron microscopyFor the transmission electron microscopy studies, the L02 cells
were treated with CdCl2 (12 μM) for 12 h, then washed twice
with PBS and fixed with 2.5% glutaraldehyde. The cells were postfixed in 2% osmium tetroxide, embedded and stained with uranyl acetate/lead citrate. The cells were observed using a Hitachi-7500 electron microscope (Japan) to detect autophagic vacuoles, as described elsewhere,75,76 and mitochondrial size in the

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 19
EM images was quantified as previously described.29 The liver sections were washed with PBS and cut into small pieces (1 mm3). Following fixation with precooled (4 °C) 2.5% glutaraldehyde, the livers were postfixed with 2% osmium tetroxide in 0.1 M PBS, rinsed, dehydrated, and embedded. Random thin sections (0.05 μm) were obtained using glass knives. The sections were collected on naked copper-mesh grids and were stained with uranyl acetate and lead citrate. The sections were examined and viewed using a Hitachi-7500 electron microscope, and the examination of mitochondrial fragmentation in liver tissues was performed as in a previous study.65
Western blot analysisThe protein samples that were isolated from the liver tissues and
L02 cells were separated by SDS-PAGE. Following protein transfer to PVDF membranes, the membranes were blocked and incubated with various primary antibodies at 4 °C overnight. The following primary antibodies were used for the western blots: rabbit anti-LC3B (Sigma, L7543), mouse anti-SQSTM1/p62 (Abcam, ab56416), rabbit anti- PPARGC1A (Abcam, ab54481), mouse anti-TFAM (Abcam, ab89818), mouse anti-COX 4I1 (Abcam, ab14744), rabbit anti-optic atrophy 1 (OPA1) (Abcam, ab90857), rabbit anti-mitofusin 1 (MFN1) (Abcam, ab104585), rabbit anti-mitofusin 2 (MFN2) (Abcam, ab50838), mouse anti-DNM1L (BD Biosciences, 611113), mouse anti-DNM1L for human (Santa Cruz, sc-271583), rabbit anti-fission 1 (FIS1) (Abcam, ab71498), rabbit anti-ATG5 (Cell Signaling Technology, 8540S), and mouse anti-ACTB (Santa Cruz, sc-81178). The membrane was then incubated with either an anti-mouse or anti-rabbit IgG antibody (1:1000) (Beyotime Company, A0208 or A0216) for 1 h. The membrane was visualized by enhanced chemiluminescence using Super Signal West Pico blotting (Pierce, 34079) detection reagents and the produced signals were visualized by exposure to Hyper Performance Chemiluminescence film. For the analysis of the
mitochondrial DNM1L, LC3-II, and SQSTM1 protein levels, the mitochondrial extracts were isolated from the L02 cells and mouse liver tissues using the Mitochondria Isolation Kit (Beyotime Company, C3601 or C3606), following the manufacturer’s instructions. The mitochondrial DNM1L, LC3-II, and SQSTM1 protein levels were then determined as described above.
Statistical analysisAll of the experimental data are expressed as the mean ± SEM,
and each experiment was performed a minimum of 3 times. Comparisons of the data between the groups were performed using one-way ANOVAs. The Student t-test was used to evaluate the significant differences between the experimental values of the 2 samples being compared, and P < 0.05 was considered to be statistically significant. All of the counting analyses were performed in a blinded manner, and the colocalization was confirmed by the 3D-image.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the National Basic Research Program of China (National 973 Program; Grant No. 2011CB503700) and the National Natural Science Foundation of China (No. 81172646). We appreciate Dr Weixia Duan and Dr Hongjuan Wu in our department for their hospitable technical assistance in animal sample collection. We also appreciate Dr Rong Xu, Dr Feng Lin in Chongqing University for their professional assistance in time-lapse microscopy analysis.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/25665
References1. Järup L, Berglund M, Elinder CG, Nordberg G,
Vahter M. Health effects of cadmium exposure--a review of the literature and a risk estimate. Scand J Work Environ Health 1998; 24(Suppl 1):1-51; PMID:9569444
2. Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect 2004; 112:1099-103; PMID:15238284; http://dx.doi.org/10.1289/ehp.6751
3. Klaassen CD, Liu J, Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol 1999; 39:267-94; PMID:10331085; http://dx.doi.org/10.1146/annurev.pharmtox.39.1.267
4. Kuester RK, Waalkes MP, Goering PL, Fisher BL, McCuskey RS, Sipes IG. Differential hepatotoxicity induced by cadmium in Fischer 344 and Sprague-Dawley rats. Toxicol Sci 2002; 65:151-9; PMID:11752694; http://dx.doi.org/10.1093/toxsci/65.1.151
5. Cannino G, Ferruggia E, Luparello C, Rinaldi AM. Cadmium and mitochondria. Mitochondrion 2009; 9:377-84; PMID:19706341; http://dx.doi.org/10.1016/j.mito.2009.08.009
6. Lasfer M, Vadrot N, Aoudjehane L, Conti F, Bringuier AF, Feldmann G, Reyl-Desmars F. Cadmium induces mitochondria-dependent apoptosis of normal human hepatocytes. Cell Biol Toxicol 2008; 24:55-62; PMID:17610031; http://dx.doi.org/10.1007/s10565-007-9015-0
7. Gobe G, Crane D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol Lett 2010; 198:49-55; PMID:20417263; http://dx.doi.org/10.1016/j.toxlet.2010.04.013
8. Takaki A, Jimi S, Segawa M, Hisano S, Takebayashi S, Iwasaki H. Long-term cadmium exposure accelerates age-related mitochondrial changes in renal epithelial cells. Toxicology 2004; 203:145-54; PMID:15363590; http://dx.doi.org/10.1016/j.tox.2004.06.005
9. Shih YL, Lin CJ, Hsu SW, Wang SH, Chen WL, Lee MT, Wei YH, Shih CM. Cadmium toxicity toward caspase-independent apoptosis through the mitochondria-calcium pathway in mtDNA-depleted cells. Ann N Y Acad Sci 2005; 1042:497-505; PMID:15965096; http://dx.doi.org/10.1196/annals.1338.043
10. Cannino G, Ferruggia E, Luparello C, Rinaldi AM. Effects of cadmium chloride on some mitochondria-related activity and gene expression of human MDA-MB231 breast tumor cells. J Inorg Biochem 2008; 102:1668-76; PMID:18534682; http://dx.doi.org/10.1016/j.jinorgbio.2008.04.002
11. Cannino G, Ferruggia E, Luparello C, Rinaldi AM. Mitochondrial compartment: a possible target of cadmium effects on breast epithelial cells. Mol Cell Biochem 2009; 328:75-84; PMID:19266167; http://dx.doi.org/10.1007/s11010-009-0076-7
12. Adiele RC, Stevens D, Kamunde C. Differential inhibition of electron transport chain enzyme complexes by cadmium and calcium in isolated rainbow trout (Oncorhynchus mykiss) hepatic mitochondria. Toxicol Sci 2012; 127:110-9; PMID:22345309; http://dx.doi.org/10.1093/toxsci/kfs091
13. Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy 2011; 7:297-300; PMID:21252623; http://dx.doi.org/10.4161/auto.7.3.14502
14. Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy 2007; 3:337-46; PMID:17404498
15. Wang X, Su B, Liu W, He X, Gao Y, Castellani RJ, Perry G, Smith MA, Zhu X. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: implications for Parkinson’s disease. Aging Cell 2011; 10:807-23; PMID:21615675; http://dx.doi.org/10.1111/j.1474-9726.2011.00721.x
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9569444&dopt=Abstract

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
20 autophagy Volume 9 issue 11
16. Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814-24; PMID:21252228; http://dx.doi.org/10.1074/jbc.M110.132514
17. Sansanwal P, Yen B, Gahl WA, Ma YW, Ying LH, Wong LJC, Sarwal MM. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 2010; 21:272-83; PMID:19959713; http://dx.doi.org/10.1681/ASN.2009040383
18. Sampaio-Marques B, Felgueiras C, Silva A, Rodrigues M, Tenreiro S, Franssens V, Reichert AS, Outeiro TF, Winderickx J, Ludovico P. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 2012; 8:1494-509; PMID:22914317; http://dx.doi.org/10.4161/auto.21275
19. Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet 2005; 14 Spec No. 2:R283-9; PMID:16244327; http://dx.doi.org/10.1093/hmg/ddi270
20. Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2002; 2:55-67; PMID:11782314; http://dx.doi.org/10.1016/S1534-5807(01)00116-2
21. Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 2005; 6:657-63; PMID:16025099; http://dx.doi.org/10.1038/nrm1697
22. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433-46; PMID:18200046; http://dx.doi.org/10.1038/sj.emboj.7601963
23. Jiang W, Ogretmen B. Ceramide stress in survival versus lethal autophagy paradox: ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Autophagy 2013; 9:258-9; PMID:23182807; http://dx.doi.org/10.4161/auto.22739
24. Bota DA, Davies KJ. Protein degradation in mitochondria: implications for oxidative stress, aging and disease: a novel etiological classification of mitochondrial proteolytic disorders. Mitochondrion 2001; 1:33-49; PMID:16120267; http://dx.doi.org/10.1016/S1567-7249(01)00005-8
25. Rodriguez-Enriquez S, Kai Y, Maldonado E, Currin RT, Lemasters JJ. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy 2009; 5:1099-106; PMID:19783904; http://dx.doi.org/10.4161/auto.5.8.9825
26. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
27. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24:8055-68; PMID:15340068; http://dx.doi.org/10.1128/MCB.24.18.8055-8068.2004
28. Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33-42; PMID:9639028; http://dx.doi.org/10.1247/csf.23.33
29. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008; 88:611-38; PMID:18391175; http://dx.doi.org/10.1152/physrev.00025.2007
30. Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A 2007; 104:1325-30; PMID:17227870; http://dx.doi.org/10.1073/pnas.0605208103
31. Cheung ZH, Ip NY. The emerging role of autophagy in Parkinson’s disease. Mol Brain 2009; 2:29; PMID:19754977; http://dx.doi.org/10.1186/1756-6606-2-29
32. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008; 14:193-204; PMID:18267088; http://dx.doi.org/10.1016/j.devcel.2007.11.019
33. Silva JP, Köhler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 2000; 26:336-40; PMID:11062475; http://dx.doi.org/10.1038/81649
34. Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A 2002; 99:15066-71; PMID:12417746; http://dx.doi.org/10.1073/pnas.232591499
35. Sörensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci 2001; 21:8082-90; PMID:11588181
36. Galindo MF, Solesio ME, Atienzar-Aroca S, Zamora MJ, Jordán Bueso J. Mitochondrial dynamics and mitophagy in the 6-hydroxydopamine preclinical model of Parkinson’s disease. Parkinsons Dis 2012; 2012:131058; PMID:22966477
37. Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170:75-86; PMID:17200184; http://dx.doi.org/10.2353/ajpath.2007.060524
38. Solesio ME, Saez-Atienzar S, Jordán J, Galindo MF. Characterization of mitophagy in the 6-hydoxydopamine Parkinson’s disease model. Toxicol Sci 2012; 129:411-20; PMID:22821850; http://dx.doi.org/10.1093/toxsci/kfs218
39. Ermak G, Sojitra S, Yin F, Cadenas E, Cuervo AM, Davies KJA. Chronic expression of RCAN1-1L protein induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis in neuronal cells. J Biol Chem 2012; 287:14088-98; PMID:22389495; http://dx.doi.org/10.1074/jbc.M111.305342
40. Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012; 55:222-32; PMID:21932416; http://dx.doi.org/10.1002/hep.24690
41. Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res 2007; 67:6314-24; PMID:17616690; http://dx.doi.org/10.1158/0008-5472.CAN-06-4217
42. Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain 2009; 2:24; PMID:19640278; http://dx.doi.org/10.1186/1756-6606-2-24
43. Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2006; 2:39-46; PMID:16874071
44. Kim EH, Choi KS. A critical role of superoxide anion in selenite-induced mitophagic cell death. Autophagy 2008; 4:76-8; PMID:17952022
45. Park MK, Ashby MC, Erdemli G, Petersen OH, Tepikin AV. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J 2001; 20:1863-74; PMID:11296220; http://dx.doi.org/10.1093/emboj/20.8.1863
46. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 2005; 280:26185-92; PMID:15899901; http://dx.doi.org/10.1074/jbc.M503062200
47. Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 2008; 65:3640-52; PMID:18850067; http://dx.doi.org/10.1007/s00018-008-8383-9
48. Yang LY, Wu KH, Chiu WT, Wang SH, Shih CM. The cadmium-induced death of mesangial cells results in nephrotoxicity. Autophagy 2009; 5:571-2; PMID:19333000; http://dx.doi.org/10.4161/auto.5.4.8311
49. Kim SJ, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog 2013; 9:e1003285; PMID:23555273; http://dx.doi.org/10.1371/journal.ppat.1003285
50. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010; 11:872-84; PMID:21102612; http://dx.doi.org/10.1038/nrm3013
51. Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva YE, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006; 25:3900-11; PMID:16874299; http://dx.doi.org/10.1038/sj.emboj.7601253
52. Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 2008; 105:19318-23; PMID:19050078; http://dx.doi.org/10.1073/pnas.0804871105
53. Gomez-Lazaro M, Bonekamp NA, Galindo MF, Jordán J, Schrader M. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free Radic Biol Med 2008; 44:1960-9; PMID:18395527; http://dx.doi.org/10.1016/j.freeradbiomed.2008.03.009
54. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589-98; PMID:21478857; http://dx.doi.org/10.1038/ncb2220
55. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 2011; 108:10190-5; PMID:21646527; http://dx.doi.org/10.1073/pnas.1107402108
56. Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta 2008; 1777:860-6; PMID:18515060; http://dx.doi.org/10.1016/j.bbabio.2008.05.442
57. Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells 2010; 15:923-33; PMID:20670274; http://dx.doi.org/10.1111/j.1365-2443.2010.01433.x

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com autophagy 21
58. Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta 2013; 1833:205-12; PMID:22406072; http://dx.doi.org/10.1016/j.bbamcr.2012.02.012
59. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589-98; PMID:21478857; http://dx.doi.org/10.1038/ncb2220
60. Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 1999; 4:815-26; PMID:10619028; http://dx.doi.org/10.1016/S1097-2765(00)80391-3
61. Legesse-Miller A, Massol RH, Kirchhausen T. Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol Biol Cell 2003; 14:1953-63; PMID:12802067; http://dx.doi.org/10.1091/mbc.E02-10-0657
62. Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 2005; 170:1021-7; PMID:16186251; http://dx.doi.org/10.1083/jcb.200506078
63. Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 2011; 301:H1924-31; PMID:21890690; http://dx.doi.org/10.1152/ajpheart.00368.2011
64. Yan J, Feng Z, Liu J, Shen W, Wang Y, Wertz K, Weber P, Long J, Liu J. Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: ameliorating effects of (-)-epigallocatechin-3-gallate. J Nutr Biochem 2012; 23:716-24; PMID:21820301; http://dx.doi.org/10.1016/j.jnutbio.2011.03.014
65. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009; 119:1275-85; PMID:19349686; http://dx.doi.org/10.1172/JCI37829
66. Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS One 2012; 7:e32388; PMID:22479323; http://dx.doi.org/10.1371/journal.pone.0032388
67. Yu W, Sun Y, Guo S, Lu B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 2011; 20:3227-40; PMID:21613270; http://dx.doi.org/10.1093/hmg/ddr235
68. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 2008; 105:1638-43; PMID:18230723; http://dx.doi.org/10.1073/pnas.0709336105
69. Xu SC, He MD, Lu YH, Li L, Zhong M, Zhang YW, Wang Y, Yu ZP, Zhou Z. Nickel exposure induces oxidative damage to mitochondrial DNA in Neuro2a cells: the neuroprotective roles of melatonin. J Pineal Res 2011; 51:426-33; PMID:21797922; http://dx.doi.org/10.1111/j.1600-079X.2011.00906.x
70. Xu SC, Chen YB, Lin H, Pi HF, Zhang NX, Zhao CC, Shuai L, Zhong M, Yu ZP, Zhou Z, et al. Damage to mtDNA in liver injury of patients with extrahepatic cholestasis: the protective effects of mitochondrial transcription factor A. Free Radic Biol Med 2012; 52:1543-51; PMID:22306509; http://dx.doi.org/10.1016/j.freeradbiomed.2012.01.007
71. Lian J, Wu X, He F, Karnak D, Tang W, Meng Y, Xiang D, Ji M, Lawrence TS, Xu L. A natural BH3 mimetic induces autophagy in apoptosis-resistant prostate cancer via modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell Death Differ 2011; 18:60-71; PMID:20577262; http://dx.doi.org/10.1038/cdd.2010.74
72. Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lämmermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem 2009; 284:22938-51; PMID:19546216; http://dx.doi.org/10.1074/jbc.M109.035774
73. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094-104; PMID:22505714; http://dx.doi.org/10.1074/jbc.M111.322933
74. Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol 2012; 8:831-8; PMID:22922758; http://dx.doi.org/10.1038/nchembio.1059
75. Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy 2008; 4:770-82; PMID:18594198
76. Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284:13843-55; PMID:19279012; http://dx.doi.org/10.1074/jbc.M808515200




















