
Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in...
8
518 Journal of Leukocyte Biology Volume 59, April 1996 Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in transgenic mice Lesley Probert, Katerina Akassoglou, Lena Alexopoulou, Eleni Douni, Sylva Haralambous, Sally Hill, George Kassiotis, Dimitris Kontoyiannis, Manolis Pasparakis, David Plows, and George Kollias Department of Molecular Genetics, Hellenic Pasteur Institute, Athens, Greece Abstract: With increasing awareness that seemingly diverse immune-mediated diseases involve similar pathogenetic mechanisms, and the identification of a growing number of key effector molecules, it is becoming possible to design and generate effective transgenic models for such diseases. Tumor necrosis factor (TNF) plays a prominent role in immune and host defense responses and there is strong evidence that abnormal TNF production contributes to disease initiation and progression in rheumatoid arthritis, systemic inflammatory response syndrome, diabetes, multiple sclerosis, and many other immune-mediated disorders. The generation of TNF transgenic mice, in which TNF production is deregulated, has pro- vided us with direct evidence that, in vivo, this cytokine can indeed trigger the development of such complex disease phenotypes. Transgenic mice that have been engineered to overexpress human or mur- me TNF molecules in peripheral joints, T cells, or neurons of the central nervous system represent important animal models for human rheumatoid ar- thritis, systemic inflammation, and multiple sclerosis, respectively. In addition to establishing a central role for TNF in such diseases, these animal models have already proved valuable for identifying additional important disease-effector molecules, and for gain- ing an insight into the complex in vivo mechanisms that are involved in disease pathogenesis. For exam- ple, in the case of arthritis, TNF has been found to transmit its pathogenic effects entirely through inter- leukin-1, which may therefore represent an addi- tional important target for therapeutic intervention in the human disease. In summary, TNF transgenic models of human disease are expected to serve as important in vivo tools for defining details of disease pathogenesis, potential targets for therapeutic inter- vention, and for evaluating the possible involvement of additional genetic and environmental factors on the disease state. J. Leukoc. Riot. 59: 518-525; 1996. Key Words: autoinmuniuy . disease models . inflammation neuroimmunology arthritis cytokine INTRODUCTION Tumor necrosis factor a (TNF-a) is a proinflammatory cytokine produced mainly by activated macrophages, but also by many other cell types including T cells, pre-B cells, natural killer cells, mast cells, neutrophils, fi- broblasts, keratinocytes, and microglial cells. It is pro- duced during immune and host defense responses and, under physiological conditions, acts to preserve architec- tural and biochemical homeostasis [1]. Thus, TNF-a is involved in tissue remodeling, defense against infection, and inflammation. In contrast to this, overproduction of TNF-a can cause severe pathological changes leading to tissue injury, shock, and even death. TNF-a has been directly implicated in the pathogenesis of several disease states such as the systemic inflammatory response syn- drome, graft-versus-host reactions, and autoimmune dis- eases including diabetes, rheumatoid arthritis, and multiple sclerosis. The pleiotropism of TN F-a action is a result of the mul- tilevel mechanisms that operate to regulate its expression and functional potency. In addition to its diverse cellular source, TNF-a occurs naturally in two biologically active molecular forms, a 26-kDa membrane-anchored precursor protein (pro-TNF-a) acting locally through cell-to-cell contact, and a secreted, mature 17-kDa form that is cleaved from pro-TNF-a by proteolytic enzymes and is capable of acting on distant targets [2]. Evidence is accu- mulating that the two molecular forms of TNF-a can medi- ate distinct biological functions. For example, transmembrane TNF-a has been associated with contact- dependent lymphocyte and monocyte-mediated cytotoxic- ity [3] and may play a role in B cell activation [4, 5]. Diversity of TNF-a action is further acheived at the recep- tor level. TNF-a signaling is mediated by two specific high-affinity receptors, the p55 TNF-a-R (CD12Oa) and p75 TNF-a-R (CD12Ob) [see ref. 6]. The two receptors are co-expressed in most tissues but contain very different Abbreviations: TNF-a, tumor necrosis factor-a; IL, interleukin; CNS, central nervous system; MS. multiple sclerosis; RAE, experimental autoimmune encepha- lomyelitis. Reprint requests: Dr. George Kollias, Department of Molecular Cenetics, Hel- lenic Pasteur Institute, 127 Vass Sofias Avenue, 11521 Athens, Hellas. Received November29, 1995; revised January 19, 1996; accepted January 22, 1996.
-
Upload
agriculturalathensu -
Category
Documents
-
view
5 -
download
0
Transcript of Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in...

518 Journal of Leukocyte Biology Volume 59, April 1996
Dissection of the pathologies induced by transmembraneand wild-type tumor necrosis factor in transgenic mice
Lesley Probert, Katerina Akassoglou, Lena Alexopoulou, Eleni Douni, Sylva Haralambous,Sally Hill, George Kassiotis, Dimitris Kontoyiannis, Manolis Pasparakis, David Plows, andGeorge KolliasDepartment of Molecular Genetics, Hellenic Pasteur Institute, Athens, Greece
Abstract: With increasing awareness that seemingly
diverse immune-mediated diseases involve similar
pathogenetic mechanisms, and the identification of a
growing number of key effector molecules, it is
becoming possible to design and generate effective
transgenic models for such diseases. Tumor necrosis
factor (TNF) plays a prominent role in immune and
host defense responses and there is strong evidence
that abnormal TNF production contributes to disease
initiation and progression in rheumatoid arthritis,
systemic inflammatory response syndrome, diabetes,
multiple sclerosis, and many other immune-mediated
disorders. The generation of TNF transgenic mice,
in which TNF production is deregulated, has pro-
vided us with direct evidence that, in vivo, this
cytokine can indeed trigger the development of such
complex disease phenotypes. Transgenic mice that
have been engineered to overexpress human or mur-
me TNF molecules in peripheral joints, T cells, or
neurons of the central nervous system represent
important animal models for human rheumatoid ar-
thritis, systemic inflammation, and multiple sclerosis,respectively. In addition to establishing a central role
for TNF in such diseases, these animal models have
already proved valuable for identifying additional
important disease-effector molecules, and for gain-
ing an insight into the complex in vivo mechanisms
that are involved in disease pathogenesis. For exam-
ple, in the case of arthritis, TNF has been found to
transmit its pathogenic effects entirely through inter-
leukin-1, which may therefore represent an addi-
tional important target for therapeutic interventionin the human disease. In summary, TNF transgenic
models of human disease are expected to serve as
important in vivo tools for defining details of disease
pathogenesis, potential targets for therapeutic inter-
vention, and for evaluating the possible involvement
of additional genetic and environmental factors on
the disease state. J. Leukoc. Riot. 59: 518-525;
1996.
Key Words: autoin�muniuy . disease models . inflammation
neuroimmunology arthritis cytokine
INTRODUCTION
Tumor necrosis factor a (TNF-a) is a proinflammatory
cytokine produced mainly by activated macrophages, but
also by many other cell types including T cells, pre-B
cells, natural killer cells, mast cells, neutrophils, fi-
broblasts, keratinocytes, and microglial cells. It is pro-
duced during immune and host defense responses and,
under physiological conditions, acts to preserve architec-
tural and biochemical homeostasis [1]. Thus, TNF-a is
involved in tissue remodeling, defense against infection,
and inflammation. In contrast to this, overproduction of
TNF-a can cause severe pathological changes leading to
tissue injury, shock, and even death. TNF-a has been
directly implicated in the pathogenesis of several disease
states such as the systemic inflammatory response syn-
drome, graft-versus-host reactions, and autoimmune dis-
eases including diabetes, rheumatoid arthritis, and
multiple sclerosis.
The pleiotropism of TN F-a action is a result of the mul-
tilevel mechanisms that operate to regulate its expression
and functional potency. In addition to its diverse cellular
source, TNF-a occurs naturally in two biologically active
molecular forms, a 26-kDa membrane-anchored precursor
protein (pro-TNF-a) acting locally through cell-to-cell
contact, and a secreted, mature 17-kDa form that is
cleaved from pro-TNF-a by proteolytic enzymes and is
capable of acting on distant targets [2]. Evidence is accu-
mulating that the two molecular forms of TNF-a can medi-
ate distinct biological functions. For example,
transmembrane TNF-a has been associated with contact-
dependent lymphocyte and monocyte-mediated cytotoxic-
ity [3] and may play a role in B cell activation [4, 5].
Diversity of TNF-a action is further acheived at the recep-
tor level. TNF-a signaling is mediated by two specific
high-affinity receptors, the p55 TNF-a-R (CD12Oa) and
p75 TNF-a-R (CD12Ob) [see ref. 6]. The two receptors are
co-expressed in most tissues but contain very different
Abbreviations: TNF-a, tumor necrosis factor-a; IL, interleukin; CNS, central
nervous system; MS. multiple sclerosis; RAE, experimental autoimmune encepha-lomyelitis.
Reprint requests: Dr. George Kollias, Department of Molecular Cenetics, Hel-lenic Pasteur Institute, 127 Vass Sofias Avenue, 11521 Athens, Hellas.
Received November29, 1995; revised January 19, 1996; accepted January 22,1996.

Probert et al. Tranamembrane and wild-type TNF tranagenic mice 519
intracellular domains associated with distinct cytoplasmic
proteins [7, 8], and thus mediate distinct cellular re-
sponses in vivo. The murine p55 TNF-a-R appears to mo-
nopolize TNF-a-mediated signaling and can signal almost
all reported TNF-a activities including cytotoxicity [9, 10],
apoptosis [11], and proliferation [12]. In contrast, p75
TNF-a-R signaling is limited to few cell systems, and pro-
motes proliferation of thymocytes, T lymphocytes [13], and
other cells of hemopoietic origin, as well as apoptotic sig-
nals in mature, activated CD8� T cells [14]. Recent evi-
dence indicates that the 26-kDa transmembrane TNF-a
molecule is superior to soluble TNF-cx in activating the
p’75 TNF-ct-R in various systems such as T cell activation
and thymocyte proliferation [15] and implies an important
physiological role for this receptor in local inflammatory
responses. Both the p55 and p7S TNF-a-R exist as trans-
membrane proteins that are inducibly sheddable. The re-
sulting soluble receptors can compete for TNF-ct and thus
modulate its bioavailability either inhibiting or enhancing
its action depending upon the microenvironment [16].
Considering the multiple and potent actions of TNF-a it is
not surprising that the biological availability of this protein
is strictly regulated. As with other pro-inflammatory cytok-
ines, TNF-a mRNA contains a 3’ UTR region that is
thought to be critical in the regulation of both mRNA sta-
bility [17] and translational efficiency [18, 19].
Although we are now beginning to gain insight into sev-
eral aspects of TNF-cx biology, for example TNF-a-R sig-
naling mechanisms, the regulation of TNF-a production,
and the relative roles of membrane-associated and soluble
TNF-a and TNF-a-R proteins, we know little of their bio-
logical significance in the in vivo context during physi-
ological and pathophysiological processes. Our laboratory
has been using transgenic technology to investigate the in
vivo biological potential of TNF-a during health and dis-
ease, and to analyze the multiple mechanisms that regulate
its expression and functional potency. It is possible, for
example, to differentiate between the two TNF-a receptors
in vivo, by expressing huTNF-a transgenes in transgenic
mice. In this way, the function of the p55 TNF-a-R is
specifically probed by virtue of its wide species cross-re-
activity, while the species-specific mouse p75 TNF-a-R is
unable to mediate signals from huTNF-a [20]. In addition,
analysis of the role of 26-kDa transmembrane huTNF-a in
vivo can be examined by use of a mutant huTNF-a trans-
gene in which the triplets encoding the first to twelfth
amino acids containing the putative cleavage site for ma-
ture soluble TNF-a, have been deleted (iM-12 TNF-a).
This i�1-12 mutant TNF-a gene has been shown to pro-
duce a transmembrane huTNF-a protein that remains
bioactive against cellular targets in vitro [21].
A TRANSGENIC MOUSE MODEL OF CHRONICINFLAMMATORY ARTHRITIS
Rheumatoid arthritis is an autoimmune disease involving
localized inflammation of the synovial joint. Immunoge-
Fig. 1. Sections taken through the knee joint of an arthritic Tg197(huTNF-a-globin) mouse. Immunocytochemical demonstration of largenumbers of 132 integrin (CD18)-immunoreactive cells adhering to vascular
endothelium and infiltrating the proliferated synovium. The anti-CD18
antibody was kindly provided by Dr. Leslie Moloney, Tanabe Research
Laboratories, San Diego, CA. Scale bar, 200 Ltm.
netic susceptibility, physical stress or injury, infectious
agents, and endogenous substances have been implicated
in triggering the primary immune response in arthritis. The
subsequent amplification and perpetuation of joint inflam-
mation involves a complex interplay between pro-inflam-
matory and immunomodulatory cytokines, neuropeptides,
metalloproteinases, adhesion molecules, and other inflam-
matory mediators. Elevated levels of pro-inflammatory cy-
tokines such as TNF-cz, interleukin-1 (IL-i), IL-6, IL-8,
and granulocyte/macrophage colony stimulating factor are
present in the synovial fluid and serum of patients with
rheumatoid arthritis [22-24] and TNF-a and its receptors
are expressed by rheumatoid synoviocytes [25-28].
TNF-a shows many biological activities that are consis-
tent with it playing a central role in the development of
chronic arthritic disease. TNF-a triggers the activation and
proliferation of synoviocytes in vitro [29, 30] and may
interfere with cartilage and bone metabolism through in-
duction of collagenase production by synovial cells [31,
32], inhibition of proteoglycan synthesis by articular chon-
drocytes [33, 34], and stimulation of bone resorption in
vitro [35, 36]. Other indirect activities include the induc-
tion of additional cytokines [37, 38], and the triggering of
adhesion molecule expression on endothelial cells, which
enables lymphocytes to home to the site of inflammation
[39]. These mechanisms are considered to be decisive in
the development of arthritis.
To investigate the role of TNF-a in vivo in the develop-
ment of arthritis, our laboratory has generated transgenic
mice that overexpress TNF-a. Our early work showed that
a human TNF-a transgene could be expressed efficiently
in peripheral joints of transgenic mice when its 3’ UTR and
3’ flanking sequences were replaced with corresponding
sequences from the human �3-globin gene to stabilize the
mRNA and enhance its translational efficiency. In these
mice exogenous huTNF-a was produced in both trans-
membrane (26 kDa) and soluble (17 kDa) molecular forms.
Mice expressing the huTNF-a-globin construct develop
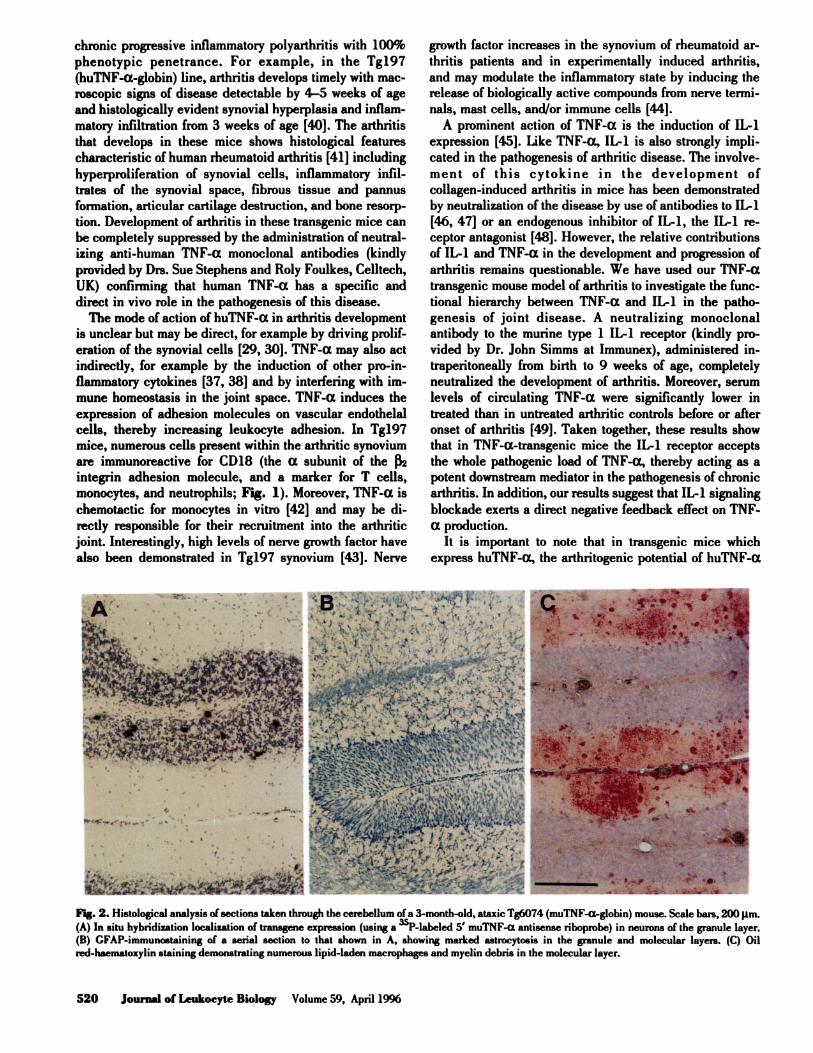
Fig. 2. Histological analysis of sections taken through the cerebellum of a 3-month-old, ataxic Tg6074 (muTNF-a-globin) mouse. Scale bars, 200 �tm.(A) In situ hybridization localization of transgene expression (using a �P-labeled 5’ muTNF-a antisense riboprobe) in neurons of the granule layer.
(B) GFAP-immunoataining of a serial section to that shown in A, showing marked astrocytosis in the granule and molecular layers. (C) Oil
red-haematoxylin staining demonstrating numerous lipid-laden macrophages and myelin debris in the molecular layer.
520 Journal of Leukocyte Biology Volume 59, April 1996
chronic progressive inflammatory polyarthritis with 100%
phenotypic penetrance. For example, in the Tg197
(huTNF-a-globin) line, arthritis develops timely with mac-
roscopic signs of disease detectable by 4-5 weeks of age
and histologically evident synovial hyperplasia and inflam-
matory infiltration from 3 weeks of age [40]. The arthritis
that develops in these mice shows histological features
characteristic of human rheumatoid arthritis [41] including
hyperproliferation of synovial cells, inflammatory infil-
trates of the synovial space, fibrous tissue and pannus
formation, articular cartilage destruction, and bone resorp-
tion. Development of arthritis in these transgenic mice can
be completely suppressed by the administration of neutral-
izing anti-human TNF-a monoclonal antibodies (kindly
provided by Drs. Sue Stephens and Roly Foulkes, Celitech,
UK) confirming that human TNF-a has a specific and
direct in vivo role in the pathogenesis of this disease.
The mode of action of huTNF-a in arthritis development
is unclear but may be direct, for example by driving prolif-
eration of the synovial cells [29, 30]. TNF-a may also act
indirectly, for example by the induction of other pro-in-
flammatory cytokines [37, 38] and by interfering with im-
mune homeostasis in the joint space. TNF-a induces the
expression of adhesion molecules on vascular endothelal
cells, thereby increasing leukocyte adhesion. In Tg197
mice, numerous cells present within the arthritic synovium
are immunoreactive for CD18 (the a subunit of the �
integrin adhesion molecule, and a marker for T cells,
monocytes, and neutrophils; Fig. 1). Moreover, TNF-a is
chemotactic for monocytes in vitro [42] and may be di-
rectly responsible for their recruitment into the arthritic
joint. Interestingly, high levels of nerve growth factor have
also been demonstrated in Tgi97 synovium [43]. Nerve
growth factor increases in the synovium of rheumatoid ar-
thritis patients and in experimentally induced arthritis,
and may modulate the inflammatory state by inducing the
release of biologically active compounds from nerve termi-
nals, mast cells, and/or immune cells [44].
A prominent action of TNF-a is the induction of IL-i
expression [45]. Uke TNF-a, IL-i is also strongly impli-
cated in the pathogenesis of arthritic disease. The involve-
ment of this cytokine in the development of
collagen-induced arthritis in mice has been demonstrated
by neutralization of the disease by use of antibodies to IL-i
[46, 47] or an endogenous inhibitor of IL-i, the IL-i re-
ceptor antagonist [48]. However, the relative contributions
of IL-i and TNF-a in the development and progression of
arthritis remains questionable. We have used our TNF-a
transgenic mouse model of arthritis to investigate the func-
tional hierarchy between TNF-a and IL-i in the patho-
genesis of joint disease. A neutralizing monoclonal
antibody to the murine type 1 IL-i receptor (kindly pro-
vided by Dr. John Simms at Immunex), administered in-
traperitoneally from birth to 9 weeks of age, completely
neutralized the development of arthritis. Moreover, serum
levels of circulating TNF-a were significantly lower in
treated than in untreated arthritic controls before or after
onset of arthritis [49]. Taken together, these results show
that in TNF-a-transgenic mice the IL-i receptor accepts
the whole pathogenic load of TNF-ct, thereby acting as a
potent downstream mediator in the pathogenesis of chronic
arthritis. In addition, our results suggest that IL-i signaling
blockade exerts a direct negative feedback effect on TNF-
a production.
It is important to note that in transgenic mice which
express huTNF-a, the arthritogenic potential of huTNF-a
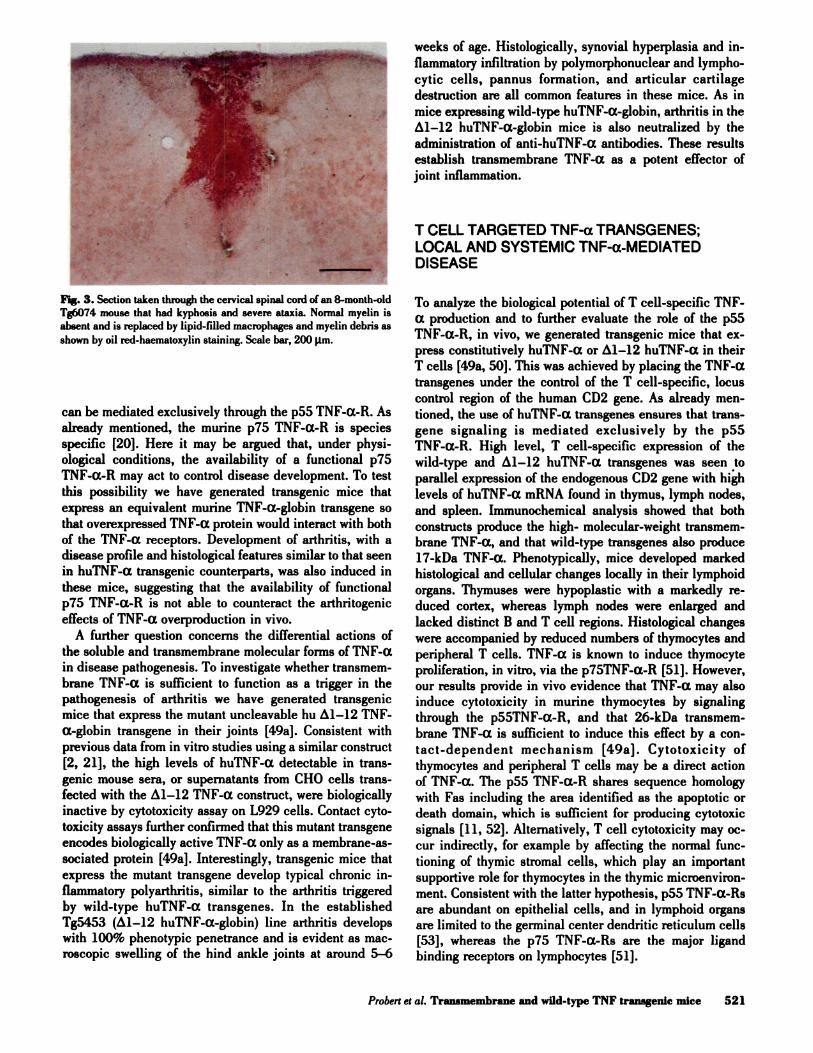
Fig. 3. Section taken through the cervical spinal cord of an 8-month-old
Tg6074 mouse that had kyphosis and severe ataxia. Normal myelin is
absent and is replaced by lipid-filled macrophages and myelin debris as
shown by oil red-haematoxylin staining. Scale bar, 200 l,tm.
Probert et al. Tranamembrane and wild-type TNF tranagenic mice 521
can be mediated exclusively through the pSS TNF-a-R. As
already mentioned, the murine p75 TNF-a-R is species
specific [20]. Here it may be argued that, under physi-
ological conditions, the availability of a functional p7S
TNF-a-R may act to control disease development. To test
this possibility we have generated transgenic mice that
express an equivalent murine TNF-a-globin transgene so
that overexpressed TNF-a protein would interact with both
of the TNF-a receptors. Development of arthritis, with a
disease profile and histological features similar to that seen
in huTNF-a transgenic counterparts, was also induced in
these mice, suggesting that the availability of functional
p’ZS TNF-ct-R is not able to counteract the arthritogenic
effects of TNF-a overproduction in vivo.
A further question concerns the differential actions of
the soluble and transmembrane molecular forms of TNF-a
in disease pathogenesis. To investigate whether transmem-
brane TNF-a is sufficient to function as a trigger in the
pathogenesis of arthritis we have generated transgenic
mice that express the mutant uncleavable hu z�i-i2 TNF-
cz-globin transgene in their joints [49a]. Consistent with
previous data from in vitro studies using a similar construct
[2, 21], the high levels of huTNF-cz detectable in trans-
genic mouse sera, or supematants from CHO cells trans-
fected with the Ai-12 TNF-a construct, were biologically
inactive by cytotoxicity assay on L929 cells. Contact cyto-
toxicity assays further confirmed that this mutant transgene
encodes biologically active TNF-a only as a membrane-as-
sociated protein [49a]. Interestingly, transgenic mice that
express the mutant transgene develop typical chronic in-
flammatory polyarthritis, similar to the arthritis triggered
by wild-type huTNF-a transgenes. In the established
Ig5453 (A1-i2 huTNF-ct-globin) line arthritis develops
with 100% phenotypic penetrance and is evident as mac-
roscopic swelling of the hind ankle joints at around 5-6
weeks of age. Histologically, synovial hyperplasia and in-
flammatory infiltration by polymorphonuclear and lympho-
cytic cells, pannus formation, and articular cartilage
destruction are all common features in these mice. As in
mice expressing wild-type huTNF-a-globin, arthritis in the
�1-i2 huTNF-ct-globin mice is also neutralized by the
administration of anti-huTNF-a antibodies. These results
establish transmembrane TNF-a as a potent effector of
joint inflammation.
T CELL TARGETED TNF-a TRANSGENES;LOCAL AND SYSTEMIC TNF-a-MEDIATEDDISEASE
To analyze the biological potential of T cell-specific TNF-
a production and to further evaluate the role of the p55
TNF-cZ-R, in vivo, we generated transgenic mice that ex-
press constitutively huTNF-a or �1-i2 huTNF-a in their
T cells [49a, 50]. This was achieved by placing the TNF-a
transgenes under the control of the T cell-specific, locus
control region of the human CD2 gene. As already men-
tioned, the use of huTNF-a transgenes ensures that trans-
gene signaling is mediated exclusively by the p55
TNF-a-R. High level, T cell-specific expression of the
wild-type and M-12 huTNF-a transgenes was seen to
parallel expression of the endogenous CD2 gene with high
levels of huTNF-a mRNA found in thymus, lymph nodes,
and spleen. Immunochemical analysis showed that both
constructs produce the high- molecular-weight transmem-
brane TNF-a, and that wild-type transgenes also produce
i7-kDa TNF-a. Phenotypically, mice developed marked
histological and cellular changes locally in their lymphoid
organs. Thymuses were hypoplastic with a markedly re-
duced cortex, whereas lymph nodes were enlarged and
lacked distinct B and T cell regions. Histological changes
were accompanied by reduced numbers of thymocytes and
peripheral T cells. TNF-a is known to induce thymocyte
proliferation, in vitro, via the p75TNF-a-R [51]. However,
our results provide in vivo evidence that TNF-a may also
induce cytotoxicity in murine thymocytes by signaling
through the p55TNF-a-R, and that 26-kDa transmem-
brane TNF-a is sufficient to induce this effect by a con-
tact-dependent mechanism [49a]. Cytotoxicity of
thymocytes and peripheral T cells may be a direct action
of TNF-a. The p55 TNF-a-R shares sequence homology
with Fas including the area identified as the apoptotic or
death domain, which is sufficient for producing cytotoxic
signals [ii, 52]. Alternatively, T cell cytotoxicity may oc-
cur indirectly, for example by affecting the normal func-
tioning of thymic stromal cells, which play an important
supportive role for thymocytes in the thymic microenviron-
ment. Consistent with the latter hypothesis, pSS TNF-a-Rs
are abundant on epithelial cells, and in lymphoid organs
are limited to the germinal center dendritic reticulum cells
[53], whereas the p75 TNF-a-Rs are the major ligand
binding receptors on lymphocytes [51].

522 Journal of Leukocyte Biology Volume 59, April 1996
High-level T cell-specific expression of the wild-type
TNF-a transgene resulted in spillover of soluble TNF-a
protein into the circulation. In the established Tg2ii
(CD2-huTNF-a-globin) line, for example, serum levels of
huTNF-a ranged between 100 and 1300 pg/mI. The pres-
ence of circulating TNF-cx protein was correlated with de-
velopment of a lethal wasting syndrome characterized by
widespread vascular necrosis and ischemic tissue necrosis
in peripheral organs, particularly the liver, pancreas, and
lymph nodes. As for the lymphoid organ abnormalities, the
wasting disease could be completely neutralized by anti-
huTNF-a antibodies [50, 54]. TNF-a is known to exert a
variety of in vitro effects on vascular endothelial cells,
altering morphology and growth rates [55], and stimulating
adhesion molecule expression [56, 57]. In addition, TNF-a
acts to induce IL-i production by vascular endothelial
cells [58, 59] and both cytokines act to produce procoagu-
lant activity [60-62]. As a consequence, TNF-a may cre-
ate a situation in which both leukocytes and fibrin
accumulate along the walls of vessels and hemostasis is
induced. Confirmation that the systemic pathology was ef-
fected by soluble circulating TNF-a, rather than by trans-
membrane TNF-a present on the surface of circulating T
cells, was obtained in transgenic mice expressing only the
uncleavable huTNF-a protein. These animals, despite
similar overall levels of TNF-a mRNA production, do not
develop systemic pathology or wasting syndrome [49a].
The development of wasting disease in TNF-a trans-
genic mice fits well with some of the documented effects of
TNF-a in vivo. TNF-a has been shown to be directly in-
volved in the pathogenesis of endotoxic shock [63, 64] and
cachexia induced in mice bearing tumors that secrete
TNF-a [65]. Similarly, the chronic administration of
sublethal doses of TNF-a to experimental animals leads to
wasting accompanied by specific inflammatory changes in
several tissues [66], whereas single lethal doses induce
intravascular thrombosis with hemorrhagic necrosis of tis-
sues [67]. Our finding that the overexpression of huTNF-a
in transgenic mice leads to vascular obstruction, hemosta-
sis, and resulting ishemic tissue necrosis and wasting dis-
ease, demonstrates that such effects can be mediated by
the pSS TNF-a-R and that specifically blocking the action
of this receptor might find therapeutical application to aid
the management of systemic inflammation and its conse-
quences.
CNS-SPECIFIC EXPRESSION OF TNF-aINDUCES INFLAMMATORY DEMYELINATINGDISEASE
In addition to their classical role in peripheral immune
function and host defense, cytokines are now emerging as
important mediators of central nervous system (CNS)
physiology and pathophysiology. TNF-a is produced byneurons in the normal murine brain [68] and exerts neuro-
modulatory effects [69]. The expression of TNF-a is up-
regulated in a wide range of CNS disorders, including mul-
tiple sclerosis (MS) [70, 71], AIDS dementia complex [72],
bacterial meningitis [73], Parkinson’s disease [74], and
human and murine cerebral malaria [75]. This increased
expression might derive from activated infiltrating T cells
or macrophages [1], activated astrocytes [76], and micro-
glial cells [77]. The contribution to disease pathogenesis of
increased TNF-a production in the CNS and its mecha-
nism of action are at present ill-defined. However, there is
strong in vitro evidence that in the CNS, TNF-a acts as a
potent pro-inflammatory cytokine and a major effector of
immune-mediated demyelination. TNF-a can induce oh-
godendrocyte degeneration and myelin vesiculation in
murine CNS explants [78, 79] and causes selective cyto-
toxicity to primary oligodendrocytes [80]. Moreover, stud-
ies in experimental autoimmune encephalomyelitis (EAE),
which is often used as a model for human MS, have pro-
vided additional in vivo evidence supporting the impor-
tance of TNF-a in disease pathogenesis. The adoptive
transfer of this disease by MBP-reactive T cells can be
prevented by the administration of neutralizing antibody to
TNF-a [81] or by treatment with soluble pSS TNF-a R
[82].
To investigate the in vivo role of TNF-a overproduction
in the CNS, we have generated transgenic mice that ex-
press specifically in neurons of their CNS (see Fig. 2A), a
3’-untranslated region (UTR)-modified murine TNF-a
transgene under the control of its own promoter, (Tg6074
line, muTNF-cx-globin) [83]. From between 3 and 8 weeks
of age, mice spontaneously develop chronic inflammatory
demyelinating disease with 100% phenotypic penetrance.
Mice die prematurely within -8 months after developing
progressively severe neurological symptoms ranging from
mild tremors and ataxia to severe imbalances and seizures.
Other symptoms include loss of the limb flexion reflex,
hind limb paresis, and kyphosis. Histopathological fea-
tures were reactive astrocytosis (Fig. 2B) and microgliosis,
infiltration of the meninges and CNS parenchyma with
macrophages (Fig. 2C) and CD4� and CD8� T lympho-
cytes, and focal demyelination (Fig. 3). The direct action
of TNF-a in the pathogenesis of this disease was confirmed
by peripheral administration of a neutralizing anti-murine
TNF-a antibody. This treatment completely prevented the
development of neurological symptoms, T cell infiltration
into the CNS parenchyma, astrocytosis, and demyeiination,
and greatly reduced the severity of reactive microgliosis.
One of the earliest histopathological changes in the CNS
of Tg6074 mice, and one that is prominent by 3 weeks of
age, is the accumulation of T lymphocytes at the meningeal
surface and their subsequent infiltration into the CNS pa-
renchyma. T cell presence in the CNS is low under normal
conditions, but is markedly increased in inflammatory dis-
eases such as MS [84] and EAE [85] and is thought to be
central to disease pathogenesis [84, 86]. The prevailing
belief is that MS is primarily autoimmune in nature, in-
volving an integrated attack by T cells, B cells, and macro-
phages on the myelin sheath. In both MS and EAE,

Probert et al. Transmembrane and wild-type TNF transgenic mice 523
transgression of the blood-brain barrier by inflammatory
cells is a key initial event in disease initiation [86, 87],
and a similar pathogenetic mechanism may take place in
1g6074 mice. Antibody neutralization experiments show
that disease in Tg6074 mice can be completely prevented
if infiltration of the CNS by inflammatory cells is blocked.
TNF-a overexpression in the CNS of Tg6074 transgenic
mice may directly influence inflammatory cell trafficking
at the blood-brain barrier. TNF-a is known to induce the
expression of adhesion molecules on vascular endothe-
hum, thereby selectively enhancing their adhesiveness for
inflammatory cells [see ref. i]. Moreover, TNF-a is chemo-
tactic for monocytes [42]. It remains to be determined
whether the T cells that occupy the CNS in Tg6074 mice
show specificity for neuronal antigens and whether they
have a pathogenic or immunomodulatory role during the
development of disease.
The demyehination that occurs in Tg6074 mice substan-
tiates a wealth of previous in vitro evidence that has impli-
cated TNF-a as an effector of immune-mediated
demyehination. In addition to its selective cytotoxic effect
on oligodendrocytes [79, 80], TNF-cx-immunoreactive in-
flammatory cells and astrocytes are present in MS plaques
[70], and disease progression in MS has been correlated
with high cerebrospinal fluid levels of TNF-a [88]. There-
fore, demyehination in Tg6074 mice might occur directly
from transgene-expressed TNF-a or as a bystander effect
of locally activated astrocytes, macrophages/microglia, and
activated infiltrating T cells. Alternatively, TNF-a may act
through the induction of further inflammatory molecules
such as nitric oxide, which in turn induces oligodendrocyte
death [89, 90] and affects the normal differentiation and
growth of neuronal precursors [90].
The evidence for a major involvement of TNF-a in the
pathogenesis of inflammatory demyelinating diseases such
as MS and EAE is now strong, and many therapeutical
approaches target the action of this cytokine. EAE can be
inhibited by several anti-TNF-a strategies [81, 82, 91], 1
cell class switching from Thi (TNF-a-producing) to immu-
nomodulatory Th2 cells [92], and the anti-depressant
rohipram, which inhibits T cell activation and TNF-a se-
cretion [93]. Transgenic mice that express TNF-cz in their
CNS, such as the Tg6074 mice described here, represent
important model systems for inflammatory and/or demyeli-
nating diseases in humans, and are expected to aid patho-
genetic studies and the identification of potential targets
for therapeutic intervention.
ACKNOWLEDGMENTS
We thank Drs. Sue Stephens and Roly Foulkes (Celltech
Ltd. UK) for providing us with the anti-human and anti-
murine TNF-a antibodies we use to maintain our trans-
genic lines and Dr. John Simms (Immunex, USA) for the
anti-murine type I IL-i receptor antibody. This work was
supported by the Greek Secretariat for Research and Tech-
nology and European Commission Grants, BIO-CT94-2092
and ERBCHRX-CT930182.
REFERENCES
1. Vassalli, P. (1992) The pathophyaiology of tumor necrosis factors.Annu. Rev.
Immunol. 10,411-452.2. Kriegler, M., Perez, C., Defay, K., Albert, I., Lu, S. D. (1988) A novel form
of TNF-cx/cachectin is a cell surface cytotoxic transmembrane protein:
ramifications for the complex physiology of TNF-u.. Cell 53,45-53.3. Peck, R., Brockhaus, M., Frey, J. R. (1989) Cell surface tumor necrosis factor
(TNF-a) accounts for monocyte- and lymphocyte-mediated killing of TNF-a-resistant target cells. Cell. Immunol. 122, 1-10.
4. Aversa, C., Punnonen, J., De Vries, J. E. (1993) The 26-kD transmembraneform of tumour necrosis factor a on activated CD4� T cell clones provides a
coatimulatory signal for human B cell activation. J. Exp. Med. 177,1575-1585.
5. Macchia, D., Almerigogna, F., Parronchi, P., Ravina, A., Maggi, E., Romag.nani, S. (1993) Membrane tumour necrosis factor-a is involved in thepolyclonsl B-cell activation induced by HI V-infected human T cells. Nature363,464.
6. Vandenbeele, P., Declercq, W., Beyaert, R., Fiers, W. (1995) Two tumournecrosis factor receptors: structure and function Trends Cell Biol. 5,392-399.
7. Hsu, H., Xiong, J., Goeddel, D. V. (1995) The TNF-a receptor 1-associatedprotein TRADD signals cell death and NF-icB activation. Cell 81,495-504.
8. Rothe, M., Wong, S. C., Henzel, W. J., Goeddel, D. V. (1994) A novel familyof putative signal transducers associated with the cytoplasmic domain of the75 kDa tumor necrosis factor receptor. Cell 78, 681-692.
9. Tartaglia, L A., Rothe, M., Hu, Y. F., Goeddel, D. V. (1993) Tumor necrosisfactor’s cytotoxic activity is signaled by the p55 TNF-a receptor. Cell 73,213-216.
10. Rothe, J., Leaslauer, W., Lotacher, H., Lang, Y., Koebel, P., Kontgen, F.,Althage, A., Zinkernagel, R., Steinmetz, M., Bluethmann, H. (1993) Micelacking the tumour necrosis factor receptor fare resistant to TNF.a-mediatedtoxicity but highly susceptible to infection by Li.steria monocytogenes. Nature[London] 364,798-802.
11. Tartaglia, L A., Ayres, T. M., Wong, C. H., Goeddel, D. V. (1993) A noveldomain within the 55 kd TNF.a receptor signals cell death. Cell 74,845-853.
12. Eapevik, T., Brockhsus, M., Loetscher, H., Nonatad, U., Shalaby, R. (1990)Characterization of binding and biological effects of monoclonal antibodiesagainst a human tumor necrosis factor receptor.). Exp. Med. 171, 415-426.
13. Tartaglia, L A., Goeddel, D. V. (1992) Two TNF-a receptors. Immuno).
Today 13, 151-153.14. Zheng, L., Fisher, C., Miller, R. E., Pesehon, J., Lynch, D. H., Lenardo, M.
J. (1995) Induction of apoptosis in mature T cells by tumour necrosis factor.Nature [Lond] 377,348-351.
15. Grell, M., Douni, E., Wajant, H., Lohden, M., Clausa, M., Maxeiner, B.,Georgopoulos, S., Lesalauer, W., Kolliaa, C., Pfizenmaier, K., Scheurich, P.The transmembmne form of tumor necrosis factor (TNF-a) is the primeactivating ligand of the 80 kDa TNF.a receptor Cell, 83, 793-802.
16. Aderka, D., Engelmann, H., Maor, Y., Brakebusch, C., Wallach, D. (1992)Stabilisation of the bioactivity of tumor necrosis factor by its soluble recep-tors. J. Exp. Med. 175,323-329.
17. Shaw, C., Kamen, R. (1986) A conserved AU sequence from the 3’ untrans-lated region of CM-CSF mRNA mediates selective mRNA degradation. Cell46,659-667.
18. Han, J., Brown, T., Beutler, B. (1990) Endotoxin-responsive sequencescontrol cachectin/tumor necrosis factor at the translational level.J. Exp. Med.171,465-475.
19. Kruys, V., Marinx, 0., Shaw, C., Deschamps, J., Huez, C. (1989) Transla-tional blockade imposed by cytokine-derived UA-rich sequences. Science245,852-855.
20. Lewis, M., Tartaglia, L.A., Lee, A., Bennett, C. L, Rice, C. C., Wong, C. H.W., Chen, E. Y., Goeddel, D. V. (1991) Cloning and expression of cDNAafor two distinct murine tumor necrosis factor receptors demonstrate onereceptor is species specific. Proc. Nod. Aced. Sci. USA 88,2830-2834.
21. Perez, C., Albert, I., DeFay, K., Zachariadea, N., Cooding, L, Kriegler, M.(1990) A non-secretable cell surface mutant of tumour necrosis factor(‘FNF.a) kills by cell to cell contact. Cell 63, 251-258.
22. Saxne, T., Palladino, M. A., Heinegard, D., Talal, N., Wollheim, F. A. (1988)Detection of tumor necrosis factor a but not tumor necrosis factor 13 inrheumatoid arthritis synovial fluid and serum. Arthr. Rheumotol. 31,1041-1045.
23. Hopkins, S. J., Meager, A. (1988) Cytokines in synovial fluid: II. The
presence of tumour necrosis factor and interferon. Clin. Exp. Immunol. 73,88-92.
24. Yocum, D. E., Esparza, L, Dubry, S., Benjamin, J. B., Volz, R., Scuderi, P.(1989) Characteristics of tumor necrosis factor production in rheumatoidarthritis. Cell. Immunol. 122, 131-145.
25. Husby, C., Williams, R. C. (1988) Synovial localization of tumor necrosisfactor in patients with rheumatoid arthritis.). Autoimmun. 1,363-371.

524 Journal of Leukocyte Biology Volume 59, April 1996
26. Buchan, C., Barrett, K., Turner, M., Chantry, D., Maini, R. N., Feldmann,M. (1988) Interleukin-1 and tumour necrosis factor mRNA expression inrheumatoid arthritis: prolonged production of IL-i a. Clin. Exp. Immunol.73,449-455.
27. Macnaul, K. W., Hutchinson, N. I., Parsons, J. N., Bayne, E. K., Tocci, M.i. (1990) Analysis of IL-i and TNF-a gene expression in human rheumatoidaynoviocytes and normal monocytes by in situ hybridization. I. Immunol.
145,4154-4166.28. Brennan, F. M., Gibbons, D. L., Mitchell, T., Cope, A. P., Maini, R. N.,
Feldmann, M. (1992) Enhanced expression of tumor necrosis factor receptormRNA and protein in mononuclear cells isolated from rheumatoid arthritissynovial joints. Eur. J. Immunol. 22, 1907-1912.
29. Butler, D. M., Piccoli, D. S., Hart, P. H., Hamilton, J. A. (1988) Stimulation
of human synovial fibroblast DNA synthesis by recombinant human cytoki-nes.). Rheumatol. 15, 1463-1470.
30. Citter, B. D., Labus, J. M., Lees, S. L., Scheetz, M. E. (1989) Characteristicsof human synovial fibroblast activation by IL-113 and TNF-a. Imnwnology66, 196-200.
31. Dayer, J. -M., Beutler, B., Cerami, A. (1985) Cachecti n/tumor necrosis factorstimulates collagenase and prostaglandin E2 production by human synovialcells and dermal fibroblasts. J. Exp. Med. 162,2163-2168.
32. Dayer, J.-M., DeRochemoneix, B., Buruus, B., Demezuk, S., Dinarello, C. A.(1986) Human recombinant interleukin I stimulates collagenase and pro-staglandin E2 production by synovial cells.). Clin. Invest. 77,645-648.
33. Saklatvala, J., Sarsfield, S. J., Townsend, Y. (1985) Pig interleukin 1.
Purification of two immunologically different leukocyte proteins that causecartilage resorption, lymphocyte activation and fever. J. Exp. Med. 162,1208-1222.
34. Saklatvala, J. (1986) Tumour necrosis factor a stimulates resorption andinhibits synthesis of proteoglycan in cartilage. Nature 322,547-549.
35. Bertolini, D. R., Nedwin, C. E., Bringman, T. S., Smith, D. D., Mundy, C. R.(1986) Stimulation of bone resorption and inhibition of bone formation invitro by human tumour necrosis factors. Nature [London] 319, 516-518.
36. Cowan, M., Wood, D. D., Ihrie, E. J., McCuire, M. K. B., Russell, R. C. C.(1983) An interleukin 1-like factor stimulates bone resorption in vitro. Nature[London] 306,378-380.
37. Brennan, F. NI., Chantry, D., Jackson, A., Maini, R., Feldmann, M. (1989)Inhibitory effect of TNF-a antibodies on synovial cell interleukin.1 produc-tion in rheumatoid arthritis. Lancet 2,244-247.
38. Haworth, C., Brennan, F. M., Chantry, D., Turner, M., Maini, R. N., Feld-mann, M. (1991) Expression of granulocyte-macrophage colony-stimulatingfactor in rheumatoid arthritis: regulation by tumor necrosis factor-a. Eur. J.Immunol. 21,2575-2579.
39. Issekutz, A. C., Meager, A., Otterness, I., Issekutz, T. B. (1994) The role oftumour necrosis factor-alpha and IL-i in polymorphonuclear leucocyte andT lymphocyte recruitment to joint inflammation in adjuvant arthritis. Clin.Exp. Immunol. 97,26-32.
40. Keffer, j., Probert, L, Cazlaris, H., Ceorgopoulos, S., Kaslaris, E., Kioussis,D., Kollias, C. (1991) Transgenic mice expressing human tumour necrosisfactor: a predictive genetic model of arthritis. EMBO). 10,4025-4031.
41. Harris, E. D. (1990) Rheumatoid arthritis: pathophysiology and implications
for therapy. N. Engl. J. Med. 322, 1277-1289.42. Wang, J. M., Walter, S., Mantovani, A. (1990) Re-evaluation of the chemo-
tactic activity of tumour necrosis factor for monocytes. Immunology 71,364-367.
43. Aloe, L., Probert, L., Kollias, C., Bracci-Laudiero, L., Spillantini, M. C.,
Levi-Montalcini, R. (1993) The synovium of tranagenic arthritic mice ex-pressing human tumor necrosis factor contains a high level of nerve growthfactor. Growth Factors 9, 149-155.
44. Aloe, L, Probert, L, Kollias, C., Bracci-Laudiero, L, Micera, A., Mollinari,C., Levi-Montalcini, R. (1993) mt. I. Tiss. Reac. 15, 139-143.
45. Akira, S., Hirano, T., Taga, T., Kishimoto, T. (1990) Biology of multifunc-tional cytokines:IL-6 and related molecules (IL-i and TNF-a). FASEB J. 4,2860-2867.
46. Geiger, T., Towbin, H., Cosenti-Vargas, A., Zingel, 0., Arnold, J., Rordorf,C., Clatt, M., Vosbeck, K. (1993) Neutralization of interleukin-1 13activityin vivo with a monoclonal antibody alleviates collagen-induced arthritis inDBA/1 mice and prevents the associated acute-phase response. Clin. Exp.
Rheurnatol. 11,515-522.
47. van den Berg, W. B.,Joosten, L.A. B., Helsen, M., van de Leo, F. A. J. (1994)Amelioration of established murine collagen-induced arthritis with anti-IL-itreatment. Clin. Exp. Immunol. 95, 237-243.
48. Wooley, P. H., Whalen, J. D., Chapman, D. L, Berger, A. E., Richard, K.A., Aspar, D.C., Staite, N. D. (1993) The effect of an interleukin-i receptorantagonist protein on type II collagen-induced arthritis in mice. Arthr.Rheusnatol. 36, 1305-1313.
49. Pmbert, L, Plows, D., Kontogeorgos, C., Kollias, C. (1995) The type Iinterleukin-i receptor acts in series with tumor necrosis factor (TNF-a) toinduce arthritis in TNF-a-transgenic mice. Eur. J. Immunol. 25,1794-1797.
49a. Georgopoulos, S., Plows, D., Kolliaa, C. (1996) Transmembrane TNF issufficient to induce localised tissue toxicity and chronic inflammatory arthri-tis in transgenic mice.). Inflamin, in press.
50. Probert, L., Keffer, J., Corbella, P., Cazlaria, H., Patsavoudi, E., Stephens,S., Kaslaris, E., Kioussis, D., Kollias, C. (1993) Wasting, ischaemia, and
lymphoid abnormalities in mice expressing T cell-targeted human tumornecrosis factor transgenes.). Immunol. 151, 1894.-1906.
51. Sheurich, P., Thoma, B., Ucer, U., Pfizenmaier, K. (1987) lmmunoregulatoiyactivity of recombinant human tumor necrosis factor (‘I’NF-a): induction ofTNF-a receptors on human T cells and TNF-a-mediated enhancement of Tcell responses.). Immunol. 138, 1786-1790.
52. Itoh, N., Nagata, S. (1993) A novel protein domain required for apoptosis.Mutational analysis of human Fas antigen. J. Biol. Chem. 268,10932-10937.
53. Ryffel, B., Brockhaua, M., Durmuller, U., Cudat, F. (1991) Tumor necrosisfactor receptors in lymphoid tissues and lymphomas. Am.). Pathol. 139,7-15.
54. Siegel, S. A., Shealy, D. J., Nakada, M. T., Le, J., Woulfe, D. S., Probed, L.,Kollias, C., Chrayeb, J., Vilcek, J., Daddona, P. E. (1995) The mouse/humanchimeric monoclonal antibody cA2 neutralizes TNF-a in vitro and protects
tranagenic mice from cachexia and TNF-a lethality in vivo. Cytokine 7,15-25.
55. Stolpen, A. H., Cuinan, E. C., Fiers, W., Pober, J. S. (1986) Recombinanttumor necrosis factor and immune interferon act singly and in combinationto reorganize human vascular endothelial cell monolayers. Am. I. Pathol.
123, 16-24.56. Gamble,J. R., Harlan,J. M., Klebanoff, S.J., Vadas, M. A. (1985) Stimulation
of the adherence of neutrophils to umbilical vein endothelium by humanrecombinant tumor necrosis factor. Proc. Nod. Acad. Sci. USA 82,8667-8671.
57. Messadi,D.V.,Pober,J.S.,Fiers,W.,Gimbrone,M.A.,Murphy,G.F.(1987)Induction of an activation antigen on postcapillary venular endothelium inhuman skin organ culture.). Immunol. 139, 1557-1562.
58. Libby, P., Ordovas, J. M., Auger, K. R., Robbins, A. H., Birinyi, L K.,Dinarello, C. A. (1986) Endotoxin and tumor necrosis factor induce inter-leukin-i gene expression in adult human vascular endothelial cells. Am. J.Pathol. 124, 179-185.
59. Nawmth, P. P., Bank, I., Handley, D., Cassimeris, J., Chess, L., Stern, D.(1986) Tumor necrosis factor/cachectin interacts with endothelial cell recep-tors to induce release of interleukin 1.). Ezp. Med. 163, 1363.
60. Nawroth, P. P., Stem, D. M. (1986) Modulation of endothelial cell hemostaticproperties by tumor necrosis factor. J. Exp. Med. 163, 740-745.
61. Bevilacqua, NI. P., Pober, J. S., Majeau, C. R., Fiets, W., Cotran, R. S.,Cimbrone, NI. A. (1986) Recombinant tumor necrosis factor induces proco-agulant activity in cultured human vascular endothelium: characterizationand comparison with the actions of interleukin 1. Proc. Nail. Acad. Sci. USA83,4533-4537.
62. van der Poll, T., Buller, H. R., ten Cate, H., Wortel, C. H., Bauer, K. A., vanDeventer, S. J. H., Hack, C. E., Sauerwein, H. P., Rosenberg, R. D., ten Cate,J. W. (1990) Activation of cosgulation after administration of tumor necrosis
factor to normal subjects. N. Engl. I. Med. 322, 1622-1627.63. Beutler, B., Milsark, I. W., Cerami, A. C. (1985) Passive immunization
against cachectin/tumor necrosis factor protects mice from lethal effect ofendotoxin. Science 229,869-871.
64. Tracey, K. J., Beutler, B., Lowry, S. F., Merryweather, J., Wolpe, S., Milsark,I. W., Hariri, R. J., Fahey, T. J., Zentella, A., Albert, J. D., Shires, C. T.,Cerami, A. (1986) Shock and tissue injury induced by recombinant humancachectin. Science 234,470-473.
65. Oliff, A., Defeo-Jones, D., Boyer, NI., Martinez, U., Kiefer, U., Voucolo, C.,Wolfe, A., Socher, S. H. (1987) Tumors secreting human TNF-a/cachectininduce cachexia in mice. Cell 50, 555-563.
66. Tracey, K. J., Wei, H., Manogue, K. R., Fong, Y., Hesse, D. C., Nguyen, H.T., Kuo, C. C., Beutler, B., Cotran, R. S., Cerami, A., Lowry, S. F. (1988)Cachecti n/tumor necrosis factor induces cachexia, anemia, and inflamma-tion.). Exp. Med. 167, 121 1-1227.
67. Tracey, K. J., Lowry, S. F., Fahey, T. J., Albert, J. D., Fong, Y., Hesse, D.,Beutler, B., Manogue, K. R., Calvano, S., Wei, H., Cerami, A., Shires, C. T.(1987) Cachectin/tumor necrosis factor induces lethal shock and stresshormone responses in the dog. Surg. Gynecol. Obstet. 164,415-422.
68. Breder, C. D., Tsujimoto, M., Terano, Y., Scott, U. W., Saper, C. B. (1993)Distribution and characterization of tumor necrosis factor-a-like immunore-activity in the murine central nervous system. J. Comp. Neural. 337,543-567.
69. Plata-Salaman, C. R., Oomura, Y., Kai, Y. (1988) Tumor necrosis factor andinterleukin-i beta: suppression of food intake by direct action within thecentral nervous system. Brain Ret. .448, 106-i 14.
70. Hofman, F. M., Hinton, D. R., Johnson, K., Merrill, J. E. (1989) Tumornecrosis factor identified in multiple sclerosis brain. J. Exp. Med. 170,607-6i2.
71. Selmaj, K., Raine, C. S., Cannella, B., Bmsnan, C. F. (1991) Identificationof lymphotoxin and tumor necrosis factor in multiple sclerosis lesions.). Clin.Invest. 87,949-954.
72. Tyor, W. R., Class, J. D., Criffin, J. W., Becker, P. S., McArthur, J. C.,Bezman, L, Griffin, D. E. (1992) Ann. Neurol. 31, 349-360.
73. Leist,T. P., Frei, K., Kam-Hansen, S., Zinkernagel, R. M., Fontana, A. (1988)Tumor necrosis factor a in cerebrospinal fluid during bacterial, but not viral,meningitis.). Exp. Med. 167, 1743-1748.
74. Mogi, M., Harada, NI., Riederer, P., Narabayashi, H., Fujita, K., Nagatsu, T.(1994) Tumor necrosis factor- a (TNF-a) increases both in the brain and inthe cerebrospinal fluid from parkinsonian patients. Neurosci. Leu. 165,208-210.

Probert et al. Transmemhrane and wild-type TNF transgenic mice 525
75. Grau, C. E., Piguet, P. -F., Vassalli, P., Lambert, P.-H. (1989) Tumor-ne-crosis factor and other cytokines in cerebral malaria: experimental andclinical data. Immunol. Rev. 189, 49-70.
76. Liebennan, A. P., Pitha, P. NI., Shin, H. S., Shin, M. L. (1989) Production oftumor necrosis factor and other cytokines by astrocytes stimulated withlipopolysaccharide or a neurotropic virus. Proc. Nail. Aced. Sci. USA 86,6348-6352.
77. Frei, K., Siepl, C., Groscurth, P., Bodmer, S., Schwerdel, C., Fontana, A.(1989) Antigen presentation and tumor cytotoxicity by interferon gammatreated microglial cells. Eur. J. Immunol. 19, 127 1-1278.
78. Selmaj, K., Raine, C. S. (1988) Tumor necrosis factor mediates myelin andoligodendmcyte damage in vitro. Ann. Neural. 23,339-346.
79. Selmaj, K., Raine. C. S., Farooq, NI., Norton, W. T., Brosnan, C. F. (1991)Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lym-photoxin. J. Immunol. 147, 1522-1529.
80. Robbins, U.S., Shirazi, Y., Drysdale, B.-E., Lieberman, A., Shin, H. S., Shin,NI. L. (1987) Production of cytotoxic factor foroligodendrocytes by stimulatedastrocytes.). Immunol. 139,2593-2597.
81. Ruddle, N.H., Bergman, C. NI., McGrath, K. NI., Lingenheld, E.G., Cnsnnet,M. L., Padula, S. J., Clark, R. B. (1990) An antibody to lymphotoxin andtumor necrosis factor prevents transfer of experimental allergic encephalo-
myelitia. I. Exp. Med. 173, 1193-1200.82 Selmaj, K., Papierz, W., Glabinski, A., Kohno, T. (1995) Prevention of
chronic relapsing experimental autoimmune encephalomyelitis by solubletumor necrosis factor receptor 1.1. Neuroimmunol. 56, 135-141.
83. Prohert, L, Akassoglou, K., Pasparakis, M., Kontogeorgos, C., Kollias, C.(1995) Spontaneous inflammatory demyelinating disease in transgenic miceshowing CNS-specific expression of tumor necrosis factor a Proc. Nod. Aced.Sci. USA 92, 11294-11298.
84. Raine, C. S. (1991) Multiple sclerosis: a pivotal role for the T cell in lesion
development. Neuropathol. AppI. Neurobiol. 17,265-274.85. Zamvil, S. S., Steinman, L. (1990) The T lymphocyte in experimental allergic
encephalomyelitis. Annu. Rev. Immunol. 8, 579-621.86. Williams, K. C., Ulvestad, E., Hickey, W. F. (1994) Immunology of multiple
sclerosis. Clin. Neurosci. 2, 229-245.87. Wekerle, H., Linington, H., Lassman, H., Meyermann, R. (1986) Cellular
immune reactivity within the CNS. Trends Neurosci. 9,271-277.88. Sharief, M. K., Hentges, R. (1991) Association between tumor necrosis
factor- and disease progression in patients with multiple sclerosis. N. EngI.J. Med. 325,467-472.
89. Merrill,J. E., Ignarm, L. J., Sherman, NI. P., Melinek, J., Lane, T. E. (1993)Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric
oxide.). Immunol, 151 2132-2141.90. Peunova, N., Enikolopov, C. (1995) Nitric oxide triggers a switch to growth
arrest during differentiation of neuronal cells. Nature [London] 375,68-73.91. Baker, D., Butler, D., Scallon, B. J., O’Neill, J.K., Turk, J. L, Feldmann, M.
(1994) Control of established experimental allergic encephalomyelitis by
inhibition of tumor necrosis factor (TNF-a) activity within the central nervoussystem using monoclonal antibodies and TNF-a receptor-immunoglobulinfusion proteins. Eur. I. Immunol. 24,2040-2048.
92. Racke, NI. K., Bonomo, A., Scott, U. E., Cannella, B., Levine, A., Raine, C.S., Shevach, E. NI., Rocken, M. (1994) Cytokine-induced immune deviationas a therapy for inflammatory autoimmune disease. I. Exp. Med. 180,1961-1966.
93. Sommer, N., Loschmann, P. A., Northoff, C. H., Weller, M., Steinhrecher,A., Steinbach, J. P., Lichtenfels, R., Meyennann, R., Riethmuller, A.,Fontana, A., Dichgans, J,, Martin, R. (1995) Nature Med. [London] 244-248.




















