
Dermatofibrosarcoma protuberans: a comprehensive review and update on diagnosis and management
16
Dermatofibrosarcoma protuberans: a comprehensive review and update on diagnosis and management Beatriz Llombart, MD, a Carlos Serra-Guillén, MD, a Carlos Monteagudo, MD, PhD, b José Antonio López Guerrero, PhD, c and Onofre Sanmartín, MD a From the a Dermatology Department, Instituto Valenciano de Oncología, Valencia, Spain; b Pathology Department. Hospital Clínico Universitario de Valencia, Universidad de Valencia, Valencia, Spain; and the c Laboratory of Molecular Biology, Instituto Valenciano de Oncología, Valencia, Spain. Dermatofibrosarcoma protuberans (DFSP) is a rare superficial tumor characterized by high rates of local recurrence and low risk of metastasis. DFSP occurs most commonly on the trunk and proximal extremities, affects all races, and often develops between the second and fifth decade of life. The tumor grows slowly, typically over years. Histologically, several variants of DFSP have been described and should be well characterized to avoid misdiagnosis with other tumors. These include pigmented (Bednar tumor), myxoid, myoid, granular cell, sclerotic, atrophic DFSP, giant cell fibroblastoma, and DFSP with fibrosarcomatous areas. Of all these variants, only the DFSP with fibrosarcomatous areas is high grade, with a higher rate of local recurrence and distant metastasis. DFSP is genetically characterized by the t(17;22)(q22;q13), resulting in the fusion of alpha chain type 1 of collagen gene and platelet-derived growth factor beta gene. This translocation is present in 90% of DFSP and represents a very useful tool in the differential diagnosis of DFSP with other tumors with similar histology. The standard treatment is wide local excision with at least a 2-cm margin. However, local recurrence after apparently adequate surgical excision is well recognized. Mohs micrographic surgery would be the treatment of choice with a better cure rate and maximal conservation of tissue. When surgery is insufficient, clinical evidence has suggested that imatinib mesylate is a safe and effective treatment in DFSP, especially in cases of local advanced or metastatic disease. This article presents an overview of the state of the art in the clinicopathological management of this disease. © 2013 Elsevier Inc. All rights reserved. KEYWORDS Dermatofibrosarcoma protuberans; Fibrosarcomatous; Imatinib; Mohs micrographic surgery; Review; Genetic Dermatofibrosarcoma protuberans (DFSP) is a relatively unusual, locally aggressive cutaneous tumor, characterized by high rates of local recurrence, but low risk of metasta- sis. 1,2 The first descriptions of this entity were made inde- pendently in 1890 by Sherwell 3 and Taylor. 4 In 1924, Darier and Ferrand 5 designated this tumor as a progressive and recurrent dermatofibroma. One year later, based on the tendency of the tumor to develop protruding nodules, Hoff- man 6 coined the term DFSP. Most early reports of DFSP described the clinical characteristics and the tendency for recurrence after surgical excision. In 1962, Taylor and Helwig, 7 in a review of 115 cases, described the histologic characteristics of the neoplasia in detail and characterized a fibroblastic growth appearing as a low-grade sarcoma in which the tumor cells were organized in fascicles with a spiral or cartwheel arrangement. Gener- ally, the neoplastic cells show little or no pleomorphism, and the mitotic rate is low. Histologically, several variants have been described that include pigmented (Bednar tu- Address reprint requests and correspondence: Beatriz Llombart, MD, Departamento de dermatología, Instituto Valenciano de Oncología, C/Profesor Beltrán Báguena N°8, 46009 Valencia, Spain. E-mail address: [email protected]. 0740-2570/$ -see front matter © 2013 Elsevier Inc. All rights reserved. doi:10.1053/j.semdp.2012.01.002 Seminars in Diagnostic Pathology (2013) 30, 13-28
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Dermatofibrosarcoma protuberans: a comprehensive review and update on diagnosis and management

pa
Seminars in Diagnostic Pathology (2013) 30, 13-28
Dermatofibrosarcoma protuberans: a comprehensivereview and update on diagnosis and management
Beatriz Llombart, MD,a Carlos Serra-Guillén, MD,a Carlos Monteagudo, MD, PhD,b
José Antonio López Guerrero, PhD,c and Onofre Sanmartín, MDa
From the aDermatology Department, Instituto Valenciano de Oncología, Valencia, Spain;bPathology Department. Hospital Clínico Universitario de Valencia, Universidad de Valencia, Valencia, Spain; and the
cLaboratory of Molecular Biology, Instituto Valenciano de Oncología, Valencia, Spain.Dermatofibrosarcoma protuberans (DFSP) is a rare superficial tumor characterized by high rates of localrecurrence and low risk of metastasis. DFSP occurs most commonly on the trunk and proximalextremities, affects all races, and often develops between the second and fifth decade of life. The tumorgrows slowly, typically over years. Histologically, several variants of DFSP have been described andshould be well characterized to avoid misdiagnosis with other tumors. These include pigmented(Bednar tumor), myxoid, myoid, granular cell, sclerotic, atrophic DFSP, giant cell fibroblastoma, andDFSP with fibrosarcomatous areas. Of all these variants, only the DFSP with fibrosarcomatous areas ishigh grade, with a higher rate of local recurrence and distant metastasis. DFSP is geneticallycharacterized by the t(17;22)(q22;q13), resulting in the fusion of alpha chain type 1 of collagen geneand platelet-derived growth factor beta gene. This translocation is present in 90% of DFSP andrepresents a very useful tool in the differential diagnosis of DFSP with other tumors with similarhistology. The standard treatment is wide local excision with at least a 2-cm margin. However, localrecurrence after apparently adequate surgical excision is well recognized. Mohs micrographic surgerywould be the treatment of choice with a better cure rate and maximal conservation of tissue. Whensurgery is insufficient, clinical evidence has suggested that imatinib mesylate is a safe and effectivetreatment in DFSP, especially in cases of local advanced or metastatic disease. This article presents anoverview of the state of the art in the clinicopathological management of this disease.© 2013 Elsevier Inc. All rights reserved.
KEYWORDSDermatofibrosarcomaprotuberans;Fibrosarcomatous;Imatinib;Mohs micrographicsurgery;Review;Genetic
dr
Dermatofibrosarcoma protuberans (DFSP) is a relativelyunusual, locally aggressive cutaneous tumor, characterizedby high rates of local recurrence, but low risk of metasta-sis.1,2 The first descriptions of this entity were made inde-endently in 1890 by Sherwell3 and Taylor.4 In 1924, Dariernd Ferrand5 designated this tumor as a progressive and
recurrent dermatofibroma. One year later, based on the
Address reprint requests and correspondence: Beatriz Llombart,MD, Departamento de dermatología, Instituto Valenciano de Oncología,C/Profesor Beltrán Báguena N°8, 46009 Valencia, Spain.
E-mail address: [email protected].
0740-2570/$ -see front matter © 2013 Elsevier Inc. All rights reserved.doi:10.1053/j.semdp.2012.01.002
tendency of the tumor to develop protruding nodules, Hoff-man6 coined the term DFSP. Most early reports of DFSPescribed the clinical characteristics and the tendency forecurrence after surgical excision.
In 1962, Taylor and Helwig,7 in a review of 115 cases,described the histologic characteristics of the neoplasia indetail and characterized a fibroblastic growth appearing as alow-grade sarcoma in which the tumor cells were organizedin fascicles with a spiral or cartwheel arrangement. Gener-ally, the neoplastic cells show little or no pleomorphism,and the mitotic rate is low. Histologically, several variants
have been described that include pigmented (Bednar tu-
ipsnf
hacrapso
Dnfepcbtl
Dv1dg
mopD(dp
14 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
mor), myxoid, granular cell, atrophic DFSP, DFSP withfibrosarcomatous areas (DFSP-FS), DFSP with areas ofgiant cell fibroblastoma (GCF), DFSP/DFSP-FS with fociof myoid/myofibroblastic differentiation, and sclerosing/sclerotic DFSP.
In 1993, immunoreactivity for CD34 in DFSP was de-scribed for the first time,8-10 and continues to be the mainmmunohistochemical marker for diagnosis of the DFSP,articularly when associated with the absence of immuno-taining for factor XIIIa. Nevertheless, 10% of DFSP areegative for CD34, and 25% of DFSP can be positive foractor XIIIa.11,12
Cytogenetic analysis of DFSP dates back to 1990, withinitial descriptions showing the presence of a recurrentt(17;22)(q22;q13) translocation or of supernumerary ringchromosomes containing material from chromosomal re-gions 17q22 and 22q13 accompanied by simple chromo-some trisomies.13,14 The combination of fluorescent in situybridization (FISH), comparative genomic hybridization,nd molecular techniques has been valuable in further de-iphering the composition of the DFSP chromosomal rear-angements, showing that these result in the fusion of thelpha chain type 1 of collagen (COL1A1) gene with thelatelet-derived growth factor beta (PDGFB) gene, the tran-criptional upregulation of the PDGFB gene being the resultf this fusion gene.15-18
In recent years, a number of advances have been maderegarding the immunohistochemical, chromosomal, andmolecular features of this tumor. Furthermore, innovativesurgical approaches and emerging targeted pharmacologictreatments have piqued new research and clinical interest inDFSP. This article presents a review and update on theepidemiology, histology, immunohistochemistry, cytoge-netics, and management of this tumor. Special emphasiswill be placed on describing the histology, molecular biol-ogy, and the treatment options through Mohs micrographicsurgery (MMS) and the use of PDGF receptor inhibitors.
Epidemiology
DFSP is a rare tumor that constitutes �0.1% of all malig-nancies and 1% of all soft-tissue sarcomas.1,19 Its incidencein the United States has been calculated to be between 0.8and 4.5 cases per million individuals per year.20,21 Never-theless, DFSP is the most common sarcoma of cutaneousorigin.2
DFSP most commonly occurs between 20 and 50 yearsof age, although its can appear at any age. The age spectrumvaries from congenital cases to patients �90 years. Al-though the proportion of pediatric cases in published seriesof DFSP ranges between 6% and 20%,7,22 DFSP is anasymptomatic tumor with a slow growth, and we believe, asother authors,22-25 that many cases diagnosed in adultsbegin during childhood. In addition, it should be taken intoaccount that GCF is currently considered to be the juvenile
form of DFSP.Although DFSP has been described in all races, it isdifficult to draw specific conclusions regarding the racialincidence of DFSP because race is not mentioned in manyof the larger series of patients. In a recent epidemiologicstudy of 2885 cases,21 the incidence of DFSP in blackindividuals was observed to be approximately twice that ofwhites.
The literature reveals an equal sex distribution, with aslight male predominance in some series21,26,27 and slightfemale predominance in others.21,28,29
DFSP is preferentially located on the trunk. In 40%-50%of cases, the tumor is located in this area, generally on thechest and shoulders; in 30%-40% of cases, the tumor islocated in the proximal portion of the limbs (more often onthe arms than the legs); and in 10%-15% of cases, DFSPaffects the head and neck, generally the scalp, cheek, andsupraclavicular area.1,2 It has been reported that childhood
FSP has a greater tendency toward acral location. Rabi-owitz et al30 reviewed 27 cases of childhood DFSP andound that 14.8% were located on the hands or feet. How-ver, in our review of the 150 pediatric cases of DFSPublished till 2006,23 acral location was reported in �9% ofases. In our experience, acral DFSP is infrequent. Weelieve that some CD34 positive acral lesions described inhe literature as DFSP were in fact other fibroblastic-likeesions, for example, superficial acral fibromyxoma.
A history of trauma as a possible etiologic factor inFSP has been debated. Such events might favor the de-elopment of the tumor, as a history of trauma is reported in0%-20% of cases.2, 7 Likewise, cases of DFSP have beenescribed in which tumors are located on the sites of sur-ical scars,31 burns,32 radiodermatitis,33 vaccination scars,34
and sites of central venous lines.35
Clinical features
The appearance of the tumor depends on the stage of dis-ease, as the tumor progresses slowly over a long periodbefore entering a rapid growth phase.36 DFSP initially ap-pears as an asymptomatic, indurate plaque that may have aviolaceous, red-blue, or brown appearance, with a hardconsistency and fixed to the skin but not the deep layers(Figure 1A).1,7 Over a period, which can vary from a few
onths to decades, the DFSP grows with the developmentf multiple nodules within the plaque, from which its namerotuberans is derived (Figure 1B and C). Less commonly,FSP presents initially as a unique firm cutaneous nodule
Figure 1D). In the initial stages of DFSP (Figure 1A),iagnostic errors are common, with the lesion being inter-reted as a scar, morphea,37 morpheaform basal cell carci-
noma, atrophoderma, or vascular malformations.24 Whenthe tumor progresses (Figure 1B and C) or starts as aprotruding mass (Figure 1D), DFSP may be confused withanother tumor type; however, in our experience, the morefrequent mistaken diagnoses are sebaceous cyst, lipoma, or
dermatofibroma.
huia
sa
15Llombart et al Dermatofibrosarcoma Protuberans
DFSP typically ranges in size from 2 to 5 cm, althoughon some occasions, and if not remedied earlier, these lesionsmay grow as large as 20 cm in diameter (Figure 2) and havemultiple satellite nodules. The tumor is usually fixed tooverlying skin, but not to deeper structures. However, re-current or long-standing tumors may invade fascia, striatedmuscle, periosteum, and bone (Figure 2).38-40
Figure 1 Clinical appearance. (A) Dermatofibrosarcoma protunodules within plaque of DFSP in the supraclavicular area. (C)scalp.
Figure 2 Giant recurrent dermatofibrosarcoma protuberans(DFSP) on the upper back. The histology of this case contained
areas of fibrosarcomatous transformation.Histology
DFSP appears as a poorly circumscribed tumor that infil-trates the whole dermis destroying the preexisting structuresand spreading into the cellular subcutaneous tissue (Figure3A–D). The tumor is composed predominantly of a dense,uniform array of cells with spindle-shaped nuclei embeddedin varying amounts of collagen. This fibroblast-like prolif-eration is typically arranged into irregular, interwoven fas-cicles, resulting in a storiform pattern, as is seen in manyother fibrous proliferations. In some areas, the tumor cellsappear to be arranged radially about a central “hub,” pro-ducing a pattern resembling the spokes of a wheel or whirl-igig (Figure 3D). This pattern is most readily observed inthe more cellular areas and was reported in 1962 by Taylorand Helwig7 as being of great diagnostic value. Tumor cellsave large nuclei with low pleomorphism, and mitotic fig-res are infrequent, even in cellular areas. Inflammatorynfiltrates, hemosiderin deposits, multinucleated giant cells,nd foamy histiocytes are uncommon.41 DFSP may contain
small amounts of stromal mucin, which is often seen justbelow the epidermis.1 Cystic changes, dilated vascularpaces, and hemorrhage are sometimes present, but necrosisnd lymphovascular invasion are rare.7 The epidermis over
the tumors is usually thin, with flattened rete ridges. Lessoften, there is a slight-to-moderate acanthosis, although notto the extent that can be seen in dermatofibromas.7
The main histologic characteristic of DFSP is its capacity
(DFSP) as an asymptomatic, indurated plaque. (B) Protuberantnodular DFSP in the back. (D) DFSP as a unique nodule in the
beransMulti
to invade surrounding tissues to a considerable distance

tutcinp
nr
ctdnmms
a
f(
16 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
from the central focus of the tumor. The cellularity is greaterin the central zone than in the peripheral part of the tumor,where the edges invade the surrounding dermis and subcu-tis. The tumor cells invade the subcutaneous tissue in theform of tentacle-like projections through the septa and fatlobules. These tumor extensions contain few cells and, atfirst sight, can appear similar to normal fibrous tracts. Thismakes it difficult to determine the true extent of the lesionand may be why recurrences appear after excision withapparently wide margins. DFSP invades the fat with aparticular “honeycomb” pattern (Figure 3D) or, more fre-quently, with a multilayered pattern or “sandwich” involv-ing spindle cell layers oriented parallel to the skin sur-face.42,43 Involvement of the fascia, underlying muscles,periosteum, and bone is a late event.
Histologic subtypes
Pigmented DFSP (Bednar tumor)In 1957, Bednar44 originally described a specific tumor
hat showed a characteristic storiform pattern in the dermisnder the name “storiform neurofibroma.” He also dividedhis neoplasm into pigmented and nonpigmented types, ac-ording to the presence of melanin-containing cells. Later,mmunohistochemical studies performed in the cases, origi-ally reported by Bednar, observed that the tumor cells ex-
Figure 3 Histologic appearance of classic dermatofibrosarcomreplacing the dermis (H&E stain, 4�). (B) Dermal locationatrophic epidermis (H&E stain, 40�). (C) Deep aspect of that surrounded by infiltrating tumor cells (H&E stain, 40�H&E stain, 100�).
ressed CD34, but not protein S-100, rejecting their Schwan-
ian origin, and concluded that this neoplasm should beegarded as pigmented DFSP or Bednar tumor.45,46
Pigmented DFSP occurs predominantly in black personsand accounts for approximately only 1%-5% of all cases ofDFSP.45,47 Histologically, however, this unusual tumor isharacterized by spindle cells arranged in a storiform pat-ern admixed with a variable population of melanin-bearingendritic cells (Figure 4A and B). The presence of largeumbers of melanin-containing cells may cause some tu-ors to appear clinically blue or black. In other lesions,elanin is so scant that it can only be appreciated micro-
copically.1
The biological behavior of Bednar tumor is compara-ble with cases of conventional DFSP with very unusualmetastases.48 The fact that conventional DFSP may recurs pigmented DFSP,49 as well as the description of hybrid
pigmented DFSP with either fibroblastoma of giantcells50 or with fibrosarcoma,48,51,52 support the idea thatall these subtypes are different morphologic expressionsof the same neoplasm.
The histogenesis of Bednar tumor remains controver-sial. The presence of dendritic melanocytes and cellssuggestive of Schwannian differentiation has led someauthors to regard these tumors as being of neuroectoder-mal origin.46,53 Others have found no solid evidence ofneuroectodermal differentiation because the pigment
uberans. (A) Low-power view of tumor cells almost completelymatofibrosarcoma protuberans with uninvolved zone beneathor exhibits a honeycomb pattern of entrapped subcutaneous) Interwoven fascicles of cells forming storiform pattern
a protof dere tum). (D
cells are S-100 protein negative, and postulated that

GiydmtotDa
tmda
v
ccDa
�).
17Llombart et al Dermatofibrosarcoma Protuberans
pigmentation may simply reflect secondary melanocytecolonization from the epidermis and not constitute a realtumor component.47,54
GCFShmookler and Enzinger55 presented the original series of
CF as an abstract at the International Academy of Pathologyn Boston in 1982. In this study of 20 cases, 85% were �10ears old. The tumor was moderately cellular, involved theermis and subcutis, and consisted of ovoid fibroblastic cells,ultinucleated giant cells, and pseudovascular spaces. In 1989,
he same authors published a study of 28 cases including theriginal cases.56 The authors suggested a relationship betweenhis childhood tumor and DFSP because they observed cases ofFSP with GCF-likes areas (Hybrid cases) and proposed GCF
s the juvenile variant of DFSP.Histologically, GCF may demonstrate a unique combina-
ion of spindle cell patterns (including the storiform pattern),yxoid areas, pleomorphic and multinucleated giant cells,
istinctive sinusoid-like spaces, and tentacular infiltration ofdjacent subcutaneous tissue (Figure 4C and D).57 The sinu-
soidal spaces of vascular appearance do not constitute authen-tic vascular structures, as these sinusoidal spaces are not cov-ered by endothelial cells but by the neoplastic cells.55,56,58
Ultrastructural studies have demonstrated that the apparentmultinucleated giant cells under light microscope are reallycells with a polysegmented unique nucleus.59,60
The relationship between GCF and DFSP has been sus-
Figure 4 (A) The Bednar tumor is a pigmented variant of DFmelanin-bearing dendritic cells (H&E stain, 40�). (B) This view100�). (C) Giant cell fibroblastoma (GCF) is often myxoid and putissue spaces lined by multinucleated giant cells (H&E stain, 100
pected since the description of numerous cases of hybrid le-
sions,61,62 recurrent cases of GCF as DFSP,56,63 and viceersa,64 as well as recurrent GCF as Bednar tumor.65 Addi-
tional support for this relationship includes the same clinicalappearance, immunohistochemical and molecular features.57
Atrophic DFSPAtrophic DFSP is a depressed plaque, of soft or hard
consistency, which can be clinically confused with mor-phea, anetoderma, morpheaform basal cell carcinoma, andscar.22 The thickness of the dermis is reduced by �50% inomparison with the surrounding dermis, placing the sub-utis close to the epidermis, and, as in other subtypes ofFSP, a spindle cell proliferation replaces the dermis and
lmost invariably extends into the subcutis.22,66-73 The epi-dermis is usually normal or mildly atrophic, although slightand/or focal epidermal hyperplasia may be present. Atro-phic DFSP is usually confused with non-nodular (plaque)DFSP. In fact, all atrophic DFSP are clinically in plaque, butnot all of DFSP in plaque are atrophic at histologic level.74
This variant usually occurs in the third decade of life,affecting both men and women equally. Most cases presentin the trunk, with no differences with conventional DFSPregarding prognosis.75
Sclerosing DFSPSclerosing or sclerotic DFSP is characterized by the
existence of paucicellular or even acellular areas with a
aracterized by spindle cells mixed with a variable population ofs spindled character of cells and abundant pigment (H&E stain,d by giant cells (H&E stain, 40�). (D) GCF with pseudovascular
SP chshow
nctuate
sclerotic component of thick homogenous collagen bundles

lDDsV
oapN
D
hcoF
a
ttm
appbi
crss
18 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
that represent at least 50% of the tumor mass.76-79 However,arge areas of the neoplasm showing typical features ofFSP are always present, and a transition between typicalFSP and sclerotic areas is also observed. In addition, in
ome sclerosing DFSP, prominent nuclear palisade anderocay body formation has been described.80
The cause of sclerosis is unclear. No external injury,radiotherapy, or any other known cause can explain thishistology, suggesting a spontaneous phenomenon, althoughsome authors believe that these sclerosing DFSP are theresult of a long-term evolution of the tumor and the pro-duction of great amounts of collagen by neoplastic cells.77
Granular cell variant of DFSPThere are only 2 reports of 3 cases each describing this
variant,81,82 which is characterized by an intimate mixturef spindle cells with a significant population of cells havingbundant lysosomal granules, round eccentric nuclei, androminent nucleoli. Granular cells are strongly positive forK inhibitory factor (NK1C3) but negative for S-100.
FSP-FSFS change is defined by its characteristic fascicular, often
erringbone architecture, hypercellularity, abundant atypi-al cells, increased mitotic rate, and focal or complete lossf CD34 expression (Figure 5A–C).83-85 The incidence of
Figure 5 Histologic appearance of fibrosarcomatous dermatoells almost completely replacing the dermis and (H&E stain, 4esembling the herringbone pattern (H&E stain, 40�). (C) In Dtain, 100�). (D) Myxoid DFSP characterized by moderately ctain, 100�).
S changes in DFSP has been reported to be between 10%
nd 20% of all DFSP,86-89 the proportion of FS areasranging from 5% to �75% of the tumor.
This histologic variant is characterized by a more aggres-sive behavior, with a higher rate of local recurrence aftersurgery and an increased risk of distant metastasis.83,85,86,90
However, there is no full consensus on the clinical behaviorof DFSP-FS compared with classic DFSP.90,91
DFSP always has infiltrative borders, and a typical dif-fuse honeycomb infiltration into underlying fatty tissue isseen. However, in our experience,89 and also described byothers,88,92 the DFSP-FS often exhibits an expansive pat-ern, which can also be CD34 negative, in some parts of theumor. We believe that recognition of these features isandatory to avoid misdiagnosis.There are no differences in age, gender, location, clinical
ppearance, previous trauma, or presentation status betweenatients with DFSP-FS and DFSP. Clinically, DFSP-FS is aotentially more aggressive tumor than low-grade DFSPecause of the larger tumor size (Figure 2) and frequentnfiltration of the neighboring fascia and muscle.89
TP53 mutations and p53 overexpression have been re-lated to tumor progression in the FS change of DFSP. Infact, p53 expression in the FS areas ranges from 60% to92% compared with the practically null expression of thisprotein in the conventional areas of DFSP.90,93,94 Interest-ingly, at least 50% of cases with p53 expression are asso-
rcoma protuberans (DFSP-FS). (A) Low-power view of tumor) In DFSP-FS, the proliferation of spindled arrange in fasciclesFS, cells show mild-to-moderate nuclear pleomorphism (H&E
areas with abundant interstitial accumulation of mucin (H&E
fibrosa�). (B
FSP-ellular
ciated with TP53 mutations.95

ccG
vma
bm
19Llombart et al Dermatofibrosarcoma Protuberans
Myxoid DFSP. Conventional Dfsp Often Contains Small Ar-eas of Myxoid Degeneration. However, on occasions, thepredominant histologic pattern throughout the tumor can bemyxoid (�50%). This variant is characterized by the pres-ence of moderately cellular areas made up of stellate orfusiform cells with abundant accumulation of hyaluroni-dase-sensitive mucin in the intercellular space (Figure5D).96 The tumor tends to be well vascularized, and areas oflassic DFSP with dense cellularity and a storiform patternan be found. Some cases show focal areas of FS andCF-like differentiation.97-99
Myxoid DFSP does not differ from conventional DFSPin terms of clinical characteristics or prognosis, and itsrecognition is only of use in the differential diagnosis withother myxoid tumors such as myxoid neurofibroma, super-ficial acral fibromyxoma, superficial angiomyxoma, low-grade myxofibrosarcoma, and myxoid liposarcoma. Almostall cases are positive for CD34, typically in a diffuse pat-tern, and negative for the neural marker S-100 protein.99,100
Myoid DFSP. Myoid Differentiation in Dfsp and Dfsp-fsSeems to Be a Rare Phenomenon. Calonje and Fletcher101
reported 5 DFSP with myoid areas distributed throughoutthe tumor and having no consistent relation to hair folliclesor blood vessel walls; the authors interpreted this as myoiddifferentiation of neoplastic cells of DFSP. Histologically,these tumors are typical DFSP-FS or ordinary DFSP exceptfor the presence of scattered to confluent nodules and bun-dles of eosinophilic spindle cells associated with well-de-fined cytoplasmic margins and vesicular nuclei associatedwith focal stromal hyalinization. However, Díaz-Cascajo102
Figure 6 (A, B) Strong CD34 expression in tumor cells of dermaDFSP area (bottom) stains strongly for CD34, whereas the fibrosa
Complete absence of CD34 staining in fibrosarcomatous DFSP (H&E stbriefly introduced 2 independent cases containing myoidareas and commented that in several sections, tumors revealeda close relationship between myoid areas and the wall ofintraneoplastic blood vessels. In fact, Sanz-Trelles et al92 statedthat the origin of the myomatous component areas could be ahyperplasia of the vascular muscle cells or proliferation ofpericytes with differentiation toward smooth muscle cells.However, other authors83 agreed that myoid differentiation is aariant of DFSP and DFSP-FS. Immunohistochemically, theyoid areas are typically negative for CD34 and desmin and
re positive for smooth muscle actin.
Immunohistochemical profile
The definitive diagnosis of DFSP is usually estab-lished on the basis of routine histopathological and im-munohistochemical features. Immunohistochemical ex-pression of CD34 has been considered characteristic forthe diagnosis of DFSP. Approximately 80%-100% ofDFSP express this marker (Figure 6A and B), althoughbetween 10% and 20% are negative, most commonly, thefibrosarcomatous variant (Figure 6C and D).10-12,84 Nonethe-less, CD34 expression has been increasingly reported in othersarcomas,103,104 such as inflammatory myofibroblastic tumor,myofibrosarcoma, epithelioid sarcoma, or angiosarcoma, andeven in some benign fibrohistiocytic lesions, such as soli-tary fibrous tumor, sclerotic fibroma,105 cellular digital fi-romas,106 nuchal-type fibroma,107 superficial acral fibro-yxomas,104,108 and dermatofibromas.12,109 Consequently,
this marker should now be considered less specific for DFSP.
sarcoma protuberans (DFSP) (H&E stain, 4� and100�). (C) Thetous area (top) is negative for this antigen (H&E stain, 4�). (D)
tofibrorcoma
ain, 100�).
cmbpsc
f3
fsCs
rft
d(
20 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
Factor XIIIa is very useful in the differential diagnosisbetween DFSP and cellular fibrous histiocytomas, as it isusually negative in DFSP. However, a fraction of between10% and 15% of cutaneous fibrous histiocytomas are neg-ative for this marker, and, approximately, the same propor-tion of DFSP shows some level of expression.12,109 As aonsequence, in recent years, new immunohistochemicalarkers have been described for the differential diagnosis
etween these 2 entities, including stromelysin III, apolipo-rotein D, nestin, and CD163, although most of these aretill under discussion. In summary, we could say that DFSPells usually express CD34 antigen, apolipoprotein D,110
and nestin,111 whereas they are negative for factor XIIIa,stromelysin III,112,113 HMGA1, HMGA2,109 tenascin,12
D2-40,114 and CD163.115
Cytogenetic and molecular biology of DFSP
Genetically, DFSP is characterized by a reciprocal trans-location t(17;22)(q22;q13) or more often as a supernumer-ary ring chromosome involving also chromosomes 17 and22.15,17,116,117 These chromosomal rearrangements lead to ausion of the COL1A1 gene in chromosome band 17q21 and3 and the PDGFB gene in chromosome 22q13.1.118 The
main consequence of the t(17;22)(q22;q13) is the overpro-duction of PDGFB by the tumor cells, which leads to theconstitutive activation of the PDGFB receptor, a type IIItyrosine kinase receptor.119,120 The recognition of this au-tocrine and paracrine action mechanism has given rise to thesuggestion that the tyrosine kinase inhibitors (TKI) such asimatinib mesylate could be therapeutic options for DFSPpatients as neoadjuvant treatment to reduce tumor size inlocally advanced tumors or in cases presenting metastaticdisease.121,122
The t(17;22)(q22;q13) can be detected either by FISH oninterphase nuclei and/or by multiplex reverse transcription
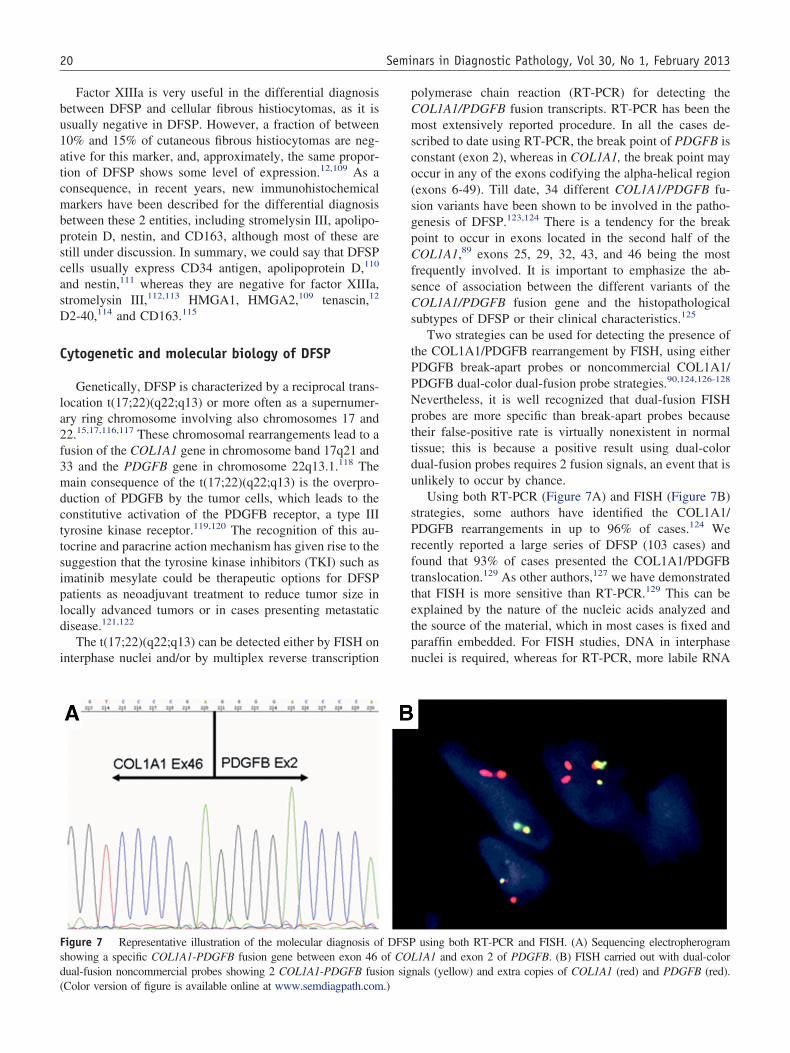
Figure 7 Representative illustration of the molecular diagnosis oshowing a specific COL1A1-PDGFB fusion gene between exon 46ual-fusion noncommercial probes showing 2 COL1A1-PDGFB fus
Color version of figure is available online at www.semdiagpath.com.)polymerase chain reaction (RT-PCR) for detecting theCOL1A1/PDGFB fusion transcripts. RT-PCR has been themost extensively reported procedure. In all the cases de-scribed to date using RT-PCR, the break point of PDGFB isconstant (exon 2), whereas in COL1A1, the break point mayoccur in any of the exons codifying the alpha-helical region(exons 6-49). Till date, 34 different COL1A1/PDGFB fu-sion variants have been shown to be involved in the patho-genesis of DFSP.123,124 There is a tendency for the breakpoint to occur in exons located in the second half of theCOL1A1,89 exons 25, 29, 32, 43, and 46 being the mostrequently involved. It is important to emphasize the ab-ence of association between the different variants of theOL1A1/PDGFB fusion gene and the histopathological
ubtypes of DFSP or their clinical characteristics.125
Two strategies can be used for detecting the presence ofthe COL1A1/PDGFB rearrangement by FISH, using eitherPDGFB break-apart probes or noncommercial COL1A1/PDGFB dual-color dual-fusion probe strategies.90,124,126-128
Nevertheless, it is well recognized that dual-fusion FISHprobes are more specific than break-apart probes becausetheir false-positive rate is virtually nonexistent in normaltissue; this is because a positive result using dual-colordual-fusion probes requires 2 fusion signals, an event that isunlikely to occur by chance.
Using both RT-PCR (Figure 7A) and FISH (Figure 7B)strategies, some authors have identified the COL1A1/PDGFB rearrangements in up to 96% of cases.124 Weecently reported a large series of DFSP (103 cases) andound that 93% of cases presented the COL1A1/PDGFBranslocation.129 As other authors,127 we have demonstrated
that FISH is more sensitive than RT-PCR.129 This can beexplained by the nature of the nucleic acids analyzed andthe source of the material, which in most cases is fixed andparaffin embedded. For FISH studies, DNA in interphasenuclei is required, whereas for RT-PCR, more labile RNA
using both RT-PCR and FISH. (A) Sequencing electropherogram1A1 and exon 2 of PDGFB. (B) FISH carried out with dual-color
nals (yellow) and extra copies of COL1A1 (red) and PDGFB (red).
f DFSPof COLion sig

sHiP
ntspuoi
u
21Llombart et al Dermatofibrosarcoma Protuberans
molecules are necessary, especially keeping in mind thatRNA is more affected by fixation processes than DNA. Inaddition, it has been demonstrated that the use of frozentissue for the RT-PCR could improve the success rate of thistechnique,125 although this material is not always availablein a routine pathology laboratory.
For a rapid molecular result, the method of choice isusually FISH. Although RT-PCR analysis is useful forretrospective studies or to confirm a doubtful FISH result,in daily practice, it is both laborious and time consuming.
In our opinion, although molecular studies are not nec-essary for routine pathologic diagnosis, the identification ofthe COL1A1/PDGFB rearrangements is mandatory in casesin which the differential diagnosis is inconclusive, such asunusual or rare histopathological subtypes, poorly informa-tive immunohistochemical features (CD34 negative), or be-fore starting treatment with imatinib mesylate in metastaticor locally advanced cases.
The COL1A1/PDGFB is present in all histologic DFSPubtypes, indicating a common pathogenic mechanism.owever, not all DFSP express the translocation,125 and
n a small percentage of DFSP (�10%), the COL1A1/DGFB fusion gene is absent. Bianchini et al130 reported
a case of DFSP in which they used FISH, RT-PCR, andkaryotyping to demonstrate that the COL1A1/PDGFBrearrangement was not present, but identified a newtranslocation between chromosomes 5 and 8 involvingthe CSPG2 and PTK2B genes at 5q14.3 and 8p21.2,respectively. Thus, the report confirmed the hypothesisthat a small percentage of DFSP can display differentgenetic abnormalities.
Diagnostic evaluation
After a diagnosis of DFSP, a complete history, reviewof systems, and physical examination of the patientsshould be performed. DFSP rarely exhibits lymphatic orhematogenous dissemination.1,2 Before surgery, mag-
etic resonance imaging is useful, which is more sensi-ive than palpation for ascertaining depth of tumor inva-ion. Magnetic resonance imaging seems to be useful inrimary DFSP in locations other than the head, neck, andpper part of the thorax.29 Computed tomography (CT) isnly indicated in rare cases in which underlying bonenvolvement is suspected.
Treatment
Surgery
Complete surgical resection is accepted as the optimaltreatment for local DFSP. However, the minimum resectionmargin needed to achieve local control remains undefined.Achieving local control of the tumor is difficult evidenced
by the fact that after conventional surgery, DFSP recurs ina mean of 20% of cases, whereas with Mohs surgery,recurrence rates are reduced to �1%.
The high recurrence rate for conventional surgery can beexplained by the eccentric growth of the tumor when itinvades the subcutaneous cellular tissue. At this level, thetumor invades in the form of tentacle-like projections at adistance from the initial focus. These projections can passclinically unnoticed and can remain undetected if an ex-haustive histologic study of the surgical margins is notperformed.1,2,131 Studies in which DFSP had been excisedwith undefined or conservative surgical margins found localrecurrences rates ranging from 26% to 60%.27,132 In con-trast, after wide local excision (2-3 cm), the reported totallocal recurrence rate is much lower and varies from 0% to30%.27,86,89,132-135 Series in which margins of 5 cm weresed reported rates of recurrence of �5%.132,136 Therefore,
increasingly wider margins have resulted in lower recur-rence rates. Nevertheless, obtaining generous margins is notalways possible in patients when the tumors involve the faceor neck or in pediatric cases. In addition, as the surgicalmargins are extended, the risk of complications after sur-gery increases (infection, or bleeding), the closing of theresultant wound is more complex and may leave an impor-tant cosmetic defect.89
MMS uses the microscope to trace out the tentacle-likeprojections and a map to guide residual tumor excision. Incontrast to wide local excision, which uses representativevertical sectioning, the Mohs technique requires continuingsequential horizontal sectioning during resection (Figure 8).The efficacy of MMS in the treatment of DFSP has beendocumented in several studies, with a median recurrencerate of �1% and a range of 0% to 8.3%.89,131-133,135,137-139
A modified MMS technique is usually used in DFSP, withthe tissue fixed in formalin and embedded in paraffin. Thismodification slows the procedure but allows improved di-agnosis of fat invasion by the tumor, which can pass unrec-ognized in frozen sections. At our institution, modifiedMMS is routinely performed for the treatment of DFSP.89
First, the tumor is debulked by excision (Figure 8). Thisexcision margin is made just beyond the visible tumormargin and beneath the tumor mass. The Mohs layer istaken as a 0.5- to 1-cm margin of tissue excision around andunder the debulked excision wound positioning the scalpelat an angle of 45°. The Mohs specimen is oriented using silksuture thread and photographed. The tissue is divided intomultiple specimens, mapped for precise anatomic orienta-tion, and sent to the Mohs histotechnician for formalinfixation and paraffin embedding before taking horizontalsections. The sections are stained with hematoxylin andeosin and confirmed by CD34 immunostaining. In patientswith a positive margin(s), the residual tumor is excised withadditional 0.5-cm margins. The cyclic process of excision,mapping, and microscopic examination is repeated till notumor is microscopically detected. The skin defects aretemporarily covered with synthetic wound dressings till
complete excision is proven.89
ln
22 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
We agree with other authors89,131-133,135,137-139 that themodified Mohs surgery is the treatment of choice forDFSP. Mohs surgery offers the advantage of accurate andcomplete evaluation of the entire peripheral and deepmargins, resulting in extremely low reported rates oflocal recurrence.
Molecular target therapy
The COL1A1-PDGFB fusion protein is processed extra-cellularly to produce fully mature and functional PDGFB.This growth factor acts as a potent mitogen through theactivation of its PDGFB receptor,119 which has tyrosinekinase activity, and activates intracellular signaling path-ways, especially those mediated by PI3 kinase and Ras-MAP kinases. In turn, this activity controls vital cell
Figure 8 Mohs micrographic surgery. (A) DFSP located on sup(C) The tumor is debulked by excision. This excision margin is m(D) Defect after debulking. (E) The Mohs layer is taken as a 1-cm m(F) The Mohs specimen is oriented using silk suture thread and phofor precise anatomic orientation. (H) Wound after the first stage oavailable online at www.semdiagpath.com.)
functions, such as proliferation, cell adhesion, and apo- w
ptosis.119,140 The recognition of this action mechanism ofthe COL1A1-PDGFB fusion protein in DFSP has led tothe suggestion that TKI such as imatinib mesylate can beused as therapy in DFSP.121,122 Imatinib mesylate(STI571, Gleevec, Novartis Pharmaceuticals, Basel,Switzerland), a TKI, whose targets include ABL, BCR-ABL, KIT, and PDGFR has been postulated to be usefulin cases of DFSP that are not amenable to radical surgicaltreatment, thus offering a previously unavailable thera-peutic alternative for use in metastatic or locally ad-vanced DFSP.121,122
The first studies with imatinib mesylate involved pal-liative use of the drug in 4 patients with metastaticDFSP.121,141,142 In 2 cases, the effect was temporary andung metastases were reduced in size and number, but didot disappear, and in the remaining 2 cases, the treatment
cular area before surgery. (B) Drawing and measuring the tumor.st beyond the visible tumor margin and beneath the tumor mass.of tissue excision around and under the debulked excision wound.
hed. (G) The tissue is divided into multiple specimens and mappeds. (I) The defect is closed with a graft. (Color version of figure is
raclaviade juargin
tograpf Moh
as successful. These results led to larger clinical trials.

tvasc
vtfm(mtkpmaaddiDdgrsw
ldand
ntsD
miRa
Dpw
23Llombart et al Dermatofibrosarcoma Protuberans
In 2005, McArthur et al122 assessed the clinical responseo imatinib mesylate in 10 patients— 8 with locally ad-anced DFSP and 2 with metastasis. Of these, 5 showed
partial response (50% reduction in tumor size), 4howed complete response, and only 1 showed nohange.
To our knowledge, around 100 patients with local ad-anced DFSP have been reported to be treated with ima-inib,122,143-157 with doses between 400 and 800 mg dailyor a period ranging from 2 to 24 months (median, 4onths), producing an average tumor reduction of 50%
range, 19%-100%) after a median follow-up time of 24onths (range, 88 days to 72 months). The toxicity of this
herapy is minimal, and the most common side effectsnown include dyspepsia, nausea, vomiting, and myelosup-ression. After treatment with imatinib, the surgical speci-en showed hypocellular fibrovascular tissue and even
cellular areas as a scar (Figure 9), whereas other tumorreas remained intact. Some authors have suggested that thisrug causes apoptosis of the tumor cells and completeestruction of the tumor,158,159 whereas others believe thatmatinib mesylate causes a change in the phenotype ofFSP involving reduced proliferation and, as a result, re-uced tumor size,141,160 thus facilitating more optimal sur-ery. It is not clear whether imatinib therapy before surgeryeduces the need for wide excision margins or for Mohsurgery.122 In our experience, imatinib therapy almost al-
Figure 9 Histologic images before and after surgery of residermatofibrosarcoma protuberans (DFSP) before treatment with iaucicellular histologic appearance with an abundant collagen dww.semdiagpath.com.)
ays reduces the clinical appearance and histologic cellu-
arity of tumor, but we cannot confirm that this treatmentecreases the overall breadth of tissue involvement. Hence,s other authors,146 we believe that the clinical use ofeoadjuvant therapy before complete excision remains to beetermined.
Several questions remain regarding the action mecha-ism of imatinib and possible resistance to this targetherapy in DFSP. However, imatinib is currently the goldtandard in the treatment of local advanced or metastaticFSP.Another consideration to remember is the fact that tu-
ors lacking the t(17;22) translocation may not respond tomatinib;122 thus, molecular analysis of the tumor usingT-PCR or FISH may be useful before the administration ofn imatinib-based therapy.
Radiotherapy
DFSP is considered to be radiosensitive, although therole of radiotherapy in treating this neoplasm remains un-certain. Radiation has been used as an adjuvant therapy aftersurgery, and can be considered in cases where there is aconcern about the adequacy of surgical margins, when pos-itive surgical margins are found after resection and furthersurgery is not feasible, or for negative surgical margins forlarge lesions. Radiation has occasionally been used as a
sease and after partial response to therapy with imatinib. (A). (B–D) DFSP after treatment with imatinib. The tumor shows a
on in the stroma. (Color version of figure is available online at
ual dimatinibepositi
primary treatment.161-166

dtcCp
lhatccma
24 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
Follow-up recommendations andprognostic factors
After surgery, patients should be examined every 6 monthsfor the first 3 years and annually for the rest of the life.Physical examination should pay particular attention tocareful inspection and palpation of the scar because DFSP ischaracterized by its capacity for local recurrence. In thiscontext, most local recurrences appear within 3 years ofsurgery, although later recurrences can also occur.31,167
As DFSP metastasizes in only 2%-5% of cases, extensiveevaluations with CT scans, blood cell counts, and liverfunction tests are not indicated.1,2 DFSP most commonlyisseminates hematogenously to the lungs, particularly ifhe lesion is advanced, recurrent, or DFSP-FS; therefore, ahest x-ray should be performed for all patients, and chestT should be performed only for patients with suspicion ofulmonary metastases.
As stated earlier, metastases preferentially localize to theung, but have also been reported in the brain, bone, andeart. Although, it is difficult to determine which cases aret risk of metastasis, they generally involve recurrent lesionshat have progressed for many years and when a fibrosar-omatous component is seen by histology.86,90 The in-reased age, tumor size �5 cm, head or neck location, highitotic index, p53 mutations, and increased cellularity are
lso predictors of poor clinical outcome.86,91,95,168
References
1. Gloster HM, Jr: Dermatofibrosarcoma protuberans. J Am Acad Der-matol 35:355-374, 1996; quiz:75-76
2. Sanmartín O, Llombart B, López-Guerrero JA, et al: [Dermatofibro-sarcoma protuberans]. Actas Dermosifiliogr 98:77-87, 2007
3. Sherwell S: Morphea. Arch Dermatol 8:72-73, 18904. Taylor RW: Sarcomatous tumors resembling in some respects ke-
loids. Arch Dermatol 8:384-387, 18905. Darier J, Ferrand M: Dermatofibromes progressifs et récidivants ou
fibrosarcomes de la peu. Ann Dermatol Syph 5:545-562, 19246. Hoffman E: Ueber das knollentribende fibrosarkam der haut (der-
matofibrosarcoma protuberans). Dermatol Z 43:1-28, 19257. Taylor HB, Helwig EB: Dermatofibrosarcoma protuberans. A study
of 115 cases. Cancer 15:717-725, 19628. Altman DA, Nickoloff BJ, Fivenson DP: Differential expression of
factor XIIIa and CD34 in cutaneous mesenchymal tumors. J CutanPathol 20:154-158, 1993
9. Kutzner H: Expression of the human progenitor cell antigen CD34(HPCA-1) distinguishes dermatofibrosarcoma protuberans from fi-brous histiocytoma in formalin-fixed, paraffin-embedded tissue. J AmAcad Dermatol 28:613-617, 1993
10. Abenoza P, Lillemoe T: CD34 and factor XIIIa in the differentialdiagnosis of dermatofibroma and dermatofibrosarcoma protuberans.Am J Dermatopathol 15:429-434, 1993
11. Cohen PR, Rapin RP, Farhood AI: Dermatofibroma and dermatofi-brosarcoma protuberans: differential expression of CD34 and factorXIIIa. Am J Dermatopathol 16:573-574, 1994
12. Kahn HJ, Fekete E, From L: Tenascin differentiates dermatofibromafrom dermatofibrosarcoma protuberans: comparison with CD34 andfactor XIIIa. Hum Pathol 32:50-56, 2001
13. Bridge JA, Neff JR, Sandberg AA: Cytogenetic analysis of dermato-
fibrosarcoma protuberans. Cancer Genet Cytogenet 49:199-202, 199014. Mandahl N, Heim S, Willén H, et al: Supernumerary ring chromo-some as the sole cytogenetic abnormality in a dermatofibrosarcomaprotuberans. Cancer Genet Cytogenet 49:273-275, 1990
15. Pedeutour F, Simon MP, Minoletti F, et al: Ring 22 chromosomes indermatofibrosarcoma protuberans are low-level amplifiers of chro-mosome 17 and 22 sequences. Cancer Res 55:2400-2403, 1995
16. Minoletti F, Miozzo M, Pedeutour F, et al: Involvement of chromo-somes 17 and 22 in dermatofibrosarcoma protuberans. Genes Chro-mosomes Cancer 13:62-65, 1995
17. Naeem R, Lux ML, Huang SF, et al: Ring chromosomes in dermato-fibrosarcoma protuberans are composed of interspersed sequencesfrom chromosomes 17 and 22. Am J Pathol 147:1553-1558, 1995
18. Mandahl N, Limon J, Mertens F, et al: Ring marker containing 17qand chromosome 22 in a case of dermatofibrosarcoma protuberans.Cancer Genet Cytogenet 89:88-91, 1996
19. Bendix-Hansen K, Myhre-Jensen O, Kaae S: Dermatofibrosarcomaprotuberans. A clinico-pathological study of nineteen cases and re-view of world literature. Scand J Plast Reconstr Surg 17:247-252,1983
20. Chuang TY, Su WP, Muller SA: Incidence of cutaneous T celllymphoma and other rare skin cancers in a defined population. J AmAcad Dermatol 23:254-256, 1990
21. Criscione VD, Weinstock MA: Descriptive epidemiology of der-matofibrosarcoma protuberans in the United States, 1973 to 2002.J Am Acad Dermatol 56:968-973, 2007
22. Martin L, Combemale P, Dupin M, et al: The atrophic variant ofdermatofibrosarcoma protuberans in childhood: a report of six cases.Br J Dermatol 139:719-725, 1998
23. Llombart B, Sanmartin O, López-Guerrero JA: Dermatofibrosarcomaprotuberante en la infancia. Piel 21:435-441, 2006
24. Martin L, Piette F, Blanc P, et al: Clinical variants of the preprotu-berant stage of dermatofibrosarcoma protuberans. Br J Dermatol153:932-936, 2005
25. Strauss RM, Merchant WJ, Roberts P, et al: A case of childhooddermatofibrosarcoma protuberans without detected cytogenetic ab-normality. Br J Dermatol 148:1051-1055, 2003
26. Pack GT, Tabah EJ: Dermato-fibrosarcoma protuberans. A report of39 cases. AMA Arch Surg 62:391-411, 1951
27. Rutgers EJ, Kroon BB, Albus-Lutter CE, et al: Dermatofibrosarcomaprotuberans: treatment and prognosis. Eur J Surg Oncol 18:241-248,1992
28. Tan AW, Tan SH: Dermatofibrosarcoma protuberans: a clinicopath-ological analysis of 10 cases in Asians. Australas J Dermatol 45:29-33, 2004
29. Serra-Guillen C, Sanmartin O, Llombart B, et al: Correlation betweenpreoperative magnetic resonance imaging and surgical margins withmodified Mohs for dermatofibrosarcoma protuberans. Dermatol Surg37:1638-1645, 2011
30. Rabinowitz LG, Luchetti ME, Segura AD, et al: Acrally occurringdermatofibrosarcoma protuberans in children and adults. J DermatolSurg Oncol 20:655-659, 1994
31. McPeak CJ, Cruz T, Nicastri AD: Dermatofibrosarcoma protuberans:an analysis of 86 cases—five with metastasis. Ann Surg 166:803-816,1967
32. Petoin DS, Baruch J, Raulo Y, et al: Darier–Ferrand progressive andrecurrent dermatofibroma. Anatomo-clinical study of 17 cases]. AnnChir Plast Esthet 30:338-344, 1985
33. McLoughlin PM, Girach M, Wood GA: Dermatofibrosarcoma pro-tuberans of the scalp. Br J Oral Maxillofac Surg 30:401-403, 1992
34. McLelland J, Chu T: Dermatofibrosarcoma protuberans arising in aBCG vaccination scar. Arch Dermatol 124:496-497, 1988
35. Bukhari I, Al Akloby O, Bedaiwi Y: Dermatofibrosarcoma protuber-ans at the site of a central venous line. Case report. Am J ClinDermatol 6:61-64, 2005
36. Laskin WB: Dermatofibrosarcoma protuberans. CA Cancer J Clin42:116-125, 1992
37. Lambert WC, Abramovits W, Gonzalez-Sevra A, et al: Dermatofi-
brosarcoma non-protuberans: description and report of five cases of a
25Llombart et al Dermatofibrosarcoma Protuberans
morpheaform variant of dermatofibrosarcoma. J Surg Oncol 28:7-11,1985
38. Barnes L, Coleman JA, Jr, Johnson JT: Dermatofibrosarcoma protu-berans of the head and neck. Arch Otolaryngol 110:398-404, 1984
39. Rockley PF, Robinson JK, Magid M, et al: Dermatofibrosarcomaprotuberans of the scalp: a series of cases. J Am Acad Dermatol21:278-283, 1989
40. Rich JD, Zbylski JR, LaRossa DD: Dermatofibrosarcoma protuberansof the head and neck. Am Surg 46:208-215, 1980
41. Kamino H, Jacobson M: Dermatofibroma extending into the subcu-taneous tissue. Differential diagnosis from dermatofibrosarcoma pro-tuberans. Am J Surg Pathol 14:1156-1164, 1990
42. Bague S, Folpe AL: Dermatofibrosarcoma protuberans presenting asa subcutaneous mass: a clinicopathological study of 15 cases withexclusive or near-exclusive subcutaneous involvement. Am J Der-matopathol 30:327-332, 2008
43. Zelger B, Sidoroff A, Stanzl U, et al: Deep penetrating dermatofi-broma versus dermatofibrosarcoma protuberans. A clinicopathologiccomparison. Am J Surg Pathol 18:677-686, 1994
44. Bednar B: Storiform neurofibromas of the skin, pigmented and non-pigmented. Cancer 10:368-376, 1957
45. Dupree WB, Langloss JM, Weiss SW: Pigmented dermatofibrosar-coma protuberans (Bednar tumor). A pathologic, ultrastructural, andimmunohistochemical study. Am J Surg Pathol 9:630-639, 1985
46. Kaburagi Y, Hatta N, Kawara S, et al: Pigmented dermatofibrosar-coma protuberans (Bednár tumor) occurring in a Japanese infant.Dermatology 197:48-51, 1998
47. Fletcher CD: Giant cell fibroblastoma of soft tissue: a clinicopatho-logical and immunohistochemical study. Histopathology 13:499-508,1988
48. Suehara Y, Yazawa Y, Hitachi K: Metastatic Bednar tumor (pig-mented dermatofibrosarcoma protuberans) with fibrosarcomatouschange: a case report. J Orthop Sci 9:662-665, 2004
49. Rytina ER, Ball RY: Transformation of recurrent dermatofibrosar-coma protuberans to its pigmented variant (Bednar tumour). Histo-pathology 32:384-385, 1998
50. Zámecník M, Michal M: Giant-cell fibroblastoma with pigmenteddermatofibrosarcoma protuberans component. Am J Surg Pathol 18:736-740, 1994
51. Kini H, Raghuveer CV, Pai MR, et al: Fibrosarcomatous Bednartumor with distant metastases—a case report. Indian J Pathol Micro-biol 47:26-29, 2004
52. Bisceglia M, Vairo M, Calonje E, et al: Pigmented fibrosarcomatousdermatofibrosarcoma protuberans (Bednar tumor). 3 case reports,analogy with the “conventional” type and review of the literature].Pathologica 89:264-273, 1997
53. Kobayashi T, Hasegawa Y, Konohana A, et al: A case of Bednartumor. Immunohistochemical positivity for CD34. Dermatology 195:57-59, 1997
54. Goncharuk V, Mulvaney M, Carlson JA: Bednár tumor associatedwith dermal melanocytosis: melanocytic colonization or neuroecto-dermal multidirectional differentiation? J Cutan Pathol 30:147-151,2003
55. Shmookler BM, Enzinger FM: Giant cell fibroblastoma: a peculiarchildhood tumor. Lab Invest 46:76A, 1982
56. Shmookler BM, Enzinger FM, Weiss SW: Giant cell fibroblastoma.A juvenile form of dermatofibrosarcoma protuberans. Cancer 64:2154-2161, 1989
57. Jha P, Moosavi C, Fanburg-Smith JC: Giant cell fibroblastoma: anupdate and addition of 86 new cases from the Armed Forces Instituteof Pathology, in honor of Dr. Franz M. Enzinger. Ann Diagn Pathol11:81-88, 2007
58. Maeda T, Hirose T, Furuya K, et al: Giant cell fibroblastoma asso-ciated with dermatofibrosarcoma protuberans: a case report. ModPathol 11:491-495, 1998
59. Michal M, Zámecník M: Ultrastructure of composite tumours con-sisting of giant cell fibroblastoma and dermatofibrosarcoma protuber-
ans. Zentralbl Pathol 140:415-420, 199560. Pinto A, Hwang WS, Wong AL, et al: Giant cell fibroblastoma inchildhood immunohistochemical and ultrastructural study. ModPathol 5:639-642, 1992
61. Beham A, Fletcher CD: Dermatofibrosarcoma protuberans with areasresembling giant cell fibroblastoma: report of two cases. Histopathol-ogy 17:165-167, 1990
62. Michal M, Zamecnik M: Giant cell fibroblastoma with a dermatofi-brosarcoma protuberans component. Am J Dermatopathol 14:549-552, 1992
63. Allen PW, Zwi J: Giant cell fibroblastoma transforming into dermato-fibrosarcoma protuberans. Am J Surg Pathol 16:1127-1129, 1992
64. Coyne J, Kaftan SM, Craig RD: Dermatofibrosarcoma protuberansrecurring as a giant cell fibroblastoma. Histopathology 21:184-187,1992
65. De Chadarévian JP, Coppola D, Billmire DF: Bednar tumor pattern inrecurring giant cell fibroblastoma. Am J Clin Pathol 100:164-166,1993
66. Zelger BW, Ofner D, Zelger BG: Atrophic variants of dermatofi-broma and dermatofibrosarcoma protuberans. Histopathology 26:519-527, 1995
67. Marini M, Saponaro A, Magariños G, et al: Congenital atrophicdermatofibrosarcoma protuberans. Int J Dermatol 40:448-450, 2001
68. See AC, Kossard SS, Murrell DF: Guess what. Dermatofibrosarcomaprotuberans presenting as an atrophic red plaque. Eur J Dermatol11:147-149, 2001
69. Davis DA, Sánchez RL: Atrophic and plaquelike dermatofibrosar-coma protuberans. Am J Dermatopathol 20:498-501, 1998
70. Fujimoto M, Kikuchi K, Okochi H, et al: Atrophic dermatofibrosar-coma protuberans: a case report and review of the literature. Derma-tology 196:422-424, 1998
71. Ashack RJ, Tejada E, Parker C, et al: A localized atrophic plaque onthe back. Dermatofibrosarcoma protuberans (DFSP) (atrophic vari-ant). Arch Dermatol 128:549,552, 1992
72. Page EH, Assaad DM: Atrophic dermatofibroma and dermatofibro-sarcoma protuberans. J Am Acad Dermatol 17:947-950, 1987
73. Chuan MT, Tsai TF, Wu MC, et al: Atrophic pigmented dermatofi-brosarcoma presenting as infraorbital hyperpigmentation. Dermatol-ogy 194:65-67, 1997
74. Llombart B, Sanmartin O, Requena C, et al: Atrophic dermatofibro-sarcoma protuberans with the fusion gene COL1A1-PDGFB. J EurAcad Dermatol Venereol 22:371-374, 2008
75. Young CR, 3rd, Albertini MJ: Atrophic dermatofibrosarcoma protu-berans: case report, review, and proposed molecular mechanisms.J Am Acad Dermatol 49:761-764, 2003
76. Barr FJ, Golitz LE, Siongco A: Sclerotic dermatofibrosarcoma pro-tuberans–a lesion which may confused with sclerotic fibroma [ab-stract]. In: 31th Annual Meeting American Society of Dermatopa-thology; March 1997
77. Díaz-Cascajo C, Weyers W, Borghi S: Sclerosing dermatofibrosar-coma protuberans. J Cutan Pathol 25:440-444, 1998
78. Hattori H: Nodular sclerotic change in dermatofibrosarcoma protu-berans: a potential diagnostic problem. Br J Dermatol 148:357-360,2003
79. Sabater-Marco V, Pérez-Vallés A, Berzal-Cantalejo F, et al: Scleros-ing dermatofibrosarcoma protuberans (DFSP): an unusual variantwith focus on the histopathologic differential diagnosis. Int J Der-matol 45:59-62, 2006
80. Llatjós R, Fernández-Figueras MT, Díaz-Cascajo C, et al: Palisadingand verocay body-prominent dermatofibrosarcoma protuberans: areport of three cases. Histopathology 37:452-455, 2000
81. Banerjee SS, Harris M, Eyden BP, et al: Granular cell variant ofdermatofibrosarcoma protuberans. Histopathology 17:375-378, 1990
82. Maire G, Pédeutour F, Coindre JM: COL1A1-PDGFB gene fusiondemonstrates a common histogenetic origin for dermatofibrosarcomaprotuberans and its granular cell variant. Am J Surg Pathol 26:932-937, 2002
83. Mentzel T, Beham A, Katenkamp D, et al: Fibrosarcomatous (“high-
grade”) dermatofibrosarcoma protuberans: clinicopathologic and im-
26 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
munohistochemical study of a series of 41 cases with emphasis onprognostic significance. Am J Surg Pathol 22:576-587, 1998
84. Goldblum JR: CD34 positivity in fibrosarcomas which arise in der-matofibrosarcoma protuberans. Arch Pathol Lab Med 119:238-241,1995
85. Diaz-Cascajo C, Weyers W, Borrego L, et al: Dermatofibrosarcomaprotuberans with fibrosarcomatous areas: a clinico-pathologic andimmunohistochemic study in four cases. Am J Dermatopathol 19:562-567, 1997
86. Bowne WB, Antonescu CR, Leung DH, et al: Dermatofibrosarcomaprotuberans: a clinicopathologic analysis of patients treated and fol-lowed at a single institution. Cancer 88:2711-2720, 2000
87. Wrotnowski U, Cooper PH, Shmookler BM: Fibrosarcomatouschange in dermatofibrosarcoma protuberans. Am J Surg Pathol 12:287-293, 1988
88. Connelly JH, Evans HL: Dermatofibrosarcoma protuberans. A clin-icopathologic review with emphasis on fibrosarcomatous areas. Am JSurg Pathol 16:921-925, 1992
89. Llombart B, Monteagudo C, Sanmartín O, et al: Dermatofibrosar-coma protuberans: a clinicopathological, immunohistochemical, ge-netic (COL1A1-PDGFB), and therapeutic study of low-grade versushigh-grade (fibrosarcomatous) tumors. J Am Acad Dermatol 65:564-575, 2011
90. Abbott JJ, Oliveira AM, Nascimento AG: The prognostic significanceof fibrosarcomatous transformation in dermatofibrosarcoma protuber-ans. Am J Surg Pathol 30:436-443, 2006
91. Goldblum JR, Reith JD, Weiss SW: Sarcomas arising in dermatofi-brosarcoma protuberans: a reappraisal of biologic behavior in eigh-teen cases treated by wide local excision with extended clinicalfollow up. Am J Surg Pathol 24:1125-1130, 2000
92. Sanz-Trelles A, Ayala-Carbonero A, Rodrigo-Fernández I, et al:Leiomyomatous nodules and bundles of vascular origin in the fibro-sarcomatous variant of dermatofibrosarcoma protuberans. J CutanPathol 25:44-49, 1998
93. Hisaoka M, Okamoto S, Morimitsu Y, et al: Dermatofibrosarcomaprotuberans with fibrosarcomatous areas. Molecular abnormalities ofthe p53 pathway in fibrosarcomatous transformation of dermatofibro-sarcoma protuberans. Virchows Arch 433:323-329, 1998
94. Sasaki M, Ishida T, Horiuchi H, et al: Dermatofibrosarcoma protu-berans: an analysis of proliferative activity, DNA flow cytometry andp53 overexpression with emphasis on its progression. Pathol Int49:799-806, 1999
95. Takahira T, Oda Y, Tamiya S, et al: Microsatellite instability and p53mutation associated with tumor progression in dermatofibrosarcomaprotuberans. Hum Pathol 35:240-245, 2004
96. Orlandi A, Bianchi L, Spagnoli LG: Myxoid dermatofibrosarcomaprotuberans: morphological, ultrastructural and immunohistochemi-cal features. J Cutan Pathol 25:386-393, 1998
97. Sato N, Kimura K, Tomita Y: Recurrent dermatofibrosarcoma pro-tuberans with myxoid and fibrosarcomatous changes paralleled byloss of CD34 expression. J Dermatol 22:665-672, 1995
98. Zámecnik M, Michal M: Myxoid variant of dermatofibrosarcomaprotuberans with fibrosarcomatous areas. Zentralbl Pathol 139:373-376, 1993
99. Mentzel T, Schärer L, Kazakov DV, et al: Myxoid dermatofibrosar-coma protuberans: clinicopathologic, immunohistochemical, and mo-lecular analysis of eight cases. Am J Dermatopathol 29:443-448,2007
100. Reimann JD, Fletcher CD: Myxoid dermatofibrosarcoma protuber-ans: a rare variant analyzed in a series of 23 cases. Am J Surg Pathol31:1371-1377, 2007
101. Calonje E, Fletcher CD: Myoid differentiation in dermatofibrosar-coma protuberans and its fibrosarcomatous variant: clinicopathologicanalysis of 5 cases. J Cutan Pathol 23:30-36, 1996
102. Diaz-Cascajo C: Myoid differentiation in dermatofibrosarcoma pro-tuberans and its fibrosarcomatous variant. J Cutan Pathol 24:197-198,
1997103. Fisher C: Low-grade sarcomas with CD34-positive fibroblasts andlow-grade myofibroblastic sarcomas. Ultrastruct Pathol 28:291-305,2004
104. Tardío JC: CD34-reactive tumors of the skin. An updated review ofan ever-growing list of lesions. J Cutan Pathol 35:1079-1092, 2008
105. Hanft VN, Shea CR, McNutt NS, et al: Expression of CD34 insclerotic (“plywood”) fibromas. Am J Dermatopathol 22:17-21, 2000
106. McNiff JM, Subtil A, Cowper SE, et al: Cellular digital fibromas:distinctive CD34-positive lesions that may mimic dermatofibrosar-coma protuberans. J Cutan Pathol 32:413-418, 2005
107. Diwan AH, Horenstein MG: Dermatofibrosarcoma protuberans asso-ciation with nuchal-type fibroma. J Cutan Pathol 31:62-66, 2004
108. Tardío JC, Butrón M, Martín-Fragueiro LM: Superficial acral fibro-myxoma: report of 4 cases with CD10 expression and lipomatouscomponent, two previously underrecognized features. Am J Dermato-pathol 30:431-435, 2008
109. Li N, McNiff J, Hui P, et al: Differential expression of HMGA1 andHMGA2 in dermatofibroma and dermatofibrosarcoma protuberans:potential diagnostic applications, and comparison with histologicfindings, CD34, and factor XIIIa immunoreactivity. Am J Dermato-pathol 26:267-272, 2004
110. West RB, Harvell J, Linn SC, et al: Apo D in soft tissue tumors: anovel marker for dermatofibrosarcoma protuberans. Am J SurgPathol 28:1063-1069, 2004
111. Mori T, Misago N, Yamamoto O, et al: Expression of nestin indermatofibrosarcoma protuberans in comparison to dermatofibroma.J Dermatol 35:419-425, 2008
112. Cribier B, Noacco G, Peltre B, et al: Stromelysin 3 expression: auseful marker for the differential diagnosis dermatofibroma versusdermatofibrosarcoma protuberans. J Am Acad Dermatol 46:408-413,2002
113. Kim HJ, Lee JY, Kim SH, et al: Stromelysin-3 expression in thedifferential diagnosis of dermatofibroma and dermatofibrosarcomaprotuberans: comparison with factor XIIIa and CD34. Br J Dermatol157:319-324, 2007
114. Bandarchi B, Ma L, Marginean C, et al: D2-40, a novel immunohis-tochemical marker in differentiating dermatofibroma from dermato-fibrosarcoma protuberans. Mod Pathol 23:434-438, 2010
115. Sachdev R, Sundram U: Expression of CD163 in dermatofibroma,cellular fibrous histiocytoma, and dermatofibrosarcoma protuberans:comparison with CD68, CD34, and factor XIIIa. J Cutan Pathol33:353-360, 2006
116. Pedeutour F, Coindre JM, Nicolo G, et al: Ring chromosomes indermatofibrosarcoma protuberans contain chromosome 17 sequen-ces: fluorescence in situ hybridization. Cancer Genet Cytogenet67:149, 1993
117. Pedeutour F, Simon MP, Minoletti F, et al: Translocation, t(17;22)(q22;q13), in dermatofibrosarcoma protuberans: a new tumor-associated chromosome rearrangement. Cytogenet Cell Genet 72:171-174, 1996
118. Simon MP, Pedeutour F, Sirvent N, et al: Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen geneCOL1A1 in dermatofibrosarcoma protuberans and giant-cell fibro-blastoma. Nat Genet 15:95-98, 1997
119. Shimizu A, O’Brien KP, Sjöblom T, et al: The dermatofibrosarcomaprotuberans-associated collagen type Ialpha1/platelet-derived growthfactor (PDGF) B-chain fusion gene generates a transforming proteinthat is processed to functional PDGF-BB. Cancer Res 59:3719-3723,1999
120. Sjöblom T, Shimizu A, O’Brien KP, et al: Growth inhibition ofdermatofibrosarcoma protuberans tumors by the platelet-derivedgrowth factor receptor antagonist STI571 through induction of apo-ptosis. Cancer Res 61:5778-5783, 2001
121. Rubin BP, Schuetze SM, Eary JF, et al: Molecular targeting ofplatelet-derived growth factor B by imatinib mesylate in a patientwith metastatic dermatofibrosarcoma protuberans. J Clin Oncol 20:
3586-3591, 2002
27Llombart et al Dermatofibrosarcoma Protuberans
122. McArthur GA, Demetri GD, van Oosterom A, et al: Molecular andclinical analysis of locally advanced dermatofibrosarcoma protuber-ans treated with imatinib: imatinib target exploration consortiumstudy B2225. J Clin Oncol, 2005 23:866-73
123. Giacchero D, Maire G, Nuin PA, et al: No correlation between themolecular subtype of COL1A1-PDGFB fusion gene and the clinico-histopathological features of dermatofibrosarcoma protuberans. J In-vest Dermatol 130:904-907, 2010
124. Patel KU, Szabo SS, Hernandez VS, et al: Dermatofibrosarcomaprotuberans COL1A1-PDGFB fusion is identified in virtually alldermatofibrosarcoma protuberans cases when investigated by newlydeveloped multiplex reverse transcription polymerase chain reactionand fluorescence in situ hybridization assays. Hum Pathol 39:184-193, 2008
125. Llombart B, Sanmartín O, López-Guerrero JA, et al: Dermatofibro-sarcoma protuberans: clinical, pathological, and genetic (COL1A1-PDGFB) study with therapeutic implications. Histopathology 54:860-872, 2009
126. Craver R, Dewenter T, Ebran N, et al: COL1A1-PDGFB fusion in apediatric Bednar tumor with 2 copies of a der(22)t(17;22). CancerGenet Cytogenet 168:155-157, 2006
127. Maire G, Fraitag S, Galmiche L, et al: A clinical, histologic, andmolecular study of 9 cases of congenital dermatofibrosarcoma pro-tuberans. Arch Dermatol 143:203-210, 2007
128. Segura S, Salgado R, Toll A, et al: Identification of t(17;22)(q22;q13)(COL1A1/PDGFB) in dermatofibrosarcoma protuberans by fluores-cence in situ hybridization in paraffin-embedded tissue microarrays.Hum Pathol 42:176-184, 2011
129. Salgado R, Llombart B, M Pujol R, et al: Molecular diagnosis ofdermatofibrosarcoma protuberans: a comparison between reversetranscriptase-polymerase chain reaction and fluorescence in situ hy-bridization methodologies. Genes Chromosomes Cancer 50:510-517,2011
130. Bianchini L, Maire G, Guillot B, et al: Complex t(5;8) involving theCSPG2 and PTK2B genes in a case of dermatofibrosarcoma protu-berans without the COL1A1-PDGFB fusion. Virchows Arch 452:689-696, 2008
131. Gloster HM, Jr, Harris KR, Roenigk RK: A comparison betweenMohs micrographic surgery and wide surgical excision for the treat-ment of dermatofibrosarcoma protuberans. J Am Acad Dermatol35:82-87, 1996
132. Lemm D, Mügge LO, Mentzel T, et al: Current treatment options indermatofibrosarcoma protuberans. J Cancer Res Clin Oncol 135:653-665, 2009
133. DuBay D, Cimmino V, Lowe L, et al: Low recurrence rate aftersurgery for dermatofibrosarcoma protuberans: a multidisciplinaryapproach from a single institution. Cancer 100:1008-1016, 2004
134. Fiore M, Miceli R, Mussi C, et al: Dermatofibrosarcoma protuberanstreated at a single institution: a surgical disease with a high cure rate.J Clin Oncol 23:7669-7675, 2005
135. Paradisi A, Abeni D, Rusciani A, et al: Dermatofibrosarcoma protu-berans: wide local excision vs. Mohs micrographic surgery. CancerTreat Rev 34:728-736, 2008
136. Arnaud EJ, Perrault M, Revol M, et al: Surgical treatment of der-matofibrosarcoma protuberans. Plast Reconstr Surg 100:884-895,1997
137. Ratner D, Thomas CO, Johnson TM, et al: Mohs micrographicsurgery for the treatment of dermatofibrosarcoma protuberans. Re-sults of a multiinstitutional series with an analysis of the extent ofmicroscopic spread. J Am Acad Dermatol 37:600-613, 1997
138. Snow SN, Gordon EM, Larson PO, et al: Dermatofibrosarcomaprotuberans: a report on 29 patients treated by Mohs micrographicsurgery with long-term follow-up and review of the literature. Cancer101:28-38, 2004
139. Love WE, Keiler SA, Tamburro JE, et al: Surgical management ofcongenital dermatofibrosarcoma protuberans. J Am Acad Dermatol
61:1014-1023, 2009140. Leevers SJ, Vanhaesebroeck B, Waterfield MD: Signalling throughphosphoinositide 3-kinases: the lipids take centre stage. Curr OpinCell Biol 11:219-225, 1999
141. Maki RG, Awan RA, Dixon RH, et al: Differential sensitivity toimatinib of 2 patients with metastatic sarcoma arising from dermato-fibrosarcoma protuberans. Int J Cancer 100:623-626, 2002
142. Mizutani K, Tamada Y, Hara K, et al: Imatinib mesylate inhibits thegrowth of metastatic lung lesions in a patient with dermatofibrosar-coma protuberans. Br J Dermatol 151:235-237, 2004
143. Serra-Guillen C, Llombart B, Sanmartin O, et al: Dermatofibro-sacoma protubernas Actas Dermosifiliogr (in press)
144. Lemm D, Muegge LO, Hoeffken K, et al: Remission with imatinibmesylate treatment in a patient with initially unresectable dermato-fibrosarcoma protuberans—a case report. Oral Maxillofac Surg 12:209-213, 2008
145. Price VE, Fletcher JA, Zielenska M, et al: Imatinib mesylate: anattractive alternative in young children with large, surgically chal-lenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 44:511-515, 2005
146. Mehrany K, Swanson NA, Heinrich MC, et al: Dermatofibrosarcomaprotuberans: a partial response to imatinib therapy. Dermatol Surg32:456-459, 2006
147. Savoia P, Ortoncelli M, Quaglino P, et al: Imatinib mesylate in thetreatment of a large unresectable dermatofibrosarcoma protuberans: acase study. Dermatol Surg 32:1097-1102, 2006
148. Wright TI, Petersen JE: Treatment of recurrent dermatofibrosarcomaprotuberans with imatinib mesylate, followed by Mohs micrographicsurgery. Dermatol Surg 33:741-744, 2007
149. Heinrich MC, Joensuu H, Demetri GD, et al: Phase II, open-labelstudy evaluating the activity of imatinib in treating life-threateningmalignancies known to be associated with imatinib-sensitive tyrosinekinases. Clin Cancer Res 14:2717-2725, 2008
150. Thomison J, McCarter M, McClain D, et al: Hyalinized collagen in adermatofibrosarcoma protuberans after treatment with imatinib me-sylate. J Cutan Pathol 35:1003-1006, 2008
151. Han A, Chen EH, Niedt G, et al: Neoadjuvant imatinib therapy fordermatofibrosarcoma protuberans. Arch Dermatol 145:792-796, 2009
152. Rutkowski P, Van Glabbeke M, Rankin CJ, et al: Imatinib mesy-late in advanced dermatofibrosarcoma protuberans: pooled analy-sis of two phase II clinical trials. J Clin Oncol 28:1772-1779, 2010
153. Gooskens SL, Oranje AP, van Adrichem LN, et al: Imatinib mesylatefor children with dermatofibrosarcoma protuberans (DFSP). PediatrBlood Cancer 55:369-373, 2010
154. Kérob D, Porcher R, Vérola O, et al: Imatinib mesylate as a preop-erative therapy in dermatofibrosarcoma: results of a multicenter phaseII study on 25 patients. Clin Cancer Res 16:3288-3295, 2010
155. Edelweiss M, Malpica A: Dermatofibrosarcoma protuberans of thevulva: a clinicopathologic and immunohistochemical study of 13cases. Am J Surg Pathol 34:393-400, 2010
156. Stacchiotti S, Pedeutour F, Negri T, et al: Dermatofibrosarcomaprotuberans-derived fibrosarcoma: clinical history, biological pro-file and sensitivity to imatinib. Int J Cancer 129:1761-1772, 2011
157. Rutkowski P, Wozniak A, Switaj T: Advances in molecular charac-terization and targeted therapy in dermatofibrosarcoma protuberans.Sarcoma, 2011:959132, 2011
158. Liu YC, Chen SC, Chang C, et al: Platelet-derived growth factor is anautocrine stimulator for the growth and survival of human esophagealcarcinoma cell lines. Exp Cell Res 228:206-211, 1996
159. Funa K, Ahgren A: Characterization of platelet-derived growth factor(PDGF) action on a mouse neuroblastoma cell line, NB41, by intro-duction of an antisense PDGF beta-receptor RNA. Cell GrowthDiffer 8:861-869, 1997
160. Greco A, Roccato E, Miranda C, et al: Growth-inhibitory effect ofSTI571 on cells transformed by the COL1A1/PDGFB rearrangement.Int J Cancer 92:354-360, 2001
161. Suit H, Spiro I, Mankin HJ, et al: Radiation in management ofpatients with dermatofibrosarcoma protuberans. J Clin Oncol 14:
2365-2369, 1996
28 Seminars in Diagnostic Pathology, Vol 30, No 1, February 2013
162. Ballo MT, Zagars GK, Pisters P, et al: The role of radiation therapyin the management of dermatofibrosarcoma protuberans. Int J RadiatOncol Biol Phys 40:823-827, 1998
163. Sun LM, Wang CJ, Huang CC, et al: Dermatofibrosarcoma protuberans:treatment results of 35 cases. Radiother Oncol 57:175-181, 2000
164. Dagan R, Morris CG, Zlotecki RA, et al: Radiotherapy in the treat-ment of dermatofibrosarcoma protuberans. Am J Clin Oncol 28:537-539, 2005
165. McArthur G: Dermatofibrosarcoma protuberans: recent clinical prog-
ress. Ann Surg Oncol 14:2876-2886, 2007166. National Comprehensive Cancer Network Clinical practice guide-lines in oncology. Dermatofibrosarcoma Protuberans. Version 1.Available at: http://www.nccn.org/professionals/physicians_gls/PDF/dfsp.pdf. Accessed December 2009
167. Smola MG, Soyer HP, Scharnagl E: Surgical treatment of dermato-fibrosarcoma protuberans. A retrospective study of 20 cases withreview of literature. Eur J Surg Oncol 17:447-453, 1991
168. Gayner SM, Lewis JE, McCaffrey TV: Effect of resection margins ondermatofibrosarcoma protuberans of the head and neck. Arch Oto-
laryngol Head Neck Surg 123:430-433, 1997



















