
Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of...
18
Department of Neurotoxicology, Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland Read the full article ‘Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of synaptic proteins: in vitro and in vivo studies’ on doi: 10.1111/jnc.12810 The term hepatic encephalopathy (HE) defines a complex neurological syndrome associated with liver failure. HE is related to accumulation in brain of toxic metabolites, among which ammonia plays a major role. HE is charac- terized by motor and cognitive dysfunctions progressing towards coma, events reflecting development of imbalance between excitatory (mostly glutamatergic) and inhibitory (mostly GABAergic) neurotransmission in disfavor of the former (Albrecht and Jones 1999; Prakash and Mullen 2010, and references therein). Impaired neurotransmission in HE is in a great part because of the disturbances in astrocytic functions (Albrecht 2005). HE, hyperammonemia or exposure of astrocytes to ammonia, impairs the expres- sion and the function of crucial ion and neurotransmitter transporting moieties in the astrocytic membrane, among these the glutamate transporters GLT-1 (Norenberg et al. 1997) and GLAST (Chan et al. 2000), the inward rectifying potassium channel Kir4.1 (Obara-Michlewska et al. 2011), and the Na–K–Cl cotransporter (Jayakumar et al. 2008). These changes elicit neurotransmitter and ion imbalance in the synaptic cleft resulting in synaptic dysfunction, best exemplified by disinhibition by increased extracellular K + of neuronal circuits, which is mediated by overactivation of neuronal Na–K–Cl cotransporter (Rangroo Thrane et al. 2013). Collectively, the current view is that dysfunctional astrocytes affect the fluxes of molecules directly involved in the neurotransmission process that is, the neurotransmis- sion software. An intriguing, yet unanswered question is whether and to what degree, the HE-affected astrocytes impair synthesis, assembly or function of proteins shaping the synaptic structure that is, the neurotransmission hard- ware? Thrombospondin 1 (TSP-1), a member of a family of astrocyte-secreted extracellular matrix proteins participates in synaptogenesis (Christopherson et al. 2005). In adult brain, TSP-1 promotes structural and functional recovery of synapses compromised by stroke (Liauw et al. 2008), by a mechanism involving up-regulation of a neuroprotective cytokine Tumor growth factor-b1 (Cekanaviciute et al. 2014). Recently, impaired TSP-1 delivery from astrocytes to neurons has been implicated in the synaptic pathology associated with Down’s syndrome (Garcia et al. 2010). In a study reported in this issue, Michael Norenberg’s group presents data indicating that shortage of astroglia-derived TSP-1 may contribute to synaptic dysfunction in HE (Jayakumar et al. 2014). In their hands, incubation of astrocytes with ammonia evoked a decrease in TSP-1 liberation to the incubation media. Next, media conditioned by ammonia-exposed astrocytes when added to cultured neurons, decreased the neuronal content of three proteins critically involved in maintaining structural and functional integrity of the glutamatergic synapse: the glutamate release-controlling pre-synaptic proteins synaptophysin and synaptostagmin, and PSD-95, a component of the post- synaptic PDZ domain which organizes glutamate receptors and their associated signaling proteins in the glutamatergic synapse. Increasing the TSP-1 content in astrocytes by Received June 24, 2014; accepted June 30, 2014. Address correspondence and reprint requests to Jan Albrecht, Department of Neurotoxicology, Mossakowski Medical Research Cen- tre, Polish Academy of Sciences, Pawi nskiego 5, 02-106 Warszawa, Poland. E-mail: [email protected] Abbreviations used: HE, hepatic encephalopathy; TSP-1, thrombo- spondin 1. © 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12823 1 JOURNAL OF NEUROCHEMISTRY | 2014 doi: 10.1111/jnc.12823
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of...

Department of Neurotoxicology, Mossakowski Medical Research Centre, Polish Academy of Sciences,
Warsaw, Poland
Read the full article ‘Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of synaptic proteins: invitro and in vivo studies’ on doi: 10.1111/jnc.12810
The term hepatic encephalopathy (HE) defines a complexneurological syndrome associated with liver failure. HE isrelated to accumulation in brain of toxic metabolites,among which ammonia plays a major role. HE is charac-terized by motor and cognitive dysfunctions progressingtowards coma, events reflecting development of imbalancebetween excitatory (mostly glutamatergic) and inhibitory(mostly GABAergic) neurotransmission in disfavor of theformer (Albrecht and Jones 1999; Prakash and Mullen2010, and references therein). Impaired neurotransmissionin HE is in a great part because of the disturbances inastrocytic functions (Albrecht 2005). HE, hyperammonemiaor exposure of astrocytes to ammonia, impairs the expres-sion and the function of crucial ion and neurotransmittertransporting moieties in the astrocytic membrane, amongthese the glutamate transporters GLT-1 (Norenberg et al.1997) and GLAST (Chan et al. 2000), the inward rectifyingpotassium channel Kir4.1 (Obara-Michlewska et al. 2011),and the Na–K–Cl cotransporter (Jayakumar et al. 2008).These changes elicit neurotransmitter and ion imbalance inthe synaptic cleft resulting in synaptic dysfunction, bestexemplified by disinhibition by increased extracellular K+
of neuronal circuits, which is mediated by overactivation ofneuronal Na–K–Cl cotransporter (Rangroo Thrane et al.2013). Collectively, the current view is that dysfunctionalastrocytes affect the fluxes of molecules directly involvedin the neurotransmission process that is, the neurotransmis-sion software. An intriguing, yet unanswered question iswhether and to what degree, the HE-affected astrocytesimpair synthesis, assembly or function of proteins shapingthe synaptic structure that is, the neurotransmission hard-ware?
Thrombospondin 1 (TSP-1), a member of a family ofastrocyte-secreted extracellular matrix proteins participatesin synaptogenesis (Christopherson et al. 2005). In adultbrain, TSP-1 promotes structural and functional recovery ofsynapses compromised by stroke (Liauw et al. 2008), by amechanism involving up-regulation of a neuroprotectivecytokine Tumor growth factor-b1 (Cekanaviciute et al.2014). Recently, impaired TSP-1 delivery from astrocytesto neurons has been implicated in the synaptic pathologyassociated with Down’s syndrome (Garcia et al. 2010). In astudy reported in this issue, Michael Norenberg’s grouppresents data indicating that shortage of astroglia-derivedTSP-1 may contribute to synaptic dysfunction in HE(Jayakumar et al. 2014). In their hands, incubation ofastrocytes with ammonia evoked a decrease in TSP-1liberation to the incubation media. Next, media conditionedby ammonia-exposed astrocytes when added to culturedneurons, decreased the neuronal content of three proteinscritically involved in maintaining structural and functionalintegrity of the glutamatergic synapse: the glutamaterelease-controlling pre-synaptic proteins synaptophysin andsynaptostagmin, and PSD-95, a component of the post-synaptic PDZ domain which organizes glutamate receptorsand their associated signaling proteins in the glutamatergicsynapse. Increasing the TSP-1 content in astrocytes by
Received June 24, 2014; accepted June 30, 2014.Address correspondence and reprint requests to Jan Albrecht,
Department of Neurotoxicology, Mossakowski Medical Research Cen-tre, Polish Academy of Sciences, Pawi�nskiego 5, 02-106 Warszawa,Poland. E-mail: [email protected] used: HE, hepatic encephalopathy; TSP-1, thrombo-
spondin 1.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12823 1
JOURNAL OF NEUROCHEMISTRY | 2014 doi: 10.1111/jnc.12823

transfection with TSP-1 DNA, or by stimulation of itssynthesis with metformin, prevented the deleterious effectsof astrocyte-conditioned media. In the same study, braindeficit of TSP-1 and synaptophysin have been recorded inthe brain of rats with chronic HE related to thioacetamide-induced liver failure (Jayakumar et al. 2014). Of note, theresponses of neurons to media derived from ammonia-treated astrocytes strikingly resembled those previouslyrecorded with amyloid b-treated astrocytes (Rama Raoet al. 2013), suggesting that decreased astrocyte-to-neuron
transport of TSP-1 may be a common feature of Alzhei-mer’s disease and HE.Chronic HE is associated with a loss of the cerebral
cortical ionotropic glutamate receptors, mainly the NMDAreceptors, a phenomenon likely to contribute to the decreasedglutamatergic tone (Peterson et al. 1990; Saransaari et al.1997). However, the mechanism underlying the loss has sofar remained obscure. The study of Jayakumar et al. (2014)tempts one to link the receptor loss to the astroglia-mediatedchanges in PSD-95 and/or other post-synaptic proteins (for
Scaffolding proteins
SynaptophysinSynaptostagmin
AMPANR1 NR2A NR2B
Thrombospondin-1
Vesicles
PresynapticNeuron
NR1/NR2ANR1/NR2B
PSD-95 PSD-95
PSD
PostsynapticNeuron
Astrocyte
TSP1
Vesicles
PresynapticNeuron
NR1/NR2ANR1/NR2B
PSD-95
PSD
PostsynapticNeuron
Astrocyte
TSP1
Control CHEAMMONIA
ION IMBALANCE
ROS
CELL MEMBRANE
DYSFUNCTION
SAP-102
PSD-95
SAP-102
SAP-102
PSD-95
Fig. 1 Illustration of a hypothesis linking ammonia-induced deficit ofastroglia-derived thrombospondin-1 (TSP-1) to synaptic dysfunction in
chronic hepatic encephalopathy (CHE). Astrocytes secrete thrombo-spondins, among these TSP-1, which are thought to influence thepositioning and assembly of different synaptic proteins, in this way
ascertaining structural and functional integrity of the glutamatergicsynapse. Ammonia, by an as yet not fully defined mechanism includingionic imbalance and accumulation of reactive oxygen species (ROS),perturbs the astrocytic cell membrane and limits TSP-1 release from
(increases its retention in) astrocytes. TSP-1 deficiency limits theexpression of the pre-synaptic proteins, synaptophysin and synapto-stagmin, and of the post-synaptic protein, PSD-95, which are constit-
uents of the ‘synaptic hardware’. Shortage of pre-synaptic proteinsimpairs synaptic vesicle assembly and release, whereas decreased
availability of PSD-95, a constituent of the post-synaptic density (PSD)scaffolding complex, impairs positioning of the NMDA receptorsubunits (NR1, NR2A, NR2B) and may contribute to selective loss of
NMDA receptors in CHE. The fate of other constituents of the synapticmachinery (among these the other scaffolding protein, SAP-102 andthe AMPA receptor), as well as the roles of TSPs other than TSP-1remain to be established. Collectively, the changes outlined in this
figure are likely to contribute to the decrease in the glutamatergic toneassociated with CHE.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12823
2 Review

elaboration of the hypothesis see Fig. 1), and to initiateexperiments aiming at testing this hypothesis. A questionworth addressing in the pathophysiological context iswhether the incomplete reversibility of neurological deficitsfrequently noted in HE patients who underwent successfulliver transplantation (Sotil et al. 2009) could be attributed tothe HE-induced derangement of the synaptic hardware.Detailed examination of the synaptic changes pre- and post-transplantation should allow to assess their relative roleversus that of a plethora of pre-existent or transplantation-related systemic and neurological impairments occurringindependently of HE.The work of Jayakumar et al. (2014) heralds the onset of a
much desired new line of investigations that eventuallyshould disentangle the role of synaptic protein deficit in theimpairment of neurotransmission associated with HE, and, inparticular, the role of astrocytic–neuronal interaction inproducing the deficit. One of the central questions is whetherthe loss of synaptic proteins goes far enough to be translatedto durable, functionally significant derangement of synaptichardware. Further studies along the line initiated by Jayaku-mar et al. (2014) are likely challenge the long prevailingview that neurophysiologic manifestations of astrocyticdysfunction in HE exclusively reflect mishandling byastrocytes of the neurotransmission software. The emergingvision is that, attempts at repairing the synaptic hardwareeither directly or by correcting astrocytic dysfunction mayoffer new treatment modalities of HE. The vision appears tobe open to experimental verification.
Acknowledgments and conflict of interestdisclosure
The authors’ work is sponsored by the National Centre for Researchand Development (NCBiR), Polish-Norwegian grant “CORE” (Pol-Nor/196190/26/2013) (JA) and the National Science Centre grantno. 2013/09/B/NZ4/00536M (MZ). JA is editor at Journal ofNeurochemistry.
The authors have no conflict of interest to declare.
References
Albrecht J. (2005) Astrocytes in ammonia neurotoxicity: a target, amediator and a shield, in The Role of Glia in Neurotoxicity(Aschner M. and Costa L. G., eds), pp. 329–342. CRC Press, BocaRaton.
Albrecht J. and Jones E. A. (1999) Hepatic encephalopathy: molecularmechanisms underlying the clinical syndrome. J. Neurol. Sci. 170,138–146.
Cekanaviciute E., Fathali N., Doyle K. P., Williams A. M., Han J. andBuckwalter M. S. (2014) Astrocytic transforming growth factor-
beta signaling reduces subacute neuroinflammation after stroke inmice. Glia 62, 1227–1240.
Chan H., Hazell A. S., Desjardins P. and Butterworth R. F. (2000) Effectsof ammonia on glutamate transporter (GLAST) protein and mRNAin cultured rat cortical astrocytes. Neurochem. Int. 37, 243–248.
Christopherson K. S., Ullian E. M., Stokes C. C., Mullowney C. E., HellJ. W., Agah A., Lawler J., Mosher D. F., Bornstein P. and BarresB. A. (2005) Thrombospondins are astrocyte-secreted proteins thatpromote CNS synaptogenesis. Cell 120, 421–433.
Garcia O., Torres M., Helguera P., Coskun P. and Busciglio J. (2010) Arole for thrombospondin-1 deficits in astrocyte-mediated spine andsynaptic pathology in Down’s syndrome. PLoS ONE 5, e14200.
Jayakumar A. R., Liu M., Moriyama M., Ramakrishnan R., Forbush B.,3rd, Reddy P. V. and Norenberg M. D. (2008) Na-K-ClCotransporter-1 in the mechanism of ammonia-induced astrocyteswelling. J. Biol. Chem. 283, 33874–33882.
Jayakumar A. R., Tong X. Y, Curtis K. M., Ruiz-Cordero R.,Shamaladevi N., Abuzamel M., Johnstone J., Gaidosh G., RamaRao K. V. and Norenberg M. D. (2014) Decreased astrocyticthrombospondin-1 secretion after chronic ammonia treatmentreduces the level of synaptic proteins: in vitro and in vivostudies. J. Neurochem.
Liauw J., Hoang S., Choi M. et al. (2008) Thrombospondins 1 and 2 arenecessary for synaptic plasticity and functional recovery afterstroke. J. Cereb. Blood Flow Metab. 28, 1722–1732.
Norenberg M. D., Hugo Z., Neary J. T. and Roig-Cantesano A. (1997)The glial glutamate transporter in hyperammonemia and hepaticencephalopathy: relation to energy metabolism and glutamatergicneurotransmission. Glia 21, 124–133.
Obara-Michlewska M., Pannicke T., Karl A., Bringmann A.,Reichenbach A., Szeliga M., Hilgier W., Wrzosek A., SzewczykA. and Albrecht J. (2011) Down-regulation of Kir4.1 in thecerebral cortex of rats with liver failure and in cultured astrocytestreated with glutamine: implications for astrocytic dysfunction inhepatic encephalopathy. J. Neurosci. Res. 89, 2018–2027.
Peterson C., Giguere J. -F., Cotman C. W. and Butterworth R. F. (1990)Selective loss of N-methyl-D-aspartate binding sites in rat brainfollowing portacaval anastomosis. J. Neurochem. 55, 386–390.
Prakash R. and Mullen K. D. (2010) Mechanisms, diagnosis andmanagement of hepatic encephalopathy. Nat. Rev. Gastroenterol.Hepatol. 7, 515–525.
Rama Rao K. V., Curtis K. M., Johnstone J. T. and Norenberg M. D.(2013) Amyloid-[beta] inhibits thrombospondin 1 releasefrom cultured astrocytes: effects on synaptic protein expression.J. Neuropathol. Exp. Neurol. 72, 735–744.
Rangroo Thrane V., Thrane A. S., Wang F. et al. (2013) Ammoniatriggers neuronal disinhibition and seizures by impairing astrocytepotassium buffering. Nat. Med. 19, 1643–1648.
Saransaari P., Oja S. S., Borkowska H. D., Kostinaho J., Hilgier W. andAlbrecht J. (1997) Effects of thioacetamide-induced hepatic failureon the N-methyl-D-aspartate receptor complex in the rat cerebralcortex, striatum and hippocampus: binding of different ligands andexpression of receptor subunit mRNA’s. Mol. Chem. Neuropathol.32, 179–194.
Sotil E. U., Gottstein J., Ayala E., Randolph C. and Blei A. T. (2009)Impact of preoperative overt hepatic encephalopathy onneurocognitive function after liver transplantation. Liver Transpl.15, 184–192.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12823
Review 3

, ,
*Laboratory of Neuropathology, Veterans Affairs Medical Center, Miami, Florida, USA
†Department of Pathology, University of Miami School of Medicine, Miami, Florida, USA
‡Geriatric Research, Education, and Clinical Center and Research Service, Miami, Florida, USA
§Department of Urology, University of Miami School of Medicine, Miami, Florida, USA
¶Bascom Palmer Eye Institute, University of Miami School of Medicine, Miami, Florida, USA
**Departments of Biochemistry & Molecular Biology, University of Miami School of Medicine, Miami,
Florida, USA
AbstractChronic hepatic encephalopathy (CHE) is a major complica-tion in patients with severe liver disease. Elevated blood andbrain ammonia levels have been implicated in its pathogen-esis, and astrocytes are the principal neural cells involved inthis disorder. Since defective synthesis and release ofastrocytic factors have been shown to impair synaptic integrityin other neurological conditions, we examined whether throm-bospondin-1 (TSP-1), an astrocytic factor involved in themaintenance of synaptic integrity, is also altered in CHE.Cultured astrocytes were exposed to ammonia (NH4Cl, 0.5–2.5 mM) for 1–10 days, and TSP-1 content was measured incell extracts and culture media. Astrocytes exposed toammonia exhibited a reduction in intra- and extracellularTSP-1 levels. Exposure of cultured neurons to conditionedmedia from ammonia-treated astrocytes showed a decrease in
synaptophysin, PSD95, and synaptotagmin levels. Condi-tioned media from TSP-1 over-expressing astrocytes that weretreated with ammonia, when added to cultured neurons,reversed the decline in synaptic proteins. Recombinant TSP-1similarly reversed the decrease in synaptic proteins. Metfor-min, an agent known to increase TSP-1 synthesis in other celltypes, also reversed the ammonia-induced TSP-1 reduction.Likewise, we found a significant decline in TSP-1 level incortical astrocytes, as well as a reduction in synaptophysincontent in vivo in a rat model of CHE. These findings suggestthat TSP-1 may represent an important therapeutic target forCHE.Keywords: ammonia, astrocytes, chronic hepatic encephalo-pathy, synaptic proteins, thrombospondin-1.J. Neurochem. (2014) 10.1111/jnc.12810
Hepatic encephalopathy (HE) is a major neurologicalcomplication in patients with severe liver disease. It ischaracterized by impaired neurological function and occursin acute and chronic forms. The encephalopathy associatedwith acute HE (acute liver failure) generally occurs followingmassive liver necrosis, generally because of viral hepatitis,hepatic neoplasms, vascular causes, or exposure to varioushepatotoxins. It presents with the abrupt onset of delirium,seizures, and coma, and has an extremely poor prognosis
Received April 26, 2014; revised manuscript received June 16, 2014;accepted June 22, 2014.Address correspondence and reprint requests to Michael D. Noren-
berg, Department of Pathology (D-33), P.O. Box 016960, University ofMiami School of Medicine, Miami, FL 33101, USA. E-mail:[email protected] used: CHE, chronic hepatic encephalopathy; CM,
conditioned media; GFAP, glial fibrillary acidic protein; NH4Cl,ammonium chloride (ammonia); TAA, thioacetamide; TSP-1, thrombo-spondin-1.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810 1
JOURNAL OF NEUROCHEMISTRY | 2014 doi: 10.1111/jnc.12810

(70%mortality) (Lee 2012; Shawcross andWendon2012).HEin the setting of chronic liver disease [chronic hepaticencephalopathy (CHE)] generally occurs as a consequenceof cirrhosis of the liver, usually secondary to alcoholism(Wakim-Fleming 2011). CHE is characterized by confusion,disorientation, behavioral changes, impaired cognition,inverted sleep–wake cycles, and motor disturbances. Themolecular basis for the neuropsychiatric disorder in CHEremains elusive (Mullen and Prakash 2012; Patel et al. 2012).Neuronal dysfunction is a well-established finding in CHE
(Cauli et al. 2009). Increased synthesis of gamma-aminobu-tyric acid (GABA) and subsequent alterations in GABA-ergic neurotransmission, as well as an increase in endoge-nous benzodiazepines (which modulate GABA-mediatedneurotransmission), have been proposed to be involved inCHE (Llansola et al. 2012). Inhibition of cortical functionand the subsequent behavioral defects from altered GABA-ergic signaling have also been postulated as major mecha-nisms leading to CHE (Bismuth et al. 2011).HE is one neurological disorder in which, early on,
astrocytes were suggested to play a vital role (Norenberg1986). A major factor in the pathogenesis of HE is elevatedblood and brain ammonia levels because of the inability ofthe injured liver to detoxify ammonia by the synthesis ofurea. Once in brain, ammonia is metabolized to glutamine bythe action of glutamine synthetase, a process that only occursin astrocytes (Norenberg 1979). It is therefore of interest thatthe principal histopathological finding in CHE is the presenceof Alzheimer type II astrocytosis. These astrocytes arecharacterized by the presence of pale and enlarged nuclei(often found in pairs), along with margination of nuclearchromatin and the presence of prominent nucleoli (Noren-berg 1979). Although the significance of this astrocyticchange is incompletely understood, abundant data stronglysuggest that Alzheimer type II astrocytes are dysfunctionalcells (Norenberg 1987; Norenberg et al. 1998). This has ledto the concept that HE fundamentally represents a primaryastrogliopathy (Norenberg 1987; Norenberg et al. 1998).Astrocytes play a crucial role in the central nervous system
by regulating a number of critical processes, includingsynaptogenesis, synaptic function, modulation of neurotrans-mission, regulation of pH, ion and water homoeostasis,energy metabolism, defense against oxidative stress and thedetoxification of ammonia, metals, and other toxins (Noren-berg 1987; Wang and Bordey 2008; B�elanger and Magistretti2009). Astrocytes are also involved in the provision ofgrowth factors and nutrients to neurons and other neural cells(Wang and Bordey 2008), as well as in the formation andmaintenance of the blood–brain barrier (Abbott et al. 2006).One potential mechanism by which defective astrocytes
may impact neuronal integrity is through a reduction inthrombospondin-1 (TSP-1) levels. Astrocytes are known tosynthesize and secrete TSP-1 (Christopherson et al. 2005;Tran and Neary 2006), and a reduction in TSP-1 expression
by siRNA silencing was reported to decrease neuronalsynaptophysin protein expression (Yu et al. 2008). Suchreduction in TSP-1 and synaptophysin protein levels wasassociated with behavioral abnormalities in experimentalmodels of stroke (Lin et al. 2003; Liauw et al. 2008),Alzheimer’s disease (Bu�ee et al. 1992), and Down’s syn-drome (Garcia et al. 2010).This study examined intra- and extracellular levels of TSP-
1 in ammonia-treated cultured astrocytes, and the effect ofconditioned media (CM) from ammonia-treated astrocytes onsynaptic protein levels in cultured neurons. A significantdecrease in TSP-1 protein level was observed in ammonia-treated astrocyte cultures. We also found a reduction insynaptophysin protein levels when cultured neurons wereexposed to CM from ammonia-treated astrocytes, and thatenhancing intra- and extracellular levels of astrocytic TSP-1reversed the synaptophysin loss. We further observed asignificant decline in astrocytic TSP-1, and in neuronalsynaptophysin levels in a rat model of CHE. Our findingssuggest that dysfunctional astrocytes resulting from ammoniatreatment negatively impacts neuronal synaptic integrity,thereby contributing to the neurological abnormalities asso-ciated with CHE.
Materials and methods
Astrocyte cultures
Primary cultures of cortical astrocytes were prepared from brains of1- to 2-day-old rat pups by the method of Ducis et al. (1990).Briefly, cerebral cortices were freed of meninges, minced, dissoci-ated by trituration and vortexing, and were seeded onto 35-mmculture dishes in Dulbecco’s modified Eagle’s medium containingpenicillin, streptomycin, and 15% fetal bovine serum. The cultureplates were incubated at 37°C with 5% CO2 and 95% air. Culturemedia were changed twice weekly. On day 10 post seeding, fetalbovine serum was replaced with 10% horse serum. After 14 days,cultures were treated with 0.5 mM dibutyryl cAMP (Sigma, St.Louis, MO, USA) to enhance cellular differentiation (Juurlink andHertz 1985). Cultures consisted of at least 95% astrocytes asdetermined by glial fibrillary acidic protein (GFAP) immunohisto-chemistry. All cultures used were 21–23 days old.
All animal procedures followed the guidelines established by theNational Institutes of Health Guide for the Care and Use ofLaboratory Animals and were approved by our Institutional AnimalCare and Use Committee (IACUC).
Neuronal cultures
Cortical neuronal cultures were prepared by a modification of themethod described by Schousboe et al. (1989). Briefly, cortices wereremoved from 16- to 18-day-old rat fetuses and placed inDulbecco’s modified Eagle’s medium (30 mM glucose) with25 mM KCl and 10% horse serum. The tissue was minced andmechanically dissociated with a pipette. Approximately 1–2 9 106
cells per mL were seeded onto poly-D-lysine-coated 35-mm culturedishes. To prevent the proliferation of astrocytes, cytosine arabino-side (10 lM) was added to the culture medium 48 h after seeding.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
2 A. R. Jayakumar et al.

These cultures consist of at least 90% neurons as determined byimmunohistochemical staining for neurofilament protein; theremaining cells were chiefly astrocytes. Experiments were per-formed on cultures that were 7–8 days old.
TSP-1 over-expression in cultured astrocytes
To examine whether the exposure of cultured neurons to CM fromammonia-treated astrocytes in which TSP-1 is over-expresseddiminishes or prevents the reduction in synaptic proteins, we over-expressed TSP-1 in cultured astrocytes. Briefly, astrocytes weretransfected with TSP-1 cDNA (tagged with the pIRES2-AcGFP1vector; GENEWIZ Inc, South Plainfield, NJ, USA). Cultures wereexposed to different concentrations of TSP-1 cDNA (50, 100, and250 ng/2.5 9 105 cells) for 72 h. Mirus TransIT-TKO transfectionreagent was used to transfect TSP-1 following the manufacturer’sinstructions (#MIR 2150; Mirus, Madison, WI, USA). At the end oftransfection, the culture media were replaced with normal media.These cultures were treated with and without ammonia for 10 days,and at the end of treatment, intra- and extracellular levels of TSP-1were measured by western blots. The empty vector, as well as thetransfection reagent alone, was used as controls for all experiments.
Immunohistochemistry of TSP-1 and synaptophysin in CHE
Control and thioacetamide (TAA)-treated rats (four animals each)were anesthetized at the end of TAA treatment with a mixture ofketamine (80 mg/kg) and xylacine (20 mg/kg), and were thentranscardially perfused with heparinized saline for 1 min, followedby fixation in 4% paraformaldehyde for 15 min. After decapitation,heads were left in the same fixative for an additional 24 h at 22°C.Brains were cryoprotected (12–24 h) in 30% sucrose. Corticalsections (20 lm thick) were obtained with a cryostat; sections wereblocked with 10% goat serum and incubated overnight at 4°C withanti-synaptophysin antibody (rabbit monoclonal, YE269, 1 : 150dilution; cat# 32127; Abcam, Cambridge, MA, USA), BA24 anti-thrombospondin 1 antibody (mouse monoclonal A6.1, 1 : 100dilution; Millipore, Billerica, MA, USA), and anti-GFAP antibody(astrocyte marker, 1 : 150 dilution, cat# ab7260; Abcam), asdescribed previously (Jayakumar et al. 2014). Following incubationwith primary antibodies, sections were washed with tris-bufferedsaline containing 1% Tween-20 and incubated with fluorescenthorseradish peroxidase-conjugated secondary antibodies [(1 : 500;Alexa Flour-546 goat anti-rabbit IgG (H + L) (Life Technologies,Grand Island, NY, USA) for GFAP, Alexa Flour-488 goat anti-mouse IgG (H + L) (Life Technologies) for TSP-1, and AlexaFlour-488 goat anti-rabbit IgG (H + L) (Life Technologies) forsynaptophysin)], for 2 h. Sections were then covered with com-mercial mounting media (Vector Laboratories, Burlingame, CA,USA) containing 40,6-diamidino-2-phenylindole (DAPI) (nuclearstain). Immunofluorescent images were acquired with a ZeissLSM510/UV Axiovert 200M confocal microscope (Carl ZeissMicroscopy, LLC, Thornwood, NY, USA) with a plan apochromat409 objective lens, and a 29 zoom resulting in images125 9 125 lm in area and 1.0 lm optical slice thickness (1.0 Airyunits for Alexa Fluor 546 or 568 emission channel). Randomcollection of images from sections of control and TAA-treated ratswas achieved by systematically capturing each image in a ‘blinded’manner by moving the microscope stage approximately 5 mm infour different directions. At least 17 fluorescent images were
captured per rat, and the images merged to localize astrocytic TSP-1protein. TSP-1 and synaptophysin levels in cultured neurons and inbrain sections were quantified using the Volocity 6.0 HighPerformance Cellular Imaging Software (PerkinElmer, Waltham,MA, USA) as described previously (Rick et al. 2013; Jayakumaret al. 2014), and normalized to the number of DAPI-positive cells,as well as to the intensity of DAPI.
Statistical analysis
All experiments were performed and repeated four to six times usingcells derived from different batches of astrocyte and neuronalcultures. Five to six individual culture plates were used in eachexperimental group for TSP-1, and four to six for synaptophysinmeasurements. Six animals from each group were used for in vivostudies. Data of all experiments were subjected to analysis ofvariance followed by Tukey’s post hoc comparisons. A value ofp < 0.05 was considered significant. Error bars, mean � SE.
Results
Intra- and extracellular TSP-1 in cultured astrocytes after
treatment with ammonia
Primary cultures of rat cortical astrocytes treated withammonia were used in this study. The use of such culturesas a model of HE is highly appropriate since substantialevidence invokes a crucial role of ammonia in the patho-genesis of HE, and astrocytes are the principal cells affectedin this condition (Norenberg et al. 2009). Moreover, many ofthe findings occurring in HE in vivo are also observed inammonia-treated astrocyte cultures, including characteristicmorphologic changes, cell swelling, defects in glutamatetransport, up-regulation of the 18-kDa translocator protein,reduction in levels of glial fibrillary acidic protein (Sobelet al. 1981; Kretzschmar et al. 1985; Kimura and Budka1986) and myo-inositol (Norenberg et al. 2009), disturbancein energy metabolism, and evidence of oxidative/nitrativestress (ONS) (Lange et al. 2012).Pathophysiological concentrations of ammonia (0.5, 1.0,
and 2.5 mM NH4Cl) (Dejong et al. 1993; Singh and Trigun2010; Carbonero-Aguilar et al. 2011) (also see Table 1)were added to astrocyte cultures for different time periods (1,5, and 10 days with regular media changes, once in 2 daysfor both 5- and 10-day treatment). At the end of treatment(24 h after the addition of last ammonia exposure), TSP-1
Table 1 Blood and brain ammonia levels of rats treated with thioac-
etamide (TAA)
Control TAA
Blood ammonia (lM) 191.6 � 11.7 611.2 � 32.1*Brain ammonia (mM) 0.34 � 0.07 0.96 � 0.16*
Control n = 5; TAA n = 5. Blood and brain ammonia levels weredetermined 24 h after the last injection of TAA (100 mg/kg bw).
*p < 0.05 versus control.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 3

protein levels in the cell extracts and culture media weremeasured by western blots. Fresh addition of inhibitors andantioxidants was performed with each change of culturemedium during the entire period of incubation.Exposure of astrocytes to 0.5, 1.0, and 2.5 mM NH4Cl for
10 days caused a reduction in extracellular TSP-1 levels(58.6, 43.9, and 67.5%, respectively) (Fig. 1). IntracellularTSP-1 levels were also reduced (16.2, 57.4, and 40.7%,respectively) (Fig. 2). We additionally found a significantdecline in TSP-1 mRNA level when astrocytes were exposedto 0.5, 1.0, and 2.5 mM ammonia for 10 days (Fig. 2c).Exposure of astrocytes to 0.5 and 1.0 mM ammonia for
5 days had no effect on extra- (data not shown) orintracellular TSP-1 levels (Figure S2a). However, exposureto 2.5 mM ammonia significantly reduced TSP-1 levels by51.6% (Figure S2a and b). Although TSP-1 protein levelswere not altered in 0.5 and 1.0 mM ammonia-treatedastrocytes, a significant decline in TSP-1 mRNA level wasobserved at these ammonia concentrations (Figure S2c).
Hevin is another matricellular protein that is known toinfluence neuronal synaptic integrity in other conditions(Kucukdereli et al. 2011). Its level also decreased inammonia-treated cultured astrocytes (intra- and extracellu-lar levels by 35.7% and 31.1%, respectively, n = 5,p < 0.05 vs. control). As decreased transforming growthfactor beta (TGF-b1) was shown to influence TSP-1 levels(Okamoto et al. 2002; Mimura et al. 2005; McGillicuddy
(a)
(b)
Fig. 1 Extracellular thrombospondin-1 (TSP-1) level after a 10-daytreatment of cultured astrocytes with 0.5–2.5 mM ammonia (NH4Cl).(a) Representative western blots from cell culture media of ammonia-treated astrocytes show a significant decrease in TSP-1 levels. (b)
Quantification of ammonia-induced changes in TSP-1 protein levels.*p < 0.05 versus control. C, control; NH4
+, ammonia.
(a)
(b)
(c)
Fig. 2 Intracellular thrombospondin-1 (TSP-1) level after a 10-day
treatment of cultured astrocytes with 0.5–2.5 mM ammonia. (a)Representative western blots from ammonia-treated astrocytes(1 and 2.5 mM) show a significant decrease in intracellular TSP-1levels. (b) Quantification of NH4Cl-induced changes in TSP-1 protein.
TSP-1 levels were normalized against a-tubulin. (c) TSP-1 mRNAexpression after ammonia treatment. *p < 0.05 versus control. C,control; N, NH4Cl.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
4 A. R. Jayakumar et al.

et al. 2006), we examined whether ammonia also influ-enced TGF-b1 levels. A reduction (57.8%) in intracellularlevels of TGF-b1 was detected when astrocytes weretreated with ammonia (Figure S3). The reduction in thelevel of hevin, however, was of a lesser magnitude thanthat observed with TSP-1.
Effect of conditioned media from ammonia-treated
astrocytes on neuronal synaptic proteins
We examined whether an ammonia-induced decline inextracellular TSP-1 concentration contributes to a reductionin neuronal synaptophysin. Accordingly, cultured astrocyteswere treated with ammonia (0.5, 1.0, and 2.5 mM, NH4Cl)for 10 days, and at the end of treatment, 1 mL of CM fromammonia-treated astrocytes was added to neuronal cultures.Synaptophysin level was determined 24 h later by immuno-
fluorescence and western blots. Cultured neurons exposed toCM from 0.5–2.5 mM NH4Cl-treated astrocytes showed asignificant reduction in synaptophysin levels in a dose-dependent manner, as measured both by immunofluores-cence (Figure S4a–e) and western blots (Fig. 3a), whichcorresponded well with levels of TSP-1 reduction. Quanti-fication of the immunocytochemistry data also correspondedwell with findings in western blots (Figure S4e and Fig. 3a).While CM from ammonia-treated astrocytes (0.5–2.5 mM)reduced synaptophysin levels, the extent of reduction amongthe three ammonia-treated groups was not statisticallysignificantly different from each other. Exposed neuroncultures to CM from control astrocytes had no effect onsynaptophysin protein levels (data not shown).It should be emphasized that the direct exposure of
cultured cortical neurons to 0.5–2.5 mM ammonia for 24 h
(a)
(b) (c)
Fig. 3 Synaptophysin, PSD95 andsynaptotagmin protein levels in cultured
neurons. (a) Representative western blotsfrom astrocytes treated with ammonia (0.5–2.5 mM) for 10 days and the conditioned
media (CM) added to neurons for 24 h.Such treatment led to a significant reductionin synaptophysin in a dose-dependent
manner. (b) CM from ammonia-treatedastrocytes also decreased PSD95, andsynaptotagmin levels (c). *p < 0.05 versus
control. C, control.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 5

had no significant effect on synaptophysin level. Addition-ally, we found no residual ammonia in the ammonia-treatedastrocytic CM (at 24 h) that was subsequently applied tocultured neurons. We also found no detectable levels ofammonia 2 h after exposure of cultured astrocytes toammonia (Jayakumar et al. 2006). This absence of ammoniais likely caused by a rapid conversion of ammonia intoglutamine via glutamine synthetase activity (Cooper et al.1979). These findings indicate that the ammonia-mediatedreduction in neuronal synaptophysin levels is caused by aneffect of ammonia on astrocytes, and not to a direct effect ofammonia on neurons.We also examined whether PSD95, a post-synaptic density
protein, and synaptotagmin-1, a Ca2+ sensor in the membraneof the pre-synaptic axon terminal, which has also beenimplicated in the maintenance of synaptic integrity, weresimilarly affected by CM from ammonia-treated astrocytes.We indeed found that when cultured neurons were exposedto CM from ammonia-treated astrocytes (1.0 mM, 10 days),decreased levels of PSD95 (32.7%) and synaptotagmin-1(26.8%) were observed (Fig. 3b and c). However, the extentof reduction in PSD95 and synaptotagmin-1 levels was lessthan that observed in synaptophysin. The direct exposure ofcultured neurons to ammonia (1 mM, 24 h) also had nosignificant effect on PSD95 protein levels (data not shown).
CM from TSP-1 over-expressing cultured astrocytes that
were treated with ammonia, when added to cultured
neurons, reversed the decrease in synaptic proteins
We then examined whether the effect of CM from ammonia-treated cultured astrocytes on the decline in levels ofneuronal synaptic proteins is indeed caused by diminishedastrocytic TSP-1 levels. Accordingly TSP-1 was over-expressed (with 100 and 250 ng/mL TSP-1 cDNA) incultured astrocytes. TSP-1 over-expressed cells that weretreated with ammonia (1 mM, 10 days) showed a significantincrease in extracellular levels of TSP-1 (1.2- to 1.5-foldmore than the effect of CM from ammonia-treated astrocytesthat were exposed to an ‘empty’ vector or with thetransfection reagent alone). Exposure of cultured corticalneurons to CM from ammonia-treated astrocytes showed areduction in synaptic proteins. On the other hand, exposureof neurons to CM from ammonia-treated astrocyte culturesthat had been transfected with 250 ng TSP-1 cDNA led to alesser reduction in synaptophysin, PSD95, and synaptotag-min content by 73.5%, 65.9%, and 78.2%, respectively(n = 4, p < 0.05 vs. respective controls).We also examined whether the addition of recombinant
TSP-1 (rTSP-1) also prevented the effect of CM fromammonia-treated (1.0 mM) cultured astrocytes on the syn-aptophysin loss in neurons. For this purpose, cultured corticalneurons were exposed to CM from ammonia-treated(1.0 mM) cultured astrocytes, along with recombinantTSP-1 (rTSP-1; 50, 100, 200 ng/mL) for 24 h, and levels
of synaptophysin were measured by western blots. Neuronsexposed to CM from ammonia-treated astrocytes showed a43.6% reduction in synaptophysin content, and such effectwas significantly reversed by 50, 100, and 200 ng/mL rTSP-1 (39.5%, 68.7%, and 35.6%, respectively) (Fig. 4).In addition, we investigated whether depletion of TSP-1 in
the astrocytic CM affects neuronal synaptophyin content. Wefound that TSP-1 levels in the CM of astrocytes weredepleted by immunoprecipitation. The resulting CM whenadded to cultured neurons (for 24 h) exhibited a significantloss of synaptophysin content (43.6%), supporting theconcept that TSP-1 is critical for the maintenance ofsynaptophysin levels in CHE.
Effect of c-Myc on TSP-1 level after ammonia treatment to
cultured astrocytes
c-Myc is a transcription factor expressed in astrocytes in vitroand in vivo (Saljo et al. 2002; Liu et al. 2006), and its over-expression was shown to cause a decrease in TSP-1 proteinby exerting a repressor effect on TSP-1 mRNA (Watnicket al. 2003). We therefore examined whether c-Myccontributes to the ammonia-induced reduction in astrocytic
Fig. 4 Effect of recombinant thrombospondin-1 (rTSP-1) on synapto-
physin level. Representative western blots from cultured astrocytestreated with ammonia (1.0 mM) for 10 days and the CM was thenadded to cultured neurons for 24 h, along with rTSP-1. Such treatmentcaused a reversal of the synaptophysin loss. *p < 0.05 versus control.
†p < 0.05 versus CM from ammonia-treated (1.0 mM) astrocytes. C,control; CM, conditioned medium.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
6 A. R. Jayakumar et al.

TSP-1. Exposure of cultured astrocytes to ammonia (1 mM,10 days) increased levels of c-Myc protein by 86.2% (Fig. 5a).We then examined whether silencing c-Myc with siRNA
prevents or diminishes the reduction in TSP-1 levels byammonia. c-Myc was silenced in cultured astrocytes withsiRNA as described previously (Lu and Hong 2009), andfound that transfection with 40, 60, and 80 nM of c-MycsiRNA significantly reduced the concentration of c-Myc (by47.8%, 69.4%, and 76.5%, respectively) (Fig. 5b). Exposureof c-Myc-silenced cells (80 nM siRNA) to 1 mM ammonia(10 days) caused a 64% and 60% recovery in intra- andextracellular levels of TSP-1, respectively (Fig. 5c and d)(n = 5, p < 0.05 vs. respective controls) as compared to theeffect of ammonia on scrambled siRNA-treated cells, whichshowed a 40–45% reduction in TSP-1. Exposure of culturedneurons (24 h) to CM from ammonia-treated astrocytes(10 days), in which the c-Myc-gene was silenced, resulted ina lesser reduction in the level of synaptophysin (by 61%)(Fig. 6a).
We also examined whether a pharmacological inhibition ofc-Myc prevents or diminishes the reduction in TSP-1 levelsby ammonia. We found that treatment of astrocytes withammonia plus 10058-F4 (50 lM), an inhibitor of c-Myc, for10 days enhanced intra- and extracellular TSP-1 levels by64.2% and 61.2%, respectively (Fig. 6b). Higher doses of10058-F4 (100 and 150 lM) showed no additional effect(data not shown). Furthermore, exposure of cultured neurons(24 h) to CM from ammonia-treated astrocytes (10 days), inwhich c-Myc was inhibited by 10058-F4, caused a reversalof the synaptophysin loss (59.4%), as compared to the effectof CM only from ammonia-treated astrocytes (Fig. 6c).
Attenuation of the ammonia-induced inhibition of TSP-1 byantioxidants and an inhibitor of nuclear factor kappa B
A considerable body of evidence suggests that the formationof reactive oxygen/nitrogen species and the resulting ONSplays a major role in HE (Jayakumar and Norenberg 2012).In addition, activation of the transcription factor nuclear
(a) (b)
(c) (d)
Fig. 5 Effect of c-Myc on thrombospondin-1(TSP-1) expression. (a) Representative
western blot from ammonia-treatedastrocytes shows a significant increase in c-Myc protein. (b) Treatment with c-Myc siRNA
significantly reduced c-Myc concentration.(c and d) Inhibition of c-Myc by siRNAsignificantly increased intra- and
extracellular TSP-1 protein level. *p < 0.05versus control; †p < 0.05 versus scrambledcontrol. C, control; NH4
+, ammonia; Scr,
scrambled control (control siRNA).
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 7

factor kappa B (NF-кB) was identified in ammonia-treatedcultured astrocytes (Schliess et al. 2002; Sinke et al. 2008).Since ONS and NF-кB have been strongly implicated in themechanism of TSP-1 down-regulation in other conditions(De Stefano et al. 2009; Tan et al. 2009; Chen et al. 2011),we examined whether ONS and NF-кB are likewise involvedin the mechanism of the ammonia-induced reduction in TSP-1 levels in cultured astrocytes. Astrocyte cultures weretreated with the antioxidants Mn(III) tetrakis (4-benzoic acid)porphyrin (MnTBAP, 10 lM), dimethylthiourea (DMTU,100 lM), and the nitric oxide synthase inhibitor N-nitro-L-arginine methyl ester (L-NAME) (250 lM), as well as SN50(0.5–1.0 lM), an inhibitor of NF-кB, along with ammonia
(1.0 mM) for 10 consecutive days (with regular mediachanges), and the level of TSP-1 in the culture media wasdetermined by western blots. MnTBAP, DMTU, and L-NAME significantly reduced the inhibition of TSP-1 level inthe culture media after the exposure to 1.0 mM ammonia(Figure S5a). Similarly, SN50 significantly reduced theinhibition of extracellular TSP-1 level after the exposure to1.0 mM ammonia (Figure S5b). These findings indicate thatONS and the activation of NF-кB indeed contribute to theammonia-induced inhibition of TSP-1 release.The effect of antioxidants on the ammonia-induced
reduction in TSP-1 mRNA expression was also examinedin cultured astrocytes. Treatment of astrocytes with MnT-
(a) (b)
(c)
Fig. 6 Effect of astrocytic c-Myc inhibition
on neuronal synaptophysin levels. (a–c)Astrocytes in which the c-Myc-gene wassilenced, or exposed to a c-Myc inhibitor,10058-F4, along with ammonia and the CM
then added to cultured neurons (for 24 h)resulted in a lesser reduction insynaptophysin level. *p < 0.05 versus
control; †p < 0.05 versus scrambledcontrol. C, control; NH4
+, ammonia; Scr,scrambled control (control siRNA); CM,
conditioned media.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
8 A. R. Jayakumar et al.

BAP (10 lM), DMTU (100 lM), and L-NAME (250 lM)significantly diminished the ammonia-induced reduction inTSP-1 mRNA (Figure S5c), suggesting that the reduction inTSP-1 levels in ammonia-treated astrocytes is caused by adefective transcriptional regulation of TSP-1 by ONS.We further examined whether an agent known to enhance
TSP-1 synthesis and release is capable of reversing theammonia-induced reduction in TSP-1. For this purpose, weinvestigated the effect of metformin, a commonly used anti-diabetic agent that is known to enhance the release of TSP-1(Tan et al. 2009). Metformin was also shown to exert aprotective effect in diabetic patients who also had HE(Ampuero et al. 2012). We found that metformin (25 lM)diminished the ammonia-induced reduction in intra- andextracellular levels of TSP-1 in astrocytes (by 77.3% and68.9%, respectively, p < 0.05 vs. ammonia group). Further-more, exposure of cultured neurons (24 h) to CM fromammonia-treated astrocytes (10 days), in which TSP-1 levelswere enhanced by metformin, caused an elevation insynaptophysin levels by 65.8% (p < 0.05), as compared tothe effect of CM astrocytes treated only with ammonia.
TSP-1 and synaptophysin levels in cerebral cortex of rats
with CHE
To examine whether comparable alterations in TSP-1 andsynaptophysin protein level also occur in an in vivo model ofCHE, rats were treated with the liver toxin thioacetamide(TAA, 100 mg/kg bw) for 10 days, and the extent of TSP-1and synaptophysin protein expression in cortical sectionswere examined. To identify changes in TSP-1 in astrocytes,sections were co-immunostained with GFAP (astrocytemarker) and DAPI (nuclear marker), and the degree ofimmunofluorescence examined with a confocal laser scan-ning microscope. A significant reduction (52.4%) in TSP-1fluorescence was detected in astrocytes from TAA-treatedrats as compared to control rats (Fig. 7a–c). We also found acomparable reduction in synaptophysin protein in corticalsections of rats treated with TAA (Fig. 8). A slight reductionin GFAP fluorescence was identified in TAA-treated rats(Fig. 7). Such reduction was previously reported in humanswith HE, as well as in ammonia-treated cultured astrocytes(Sobel et al. 1981; Kretzschmar et al. 1985; Kimura andBudka 1986). However, we did not observe a significantchange in the total number of astrocytes in this in vivo modelof CHE as measured by counting the astrocyte nuclei (stainedwith DAPI) that were co-stained with GFAP. In addition, itshould be noted that a decrease in the number of astrocytes isnot a feature of humans with chronic HE (Norenberg et al.1992). We also measured TSP-1 level in cerebral cortex ofTAA-treated rats by western blots. Cortical tissues from ratstreated with TAA for 3 days showed a significant decline inTSP-1 content (42.3% decrease, as compared to control;n = 5) (Fig. 7d and e), which corresponded well with dataobtained by immunofluorescence.
Discussion
Our results demonstrate that chronic ammonia toxicity incultured astrocytes results in decreased extracellular levels ofTSP-1. In addition, synaptophysin levels declined in culturedneurons after exposure to CM derived from ammonia-treatedastrocytes, and such effect was diminished when recombi-nant TSP-1 (rTSP-1) was added to the CM of ammonia-treated astrocytes. We also found decreased levels of othersynaptic proteins (PSD95 and synaptotagmin-1), whenneurons were exposed to CM derived from ammonia-treatedastrocytes. Increased c-Myc protein content was detected inammonia-treated astrocytes, while silencing c-Myc or phar-macological inhibition of c-Myc significantly enhanced intra-and extracellular levels of TSP-1 after ammonia treatment incultured astrocytes. Furthermore, exposure of cultured neu-rons (24 h) to CM from ammonia-treated astrocytes, inwhich c-Myc was silenced or inhibited by 10058-F4, causeda reversal of the synaptophysin loss. A reduction in TSP-1and neuronal synaptophysin was also detected in an in vivorat model of CHE. Altogether, these findings stronglysuggest that an ammonia-induced reduction in TSP-1, andpossibly other factors in astrocytes (see below), results in adecline in synaptic protein levels, which likely contributes tothe pathogenesis of CHE.TSP-1, also known as THBS1 or THP1 protein, is a
member of the thrombospondin family that in humans isencoded by the THBS1 gene. This protein can bind tofibrinogen, fibronectin, laminin, type V collagen, andintegrins alpha-V/beta-1, and thereby initiate cell–cell andcell–matrix interactions (Li et al. 2002). TSP-1 expressionwas identified in post-natal and young adult animal brains(Lu and Kipnis 2010; Yonezawa et al. 2010), as well as innormal human cortical astrocytes (Asch et al. 1986).Furthermore, cultured astrocytes are known to synthesizeand secrete TSP-1 (Christopherson et al. 2005; Tran andNeary 2006; Yonezawa et al. 2010). Under normal condi-tions, TSP-1 causes an increase in the total number ofsynapses (Christopherson et al. 2005; Eroglu et al., 2009),as well as accelerates synaptogenesis (Xu et al., 2010).Astrocyte-derived TSP-1 was also shown to mediate thedevelopment of pre-synaptic plasticity in vitro (Crawfordet al. 2012). Conversely, defective astrocytic TSP-1 releaseis known to be associated with neuronal dysfunction (Garciaet al. 2010).Yu et al. (2008) demonstrated that retinal ganglion cell
survival and neurite outgrowth were improved whenco-cultured with bone marrow stromal cells (which areknown to release TSP-1), a process mediated by the up-regulation of synaptophysin. On the other hand, decreasingTSP-1 expression by siRNA silencing led to a reduction inneurite outgrowth and a decrease in the expression ofsynaptophysin in retinal ganglion cells (Yu et al. 2008). Incomparable studies, Rama Rao et al. (2013) showed that
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 9

exposure of cultured astrocytes to b-amyloid peptide signif-icantly reduced extracellular TSP-1, and that CM from theseb-amyloid peptide-treated astrocytes when added to culturedneurons led to a decrease in synaptophysin protein levels.In the present study, we observed a significant decrease in
both TSP-1 synthesis and release when cultured astrocyteswere exposed to a pathophysiologically relevant concentra-tion of ammonia (1 mM), and that inhibition of c-Mycreversed the ammonia-induced reduction in TSP-1 (seebelow), suggesting the involvement of a transcriptionalregulatory mechanism in the effect of ammonia on TSP-1.
However, as noted just above, b-amyloid peptide reducedonly the extracellular, but not intracellular TSP-1 levels,suggesting that the mode of action of ammonia on astrocyticTSP-1 content is different from the effect of b-amyloidpeptide. We also found that exposure of cultured neurons toCM from ammonia-treated astrocytes led to a decrease insynaptophysin protein levels, while the addition of recombi-nant TSP-1 diminished this effect. These findings indicatethat a decrease in both intra- and extracellular TSP-1contributes to the reduction of neuronal synaptophysinobserved in CHE. It is, however, possible that a diminution
(a) (b) (c)
(d)
(e)
Fig. 7 Thrombospondin-1 (TSP-1) protein levels in astrocytes fromcerebral cortex of rats with chronic hepatic encephalopathy (CHE)following the administration of the hepatotoxin thioacetamide (TAA,
100 mg/kg) for 10 days. (a) Control brain: glial fibrillary acidic protein(GFAP, red, astrocytes), TSP-1 (green), and DAPI (blue, nuclei). Theco-localization (merged) image of TSP-1 and GFAP illustrates the
enrichment of TSP-1 in astrocytes. (b) TAA-treated rat brain showed a
reduction in astrocytic TSP-1 content. (c) Relative quantification ofTSP-1 immunofluorescence staining. Scale bar = 20 lm. (d). CorticalTSP-1 level after a 10-day treatment of rats with TAA. Representative
western blots from TAA-treated rats show a significant decrease inintracellular TSP-1 level. (e) Quantification of TSP-1 immunoblots.*p < 0.05 versus control. C, control.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
10 A. R. Jayakumar et al.
in astrocytic factors other than TSP-1 (e.g., secreted proteinacidic and rich in cysteine, glypicans, nerve growth factor,basic fibroblast growth factor, as well as cholesterol) mayalso have contributed to the reduction in synaptophysinlevels. In this regard, it is noteworthy that we found areduction in hevin in ammonia-treated astrocytes, which mayalso have contributed to the reduction in synaptophysin.As noted above and shown in Figure 2, TSP-1 protein
levels were not changed at day 10 after 0.5 mM ammoniatreatment, although a significant decline in TSP-1 mRNAlevel was detected at that time. Similar findings were alsoobserved with 0.5 and 1 mM ammonia at day 5 aftertreatment (Figure S2b and c). The reason for the delayeddecrease in TSP-1 protein compared to mRNA may resultfrom a slow turnover rate of TSP-1 protein. Further, exposureof astrocytes to 1 mM ammonia for 10 days showed areduction in intra- and extracellular levels of TSP-1 as well asTSP-1 mRNA (Fig. 2), and that inhibitors of ONS, c-Myc orNF-кB prevented such effects (Figs 5 and 6 and Figure S5a,b and c). These findings suggest that the reduction in TSP-1levels in 1 mM ammonia-treated astrocytes may be due to adefective transcriptional regulation of TSP-1. On the otherhand, exposure of astrocytes to 0.5 mM ammonia for 10days showed a reduction in extra-cellular, but not intracel-lular, TSP-1 levels (Figs 1b and 2b), suggesting animpairment in the release of TSP-1 or that enhanced extra-cellular degradation mechanisms are likely involved.While the mechanism by which ammonia reduces the
synthesis and secretion of TSP-1 in CHE is not known, it islikely that ONS and the activation of NF-кB are involved (DeStefano et al. 2009; Tan et al. 2009; Chen et al. 2011).Markers of ONS have been identified in animal models ofCHE, and in humans with CHE (Jayakumar and Norenberg2012). In addition, activation of NF-кB was identified inammonia-treated cultured astrocytes (Schliess et al. 2002;Sinke et al. 2008; Jayakumar et al. 2011). In this study, thedecrease in intra- and extracellular TSP-1 levels whenastrocytes were treated with ammonia was inhibited by
antioxidants, as well as by an NF-кB inhibitor, implicatingONS and NF-кB in the reduction of astrocytic TSP-1 byammonia. In agreement with these findings, it was shownthat exposure of astrocyte cultures to cobalt chloride (aninducer of reactive oxygen species) inhibited TSP-1 mRNAexpression (Chen et al. 2011), whereas the NF-кB inhibitorSN50 reduced the TSP-1 down-regulation in cultured humanretinal glial cells following viral infection (Cinatl et al.2000).c-Myc is a transcription factor that is expressed in
astrocytes in vitro and in vivo (Saljo et al. 2002; Liu et al.2006). Over-expression of c-Myc protein was shown to causea decrease in TSP-1 protein level by exerting a repressoreffect on TSP-1 mRNA (Watnick et al. 2003). Our study alsoidentified an increase in c-Myc protein level in ammonia-treated astrocytes (1.0 mM for 10 days), while the pharma-cological inhibition of c-Myc, or silencing c-Myc by siRNAprevented the reduction in TSP-1 levels by ammonia (Figs 5and 6). In addition to c-Myc inhibition, antioxidants and NF-кB inhibitors also prevented the reduction in TSP-1 levels byammonia (see above), suggesting that ONS and NF-кB alsocontribute to the regulation of TSP-1 in CHE.The activation of ONS, NF-кB, and c-Myc were identified
in astrocytes at similar time points (8–10 days) afterammonia treatment. Since all of these factors appear to havecontributed to the reduction in TSP-1 synthesis and release, itis possible that these factors acted independently to reduceTSP-1 synthesis and release. However, interaction amongthese factors may also have occurred. For example, ONS isknown to activate NF-кB (Bowie and O’Neill 2000), NF-кBis known to stimulate c-Myc activation (Ji et al. 1994; LaRosa et al. 1994), and ONS is known to increase c-Mycexpression (H€ocker et al. 1998; Joseph et al. 2001). Never-theless, the precise mechanisms by which these factorsultimately lead to a reduction in TSP-1 levels in ammonia-treated astrocytes remains to be determined.It should also be noted that ONS and NF-кB can inhibit
TGF-b (Bitzer et al., 2000; Choi et al. 2013), which is
(a) (b) (c)
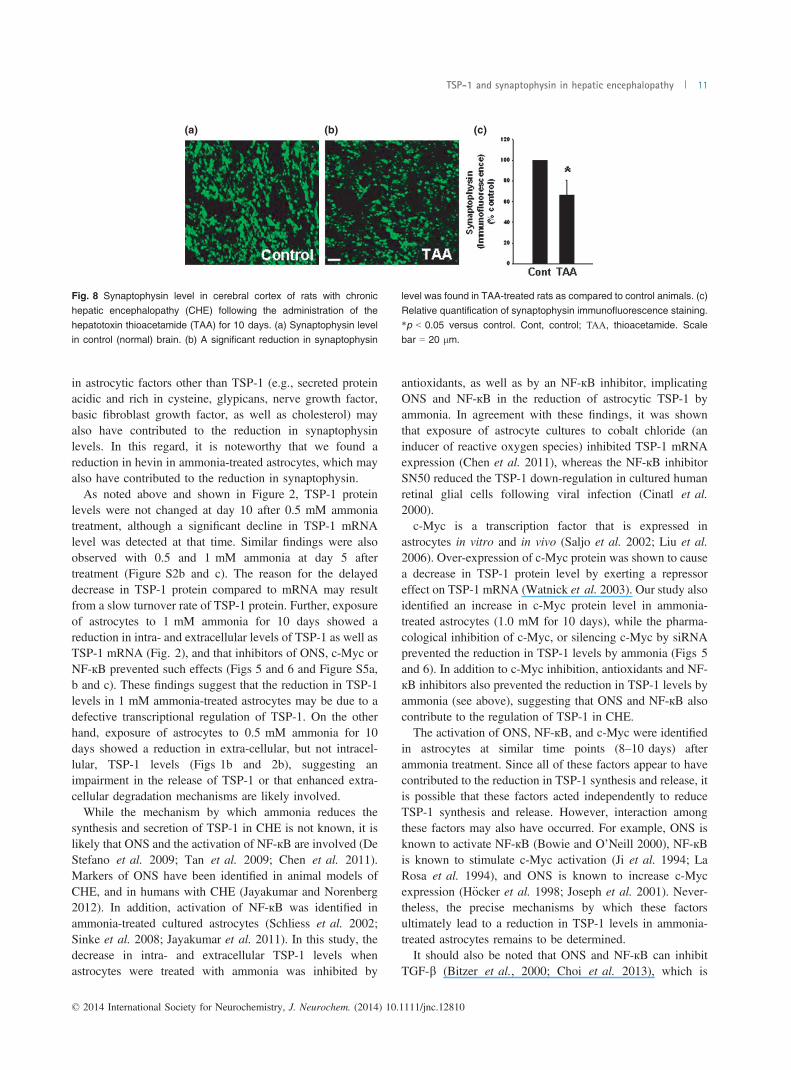
Fig. 8 Synaptophysin level in cerebral cortex of rats with chronic
hepatic encephalopathy (CHE) following the administration of thehepatotoxin thioacetamide (TAA) for 10 days. (a) Synaptophysin levelin control (normal) brain. (b) A significant reduction in synaptophysin
level was found in TAA-treated rats as compared to control animals. (c)
Relative quantification of synaptophysin immunofluorescence staining.*p < 0.05 versus control. Cont, control; ΤΑΑ, thioacetamide. Scalebar = 20 lm.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 11

known to stimulate the production of TSP-1 in astrocytes(Cambier et al. 2005; Yonezawa et al. 2010). Furthermore,TGF-b has been shown to reduce c-Myc expression (Pieten-pol et al. 1990; Warner et al. 1999). Since we founddecreased levels of TGF-b in ammonia-treated astrocytes, itis possible that the ONS- and NF-кB-mediated decrease inTGF-b may have enhanced c-Myc expression resulting inreduced levels of TSP-1.In addition to ONS, NF-кB, and c-Myc, a number of other
signaling factors, including p53, specificity protein 1, activat-ing transcription factor-1, activator protein 1, Forkhead box O1, E2F transcription factor 1, Runx2/3, as well as TGF-b, havebeen implicated in the mechanism of TSP-1 regulation in otherconditions (Salnikow et al. 1997; Janz et al. 2000; Li andRossman 2001; Ji et al. 2010; Roudier et al. 2013; Shi et al.2013).Whether these factors are also involved in the ammonia-induced reduction of TSP-1 in astrocytes, and the associatedloss of synaptophysin, is not known.Synaptophysin is an abundant integral membrane protein
of pre-synaptic vesicles involved in the regulation ofneurotransmitter release and synaptic plasticity (Alder et al.1995; Janz et al. 1999). It also participates in the biogenesisand recycling of synaptic vesicles, as mice lacking synapt-ophysin exhibit behavioral alterations and learning deficits(Schmitt et al. 2009). In addition, the loss of synaptophysinin the hippocampus was shown to correlate with a cognitivedecline in patients with Alzheimer’s disease (Sze 1997), inkeeping with the involvement of synaptophysin in themaintenance of synaptic integrity. Our study likewiseidentified a decrease in synaptophysin level when culturedneurons were exposed to CM derived from ammonia-treatedastrocytes. We similarly found decreased synaptophysinlevels in brains from rats with experimental CHE. Further-more, PSD95, a post-synaptic density protein and synapto-tagmin-1, a Ca2+ sensor in the plasma membrane ofpre-synaptic axon terminals which is involved in themaintenance of synaptic integrity, were similarly negativelyaffected by ammonia.It should be emphasized that a reduction in synaptic
proteins in CHE was not caused by neuronal loss, as suchloss is not a feature of CHE (Norenberg 1981). Moreover, noneuronal loss or reactive glial changes were observed in theserats. Instead, the presence of Alzheimer type II astrocyteswas the major neuropathological abnormality observed inthis condition (Figure S1), consistent with the concept thatdefective astrocytes are a major factor in the loss of synapticintegrity in CHE.The means by which a reduction in TSP-1 decreases
synaptic protein levels in CHE is not known. Studies haveshown that TSP-1 binds and activates integrin alpha-V/beta-1, leading to the stability of synaptophysin (DeFreitas et al.1995), and that a defect in this process may lead to areduction in synaptophysin levels, with or without synapticloss. Whether integrins alpha-V/beta-1 or other integrin
receptors are involved in the reduction in synaptic proteins inCHE remains to be established.In conclusion, a significant reduction in TSP-1 protein
level was observed in ammonia-treated astrocyte cultures.Such reduction was associated with a decrease in neuronalsynaptic proteins when conditioned media from ammonia-treated astrocytes were added to cultured neurons. We furtherobserved a significant reduction in astrocytic TSP-1 and inneuronal synaptophysin protein in a rat model of CHE. Ourfindings suggest that dysfunctional astrocytes (a glyopathy)resulting from ammonia treatment negatively impacts neu-ronal synaptic integrity, which may contribute to theneurological abnormalities known to occur in patients withCHE. Targeting astrocytic TSP-1 may provide a noveltherapeutic strategy for the neurological abnormalities asso-ciated with CHE.
Acknowledgments and conflict of interestdisclosure
This work was supported by a Merit Review from the Department ofVeterans Affairs and by the National Institutes of Health grantDK063311. KMC was supported by an NIH-NIAMS post-doctoralfellowship (7F32AR062990-02). The authors thank Alina Fernan-dez-Revuelta for the preparation of cell cultures. The authors declareno competing financial interests.
All experiments were conducted in compliance with the ARRIVEguidelines.
Supporting information
Additional supporting information may be found in the onlineversion of this article at the publisher's web-site:
Appendix S1. Supplementary Materials and methods.Figure S1. Histopathological changes in the cerebral cortex of
rats treated with thioacetamide (TAA) for 10 days.Figure S2. Intracellular TSP-1 level after a 5-day treatment of
cultured astrocytes with 0.5–2.5 mM ammonia.Figure S3. TGF-b1 level after a 10-day treatment of cultured
astrocytes with 1.0 mM ammonia.Figure S4. Immunocytochemistry of synaptophysin in cultured
neurons.Figure S5. Effect of antioxidants and the NF-кB inhibitor (SN50)
on extracellular TSP-1 levels in cultured astrocytes.
References
Abbott N. J., R€onnb€ack L. and Hansson E. (2006) Astrocyte-endothelialinteractions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53.
Alder J., Kanki H., Valtorta F., Greengard P. and Poo M. M. (1995)Overexpression of synaptophysin enhances neurotransmittersecretion at Xenopus neuromuscular synapses. J. Neurosci. 15,511–519.
Ampuero J., Ranchal I., Nu~nez D. et al. (2012) Metformin inhibitsglutaminase activity and protects against hepatic encephalopathy.PLoS ONE 7, e49279.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
12 A. R. Jayakumar et al.

Asch A. S., Leung L. L., Shapiro J. and Nachman R. L. (1986) Humanbrain glial cells synthesize thrombospondin. Proc. Natl Acad. Sci.USA 83, 2904–2908.
B�elanger M. and Magistretti P. J. (2009) The role of astroglia inneuroprotection. Dialogues Clin. Neurosci. 11, 281–295.
Bismuth M., Funakoshi N., Cadranel J. F. and Blanc P. (2011) Hepaticencephalopathy: from pathophysiology to therapeutic management.Eur. J. Gastroenterol. Hepatol. 23, 8–22.
Bitzer M., von Gersdorff G., Liang D., Dominguez-Rosales A., Beg A.A., Rojkind M. and B€ottinger E. P. (2000) A mechanism ofsuppression of TGF-beta/SMAD signaling by NF-kappa B/RelA.Genes. Dev. 14, 187–197.
Bowie A. and O’Neill L. A. (2000) Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light ofrecent discoveries. Biochem. Pharmacol. 59, 13–23.
Bu�ee L., Hof P. R., Roberts D. D., Delacourte A., Morrison J. H. andFillit H. M. (1992) Immunohistochemical identification ofthrombospondin in normal human brain and in Alzheimer’sdisease. Am. J. Pathol. 141, 783–788.
Cambier S., Gline S., Mu D., Collins R., Araya J., Dolganov G.,Einheber S., Boudreau N. and Nishimura S. L. (2005) Integrinalpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am.J. Pathol. 166, 1883–1894.
Carbonero-Aguilar P., Diaz-Herrero Mdel M., Cremades O., Romero-G�omez M. and Bautista J. (2011) Brain biomolecules oxidation inportacaval shunted rats. Liver Int. 31, 964–969.
Cauli O., Rodrigo R., Llansola M., Montoliu C., Monfort P., PiedrafitaB., El Mlili N., Boix J., Agust�ı A. and Felipo V. (2009)Glutamatergic and gabaergic neurotransmission and neuronalcircuits in hepatic encephalopathy. Metab. Brain Dis. 24, 69–80.
Chen J., Zhan Y. J., Yang C. S. and Tzeng S. F. (2011) Oxidative stress-induced attenuation of thrombospondin-1 expression in primary ratastrocytes. J. Cell. Biochem. 112, 59–70.
Choi J., Park S. J., Jo E. J., Lee H. Y., Hong S., Kim S. J. and Kim B. C.(2013) Hydrogen peroxide inhibits transforming growth factor-b1-induced cell cycle arrest by promoting Smad3 linkerphosphorylation through activation of Akt-ERK1/2-linkedsignaling pathway. Biochem. Biophys. Res. Commun. 435, 634–639.
Christopherson K. S., Ullian E. M., Stokes C. C., Mullowney C. E., HellJ. W., Agah A., Lawler J., Mosher D. F., Bornstein P. and BarresB. A. (2005) Thrombospondins are astrocyte-secreted proteins thatpromote CNS synaptogenesis. Cell 120, 421–433.
Cinatl J., Jr, Bittoova M., Margraf S., Vogel J. U., Cinatl J., Preiser W.and Doerr H. W. (2000) Cytomegalovirus infection decreasesexpression of thrombospondin-1 and -2 in cultured human retinalglial cells: effects of antiviral agents. J. Infect. Dis. 182, 643–651.
Cooper A. J., McDonald J. M., Gelbard A. S., Gledhill R. F. and DuffyT. E. (1979) The metabolic fate of 13N-labeled ammonia in ratbrain. J. Biol. Chem. 254, 4982–4992.
Crawford D. C., Jiang X., Taylor A. and Mennerick S. (2012) Astrocyte-derived thrombospondins mediate the development of hippocampalpresynaptic plasticity in vitro. J. Neurosci. 32, 13100–13110.
De Stefano D., Nicolaus G., Maiuri M. C., Cipolletta D., Galluzzi L.,Cinelli M. P., Tajana G., Iuvone T. and Carnuccio R. (2009) NF-kappaB blockade upregulates Bax, TSP-1, and TSP-2 expression inrat granulation tissue. J. Mol. Med. 87, 481–492.
DeFreitas M. F., Yoshida C. K., Frazier W. A., Mendrick D. L., KyptaR. M. and Reichardt L. F. (1995) Identification of integrin alpha 3beta 1 as a neuronal thrombospondin receptor mediating neuriteoutgrowth. Neuron 15, 333–343.
Dejong C. H., Deutz N. E. and Soeters P. B. (1993) Cerebral cortexammonia and glutamine metabolism in two rat models of chronic
liver insufficiency-induced hyperammonemia: influence of pair-feeding. J. Neurochem. 60, 1047–1057.
Ducis I., Norenberg L. O. and Norenberg M. D. (1990) Thebenzodiazepine receptor in cultured astrocytes from geneticallyepilepsy-prone rats. Brain Res. 531, 318–321.
Eroglu C., Allen N. J., Susman M. W., et al. (2009) Gabapentin receptoralpha2delta-1 is a neuronal thrombospondin receptor responsiblefor excitatory CNS synaptogenesis. Cell 139, 380–392.
Garcia O., Torres M., Helguera P., Coskun P. and Busciglio J. (2010) Arole for thrombospondin-1 deficits in astrocyte-mediated spine andsynaptic pathology in Down’s syndrome. PLoS ONE 5, e14200.
H€ocker M., Rosenberg I., Xavier R., Henihan R. J., Wiedenmann B.,Rosewicz S., Podolsky D. K. and Wang T. C. (1998) Oxidativestress activates the human histidine decarboxylase promoter inAGS gastric cancer cells. J. Biol. Chem. 273, 23046–23054.
Janz R., S€udhof T. C., Hammer R. E., Unni V., Siegelbaum S. A. andBolshakov V. Y. (1999) Essential roles in synaptic plasticity forsynaptogyrin I and synaptophysin I. Neuron 24, 687–700.
Janz A., Sevignani C., Kenyon K., Ngo C. V. and Thomas-TikhonenkoA. (2000) Activation of the myc oncoprotein leads to increasedturnover of thrombospondin-1 mRNA. Nucleic Acids Res. 28,2268–2275.
Jayakumar A. R. and Norenberg M. D. (2012) Oxidative stress in hepaticencephalopathy, in Hepatic Encephalopathy, ClinicalGastroenterology (Mullen K. D. and Prakash R. K., eds), pp.47–70. Springer, New York.
Jayakumar A. R., Rao K. V., Murthy Ch R and Norenberg M. D. (2006)Glutamine in the mechanism of ammonia-induced astrocyteswelling. Neurochem. Int. 48, 623–628.
Jayakumar A. R., Bethea J. R., Tong X. Y., Gomez J. and Norenberg M.D. (2011) NF-jB in the mechanism of brain edema in acute liverfailure: studies in transgenic mice. Neurobiol. Dis. 41, 498–507.
Jayakumar A. R., Tong X. Y., Curtis K. M., Ruiz-Cordero R., Abreu M.T. and Norenberg M. D. (2014) Increased toll-like receptor 4 incerebral endothelial cells contributes to the astrocyte swelling andbrain edema in acute hepatic encephalopathy. J. Neurochem. 128,890–903.
Ji L., Arcinas M. and Boxer L. M. (1994) NF-kappa B sites function aspositive regulators of expression of the translocated c-myc allele inBurkitt’s lymphoma. Mol. Cell. Biol. 14, 7967–7974.
Ji W., Zhang W. and Xiao W. (2010) E2F-1 directly regulatesthrombospondin 1 expression. PLoS ONE 5, e13442.
Joseph P., Muchnok T. K., Klishis M. L., Roberts J. R., Antonini J. M.,Whong W. Z. and Ong T. (2001) Cadmium-induced celltransformation and tumorigenesis are associated withtranscriptional activation of c-fos, c-jun, and c-myc proto-oncogenes: role of cellular calcium and reactive oxygen species.Toxicol. Sci. 61, 295–303.
Juurlink B. H. and Hertz L. (1985) Plasticity of astrocytes in primarycultures: an experimental tool and a reason for methodologicalcaution. Dev. Neurosci. 7, 263–277.
Kimura T. and Budka H. (1986) Glial fibrillary acidic protein and S-100protein in human hepatic encephalopathy: immunocytochemicaldemonstration of dissociation of two glia-associated proteins. ActaNeuropathol. 70, 17–21.
Kretzschmar H. A., DeArmond S. J. and Forno L. S. (1985)Measurement of GFAP in hepatic encephalopathy by ELISA andtransblots. J. Neuropathol. Exp. Neurol. 44, 459–471.
Kucukdereli H., Allen N. J., Lee A. T. et al. (2011) Control of excitatoryCNS synaptogenesis by astrocyte-secreted proteins Hevin andSPARC. Proc. Natl Acad. Sci. USA 108, E440–E449.
La Rosa F. A., Pierce J. W. and Sonenshein G. E. (1994) Differentialregulation of the c-myc oncogene promoter by the NF-kappa B relfamily of transcription factors. Mol. Cell. Biol. 14, 1039–1044.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 13

Lange S. C., Bak L. K., Waagepetersen H. S., Schousboe A. andNorenberg M. D. (2012) Primary cultures of astrocytes: their valuein understanding astrocytes in health and disease. Neurochem. Res.37, 2569–2588.
Lee W. M. (2012) Acute liver failure. Semin. Respir. Crit. Care Med. 33,36–45.
Li P. and Rossman T. G. (2001) Genes upregulated in lead-resistantglioma cells reveal possible targets for lead-induced developmentalneurotoxicity. Toxicol. Sci. 64, 90–99.
Li Z., Calzada M. J., Sipes J. M., Cashel J. A., Krutzsch H. C., Annis D.S., Mosher D. F. and Roberts D. D. (2002) Interactions ofthrombospondins with alpha4beta1 integrin and CD47differentially modulate T cell behavior. J. Cell Biol. 157, 509–519.
Liauw J., Hoang S., Choi M. et al. (2008) Thrombospondins 1 and 2 arenecessary for synaptic plasticity and functional recovery afterstroke. J. Cereb. Blood Flow Metab. 28, 1722–1732.
Lin T. N., Kim G. M., Chen J. J., Cheung W. M., He Y. Y. and Hsu C.Y. (2003) Differential regulation of thrombospondin-1 andthrombospondin-2 after focal cerebral ischemia/reperfusion.Stroke 34, 177–186.
Liu J., Narasimhan P., Lee Y. S., Song Y. S., Endo H., Yu F. and ChanP. H. (2006) Mild hypoxia promotes survival and proliferation ofSOD2-deficient astrocytes via c-Myc activation. J. Neurosci. 26,4329–4337.
Llansola M., Montoliu C., Cauli O., Hern�andez-Rabaza V., Agust�ı A.,Cabrera-Pastor A., Gim�enez-Garz�o C., Gonz�alez-Usano A. andFelipo V. (2012) Chronic hyperammonemia, glutamatergicneurotransmission and neurological alterations. Metab. Brain Dis.28, 151–154.
Lu Q. and Hong W. (2009) Bcl2 enhances c-Myc-mediated MMP-2expression of vascular smooth muscle cells. Cell. Signal. 21, 1054–1059.
Lu Z. and Kipnis J. (2010) Thrombospondin 1-a key astrocyte-derivedneurogenic factor. FASEB J. 24, 1925–1934.
McGillicuddy F. C., O’Toole D., Hickey J. A., Gallagher W. M.,Dawson K. A. and Keenan A. K. (2006) TGF-beta1-inducedthrombospondin-1 expression through the p38 MAPK pathway isabolished by fluvastatin in human coronary artery smooth musclecells. Vascul. Pharmacol. 44, 469–475.
Mimura Y., Ihn H., Jinnin M., Asano Y., Yamane K. and Tamaki K.(2005) Constitutive thrombospondin-1 overexpression contributesto autocrine transforming growth factor-beta signaling in culturedscleroderma fibroblasts. Am. J. Pathol. 166, 1451–1463.
Mullen K. D. and Prakash R. K. (2012) New perspectives in hepaticencephalopathy. Clin. Liver Dis. 16, 1–5.
Norenberg M. D. (1979) Distribution of glutamine synthetase in the ratcentral nervous system. J. Histochem. Cytochem. 27, 756–762.
Norenberg M. D. (1981) The astrocyte in liver disease, in Advances inCellular Neurobiology (Fedoroff S. and Hertz L., eds), pp. 303–352. Academic Press, New York.
Norenberg M. D. (1986) Hepatic encephalopathy: a disorder ofastrocytes, in Astrocytes (Fedoroff S. and Vernadakis A., eds),Vol. 3, pp. 425–460. Academic Press, Orlando.
Norenberg M. D. (1987) The role of astrocytes in hepaticencephalopathy. Neurochem. Pathol. 6, 13–33.
Norenberg M. D., Neary J. T., Bender A. S. and Dombro R. S. (1992)Hepatic encephalopathy: a disorder in glial–neuronalcommunication, in Neuronal–Astrocytic Interactions. Implicationsfor Normal and Pathological CNS Function (Yu A. C. H., Hertz L.,Norenberg M. D., Sykova E. and Waxman S. G. eds), Vol. 94, pp.261–269. Progress in Brain Research, Elsevier, Amsterdam.
Norenberg M. D., Itzhak Y., Bender A. S., Baker L., Aguila-Mansilla H.N., Zhou B. G. and Isaacks R. (1998) Ammonia and astrocytefunction, in liver and nervous system, in Liver and Nervous System
(H€aussinger D., Jungermann K., eds.), pp. 276–293. Kluwer AcadPubl, Lancaster, England.
Norenberg M. D., Rama Rao K. V. and Jayakumar A. R. (2009)Signaling factors in the mechanism of ammonia neurotoxicity.Metab. Brain Dis. 24, 103–117.
Okamoto M., Ono M., Uchiumi T., Ueno H., Kohno K., Sugimachi K.and Kuwano M. (2002) Up-regulation of thrombospondin-1 geneby epidermal growth factor and transforming growth factor beta inhuman cancer cells–transcriptional activation and messenger RNAstabilization. Biochim. Biophys. Acta 1574, 24–34.
Patel D., McPhail M. J., Cobbold J. F. and Taylor-Robinson S. D. (2012)Hepatic encephalopathy. Br. J. Hosp. Med. 73, 79–85.
Pietenpol J. A., Holt J. T., Stein R. W. and Moses H. L. (1990)Transforming growth factor beta 1 suppression of c-myc genetranscription: role in inhibition of keratinocyte proliferation. Proc.Natl Acad. Sci. USA 87, 3758–3762.
Rama Rao K. V., Curtis K. M., Johnstone J. T. and Norenberg M. D.(2013) Amyloid-b inhibits thrombospondin 1 release from culturedastrocytes: effects on synaptic protein expression. J. Neuropathol.Exp. Neurol. 72, 735–744.
Rick F. G., Abi-Chaker A., Szalontay L. et al. (2013) Shrinkage ofexperimental benign prostatic hyperplasia and reduction ofprostatic cell volume by a gastrin-releasing peptide antagonist.Proc. Natl Acad. Sci. USA 12, 2617–2622.
Roudier E., Milkiewicz M., Birot O. et al. (2013) Endothelial FoxO1 isan intrinsic regulator of thrombospondin 1 expression that restrainsangiogenesis in ischemic muscle. Angiogenesis 16, 759–772.
Saljo A., Bao F., Shi J., Hamberger A., Hansson H. A. and Haglid K. G.(2002) Expression of c-Fos and c-Myc and deposition of beta-APPin neurons in the adult rat brain as a result of exposure to short-lasting impulse noise. J. Neurotrauma 19, 379–385.
Salnikow K., Wang S. and Costa M. (1997) Induction of activatingtranscription factor 1 by nickel and its role as a negative regulatorof thrombospondin I gene expression. Cancer Res. 57, 5060–5066.
Schliess F., G€org B., Fischer R., Desjardins P., Bidmon H. J., HerrmannA., Butterworth R. F., Zilles K. and H€aussinger D. (2002)Ammonia induces MK-801-sensitive nitration andphosphorylation of protein tyrosine residues in rat astrocytes.FASEB J. 16, 739–741.
Schmitt U., TanimotoN., SeeligerM., Schaeffel F. and LeubeR. E. (2009)Detection of behavioral alterations and learning deficits in micelacking synaptophysin. Neuroscience 162, 234–243.
Schousboe A., Meier E., Drejer J. and Hertz L. (1989) Preparation ofprimary cultures of mouse (rat) cerebellar granule cells, in ADissection and Tissue Culture Manual for The Nervous System(Shahar A., de Vellis J., Vernadakis A. and Haber S., eds), pp.203–206. Alan R. Liss, New York.
Shawcross D. L. and Wendon J. A. (2012) The neurologicalmanifestations of acute liver failure. Neurochem. Int. 60, 662–671.
Shi X., Deepak V., Wang L., Ba X., Komori T., Zeng X. and Liu W.(2013) Thrombospondin-1 is a putative target gene of runx2 andrunx3. Int. J. Mol. Sci. 14, 14321–14332.
Singh S. and Trigun S. K. (2010) Activation of neuronal nitric oxidesynthase in cerebellum of chronic hepatic encephalopathy rats isassociated with up-regulation of NADPH-producing pathway.Cerebellum 9, 384–397.
Sinke A. P., Jayakumar A. R., Panickar K. S., Moriyama M., Reddy P.V. and Norenberg M. D. (2008) NFkappaB in the mechanism ofammonia-induced astrocyte swelling in culture. J. Neurochem.106, 2302–2311.
Sobel R. A., DeArmond S. J., Forno L. S. and Eng L. F. (1981) Glialfibrillary acidic protein in hepatic encephalopathy. An immunohisto-chemical study. J. Neuropathol. Exp. Neurol. 40, 625–632.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
14 A. R. Jayakumar et al.

Sze C. I. (1997) Loss of the presynaptic vesicle protein synaptophysin inhippocampus correlates with cognitive decline in Alzheimerdisease. J. Neuropathol. Exp. Neurol. 56, 933–944.
Tan B. K., Adya R., Chen J., Farhatullah S., Heutling D., Mitchell D.,Lehnert H. and Randeva H. S. (2009) Metformin decreasesangiogenesis via NF-kappaB and Erk1/2/Erk5 pathways byincreasing the antiangiogenic thrombospondin-1. Cardiovasc.Res. 83, 566–574.
Tran M. D. and Neary J. T. (2006) Purinergic signaling inducesthrombospondin-1 expression in astrocytes. Proc. Natl Acad. Sci.USA 103, 9321–9326.
Wakim-Fleming J. (2011) Hepatic encephalopathy: suspect it early inpatients with cirrhosis. Cleve. Clin. J. Med. 78, 597–605.
Wang D. D. and Bordey A. (2008) The astrocyte odyssey. Prog.Neurobiol. 86, 342–367.
Warner B. J., Blain S. W., Seoane J. and Massagu�e J. (1999) Mycdownregulation by transforming growth factor beta required for
activation of the p15(Ink4b) G(1) arrest pathway. Mol. Cell. Biol.19, 5913–5922.
Watnick R. S., Cheng Y. N., Rangarajan A., Ince T. A. and Weinberg R.A. (2003) Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell 3,219–231.
Xu J., Xiao N. and Xia J. (2010) Thrombospondin 1 acceleratessynaptogenesis in hippocampal neurons through neuroligin 1. NatNeurosci. 13, 22–24.
Yonezawa T., Hattori S., Inagaki J., Kurosaki M., Takigawa T., HirohataS., Miyoshi T. and Ninomiya Y. (2010) Type IV collagen inducesexpression of thrombospondin-1 that is mediated by integrinalpha1beta1 in astrocytes. Glia 58, 755–767.
Yu K., Ge J., Summers J. B., Li F., Liu X., Ma P., Kaminski J. andZhuang J. (2008) TSP-1 secreted by bone marrow stromal cellscontributes to retinal ganglion cell neurite outgrowth and survival.PLoS ONE 3, e2470.
© 2014 International Society for Neurochemistry, J. Neurochem. (2014) 10.1111/jnc.12810
TSP-1 and synaptophysin in hepatic encephalopathy 15

![Astrocytic tracer dynamics estimated from [1-11C]-acetate PET measurements](https://static.fdokumen.com/doc/165x107/6334cca03e69168eaf070c95/astrocytic-tracer-dynamics-estimated-from-1-11c-acetate-pet-measurements.jpg)


















