
Deconvolution of Combinatorial Libraries for Drug Discovery: Experimental Comparison of Pooling...
10
Deconvolution of Combinatorial Libraries for Drug Discovery: Theoretical Comparison of Pooling Strategies Danielle A. M. Konings, ‡,§ Jacqueline R. Wyatt, ² David J. Ecker, ² and Susan M. Freier* ,² ISIS Pharmaceuticals, 2292 Faraday Avenue, Carlsbad, California 92008, and Department of Molecular, Cellular and Developmental Biology, University of Colorado, Boulder, Colorado 80309 Received March 4, 1996 X Synthesis and testing of mixtures of compounds in a combinatorial library allow much greater throughput than synthesis and testing of individual compounds. When mixtures of compounds are screened, however, the possibility exists that the most active compound will not be identified. The specific strategies employed for pooling and deconvolution will affect the likelihood of success. We have used a nucleic acid hybridization example to develop a theoretical model of library deconvolution for a library of more than 250 000 compounds. This model was used to compare various strategies for pooling and deconvolution. Simulations were performed in the absence and presence of experimental error. We found iterative deconvolution to be most reliable when active molecules were assigned to the same subset in early rounds. Reliability was reduced only slightly when active molecules were assigned randomly to all subsets. Iterative deconvolution with as many as 65 536 compounds per subset did not drastically reduce the reliability compared to one-at-a-time testing. Pooling strategies compared using this theoretical model are compared experimentally in an accompanying paper. Introduction Advances in chemical synthesis of combinatorial libraries have enabled preparation of an unprecedented number of novel compounds for drug screening. 1-7 Synthesis and testing of mixtures of compounds in a combinatorial library offer the potential of much greater throughput than the “one compound, one well” ap- proach. When mixtures of compounds are screened, however, a “deconvolution” method must be used to determine which molecule(s) in the library is responsible for the activity. Iterative deconvolution begins by dividing the library into nonoverlapping subsets. The subsets are tested separately, and the one with greatest activity is identi- fied. The compounds in the most active subset are divided into a new set of subsets and retested for activity. The process of dividing the most active subset into smaller subsets for retesting is continued until a unique molecule is identified. There are many ways of organizing subsets or “pooling strategies” for iterative deconvolution. Most common is pooling by fixed position where, at each round, molecules with a common func- tionality at a single position are grouped together. For example, in round 1, the subsets could consist of all molecules, NXN, where X is a single functionality unique to each subset and N is an equimolar mixture of all the functionalities at that position. In subsequent rounds, additional positions would be fixed. Fixed position pooling has been used for libraries synthesized using a variety of chemistries (Figure 1), and iterative deconvolution strategies have been used to identify a single active compound from a mixture. 8-20 Iterative strategies based on pooling by fixed position can differ from one another in which positions are fixed at each round (order of deconvolution) or the number of posi- tions fixed at each round. Iterative deconvolution does not require fixed position pooling; any method for dividing the compounds into nonoverlapping subsets is allowed, although it may not always be feasible. We have previously used computer simulations to ask if the deconvolution procedure can identify the most active molecule(s) in a combinatorial library and to determine the effects of deconvolution order and experi- mental error on the outcome. 21 We found that even when the library contained many molecules with sub- optimal activity, iterative deconvolution almost always selected the most active molecule in the library. When reasonable experimental error was included in the * Corresponding author: 619-603-2345 (phone), 619-431-2768 (fax), [email protected] (internet). ² ISIS Pharmaceuticals. ‡ University of Colorado. § Present address: Department of Microbiology, Southern Illinois University, Carbondale, IL 62901. X Abstract published in Advance ACS Abstracts, June 15, 1996. Figure 1. Examples of pooling by fixed position for libraries of (a) N-methyl peptides, 48 (b) phosphoryl-linked compounds, 41 and (c) (mercaptoacyl)proline derivatives. 49 Each subset con- sists of a single fixed functionality at one or more positions (X 1 ,X 2 ) and an equimolar mix of several functionalities at the other positions (R 1 ,R 2 ,R 3 ). 2710 J. Med. Chem. 1996, 39, 2710-2719 S0022-2623(96)00168-9 CCC: $12.00 © 1996 American Chemical Society
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Deconvolution of Combinatorial Libraries for Drug Discovery: Experimental Comparison of Pooling...

Deconvolution of Combinatorial Libraries for Drug Discovery: TheoreticalComparison of Pooling Strategies
Danielle A. M. Konings,‡,§ Jacqueline R. Wyatt,† David J. Ecker,† and Susan M. Freier*,†
ISIS Pharmaceuticals, 2292 Faraday Avenue, Carlsbad, California 92008, and Department of Molecular, Cellular andDevelopmental Biology, University of Colorado, Boulder, Colorado 80309
Received March 4, 1996X
Synthesis and testing of mixtures of compounds in a combinatorial library allow much greaterthroughput than synthesis and testing of individual compounds. When mixtures of compoundsare screened, however, the possibility exists that the most active compound will not be identified.The specific strategies employed for pooling and deconvolution will affect the likelihood ofsuccess. We have used a nucleic acid hybridization example to develop a theoretical model oflibrary deconvolution for a library of more than 250 000 compounds. This model was used tocompare various strategies for pooling and deconvolution. Simulations were performed in theabsence and presence of experimental error. We found iterative deconvolution to be mostreliable when active molecules were assigned to the same subset in early rounds. Reliabilitywas reduced only slightly when active molecules were assigned randomly to all subsets.Iterative deconvolution with as many as 65 536 compounds per subset did not drastically reducethe reliability compared to one-at-a-time testing. Pooling strategies compared using thistheoretical model are compared experimentally in an accompanying paper.
Introduction
Advances in chemical synthesis of combinatoriallibraries have enabled preparation of an unprecedentednumber of novel compounds for drug screening.1-7
Synthesis and testing of mixtures of compounds in acombinatorial library offer the potential of much greaterthroughput than the “one compound, one well” ap-proach. When mixtures of compounds are screened,however, a “deconvolution” method must be used todetermine which molecule(s) in the library is responsiblefor the activity.Iterative deconvolution begins by dividing the library
into nonoverlapping subsets. The subsets are testedseparately, and the one with greatest activity is identi-fied. The compounds in the most active subset aredivided into a new set of subsets and retested foractivity. The process of dividing the most active subsetinto smaller subsets for retesting is continued until aunique molecule is identified. There are many ways oforganizing subsets or “pooling strategies” for iterativedeconvolution. Most common is pooling by fixed positionwhere, at each round, molecules with a common func-tionality at a single position are grouped together. Forexample, in round 1, the subsets could consist of allmolecules, NXN, where X is a single functionalityunique to each subset and N is an equimolar mixtureof all the functionalities at that position. In subsequentrounds, additional positions would be fixed. Fixedposition pooling has been used for libraries synthesizedusing a variety of chemistries (Figure 1), and iterativedeconvolution strategies have been used to identify asingle active compound from a mixture.8-20 Iterativestrategies based on pooling by fixed position can differ
from one another in which positions are fixed at eachround (order of deconvolution) or the number of posi-tions fixed at each round. Iterative deconvolution doesnot require fixed position pooling; any method fordividing the compounds into nonoverlapping subsets isallowed, although it may not always be feasible.We have previously used computer simulations to ask
if the deconvolution procedure can identify the mostactive molecule(s) in a combinatorial library and todetermine the effects of deconvolution order and experi-mental error on the outcome.21 We found that evenwhen the library contained many molecules with sub-optimal activity, iterative deconvolution almost alwaysselected the most active molecule in the library. Whenreasonable experimental error was included in the
* Corresponding author: 619-603-2345 (phone), 619-431-2768 (fax),[email protected] (internet).
† ISIS Pharmaceuticals.‡ University of Colorado.§ Present address: Department of Microbiology, Southern Illinois
University, Carbondale, IL 62901.X Abstract published in Advance ACS Abstracts, June 15, 1996.
Figure 1. Examples of pooling by fixed position for librariesof (a)N-methyl peptides,48 (b) phosphoryl-linked compounds,41and (c) (mercaptoacyl)proline derivatives.49 Each subset con-sists of a single fixed functionality at one or more positions(X1, X2) and an equimolar mix of several functionalities at theother positions (R1, R2, R3).
2710 J. Med. Chem. 1996, 39, 2710-2719
S0022-2623(96)00168-9 CCC: $12.00 © 1996 American Chemical Society
simulations, a molecule with activity nearly as great asthat of the most active compound was usually selected.In this report, we have extended these computer
simulations to include a large variety of pooling strate-gies (Figure 2). Using two different library-target pairswith very different affinity profiles, we found thatiterative strategies using nonoverlapping subsets werequite successful at identifying one of the most activemolecules in the library. Simulations which includedexperimental error suggested pooling strategies thatconcentrated active molecules in a single subset weresomewhat more reliable than strategies that separatedthe most active molecules into different subsets in theearly rounds. In an accompanying paper we reportexperimental evaluation of several of the pooling strate-gies using a library of 810 chemically synthesizedcompounds and an in vitro assay for inhibition ofphospholipase A2 (PLA2).
ResultsCharacterization of TwoMolecular Landscapes.
RNA hybridization was chosen as the molecular inter-action for our model because calculations based uponexperimentally determined parameters can accuratelypredict the association constants of very large numbersof molecules.22 We were able to calculate the bindingaffinity for several hundred thousand different mol-ecules for a specific target to create a “molecularlandscape”.23 We created two different landscapes byselecting two RNA targets, a 9-mer and a 6-mer, and alibrary of all possible 262 144 RNA 9-mers. The energyprofiles for these two library-target pairs have beenpreviously described in detail.21 We call the two land-scapes A (9-mer target) and B (6-mer target). Activitiesare reported relative to that of the most active com-pound in the library. Thus the most active compoundhas an activity of 1.0. Any activity greater than 0.2 iscalled “good” activity.
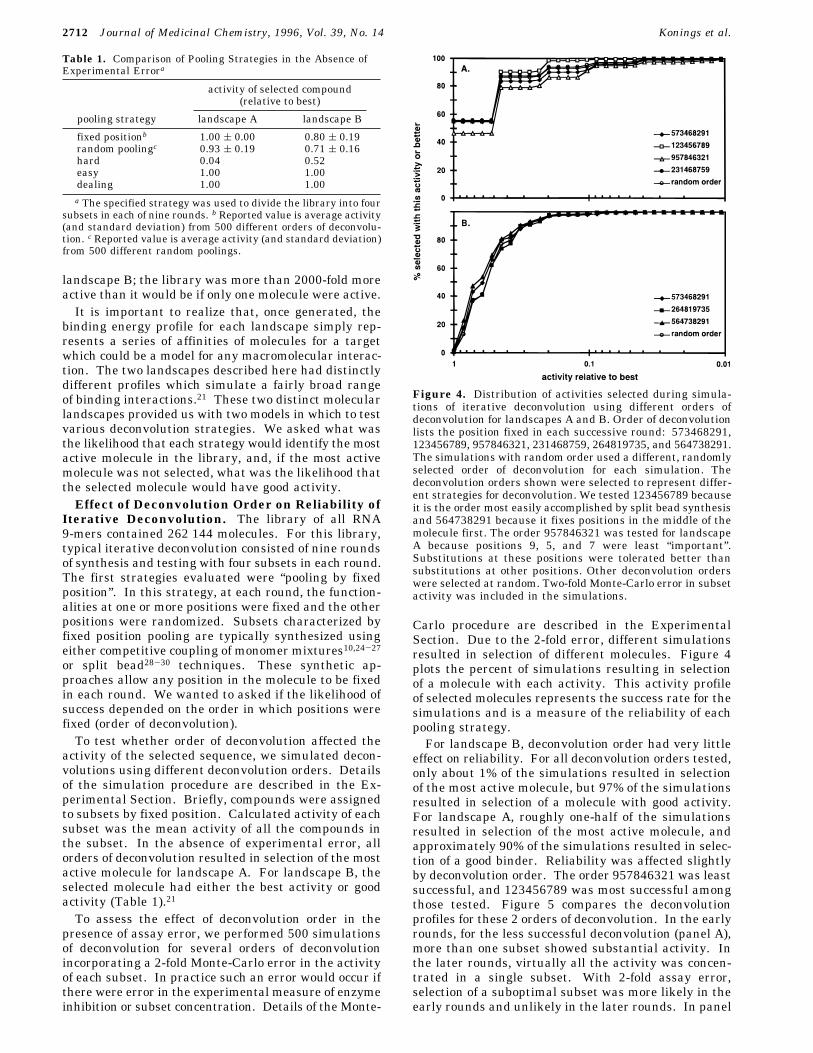
Figure 3 plots the cumulative frequency distributionfor activities in these two molecular landscapes. Land-scape A contained relatively few compounds with sig-nificant activity. There were two molecules with thehighest affinity (“best” molecules) and only 12 molecules(0.005% of the library) with good activity. Almost allof the molecules (98.7%) had activities less than 10-5
relative to the best. In contrast, landscape B containedmany compounds with significant activity. There were16 molecules with the best activity and 2414 molecules(0.9%) with good activity. Only 37% of the moleculeshad activities less than 10-5. Relatively few moleculescontributed to activity of the library in landscape A; thelibrary was only 8 times more active than it would be ifonly one molecule were active. In contrast, manymolecules contributed to activity of the library in
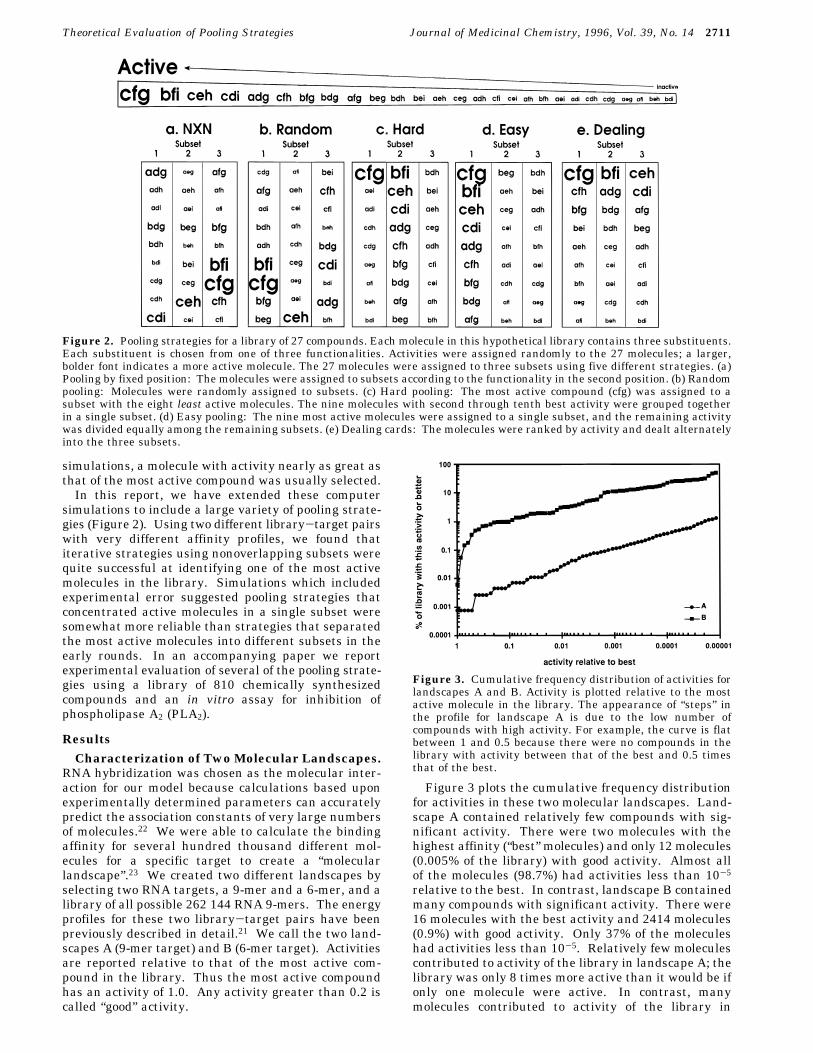
Figure 2. Pooling strategies for a library of 27 compounds. Each molecule in this hypothetical library contains three substituents.Each substituent is chosen from one of three functionalities. Activities were assigned randomly to the 27 molecules; a larger,bolder font indicates a more active molecule. The 27 molecules were assigned to three subsets using five different strategies. (a)Pooling by fixed position: The molecules were assigned to subsets according to the functionality in the second position. (b) Randompooling: Molecules were randomly assigned to subsets. (c) Hard pooling: The most active compound (cfg) was assigned to asubset with the eight least active molecules. The nine molecules with second through tenth best activity were grouped togetherin a single subset. (d) Easy pooling: The nine most active molecules were assigned to a single subset, and the remaining activitywas divided equally among the remaining subsets. (e) Dealing cards: The molecules were ranked by activity and dealt alternatelyinto the three subsets.
Figure 3. Cumulative frequency distribution of activities forlandscapes A and B. Activity is plotted relative to the mostactive molecule in the library. The appearance of “steps” inthe profile for landscape A is due to the low number ofcompounds with high activity. For example, the curve is flatbetween 1 and 0.5 because there were no compounds in thelibrary with activity between that of the best and 0.5 timesthat of the best.
Theoretical Evaluation of Pooling Strategies Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 2711
landscape B; the library was more than 2000-fold moreactive than it would be if only one molecule were active.It is important to realize that, once generated, the
binding energy profile for each landscape simply rep-resents a series of affinities of molecules for a targetwhich could be a model for any macromolecular interac-tion. The two landscapes described here had distinctlydifferent profiles which simulate a fairly broad rangeof binding interactions.21 These two distinct molecularlandscapes provided us with two models in which to testvarious deconvolution strategies. We asked what wasthe likelihood that each strategy would identify the mostactive molecule in the library, and, if the most activemolecule was not selected, what was the likelihood thatthe selected molecule would have good activity.Effect of Deconvolution Order on Reliability of
Iterative Deconvolution. The library of all RNA9-mers contained 262 144 molecules. For this library,typical iterative deconvolution consisted of nine roundsof synthesis and testing with four subsets in each round.The first strategies evaluated were “pooling by fixedposition”. In this strategy, at each round, the function-alities at one or more positions were fixed and the otherpositions were randomized. Subsets characterized byfixed position pooling are typically synthesized usingeither competitive coupling of monomer mixtures10,24-27
or split bead28-30 techniques. These synthetic ap-proaches allow any position in the molecule to be fixedin each round. We wanted to asked if the likelihood ofsuccess depended on the order in which positions werefixed (order of deconvolution).To test whether order of deconvolution affected the
activity of the selected sequence, we simulated decon-volutions using different deconvolution orders. Detailsof the simulation procedure are described in the Ex-perimental Section. Briefly, compounds were assignedto subsets by fixed position. Calculated activity of eachsubset was the mean activity of all the compounds inthe subset. In the absence of experimental error, allorders of deconvolution resulted in selection of the mostactive molecule for landscape A. For landscape B, theselected molecule had either the best activity or goodactivity (Table 1).21
To assess the effect of deconvolution order in thepresence of assay error, we performed 500 simulationsof deconvolution for several orders of deconvolutionincorporating a 2-fold Monte-Carlo error in the activityof each subset. In practice such an error would occur ifthere were error in the experimental measure of enzymeinhibition or subset concentration. Details of the Monte-
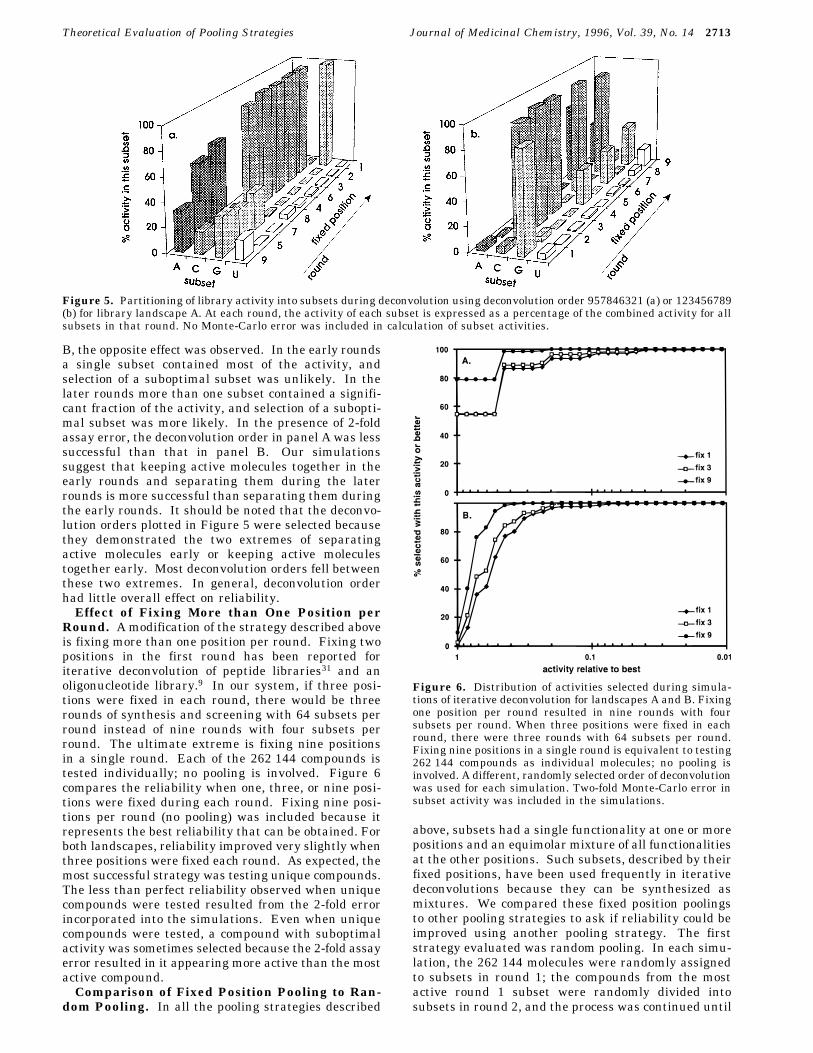
Carlo procedure are described in the ExperimentalSection. Due to the 2-fold error, different simulationsresulted in selection of different molecules. Figure 4plots the percent of simulations resulting in selectionof a molecule with each activity. This activity profileof selected molecules represents the success rate for thesimulations and is a measure of the reliability of eachpooling strategy.For landscape B, deconvolution order had very little
effect on reliability. For all deconvolution orders tested,only about 1% of the simulations resulted in selectionof the most active molecule, but 97% of the simulationsresulted in selection of a molecule with good activity.For landscape A, roughly one-half of the simulationsresulted in selection of the most active molecule, andapproximately 90% of the simulations resulted in selec-tion of a good binder. Reliability was affected slightlyby deconvolution order. The order 957846321 was leastsuccessful, and 123456789 was most successful amongthose tested. Figure 5 compares the deconvolutionprofiles for these 2 orders of deconvolution. In the earlyrounds, for the less successful deconvolution (panel A),more than one subset showed substantial activity. Inthe later rounds, virtually all the activity was concen-trated in a single subset. With 2-fold assay error,selection of a suboptimal subset was more likely in theearly rounds and unlikely in the later rounds. In panel
Table 1. Comparison of Pooling Strategies in the Absence ofExperimental Errora
activity of selected compound(relative to best)
pooling strategy landscape A landscape B
fixed positionb 1.00 ( 0.00 0.80 ( 0.19random poolingc 0.93 ( 0.19 0.71 ( 0.16hard 0.04 0.52easy 1.00 1.00dealing 1.00 1.00a The specified strategy was used to divide the library into four
subsets in each of nine rounds. b Reported value is average activity(and standard deviation) from 500 different orders of deconvolu-tion. c Reported value is average activity (and standard deviation)from 500 different random poolings.
Figure 4. Distribution of activities selected during simula-tions of iterative deconvolution using different orders ofdeconvolution for landscapes A and B. Order of deconvolutionlists the position fixed in each successive round: 573468291,123456789, 957846321, 231468759, 264819735, and 564738291.The simulations with random order used a different, randomlyselected order of deconvolution for each simulation. Thedeconvolution orders shown were selected to represent differ-ent strategies for deconvolution. We tested 123456789 becauseit is the order most easily accomplished by split bead synthesisand 564738291 because it fixes positions in the middle of themolecule first. The order 957846321 was tested for landscapeA because positions 9, 5, and 7 were least “important”.Substitutions at these positions were tolerated better thansubstitutions at other positions. Other deconvolution orderswere selected at random. Two-fold Monte-Carlo error in subsetactivity was included in the simulations.
2712 Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 Konings et al.
B, the opposite effect was observed. In the early roundsa single subset contained most of the activity, andselection of a suboptimal subset was unlikely. In thelater rounds more than one subset contained a signifi-cant fraction of the activity, and selection of a subopti-mal subset was more likely. In the presence of 2-foldassay error, the deconvolution order in panel A was lesssuccessful than that in panel B. Our simulationssuggest that keeping active molecules together in theearly rounds and separating them during the laterrounds is more successful than separating them duringthe early rounds. It should be noted that the deconvo-lution orders plotted in Figure 5 were selected becausethey demonstrated the two extremes of separatingactive molecules early or keeping active moleculestogether early. Most deconvolution orders fell betweenthese two extremes. In general, deconvolution orderhad little overall effect on reliability.Effect of Fixing More than One Position per
Round. A modification of the strategy described aboveis fixing more than one position per round. Fixing twopositions in the first round has been reported foriterative deconvolution of peptide libraries31 and anoligonucleotide library.9 In our system, if three posi-tions were fixed in each round, there would be threerounds of synthesis and screening with 64 subsets perround instead of nine rounds with four subsets perround. The ultimate extreme is fixing nine positionsin a single round. Each of the 262 144 compounds istested individually; no pooling is involved. Figure 6compares the reliability when one, three, or nine posi-tions were fixed during each round. Fixing nine posi-tions per round (no pooling) was included because itrepresents the best reliability that can be obtained. Forboth landscapes, reliability improved very slightly whenthree positions were fixed each round. As expected, themost successful strategy was testing unique compounds.The less than perfect reliability observed when uniquecompounds were tested resulted from the 2-fold errorincorporated into the simulations. Even when uniquecompounds were tested, a compound with suboptimalactivity was sometimes selected because the 2-fold assayerror resulted in it appearing more active than the mostactive compound.Comparison of Fixed Position Pooling to Ran-
dom Pooling. In all the pooling strategies described
above, subsets had a single functionality at one or morepositions and an equimolar mixture of all functionalitiesat the other positions. Such subsets, described by theirfixed positions, have been used frequently in iterativedeconvolutions because they can be synthesized asmixtures. We compared these fixed position poolingsto other pooling strategies to ask if reliability could beimproved using another pooling strategy. The firststrategy evaluated was random pooling. In each simu-lation, the 262 144 molecules were randomly assignedto subsets in round 1; the compounds from the mostactive round 1 subset were randomly divided intosubsets in round 2, and the process was continued until
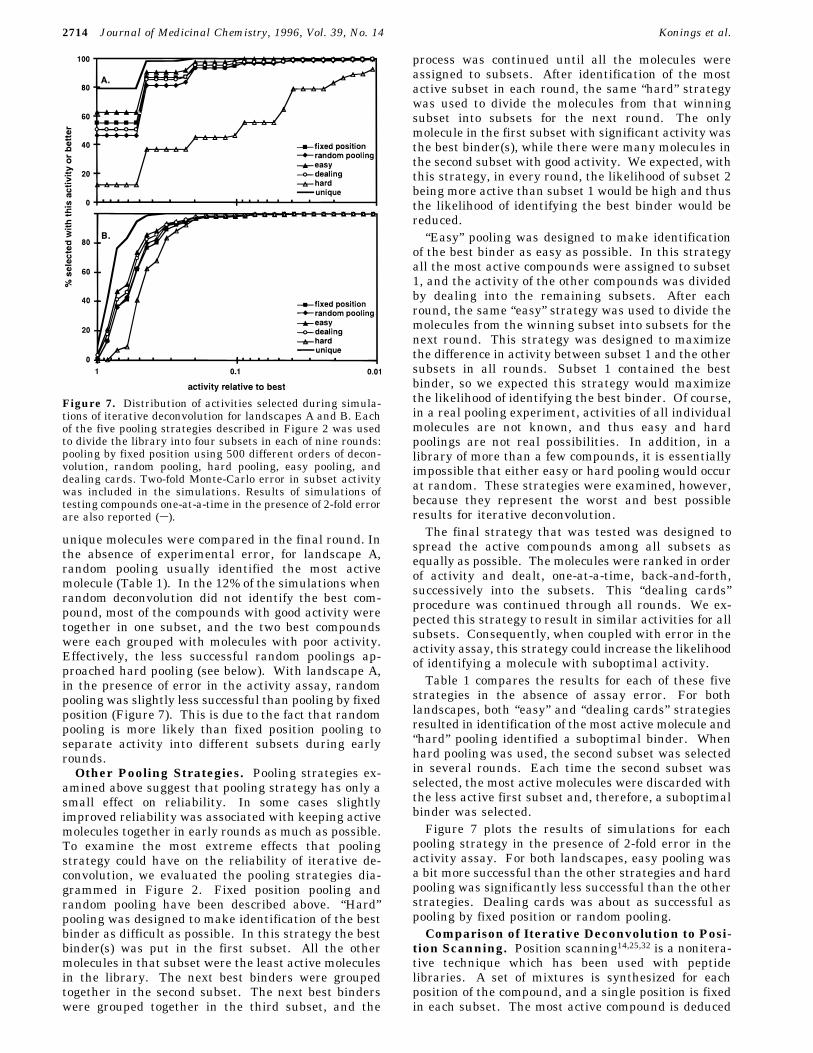
Figure 5. Partitioning of library activity into subsets during deconvolution using deconvolution order 957846321 (a) or 123456789(b) for library landscape A. At each round, the activity of each subset is expressed as a percentage of the combined activity for allsubsets in that round. No Monte-Carlo error was included in calculation of subset activities.
Figure 6. Distribution of activities selected during simula-tions of iterative deconvolution for landscapes A and B. Fixingone position per round resulted in nine rounds with foursubsets per round. When three positions were fixed in eachround, there were three rounds with 64 subsets per round.Fixing nine positions in a single round is equivalent to testing262 144 compounds as individual molecules; no pooling isinvolved. A different, randomly selected order of deconvolutionwas used for each simulation. Two-fold Monte-Carlo error insubset activity was included in the simulations.
Theoretical Evaluation of Pooling Strategies Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 2713
unique molecules were compared in the final round. Inthe absence of experimental error, for landscape A,random pooling usually identified the most activemolecule (Table 1). In the 12% of the simulations whenrandom deconvolution did not identify the best com-pound, most of the compounds with good activity weretogether in one subset, and the two best compoundswere each grouped with molecules with poor activity.Effectively, the less successful random poolings ap-proached hard pooling (see below). With landscape A,in the presence of error in the activity assay, randompooling was slightly less successful than pooling by fixedposition (Figure 7). This is due to the fact that randompooling is more likely than fixed position pooling toseparate activity into different subsets during earlyrounds.Other Pooling Strategies. Pooling strategies ex-
amined above suggest that pooling strategy has only asmall effect on reliability. In some cases slightlyimproved reliability was associated with keeping activemolecules together in early rounds as much as possible.To examine the most extreme effects that poolingstrategy could have on the reliability of iterative de-convolution, we evaluated the pooling strategies dia-grammed in Figure 2. Fixed position pooling andrandom pooling have been described above. “Hard”pooling was designed to make identification of the bestbinder as difficult as possible. In this strategy the bestbinder(s) was put in the first subset. All the othermolecules in that subset were the least active moleculesin the library. The next best binders were groupedtogether in the second subset. The next best binderswere grouped together in the third subset, and the
process was continued until all the molecules wereassigned to subsets. After identification of the mostactive subset in each round, the same “hard” strategywas used to divide the molecules from that winningsubset into subsets for the next round. The onlymolecule in the first subset with significant activity wasthe best binder(s), while there were many molecules inthe second subset with good activity. We expected, withthis strategy, in every round, the likelihood of subset 2being more active than subset 1 would be high and thusthe likelihood of identifying the best binder would bereduced.“Easy” pooling was designed to make identification
of the best binder as easy as possible. In this strategyall the most active compounds were assigned to subset1, and the activity of the other compounds was dividedby dealing into the remaining subsets. After eachround, the same “easy” strategy was used to divide themolecules from the winning subset into subsets for thenext round. This strategy was designed to maximizethe difference in activity between subset 1 and the othersubsets in all rounds. Subset 1 contained the bestbinder, so we expected this strategy would maximizethe likelihood of identifying the best binder. Of course,in a real pooling experiment, activities of all individualmolecules are not known, and thus easy and hardpoolings are not real possibilities. In addition, in alibrary of more than a few compounds, it is essentiallyimpossible that either easy or hard pooling would occurat random. These strategies were examined, however,because they represent the worst and best possibleresults for iterative deconvolution.The final strategy that was tested was designed to
spread the active compounds among all subsets asequally as possible. The molecules were ranked in orderof activity and dealt, one-at-a-time, back-and-forth,successively into the subsets. This “dealing cards”procedure was continued through all rounds. We ex-pected this strategy to result in similar activities for allsubsets. Consequently, when coupled with error in theactivity assay, this strategy could increase the likelihoodof identifying a molecule with suboptimal activity.Table 1 compares the results for each of these five
strategies in the absence of assay error. For bothlandscapes, both “easy” and “dealing cards” strategiesresulted in identification of the most active molecule and“hard” pooling identified a suboptimal binder. Whenhard pooling was used, the second subset was selectedin several rounds. Each time the second subset wasselected, the most active molecules were discarded withthe less active first subset and, therefore, a suboptimalbinder was selected.Figure 7 plots the results of simulations for each
pooling strategy in the presence of 2-fold error in theactivity assay. For both landscapes, easy pooling wasa bit more successful than the other strategies and hardpooling was significantly less successful than the otherstrategies. Dealing cards was about as successful aspooling by fixed position or random pooling.Comparison of Iterative Deconvolution to Posi-
tion Scanning. Position scanning14,25,32 is a nonitera-tive technique which has been used with peptidelibraries. A set of mixtures is synthesized for eachposition of the compound, and a single position is fixedin each subset. The most active compound is deduced
Figure 7. Distribution of activities selected during simula-tions of iterative deconvolution for landscapes A and B. Eachof the five pooling strategies described in Figure 2 was usedto divide the library into four subsets in each of nine rounds:pooling by fixed position using 500 different orders of decon-volution, random pooling, hard pooling, easy pooling, anddealing cards. Two-fold Monte-Carlo error in subset activitywas included in the simulations. Results of simulations oftesting compounds one-at-a-time in the presence of 2-fold errorare also reported (s).
2714 Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 Konings et al.
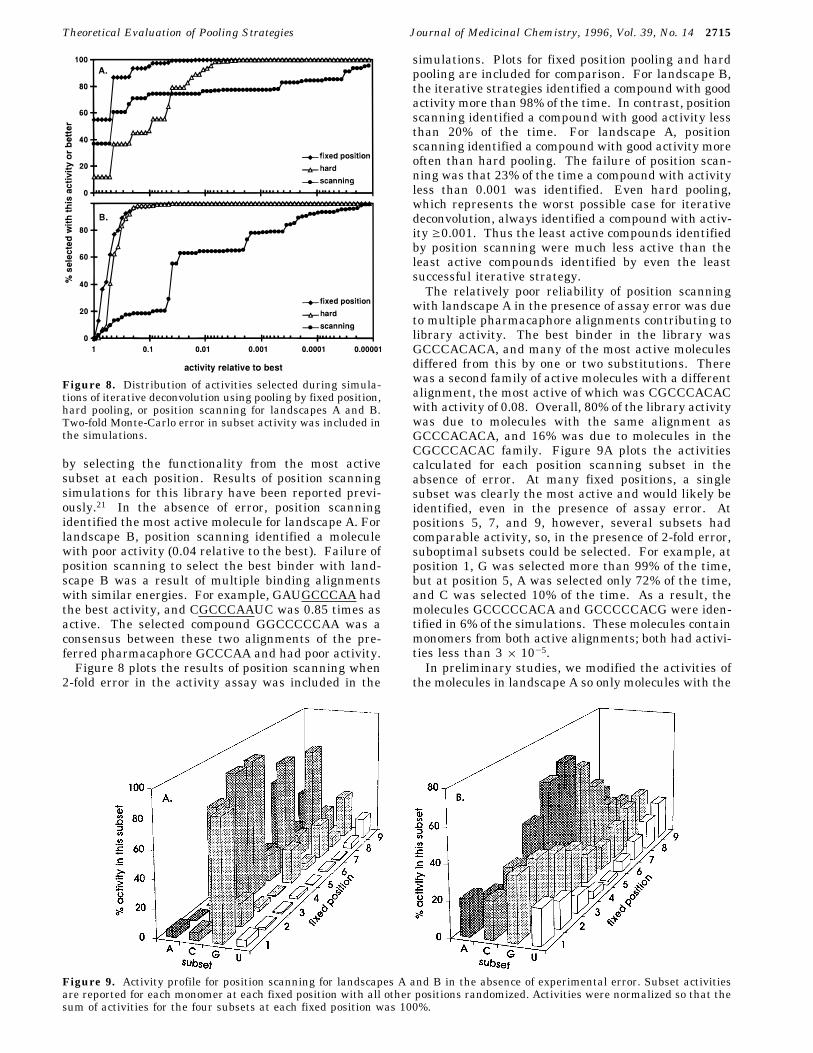
by selecting the functionality from the most activesubset at each position. Results of position scanningsimulations for this library have been reported previ-ously.21 In the absence of error, position scanningidentified the most active molecule for landscape A. Forlandscape B, position scanning identified a moleculewith poor activity (0.04 relative to the best). Failure ofposition scanning to select the best binder with land-scape B was a result of multiple binding alignmentswith similar energies. For example, GAUGCCCAA hadthe best activity, and CGCCCAAUC was 0.85 times asactive. The selected compound GGCCCCCAA was aconsensus between these two alignments of the pre-ferred pharmacaphore GCCCAA and had poor activity.Figure 8 plots the results of position scanning when
2-fold error in the activity assay was included in the
simulations. Plots for fixed position pooling and hardpooling are included for comparison. For landscape B,the iterative strategies identified a compound with goodactivity more than 98% of the time. In contrast, positionscanning identified a compound with good activity lessthan 20% of the time. For landscape A, positionscanning identified a compound with good activity moreoften than hard pooling. The failure of position scan-ning was that 23% of the time a compound with activityless than 0.001 was identified. Even hard pooling,which represents the worst possible case for iterativedeconvolution, always identified a compound with activ-ity g0.001. Thus the least active compounds identifiedby position scanning were much less active than theleast active compounds identified by even the leastsuccessful iterative strategy.The relatively poor reliability of position scanning
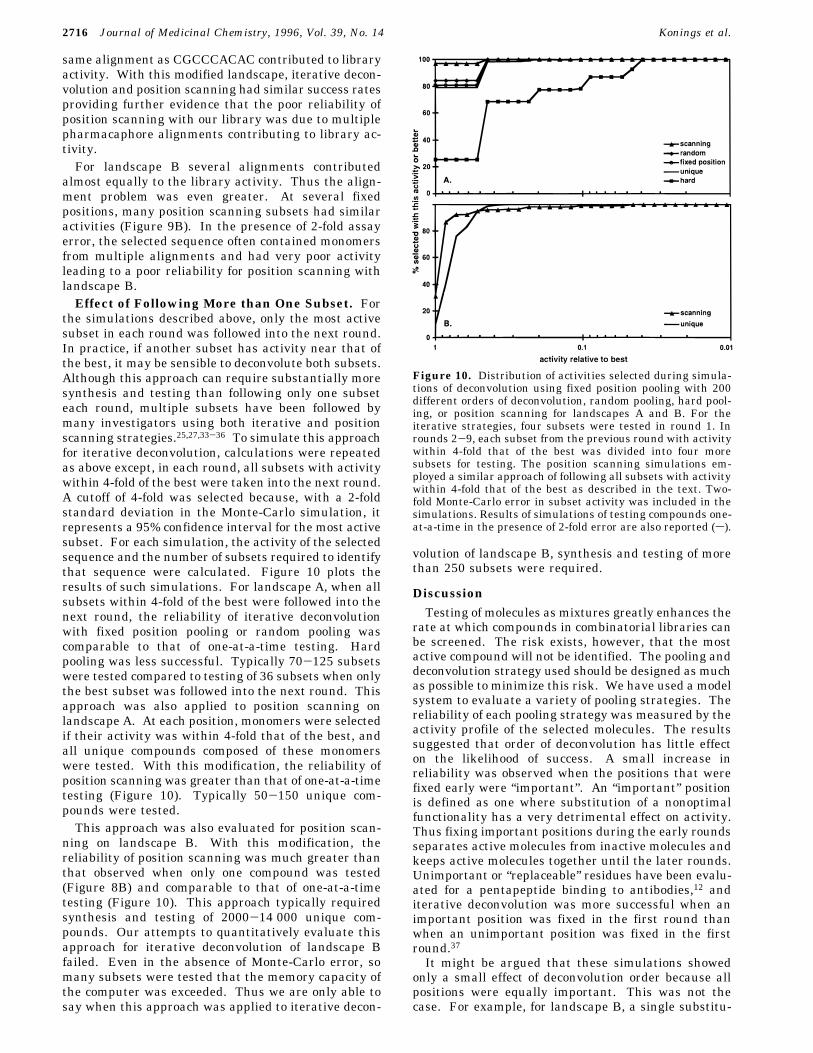
with landscape A in the presence of assay error was dueto multiple pharmacaphore alignments contributing tolibrary activity. The best binder in the library wasGCCCACACA, and many of the most active moleculesdiffered from this by one or two substitutions. Therewas a second family of active molecules with a differentalignment, the most active of which was CGCCCACACwith activity of 0.08. Overall, 80% of the library activitywas due to molecules with the same alignment asGCCCACACA, and 16% was due to molecules in theCGCCCACAC family. Figure 9A plots the activitiescalculated for each position scanning subset in theabsence of error. At many fixed positions, a singlesubset was clearly the most active and would likely beidentified, even in the presence of assay error. Atpositions 5, 7, and 9, however, several subsets hadcomparable activity, so, in the presence of 2-fold error,suboptimal subsets could be selected. For example, atposition 1, G was selected more than 99% of the time,but at position 5, A was selected only 72% of the time,and C was selected 10% of the time. As a result, themolecules GCCCCCACA and GCCCCCACG were iden-tified in 6% of the simulations. These molecules containmonomers from both active alignments; both had activi-ties less than 3 × 10-5.In preliminary studies, we modified the activities of
the molecules in landscape A so only molecules with the
Figure 8. Distribution of activities selected during simula-tions of iterative deconvolution using pooling by fixed position,hard pooling, or position scanning for landscapes A and B.Two-fold Monte-Carlo error in subset activity was included inthe simulations.
Figure 9. Activity profile for position scanning for landscapes A and B in the absence of experimental error. Subset activitiesare reported for each monomer at each fixed position with all other positions randomized. Activities were normalized so that thesum of activities for the four subsets at each fixed position was 100%.
Theoretical Evaluation of Pooling Strategies Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 2715
same alignment as CGCCCACAC contributed to libraryactivity. With this modified landscape, iterative decon-volution and position scanning had similar success ratesproviding further evidence that the poor reliability ofposition scanning with our library was due to multiplepharmacaphore alignments contributing to library ac-tivity.For landscape B several alignments contributed
almost equally to the library activity. Thus the align-ment problem was even greater. At several fixedpositions, many position scanning subsets had similaractivities (Figure 9B). In the presence of 2-fold assayerror, the selected sequence often contained monomersfrom multiple alignments and had very poor activityleading to a poor reliability for position scanning withlandscape B.Effect of Following More than One Subset. For
the simulations described above, only the most activesubset in each round was followed into the next round.In practice, if another subset has activity near that ofthe best, it may be sensible to deconvolute both subsets.Although this approach can require substantially moresynthesis and testing than following only one subseteach round, multiple subsets have been followed bymany investigators using both iterative and positionscanning strategies.25,27,33-36 To simulate this approachfor iterative deconvolution, calculations were repeatedas above except, in each round, all subsets with activitywithin 4-fold of the best were taken into the next round.A cutoff of 4-fold was selected because, with a 2-foldstandard deviation in the Monte-Carlo simulation, itrepresents a 95% confidence interval for the most activesubset. For each simulation, the activity of the selectedsequence and the number of subsets required to identifythat sequence were calculated. Figure 10 plots theresults of such simulations. For landscape A, when allsubsets within 4-fold of the best were followed into thenext round, the reliability of iterative deconvolutionwith fixed position pooling or random pooling wascomparable to that of one-at-a-time testing. Hardpooling was less successful. Typically 70-125 subsetswere tested compared to testing of 36 subsets when onlythe best subset was followed into the next round. Thisapproach was also applied to position scanning onlandscape A. At each position, monomers were selectedif their activity was within 4-fold that of the best, andall unique compounds composed of these monomerswere tested. With this modification, the reliability ofposition scanning was greater than that of one-at-a-timetesting (Figure 10). Typically 50-150 unique com-pounds were tested.This approach was also evaluated for position scan-
ning on landscape B. With this modification, thereliability of position scanning was much greater thanthat observed when only one compound was tested(Figure 8B) and comparable to that of one-at-a-timetesting (Figure 10). This approach typically requiredsynthesis and testing of 2000-14 000 unique com-pounds. Our attempts to quantitatively evaluate thisapproach for iterative deconvolution of landscape Bfailed. Even in the absence of Monte-Carlo error, somany subsets were tested that the memory capacity ofthe computer was exceeded. Thus we are only able tosay when this approach was applied to iterative decon-
volution of landscape B, synthesis and testing of morethan 250 subsets were required.
DiscussionTesting of molecules as mixtures greatly enhances the
rate at which compounds in combinatorial libraries canbe screened. The risk exists, however, that the mostactive compound will not be identified. The pooling anddeconvolution strategy used should be designed as muchas possible to minimize this risk. We have used a modelsystem to evaluate a variety of pooling strategies. Thereliability of each pooling strategy was measured by theactivity profile of the selected molecules. The resultssuggested that order of deconvolution has little effecton the likelihood of success. A small increase inreliability was observed when the positions that werefixed early were “important”. An “important” positionis defined as one where substitution of a nonoptimalfunctionality has a very detrimental effect on activity.Thus fixing important positions during the early roundsseparates active molecules from inactive molecules andkeeps active molecules together until the later rounds.Unimportant or “replaceable” residues have been evalu-ated for a pentapeptide binding to antibodies,12 anditerative deconvolution was more successful when animportant position was fixed in the first round thanwhen an unimportant position was fixed in the firstround.37It might be argued that these simulations showed
only a small effect of deconvolution order because allpositions were equally important. This was not thecase. For example, for landscape B, a single substitu-
Figure 10. Distribution of activities selected during simula-tions of deconvolution using fixed position pooling with 200different orders of deconvolution, random pooling, hard pool-ing, or position scanning for landscapes A and B. For theiterative strategies, four subsets were tested in round 1. Inrounds 2-9, each subset from the previous round with activitywithin 4-fold that of the best was divided into four moresubsets for testing. The position scanning simulations em-ployed a similar approach of following all subsets with activitywithin 4-fold that of the best as described in the text. Two-fold Monte-Carlo error in subset activity was included in thesimulations. Results of simulations of testing compounds one-at-a-time in the presence of 2-fold error are also reported (s).
2716 Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 Konings et al.

tion at position 4 reduced activity of the best compoundto 1.6 × 10-5. In contrast, a single substitution atposition 8 resulted in an activity of 0.63. Thus theimportance of positions in this library differed by morethan 104-fold.Although our results suggested important positions
should be fixed first, it is often not known whichpositions are important. Random pooling and dealingcards were tested to examine the reliability of iterativedeconvolution when no information was available aboutthe activity profile and active molecules were not kepttogether in early rounds. The results with randompooling were somewhat less reliable than fixed positionpooling; pooling by fixed position keeps active moleculestogether a bit more than random pooling. Pooling bydealing cards was designed to systematically distributethe active molecules into all subsets. The reliability fordealing cards was similar to that for random poolingbecause random pooling, on average, also distributes theactivity equally into all the subsets.An improvement in success was observed when there
were more subsets per round and fewer rounds. Themodest difference between curves in Figure 6 demon-strates that pooling even as many as 65 536 compoundsper subset did not drastically reduce the reliabilitycompared to one-at-a-time testing. The slight increasein reliability with fewer subsets may not be worth theincreased effort involved. For example, with our 9-merlibrary, fixing three positions per round would requiresynthesis and testing of 192 (3 rounds × 64 subsets perround) samples compared to only 36 (9 × 4) samples ifone position is fixed each round. If subsets are preparedusing manual mixing and splitting of beads at eachsynthesis position, then the small increase in successrate observed in Figure 6 when three positions werefixed per round would not be worth the increasedsynthetic effort required. The effort required for strate-gies using fewer rounds and more subsets per round canbe reduced if robotic synthesis38-41 and high throughputscreening are employed. Libraries can be divided intothousands of subsets which should increase the likeli-hood of identifying the most active compound in thelibrary.In addition to an increased likelihood of success, fewer
rounds with more subsets per round result in fewercompounds per subset in the first round so the concen-tration of each individual compound in the subset willbe greater. Thus a practical consequence of moresubsets in the first round is increased subset activityfor the subset containing the most active compound. Ifidentification of the most active subset in round 1 islimited by assay sensitivity, then more subsets andfewer compounds per subset should improve the likeli-hood of finding an active compound in the library.To estimate how much pooling strategy could help or
hurt reliability we examined two extreme cases: easyand hard pooling. We found that the reliability of easypooling was a bit greater than pooling by fixed positionor random pooling. Hard pooling was substantially lessreliable, especially for landscape A. Fortunately, thisresult is highly unlikely. The poor reliability of hardpooling occurred only when the most active molecule(s)was intentionally and repeatedly put in a subset withinactive molecules and the remaining active moleculeswere grouped together in another subset. Practical
implementation of easy or hard pooling would requireknowledge of the activities of all the molecules in thelibrary, and if this information were available, decon-volution would be unnecessary. Easy and hard poolingdo, however, represent the extremes in reliability thatcan result from different pooling strategies.Position scanning was less successful than iterative
deconvolution. When position scanning was unsuccess-ful, a molecule which was a combination of activemolecules from two different alignments was typicallyselected. Activity of this blended molecule was muchworse than the activity of the active molecule fromeither alignment. This phenomenon did not occur withiterative deconvolution resulting in a higher reliabilityfor iterative deconvolution.Selection of an inhibitor of antibody binding from a
library of hexapeptides25 provides an experimentalexample of the effect of multiple alignments on positionscanning. Two hexapeptides with two different align-ments, Ac-YPYPNL-NH2 and Ac-PYPNLS-NH2, arepotent inhibitors of binding of a monoclonal antibody.42When position scanning was applied to this library,multiple subsets were active for some fixed positions.25Twelve sequences were selected for synthesis andscreening as unique compounds. One-half of these,including Ac-PYPPLL-NH2, represented a combinationof the two active alignments and had poor activity.In this peptide example, judicious selection of more
than one monomer at some positions allowed identifica-tion of a correctly aligned compound with good activity.25A similar approach also improved the reliability in oursystem. For landscape A, two or three monomerstypically showed activity within 4-fold that of the bestat three to six of the nine positions. Subsequent testingof the 50-150 unique compounds defined by thesemonomers resulted in a large improvement in reliabilitycompared to testing of the single compound defined bythe best monomer at each position (compare trianglesin Figure 10A to circles in Figure 8A). Reliability ofiterative strategies also improved when more than onesubset was followed into subsequent rounds (Figure10A). For landscape A, this iterative approach requiredsynthesis and testing of 70-125 subsets. Synthesis ofcompound mixtures is almost always more difficult thansynthesis of single compounds. Thus, position scanningfollowing all monomers with activity near that of thebest may be the most efficient strategy for a landscapesimilar to landscape A.For landscape B, the reliability of position scanning
also improved when multiple monomers were pursued.This improvement required synthesis and testing of2000-14 000 compounds and is therefore impractical.Following more subsets would likely also improve thereliability of iterative deconvolution for landscape B.Unfortunately, the large number of subsets requiredmade even simulation of this strategy impossible. Thus,for landscapes similar to landscape B, following morethan one subset is impractical and iterative strategiesfollowing only the best subset appear to be mostefficient.When searching for lead compounds from chemical
libraries, the most important question is whether or notthe selected compound will have activity sufficient foroptimization to a drug candidate. These simulationssuggest that most iterative deconvolution procedures arelikely to identify the most active compound in a chemical
Theoretical Evaluation of Pooling Strategies Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 2717

library or at least a compound with activity near thebest. They do not, however, address the activity of thismost active compound. We have previously reportedguidelines for predicting activity of the final selectedcompound using the suboptimal binding factor.21 Val-ues of the suboptimal binding factor reported previ-ously21,23 and those from more recently published de-convolutions27,36,43,44 can be used to predict activity ofthe most active compound.In summary, we used nucleic acid hybridization to
model deconvolution of combinatorial libraries and totest pooling strategies. The model system provided arapid and inexpensive method for exploring a largenumber of deconvolution strategies. The simulationssuggested iterative deconvolution is most successfulwhen the most active molecules are pooled togetherduring early rounds. Reliability was reduced onlyslightly when active molecules were assigned randomlyto all subsets. Clearly the merits of different deconvo-lution approaches depend on the relative activities ofthe compounds in the libraries. Thus validity of thesepredictions for libraries, such as those in Figure 1,depends on the similarity between activity distributionsin our model systems and those in real libraries. Weused two library-target pairs which differ greatly inthe distribution of activities in the library. The activityprofiles of these two systems span those of manycombinatorial libraries.3,21 In addition, the model useddoes not assume additivity between monomer units45,46and thus does not oversimplify molecular interactions.We believe, therefore, that this model is representativeof many types of libraries. In an accompanying paperwe provide an experimental comparison of several of thepooling strategies using a library of 810 chemicallysynthesized compounds and an in vitro assay for inhibi-tion of PLA2. The experimental results support thepredictions of the theoretical model that nearly allstrategies of iterative deconvolutions are successful. Inthe experimental studies, only hard pooling failed toidentify the most active compound.41
Experimental SectionSimulations of Pooling and Deconvolution. The tar-
gets for landscapes A and B were respectively 5′-GUGUGGGCA-3′ and 5′-UGGGCA-3′. Methods for calculation of free energiesfor library sequences binding to target RNA have beendescribed previously.21 We define the activity of a moleculeas the reciprocal of the concentration needed to bind 50% ofthe target molecules:
where KA is the association constant for the molecule, -∆G°37is the binding free energy, R is the gas constant (0.001 987kcal/mol/K), and T is temperature (310.15 K).Pooling strategies were simulated by dividing the library
into subsets according to each pooling scheme. Activities ofeach subset were calculated as the average activity of thecompounds in the subset.21,27 This calculation assumes nosynergism or antagonism between compounds within a subset.A result of this averaging procedure is that the reciprocal ofthe activity of a mixture is the total concentration of com-pounds in the mixture needed for 50% binding.Two-fold experimental error in subset activity was included
in the simulations by assuming the observed activities had alog normal distribution about the true activity. We assumedlog(activity) had a normal distribution with a mean equal tolog(true activity) and a standard deviation equal to log(2).Observed activities for each subset were generated using
standard Monte-Carlo techniques.47 Typically 500 simulationswere performed for each set of conditions. When more thanone subset was followed, only 100-200 simulations wereperformed. Reproducibility of the results of Monte-Carlosimulations was assessed by performing two sets of 500simulations using identical conditions. When the two sets ofsimulations were compared, percent selected at each activity(see, for example, Figure 4) differed by 2% or less at allactivities. Thus, we estimate the inaccuracy in percentselected during our simulations to be 2% or less.
Acknowledgment. The authors thank Dr. C. Pinillafor valuable discussions. Computing resources weregenerously provided by the University of Colorado.
References(1) Gallop, M. A.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.;
Gordon, E. M. Applications of combinatorial technologies to drugdiscovery. 1. Background and peptide combinatorial libraries.J. Med. Chem. 1994, 37, 1233-1251.
(2) Gordon, E. M.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.;Gallop, M. A. Applications of combinatorial technologies to drugdiscovery. 2. Combinatorial organic synthesis, library screeningstrategies, and future directions. J. Med. Chem. 1994, 37, 1385-1401.
(3) Terrett, N. K.; Gardner, M.; Gordon, D. W.; Kobylecki, R. J.;Steele, J. Combinatorial synthesis--The design of compoundlibraries and their application to drug discovery. Tetrahedron1995, 51, 8135-8173.
(4) Eichler, J.; Houghten, R. A. Generation and utilization ofsynthetic combinatorial libraries. Mol. Med. Today 1995, 174-180.
(5) Janda, K. D. Tagged versus untagged libraries: Methods for thegeneration and screening of combinatorial chemical libraries.Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 10779-10785.
(6) Houghten, R. A. Soluble Combinatorial Libraries: Extending theRange and Repertoire of Chemical Diversity. Methods: Com-panion Methods Enzym. 1994, 6, 354-360.
(7) Pinilla, C.; Appel, J.; Blondelle, S.; Dooley, C.; Dorner, B.;Eichler, J.; Ostresh, J.; Houghten, R. A. A Review of the Utilityof Soluble Peptide Combinatorial Libraries. Biopolymers (Pept.Sci.) 1995, 37, 221-240.
(8) Ecker, D. J.; Vickers, T. A.; Hanecak, R.; Driver, V.; Anderson,K. Rational screening of oligonucleotide combinatorial librariesfor drug discovery. Nucleic Acids Res. 1993, 21, 1853-1856.
(9) Wyatt, J. R.; Vickers, T. A.; Roberson, J. L.; Buckheit, R. W.,Jr.; Klimkait, T.; DeBaets, E.; Davis, P. W.; Rayner, B.; Imbach,J. L.; Ecker, D. J. Combinatorially selected guanosine-quartetstructure is a potent inhibitor of human immunodeficiency virusenvelope-mediated cell fusion. Proc. Natl. Acad. Sci. U.S.A. 1994,91, 1356-1360.
(10) Geysen, H. M.; Rodda, S. J.; Mason, T. J. A priori delineation ofa peptide which mimics a discontinuous antigenic determinant.Mol. Immunol. 1986, 23, 709-715.
(11) Blake, J.; Litzi-Davis, L. Evaluation of peptide libraries: Aniterative strategy to analyze the reactivity of peptide mixtureswith antibodies. Bioconjugate Chem. 1992, 3, 510-513.
(12) Geysen, H. M.; Rodda, S. J.; Mason, T. J.; Tribbick, G.; Schoofs,P. G. Strategies for epitope analysis using peptide synthesis. J.Immunol. Methods 1987, 102, 259-274.
(13) Houghten, R. A.; Pinilla, C.; Blondelle, S. E.; Appel, J. R.; Dooley,C. T.; Cuervo, J. H. Generation and use of synthetic peptidecombinatorial libraries for basic research and drug discovery.Nature 1991, 354, 84-86.
(14) Houghten, R. A.; Appel, J. R.; Blondelle, S. E.; Cuervo, J. H.;Dooley, C. T.; Pinilla, C. The use of synthetic peptide combina-torial libraries for the identification of bioactive peptides.BioTechniques 1992, 13, 412-421.
(15) Edmundson, A. B.; Harris, D. L.; Fan, Z.-C.; Guddat, L. W.;Schley, B. T.; Hanson, B. L.; Tribbick, G.; Geysen, H. M.Principles and pitfalls in designing site-directed peptide ligands.Proteins 1993, 16, 246-267.
(16) Owens, R. A.; Gesellchen, P. D.; Houchins, B. J.; DiMarchi, R.D. The rapid identification of HIV protease inhibitors throughthe synthesis and screening of defined peptide mixtures. Bio-chem. Biophys. Res. Commun. 1991, 181, 402-408.
(17) Eichler, J.; Houghten, R. A. Identification of substrate-analogtrypsin inhibitors through the screening of synthetic peptidecombinatorial libraries. Biochemistry 1993, 32, 11035-11041.
(18) Buckheit, R. W., Jr.; Roberson, J. L.; Lackman-Smith, C.; Wyatt,J. R.; Vickers, T. A.; Ecker, D. J. Potent and specific inhibitionof HIV envelope-mediated cell fusion and virus binding by Gquartet-forming oligonucleotide (ISIS 5320.. AIDS Res. Hum.Retroviruses 1994, 10, 1497-1506.
activity ) 1/IC50 ) KA ) exp (-∆G°37/(RT))
2718 Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 Konings et al.

(19) Ecker, D. J.; Wyatt, J. R.; Vickers, T. Novel guanosine quartetstructure binds to the HIV envelope and inhibits envelopemediated cell fusion. Nucleosides Nucleotides 1995, 14, 1117-1127.
(20) Davis, P. W.; Vickers, T. A.; Wilson-Lingardo, L.; Wyatt, J. R.;Guinosso, C. J.; Sanghvi, Y. S.; DeBaets, E. A.; Acevedo, O. L.;Cook, P. D.; Ecker, D. J. Drug leads from combinatorial phos-phorodiester libraries. J. Med. Chem. 1995, 38, 4363-4366.
(21) Freier, S. M.; Konings, D. A. M.; Wyatt, J. R.; Ecker, D. J.Deconvolution of Combinatorial Libraries for Drug Discovery:A Model System. J. Med. Chem. 1995, 38, 344-352.
(22) Turner, D. H.; Sugimoto, N.; Jaeger, J. A.; Longfellow, C. E.;Freier, S. M.; Kierzek, R. Improved parameters for predictionof RNA structure. Cold Spring Harb. Symp. Quant. Biol. 1987,52, 123-133.
(23) Kauffman, S. A. The Origins of Order, Self-Organization andSelection in Evolution; Oxford University Press: Oxford, U.K.,1993.
(24) Carell, T.; Wintner, E. A.; Bashir-Heshemi, A.; Rebek, J., Jr. Anovel procedure for the synthesis of libraries containing smallorganic molecules. Angew. Chem., Int. Ed. Engl. 1994, 33, 2059-2061.
(25) Pinilla, C.; Appel, J. R.; Blanc, P.; Houghten, R. A. Rapididentification of high affinity peptide ligands using positionalscanning synthetic peptide combinatorial libraries. BioTech-niques 1992, 13, 901-905.
(26) Smith, P. W.; Lai, J. Y. Q.; Whittington, A. R.; Cox, B.; Houston,J. G.; Stylli, C. H.; Banks, M. N.; Tiller, P. R. Synthesis andBiological Evaluation of a Library Containing Potentially 1600Amides/Esters. A Strategy for Rapid Compound Generation andScreening. Bioorg. Med. Chem. Lett. 1994, 4, 2821-2824.
(27) Pirrung, M. C.; Chen, J. Preparation and screening againstacetylchoinesterase of a non-peptide “indexed” combinatoriallibrary. J. Am. Chem. Soc. 1995, 117, 1240-1245.
(28) Furka, A.; Sebestyn, F.; Asgedom, M.; Dibo, G. General methodfor rapid synthesis of multicomponent peptide mixtures. Int. J.Pept. Protein Res. 1991, 37, 487-493.
(29) Lebl, M.; Krchnak, V.; Sepetov, N. F.; Seligmann, B.; Strop, P.;Felder, S.; Lam, K. S. One-bead-one-structure combinatoriallibraries. Biopolymers 1995, 37, 177-198.
(30) Stankova, M.; Issakova, O.; Sepetov, N. F.; Krchnak, V.; Lam,K. S.; Lebl, M. Application of one-bead one-structure approachto identification of nonpeptidic ligands. Drug Dev. Res. 1994,33, 146-156.
(31) Dooley, C. T.; Chung, N. N.; Schiller, P. W.; Houghten, R. A.Acetalins: Opioid receptor antagonists determined through theuse of synthetic peptide combinatorial libraries. Proc. Natl. Acad.Sci. U.S.A. 1993, 90, 10811-10815.
(32) Dooley, C. T.; Houghten, R. A. The use of positional scanningsynthetic peptide combinatorial libraries for the rapid determi-nation of opioid receptor ligands. Life Sci. 1993, 52, 1509-1517.
(33) Houghten, R. A.; Dooley, C. T. Biorg. Med. Chem. Lett. 1993, 3,405-412 (abstract).
(34) Blondelle, S. E.; Takahashi, E.; Weber, P. A.; Houghten, R. A.Identification of antimicrobial peptides by using combinatoriallibraries made up of unnatural amino acids. Antimicrob. AgentsChemother. 1994, 38, 2280-2286.
(35) Pinilla, C.; Appel, J. R.; Houghten, R. A. Synthetic peptidecombinatorial libraries (SPCLs.:identification of the antigenicdeterminant of B-endorphin recognized by monoclonal antibody3E7. Gene 1993, 128, 71-76.
(36) Terrett, N. K.; Bojanic, D.; Brown, D.; Bungay, P. J.; Gardner,M.; Gordon, D. W.; Mayers, C. J.; Steele, J. The CombinatorialSynthesis of a 30,752-compound Library: Discovery of SARAround the Endothelin Antagonist, FR-139,317. Bioorg. Med.Chem. Lett. 1995, 5, 917-922.
(37) Geysen, H. M. Combinatorial peptide libraries: A criticalevaluation. Abstract for IBC Conference on CombinatorialLibraries, San Francisco, CA, Aug. 11-12, 1994.
(38) Zuckermann, R. N.; Kerr, J. M.; Siani, M. A.; Banville, S. C.;Santi, D. V. Identification of highest-affinity ligands by affinityselection from equimolar peptide mixtures generated by roboticsynthesis. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 4505-4509.
(39) Zuckermann, R. N.; Kerr, J. M.; Siani, M. A.; Banville, S. C.Design, construction and application of a fully automatedequimolar peptide mixture synthesizer. Int. J. Pept. Protein Res.1992, 40, 497-506.
(40) Lashkari, D. A.; Hunicke-Smith, S. P.; Norgren, R. M.; Davis,R. W.; Brennan, T. An automated multiplex oligonucleotidesynthesizer: Development of high-throughput, low-cost DNAsynthesis. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7912-7915.
(41) Wilson-Lingardo, L. A.; Davis, P. W.; Ecker, D. E.; Hebert, N.;Sprankle, K.; Brennan, T.; Freier, S. M.; Wyatt, J. R. Deconvo-lution of Combinatorial Libraries for Drug Discovery: Experi-mental Comparison of Pooling Strategies. J. Med. Chem. 1996,39, 2720-2726.
(42) Appel, J. R.; Pinilla, C.; Houghten, R. A. Identification of relatedpeptides recognized by a monoclonal antibody using a syntheticpeptide combinatorial library. Immunomethods 1992, 1, 17-23.
(43) Campbell, D. A.; Bermak, J. C.; Burkoth, T. S.; Patel, D. V. Atransition state analogue inhibitor combinatorial library. J. Am.Chem. Soc. 1995, 117, 5381-5382.
(44) Carell, T.; Wintner, E. A.; Rebek, J., Jr. A solution-phasescreening procedure for the isolation of active compounds froma library of molecules. Angew. Chem., Int. Ed. Engl. 1994, 33,2061-2064.
(45) Borer, P. N.; Dengler, B.; Tinoco, I. J.; Uhlenbeck, O. C. Stabilityof ribonucleic acid double-stranded helices. J. Mol. Biol. 1974,86, 843-853.
(46) Tinoco, I., Jr.; Borer, P. N.; Dengler, B.; Levine, M.; Uhlenbeck,O. C.; Crothers, D. M.; Gralla, J. Improved Estimation ofSecondary Structure in Rionucleic Acids.Nature New Biol. 1973,246, 40-41.
(47) Bevington, P. R.; Robinson, D. K. Monte carlo techniques. DataReduction and Error Analysis for the Physical Sciences;McGraw-Hill: San Francisco, 1992; pp 75-95.
(48) Ostresh, J. M.; Husar, G. M.; Blondelle, S. E.; Dorner, B.; Weber,P. A.; Houghten, R. A. “Libraries from libraries”: Chemicaltransformation of combinatorial libraries to extend the rangeand repertoire of chemical diversity. Proc. Natl. Acad. Sci. U.S.A.1994, 91, 11138-11142.
(49) Murphy, M. M.; Schullek, J. R.; Gordon, E. M.; Gallop, M. A.Combinatorial organic synthesis of highly functionalizedpyrrolidines: Identification of a potent angiotensin convertingenzyme inhibitor from a mercaptoacyl proline library. J. Am.Chem. Soc. 1995, 117, 7029-7030.
JM960168O
Theoretical Evaluation of Pooling Strategies Journal of Medicinal Chemistry, 1996, Vol. 39, No. 14 2719




















