
Design and Synthesis of a γ 1 β 8 -Cyclodextrin Oligomer: A New Platform with Potential...
8
Cyclodextrin Oligomers A. Barge, M. Caporaso, G. Cravotto,* K. Martina,P. Tosco, S. Aime, C. Carrera, E. Gianolio, G. Pariani, D. Corpillo .................... &&&&—&&&& Design and Synthesis of a g 1 b 8 -Cyclo- dextrin Oligomer: A New Platform with Potential Application as a Dendrimeric Multicarrier Star-shaped poly-CD: A water-soluble g 1 b 8 -polycyclodextrin (see graphic) has been obtained by azidation of cyclo- dextrin followed by microwave-assisted CuAAC reactions. High ligand hosting capability was indicated by relaxomet- ric titration and nuclear magnetic relaxation dispersion profiling, sup- ported by molecular dynamics simula- tions. The properties of this dendri- meric multicarrier make it suitable for pharmaceutical and diagnostic applica- tions, ranging from targeted drug deliv- ery to molecular imaging. Chem. Eur. J. 2013, 00,0–0 # 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim These are not the final page numbers! ÞÞ &1& FULL PAPER
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Design and Synthesis of a γ 1 β 8 -Cyclodextrin Oligomer: A New Platform with Potential...

Cyclodextrin Oligomers
A. Barge, M. Caporaso, G. Cravotto,*K. Martina, P. Tosco, S. Aime,C. Carrera, E. Gianolio, G. Pariani,D. Corpillo . . . . . . . . . . . . . . . . . . . . &&&&—&&&&
Design and Synthesis of a g1b8-Cyclo-dextrin Oligomer: A New Platformwith Potential Application as aDendrimeric Multicarrier
Star-shaped poly-CD : A water-solubleg1b8-polycyclodextrin (see graphic) hasbeen obtained by azidation of cyclo-dextrin followed by microwave-assistedCuAAC reactions. High ligand hostingcapability was indicated by relaxomet-ric titration and nuclear magneticrelaxation dispersion profiling, sup-ported by molecular dynamics simula-tions. The properties of this dendri-meric multicarrier make it suitable forpharmaceutical and diagnostic applica-tions, ranging from targeted drug deliv-ery to molecular imaging.
Chem. Eur. J. 2013, 00, 0 – 0 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
These are not the final page numbers! ��&1&
FULL PAPER

DOI: 10.1002/chem.201301215
Design and Synthesis of a g1b8-Cyclodextrin Oligomer: A New Platform withPotential Application as a Dendrimeric Multicarrier
Alessandro Barge,[a] Marina Caporaso,[a] Giancarlo Cravotto,*[a] Katia Martina,[a]
Paolo Tosco,[a] Silvio Aime,[b] Carla Carrera,[b] Eliana Gianolio,[b] Giorgio Pariani,[b] andDavide Corpillo[c]
Introduction
The search for new polyvalent and dendritic compoundswith a well-defined chemical architecture is a hot topic indrug delivery and theranostic applications. Although signifi-cant progress has been achieved, the development of alter-native template molecules as efficient carriers is still verymuch a work in progress.[1] An ideal vehicle requires specificstructural features: first of all, a highly functionalized sur-face, which facilitates selective chemical and biological inter-action; secondly, a propensity for self-order and assembly;and thirdly, predictable and reproducible pharmacokinetics(PK) and pharmacodynamics (PD), which ideally requiresize monodispersity and well-established loading ratio.[2]
Precise knowledge of the loading stoichiometry is mandato-
ry when such carriers are employed for the delivery of mul-tiple therapeutic agents in a single formulation, or for thecombination of imaging and drug therapy to monitor effectsin real time. Functionalized liposomes[3] and nanoparticles[4]
that allow molecular recognition of the target tissue havebeen widely used for active or triggered delivery of drugs.[5]
Unfortunately, the key design parameters that govern theperformance of nanocarriers and liposomes (size, surfaceproperties, modulus, and shape) suffer from a lack of prepa-rative reproducibility.[6] Since particle size and surface chem-istry properties strongly affect in vivo distribution, metabo-lism, and drug release,[7] PK/PD reproducibility represents amajor issue. Further requirements for the new generation ofcarriers are very low toxicity, compatible immunogenicity,and easy excretion.[8]
Cyclodextrins (CDs) are natural cyclic oligosaccharidesthat constitute exceptional building blocks for dendrimericarchitectures, as they are essentially non-immunogenic andnon-toxic in animals and humans. CDs can accommodatevarious organic molecules within their truncated cone-shaped hydrophobic cavity, generating host–guest supramo-lecular species in aqueous solution. This property has beenexploited in pharmaceutical applications, and CDs havebeen incorporated into polymers or nanoparticles[9] to bind,stabilize, solubilize, and/or reduce the toxicity of drugs, oli-gonucleotides, and proteins. The synthetic design of CD-based oligomers such as trimers[10] and tetramers[11] has al-ready been described; however, there are very few examplesof larger constructs.[12] The assembly of large CD constructsrequires the use of reactions capable of efficiently linking
Abstract: We report the synthesis andcharacterization of a water-soluble,star-shaped macromolecular platformconsisting of eight b-cyclodextrin (b-CD) units anchored to the narrowerrim of a g-CD core through bis(triazo-lyl)alkyl spacers. The efficient syntheticprotocol is based on the microwave(MW)-promoted Cu-catalyzed 1,3-di-polar cycloaddition of CD monoazidesto CD monoacetylenes. The ligand-hosting capability of the construct hasbeen assessed by relaxometric titration
and nuclear magnetic relaxation disper-sion (NMRD) profiling, which showedit to be good, and this was supportedby molecular dynamics simulations. Todemonstrate the feasibility of obtainingsupramolecular structures with highhosting ability, we designed a dimeric
platform, formed by joining two non-amers through the g-CD cores througha bis(lithocholic acid) linker. With aview to the potential biological applica-tions, cytotoxicity and extent of bindingto human serum albumin were as-sessed. The properties of this dendri-meric multicarrier make it suitable forpharmaceutical and diagnostic purpos-es, ranging from targeted drug deliveryto molecular imaging.
Keywords: click chemistry · cyclo-dextrin oligomers · Gd-based con-trast agents · imaging agents · mi-crowave chemistry
[a] Dr. A. Barge, M. Caporaso, Prof. G. Cravotto, Dr. K. Martina,Dr. P. ToscoDepartment of Drug Science and TechnologyUniversity of Turin, Via Pietro Giuria 9, 10125 Torino (Italy)Fax: (+39) 011-6707687E-mail : [email protected]
[b] Prof. S. Aime, Dr. C. Carrera, Dr. E. Gianolio, Dr. G. ParianiDepartment of Chemistry IFM andMolecular & Preclinical Imaging CentersUniversity of Turin, Via Nizza 52, 10126 Torino (Italy)
[c] Dr. D. CorpilloABLE Biosciences, Bioindustry Park Silvano Fumero S.p.A.Via Ribes 5, 10010 Colleretto Giacosa (Italy)
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.201301215.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 0000, 00, 0 – 0
�� These are not the final page numbers!&2&

two or more CD units. It is well established that the micro-wave (MW)-promoted Cu-catalyzed 1,3-dipolar cycloaddi-tion (CuAAC) between CD monoazides and monoacetylenemoieties, which results in the formation of a triazole bridge,is the most efficient way of modifying the CD surface.[13]
Using the aforementioned procedure, we have previouslypublished the preparation of water-soluble CD homo- andheterodimers and trimers of a-, b-, and g-CD as host mole-cules.[10b, 14]
The aim of this work was the design, preparation, andcharacterization of a new water-soluble oligocyclodextrinheterononamer for use as a dendrimeric multicarrier. Theloading ratio of this nanostructure was first predicted insilico through molecular dynamics (MD) simulations com-bined with solvent-accessible surface (SAS) calculations,then measured through the use of a paramagnetic Gd com-plex. The possibility of assembling multiple nonamers intosupramolecular structures with high hosting ability was ex-plored by preparing a dimeric platform through a bis(litho-cholic acid) linker. Adducts between Gd complexes and thedendrimeric platforms were studied by relaxometric titrationto assess their potential as contrast agents for MRI diagnos-tics; finally, their cytotoxicity and binding affinity towardsalbumin were evaluated.
Results and Discussion
The designed platform consists of eight b-CD units anch-ored to a g-CD core through bis(triazolyl)alkyl spacers togive a star-shaped molecule (Figure 1). The difference incavity size between b- and g-CDs allows for selective com-plexation. For instance, the eight b-CD units could host adrug or a contrast agent, while the g-CD could form a non-covalent adduct with a vector unit capable of guiding thewhole platform to the target site. Alternatively, the g-CD
cavity could be exploited to form supramolecular adductsbetween multiple CD dendrimers through a linker, thusmultiplying the number of b-CD cavities available for theinclusion of drugs or diagnostic agents.
Chemistry : The first step in the synthetic pathway(Scheme 1) was the selective monosubstitution of b-CD toafford the 6-monoazido-b-CD 1.[15] This was followed by se-lective per-substitution of the primary -OH groups on the g-
CD to give the 6-perazido-g-CD 3.[16] Two MW-assisted clickreactions with a linear bis ACHTUNGTRENNUNG(alkyne), carried out in succession,generated the g1b8-nonamer 4 (Figure 1). In the firstCuAAC, the use of an excess of 1,8-nonadiyne preventedthe formation of the b-CD dimer and allowed the 6-hepty-nyltriazolyl-b-CD 2 to be isolated in good yield. The pro-moting effect of MW irradiation on this reaction was strik-ing both in terms of reaction time and yield. Derivative 2was precipitated due to its poor solubility in water, washedwith water, and used without further purification. Thehigher water solubility of 4, compared to its precursors, fa-cilitated its purification after the second CuAAC. Nonamer4 was further purged of residual copper salts and other im-purities by preparative HPLC and the product was obtainedat a purity greater than 90 % (by analytical HPLC-ELSD).With a view to possible scale-up, an alternative, faster purifi-cation could be accomplished through the addition of ethyl-enediaminetetraacetic acid disodium salt dehydrate(Na2H2EDTA) to the reaction mixture, followed by ultrafil-tration through a 3 kDa cut-off membrane (purity 72–75 %by analytical HPLC-ELSD). The final product did not con-tain impurities, but contained less-substituted analogues,from which one or two b-CD units were missing. Nonamer 4Figure 1. Three-dimensional model of g1b8-cyclodextrin-nonamer 4.
Scheme 1. Synthetic route leading to the CD-nonamer 4.
Chem. Eur. J. 2013, 00, 0 – 0 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org
These are not the final page numbers! ��&3&
FULL PAPERA g1b8-Cyclodextrin Oligomer as a Dendrimeric Multicarrier
was characterized by MALDI-TOF MS, 1H NMR, 2D-COSY NMR, 2D-1H–13C HMQC NMR, and2D-1H–13C HMBC NMR spectroscopies (Figures S1–S4,Supporting Information). All spectroscopic data were con-sistent with the proposed structure.
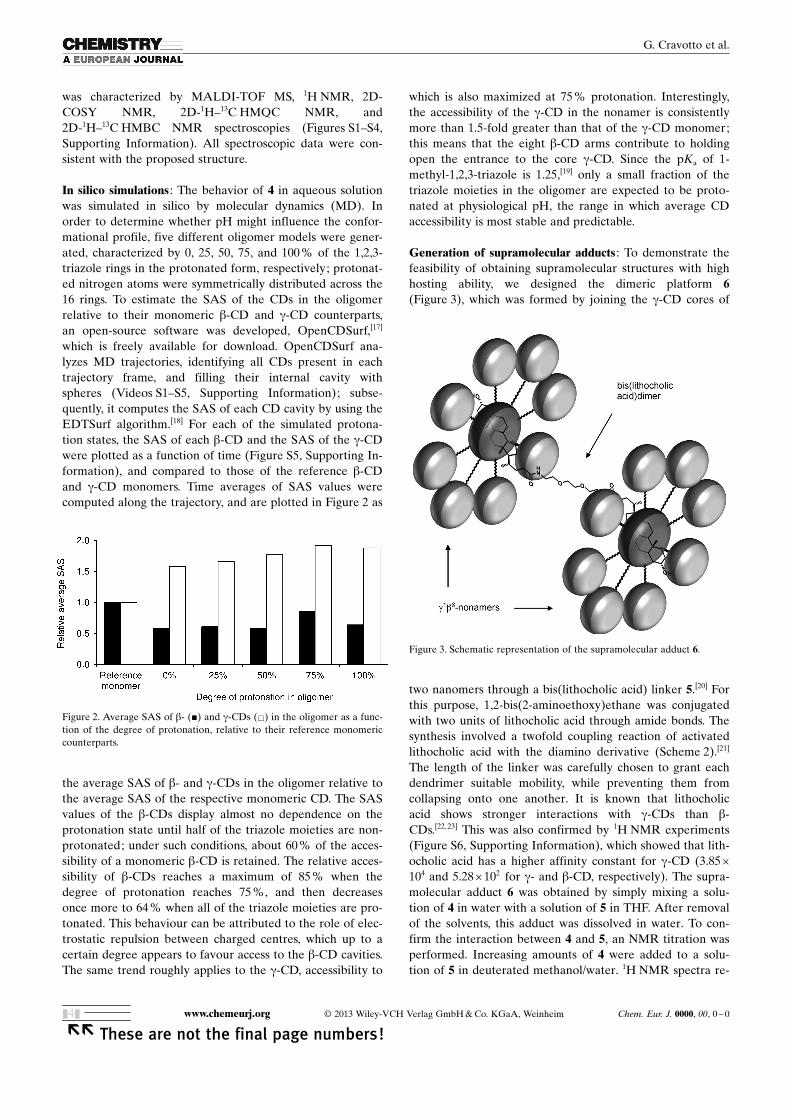
In silico simulations : The behavior of 4 in aqueous solutionwas simulated in silico by molecular dynamics (MD). Inorder to determine whether pH might influence the confor-mational profile, five different oligomer models were gener-ated, characterized by 0, 25, 50, 75, and 100 % of the 1,2,3-triazole rings in the protonated form, respectively; protonat-ed nitrogen atoms were symmetrically distributed across the16 rings. To estimate the SAS of the CDs in the oligomerrelative to their monomeric b-CD and g-CD counterparts,an open-source software was developed, OpenCDSurf,[17]
which is freely available for download. OpenCDSurf ana-lyzes MD trajectories, identifying all CDs present in eachtrajectory frame, and filling their internal cavity withspheres (Videos S1–S5, Supporting Information); subse-quently, it computes the SAS of each CD cavity by using theEDTSurf algorithm.[18] For each of the simulated protona-tion states, the SAS of each b-CD and the SAS of the g-CDwere plotted as a function of time (Figure S5, Supporting In-formation), and compared to those of the reference b-CDand g-CD monomers. Time averages of SAS values werecomputed along the trajectory, and are plotted in Figure 2 as
the average SAS of b- and g-CDs in the oligomer relative tothe average SAS of the respective monomeric CD. The SASvalues of the b-CDs display almost no dependence on theprotonation state until half of the triazole moieties are non-protonated; under such conditions, about 60 % of the acces-sibility of a monomeric b-CD is retained. The relative acces-sibility of b-CDs reaches a maximum of 85 % when thedegree of protonation reaches 75 %, and then decreasesonce more to 64 % when all of the triazole moieties are pro-tonated. This behaviour can be attributed to the role of elec-trostatic repulsion between charged centres, which up to acertain degree appears to favour access to the b-CD cavities.The same trend roughly applies to the g-CD, accessibility to
which is also maximized at 75 % protonation. Interestingly,the accessibility of the g-CD in the nonamer is consistentlymore than 1.5-fold greater than that of the g-CD monomer;this means that the eight b-CD arms contribute to holdingopen the entrance to the core g-CD. Since the pKa of 1-methyl-1,2,3-triazole is 1.25,[19] only a small fraction of thetriazole moieties in the oligomer are expected to be proto-nated at physiological pH, the range in which average CDaccessibility is most stable and predictable.
Generation of supramolecular adducts : To demonstrate thefeasibility of obtaining supramolecular structures with highhosting ability, we designed the dimeric platform 6(Figure 3), which was formed by joining the g-CD cores of
two nanomers through a bis(lithocholic acid) linker 5.[20] Forthis purpose, 1,2-bis(2-aminoethoxy)ethane was conjugatedwith two units of lithocholic acid through amide bonds. Thesynthesis involved a twofold coupling reaction of activatedlithocholic acid with the diamino derivative (Scheme 2).[21]
The length of the linker was carefully chosen to grant eachdendrimer suitable mobility, while preventing them fromcollapsing onto one another. It is known that lithocholicacid shows stronger interactions with g-CDs than b-CDs.[22,23] This was also confirmed by 1H NMR experiments(Figure S6, Supporting Information), which showed that lith-ocholic acid has a higher affinity constant for g-CD (3.85 �104 and 5.28 � 102 for g- and b-CD, respectively). The supra-molecular adduct 6 was obtained by simply mixing a solu-tion of 4 in water with a solution of 5 in THF. After removalof the solvents, this adduct was dissolved in water. To con-firm the interaction between 4 and 5, an NMR titration wasperformed. Increasing amounts of 4 were added to a solu-tion of 5 in deuterated methanol/water. 1H NMR spectra re-
Figure 2. Average SAS of b- (&) and g-CDs (&) in the oligomer as a func-tion of the degree of protonation, relative to their reference monomericcounterparts.
Figure 3. Schematic representation of the supramolecular adduct 6.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 0000, 00, 0 – 0
�� These are not the final page numbers!&4&
G. Cravotto et al.

corded after each addition showed a characteristic shift andline broadening of the signals of the methyl protons of 5(Figure S7, Supporting Information) owing to the interactionbetween 4 and 5. In fact, upon interaction with the CD den-drimer, the lithocholic acid residue increased its reorienta-tional correlation time, resulting in a reduction of the trans-versal relaxation time and an increase in line width.
Assessment of ligand binding affinity and loading ratio : Thecarrier abilities of CD nonamer 4 and bis(CD-nonamer)adduct 6 were assessed by loading their respective b-CDcavities with an appropriate paramagnetic metal complex.For this purpose, a Gd complex was functionalized with anadamantyl moiety, which is well-known to have high affinitytoward the b-CD cavity (7, Scheme 3).[24] The strengths ofthe interactions between 7 and dendrimers 4 and 6 werestudied by relaxometric titration. Binding parameters (affin-ity constant KA, number of equivalent and independentbinding sites n, and relaxivity of the supramolecular adductrb
1) were determined by using the proton relaxation enhance-ment (PRE) method, which considers the relaxation en-hancement due to the formation of a slow-moving macro-molecular adduct.[25] The binding parameters were deter-mined by two different experiments (Figure 4). In the firstexperiment, the water proton relaxation rate (r1 =1/T1;20 MHz and 25 8C) of a 0.1 mm solution of 7 was measuredas a function of the concentration of 4 (or 6, data notshown). Analysis of the experimental data gave very similaraffinity constants (nKA =3.7 � 105
m�1 for 4 ; 2.2 � 105
m�1 for
6) and rb1 values (12.4 mm
�1 s�1 for 4 ; 12.5 mm�1 s�1 for 6).
The second experiment involved a relaxometric titration ofa fixed concentration (0.05 mm) of 4 (or 6, data not shown)with 7. The water proton relaxation rate increased linearly
Scheme 2. Synthetic route leading to the lithocholic acid dimer 5 (DCC =
N,N-dicyclohexylcarbodiimide; NHS=N-hydroxysuccinimide).
Scheme 3. Synthesis of the adamantyl Gd complex 7 (DIPEA= N,N-di-ACHTUNGTRENNUNGisopropylethylamine).
Figure 4. a) Observed proton relaxation rate of a 0.1 mm solution of 7 inPBS as a function of increasing concentration of 4, measured at 20 MHz
and 25 8C. b) Observed proton relaxation rate of a 0.05 mm solution of 4in PBS as a function of increasing concentration of 7, measured at20 MHz and 25 8C.
Chem. Eur. J. 2013, 00, 0 – 0 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org
These are not the final page numbers! ��&5&
FULL PAPERA g1b8-Cyclodextrin Oligomer as a Dendrimeric Multicarrier
with the concentration of 7 until a discontinuity in the slope,which indicated that the oligomer binding sites had been sa-turated. After this point, an additional contribution to theoverall relaxation rate was observed, which could be attrib-uted to the free Gd complex. The 7/4 ratio of 8 and 7/6 ratioof 16, which were found at this point, roughly correspond tothe numbers of binding sites (n). These data indicate thatthe eight b-CD cavities present on the surface of 4 and thesixteen cavities of the supramolecular adduct 6 are all fullyloaded with 7. In the case of 4, the average affinity decreas-es slightly if the g-CD cavity is considered as a ninth interac-tion site. This was corroborated by MD simulations, whichindicated the presence of a larger than expected cavity inthe g-CD unit, resulting in negligible interaction with theGd complex.
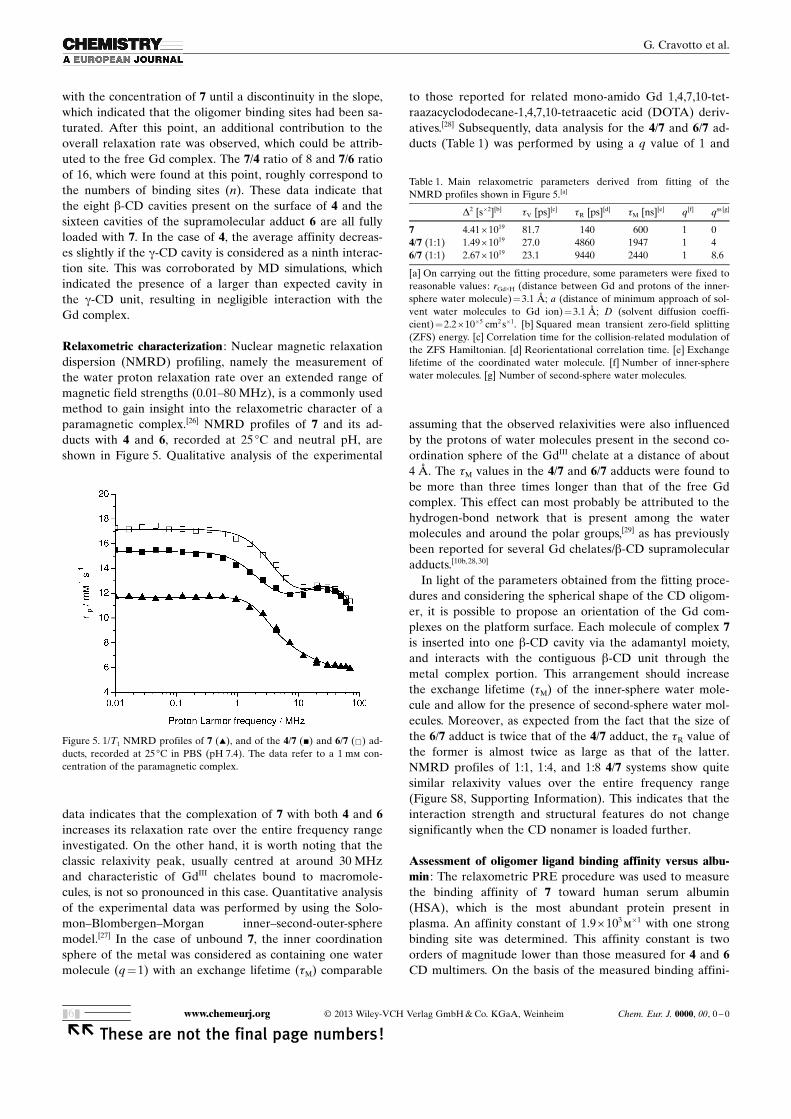
Relaxometric characterization : Nuclear magnetic relaxationdispersion (NMRD) profiling, namely the measurement ofthe water proton relaxation rate over an extended range ofmagnetic field strengths (0.01–80 MHz), is a commonly usedmethod to gain insight into the relaxometric character of aparamagnetic complex.[26] NMRD profiles of 7 and its ad-ducts with 4 and 6, recorded at 25 8C and neutral pH, areshown in Figure 5. Qualitative analysis of the experimental
data indicates that the complexation of 7 with both 4 and 6increases its relaxation rate over the entire frequency rangeinvestigated. On the other hand, it is worth noting that theclassic relaxivity peak, usually centred at around 30 MHz
and characteristic of GdIII chelates bound to macromole-cules, is not so pronounced in this case. Quantitative analysisof the experimental data was performed by using the Solo-mon–Blombergen–Morgan inner–second-outer-spheremodel.[27] In the case of unbound 7, the inner coordinationsphere of the metal was considered as containing one watermolecule (q=1) with an exchange lifetime (tM) comparable
to those reported for related mono-amido Gd 1,4,7,10-tet-raazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) deriv-atives.[28] Subsequently, data analysis for the 4/7 and 6/7 ad-ducts (Table 1) was performed by using a q value of 1 and
assuming that the observed relaxivities were also influencedby the protons of water molecules present in the second co-ordination sphere of the GdIII chelate at a distance of about4 �. The tM values in the 4/7 and 6/7 adducts were found tobe more than three times longer than that of the free Gdcomplex. This effect can most probably be attributed to thehydrogen-bond network that is present among the watermolecules and around the polar groups,[29] as has previouslybeen reported for several Gd chelates/b-CD supramolecularadducts.[10b, 28,30]
In light of the parameters obtained from the fitting proce-dures and considering the spherical shape of the CD oligom-er, it is possible to propose an orientation of the Gd com-plexes on the platform surface. Each molecule of complex 7is inserted into one b-CD cavity via the adamantyl moiety,and interacts with the contiguous b-CD unit through themetal complex portion. This arrangement should increasethe exchange lifetime (tM) of the inner-sphere water mole-cule and allow for the presence of second-sphere water mol-ecules. Moreover, as expected from the fact that the size ofthe 6/7 adduct is twice that of the 4/7 adduct, the tR value ofthe former is almost twice as large as that of the latter.NMRD profiles of 1:1, 1:4, and 1:8 4/7 systems show quitesimilar relaxivity values over the entire frequency range(Figure S8, Supporting Information). This indicates that theinteraction strength and structural features do not changesignificantly when the CD nonamer is loaded further.
Assessment of oligomer ligand binding affinity versus albu-min : The relaxometric PRE procedure was used to measurethe binding affinity of 7 toward human serum albumin(HSA), which is the most abundant protein present inplasma. An affinity constant of 1.9 � 103
m�1 with one strong
binding site was determined. This affinity constant is twoorders of magnitude lower than those measured for 4 and 6CD multimers. On the basis of the measured binding affini-
Figure 5. 1/T1 NMRD profiles of 7 (~), and of the 4/7 (&) and 6/7 (&) ad-ducts, recorded at 25 8C in PBS (pH 7.4). The data refer to a 1 mm con-centration of the paramagnetic complex.
Table 1. Main relaxometric parameters derived from fitting of theNMRD profiles shown in Figure 5.[a]
D2 [s�2][b] tV [ps][c] tR [ps][d] tM [ns][e] q[f] qss [g]
7 4.41 � 1019 81.7 140 600 1 04/7 (1:1) 1.49 � 1019 27.0 4860 1947 1 46/7 (1:1) 2.67 � 1019 23.1 9440 2440 1 8.6
[a] On carrying out the fitting procedure, some parameters were fixed toreasonable values: rGd�H (distance between Gd and protons of the inner-sphere water molecule)=3.1 �; a (distance of minimum approach of sol-vent water molecules to Gd ion)=3.1 �; D (solvent diffusion coeffi-cient) =2.2 � 10�5 cm2 s�1. [b] Squared mean transient zero-field splitting(ZFS) energy. [c] Correlation time for the collision-related modulation ofthe ZFS Hamiltonian. [d] Reorientational correlation time. [e] Exchangelifetime of the coordinated water molecule. [f] Number of inner-spherewater molecules. [g] Number of second-sphere water molecules.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 0000, 00, 0 – 0
�� These are not the final page numbers!&6&
G. Cravotto et al.

ties toward HSA and 6, it was calculated that, when the con-centrations of 7 and 6 are 1 mm and 62 mm (1/16 of 7), re-spectively, and considering HSA to be present in humanplasma at a concentration of 0.58 mm, 68 % of 7 is bound to6, 15 % is bound to albumin, and 17 % is free. These data in-dicate that, once administered in vivo, most of the paramag-netic probe should be loaded on the CD supramolecularstructure.
Assessment of cytotoxicity: Finally, with a view to the po-tential application of these macromolecular carriers in drugdelivery, cell labeling, and/or in vivo applications, a cytotox-icity test against a glioblastoma cell line (U87 MG) was per-formed (Figure S9, Supporting Information). Cell viabilitywas evaluated by using the Trypan Blue exclusion test andreferenced to untreated cells as a control (100% viable). Anacceptable viable cell value (>80 %) was recovered whenusing a carrier concentration lower than 0.1 mm, whichwould in turn enable the transport of drug (or diagnostic)molecules at a concentration of 0.8–1.6 mm in the case of 4and 6, respectively. An inherent challenge with CD plat-forms for in vivo host–guest applications is the competitionfrom binding of the included ligands to plasma proteins.
Conclusion
A new g1b8-CD platform has been designed and synthesized.It fulfils the main requirements for use as a multi-carrier,namely water solubility, surface accessibility, and hosting ca-pability. Exploiting the different hosting abilities of b- andg-CDs, we have shown that it is possible to assemble supra-molecular adducts by joining multiple platforms through ap-propriate linkers. These findings may open up access to awide range of potential pharmaceutical and diagnostic appli-cations.
Experimental Section
Chemistry : Commercially available reagents and solvents were used with-out further purification. Native CDs were kindly provided by WackerChemie Italia srl (Italy). Reactions were monitored by TLC on Merck 60F254 (0.25 mm) plates. Spot detection was carried out by staining with5% H2SO4 in ethanol. IR spectra were recorded on a Shimadzu FTIR8001 spectrophotometer. NMR spectra were recorded at 25 8C on aBruker Avance 300 spectrometer operating at 7 T; chemical shifts (d) aregiven in ppm, coupling constants (J) in Hz. MALDI-TOF mass spectrawere recorded on a Bruker Ultraflex TOF mass spectrometer. MW-pro-moted reactions were carried out in a professional oven (MicroSYNTH-Milestone, Italy). The reaction temperature was monitored by a fibre-optic thermometer directly inserted into the reaction vessel. HPLC analy-ses were carried out at 25 8C with a flow rate of 1 mL min�1 on a WatersXBridge BEH 130 column (4.6/100, 5 mm) by using an evaporative lightscatter detector (ELSD), with H2O/trifluoroacetic acid (TFA) 0.1% v/v(phase A) and MeOH/TFA 0.1% v/v (phase B) as mobile phases. HPLCmethod (time (min), %B): 0, 10; 1.68, 10; 8.32, 35; 13.30, 35; 23.28, 100;29.00, 100. The injection volume was 10 mL. Separations were carried outat 25 8C with a flow rate of 20 mL min�1 by using a Waters XBridge BEH130 column (19/100, 5 mm) with H2O/TFA 0.1% v/v (phase A) and
MeOH/TFA 0.1% v/v (phase B) as mobile phases. HPLC method (time(min), %B): 0, 20; 7.10, 20; 8.51, 30; 15.60, 30; 17.02, 40; 23.00, 40.
6I-Deoxy-6I-[4-(hept-6-ynyl)-1H-1,2,3-triazolyl]-b-CD (2): The reactionbetween 1 (1.00 g, 0.862 mmol) and 1,8-nonadiyne (670 mL, 4.48 mmol)was carried out in a professional MW oven by using a pressure-resistantclosed vessel equipped with a fibre-optic thermometer and a magneticstirring bar. The reaction mixture, which also contained CuSO4 (43 mg,0.172 mmol) and l-ascorbic acid (60.6 mg, 0.345 mmol), was dissolved inDMF (10 mL) and heated at 70 8C (70 W) for 2 h. After concentrationunder vacuum to half of the original volume and addition of acetone(10 mL), a solid product was collected by filtration on a Hirsch funnel.The product was washed with water (3 � 5 mL) and used without furtherpurification (white powder; yield 86 %). Purity: 97.4 % by HPLC-ELSD.Rf : 0.36 (silica; iPrOH/H2O/EtOAc/NH4OH, 5:3:1:1); IR (KBr): n=
3420, 2928, 1460, 1369, 1032, 1157, 1030, 945, 756 cm�1; NMR and MSdata are reported in the Supporting Information (Figures S1–S3 and re-spective captions).
g1b8-CD oligomer (4): The reaction between 2 (300 mg, 0.234 mmol) andper-6-azido-per-6-deoxy-g-CD 3 (29.2 mg, 0.0195 mmol) was carried outin a two-necked round-bottomed flask (25 mL) equipped with a stirringbar and a fibre-optic thermometer in a professional MW oven. The reac-tion mixture, which also contained CuSO4 (58.8 mg, 0.234 mmol) and l-ascorbic acid (82.5 mg, 0.468 mmol), was suspended in a DMF/H2O mix-ture (1:5) and heated at 100 8C (120 W) for 3 h. The crude product waseither purified by semi-preparative HPLC (pale-yellow powder; yield22%) or by the addition of Na2H2EDTA and ultrafiltration (3 kDa disc)in a stirred cell (freeze-dried, pale-yellow powder; yield 79 %). NMR andMS data are reported in the Supporting Information (Figure S4 and cap-tion).
Bis(deoxycholamide) of 1,2-bis(2-aminoethoxy)ethane (5): N-Hydroxy-succinimide (345 mg, 3 mmol) and N,N-dicyclohexylcarbodiimide (DCC;621 mg, 3.04 mmol) were added to a solution of lithocholic acid (1.0 g,2.66 mmol) in THF (20 mL). The mixture was stirred for 4 h at roomtemperature and filtered to remove the insoluble urea. A solution pre-pared from 1,2-bis(2-aminoethoxy)ethane (148 mL, 1.0 mmol), triethyla-mine (1 mL), and water (5 mL) was added to the filtrate. After being stir-red overnight, the mixture was poured into 1 m aqueous HCl (25 mL).The resulting solid was collected by filtration and purified by columnchromatography (silica gel; CH2Cl2/CH3OH/NH4OH, 95:5:0.3 v/v/v) togive the bis(deoxycholamide) of 1,2-bis(2-aminoethoxy)ethane (yield51%). Rf : 0.59 (silica; CHCl3/MeOH/H2O, 6:1:0.05); 1H NMR([D6]DMSO, 300 MHz): d =0.64 (s, 6H), 0.91–2.13 (m, 68H), 3.2–3.23(m, 4H), 3.35–3.44 (m, overlay with water signal), 3.53 (s, 4 H), 4.49 (d,2H), 7.84–7.88 ppm (m, 2 H); MS (ESI): m/z calcd for C54H92N2O6:866.32 [M+H]+ ; found: 866.09.
g1b8-Nonamer/2,2’-(ethylenedioxy)bis(ethylamine)dideoxycholamidecomplex (6): Nonamer 4 (50 mg, 0.00426 mmol) was dissolved in water(1 mL), and then a solution of 2,2’-(ethylenedioxy)bis(ethylamine)dideox-ycholamide (1.84 mg, 0.00213 mmol) in THF (500 mL) was added. Afterevaporation of the THF, the solution was freeze-dried.
1-[9-(1-Adamantyl)carbonylamino-3-aza-2-oxononyl]-1,4,7,10-tetraazacy-clododecane-4,7,10-triacetate gadolinium (7): The complexation reactionwas performed with GdCl3 in aqueous solution at pH 7 by the ligand ad-dition method.[31] An equimolar amount of aqueous GdCl3 (524 mg,1.41 mmol) solution was slowly added to an aqueous solution of 7 c(1.00 g, 1.41 mmol), maintaining the pH value at 7 with 0.1 n NaOH. Themixture was stirred at room temperature until the pH remained constant.When a slight excess of the metal cation was reached, as monitored withthe orange xylenol assay,[32] a small excess of ligand was added (<0.13 %). The complex was then desalted by size-exclusion chromatogra-phy on Sephadex G10 resin and freeze-dried (yield 53%).
Computational methods : Molecular simulations were carried out byusing MOE,[33] AMBER 10,[34] and our in-house software OpenCDSurf.[17]
Full methodological details are reported in the Supporting Information.
Chem. Eur. J. 2013, 00, 0 – 0 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org
These are not the final page numbers! ��&7&
FULL PAPERA g1b8-Cyclodextrin Oligomer as a Dendrimeric Multicarrier

Acknowledgements
The computational work was supported by the Chemical ComputingGroup.
[1] a) M. A. Mintzer, M. W. Grinstaff, Chem. Soc. Rev. 2011, 40, 173 –190; b) K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M.Ferrari, H. Fuchs, Angew. Chem. 2009, 121, 886 – 913; Angew. Chem.Int. Ed. 2009, 48, 872 –897; c) W. J. Stark, Angew. Chem. 2011, 123,1276 – 1293; Angew. Chem. Int. Ed. 2011, 50, 1242 –1258; d) C. C.Lee, J. A. MacKay, J. M. J. Frechet, F. C. Szoka, Nat. Biotechnol.2005, 23, 1517 –1526.
[2] L. Rçglin, E. H. M. Lempens, E. W. Meijer, Angew. Chem. 2011,123, 106 –117; Angew. Chem. Int. Ed. 2011, 50, 102 – 112.
[3] a) A. Samad, Y. Sultana, M. Aqil, Curr. Drug Delivery 2007, 4, 297 –305; b) W. T. Al-Jamal, K. Kostarelos, Acc. Chem. Res. 2011, 44,1094 – 1104.
[4] a) D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit, R.Langer, Nat. Nanotechnol. 2007, 2, 751 –760; b) J. Shi, Z. Xiao, N.Kamaly, O. C. Farokhzad, Acc. Chem. Res. 2011, 44, 1123 – 1134.
[5] Y.-W. Jun, J.-H. Lee, J. Cheon, Angew. Chem. 2008, 120, 5200 –5213;Angew. Chem. Int. Ed. 2008, 47, 5122 –5135.
[6] a) J. L. Perry, K. P. Herlihy, M. E. Napier, J. M. Desimone, Acc.Chem. Res. 2011, 44, 990 – 998; b) S. E. A. Gratton, S. S. Williams,M. E. Napier, P. D. Pohlhaus, Z. Zhou, K. B. Wiles, B. W. Maynor,C. Shen, T. Olafsen, E. T. Samulski, J. M. Desimone, Acc. Chem.Res. 2008, 41, 1685 – 1695.
[7] S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer, W. C. W. Chan,Nano Lett. 2009, 9, 1909 –1915.
[8] a) Y. Teow, P. V. Asharani, M. P. Handec, S. Valiyaveettil, Chem.Commun. 2011, 47, 7025 – 7038; b) K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland, M. L. Forrest, Adv. Drug Delivery Rev. 2009,61, 457 – 466; c) K. R. Vega-Villa, J. K. Takemoto, J. A. Y�Çez, C. M.Remsberg, M. L. Forrest, N. M. Davies, Adv. Drug Delivery Rev.2008, 60, 929 –938.
[9] a) M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tol-cher, C. A. Alabi, Y. Yen, J. D. Heidel, A. Ribas, Nature 2010, 464,1067 – 1070; b) Y. Zhu, L. Che, H. He, Y. Jia, J. Zhang, X. Li, J. Con-trolled Release 2011, 152, 317 –324; c) M. Adeli, F. Hakimpoor, M.Parsamanesh, M. Kalantari, Z. Sobhani, F. Attyabi, Polymer 2011,52, 2401 –2413.
[10] a) H. S. Christensen, B. W. Sigurskjold, T. G. Frihed, L. G. Marine-scu, C. M. Pedersen, M. Bols, Eur. J. Org. Chem. 2011, 5279 –5290;b) S. Aime, E. Gianolio, F. Arena, A. Barge, K. Martina, G. Hero-poulos, G. Cravotto, Org. Biomol. Chem. 2009, 7, 370 – 379.
[11] a) T. Lecourt, Y. Bl�riot, R. Auz�ly-Velty, M. Sollogoub, Chem.Commun. 2010, 46, 2238 – 2240; b) T. Jiang, M. Li, D. S. Lawrence, J.Org. Chem. 1995, 60, 7293 – 7297; c) R. Breslow, X. Zhang, R. Xu,M. Maletic, R. Merger, J. Am. Chem. Soc. 1996, 118, 11678 –11679.
[12] a) M. Fathalla, A. Neuberger, S.-C. Li, R. Schmehl, U. Diebold, J.Jayawickramarajah, J. Am. Chem. Soc. 2010, 132, 9966 –9967;b) G. K. Rawal, P. Zhang, C.-C. Ling, Org. Lett. 2010, 12, 3096 –3099; c) Z. Kotkov�, J. Kotek, D. Jir�k, P. Jendelov�, V. Herynek, Z.
Berkov�, P. Hermann, I. Lukes, Chem. Eur. J. 2010, 16, 10094 –10102.
[13] a) G. Cravotto, V. V. Fokin, D. Garella, A. Binello, L. Boffa, A.Barge, J. Comb. Chem. 2010, 12, 13 –15; b) A. Barge, S. Tagliapietra,A. Binello, G. Cravotto, Curr. Org. Chem. 2011, 15, 189 – 203.
[14] G. Cravotto, F. Mendicuti, K. Martina, S. Tagliapietra, B. Robaldo,A. Barge, Synlett 2008, 2642 – 2646.
[15] K. Martina, F. Trotta, B. Robaldo, N. Belliardi, L. Jicsinszky, G. Cra-votto, Tetrahedron Lett. 2007, 48, 9185 – 9189.
[16] S. Srinivasachari, K. M. Fichter, T. M. Reineke, J. Am. Chem. Soc.2008, 130, 4618 –4627.
[17] P. Tosco, OpenCDSurf, http://opencdsurf.sourceforge.net, 2012.[18] D. Xu, Y. Zhang, PLoS One 2009, 4, e8140.[19] J.-L. M. Abboud, C. Foces-Foces, R. Notario, R. E. Trifonov, A. P.
Volovodenko, V. A. Ostrovskii, I. Alkorta, J. Elguero, Eur. J. Org.Chem. 2001, 3013 –3024.
[20] S. Aime, E. Gianolio, F. Uggeri, S. Tagliapietra, A. Barge, G. Cra-votto, J. Inorg. Biochem. 2006, 100, 931 –938.
[21] V. Janout, B. Jing, S. L. Regen, J. Am. Chem. Soc. 2005, 127, 15862 –15870.
[22] Y. Shakalisava, F. Regan, Electrophoresis 2006, 27, 3048 – 3056.[23] M. Narita, F. Hamada, J. Chem. Soc. Perkin Trans. 2 2000, 4, 823 –
832.[24] J. Carrazana, A. Jover, F. Meijide, V. H. Soto, J. Vasquez Tato, J.
Phys. Chem. B 2005, 109, 9719.[25] S. Aime, M. Botta, M. Fasano, E. Terreno in Chemistry of Contrast
Agents in Medical Magnetic Resonance Imaging (Eds.: A. E. Mer-bach, �. T�th), Wiley, Chichester, 2001, pp. 193 – 241.
[26] D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebe-dev, W. Schlaepfer, A. E. Merbach, J. Am. Chem. Soc. 1996, 118,9333 – 9346.
[27] a) N. Bloembergen, J. Chem. Phys. 1957, 27, 572 – 573; b) I. Solo-mon, Phys. Rev. 1955, 99, 559 –565.
[28] a) L. Tei, A. Barge, S. Geninatti Crich, R. Pagliarin, V. Negri, D. Ra-mella, G. Cravotto, S. Aime, Chem. Eur. J. 2010, 16, 8080 –8087;b) E. Battistini, E. Gianolio, R. Gref, P. Couvreur, S. Fuzerova, M.Othman, S. Aime, B. Badet, P. Durand, Chem. Eur. J. 2008, 14,4551 – 4561; c) P. Caravan, J. J. Ellison, T. J. McMurry, R. B. Lauffer,Chem. Rev. 1999, 99, 2293 –2352.
[29] M. Botta, Eur. J. Inorg. Chem. 2000, 399 –407.[30] S. Aime, M. Botta, F. Fedeli, E. Gianolio, E. Terreno, P. Anelli,
Chem. Eur. J. 2001, 7, 5261 – 5269.[31] S. Geninatti Crich, C. Cabella, A. Barge, S. Belfiore, C. Ghirelli, L.
Lattuada, S. Lanzardo, A. Mortillaro, L. Tei, M. Visigalli, G. Forni,S. Aime, J. Med. Chem. 2006, 49, 4926 –4936.
[32] A. Barge, G. Cravotto, E. Gianolio, F. Fedeli, Contrast Media Mol.Imaging 2006, 1, 184 – 188.
[33] MOE version 2010.11, Chemical Computing Group Inc., Montreal,Quebec, 2011.
[34] D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo,K. M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang, R. Woods, J.Comput. Chem. 2005, 26, 1668 –1688.
Received: March 30, 2013Revised: June 16, 2013
Published online: && &&, 0000
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 0000, 00, 0 – 0
�� These are not the final page numbers!&8&
G. Cravotto et al.














![Chiral separation by a monofunctionalized cyclodextrin derivative: From selector to permethyl-[beta]-cyclodextrin bonded stationary phase](https://static.fdokumen.com/doc/165x107/63327b24576b626f850d70ad/chiral-separation-by-a-monofunctionalized-cyclodextrin-derivative-from-selector.jpg)





