
CIRCONSTANCES DE DÉCOUVERTE DES PATHOLOGIES ...
178
ROYAUME DU MAROC UNIVERSITE MOHAMMED V DE RABAT FACULTE DE MEDECINE ET DE PHARMACIE RABAT CIRCONSTANCES DE DÉCOUVERTE DES PATHOLOGIES HÉMORRAGIQUES CONSTITUTIONNELLES DE L’HÉMOSTASE A PROPOS DE 63 CAS Année : 2019 Présentée est soutenue publiquement le ../06/2019 par Née le 25/02/1993 à Rabat POUR L’OBTENTION DU DIPLÔME DE DOCTEUR EN MEDECINE MOTS CLÉ : ANTÉCÉDENTS FAMILIAUX HÉMORRAGIQUES, HISTOIRE HÉMORRAGIQUE PERSONNELLE, CONSANGUINITÉ, SYMPTOMATOLOGIE HÉMORRAGIQUE, SYMPTOMATOLOGIE NON SPÉCIFIQUE, DÉFICITS EN FACTEURS. Professeur de Pédiatrie Professeur de Pédiatrie Professeur de réanimation médicale Professeur de pédiatrie Professeur de Biologie Moléculaire & Biochimie Mlle Nour Al Houda HAJJI Mme BENJELLOUN DAKHAMA Badr Sououd PRÉSIDENTE RAPPORTEUR JUGES M. EL KHORASSANI Mohamed Mme BELAYACHI Jihane Mme KARBOUBI Lamya M. MASRAR Azlarab MEMBRES DU JURY THÈSE Thèse N° : 300/2019
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of CIRCONSTANCES DE DÉCOUVERTE DES PATHOLOGIES ...

ROYAUME DU MAROCUNIVERSITE MOHAMMED V DE RABAT
FACULTE DE MEDECINE ET DE PHARMACIERABAT
CIRCONSTANCES DE DÉCOUVERTE DES PATHOLOGIES HÉMORRAGIQUES
CONSTITUTIONNELLES DE L’HÉMOSTASE
A PROPOS DE 63 CAS
Année : 2019
Présentée est soutenue publiquement le ../06/2019par
Née le 25/02/1993 à RabatPOUR L’OBTENTION DU DIPLÔME DE DOCTEUR EN MEDECINE
Mots clé : Antécédents fAMiliAux héMorrAgiques, histoire héMorrAgique personnelle, consAnguinité, syMptoMAtologie héMorrAgique, syMptoMAtologie non
spécifique, déficits en fActeurs.
Professeur de Pédiatrie
Professeur de Pédiatrie
Professeur de réanimation médicale
Professeur de pédiatrie
Professeur de Biologie Moléculaire & Biochimie
Mlle Nour Al Houda HAJJI
Mme BENJELLOUN DAKHAMA Badr Sououd PRÉSIDENTE
RAPPORTEUR
JUGES
M. EL KHORASSANI Mohamed
Mme BELAYACHI Jihane
Mme KARBOUBI Lamya
M. MASRAR Azlarab
MEMBRES DU JURY
Thèse
Thèse N° : 300/2019

I don’t believe that life can be counted by years or days, but by experience and how you
leave the world by the way you live.
Jerri nielsen
Ice Bound: A Doctor’s Incredible Battle For Survival at
the South Pole (2001)

DOYENS HONORAIRES :
1962 – 1969 : ProfesseurAbdelmalek FARAJ
1969 – 1974 : Professeur Abdellatif BERBICH
1974 – 1981 : Professeur Bachir LAZRAK
1981 – 1989 : Professeur Taieb CHKILI
1989 – 1997 : Professeur Mohamed Tahar ALAOUI
1997 – 2003 : Professeur Abdelmajid BELMAHI
2003 – 2013 : Professeur Najia HAJJAJ – HASSOUNI
ADMINISTRATION :
Doyen
Professeur Mohamed ADNAOUI
Vice Doyen chargé des Affaires Académiques et estudiantines
Professeur Brahim LEKEHAL
Vice Doyen chargé de la Recherche et de la Coopération
Professeur Taoufiq DAKKA
Vice Doyen chargé des Affaires Spécifiques à la Pharmacie
Professeur Jamal TAOUFIK
Secrétaire Général
Mr. Mohamed KARRA
UNIVERSITE MOHAMMED V
FACULTE DE MEDECINE ET DE PHARMACIE
RABAT

1- ENSEIGNANTS-CHERCHEURS MEDECINS ET PHARMACIENS
PROFESSEURS :
Décembre 1984
Pr. MAAOUNI Abdelaziz Médecine Interne – Clinique Royale
Pr. MAAZOUZI Ahmed Wajdi Anesthésie -Réanimation
Pr. SETTAF Abdellatif pathologie Chirurgicale
Novembre et Décembre 1985
Pr. BENSAID Younes Pathologie Chirurgicale
Janvier, Février et Décembre 1987
Pr. LACHKAR Hassan Médecine Interne
Pr. YAHYAOUI Mohamed Neurologie
Décembre 1989
Pr. ADNAOUI Mohamed Médecine Interne –Doyen de la FMPR
Pr. OUAZZANI Taïbi Mohamed Réda Neurologie
Janvier et Novembre 1990
Pr. HACHIM Mohammed* Médecine-Interne
Pr. KHARBACH Aîcha Gynécologie -Obstétrique
Pr. TAZI Saoud Anas Anesthésie Réanimation
Février Avril Juillet et Décembre 1991
Pr. AZZOUZI Abderrahim Anesthésie Réanimation Doyen de la FMPO
Pr. BAYAHIA Rabéa Néphrologie
Pr. BELKOUCHI Abdelkader Chirurgie Générale
Pr. BENCHEKROUN Belabbes Abdellatif Chirurgie Générale
Pr. BENSOUDA Yahia Pharmacie galénique
Pr. BERRAHO Amina Ophtalmologie
Pr. BEZZAD Rachid Gynécologie Obstétrique Méd Chef Maternité des Orangers
Pr. CHERRAH Yahia Pharmacologie
Pr. CHOKAIRI Omar Histologie Embryologie
Pr. KHATTAB Mohamed Pédiatrie
Pr. SOULAYMANI Rachida Pharmacologie Dir. du Centre National PV Rabat
Pr. TAOUFIK Jamal Chimie thérapeutique V.D à la pharmacie+Dir duCEDOC+Directeur
du Médicament
Décembre 1992
Pr. AHALLAT Mohamed Chirurgie Générale Doyen de FMPT
Pr. BENSOUDA Adil Anesthésie Réanimation
Pr. CHAHED OUAZZANI Laaziza Gastro-Entérologie

Pr. CHRAIBI Chafiq Gynécologie Obstétrique
Pr. EL OUAHABI Abdessamad Neurochirurgie
Pr. FELLAT Rokaya Cardiologie
Pr. GHAFIR Driss* Médecine Interne
Pr. JIDDANE Mohamed Anatomie
Pr. TAGHY Ahmed Chirurgie Générale
Pr. ZOUHDI Mimoun Microbiologie
Mars 1994
Pr. BENJAAFAR Noureddine Radiothérapie
Pr. BEN RAIS Nozha Biophysique
Pr. CAOUI Malika Biophysique
Pr. CHRAIBI Abdelmjid Endocrinologie et Maladies Métaboliques Doyen de la FMPA
Pr. EL AMRANI Sabah Gynécologie Obstétrique
Pr. EL BARDOUNI Ahmed Traumato-Orthopédie
Pr. EL HASSANI My Rachid Radiologie
Pr. ERROUGANI Abdelkader Chirurgie Générale Directeur CHIS -Rabat
Pr. ESSAKALI Malika Immunologie
Pr. ETTAYEBI Fouad Chirurgie Pédiatrique
Pr. HASSAM Badredine Dermatologie
Pr. IFRINE Lahssan Chirurgie Générale
Pr. MAHFOUD Mustapha Traumatologie – Orthopédie
Pr. RHRAB Brahim Gynécologie –Obstétrique
Pr. SENOUCI Karima Dermatologie
Mars 1994
Pr. ABBAR Mohamed* Urologie Directeur Hôpital My Ismail Meknès Pr. ABDELHAK M’barek Chirurgie – Pédiatrique
Pr. BENTAHILA Abdelali Pédiatrie
Pr. BENYAHIA Mohammed Ali Gynécologie – Obstétrique
Pr. BERRADA Mohamed Saleh Traumatologie – Orthopédie
Pr. CHERKAOUI Lalla Ouafae Ophtalmologie
Pr. LAKHDAR Amina Gynécologie Obstétrique
Pr. MOUANE Nezha Pédiatrie
Mars 1995
Pr. ABOUQUAL Redouane Réanimation Médicale
Pr. AMRAOUI Mohamed Chirurgie Générale
Pr. BAIDADA Abdelaziz Gynécologie Obstétrique
Pr. BARGACH Samir Gynécologie Obstétrique
Pr. DRISSI KAMILI Med Nordine* Anesthésie Réanimation
Pr. EL MESNAOUI Abbes Chirurgie Générale
Pr. ESSAKALI HOUSSYNI Leila Oto-Rhino-Laryngologie
Pr. HDA Abdelhamid* Cardiologie

Directeur du Service de Santé des FAR Pr. IBEN ATTYA ANDALOUSSI Ahmed Urologie
Pr. OUAZZANI CHAHDI Bahia Ophtalmologie
Pr. SEFIANI Abdelaziz Génétique
Pr. ZEGGWAGH Amine Ali RéanimationMédicale
Décembre 1996
Pr. AMIL Touriya* Radiologie
Pr. BELKACEM Rachid Chirurgie Pédiatrie
Pr. BOULANOUAR Abdelkrim Ophtalmologie
Pr. EL ALAMI EL FARICHA EL Hassan Chirurgie Générale
Pr. GAOUZI Ahmed Pédiatrie
Pr. MAHFOUDI M’barek* Radiologie
Pr. OUZEDDOUN Naima Néphrologie
Pr. ZBIR EL Mehdi* Cardiologie Directeur Hôp. Mil.d’Instruction Med V Rabat
Novembre 1997
Pr. ALAMI Mohamed Hassan Gynécologie-Obstétrique
Pr. BEN SLIMANE Lounis Urologie
Pr. BIROUK Nazha Neurologie
Pr. ERREIMI Naima Pédiatrie
Pr. FELLAT Nadia Cardiologie
Pr. KADDOURI Noureddine Chirurgie Pédiatrique
Pr. KOUTANI Abdellatif Urologie
Pr. LAHLOU Mohamed Khalid Chirurgie Générale
Pr. MAHRAOUI CHAFIQ Pédiatrie
Pr. TAOUFIQ Jallal Psychiatrie Directeur Hôp. Arrazi Salé
Pr. YOUSFI MALKI Mounia Gynécologie Obstétrique
Novembre 1998
Pr. BENOMAR ALI Neurologie – Doyen de la FMP Abulcassis
Pr. BOUGTAB Abdesslam Chirurgie Générale
Pr. ER RIHANI Hassan Oncologie Médicale
Pr. BENKIRANE Majid* Hématologie
Janvier 2000
Pr. ABID Ahmed* Pneumophtisiologie
Pr. AIT OUMAR Hassan Pédiatrie
Pr. BENJELLOUN Dakhama Badr.Sououd Pédiatrie
Pr. BOURKADI Jamal-Eddine Pneumo-phtisiologie Directeur Hôp. My Youssef
Pr. CHARIF CHEFCHAOUNI Al Montacer Chirurgie Générale
Pr. ECHARRAB El Mahjoub Chirurgie Générale
Pr. EL FTOUH Mustapha Pneumo-phtisiologie
Pr. EL MOSTARCHID Brahim* Neurochirurgie
Pr. MAHMOUDI Abdelkrim* Anesthésie-Réanimation
Pr. TACHINANTE Rajae Anesthésie-Réanimation
Pr. TAZI MEZALEK Zoubida Médecine Interne

Novembre 2000
Pr. AIDI Saadia Neurologie
Pr. AJANA Fatima Zohra Gastro-Entérologie
Pr. BENAMR Said Chirurgie Générale
Pr. CHERTI Mohammed Cardiologie
Pr. ECH-CHERIF EL KETTANI Selma Anesthésie-Réanimation
Pr. EL HASSANI Amine Pédiatrie Directeur Hôp. Chekikh Zaied
Pr. EL KHADER Khalid Urologie
Pr. EL MAGHRAOUI Abdellah* Rhumatologie
Pr. GHARBI Mohamed El Hassan Endocrinologie et Maladies Métaboliques
Pr. MDAGHRI ALAOUI Asmae Pédiatrie
Pr. ROUIMI Abdelhadi* Neurologie
Décembre 2000
Pr. ZOHAIR ABDELAH* ORL
Décembre 2001
Pr. BALKHI Hicham* Anesthésie-Réanimation
Pr. BENABDELJLIL Maria Neurologie
Pr. BENAMAR Loubna Néphrologie
Pr. BENAMOR Jouda Pneumo-phtisiologie
Pr. BENELBARHDADI Imane Gastro-Entérologie
Pr. BENNANI Rajae Cardiologie
Pr. BENOUACHANE Thami Pédiatrie
Pr. BEZZA Ahmed* Rhumatologie
Pr. BOUCHIKHI IDRISSI Med Larbi Anatomie
Pr. BOUMDIN El Hassane* Radiologie
Pr. CHAT Latifa Radiologie
Pr. DAALI Mustapha* Chirurgie Générale
Pr. DRISSI Sidi Mourad* Radiologie
Pr. EL HIJRI Ahmed Anesthésie-Réanimation
Pr. EL MAAQILI Moulay Rachid Neuro-Chirurgie
Pr. EL MADHI Tarik Chirurgie-Pédiatrique
Pr. EL OUNANI Mohamed Chirurgie Générale
Pr. ETTAIR Said Pédiatrie Directeur. Hôp.d’Enfants Rabat
Pr. GAZZAZ Miloudi* Neuro-Chirurgie
Pr. HRORA Abdelmalek Chirurgie Générale
Pr. KABBAJ Saad Anesthésie-Réanimation
Pr. KABIRI EL Hassane* Chirurgie Thoracique
Pr. LAMRANI Moulay Omar Traumatologie Orthopédie
Pr. LEKEHAL Brahim Chirurgie Vasculaire Périphérique
Pr. MAHASSIN Fattouma* Médecine Interne
Pr. MEDARHRI Jalil Chirurgie Générale
Pr. MIKDAME Mohammed* Hématologie Clinique
Pr. MOHSINE Raouf Chirurgie Générale
Pr. NOUINI Yassine Urologie Directeur Hôpital Ibn Sina

Pr. SABBAH Farid Chirurgie Générale
Pr. SEFIANI Yasser Chirurgie Vasculaire Périphérique
Pr. TAOUFIQ BENCHEKROUN Soumia Pédiatrie
Décembre 2002
Pr. AL BOUZIDI Abderrahmane* Anatomie Pathologique
Pr. AMEUR Ahmed * Urologie
Pr. AMRI Rachida Cardiologie
Pr. AOURARH Aziz* Gastro-Entérologie
Pr. BAMOU Youssef * Biochimie-Chimie
Pr. BELMEJDOUB Ghizlene* Endocrinologie et Maladies Métaboliques
Pr. BENZEKRI Laila Dermatologie
Pr. BENZZOUBEIR Nadia Gastro-Entérologie
Pr. BERNOUSSI Zakiya Anatomie Pathologique
Pr. BICHRA Mohamed Zakariya* Psychiatrie
Pr. CHOHO Abdelkrim * Chirurgie Générale
Pr. CHKIRATE Bouchra Pédiatrie
Pr. EL ALAMI EL FELLOUS Sidi Zouhair Chirurgie Pédiatrique
Pr. EL HAOURI Mohamed * Dermatologie
Pr. FILALI ADIB Abdelhai Gynécologie Obstétrique
Pr. HAJJI Zakia Ophtalmologie
Pr. IKEN Ali Urologie
Pr. JAAFAR Abdeloihab* Traumatologie Orthopédie
Pr. KRIOUILE Yamina Pédiatrie
Pr. MABROUK Hfid* Traumatologie Orthopédie
Pr. MOUSSAOUI RAHALI Driss* Gynécologie Obstétrique
Pr. OUJILAL Abdelilah Oto-Rhino-Laryngologie
Pr. RACHID Khalid * Traumatologie Orthopédie
Pr. RAISS Mohamed Chirurgie Générale
Pr. RGUIBI IDRISSI Sidi Mustapha* Pneumophtisiologie
Pr. RHOU Hakima Néphrologie
Pr. SIAH Samir * Anesthésie Réanimation
Pr. THIMOU Amal Pédiatrie
Pr. ZENTAR Aziz* Chirurgie Générale
Janvier 2004
Pr. ABDELLAH El Hassan Ophtalmologie
Pr. AMRANI Mariam Anatomie Pathologique
Pr. BENBOUZID Mohammed Anas Oto-Rhino-Laryngologie
Pr. BENKIRANE Ahmed* Gastro-Entérologie
Pr. BOUGHALEM Mohamed* Anesthésie Réanimation
Pr. BOULAADAS Malik Stomatologie et Chirurgie Maxillo-faciale
Pr. BOURAZZA Ahmed* Neurologie
Pr. CHAGAR Belkacem* Traumatologie Orthopédie
Pr. CHERRADI Nadia Anatomie Pathologique

Pr. EL FENNI Jamal* Radiologie
Pr. EL HANCHI ZAKI Gynécologie Obstétrique
Pr. EL KHORASSANI Mohamed Pédiatrie
Pr. EL YOUNASSI Badreddine* Cardiologie
Pr. HACHI Hafid Chirurgie Générale
Pr. JABOUIRIK Fatima Pédiatrie
Pr. KHARMAZ Mohamed Traumatologie Orthopédie
Pr. MOUGHIL Said Chirurgie Cardio-Vasculaire
Pr. OUBAAZ Abdelbarre* Ophtalmologie
Pr. TARIB Abdelilah* Pharmacie Clinique
Pr. TIJAMI Fouad Chirurgie Générale
Pr. ZARZUR Jamila Cardiologie
Janvier 2005
Pr. ABBASSI Abdellah Chirurgie Réparatrice et Plastique
Pr. AL KANDRY Sif Eddine* Chirurgie Générale
Pr. ALLALI Fadoua Rhumatologie
Pr. AMAZOUZI Abdellah Ophtalmologie
Pr. AZIZ Noureddine* Radiologie
Pr. BAHIRI Rachid Rhumatologie Directeur. Hôp. Al Ayachi Salé
Pr. BARKAT Amina Pédiatrie
Pr. BENYASS Aatif Cardiologie
Pr. DOUDOUH Abderrahim* Biophysique
Pr. EL HAMZAOUI Sakina* Microbiologie
Pr. HAJJI Leila Cardiologie (mise en disponibilité)
Pr. HESSISSEN Leila Pédiatrie
Pr. JIDAL Mohamed* Radiologie
Pr. LAAROUSSI Mohamed Chirurgie Cardio-vasculaire
Pr. LYAGOUBI Mohammed Parasitologie
Pr. RAGALA Abdelhak Gynécologie Obstétrique
Pr. SBIHI Souad Histo-Embryologie Cytogénétique
Pr. ZERAIDI Najia Gynécologie Obstétrique
Avril 2006
Pr. ACHEMLAL Lahsen* Rhumatologie
Pr. AKJOUJ Said* Radiologie
Pr. BELMEKKI Abdelkader* Hématologie
Pr. BENCHEIKH Razika O.R.L
Pr. BIYI Abdelhamid* Biophysique
Pr. BOUHAFS Mohamed El Amine Chirurgie - Pédiatrique
Pr. BOULAHYA Abdellatif* Chirurgie Cardio – Vasculaire
Pr. CHENGUETI ANSARI Anas Gynécologie Obstétrique
Pr. DOGHMI Nawal Cardiologie
Pr. FELLAT Ibtissam Cardiologie
Pr. FAROUDY Mamoun Anesthésie Réanimation
Pr. HARMOUCHE Hicham Médecine Interne

Pr. HANAFI Sidi Mohamed* Anesthésie Réanimation
Pr. IDRISS LAHLOU Amine* Microbiologie
Pr. JROUNDI Laila Radiologie
Pr. KARMOUNI Tariq Urologie
Pr. KILI Amina Pédiatrie
Pr. KISRA Hassan Psychiatrie
Pr. KISRA Mounir Chirurgie – Pédiatrique
Pr. LAATIRIS Abdelkader* Pharmacie Galénique
Pr. LMIMOUNI Badreddine* Parasitologie
Pr. MANSOURI Hamid* Radiothérapie
Pr. OUANASS Abderrazzak Psychiatrie
Pr. SAFI Soumaya* Endocrinologie
Pr. SEKKAT Fatima Zahra Psychiatrie
Pr. SOUALHI Mouna Pneumo – Phtisiologie
Pr. TELLAL Saida* Biochimie
Pr. ZAHRAOUI Rachida Pneumo – Phtisiologie
Decembre 2006
Pr SAIR Khalid Chirurgie générale Dir. Hôp.Av.Marrakech
Octobre 2007
Pr. ABIDI Khalid Réanimation médicale
Pr. ACHACHI Leila Pneumo phtisiologie
Pr. ACHOUR Abdessamad* Chirurgie générale
Pr. AIT HOUSSA Mahdi* Chirurgie cardio vasculaire
Pr. AMHAJJI Larbi* Traumatologie orthopédie
Pr. AOUFI Sarra Parasitologie
Pr. BAITE Abdelouahed* Anesthésie réanimation Directeur ERSSM
Pr. BALOUCH Lhousaine* Biochimie-chimie
Pr. BENZIANE Hamid* Pharmacie clinique
Pr. BOUTIMZINE Nourdine Ophtalmologie
Pr. CHARKAOUI Naoual* Pharmacie galénique
Pr. EHIRCHIOU Abdelkader* Chirurgie générale Pr. EL BEKKALI Youssef * Chirurgie cardio-vasculaire
Pr. ELABSI Mohamed Chirurgie générale
Pr. EL MOUSSAOUI Rachid Anesthésie réanimation
Pr. EL OMARI Fatima Psychiatrie
Pr. GHARIB Noureddine Chirurgie plastique et réparatrice
Pr. HADADI Khalid* Radiothérapie
Pr. ICHOU Mohamed* Oncologie médicale
Pr. ISMAILI Nadia Dermatologie
Pr. KEBDANI Tayeb Radiothérapie
Pr. LALAOUI SALIM Jaafar* Anesthésie réanimation
Pr. LOUZI Lhoussain* Microbiologie
Pr. MADANI Naoufel Réanimation médicale
Pr. MAHI Mohamed* Radiologie

Pr. MARC Karima Pneumo phtisiologie
Pr. MASRAR Azlarab Hématologie biologique
Pr. MRANI Saad* Virologie
Pr. OUZZIF Ez zohra* Biochimie-chimie
Pr. RABHI Monsef* Médecine interne
Pr. RADOUANE Bouchaib* Radiologie
Pr. SEFFAR Myriame Microbiologie
Pr. SEKHSOKH Yessine* Microbiologie
Pr. SIFAT Hassan* Radiothérapie
Pr. TABERKANET Mustafa* Chirurgie vasculaire périphérique
Pr. TACHFOUTI Samira Ophtalmologie
Pr. TAJDINE Mohammed Tariq* Chirurgie générale
Pr. TANANE Mansour* Traumatologie orthopédie
Pr. TLIGUI Houssain Parasitologie
Pr. TOUATI Zakia Cardiologie
Décembre 2008
Pr TAHIRI My El Hassan* Chirurgie Générale
Mars 2009
Pr. ABOUZAHIR Ali* Médecine interne
Pr. AGDR Aomar* Pédiatre
Pr. AIT ALI Abdelmounaim* Chirurgie Générale
Pr. AIT BENHADDOU El hachmia Neurologie
Pr. AKHADDAR Ali* Neuro-chirurgie
Pr. ALLALI Nazik Radiologie
Pr. AMINE Bouchra Rhumatologie
Pr. ARKHA Yassir Neuro-chirurgie Directeur Hôp.des Spécialités
Pr. BELYAMANI Lahcen* Anesthésie Réanimation
Pr. BJIJOU Younes Anatomie
Pr. BOUHSAIN Sanae* Biochimie-chimie
Pr. BOUI Mohammed* Dermatologie
Pr. BOUNAIM Ahmed* Chirurgie Générale
Pr. BOUSSOUGA Mostapha* Traumatologie orthopédique
Pr. CHTATA Hassan Toufik* Chirurgie vasculaire périphérique
Pr. DOGHMI Kamal* Hématologie clinique
Pr. EL MALKI Hadj Omar Chirurgie Générale
Pr. EL OUENNASS Mostapha* Microbiologie
Pr. ENNIBI Khalid* Médecine interne
Pr. FATHI Khalid Gynécologie obstétrique
Pr. HASSIKOU Hasna * Rhumatologie
Pr. KABBAJ Nawal Gastro-entérologie
Pr. KABIRI Meryem Pédiatrie
Pr. KARBOUBI Lamya Pédiatrie
Pr. LAMSAOURI Jamal* Chimie Thérapeutique

Pr. MARMADE Lahcen Chirurgie Cardio-vasculaire
Pr. MESKINI Toufik Pédiatrie
Pr. MESSAOUDI Nezha * Hématologie biologique
Pr. MSSROURI Rahal Chirurgie Générale
Pr. NASSAR Ittimade Radiologie
Pr. OUKERRAJ Latifa Cardiologie
Pr. RHORFI Ismail Abderrahmani * Pneumo-phtisiologie
Octobre 2010
Pr. ALILOU Mustapha Anesthésie réanimation
Pr. AMEZIANE Taoufiq* Médecine interne
Pr. BELAGUID Abdelaziz Physiologie
Pr. CHADLI Mariama* Microbiologie
Pr. CHEMSI Mohamed* Médecine aéronautique
Pr. DAMI Abdellah* Biochimie chimie
Pr. DARBI Abdellatif* Radiologie
Pr. DENDANE Mohammed Anouar Chirurgie pédiatrique
Pr. EL HAFIDI Naima Pédiatrie
Pr. EL KHARRAS Abdennasser* Radiologie
Pr. EL MAZOUZ Samir Chirurgie plastique et réparatrice
Pr. EL SAYEGH Hachem Urologie
Pr. ERRABIH Ikram Gastro entérologie
Pr. LAMALMI Najat Anatomie pathologique
Pr. MOSADIK Ahlam Anesthésie Réanimation
Pr. MOUJAHID Mountassir* Chirurgie générale
Pr. NAZIH Mouna* Hématologie biologique
Pr. ZOUAIDIA Fouad Anatomie pathologique
Decembre 2010 Pr.ZNATI Kaoutar Anatomie Pathologique
Mai 2012
Pr. AMRANI Abdelouahed Chirurgie Pédiatrique
Pr. ABOUELALAA Khalil* Anesthésie Réanimation
Pr. BENCHEBBA Driss* Traumatologie Orthopédique
Pr. DRISSI Mohamed* Anesthésie Réanimation
Pr. EL ALAOUI MHAMDI Mouna Chirurgie Générale
Pr. EL KHATTABI Abdessadek* Médecine Interne
Pr. EL OUAZZANI Hanane* Pneumophtisiologie
Pr. ER-RAJI Mounir Chirurgie Pédiatrique
Pr. JAHID Ahmed Anatomie pathologique
Pr. MEHSSANI Jamal* Psychiatrie
Pr. RAISSOUNI Maha* Cardiologie *Enseignants Militaires

Février 2013
Pr. AHID Samir Pharmacologie – Chimie
Pr. AIT EL CADI Mina Toxicologie
Pr. AMRANI HANCHI Laila Gastro-Entérologie
Pr. AMOUR Mourad Anesthésie Réanimation
Pr. AWAB Almahdi Anesthésie Réanimation
Pr. BELAYACHI Jihane Réanimation Médicale
Pr. BELKHADIR Zakaria Houssain Anesthésie Réanimation
Pr. BENCHEKROUN Laila Biochimie-Chimie
Pr. BENKIRANE Souad Hématologie biologique
Pr. BENNANA Ahmed* Informatique Pharmaceutique
Pr. BENSGHIR Mustapha* Anesthésie Réanimation
Pr. BENYAHIA Mohammed* Néphrologie
Pr. BOUATIA Mustapha Chimie Analytique et Bromatologie
Pr. BOUABID Ahmed Salim* Traumatologie Orthopédie
Pr. BOUTARBOUCH Mahjouba Anatomie
Pr. CHAIB Ali* Cardiologie
Pr. DENDANE Tarek Réanimation Médicale
Pr. DINI Nouzha* Pédiatrie
Pr. ECH-CHERIF EL KETTANI Mohamed Ali Anesthésie Réanimation
Pr. ECH-CHERIF EL KETTANI Najwa Radiologie
Pr. ELFATEMI Nizare Neuro-Chirurgie
Pr. EL GUERROUJ Hasnae Médecine Nucléaire
Pr. EL HARTI Jaouad Chimie Thérapeutique
Pr. EL JOUDI Rachid* Toxicologie
Pr. EL KABABRI Maria Pédiatrie
Pr. EL KHANNOUSSI Basma Anatomie Pathologie
Pr. EL KHLOUFI Samir Anatomie
Pr. EL KORAICHI Alae Anesthésie Réanimation
Pr. EN-NOUALI Hassane* Radiologie
Pr. ERRGUIG Laila Physiologie
Pr. FIKRI Meryim Radiologie
Pr. GHFIR Imade Médecine Nucléaire
Pr. IMANE Zineb Pédiatrie
Pr. IRAQI Hind Endocrinologie et maladies métaboliques
Pr. KABBAJ Hakima Microbiologie
Pr. KADIRI Mohamed* Psychiatrie
Pr. LATIB Rachida Radiologie
Pr. MAAMAR Mouna Fatima Zahra Médecine Interne
Pr. MEDDAH Bouchra Pharmacologie
Pr. MELHAOUI Adyl Neuro-chirurgie
Pr. MRABTI Hind Oncologie Médicale
Pr. NEJJARI Rachid Pharmacognosie
Pr. OUBEJJA Houda Chirurgie Pédiatrique
Pr. OUKABLI Mohamed* Anatomie Pathologique

Pr. RAHALI Younes Pharmacie Galénique
Pr. RATBI Ilham Génétique
Pr. RAHMANI Mounia Neurologie
Pr. REDA Karim* Ophtalmologie
Pr. REGRAGUI Wafa Neurologie
Pr. RKAIN Hanan Physiologie
Pr. ROSTOM Samira Rhumatologie
Pr. ROUAS Lamiaa Anatomie Pathologique
Pr. ROUIBAA Fedoua* Gastro-Entérologie
Pr. SALIHOUN Mouna Gastro-Entérologie
Pr. SAYAH Rochde Chirurgie Cardio-Vasculaire
Pr. SEDDIK Hassan* Gastro-Entérologie
Pr. ZERHOUNI Hicham Chirurgie Pédiatrique
Pr. ZINE Ali* Traumatologie Orthopédie
Avril 2013
Pr. EL KHATIB Mohamed Karim* Stomatologie et Chirurgie Maxillo-faciale
MAI 2013
Pr.BOUSLIMAN Yassir Toxicologie
MARS 2014
Pr. ACHIR Abdellah Chirurgie Thoracique
Pr. BENCHAKROUN Mohammed * Traumatologie- Orthopédie
Pr. BOUCHIKH Mohammed Chirurgie Thoracique
Pr. EL KABBAJ Driss * Néphrologie
Pr. EL MACHTANI IDRISSI Samira * Biochimie-Chimie
Pr. HARDIZI Houyam Histologie- Embryologie-Cytogénétique
Pr. HASSANI Amale * Pédiatrie
Pr. HERRAK Laila Pneumologie
Pr. JANANE Abdellah * Urologie
Pr. JEAIDI Anass * Hématologie Biologique
Pr. KOUACH Jaouad* Génycologie-Obstétrique
Pr. LEMNOUER Abdelhay* Microbiologie
Pr. MAKRAM Sanaa * Pharmacologie
Pr. OULAHYANE Rachid* Chirurgie Pédiatrique
Pr. RHISSASSI Mohamed Jaafar CCV
Pr. SABRY Mohamed* Cardiologie
Pr. SEKKACH Youssef* Médecine Interne
Pr. TAZI MOUKHA Zakia Génécologie-Obstétrique
AVRIL 2014
Pr.ZALAGH Mohammed ORL

PROFESSEURS AGREGES :
DECEMBRE 2014
Pr. ABILKASSEM Rachid* Pédiatrie
Pr. AIT BOUGHIMA Fadila Médecine Légale
Pr. BEKKALI Hicham * Anesthésie-Réanimation
Pr. BENAZZOU Salma Chirurgie Maxillo-Faciale
Pr. BOUABDELLAH Mounya Biochimie-Chimie
Pr. BOUCHRIK Mourad* Parasitologie
Pr. DERRAJI Soufiane* Pharmacie Clinique
Pr. DOBLALI Taoufik* Microbiologie
Pr. EL AYOUBI EL IDRISSI Ali Anatomie
Pr. EL GHADBANE Abdedaim Hatim* Anesthésie-Réanimation
Pr. EL MARJANY Mohammed* Radiothérapie
Pr. FEJJAL Nawfal Chirurgie Réparatrice et Plastique
Pr. JAHIDI Mohamed* O.R.L
Pr. LAKHAL Zouhair* Cardiologie
Pr. OUDGHIRI Nezha Anesthésie-Réanimation
Pr. RAMI Mohamed Chirurgie Pédiatrique
Pr. SABIR Maria Psychiatrie
Pr. SBAI IDRISSI Karim* Médecine préventive, santé publique et Hyg.
AOUT 2015
Pr. MEZIANE Meryem Dermatologie
Pr. TAHRI Latifa Rhumatologie
JANVIER 2016
Pr. BENKABBOU Amine Chirurgie Générale
Pr. EL ASRI Fouad* Ophtalmologie
Pr. ERRAMI Noureddine* O.R.L
Pr. NITASSI Sophia O.R.L
JUIN 2017
Pr. ABI Rachid* Microbiologie
Pr. ASFALOU Ilyasse* Cardiologie
Pr. BOUAYTI El Arbi* Médecine préventive, santé publique et Hyg.
Pr. BOUTAYEB Saber Oncologie Médicale
Pr. EL GHISSASSI Ibrahim Oncologie Médicale
Pr. OURAINI Saloua* O.R.L
Pr. RAZINE Rachid Médecine préventive, santé publique et Hyg.
Pr. ZRARA Abdelhamid* Immunologie
* Enseignants Militaires

2- ENSEIGNANTS – CHERCHEURS SCIENTIFIQUES
PROFESSEURS / PRs. HABILITES
Pr. ABOUDRAR Saadia Physiologie
Pr. ALAMI OUHABI Naima Biochimie – chimie
Pr. ALAOUI Katim Pharmacologie
Pr. ALAOUI SLIMANI Lalla Naïma Histologie-Embryologie
Pr. ANSAR M’hammed Chimie Organique et Pharmacie Chimique Pr. BARKIYOU Malika Histologie-Embryologie
Pr. BOUHOUCHE Ahmed Génétique Humaine
Pr. BOUKLOUZE Abdelaziz Applications Pharmaceutiques
Pr. CHAHED OUAZZANI Lalla Chadia Biochimie – chimie
Pr. DAKKA Taoufiq Physiologie Pr. FAOUZI Moulay El Abbes Pharmacologie
Pr. IBRAHIMI Azeddine Biologie moléculaire/Biotechnologie
Pr. KHANFRI Jamal Eddine Biologie
Pr. OULAD BOUYAHYA IDRISSI Med Chimie Organique
Pr. REDHA Ahlam Chimie
Pr. TOUATI Driss Pharmacognosie
Pr. ZAHIDI Ahmed Pharmacologie
Mise à jour le 10/10/2018
Khaled Abdellah
Chef du Service des Ressources Humaines

DÉDICACES

À ma mère, la lumière de ma vie et mon cher soutien.
À mon père, le pilier de notre famille et sa force.
À ma sœur Hana, ma force et ma faiblesse.
À mon frère Jamal, mon héros et ami.À Salma Majidi, le nouveau sourire de la famille et ma chère belle-sœur.À toute ma famille maternelle, qui m’a vu grandir et qui m’a bercé d’amour et de joie pendant toute ma
vie.À toute ma famille paternelle, qui est un rappel de bonté, une terre de compassion et une main tendue
quand j’en ai besoin.À ma meilleure amie Lina, la douce
et la confidente fidèle.À Khaoula, Lhajja et à toute sa famille, mon second foyer et refuge.À toutes mes ami(e)s : Khadija, Sara, Maryam, Imane, Jihane, Mouna, Monya, Meryem, Hind, Intissar, Badiaa, Rihab, Ikrame, Hamza, Nawfel, Anas, mes compagnons de
route.

REMERCIEMENTS

À Notre Maître, Présidente du jury Madame le Professeur BENJELLOUN Dakhama
Badr Sououd Pour avoir accepté d’être la présidente du jury de ce mémoire. Vous nous avez marqué autant sur le
niveau académique que humain; Votre compétence et savoir, et vos qualités humaines, sont incontestablement un exemple à suivre pour tout médecin désirant atteindre le niveau élevé où vous
vous tenez aujourd’hui. Veuillez accepter, chère Maître, l’assurance de notre profond respect.

À Notre Maître, Rapporteur de thèse Monsieur le Professeur EL KHORASSANI Mohamed Professeur de Pédiatrie Pour avoir encadré, avec professionnalisme et passion, ce
travail de recherche. Vous m’avez été d’une grande aide, et sans votre chère et précieuse contribution, ce travail n’aurait pas atteint la maturité nécessaire à son
accomplissement. Veuillez trouver ici, cher Maître, l’expression de ma respectueuse
considération

À Nos Maîtres et Juges de thèse Monsieur le professeur MASRAR
Azlarab Madame la professeure KARBOUBI
LamyaMadame la professeure BELAYACHI
Jihane Pour nous avoir honorer de leur présence et de leur contribution à l’évaluation de ce travail de fin d’étude. Votre apport à ce travail est indéniablement d’une grande valeur. Veuillez accepter mon respect, mon admiration et remerciements Envers votre compétence et votre qualité
d’enseignement

SOMMAIRE INTRODUCTION GÉNÉRALE 1RAPPEL PHYSIOLOGIQUE DE L’HÉMOSTASE 3 I. Généralités 4 II. Physiologie de l’hémostase primaire 6 1. Facteurs de l’hémostase primaire 6 1.1. La paroi vasculaire 6 1.2. Les plaquettes 7 1.3. Le facteur Von Willebrand 8 1.4. Le fibrinogène 9 2. Mise en jeu des différents paramètres de l’hémostase primaire 9 2.1. Le spasme vasculaire 9 2.2. L’adhésion plaquettaire 9 2.3. L’activation plaquettaire 9 2.4. L’agrégation plaquettaire 9 III. Physiologie de la coagulation 12 1. Les cellules et facteurs impliqués 12 2. Facteurs de la coagulation et leurs inhibiteurs 13 3. Activation de la coagulation 14 3.1. Représentation classique 14 3.2. Représentation moderne 14 4. Mécanismes de régulation 17 IV. Physiologie de la fibrinolyse 19PARTICULARITÉS DE L’HÉMOSTASE EN PÉDIATRIE 20 I. Généralités : 21 II. Particularités physiologiques de l’hémostase du nouveau-né, du nourrisson et de l’enfant 23 1. L’hémostase primaire 23 2. La coagulation plasmatique 24 3. Les facteurs de la coagulation 24 4. Les inhibiteurs de la coagulation 27 5. Les composantes de la fibrinolyse 27 III. Implications cliniques 28
PREMIÈRE PARTIE LES PATHOLOGIES HÉMORRAGIQUES CONSTITUTIONNELLES DE L’HÉMOSTASE ET LEURS CIRCONSTANCES DE DÉCOUVERTE I. Les pathologies de l’hémostase primaire 30 1. La maladie de Willebrand 30 1.1. Historique 30 1.2. Définition 31 1.3. La classification 31 1.4. Epidémiologie 32 1.5. Le diagnostic positif 32 2. Les thrombopathies constitutionnelles 33 2.1. La thrombasthénie de Glanzmann 34

2.1.1. Historique 34 2.1.2. Définition 35 2.1.3. Le diagnostic positif 35 2.2. Le syndrome de Bernard-soulier 37 2.2.1. Historique 37 2.2.2. Définition 38 2.2.3. Le diagnostic positif 39 2.3. Les autres thrombopathies 40 3. Les thrombopénies constitutionnelles 40 3.1. Généralités 40 3.2. La classification 41 3.3. À propos d’un cas de la littérature 43 3.4. Démarche diagnostique 44 II. Pathologies de la coagulation 47 1. L’hémophilie 47 1.1. Historique 47 1.2. Définition 48 1.3. Epidémiologie 48 1.4. Génétique 49 1.5. Le diagnostic biologique 50 2. Les déficits rares de la coagulation 50 III. Pathologies de la fibrinolyse 55 1. Déficit en α2-antiplasmine 55 2. Déficit en PAI-1 56 IV. Circonstances de découverte des pathologies constitutionnelles de l’hémostase 57 1. Symptomatique 57 1.1. Symptomatologie hémorragique 57 1.2. Symptomatologie non spécifique 69 2 .Découverte fortuite 72 V. Démarche diagnostique devant un syndrome hémorragique de l’enfant 73 1. Syndrome hémorragique clinique 73 1.1. L’interrogatoire 73 1.2. L’Examen physique 75 1.3. Les éléments cliniques de gravité 75 2. Particularités des phases de traitement biologique 76 2.1. L’étape pré-analytique 76 2.2. L’étape analytique 77 2.3. L’étape post analytique 78 3. Exploration d’un syndrome hémorragique 78 3.1. L’exploration de l’hémostase primaire 79 3.2. Exploration de la coagulation 81
DEUXIÈME PARTIE ETUDE RÉTROSPECTIVE DESCRIPTIVE ET ANALYTIQUE: À PROPOS DE 63 CAS
OBJECTIFS, MATÉRIELS ET MÉTHODES 85 1. Objectifs de l’étude 86

2. Matériels et méthodes 86 2.1. Critères d’inclusion 86 2.2. Critères d’exclusion 87 2.3. Matériel 87 2.4. Le circuit des patients 87
RÉSULTATS 92 I. Données épidémiologiques 95 1. Répartition selon l’année d’admission 95 2. Répartition selon le sexe 95 3. Répartition selon l’âge 96 3.1. Age à l’admission 96 3.2. Age au diagnostic 97 3.3. Age des premiers symptômes hémorragiques 98 4. Répartition selon l’origine géographique 99 5. Répartition selon le niveau socio-économique 100 II. Antécédents familiaux 101 1. Consanguinité 101 2. Maladie hématologique hémorragique au sein de la famille 102 3. Décès dans la famille lié à une pathologie de l’hémostase 104 III. Antécédents personnels 106 1. Conduite des parents face à ces premiers événements hémorragiques 106 2. Age de survenue du premier antécédent hémorragique 107 3. Nombre d’épisodes hémorragiques 108 4. Evénement déclenchant 109 5. Type de saignement 110 IV. Circonstances de découverte 111 1. Symptomatologie hémorragique 112 1.1. Type de saignement 112 1.2. le mode d’apparition 114 1.3. Gravité du saignement 115 1.3.1. Degré du saignement 115 1.3.2. Transfusion 116 1.3.3. Etat hémodynamique à l’admission 116 2. Découverte fortuite 117 3. Enquête familiale 118 4. Symptomatologie non spécifique 118
DISCUSSION 119
LIMITES DE L’ÉTUDE 132
CONCLUSION 134
BIBLIOGRAPHIE 138

LISTE DES FIGURESFigure 1 : Caillot sanguinFigure 2 : Étapes de l’hémostase primaireFigure 3: Coupe histologique transversale d’une artèreFigure 4 : Image reconstituée en 3D des plaquettes sanguinesFigure 5: Facteur de Von WillebrandFigure 6 : Rôle des plaquettes dans l’hémostase primaireFigure 7 : FibrinoformationFigure 8 : Caillot d’hémostaseFigure 9 : La cascade de la coagulationFigure10 : Portrait de Erik Adolf Von WillebrandFigure 11 : Portrait d’Edouard GlanzmannFigure 12 : Platelet function analyser (PFA)Figure13 : Aspect d’une Thrombasthénie de Glanzmann sur un test fonctionnel plaquettaireFigure14 : Quatre hématologistes au congrès de Buffalo (1949) de gauche à droite: Jean Pierre Soulier, Marcel Bessis, Jean Bernard, Jean Dausset (DR).Figure 15 : Pseudocorps de Döhle (B).Figure16 : Portrait de la reine Victoria.Figure 17 : Hématome suite à une ponction veineuse chez un enfant hémophile de3ans.Figure 18 : Hémarthrose de la cheville.Figure 19 : Hémarthrose du genou.Figure 20 : Ecchymoses (stades de la biligénie).Figure 21 : TDM cérébrale d’un enfant de 5ans qui a présenté des troubles de la conscience. Elle montre des hématomes sous duraux : Frontopariétal droit mesurant 11mm et Basitemporal droit mesurant 4,5mm. Déficit en facteur VI.Figure 22 : Hématome extradural mesurant 5 mm d’épaisseur associé à une fracture pariétale gauche chez un nourrisson de 7 mois qui a eu un traumatisme crânien. Diagnostic retenu: hémophilie B.Figure 23 : Tube miniCollect®, de chez Greiner.Figure 24 : Conduite à tenir devant un allongement du temps de saignement.Figure 25 : Déficits en facteurs selon les résultats de la TCA et du temps de Quick.Figure 26 : Arbre décisionnel devant la diminution du TQ et l’allongement du TCA.Figure 27 : Graphe montrant la répartition des cas selon leur provenance.Figure 28 : Répartition des cas selon la pathologie.Figure 29 : Répartition des cas selon le sexe.Figure 30 : Carte du Maroc montrant la répartition des cas étudiés sur les 12 régions.Figure 31 : Distribution des cas selon leur couverture sociale.Figure 32 : Pourcentage de consanguinité dans notre série.Figure 33 : Répartition selon le degré de consanguinité.Figure 34 : Le nombre de cas ayant une fratrie atteinte de la même pathologie.Figure35 : Répartition de la maladie hémorragique au sein de la famille (en dehors de la fratrie).

Figure 36 : Le pourcentage de décès lié à une pathologie de l’hémostase dans la famille.Figure 37 : Distribution des patients selon la présence ou non d’antécédents hémorragiques personnels.Figure 38 : Pourcentage de patients ayant consultés devant le premier symptôme hémorragique.Figure 39 : Répartition selon le nombre d’épisodes hémorragiques antécédents.Figure 40 : La distribution selon le mécanisme déclenchant.Figure 41 : Pourcentage de chaque type de saignement.Figure 42 : Circonstances de découverte des pathologies hémorragiques héréditaires en pourcentage.Figure 43 : Distribution des patients selon le type de saignement.Figure 44 : Pourcentage des différents types de saignements muqueux.Figure 45 : Répartition selon le mode d’apparition.Figure 46 : Répartition des cas selon la gravité de l’hémorragie.Figure 47 : Distribution des cas selon la nécessité d’une transfusion.Figure 48 : Répartition des cas selon leur état hémodynamique.Figure 49 : Distribution selon le type de bilan fortuit.LISTE DES FIGURES Figure 1 : Caillot sanguinFigure 2 : Étapes de l’hémostase primaireFigure 3: Coupe histologique transversale d’une artèreFigure 4 : Image reconstituée en 3D des plaquettes sanguinesFigure 5: Facteur de Von WillebrandFigure 6 : Rôle des plaquettes dans l’hémostase primaireFigure 7 : FibrinoformationFigure 8 : Caillot d’hémostaseFigure 9 : La cascade de la coagulationFigure10 : Portrait de Erik Adolf Von WillebrandFigure 11 : Portrait d’Edouard GlanzmannFigure 12 : Platelet function analyser (PFA)Figure13 : Aspect d’une Thrombasthénie de Glanzmann sur un test fonctionnel plaquettaireFigure14 : Quatre hématologistes au congrès de Buffalo (1949) de gauche à droite: Jean Pierre Soulier, Marcel Bessis, Jean Bernard, Jean Dausset (DR).Figure 15 : Pseudocorps de Döhle (B).Figure16 : Portrait de la reine Victoria.Figure 17 : Hématome suite à une ponction veineuse chez un enfant hémophile de3ans.Figure 18 : Hémarthrose de la cheville.Figure 19 : Hémarthrose du genou.Figure 20 : Ecchymoses (stades de la biligénie).Figure 21 : TDM cérébrale d’un enfant de 5ans qui a présenté des troubles de la conscience. Elle montre des hématomes sous duraux : Frontopariétal droit mesurant 11mm et Basitemporal droit mesurant 4,5mm. Déficit en facteur VI.Figure 22 : Hématome extradural mesurant 5 mm d’épaisseur associé à une fracture pariétale gauche chez un nourrisson de 7 mois qui a eu un traumatisme crânien. Diagnostic retenu:

hémophilie B.Figure 23 : Tube miniCollect®, de chez Greiner.Figure 24 : Conduite à tenir devant un allongement du temps de saignement.Figure 25 : Déficits en facteurs selon les résultats de la TCA et du temps de Quick.Figure 26 : Arbre décisionnel devant la diminution du TQ et l’allongement du TCA.Figure 27 : Graphe montrant la répartition des cas selon leur provenance.Figure 28 : Répartition des cas selon la pathologie.Figure 29 : Répartition des cas selon le sexe.Figure 30 : Carte du Maroc montrant la répartition des cas étudiés sur les 12 régions.Figure 31 : Distribution des cas selon leur couverture sociale.Figure 32 : Pourcentage de consanguinité dans notre série.Figure 33 : Répartition selon le degré de consanguinité.Figure 34 : Le nombre de cas ayant une fratrie atteinte de la même pathologie.Figure35 : Répartition de la maladie hémorragique au sein de la famille (en dehors de la fratrie).Figure 36 : Le pourcentage de décès lié à une pathologie de l’hémostase dans la famille.Figure 37 : Distribution des patients selon la présence ou non d’antécédents hémorragiques personnels.Figure 38 : Pourcentage de patients ayant consultés devant le premier symptôme hémorragique.Figure 39 : Répartition selon le nombre d’épisodes hémorragiques antécédents.Figure 40 : La distribution selon le mécanisme déclenchant.Figure 41 : Pourcentage de chaque type de saignement.Figure 42 : Circonstances de découverte des pathologies hémorragiques héréditaires en pourcentage.Figure 43 : Distribution des patients selon le type de saignement.Figure 44 : Pourcentage des différents types de saignements muqueux.Figure 45 : Répartition selon le mode d’apparition.Figure 46 : Répartition des cas selon la gravité de l’hémorragie.Figure 47 : Distribution des cas selon la nécessité d’une transfusion.Figure 48 : Répartition des cas selon leur état hémodynamique.Figure 49 : Distribution selon le type de bilan fortuit.

LISTE DES TABLEAUXTableau 1 : Facteurs et protéines de la coagulationTableau 2 : Principales études définissant les valeurs de référence pour les paramètres d’hé-mostase dans des populations pédiatriques. Principales caractéristiques démographiques des populations étudiées.Tableau 3 : valeurs de référence en pédiatrie selon Andrew (A) et Monagle (M).Tableau 4 : La classification SSC-ISTH de la maladie de WillebrandTableau 5 : Thrombopénies constitutionnelles. Classification en fonction de l’anomalie génétique.Tableau 6 : Principales thrombopénies constitutionnelles. Classification en fonction de la taille des plaquettes et du caractère isolé ou syndromique.Tableau 7 : Some features of rare coagulation disorders.Tableau 8 : Caractéristiques différenciant les déficits rares de la coagulation d’une hémo-philie d’après S.S. Acharya.Tableau 9 : Initial features suggestives of pathologic bleeding in children.Tableau 10 : Nombre et Pourcentage de chaque pathologie.Tableau 11 : Répartition des cas selon leur année d’admission.Tableau 12 : Répartition selon l’âge à l’admission au CHOP.Tableau 13 : Répartition selon l’âge des patients au moment du diagnostic.Tableau 14 : Tableau montrant l’âge des patients aux premiers symptômes hémorragiques.Tableau 15 : Âge de survenue du 1er antécédent de nature hémorragique.Tableau 16 : Les mécanismes déclenchant les hémorragies et leur effectif.Tableau 17 : Anomalies biologiques de l’hémostase rencontrées.Tableau 18 : Comparaison de la fréquence des pathologies hémorragiques héréditaires dans 3 études différentes.Tableau 19 : Comparaison des Antécédents hémorragiques de nos patients avec ceux d’une étude faite au Caire.Tableau 20 : Présentation clinique hémorragique dans notre étude comparée à deux autres études.Tableau 21 : Traductions cliniques non spécifiques de certains troubles de l’hémostase (cas cliniques du CHOP).

LISTE DES ABRÉVIATIONS
AT: AntithrombineATCD: Antécédents ATP : Adénosine triphosphateBSS: Bernard Soulier SyndromCHOP: centre d’hématologie et d’oncologie pédiatriqueDHPC: Déficit rare en protéine coagulanteFT: Facteur tissulaireFVW: Facteur de Von WillebrandHER: Hôpital d’enfants de RabatMAIPA: Antibody-specific Immobilization of Platelet AntigensMW : maladie de WillebrandNO: Monoxyde d’azotePC: protéine CPDF: Produits de dégradation de la fibrine PFA : Platelet Function AnalyserPS : protéine SPTAI: Purpura thrombopénique auto-immunRFC: Réseau FranceCoagRIPA : Ristocetin-Induced Platelet AgregationSBS: Syndrome de Bernard SoulierTC: thrombopénies constitutionnellesTCA: Temps de Céphaline + ActivateurTCK: Temps de Céphaline Kaolin TDM: Tomodensitométrie TFPI: Tissue Factor Pathway inhibitorTG : Thrombasthénie de GlanzmannTP: Temps de prothrombinet-PA: tissue Plasminogen ActivatorTS: Temps de saignementTXA2: Thromboxane A2VIH: Virus d’immunodéficience humaine

1
INTRODUCTION GÉNÉRALE
Le XXe siècle est sans doute le siècle des découvertes et travaux scientifiques les plus récents en hématologie. D’énormes progrès dans la compréhension des phénomènes physiologiques de l’hémostase et de leurs acteurs (facteurs de la coagulation, facteur de Willebrand, facteur VIII…) ont été accomplis. Il s’agit d’un grand tournent historique où furent découvertes toutes les pathologies hémorragiques constitutionnelles de l’hémostase que nous connaissons aujourd’hui à l’exception de l’hémophilie qui est connue depuis des millénaires.
Le domaine de l’hémostase pédiatrique, à l’instar de la pédiatrie, a bénéficié ces quarante dernières années de progrès remarquables et du développement des méthodes d’exploration biologique et d’imagerie adaptés à l’enfant, au petit enfant et au fœtus. Ceci dans un premier temps a permis de mettre en évidence les particularités d’un système hémostatique dit en développement et de publier des valeurs de référence des paramètres de la coagulation suivant le terme et l’âge de l’enfant. Ce qui a aidé à mieux aborder une seconde étape qui est la compréhension et le diagnostic des principales pathologies hémorragiques de l’enfant.
Les pathologies constitutionnelles de l’hémostase forment un groupe de pathologies pouvant prédisposer à des saignements parfois très graves. Elles sont liées à une anomalie génétique et sont donc présentes dès la naissance (anomalies constitutionnelles) et plusieurs membres de la famille peuvent être atteints (anomalies héréditaires). De ce fait, elles peuvent toucher aussi la descendance.
Nous nous sommes intéressés dans ce travail de thèse aux circonstances de découverte de ces pathologies constitutionnelles. Nous avons donc mené dans ce sens une étude rétrospective sur 3 années afin de déterminer les conditions dans lesquelles les patients porteurs de ce type d’affection sont diagnostiqués. Ceci pour une meilleure approche diagnostique et prise en charge de ces maladies rares mais invalidantes de l’enfant.

2
Quelles sont donc ces pathologies hémorragiques constitutionnelles de l’hémostase ?
Dans quelles circonstances sont-elles découvertes ? S’agit -t-il toujours d’une symptomatologie hémorragique spécifique à ce type d’anomalies ?
Quelle est la démarche diagnostique à suivre devant la suspicion d’une maladie hémorragique congénitale de l’hémostase chez l’enfant ?

3
RAPPEL PHYSIOLOGIQUE DE L’HÉMOSTASE
Fig1 : Caillot sanguin

4
I. GÉNÉRALITÉS1. QU’EST-CE L’HÉMOSTASE ? (QUOI ?)
3. COMMENT ? [3]
2. QUAND EST CE QUE CE PHÉNOMÈNE PHYSIOLOGIQUE EST-IL DÉCLENCHÉ? (QUAND ?)
L’hémostase (du grec aima, sang et stasis, arrêt) est définie, étymologiquement, par l’arrêt d’un saignement et, en Hématologie par l’ensemble des mécanismes physiologiques qui permet la prévention et l’arrêt des hémorragies en cas de lésion de la paroi vasculaire. Elle joue également un rôle dans le maintien de la fluidité du sang par la mise en jeu de systèmes inhibiteurs de la coagulation. [1]
L’hémostase peut être subdivisée en trois étapes distinctes mais imbriquées et dépendantes l’une de l’autre :
1. l’hémostase primaire qui est un mécanisme d’urgence qui aboutit à la formation du clou plaquettaire ou thrombus blanc formé par un agrégat de plaquettes adhérés à l’endothélium et qui dure de 3 à 5 minutes. 2. La cascade de coagulation dont le produit final est la fibrine qui vient renforcer le clou plaquettaire formant ainsi le caillot en une durée de 5 à 10 minutes. 3. La fibrinolyse assurant secondairement la dégradation enzymatique de la masse
L’hémostase s’oppose à la perte de sang consécutive à la lésion de petits vaisseaux. Pour qu’il y ait saignement il faut qu’il y ait une brèche dans la paroi des vaisseaux par où le sang sort sous l’effet de la différence de pression entre leur intérieur et leur extérieur. Les mécanismes naturels de l’hémostase sont normalement adéquats pour obturer la brèche et arrêter le saignement des petits vaisseaux, artérioles, capillaires et veinules qui sont fréquemment lésés par les traumatismes de la vie quotidienne qui sont la cause la plus courante de saignements alors même que l’on ne s’est pas rendu compte dans tous les cas que l’on s’est blessé. L’hémostase réduit à un minimum le saignement de ces petites lésions vasculaires. Les plus rares saignements provenant de vaisseaux de moyen ou grand diamètre ne sont pas en règle générale arrêtés par les seuls mécanismes hémostatiques.[2]

5
Fig2 : Étapes de l’hémostase primaire
fibrinoplaquettaire initialement formée à l’issue de la réparation vasculaire qui peut durer de 48 à 72 heures.Ces différentes phases de l’hémostase sont hautement régulées par un système d’activateurs et d’inhibiteurs plasmatiques assurant un contrôle local de la constitution du caillot et évitant l’activation de la coagulation à distance de la brèche vasculaire.
Nous allons donc décrire en détails chaque étape dans ce qui suit.
6
II. PHYSIOLOGIE DE L’HÉMOSTASE PRIMAIRE [4], [5]
1. FACTEURS DE L’HÉMOSTASE PRIMAIRE
1.1. La paroi vasculaire
Pour étudier la physiologie de cette première étape il est primordial de détailler les éléments qui entrent en jeu dans ce processus naturel, et qui sont : la paroi vasculaire, les plaquettes, le facteur de Willebrand et le fibrinogène.
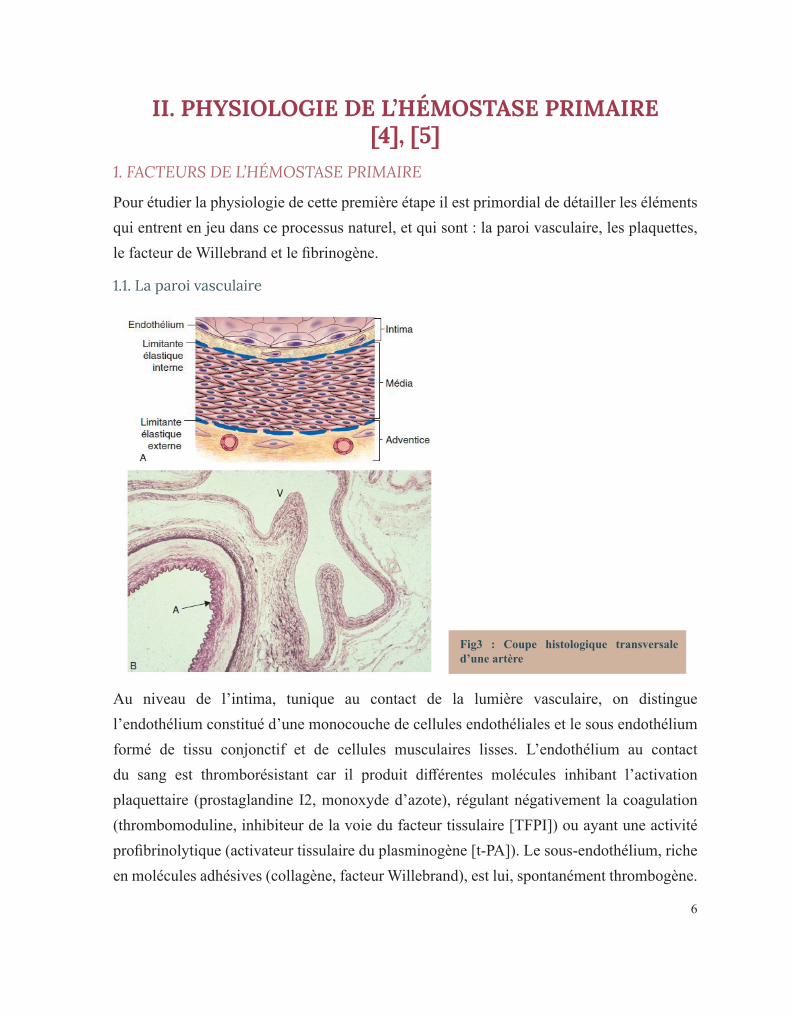
Au niveau de l’intima, tunique au contact de la lumière vasculaire, on distingue l’endothélium constitué d’une monocouche de cellules endothéliales et le sous endothélium formé de tissu conjonctif et de cellules musculaires lisses. L’endothélium au contact du sang est thromborésistant car il produit différentes molécules inhibant l’activation plaquettaire (prostaglandine I2, monoxyde d’azote), régulant négativement la coagulation (thrombomoduline, inhibiteur de la voie du facteur tissulaire [TFPI]) ou ayant une activité profibrinolytique (activateur tissulaire du plasminogène [t-PA]). Le sous-endothélium, riche en molécules adhésives (collagène, facteur Willebrand), est lui, spontanément thrombogène.
Fig3 : Coupe histologique transversale d’une artère
7
1.2. Les plaquettes
Dans une communication en 1881 adressée à l’académie royale de médecine de Turin, le médecin italien Giulio Bizzozero révéla la présence dans le sang humain d’éléments discrets qu’il nomma («bluttplatchen» dans un journal Allemand et «petites plaques» dans une communication en Français). Décrites avant comme étant de simples «agrégats granulaires» non fonctionnels, ils seront depuis au cœur de l’explication des phénomènes hémostatiques et thrombotiques.
Les plaquettes sanguines ne sont pas des cellules proprement dites mais des fragments cellulaires qui se sont détachés par bourgeonnement de très grandes cellules résidentes de la moelle osseuse appelées mégacaryocytes qui proviennent des mêmes cellules souches indifférenciées que les érythrocytes et les lignées de leucocytes. Dans le sang, les plaquettes sont au nombre de 150 000 à 450000/mm3. Leur durée de vie est de 8 à 10 jours. La plaquette est une cellule anucléée, ayant une forme de disque à l’état de repos. Cette cellule est délimitée par une membrane plasmique composée de glycoprotéines et de phospholipides. Dans le cytoplasme, on retrouve des granules denses et des granules alpha. Ces granules contiennent des composés importants qui interviennent dans la phase d’agrégation plaquettaire[1].
Fig4 : Image reconstituée en 3D des plaquettes sanguines
8
1.3. Le facteur Von Willebrand
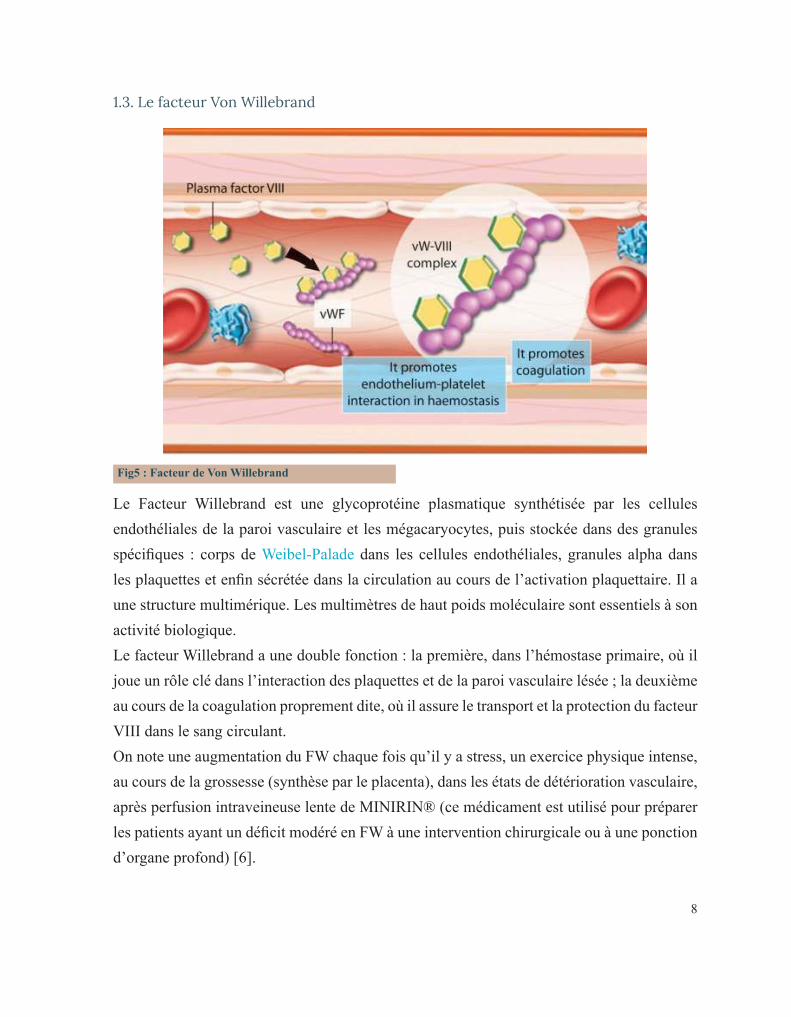
Le Facteur Willebrand est une glycoprotéine plasmatique synthétisée par les cellules endothéliales de la paroi vasculaire et les mégacaryocytes, puis stockée dans des granules spécifiques : corps de Weibel-Palade dans les cellules endothéliales, granules alpha dans les plaquettes et enfin sécrétée dans la circulation au cours de l’activation plaquettaire. Il a une structure multimérique. Les multimètres de haut poids moléculaire sont essentiels à son activité biologique.Le facteur Willebrand a une double fonction : la première, dans l’hémostase primaire, où il joue un rôle clé dans l’interaction des plaquettes et de la paroi vasculaire lésée ; la deuxième au cours de la coagulation proprement dite, où il assure le transport et la protection du facteur VIII dans le sang circulant. On note une augmentation du FW chaque fois qu’il y a stress, un exercice physique intense, au cours de la grossesse (synthèse par le placenta), dans les états de détérioration vasculaire, après perfusion intraveineuse lente de MINIRIN® (ce médicament est utilisé pour préparer les patients ayant un déficit modéré en FW à une intervention chirurgicale ou à une ponction d’organe profond) [6].
Fig5 : Facteur de Von Willebrand

9
1.4. Le fibrinogène
2.1. Le spasme vasculaire réduit l’écoulement de sang dans le vaisseau lésé
2.2. L’adhésion plaquettaire
Le fibrinogène est une glycoprotéine plasmatique synthétisée par le foie. Il joue un rôle très important dans la coagulation mais c’est aussi le cofacteur de l’agrégation plaquettaire.
Un vaisseau sectionné ou déchiré se contracte aussitôt ce qui explique pourquoi le saignement est réduit pendant les 30 premières secondes avant d’augmenter par la suite. Le mécanisme sous-jacent n’est pas bien élucidé, mais on pense qu’il s’agit d’une réponse intrinsèque déclenchée par un médiateur paracrine libéré par l’endothélium du vaisseau lésé qui déclenche une brève vasoconstriction des muscles lisses de la paroi en amont de la lésion. Cette vasoconstriction favorise l’accumulation locale de substances hémostatiques. La sérotonine et le thromboxane A2 (TXA2) libérés par les plaquettes activées, sont de puissants agents vasoconstricteurs. En même temps, il y a mise en contact du facteur tissulaire qui provient de l’extérieur du vaisseau, avec les facteurs plasmatiques de la coagulation. C’est le point de départ de la coagulation qui aboutit à la formation précoce de thrombine.
Normalement, les Plaquettes n’adhèrent pas à l’endothélium des vaisseaux. Quand il y a rupture de continuité de l’endothélium à la suite d’une lésion vasculaire, les structures sous endothéliales hautement thrombogènes sont mises à nu et mises en contact avec les plaquettes. Ces dernières adhèrent au sous endothélium par l’intermédiaire du facteur de Willebrand. Ce dernier, comme précédemment cité est une molécule multimérique de très haut poids moléculaire synthétisée par la cellule endothéliale et le mégacaryocyte. Sa liaison au collagène induit sa modification conformationnelle, permettant ainsi sa liaison à la glycoprotéine plaquettaire GPIb-IX-V.
2-MISE EN JEU DES DIFFÉRENTS PARAMÈTRES DE L’HÉMOSTASE PRIMAIRE :[2], [7]
Cette mise en jeu est rapide, elle aboutit à la formation d’un thrombus blanc de nature plaquettaire qui va colmater la brèche vasculaire. Les diverses phases sont les suivantes :

10
2.4. L’agrégation plaquettaire
L’agrégat plaquettaire va croître par apposition successive de nouvelles plaquettes. Au niveau de la membrane plaquettaire, le complexe glycoprotéinique IIb/IIIa est indispensable. Grâce à ce site, le fibrinogène va se fixer sur la membrane pour former avec le Ca++ des ponts interplaquettaires qui permettent la formation de l’agrégat. Une fois que leur agrégation a
2.3. L’activation plaquettaire
L’activation des cellules plaquettaires est caractérisée par deux phénomènes principaux, leur changement de forme et leur activation métabolique. Il s’agit de processus actifs nécessitant de l’énergie, sous forme d’ATP dérivant du métabolisme du glucose, et la disponibilité intracytoplasmique des ions calcium (Ca++) indispensables à l’activation du système contractile actine-myosine. Discoïdes à l’état de repos, les plaquettes activées deviennent sphériques, émettent des pseudopodes et s’étalent sur la surface d’adhésion. Les granules intra cytoplasmiques fusionnent avec le système canaliculaire ouvert et y libèrent leur contenu, qui se déverse ainsi dans le plasma environnant. Ce phénomène de sécrétion plaquettaire, libère de nombreuses substances proagrégantes (ADP, fibrinogène, sérotonine), pro coagulantes (facteur V, VWF, fibrinogène) ou vasomotrices (sérotonine, NO, TXA2) contribuant à l’amplification du processus d’hémostase primaire et créant les conditions favorables à la coagulation plasmatique.Par ailleurs, la plaquette activée génère de nombreuses substances pharmacologiquement actives à partir de ses phospholipides membranaires comme l’acide arachidonique. Celui-ci est métabolisé par la phospholipase A2 pour aboutir à la TXA2, puissant agent vasoconstricteur (L’Aspirine* exerce un effet anti-agrégant plaquettaire en bloquant la synthèse des précurseurs du TXA2) et proagrégant, et à d’autres prostaglandines modulant les activités plaquettaire et vasculaire.Un autre phénomène essentiel se déroule au cours de la phase d’activation plaquettaire est le phénomène de «flip-flop» membranaire, permettant aux structures internes de la membrane de se repositionner vers l’extérieur en contact avec le plasma. Cette modification permet aux phospholipides chargés négativement, et notamment la phosphatidylsérine, de s’extérioriser et de devenir disponibles pour la fixation des facteurs de la coagulation vitamine K-dépendants, amplifiant par-là considérablement les processus enzymatiques de la cascade de la coagulation.

11
Fig6 : Rôle des plaquettes dans l’hémostase primaire
commencé, les plaquettes libèrent plusieurs substances chimiques importantes notamment le diphosphate d’adénosine (ADP). L’ADP rend adhésive la surface des plaquettes circulant à proximité de sorte qu’elles adhèrent à la première couche de plaquettes agrégées et produisent à leur tour ces mêmes substances chimiques ce qui cause encore plus de plaquettes à s’empiler et ainsi de suite. C’est ainsi que ce constitue rapidement le clou plaquettaire par un mécanisme de rétroaction positive.
L’agrégation des plaquettes étant auto-entretenue, comment se fait-il qu’elle soit limitée au siège de la lésion vasculaire et ne s’étende pas à l’endothélium voisin? Une raison cruciale est que l’ADP ainsi que d’autres substances libérées par les plaquettes activées simulent la production de prostacycline et de monoxyde d’azote par l’endothélium sain voisin et inhibent fortement l’agrégation dans leur territoire, ce qui confine le clou plaquettaire à la zone lésée et s’oppose à son extension aux zones saines.
Il se forme finalement un gros amas plaquettaire, l’agrégat plaquettaire hémostatique, qui arrête en partie l’hémorragie mais qui doit être consolidé par le réseau de fibrine créé par la coagulation.

12
III. PHYSIOLOGIE DE LA COAGULATION La 1ère description de la coagulation remonte à 1905 par PAUL MORAWITZ, médecin allemand, qui proposa une théorie afin d’expliquer le phénomène de la coagulation dans laquelle quatre acteurs sont cités comme étant nécessaires et suffisants : la thrombokinase, la prothrombine, le fibrinogène et le calcium. Bien qu’incomplète cette théorie pose les bases de la cascade de la coagulation. Au cours des années 1950, les chercheurs ont identifié les facteurs de la coagulation. Dans un article publié en 1964 dans la revue Nature, on décrivait en détails le processus de la coagulation. L’interaction des différents facteurs nécessaires à la coagulation sanguine a alors pris le nom de réactions en cascade de la coagulation.[8] La coagulation est le plus puissant des mécanismes de l’hémostase et est nécessaire à l’arrêt du saignement, l’hémostase obtenue par le clou plaquettaire étant fragile et temporaire, elle doit être consolidée par la génération d’un réseau protéique qui réalise ainsi une hémostase permanente.
La coagulation correspond à la conversion du fibrinogène, protéine soluble, en fibrine, une molécule filamenteuse insoluble, qui constitue l’armature du caillot. Cette conversion est la conséquence d’une cascade de réactions enzymatiques à laquelle participent plusieurs protéines plasmatiques appelées facteurs de la coagulation.
La coagulation doit être appréhendée de manière dynamique. Après son initiation, elle s’amplifie. Mais elle doit rester localisée à la brèche vasculaire et ne pas être associée à une hypercoagulabilité circulante ou systémique. À cet effet, des mécanismes régulateurs importants sont mis en jeu. Les principaux inhibiteurs de la coagulation sont le tissue factor pathway inhibitor (TFPI), l’Antithrombine (AT), le système de la protéine C (protéines C et S).
1. LES CELLULES ET FACTEURS IMPLIQUÉS
La coagulation ne peut se dérouler qu’en présence de cellules ou des composants qui en sont issus. Les cellules les plus importantes dans la coagulation sont les cellules endothéliales, les monocytes, les plaquettes et les cellules périvasculaires. La coagulation a lieu à la surface
13
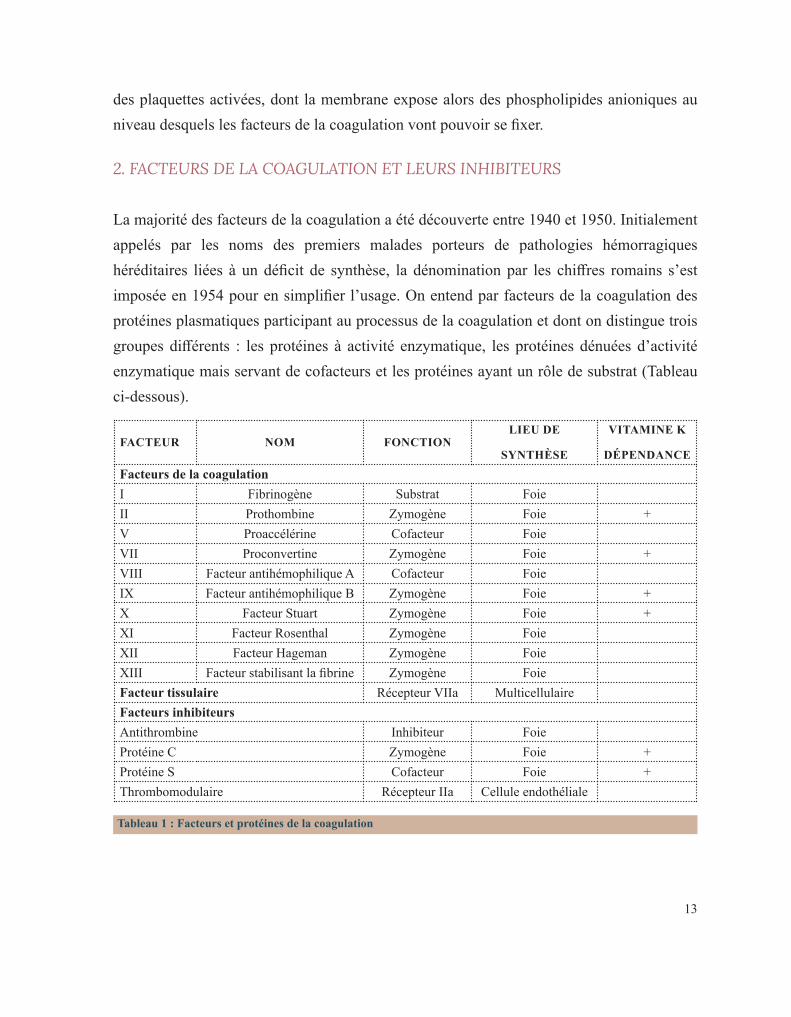
Tableau 1 : Facteurs et protéines de la coagulation
FACTEUR NOM FONCTIONLIEU DE
SYNTHÈSE
VITAMINE K
DÉPENDANCEFacteurs de la coagulationI Fibrinogène Substrat FoieII Prothombine Zymogène Foie +V Proaccélérine Cofacteur FoieVII Proconvertine Zymogène Foie +VIII Facteur antihémophilique A Cofacteur FoieIX Facteur antihémophilique B Zymogène Foie +X Facteur Stuart Zymogène Foie +XI Facteur Rosenthal Zymogène FoieXII Facteur Hageman Zymogène FoieXIII Facteur stabilisant la fibrine Zymogène FoieFacteur tissulaire Récepteur VIIa MulticellulaireFacteurs inhibiteursAntithrombine Inhibiteur FoieProtéine C Zymogène Foie +Protéine S Cofacteur Foie +Thrombomodulaire Récepteur IIa Cellule endothéliale
des plaquettes activées, dont la membrane expose alors des phospholipides anioniques au niveau desquels les facteurs de la coagulation vont pouvoir se fixer.
2. FACTEURS DE LA COAGULATION ET LEURS INHIBITEURS
La majorité des facteurs de la coagulation a été découverte entre 1940 et 1950. Initialement appelés par les noms des premiers malades porteurs de pathologies hémorragiques héréditaires liées à un déficit de synthèse, la dénomination par les chiffres romains s’est imposée en 1954 pour en simplifier l’usage. On entend par facteurs de la coagulation des protéines plasmatiques participant au processus de la coagulation et dont on distingue trois groupes différents : les protéines à activité enzymatique, les protéines dénuées d’activité enzymatique mais servant de cofacteurs et les protéines ayant un rôle de substrat (Tableau ci-dessous).

14
3. ACTIVATION DE LA COAGULATION :[7], [9]
3.1. Représentation classique
Le schéma classique et historique de la coagulation comporte deux voies d’activation : -La voie intrinsèque dans laquelle la coagulation est déclenchée par un activateur de la phase contact. Physiologiquement, le système du « contact », appelé ainsi car activé lors du contact du sang avec une surface mouillable comme le verre (ou le Kaolin, la silice ou l’acide ellagique utilisés dans les tests de laboratoire), et comprenant notamment le facteur FXII, ne joue pas de rôle significatif. En effet le FXII n’est associé à aucun risque de saignement. -La voie extrinsèque qui est activée par le récepteur cellulaire du FVII; le facteur tissulaire (FT), correspondant à la thromboplastine utilisée par les laboratoires.Cette conception duelle de la coagulation reflète assez justement les mécanismes mis en jeu in vitro, c’est-à-dire lors de l’exploration de la coagulation au laboratoire, c’est donc en se fondant sur ce schéma qu’on raisonne pour interpréter les tests de la coagulation usuels, qui sont le Temps de céphaline+ Activateur pour l’exploration de la voie intrinsèque et le temps de prothrombine (TP) pour la voie extrinsèque. En revanche, cela ne correspond pas réellement à ce qui se passe in vivo au décours d’une lésion vasculaire.
3.2. Représentation moderne
La conception actuelle de la coagulation est plus dynamique que la précédente, elle est aussi plus représentative des phénomènes in vivo initiés par la mise à nu du FT (composant de la thromboplastine). En effet, il est aujourd’hui admis que l’élément déclenchant de la coagulation in vivo est l’expression à la surface des cellules d’une protéine membranaire appelée «facteur tissulaire» (FT). Certaines cellules en contact permanent avec le flux sanguin, n’expriment le FT que lorsqu’elles sont activées c’est le cas des monocytes et des cellules endothéliales. D’autres l’expriment de façon constitutive et donc permanente : ce sont des cellules périvasculaires (fibroblastes, myocytes, cellules mésenchymateuses) qui ne sont pas en contact avec le flux sanguin en l’absence de rupture de la continuité vasculaire.Plusieurs étapes sont identifiées :

15
• 1ère étape : déclenchement de la coagulation par activation du facteur VII ;• 2éme étape : activation du facteur X et formation du complexe enzymatique prothrombinase;• 3ème étape : formation de la thrombine ; • 4ème étape : formation du réseau de fibrine insoluble.
3.2.1. Déclenchement de la coagulation par activation du facteur VII
La rupture de la tunique endothéliale thromborésistante, secondaire à une lésion vasculaire, permet le contact du sang circulant avec les structures sous-endothéliales. La fixation du facteur VII plasmatique au facteur tissulaire, qui est exprimée de façon constitutive par les cellules musculaires lisses et les fibroblastes, représente le signal du déclenchement de la cascade enzymatique. La liaison du facteur VII permet en outre son auto activation, amplifiant considérablement l’activité du complexe facteur tissulaire-facteur VII (FT-FVII).
3.2.2. Activation du facteur X et formation du complexe enzymatique prothrombinase
Le complexe FT-FVII active très rapidement par protéolyse le facteur X en facteur Xa. Celui-ci active en retour le facteur VII, rendant le complexe beaucoup plus actif et amplifiant ainsi sa propre production. Le facteur Xa forme, en association avec les phospholipides plaquettaires, le calcium et le cofacteur Va, un complexe enzymatique assurant le clivage protéolytique de la prothrombine(FII) qui génère ainsi la molécule de thrombine, d’où son nom de complexe prothrombinase.Par ailleurs, le complexe FT-FVII active, mais beaucoup plus lentement, le facteur IX (facteur antihémophilique B) en facteur IXa. Il se forme de la même façon un complexe enzymatique, appelé complexe tenase, associant facteur IXa, phospholipides plaquettaires, calcium, et le cofacteur VIIIa, qui active le facteur X en facteur Xa, amplifiant considérablement le rendement de la production de prothrombinase. Il existe donc deux voies d’activation protéolytique du facteur X qui sont distinctes dans leur cinétique. L’activation directe par le complexe FT-FVII est très rapide, et constitue le starter de la cascade enzymatique, pour aboutir précocement aux premières molécules de thrombine, alors que la voie indirecte passant par l’activation du facteur IX est beaucoup plus lente à se mettre en place mais est

16
quantitativement prépondérante. Il existe une autre voie d’activation passant par le facteur XI qui est activé lentement par la thrombine nouvellement formée. Le facteur XIa active en retour le facteur IX pour renforcer la génération du complexe tenase. Le facteur XI peut également être activé par les facteurs contacts après exposition des composants du sous-endothélium, mais l’importance de cette voie d’activation est mineure.
3.2.3. Formation de la thrombine
Le complexe prothrombinase assure la protéolyse de la prothrombine (facteur II) en thrombine (facteur IIa), protéine clé de la coagulation responsable de la génération du caillot de fibrine. En outre, la thrombine assure une amplification du rendement de la cascade enzymatique en activant les cofacteurs V et VIII qui accélèrent considérablement l’activité des complexes de la prothrombinase (Va) et de la tenase (VIIIa), conduisant à un accroissement explosif de la production de la thrombine. On considère en effet que la présence du cofacteur activé au sein du complexe enzymatique accroît son rendement par un facteur 10 6. Ce phénomène est nommé double boucle de rétroactivation de la génération de thrombine sur laquelle repose toute l’efficacité et la puissance du système.
3.2.4. Fibrinoformation
La dernière étape repose sur la transformation du fibrinogène soluble par l’hydrolyse de
Fig7 : Fibrinoformation
ces différentes chaînes polypeptidiques en monomères de fibrine, qui s’associent les unes aux autres grâce à des liaisons hydrogène de faible affinité pour former un gel de fibrine,

17
ou le caillot de fibrine, qui est tout d’abord instable. Le facteur XIII, facteur de stabilisation de la fibrine, préalablement activé par la thrombine, solidifie alors les molécules de fibrine par l’établissement de liaisons covalentes entre les différentes molécules conduisant à une polymérisation des monomères de fibrine.
4. MÉCANISMES DE RÉGULATION
Un système physiologique très complexe de régulation de la coagulation est mis en œuvre, afin de limiter l’extension locale du caillot et d’éviter la diffusion à distance de la fibrinoformation. Celui-ci a été démembré par l’identification de protéines déficitaires chez des sujets présentant une pathologie thrombotique récidivante dans un contexte familial.L’antithrombine inhibe les protéines activées de la coagulation : IIa, IXa, Xa, XIa, XIIa. La thrombomoduline capte la thrombine libre et inhibe ses fonctions coagulantes. De plus, ce complexe active la protéine C. La protéine C activée, en présence de son cofacteur, la protéine S, inhibe par protéolyse les facteurs Va et VIIIa. La protéine C et la protéine S sont vitamine K-dépendantes.La voie extrinsèque de la coagulation est régulée par le TFPI (tissue factor pathway inhibitor). Le TFPI forme un complexe avec le complexe FT/FVIIa et le facteur Xa, limitant ainsi la génération de facteur Xa.
Fig8 : Caillot d’hémostase
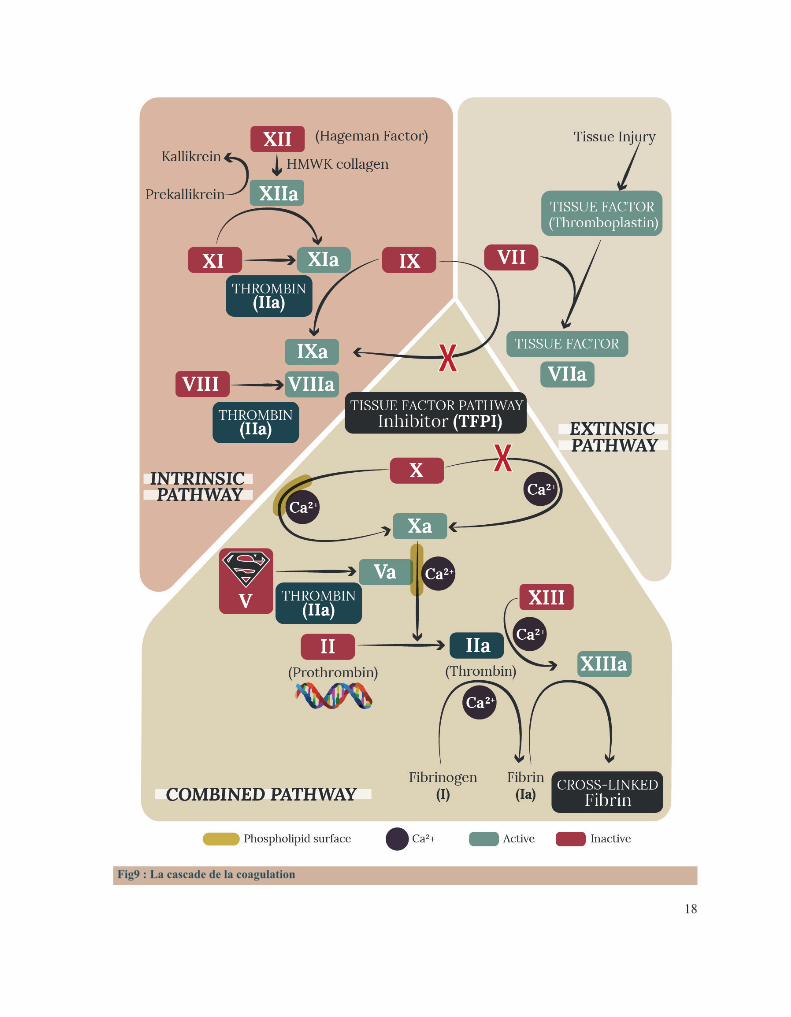
18
Fig9 : La cascade de la coagulation

19
IV. PHYSIOLOGIE DE LA FIBRINOLYSE :[1]
Le caillot n’est pas la solution définitive d’une lésion vasculaire. C’est un dispositif transitoire d’arrêt du saignement jusqu’à ce que le vaisseau soit réparé.
La fibrinolyse est un processus physiologique qui empêche l’installation mais surtout l’extension du caillot en détruisant les polymères de fibrine une fois l’endothélium réparé permettant ainsi la restitution de la perméabilité vasculaire. La fibrinolyse est bâtie selon la même conception que le système de la coagulation comprenant des molécules à activité protéolytique, qui agissent sur un substrat, contrôlées par un système d’activateurs et d’inhibiteurs permettant une régulation physiologique très précise.
L’enzyme centrale de la fibrinolyse est la plasmine qui dérive d’un précurseur plasmatique inactif, le plasminogène, glycoprotéine d’origine hépatique. Le plasminogène possède une grande affinité pour la fibrine, et s’y fixe par un récepteur spécifique aux côtés de son activateur, permettant ainsi la génération locale de plasmine via le démasquage des sites protéolytiques. La plasmine protéolyse le fibrinogène et la fibrine en divers fragments de tailles variables, identifiés comme les produits de dégradation de la fibrine, ou PDF, qui sont quantifiables dans le plasma. Les activateurs principaux du plasminogène sont le t-PA (activateur tissulaire du plasminogène) et la pro-urokinase.
La régulation très précise de la fibrinolyse nécessite la mise en jeu d’inhibiteurs. Le principal est représenté par l’alpha-2-anti-plasmine qui est capable de neutraliser très rapidement la plasmine libre non fixée sur le caillot de fibrine. Le second inhibiteur, l’alpha-2-macroglobuline, est d’action plus modeste.[10]

20
PARTICULARITÉS DE L’HÉMOSTASE
EN PÉDIATRIE

21
I. GÉNÉRALITÉS
Au cours des dernières décennies, et depuis les travaux de Maureen Andrew à la fin des années 1980, de grands progrès ont été accomplis dans la compréhension de la physiologie du système hémostatique pédiatrique et de ses particularités. En effet l’hémostase pédiatrique, et tout particulièrement l’hémostase du nouveau-né, est différente de celle de l’adulte. Elle est décrite dans les travaux pionniers de Hathaway et Andrew comme un système en développement, reflet des changements quantitatifs et qualitatifs qui s’opèrent durant le dernier mois de la vie intra-utérine, dans les jours qui suivent l’accouchement et qui se poursuivent dans la première année de vie [10], [11].
Malgré cela on peut considérer que le système hémostatique du nouveau-né et de l’enfant, bien que particulier, reste équilibré, sans saignement ni thrombose. Néanmoins cet équilibre est instable et expose le nouveau-né et le nourrisson à des pathologies acquises et constitutionnelles de l’hémostase parfois sévères, dont le diagnostic est essentiel pour garantir un traitement efficace.
Les travaux originaux de M. Andrew ont permis de déterminer des valeurs de référence en fonction de l’âge pour les facteurs coagulants mais aussi anticoagulants[10], [12]. Ils ont été confirmés par plusieurs autres études ayant évalué différentes populations pédiatriques dans différents environnements techniques. Le tableau I résume les principales études faites jusqu’à ce jour et les caractéristiques des populations étudiées[13].
Ce concept assez récent d’une hémostase dynamique est une grande avancée médicale dans le domaine de l’hématologie pédiatrique. Sa compréhension est donc fondamentale pour assurer une prévention, un diagnostic et un traitement approprié des maladies hémorragiques et thrombotiques chez les enfants[13].
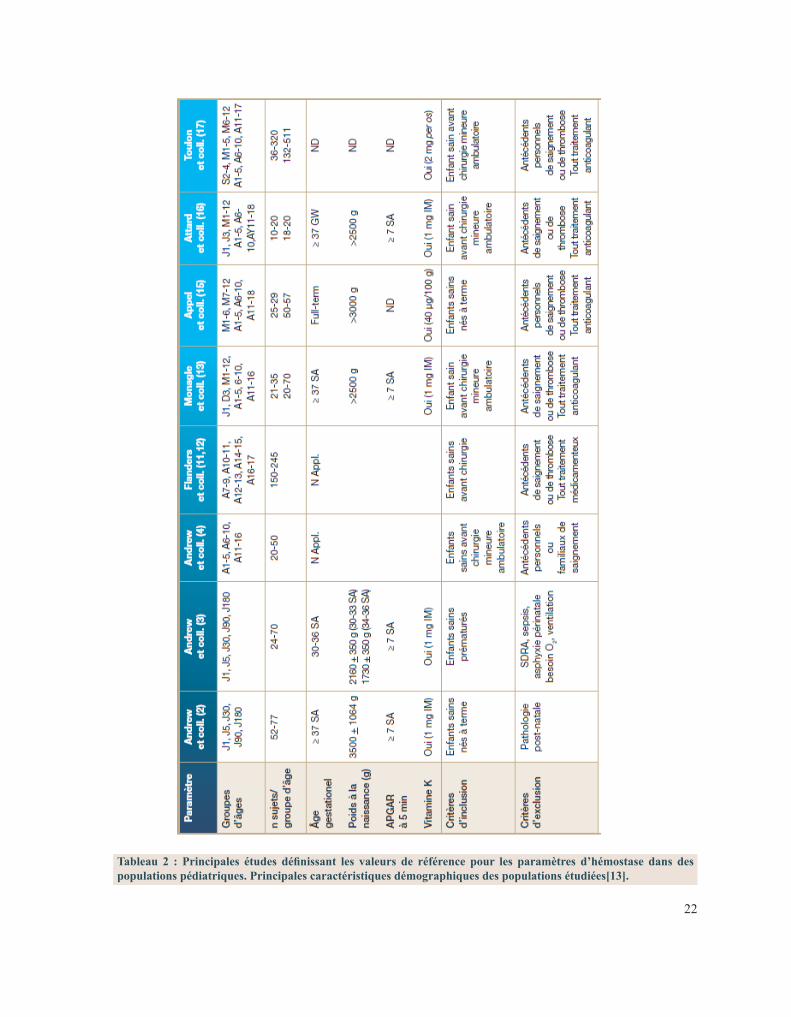
22
Tableau 2 : Principales études définissant les valeurs de référence pour les paramètres d’hémostase dans des populations pédiatriques. Principales caractéristiques démographiques des populations étudiées[13].

23
II. PARTICULARITÉS PHYSIOLOGIQUES DE L’HÉMOSTASE DU NOUVEAU-NÉ, DU NOURRISSON
ET DE L’ENFANT
1. L’HÉMOSTASE PRIMAIRE :[14]
Chez le fœtus normal, la mégacaryocytopoïèse permet, dès la quinzième semaine de vie intra-utérine, de libérer dans la circulation un nombre de plaquettes comparable à celui de l’adulte. Ainsi, le nombre de plaquettes est normal entre 18 et 35 semaines de grossesse, compris entre 200 et 300 Giga/l, et stable jusqu’à la naissance. Le taux de plaquettes durant les premières semaines de vie s’élève régulièrement et présente 2 pics d’hyperplaquettose, le premier vers 2 à 3 semaines, en rapport probablement avec des concentrations élevées de la thrombopoïétine, protéine régulatrice de la mégacaryocytopoièse et le deuxième vers 6 à 7 semaines, le plus souvent réactionnel. Le nombre de plaquettes dans ce cas peut atteindre 750 Giga/l.A la naissance, l’étude des fonctions plaquettaires a montré une diminution de l’agrégation plaquettaire chez le nouveau-né à terme et le prématuré, sans différence entre ces deux groupes de patient, et ce jusqu’à le 2ème mois de vie. Ces particularités n’ont pas de traduction clinique, les capacités d’adhésion des plaquettes semblant compenser le défaut d’agrégation.En période néonatale, l’expression des complexes membranaires plaquettaires GPIIbIIIa (récepteur fibrinogène) et GPIb (récepteur du facteur Willebrand) présente certaines particularités observées avant et après activation des plaquettes en cytométrie en flux et témoigne d’un défaut d’activation des plaquettes à cette période de la vie. Le complexe glycoprotéique GPIIbIIIa (CD41a et CD61) est diminué sur la plaquette au repos, mais son externalisation est satisfaisante après activation plaquettaire. Le diagnostic prénatal de Thrombasthénie de Glanzmann (GPIIbIIIa) ou du syndrome de Jean Bernard et Soulier (GP Ib) sont donc possibles dès ce terme précoce.
Le facteur Willebrand (VWF) (antigène et activité) est élevé chez le nouveau-né à terme et le prématuré. Il diminue progressivement jusqu’à l’âge de 6 à 12 mois, puis augmente de façon progressive jusqu’à l’âge adulte Le taux n’est pas influencé par le groupe sanguin chez

24
l’enfant de moins de 1 an. De plus, chez le fœtus et le nouveau-né, les multimères de très haut poids moléculaire (THPM) sont présents en circulation. Leur présence s’accompagne d’un effet pro-hémostatique puissant en induisant une augmentation significative de la capacité d’adhésion des plaquettes au collagène. Elle se traduit in vitro par une augmentation de l’agglutination des plaquettes en présence de Ristocétine et un raccourcissement du temps d’occlusion plaquettaire, sur l’automate PFA-100(Siemens).
Au total, l’hémostase primaire est efficace voire même accélérée, en période néonatale, avec un rôle essentiel exercé par le facteur Willebrand.
2. LA COAGULATION PLASMATIQUE
Le développement de la coagulation est très influencé par l’âge. Les taux d’activateurs et d’inhibiteurs de la coagulation plasmatique évoluent, effectivement, tout au long de la vie intra-utérine de façon dynamique jusqu’à la naissance. Ce développement se poursuit encore pendant la période néonatale et l’enfance avant d’atteindre le système achevé de l’adulte. A la naissance, les temps de coagulation, le temps de Quick(TQ) et le temps de céphaline avec activateur (TCA), sont allongés. Le temps de Quick étant proche de celui d’un adulte sous antivitamine K et le TCA étant comparable à celui obtenu lors d’un traitement par l’héparine. Ces résultats sont le reflet chez le fœtus et le nouveau-né d’une synthèse hépatique réduite de la majorité des facteurs de la coagulation et de leur clairance élevée. La génération de la thrombine dans le plasma du nouveau-né est également diminuée par rapport à celle d’un adulte. Toutefois, étudiée en présence de thrombomoduline (récepteur endothélial de la thrombine activateur de la protéine C), elle est comparable chez le nouveau-né sain et l’adulte, prouvant que l’équilibre hémostatique est préservé à cet âge de la vie[15].
3. LES FACTEURS DE LA COAGULATION
Entre la 20e et 30e semaine de gestation, les taux des facteurs vitamine K dépendants (II, VII, IX et X) sont compris entre 10 % (IX) et 30 % (VII). Ils augmentent ensuite de façon progressive durant les dernières semaines de la vie fœtale, avec, à la naissance, des valeurs comprises entre 30 et 50 %. Le déficit est d’autant plus important que la prématurité

25
est grande. Il est principalement le reflet de l’immaturité hépatique, à laquelle s’associe une carence en vitamine K, cofacteur d’une carboxylase hépatique assurant la gamma-carboxylation des FII, VII, IX et X et des inhibiteurs de la coagulation, les protéines C (PC) et S (PS). Le transfert materno-foetal de la vitamine K étant faible, le nouveau-né présente des réserves diminuées en vitamine K1. La normalisation des facteurs est progressive, le FVII est le premier à atteindre les valeurs de l’adulte, puis le X, le II et le IX. Entre 6 et 12 mois, la limite inférieure des valeurs normales de l’adulte est atteinte. La correction est plus lente pour le FIX, (vers l’âge de 12 mois)[16], [17]. Les facteurs de la phase contact (XI, XII, prékallicréine et kininogène de haut poids moléculaire) sont bas à la naissance, entre 20 et 40 % chez le prématuré et entre 30 et 50 % chez le nouveau-né à terme avec une normalisation vers 6 à 12 mois. Le déficit en l’un de ces facteurs (excepté le FXI) ne s’associe pas à un risque hémorragique, mais leurs taux bas contribuent largement à l’allongement du Temps de Céphaline Activateur (TCA) chez le nouveau-né et le nourrisson. Les concentrations des FV et VIII sont voisines de celles de l’adulte (tableau 1). Chez le prématuré, le taux de FV est discrètement abaissé, voisin de 60 %. Chez le nouveau-né à terme et le prématuré, les valeurs de référence du FVIII sont très larges, mais les taux sont le plus souvent supérieurs aux valeurs normales de l’adulte dans les premiers jours de vie, avec une normalisation à partir du 15e jour de vie. À la naissance, la concentration du fibrinogène est normale, comparable aux valeurs basses de l’adulte sain. Cependant, l’activité mesurée par la méthode de Clauss est inférieure à la concentration antigénique, faisant penser qu’il existe un fibrinogène « fœtal », responsable de l’allongement modéré des temps de thrombine et de reptilase observé chez le nouveau-né. Le taux de FXIII est normal dès la naissance [14].

26
Tableau 3 : valeurs de référence en pédiatrie selon Andrew (A) et Monagle (M) [110]

27
4. LES INHIBITEURS DE LA COAGULATION
Il existe chez le nouveau-né et le nourrisson, tout comme chez l’enfant et l’adulte, un équilibre entre les activateurs de la coagulation et leurs inhibiteurs. L’inhibition de la thrombine par l’antithrombine, principal inhibiteur de la thrombine, est très faible chez le nouveau-né, due au taux bas de cet inhibiteur à la naissance qui est de 50% chez le nouveau-né à terme, et qui n’atteint les valeurs adultes qu’à 3 mois de vie. Cette action inhibitrice minime est compensée par l’α2-macroglobuline qui est significativement élevée pendant dans les six premiers mois de vie, comparé à l’adulte. Sa concentration reste élevée jusqu’à l’âge de 16 ans, ce qui pourrait conférer un rôle protecteur important contre la thrombose chez le nourrisson et l’enfant.
En revanche, le système inhibant la génération de thrombine est faiblement représenté durant la vie fœtale et en période néonatale. Les taux de PC et PS, synthétisées par le foie en présence de vitamine K, sont bas à la naissance. La diminution est particulièrement importante pour la PC, dont les taux à la naissance varient entre 18 % et 50 % chez le nouveau-né à terme et prématuré et restent significativement diminués jusqu’à l’âge de 16 ans, comparés aux valeurs normales de l’adulte. Les taux de PS restent significativement diminués jusqu’à l’âge de 6 mois mais la PS totale atteint des taux comparables à ceux de l’adulte vers l’âge de 3 mois. Chez l’adulte, cet inhibiteur est présent sous une forme libre, active, et sous une forme liée à une protéine du système du complément, la C4b-binding protein (C4b-BP). Chez le nouveau-né, seule la PS libre, active, est présente dans le plasma en raison de taux diminués de C4b-BP [18].
5. LES COMPOSANTES DE LA FIBRINOLYSE
La fibrinolyse, système permettant la dissolution du caillot de fibrine formé lors de la coagulation plasmatique, est immature à la naissance. Le taux de plasminogène est de 30 à 50 % chez le nouveau-né à terme. Il est encore plus bas chez le prématuré. Le taux d’alpha2-antiplasmine, inhibiteur principal de la plasmine, est comparable à la naissance à celui de l’adulte. En revanche, les concentrations de l’activateur plasmatique du plasminogène (t-PA) et de son inhibiteur de type I (PAI-1) sont significativement augmentées. L’augmentation

28
du t-PA est transitoire et très rapidement son taux est d’environ 50 % par rapport à la valeur moyenne de l’adulte. À l’opposé, la concentration plasmatique du PAI-1 reste supérieure aux valeurs adultes durant l’enfance. L’inhibiteur de l’activateur du plasminogène de type 2 (PAI-2) est présent dans le sang de cordon à la naissance.
En résumé, l’équilibre entre génération de thrombine (coagulation) et génération de plasmine est singulier en période néonatale mais adaptée à cette tranche de vie [19].
III. IMPLICATIONS CLINIQUES
L’enfant n’est pas simplement un « adulte en miniature », du moins en ce qui concerne la balance hémostatique, très différente de celle de l’adulte. La compréhension du système hémostatique et l’établissement de valeurs de référence pour les paramètres de la coagulation, dans l’enfance, sont fondamentales pour assurer dans des conditions optimales la prévention, le diagnostic et le traitement des maladies hémorragiques ou thrombotiques en pédiatrie [20].
La connaissance des particularités quantitatives et qualitatives de l’hémostase, spécialement chez le nouveau-né et le nourrisson, est donc élémentaire pour une interprétation correcte des principaux tests d’hémostase. Une interprétation qui se basera sur les valeurs de référence adaptés à l’âge et définis par chaque laboratoire selon son environnement technique (réactif/automate). La qualité du résultat va être dépendante de la qualité du prélèvement, étape rendue difficile à cet âge de la vie par les difficultés techniques de prélèvement et d’analyse et par l’hypercoagulabilité physiologique du nouveau-né. L’histoire clinique de l’enfant avant l’âge de la marche étant insuffisante, l’exploration biologique de l’hémostase va être capitale pour apprécier un éventuel risque hémorragique avant un geste invasif [21]. L’évaluation de l’hémostase est donc, en pédiatrie, une étape critique dans la prise en charge des patients.

29
LES PATHOLOGIES HÉMORRAGIQUES
CONSTITUTIONNELLES DE L’HÉMOSTASE
ET LEURS CIRCONSTANCES DE
DÉCOUVERTE
PREMIÈRE PARTIE

30
I. LES PATHOLOGIES DE L’HÉMOSTASE PRIMAIRE
1. LA MALADIE DE WILLEBRAND
1.1. Historique :[22], [23]
Fig10 : Portrait de Erik Adolf Von Willebrand
La maladie de Willebrand a été décrite pour la première fois en 1926 par Erik Adolf Von Willebrand, médecin interniste à Helsinki, chez une fille de l’Archipel Åland en Finlande. Cette jeune fille était âgée de 05ans, et souffrait d’épistaxis et de gingivorragies, elle était issue d’un mariage consanguin et onze membres de sa famille avaient des antécédents hémorragiques dont quatre en avaient péri. L’auteur suggéra une anomalie fonctionnelle des plaquettes et une lésion systémique vasculaire comme cause possible de cette affection qu’il publia en 1926 sous l’appellation de pseudohémophilie héréditaire.
Dans le temps, cette nouvelle maladie hémorragique qui touche les deux sexes et qui se manifeste par des saignements muqueux avec un allongement du temps de saignement et un taux de plaquettes normales ne sera vraiment élucidée qu’en 1971 grâce à Zimmerman et un autre groupe d’enquêteur. Ces derniers ont fait des progrès significatifs et ont montré, pour la première fois à l’aide de tests immunologiques, que le FVIII et le facteur de Von Willebrand étaient des protéines distinctes. La nature distincte du VWF a été démontrée par la caractérisation du gène du VWF en 1985 par quatre groupes d’enquêteurs indépendants. Cette découverte a permis de mieux comprendre la base génétique de la maladie de Von Willebrand.
Aujourd’hui et après plusieurs observations cliniques, et grâce aux études génétiques et aux efforts soutenus des scientifiques durant ce dernier quart de siècle, la maladie de Willebrand est mieux connue.

31
1.2. Définition :
La maladie de Willebrand est une pathologie de l’hémostase primaire. Elle désigne toute pathologie hémorragique constitutionnelle due à un déficit quantitatif (défaut de la concentration) ou qualitatif (défaut de la fonction) en facteur Willebrand, protéine plasmatique indispensable pour l’adhésion des plaquettes aux structures sous endothéliales en cas de lésion de la paroi vasculaire. C’est une anomalie génétique touchant le gène codant pour le facteur Willebrand qui est situé sur le bras court du chromosome 12, de transmission autosomique, généralement dominante, ce qui explique que garçons et filles peuvent être touchés contrairement à l’hémophilie.[22]
1.3. La classification :[6]
La classification de la maladie de Willebrand a été considérablement simplifiée par l’International Society on Thrombosis and Haemostasis (ISTH), il y a quelques années permettant de la classer en 3 grands types correspondant à des mécanismes physiopathologiques distincts, et présentant des caractéristiques cliniques spécifiques. La classification internationale repose sur une approche phénotypique et distingue les déficits quantitatifs partiels (type 1) ou complets (type 3), et les déficits qualitatifs (type 2). Le type 2 regroupe quatre sous-types : 2A, 2B, 2M et 2N. La transmission du déficit se fait le plus souvent sur un mode autosomal dominant, à l’exception des types 3, des types 2N ainsi que de rares variants 2A qui sont à transmission récessive. La distribution des mutations sur le gène VWF est répartie sur l’ensemble du gène dans les types 1 et 3 alors que pour les types 2 les mutations sont localisées à proximité des sites fonctionnels d’interaction.

32
1.4. Epidémiologie:[24]
La maladie de Willebrand est la plus fréquente des maladies hémorragiques constitutionnelles se plaçant avant l’hémophilie. Toutefois l’estimation de sa prévalence (classiquement 1%) est largement débattue aujourd’hui. Le nombre de patients qui relèvent d’un traitement est beaucoup plus faible : il est estimé à 1 pour 10 000 habitants. La prévalence réelle de la maladie est difficile à déterminer tant sur le plan biologique –certains déficits modérés n’étant pas associés à des anomalies du gène du VWF –que sur le plan clinique –la plupart des symptômes hémorragiques n’étant pas spécifiques de la maladie et pouvant être retrouvés dans une population saine. Le diagnostic de maladie de Willebrand est donc vraisemblablement souvent surestimé, particulièrement quand le déficit est quantitatif et modéré.
1.5. Le diagnostic positif
Le diagnostic de la maladie de Willebrand doit, du fait de sa fréquence, être évoqué facilement
Tableau 4 : La classification SSC-ISTH de la maladie de Willebrand

33
devant toute tendance hémorragique.L’évaluation clinique doit préciser les antécédents hémorragiques personnels et/ou familiaux, spontanés et postopératoires. La MW se manifeste le plus souvent par des hémorragies spontanées, cutanéomuqueuses ou provoquées après un geste invasif ou une intervention chirurgicale.
Le diagnostic de cette pathologie est biologique. Le bilan de 1ère intention en cas de suspicion de MW associe au minimum un dosage de FVIII, une exploration du VWF plasmatique associant dosage antigénique (VWF : Ag) et la mesure de son activité fonctionnelle (mesure de l’activité cofacteur de la Ristocétine ou test apparenté), et la réalisation d’une agrégation plaquettaire aux faibles concentrations de Ristocétine (RIPA) si ce test est disponible. Le calcul des ratios FVIII/VWF : Ag et VWF : RCo/VWF: Ag fait partie intégrante du bilan de dépistage ce qui implique que les dosages de FVIII, VWF Ag et VWF:RCo sont indissociables.
La réalisation d’un temps d’occlusion sur PFA-100® peut s’avérer utile en raison de sa sensibilité au déficit en VWF, à l’exception du type 2N où il est normal. La réalisation d’une numération globulaire, d’un bilan de coagulation standard et d’un groupage ABO sont par ailleurs indispensables au diagnostic différentiel et à l’interprétation des dosages de VWF. Les résultats de FVIII, VWF:Ag et VWF:RCo sont exprimés en % d’un plasma normal ou en (UI/dl) si le plasma standard est titré par rapport au standard international [plasma standard FVIII/VWF (07/316)].[6]
2. LES THROMBOPATHIES CONSTITUTIONNELLES
Les thrombopathies constitutionnelles représentent un groupe de pathologies très hétérogènes, rares, dont la fréquence est estimée à 1/10.000 individus en France. Elles sont dues à des anomalies génétiques affectant l’expression ou la fonctionnalité des glycoprotéines (GP) impliquées dans les différentes étapes de l’activation des plaquettes (sécrétion, adhésion, agrégation, activité pro coagulante)[14]. Les thrombopathies que nous allons aborder en raison de leur gravité et de leur fréquence sont celles impliquant deux complexes protéiques majeur, les complexes GPIIbIIIa et GPIb-IX-V.

34
2.1. La thrombasthénie de Glanzmann
2.1.1. Historique
La thrombasthénie de Glanzmann a été découverte en 1918, à Berne, en Suisse, par un pédiatre du nom d’Édouard Glanzmann. Les enfants atteints de cette maladie provenaient tous d’un petit village, appelé Le Valais, situé en altitude dans les Alpes suisses. Dans ce village, les mariages entre proches étaient fréquents. Glanzmann décrivit un nouveau type de purpura, différent du purpura anaphylactoïde et de la maladie de Wehrlof, un purpura avec une numération plaquettaire normale et un temps de saignement allongé. Cette nouvelle maladie fut nommée «thrombopathie hémorragique héréditaire» ou «maladie des bleus»[25].
Fig11 : Portrait d’Edouard Glanzmann
En 1956, Braunsteiner et Pakesch passèrent en revue les troubles de la fonction plaquettaire et décrivirent cette thrombopathie comme une maladie héréditaire caractérisée par des plaquettes de taille normale et un temps de saignement allongé et une étude de rétraction du caillot altérée. Les caractéristiques diagnostiques de la TG, y compris l’absence d’agrégation plaquettaire comme caractéristique principale, ont été clairement établies en 1964 dans le rapport d’une étude faite sur 15 patients français de Caen et al[25].L’importance historique de cette maladie rare réside dans le fait que l’analyse de sa physiopathologie a permis le développement de médicaments antiplaquettaires.
2.1.2. Définition
La thrombasthénie de Glanzmann (TG) est une maladie hémorragique héréditaire de transmission autosomique récessive (dans la majorité des cas), liée à une anomalie quantitative

35
ou qualitative du récepteur membranaire plaquettaire αIIbβ3 (GPIIbIIIa), impliqué dans l’agrégation des plaquettes, induisant un trouble de l’hémostase primaire[26]Il s’agit d’une maladie rare qui touche environ 500 patients en France, toutefois elle est relativement fréquente dans certaines régions du globe où les mariages consanguins sont communs telle la population gitane[28]. Certaines régions du Maroc figurent parmi les foyers géographiques de la TG[28].
Il existe plusieurs types de TG : les types I, II et les variants. Les types I et II présentent des anomalies quantitatives du récepteur αIIbβ3 (GPIIb-IIIa) : dans le type I, le déficit est majeur et le récepteur est absent ou n’est présent qu’à l’état de traces (< 5 %) ; dans le type II, le taux résiduel est de l’ordre de 5 à 20 %. Les variants sont des anomalies qualitatives du récepteur, présent à des taux proches de la normale.
2.1.3. Le diagnostic positif
L’examen clinique soigneux s’attachera à caractériser le type d’atteinte cutanéomuqueuse, l’existence de signes évocateurs d’une affection associée et/ou d’une atteinte organique induisant une pathologie acquise de l’hémostase. L’interrogatoire orienté devra caractériser le type de manifestation hémorragique, sa nature et sa fréquence, déterminer le mécanisme spontané ou provoqué, apprécier la sévérité du syndrome hémorragique et le retentissement global de cette atteinte de l’hémostase. Stéréotypé et informatif, cet interrogatoire devra aussi relever les antécédents chirurgicaux et médicaux, l’histoire clinique personnelle et familiale, les traitements récents ou non, et même établir un arbre généalogique et l’existence d’une éventuelle consanguinité.
Biologiquement la Thrombasthénie de Glanzmann est facile à diagnostiquer :
-La numération plaquettaire : Dans la Thrombasthénie de Glanzmann le nombre de plaquettes est normal. Cette méthode est un examen de première intention devant tout désordre hémorragique. Elle reflète l’équilibre entre la production au niveau de la moelle osseuse et la destruction périphérique. Le taux physiologique est compris entre 150 à 400 G/L.

36
-Le temps de saignement est allongé TS > 20min [29]. Plusieurs techniques ont été utilisées mais la technique d’Ivy incision est considérée comme la technique de référence. Avant toute pratique d’un TS, l’interrogatoire doit rechercher la prise de salicylés ou d’anti-inflammatoires non stéroïdiens, qui allongent le TS par l’inhibition pharmacologique des fonctions plaquettaires.[30]
- Le temps d’occlusion : L’utilisation in-vitro du platelet function analyseur (PFA-100) est proposée pour remplacer la réalisation d’un TS dans le diagnostic des troubles de l’hémostase [31]. Le PFA-100 se révèle être un excellent outil de diagnostic grâce à sa grande sensibilité de détection des dysfonctionnements plaquettaires.
Fig12 : Platelet function analyser
- L’agrégation plaquettaire : L’agrégation plaquettaire est nulle [29]. Les tests d’agrégation sont réalisés en première intention avec un plasma riche en plaquettes préparé par centrifugation lente de sang total anticoagulé. Le sang total est recueilli sur citrate, dans les conditions habituelles pour les études de la coagulation. On utilise pour stimuler l’agrégation plaquettaire, des inducteurs physiologiques tels que l’ADP, le collagène, l’acide arachidonique, l’adrénaline, la thrombine. Quel que soit l’inducteur utilisé, les plaquettes ne s’agrègent pas entre elles. La Ristocétine, qui n’est pas un activateur plaquettaire physiologique, provoque l’agglutination des plaquettes par la fixation du facteur Von Willebrand plasmatique à la Gp Ib plaquettaire. L’absence d’agrégation quel que soit l’inducteur, mais avec persistance d’un
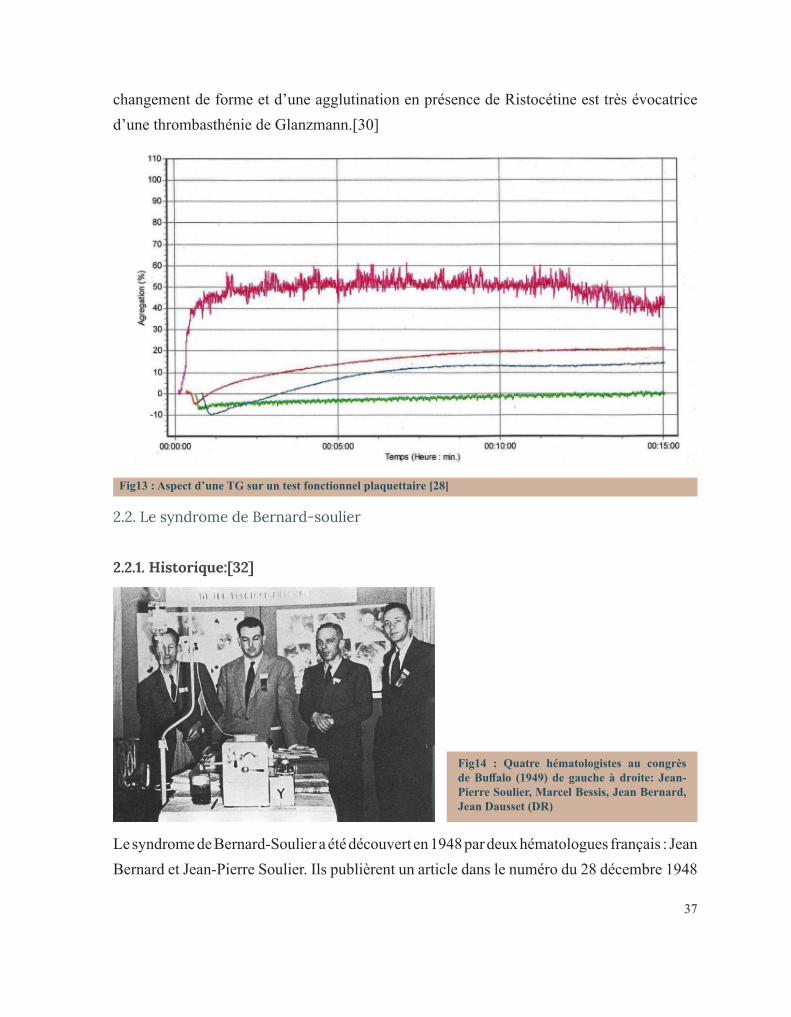
37
changement de forme et d’une agglutination en présence de Ristocétine est très évocatrice d’une thrombasthénie de Glanzmann.[30]
Fig13 : Aspect d’une TG sur un test fonctionnel plaquettaire [28]
Fig14 : Quatre hématologistes au congrès de Buffalo (1949) de gauche à droite: Jean-Pierre Soulier, Marcel Bessis, Jean Bernard, Jean Dausset (DR)
2.2. Le syndrome de Bernard-soulier
2.2.1. Historique:[32]
Le syndrome de Bernard-Soulier a été découvert en 1948 par deux hématologues français : Jean Bernard et Jean-Pierre Soulier. Ils publièrent un article dans le numéro du 28 décembre 1948

38
de La Semaine des hôpitaux, intitulé : «Sur une nouvelle variété de dystrophie thrombocytaire hémorragipare congénitale». Cette publication rapportait le cas d’un jeune garçon affecté dès le début de sa vie par une diathèse hémorragique dont la sœur aînée était elle-même décédée par une hémorragie mortelle. Les plaquettes de cet enfant étaient géantes et le temps de saignement allongé. Une nouvelle maladie venait d’être reconnue. Son étude pendant plusieurs décennies allait se montrer particulièrement fructueuse pour la compréhension des modalités de mise en œuvre des glycoprotéines de membrane dans l’activation des plaquettes et pour l’exploration des mécanismes complexes de l’interaction des plaquettes avec les endothéliums vasculaires lésés. Le syndrome était rare mais sa découverte d’importance. La place de cette première description est aujourd’hui universellement reconnue sous le nom de « syndrome de Bernard-Soulier » (BSS pour les Anglo-Saxons), honorant ainsi le nom de ses découvreurs, et soulignant une des nombreuses contributions françaises aux progrès dans ce domaine. Ce syndrome continue à être au centre de nombreux travaux sur la glycoprotéine Ib-IX, que l’on sut, une trentaine d’années plus tard, être le chaînon défaillant dans cette maladie. Le flambeau de l’étude des plaquettes, de leurs fonctions et de leurs interactions avec les endothéliums vasculaires fut ensuite repris par Jacques Caen et son équipe et porté au niveau de reconnaissance médicale et scientifique internationale.
2.2.2. Définition
Le syndrome de Bernard Soulier est une maladie hémorragique héréditaire autosomique récessive caractérisée par des plaquettes géantes, une numération plaquettaire normale ou une thrombopénie habituellement modérée, un temps de saignement allongé et une absence d’agglutination à la Ristocétine. Les plaquettes des patients BSS présentent généralement un défaut d’expression en surface de la glycoprotéine Ib-IX-V, un complexe de signalisation et d’adhérence spécifique des plaquettes, composé de disulfure de GPIba lié à GPIbβ et associé de manière non covalente à GPIX et à GPV[33]. C’est une maladie rare. La prévalence est inférieure à un par million d’habitants dans les populations les plus étudiées : Europe, Amérique du nord et Japon, mais semble sous-estimée. En effet, de 1948 à 1998, seulement 88 cas ont été décrits dans la littérature[34].

39
2.2.3. Le diagnostic positif
La thrombopénie est variable dans le SBS et le nombre de plaquettes varie généralement entre moins de 30 et 200 × 103 /μL. Le temps de saignement est d’ordinaire nettement prolongée[35]. L’évaluation du frottis sanguin périphérique révélera des plaquettes de grande taille; généralement plus du tiers des plaquettes ont environ la moitié de la taille d’un globule rouge (3,5 μm), et certaines plaquettes sont aussi grosses ou plus grosses qu’un lymphocyte[36].
Les échantillons de biopsie de moelle osseuse présentent un nombre normal de mégacaryocytes sans anomalies morphologiques significatives. Des tests modernes de la fonction plaquettaire, tels que le PFA-100, peuvent être utiles pour identifier des troubles plaquettaires qualitatifs tels que le SBS, mais avec une sensibilité variable, en fonction de la gravité du défaut. Actuellement, leur utilisation est limitée au dépistage d’un dysfonctionnement plaquettaire, et des tests supplémentaires, tels que l’agrégométrie ou la cytométrie en flux, sont nécessaires pour confirmation[37]. Les études d’agrégation plaquettaire in vitro montrent de manière caractéristique un échec d’agrégation avec la Ristocétine et une réponse lente avec de faibles doses de thrombine. Cette absence d’agrégation ne peut pas être corrigée par l’ajout de plasma normal, ce qui distingue le SBS de la maladie de Von Willebrand. Les plaquettes montrent une agrégation normale avec l’épinéphrine, l’adénosine diphosphate, le collagène et l’acide arachidonique[36].
La cytométrie en flux peut être utilisée pour confirmer les défauts du complexe GPIb-IX-V par des anticorps dirigés contre l’antigène CD42b de l’antigène plaquettaire de surface, révélant une réduction ou un déficit sévère de GPIba. Les autres antigènes plaquettaires, CD41 (GPIIb) et CD61 (GPIIIa), sont normaux. La cause de la thrombocytopénie dans le SBS est inconnue, et des études préliminaires ont suggéré une diminution de la survie plaquettaire. Cependant, une étude ultérieure utilisant des plaquettes marquées à la 111-oxyde d’indium montre une diminution faible ou nulle de la durée de survie des plaquettes, suggérant une thrombopoïèse inefficace ou en diminution [38], [39].

40
2.3. Les autres thrombopathies:[40]
Il existe d’autres types d’anomalies héréditaires touchant la fonction plaquettaire, nous citons :
- Maladie de von Willebrand de type plaquettaire : gain de fonction de la glycoprotéine Ib-IX-V.- Anomalies des récepteurs agonistes des plaquettes : les mutations rares du récepteur de l’ADP, P2Y12 et du récepteur de la TxA2.- Anomalies de la sécrétion plaquettaire : _ Delta-granules (déficit en pool de stockage , syndrome de Hermansky – Pudlak, syndrome de Chediak – Higashi, thrombocytopénie avec syndrome des rayons absents) _ Alpha-granules (syndrome des plaquettes grises, maladie des plaquettes du Québéc, Syndrome de Paris – Trousseau – Jacob) _ Granules alpha et delta.- Syndrome de Wiskott – Aldrich- Thrombocytopénie liée à l’X- Anomalies des phospholipides membranaires- Syndrome de Scott- Syndrome de Stormorken- Anomalies des voies de transduction du signal;
3. LES THROMBOPÉNIES CONSTITUTIONNELLES
3.1. Généralités
Les thrombopénies acquises telles que les purpuras thrombopéniques immunologiques ou idiopathiques (PTI) sont beaucoup plus fréquentes que les thrombopénies constitutionnelles ou génétiques. En effet, ces dernières sont des entités rares et hétérogènes mais certainement sous-diagnostiquées car souvent étiquetées thrombopénies auto-immunes. Ce sont des thrombopénies chroniques définies par un taux de plaquettes inférieur à 150 G/l. Une vingtaine de gènes ont été décrits comme responsables de ces thrombopénies. Leur

41
diagnostic précis est nécessaire car elles n’ont pas toutes le même pronostic, certaines pouvant notamment se compliquer d’hémopathies. Dans un premier temps, il est important de réunir un faisceau d’arguments orientant vers l’origine constitutionnelle : l’existence dès l’enfance de la thrombopénie et d’autres cas dans la famille constitue un argument fort. La deuxième difficulté est d’orienter l’étude génétique permettant de poser un diagnostic précis. Les variantes à l’origine de ces thrombopénies agissent à des phases distinctes de la mégacaryocytopoïèse et entraînent des thrombopénies aux caractéristiques différentes[41]
Les signes cliniques hémorragiques seront présents ou absents suivant qu’une thrombopathie est associée ou non. II n’y a pas de proportionnalité entre l’intensité de la thrombopénie et la présence de signes hémorragiques. La notion de dissociation clinique entre l’importance des signes hémorragiques et le taux de plaquettes, c’est-à-dire une symptomatologie hémorragique marquée avec un taux de plaquettes modérément abaisse, doit faire rechercher une anomalie de fonction surajoutée.
3.2. La Classification
Plus de 20 gènes ont été décrits comme responsables de thrombopénies constitutionnelles (Tableau 5 ci-dessous). Le mode de transmission peut être lié à l’X, autosomique dominant ou autosomique récessif (homozygote ou hétérozygote composite). Les formes à transmission autosomique récessive sont très souvent liées à un contexte de consanguinité. La classification la plus répandue des thrombopénies constitutionnelles est celle basée sur la taille plaquettaire évaluée par la mesure du volume plaquettaire moyen (VPM) par les automates ou la mesure de la taille des plaquettes en microscopie sur frottis sanguin coloré au May-Grünwald-Giemsa (tableau 6 ci-dessous). On distingue trois groupes : le groupe 1 avec des macroplaquettes ou des plaquettes géantes (taille entre 4 et 8 µm ou > 8 µm respectivement), le groupe 2 avec des plaquettes de taille normale (entre 4 et 8 µm) et le groupe 3 avec des plaquettes de petite taille (< 4 µm).D’autres classifications prennent en considération la présence ou non de manifestations extra-hématologiques (thrombopénies syndromiques), et puis d’autres en fonction du niveau d’atteinte de la mégacaryocytopoièse[42].
Les plus fréquentes des TC sont les macrothrombopénies constitutionnelles liées au
42
Tableau 5 : Thrombopénies constitutionnelles. Classification en fonction de l’anomalie génétique [41]
Tableau 6 : Principales thrombopénies constitutionnelles. Classification en fonction de la taille des plaquettes et du caractère isolé ou syndromique[42].

43
syndrome MYH9, de transmission autosomique dominante, conséquence de mutations localisées sur le gène Myosine heavy chain MYH9, gène découvert en 2000. Ce groupe de thrombopénies constitutionnelles avec macroplaquettes et plaquettes géantes associées à des inclusions leucocytaires ou pseudocorps de Döhle comprend les syndromes de May-Hegglin, de Fechtner, d’Epstein, de Sebastian, et le syndrome « Alport-like » avec macrothrombocytopénie regroupés dans une même entité. Les anomalies caractéristiques de ces syndromes ont d’abord été rapportées par May (en 1909) et Hegglin (en 1945) puis les différents syndromes associés ont été décrits successivement par Epstein (en 1972), Fechtner (en 1985) et Sebastian (en 1990). Ces entités présentent des signes cliniques variés mais sont toutes associées à la présence d’une macrothrombopénie (20–120 G/L). Les signes hémorragiques sont inconstants, habituellement modérés, surtout provoqués lors de traumatismes ou d’interventions chirurgicales. Dans certains cas les hémorragies sont plus sévères. Les syndromes de May-Hegglin et de Sebastian sont caractérisés exclusivement par la présence d’inclusions leucocytaires de différentes tailles appelées corps de Döhle alors que les syndromes d’Epstein et de Fechtner associent aux corps de Döhle la présence d’une surdité, d’une néphrite et/ou d’une cataracte.[43]
3.3. A propos d’un cas de la littérature :[44]
Une jeune fille âgée de 15 ans, d’origine comorienne, est hospitalisée dans le service pour une fièvre à 38,7 °C associée à une éruption cutanée évocatrice de varicelle. Elle séjourne en France depuis deux ans et ne rapporte aucun antécédent médical ou chirurgical particulier. Une thrombopénie à 20 G/l est détectée lors d’un hémogramme réalisé avant l’admission par son médecin traitant. Elle n’avait jamais eu d’analyse sanguine auparavant. Le tableau clinique à l’admission retrouve des lésions cutanées diffuses, vésiculeuses typiques d’une varicelle. Les lésions n’ont pas de caractère hémorragique et il n’existe pas de purpura. La patiente ne signale par ailleurs aucune hémorragie extériorisée. Devant une dyspnée et une toux, la radiographie thoracique confirme l’existence d’un syndrome interstitiel évocateur de pneumopathie varicelleuse. Il n’existe pas d’hépatosplénomégalie. Le bilan biologique constate : hémoglobine 11,7 g/dl, leucocytes 3,73 G/l avec 1,19 G/l polynucléaires neutrophiles et 2,35 G/l lymphocytes. Le chiffre des plaquettes est à 17 G/l avec un volume plaquettaire moyen (VPM) à 20,2 fl. L’analyse cytologique du frottis

44
sanguin confirme la présence de macroplaquettes et détecte aussi des pseudocorps de Döhle dans le cytoplasme des polynucléaires neutrophiles. Le bilan auto-immun met en évidence des anticorps antinucléaires (titre 1/400 d’aspect moucheté sans spécificité) sans anti-DNA natifs. La créatinine sérique est normale et la recherche d’une protéinurie des 24 heures négative. Les tests de Coombs érythrocytaire et la recherche d’anticorps antiplaquettes par la technique du MAIPA (monoclonal antibody specific immobilization of platelets antigen) sont négatifs. Le diagnostic retenu est celui d’une thrombopénie à plaquettes géantes dans le cadre d’une maladie de May-Hegglin, découverte fortuitement lors d’une varicelle. Cette jeune patiente n’a jamais présenté de manifestations hémorragiques malgré un taux de plaquettes stable oscillant entre 30 et 14 G/l.
Fig15 : Pseudocorps de Döhle (B) [44]
3.4. Démarche diagnostique
Les thrombopénies constitutionnelles ou héréditaires ont longtemps fait partie de groupe d’affections hématologiques méconnues, réservées aux spécialistes et aux fondamentalistes dont la thématique de recherche était la mégacaryocytopoièse. Cependant, la pratique systématique de la numération plaquettaire, et donc l’augmentation de détection en fréquence de ces anomalies, ont impliqué le biologiste plus activement dans le rôle d’orientation diagnostique en cas de thrombopénie dont l’étiologie et les mécanismes n’apparaissent pas identifiables. Aujourd’hui et grâce aux données cliniques, aux aspects biologiques et aux connaissances actuelles moléculaires et génétiques en matière de thrombopénies constitutionnelles, l’approche diagnostique à ce groupe de pathologies est plus facile et mieux établie.

45
La découverte d’une thrombopénie isolée, sans anémie ni leucopénie, et sans syndrome tumoral, évoque le plus souvent chez l’enfant le diagnostic de purpura thrombopénique immunologique (PTI). Nonobstant dans certains cas, il faut savoir évoquer d’autres diagnostics tels qu’une hypoplasie médullaire idiopathique ou constitutionnelle (anémie de Fanconi, dyskératose congénitale), une myélodysplasie ou une thrombopénie d’origine génétique. De même, devant certaines thrombopénies prolongées dans le temps, il faut savoir remettre en question le diagnostic souvent posé de PTI persistant ou chronique et évoquer l’hypothèse d’une thrombopénie constitutionnelle. Une thrombopénie chez l’enfant reste définie par un chiffre de plaquettes inférieur à 150 G/L, les valeurs normales avant l’âge de 15 ans se situent pour 95 % des enfants entre 165 G/L et 473 G/L avec une valeur médiane à 299 G/L[45].
Les principaux éléments cliniques qui doivent évoquer, chez l’enfant, la possibilité d’une thrombopénie constitutionnelle sont les suivants :
• Des antécédents familiaux de thrombopénie et/ou de manifestations hémorragiques, d’hémopathie myéloïde et/ou de myélodysplasie ; • Des anomalies cliniques associées, morphologiques ou fonctionnelles : syndrome dysmorphique, fente vélopalatine, eczéma, anomalies osseuses, malformations, infections à répétition, troubles auditifs, ophtalmologiques, rénaux, en sachant que certains de ces signes peuvent apparaître secondairement; • Un syndrome hémorragique plus sévère que ne le laisserait prévoir le nombre de plaquettes évoquant une thrombopathie associée ; • L’absence de modification ou une remontée faible du nombre de plaquettes après traitement, par gammaglobulines ou corticoïdes, dans l’hypothèse d’un PTI.[46]
Sur le plan hématologique, la découverte d’une thrombopénie impose de vérifier les autres paramètres de l’hémogramme : volume plaquettaire (en sachant que le résultat de l’automate est une moyenne), leucocytose, aspect des polynucléaires neutrophiles, et formule sanguine, taux d’hémoglobine, volume globulaire moyen, absence de schizocytes et de cellules anormales. Il faut également regarder la morphologie plaquettaire sur le frottis sanguin.

46
Il peut exister des anomalies de la morphologie (inclusions intraplaquettaires, couleur des plaquettes) ou de la taille des plaquettes (macrocytaires ou microcytaires), ou la présence d’inclusions intraleucocytaires (pseudo-corps de Dohle).
Toute thrombopénie « atypique » ou prolongée justifie un bilan simple comportant : • Une numération plaquettaire des parents ; • Une recherche d’autoanticorps sériques ou fixés à la surface des plaquettes (MAIPA) (surtout si la thrombopénie est modérée) ; • Un dosage du facteur Willebrand antigène et du cofacteur de la ristocétine dans l’hypothèse d’une maladie de Willebrand de type 2B ; • Un myélogramme doit être discuté.
Ce bilan peut être complété en fonction du contexte clinique et hématologique, par : • Une étude des fonctions plaquettaires à la recherche d’une thrombopathie associée. • L’étude biochimique des glycoprotéines de membrane plaquettaire, ou de constituants intracytoplasmiques (protéine WASP) ou granulaires (fibrinogène, facteur Willebrand). L’analyse par immunofluorescence de la distribution de la myosine intraplaquettaire et leucocytaire. • Un caryotype, avec technique d’hybridation in situ fluorescence (FISH), voire une analyse par CGH array ; • Et surtout, une analyse moléculaire génétique en fonction des orientations cliniques et hématologiques avec l’étude des gènes connus et identifiés actuellement dans des entités cliniques syndromiques bien individualisées : GPIb (BSS), WAS, MYH9, c-Mpl, AML-1, GATA1, FlI-1[47].

47
II- LES PATHOLOGIES DE LA COAGULATION :
1. L’HÉMOPHILIE
1.1. Historique :[48], [49]
Les premiers écrits sur ce qui aurait pu être l’hémophilie remontent au IIe siècle après Jésus-Christ et sont retrouvés dans le talmud, recueil d’écrits hébraïques, où l’on peut lire des recommandations sur la circoncision telle que «tout garçon dont au moins 2 frères aînés étaient décédés de suites hémorragiques d’une circoncision devait en être exempté…». Au Xème siècle, des cas d’hommes d’un certain village saignant jusqu’à la mort à la suite de petites blessures ont été décrits par Khalaf ibn Abbas, que nous connaissons sous le nom d’Albucasis.Elle sera nommée pour un moment hémorraphilie Fig16 : Portrait de la reine Victoria
« affinité à saigner » toutefois c’est le nom d’hémophilie proposé par F. HOPFF en 1828 qui sera retenu, même si sa signification « amour du sang » est sans lien étymologique avec la maladie. Plus près de notre époque, «la maladie des rois », affection incurable qui sévit dans les cours d’Europe au début du XXe siècle restait encore mal connu. La Reine Victoria transmet l’hémophile à son fils Léopold qui décédera à 31 ans d’une hémorragie cérébrale. Par l’intermédiaire du mariage de ses deux filles, Alice et Béatrice, la maladie est disséminée aux autres dynasties européennes (Allemagne, Espagne et Russie).
Ainsi l’hémophile est connue depuis des millénaires comme une maladie hémorragique héréditaire qui ne touche que les garçons, mais n’a bénéficié d’un traitement efficace que depuis la deuxième moitié du XXème siècle. De fait que la découverte de la cryoprécipitation par Judith Pool en 1960 a permis d’isoler un précipité riche en facteur VIII « cryoprécipité»

48
qui va être utilisé par les établissements de transfusion pour soigner les hémophiles A. Cet énorme pas en avant dans la prise en charge de cette pathologie conduit à abandonner la transfusion de sang total et de plasma frais congelé.
Par la suite, vers la fin des années 1960 et au début des années 1970, les concentrés de facteur VIII et de facteur IX ont fait leur apparition. Les concentrés lyophilisés pouvaient, dès lors être utilisés au besoin et ont révolutionné le traitement de l’hémophilie. Or, le drame des contaminations sur la décade 1980-1990 où de nombreux lots de concentrés de facteurs furent infectés par le VIH, le virus de l’hépatite B et C et furent utilisés chez les patients, met fin à l’âge d’or de la prise en charge de l’hémophilie.En 1987, le développement de méthodes d’inactivation virales efficaces ont permis la production de concentrés de facteurs d’origine plasmatique sans risque de contamination et ont permis à l’espoir de renaitre. En 1989, un médicament d’un genre nouveau est mis à la disposition des patients atteints de l’hémophilie A, c’est le traitement par facteur VIII recombinant, exempt d’agents pathogènes transmissibles.
Aujourd’hui, en ce début du XXIème siècle, trois défis restent à relever : la prévention efficace du handicap fonctionnel secondaire à l’arthropathie hémophilique, la disparition des inhibiteurs anti-facteurs VIII ou anti-facteur IX, l’accessibilité au traitement substitutif pour les hémophiles du monde entier.
1.2. Définition
L’hémophilie est une maladie hémorragique constitutionnelle et chronique. C’est une pathologie héréditaire récessive liée à l’X. On distingue deux types d’hémophilie selon la protéine déficitaire: l’hémophilie A si le déficit est lié au facteur VIII, hémophilie B s’il est lié au facteur XI.
1.3. Epidémiologie
Selon la définition de l’organisation mondiale de la santé (OMS), l’hémophilie est une maladie

49
rare. En effet son incidence dans la population générale est inférieure à 0,65 - 1‰. Un garçon sur 5000 naissances de sexe masculin naît atteint d’hémophilie A dans le Monde tandis que 1 garçon sur 25 000 naît avec une hémophilie B (il y a environ un cas d’hémophilie B pour 5 cas d’hémophilie A). La prévalence (nombre des cas dans une population donnée) varie de 1 sur 18 000 à 1 sur 7 000 personnes du sexe masculin selon les pays pour l’hémophilie A. Ces chiffres varient entre 1 sur 100 000 et 1 sur 30 000 personnes du sexe masculin pour l’hémophilie B.[50]Il n’y a pas de différence significative entre les différentes races. Cependant une étude a démontré que dans les pays où les mariages consanguins sont fréquents comme les pays musulmans ou le sud de l’Inde, les déficits héréditaires de la coagulation ont des prévalences plus élevées au point de représenter un problème de santé publique [51].
1.4. Génétique
L’hémophilie est une maladie héréditaire, récessive, liée à des gènes situés sur le chromosome X. Les gènes responsables de la synthèse des protéines facteur VIII et facteur IX sont situés sur le bras long du chromosome X à deux endroits distincts : Xq28 pour le facteur VIII, Xq27 pour le facteur IX. Différentes anomalies génétiques à l’origine de l’hémophilie aboutissent à l’absence totale de facteur VIII ou de facteur IX fonctionnels et sont à l’origine de la plupart des formes sévères de la maladie. D’autres permettent la synthèse d’un taux faible de facteurs VIII ou IX circulant à l’origine des formes modérées ou mineures. La transmission par le chromosome X entraîne une expression clinique de la maladie chez l’homme et une transmission de la maladie pour les hommes et les femmes. Dans une même famille, le type de l’hémophilie (A ou B) et le degré de sévérité de la maladie sont toujours les mêmes. Dans 30 % des cas, il n’est pas possible de retrouver un antécédent d’hémophilie dans la famille, on parle alors d’hémophilie sporadique. L’hémophilie féminine existe, mais elle est exceptionnelle. Le recours au conseil génétique est indispensable dès le diagnostic établi pour informer l’hémophile et sa famille et préparer à la prise en charge ultérieure de l’ensemble de la famille sur le plan génétique, en particulier pour aborder la transmission de la maladie[52].

50
1.5. Le diagnostic biologique :[53]
Ce diagnostic repose d’une part, sur un interrogatoire minutieux à la recherche d’une histoire hémorragique personnelle et/ou familiale qui permettra d’identifier les accidents hémorragiques et les complications survenues dans certaines situations cliniques caractéristiques de cette maladie constitutionnelle de l’hémostase telles que les hémarthroses, les extractions dentaires, les amygdalectomies et adénoïdectomies ; et d’autre part, sur le bilan d’hémostase qui permet, devant un allongement isolé du temps de céphaline activateur (TCA), la caractérisation du déficit en FVIII ou FIX et la présence éventuelle d’un inhibiteur. Le temps de saignement, la numération plaquettaire, le temps de Quick et le temps de thrombine sont normaux [42].
Les examens d’hémostase de première intention effectués chez un hémophile sont : temps de céphaline + activateur (TCA), temps de Quick, taux de fibrinogène, et la numération plaquettaire. L’hémophilie est suspectée devant un allongement isolé du TCA. Cependant, le TCA peut être faussement raccourci par une activation in vitro en cas de prélèvement difficile. Dans tous les cas le diagnostic n’est confirmé qu’après des tests complémentaires.Le diagnostic de confirmation repose sur les dosages des activités FVIII dans l’hémophilie A, et FIX dans l’hémophilie B. Ces dosages permettent d’évaluer la sévérité du déficit. Celui-ci s’exprime en pourcentage ; et suivant le taux du facteur concerné on distingue les hémophilies sévères (taux inférieurs à 1 %), modérées (1 à 5 %) et mineures (5 à 30 %).
2. LES DÉFICITS RARES DE LA COAGULATION
Les déficits rares de la coagulation représentent 3 à 5 % des déficits congénitaux de la coagulation. Ils regroupent les déficits constitutionnels isolés en FII, V, VII, X, XI, XIII, fibrinogène et les déficits combinés en FV et VIII et en facteurs vitamine K dépendants. Les informations recueillies auprès de plusieurs registres (World Federation of Hemophilia global survey, and the European registry[54]) confirment que le déficit en FVII et le déficit en FXI sont les plus fréquents et le déficit en prothrombine et en facteur XIII sont les moins fréquents. Ils ont généralement une transmission autosomique récessive. Les formes homozygotes ont une expression hémorragique plus marquée, essentiellement dans un

51
contexte de traumatisme ou de chirurgie. Les déficits sévères en FVII, FX, FXIII, ou en fibrinogène peuvent avoir une révélation précoce en période néonatale, avec un risque élevé d’hémorragies intracrâniennes. Une des caractéristiques de ces déficits rares est une mauvaise corrélation entre le risque hémorragique et le taux du facteur déficitaire, particulièrement pour le FXI [14].
Tableau 7 : Some features of rare coagulation disorders [54]
Les déficiences héréditaires sont décrites pour tous les facteurs de la coagulation connus. En outre, il existe des personnes présentant une tendance hémorragique anormale pour lesquelles aucune cause n’a encore été identifiée. Le concept d’équilibre hémostatique global est de plus en plus reconnu, selon lequel ce n’est pas uniquement le niveau d’un facteur qui compte, mais le contrôle général de l’hémostase qui peut déterminer le risque de saignement. Cela peut expliquer pourquoi le risque de saignement ne peut être prédit à partir d’un seul facteur, comme par exemple dans le déficit en facteur VII ou en facteur XI. Les carences en facteurs rares ont plusieurs propriétés communes : elles sont héritées de manière autosomique, bien que des symptômes de saignement aient été décrits chez des individus hétérozygotes partiellement déficients. Ces carences rares sont plus courantes dans les populations et les pays où les mariages consanguins sont plus fréquents [55].
Le but n’étant pas de passer en revue tous les déficits de la coagulation, nous présenterons brièvement certains d’entre eux dans ce qui suit :

52
Le déficit en facteur V : Le déficit congénital en facteur V se transmet selon le mode autosomique récessif. Il se traduit par des hémorragies de sévérité variable et peut se déclarer à tout âge. Aucune prédisposition ethnique n’a été rapporté [13]. Le déficit en facteur V est 10 fois plus fréquent dans les pays à consanguinité élevée [10]. Plus de 200 cas ont été rapportés dans la littérature [5,14]. Le facteur V est synthétisé principalement par le foie, mais également par les plaquettes. Vingt pour cent du taux du facteur V se trouvent dans les mégacaryocytes, le reste circulant dans le plasma [5]. Le facteur V activé (FVa) est un cofacteur de la prothrombinase qui active la prothrombine en thrombine [5], sa production est contrôlée par le gène F5 (1q24.2). Vingt-six mutations responsables du déficit en facteur V ont été isolées à ce jour [6]. Les déficits sont de 2 types : type 1 (quantitatif) avec un taux très bas de facteur V fonctionnel et type 2 (qualitatif) avec un taux normal ou légèrement diminué de facteur V mais une réduction de l’activité coagulante.
Le déficit en facteur VII : Le déficit congénital en facteur VII a été décrit pour la première fois en 1951 par Alexander. Sa prévalence est estimée à 1/1 000 000. Il est transmis selon le mode autosomique récessif. Seuls les patients homozygotes ou hétérozygotes composites peuvent présenter un syndrome hémorragique, les sujets hétérozygotes étant asymptomatiques. Le gène responsable est localisé sur le chromosome 13. Plus de 130 mutations différentes ont été actuellement identifiées. Les patients homozygotes ont un déficit majeur, avec un taux de facteur VII inférieur à 10 %. Ce déficit profond peut rester asymptomatique ou se traduire par un syndrome hémorragique plus ou moins sévère. La corrélation entre la sévérité du saignement et l’importance du déficit n’est pas prouvée, et la symptomatologie est variable [56].
Le déficit en facteur XI : Le déficit congénital en facteur XI appelé aussi maladie de Rosenthal ou hémophilie C, est une maladie hémorragique rare par déficit constitutionnel en facteur de coagulation qui touche surtout la population d’origine juive ashkénaze. Le déficit en facteur XI a été décrit en 1953 par Rosenthal et al. C’est un déficit génétique rare avec une prévalence de 1/106 millions, découvert surtout à l’âge adulte[57]. Il se différencie de l’hémophilie A et B par le fait qu’il ne provoque pas de saignements articulaires ou musculaires. Il se transmet selon un mode autosomique récessif avec une pénétrance variable

53
et le gène est localisé sur le chromosome 4. Deux types de mutations ont été individualisés : type II (Glu117stop) observé chez les juifs iraquiens, et type III (Phe283Leu) chez les juifs ashkénazes. Le risque hémorragique est beaucoup plus important chez les sujets ayant un génotype II/II ou hétérozygotes composites type II/III que chez les homozygotes III/III et les hétérozygotes[58]D’autres mutations ont été identifiées chez les non-juifs : Cys128stop décrite en Grande-Bretagne et Cys38Arg en France.
Le déficit en facteur XIII : Le déficit congénital en facteur XIII (FXIII) de la coagulation (facteur stabilisant de la fibrine), découvert par Duckert et al en 1960, est un défaut génétique exceptionnel de l’hémostase dont la fréquence dans le monde est estimée à un cas pour deux millions d’habitants. En 2011, plus de 200 cas sont rapportés dans le Monde. Il s’agit d’une affection autosomique récessive s’exprimant à l’état homozygote ou double hétérozygote par un taux de facteur XIII inférieur à 1 % dans le plasma et l’apparition d’hémorragie dès le jeune âge. Le diagnostic du déficit en FXIII peut être difficile en l’absence d’une forte suspicion clinique. Les tests standards d’hémostase tels que temps de prothrombine, taux de plaquette, taux de fibrinogène et temps de saignement sont tous normaux. La suspicion d’une anomalie génétique de l’hémostase doit conduire au dosage d’activité du FXIII.
L’afibrinogénémie congénitale : Décrite pour la première fois en 1920, quelques centaines de cas seulement ont été rapportés depuis faisant d’elle une des affections rares de la coagulation. L’afibrinogénémie congénitale est une maladie rare dont la prévalence est estimée actuellement à 1/1 million d’habitant, due à un déficit constitutionnel en fibrinogène. La transmission se fait selon le mode autosomique récessif et la maladie est caractérisée par des saignements susceptibles d’engager le pronostic vital.Le diagnostic de la maladie est confirmé par la biologie. Trois classes de défauts héréditaires du fibrinogène sont reconnues : les dysfibrinogénémies, les hypofibrinogénémies et les afibrinogénémies. Les dysfibrinogénémies sont des anomalies qualitatives, dues à la présence d’un fibrinogène anormal ; la plupart des sujets atteints sont asymptomatiques et la découverte est souvent fortuite. La symptomatologie est faite de signes hémorragiques dans 25 % des cas ou de manifestations thromboemboliques (thrombophlébite, embolie pulmonaire...) dans 20 % des cas, faisant parfois la gravité de l’affection. Les hypofibrinogénémies sont

54
caractérisées par un déficit partiel en fibrinogène souvent asymptomatique et peuvent s’exprimer par une hémorragie après un traumatisme ou bien en cas de la coexistence d’un autre trouble de l’hémostase. Dys et hypofibrinogénémie sont des diagnostics différentiels de l’afibrinogénémie qui est la forme la plus extrême de la déficience quantitative en fibrinogène. Le bilan d’hémostase met en évidence un sang incoagulable avec un taux de plaquettes normal, un TS, un temps de céphaline activé(TCA) très allongés et un TP très bas. En cas d’afibrinogénémie, le fibrinogène est indosable, alors que dans les hypofibrinogénémies, son taux est faible, détectable par des techniques de dosage immunologique[59].
Tableau 8 : Caractéristiques différenciant les déficits rares de la coagulation d’une hémophilie d’après S.S. Acharya [14]

55
III-LES PATHOLOGIES DE LA FIBRINOLYSE
La fibrinolyse est l’ensemble des mécanismes cellulaires et plasmatiques permettant la destruction du thrombus, mais elle intervient aussi dans le développement tissulaire. Son dérèglement contribue à la survenue de pathologies thrombotiques et hémorragiques. Au niveau hémorragique, deux déficits en inhibiteur cliniquement pertinents ont pu être identifiés, le déficit en α2-antiplasmine et le déficit en PAI-1. Bien que leur symptomatologie soit variable, elle est assez similaire, tout comme les options thérapeutiques. Ils sont caractérisés par une fréquence très faible, ne sont pas repérés par des tests usuels ou simples et nécessitent donc une mesure spécifique qui n’est pas toujours effectuée. Ils se doivent d’être recherchés au moins dans les cas bien documentés de pathologie hémorragique persistante, après élimination des causes biologiques plus fréquentes comme une maladie de Willebrand, un déficit en facteur de la coagulation, une thrombocytopathie. En effet la sanction thérapeutique est simple, ainsi que la recherche chez les apparentés.
1. DÉFICIT EN α2-ANTIPLASMINE
Le déficit constitutionnel en α2-antiplasmine est une maladie rare dont la prévalence exacte est inconnue, mais très faible[60]. Une quarantaine de cas ont été décrits jusqu’à l’année de 2012, correspondant à des anomalies hétérozygotes pour la plupart. Lors d’anomalie homozygote, la consanguinité est habituelle. Le déficit se traduit par une symptomatologie hémorragique.[61]Le diagnostic biologique est habituellement un diagnostic d’élimination, parfois difficile à réaliser. En effet, ces patients sont investigués à plusieurs reprises à la recherche d’une étiologie à un syndrome hémorragique inhabituel, par des tests classiques de l’hémostase qui restent normaux (temps de Quick, temps de céphaline activé, temps de thrombine, mesure du fibrinogène, agrégabilité plaquettaire, facteur Willebrand,). La mise en évidence du déficit nécessite la mesure spécifique de l’α2-antiplasmine, ce qui n’est pas souvent réalisé étant donné la rareté des occurrences, et peut conduire à des absences de diagnostic[62], [63]
L’activité inhibitrice est quantifiée à l’aide d’un substrat chromogène sensible à la

56
plasmine[64]. L’activation de la réaction est réalisée par l’ajout de plasmine en quantité connue dans le milieu plasmatique testé. La plasmine résiduelle est mesurée après action de l’α2-antiplasmine présente dans le plasma. Cependant, d’autres molécules sont susceptibles d’interférer et il est important de respecter les critères recommandés pour l’utilisation de technique enzymatique lors de la mesure spécifique de cet inhibiteur. Une caractérisation du type de déficit peut être réalisée par la mesure de l’antigène (quantifiable par les techniques immunoprécipitantes) et la recherche d’une fonctionnalité de la molécule variante. Le déficit est alors répertorié de type I ou de type II selon l’absence ou la présence d’une molécule reconnue immunologiquement et à faible activité. Les mesures plus générales de la fibrinolyse peuvent être perturbées, avec par exemple un raccourcissement des temps de lyse du caillot de sang total ou du caillot des euglobulines, mais de façon non systématique [65] posant le problème de l’absence de dépistage de valeur sur un test autre que la mesure directe de l’activité.
2. DÉFICIT EN PAI-1:
L’inhibiteur de l’activateur du plasminogène de type 1 (PAI-1) est un composant important du système de coagulation qui régule la fibrinolyse dans la circulation. Une diminution des taux de PAI-1 peut entraîner une fibrinolyse accrue et une diathèse hémorragique associée. La documentation claire du déficit en PAI-1 en tant que cause d’un trouble de la coagulation est rare. Le PAI-1 a été identifié pour la première fois dans les années 1980 et le premier cas signalé de déficit en PAI-1 est apparu en 1989. Plusieurs rapports ont suivi, bien que deux seulement aient identifié un défaut génétique sous-jacent. Ces rapports de déficit en PAI-1 suggèrent que les personnes touchées présentent des symptômes de saignement légers à modérés, y compris épistaxis, ménorragie et saignement retardé après un traumatisme ou une intervention chirurgicale. Les individus affectés présentent rarement des saignements spontanés fréquemment observés dans les autres carences en procoagulants. [66].
Comme pour le déficit en α2-antiplasmine, étant donné sa rareté, le diagnostic biologique est un diagnostic d’élimination. Par ailleurs, il existe des difficultés lors du diagnostic biologique, correspondant au fait que si la mesure de l’activité du PAI-1 ne pose pas de difficulté pour rechercher une élévation (comme cela existe en association avec les risques thrombotiques

57
artériels et veineux), la sensibilité des méthodes d’activité est médiocre. La mesure d’activité est basée sur la réaction d’inhibition de t-PA actif ajouté en excès, repéré par son action sur du plasminogène et un substrat sensible à la plasmine. Mais cette technique pose des problèmes de standardisation et de bruit de fond. Les mesures antigéniques par technique Elisa ne permettent de confirmer le diagnostic que dans les cas de déficit quantitatif, ce qui n’est pas systématique. La recherche d’un déficit en PAI-1 plaquettaire peut renforcer la présomption diagnostique, mais il n’est pas toujours présent. Le respect de conditions pré-analytiques très contraignantes (afin de limiter les artefacts liés au cycle nycthéméral et à la libération du pool plaquettaire d’inhibiteur) qui sont de rigueur lors du prélèvement pour la recherche d’une élévation du taux plasmatique de PAI-1 corrélée au risque thrombotique artériels ou veineux, est habituellement élidé. Seule la mise en évidence d’une anomalie génique permettra de confirmer le diagnostic de façon certaine. À ce jour uniquement 3 mutations ont été décrites, dont celle retrouvée dans la population Amish [61], [66].
IV. CIRCONSTANCES DE DÉCOUVERTE DES PATHOLOGIES CONSTITUTIONNELLES DE
L’HÉMOSTASE
1. SYMPTOMATIQUE
1.1. Symptomatologie hémorragique
Les saignements bénins sont fréquents chez les enfants en bonne santé, mais peuvent occasionnellement indiquer la présence d’une diathèse hémorragique congénitale ou acquise. Ils n’indiquent donc pas toujours un trouble de saignement sous-jacent. Des épistaxis et des ecchymoses légères ont été rapportés chez 39% et 24% des enfants, respectivement[67]. Les ecchymoses peuvent être liées à une activité «normale» de l’enfance et leur prévalence varie selon le stade de développement moteur, elles sont retrouvées chez 61% à 90% des enfants âgés de 2 à 10 ans [68]. Les autres facteurs de saignement chez les enfants incluent les anomalies anatomiques qui contribuent à des saignements graves, tels que des saignements intracrâniens, gastro-intestinaux ou génito-urinaires, et des saignements menstruels abondants

58
chez les adolescentes [69], résultant de l’immaturité l’axe hypothalamo-hypophyso-ovarien et l’anovulation. Le taux global de saignements après une amygdalectomie ou une procédure pédiatrique courante, a été rapporté à 3,3% chez les patients ne présentant pas de trouble de la coagulation[70].
L’identification de l’enfant présentant un saignement pathologique peut donc s’avérer difficile compte tenu de la fréquence relative des saignements non pathologiques et des similitudes entre les symptômes pathologiques et non pathologiques. Les sites de saignement courants (la peau et les muqueuses) sont similaires et, bien que les saignements se produisent souvent de manière spontanée en cas de troubles de la coagulation, les hémorragies peuvent se présenter initialement après un traumatisme ou une intervention chirurgicale ou, chez les filles, à la ménarche.Inversement, les enfants atteints de troubles de la coagulation peuvent tolérer certains problèmes hémostatiques sans présenter un saignement excessif en apparence, avant de présenter un saignement pathologique plus tard dans la vie. Les facteurs pouvant aider à différencier le saignement pathologique par rapport au saignement non pathologique comprennent : l’âge au premier épisode, fréquence, étendue et durée du saignement, antécédents personnels de récidive spontanée ou provoquée ou saignements menstruels abondants chez les filles, en particulier s’ils sont accompagnés d’autres saignements ; et antécédents familiaux d’hémorragie ou de diathèses hémorragiques connues (tableau ci-dessous) [71].

59
Les traumatismes non accidentels doivent également être pris en compte chez les enfants présentant des symptômes de saignement inexpliqués ou excessifs. En particulier, toute contusion chez un enfant non mobile doit être considérée comme pathologique et peut indiquer soit un trouble de la coagulation, soit un traumatisme non accidentel [72]. Des ecchymoses loin des protubérances osseuses, en particulier du visage, de la tête et du cou, sont particulièrement évocatrices d’un traumatisme non accidentel.Les troubles hémorragiques congénitaux surviennent lorsque les protéines responsables de la coagulation, de la fonction plaquettaire ou de la fibrinolyse sont déficientes. À l’exception des facteurs de contact, dans lesquels aucun état de déficience clinique n’a été reconnu, les déficiences héréditaires de l’une des protéines hémostatiques connues provoquent des troubles de saignement ou de coagulation. Les manifestations de la maladie n’apparaissent généralement que lors d’une épreuve hémostatique, bien qu’une hémorragie spontanée puisse survenir chez les patients atteints d’hémophilie ou de déficits graves. Chez les enfants, moins
Tableau 9 : Initial features suggestives of pathologic bleeding in children [71]

60
susceptibles que les adultes d’avoir subi une chirurgie ou un traumatisme, l’identification des symptômes de saignement nécessite des antécédents médicaux minutieux, un examen physique et des antécédents familiaux afin de détecter un éventuel trouble de la coagulation.
Révélation en période néonatale Les troubles hémorragiques congénitaux peuvent se révéler pendant la période néonatale. L’hémophilie, la déficience en fibrinogène (hypofibrinogénémie ou afibrinogénémie), déficit en facteur VII, déficit homozygote en α2-antiplasmine et déficit en facteur XIII peuvent être associées à une cicatrisation retardée du tronc ombilical ou à des saignements à la chute du cordon. Effectivement, le saignement à la chute du cordon ombilical reste la manifestation la plus précoce, la plus fréquente et la plus caractéristique du déficit congénital en facteur XIII, constaté dans 87% des cas de la littérature [73] et peut-être révélateur de l’afibrinogénémie dans 50 % des cas[59].Une hémorragie intracrânienne (ICH) peut être également le symptôme révélateur d’un trouble de la coagulation en période néonatale. L’incidence de l’hémorragie intracrânienne néonatale chez les garçons atteints d’hémophilie est estimée à 3% [74], mais elle est particulièrement élevée dans le déficit en facteur XIII (25% à 60%) [75]. Dans le déficit en facteur VII, la révélation précoce au cours des premiers mois de vie traduit souvent une forme grave de la maladie et selon certaines études, un nouveau-né sur 6 atteint de ce déficit présente un saignement intracrânien [76]. Le saignement intracrânien secondaire à des troubles de la coagulation peut être sous-dural, sous-arachnoïdien, intraventriculaire, épidural ou intra-parenchymateux et est associé aux mêmes signes et symptômes que chez les patients ne présentant pas de trouble de la coagulation. Le diagnostic d’un trouble de la coagulation, dans ce contexte est urgent, car un traitement de substitution spécifique est nécessaire pour éviter une extension du saignement. Les hémorragies intracrâniennes sont en règle générale rares mais graves et affectent le pronostic vital.Les saignements prolongés du talon du nouveau-né annoncent généralement un trouble de la coagulation qui peut être congénital ou acquis. Ces symptômes doivent être étudiés pour diagnostiquer avec précision la cause du saignement et prévenir d’autres blessures.

61
Hémorragie en post circoncisionLes saignements excessifs dus à la circoncision sont souvent un indicateur d’un trouble de l’hémostase, en particulier lorsqu’ils se produisent de manière diffuse sur le gland et ne sont pas dus à un vaisseau saignant. L’incidence exacte des saignements résultant de la circoncision chez les patients ayant un trouble de la coagulation est variable, chez les hémophiles par exemple elle est estimée par les auteurs entre 0,1 et 35% [77]. L’hémorragie en post circoncision est assez fréquente et peut révéler l’existence de n’importe quelle anomalie héréditaire de l’hémostase, entre autres le déficit en facteur XI et l’hémophilie mineure où les manifestations hémorragiques ne sont généralement que légères à modérées, et ne surviennent qu’après une chirurgie ou une intervention post-invasive.
Les ecchymoses, les autres saignements cutanés et sous cutanésLes ecchymoses chez les enfants doivent d’abord être différenciées des causes extrinsèques telles que les abus envers les enfants, qui sont malheureusement plus fréquents que l’hémophilie. Les ecchymoses causées par un traumatisme sont plus susceptibles de se produire au calvarium, à la poitrine, au dos et aux os longs. Les ecchymoses peuvent conserver les contours de l’impression de la main ou de la ceinture incriminées. Les ecchymoses associées aux troubles plaquettaires surviennent généralement dans les zones de traumatismes infantiles typiques, telles que les protubérances osseuses des extrémités et les processus épineux le long du dos. Les ecchymoses de type plaquettaire peuvent être étendues, mais sont généralement superficielles et auront plusieurs étapes de résolution spontanée en passant par les stades de la biligénie (bleu-violet, vert-jaune puis brun). Les ecchymoses qui surviennent dans les troubles de la coagulation ont tendance à être profondes et larges et peuvent être surélevées ou calcifiées [78]. Les nourrissons présentant un trouble grave de la fonction plaquettaire peuvent présenter un purpura palpable ou des « tâches de muffins aux bleuets ». Les pétéchies représentent un dysfonctionnement plaquettaire ou une fragilité capillaire, mais peuvent être induites par un traumatisme. L’hématome intramusculaire peut être plus difficile à visualiser, mais provoque un gonflement du groupe musculaire et une douleur liée à l’utilisation du muscle. Des ecchymoses, en particulier des tissus mous profonds et des hématomes intramusculaires, peuvent se développer au cours des premiers mois de la vie, ou plus tard dans l’enfance

62
notamment à la suite d’un traumatisme chez les hémophiles ayant un déficit sévère [79], mais peuvent se voir également chez les patients porteurs de thrombopathies (Thrombasthénie de Glanzmann, syndrome de Bernard Soulier) de thrombopénies constitutionnelles ou de déficits rares de la coagulation. Les hématomes sont redoutées pour leur complications. Leur gravité est liée soit à leur volume (risque de spoliation sanguine), soit à leur localisation. Certains hématomes mettent en jeu le pronostic vital : hématome intracérébral ou rétropéritonéal, localisation dans la sphère ORL. D’autres localisations compromettent le pronostic fonctionnel comme les hématomes compressifs des membres avec compression vasculaire (risque ischémique) ou nerveuse (risque de paralysie, hématome péri rachidien, rétro-orbitaire, etc. Un hématome mal résorbé ou insuffisamment traité peut également évoluer vers une pseudotumeur hémophilique avec formation d’une coque fibreuse, adhérente aux plans adjacents qui peut ultérieurement évoluer pour son propre compte.
L’hémarthroseL’hémarthrose caractéristique des hémophiles, saigne dans l’articulation, et provoque un épanchement articulaire et des douleurs lors du mouvement de l’articulation. Les hémorragies intra-articulaires sont plus fréquentes dans l’hémophilie sévère où elles sont spontanées, récidivantes et douloureuses, leur nombre varie entre 20 à 25 saignements par an. Elles apparaissent vers l’âge de 1-3 ans lorsque la marche commence. Toutes les articulations peuvent être touchées, mais les plus souvent atteintes sont le genou, la cheville et le coude. L’hémarthrose aigüe, de diagnostic évident, doit être traitée le plus précocement possible. La répétition des hémarthroses sur une même articulation entraîne très tôt des lésions irréversibles de la synoviale et du cartilage qui aboutissent à la destruction de l’articulation, avec rétractions tendineuses, amyotrophie des muscles adjacents (arthropathie hémophilique), source de handicap majeur dans l’hémophilie [52]. Néanmoins l’hémarthrose peut se manifester dans d’autres pathologies constitutionnelles : la maladie de Von Willebrand de type 3, le déficit en facteur II, XIII, VII et les déficits combinés en facteurs. Elle est exceptionnelle dans les déficits en fibrinogène, le déficit en facteur XI et X [57].

63
Fig17 : Hématome suite à une ponction veineuse chez un enfant hémophile de 3ans.
Fig18 : Hémarthrose de la cheville
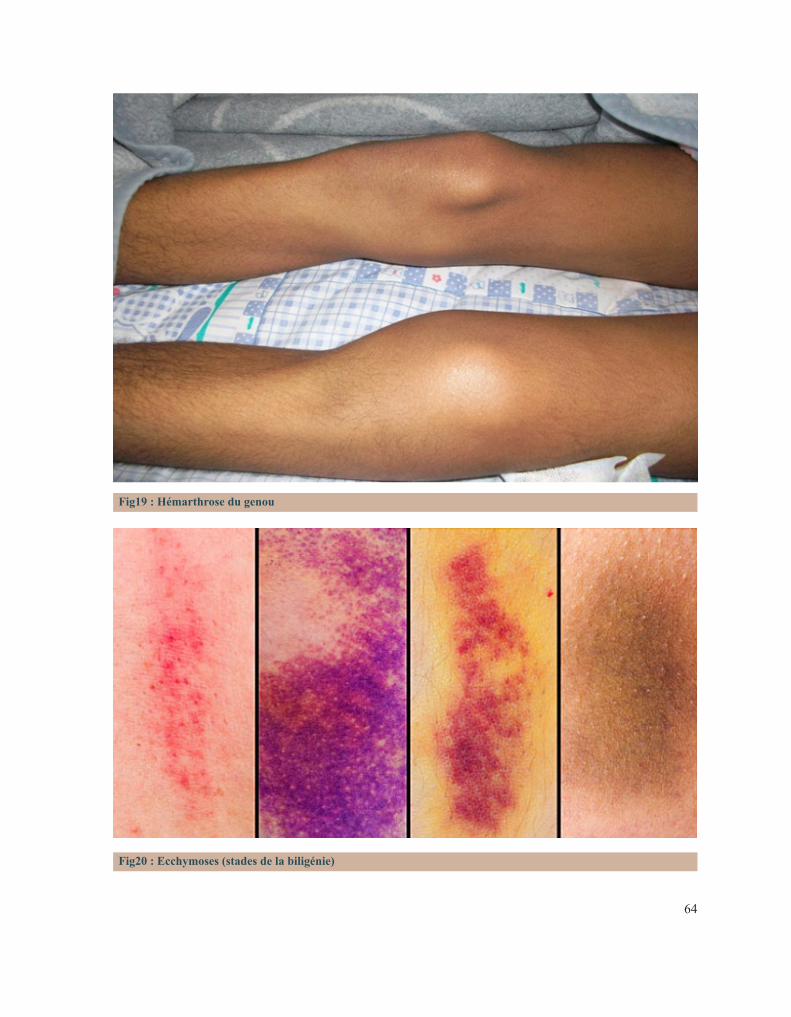
64
Fig19 : Hémarthrose du genou
Fig20 : Ecchymoses (stades de la biligénie)

65
Les saignements muqueux Les saignements cutanéomuqueux sont caractéristiques des troubles de l’hémostase primaire et sont de gravité variable selon le type du trouble et selon l’importance du déficit.
Dans la Thrombasthénie de Glanzmann qui est la plus sévère des thrombopathies constitutionnelles la révélation de la maladie peut se faire en période néonatale, parfois dès la naissance par un syndrome hémorragique grave, principalement cutanéomuqueux (épistaxis, purpura, gingivorragies). Mais le plus souvent, la TG est rarement hémorragique en période néonatale, même lorsque les patients naissent par voie basse sans précaution particulière et se manifeste plutôt dans la petite enfance et l’épistaxis est alors une cause fréquente de saignements graves [80].
La maladie de Von Willebrand de type 1 et 2N se manifeste essentiellement par des hémorragies cutanéomuqueuses, en rapport avec le rôle du facteur Von Willebrand dans l’hémostase primaire, et qui sont le plus souvent modérées, ubiquitaires et d’apparition spontanée ou provoquée par des traumatismes sous forme : d’épistaxis récidivantes souvent bilatérales, de gingivorragies, d’ecchymoses et de ménorragies chez la femme (alors que les pétéchies sont rares) [81].
Dans le syndrome de Bernard Soulier: les manifestations cliniques comprennent généralement des épisodes d’épistaxis fréquents, des saignements gingivaux et cutanés et des hémorragies associées à un traumatisme. Les ecchymoses sans traumatisme important sont relativement courantes, tout comme les épisodes d’épistaxis spontané et les saignements gingivaux. Bien que ces caractéristiques soient typiques, les comparaisons des profils cliniques des patients ayant un SBS révèlent des variations considérables entre les individus [82].
L’épistaxis est une plainte courante chez les enfants et est probablement due à des facteurs locaux tels que le dessèchement de la muqueuse nasale, la rhinite allergique ou un traumatisme[83]. Parmi les patients référés à un service de pédiatrie clinique d’hématologie pour épistaxis récurrent, 25 à 33% des patients présentent une coagulopathie ou un dysfonctionnement plaquettaire[84], [85]. Lors d’une analyse rétrospective, la durée et la

66
gravité de l’épistaxis et la présence d’autres symptômes de saignement n’avaient aucune valeur prédictive, cependant, les antécédents familiaux de symptômes hémorragiques étaient significativement associés à la coagulopathie lors des tests de laboratoire.
Le diagnostic de ménométrorragies est difficile chez les jeunes adolescentes, quand les règles sont d’installation récente. Ce sont les saignements à répétition ou l’importance de l’hémorragie qui conduisent les adolescentes en consultation. Les ménométrorragies sont le plus souvent dues à des troubles fonctionnels. Les anomalies de l’hémostase représentent la 2ème cause, évoquées lorsque l’adolescente a dans ses antécédents personnels des épisodes de saignements (épistaxis, gingivorragies, hémorragie postopératoire, hématomes fréquents) ou lorsqu’une anomalie est déjà connue dans la famille. Une origine organique est rare mais il faut la rechercher en priorité quand le saignement est caractéristique : peu abondant, non concomitant aux premières règles, anarchique, avec conservation de règles normales. La présence de douleurs pelvi-abdominales ou l’existence d’une activité sexuelle ou d’une contraception sont également des éléments en faveur d’une origine organique. Les ménométrorragies fonctionnelles sont souvent dues à une anovulation en rapport avec une immaturité de l’axe hypothalamo-hypophyso-ovarien. Les causes organiques peuvent être tumorales (tumeurs cervico-vaginales ou tumeurs sécrétantes de l’ovaire) ou secondaires à des infections génitales hautes, comme elles peuvent compliquer une grossesse ou une contraception. Les anomalies de l’hémostase peuvent être secondaires à une insuffisance hépatique ou rénale ou constitutionnelles par déficit congénital en facteurs de coagulation. Les principaux déficits sont la maladie de Willebrand, le déficit en facteur XI et la Thrombasthénie de Glanzmann. Les déficits en facteurs X, VII ou V sont plus rares[86]
Hémorragie provoquée par une intervention chirurgicaleLes interventions chirurgicales courantes chez les enfants qui posent un problème hémostatique majeur sont : la circoncision, l’amygdalectomie et les extractions dentaires. En plus des saignements incontrôlés, une intervention chirurgicale non planifiée peut entraîner la formation d’un hématome avec une guérison et une infection médiocres. Les lacérations peuvent saigner excessivement et retarder la guérison. Dans le déficit en α2-antiplasmine par exemple les hémorragies surviennent dans les suites d’un traumatisme ou d’une chirurgie,

67
lors de la résorption du caillot, et peuvent persister plusieurs semaines. Cette apparition retardée et post-traumatique des saignements est une caractéristique essentielle, traduisant vraisemblablement le caractère retardé de la fibrinolyse. Les saignements excessifs après des blessures mineures sont une plainte subjective qui différencie de manière significative les femmes atteintes de la maladie de von Willebrand des témoins normaux.
En résumé : Les troubles hémorragiques congénitaux surviennent lorsque les protéines responsables de la coagulation, de la fonction plaquettaire ou de la fibrinolyse sont déficientes. Les ecchymoses, les hématomes superficiels et profonds, les saignements gastro-intestinaux, l’épistaxis, l’hématurie, l’hémarthrose et les hémorragies cérébrales sont les manifestations hémorragiques les plus fréquemment observées chez les patients atteints de troubles hémorragiques congénitaux. Ces manifestations hémorragiques peuvent être observées dans presque tous les défauts de la coagulation. Cependant, il est bien connu que certaines manifestations hémorragiques sont plus fréquentes dans un état donné que dans d’autres. Ainsi, les hémophilies sont généralement associées à une hémarthrose, alors que les patients présentant un trouble de von Willebrand (vW) présentent plus fréquemment des saignements de la muqueuse et de l’appareil gastro-intestinal. Les thrombopénies constitutionnelles ont une symptomatologie hémorragique inconstante, habituellement modérée, surtout provoquée par des traumatismes ou des interventions chirurgicales.
D’autre part, on observe parfois chez certains patients des saignements rares, dont l’importance peut être négligée.
Les manifestations hémorragiques inhabituellesDéficit en facteur II : Une manifestation inhabituelle de saignement a été rapportée par Lee et al. Chez un patient de 14 mois présentant un déficit en prothrombine (niveau de prothrombine correspondant à 6% de la normale) et une gale[87] La coexistence de ces deux affections était responsable d’une forme hémorragique de gale caractérisée par hématomes de la paume gauche avec suintement de sang intermittent.
Déficit en facteur VII: Une manifestation hémorragique rare est représentée par les

68
hématomes de la gaine du muscle droit. Cela peut se produire spontanément ou après un traumatisme, même léger. Si rapidement diagnostiqué et traité, la maladie évolue bien. Au contraire, si le diagnostic est retardé et que l’hématome est devenu important, le risque de syndrome de compression intra-abdominale est à envisager. Des larmes sanglantes ont été rapportées dans au moins un cas[88].Il s’agissait d’un nourrisson de 3 mois qui avait également des saignements intracrâniens et des saignements du conduit auditif externe droit.
L’hémophilie : On a découvert de l’hémophilie B chez une patiente présentant une hémorragie spontanée au niveau de la columelle nasale[89]. Un saignement orbitaire sous-périosté a été signalé comme symptôme initial chez un patient atteint d’hémophilie B. Des hémarthroses des petites mains et des pieds ont été décrites chez des patients hémophiles infectés par le VIH lors d’un traitement par des inhibiteurs de protéase, notamment le Ritanavir [90]. Ceci est particulier car chez les patients hémophiles ce sont les grosses articulations qui sont généralement impliqués. La douleur, le gonflement et les limitations dans le fonctionnement des mains et des pieds sont les signes les plus importants. La maladie est souvent associée à des saignements dans les tissus mous des paumes et des plantes. Il a été démontré que l’hémoptysie était rare dans l’hémophilie et d’autres troubles de la coagulation. Les piqûres de moustiques peuvent parfois provoquer des saignements spontanés dans l’hémophilie ou dans d’autres troubles de la coagulation. La lésion apparaît comme un gonflement sanglant rond localisé.
Le déficit en facteur X : Les saignements inhabituels sont représentés par des saignements dus à la rupture du follicule au moment de l’ovulation[91], [92] De la même manière, pour ce qui a été observé pour la déficience en FVII, au moins un cas d’hématome du muscle droit a été décrit[93].
La maladie de Von Willebrand: Une hémorragie intra myométriale a été décrite dans au moins un cas[94]. Un hémopéritoine par rupture du follicule au moment de l’ovulation et une hémorragie intracholécystique spontanée ont également été décrits[95].

69
1.2. Symptomatologie non spécifique
Les pathologies constitutionnelles de l’hémostase peuvent avoir un mode de révélation atypique non hémorragique mais qui cache un saignement profond inhabituel. Certains cas ont été rapporté dans la littérature.
Déficit en fibrinogène : Apparemment, très peu de saignements particuliers ont été rapportés. L’un d’eux fait référence à une rupture de la rate avec hémopéritoine massif conséquent. Cela a été signalé dans plusieurs cas d’afibrinogénémie et l’on maintient qu’il est assez typique du défaut [96]. Il peut être post-traumatique ou même, apparemment, spontané. La persistance d’un processus de brevet vaginalis peut occasionnellement provoquer chez l’homme une hématocèle, accompagnée d’un gonflement douloureux du testicule gauche.
L’hémophilie : Ces présentations cliniques sont rares, probablement parce que toute l’attention est concentrée sur les épisodes hémorragiques majeurs. Des cas d’hématome intra-surrénalien ont été rapportés chez un nouveau-né atteint d’hémophilie A. Une rupture splénique spontanée ou provoquée a été observée dans l’hémophilie A et B [97]. Les hématomes épiduraux spinaux sont des manifestations hémorragiques extrêmement rares chez les patients hémophiles. Ils nécessitent généralement une intervention chirurgicale rapide pour éviter la compression de la moelle épinière, entraînant de graves séquelles neurologiques[98]. L’hématome intra-mural de la paroi intestinale est un événement hémorragique important. En raison de l’image aiguë de la douleur abdominale, de la masse palpable et même de l’obstruction intestinale, plusieurs problèmes de diagnostic sont soulevés. Des hématomes isolés dans le foie ont été rapportés chez quelques nouveau-nés atteints d’hémophilie A. Les saignements hépatiques peuvent être subdivisés en 3 types, à savoir l’hématome parenchymateux, l’hématome sous-capsulaire et l’hémobilie. Toutes ces formes de saignement ont été décrites chez des patients atteints d’hémophilie A. L’hématome parenchymateux a été décrit principalement chez les nourrissons ou les jeunes garçons. Les hématomes sous-capsulaires, qui peuvent être associés à un hématome parenchymateux, sont généralement secondaires à une biopsie du foie. Les symptômes les plus fréquents sont la douleur, les nausées, les vomissements et la fièvre. L’hémobilie est une manifestation rare qui donne un tableau de douleurs abdominales, jaunisse, nausée et vomissements.

70
Contrairement à ce qui s’est produit pour l’hémophilie A, l’hémorragie hépatique n’a été décrite que rarement chez l’hémophilie B. Que cela indique une préférence pathogénique ou simplement en raison de la prévalence plus élevée de l’hémophilie A par rapport à l’hémophilie B reste à clarifier[99].
Le déficit en facteur XIII : Parmi les manifestations inhabituelles, une rupture splénique a été décrite. Depuis que ce type de manifestations hémorragiques a été décrit chez quelques patients, ce saignement est a été considéré comme typique de la maladie. La distension abdominale, la douleur, la sensibilité au toucher et l’anémie constituent le tableau clinique typique[100].
La maladie de Willebrand : Parmi les présentations inhabituelles, on peut citer les hématomes intra-muraux duodénaux ou de l’intestin grêle. Le tableau se caractérise par une masse abdominale, une douleur et une obstruction ou une sub-occlusion intestinale imitant plusieurs formes d’abdomen aigu (abcès, appendicite aiguë, salpingite, etc) [101].
Les thrombopathies : Il existe peu de cas de saignements particuliers. Le plus intéressant est l’hemobilia qui a été occasionnellement signalés dans le syndrome de Bernard-Soulier [71] Comme nous l’avons vu précédemment, l’hémobilie est plus fréquente chez les patients hémophiliques A que ce soit spontanément ou après une biopsie du foie par accès de la veine jugulaire. On ignore si l’apparition d’une hémobilie dans le syndrome de BS représente une association fortuite ou si elle a une signification pathogénique. Cette manifestation de saignement particulière a n’a jamais été signalé dans une thrombasténie de Glanzman.

71
Fig21 : TDM cérébrale d’un enfant de 5ans qui a présenté des troubles de la conscience. Elle montre des hématomes sous duraux : Frontoparietal droit mesurant 11mm et Basitemporale droit mesurant 4,5mm. Diagnostic retenu:le déficit en facteur VII.
Fig22 : Hématome extradural mesurant 5 mm d’épaisseur associé à une fracture pariétale gauche chez un nourrisson de 7 mois qui a eu un traumatisme crânien. Diagnostic retenu: hémophilie B.

72
2. DÉCOUVERTE FORTUITE
L’incidence des comorbidités étant plus faible chez l’enfant, les indications des bilans préopératoires sont surtout focalisées sur le diagnostic des pathologies congénitales ou acquises de l’hémostase. Dans toutes circonstances, l’examen clinique et l’anamnèse des antécédents personnels et familiaux sont indispensables. Avant l’acquisition de la marche, cette évaluation clinique est néanmoins très limitée par la courte histoire de l’enfant. C’est donc la seule indication retenue par les recommandations formalisées d’expert pour la prescription d’un bilan systématique (TP, TCK, numération plaquettaire) avant un acte invasif. Cependant, la maturation physiologique de l’hémostase en complexifie sérieusement l’interprétation chez le nouveau-né et le très jeune nourrisson. Au-delà de l’acquisition de la marche, quelle que soit la nature de la chirurgie, les indications du bilan biologique sont comme chez l’adulte, exclusivement orientées par l’anamnèse et l’examen clinique. Aucune étude n’a pu en effet mettre en évidence une quelconque contribution d’un bilan biologique d’hémostase chez un enfant asymptomatique sans antécédent personnel ou familial. Il en est de même pour la numération, l’ionogramme, la créatininémie et la glycémie, en l’absence de signe d’appel. En revanche, un TCA normal n’élimine pas toutes les coagulopathies. Une histoire clinique significative nécessite l’exploration de l’hémostase primaire et un conseil spécialisé[102]. Cela dit, il n’est pas rare que dans les formes frustes, la découverte de la pathologie constitutionnelle de l’hémostase se fasse lors d’un bilan préopératoire, ou à l’occasion d’un bilan d’hémostase systématique.

73
V. DÉMARCHE DIAGNOSTIQUE DEVANT UN SYNDROME HÉMORRAGIQUE DE L’ENFANT
Un syndrome hémorragique peut revêtir plusieurs aspects cliniques et être la traduction d’étiologies très variées. Le diagnostic d’un syndrome hémorragique repose sur deux étapes essentielles et étroitement liées : l’examen clinique et l’exploration biologique. Le premier doit toujours précéder le second et l’orienter [103].
1. SYNDROME HÉMORRAGIQUE CLINIQUE
1.1. L’interrogatoire :[104]
L’interrogatoire constitue une étape essentielle au diagnostic. Il convient de préciser plusieurs éléments anamnestiques qui vont contribuer à poser un diagnostic :
- L’âge et le sexe du patient ;
- La localisation et la nature du saignement : épistaxis, gingivorragies, ecchymoses, purpura, hématomes, hémorragies digestives, hémarthroses et /ou ménométrorragies chez la jeune fille. La répétition des saignements dans des territoires différents, ou l’association de plusieurs types de saignements orientent vers une diathèse hémorragique. Les hémorragies cutanéomuqueuses orientent vers une anomalie de l’hémostase primaire tandis que des saignements profonds orientent plutôt vers un trouble de la coagulation ;
- L’âge de début des signes hémorragiques : un âge précoce oriente vers un déficit constitutionnel, et le mode de révélation est d’autant plus précoce que le déficit est sévère : premières manifestations hémorragiques dans les premiers mois de vie (6-12 mois) chez l’hémophile sévère par exemple. Un âge plus tardif oriente vers un trouble modéré de l’hémostase ou une pathologie acquise ;
- L’existence d’antécédents familiaux ou la notion de consanguinité peuvent orienter vers une anomalie constitutionnelle de l’hémostase. La réalisation d’un arbre généalogique

74
trouve ici tout son intérêt dans l’identification d’un mode de transmission autosomique dominant, récessif ou lié à l’X.
- L’évaluation de l’importance des saignements, précisant les antécédents d’anémie et leur tolérance clinique, le recours à un traitement martial (durée du traitement, fréquence de la supplémentation), voire des transfusions érythrocytaires, et la nécessité éventuelle d’un traitement complémentaire (acide tranexamique, desmopressine, transfusion de plasma) ;
- Le mode d’apparition : saignement spontané ou déclenché par un traumatisme ou geste invasif (choc, point de ponction intraveineuse ou intramusculaire, avulsion dentaire, geste chirurgical ou endoscopique, accouchement);
- L’existence d’un contexte thérapeutique particulier (prise d’aspirine ou d’AINS) ;
- L’association éventuelle à une affection organique responsable d’un risque hémorragique accru (hémopathie, insuffisance hépatique, insuffisance rénale) ;
- Le caractère récidivant ;
- La connaissance des résultats d’examens antérieurs (hémogramme, bilan d’hémostase) ;
A l’issue de l’interrogatoire, un score hémorragique peut être établi. Ce score permet de quantifier la symptomatologie hémorragique. Il est établi en cotant chaque item correspondant à un symptôme hémorragique en fonction de son intensité et de la nécessité d’un recours au soin, voire d’un traitement substitutif ou d’une transfusion érythrocytaire. Le score souvent utilisé est le score de Tosetto qui cote 12 items, attribuant des points à chacun de -1 à +4 selon l’importance du syndrome hémorragique. Le score final est obtenu en sommant les scores de tous les items. Plus le score est élevé plus la symptomatologie est marquée. Chez les sujets sains le score est inférieur à 4. Un score similaire a récemment été établi pour l’enfant[105].

75
1.2. L’examen physique :[78]
Il permet dans un premier lieu d’évaluer l’intensité du syndrome hémorragique et puis dans un deuxième temps d’analyser les différents éléments hémorragiques qui peuvent orienter vers une anomalie de l’hémostase primaire, de la coagulation ou des deux. Il doit rechercher la présence d’ecchymoses, de pétéchies ou de purpura et noter l’existence de bulles hémorragiques ou d’hématomes. Il doit aussi comprendre l’examen des conjonctives et des muqueuses, rechercher des déformations articulaires en faveur d’antécédents d’hémarthroses, et dépister une hépatosplénomégalie ou la présence d’adénopathies.
La nature du saignement peut orienter le diagnostic. Des antécédents d’hémarthroses sont très spécifiques d’un trouble sévère de la coagulation. Un syndrome hémorragique cutanéomuqueux avec le plus souvent des épistaxis ou des gingivorragies, et parfois un purpura, est le signe d’une anomalie de l’hémostase primaire. Une hémorragie cérébroméningée peut être également présente dans ce genre de trouble.La présence d’hématomes collectés est le signe d’un trouble de l’hémostase secondaire. Ces hématomes correspondent à un saignement actif qui est de topographie variable : sous cutanée, intramusculaire, intra-articulaire, ou cérébrale.
Dans le cadre d’une éventuelle anomalie plaquettaire constitutionnelle, il convient de rechercher des signes cliniques extra hématologiques : éléments dysmorphiques, aplasie radiale, surdité, atteinte ophtalmologique de type cataracte, atteinte rénale, cardiaque, cutanée de type eczéma, déficits immunitaires et susceptibilité aux infections.
1.3. Les éléments cliniques de gravité :[103]
La sévérité du syndrome hémorragique s’apprécie par l’évaluation :De son abondance et de sa dynamique d’extension : rapidité d’extension d’un purpura, présence de bulles hémorragiques, notamment endo-buccales, d’un saignement digestif extériorisé, d’une atteinte conjonctivale faisant craindre une hémorragie rétinienne et donc éventuellement cérébroméningée associée.De l’état hémodynamique en déterminant : le temps de recoloration cutanée, présence de

76
marbrures, fréquence cardiaque, pression artérielle. Une instabilité hémodynamique doit faire craindre d’une part une anémie sévère mal tolérée et d’autre part un sepsis, d’autant plus grave qu’il est associé à la présence d’un purpura. La température du patient est donc un élément essentiel de l’examen.
Les signes de gravité sont alors un purpura fébrile, des signes de défaillance hémodynamique ou encore des signes neurologiques. En effet, l’examen neurologique, doit être précis, à la recherche d’un syndrome méningé, de signes de localisations sensitivomoteurs, de troubles visuels, d’une altération de la conscience. Un examen neurologique pathologique impose la réalisation d’une imagerie cérébrale en urgence.
2. PARTICULARITÉS DES PHASES DE TRAITEMENT BIOLOGIQUE
L’évaluation de l’hémostase est, en pédiatrie, une phase critique de la prise en charge des patients. La problématique des valeurs de référence et les particularités physiologiques sont des éléments clefs. Le rôle du laboratoire dans cette démarche inclut la gestion des étapes pré per et post analytique. Nous allons donc détailler ces étapes afin de démontrer le rôle primordial du laboratoire et du biologiste dans l’optimisation spécifique des procédures diagnostiques sur des prélèvements précieux.
2.1 . L’étape pré-analytique
Cette phase est délicate chez l’enfant est surtout le nouveau-né, et conditionne la qualité des résultats. En pédiatrie, le clinicien souhaite limiter le volume sanguin prélevé et éviter tout geste invasif douloureux en particulier les ponctions veineuses répétées. Le débit sanguin est souvent lent et le risque de la coagulation du prélèvement in vitro est élevé. Aussi le prélèvement doit être traité le plus rapidement possible. Le volume est souvent faible limitant la liste Fig23 : Tube miniCollect®, de chez Greiner

77
d’examens réalisable. Ceci implique que ces prélèvements difficiles et soustrayant un volume de sang non négligeable relativement à l’enfant doivent être considérés comme précieux. En période néonatale, les non-conformités pré-analytiques les plus fréquentes sont le remplissage insuffisant du tube et le prélèvement coagulé. Elles s’expliquent par les difficultés techniques de prélèvement et d’abord veineux, aggravés par l’hypercoagulabilité physiologique du nouveau-né et son hématocrite élevé, souvent supérieur à 55% [14]. En accord avec les recommandations pré-analytiques émises par le GFHT (Groupe Français d’études sur l’Hémostase et la Thrombose)[106], les principales règles à respecter pour un prélèvement d’hémostase de qualité en pédiatrie sont les suivantes : privilégier le recueil du sang à partir d’un prélèvement veineux, parfois artériel, jamais capillaire, utiliser des aiguilles de prélèvement dont le diamètre ne doit pas excéder 25 gauge et des microtubes de 1 ml minimum, éviter l’utilisation de tubes « sous vide » chez le nouveau-né (risque de remplissage insuffisant), acheminer rapidement le prélèvement au laboratoire. La quantité de sang minimum requise est de 0,9 ml, soit en capillaire soit en veineux (tube MiniCollect®, de chez Greiner). Le ratio citrate/sang doit être respecté. Les tubes doivent être rejetés si le taux de remplissage est inférieur à 80 % du volume total. Ils sont dits «acceptables» entre 80 et 90 %. La concentration de l’anticoagulant recommandée est celle de l’adulte 0,109 M ou 3,2 %[107]. Les micro-prélèvements capillaires sont réservés aux cas très particuliers des enfants prématurés de très petit poids et sont de moins en moins pratiqués.
2.2. L’étape analytique
Les analyseurs d’hémostase permettent désormais la prise en charge des prélèvements pédiatriques par une miniaturisation des techniques. Le biologiste doit adapter son automate à une activité pédiatrique et privilégier les tests ayant une prise d’essai faible (tests dilués, dosages des facteurs), par rapport aux tests nécessitant du plasma pur. En raison du faible volume de plasma recueilli, il réalise les examens ayant le plus de pertinence clinique à cet âge de la vie.[14]
La phase analytique s’est grandement améliorée avec les automates actuels. Les volumes morts de plasma sont restreints et les tests semi-globaux nécessitent une prise d’essai moindre, en règle générale 50 μl au lieu de 100 μl autrefois. Dans ce contexte parcimonieux,

78
on est amené à sélectionner les tests les plus utiles. Le TP, souvent plus ou moins activé, peut être remplacé, dans le cadre d’un bilan de dépistage, par la mesure des facteurs (II, V, VII et X ou VII+X) qui sont plus informatifs sauf en cas de traitement antivitamine K où l’on a besoin de l’INR [108].Pour le TCA, la sélection d’un réactif sensible aux déficits, surtout des facteurs VIII et IX, est préférable à des céphalines sensibles surtout aux antiprothrombinases et à l’héparine. Pour la mesure de l’héparinémie (activité anti-Xa), il est recommandé de ne pas utiliser un système réactionnel utilisant un apport d’antithrombine in vitro chez le très jeune enfant; il existe en effet un déficit de cette protéine qui se normalise vers l’âge de 3 mois[10].
2.3. L’étape post analytique
L’étape post-analytique impose en premier lieu de confronter les résultats aux valeurs de référence qui varient avec l’âge. En effet la maturation de l’enfant vers l’âge adulte est associée à une évolution du système de l’hémostase, affectant notamment les concentrations protéiques et leur structure qualitative. L’établissement de ces valeurs de référence constitue une difficulté spécifique, car les prélèvements provenant de sujets non pathologiques sont, pour des raisons éthiques, exceptionnels. La phase post-analytique devra être réalisée avec les valeurs de référence les plus pertinentes possibles. Pour l’interprétation, la prudence afférente aux spécificités que nous avons évoquées est à prendre en compte. L’implémentation des références en fonction de l’état civil peut être réalisée dans les systèmes informatiques habituels, mais l’âge réel n’est pas pris en compte en cas de prématurité. Entre 1 et 5 ans, l’exemple du TCA est caractéristique des difficultés rencontrées ; si l’on utilise des valeurs de l’adulte, jusqu’à 30 % des enfants sont retenus comme ayant un TCA anormal [109]. Chez l’enfant, le TCA est souvent allongé du fait de la présence d’un anticoagulant circulant de type lupique transitoire. Enfin, il n’est pas rare d’avoir un hématocrite supérieur à 55 % et d’être obligé alors de rendre les résultats sous réserve et/ou de pratiquer des tests complémentaires, notamment pour le TCA qui a une forte probabilité d’être allongé dans ces cas[110]. 3. EXPLORATION D’UN SYNDROME HÉMORRAGIQUE
En pédiatrie comme chez l’adulte les bilans de la coagulation sont indiqués dans quatre

79
situations : exploration d’un syndrome hémorragique, détection d’un risque thrombotique, dépistage préopératoire et surveillance d’un traitement anticoagulant[111].
3.1. L’exploration de l’hémostase primaire
Physiopathologie : L’hémostase primaire correspond à l’ensemble des interactions entre la paroi vasculaire, les plaquettes et les protéines d’adhésion, qui aboutissent à la formation du thrombus plaquettaire. En cas de lésion endothéliale vasculaire, les plaquettes adhèrent aux molécules de la membrane basale sous-endothéliale et activent une cascade de réactions qui aboutissent à la formation du thrombus plaquettaire.
Ce phénomène fait notamment intervenir une protéine d’adhésion essentielle : le facteur de Von Willebrand, synthétisé par les cellules endothéliales mais aussi par les plaquettes elles-mêmes. Une anomalie de l’hémostase primaire peut donc correspondre à une anomalie des plaquettes, à un déficit en facteur de Von Willebrand ou à une pathologie vasculaire.
Examens complémentaires :La numération plaquettaire est un examen essentiel et de première intention la thrombopénie étant une des étiologies les plus fréquentes des troubles de l’hémostase primaire. L’étude du frottis sanguin est également importante ; Cet examen permet de rechercher des éléments d’orientation comme la taille et l’aspect des plaquettes, l’aspect des autres cellules sanguines, la présence d’inclusions intracytoplasmiques, la présence de cellules pathologiques (blastes par exemple). L’hémostase primaire peut être explorée par la réalisation d’un temps de saignement, qui est un test global de mesure in vivo de temps nécessaire à l’arrêt du saignement induit après incision superficielle de la peau du patient. Cependant, ce test est actuellement considéré comme obsolète car invasif, peu reproductible, ayant une mauvaise sensibilité et spécificité, et un manque de prédictibilité. Il n’est plus recommandé[103]. Il a été proposé de le remplacer par la mesure d’un temps d’occlusion plaquettaire in vitro, réalisé à l’aide d’un automate (PFA-100)[31].
Le PFA100 TM (Dade Behring) est un nouvel automate qui permet d’évaluer la capacité fonctionnelle des plaquettes, en sang citraté, sans aucune préparation préalable de l’échantillon.

80
D’utilisation simple, le PFA100TM simule in vitro les conditions rencontrées lors d’une brèche de la paroi artériolaire. Dans le cadre de l’exploration de troubles hémorragiques évoquant un trouble de l’hémostase primaire au cours d’un interrogatoire systématique, il est capable de détecter de manière sensible les anomalies les plus fréquentes, la prise d’aspirine et un déficit en facteur Willebrand. Le système utilise une membrane de nitrocellulose biologiquement active recouverte de collagène de type 1 et un autre agoniste plaquettaire, soit l’adénosine diphosphate (ADP), soit l’épinéphrine. Cette membrane possède un Orifice central au travers duquel le sang s’écoule dans des conditions de flux contrôlé. Le système mesure le TO, temps nécessaire à l’écoulement du sang correspondant à la formation du clou plaquettaire au niveau de l’orifice de la membrane. Les études ont montré une bonne sensibilité du PFA100TM pour le dépistage de la maladie de Willebrand (allongement du TO avec les cartouches ADP et épinéphrine) et le dépistage des patients ayant ingéré de l’aspirine (allongement du TO avec la cartouche épinéphrine). En revanche, une mauvaise sensibilité du PFA100TM à la prise de ticlopidine et de clopidogrel a été rapportée. Sa sensibilité aux autres AINS que l’aspirine et aux anti-IIb/IIIa n’a pas encore été étudiée.[112]
Dans un deuxième temps, des examens plus spécialisés peuvent être nécessaires : dosage du facteur Willebrand (vWF), du fibrinogène, étude des fonctions plaquettaires. Le dosage du facteur de von Willebrand doit comporter un dosage antigénique (évaluation quantitative) et un dosage de l’activité du facteur (évaluation qualitative), associés à un dosage du facteur VIII. Les taux sont à interpréter en fonction du groupe sanguin, de l’âge du patient, de la présence d’un éventuel syndrome inflammatoire biologique, de la prise d’un traitement hormonal. L’étude des fonctions plaquettaires est faite grâce à l’étude de l’agrégation plaquettaire à différents agonistes et l’étude des glycoprotéines plaquettaires en cytométrie de flux. Les principales étapes du diagnostic étiologique devant un allongement du temps de saignement sont résumées dans le schéma ci-dessous :
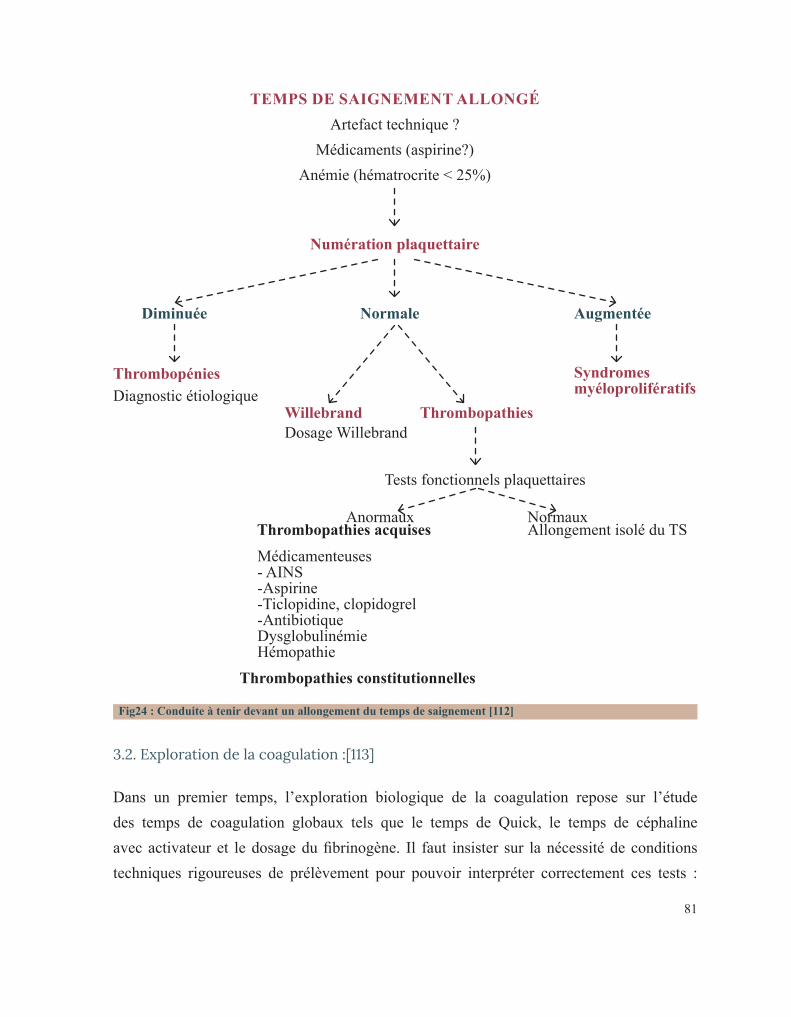
81
TEMPS DE SAIGNEMENT ALLONGÉArtefact technique ?
Médicaments (aspirine?)Anémie (hématrocrite < 25%)
Numération plaquettaire
Diminuée Normale
Willebrand
Augmentée
Thrombopénies
Thrombopathies
Thrombopathies acquises
Thrombopathies constitutionnelles
Médicamenteuses- AINS-Aspirine-Ticlopidine, clopidogrel-AntibiotiqueDysglobulinémieHémopathie
Allongement isolé du TS
Tests fonctionnels plaquettaires
Dosage Willebrand
Anormaux Normaux
Syndromes myéloprolifératifsDiagnostic étiologique
Fig24 : Conduite à tenir devant un allongement du temps de saignement [112]
3.2. Exploration de la coagulation :[113]
Dans un premier temps, l’exploration biologique de la coagulation repose sur l’étude des temps de coagulation globaux tels que le temps de Quick, le temps de céphaline avec activateur et le dosage du fibrinogène. Il faut insister sur la nécessité de conditions techniques rigoureuses de prélèvement pour pouvoir interpréter correctement ces tests :

82
ponction veineuse franche afin d’éviter toute activation de la coagulation in vitro, absence de contamination par l’héparine, respect strict de la proportion entre le sang et l’anticoagulant, rapidité d’exécution des tests à partir du moment où le prélèvement est effectué. Le temps de Quick (TQ) explore les facteurs de la voie extrinsèque et du tronc commun de la coagulation (II, V, VII et X) et est également perturbé en cas d’anomalies quantitatives et qualitatives sévères du fibrinogène (concentration inferieure a 0,5 g/L). Le temps de céphaline avec activateur explore la voie intrinsèque et le tronc commun (tous les facteurs plasmatiques de la coagulation a l’exclusion du VII) ; il est influence uniquement par une anomalie sévère du fibrinogène (taux inférieur à 0,5 g/L). Le temps de thrombine explore la fibrinoformation. En dehors de la période néonatale, le temps de Quick est compris entre 70 et 120 %, et le temps de céphaline avec activateur ne doit pas être supérieur à 1,2 fois le temps du témoin.
Le dosage de chacun des facteurs est un examen de seconde intention. En cas d’allongement du TQ, il convient de doser les facteurs II, V, VII et X. En cas d’allongement du TCA, il faut doser les facteurs VIII, IX, XI et XII (le déficit en facteur XII n’expose pas à un risque hémorragique).
Fig25 : Déficits en facteurs selon les résultats de la TCA et le temps de Quick
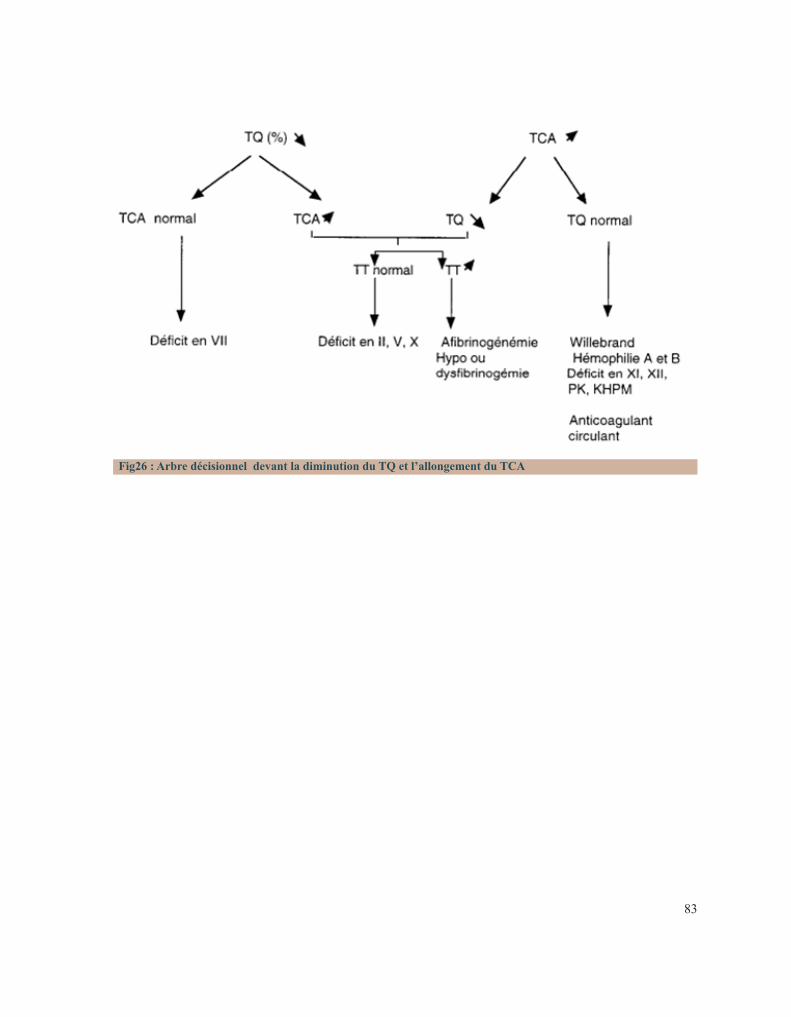
83
Fig26 : Arbre décisionnel devant la diminution du TQ et l’allongement du TCA

84
ETUDE RÉTROSPECTIVE DESCRIPTIVE ET
ANALYTIQUE : À PROPOS DE 63CAS
DEUXIÈME PARTIE

85
OBJECTIFS, MATÉRIELS ET
MÉTHODES

86
1. OBJECTIFS DE L’ÉTUDE
• Étudier les aspects épidémiologiques, socio-économiques, et cliniques des pathologies constitutionnelles de l’hémostase en pédiatrie au CHOP ;
• Rechercher la présence d’événements hémorragiques à l’enfance et d’antécédents hémorragiques familiaux.
• Définir et décrire la symptomatologie et les circonstances révélatrices des pathologies hémorragiques constitutionnelles de l’hémostase chez l’enfant.
• Evaluer la gravité de la symptomatologie hémorragique lors de la première consultation.• Suivre le circuit par lequel passe le patient avant d’être pris en charge au CHOP et
déterminer le délai entre le premier événement hémorragique et le diagnostic.• Comparer les résultats obtenus avec les données de la littérature sur trois volets
essentiels : épidémiologie, histoire hémorragique familiale et personnelle et motif de consultation (syndrome hémorragique, bilan préopératoire, symptomatologie non spécifique….).
• Discuter de l’intérêt d’une prise en charge précoce et d’une démarche diagnostique bien conduite pour une meilleure approche et gestion des pathologies constitutionnelles de l’hémostase.
2. MATÉRIELS ET MÉTHODES DE L’ÉTUDE
Afin de répondre à ces objectifs une étude rétrospective descriptive et analytique a été menée au centre de traitement de l’hémophilie et des maladies hémorragiques constitutionnelles à l’hôpital d’enfants de Rabat, et qui a permis de collecter les données de 63 cas, pendant la période s’étendant du mois de janvier de l’année 2016 au mois de décembre de l’année 2018.Une fiche d’exploitation a été réalisée dans le but de recueillir l’ensemble des informations nécessaires à cette étude. Le choix des dossiers s’est fait sur une base de critères d’inclusion et d’exclusions précis.
2.1. Critères d’inclusion
Ont été inclus dans notre étude tous les dossiers regroupant tous les critères suivants : - Les enfants âgés de 15 ans ou moins,

87
- Les enfants diagnostiqués porteurs d’une maladie hémorragique constitutionnelle de l’hémostase ; - Dossiers contenant suffisamment d’informations pour être exploité ;Les dossiers ayant répondu à cette condition sont les patients hospitalisés entre la période s’étendant du mois de janvier 2016 au mois de décembre 2018, ce qui correspond à 3 années.
2.2. Critères d’exclusion
Ont été exclus de notre étude tous les dossiers répondant à un ou plusieurs des critères suivants : - Dossiers ne contenant pas assez d’informations et donc inexploitables. - Les pathologies constitutionnelles de l’hémostase thrombotiques et les pathologies hémorragiques acquises. - Les thrombopénies d’origine auto-immune.
En respectant donc ces critères, et avec une base de dossiers initiale concernant 73 cas, nous avons retiré 10 dossiers inexploitables (manque d’informations ou dossier vide).Nous avons donc finalement effectué notre étude rétrospective sur 63 dossiers de patients, tous issus des archives du CHOP.
2.3. Matériel
Nous avons recueilli les informations selon une fiche d’exploitation précise (voir page suivante) et nous avons traité et analysé les données grâce aux logiciels Microsoft Excel 2010 et IBM SPSS Statistics 23.
2.4. Le circuit des patients
Les cas reçus au service de CHOP au cours des 3 dernières années (de 2016 à 2018) proviennent soit :
• Des urgences de l’HER et sont adressés après avoir séjourné dans un service de pédiatrie (25.4%).
• Des urgences ou service de pédiatrie d’un hôpital périphérique ou régional 25.4%
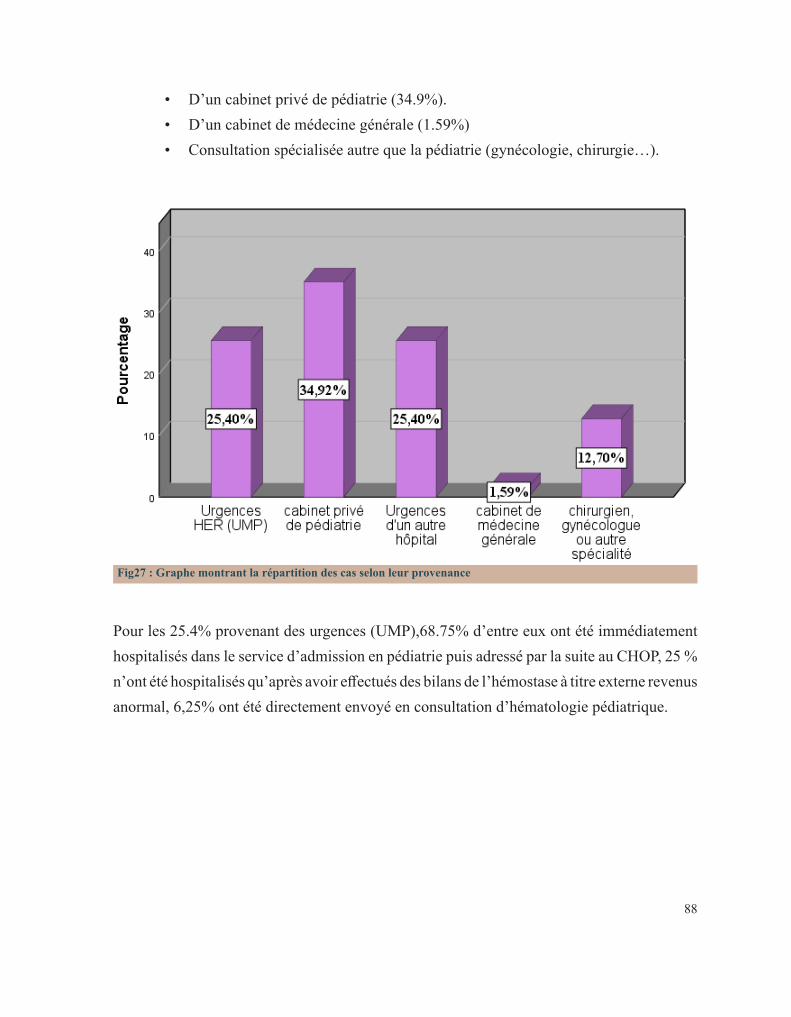
88
• D’un cabinet privé de pédiatrie (34.9%).• D’un cabinet de médecine générale (1.59%)• Consultation spécialisée autre que la pédiatrie (gynécologie, chirurgie…).
Fig27 : Graphe montrant la répartition des cas selon leur provenance
Pour les 25.4% provenant des urgences (UMP),68.75% d’entre eux ont été immédiatement hospitalisés dans le service d’admission en pédiatrie puis adressé par la suite au CHOP, 25 % n’ont été hospitalisés qu’après avoir effectués des bilans de l’hémostase à titre externe revenus anormal, 6,25% ont été directement envoyé en consultation d’hématologie pédiatrique.

89
FICHE D’EXPLOITATION
Données du patient : Numéro d’hospitalisation : Date de prise en charge :Nom et prénom: Date de naissance: / / Sexe: masculin féminin Provenance: Urgences (UMP) Autre service Autre hôpital Cabinet privé Fratrie : Adresse: Téléphone :Couverture sociale: RAMED CNOPS CNSS MAFAR
Antécédents : Notion de consanguinité: Oui NonSi consanguinité : 1er degré 2ème degré Hémorragie à : la chute du cordon : Oui Non la vaccination : Oui Non la circoncision : Oui Non
ATCD personnels hémorragiques: Date de début : Type de saignement: Elément déclenchant : Nombre d’épisodes :
ATCD hémorragiques familiaux:Autres:

90
Motif de consultation : 1 . Syndrome hémorragique:Date de début :Le mode d’apparition: Spontané Provoqué Traumatisme ou chute prélèvement ou injection Circoncision extraction dentaire vaccination Plaie ou blessure chute du cordon Autres: Localisation: généralisée localisée Aspect clinique: syndrome hémorragique cutané: purpura ecchymoses
Hématome pétéchies Muqueux: Epistaxis gingivorragies MénométrorragiesViscéral: hématémèse hémoptysie Rectorragie hématurie méléna Articulaire : hémarthrose Hémorragie active au site de plaie ou blessure Gravité: Abondance : petite moyenne grande Choc hémorragique Etat hémodynamique : TA= Pouls= FR= Présence de bulles hémorragiques buccales : oui non Etat neurologique : Nécessité d’une transfusion :Oui Non
2- Autre motif de consultation ( à détailler ) [ Non hémorragique ]

91
3- Bilan biologique de l’hémostase perturbé:A quelle occasion ce bilan a été fait ? Bilan préopératoire Lors de l’exploration d’une autre pathologie Lors d’une enquête familiale Autres: Numération formule sanguine : Taux de plaquettes : Taux d’hémoglobine : VGM : CCMH : Taux de Globules blancs : TP : TCA : FIBRINOGENE : Temps de saignement :

92
RÉSULTATS
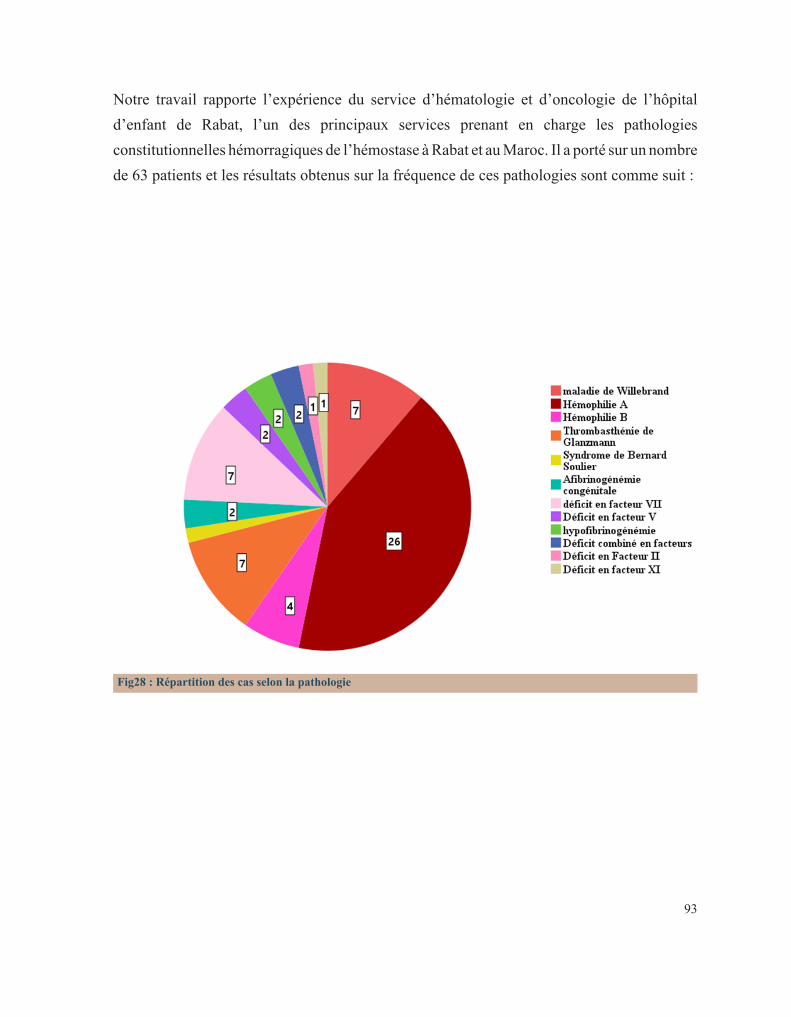
93
Notre travail rapporte l’expérience du service d’hématologie et d’oncologie de l’hôpital d’enfant de Rabat, l’un des principaux services prenant en charge les pathologies constitutionnelles hémorragiques de l’hémostase à Rabat et au Maroc. Il a porté sur un nombre de 63 patients et les résultats obtenus sur la fréquence de ces pathologies sont comme suit :
Fig28 : Répartition des cas selon la pathologie

94
L’anomalie congénitale dominante est l’hémophilie A avec 26 cas, vient ensuite la maladie de Willebrand, le déficit en facteur VII et la Thrombasthénie de Glanzmann avec 7 cas chacun. Les autres déficits en facteurs de la coagulation, les anomalies du fibrinogène ainsi que la thrombopathie de Bernard soulier ont aussi été diagnostiqués.
Fréquence Pourcentage Hémophilie A 26 41,90%Maladie de Willebrand 7 11,30%Thrombasthénie de Glanzmann 7 11,30%Déficit en facteur VII 7 11,30%Hémophilie B 4 6,50%Hypofibrinogénémie 2 3,20%Afibrinogénémie congénitale 2 3,20%
Déficit en facteur V 2 3,20%Déficit combiné en facteurs 2 3,20%Syndrome de Bernard soulier 1 1,60%
Déficit en facteur II 1 1,60%
Déficit en facteur XI 1 1,60%
Les deux cas du déficit combiné en facteurs sont un cas associant un déficit en facteurs VII+XII et un autre en facteurs V+VIII.
Tableau 10 : Nombre et Pourcentage de chaque pathologie
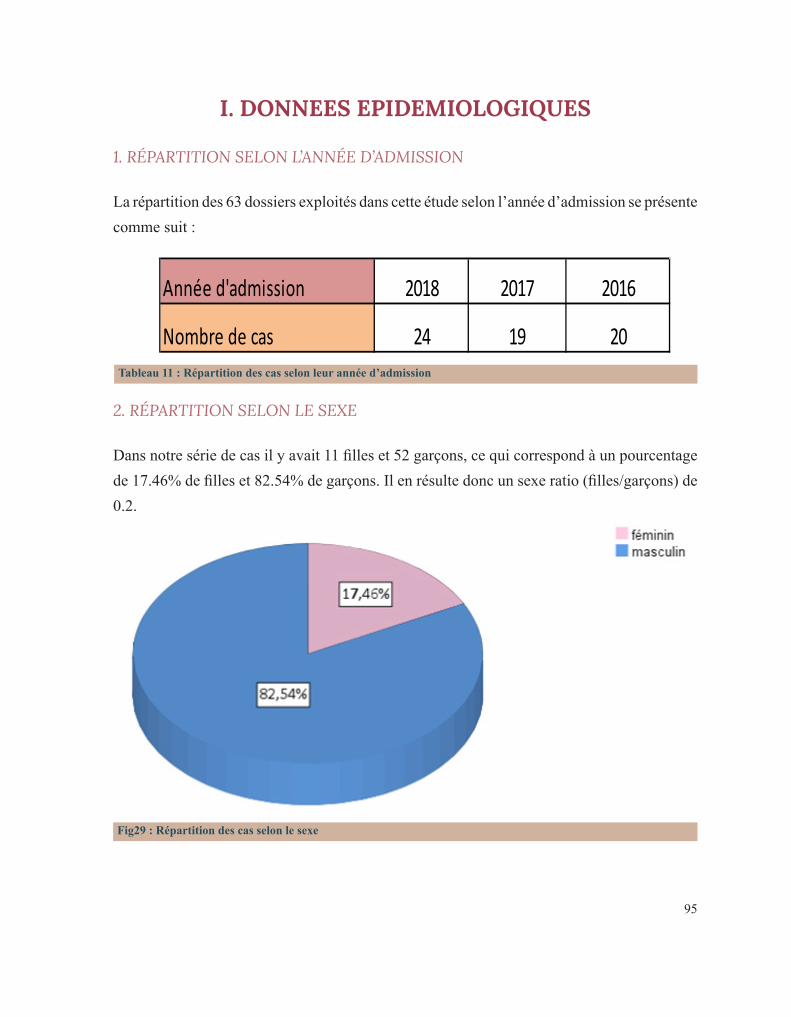
95
I. DONNEES EPIDEMIOLOGIQUES
1. RÉPARTITION SELON L’ANNÉE D’ADMISSION
La répartition des 63 dossiers exploités dans cette étude selon l’année d’admission se présente comme suit :
Année d'admission 2018 2017 2016
Nombre de cas 24 19 20Tableau 11 : Répartition des cas selon leur année d’admission
2. RÉPARTITION SELON LE SEXE
Dans notre série de cas il y avait 11 filles et 52 garçons, ce qui correspond à un pourcentage de 17.46% de filles et 82.54% de garçons. Il en résulte donc un sexe ratio (filles/garçons) de 0.2.
Fig29 : Répartition des cas selon le sexe

96
3. RÉPARTITION SELON L’ÂGE
Dans notre étude nous avons distingué l’âge d’apparition des premiers symptômes de l’âge des patients à leur admission au CHOP.
3.1. Age à l’admission
L’âge dont il est question dans ce chapitre est l’âge des patients au moment de la prise en charge au CHOP pour la première fois. Il a varié d’un minimum d’un mois de vie à un maximum de 14 ans avec un âge médian de 7 ans et 4 mois.
Age Fréquence Age Fréquence 1 mois 1 5 ans 43 mois 3 5 ans et 3 mois 14 mois 1 5 ans et 6 mois 16 mois 2 5ans et 10 mois 27 mois 2 6 ans 18 mois 2 6 ans et 3 mois 11 an 1 6 ans et 4 mois 113 mois 2 6 ans et 7 mois 114 mois 1 7 ans 115 mois 1 7 ans et 6 mois 118 mois 2 8 ans 121 mois 1 8 ans et 5 mois 123 mois 1 8 ans et 6 mois 12 ans 5 8 ans et 9 mois 12 ans et 2 mois 1 9 ans 22 ans et 4 mois 1 9 ans et 5 mois 12 ans et 5 mois 1 11 ans 22 ans et 9 mois 1 11ans et 6 mois 13 ans et 6 mois 2 11 ans et 9 mois 14 ans 3 12 ans 14 ans et 3 mois 1 14 ans 2
Tableau 12 : Répartition selon l’âge à l’admission au CHOP

97
3.2. Age au diagnostic
Age au diagnostic Fréquence Age au diagnostic Fréquence10 jours 1 4 ans 53 mois 4 4 ans et 3 mois 14 mois 1 5 ans 45 mois 1 5ans et 3 mois 16 mois 1 5 ans et 6 mois 17 mois 2 5ans et 10 mois 18 mois 2 6 ans 29 mois 1 6ans et 2 mois 112 mois 2 6ans et 4 mois 113 mois 2 7ans 114 mois 1 8ans 115 mois 1 8ans et 5 mois 118 mois 3 8ans et 6 mois 121 mois 1 9ans 223 mois 1 9 ans et 5 mois 12 ans 5 11 ans 12 ans et 4 mois 1 11 ans et 6 mois 12ans et 9 mois 1 11 ans et 9 mois 13 ans et 6 mois 2 14 ans 2
L’âge au moment du diagnostic est à différencier de l’âge des patients à leur admission, puisque certains patients ont été diagnostiqués avant leur prise en charge au service. Ainsi, notre série nous a permis de trouver des valeurs extrêmes concernant cet âge allant d’un minimum de 10 jours à un maximum de 14ans, avec un âge médian de diagnostic d’approximativement 7ans et 1 mois.
Tableau 13 : Répartition selon l’âge des patients au moment du diagnostic
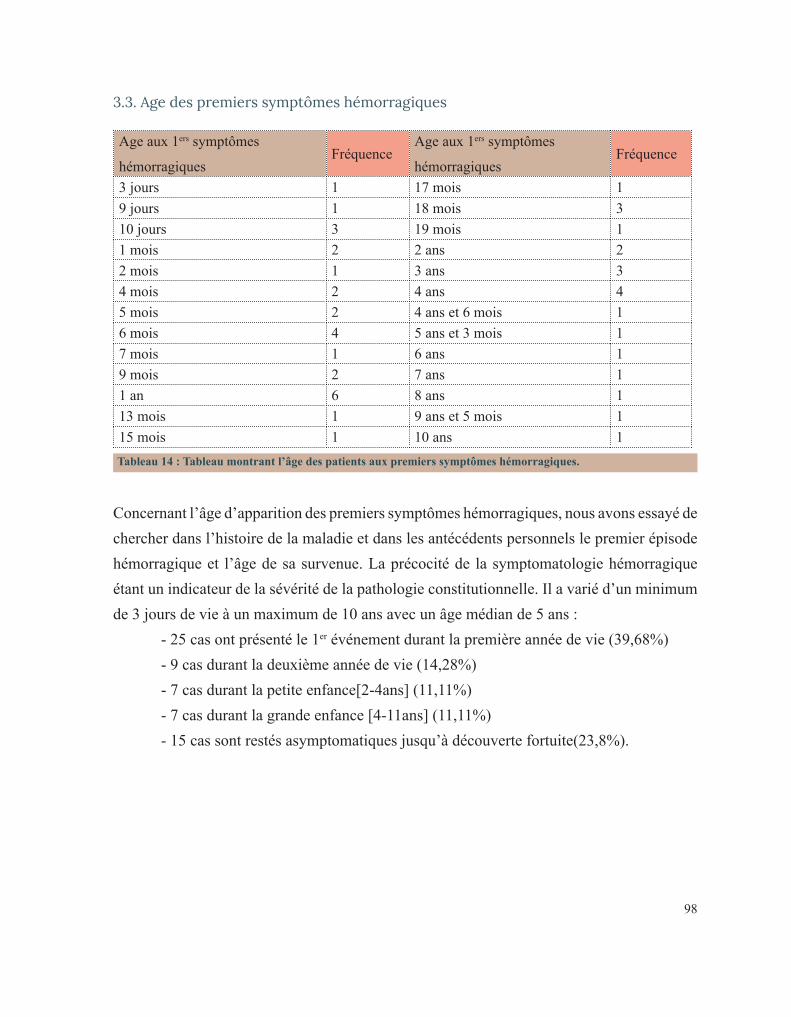
98
3.3. Age des premiers symptômes hémorragiques
Age aux 1ers symptômes
hémorragiquesFréquence
Age aux 1ers symptômes
hémorragiquesFréquence
3 jours 1 17 mois 19 jours 1 18 mois 310 jours 3 19 mois 11 mois 2 2 ans 22 mois 1 3 ans 34 mois 2 4 ans 45 mois 2 4 ans et 6 mois 16 mois 4 5 ans et 3 mois 17 mois 1 6 ans 19 mois 2 7 ans 11 an 6 8 ans 113 mois 1 9 ans et 5 mois 115 mois 1 10 ans 1
Concernant l’âge d’apparition des premiers symptômes hémorragiques, nous avons essayé de chercher dans l’histoire de la maladie et dans les antécédents personnels le premier épisode hémorragique et l’âge de sa survenue. La précocité de la symptomatologie hémorragique étant un indicateur de la sévérité de la pathologie constitutionnelle. Il a varié d’un minimum de 3 jours de vie à un maximum de 10 ans avec un âge médian de 5 ans : - 25 cas ont présenté le 1er événement durant la première année de vie (39,68%) - 9 cas durant la deuxième année de vie (14,28%) - 7 cas durant la petite enfance[2-4ans] (11,11%) - 7 cas durant la grande enfance [4-11ans] (11,11%) - 15 cas sont restés asymptomatiques jusqu’à découverte fortuite(23,8%).
Tableau 14 : Tableau montrant l’âge des patients aux premiers symptômes hémorragiques.

99
4. RÉPARTITION SELON L’ORIGINE GÉOGRAPHIQUE
Fig30 : Carte du Maroc montrant la répartition des cas étudiés sur les 12 régions
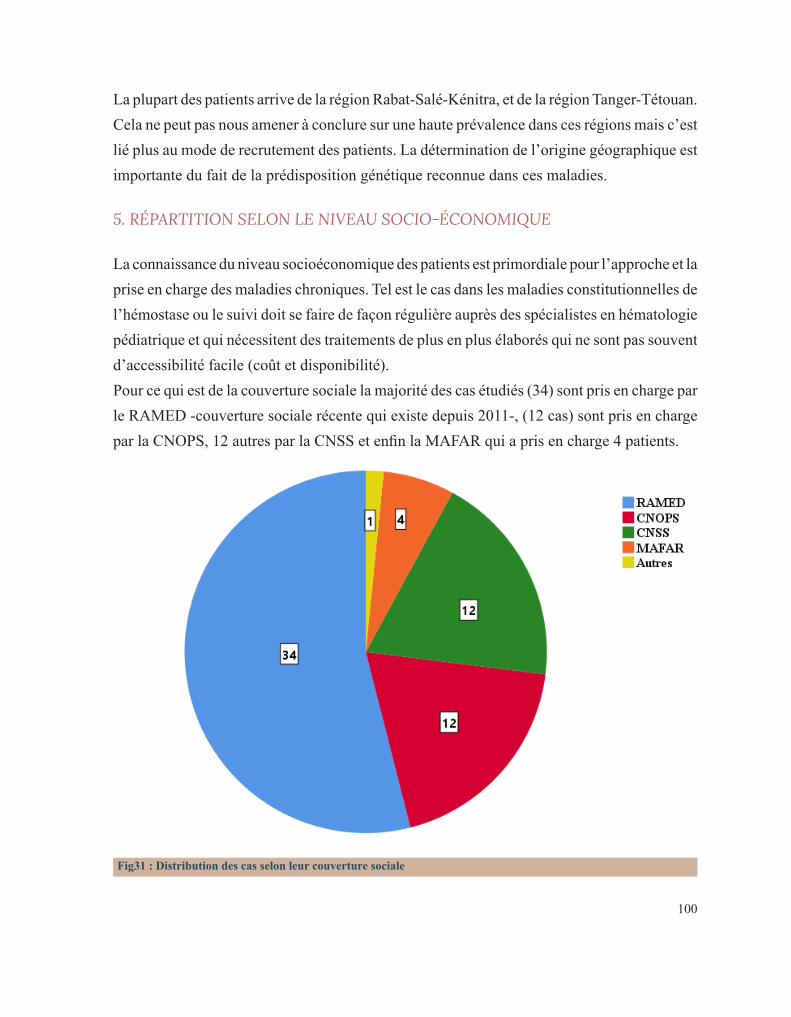
100
La plupart des patients arrive de la région Rabat-Salé-Kénitra, et de la région Tanger-Tétouan. Cela ne peut pas nous amener à conclure sur une haute prévalence dans ces régions mais c’est lié plus au mode de recrutement des patients. La détermination de l’origine géographique est importante du fait de la prédisposition génétique reconnue dans ces maladies.
5. RÉPARTITION SELON LE NIVEAU SOCIO-ÉCONOMIQUE
La connaissance du niveau socioéconomique des patients est primordiale pour l’approche et la prise en charge des maladies chroniques. Tel est le cas dans les maladies constitutionnelles de l’hémostase ou le suivi doit se faire de façon régulière auprès des spécialistes en hématologie pédiatrique et qui nécessitent des traitements de plus en plus élaborés qui ne sont pas souvent d’accessibilité facile (coût et disponibilité). Pour ce qui est de la couverture sociale la majorité des cas étudiés (34) sont pris en charge par le RAMED -couverture sociale récente qui existe depuis 2011-, (12 cas) sont pris en charge par la CNOPS, 12 autres par la CNSS et enfin la MAFAR qui a pris en charge 4 patients.
Fig31 : Distribution des cas selon leur couverture sociale
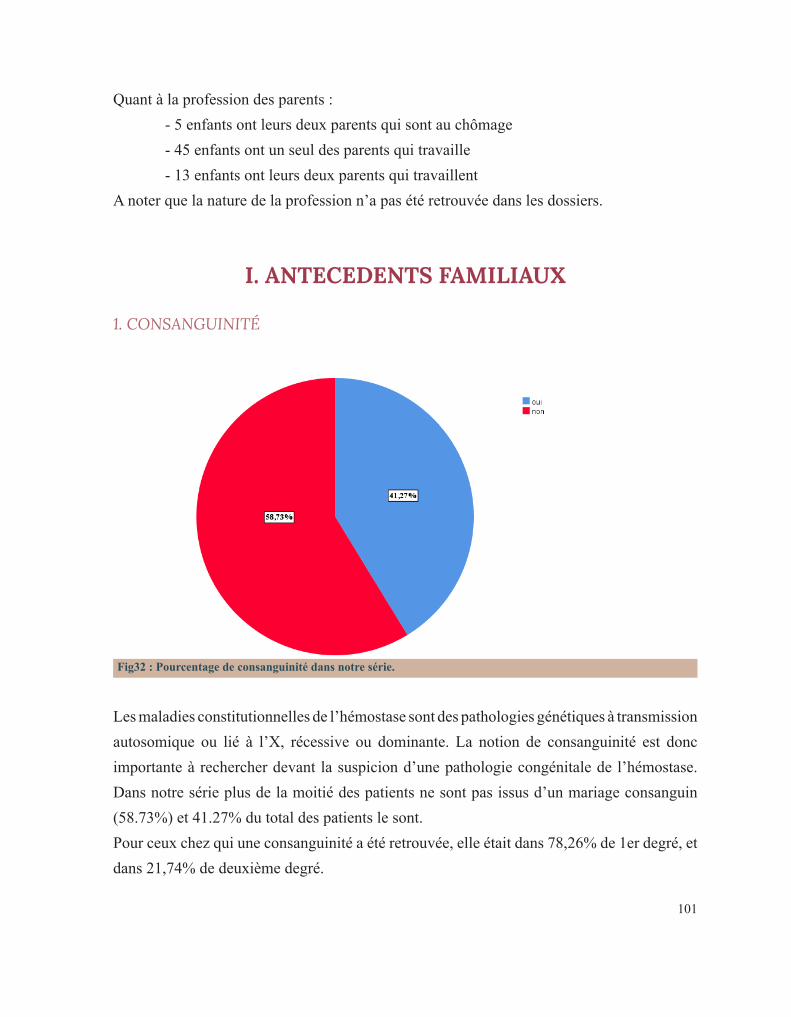
101
Quant à la profession des parents : - 5 enfants ont leurs deux parents qui sont au chômage - 45 enfants ont un seul des parents qui travaille - 13 enfants ont leurs deux parents qui travaillent A noter que la nature de la profession n’a pas été retrouvée dans les dossiers.
I. ANTECEDENTS FAMILIAUX
1. CONSANGUINITÉ
Fig32 : Pourcentage de consanguinité dans notre série.
Les maladies constitutionnelles de l’hémostase sont des pathologies génétiques à transmission autosomique ou lié à l’X, récessive ou dominante. La notion de consanguinité est donc importante à rechercher devant la suspicion d’une pathologie congénitale de l’hémostase. Dans notre série plus de la moitié des patients ne sont pas issus d’un mariage consanguin (58.73%) et 41.27% du total des patients le sont.Pour ceux chez qui une consanguinité a été retrouvée, elle était dans 78,26% de 1er degré, et dans 21,74% de deuxième degré.
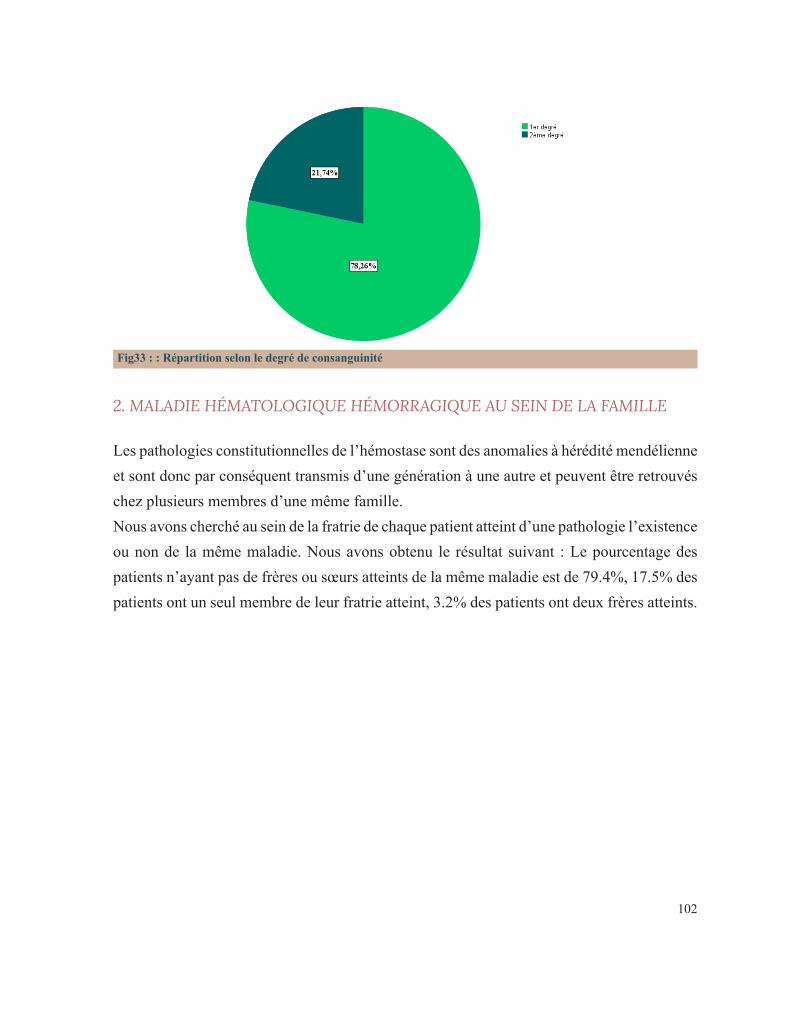
102
2. MALADIE HÉMATOLOGIQUE HÉMORRAGIQUE AU SEIN DE LA FAMILLE
Les pathologies constitutionnelles de l’hémostase sont des anomalies à hérédité mendélienne et sont donc par conséquent transmis d’une génération à une autre et peuvent être retrouvés chez plusieurs membres d’une même famille.Nous avons cherché au sein de la fratrie de chaque patient atteint d’une pathologie l’existence ou non de la même maladie. Nous avons obtenu le résultat suivant : Le pourcentage des patients n’ayant pas de frères ou sœurs atteints de la même maladie est de 79.4%, 17.5% des patients ont un seul membre de leur fratrie atteint, 3.2% des patients ont deux frères atteints.
Fig33 : : Répartition selon le degré de consanguinité
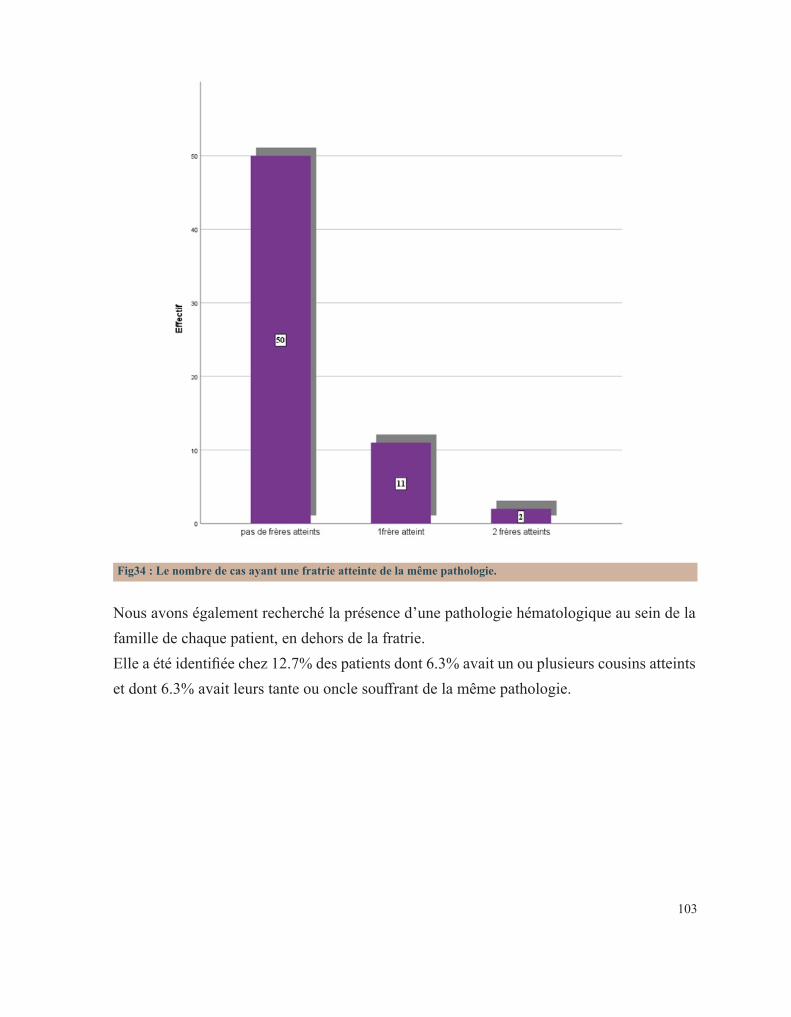
103
Nous avons également recherché la présence d’une pathologie hématologique au sein de la famille de chaque patient, en dehors de la fratrie.Elle a été identifiée chez 12.7% des patients dont 6.3% avait un ou plusieurs cousins atteints et dont 6.3% avait leurs tante ou oncle souffrant de la même pathologie.
Fig34 : Le nombre de cas ayant une fratrie atteinte de la même pathologie.

104
3. DÉCÈS DANS LA FAMILLE LIÉ À UNE PATHOLOGIE DE L’HÉMOSTASE
La notion de décès à la suite d›un syndrome hémorragique ou dans des circonstances indéterminées au sein de la fratrie ont également été recherchées dans les informations recueillies lors de l’interrogatoire des patients. Cette situation a été identifiée chez 10 cas ce qui correspond à un pourcentage de 15,87%.
1 cas d’hémophilie A : (2 frères décédés : un frère décédé à 03 jours de vie à domicile, un autre est mort à 9 mois de vie par un syndrome hémorragique en post circoncision) ;
1 cas de Thrombasthénie de Glanzmann : sœur décédée à l’âge de 3 ans et qui était suivi pour la même maladie.
1 cas d’hémophilie A : 2 frères décédés suite un syndrome hémorragique post circoncision. 1 cas d’hypofibrinogénémie : deux frères décédés : un à J7 de vie suite à une hémorragie ombilicale, un autre décédé à J2 de vie.
Fig35 : La répartition de la maladie hémorragique au sein de la famille( en dehors de la fratrie) .

105
1 cas de Thrombasthénie de Glanzmann : frère décédé à l’âge de 06 mois qui a succombé à une hémorragie cérébrale.
1 cas d’hémophilie A: un frère décédé qui était atteint de la même pathologie.
1 cas de Thrombasthénie de Glanzmann : frère décédé à l’âge de de 02 ans de la même pathologie.
1 cas d’hémophilie B : frère décédé à l’âge d’un an au cours de sa circoncision après une hémorragie importante.
1 cas d’hémophilie A : un frère de 14 ans qui a perdu la vie dans un accident de voiture.
1 cas de Thrombasthénie de Glanzmann : un frère de 02 ans qui est mort après un état de choc hémorragique.
Fig36 : Le pourcentage de décès lié à une pathologie de l’hémostase dans la famille.

106
III. ANTÉCÉDENTS PERSONNELS
Ce qui nous intéresse dans notre étude ce sont les antécédents hémorragiques. Ils ont été retrouvés chez 52,38% des patients, alors que 47,62% n’ont présenté aucun événement hémorragique antérieur.
Fig37: Distribution des patients selon la présence ou non d’antécédents hémorragiques personnels
1. CONDUITE DES PARENTS FACE À CES PREMIERS ÉVÉNEMENTS HÉMORRAGIQUES
Le pourcentage des patients ayant consulté devant la survenue du premier événement hémorragique est de 40 % contre 60% qui n’ont pas consulté. L’intérêt de cette question est double : apprécier le vécu parental devant une symptomatologie hémorragique et la conduite adoptée face à cette dernière (alerte et angoisse, banalité du signe)Comprendre les causes du retard diagnostic même devant l’apparition de signes hémorragiques.
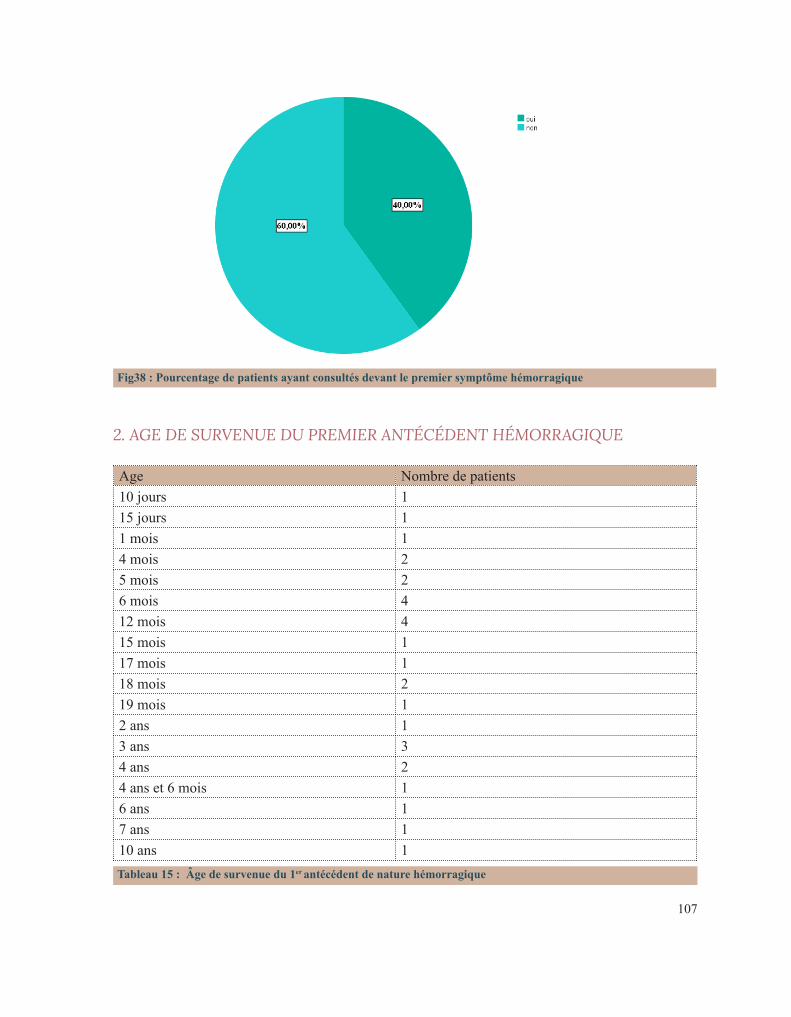
107
2. AGE DE SURVENUE DU PREMIER ANTÉCÉDENT HÉMORRAGIQUE
Age Nombre de patients 10 jours 115 jours 11 mois 14 mois 25 mois 26 mois 412 mois 415 mois 117 mois 118 mois 219 mois 12 ans 13 ans 34 ans 24 ans et 6 mois 16 ans 17 ans 110 ans 1
Fig38 : Pourcentage de patients ayant consultés devant le premier symptôme hémorragique
Tableau 15 : Âge de survenue du 1er antécédent de nature hémorragique

108
L’âge de survenue du 1er antécédent hémorragique a différé d’un âge minimum de 10 jours de vie à un âge maximum de 10 ans. La moitié des patients (15) a présenté le 1 er symptôme durant la 1 ère année de vie, 6 durant la deuxième année de vie, 5 durant la petite enfance, et finalement 4 durant la grande enfance.
3. NOMBRE D’ÉPISODES HÉMORRAGIQUES
La récurrence des épisodes de saignements n’est pas la même chez tous les enfants. Tandis que quelques-uns ont présenté plusieurs épisodes, d’autres n’ont présenté qu’un seul incident de nature hémorragique.
Fig39 : Répartition selon le nombre d’épisodes hémorragiques antécédents
Les Epistaxis ou gingivorragies récidivantes et les Ecchymoses généralisées d’apparition spontanée sont les épisodes récurrents les plus retrouvés chez nos patients.
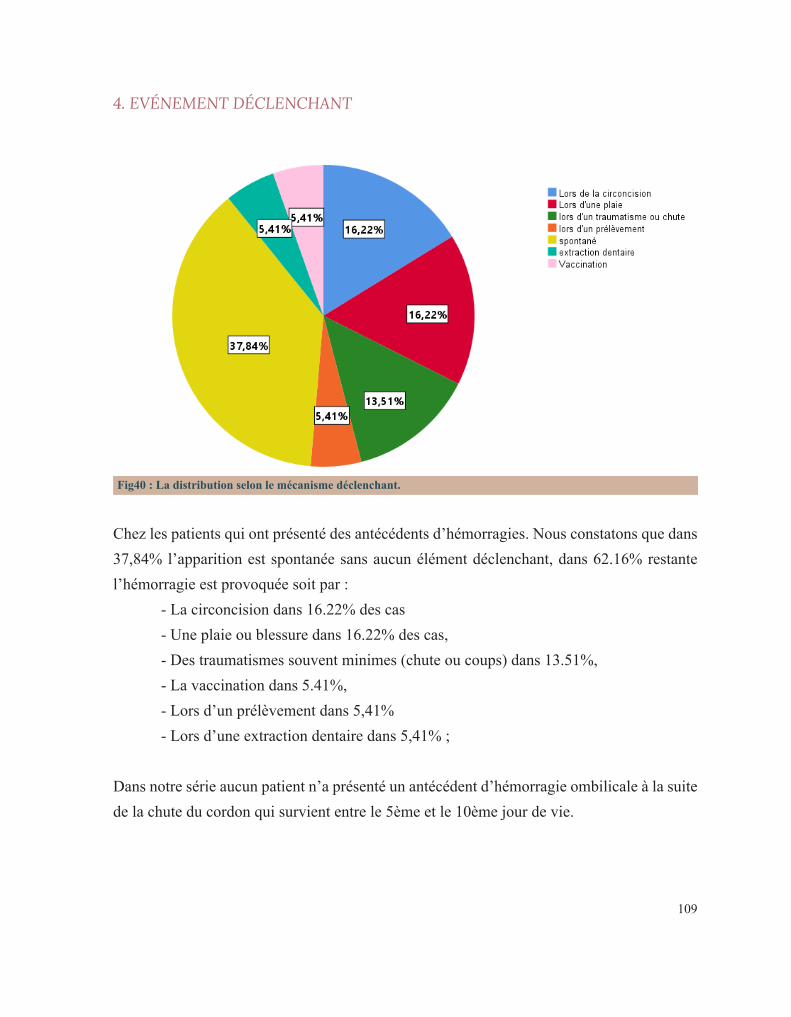
109
4. EVÉNEMENT DÉCLENCHANT
Fig40 : La distribution selon le mécanisme déclenchant.
Chez les patients qui ont présenté des antécédents d’hémorragies. Nous constatons que dans 37,84% l’apparition est spontanée sans aucun élément déclenchant, dans 62.16% restante l’hémorragie est provoquée soit par : - La circoncision dans 16.22% des cas - Une plaie ou blessure dans 16.22% des cas, - Des traumatismes souvent minimes (chute ou coups) dans 13.51%, - La vaccination dans 5.41%, - Lors d’un prélèvement dans 5,41% - Lors d’une extraction dentaire dans 5,41% ;
Dans notre série aucun patient n’a présenté un antécédent d’hémorragie ombilicale à la suite de la chute du cordon qui survient entre le 5ème et le 10ème jour de vie.

110
5. TYPE DE SAIGNEMENT
Fig41 : Pourcentage de chaque type de saignement
Les saignements muqueux d’apparition spontanée comme les épistaxis ou provoquée par une extraction dentaire comme les gingivorragies sont les plus fréquentes (30%). L’hémorragie secondaire à une plaie ou blessure cutanée est retrouvée dans 22,5% des cas.
L’ecchymose est un symptôme retrouvé couramment dans l’histoire hémorragique des enfants affecté par une pathologie de l’hémostase, elles sont souvent spontanées ou causées par des traumatismes minimes. Elle représente 20% de la symptomatologie dans notre série.
La circoncision est une tradition historique de longue date dans les religions juive et musulmane. Elle est le plus souvent pratiquée au Maroc à un âge précoce. Elle a été responsable dans notre étude d’une hémorragie dans 12,5% des cas.
Le syndrome cutanéomuqueux associant le plus souvent des ecchymoses + des épistaxis dans notre série est retrouvé chez 2,5% du total des enfants.

111
IV. CIRCONSTANCES DE DECOUVERTE
Les circonstances de découverte proprement dit et dont il est question dans ce chapitre regroupent les conditions révélatrices de la pathologie hémorragique constitutionnelle de l’hémostase et qui ont permis de poser le diagnistic exclus tous les événements hémorragiques antérieurs qu’on a détaillé dans le chapitre précédent ;
Fig42 : Circonstances de découverte des pathologies hémorragiques héréditaires en pourcentage.
La symptomatologie hémorragique est la condition révélatrice de la pathologie constitutionnelle de l’hémostase dans la majorité des cas avec un pourcentage de 58,73%, la découverte est fortuite dans 34.92% des cas étudiés. Le diagnostic a été posé dans 9,52% lors d’une enquête familiale. Et la symptomatologie non spécifique a été retrouvée dans 6.35% des cas.

112
1. SYMPTOMATOLOGIE HÉMORRAGIQUE
1.1. Type de saignement
Fig43 : Distribution des patients selon le type de saignement
La présentation clinique du syndrome hémorragique dans les anomalies de l’hémostase est polymorphe et variable selon la pathologie en cause et selon le degré de sévérité de cette dernière. Elle a une grande valeur dans l’orientation des examens complémentaires. Les saignements muqueux sont les plus fréquents (29.73%). Les gingivorragies sont les saignements muqueux les plus communs, suivis par les épistaxis et enfin les ménométrorragies chez les jeunes filles.
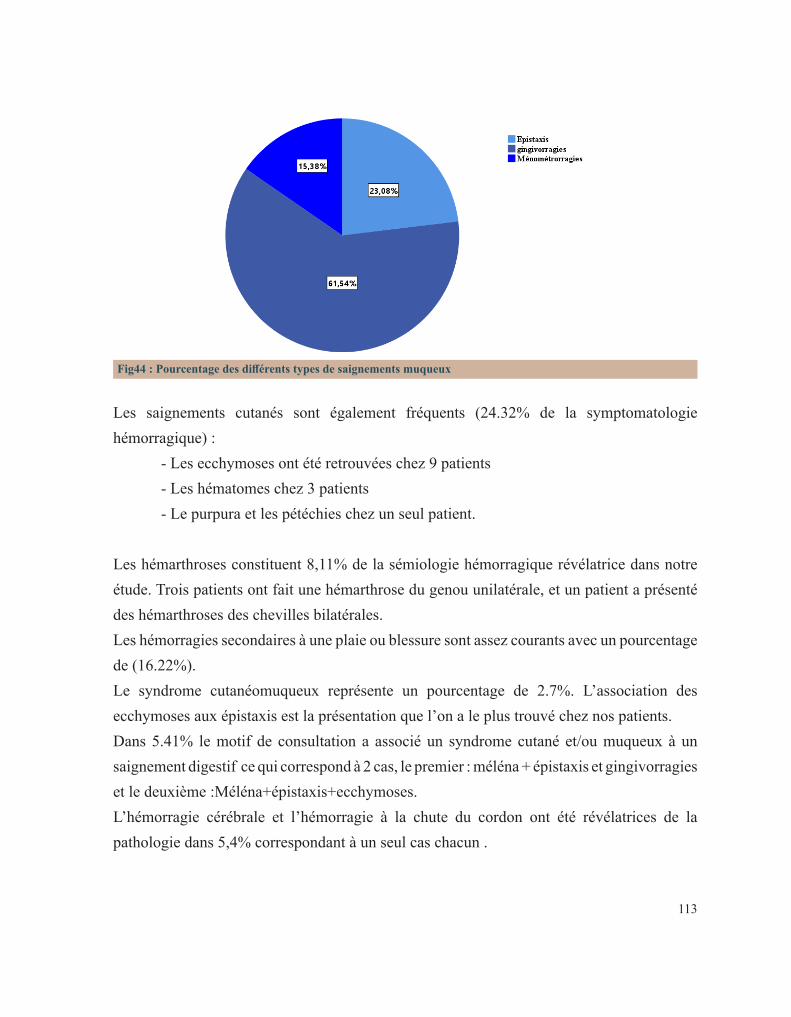
113
Fig44 : Pourcentage des différents types de saignements muqueux
Les saignements cutanés sont également fréquents (24.32% de la symptomatologie hémorragique) : - Les ecchymoses ont été retrouvées chez 9 patients - Les hématomes chez 3 patients - Le purpura et les pétéchies chez un seul patient.
Les hémarthroses constituent 8,11% de la sémiologie hémorragique révélatrice dans notre étude. Trois patients ont fait une hémarthrose du genou unilatérale, et un patient a présenté des hémarthroses des chevilles bilatérales.Les hémorragies secondaires à une plaie ou blessure sont assez courants avec un pourcentage de (16.22%). Le syndrome cutanéomuqueux représente un pourcentage de 2.7%. L’association des ecchymoses aux épistaxis est la présentation que l’on a le plus trouvé chez nos patients.Dans 5.41% le motif de consultation a associé un syndrome cutané et/ou muqueux à un saignement digestif ce qui correspond à 2 cas, le premier : méléna + épistaxis et gingivorragies et le deuxième :Méléna+épistaxis+ecchymoses.L’hémorragie cérébrale et l’hémorragie à la chute du cordon ont été révélatrices de la pathologie dans 5,4% correspondant à un seul cas chacun .

114
1.2. Le mode d’apparition
La symptomatologie hémorragique qui a permis de poser le diagnostic dans notre série est apparue spontanément dans 62.16% des cas, en revanche dans 37.84% des cas elle a été provoquée par les mécanismes suivants :
Mécanisme déclenchant Fréquence Choc ou traumatisme 6Extraction dentaire 4Circoncision 3Plaie cutanée 3Prélèvement ou injection 2Ménarches 2Chute du cordon 2Vaccination 1Chirurgie autre que la circoncision 1
Fig45 : Répartition selon le mode d’apparition
Tableau 16 : Les mécanismes déclenchant les hémorragies et leur effectif.
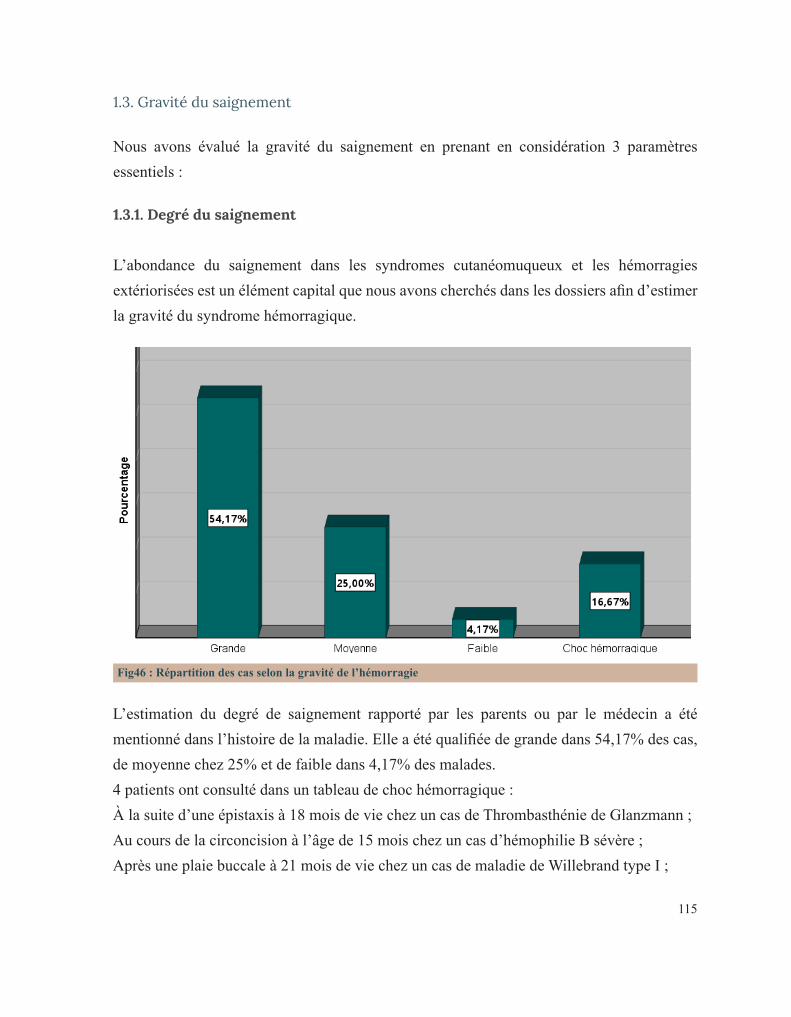
115
1.3. Gravité du saignement
Nous avons évalué la gravité du saignement en prenant en considération 3 paramètres essentiels :
1.3.1. Degré du saignement
L’abondance du saignement dans les syndromes cutanéomuqueux et les hémorragies extériorisées est un élément capital que nous avons cherchés dans les dossiers afin d’estimer la gravité du syndrome hémorragique.
Fig46 : Répartition des cas selon la gravité de l’hémorragie
L’estimation du degré de saignement rapporté par les parents ou par le médecin a été mentionné dans l’histoire de la maladie. Elle a été qualifiée de grande dans 54,17% des cas, de moyenne chez 25% et de faible dans 4,17% des malades. 4 patients ont consulté dans un tableau de choc hémorragique :À la suite d’une épistaxis à 18 mois de vie chez un cas de Thrombasthénie de Glanzmann ;Au cours de la circoncision à l’âge de 15 mois chez un cas d’hémophilie B sévère ;Après une plaie buccale à 21 mois de vie chez un cas de maladie de Willebrand type I ;

116
À la suite de ménorragies lors des ménarches à l’âge de 14 ans chez un cas de syndrome de Bernard Soulier ;
1.3.2. Transfusion
Le pourcentage des patients n’ayant pas été transfusés est supérieur à celui qui ont été transfusé.
1.3.3. Etat hémodynamique à l’admission
Fig47 : Distribution des cas selon la nécessité d’une transfusion
Fig48 : Répartition des cas selon leur état hémodynamique
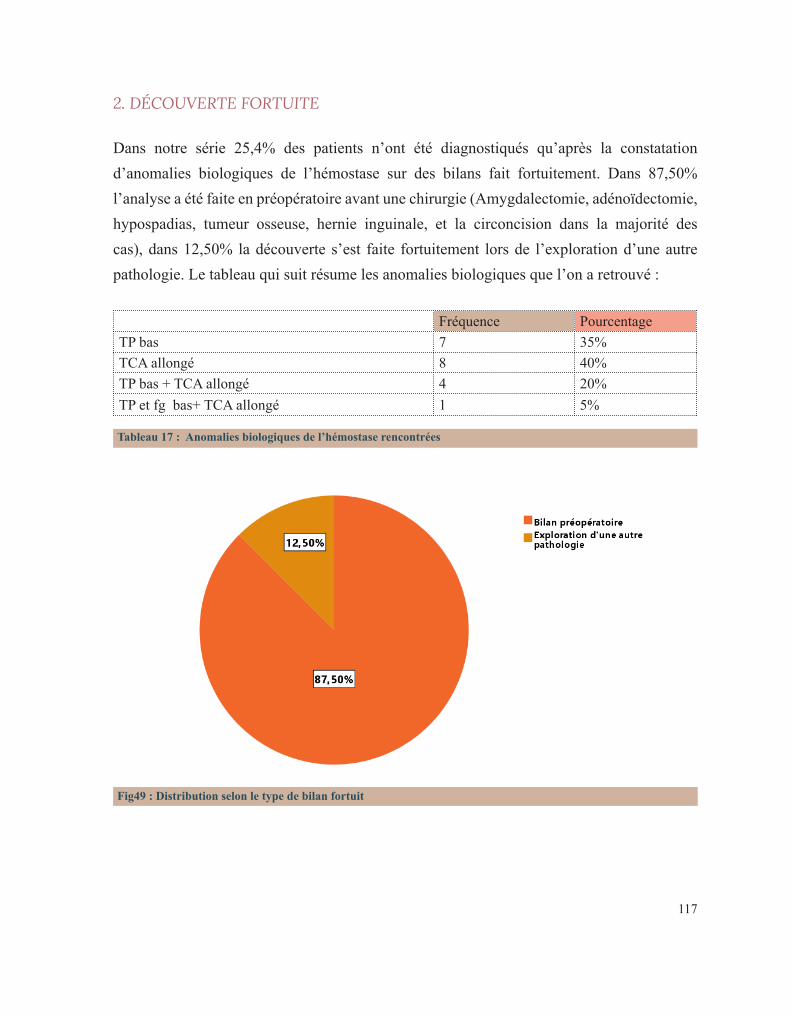
117
2. DÉCOUVERTE FORTUITE
Dans notre série 25,4% des patients n’ont été diagnostiqués qu’après la constatation d’anomalies biologiques de l’hémostase sur des bilans fait fortuitement. Dans 87,50% l’analyse a été faite en préopératoire avant une chirurgie (Amygdalectomie, adénoïdectomie, hypospadias, tumeur osseuse, hernie inguinale, et la circoncision dans la majorité des cas), dans 12,50% la découverte s’est faite fortuitement lors de l’exploration d’une autre pathologie. Le tableau qui suit résume les anomalies biologiques que l’on a retrouvé :
Fréquence Pourcentage TP bas 7 35%TCA allongé 8 40%TP bas + TCA allongé 4 20%TP et fg bas+ TCA allongé 1 5%
Tableau 17 : Anomalies biologiques de l’hémostase rencontrées
Fig49 : Distribution selon le type de bilan fortuit

118
3. ENQUÊTE FAMILIALE
Il s’agit d’un dépistage orienté par l’existence d’une histoire familiale. Il a permis de poser le diagnostic d’une pathologie constitutionnelle dans 9,52%.5 cas d’hémophilie A et un cas de Thrombasthénie de Glanzmann
4 SYMPTOMATOLOGIE NON SPÉCIFIQUE ( NON HÉMORRAGIQUE)
6,35% des patients ce qui correspond à 4 cas ont été diagnostiqué atteint d’une pathologie congénitale de l’hémostase à la suite d’une symptomatologie non hémorragique. Les 3 motifs de consultation étaient :
- Arthrite septique ne s’améliorant pas sous antibiothérapie - Douleur et rougeur oculaire - Impotence fonctionnelle de la hanche gauche ; - Ostéoarthrite du genou.

119
DISCUSSION

120
Les troubles héréditaires de la coagulation sont causés par des altérations quantitatives et qualitatives des protéines plaquettaires ou plasmatiques impliquées dans la coagulation et la fibrinolyse. Les données sur leur épidémiologie et leur impact dans les pays en développement sont limités. L’objectif de cette étude est d’évaluer la prévalence locale de ces pathologies et d’établir les variables cliniques et historiques prédictives de ces troubles en pédiatrie.
Environ 84% de la population mondiale vit dans des pays à revenu faible ou moyen et souffre de 93% des maladies dans le monde, alors qu’ils touchent 18% du revenu total et 11% des dépenses de santé mondiales. Les troubles de la coagulation représentent une part notable des diverses maladies dans les pays en développement, où le taux de mariages consanguins est élevé. En effet, la prévalence des troubles rares de la coagulation est 10 fois plus élevée dans ces pays que dans les pays développés [51].
L’hémophilie est la pathologie héréditaire hémorragique la plus fréquente. Cependant, les données accumulées provenant de diverses études ont montré que la maladie de von Willebrand (MVW) était la cause la plus courante, avec une augmentation également de l’incidence des désordres de la fonction plaquettaire, principalement en raison de l’augmentation du taux de consanguinité dans certaines communautés [114], [115].
Dans notre étude l’hémophilie A est la maladie constitutionnelle hémorragique de l’hémostase dominante (41,9%), elle l’est mêmement dans 2 autres études :• Le Réseau FranceCoag (RFC) qui est un dispositif national français reposant sur un suivi prospectif de cohorte de patients porteurs d’un DHPC. Mis en place par les pouvoirs publics en 2003 en amont du Plan National Maladies Rares 2003-2007, RFC est coordonné par l’Institut de Veille Sanitaire (InVS) depuis janvier 2004. Au 20 novembre 2009, soit au terme de 7 années de fonctionnement, 6622 patients au total avaient été inclus dans la cohorte. Dont 1963 patients étaient âgés de moins de 18 ans (30,4 % de la cohorte) [116].• Une Enquête prospective réalisée de septembre 2009 à mars 2011. Les dossiers médicaux de trois grands hôpitaux de référence (hôpital Ghaem, hôpital Imam Reza et hôpital pédiatrique Dr. Sheikh) ont été examinés et tous les cas diagnostiqués de troubles de

121
la coagulation et de l’hémostase primaire ont été inclus dans cette étude. Nous regroupons dans le tableau qui suit les résultats des 3 études [117].
Pathologie Etude CHOP FranceCoag Investigation in North Eastern Iran Maladie de Willebrand 11,3% 14,3% 8,5%Hémophilie A 41,9% 65,7% 53,9%Hémophilie B 6,5% 14,9% 16,6%Déficit en Facteur II 1,6% 0,05% 0%Déficit en facteur V 3,2% 0,6% 3,6%Déficit en facteur VII 11,3% 1,5% 4%
Déficit en facteur X 0% 0,15% 0,38%Déficit en facteur XI 1,6% 1,3% 0,38%Déficit en facteur XIII 0% 0,6% 2%Afibrinogénémie congénitale 3,2% 0,7% 0,38%Hypofibrinogénémie 3,2% 0% 0%Thrombasthénie de Glanz-mann 11,3% 0% 6,9%Syndrome de Bernard Soulier 1,6% 0%
La maladie de Willebrand vient en deuxième position dans notre étude (11,3%), elle occupe la troisième position dans les deux autres études, devancée par l’hémophilie B, qui occupe la 4ème position dans notre étude. Le déficit en facteur VI est le déficit le plus fréquent parmi les déficits rares de la coagulation dans les 3 études, les autres déficits sont rarissimes. Les thrombopathies sont également fréquentes chez nous (11,3% des patients pour la thrombasthénie de Glanzmann et 1,6% pour le syndrome de bernard-soulier).
Les pathologies hémorragiques constitutionnelles peuvent survenir à tout âge, mais s’ils sont graves, ils surviennent généralement à un âge plus précoce [105], les désordres modérés et surtout mineurs peuvent également survenir à n’importe quel âge mais nécessite le plus souvent un important défi hémostatique pour se révéler, la plupart du temps une intervention chirurgicale ou une extraction dentaire [118]. Les femmes atteintes de troubles de la coagulation héréditaires légers, modérés ou graves peuvent présenter des saignements gênants au début des règles ou des saignements liés à l’accouchement.
Tableau 18 : Comparaison de la fréquence des pathologies hémorragiques héréditaires dans 3 études différentes.

122
L’âge médian au diagnostic des troubles plaquettaires et de la coagulation était de 7ans et 1 mois dans notre étude avec des valeurs extrêmes allant d’un minimum de 10 jours (Afibrinogénémie et Hypofibrinogénémie) à un maximum de 14ans (syndrome de Bernard soulier, hémophilie A mineure et maladie de Von Willebrand). L’âge médian dans notre série reste assez élevé par rapport à d’autres études, en effet dans une étude prospective menée (du 1er janvier 1994 au 31 décembre 2009) sur des enfants atteints de troubles de la coagulation dans le département de pédiatrie de l’hôpital Ain Shams au Caire en Égypte, l’âge médian au diagnostic était de 33 mois dans les déficits en facteurs de la coagulation et de 72 mois(6ans) dans les anomalies de l’hémostase primaire [119].
En ce qui la concerne la distribution de nos patients selon le sexe, le sexe féminin était significativement plus présent dans les troubles plaquettaires (62,5%) que dans les troubles rares de la coagulation (29,4%). Chez les patients atteints de la maladie de von Willebrand, 14,28% étaient des femmes. Tous les patients atteints d’hémophilie A et d’hémophilie B étaient des hommes. Contrairement à l’hémophilie, la plupart des troubles héréditaires liés aux plaquettes ne sont pas liés à l’X et sont également répartis entre les deux sexes [36]. Dans la littérature environ 75% de la MVW a été diagnostiquée chez des femmes. De même Sadler et al. [120] ont rapporté que la maladie de von Willebrand était diagnostiquée plus souvent chez la femme, ce qui se manifestait principalement par une ménorragie. L’analyse de diverses données anamnestiques (l’histoire familiale et personnelle et la sémiologie hémorragiques) indique que les antécédents familiaux d’ecchymoses et d’hémorragies sont le facteur prédictif le plus significatif de la survenue de pathologies hémorragiques congénitales, à l’appui de résultats similaires de l’étude de Jayasinghe et al. [121]. La recherche de l’existence d’une histoire hémorragique familiale et d’une consanguinité est donc un élément capital dans le diagnostic du type de pathologies que nous traitons dans la présente étude. La consanguinité augmente le risque d’apparition des troubles hémorragiques[122] ,plus répandus au Moyen-Orient [123]. Elle est définie par le mariage entre personnes qui sont cousines ou proches, et au moins 20% de la population humaine vit dans des

123
communautés où le mariage consanguin est une préférence. Au Maroc, les mariages consanguins sont fréquents avec une prévalence élevée de troubles autosomiques récessifs. Les anomalies de l’hémostase héréditaires ont pour la majorité un mode de transmission autosomique récessif (Maladie de Willebrand type 2N et type 3, Thrombasthénie de Glanzmman, Syndrome de Bernard soulier, déficits rares en facteurs de la coagulation (FII, V, VII, XI) et hypo et afibrinogénémie congénitale). De ce fait ces pathologies sont plus fréquentes dans les pays où la consanguinité est élevée, la Thrombasthénie de Glanzmann par exemple est plus courante chez les arabes et les juifs [28].Dans notre série la consanguinité a été retrouvée dans 58,73% des cas et les antécédents hémorragiques familiaux chez 33,4% des patients. En ce qui concerne la consanguinité, nos résultats s’approchent des résultats d’une étude prospective faite durant la période s’étendant du mois de Janvier 2005 à décembre 2006 sur un groupe de 43 enfants et adolescents âgés de 1 à 18 ans traités dans des cliniques d’hématologie pédiatrique du Centre national de recherche et des hôpitaux Sausan Mubarek en Égypte et King Abdulaziz en Arabie saoudite dans laquelle la consanguinité positive a été documentée dans 55,8% des cas. Alors que Vingt-six cas sur 43 (60,5%) avaient des antécédents familiaux de saignement positifs dans cette étude [124]. La transmission de l’hémophilie obéit à la loi de l’hérédité récessive liée au chromosome X. Cependant, l’absence d’antécédents connu par la famille est une situation fréquente lors du diagnostic d’un nouveau cas d’hémophilie. Ces cas isolés dit sporadiques sont majoritaires dans la cohorte française (55%) et dans le registre national suédois (58%) [69], en revanche ils sont minoritaires dans notre étude 46,15% [125].
A côté des antécédents hémorragiques familiaux, les antécédents hémorragiques cliniques des patients sont un outil important pour diagnostiquer correctement les pathologies constitutionnelles de l’hémostase. Par conséquent, la recherche des premiers symptômes hémorragiques et la détermination de leur âge d’apparition ainsi que leur mode d’apparition a une grande valeur diagnostique.Pollmann et al. [126] ont rapporté que dans les troubles héréditaires de la coagulation, environ 44% des cas signalaient un saignement au cours de la première année de vie, alors que d’autres n’avaient eu leur premier épisode de saignement qu’à l’âge de 4 ans. Cela
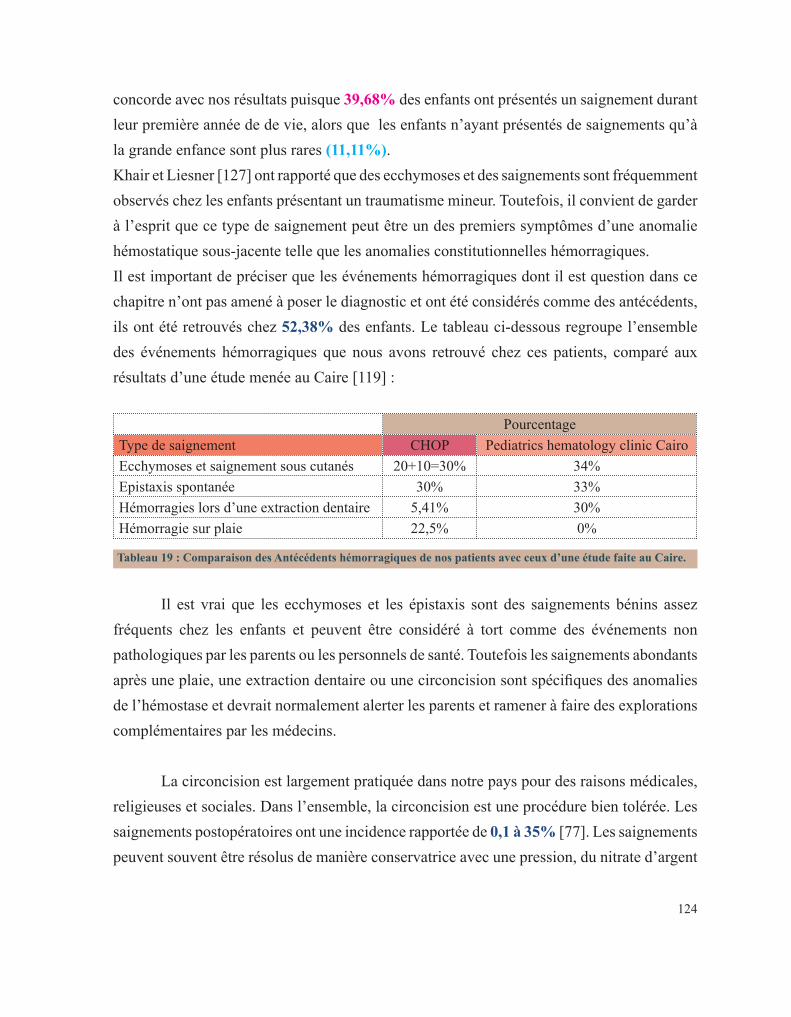
124
concorde avec nos résultats puisque 39,68% des enfants ont présentés un saignement durant leur première année de de vie, alors que les enfants n’ayant présentés de saignements qu’à la grande enfance sont plus rares (11,11%).Khair et Liesner [127] ont rapporté que des ecchymoses et des saignements sont fréquemment observés chez les enfants présentant un traumatisme mineur. Toutefois, il convient de garder à l’esprit que ce type de saignement peut être un des premiers symptômes d’une anomalie hémostatique sous-jacente telle que les anomalies constitutionnelles hémorragiques. Il est important de préciser que les événements hémorragiques dont il est question dans ce chapitre n’ont pas amené à poser le diagnostic et ont été considérés comme des antécédents, ils ont été retrouvés chez 52,38% des enfants. Le tableau ci-dessous regroupe l’ensemble des événements hémorragiques que nous avons retrouvé chez ces patients, comparé aux résultats d’une étude menée au Caire [119] :
Pourcentage Type de saignement CHOP Pediatrics hematology clinic CairoEcchymoses et saignement sous cutanés 20+10=30% 34%Epistaxis spontanée 30% 33%Hémorragies lors d’une extraction dentaire 5,41% 30%Hémorragie sur plaie 22,5% 0%
Il est vrai que les ecchymoses et les épistaxis sont des saignements bénins assez fréquents chez les enfants et peuvent être considéré à tort comme des événements non pathologiques par les parents ou les personnels de santé. Toutefois les saignements abondants après une plaie, une extraction dentaire ou une circoncision sont spécifiques des anomalies de l’hémostase et devrait normalement alerter les parents et ramener à faire des explorations complémentaires par les médecins.
La circoncision est largement pratiquée dans notre pays pour des raisons médicales, religieuses et sociales. Dans l’ensemble, la circoncision est une procédure bien tolérée. Les saignements postopératoires ont une incidence rapportée de 0,1 à 35% [77]. Les saignements peuvent souvent être résolus de manière conservatrice avec une pression, du nitrate d’argent
Tableau 19 : Comparaison des Antécédents hémorragiques de nos patients avec ceux d’une étude faite au Caire.
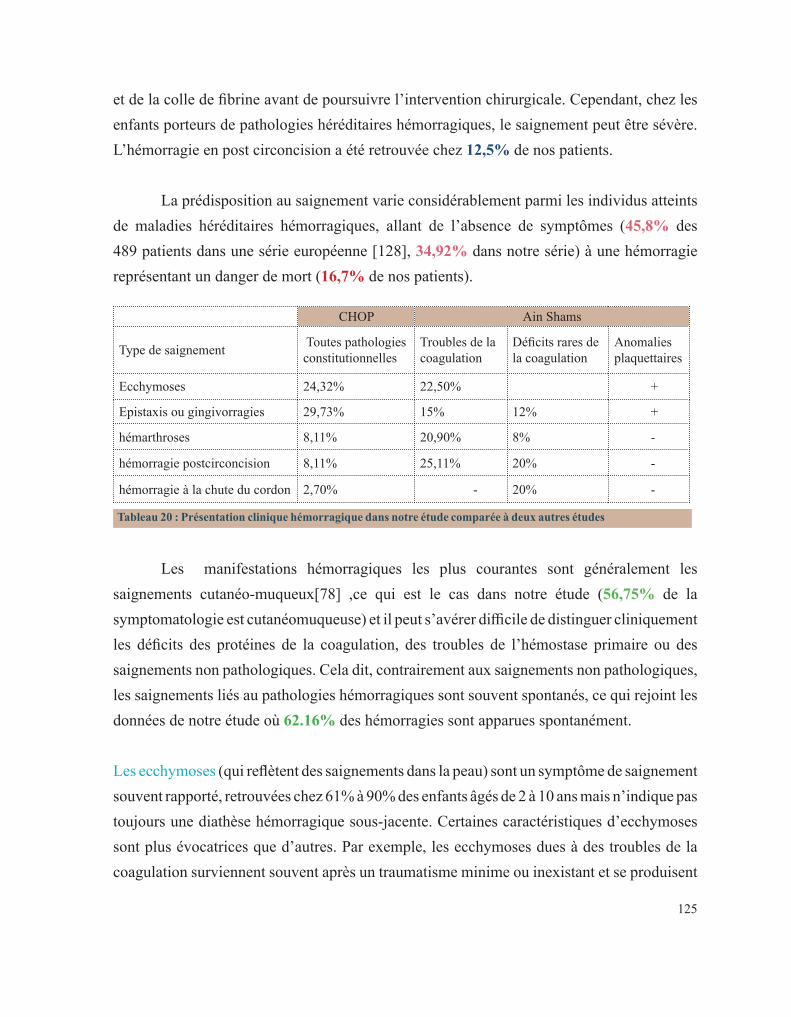
125
et de la colle de fibrine avant de poursuivre l’intervention chirurgicale. Cependant, chez les enfants porteurs de pathologies héréditaires hémorragiques, le saignement peut être sévère. L’hémorragie en post circoncision a été retrouvée chez 12,5% de nos patients. La prédisposition au saignement varie considérablement parmi les individus atteints de maladies héréditaires hémorragiques, allant de l’absence de symptômes (45,8% des 489 patients dans une série européenne [128], 34,92% dans notre série) à une hémorragie représentant un danger de mort (16,7% de nos patients).
CHOP Ain Shams
Type de saignement Toutes pathologies constitutionnelles
Troubles de la coagulation
Déficits rares de la coagulation
Anomalies plaquettaires
Ecchymoses 24,32% 22,50% +
Epistaxis ou gingivorragies 29,73% 15% 12% +
hémarthroses 8,11% 20,90% 8% -
hémorragie postcirconcision 8,11% 25,11% 20% -
hémorragie à la chute du cordon 2,70% - 20% -
Les manifestations hémorragiques les plus courantes sont généralement les saignements cutanéo-muqueux[78] ,ce qui est le cas dans notre étude (56,75% de la symptomatologie est cutanéomuqueuse) et il peut s’avérer difficile de distinguer cliniquement les déficits des protéines de la coagulation, des troubles de l’hémostase primaire ou des saignements non pathologiques. Cela dit, contrairement aux saignements non pathologiques, les saignements liés au pathologies hémorragiques sont souvent spontanés, ce qui rejoint les données de notre étude où 62.16% des hémorragies sont apparues spontanément.
Les ecchymoses (qui reflètent des saignements dans la peau) sont un symptôme de saignement souvent rapporté, retrouvées chez 61% à 90% des enfants âgés de 2 à 10 ans mais n’indique pas toujours une diathèse hémorragique sous-jacente. Certaines caractéristiques d’ecchymoses sont plus évocatrices que d’autres. Par exemple, les ecchymoses dues à des troubles de la coagulation surviennent souvent après un traumatisme minime ou inexistant et se produisent
Tableau 20 : Présentation clinique hémorragique dans notre étude comparée à deux autres études

126
généralement dans des zones fréquemment exposées à un traumatisme (par exemple, les membres inférieurs, les hanches externes, les bras). Dans notre étude ce type de saignement a été révélateur dans 24,32%. Les autres types de saignements cutanés, les pétéchies et/ou le purpura, sont moins fréquents et sont typiques d’une thrombocytopénie (PTI). Ils sont moins fréquemment observés dans d’autres conditions, notamment les troubles graves de la fonction plaquettaire.
L’épistaxis comme l’ecchymose est un symptôme de saignement souvent rapporté, qui ne reflète pas toujours un trouble de la coagulation, même lorsque les saignements sont fréquents et/ou nécessitent des interventions médicales. Elle est causée ordinairement par des anomalies de l’hémostase primaire (problèmes de plaquettes ou de facteur de Willebrand, par exemple). Elle peut également résulter d’une déficience grave en facteurs de la coagulation de la voie commune (déficience congénitale en facteur V, facteur X ou prothrombine, par exemple). Les saignements du nez dus aux troubles de la coagulation sont souvent aggravés dans l’enfance et suscitent de l’intérêt par leur fréquence et leur aggravation. Lorsque le saignement du nez est abondant, le passage des caillots sanguins au tube digestif peut être responsable de l’apparition de méléna. Il s’agit en effet, d’une présentation que nous avons retrouvée chez 2 patients(/63). Bien que les saignements de nez ne soient pas spécifiques aux troubles de la coagulation, il est important de poser des questions à ce sujet, car les saignements de nez graves peuvent être invalidants pour certaines personnes atteintes de troubles de la coagulation, telles que la maladie de von Willebrand [129].
Le saignement des gencives peut être problématique avec les troubles hémorragiques héréditaires ou acquis qui affectent les plaquettes ou le facteur de von Willebrand, bien qu’il reflète plus souvent la maladie des gencives. Les saignements sévères avec perte de dents primaires sont également suspects d’un trouble hémorragique sous-jacent.
Les symptômes de saignement réservés aux femmes : La ménorragie, est souvent associée à une anémie et à une qualité de vie réduite. La ménorragie peut être le symptôme d’une adolescente présentant un trouble de la coagulation. Trente-cinq pour cent des femmes s’identifient comme ayant des saignements menstruels abondants, mais la proportion de

127
ménorragies quantifiables est de 10% [130]. Un tableau d’évaluation graphique du débit sanguin peut être utilisé dans le bureau pour fournir une évaluation semi-quantitative de la perte de sang menstruel. La ménorragie est plus susceptible d’être associée à un trouble de la coagulation si elle se produit au cours du premier cycle de la ménarche, ou si elle est associée à une inondation ou à une anémie [131].
En 2000, le comité américain d’obstétrique et de gynécologie recommande que les femmes atteintes de ménorragie soient évaluées pour la maladie de von Willebrand, mais les troubles de la fonction plaquettaire et d’autres coagulopathies sont également des causes fréquentes de saignements menstruels abondants chez les adolescentes et les femmes adultes[132].
Les Saignements articulaires et saignements musculaires (par exemple, saignements du muscle iliopsoas), qui sont des symptômes saignants peu communs, suggèrent un défaut de coagulation grave ou un trouble fibrinolytique. Cependant, un saignement dans une articulation après une blessure ou une blessure orthopédique (par exemple, une arthropathie) peut se voir chez les patients présentant d’autres types de troubles de la coagulation. L’hémarthrose est caractéristique des hémophiles et doit faire évoquer en premier lieu des pathologies de la coagulation. Toutefois, elle peut annoncer un autre type de trouble hémostatique. Elle a été révélatrice dans 8,11% des cas dans notre série.
L’Hémorragie intracrânienne : Un nouveau-né ou un enfant présentant une hémorragie intracrânienne spontanée ou provoquée par un traumatisme doit faire l’objet d’une recherche de troubles hémorragiques sous-jacents graves, tels que thrombocytopénie, hémophilie majeure du nourrisson, déficit en facteur XIII, ou défaut grave des plaquettes ou du facteur von Willebrand. Elle a permis de poser le diagnostic d’hémophile A sévère chez un seul patient dans notre étude.
Le saignement gastro-intestinal peut compliquer un trouble de la coagulation, mais il s’agit rarement d’un symptôme révélateur. Dans notre étude 2 cas se sont déclarés par des mélénas associées à un saignement muqueux.

128
Les pathologies constitutionnelles hémorragiques de l’hémostase peuvent également présenter un saignement excessif après une épreuve hémostatique. Les saignements après des procédures telles que la circoncision lors de la première ou la seconde année de vie ou les extractions dentaires plus tard dans l’enfance sont des symptômes communs à toutes ces pathologies, bien que l’absence de saignement après ce type d’intervention n’exclue pas nécessairement une pathologie de l’hémostase.
La gravité du trouble de la coagulation affecte le nombre, le degré et le type de manifestations hémorragiques. Les scores numériques, dérivés d’outils d’évaluation standardisés des antécédents de saignement, montrent un chevauchement considérable entre des sujets de gravité différente [133]. L’utilisation d’un outil standardisé pour quantifier les symptômes d’hémorragie a été recommandée par la Société internationale de thrombose et d’hémostase [105], l’outil nécessite une validation prospective supplémentaire avant son utilisation dans la pratique clinique de routine.
La présentation individuelle de la symptomatologie hémorragique dans ce type de pathologie est variable. En général, les saignements graves (par exemple, les hémorragies intracrâniennes, les saignements musculosquelettiques ou du cordon ombilical) sont rares dans les carences en FV et en FXI et dans les anomalies de l’hémostase primaire et contrairement à l’hémophilie A et B, il existe une faible corrélation entre le degré du saignement et le niveau d’activité du facteur. En effet, une revue récente a mis en évidence une faible corrélation entre la sévérité clinique et l’activité coagulante dans les déficiences en FV et FVII et l’absence de corrélation entre la sévérité clinique et l’activité coagulante dans la déficience en FXI, même à des niveaux indétectables ou modérément réduits (20%). La carence grave en FVII peut être asymptomatique ou, au contraire, présenter des événements hémorragiques graves (par exemple, ICH) pendant la petite enfance. En revanche, dans les déficiences en FI, FII, FX et FXIII, la sévérité clinique est fortement corrélée aux niveaux de facteur. Le phénotype de saignement en déficit en FI (afibrinogénémie) peut être similaire à celui observé dans les cas d’HA ou d’HB modérés à sévères.Bien que cela soit plus fréquent chez certains patients souffrant de pathologies hémorragiques, des épisodes hémorragiques graves peuvent survenir sur tous les types d’anomalies de

129
l’hémostase et ne permettent donc pas toujours de distinguer un trouble particulier d’un autre. Par exemple, un saignement du cordon ombilical peut survenir dans des conditions autres que les déficiences en FX et FXIII, notamment dans l’Hémophilie A, et l’Hémophilie B et d’autres déficits rares en facteurs de la coagulation. De même, l’hémorragie intracrânienne a été décrite dans la période néonatale dans les déficiences sévères en FII, FV et FVII, et elle est particulièrement commune dans les déficiences en FX et FXIII. L’hémorragie cérébrale a été signalé chez 20% des individus avec un déficit en FX et jusqu’à 30% de ceux présentant un déficit en FXIII. Elle est la principale cause de décès et d’invalidité [23,49]. D’autres saignements mettant en danger la vie et les membres, tels que l’hémopéritoine ovulatoire et hémarthroses le plus souvent rapportés dans les cas de déficit en FII, FX et FXIII.
Il n’est pas rare, dans certains cas, que le diagnostic de pathologie constitutionnelle hémorragique ne soit posé qu’à l’occasion d’un bilan préopératoire ou lors d’un bilan systématique fait fortuitement. Ça a été le cas dans notre étude chez 25,4% des patients. En effet et comme décrit dans la littérature toutes les pathologies héréditaires de l’hémostase et surtout dans les déficits mineurs en hémophilie et d’autres déficits en facteurs de la coagulation (II , X ) et la maladie de Willebrand type 1, les patients peuvent restés asymptomatiques pendant un longue durée et n’être diagnostiqués qu’après la constatation d’anomalies biologiques sur un bilan d’hémostase . Toutefois dans notre série 6 cas sur 16 ont une histoire hémorragique personnelle mais n’ont été diagnostiqués qu’au décours d’un bilan réalisé fortuitement.
Finalement, chez les patients dont la pathologie hémorragique n’est pas encore connue, la traduction initiale peut ne pas être spécifique et n’est pas donc hémorragique. Ceci est plus fréquent chez le nouveau-né, le nourrisson et chez le grand enfant ou adulte jeune ayant une affection constitutionnelle mineure. A travers certaines observations collectées au centre de traitement de l’hémophilie de l’HER, nous avons retenu quelques manifestations cliniques non spécifiques que nous résumons dans le tableau ci-dessous:

130
Patie
nt 1
Pa
tient
2
Patie
nt 3
Pa
tient
4
Patie
nt 5
Pa
tient
6
Age
du
patie
nt
5ans
6a
ns
7 m
ois
8 m
ois
15 m
ois
3 m
ois
Ant
écéd
ents
hém
orra
giqu
es
pers
onne
ls
Oui
Oui
Non
N
on
Oui
Non
Ant
écéd
ents
hém
orra
giqu
es
fam
iliau
xN
onO
uiO
ui
Non
O
uiN
on
Tabl
eau
clin
ique
initi
ale
Trou
bles
de
la
cons
cien
ce
Torti
colis
Fi
èvre
et r
efus
de
téte
r O
stéo
arth
rite
du g
enou
dro
it To
rtico
lis
Cris
es
conv
ulsi
ves
toni
cocl
oniq
ues
foca
les
Dia
gnos
tic
Hém
atom
e fr
onto
parié
tal
révé
lant
un
défic
it en
fact
eur
VII
Hém
atom
e m
édul
laire
ré
véla
nt u
ne
hém
ophi
lie A
hém
atom
e ex
tradu
ral
révé
lant
une
hé
mop
hilie
B
Hém
ophi
lie A
Hém
atom
e ép
idur
al
cerv
ical
ré
véla
nt u
ne
hém
ophi
lie A
Hém
atom
e so
us
dura
l rév
élan
t un
e hé
mop
hilie
A
Tableau 21 : Traductions cliniques non spécifiques de certains troubles de l’hémostase (cas cliniques du CHOP)

131
L’anémie hypochrome microcytaire ferriprive peut parfois cacher une affection congénitale de l’hémostase mineure (c’est le cas des thrombopathies). Des antécédents d’anémie et / ou de traitement antérieur par remplacement du fer sont souvent rapportés par les jeunes filles atteintes de troubles de la coagulation. On observe souvent une pâleur des paumes lorsque le taux d’hémoglobine est inférieur à 10 g / dL. Elle est retrouvée en cas de saignement aigu ou de saignement persistant chronique conduisant à une carence en fer qui compromet la production de globules rouges. Les réserves de fer faibles peuvent également refléter des saignements gastro-intestinaux en cours (manifestes ou occultes) qui, s’ils sont présents, doivent être examinés même s’il existe un problème connu de saignements congénitaux ou acquis. Certains troubles de la coagulation (par exemple, des troubles plaquettaires dus à des mutations de GATA1) sont associés à une anémie et une thrombocytopénie[78].

132
LIMITES DE L’ÉTUDE

133
Durant l’élaboration de notre thèse, nous avons rencontré quelques difficultés :
• Liées au caractère rétrospectif de l’étude puisque ce type d’étude repose sur la récolte de données à partir des dossiers archivés au CHOP. Ces derniers sont retrouvés parfois vides au incomplets et manquent d’informations nécessaires à l’étude : antécédents personnels ou familiaux, données de l’interrogatoire, date de naissance, …
• Liées au diagnostic étiologique des thrombopénies, en dehors de la cause auto-immune. Un nombre important de dossiers ne contient pas de diagnostic étiologique de la thrombopénie, et aucun cas d’une origine constitutionnelle n’a été retrouvé.

134
CONCLUSION
La pathologie constitutionnelle de l’hémostase reste méconnue. Le problème ne se pose généralement pas lorsque cette pathologie est diagnostiquée et connue chez la famille. La problématique se pose lorsque celle-ci doit être diagnostiquée De Novo. La traduction clinique initiale est certes la symptomatologie hémorragique, mais celle-ci peut passer inaperçue quand elle est minime ou épisodique, jusqu’à ce qu’elle se déclare dans un tableau d’hémorragie grave. La situation est plus compliquée quand le symptôme révélateur est non hémorragique, le trouble de l’hémostase est alors difficilement évoqué. Ceci dit il faut garder en tête que la pathologie constitutionnelle de l’hémostase est d’abord une histoire hémorragique et que l’interrogatoire reste l’étape fondamentale. L’objectif principal est l’identification précoce des affections hémorragiques congénitales chez l’enfants pour assurer un traitement optimal de tout événement hémorragique et pour atténuer tout risque d’hémorragie. Les saignements peuvent être graves, voire mortels, et étant donné la nature congénitale de ces troubles, les symptômes peuvent apparaître au cours de la période néonatale ou de la petite enfance. La mise au point d’un outil d’évaluation du saignement suffisamment utile pour distinguer les personnes atteintes de troubles de la coagulation de ceux qui n’ont pas de problème de saignement important pourrait améliorer l’évaluation clinique des troubles de la coagulation. Il est nécessaire de disposer d’informations supplémentaires sur les causes génétiques des troubles courants (par exemple, la maladie de von Willebrand de type 1, les troubles de la fonction plaquettaire courants tels que les défauts de sécrétion) afin de mieux comprendre la pathogenèse du trouble de la coagulation et les risques de saignement.

135
RÉSUMÉCIRCONSTANCES DE DÉCOUVERTE DES PATHOLOGIES HÉMORRAGIQUES CONSTITUTIONNELLES DE L’HÉMOSTASE CHE L’ENFANT : À PROPOS DE 63
CAS.
Auteur : Mademoiselle HAJJI Nour Al Houda
Rapporteur : Pr. EL KHORASSANI Mohamed
Mots-clés : Antécédents familiaux hémorragiques, Histoire hémorragique personnelle,
symptomatologie hémorragique, symptomatologie non spécifique,déficits en facteurs.
Les pathologies hémorragiques constitutionnelles de l’hémostase regroupent l’ensemble des
anomalies quantitatives et qualitatives des protéines plaquettaires ou plasmatiques impliquées dans
l’hémostase primaire, la coagulation et la fibrinolyse. Ce sont des pathologies génétiques rares, dont
l’hémophilie A et la maladie de Von Willebrand sont les plus fréquentes. Elles peuvent se manifester
au cours de la période néonatale ou de la petite enfance par des saignements qui peuvent être graves,
voire mortels. Dans certains cas ces troubles congénitaux restent asymptomatiques jusqu’à la grande
enfance et sont découverts fortuitement lors d’un bilan préopératoire ou devant une symptomatologie
non spécifique (non hémorragique).
Notre étude a concerné 63 cas de pathologies héréditaires de l’hémostase, tous hospitalisés au centre
de traitement de l’hémophilie et des maladies hémorragiques constitutionnelles durant la période
s’étendant du mois de Janvier 2016 à Décembre 2018. Notre objectif est d’évaluer l’histoire clinique
et les données anamnestiques prédictives de ces pathologies ainsi que les circonstances révélatrices
de ces dernières dans les données de la littérature afin de les comparer à nos résultats. L’âge médian
de nos patients était de 7 ans et 1 mois avec un sex-ratio de 0,2. Le mode de révélation dominant est
hémorragique de présentation et de gravité variable selon la sévérité et la nature de la pathologie sous-
jacente. Le bilan préopératoire d’une circoncision est une situation qui a permis de diagnostiquer un
trouble de l’hémostase chez un grand nombre de patients. La symptomatologie non spécifique est
particulière au nouveau-né et au nourrisson.
L’identification et le diagnostic précoce de ces maladies chez les enfants est un véritable challenge,
rendu difficile par les particularités physiologiques de l’hémostase chez l’enfant et la présentation
clinique atypique en particulier chez le nouveau-né et le nourrisson.

136
ABSTRACTCIRCUMSTANCES OF DISCOVERY OF INHERITED BLEEDING
DISORDERS IN PEDIATRICS: ABOUT 63 CASES.
Author: Miss HAJJI Nour Al Houda Supervisor : Pr. EL KHORASSANI Mohamed
Keywords: family history,personal haemorrhagic history, consanguinity, haemorrhagic
symptomatology, non specific symptomatology, foctor deficiency.
The inherited bleeding disorders include all the quantitative and qualitative abnormalities of platelet or plasma proteins involved in primary haemostasis, coagulation and fibrinolysis. These are rare genetic conditions, of which Haemophilia A and von Willebrand disease are the most common. They can occur during the neonatal period or early childhood with bleeding that can be serious or even fatal. In some cases, these congenital disorders remain asymptomatic until early childhood and are accidentally discovered during a preoperative assessment or in the presence of non-specific (non-haemorrhagic) symptomatology.
Our study concerned 63 cases of inherited bleeding disorders, which were hospitalized at the Hemophilia treatment center and bleeding disorders during the period from January 2016 to December 2018. Our objective is to establich the clinical history and anamnestic data that are predictive for those disorders in pediatrics, as well as the revealing circumstances of these pathologies in the data of the literature in order to compare them to our results. The median age of our patients was 7 years and 1 month with a sex ratio of 0,2. The dominant mode of revelation is haemorrhagic whose presentation and severity varies according to the severity and nature of the underlying pathology. The preoperative assessment of a circumcision is a situation that has made it possible to diagnose a haemostasis disorder in a large number of patients. Non specific symptomatology is peculiar to newborns and infants. The identification and early diagnosis of these diseases in children is a real challenge, made more difficult by the physiological specificities of the hemostasis in childhood, particularly the newborn and the infant.

137
ملخص
ظروف اكتشاف األمراض النزيفية الخلقية : دراسة حول 63 حالة.
املؤلف: اآلنسة حجي نور الهدى
إرشاف: األستاذ الخرساين محمد
كلامت البحث: : تاريخ العائلة، تاريخ النزيف الشخيص، القرابة، األعراض النزيفية، أعراض غري محددة.
تشمل األمراض النزيفية الخلقية جميع االضطرابات الكمية والنوعية لربوتينات الصفائح الدموية أو البالزما املسؤولة عن تخرث
والفيلربوند األكرث شيوًعا، والتي ميكن أن يتم اكشافها A الدم وانحالل الفيربين. هذه الحاالت الوراثية نادرة، منها مرض الهيموفيليا
خالل فرتة حديثي الوالدة أو الطفولة املبكرة مع نزيف قد يكون خطريًا أو حتى قاتل. يف بعض الحاالت، تظل هذه االضطرابات
الخلقية بدون أعراض ويتم اكتشافها عن طريق الصدفة خالل تقييم ما قبل الجراحة أو يف وجود أعراض غري نزفية
شملت دراستنا63 حالة من أمراض الدموية لتخرث الدم، والتي تم عالجها جميعها يف مصلحة عالج االموفيليا و رسطان والدم
بالرباط خالل الفرتة املمتدة من يناير 2016 إىل ديسمرب 2018. هدفنا هو تقييم تاريخ النزيف والفحص الطبي الرسيري والبيانات
التي تنبئ عن تلك االضطرابات يف طب األطفال، وكذلك الظروف التي تكشف عن هذه األمراض يف بيانات األدبيات من أجل
مقارنتها بنتائجنا. كان متوسط عمر مرضانا 7 سنوات و1 شهر مع نسبة الجنس 0,2. تم التعرف عىل هاته االمراض للمرة األوىل من
خالل اعراض نزفية يف اغلب األحيان
يعد تحديد هذه األمراض وتشخيصها مبكرًا عند األطفال تحديًا حقيقيًا وخاصًة أمام أعراض غري محددة، تزداد صعوبتها بسبب
الخصائص الفسيولوجية للتخرث الدموي عند الطفل، خاصًة عند حديث الوالدة والرضيع
.
.
.

BIBLIOGRAPHIE

139
[1] S. Dubœuf et F. Pillon, « L’hémostase, quelques notions de physiologie », Actual. Pharm., vol. 49, no 501, p. 14-15, déc. 2010.[2] L. SHERWOOD, « physiologie humaine, 2ème édition », 2ème édition., p. 321,322,323.[3] M. M. Samama, I. Elalamy, J. Conard, A. Achkar, et M.-H. Horellou, « Rappels de la Physiopathologie et de la Sémiologie Clinicobiologique », in Hémorragies et thromboses, Elsevier, 2009, p. 3-35.[4] A. Rauch et C. Paris, « Hémostase primaire », p. 12.[5] D. J. Cambus, « PHYSIOLOGIE DE L’HEMOSTASE », p. 5.[6] A. Rauch, C. Caron, S. Susen, et J. Goudemand, « Facteur von Willebrand et maladie de Willebrand : nouvelles approches », Rev. Francoph. Lab., vol. 2014, no 463, p. 53-63, juin 2014.[7] T. de Revel, « Physiologie de l’hémostase The Normal Haemostatic Process », p. 11.[8] B. Dahlbäck, « Blood coagulation », The Lancet, vol. 355, no 9215, p. 1627-1632, mai 2000.[9] E. W. Davie, K. Fujikawa, et W. Kisiel, « The coagulation cascade: initiation, maintenance, and regulation », Biochemistry, vol. 30, no 43, p. 10363-10370, oct. 1991.[10] M. Andrew et al., « Development of the human coagulation system in the full-term infant », Blood, vol. 70, no 1, p. 165-172, juill. 1987.[11] W. Hathaway et J. Corrigan, « Report of Scientific and Standardization Subcommittee on Neonatal Hemostasis: Normal coagulation Data for Fetuses and Newborn Infants », Thromb. Haemost., vol. 65, no 03, p. 323-325, mars 1991.[12] M. Andrew, P. Vegh, M. Johnston, J. Bowker, F. Ofosu, et L. Mitchell, « Maturation of the Hemostatic System During Childhood », p. 9.[13] P. Toulon et N. De Pooter, « Hémostase pédiatrique : conséquences biologiques », Rev. Francoph. Lab., vol. 2017, no 494, p. 54-59, juill. 2017.[14] M.-F. Hurtaud-Roux, A. Vincenot, et D. Lasne, « L’hémostase en pédiatrie, ses particularités, les principales pathologies hémorragiques et leur gestion », Anesth. Réanimation, vol. 4, no 4, p. 290-299, juill. 2018.[15] V. Ignjatovic et al., « Differences in the mechanism of blood clot formation and nanostructure in infants and children compared with adults », Thromb. Res., vol. 136, no 6, p. 1303-1309, déc. 2015.[16] I. M. Appel, B. Grimminck, J. Geerts, R. Stigter, M. H. Cnossen, et A. Beishuizen, « Age dependency of coagulation parameters during childhood and puberty: Pediatric reference levels of coagulation parameters », J. Thromb. Haemost., vol. 10, no 11, p. 2254-2263, nov. 2012.[17] P. Toulon et al., « Age dependency for coagulation parameters in paediatric populations: Results of a multicentre study aimed at defining the age-specific reference ranges », Thromb. Haemost., vol. 116, no 07, p. 9-16, janv. 2016.[18] R. Favier, « Hémostase pédiatrique : de la physiologie à la pathologie », EMC - Biol. Médicale, vol. 7, no 2, p. 1-14, juin 2012.[19] D. Lasne et M.-F. Hurtaud, « Particularités de l’hémostase du nouveau-né », Rev. Francoph. Lab., vol. 2019, no 508, p. 72-80, janv. 2019.[20] G. Lippi, M. Franchini, M. Montagnana, et G. Guidi, « Coagulation Testing in Pediatric Patients: The Young Are Not Just Miniature Adults », Semin. Thromb. Hemost., vol. 33, no 8, p. 816-820, nov. 2007.[21] C. Emile, « Hémostase en pédiatrie », Option/Bio, vol. 25, no 511, p. 19-20, juin 2014.[22] L. Calmette et S. Clauser, « La maladie de Willebrand », Rev. Médecine Interne, vol. 39, no 12, p. 918-924, déc. 2018.

140
[23] I. M. Nilsson, « The history of von Willebrand disease », Haemophilia, vol. 5, no s2, p. 7-11, mai 1999.[24] V. Seravalli, S. Linari, E. Peruzzi, M. Dei, E. Paladino, et V. Bruni, « Prevalence of Hemostatic Disorders in Adolescents with Abnormal Uterine Bleeding », J. Pediatr. Adolesc. Gynecol., vol. 26, no 5, p. 285-289, oct. 2013.[25] J. P. Caen, « 5 Glanzmann’s thrombasthenia », Baillières Clin. Haematol., vol. 2, no 3, p. 609-625, juill. 1989.[26] A. T. Nurden, « Glanzmann thrombasthenia », Orphanet J. Rare Dis., vol. 1, no 1, déc. 2006.[27] J. N. George, « Glanzmann’s Thrombasthenia: The Spectrum of Clinical Disease », p. 14.[28] J.-L. N. Mukendi, S. Benkirane, et A. Masrar, « Thrombasthénie de Glanzmann: à propos de 11 cas », Pan Afr. Med. J., vol. 21, 2015.[29] D. Sherer et R. Lerner, « Glanzmann’s Thrombasthenia in Pregnancy: A Case and Review of the Literature », Am. J. Perinatol., vol. 16, no 06, p. 297-301, 1999.[30] M. M. Nazih, « Pour l’Obtention du Doctorat en pharmacie », p. 106.[31] R. Karger, N. Donner-Banzhoff, H.-H. Müller, V. Kretschmer, et M. Hunink, « Diagnostic performance of the platelet function analyzer (PFA-100®) for the detection of disorders of primary haemostasis in patients with a bleeding history–a systematic review and meta-analysis », Platelets, vol. 18, no 4, p. 249-260, janv. 2007.[32] J.-Y. Muller, « Jean-Pierre Soulier (1915–2003) », Transfus. Clin. Biol., vol. 11, no 1, p. 57-64, févr. 2004.[33] A. T. NURDEN et J. P. CAEN, « Specific roles for platelet surface glycoproteins in platelet function », Nature, vol. 255, no 5511, p. 720-722, juin 1975.[34] J. A. López, R. K. Andrews, V. Afshar-Kharghan, et M. C. Berndt, « Bernard-Soulier Syndrome », Blood, vol. 91, no 12, p. 4397-4418, juin 1998.[35] P. Mhawech et A. Saleem, « Inherited Giant Platelet Disorders », Am. J. Clin. Pathol., vol. 113, no 2, p. 176-190, févr. 2000.[36] C. L. Balduini, A. Iolascon, et A. Savoia, « Inherited thrombocytopenias: from genes to therapy », Haematologica, vol. 87, no 8, p. 860-880, janv. 2002.[37] M. Franchini, « The platelet-function analyzer (PFA-100 ® ) for evaluating primary hemostasis », Hematology, vol. 10, no 3, p. 177-181, juin 2005.[38] A. du P. Heyns, P. N. Badenhorst, P. Wessels, H. Pieters, et M. G. Lötter, « Kinetics, in vivo redistribution and sites of sequestration of indium-111-labelled platelets in giant platelet syndromes», Br. J. Haematol., vol. 60, no 2, p. 323-330, juin 1985.[39] R. E. Scharf, R. McMillan, Z. M. Ruggeri, et L. A. Harkers, « Bernard-Soulier syndrome: quantitative characterization of megakaryocytes and platelets by flow cytometric and platelet kinetic measurements », Eur. J. Haematol., vol. 52, no 4, p. 193-200, avr. 2009.[40] S. J. Israels, M. El-Ekiaby, T. Quiroga, et D. Mezzano, « Inherited disorders of platelet function and challenges to diagnosis of mucocutaneous bleeding: INHERITED DISORDERS OF PLATELET FUNCTION », Haemophilia, vol. 16, p. 152-159, juin 2010.[41] V. Baccini et M. C. Alessi, « Les thrombopénies constitutionnelles : démarche diagnostique», Rev. Médecine Interne, vol. 37, no 2, p. 117-126, févr. 2016.[42] G. Leverger, A. Petit, S. Fasola, J. Landman-Parker, et R. Favier, « Les thrombopénies génétiques », Arch. Pédiatrie, vol. 17, no 8, p. 1185-1191, août 2010.[43] H. Boutroux, M.-D. Tabone, H. Lapillonne, P. Ballerini, R. Favier, et G. Leverger, « Les thrombopénies constitutionnelles. De la clinique aux actualités génétiques », Rev. Oncol. Hématologie

141
Pédiatrique, vol. 1, no 2, p. 89-97, oct. 2013.[44] N. Schleinitz et al., « Le syndrome MYH9 : à propos d’une nouvelle observation et de la mise en évidence d’une nouvelle mutation du gène MYH9 », Rev. Médecine Interne, vol. 27, no 10, p. 783-786, oct. 2006.[45] G. Biino et al., « Age- And Sex-Related Variations in Platelet Count in Italy: A Proposal of Reference Ranges Based on 40987 Subjects’ Data », PLoS ONE, vol. 8, no 1, p. e54289, janv. 2013.[46] C. Pondarre, « Thrombopénies constitutionnelles: orientation diagnostique », Arch. Pédiatrie, vol. 14, no 6, p. 676-678, juin 2007.[47] J.-F. Viallard, « Ces purpuras trombopéniques idiopathiques qui n’en sont pas : quand penser à une thrombopénie constitutionnelle ? », Rev. Médecine Interne, vol. 28, p. S312-S314, déc. 2007.[48] G. I. C. Ingram, « The history of haemophilia », p. 11.[49] R. Stevens, « THE HISTORY OF HAEMOPHILIA IN THE ROYAL FAMILIES OF EUROPE. », Br. J. Haematol., vol. 105, no 1, p. 25-32, avr. 1999.[50] M. Napolitano, « 3 - Hemophilia A and Hemophilia B », p. 21.[51] F. Peyvandi, S. Duga, S. Akhavan, et P. M. Mannucci, « Rare coagulation deficiencies », Haemoph. Off. J. World Fed. Hemoph., vol. 8, no 3, p. 308-321, mai 2002.[52] C. Guérois, « L’hémophilie aujourd’hui », Kinésithérapie Rev., vol. 9, no 88, p. 32-36, avr. 2009.[53] A. MASRAR et N. B. AGOUMI, « ASPECTS BIOLOGIQUES ET MOLÉCULAIRES DE L’HÉMOPHILIE A », Maroc Méd., vol. 27, no 3, mai 2013.[54] F. PEYVANDI et M. SPREAFICO, « National and international registries of rare bleeding disorders », Blood Transfus., no 6, p. s45–s48, 2008.[55] P. H. B. Bolton-Maggs, « The rare inherited coagulation disorders », Pediatr. Blood Cancer, vol. 60, no S1, p. S37-S40, 2013.[56] M. G. Blaizot et al., « Inherited factor VII deficiency and surgery: clinical data are the best criteria to predict the risk of bleeding », Br. J. Haematol., vol. 117, no 1, p. 172-175, avr. 2002.[57] L. Souabni, N. Meddeb, H. Ajlani, N. B. Romdhane, et S. Sellami, « Hémarthrose révélatrice d’une maladie de Rosenthal (déficit en facteurXI) », Rev. Rhum., vol. 75, no 5, p. 486-487, mai 2008.[58] H. Peretz et al., « The Two Common Mutations Causing Factor XI Deficiency in Jews Stem From Distinct Founders: One of Ancient Middle Eastern Origin and Another of More Recent European Origin », Blood, vol. 90, no 7, p. 2654-2659, oct. 1997.[59] Y. E. Boussaadni, N. Benajiba, A. E. Ouali, R. Amrani, et M. Rkain, « Afibrinogénémie congénitale : à propos d’une nouvelle observation », Arch. Pédiatrie, vol. 22, no 1, p. 50-52, janv. 2015.[60] N. Aoki, « Discovery of α2-plasmin inhibitor and its congenital deficiency: α2-Plasmin inhibitor and its congenital deficiency », J. Thromb. Haemost., vol. 3, no 4, p. 623-631, nov. 2004.[61] M. Hanss, « Anomalies constitutionnelles de la fibrinolyse et syndromes hémorragiques », Rev. Francoph. Lab., vol. 2012, no 443, p. 39-45, juin 2012.[62] F. Leebeek, J. Stibbe, E. Knot, C. Kluft, M. Gomes, et M. Beudeker, « Mild Haemostatic Problems Associated with Congenital Heterozygous α2-Antiplasmin Deficiency », Thromb. Haemost., vol. 59, no 01, p. 096-100, févr. 1988.[63] G. C. Griffin, E. F. Mammen, R. J. Sokol, A. L. Perrotta, A. Stoyanovich, et C. F. Abildgaard, « Alpha 2-antiplasmin deficiency. An overlooked cause of hemorrhage », Am. J. Pediatr. Hematol. Oncol., vol. 15, no 3, p. 328-330, août 1993.[64] A.-C. Teger-Nilsson, P. Friberger, et E. Gyzander, « Determination of a new rapid plasmin

142
inhibitor in human blood by means of a plasmin specific tripeptide substrate », Scand. J. Clin. Lab. Invest., vol. 42, no sup162, p. 403-409, janv. 1982.[65] K. Koie, K. Ogata, T. Kamiya, J. Takamatsu, et M. Kohakura, « α2-PLASMIN-INHIBITOR DEFICIENCY (MIYASATO DISEASE) », The Lancet, vol. 312, no 8104, p. 1334-1336, déc. 1978.[66] R. Mehta et A. D. Shapiro, « Plasminogen activator inhibitor type 1 deficiency: PAI-1 DEFICIENCY », Haemophilia, vol. 14, no 6, p. 1255-1260, oct. 2008.[67] B. Nosek-Cenkowska, M. Cheang, N. Pizzi, E. Israels, et J. Gerrard, « Bleeding/Bruising Symptomatology in Children with and without Bleeding Disorders », Thromb. Haemost., vol. 65, no 03, p. 237-241, mars 1991.[68] S. Maguire, M. K. Mann, J. Sibert, et A. Kemp, « Are there patterns of bruising in childhood which are diagnostic or suggestive of abuse? A systematic review », Arch. Dis. Child., vol. 90, no 2, p. 182-186, févr. 2005.[69] P. E. Warner, H. O. D. Critchley, M. A. Lumsden, M. Campbell-Brown, A. Douglas, et G. D. Murray, « Menorrhagia I: measured blood loss, clinical features, and outcome in women with heavy periods: a survey with follow-up data », Am. J. Obstet. Gynecol., vol. 190, no 5, p. 1216-1223, mai 2004.[70] P. Krishna et D. Lee, « Post-Tonsillectomy Bleeding: A Meta-Analysis »:, The Laryngoscope, vol. 111, no 8, p. 1358-1361, août 2001.[71] S. S. Acharya, « Rare Bleeding Disorders in Children: Identification and Primary Care Management », PEDIATRICS, vol. 132, no 5, p. 882-892, nov. 2013.[72] J. D. Anderst, S. L. Carpenter, T. C. Abshire, et the S. O. H. and C. O. C. A. A. Neglect, « Evaluation for Bleeding Disorders in Suspected Child Abuse », Pediatrics, vol. 131, no 4, p. e1314-e1322, avr. 2013.[73] M. Medhaffar et al., « Déficit congénital en facteur XIII de la coagulation dans le sud tunisien», Pathol. Biol., vol. 54, no 6, p. 349-352, juill. 2006.[74] E. A Chalmers, « Haemophilia and the newborn », Blood Rev., vol. 18, no 2, p. 85-92, juin 2004.[75] A. El koraichi, B. Armel, T. Nebhani, Y. Machkour, M. El Haddoury, et S. E. El Kettani, « Hémorragie intracérébrale spontanée secondaire à un déficit congénital en facteur XIII », Ann. Fr. Anesth. Réanimation, vol. 31, no 5, p. 475-477, mai 2012.[76] L. Sfaihi Ben Mansour et al., « Déficit congénital en facteur VII de la coagulation, révélé par une hémorragie cérébrale », Arch. Pédiatrie, vol. 16, no 7, p. 1024-1027, juill. 2009.[77] J. L. Liu, J. E. Michaud, W. W. Ludwig, et J. P. Gearhart, « Diagnosis of hemophilia in newborn circumcision: A case presentation », Urol. Case Rep., vol. 23, p. 101-102, mars 2019.[78] C. P. M. Hayward, « Clinical Approach to the Patient With Bleeding or Bruising », in Hematology, Elsevier, 2018, p. 1912-1921.[79] R. Bhat et W. Cabey, « Evaluation and Management of Congenital Bleeding Disorders », Hematol. Oncol. Clin. North Am., vol. 31, no 6, p. 1105-1122, déc. 2017.[80] S. M. Jobe, « Glanzmann Thrombasthenia », in Transfusion Medicine and Hemostasis, Elsevier, 2019, p. 599-600.[81] E. M. de Wee et al., « Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease », Thromb. Haemost., vol. 108, no 10, p. 683-692, 2012.[82] G. Toogeh, M. Keyhani, R. Shari, A. Emami, et H. Dalili, « A Study of Bernard-Soulier Syndrome in Tehran, Iran », p. 3.[83] D. L. Brown, « Congenital bleeding disorders », Curr. Probl. Pediatr. Adolesc. Health Care,

143
vol. 35, no 2, p. 38-62, févr. 2005.[84] V. Kiley, J. J. Stuart, et C. A. Johnson, « Coagulation studies in children with isolated recurrent epistaxis », J. Pediatr., vol. 100, no 4, p. 579-581, avr. 1982.[85] C. Sandoval, S. Dong, P. Visintainer, M. F. Ozkaynak, et S. Jayabose, « Clinical and Laboratory Features of 178 Children With Recurrent Epistaxis », J. Pediatr. Hematol. Oncol., vol. 24, no 1, p. 47, janv. 2002.[86] A. Chemaou, M. Ayachi, O. Benjelloun, et A. Zineddine, « Ménorragies par déficit congénital en facteur V chez une adolescente », Arch. Pédiatrie, vol. 20, no 1, p. 33-36, janv. 2013.[87] A. C. W. Lee et C. H. Li, « Bullous hematoma of the palm: an unusual complication of scabies in a child with congenital prothrombin deficiency », Pediatr. Dermatol., vol. 19, no 6, p. 567-568, nov. 2002.[88] G. Slem et M. Kumi, « Bloody tears due to congenital factor VII deficiency », Ann. Ophthalmol., vol. 10, no 5, p. 593-594, avr. 1978.[89] M. Kuwahara, S. Yurugi, M. Takeda, K. Fukuda, et A. Yoshioka, « Hemophilia B Diagnosed by Hematoma at the Columella Base »:, Plast. Reconstr. Surg., vol. 117, no 5, p. 1647-1648, avr. 2006.[90] J. T. Wilde, C. A. Lee, P. Collins, P. L. F. Giangrande, M. Winter, et C. R. Shiach, « Increased bleeding associated with protease inhibitor therapy in HIV-positive patients with bleeding disorders», Br. J. Haematol., vol. 107, no 3, p. 556-559, déc. 1999.[91] A. Girolami, S. Vettore, R. Scandellari, E. Allemand, et B. Girolami, « About the rarity of haemoptysis in congenital bleeding disorders. A report of five cases », Haemophilia, vol. 15, no 3, p. 825-827, mai 2009.[92] K. Dafopoulos et al., « Two Episodes of Hemoperitoneum from Luteal Cysts Rupture in a Patient with Congenital Factor X Deficiency », Gynecol. Obstet. Invest., vol. 55, no 2, p. 114-115, 2003.[93] A. Girolami, E. Allemand, F. Tezza, D. Pellati, et R. Scandellari, « Rectus muscle sheath haematoma in a patient with congenital FX deficiency and in another with congenital FVII deficiency», Haemophilia, vol. 16, no 1, p. 182-185, janv. 2010.[94] A. Biri, M. Kurdoglu, Z. Kurdoglu, S. Gultekin, et T. Gursel, « Acute Abdominal Pain Caused by a Spontaneous Intramyometrial Hematoma in Type III von Willebrand Disease »:, Pediatr. Emerg. Care, vol. 22, no 9, p. 650-652, sept. 2006.[95] Gomez, Lucia, Perella, et Aguilar, « Heamoperitoneum caused by heamorrhagic corpus luteum in a patient with type 3 von Willebrand’s disease », Haemophilia, vol. 4, no 1, p. 60-62, janv. 1998.[96] S. Gallet, V. Tran Minh, D. Louis, J. B. Cotton, J. C. Berthier, et E. Hartemann, « [Massive hemoperitoneum caused by rupture of the spleen, a complication of congenital afibrinogenemia. Conservative treatment] », Pediatrie, vol. 40, no 5, p. 385-391, août 1985.[97] N. E. Terry et W. C. Boswell, « Nonoperative management of delayed splenic rupture in a patient with hemophilia B », J. Pediatr. Surg., vol. 41, no 9, p. 1607-1609, sept. 2006.[98] C. Balkan, K. Kavakli, et D. Karapinar, « Spinal epidural haematoma in a patient with haemophilia B », Haemophilia, vol. 12, no 4, p. 437-440, juill. 2006.[99] M. Hamilton, W. French, N. Rhymes, et P. Collins, « Liver haemorrhage in haemophilia - a case report and review of the literature », Haemophilia, vol. 12, no 4, p. 441-443, juill. 2006.[100] N. Gupta, V. Dadhwal, D. Deka, S. K. Jain, et S. Mittal, « Corpus luteum hemorrhage: Rare complication of congenital and acquired coagulation abnormalities », J. Obstet. Gynaecol. Res., vol.

144
33, no 3, p. 376-380, juin 2007.[101] J. Jarry, D. Biscay, D. Lepront, A. Rullier, et D. Midy, « Spontaneous intramural haematoma of the sigmoid colon causing acute intestinal obstruction in a haemophiliac: report of a case », Haemophilia, vol. 14, no 2, p. 383-384, mars 2008.[102] C. Lejus-Bourdeau, N. Grillot, et M. Azama, « Bilan préopératoire en pédiatrie : pour qui et pourquoi ? », Anesth. Réanimation, vol. 4, no 4, p. 282-289, juill. 2018.[103] A. Harroche et C. Rothschild, « Diagnostic d’un syndrome hémorragique de l’enfant », J. Pédiatrie Puériculture, vol. 32, no 1, p. 1-11, févr. 2019.[104] B. Delahousse, « xploration de l’hmostasechezunpatient avec ant@c@dentshmorragiques », p. 8, 1995.[105] F. Rodeghiero et al., « ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders: ISTH/SSC bleeding assessment tool », J. Thromb. Haemost., vol. 8, no 9, p. 2063-2065, sept. 2010.[106] « Groupe d’etude sur l’hemostase et la thrombose – Un site utilisant WordPress ». .[107] B. Polack, J.-F. Schved, et B. Boneu, « Preanalytical Recommendations of the ‘Groupe d’Etude sur l’Hémostase et la Thrombose’ (GEHT) for Venous Blood Testing in Hemostasis Laboratories », Pathophysiol. Haemost. Thromb., vol. 31, no 1, p. 61-68, 2001.[108] D. Lasne, C. Le Roux, et C. Lejus, « L’hémostase chez le nouveau-né : quel bilan pratiquer ?», Ann. Fr. Anesth. Réanimation, vol. 29, no 7-8, p. 560-562, juill. 2010.[109] V. Ignjatovic et al., « Age-Related Differences in Plasma Proteins: How Plasma Proteins Change from Neonates to Adults », PLoS ONE, vol. 6, no 2, p. e17213, févr. 2011.[110] M. Berruyer, « Praticabilité des tests d’hémostase en pédiatrie: limites et nécessitésa », Rev. Francoph. Lab., vol. 2012, no 443, p. 47-53, juin 2012.[111] D. Lasne, « Hémostase chez le nouveau-né : ce que le biologiste peut répondre », Arch. Pédiatrie, vol. 17, no 6, p. 864-865, juin 2010.[112] M.-H. Horellou, J. Conard, et M. Samama, « Allongement du temps de saignement », EMC - Traité Médecine AKOS, vol. 1, no 1, p. 1-3, janv. 2006.[113] B. Bader-Meunier et M. Dreyfus, « Exploration des troubles de l’hémostase de l’enfant (en dehors de la période néonatale) », Arch. Pédiatrie, vol. 6, no 10, p. 1086-1091, oct. 1999.[114] G. Lippi, M. Franchini, et G. C. Guidi, « Diagnostic approach to inherited bleeding disorders», Clin. Chem. Lab. Med., vol. 45, no 1, janv. 2007.[115] A. B. Federici, « Diagnosis of Inherited von Willebrand Disease: A Clinical Perspective », Semin. Thromb. Hemost., vol. 32, no 6, p. 555-565, sept. 2006.[116] H. Chambost et F. Suzan, « Épidémiologie des maladies hémorragiques constitutionnelles : apport de la cohorte nationale », Arch. Pédiatrie, vol. 17, no 6, p. 618-619, juin 2010.[117] H. Mansouritorghabeh et L. Manavifar, « An investigation of the spectrum of common and rare inherited coagulation disorders in North-Eastern Iran », Blood Transfus., 2013.[118] A. C. Mauer et al., « Impact of sex, age, race, ethnicity and aspirin use on bleeding symptoms in healthy adults: Bleeding symptoms in healthy individuals », J. Thromb. Haemost., vol. 9, no 1, p. 100-108, janv. 2011.[119] G. M. Mokhtar, A. A. G. Tantawy, A. A. M. Adly, M. A. S. Telbany, S. E. E. Arab, et M. Ismail, « A longitudinal prospective study of bleeding diathesis in Egyptian pediatric patients: single-center experience », Blood Coagul. Fibrinolysis, vol. 23, no 5, p. 411-418, juill. 2012.[120] J. E. Sadler et al., « Impact, Diagnosis and Treatment of von Willebrand Disease », Thromb. Haemost., vol. 84, no 8, p. 160-174, 2000.

145
[121] Y. Jayasinghe, P. Moore, S. Donath, J. Campbell, P. Monagle, et S. Grover, « Bleeding disorders in teenagers presenting with menorrhagia », Aust. N. Z. J. Obstet. Gynaecol., vol. 45, no 5, p. 439-443, oct. 2005.[122] P. M. Mannucci, « Recessively inherited coagulation disorders », Blood, vol. 104, no 5, p. 1243-1252, sept. 2004.[123] F. Peyvandi et al., « Genetic diagnosis of haemophilia and other inherited bleeding disorders», Haemophilia, vol. 12, no s3, p. 82-89, juill. 2006.[124] E. A. EL-Bostany, N. Omer, E. E. Salama, E. A. El-Ghoroury, et S. K. Al-Jaouni, « The spectrum of inherited bleeding disorders in pediatrics »:, Blood Coagul. Fibrinolysis, vol. 19, no 8, p. 771-775, déc. 2008.[125] N. Zmouli, « Découverte fortuite d’une hémophilie A mineure lors d’une circoncision », Pharm. Hosp. Clin., vol. 50, no 3, p. 350-353, sept. 2015.[126] H. Pollmann, H. Richter, H. Ringkamp, et H. Jürgens, « When are children diagnosed as having severe haemophilia and when do they start to bleed? A 10-year single-centre PUP study », Eur. J. Pediatr., vol. 158, no 3, p. S166-S170, déc. 1999.[127] K. Khair et R. Liesner, « Bruising and bleeding in infants and children - a practical approach», Br. J. Haematol., vol. 133, no 3, p. 221-231, mai 2006.[128] F. Peyvandi et al., « Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders », J. Thromb. Haemost., vol. 10, no 4, p. 615-621, 2012.[129] W. L. Nichols et al., « von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA) <sup/> », Haemophilia, vol. 14, no 2, p. 171-232, mars 2008.[130] Kadir, Sabin, Pollard, Lee, et Economides, « Quality of life during menstruation in patients with inherited bleeding disorders », Haemophilia, vol. 4, no 6, p. 836-841, nov. 1998.[131] C. A. Lee, « Women and inherited bleeding disorders: menstrual issues », Semin. Hematol., vol. 36, no 3 Suppl 4, p. 21-27, juill. 1999.[132] F. Peyvandi, I. Garagiola, et M. Menegatti, « Gynecological and obstetrical manifestations of inherited bleeding disorders in women: Women with inherited bleeding disorders », J. Thromb. Haemost., vol. 9, p. 236-245, juill. 2011.[133] N. Rydz et P. D. James, « The evolution and value of bleeding assessment tools: The evolution and value of bleeding assessment tools », J. Thromb. Haemost., vol. 10, no 11, p. 2223-2229, nov. 2012.

SERMENT D’HIPPOCRATE
Au moment d’être admis à devenir membre de la profession médicale, je m’engage solennellement à consacrer ma vie au service de l’humanité.
Je traiterai mes maîtres avec le respect et la reconnaissance qui leur sont dus.
Je pratiquerai ma profession avec conscience et dignité. La santé de mes malades sera mon premier but.
Je ne trahirai pas les secrets qui me seront confiés.Je maintiendrai par tous les moyens en mon pouvoir l’honneur et les nobles
traditions de la profession médicale.Les médecins seront mes frères.
Aucune considération de religion, de nationalité, de race, aucune considération politique et sociale ne s’interposera entre mon devoir et mon patient.
Je maintiendrai le respect de la vie humaine dés la conception.Même sous la menace, je n’userai pas de mes connaissances médicales d’une façon
contraire aux lois de l’humanité.Je m’y engage librement et sur mon honneur.

ط ا برق أ قسم

اململكة املغربية
جامعة محمد الخامس بالرباط
كلية الطب والصيدلة الرباط
ظروف اكتشاف األمراضالنزيفية الخلقيةدراسة حول 63 حالة
سنة 2019
قدمت ونوقشت علنيا يوم من طرف
الطب يف دكتور ة هشاد لنيل
../06/2019
251993المزدادة في فبراير بالرباط
كلامت البحث: تاريخ العائلة، تاريخ النزف الشخيص، القرابة، األعراض النزفية، أعراض غري محددة
اآلنسة حجي نور الهدى
السيدة: بنجلون الدخامة بدر سعود
السيدة: بلعيايش جهان
السيدة: كربويب ملياء
السيد : مرسار عز العرب
السيد: محمد الخرساين
أستاذة يف طب األطفال
أستاذة يف اإلنعاش
أستاذ يف علم األحياء الجزيئي و الكيمياء الحيوية
الرئيسة
املرشف
األعضاء
أستاذ يف طب األطفال
أستاذ يف طب األطفال
أعضاء اللجنة
أرطوحة
أطروحة رقم 2019/300




















