
Dynamical angular momentum models for rotational transfer in polyatomic molecules
8
Dynamical angular momentum models for rotational transfer in polyatomic molecules Zeyad T. AlWahabi, Nicholas A. Besley, Anthony J. McCaffery, Mark A. Osborne, and Zaid Rawi School of Molecular Sciences, University of Sussex, Brighton BN19QJ, United Kingdom ~Received 11 October 1994; accepted 21 February 1995! We propose a model for collision-induced rotational transfer ~RT! in polyatomic molecules based on the angular momentum (AM) sphere, a classical representation of the dynamical motion of the rotational AM vector in the molecular frame. The model develops further that proposed by us @AlWahabi et al., J. Chem. Soc., Faraday Trans. 85, 1003 ~1989!# in which RT probabilities are related to the AM gap linking initial and final N k a k c states. The AM sphere representation embodies the full internal motion of the molecule via its effect on the inertial axes and the trajectory of the individual rotational state vectors. In this representation there is no unique AM gap for a particular transition between states of nominally well-defined N k a k c and here we propose and test several models for obtaining the distance in AM space between initial and final trajectories. Models are evaluated from their ability to fit data on NH 2 –H collisions. We find that even the simplest approximations, such as shortest distance in AM space, give good fits to data sets but the best fits are obtained when both AM trajectory and molecular geometry are averaged over. © 1995 American Institute of Physics. I. INTRODUCTION The problems involved in rationalizing collision-induced rotational transfer ~RT! in polyatomic molecules are formi- dable. There is little guidance on what are the underlying principles that control the process, e.g., early studies that appeared to demonstrate that long-range forces were domi- nant seem less relevant in light of more recent experiments. Furthermore, the complex dependence of eigenstate energies on the internal quantum numbers indicates that simple en- ergy gap relationships are unlikely to be helpful. Of particu- lar relevance here is the angular momentum ~AM! model proposed recently by us which shows promise in providing new insight into the physical basis of RT. This was first for- mulated as an AM gap relation using data in atom–triatomic molecule collisions 1 and later developed as an alternative theoretical approach to diatomic RT. 2 Most recently this AM theory, in which the probability of linear to angular momen- tum transfer is calculated explicitly, has been used as the basis of a fitting law 3 with some indications of predictive power. In this contribution we consider in more detail the appli- cation of the AM model to RT in polyatomic molecules. We begin by pointing out that despite the success of the original AM gap model for the case of atom–triatom collisions, 1 there is a flaw in the physical assumptions upon which it is based, amplified further in Sec. II but briefly is this. The model assumes that rotational AM in a polyatomic may be represented by vectors that are fixed in the molecule frame and correspond to specific values of total AM and its projec- tion on inertial a and c axes. It is well-known that k a and k c cannot simultaneously be good quantum numbers in an asymmetric rotor, the classical origin of this being the pre- cessional motion which the AM vectors undergo in the mol- ecule frame. This motion is slow compared, say, to the time taken for a collision but an experiment will sample all re- gions of the AM vector’s trajectory. The dynamical motion of the AM vector is central to the proper description of AM change since, in effect, a collision causes the AM vector to hop from its initial trajectory to some other. The details of initial and final AM vector precessional motion will be im- portant, therefore, in determining the probability of AM change. The experimental determination of state-to-state RT rates in polyatomics can often pose severe problems arising from the presence of more than one inertial axis of rotation. This, together with vibrational modes which may couple strongly with rotations, give rise to complex energy level patterns. For these and other reasons, few rotationally resolved studies of collisional state change have appeared in the literature. Interpretation of collision experiments in polyatomic mol- ecules was strongly influenced by the work of Oka 4 whose double resonance experiments indicated a dominant role for long-range forces. Selection and propensity rules were devel- oped for collision-induced transitions. For interactions be- tween polar species, these gave a satisfactory description of the observed transition probabilities. More recently it has become apparent that long-range interactions alone may be insufficient to describe collisional RT in polyatomics. 5,6 Col- lisions of polar molecules with atoms or small molecules often yields data that is not readily explained by a simple multipolar expansion 7 and it is clear that further state re- solved experiments on polyatomic molecules are needed. RT in polyatomic molecules has particular relevance to the development of our physical picture of collisional quan- tum state change. These mostly have come from studies on diatomics. Particularly important however is the change in relationship between energy and angular momentum on go- ing to polyatomics arising from the fact that the energy of a state is determined by the magnitude and direction in the molecular frame, of the rotational angular momentum vector. 7945 J. Chem. Phys. 102 (20), 22 May 1995 0021-9606/95/102(20)/7945/8/$6.00 © 1995 American Institute of Physics
Transcript of Dynamical angular momentum models for rotational transfer in polyatomic molecules

Dynamical angular momentum models for rotational transfer in polyatomicmolecules
Zeyad T. AlWahabi, Nicholas A. Besley, Anthony J. McCaffery, Mark A. Osborne,and Zaid RawiSchool of Molecular Sciences, University of Sussex, Brighton BN19QJ, United Kingdom
~Received 11 October 1994; accepted 21 February 1995!
We propose a model for collision-induced rotational transfer~RT! in polyatomic molecules based onthe angular momentum (AM) sphere, a classical representation of the dynamical motion of therotational AM vector in the molecular frame. The model develops further that proposed by us@AlWahabi et al., J. Chem. Soc., Faraday Trans.85, 1003 ~1989!# in which RT probabilities arerelated to the AMgap linking initial and finalNkakc
states. The AM sphere representation embodiesthe full internal motion of the molecule via its effect on the inertial axes and the trajectory of theindividual rotational state vectors. In this representation there is no unique AM gap for a particulartransition between states of nominally well-definedNkakc
and here we propose and test severalmodels for obtaining the distance in AM space between initial and final trajectories. Models areevaluated from their ability to fit data on NH2–H collisions. We find that even the simplestapproximations, such as shortest distance in AM space, give good fits to data sets but the best fitsare obtained when both AM trajectoryand molecular geometry are averaged over. ©1995American Institute of Physics.
n
toof-
tesomis,lys.diesre.l-
forvel-e-n ofasbe
sle-.ton-oningo-a
or.
I. INTRODUCTION
The problems involved in rationalizing collision-inducedrotational transfer~RT! in polyatomic molecules are formi-dable. There is little guidance on what are the underlyinprinciples that control the process, e.g., early studies thappeared to demonstrate that long-range forces were donant seem less relevant in light of more recent experimenFurthermore, the complex dependence of eigenstate eneron the internal quantum numbers indicates that simple eergy gap relationships are unlikely to be helpful. Of particular relevance here is the angular momentum~AM ! modelproposed recently by us which shows promise in providinnew insight into the physical basis of RT. This was first fomulated as an AMgap relation using data in atom–triatomicmolecule collisions1 and later developed as an alternativtheoretical approach to diatomic RT.2 Most recently this AMtheory, in which the probability of linear to angular momentum transfer is calculated explicitly, has been used as tbasis of a fitting law3 with some indications of predictivepower.
In this contribution we consider in more detail the applcation of the AM model to RT in polyatomic molecules. Webegin by pointing out that despite the success of the originAM gap model for the case of atom–triatom collisions,1
there is a flaw in the physical assumptions upon which itbased, amplified further in Sec. II but briefly is this. Thmodel assumes that rotational AM in a polyatomic may brepresented by vectors that arefixed in the molecule frameand correspond to specific values of total AM and its projetion on inertiala andc axes. It is well-known thatka andkccannot simultaneously be good quantum numbers inasymmetric rotor, the classical origin of this being the precessional motion which the AM vectors undergo in the moecule frame. This motion is slow compared, say, to the timtaken for a collision but an experiment will sample all re
J. Chem. Phys. 102 (20), 22 May 1995 0021-9606/95/102(20)/
gatmi-ts.giesn--
gr-
e
-he
i-
al
isee
c-
an-l-e-
gions of the AM vector’s trajectory. The dynamical motioof the AM vector is central to the proper description of AMchange since, in effect, a collision causes the AM vectorhop from its initial trajectory to some other. The detailsinitial and final AM vector precessional motion will be important, therefore, in determining the probability of AMchange.
The experimental determination of state-to-state RT rain polyatomics can often pose severe problems arising frthe presence of more than one inertial axis of rotation. Thtogether with vibrational modes which may couple strongwith rotations, give rise to complex energy level patternFor these and other reasons, few rotationally resolved stuof collisional state change have appeared in the literatuInterpretation of collision experiments in polyatomic moecules was strongly influenced by the work of Oka4 whosedouble resonance experiments indicated a dominant rolelong-range forces. Selection and propensity rules were deoped for collision-induced transitions. For interactions btween polar species, these gave a satisfactory descriptiothe observed transition probabilities. More recently it hbecome apparent that long-range interactions alone mayinsufficient to describe collisional RT in polyatomics.5,6 Col-lisions of polar molecules with atoms or small moleculeoften yields data that is not readily explained by a simpmultipolar expansion7 and it is clear that further state resolved experiments on polyatomic molecules are needed
RT in polyatomic molecules has particular relevancethe development of our physical picture of collisional quatum state change. These mostly have come from studiesdiatomics. Particularly important however is the changerelationship between energy and angular momentum oning to polyatomics arising from the fact that the energy ofstate is determined by the magnitude anddirection in themolecular frame, of the rotational angular momentum vect
79457945/8/$6.00 © 1995 American Institute of Physics

7946 AlWahabi et al.: Rotational transfer in polyatomic molecules
FIG. 1. Energy level diagram for the~0,9,0! vibrational manifold of theA 2A1 electronic state of NH2. The energy levels are indexed byNkakc.
lee
e
o
thtste
n
s
m
isin
le.na-tathe
the
e-od-thatumongtorgy
de-o-
As a result it becomes possible to distinguish an angumomentum based dependency in RT from one based onergy change. A recent example of this is the work of Kroand Rettschnick8 who demonstrated that H2–glyoxal rota-tionally inelastic scattering is dominated by AM transferather than energy transfer and there is evidence that engap relations are inadequate for describing RT in polyatommolecules.9
We have reported results from an extensive studystate-to-state RT in the asymmetric rotor NH2.
1 The motiva-tion for this work was to address the questions raised inpreceding paragraph but also the intention was to add tosparse data base of available fully state resolved RT ratesymmetric and asymmetric rotors. Approximately 100 stato-state rates were measured10,11with full resolution of initialand finalNkakc
states, of spin branching and polarizatioNote that in asymmetric rotors with electron spin,N is thetotal rotational angular momentum andka ,kc are the projec-tions ofN on the inertiala andc axes. Spectroscopic stateare indexed in these quantum numbers though onlyN andone of the projections are simultaneously good quantunumbers as noted above. Collision-induced transfer wmeasured both within and across theka stacks within a single
J. Chem. Phys., Vol. 102
arn-s
rrgyic
f
ehein-
.
as
vibrational level and also between vibrational levels. Thdata set, together with the pioneering contribution to RTtriatomic radicals by Dixon and Field,12 represents one of themost extensive currently available on a polyatomic molecu
The experimental data revealed new insights into theture of RT.1 No correlation could be established between dasets on the basis of magnitude of energy change except intrivial sense that transferwithin ka stacks follow energy gaplaws. However, this is not diagnostic of mechanism sinceenergy levels within stacks are related toN in the same wayas are those of a diatomic molecule to rotor AM. It is thtransitionsacrossthe ka stacks that are expected to distinguish between energy- and angular momentum-based mels. For interstack transitions it is possible in some casesan increase in magnitude of rotational angular momentcan result in a decrease in rotational energy due to the strdependency of the energy on the direction of the AM vecin the molecule frame. This is evident from the set of enerlevels for the~0,9,0! vibrational manifold of theA 2A1 elec-tronic state of NH2 shown in Fig. 1. The inability of energygap models to fit this data is spectacular in some data sets10,11
where an inverse dependency on energy gap may betected. When RT rates are plotted against the angular m
, No. 20, 22 May 1995
f
t
.
eth
r
s
theld
of
er-
el
-
-
7947AlWahabi et al.: Rotational transfer in polyatomic molecules
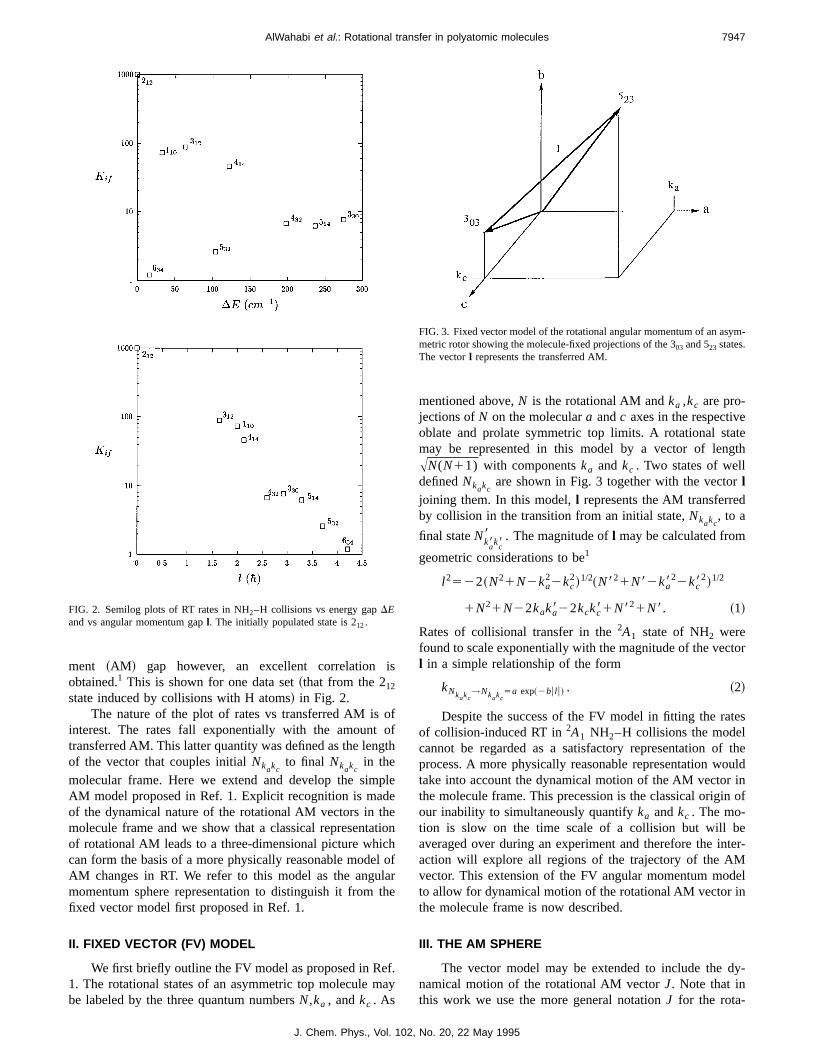
ment ~AM ! gap however, an excellent correlation isobtained.1 This is shown for one data set~that from the 212state induced by collisions with H atoms! in Fig. 2.
The nature of the plot of rates vs transferred AM is ointerest. The rates fall exponentially with the amount otransferred AM. This latter quantity was defined as the lengof the vector that couples initialNkakc
to final Nkakcin the
molecular frame. Here we extend and develop the simpAM model proposed in Ref. 1. Explicit recognition is madeof the dynamical nature of the rotational AM vectors in themolecule frame and we show that a classical representatiof rotational AM leads to a three-dimensional picture whichcan form the basis of a more physically reasonable modelAM changes in RT. We refer to this model as the angulamomentum sphere representation to distinguish it from thfixed vector model first proposed in Ref. 1.
II. FIXED VECTOR (FV) MODEL
We first briefly outline the FV model as proposed in Ref1. The rotational states of an asymmetric top molecule mabe labeled by the three quantum numbersN,ka , andkc . As
FIG. 2. Semilog plots of RT rates in NH2–H collisions vs energy gapDEand vs angular momentum gapl. The initially populated state is 212.
J. Chem. Phys., Vol. 102,
fh
le
on
ofre
y
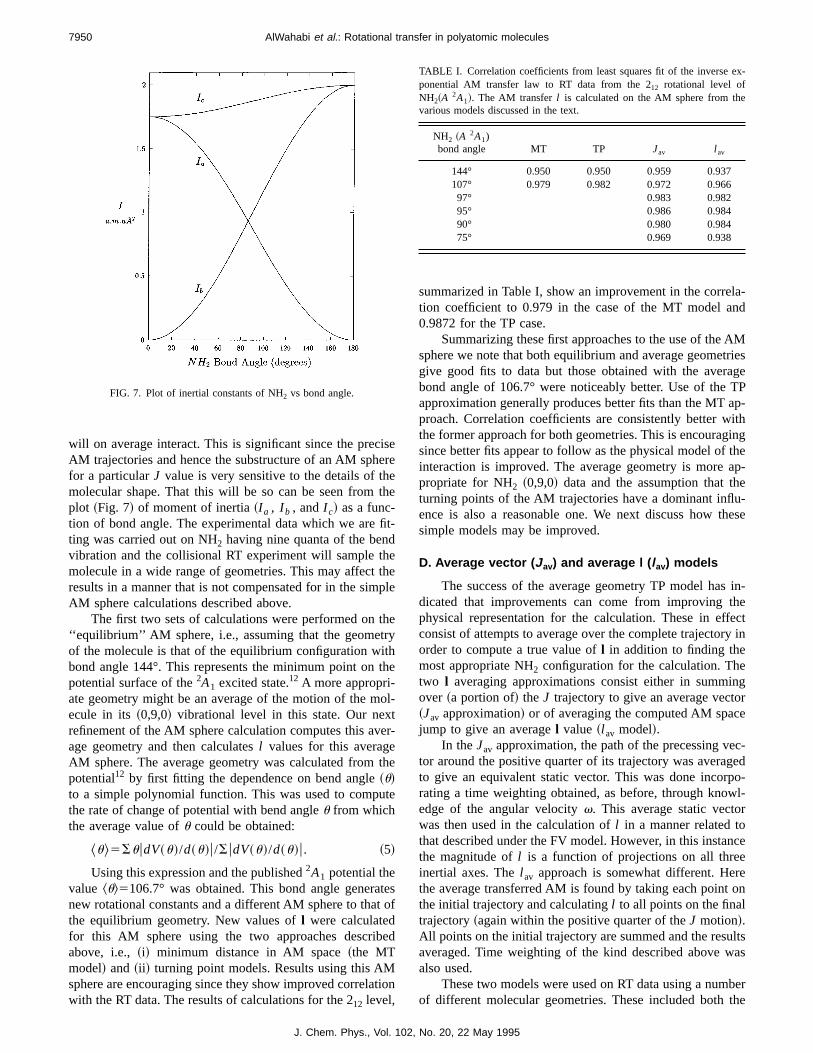
mentioned above,N is the rotational AM andka ,kc are pro-jections ofN on the moleculara andc axes in the respectiveoblate and prolate symmetric top limits. A rotational statmay be represented in this model by a vector of lengAN(N11) with componentska andkc . Two states of welldefinedNkakc
are shown in Fig. 3 together with the vectorljoining them. In this model,l represents the AM transferredby collision in the transition from an initial state,Nkakc
, to a
final stateNka8kc8
8 . The magnitude ofl may be calculated from
geometric considerations to be1
l 2522~N21N2ka22kc
2!1/2~N821N82ka822kc8
2!1/2
1N21N22kaka822kckc81N821N8. ~1!
Rates of collisional transfer in the2A1 state of NH2 werefound to scale exponentially with the magnitude of the vectol in a simple relationship of the form
kNkakc→Nkakc5a exp~2bu l u! . ~2!
Despite the success of the FV model in fitting the rateof collision-induced RT in2A1 NH2–H collisions the modelcannot be regarded as a satisfactory representation ofprocess. A more physically reasonable representation woutake into account the dynamical motion of the AM vector inthe molecule frame. This precession is the classical originour inability to simultaneously quantifyka andkc . The mo-tion is slow on the time scale of a collision but will beaveraged over during an experiment and therefore the intaction will explore all regions of the trajectory of the AMvector. This extension of the FV angular momentum modto allow for dynamical motion of the rotational AM vector inthe molecule frame is now described.
III. THE AM SPHERE
The vector model may be extended to include the dynamical motion of the rotational AM vectorJ. Note that inthis work we use the more general notationJ for the rota-
FIG. 3. Fixed vector model of the rotational angular momentum of an asymmetric rotor showing the molecule-fixed projections of the 303 and 523 states.The vectorl represents the transferred AM.
No. 20, 22 May 1995

in
e
f
ny
t
o
an
in
ofs
d
es.s
l
ge
a
7948 AlWahabi et al.: Rotational transfer in polyatomic molecules
tional AM vector where for the case of NH2, J5 AN(N11). In the rigid symmetric rotor, rotational AMtrajectories display uniform motion in the molecule-fixeframe which become nonuniform in the case of the asymmric rotor. In both casesJ follows a fixed periodic trajectorydetermined by the classical mechanics of the rotatsystem.13,14 For example, in the case of NH2 the motion ofeachNkakc
state within aJ level is derived from the rota-tional Hamiltonian
H5AJa21BJb
21CJc2. ~3!
Diagonalization of the corresponding matrix provides thegenvaluesE(Nkakc
).On expressing the Hamiltonian in terms of the spheric
polar coordinatesJ,u, andf the classical trajectory of theNkakc
state vector is obtained from those values ofu, f
which fulfill the condition
E~u,f!5E~Nkakc!. ~4!
The set of trajectories of a singleJ level form anangularmomentum sphereof radiusAN(N11). These are closelyrelated to the rotational energy trajectories described by Hter and Patterson.15 A description of the classical motion origid rotors has been given by Zare.16
An example of the trajectories that comprise theJ53level of NH2 is shown in Fig. 4. The trajectories are sensitivto bond angle and rotational constants of the triatomic aas we discuss in greater detail below, change dramaticallthese change. The example shown in Fig. 4 is for the2B1ground state of NH2 which is more bent than the excited staand consequently the component trajectories ofJ53 arewell separated. As Fig. 3 shows, the trajectories of individuNkakc
state vectors vary considerably, exhibiting uniform mtion around thea axis in the 330 but becoming markedlynonuniform as thec-axis component increases in say the 303.Motions around each inertial axis are separated by thesepa-ratrix which is shown in the figure. Trajectories close to th
FIG. 4. The AM sphere for theJ53 rotational levels of NH2 showing theclassical trajectories of the individualNkakc
state vectors in the moleculeframe. Note that for this representation, NH2 is in its ground state equilib-rium configuration since this separates individual trajectories more clethan do the excited state geometries.
J. Chem. Phys., Vol. 102
det-
g
i-
al
ar-
ed,as
e
al-
e
separatrix are less stable than those that remain close toaxis.15,16
When a series ofJ levels is considered, a set of concen-tric AM spheres is formed as shown in Fig. 5 which illus-trates the trajectories for the levels ofJ51, 3, and 5. Thereare several aspects of the AM sphere to be consideredrelation to rotational transfer which in this model consists ofa collision-induced hop from one trajectory to another, withthe appropriate change in length and direction ofJ causedby the collision. This change may take place within aJsphere and thus represent a jump between the trajectoriesFig. 5 or may involve transitions between spheres. What iapparent from the topography of theJ sphere is that unlikethe FV model, there is no unique value ofl for a givenNkakc
→Nka8kc8
8 . The length of the vectorl coupling two tra-
jectories will depend on the initial and final positions on thetrajectory that are coupled. Note that of course theenergychange in such a transition will be independent of start anend positions on the AM trajectories.
There are some other noteworthy points regarding thAM spheres and transitions between state vector trajectorieIntrastack transfer in which only one component change~e.g, Dka50,DkcÞ0) will have a direct relationship withenergy change whileinterstack transitions in which bothcomponents change will have a dependency onu, f valuesof initial and finalJ. Theminimumvalue of l between anytwo states will lie in thea,c plane. This can clearly be seenin Fig. 3 for transitions within oneJ state but also appliesbetweenJ levels. It is important to note that the precessionaangular velocity of theJ vectors is generally not uniform andhas a markedu, f dependence for certain trajectories, thosedirected along thec axis for example. This may be importantin collisional transfer out of a specified level since the turn-ing points of the trajectory may have a dominant influence inRT processes.
IV. CALCULATIONS BASED ON THE AM SPHERES
The use of the AM sphere in interpreting collision ex-periments is new and clearly there will be no universallyagreed method of use. As we have seen from the precedinsection and as is apparent from the figures, there is no uniqu
rly
FIG. 5. Angular momentum~half! spheres for theJ51, 3, and 5 rotationallevels of NH2.
, No. 20, 22 May 1995

e-s
t
tlo
ne
x
ep
i
e
t
k
h
a
t
h
s
a
-
h
nel-al-cal-sf
n
sicasine-
2efn-
-
y to
n
7949AlWahabi et al.: Rotational transfer in polyatomic molecules
value of l, the length in angular momentum space betweinitial and final precessingJ vectors. We have therefore investigated a number of approaches based on simple phyprinciples in order to determine the ways in which to best uthe AM sphere representation. The criterion used is best fiexperimental RT rates to magnitude of transferred AM~l!calculated by making certain assumptions regardingdominant processes in the collision-induced transfer. Pare on a semilogarithmic scale consistent with the notionan inverse exponential dependence on magnitude of traferred AM. It is impossible in this process to escape makia comparison with the quality of fit yielded by the FV modfor which correlation coefficients are generally excellenHowever, we should not rule out the possibility that the ecellence of fit to the FV model in this instance is misleadinand perhaps due to factors arising from the highly excitbend vibration. We are seeking a model with general apcability but have relatively little in the way of data sets forigorous testing.
A. Minimum transfer (MT) model
Our first attempt to use the AM sphere to predict Rrates was based on observation that RT scales exponentwith the inverse of themagnitudeof AM transferred. It couldtherefore be argued that theminimumAM path should be thatmost favored for RT. As described above, this would leadl vectors that lie in theac plane of Figs. 4 and 5 since thminimum distance in AM space between trajectories liesthis plane. This is the basis for the first calculation. RT rafor a number of data sets were plotted in semilog foragainst transferred AMl calculated with the ‘‘minimum dis-tance’’ model. In this calculation the geometry of NH2 wastaken to be that of the2A1 excited state in its equilibriumconfiguration in which the bond angle is 144°.12
One such plot is shown in Fig. 6~a! using data from theNkakc
level 212. Note that all data referred to in this wor~and most of that in Ref. 1! was taken on NH2 in its ~n1, n2,n3!5~0,9,0! level. We discuss the significance of this higdegree of bend vibrational excitation below. Figure 6~a! isinteresting in that the basic exponential dependence on traferred AM is reproduced but the fit to data points is notgood as the FV model. The correlation coefficient for thsame data set using the FV model is 0.990 while that forAM sphere with shortest distance in AM space is 0.95. Thalthough the physical picture is improved by allowing foJ-vector precession in the molecular frame, the fit to dausing the MT approximation is worsened compared to tFV model.
B. Turning point (TP) model
The MT model used above might be considered phycally unreasonable since the point in the AM trajectorywhich the jump takes place in this picture will be thatwhich theJ vector is moving most rapidly. The precessinvector moves mostslowlyat the turning points of the trajectory. These generally will not lie in theac plane. A morerealistic model therefore would give greater weight to tslower regions on the trajectory. Our next simple model w
J. Chem. Phys., Vol. 102
n
icalseof
hetsofns-glt.-gdli-r
Tally
to
inesm
ns-sisheusrtae
i-attg
eas
one in which the point of minimum precessional velocity othe initial trajectory was identified and the minimum distancin AM space to the final trajectory from this point was caculated. The speed of the precessional motion is readily cculated since the program that reproduces the trajectoriesculates position for fixed angular intervals which enableangular velocityv to be obtained from the rate of change oJ with respect tou andf.
The result of calculations from the AM sphere based othis turning point approximation are shown in Fig. 6~b!. Thisis also the equilibrium configuration with data from the 212level. As in the case of the shortest distance model, the baform is reproduced with reasonable fit to data, though notgood as the FV result. The correlation coefficient is aga0.95. Thus again an improved physical basis to the AM dpendency has led to a slightly poorer fit to data.
C. Average vs equilibrium geometry
At this point it is useful to consider in more detail theshape of the NH2 molecule with which the collision partner
FIG. 6. The semilog plots shown here represent fits to data from the12
level of NH2 using the different approximations described in detail in thtext. In each case data~squares! are plotted against calculated values otransferred AMl. The dotted line is a best fit assuming an inverse exponetial dependence on the length ofl. ~a! Calculations assume equilibriumgeometry for NH2 ~bond angle 144°!, AM jumps represent the shortest dis-tance between the two chosen trajectories.~b! Calculations assume equilib-rium geometry but AM jumps take place from the point on the initial trajectory that the vector spends the longest time.~c! Calculations assume theprecessing vector is averaged around the positive quarter of its trajectorgive an~averaged! equivalentstatic vectorwhich has been time-weighted.The calculations are based on the AM sphere for the NH2 bond angle of 95°.~d! Calculations assume an averagel by taking each point on the~positivequarter of! the initial trajectory and calculating the distance to all points othe final vector. The results are then averaged to give the averagel value.The calculations are based on the AM sphere for the NH2 bond angle of 95°.
, No. 20, 22 May 1995
s
-
g
-
se
-
tn
r
7950 AlWahabi et al.: Rotational transfer in polyatomic molecules
will on average interact. This is significant since the precisAM trajectories and hence the substructure of an AM sphefor a particularJ value is very sensitive to the details of themolecular shape. That this will be so can be seen from tplot ~Fig. 7! of moment of inertia~I a , I b , andI c! as a func-tion of bond angle. The experimental data which we are fiting was carried out on NH2 having nine quanta of the bendvibration and the collisional RT experiment will sample thmolecule in a wide range of geometries. This may affect thresults in a manner that is not compensated for in the simAM sphere calculations described above.
The first two sets of calculations were performed on th‘‘equilibrium’’ AM sphere, i.e., assuming that the geometryof the molecule is that of the equilibrium configuration withbond angle 144°. This represents the minimum point on tpotential surface of the2A1 excited state.
12 A more appropri-ate geometry might be an average of the motion of the mecule in its~0,9,0! vibrational level in this state. Our nextrefinement of the AM sphere calculation computes this aveage geometry and then calculatesl values for this averageAM sphere. The average geometry was calculated from tpotential12 by first fitting the dependence on bend angle~u!to a simple polynomial function. This was used to computhe rate of change of potential with bend angleu from whichthe average value ofu could be obtained:
^u&5SuudV~u!/d~u!u/SudV~u!/d~u!u. ~5!
Using this expression and the published2A1 potential thevalue ^u&5106.7° was obtained. This bond angle generatnew rotational constants and a different AM sphere to thatthe equilibrium geometry. New values ofl were calculatedfor this AM sphere using the two approaches describabove, i.e.,~i! minimum distance in AM space~the MTmodel! and~ii ! turning point models. Results using this AMsphere are encouraging since they show improved correlatwith the RT data. The results of calculations for the 212 level,
FIG. 7. Plot of inertial constants of NH2 vs bond angle.
J. Chem. Phys., Vol. 102
ere
he
t-
eeple
e
he
ol-
r-
he
te
esof
ed
ion
summarized in Table I, show an improvement in the correla-tion coefficient to 0.979 in the case of the MT model and0.9872 for the TP case.
Summarizing these first approaches to the use of the AMsphere we note that both equilibrium and average geometriegive good fits to data but those obtained with the averagebond angle of 106.7° were noticeably better. Use of the TPapproximation generally produces better fits than the MT approach. Correlation coefficients are consistently better withthe former approach for both geometries. This is encouraginsince better fits appear to follow as the physical model of theinteraction is improved. The average geometry is more appropriate for NH2 ~0,9,0! data and the assumption that theturning points of the AM trajectories have a dominant influ-ence is also a reasonable one. We next discuss how thesimple models may be improved.
D. Average vector ( J av) and average l ( l av) models
The success of the average geometry TP model has indicated that improvements can come from improving thephysical representation for the calculation. These in effecconsist of attempts to average over the complete trajectory iorder to compute a true value ofl in addition to finding themost appropriate NH2 configuration for the calculation. Thetwo l averaging approximations consist either in summingover ~a portion of! theJ trajectory to give an average vector~Jav approximation! or of averaging the computed AM spacejump to give an averagel value ~l av model!.
In theJav approximation, the path of the precessing vec-tor around the positive quarter of its trajectory was averagedto give an equivalent static vector. This was done incorpo-rating a time weighting obtained, as before, through knowl-edge of the angular velocityv. This average static vectorwas then used in the calculation ofl in a manner related tothat described under the FV model. However, in this instancethe magnitude ofl is a function of projections on all threeinertial axes. Thel av approach is somewhat different. Herethe average transferred AM is found by taking each point onthe initial trajectory and calculatingl to all points on the finaltrajectory~again within the positive quarter of theJ motion!.All points on the initial trajectory are summed and the resultsaveraged. Time weighting of the kind described above wasalso used.
These two models were used on RT data using a numbeof different molecular geometries. These included both the
TABLE I. Correlation coefficients from least squares fit of the inverse ex-ponential AM transfer law to RT data from the 212 rotational level ofNH2~A
2A1!. The AM transferl is calculated on the AM sphere from thevarious models discussed in the text.
NH2 ~A 2A1)bond angle MT TP Jav l av
144° 0.950 0.950 0.959 0.937107° 0.979 0.982 0.972 0.96697° 0.983 0.98295° 0.986 0.98490° 0.980 0.98475° 0.969 0.938
, No. 20, 22 May 1995

7t
u
n
e
a
t
n
de
oa
c
M
i
Mu
ofrkhe
t of-
ore,
cs.hehasomn.s ofin-ic-of
.tethetedtotophe
ofstsesis-acti--on.e-
totor
ve
the
l-pro-y beirter-ndgthe
e,ionon,herla-
7951AlWahabi et al.: Rotational transfer in polyatomic molecules
equilibrium, bond angle 144°, the average~106.7°!, and anumber of bent configurations to probe the effect of geometry change on the fits. Rotational constants were recalclated for each new configuration and the bond angles 995°, 90°, and 75° were used since these are close toturning point in the~0,9,0! level of 2A1 . Results of the cal-culations for theJav and l av models for the initial level 212using the turning point geometry of NH2 with a bond angleof 95° are shown in Figs. 4 and 5. The results of this calclation are summarized in Table I for the data set originatinfrom the 212 level. As seen from the table, there is an improvement in quality of fit using bothJav and l av models.These are best at the angle corresponding to the turning poof the potential surface representing the bend coordinate~ap-prox. 95°! and suggest that the interaction is dominated bthis configuration of the NH2 molecule. Physically this is notunreasonable in view of the length of time spent at the turing points relative to other positions.
V. DISCUSSION AND CONCLUSIONS
The AM gap law1 originally proposed for RT in poly-atomics represented a new approach to RT in molecules fturing an ~inverse! exponential dependence on transferreAM instead of the more usual transferred energy. Thereincreasing evidence of the unsuitability of energy gap reltions in the RT of polyatomic molecules9 and the data wereported on NH2–H collisions1 strongly indicates that theenergy gap has little influence. Notwithstanding these comments it will be evident that RTwithin k stacks, which com-prises much of the state-to-state RT data that is availablepolyatomics, may be fit with energyor AM gap laws. Asdiscussed earlier such processes, which are akin to RTdiatomics, are not diagnostic of mechanism. The exponenAM dependence noted first in Ref. 1 has been developed ina simple but powerful new theory of RT in diatomicmolecules.2,3 There is also evidence of the inadequacy of thlong-range interactions alone to account for all observatioof RT in polyatomic molecules.
It is important to recognize the dynamical nature of thAM vector in the molecule frame in the case of nonlineamolecules. Here this dynamical motion is explicitly includein the consideration of collision-induced transitions for thfirst time. This motion is completely described in the framework of the AM sphere representation that we present in thwork and use as the basis for interpreting collisional prcesses in molecules. The AM sphere representation of clsical motion of the AM vector is shown to be a very usefumeans of visualizing RT. Since it constitutes a plot of trajetories in angular momentum space defined by the individumolecule and its characteristic inertial moments, the AMsphere provides a basis for calculating the length of the Avector that causes a transition from one trajectory to anoth
The AM sphere may be adapted to include the additioncomplication of angular momenta arising from molecular vbrations, electronic orbital, or spin angular momenta as hbeen demonstrated in the case of the closely related rotional energy surfaces by Harter and co-workers15 in an ex-tensive series of applications. It is readily seen from the Atrajectories displayed in Figs. 4 and 5 that there is no uniq
J. Chem. Phys., Vol. 102
-u-°,he
-g-
int
y
-
a-dis-
-
on
inialto
es
er
-is-s-l-al
er.al-asta-
e
value of AM change when the orbital angular momentuma collision transforms one trajectory to another. In this wowe propose a number of simple models for calculating tmagnitude ofl, the transferred AM. Note thatall of thesegives a very good account of the data we use as our tesmodel quality and therefore are all a very substantial improvement on a law based on the energy gap. Furthermall will give an excellent fit to the intra-k stack data thatcomprises much of the available data on RT in polyatomi
Several approaches to calculation of the AM gap in tAM sphere representation are proposed and tested. Eacha firm basis in the physics of the interaction between an atand a molecule undergoing vibrational and rotational motioThese are not necessarily exhaustive and other methodusing the AM sphere representation may exist. A major hderance is the paucity of data with which to compare predtions. Here we test the models by their ability to fit setsdata obtained on NH2–H collisions, an extensive set inwhich wide changes ofN, ka , and kc have been detectedHowever, despite the quality and quantity of these RT raconstants this data is not ideal as an unambiguous test ofmodels we propose here since RT is of molecules exciwith nine quanta of the bend vibration. This is sufficientchange the molecular geometry from a prolate symmetricat the center of the oscillatory motion to an oblate top at textremes of the bend.
This motion in the bend coordinate may be the originthe excellent fits obtained using the FV model and suggethat this simple model may be inappropriate for moleculinitially in low vibrational states. Despite the success of thmodel in fitting NH2–H data the lack of account of the dynamical motion of the AM vector in the molecule frame isserious flaw and the AM sphere model proposed here refies this. It may be that for highly vibrationally excited species the FV model may represent a useful approximatiHowever, we feel that one or another of the AM spherbased techniques for estimatingl, the transferred AM willprove more satisfactory. We have found that the best fitdata using the AM sphere is obtained when the AM vectrajectory is averaged over via theJav or the l av approxima-tions. It is clear that the method is sensitive to the effectigeometry that the collision partner experiences, i.e., anaver-ageconfiguration rather than the equilibrium. The NH2 dataappears to be dominated by the turning point geometry inbend vibration.
As more extensive data sets of RT in polyatomic moecules become available the usefulness of the modelsposed here based on the AM sphere representation matested. Light triatomics will be particularly useful since thespectral features are often well separated and well characized. The series of measurements by Steinfeld acolleagues17 on spherical and symmetric top hydrides usininfrared double resonance techniques have demonstratedhigh selectivity of RT in such molecules. The AM spherwhich embodies the molecular symmetry and the dispositof the inertial axes in an angular momentum representatimay provide the key to the understanding of these and otcollision-induced processes where simple energy gap retions have proved to be inadequate.
, No. 20, 22 May 1995

n
J
J
,
y
e
7952 AlWahabi et al.: Rotational transfer in polyatomic molecules
ACKNOWLEDGMENTS
We thank the Science and Engineering Research Coufor financial support and for studentships~Z. T. A., M. A. O.,and Z. R.!.
1Z. T. AlWahabi, C. G. Harkin, A. J. McCaffery, and B. J. Whitaker,Chem. Soc., Faraday Trans.85, 1003~1989!.
2A. J. McCaffery, Z. T. AlWahabi, M. A. Osborne, and C. J. Williams,Chem. Phys.98, 4586~1993!.
3M. A. Osborne and A. J. McCaffery, J. Chem. Phys.101, 5604~1994!.4T. Oka, Adv. At. Mol. Phys.9, 127 ~1973!.5H. O. Everitt and F. C. DeLucia, J. Chem. Phys.92, 6480~1990!.6C. P. Bewick, J. G. Haub, R. G. Hynes, J. F. Martins, and B. J. OrrChem. Phys.88, 6350~1988!.
7K. W. Butz, H. Du, D. J. Krajnovitch, and C. S. Parmenter, J. Chem. Ph89, 4680~1988!.
8G. J. Kroes, R. P. H. Rettschnick, C. E. Dateo, and D. C. Clary, J. Ch
J. Chem. Phys., Vol. 102
cil
.
.
J.
s.
m.
Phys.93, 287 ~1990!; G. J. Kroes and R. P. H. Rettschnick,ibid. 94, 360~1991!.
9D. Harradine, B. Foy, L. Laux, M. Dubs, and J. I. Steinfeld, J. Chem.Phys.81, 4267~1984!.
10C. G. Harkin, D. Phil. thesis, University of Sussex~1989, unpublished!.11Z. T. AlWahabi, D. Phil. thesis, University of Sussex~1991, unpublished!.12R. N. Dixon and D. Field, Proc. R. Soc. London Ser. A366, 225 ~1979!.13H. Goldstein, Classical Mechanics~Addison–Wesley, Reading, MA,1953!.
14L. D. Landau and E. M. Lifshitz,Mechanics~Pergamon, New York,1969!.
15W. G. Harter and C. W. Patterson, J. Chem. Phys.80, 4241~1984!.16R. N. Zare,Angular Momentum~Wiley, New York, 1988!.17L. Laux, B. Foy, D. Harradine, and J. I. Steinfeld, J. Chem. Phys.80, 3499
~1984!; G. Foy, J. Hetzler, G. Millot, and J. I. Steinfeld,ibid. 88, 6838~1988!; J. J. Klaasen, S. L. Coy, J. I. Steinfeld, and C. Roche,ibid. 100,5519 ~1994!.
, No. 20, 22 May 1995




















