
Constraints on Viral Evolution during Chronic Hepatitis C Virus Infection Arising from a...
9
Constraints on Viral Evolution during Chronic Hepatitis C Virus Infection Arising from a Common-Source Exposure Justin R. Bailey, a Sarah Laskey, a Lisa N. Wasilewski, a Supriya Munshaw, a Liam J. Fanning, c Elizabeth Kenny-Walsh, c and Stuart C. Ray a,b Division of Infectious Diseases, Department of Medicine, a and Department of Oncology, b Johns Hopkins University, Baltimore, Maryland, USA, and Hepatitis C Unit, Department of Medicine, Cork University Hospital, National University of Ireland, Cork, United Kingdom c Extraordinary viral sequence diversity and rapid viral genetic evolution are hallmarks of hepatitis C virus (HCV) infection. Viral sequence evolution has previously been shown to mediate escape from cytotoxic T-lymphocyte (CTL) and neutralizing antibody responses in acute HCV infection. HCV evolution continues during chronic infection, but the pressures driving these changes are poorly defined. We analyzed plasma virus sequence evolution in 5.2-kb hemigenomes from multiple longitudinal time points isolated from individuals in the Irish anti-D cohort, who were infected with HCV from a common source in 1977 to 1978. We found phylogenetically distinct quasispecies populations at different plasma time points isolated late in chronic infection, sug- gesting ongoing viral evolution and quasispecies replacement over time. We saw evidence of early pressure driving net evolution away from a computationally reconstructed common ancestor, known as Bole1b, in predicted CTL epitopes and E1E2, with bal- anced evolution toward and away from the Bole1b amino acid sequence in the remainder of the genome. Late in chronic infec- tion, the rate of evolution toward the Bole1b sequence increased, resulting in net neutral evolution relative to Bole1b across the entire 5.2-kb hemigenome. Surprisingly, even late in chronic infection, net amino acid evolution away from the infecting inocu- lum sequence still could be observed. These data suggest that, late in chronic infection, ongoing HCV evolution is not random genetic drift but rather the product of strong pressure toward a common ancestor and concurrent net ongoing evolution away from the inoculum virus sequence, likely balancing replicative fitness and ongoing immune escape. A n estimated 170 million individuals are infected with hepatitis C virus (HCV) worldwide (2, 47), and approximately two- thirds of infected individuals subsequently develop chronic infec- tion, which persists throughout life without antiviral treatment. Chronic HCV infection remains the leading cause of hepatocellu- lar carcinoma and liver transplantation in the United States (30, 38, 43). Despite significant recent improvements in HCV treat- ment (16), a vaccine to prevent HCV infection is still desperately needed. HCV replicates to high viral loads using an error-prone poly- merase, generating in each host a group of related but genetically distinct viral variants called quasispecies (4, 31). This extensive genetic diversity and high rate of viral genetic evolution are major challenges for vaccine design. It has been demonstrated in simian immunodeficiency virus (SIV) and human immunodeficiency vi- rus (HIV) infection that selection from among a distribution of quasispecies variants allows viral escape from immune pressure, but that this escape is balanced by fitness constraints that drive reversion to restore replicative fitness (15, 27). Similarly, HCV has been shown to escape immune pressure from cytotoxic T lympho- cytes (CTL) and neutralizing antibody in acute infection, and studies have shown evidence of both escape and reversion, sug- gesting that intrinsic viral replicative fitness also constrains HCV escape from immune selection in vivo (8, 11, 19, 23, 37, 39, 42, 46). Most studies of HCV evolution and escape have been per- formed early in infection, as the extensive quasispecies diversity during chronic infection complicates analysis of evolution (4, 7, 9, 12, 36, 44). It has therefore remained unclear what forces drive ongoing HCV evolution during decades of chronic infection, par- ticularly given evidence that breadth and magnitude of cellular immune responses wane during this time (7, 17, 24, 41) but neu- tralizing antibody responses are maintained (11, 29, 35, 45). To analyze HCV evolution during chronic infection, we used longi- tudinal plasma samples from the Irish anti-D cohort, a group of women inadvertently infected with genotype 1b HCV from a sin- gle acutely infected source through treatment with contaminated anti-D immune globulin between May 1977 and November 1978 (Fig. 1)(20). In addition, we used the Bole1b sequence, a phylo- genetically reconstructed common ancestor of all known geno- type 1b HCV sequences (S. Munshaw and S. C. Ray, unpublished data), as a reference point in the analysis of genetic evolution in the anti-D cohort (Fig. 2A). Bole1b is analogous to the recently described genotype 1a sequence, Bole1a (5, 33). Evolution toward Bole1b is comparable to centripetal change toward a worldwide genotype 1b consensus sequence (1, 6, 23, 27, 42), except, since immune-selected changes tend to be enriched in branch tips on phylogenetic trees (3), measurement of evolution toward Bole1b may more accurately measure pressure toward optimal replicative fitness (5). Using a high-fidelity protocol, we amplified, cloned, and se- quenced 204 independent 5.2-kb hemigenomes spanning regions encoding Core through NS3 proteins from the inoculum virus and plasma of 10 anti-D cohort subjects at two chronic infection time points separated by 3 to 7 years. We analyzed the phyloge- netic relationship between clones from these three time points Received 11 June 2012 Accepted 28 August 2012 Published ahead of print 12 September 2012 Address correspondence to Stuart C. Ray [email protected]. Supplemental material for this article may be found at http://jvi.asm.org/. Copyright © 2012, American Society for Microbiology. All Rights Reserved. doi:10.1128/JVI.01440-12 12582 jvi.asm.org Journal of Virology p. 12582–12590 December 2012 Volume 86 Number 23
-
Upload
johnshopkins -
Category
Documents
-
view
0 -
download
0
Transcript of Constraints on Viral Evolution during Chronic Hepatitis C Virus Infection Arising from a...

Constraints on Viral Evolution during Chronic Hepatitis C VirusInfection Arising from a Common-Source Exposure
Justin R. Bailey,a Sarah Laskey,a Lisa N. Wasilewski,a Supriya Munshaw,a Liam J. Fanning,c Elizabeth Kenny-Walsh,c
and Stuart C. Raya,b
Division of Infectious Diseases, Department of Medicine,a and Department of Oncology,b Johns Hopkins University, Baltimore, Maryland, USA, and Hepatitis C Unit,Department of Medicine, Cork University Hospital, National University of Ireland, Cork, United Kingdomc
Extraordinary viral sequence diversity and rapid viral genetic evolution are hallmarks of hepatitis C virus (HCV) infection. Viralsequence evolution has previously been shown to mediate escape from cytotoxic T-lymphocyte (CTL) and neutralizing antibodyresponses in acute HCV infection. HCV evolution continues during chronic infection, but the pressures driving these changesare poorly defined. We analyzed plasma virus sequence evolution in 5.2-kb hemigenomes from multiple longitudinal time pointsisolated from individuals in the Irish anti-D cohort, who were infected with HCV from a common source in 1977 to 1978. Wefound phylogenetically distinct quasispecies populations at different plasma time points isolated late in chronic infection, sug-gesting ongoing viral evolution and quasispecies replacement over time. We saw evidence of early pressure driving net evolutionaway from a computationally reconstructed common ancestor, known as Bole1b, in predicted CTL epitopes and E1E2, with bal-anced evolution toward and away from the Bole1b amino acid sequence in the remainder of the genome. Late in chronic infec-tion, the rate of evolution toward the Bole1b sequence increased, resulting in net neutral evolution relative to Bole1b across theentire 5.2-kb hemigenome. Surprisingly, even late in chronic infection, net amino acid evolution away from the infecting inocu-lum sequence still could be observed. These data suggest that, late in chronic infection, ongoing HCV evolution is not randomgenetic drift but rather the product of strong pressure toward a common ancestor and concurrent net ongoing evolution awayfrom the inoculum virus sequence, likely balancing replicative fitness and ongoing immune escape.
An estimated 170 million individuals are infected with hepatitisC virus (HCV) worldwide (2, 47), and approximately two-
thirds of infected individuals subsequently develop chronic infec-tion, which persists throughout life without antiviral treatment.Chronic HCV infection remains the leading cause of hepatocellu-lar carcinoma and liver transplantation in the United States (30,38, 43). Despite significant recent improvements in HCV treat-ment (16), a vaccine to prevent HCV infection is still desperatelyneeded.
HCV replicates to high viral loads using an error-prone poly-merase, generating in each host a group of related but geneticallydistinct viral variants called quasispecies (4, 31). This extensivegenetic diversity and high rate of viral genetic evolution are majorchallenges for vaccine design. It has been demonstrated in simianimmunodeficiency virus (SIV) and human immunodeficiency vi-rus (HIV) infection that selection from among a distribution ofquasispecies variants allows viral escape from immune pressure,but that this escape is balanced by fitness constraints that drivereversion to restore replicative fitness (15, 27). Similarly, HCV hasbeen shown to escape immune pressure from cytotoxic T lympho-cytes (CTL) and neutralizing antibody in acute infection, andstudies have shown evidence of both escape and reversion, sug-gesting that intrinsic viral replicative fitness also constrains HCVescape from immune selection in vivo (8, 11, 19, 23, 37, 39, 42, 46).
Most studies of HCV evolution and escape have been per-formed early in infection, as the extensive quasispecies diversityduring chronic infection complicates analysis of evolution (4, 7, 9,12, 36, 44). It has therefore remained unclear what forces driveongoing HCV evolution during decades of chronic infection, par-ticularly given evidence that breadth and magnitude of cellularimmune responses wane during this time (7, 17, 24, 41) but neu-tralizing antibody responses are maintained (11, 29, 35, 45). To
analyze HCV evolution during chronic infection, we used longi-tudinal plasma samples from the Irish anti-D cohort, a group ofwomen inadvertently infected with genotype 1b HCV from a sin-gle acutely infected source through treatment with contaminatedanti-D immune globulin between May 1977 and November 1978(Fig. 1) (20). In addition, we used the Bole1b sequence, a phylo-genetically reconstructed common ancestor of all known geno-type 1b HCV sequences (S. Munshaw and S. C. Ray, unpublisheddata), as a reference point in the analysis of genetic evolution inthe anti-D cohort (Fig. 2A). Bole1b is analogous to the recentlydescribed genotype 1a sequence, Bole1a (5, 33). Evolution towardBole1b is comparable to centripetal change toward a worldwidegenotype 1b consensus sequence (1, 6, 23, 27, 42), except, sinceimmune-selected changes tend to be enriched in branch tips onphylogenetic trees (3), measurement of evolution toward Bole1bmay more accurately measure pressure toward optimal replicativefitness (5).
Using a high-fidelity protocol, we amplified, cloned, and se-quenced 204 independent 5.2-kb hemigenomes spanning regionsencoding Core through NS3 proteins from the inoculum virusand plasma of 10 anti-D cohort subjects at two chronic infectiontime points separated by 3 to 7 years. We analyzed the phyloge-netic relationship between clones from these three time points
Received 11 June 2012 Accepted 28 August 2012
Published ahead of print 12 September 2012
Address correspondence to Stuart C. Ray [email protected].
Supplemental material for this article may be found at http://jvi.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01440-12
12582 jvi.asm.org Journal of Virology p. 12582–12590 December 2012 Volume 86 Number 23

and, using novel computational methods, mapped and character-ized quasispecies amino acid changes from the inoculum sample(time A) to the first chronic time point (time B) as well as from thefirst chronic infection time point to the second (time C). Theseamino acid changes were characterized as to whether they repre-sented changes away from, toward, or tangential to the Bole1bamino acid sequence. We also characterized time B to time Camino acid changes as away from, toward, or tangential to theinoculum (time A) virus amino acid sequence. Finally, amino acidchanges were mapped relative to HLA-matched or unmatchedclass I epitopes.
MATERIALS AND METHODSStudy subjects. Ten women from the anti-D cohort were studied becausetime B and time C specimens were available, they provided consent, theirHLA class I genotyping was complete, and 5.2-kb hemigenomes had pre-viously been amplified from their time B plasma. Informed consent wasobtained from the subjects studied, and the research protocol was ap-proved by the Cork University Hospital Ethics Committee.
Reverse transcription-PCR (RT-PCR) amplification and sequenc-ing. Hemigenomes (5.2 kb) from time A and time B were previouslyamplified, cloned, and stored as glycerol stocks. Four previously se-quenced 5.2-kb time A clones and 2 previously sequenced time B clonesper subject were used in this analysis (39). Previous studies demonstratedthat the inoculum (time A) virus was homogeneous and time B virus washeterogeneous. To address the heterogeneity of time B virus, 8 additionaltime B clones for each subject were amplified from glycerol stocks andsequenced. Hemigenomes of 5.2 kb spanning Core-NS3 genes were alsoamplified from time C plasma according to previously described methods(39), except that PCR was performed using Accuprime Pfx (Invitrogen).The majority of samples could be amplified with outer PCR only. If nec-essary, a nested PCR was performed. Test PCRs with 1:40 diluted cDNAwere invariably positive, confirming low probability of template resam-pling in PCRs performed with undiluted cDNA. Amplicons of 5.2 kb wereTopo cloned (Invitrogen), and 10 clones from each time point in eachindividual were sequenced. Sequences were aligned using Clustal X, andalignments were manually adjusted in Bioedit. Probable PCR errors (sin-gle-nucleotide changes present in only a single clone in the entire 204clone alignment) were removed using CleanCollapse (v1.6; http://sray.med.som.jhmi.edu/SCRoftware/CleanCollapse). Single-nucleotide in-sertions or deletions in homopolymeric tracts were likewise considered tobe PCR or sequencing artifacts and removed prior to analysis.
Characterization of nucleotide and amino acid changes. The Bole1bsequence was generated using previously published methods (33). Neighbor-joining amino acid trees were constructed with bootstrapping using theJones-Taylor-Thornton (JTT) model in Mega version 5. Unifrac analysis wasperformed using FastUnifrac (http://bmf2.colorado.edu/fastunifrac/) with1,000 Monte Carlo iterations (18). Sliding window analyses of nonsynony-mous and synonymous changes were done with VarPlot (v1.7; http://sray.med.som.jhmi.edu/SCRoftware/VarPlot), with dN and dS calculated by the
Nei-Gojobori method (34). Pairwise synonymous and nonsynonymousdistances between clones were calculated by the Nei-Gojobori method inMega version 5. HVR1 was excluded from these calculations. Countingand characterization of amino acid changes from time A to time B andtime B to time C relative to Bole1b sequence or time A virus sequence wereperformed using code written for Python by S. Laskey (available on re-quest). Amino acid changes between time points at each position in the1,651-amino-acid sequence were categorized as away from, toward, ortangential to reference sequence (Bole1b or time A sequence) using thefollowing criteria: change is classified as “away” when the time A aminoacid is the same as the reference sequence amino acid and time B aminoacid at the same position is different from the reference sequenceamino acid; change is classified as “toward” when the time A amino acid isdifferent from the reference sequence amino acid and the time B aminoacid at the same position is the same as the reference sequence amino acid;and change is classified as “tangential” when the time A amino acid isdifferent from the reference sequence amino acid and the time B aminoacid at the same position is different from the reference sequence aminoacid.
CTL epitope analysis. A list of published HCV T cell epitopes wasobtained from the Immune Epitope Database (www.immuneepitope.org). All class I-restricted epitopes of known HLA restriction that wereless than or equal to 12 amino acids in length were aligned to the Bole1bamino acid sequences using PepMap at the Los Alamos Sequence Data-base (www.hiv.lanl.gov), and epitopes with less than 50% homology toBole1b were discarded. A final list of 135 unique epitopes were used forfurther analysis. Evolution in each subject was analyzed relative to class Iepitopes restricted by that individual’s HLA type (HLA matched) as wellas class I epitopes restricted by HLA types of any other study subject (HLAunmatched). Amino acid changes occurring in HLA-matched or HLA-unmatched class I epitopes were counted for all sequence comparisonsfrom time A to time B or time B to time C. These values were then dividedby the total number of amino acids in HLA-matched or HLA-unmatchedepitopes for all of the clones analyzed to give a proportion of changes/amino acids. Number of changes were also added for all comparisons of allsubjects together and then divided by the total number of amino acidsexamined to generate a proportion of all amino acids changing in HLA-matched or HLA-unmatched epitopes.
Statistical analysis. Unifrac significance was calculated using 1,000Monte Carlo iterations with Bonferroni correction for multiple compar-isons. Significance of differences between rates of evolution away from,toward, and tangential to reference sequences was calculated using pairedt tests in Excel. Significance of differences in pairwise synonymous andnonsynonymous distances was calculated using t tests with Bonferronicorrection for multiple comparisons. Significance of differences in pro-portions of amino acid changes in HLA-matched or HLA-unmatchedepitopes was calculated by comparison of proportions (z test) in Sigma-Plot with Bonferroni correction for multiple comparisons.
Nucleotide sequence accession numbers. The GenBank accessionnumbers for the sequences used in the study are JX649674 to JX649854.
RESULTSViruses isolated at longitudinal time points are phylogeneticallydistinct. We constructed neighbor-joining trees with 204 clonal,1,651-amino-acid sequences from inoculum (time A) and eachsubject (time B and time C) (Fig. 2B). The majority of subjectsshowed clear phylogenetic separation between time A, time B, andtime C amino acid sequences, suggesting ongoing replication andquasispecies replacement over time. For subjects AD01, AD05,AD07, and AD11, the phylogenetic separation between time B andtime C sequences was statistically significant by UniFrac analysis(P � 0.001) (18). For the remaining subjects, separation betweentime B and time C sequences was also statistically significant whenUniFrac analysis was repeated with a larger number of E1E2-only
FIG 1 Timeline of anti-D cohort plasma sample isolation. Hemigenomes (5.2kb) were amplified from inoculum virus (time A) and plasma virus from 10infected subjects from two time points (time B and time C) during chronicinfection.
Viral Evolution during Chronic HCV Infection
December 2012 Volume 86 Number 23 jvi.asm.org 12583

AD01
AD03
AD04
AD05
AD06
AD07
AD08
AD09
AD10
AD11
A
B
Anti-D
Inoculum
Bole 1b
0.005
0.005
**
*
*
FIG 2 Viruses isolated at longitudinal time points are phylogenetically distinct. Phylogenetic trees of anti-D HCV sequences spanning 1,651 amino acids fromthe Core through NS3 proteins. (A) Neighbor-joining amino acid tree with anti-D inoculum (purple circles), chronic anti-D sequences, unrelated genotype 1bsequences, and Bole1b (green circle). (B) Neighbor-joining amino acid trees of anti-D sequences. Center tree contains Bole 1b (green circle) and time A(inoculum) sequences (purple circles). Time B and time C sequences are shown for each study subject, with each color indicating sequences from a differentsubject. Dots at proximal nodes indicate bootstrap values of �94. For outer trees, green circles indicate Bole 1b, purple circles indicate time A sequences, bluecircles indicate sequences amplified from time B plasma, and red circles indicate sequences amplified from time C plasma. Asterisks indicate subjects withstatistically significant phylogenetic separation between time B and time C sequences.
12584 jvi.asm.org Journal of Virology

amino acid sequences (data not shown). Bootstrap analysis con-firmed that time B and C sequences from each individual weremore related to each other than to sequences from any other studysubject (bootstrap, �94), except for subject AD05, whose time Band C sequences were not clearly related by bootstrap analysis. Giventhat reinfection or cross-contamination could not be ruled out forthis subject, AD05 sequences were not used for further analyses.
Purifying selection dominates evolution late in chronic in-fection. To better characterize the genetic evolution occurring at anucleotide level among time A, time B, and time C, we quantitatedthe nonsynonymous and synonymous nucleotide changes occur-ring from time A to time B clones and from time A to time Cclones in sliding windows across the 5.2-kb hemigenome (Fig. 3).As has been previously described from analysis of a smaller num-ber of sequences from this cohort (39), from time A to time B, thenumber of synonymous changes exceeded the number of nonsyn-onymous changes for all regions except HVR1. The median pair-wise synonymous distance from time A to time C clones was greaterthan the distance from time A to time B clones (0.071 versus 0.057; P� 0.001), which was expected given continuous viral replication withan error-prone polymerase. In contrast, the median pairwise nonsyn-onymous distance from time A to time C clones was very similar tothe distance from time A to time B clones (0.011 versus 0.010; P wasnot significant), suggesting relatively little net nonsynonymous evo-lution away from the time A virus sequence over the time B to time Cperiod. These results suggest that purifying selection dominates non-synonymous nucleotide change during late chronic infection, likelydue to viral fitness constraints.
Evolution toward Bole1b accelerates later in chronic infec-tion. We next analyzed changes from time A to time B and time B
to time C at an amino acid level. Pairwise comparisons were per-formed between each clonal amino acid sequence at each timepoint (4 time A, 10 time B, and 10 time C sequences for each studysubject). We observed a total of 38,462 amino acid changes for1,260 comparisons of 204 independent clonal 1,651-amino-acidsequences: 17,294 total amino acid changes from time A to time Bsequences and 21,168 total amino acid changes from time B totime C sequences. The number of observed amino acid changeswas divided by the total number of clonal sequence comparisons,the amino acid length of the region in question, and the number ofyears between time points to give an average rate of change at eachsite per pairwise comparison. Since E1E2 could be expected toevolve differently from non-E1E2 genes due to pressure from bothcellular and humoral immunity (7, 8, 13, 19, 23, 28, 45), andbecause past studies have found different evolutionary patterns inHVR1, E1E2 excluding HVR1, and the non-E1E2 genes (Core, P7,NS2, and NS3 genes), we analyzed these regions separately in sub-sequent analyses. Here, we refer to these regions as HVR1, E1E2,and non-E1E2, respectively.
Changes in HVR1, E1E2, and non-E1E2 were characterized asaway from, toward, or tangential to the Bole1b amino acid se-quence, a computationally reconstructed ancestor representinggenotype 1b HCV sequences. As shown in Fig. 4, from time A totime B, both non-E1E2 and E1E2 exhibited significantly higherrates of amino acid evolution away from the Bole1b sequence thantoward the Bole1b sequence. From time B to time C, the rate ofamino acid change away from Bole1b in both non-E1E2 and E1E2remained constant relative to the time A to time B period. How-ever, the rate of evolution toward Bole1b increased significantlyfor both regions of the genome, resulting in net neutral evolution
FIG 3 Purifying selection dominates evolution late in chronic infection. Sliding window analysis of nonsynonymous and synonymous nucleotide changes fromtime A to time B and time A to time C. An average nonsynonymous and synonymous distance for all pairwise comparisons was calculated with a 20-nucleotidesliding window and 1-nucleotide steps. Average synonymous change from time A to all time B sequences is indicated by a light blue line and synonymous changefrom time A to time C by a light red line. Nonsynonymous change from time A to time B is indicated by a dark blue line and nonsynonymous change from timeA to time C by a dark red line. Borders of each gene and HVR1 (dashed lines) are indicated. Synonymous change exceeds nonsynonymous change in all regionsexcept HVR1.
Viral Evolution during Chronic HCV Infection
December 2012 Volume 86 Number 23 jvi.asm.org 12585
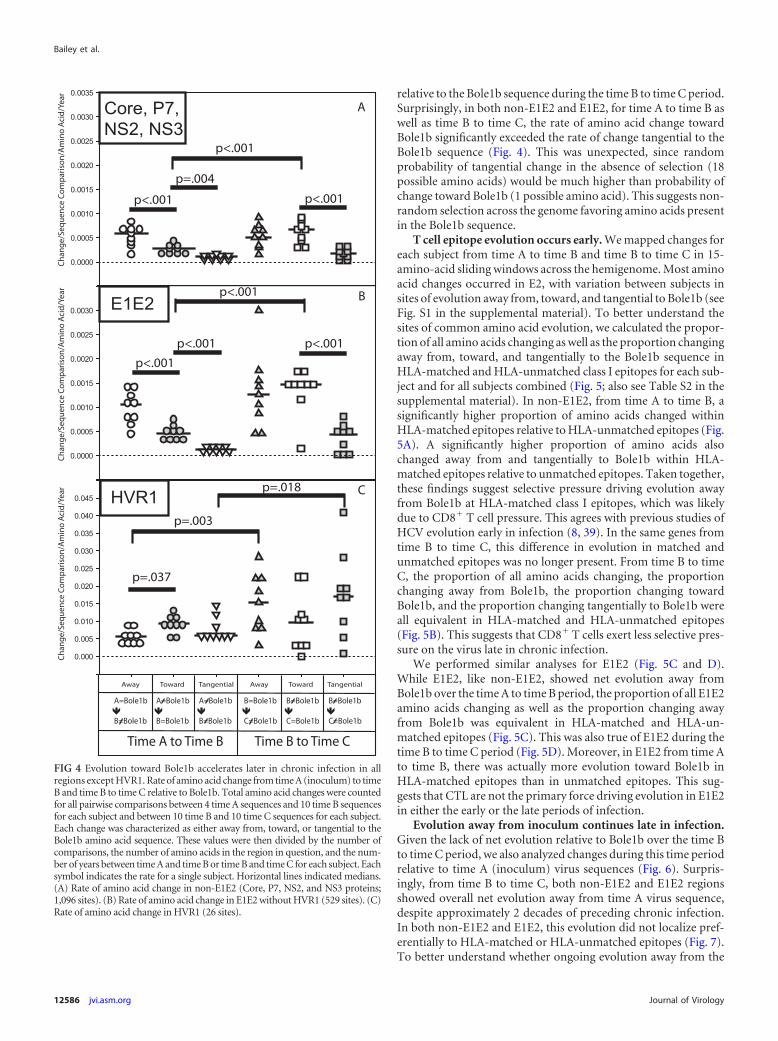
relative to the Bole1b sequence during the time B to time C period.Surprisingly, in both non-E1E2 and E1E2, for time A to time B aswell as time B to time C, the rate of amino acid change towardBole1b significantly exceeded the rate of change tangential to theBole1b sequence (Fig. 4). This was unexpected, since randomprobability of tangential change in the absence of selection (18possible amino acids) would be much higher than probability ofchange toward Bole1b (1 possible amino acid). This suggests non-random selection across the genome favoring amino acids presentin the Bole1b sequence.
T cell epitope evolution occurs early. We mapped changes foreach subject from time A to time B and time B to time C in 15-amino-acid sliding windows across the hemigenome. Most aminoacid changes occurred in E2, with variation between subjects insites of evolution away from, toward, and tangential to Bole1b (seeFig. S1 in the supplemental material). To better understand thesites of common amino acid evolution, we calculated the propor-tion of all amino acids changing as well as the proportion changingaway from, toward, and tangentially to the Bole1b sequence inHLA-matched and HLA-unmatched class I epitopes for each sub-ject and for all subjects combined (Fig. 5; also see Table S2 in thesupplemental material). In non-E1E2, from time A to time B, asignificantly higher proportion of amino acids changed withinHLA-matched epitopes relative to HLA-unmatched epitopes (Fig.5A). A significantly higher proportion of amino acids alsochanged away from and tangentially to Bole1b within HLA-matched epitopes relative to unmatched epitopes. Taken together,these findings suggest selective pressure driving evolution awayfrom Bole1b at HLA-matched class I epitopes, which was likelydue to CD8� T cell pressure. This agrees with previous studies ofHCV evolution early in infection (8, 39). In the same genes fromtime B to time C, this difference in evolution in matched andunmatched epitopes was no longer present. From time B to timeC, the proportion of all amino acids changing, the proportionchanging away from Bole1b, the proportion changing towardBole1b, and the proportion changing tangentially to Bole1b wereall equivalent in HLA-matched and HLA-unmatched epitopes(Fig. 5B). This suggests that CD8� T cells exert less selective pres-sure on the virus late in chronic infection.
We performed similar analyses for E1E2 (Fig. 5C and D).While E1E2, like non-E1E2, showed net evolution away fromBole1b over the time A to time B period, the proportion of all E1E2amino acids changing as well as the proportion changing awayfrom Bole1b was equivalent in HLA-matched and HLA-un-matched epitopes (Fig. 5C). This was also true of E1E2 during thetime B to time C period (Fig. 5D). Moreover, in E1E2 from time Ato time B, there was actually more evolution toward Bole1b inHLA-matched epitopes than in unmatched epitopes. This sug-gests that CTL are not the primary force driving evolution in E1E2in either the early or the late periods of infection.
Evolution away from inoculum continues late in infection.Given the lack of net evolution relative to Bole1b over the time Bto time C period, we also analyzed changes during this time periodrelative to time A (inoculum) virus sequences (Fig. 6). Surpris-ingly, from time B to time C, both non-E1E2 and E1E2 regionsshowed overall net evolution away from time A virus sequence,despite approximately 2 decades of preceding chronic infection.In both non-E1E2 and E1E2, this evolution did not localize pref-erentially to HLA-matched or HLA-unmatched epitopes (Fig. 7).To better understand whether ongoing evolution away from the
FIG 4 Evolution toward Bole1b accelerates later in chronic infection in allregions except HVR1. Rate of amino acid change from time A (inoculum) to timeB and time B to time C relative to Bole1b. Total amino acid changes were countedfor all pairwise comparisons between 4 time A sequences and 10 time B sequencesfor each subject and between 10 time B and 10 time C sequences for each subject.Each change was characterized as either away from, toward, or tangential to theBole1b amino acid sequence. These values were then divided by the number ofcomparisons, the number of amino acids in the region in question, and the num-ber of years between time A and time B or time B and time C for each subject. Eachsymbol indicates the rate for a single subject. Horizontal lines indicated medians.(A) Rate of amino acid change in non-E1E2 (Core, P7, NS2, and NS3 proteins;1,096 sites). (B) Rate of amino acid change in E1E2 without HVR1 (529 sites). (C)Rate of amino acid change in HVR1 (26 sites).
Bailey et al.
12586 jvi.asm.org Journal of Virology

time A virus sequence over the time B to time C period was morelikely due to immune pressure or genetic drift, we compared therate of evolution away from the time A sequence to the rate ofevolution away from an unrelated genotype 1b sequence, Con1(26) (see Fig. S2 in the supplemental material). Normalized rate ofevolution in non-E1E2 away from time A virus sequence andCon1 sequence were equivalent, but in E1E2, the rate of changeaway from time A significantly exceeded the rate of change awayfrom Con1, suggesting that the observed net evolution in E1E2away from the time A virus sequence is likely nonrandom.
Evolution away from inoculum and toward Bole1b occurssimultaneously and independently. To test the hypothesis thatevolution from time B to time C was the result of concurrent andindependent evolution away from time A (inoculum) virus aminoacid sequence and toward the Bole1b amino acid sequence, wereanalyzed amino acid changes from time B to time C, first exclud-ing all changes toward the Bole1b amino acid sequence and thenexcluding all changes away from time A virus sequence (see Fig. S3in the supplemental material). After exclusion of all time B to Cchanges that were toward Bole1b, most remaining changes wereaway from the time A virus sequence, with very low rates of evo-lution toward or tangential to the time A sequence. The medianrate of evolution away from the time A virus sequence was 13times higher than the rate of evolution toward time A sequence innon-E1E2 and was 5 times higher in E1E2 (P � 0.001 for non-
E1E2 and 0.002 for E1E2) (see Fig. S3A). These changes were notenriched in HLA-matched epitopes (see Fig. S4). After exclusionof all time B to C changes that were away from the time A virussequence, the majority of remaining changes were toward Bole1b(see Fig. S3B). The median rate of evolution toward Bole1b was 21times higher than the rate of evolution away from Bole1b in non-E1E2 and 4 times higher in E1E2 (P � 0.001 for non-E1E2 and0.001 for E1E2). These findings confirm that evolution away fromthe inoculum virus amino acid sequence and toward the Bole1bamino acid sequence contribute independently to ongoing evolu-tion late in chronic infection.
Evolution of HVR1 accelerates late in infection. HVR1showed a different pattern of evolution than non-E1E2 and theremainder of E1E2 excluding HVR1 (Fig. 4C; also see Fig. S5 in thesupplemental material). Over both time periods studied, HVR1showed an extremely high rate of amino acid change, approxi-mately 10-fold higher than the rates of non-E1E2 or E1E2 (with-out HVR1) proteins. Unlike the remainder of the hemigenome,which showed relatively constant rates of evolution away fromand tangential to Bole1b with accelerating evolution towardBole1b, HVR1 showed a relatively constant rate of evolution to-ward Bole1b and an accelerating rate of evolution away from andtangential to the Bole1b sequence, suggesting ongoing immunepressure on HVR1 late in chronic infection (Fig. 4C). Rates ofevolution tangential to and toward Bole1b were nearly constant
FIG 5 T cell epitope evolution occurs early. The location of amino acid changes relative to HLA-matched and HLA-unmatched class I epitopes. Total amino acidchanges, changes away from the Bole 1b amino acid sequence, changes toward Bole1b, and changes tangential to Bole 1b were mapped and identified as fallingwithin HLA-matched (black bars) or HLA-unmatched epitopes (gray bars) for each study subject. The total number of changes of each type was added for allstudy subjects and divided by the total number of amino acids analyzed. P values were calculated by comparison of proportions (z test). An asterisk indicatesP � 0.0001 after correction for multiple comparisons. (A) Changes in non-E1E2 (Core, P7, NS2, and NS3 proteins) from time A to time B. (B) Changes innon-E1E2 (Core, P7, NS2, and NS3 proteins) from time B to time C. (C) Changes in E1E2 (without HVR1) from time A to time B. (D) Changes in E1E2 (withoutHVR1) from time B to time C.
Viral Evolution during Chronic HCV Infection
December 2012 Volume 86 Number 23 jvi.asm.org 12587

from time A to time B and time B to time C, suggesting that thereis less constraint on evolution in this region than in the remainderof the hemigenome. However, unlike the remainder of the hemig-enome, the rate of time A to time B evolution in HVR1 towardBole1b exceeded the rate of evolution away from Bole1b, andthere was no significant net evolution in HVR1 relative to the
Bole1b sequence or time A virus sequence over the time B to timeC period (see Fig. S5), suggesting that evolution in HVR1 is notentirely unconstrained. Overall, HVR1 showed high rates ofamino acid evolution from time A to time B and from time B totime C, suggesting strong immune pressure, and high rates oftangential change, suggesting less constraint on evolution in thisregion than in the remainder of the hemigenome.
DISCUSSION
Extensive quasispecies diversity is a key feature of HCV infection,and this diversity complicates analysis of pressures driving HCVevolution during chronic infection (4, 7, 8, 13, 36, 45). In thisstudy, we amplified and sequenced 204 independent 5.2-kb clonesusing an RT-PCR spanning regions encoding Core through NS3proteins, allowing analysis of longitudinal evolution of both struc-tural and nonstructural genes. We used multiple techniques tominimize background from sporadic mutations, including use ofhigh-fidelity PCR enzymes, in silico elimination of sporadic mu-tations prior to analysis, and averaging of rates of amino acidchange across multiple clonal sequence comparisons. This analy-sis also utilized a novel computationally reconstructed ancestralgenotype 1b HCV sequence, Bole1b. This sequence inherentlycontains fewer common immune escape mutations than an arbi-trarily chosen outgroup or a consensus sequence, and amino acidchanges toward the Bole1b sequence therefore are more likely torepresent evolution toward optimal replicative fitness (3, 5).
Our analysis of the time A to time B period of infection sup-ports previous analyses suggesting that T cells play a key role incontrol of HCV early in infection and confirms previous findingsthat, early in infection, in Core and nonstructural proteins, aminoacid changes accumulate preferentially in HLA-matched class Iepitopes (39). A previous study in other subjects from the Irishanti-D cohort concluded that HLA-B*27, HLA-A*03, and HLA-Cw*01 were associated with viral clearance (32). Later studiesidentified T cell epitopes commonly targeted by HLA-A*03- orHLA-B*27-positive women in the cohort and showed that T cell
0.0000
0.0005
0.0010
0.0015
0.0020
0.0025
0.0030
0.0035
Core, P7, NS2, NS3 E1E2 (without HVR1)
Away Toward Tangential Away Toward Tangential
p=.003
p=.050
p<.001
p=.011
Cha
nge/
Sequ
ence
Com
par
ison
/Am
ino
Aci
d/Ye
ar A B
A=B
C
A=C
B
A
B C
A=B
C
A=C
B
A
B C
FIG 6 Evolution away from inoculum continues late in infection. Rate ofamino acid change from time B to time C relative to the time A (inoculum)virus amino acid sequence. Amino acid changes were counted for all pairwisecomparisons between 10 time B and 10 time C sequences for each subject, andthen each change was characterized as either away from, toward, or tangentialto time A virus amino acid sequence. These values were divided by the numberof sequence comparisons performed, the number of amino acids in the regionin question, and the number of years between time B and time C for eachsubject. Each symbol indicates the rate for a single subject. Horizontal linesindicate medians. (A) Rate of amino acid change in non-E1E2 (Core, P7, NS2,and NS3 proteins; 1,096 sites). (B) Rate of amino acid change in E1E2 (withoutHVR1) (529 sites).
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
*
Total Away Toward Tangential Total Away Toward Tangential
Core, P7, NS2, NS3 E1E2 (without HVR1)
= in HLA-matched epitopes
= in non-HLA-matched epitopes
A B
Ch
an
ge
s/A
min
o A
cid
/Co
mp
ari
so
n
FIG 7 Late evolution away from the time A virus sequence does not localize to class I-restricted epitopes. Time B to time C amino acid changes relative to timeA virus sequence mapped to HLA-matched and HLA-unmatched class I epitopes. Total amino acid changes, changes away from time A virus amino acidsequence, changes toward time A virus sequence, and changes tangential to time A virus sequence were mapped and identified as falling within HLA-matched(black bars) or HLA-unmatched epitopes (gray bars) for each study subject. The total number of changes of each type was added for all study subjects and dividedby the total number of amino acids analyzed. P values were calculated by comparison of proportions (z test). An asterisk indicates P � 0.0001 after correction formultiple comparisons. (A) Changes in non-E1E2 (Core, P7, NS2, and NS3 proteins) from time B to time C. (B) Changes in E1E2 (without HVR1) from time Bto time C.
Bailey et al.
12588 jvi.asm.org Journal of Virology

pressure drives selection of escape mutations (10, 14). In anotherstudy of HLA-B*57-positive individuals with persistent infection,mutations accumulated in HLA*B57-restricted epitopes in E2 andNS5 (21). It is likely that the majority of these amino acid changesoccur quite early, as previous longitudinal studies of acute infec-tion have shown development of mutations in the majority ofrecognized class I epitopes during the first year of infection (8).Our analysis of E1E2 from time A to time B also agrees with otheranalyses showing early net evolution away from the consensus,which has been shown to accelerate around a year after infectionas neutralizing antibody titers increase (28). This early evolutionby E1E2 away from Bole1b did not localize to HLA-matched classI epitopes. In fact, in E1E2 from time A to time B, for unclearreasons, there was more evolution toward Bole1b in HLA-matched than in HLA-unmatched epitopes. This may representreversion of mutations that developed at highly polymorphic sitesvery early in infection. Together, these results suggest that CTLlikely are not the primary force driving evolution in E1E2.
We have extended these observations by also studying evolu-tion late in chronic infection between time points approximately20 and 25 years after infection (time B to time C). We found thatmost amino acid changes late in chronic infection are negativelyselected, as shown by the greater rate of synonymous than non-synonymous nucleotide change over this time period in all regionsexcept HVR1. Surprisingly, though, at the amino acid level, wefound phylogenetically distinct populations of virus at these lon-gitudinal late chronic infection time points, suggesting ongoingviral evolution and quasispecies replacement. Strikingly, the vastmajority of amino acid changes late in chronic infection eitherrepresented evolution away from the time A (inoculum) virussequence or evolution toward the Bole1b sequence.
It is noteworthy that net evolution away from the time A (in-oculum) virus sequence continues late in chronic infection. Sinceevolution away from time A virus sequence in non-E1E2 genes latein chronic infection did not localize to known CTL epitopes, itmay represent CTL escape mutations developing at subdominantepitopes, compensatory mutations for CTL escape mutations thatdeveloped earlier in infection or genetic drift. It is not possible todistinguish between these possibilities currently, given the limitedavailability of T cells from these subjects. Targeting of new sub-dominant epitopes may be less likely given that T cell responseshave been shown to decline in magnitude and become dysfunc-tional in chronic infection (7, 8, 22, 25, 40). Given prior studiesassociating HLA-B*57 and HLA-B*27 with development of CTLescape mutations, it is possible that we would have detected addi-tional mutations if our study had included subjects with thesealleles and we had sequenced the entire genome, including NS5B(10, 21).
The majority of net evolution away from the inoculum virussequence from time B to time C occurred in E1E2, which is notsurprising given that high titers of neutralizing antibody are pres-ent in many chronically infected individuals, and a previous studyshowed some evidence of ongoing escape from neutralizing anti-body in chronic HCV infection (29, 45). The rate of evolutionaway from the inoculum virus sequence in E1E2 exceeded the rateof evolution away from an unrelated genotype 1b sequence(Con1), suggesting nonrandom immune pressure rather than ge-netic drift. The accelerating rate of HVR1 evolution late in infec-tion was surprising and also supports a key role for antibody indriving E1E2 evolution in late chronic infection. While not defin-
itive, these results argue against cell-to-cell transmission withoutantibody exposure as a major mechanism of viral persistence inchronic infection.
Even more striking than the ongoing evolution away from in-oculum was the observation of positive selective pressure towardthe Bole1b amino acid sequence. From time A to time B and fromtime B to time C, across the entire hemigenome aside from HVR1,the rate of evolution toward Bole1b significantly exceeded the rateof evolution tangential to Bole1b. This was quite unexpected,since at any position that did not initially match the Bole1b se-quence, there were 18 possible amino acids that would result intangential change and only 1 amino acid that would result inchange toward Bole1b. Therefore, evolution toward the Bole1bsequence would be extremely unlikely to occur by random chance.From time A to time B, non-E1E2 and E1E2 showed net evolutionaway from Bole1b, but by the time B to time C period, rates ofevolution toward Bole1b accelerated, resulting in net neutral evo-lution relative to Bole1b. It may be that the virus could not divergeany further from the Bole1b sequence at that point and still main-tain adequate replicative fitness.
Together, these data suggest that, late in infection, most ongo-ing HCV evolution is not random genetic drift but rather theproduct of strong pressure toward a common ancestor (Bole1b)and concurrent net ongoing evolution away from the inoculumvirus sequence. These two types of amino acid change likely bal-ance replicative fitness and ongoing immune escape.
ACKNOWLEDGMENTS
We thank David L. Thomas and members of the Center for Viral HepatitisResearch for useful discussions, Anna Snider for technical assistance, andplasma donors from the anti-D cohort.
This study was supported by NIH grants R01 DA024565 and U19AI088791-2.
REFERENCES1. Allen TM, et al. 2004. Selection, transmission, and reversion of an anti-
gen-processing cytotoxic T-lymphocyte escape mutation in human im-munodeficiency virus type 1 infection. J. Virol. 78:7069 –7078.
2. Alter MJ, et al. 1999. The prevalence of hepatitis C virus infection in theUnited States, 1988 through 1994. N. Engl. J. Med. 341:556 –562.
3. Bhattacharya T, et al. 2007. Founder effects in the assessment of HIVpolymorphisms and HLA allele associations. Science 315:1583–1586.
4. Bukh J, Miller RH, Purcell RH. 1995. Genetic heterogeneity of hepatitisC virus: quasispecies and genotypes. Semin. Liver Dis. 15:41– 63.
5. Burke KP, et al. 2012. Immunogenicity and cross-reactivity of a repre-sentative ancestral sequence in hepatitis C virus infection. J. Immunol.188:5177–5188.
6. Cabrera R, et al. 2004. An immunomodulatory role for CD4(�)CD25(�) reg-ulatory T lymphocytes in hepatitis C virus infection. Hepatology 40:1062–1071.
7. Cox AL, et al. 2005. Comprehensive analyses of CD8� T cell responsesduring longitudinal study of acute human hepatitis C. Hepatology 42:104 –112.
8. Cox AL, et al. 2005. Cellular immune selection with hepatitis C viruspersistence in humans. J. Exp. Med. 201:1741–1752.
9. Cox AL, et al. 2005. Prospective evaluation of community-acquiredacute-phase hepatitis C virus infection. Clin. Infect. Dis. 40:951–958.
10. Dazert E, et al. 2009. Loss of viral fitness and cross-recognition by CD8�T cells limit HCV escape from a protective HLA-B27-restricted humanimmune response. J. Clin. Investig. 119:376 –386.
11. Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. 2009. Selectionpressure from neutralizing antibodies drives sequence evolution duringacute infection with hepatitis C virus. Gastroenterology 136:2377–2386.
12. Farci P, Bukh J, Purcell RH. 1997. The quasispecies of hepatitis C virusand the host immune response. Springer Semin. Immunopathol. 19:5–26.
13. Farci P, et al. 2000. The outcome of acute hepatitis C predicted by theevolution of the viral quasispecies. Science 288:339 –344.
Viral Evolution during Chronic HCV Infection
December 2012 Volume 86 Number 23 jvi.asm.org 12589

14. Fitzmaurice K, et al. 2011. Molecular footprints reveal the impact of theprotective HLA-A*03 allele in hepatitis C virus infection. Gut 60:1563–1571.
15. Friedrich TC, et al. 2004. Reversion of CTL escape-variant immunode-ficiency viruses in vivo. Nat. Med. 10:275–281.
16. Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. Anupdate on treatment of genotype 1 chronic hepatitis C virus infection:2011 practice guideline by the American Association for the Study of LiverDiseases. Hepatology 54:1433–1444.
17. Gruener NH, et al. 2001. Sustained dysfunction of antiviral CD8� Tlymphocytes after infection with hepatitis C virus. J. Virol. 75:5550 –5558.
18. Hamady M, Lozupone C, Knight R. 2010. Fast UniFrac: facilitatinghigh-throughput phylogenetic analyses of microbial communities includ-ing analysis of pyrosequencing and PhyloChip data. ISME J. 4:17–27.
19. Keck ZY, et al. 2009. Mutations in hepatitis C virus E2 located outside theCD81 binding sites lead to escape from broadly neutralizing antibodiesbut compromise virus infectivity. J. Virol. 83:6149 – 6160.
20. Kenny-Walsh E. 1999. Clinical outcomes after hepatitis C infection fromcontaminated anti-D immune globulin. Irish Hepatology ResearchGroup. N. Engl. J. Med. 340:1228 –1233.
21. Kim AY, et al. 2011. Spontaneous control of HCV is associated withexpression of HLA-B 57 and preservation of targeted epitopes. Gastroen-terology 140:686 – 696.
22. Kim AY, et al. 2006. Impaired hepatitis C virus-specific T cell responsesand recurrent hepatitis C virus in HIV coinfection. PLoS Med. 3:e492.doi:10.1371/journal.pmed.0030492.
23. Kuntzen T, et al. 2007. Viral sequence evolution in acute hepatitis C virusinfection. J. Virol. 81:11658 –11668.
24. Lechner F, et al. 2000. CD8� T lymphocyte responses are induced duringacute hepatitis C virus infection but are not sustained. Eur. J. Immunol.30:2479 –2487.
25. Lechner F, et al. 2000. Analysis of successful immune responses in per-sons infected with hepatitis C virus. J. Exp. Med. 191:1499 –1512.
26. Le Pogam S, et al. 2006. In vitro selected Con1 subgenomic repliconsresistant to 2=-C-methyl-cytidine or to R1479 show lack of cross resis-tance. Virology 351:349 –359.
27. Leslie AJ, et al. 2004. HIV evolution: CTL escape mutation and reversionafter transmission. Nat. Med. 10:282–289.
28. Liu L, et al. 2010. Acceleration of hepatitis C virus envelope evolution inhumans is consistent with progressive humoral immune selection duringthe transition from acute to chronic infection. J. Virol. 84:5067–5077.
29. Logvinoff C, et al. 2004. Neutralizing antibody response during acute andchronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 101:10149 –10154.
30. Maheshwari A, Ray S, Thuluvath PJ. 2008. Acute hepatitis C. Lancet372:321–332.
31. Martell M, et al. 1992. Hepatitis C virus (HCV) circulates as a populationof different but closely related genomes: quasispecies nature of HCV ge-nome distribution. J. Virol. 66:3225–3229.
32. McKiernan SM, et al. 2004. Distinct MHC class I and II alleles are asso-ciated with hepatitis C viral clearance, originating from a single source.Hepatology 40:108 –114.
33. Munshaw S, et al. 2012. Computational reconstruction of bole1a, a rep-resentative synthetic hepatitis C virus subtype 1a genome. J. Virol. 86:5915–5921.
34. Nei M, Gojobori T. 1986. Simple methods for estimating the numbers ofsynonymous and nonsynonymous nucleotide substitutions. Mol. Biol.Evol. 3:418 – 426.
35. Netski DM, et al. 2004. The development of neutralizing antibodiesduring acute hepatitis C virus infection. Abstr. 11th Int. Symp. Hepatitis CVir. Rel. Vir., abstr. P-201.
36. Netski DM, et al. 2005. Humoral immune response in acute hepatitis Cvirus infection. Clin. Infect. Dis. 41:667– 675.
37. Neumann AU, et al. 1998. Hepatitis C viral dynamics in vivo and theantiviral efficacy of interferon-alpha therapy. Science 282:103–107.
38. Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. 2006. Thecontributions of hepatitis B virus and hepatitis C virus infections to cir-rhosis and primary liver cancer worldwide. J. Hepatol. 45:529 –538.
39. Ray SC, et al. 2005. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J. Exp. Med. 201:1753–1759.
40. Rutebemberwa A, et al. 2008. High-programmed death-1 levels on hep-atitis C virus-specific T cells during acute infection are associated withviral persistence and require preservation of cognate antigen duringchronic infection. J. Immunol. 181:8215– 8225.
41. Thimme R, et al. 2001. Determinants of viral clearance and persistenceduring acute hepatitis C virus infection. J. Exp. Med. 194:1395–1406.
42. Timm J, et al. 2004. CD8 epitope escape and reversion in acute HCVinfection. J. Exp. Med. 200:1593–1604.
43. Tong MJ, El-Farra NS, Reikes AR, Co RL. 1995. Clinical outcomes aftertransfusion-associated hepatitis C. N. Engl. J. Med. 332:1463–1466.
44. Villano SA, Vlahov D, Nelson KE, Cohn S, Thomas DL. 1999. Persis-tence of viremia and the importance of long-term follow-up after acutehepatitis C infection. Hepatology 29:908 –914.
45. von Hahn T, et al. 2007. Hepatitis C virus continuously escapes fromneutralizing antibody and T-cell responses during chronic infection invivo. Gastroenterology 132:667– 678.
46. Wang XH, et al. 2007. Progression of fibrosis during chronic hepatitis Cis associated with rapid virus evolution. J. Virol. 81:6513– 6522.
47. World Health Organization. 1997. Hepatitis C: global prevalence. Wkly.Epidemiol. Rec. 72:341–348.
Bailey et al.
12590 jvi.asm.org Journal of Virology




















