
DFT and TD-DFT Assessment of the Structural and Optoelectronic Properties of an Organic-Ag14...
35
published on J. Phys. Chem. A, Just Accepted Manuscript, DOI: 10.1021/jp507679f Publication Date (Web): September 23, 2014 Copyright 2014 American Chemical Society 1
Transcript of DFT and TD-DFT Assessment of the Structural and Optoelectronic Properties of an Organic-Ag14...

published on J. Phys. Chem. A, Just Accepted Manuscript, DOI: 10.1021/jp507679fPublication Date (Web): September 23, 2014Copyright 2014 American Chemical Society
1

DRAFT OF: “DFT and TD-DFT Assessment of
the structural and optoelectronic Properties of
Organic-Ag14 Nanocluster”
Francesco Muniz-Miranda, Maria Cristina Menziani,∗ and Alfonso Pedone
University of Modena and Reggio Emilia (UniMoRE), Department of Chemical and
Geological Sciences (DSCG), Via G. Campi 183, I-41125, Modena, Italy
E-mail: [email protected]
∗To whom correspondence should be addressed
2

Abstract
An extensive benchmark of exchange-correlation functionals on the structure of
the X-ray resolved phosphine and thiolate-protected Ag14-based nanocluster, named
XMC1, is reported. Calculations were performed both on simplified model systems,
with the complexity of the ligands greatly reduced, and on the complete XMC1 parti-
cle. Most of the density functionals that yielded good relaxed structures on analogous
calculations on gold nanoclusters (viz. those employing the generalized gradient ap-
proximation) significantly deform the structure of XMC1. On the contrary, some of
the exchange-correlation functionals including part of the exact Hartree-Fock exchange
(hybrid functionals) reproduce the experimental geometry with minimal errors. In par-
ticular, the widely adopted B3LYP yields fairly accurate structures for XMC1, while
it is outperformed by many other functionals (both hybrids and generalized gradient
corrected) in similar calculations on analogous gold-based systems. Time-dependent
density functional calculations have been employed to recover the experimental UV-Vis
spectrum. The present investigation shows that to correctly reproduce the optical fea-
ture of XMC1 the ligands cannot be omitted, since they interact with the metal core at
energies much closer to the optical gap than in the case of gold-based nanoclusters of
similar size. Due to this fact, a functional that accurately describes charge-transfer elec-
tronic transitions (such as the long-range corrected CAM-B3LYP) has to be adopted.
Keywords
TD-DFT, Ag, Nanoparticle, UV-Vis, Benchmark
Introduction
Nanoparticles with cores made up of noble metals atoms do not show metallic properties
when the size of the particles approach the single-digit nm, displaying finite and appreciable
band gaps (or, more correctly, HOMO-LUMO gaps).1 In particular, this is found in gold and
3

silver-based nanoparticles, whose HOMO-LUMO gaps increase with the reduction in size,
reaching values between about 1 and 2 eV at the sub-nanometer scale.2
Computational approaches can be used to elucidate the relationship between sizes, shapes,
and optoelectronic properties. Calculations based on density functional theory (DFT) are
nowadays one of the preferred approaches to achieve this understanding, because they of-
ten represent by far the best compromise between accuracy and feasibility of the computa-
tions.3–6 Furthermore, the time-dependent (TD) extension7 of DFT also allows the investiga-
tion of the electronic excited states (Sn , n ≥1), thus enabling the prediction and elucidation
of optical spectra of the target systems.6,8–12
While many studies probed nanosized gold particles by DFT means (e.g. Ref. 2,9,13–15),
only very recently systematic investigations of the various exchange-correlation functionals
and pseudopotentials on real nanogold experimental structures started appearing in the
literature.12,16 Yet, such studies still lack for nanosized silver, also because only recently
Ag-based clusters were resolved by means of X-ray diffraction.17,18
In this work, we investigate the Ag-based nanocluster of molecular formula Ag14(SC6H3F2)6
(SC6H2F3)6 (PPh3)8, denoted as “XMC1” in Ref. 17. This nanocluster, protected by both
aromatic thiols and phosphines, is of interest due to at least three peculiar features. First, its
crystallographic structure determined through X-ray diffraction shows the lack of so-called
“sulfur-staples”, i.e. motifs M−S−M (M being a noble metal atom), which are quite com-
mon in Au-based nanoclusters (NCs) capped by thiols.19–23 In fact, differently to gold NCs,
in this Ag14-based particle sulfur atoms are bound not to two, but to three metal atoms, as
shown in Figure 1. At the same time, Ag atoms bordering the ligands (eight metal atoms,
colored in blue in Fig.1, left) are each bound to four non-metal atoms (three S and one P).
The octahedral Ag6 inner core of XMC1 (colored in red in Fig.1, left) is another structural
feature that has also been observed18 or theoretically expected24 on other silver-based parti-
cles, but not in gold-based NCs of similar size, as for example the Au38-based particle whose
core is shown in Figure 1 (right).22 Moreover, its optical properties are characterized by a
4

Figure 1: (Left) structure of the core of XMC1; (right) structure of the core of an Au38-based nanocluster.22
Metal atoms (either Ag or Au) are colored in red or blue depending on whether they belong to the inner orouter region of the core, respectively. S and P atoms are colored in yellow and orange, respectively. Agcore−Sbonds are colored in violet. There are no Aucore−S bonds. Bonds between central metal atoms and outermetal atoms are not pictured for better clarity.
clearly structured optical absorption and emission profile, the latter giving rise to yellow
luminescence.
In order to check the reliability of current functionals to reproduce the structural features
of XMC1, ground-state DFT calculations have been carried out. Furthermore, more limited
TD-DFT calculations have been employed to recover and elucidate the optical features of
XMC1, in particular the origins of its complex UV-Vis spectrum.
The paper is organized as in the following. Details regarding the DFT calculations per-
formed on the simplified and complete model systems of XMC1 are reported in Methods and
Computational Details. Findings are presented and commented on in Results and Discussion,
with particular regard to the similarities and differences between XMC1 and previous calcu-
lations on gold nanoclusters, while the Concluding Remarks section contains final comments
and observations.
Methods and Computational Details
To choose the optimal quantum-chemistry approach to simulate XMC1, we tested several
exchange-correlation (XC) functionals, pseudopotentials (PPs), and basis set (BSs) on a
model system (XMC1core) composed by the metal Ag14-core and the simplified molecular
5

groups directly bonded to them (i.e. PH3 and SH groups in place of aromatic phosphines
and thiols, respectively). Then, more limited calculations were performed to simulate the
complete XMC1 structure. TD-DFT spectra were computed on XMC1core and a model
including part of the ligands (XMC1core+Ls). Models XMC1core and XMC1core+Ls
are pictured in the upper-central and upper-right corners of Fig. 2, respectively, along with
the complete XMC1 and the PPh3 and thiophenol ligands.
Figure 2: Three-dimensional structure of XMC1, the core of XMC1 with ligands (XMC1core+Ls), thecore of XMC1 (XMC1core), fluorurated-thiophenol (SAr), and triphenylphosphine (PPh3). StandardCPK color scheme is adopted: H atoms are white, Ag atoms are light gray, C atoms are dark gray, S atomsare yellow, P atoms are orange, F atoms are green.
All DFT calculations presented here have been carried out using the Gaussian09 suite
of programs,25 and consist of structural optimizations, single-point calculations, and time-
dependent calculations to obtain excitation energies. The ground-state (GS) calculations
have been performed adopting “tight” convergence criteria for the optimization of geometries
(corresponding to forces and atomic displacements below the 10−5 Hartree/Bohr and 4·10−5
Bohr thresholds, respectively). The experimental X-ray crystal structure of XMC117 has
been used as starting configuration for the geometry optimizations. The accuracy of the
structural relaxations has been monitored by calculating the atom-averaged absolute value
6

of the difference between Ag-Ag distances of the initial (experimental) and final (relaxed)
geometries, defined as 〈δ〉 = 〈|Rijexp−R
ijopt|〉, where Rij represents the distance between i and
j silver atoms.
We have benchmarked many combination of basis-sets and pseudopotentials (BS/PPs)
to correctly reproduce the core-valence interaction of silver electrons. We imported in the
calculations “families” of BS/PPs (LANL2, def2, SDDECP (as defined in Ref. 26), and others
that are available for silver through the Basis Set Exchange27 website. The benchmarks
consist of structural optimizations on a simple model made of two bonded Ag atoms, in
analogy to previous analysis.28 Table 1 lists the computed equilibrium distances (req) of Ag2
and their deviation (∆) from the experimental value.28,29 While the def2-TZVP combination
Table 1: Ag−Ag bond length computed with various combined BS/PPs. All calculationswere performed adopting the B3LYP40,41 XC functional. ∆ is the deviation with respect toexperimental data reported in Ref. 28.
req / A ∆ / ALANL1-DZ30 2.715 0.185LANL2-DZ31 2.611 0.081
LANL2-DZ+p31,32 2.607 0.077LANL2-TZ33 2.610 0.080
LANL2-TZ+f 33,34 2.608 0.078mWB6035 2.597 0.067
CRENBS36 2.934 0.404DGDZVP37 2.695 0.165
def2-TZVP38,39 2.596 0.066def2-TZVPPD38,39 2.589 0.059def2-QZVPPD38,39 2.582 0.052aug-cc-pV5Z-PP28 2.585 0.055
exp28,29 2.530 −
does not yield the absolute best bond lengths (def2-QZVPPD does), it represents the best
compromise between accuracy and computational burden. In fact, def2-QZVPPD and aug-
cc-pV5Z-PP are much more computational expensive. Thus, the def2 PP and the TZVP BS
have been employed in all the calculations reported in this paper to take into account core
and valence electrons of Ag atoms, respectively.
The environment given by the CH2Cl2 solvent has been accounted in all calculations by
linear response polarizable continuum model.42,43 The UV-Vis experimental spectra were
7

recorded in CH2Cl2 and the latter also co-crystallized along XMC1.17
Calculations on the Bare Metal Cluster
The selection of the best computational scheme to reproduce the relevant geometric fea-
tures of the NC has been based on calculations carried out on a reduced model particle
(XMC1core) in order to make the computations feasible and save computer time. Similar
simplifying choices have already been successfully employed on noble-metal NCs.2,9,14,16,21,23,44
DFT geometry optimizations and single point calculations have been performed adopting
a number of XC functionals, which could be sorted in at least three large families:
simple GGA or meta-GGA functionals with 0% Hartree-Fock exchange, such as BLYP,41,45
PBE,46 TPSS,47 rev-TPSS,48,49 TPSS-LYP,41,47 PBE-LYP,41,46 B-PBE,45,46 B-PW91,45,50
B-P86,45,51 VSXC,52 HCTH,53 τHCTH,54 B97D,55 MN12L;56
so called “global” hybrids with a fixed amount of Hartree -Fock exchange, like B3LYP,40,41
X3LYP,41,57 O3LYP41,58 B3P86,40,51 B3PW91,40,50 B1LYP,59 BHandH-LYP (as de-
fined in Ref. 26), PBE0,60 M05,61 M06,62 M06HF,63 mPW1-PW91,64 TPSSh,65 τHCTH-
hyb,66 SOGGA11x;67
range-separated/long-range-corrected hybrids with an Hartree-Fock exchange contribution
that changes with the interelectronic distance, namely HSE06,68 CAM-B3LYP,69 LC-
BLYP,41,45,70 LC-PBE,46,70 LC-TPSS,47,70 M11,71 N12sx,72 MN12sx.72
All the non-metal atoms of XMC1core have been simulated adopting a Gaussian 6-
311G basis set, with the exception of the S and P atoms which, being hypervalent, have
been treated with the larger 6-311G(d,p) basis set. No geometrical constraint has been
imposed on atoms during these geometry relaxations.
8

Calculations on the Complete Cluster
Both constrained and non-constrained optimizations have been performed on the full XMC1
particle. When constrains have been applied, we fixed the atomic positions of C, H, and F
atoms, i.e. the atoms belonging to the outer ligands; all P and S atoms were always let free
to relax, as well as the metal atoms.
Due to the fact that XMC1 has more than 430 atoms, the level of theory had to be
adjusted to make the computations viable. A full-electron 6-311G(d,p) BS has been employed
to describe non-metal atoms directly bonded to the metal core (sulfur and phosphorous
atoms), while the other non-metal elements have been treated with simpler STO-3G or
6-31G BSs.
As shown in Ref. 16, the DFT-optimized structure of isolated triphenyl-phosphine shows
that with 6-31G(H,C atoms)/6-311G(d,p)(P atom) and STO-3G(H,C atoms)/6-311G(d,p)(P
atom) BSs the bond-distance error with respect to optimizations carried out with 6-311++
G(d,p) basis set is at max. of order to ∼10−2A; also bond angles change of maximum 1◦ and
dihedral angles of at most 2◦.
Four exchange-correlation functionals (BPBE,45,46 B3LYP,40,41 M06HF,63 and CAM-
B3LYP69) belonging to different families have been tested to simulate complete XMC1,
each one with a different contribution of exact Hartree-Fock exchange (from 0% of BPBE to
100% of M06HF).
Electronic Spectra
The electronic spectra have been investigated computing the first 200 S0 7→Sn optical tran-
sition (n ≤200) at the TD-BLYP, TD-B3LYP, and TD-CAM-B3LYP levels of theory. These
three XC functionals share the same correlation expression41 and part of the same exchange
functional,45 thus they can be viewed as belonging to the same “family” but at very different
level of complexity and sophistication. CAM-B3LYP is known to perform particularly well
in reproducing charge-transfer optical transitions, as those that can occur between the silver
9

core and its aromatic ligands.6,69,73 It was also the reference choice to study the electronic
spectra of undecagold-based NCs.74
Due to the size of XMC1, TD-DFT calculations on excited states have been performed
on two simplified models:
one corresponding to the bare metal cluster (XMC1core) described before,
and the other one being the bare metal cluster model cupped with one complete PPH3
and three SAr ligands (with Ar=C6H3F2 and C6H2F3) labeled XMC1core+Ls in
Fig. 2, in order to reproduce the interaction between the silver core and the aromatic
molecules surrounding it, as well as the interactions between ligands.
These models and the structure of the complete XMC1 particle, the PPh3 ligands, and a
fluorurated-thiophenol ligand are displayed in Fig. 2. Computed UV-Vis spectra obtained
with XMC1core, XMC1core+Ls, PPh3 and SAr are compared and discussed.
The calculated wavelengths (λ) of the TD-DFT computed spectra have been multiplied
by a linear scaling factor of 1.5 (i.e. λscaled = λ·1.5) to achieve a better superposition with
the experimental spectrum of XMC1.17 This scaling results into a product for a multiplying
linear scaling factor of 1/1.5 ' 0.667 in the energy (E) domain:
E scaled = E · const. , const. ' 0.667 . (1)
Obviously, scaling frequencies changes the energetics of the system, including the band sep-
aration. Anyway, this change also occurs with the simple translation of the spectrum in the
wavelength domain, which is a widely adopted procedure for computed optical spectra (see
for example Ref. 5,8,74–77). In fact, when wavelengths are translated (of a factor ∆λ) we
have that
λtrans. = λ+ ∆λ , (2)
10

which can be written in terms of corresponding energies (E) as
1
E trans.=
1
E+
1
∆E
, (3)
due to the fact that λ ∝ 1/E and ∆λ ∝ 1/∆E, with E trans. being the energy corresponding
to λtrans.. The latter equation can be rearranged so to obtain
E trans. =E ·∆E
E + ∆E
. (4)
Therefore, scaling the wavelengths expressed by Eqn. 1 actually results in a much simpler
transformation in energy domain than the translation of the spectrum in the wavelength
domain expressed by Eqn. 4. The scaling of frequencies is also routinely applied in ab initio
and molecular dynamics calculations to obtain vibrational energies.78–80
The wavelengths and energies discussed in the following text and figures are all multiplied
by a factor of 1.5 and 0.667, respectively, unless explicitly stated otherwise (i.e. Table 5).
Results and Discussion
Structural properties on the bare cluster
The mean unsigned errors (defined in the previous section as 〈δ〉) yielded by the geometry
optimizations on XMC1core have been used to check the structural accuracy with respect to
the X-ray data. Figure 3 reports the 〈δ〉 values as a function of the XC functional employed.
All optimizations carried out with GGA functionals (green histograms) yield very distorted
geometries, with large mean unsigned errors (〈δ〉 ≥0.30 A). BPBE, BP86, and BPW91 give
somewhat better results than other GGA and meta-GGA XC functionals, nevertheless a
mean unsigned error of about 0.30−0.35 A denotes a severe distortion of the crystallographic
geometry. Hybrids functionals provide better results, although with many ups and downs.
Some hybrid functionals (red histograms) such as B1LYP, B3LYP, X3LYP, BHandHLYP,
11

Figure 3: Histograms showing changes in the average Ag−Ag distance (the function 〈δ〉 as defined inComputational Details) by varying DFT functionals (on the x-axis). GGA, hybrids, and functionals arerepresented by green bars, hybrid functionals are represented by red bars, and range-separated/long-range-corrected hybrids are represented by blue bars. The Hartree-Fock optimization has a black bar. Errorsgreater than 0.45 A are not shown in this scale. Structures with 〈δ〉 ≥ 0.25 A are very distorted.
M06HF, and SOGGA11x give accurate geometries, with 〈δ〉 values under 0.1A. Also O3LYP,
B98, and τ -HCTHhyb yield fairly accurate structures, with mean unsigned errors below
the 0.15 A threshold. All other hybrids tested here yielded more distorted geometries,
particularly M05, M06, and TPSSh. The only range-separated hybrids (blue histograms)
tested here that gives accurate optimized structures is CAM-B3LYP (with a 〈δ〉 value of
about 0.1A), although LC-BLYP and M11 also provide acceptable geometries (〈δ〉 '0.17
A), and all the others yielding deformed structures. Overall, the “general” hybrids tested
here have the better structural performances, closely followed by the long-range corrected
hybrids, and then lastly the GGAs.
This behavior of the various XC functionals is in striking contrast with what was observed
on gold nanoclusters16 composed of 11 and 24 metal atoms and similar ligands (thiophenols
and triphenylphosphines). In fact, in the case of Au-based NCs, GGA functionals yielded the
best structures, particularly when a PBE-like correlation was employed. Not surprisingly,
most DFT studies on Au-based particles employed GGAs, and PBE-like functionals (e.g.
PBE itself, P86, PW91) in particular.2,12,13,81 The benefits of the PBE-like correlations ex-
12

tended also to the hybrids functionals when employed to simulate gold-based clusters, with
PBE0, B3P86, and B3PW91 outperforming B3LYP, and to the range-separated hybrids,
with HSE06 (which includes the PBE exchange) outperforming CAM-B3LYP. In the case
of the Ag-based XMC1 the opposite occurs, and the LYP correlation seems to improve the
structural accuracy of the calculations, at least for hybrid functionals. This is particularly
evident in case of the BHandHLYP, which outperforms the pure BHandH for XMC1core.
For Au-based clusters instead, BHandHLYP provided deformed relaxed structures.16 Over-
all, M06HF and CAM-B3LYP seem to be the best XC functionals to reproduce the structure
of both gold and silver-based core NCs, while M05 and M06 are inadequate for both of them.
While these observations can be qualitatively understood by considering that Au atoms,
having more electrons, have a more metallic character than silver (with Au performing better
with the simpler GGA functionals), probably the binding geometry of the ligands plays its
part as well. In fact, most of the distortions occur at the eight outer Ag atoms that are each
bound to four non-metal atoms (three S and one P atoms). To the best of our knowledge,
this type of binding geometry did not occur in known X-Ray determined Au NCs, and
seems a peculiar feature of silver-based NCs,18 or at least with significant Ag presence.17
Moreover, the Au particles investigated in Ref. 16 had the gold atoms at the surface bonded
with only one non-metal element (either S, P, Br, or Cl), whereas in XMC1 every surface
Ag atom is bonded to two different non-metal elements (S and P) at the same time. The
accurate description of these many non-metallic interactions in XMC1core requires a higher
contribution of exact Hartree-Fock exchanges than for similar gold-based clusters. In fact,
even the pure Hartree-Fock calculation (black bar in Fig. 3, 〈δ〉 '0.27A) outperform all the
GGAs tested here on XMC1core, while for the undecagold-based particles it was the post
Hartree-Fock MP2 to be outperformed16 by many GGA-based relaxations.
13

Structural and electronic properties on XMC1
The complete structure of XMC1 has been investigated with a more limited set of XC func-
tionals, namely BPBE, B3LYP, CAM-B3LYP and M06HF (see Table 2). These functionals,
belonging to different families and levels of sophistication, were already employed in analo-
gous calculation on complete Au particles.16 After relaxation of the constrained geometry,
Table 2: Errors in Ag−Ag distances (the 〈δ〉 function defined in the Computational Details),of XMC1 adopting BPBE, B3LYP, M06HF, and CAM-B3LYP functionals. Some values aremissing because either structural optimizations did not converge or required too long com-putation time. Data are reported as depending on the adopted functionals (BPBE, B3LYP,CAM-B3LYP, and M06HF) and basis sets employed for outer atoms (STO-3G or 6-31G), aswell as on the presence of constraints. Results of the structural optimizations performed onXMC1core are also reported as a reference.
Complete XMC1 BPBE B3LYP CAM−B3LYP M06HFSTO-3G 6-31G STO-3G 6-31G STO-3G 6-31G STO-3G 6-31G
constrained 〈δ〉 / A 0.05 0.01 0.08 0.03 0.06 0.00 0.10 0.04unconstrained 〈δ〉 / A — 0.25 — 0.31 — 0.20 — 0.12
XMC1core BPBE B3LYP CAM−B3LYP M06HF〈δ〉 / A 0.35 0.08 0.10 0.09
accurate structures are obtained with all these functionals. Using 6-31G BS to describe the
aromatic ligands results in an even increased accuracy (particularly for M06HF), even if
atoms of the ligands are fixed to their experimental positions.
On the other hand, the unconstrained relaxations yield overall unsatisfactory results.
Only optimizations performed with M06HF provide fair accuracy, and only when 6-31G BS
for the organic ligands (C, H, and F atoms, while S and P are described by 6-31++G(d,p)
BS) is adopted in place of the simpler STO-3G. The other three functionals distorted the
experimental geometry significantly, in particular B3LYP that is one of the top performers on
the XMC1core model. This, and the fact that M06HF (the best performer on full XMC1)
is known to well mimic Van der Waals interactions,26 suggests that ligands interactions are
not well described by BPBE and B3LYP, which yield mean unsigned errors for the Ag atoms
distances ≥0.20 A.
14
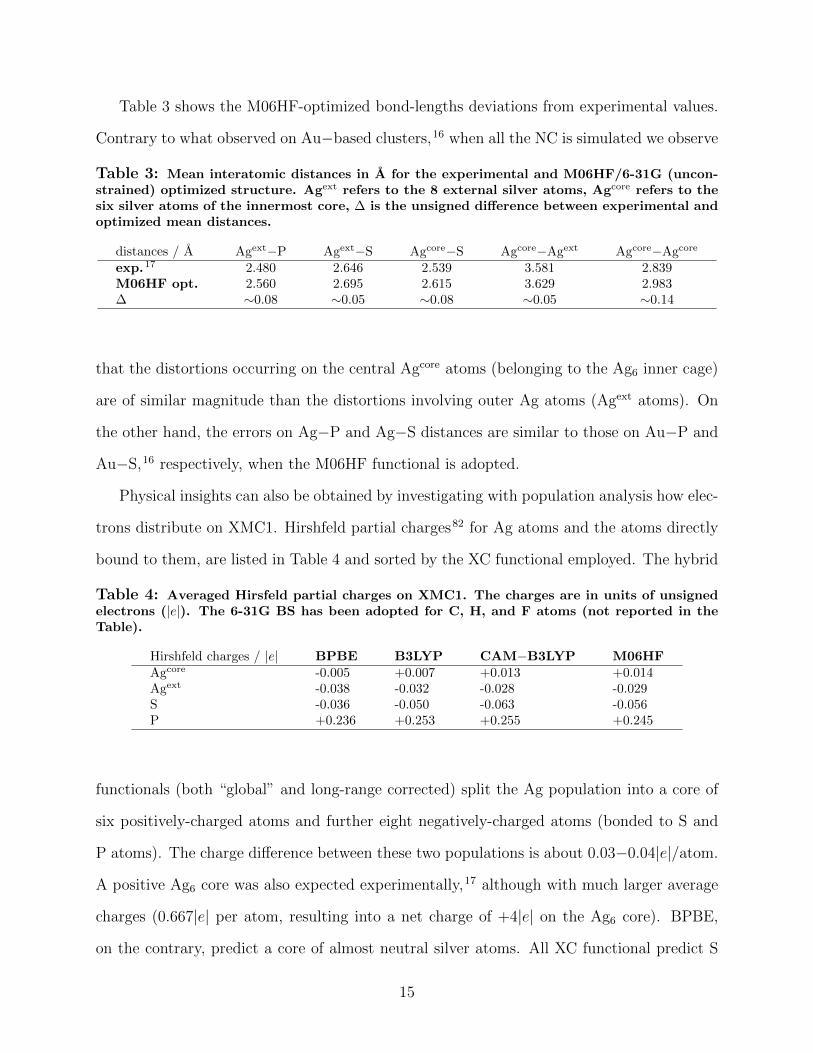
Table 3 shows the M06HF-optimized bond-lengths deviations from experimental values.
Contrary to what observed on Au−based clusters,16 when all the NC is simulated we observe
Table 3: Mean interatomic distances in A for the experimental and M06HF/6-31G (uncon-strained) optimized structure. Agext refers to the 8 external silver atoms, Agcore refers to thesix silver atoms of the innermost core, ∆ is the unsigned difference between experimental andoptimized mean distances.
distances / A Agext−P Agext−S Agcore−S Agcore−Agext Agcore−Agcore
exp.17 2.480 2.646 2.539 3.581 2.839M06HF opt. 2.560 2.695 2.615 3.629 2.983∆ ∼0.08 ∼0.05 ∼0.08 ∼0.05 ∼0.14
that the distortions occurring on the central Agcore atoms (belonging to the Ag6 inner cage)
are of similar magnitude than the distortions involving outer Ag atoms (Agext atoms). On
the other hand, the errors on Ag−P and Ag−S distances are similar to those on Au−P and
Au−S,16 respectively, when the M06HF functional is adopted.
Physical insights can also be obtained by investigating with population analysis how elec-
trons distribute on XMC1. Hirshfeld partial charges82 for Ag atoms and the atoms directly
bound to them, are listed in Table 4 and sorted by the XC functional employed. The hybrid
Table 4: Averaged Hirsfeld partial charges on XMC1. The charges are in units of unsignedelectrons (|e|). The 6-31G BS has been adopted for C, H, and F atoms (not reported in theTable).
Hirshfeld charges / |e| BPBE B3LYP CAM−B3LYP M06HFAgcore -0.005 +0.007 +0.013 +0.014Agext -0.038 -0.032 -0.028 -0.029S -0.036 -0.050 -0.063 -0.056P +0.236 +0.253 +0.255 +0.245
functionals (both “global” and long-range corrected) split the Ag population into a core of
six positively-charged atoms and further eight negatively-charged atoms (bonded to S and
P atoms). The charge difference between these two populations is about 0.03−0.04|e|/atom.
A positive Ag6 core was also expected experimentally,17 although with much larger average
charges (0.667|e| per atom, resulting into a net charge of +4|e| on the Ag6 core). BPBE,
on the contrary, predict a core of almost neutral silver atoms. All XC functional predict S
15

and P atoms negatively and positively charged, respectively. These results presents some
significant qualitative and quantitative divergences with respect to those obtained for un-
decagold clusters by the same means.16 In fact, while Au11-based NCs indeed have a central
atom with structural properties different from the other ten metal atoms, its partial charge
is negative as that of the others, hence hindering the identification of a “core” region by
population analysis only. More importantly, the M−P bonds (M being a metal, either Ag
or Au) have a much more ionic character for gold NCs than for XMC1. For example, in
undecagold particles with CAM-B3LYP and M06HF the average charge difference along the
M−P bond exceeds ∼0.48 and ∼0.50|e|, respectively.16 For silver-based XMC1 the corre-
sponding differences are only ∼0.28 and ∼0.27|e|. Analogous differences can be obtained
with B3LYP functional calculations. This means that the Ag−P bond is significantly more
covalent than Au−P, and provides some explanation of hybrid functionals outperforming
GGAs for XMC1, contrary to what was observed on gold-based particles.
Also the electronic density of states (DoS) can be helpful to characterize the XMC1
particle, particularly if it is partitioned per atomic species so to sort out contributions due
to the core or to the ligands. Figure 4 summarizes this analysis, performed with the Multiwfn
software.83 As can be seen, the relative contribution to the occupied orbital density of the
metal atoms (blue line) and the atoms directly bound to them (S and P atoms, yellow and
red lines, respectively) are of comparable magnitude, particularly in the range of energies
closer to the HOMO-LUMO gap (indicatively, the [−6, 0] eV interval). This is in contrast
with what was observed in undecagold nanoclusters,74 where the contribution due to gold
was largely predominant, and suggests an increased coupling between the energy levels of
silver atoms and the S and P atoms around them.
Optical properties
Gold-based NCs showed that often the aromatic ligands surrounding the metal cores gave lit-
tle contribution to the resulting visible spectrum.16,84 In fact, the gross shape of their optical
16

Figure 4: DoS decomposed per atomic species. Calculation has been performed on the XMC1core+Lsmodel at the cam-B3LYP level of theory. The energies are shifted so to have the center of the HOMO-LUMOgap at 0 eV.
spectrum could be recovered also if the organic ligands were completely omitted (except for
the atoms directly bound to gold, such as P and S), up to the point that Au11(PPh3)7(SPh)3
and Au11(PH3)7(SH)3 yielded basically the same excitation profile when computed with
TD-DFT.74 A further observation of the orbitals involved into the transitions of such un-
decagold particles also showed that Au→Au excitations were by far the most abundant,
while Au→ligands transitions were rare and outside the Vis region, and ligand→ligand even
rarer and blue-shifted at energies of ∼5 eV (corresponding to wavelengths of about ∼250
nm).74
This does not occur here with the silver-based XMC1. In fact, as shown in Figure 5,
the TD-DFT computed spectra on the XMC1core (left panels) and on the model including
one PPh3 and three SAr molecules (XMC1core+Ls model, right panels) are significantly
different. The distributions of excited states (red sticks) are altered by the presence of triph-
enylphosphine and of the substituted thiophenols, up to the point that the resulting spectral
shapes (blue lines) differ considerably. As was noted for gold,16,74 the BLYP XC functional,
being a GGA, yields lower frequencies,11,16,74,85 while a long-range corrected functional such
as CAM-B3LYP gives a blue-shifted spectrum. Apart from these shifts, the BLYP spec-
17

Figure 5: Calculated electronic spectra on XMC1core (left panel) and XMC1core+Ls (right panel)models at the TD-BLYP, TD-B3LYP, and TD-CAM-B3LYP levels of theory. Blue lines represent the UV-Visspectra of the different models obtained from the convolution of 200 S0 →Sn transitions (red sticks) withGaussians of half-width at half-height of 0.25 eV. The x-axis bins correspond to 25 nm.The computed TD-DFT wavelengths have been multiplied by a 1.5 scaling factor (i.e. a ∼ 0.667 scalingfactor in frequency space) for a better comparison with the experimental data.
tra appear unstructured (both for XMC1core and XMC1core+Ls), while those obtained
employing B3LYP and CAM-B3LYP are more similar to each other.
Since the spectra reported in Fig. 5 are limited to the first 200 S0 →Sn transitions,
the higher frequency maximum (λ '310 nm) in the spectrum of XMC1core+Ls model
obtained with CAM-B3LYP (represented with a blue line in the lower-right panel of Fig. 5)
is an artifact due to this truncation. To elucidate this latter feature, we repeated the TD-
CAM-B3LYP calculation computing the first 600 S0 →Sn transitions, thus reaching higher
frequencies that exceed even those investigated by UV absorption experiments, as shown in
Figure 6, Part A.
18

Figure 6: A) Calculated electronic spectrum on XMC1core+Ls model considering 600 excited states(upper panel), and the experimental17 UV-Vis spectrum (lower panel). B) Calculated electronic spectrumon the first 200 excited states of PPh3 (left) and SC6H3F3 (right).The computed spectra were obtained at the TD-CAM-B3LYP level of theory. Wavelengths have beenmultiplied by a 1.5 scaling factor (i.e. a ∼ 0.667 scaling factor in frequency space) for a better comparisonwith the experimental data. Blue lines represent the UV-Vis spectra obtained from the convolution of theS0 →Sn transitions (red sticks) with Gaussians of half-width at half-height of 0.25 eV. The x-axis binscorrespond to 25 nm.
Including more states into the calculation, the experimental profile at higher energy (the
rising of absorbance for λ ≤ 350 nm) can be correctly recovered with the CAM-B3LYP
XC functional. Thus, this XC functional can correctly describe the shape of the optical
bands, which simpler functionals of the same “family” such as B3LYP and BLYP cannot do.
This is probably due to the fact that CAM-B3LYP is proved to better reproduce charge-
transfer transitions,6,69 and these latter contribute to the rising of absorbance for λ ≤350 nm.
In fact, in this range of wavelengths the absorption of triphenylphosphine and fluorurated
19

thiophenols occurs (see Figure 6, Part B). Thus, excitations due to the silver core and the
ligands overlap significantly for XMC1, contrary to what happens in undecagold NCs, and
this overlapping prevents recovering a reliable optical spectrum with calculations performed
only on the metal part.
This is related to another issue that hinders TD-DFT calculations on the simplified
XMC1core model: while the occupied orbitals of XMC1core have a shape similar to the
occupied orbitals of XMC1core+Ls, the virtual orbitals of XMC1core (up to LUMO+20,
at least) are abnormally localized on the PH3 groups, in particular on the P atoms. The
clearly unphysical shape of the virtual orbitals seems proper of the XMC1core model, since
it was not observed on the gold cores discussed in Ref.s [ 16,74 ], nor in the XMC1core+Ls
model discussed here. We also performed calculations with a more complex simplification
scheme, putting CH3 groups in place of the H atoms of the XMC1core model, but the shape of
the virtual orbitals still appear unrealistic and very localized on the phosphorous atoms. On
the contrary, the occupied orbitals of the XMC1core model resemble the occupied orbitals
of the XMC1core+Ls model. Thus, the ground state properties (including the structural
optimizations) are not particularly affected at the orbital level by omitting or simplifying the
ligands, which anyway is a common procedure to investigate noble metal NCs.2,9,14,16,21,23,44
The orbitals of the XMC1core+Ls model that give the main contribution to the most
intense transitions (oscillator strength ≥700) are reported in Figure 7 as contour plots,
while in Table 5 these latter transitions are described in detail. Most transitions are of
the type metal→metal, as in the case of the undecagold NCs. However, transitions n=30
(HOMO→LUMO+10) and n=43 (HOMO→LUMO+6) show a clear metal→ligand charac-
ter. In particular, it has to be noticed that in the case of the Au11(PPh3)7Cl3 particle, the
first metal→triphenylphosphine transition was the n=68, occurring at 4.83 eV (un-scaled
value),74 while in the present case of XMC1 the first metal→triphenylphosphine transition
occurs at about the same energy (4.84 eV, un-scaled value) but much earlier in terms of num-
ber states (n=30). This finding helps to explain why the spectrum of XMC1 is so affected
20

Figure 7: Contour levels of some selected occupied and virtual orbitals of the XMC1core+Ls modelcomputed with the CAM-B3LYP XC functional. Blue denotes the positive lobes, while red the negativelobes. These orbitals are involved into the transitions described in Table 5.
21

Table 5: Some selected (osc.str. ≥700) optical S0 →Sn transitions of XMC1core+Ls modeland their orbital contributions. The table lists the transition number (n), the occupied(occ.orb.) and virtual (virt.orb.) orbitals involved into the transitions, their relative contri-bution (CI coeff.), oscillator strengths (osc.str.), energies (energy), and corresponding wave-lengths (λ). Calculations are performed at the TD-CAM-B3LYP level of theory.The computed TD-DFT energies and wavelengths have not been multiplied by a scaling factorin this table.
n orb.occ. → orb.virt. CI coeff. osc.str. (·104) energy/eV λ/nm1 HOMO → LUMO 0.52 2659 3.62 3422 HOMO → LUMO+1 0.54 1901 3.64 3403 HOMO → LUMO+2 0.65 2045 3.70 3357 HOMO-1 → LUMO 0.36 1042 4.40 28110 HOMO-1 → LUMO+2 0.24 1023 4.49 27630‡ HOMO → LUMO+10 0.30 731 4.84 25531 HOMO-9 → LUMO 0.22 980 4.87 25534 HOMO-10 → LUMO 0.20 1511 4.91 25236 HOMO-11 → LUMO+2 0.26 1731 4.94 25137 HOMO-11 → LUMO+1 0.21 1234 4.95 25043‡ HOMO → LUMO+6 0.24 832 5.04 246
‡ Transitions with a significant Ag→PPh3 character. All other transitions reported here havea predominant Ag→Ag character.
by ligands, while those of Au11-based particles were not: in the case of Au11(PPh3)7Cl3, the
first metal→PPh3 transition occurred at (un-scaled) energies εgap+1.9 eV (εgap being the
optical gap energy, i.e. the n=1 optical excitation11), whereas in the case of XMC1 the first
metal→PPh3 transition occurs at the (un-scaled) energy of εgap+1.2 eV, ∼0.7 eV closer to
the lower energy boundary of the XMC1 spectrum.
Concluding Remarks
Ground-state and time-dependent density functional calculations have been performed to
evaluate the most reliable approaches to reproduce the structure and electronic spectrum of
the hybrid organic-silver nanocluster named XMC1.
We have found that many exchange-correlation functionals that proved to be effective
for hybrid organic-gold nanoclusters actually perform badly for XMC1. This is probably
due to the more “metallic” character of the undecagold clusters, which favors functionals
22

belonging to the GGA family. On the contrary, XMC1 shows a much more intricate network
of metal−nonmetal bonds, which are better described by functionals that include the exact
Hartree-Fock exchange. In fact, the electronic populations analysis shows that the Ag−P
bonds are much less ionic than the corresponding Au−P bonds, thus requiring the use of
functionals better suited to describe covalent chemical bonds. The M06HF functional (with
100% of Hartree-Fock exchange) yields good optimized geometries for both the metal core
and the full nanocluster, while CAM-B3LYP (with 65% of Hartree-Fock exchange at long
distances) provides an optical spectrum in good agreement with the experimental one.
The virtual orbitals of the metal core model are very different from those obtained from
calculations in which at least one triphenylphosphine and three substituted thiophenols are
retained. This is probably due to the fact that the Ag−P bond is significantly more covalent
than Au−P, thus facilitating the occurrence of metal→ligand charge transfer transitions.
As a consequence, we have found that time-dependent density functional calculations on the
metal core yield electronic spectra significantly different from the experimental one regardless
of the functional adopted, thus suggesting that omitting the ligands is not a viable choice
to investigate the excited state of silver clusters. Charge transfer transitions of the type
Ag→ligands occur closer to the optical gap than in undecagold nanoclusters, thus affecting
a larger portion of the electronic spectrum.
In conclusion, some of the main computational schemes to simulate noble metal nan-
oclusters, such for example the simplification of the protecting organic ligands, cannot be
carelessly adopted for silver-based XMC1. On the contrary, it requires the specific approaches
described here, in particular to recover its optoelectronic properties.
Acknowledgements
This work was supported by the Italian “Ministero dell’Istruzione, dell’Universita e della
Ricerca” (MIUR) through the ‘‘Futuro in Ricerca” (FIRB) Grant RBFR1248UI 002 enti-
23

tled “Novel Multiscale Theorethical/Computational Strategies for the Design of Photo and
Thermo responsive Hybrid Organic-Inorganic Components for Nanoelectronic Circuits”, and
the “Programma di ricerca di rilevante interesse nazionale” (PRIN) Grant 2010C4R8M8 en-
titled “Nanoscale functional Organization of (bio)Molecules and Hybrids for targeted Ap-
plication in Sensing, Medicine and Biotechnology” is also acknowledged. Computation time
was granted through the CINECA project AUNANMR-HP10CJ027S.
References
(1) Chen, S.; Ingram, R. S.; Hostetler, M. J.; Pietron, J. J.; Murray, R. W.; Schaaff, T. G.;
Khoury, J. T.; Alvarez, M. M.; Whetten, R. L. Gold Nanoelectrodes of Varied Size:
Transition to Molecule-Like Charging. Science 1998, 280, 2098–2101.
(2) Walter, M.; Akola, J.; Lopez-Acevedo, O.; Jadzinsky, P. D.; Calero, G.; Ackerson, C. J.;
Whetten, R. L.; Gronbeck, H.; Hakkinen, H. A unified View of Ligand-protected Gold
Clusters as Superatom Complexes. Proceedings of the National Academy of Sciences
2008, 105, 9157–9162.
(3) Cramer, C. J.; Truhlar, D. G. Density functional theory for transition metals and
transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816.
(4) Perdew, J. P.; Ruzsinszky, A.; Constantin, L. A.; Sun, J.; Csonka, G. I. Some Funda-
mental Issues in Ground-State Density Functional Theory: A Guide for the Perplexed.
Journal of Chemical Theory and Computation 2009, 5, 902–908.
(5) Pedone, A.; Barone, V. Unraveling solvent effects on the electronic absorption spectra
of TRITC fluorophore in solution: a theoretical TD-DFT/PCM study. Phys. Chem.
Chem. Phys. 2010, 12, 2722–2729.
(6) Pedone, A. Role of Solvent on Charge Transfer in 7-Aminocoumarin Dyes: New Hints
24

from TD-CAM-B3LYP and State Specific PCM Calculations. Journal of Chemical The-
ory and Computation 2013, 9, 4087–4096.
(7) Runge, E.; Gross, E. K. U. Density-Functional Theory for Time-Dependent Systems.
Phys. Rev. Lett. 1984, 52, 997–1000.
(8) Pedone, A.; Prampolini, G.; Monti, S.; Barone, V. Absorption and emission spectra
of fluorescent silica nanoparticles from TD-DFT/MM/PCM calculations. Phys. Chem.
Chem. Phys. 2011, 13, 16689–16697.
(9) Aikens, C. M. Origin of Discrete Optical Absorption Spectra of M25(SH)18- Nanoparti-
cles (M = Au, Ag). The Journal of Physical Chemistry C 2008, 112, 19797–19800.
(10) Pedone, A.; Prampolini, G.; Monti, S.; Barone, V. Realistic Modeling of Fluorescent
Dye-Doped Silica Nanoparticles: A Step Toward the Understanding of their Enhanced
Photophysical Properties. Chemistry of Materials 2011, 23, 5016–5023.
(11) Baerends, E. J.; Gritsenko, O. V.; van Meer, R. The Kohn-Sham Gap, the Fundamental
Gap and the Optical Gap: the Physical Meaning of Occupied and Virtual Kohn-Sham
Orbital Energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425.
(12) Goh, J.-Q.; Malola, S.; Hakkinen, H.; Akola, J. Role of the Central Gold Atom in
Ligand-Protected Biicosahedral Au24 and Au25 Clusters. The Journal of Physical Chem-
istry C 2013, 117, 22079–22086.
(13) Zhu, M.; Aikens, C. M.; Hollander, F. J.; Schatz, G. C.; Jin, R. Correlating the Crystal
Structure of A Thiol-Protected Au25 Cluster and Optical Properties. Journal of the
American Chemical Society 2008, 130, 5883–5885.
(14) Ivanov, S. A.; Arachchige, I.; Aikens, C. M. Density Functional Analysis of Geometries
and Electronic Structures of Gold-Phosphine Clusters. The Case of Au4(PR3)42+ and
Au4(µ2-I)2(PR3)4. The Journal of Physical Chemistry A 2011, 115, 8017–8031.
25

(15) Lopez-Acevedo, O.; Tsunoyama, H.; Tsukuda, T.; Hakkinen, H.; Aikens, C. M. Chiral-
ity and Electronic Structure of the Thiolate-Protected Au38 Nanocluster. Journal of
the American Chemical Society 2010, 132, 8210–8218.
(16) Muniz-Miranda, F.; Menziani, M. C.; Pedone, A. Assessment of Exchange-Correlation
Functionals in Reproducing the Structure and Optical Gap of Organic-Protected Gold
Nanoclusters. The Journal of Physical Chemistry C 2014, 118, 7532–7544.
(17) Yang, H.; Lei, J.; Wu, B.; Wang, Y.; Zhou, M.; Xia, A.; Zheng, L.; Zheng, N. Crystal
structure of a luminescent thiolated Ag nanocluster with an octahedral Ag4+6 core.
Chem. Commun. 2013, 49, 300–302.
(18) Yang, H.; Wang, Y.; Zheng, N. Stabilizing subnanometer Ag(0) nanoclusters by thiolate
and diphosphine ligands and their crystal structures. Nanoscale 2013, 5, 2674–2677.
(19) Akola, J.; Walter, M.; Whetten, R. L.; Hakkinen, H.; Gronbeck, H. On the Structure of
Thiolate-Protected Au25. Journal of the American Chemical Society 2008, 130, 3756–
3757.
(20) Nunokawa, K.; Onaka, S.; Ito, M.; Horibe, M.; Yonezawa, T.; Nishihara, H.; Ozeki, T.;
Chiba, H.; Watase, S.; Nakamoto, M. Synthesis, Single Crystal X-ray analysis, and
{TEM} for a Single-sized Au11 Cluster Stabilized by {SR} Ligands: The Interface
Between Molecules and Particles. Journal of Organometallic Chemistry 2006, 691, 638
– 642.
(21) Heinecke, C. L.; Ni, T. W.; Malola, S.; Makinen, V.; Wong, O. A.; Hakkinen, H.;
Ackerson, C. J. Structural and Theoretical Basis for Ligand Exchange on Thiolate
Monolayer Protected Gold Nanoclusters. Journal of the American Chemical Society
2012, 134, 13316–13322.
(22) Qian, H.; Eckenhoff, W. T.; Zhu, Y.; Pintauer, T.; Jin, R. Total Structure Determi-
26

nation of Thiolate-Protected Au38 Nanoparticles. Journal of the American Chemical
Society 2010, 132, 8280–8281, PMID: 20515047.
(23) Das, A.; Li, T.; Nobusada, K.; Zeng, Q.; Rosi, N. L.; Jin, R. Total Structure and
Optical Properties of a Phosphine/Thiolate-Protected Au24 Nanocluster. Journal of
the American Chemical Society 2012, 134, 20286–20289.
(24) Tlahuice-Flores, A. On the structure of the thiolated Au6Ag7 cluster. Phys. Chem.
Chem. Phys. 2014, DOI:10.1039/C4CP02273D, –.
(25) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheese-
man, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A. et al. Gaussian
09, Revision D.01. Gaussian, Inc., Wallingford CT, 2013.
(26) Gaussian 09 User’s reference.
(27) Schuchardt, K.; Didier, B.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.;
Windus, T. Basis Set Exchange: A Community Database for Computational Sciences.
Journal of Chemical Information and Modeling 2007, 47, 1045–1052.
(28) Peterson, K. A.; Puzzarini, C. Systematically Convergent Basis Sets for Transition
Metals. II. Pseudopotential-based Correlation Consistent Basis Sets for the group 11
(Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theoretical Chemistry Accounts 2005,
114, 283–296.
(29) Ran, Q.; Schmude, R. W.; Gingerich, K. A.; Wilhite, D. W.; Kingcade, J. E. Dissocia-
tion energy and enthalpy of formation of gaseous silver dimer. The Journal of Physical
Chemistry 1993, 97, 8535–8540.
(30) Hay, P. J.; Wadt, W. R. Ab initio Effective Core Potentials for Molecular Calculations.
Potentials for the Transition Metal Atoms Sc to Hg. The Journal of Chemical Physics
1985, 82, 270–283.
27

(31) Dunning Jr., T. H.; Hay, P. J. Ab initio Effective Core Potentials for Molecular Cal-
culations. Potentials for the Transition Metal Atoms Sc to Hg. Journal of Chemical
Physics 82, 270.
(32) Couty, M.; Hall, M. B. Basis Sets for Transition Metals: Optimized Outer p Functions.
Journal of Computational Chemistry 1996, 17, 1359–1370.
(33) Roy, L. E.; Hay, P. J.; Martin, R. L. Revised Basis Sets for the LANL Effective Core
Potentials. Journal of Chemical Theory and Computation 2008, 4, 1029–1031.
(34) Ehlers, A.; Bohme, M.; Dapprich, S.; Gobbi, A.; Hollwarth, A.; Jonas, V.; Kohler, K.;
Stegmann, R.; Veldkamp, A.; Frenking, G. A set of f-polarization functions for pseudo-
potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au. Chemical Physics
Letters 1993, 208, 111 – 114.
(35) Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio
Pseudopotentials for the Second Row and Third Row Transition Elements. Theoretica
Chimica Acta 1990, 77, 123–141.
(36) Hurley, M. M.; Pacios, L. F.; Christiansen, P. A.; Ross, R. B.; Ermler, W. C. Ab initio
relativistic effective potentials with spin-orbit operators. II. K through Kr. The Journal
of Chemical Physics 1986, 84 .
(37) Sosa, C.; Andzelm, J.; Elkin, B. C.; Wimmer, E.; Dobbs, K. D.; Dixon, D. A. A
local density functional study of the structure and vibrational frequencies of molecular
transition-metal compounds. The Journal of Physical Chemistry 1992, 96, 6630–6636.
(38) Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedab initio
pseudopotentials for the second and third row transition elements. Theoretica chimica
acta 1990, 77, 123–141.
28

(39) Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and
quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys.
Chem. Chem. Phys. 2005, 7, 3297–3305.
(40) Becke, A. D. Density-functional Thermochemistry. III. The Role of Exact Exchange.
The Journal of Chemical Physics 1993, 98, 5648–5652.
(41) Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation-energy
Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789.
(42) Cammi, R.; Mennucci, B.; Tomasi, J. Fast Evaluation of Geometries and Properties
of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-
Dimethylaminobenzonitrile. The Journal of Physical Chemistry A 2000, 104, 5631–
5637.
(43) Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Mod-
els. Chemical Reviews 2005, 105, 2999–3094.
(44) Kruger, D.; Fuchs, H.; Rousseau, R.; Marx, D.; Parrinello, M. Interaction of Short-
chain Alkane Thiols and Thiolates with Small Gold Clusters: Adsorption Structures
and Energetics. The Journal of Chemical Physics 2001, 115, 4776–4786.
(45) Becke, A. D. Density-functional Exchange-energy Approximation with Correct Asymp-
totic Behavior. Phys. Rev. A 1988, 38, 3098–3100.
(46) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made
Simple. Phys. Rev. Lett. 1996, 77, 3865–3868.
(47) Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Climbing the Density Func-
tional Ladder: Nonempirical Meta˘Generalized Gradient Approximation Designed for
Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401.
29

(48) Perdew, J. P.; Ruzsinszky, A.; Csonka, G. I.; Constantin, L. A.; Sun, J. Workhorse
Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry.
Phys. Rev. Lett. 2009, 103, 026403.
(49) Perdew, J. P.; Ruzsinszky, A.; Csonka, G. I.; Constantin, L. A.; Sun, J. Erratum:
Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum
Chemistry [Phys. Rev. Lett. 103, 026403 (2009)]. Phys. Rev. Lett. 2011, 106, 179902.
(50) Perdew, J. P.; Burke, K.; Wang, Y. Generalized Gradient Approximation for the
Exchange-Correlation Hole of a Many-electron System. Phys. Rev. B 1996, 54, 16533–
16539.
(51) Perdew, J. P. Density-functional Approximation for the Correlation Energy of the In-
homogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822–8824.
(52) Voorhis, T. V.; Scuseria, G. E. A Novel Form for the Exchange-correlation Energy
Functional. The Journal of Chemical Physics 1998, 109, 400–410.
(53) Hamprecht, F. A.; Cohen, A. J.; Tozer, D. J.; Handy, N. C. Development and assessment
of new exchange-correlation functionals. The Journal of Chemical Physics 1998, 109 .
(54) Boese, A. D.; Handy, N. C. New exchange-correlation density functionals: The role of
the kinetic-energy density. The Journal of Chemical Physics 2002, 116 .
(55) Grimme, S. Semiempirical GGA-type density functional constructed with a long-range
dispersion correction. Journal of Computational Chemistry 2006, 27, 1787–1799.
(56) Peverati, R.; Truhlar, D. G. An improved and broadly accurate local approximation
to the exchange-correlation density functional: The MN12-L functional for electronic
structure calculations in chemistry and physics. Phys. Chem. Chem. Phys. 2012, 14,
13171–13174.
30

(57) Xu, X.; Goddard, W. A. The X3LYP extended density functional for accurate descrip-
tions of nonbond interactions, spin states, and thermochemical properties. Proceedings
of the National Academy of Sciences of the United States of America 2004, 101, 2673–
2677.
(58) Cohen, A. J.; Handy, N. C. Dynamic correlation. Molecular Physics 2001, 99, 607–15.
(59) Adamo, C.; Barone, V. Toward Reliable Adiabatic Connection Models free from Ad-
justable Parameters. Chemical Physics Letters 1997, 274, 242 – 250.
(60) Adamo, C.; Barone, V. Toward Reliable Density Functional Methods Without Ad-
justable Parameters: The PBE0 model. The Journal of Chemical Physics 1999, 110,
6158–6170.
(61) Zhao, Y.; Schultz, N. E.; Truhlar, D. G. Exchange-correlation Functional with Broad
Accuracy for Metallic and Nonmetallic Compounds, Kinetics, and Noncovalent Inter-
actions. The Journal of Chemical Physics 2005, 123, 161103.
(62) Zhao, Y.; Truhlar, D. The M06 Suite of Density Functionals for Main Group Ther-
mochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and
Transition Elements: Two New Functionals and Systematic Testing of Four M06-class
Functionals and 12 other Functionals. Theoretical Chemistry Accounts 2008, 120, 215–
241.
(63) Zhao, Y.; Truhlar, D. G. Density Functional for Spectroscopy: No Long-Range Self-
Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and
Better Performance on Average than B3LYP for Ground States. The Journal of Physical
Chemistry A 2006, 110, 13126–13130.
(64) Adamo, C.; Barone, V. Exchange Functionals with Improved Long-range Behavior
and Adiabatic Connection Methods Without Adjustable Parameters: The mPW and
mPW1PW models. The Journal of Chemical Physics 1998, 108, 664–675.
31

(65) Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Climbing the Density Func-
tional Ladder: Nonempirical Meta˘Generalized Gradient Approximation Designed for
Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401.
(66) Boese, A. D.; Martin, J. M. L. Development of density functionals for thermochemical
kinetics. The Journal of Chemical Physics 2004, 121 .
(67) Peverati, R.; Truhlar, D. G. Communication: A global hybrid generalized gradient
approximation to the exchange-correlation functional that satisfies the second-order
density-gradient constraint and has broad applicability in chemistry. The Journal of
Chemical Physics 2011, 135 .
(68) Krukau, A. V.; Vydrov, O. A.; Izmaylov, A. F.; Scuseria, G. E. Influence of the Ex-
change Screening Parameter on the Performance of Screened Hybrid Functionals. The
Journal of Chemical Physics 2006, 125, 224106.
(69) Yanai, T.; Tew, D. P.; Handy, N. C. A New Hybrid Exchange-correlation Functional Us-
ing the Coulomb-attenuating Method (CAM-B3LYP). Chemical Physics Letters 2004,
393, 51 – 57.
(70) Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A Long-range Correction Scheme for
Generalized-gradient-approximation Exchange Functionals. The Journal of Chemical
Physics 2001, 115, 3540–3544.
(71) Peverati, R.; Truhlar, D. G. Improving the Accuracy of Hybrid Meta-GGA Density
Functionals by Range Separation. The Journal of Physical Chemistry Letters 2011, 2,
2810–2817.
(72) Peverati, R.; Truhlar, D. G. Screened-exchange density functionals with broad accuracy
for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191.
32

(73) Pedone, A.; Gambuzzi, E.; Barone, V.; Bonacchi, S.; Genovese, D.; Rampazzo, E.;
Prodi, L.; Montalti, M. Understanding the photophysical properties of coumarin-based
Pluronic-silica (PluS) nanoparticles by means of time-resolved emission spectroscopy
and accurate TDDFT/stochastic calculations. Phys. Chem. Chem. Phys. 2013, 15,
12360–12372.
(74) Muniz-Miranda, F.; Menziani, M. C.; Pedone, A. On the Opto-electronic Properties of
Phosphine and Thiolate-protected Undecagold Nanoclusters. Phys. Chem. Chem.Phys.
2014, DOI:10.1039/C4CP02506.
(75) Pedone, A.; Prampolini, G.; Monti, S.; Barone, V. Realistic Modeling of Fluorescent
Dye-Doped Silica Nanoparticles: A Step Toward the Understanding of their Enhanced
Photophysical Properties. Chemistry of Materials 2011, 23, 5016–5023.
(76) Pedone, A.; Bloino, J.; Barone, V. Role of Host-Guest Interactions in Tuning the Optical
Properties of Coumarin Derivatives Incorporated in MCM-41: A TD-DFT Investiga-
tion. The Journal of Physical Chemistry C 2012, 116, 17807–17818.
(77) Tan, E. M. M.; Hilbers, M.; Buma, W. J. Excited-State Dynamics of Isolated and
Microsolvated Cinnamate-Based UV-B Sunscreens. The Journal of Physical Chemistry
Letters 2014, 5, 2464–2468.
(78) Muniz-Miranda, F.; Pagliai, M.; Cardini, G.; Schettino, V. Wavelet Transform for
Spectroscopic Analysis: Application to Diols in Water. Journal of Chemical Theory
and Computation 2011, 7, 1109–1118.
(79) VandeVondele, J.; Troster, P.; Tavan, P.; Mathias, G. Vibrational Spectra of Phosphate
Ions in Aqueous Solution Probed by First-Principles Molecular Dynamics. The Journal
of Physical Chemistry A 2012, 116, 2466–2474.
(80) Muniz-Miranda, F.; Pagliai, M.; Cardini, G.; Righini, R. Bifurcated Hydrogen Bond in
33

Lithium Nitrate Trihydrate Probed by ab Initio Molecular Dynamics. The Journal of
Physical Chemistry A 2012, 116, 2147–2153.
(81) Hadley, A.; Aikens, C. M. Thiolate Ligand Exchange Mechanisms of Au1 and Sub-
nanometer Gold Particle Au11. The Journal of Physical Chemistry C 2010, 114, 18134–
18138.
(82) Hirshfeld, F. Bonded-atom Fragments for Describing Molecular Charge Densities. The-
oretica Chimica Acta 1977, 44, 129–138.
(83) Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. Journal of Com-
putational Chemistry 2012, 33, 580–592.
(84) Provorse, M. R.; Aikens, C. M. Origin of Intense Chiroptical Effects in Undecagold
Subnanometer Particles. Journal of the American Chemical Society 2010, 132, 1302–
1310.
(85) Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized
Systems from Optimally Tuned Range-Separated Hybrid Functionals. Journal of Chem-
ical Theory and Computation 2012, 8, 1515–1531.
34
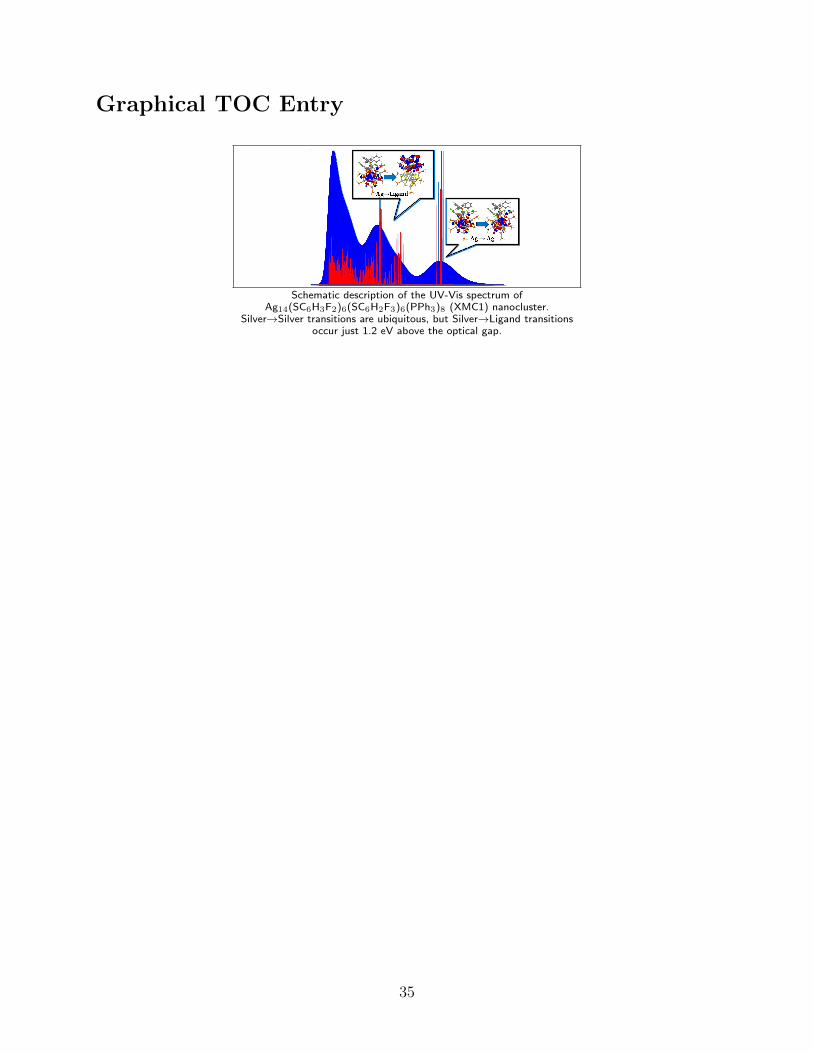
Graphical TOC Entry
Schematic description of the UV-Vis spectrum ofAg14(SC6H3F2)6(SC6H2F3)6(PPh3)8 (XMC1) nanocluster.
Silver→Silver transitions are ubiquitous, but Silver→Ligand transitionsoccur just 1.2 eV above the optical gap.
35




















